Dynamic Changes in Chromatin Accessibility Occur in CD8 + T Cells Responding to Viral Infection
|
|
|
- Ralph Nelson
- 6 years ago
- Views:
Transcription
1 Resource Dynamic Changes in Chromatin Accessibility Occur in CD8 + T Cells Responding to Viral Infection Highlights d Examined chromatin accessibility in endogenous CD8 + T cells during viral infection d d Many changes from naive T cells are stably maintained in differentiated cell types Groups of differentially accessible regions are shared between different subsets Authors James P. Scott-Browne, Isaac F. López-Moyado, Sara Trifari, Victor Wong, Lukas Chavez, Anjana Rao, Renata M. Pereira Correspondence arao@lji.org d Exhaustion-specific accessible regions are enriched for NFAT and Nr4a binding sites In Brief Scott-Browne et al. assess genome-wide changes in chromatin accessibility and gene expression in endogenous CD8 + T cells responding to acute and chronic viral infection. They describe shared patterns of differentially accessible regions between naive, effector, memory, and exhausted subsets that are associated with binding sites for known and novel transcription factors. Accession Numbers GSE88987 Scott-Browne et al., 216, Immunity 45, 1 14 December 2, 216 ª 216 Elsevier Inc.
2 Please cite this article in press as: Scott-Browne et al., Dynamic Changes in Chromatin Accessibility Occur in CD8 + T Cells Responding to Viral Infection, Immunity (216), Immunity Resource Dynamic Changes in Chromatin Accessibility Occur in CD8 + T Cells Responding to Viral Infection James P. Scott-Browne, 1 Isaac F. López-Moyado, 1 Sara Trifari, 1 Victor Wong, 1 Lukas Chavez, 1,4 Anjana Rao, 1,2,3,6, * and Renata M. Pereira 1,5 1 Division of Signaling and Gene Expression, La Jolla Institute, 942 Athena Circle, San Diego, CA 9237, USA 2 Sanford Consortium for Regenerative Medicine, La Jolla, CA 9237, USA 3 Department of Pharmacology and Moores Cancer Center, University of California San Diego, La Jolla, CA 9237, USA 4 Computational Oncoepigenomics Group, Division of Pediatric Neurooncology, German Cancer Research Center (DKFZ), Im Neuenheimer Feld 28, 6912 Heidelberg, Germany 5 Present address: Instituto de Microbiologia Prof. Paulo de Goes, Universidade Federal do Rio de Janeiro, Rio de Janeiro, RJ, Brasil 6 Lead Contact *Correspondence: arao@lji.org SUMMARY In response to acute infection, naive CD8 + T cells expand, differentiate into effector cells, and then contract to a long-lived pool of memory cells after pathogen clearance. During chronic infections or in tumors, CD8 + T cells acquire an exhausted phenotype. Here we present genome-wide comparisons of chromatin accessibility and gene expression from endogenous CD8 + T cells responding to acute and chronic viral infection using ATAC-seq and RNAseq techniques. Acquisition of effector, memory, or exhausted phenotypes was associated with stable changes in chromatin accessibility away from the naive T cell state. Regions differentially accessible between functional subsets in vivo were enriched for binding sites of transcription factors known to regulate these subsets, including E2A, BATF, IRF4, T-bet, and TCF1. Exhaustion-specific accessible regions were enriched for consensus binding sites for NFAT and Nr4a family members, indicating that chronic stimulation confers a unique accessibility profile on exhausted cells. INTRODUCTION During acute viral infection, naive antigen-specific CD8 + Tcells expand and differentiate to yield effector T cells that enable resolution of the infection (Williams and Bevan, 27). After viral clearance, most expanded cells die, but a small proportion survive as a long-lived memory population that rapidly produces cytokines and reacquires cytotoxic activity upon secondary exposure to antigen, thus providing protective immunity. These phases of the immune response are typical of acute infections where the virus is eliminated, such as with the Armstrong (Arm5) strain of lymphocytic choriomeningitis virus (LCMV) (Wherry and Ahmed, 24). When virus persists during chronic or latent infection as in mice or humans infected with LCMV clone 13, human immunodeficiency virus, hepatitis B virus, or hepatitis C virus CD8 + Tcells often enter a state of unresponsiveness to further stimulation that has been termed exhaustion (Moskophidis et al., 1993; Wherry, 211). CD8 + T cells are hyporesponsive to stimulation with decreased cytokine production, reduced ability to lyse target cells, and increased expression of several inhibitory cell surface receptors including PD-1 (programmed death 1), LAG3 (lymphocyte-activation gene 3), TIM3 (T cell immunoglobulin and mucin domain-containing protein 3), and CTLA-4 (cytotoxic T lymphocyte-associated protein 4) (Barber et al., 26; Blackburn et al., 29; Wherry et al., 23; Yamamoto et al., 211). The exhausted phenotype is also common in tumor-infiltrating CD8 + cells (Crespo et al., 213), and antibody therapy targeting inhibitory receptors ( checkpoint blockade ) has been remarkably effective in cancer immunotherapy (Pardoll, 212). Although several transcription factors (TFs), including BATF, IRF4, and the T-box family members T-bet and eomesodermin (Eomes), are known to participate in the formation and function of effector and memory CD8 + T cells (Dominguez et al., 215; Intlekofer et al., 25; Kurachi et al., 214), the molecular mechanisms that determine the exhausted phenotype are not well understood (Speiser et al., 214; Wherry and Kurachi, 215). Persistent antigen stimulation appears to be a dominant factor in inducing exhaustion in tumor-infiltrating T cells (Schietinger et al., 216); consistent with these findings, we previously implicated a gene expression program driven by the antigen-activated TF NFAT in CD8 + T cell exhaustion (Martinez et al., 215). However, given the limiting cell numbers available, it was technically difficult to confirm these findings by performing ChIP-seq (chromatin immunoprecipitation followed by sequencing) for NFAT1 and other TFs in exhausted cells. Changes in transcriptional programs are controlled not only through the action of TFs near transcription start sites (TSSs), but also through epigenetic changes in a variety of DNA and histone modifications at regulatory elements throughout the genome (Kouzarides, 27). Active regulatory elements that bind TFs can be defined operationally by their accessibility to enzymes such as DNase I and micrococcal nuclease (ENCODE Project Consortium, 212; Vierstra et al., 214). Although DNase I hypersensitivity assays are relatively cumbersome and require large numbers of cells (Pipkin et al., 21), but accessible regions Immunity 45, 1 14, December 2, 216 ª 216 Elsevier Inc. 1
3 Please cite this article in press as: Scott-Browne et al., Dynamic Changes in Chromatin Accessibility Occur in CD8 + T Cells Responding to Viral Infection, Immunity (216), of chromatin can be identified reliably even in small cell numbers by ATAC-seq (assay for transposase-accessible chromatin using sequencing), a technique that measures accessibility to an engineered Tn5 transposon containing flanking sequencing adapters (Buenrostro et al., 213). Here we have mapped accessible regulatory elements by ATAC-seq in naive, effector, memory, and exhausted CD8 + T cells from mice with acute or chronic LCMV infection. We identified dynamic changes in chromatin accessibility in CD8 + T cells, with clusters of regions with shared accessibility profiles between different subsets. By motif enrichment analysis of the regulatory elements and comparison to transcriptional profiles obtained by RNA-sequencing (RNA-seq), we have confirmed the involvement of NFAT in the exhaustion program and implicated new NFAT-induced TFs in CD8 + T cell exhaustion. Our data constitute a comprehensive analysis of the similarities and differences among functionally distinct CD8 + T cell subsets and provide a valuable resource for future comparisons of these cell types in different tissues and disease models or after genetic manipulation or antibody blockade. RESULTS Identification of Accessible Regions in CD8 + T Cells We used ATAC-seq to assess the accessible regions genomewide in CD8 + T cells responding to viral infection. Antigen-specific CD8 + T cells, defined as CD44 hi and staining with H-2D b tetramers containing a peptide from LCMV glycoprotein amino acids (H-2D b -gp33-41), were collected by fluorescenceactivated cell sorting (FACS) from mice after LMCV Arm5 or clone 13 infection (Figures 1A, S1A, and S1C S1E). Total effector cells and KLRG1 + IL7Ra (short-lived effector cells [SLECs]) or KLRG1 IL7Ra + (memory precursor effector cells [MPECs]) subsets (Figures S1A S1C) were collected 8 days after LCMV Arm5 infection. The frequencies of SLECs and MPECs were around 5% and 8% of total H-2D b -gp33-41 tetramer-positive cells, respectively (Figures S1B and S1C). At 35 days after infection with LMCV Arm5, we sorted cells expressing IL7Ra (memory) or KLRG1 (d35 KLRG1 + )(Figure S1D). Cells with an exhausted phenotype (PD-1 hi Tim3 hi ) were isolated from mice infected with LCMV clone 13 for 2 days (Figure S1E). We also compared ATAC-seq signals in naive splenic CD8 + T cells and in cultured CD8 + T cells after in vitro stimulation under various conditions or after transduction with retroviral constructs (Figure 1A). As in previous studies (Buenrostro et al., 213), ATAC-seq coverage in these samples was concentrated with high signal in narrow regions throughout the genome (illustrated for Ifng in Figure 1B). We called peaks on individual replicates (Figure S1F), merged these to 91,99 regions, counted the number of Tn5 insertions from each replicate in all peaks, and normalized these data using signals at regions with low variance with DESeq2 (Supplemental Experiment Procedures; Love et al., 214). Regions with higher signal had lower inter-replicate error (Figures S1G and S1H), so we restricted our analysis to 7,532 regions that met a minimum threshold of mean ATAC-seq signal in at least one cell type (Table S1 and Supplemental Experimental Procedures). Comparing the signal at all regions between all samples, each biological replicate clustered with the other replicates from the same group, and two technical replicates were as similar to each other as they were to independent biological replicates (Figure S1I). We also profiled transcriptional differences by RNA-seq of naive, effector, memory, and exhausted cells; each biological replicate clustered with replicates in the same cell type (Figure S1J and Table S1). In 7 kb of the Ifng locus, we identified 19 peaks that were accessible in at least one cell type (Figure 1B). The conserved region located at 22 kb relative to the transcription start site (TSS) was accessible in all samples, whereas other conserved regions were not accessible in naive T cells but were highly accessible in effector, memory, and exhausted T cells (Figure 1B). Cells that were pre-activated, cultured, and then briefly reactivated for short periods (2 hr) in vitro showed variable accessibility at these regions, as did cells transduced with a constitutively active NFAT1 that cannot cooperate with AP-1 (NFAT-CA-RIT) (Figure 1B; Martinez et al., 215). The sequences around ATAC-seq peaks were highly conserved (Figure S1K), and the peaks were frequent in promoters, introns, and distal intergenic regions (Figure S1L). Although the Ifng locus illustrates changes in accessibility near the TSS and distal conserved elements, the signal at most TSSs was less variable compared to distal elements (Figure 1C). Considering all peaks, T cells that had responded to LCMV infection were most similar to each other, whereas naive T cells or cells activated and cultured in vitro were part of a separate cluster (Figures 1D and S1I). To identify individual regions that were differentially accessible between CD8 + T cell subsets, we computed differential coverage using DESeq2. Comparing CD8 + T cells activated and cultured in vitro to effector cells at day 8 after LMCV Arm5 infection, at least 9, regions were differentially accessible (Figure S1M). Of these, differentially accessible regions that were more accessible in cells cultured in vitro than in ex vivo isolated effector cells were also more accessible in naive CD8 + T cells; conversely, regions that were less accessible in cells cultured in vitro than in ex vivo isolated effector cells also tended to be less accessible in naive cells, compared to effector, memory, and exhausted CD8 + T cells (Figure S1N). Thus, the accessibility profile of CD8 + T cells activated and cultured in vitro under these conditions remained more similar to that of naive CD8 + T cells and did not completely recapitulate the accessibility profile of effector CD8 + T cells that develop over several days of viral infection in vivo. We previously found that high (1 U/mL) or low (1 U/mL) concentrations of IL- 2 can bias CD8 + T cells cultured in vitro toward effector or memory phenotypes, respectively (Pipkin et al., 21). However, very few regions were differentially accessible between high and low IL-2 conditions (Figure S1O), indicating that exposure to IL-2 under these conditions did not alter chromatin accessibility substantially, despite clear differences in transcriptional profiles. Stable and Dynamic Changes in Accessible Chromatin of CD8 + T Cells in Acute Viral Infection We assessed chromatin accessibility changes during in vivo responses to acute viral infection by comparing naive, effector, and memory CD8 + T cells. Of the 45,439 regions that were accessible in any of these subsets, more than 12, were differentially accessible when comparing naive and effector T cells (Figure 2A); fewer (5,1) were differentially accessible between naive and memory (Figure 2B); and effector and memory 2 Immunity 45, 1 14, December 2, 216
, http://dx.doi.org/1.116/j.immuni.216.1.28 A B C D Figure 1.")
 Mean ATAC-seq coverage at the 7 kb Ifng locus with a scale of 1,2 for all tracks.")
 Pairwise euclidian distance comparison of asinh-transformed ATAC-seq signal per peak for all populations using all peaks accessible in at least one cell type.")
.")
4 Please cite this article in press as: Scott-Browne et al., Dynamic Changes in Chromatin Accessibility Occur in CD8 + T Cells Responding to Viral Infection, Immunity (216), A B C D Figure 1. High ATAC-Seq Signal in CD8 + T Cells at Conserved Regions in Promoters and Distal Regulatory Elements (A) CD8 + T cell populations collected for ATAC-seq comparison. (B) Mean ATAC-seq coverage at the 7 kb Ifng locus with a scale of 1,2 for all tracks. (C) k-means clustered heatmap of mean normalized counts or log 2 fold-change from global mean at all peaks. (D) Pairwise euclidian distance comparison of asinh-transformed ATAC-seq signal per peak for all populations using all peaks accessible in at least one cell type. Data in (B) (D) are from mean of at least two independent samples, except for a single d35 KLRG1 + replicate. See also Figure S1. T cells were the most similar with only 3, differentially accessible regions (Figure 2C). Most of these differentially accessible regions were located distal to the TSS, whereas commonly accessible regions were more evenly distributed between TSS proximal and distal elements (Figures S2A S2C). Similarly, comparison of RNA-seq data identified more than 1,5 differentially expressed genes between naive and effector cells (Figure S2D) but only 6 8 differentially expressed Immunity 45, 1 14, December 2, 216 3

 Boxplots of ATAC-seq counts per peak from the indicated samples (labeled at bottom) at common or differentially accessible regions from the comparison labeled above.")
 Mean ATAC-seq coverage at Il7r (G), Ccr7 (H), Gzma (I), Gzmk (J), and Dmrta1 (K) loci with a scale of 1,2 (left) or RNA-seq gene expression for the indicated genes (right).")
5 Please cite this article in press as: Scott-Browne et al., Dynamic Changes in Chromatin Accessibility Occur in CD8 + T Cells Responding to Viral Infection, Immunity (216), A D G H B E I J C F K L M Figure 2. Dynamic Changes in Chromatin Accessibility Occur in Antigen-Specific and CD8 + T Cells Responding to Acute Viral Infection (A C) Scatterplots of mean ATAC-seq counts per peak comparing the indicated samples. (D F) Boxplots of ATAC-seq counts per peak from the indicated samples (labeled at bottom) at common or differentially accessible regions from the comparison labeled above. Box indicates interquartile range with whiskers ±1.5 times this range and outlier points. (G K) Mean ATAC-seq coverage at Il7r (G), Ccr7 (H), Gzma (I), Gzmk (J), and Dmrta1 (K) loci with a scale of 1,2 (left) or RNA-seq gene expression for the indicated genes (right). (L and M) Venn diagrams illustrating intersection of differentially accessible regions from pairwise comparisons of naive, effector, and memory CD8 + T cells characterizing regions specific to a subset (L) or not in a subset (M) with p values and odds ratios from Fisher s test comparisons. ATAC-seq data in (A) (K) are from at least two independent replicates. RNA-seq data in (G) (K) are mean of two independent replicates for RNA-seq. See also Figure S2. 4 Immunity 45, 1 14, December 2, 216
6 Please cite this article in press as: Scott-Browne et al., Dynamic Changes in Chromatin Accessibility Occur in CD8 + T Cells Responding to Viral Infection, Immunity (216), genes between naive and memory (Figure S2E) or memory and effector (Figure S2F) cells. The differentially accessible regions were positively associated with changes in gene expression at genes located within 25 kb of the peaks (Figures S2G S2I); for example, genes near peaks with higher ATAC-seq signal in effector cells than in naive had higher expression in effector cells compared to naive cells (Figure S2G). Regions that were less accessible in effector than in naive cells also tended to be less accessible in exhausted cells (Figure 2D). In memory cells, some of these regions had a high signal similar to naive (Il7r upstream region, Figure 2G) whereas others had a low signal similar to effector (Ccr7 downstream region, Figure 2H). Similar patterns were apparent at regions more accessible in effector than naive. For example, within the Gzma locus, regions such as the +4 kb and +19 kb peaks had higher signal in effector than naive cells, but the +19 kb peak was high in memory cells and the +4 kb peak was low in memory cells (Figure 2I). The signals at some activation-induced genes, including Fasl and Prf1, were highly similar between naive, effector, and memory CD8 + T cells despite differences in gene expression (Figure S2J), suggesting that changes in expression of these genes would most likely result from differential activation by TFs binding to these commonly accessible regions. We next focused on differentially accessible regions between memory cells and either naive or effector cells. There were more differentially accessible regions between naive and memory (Figure 2B) than between effector and memory (Figure 2C) cells. Regions with lower signal in memory than naive also tended to have lower signal in effector and exhausted cells, while peaks with higher signal in memory than naive tended to have higher signal in effector and exhausted cells (Figure 2E). Taken together, these comparisons identified regions, similar to the 12 kb and +2.5 kb peaks in the Gzmk locus and the +19 kb peak in the Gzma locus, that were stably altered in naive T cells after viral infection (Figures 2I and 2J). Despite the low number of differentially accessible regions between effector and memory cells (Figure 2C), many of the regions were associated with the greatest variation among naive, effector, memory, and exhausted T cell populations (Figure 2F). In an effort to identify accessible regions that were biased toward naive, effector, or memory CD8 + T cells, we assessed the intersection of the differentially accessible regions from each comparison (Figure 2L). Although a few regions were memory specific (Figure 2L), such as the Dmrta1 TSS (Figure 2K), substantial fractions of differentially accessible regions between memory and naive or effector cells were shared with other cell types (Figure 2M) and fewer than expected were absent in memory but shared by effector and naive cells (Figure 2M). These data indicated that at least 25% of the accessible regions changed at some point during CD8 + T cell activation and differentiation during acute infection and that 5% 1% of the CD8 + T cell accessible landscape underwent a stable change from a naive T cell after response to an acute viral infection. The effector population had some unique changes, with increased accessibility at some regions and reduced accessibility at others compared to naive and memory CD8 + T cells. Finally, most of the accessibility profile of memory CD8 + T cells was shared with either naive or effector T cells, with only a small fraction of total accessible regions unique to this population. Chromatin Accessibility Profiles of SLECs and MPECs Are Similar to Those of and Cells We next compared cells with a terminal effector phenotype (SLECs) or a memory precursor phenotype (MPECs) at day 8 after LMCV Arm5 infection to assess the similarities of these subsets to effector and memory cells. Based on the signal at all accessible regions, SLECs were very similar to total effector cells and MPECs were less similar to both effector and memory cells (Figures 1D and S1I). In the direct comparison of SLECs and MPECs, we identified fewer than 1, differentially accessible regions (Figure 3A). Regions that were more accessible in SLECs than MPECs tended to have high accessibility in effector cells with progressively lower signal in exhausted, memory, and naive cells, supporting the overall similarity between effector cells and SLECs (Figure 3B). Regions that were more accessible in MPECs than SLECs tended to have high signal in naive and memory cells and lower signal in effector and exhausted cells (Figure 3B). Together these data suggested that although MPECs were very similar to effector cells, there was a partial bias toward the memory accessibility profile. This trend was also apparent when comparing the change in signal between SLECs and MPECs at differentially accessible regions between memory and effector cells. Peaks with higher signal in memory than effector cells also had higher signal in MPECs than SLECs (Figure 3C), while regions higher in effector than memory tended to have increased signal in SLECs compared to MPECs (Figure 3C), including the region near the Klrg1 TSS (Figure 3D). This bias was incomplete, as exemplified by the region 4 5 kb upstream of Aurkb, which included several peaks with comparable signal between MPECs, SLECs, and effector cells but lower in memory and naive. We also compared the accessibility between SLECs and effector cells as well as MPECs and memory cells (Figures S3A and S3B). Although SLECs were collected from female mice and effector cells were collected from male mice, there were fewer than 5 differentially accessible regions between these two subsets (Figure S3A). In addition, all but two of the regions more accessible in female SLECs than male effector cells were on the X chromosome (data not shown) and there were many female-specific peaks within the Xist/Tsix and Firre loci (Figure S3C). By comparison, there were more than 7 regions differentially accessible between female MPECs and male memory cells (Figure S3B). Those that were higher in MPECs tended to also have high signal in effector cells while those that were higher in memory tended to have high signal in memory and naive cells, indicating that these regions were largely associated with different CD8 + T cell function rather than reflecting the sex differences. Altogether, these data indicate that the accessibility assessed using the total effector cell population is highly similar to the SLEC subset, largely due to the relatively small fraction of MPECs (Figure S1B) and their similarity to SLECs (Figure 3A). MPECs were slightly biased toward the memory cell profile, but the chromatin profile of MPECs was more similar to that of SLECs (Figures 3A and S3B). Exhaustion-Specific Changes in Chromatin Accessibility We next compared effector and memory CD8 + T cells to CD8 + T cells with an exhausted phenotype, isolated from mice chronically infected with LCMV clone 13. There were approximately Immunity 45, 1 14, December 2, 216 5
7 Please cite this article in press as: Scott-Browne et al., Dynamic Changes in Chromatin Accessibility Occur in CD8 + T Cells Responding to Viral Infection, Immunity (216), A B C D E Figure 3. Precursor Cells Are Similar to Short-Lived Cells with a Slight Bias toward (A) Scatterplot of mean ATAC-seq counts per peak comparing the SLECs and MPECs. (B) Boxplot of ATAC-seq counts per peak from the indicated samples (labeled at bottom) at common or differentially accessible regions from the comparison labeled above. Box indicates interquartile range with whiskers ±1.5 times this range and outlier points. (C) Histograms of the log 2 fold-change between effector and memory cells (top) or SLECs and MPECs (bottom) at regions differentially accessible between effector and memory. (D) Mean ATAC-seq coverage at Klrg1 and Aurkb loci with a scale of 1,2. (E) RNA-seq gene expression for Klrg1 and Aurkb. Data in (A) (D) are from three independent replicates and in (E) is mean of two independent replicates. See also Figure S3. 5, and 3, differentially accessible regions in cells with an exhausted phenotype compared to memory and effector CD8 + T cells, respectively (Figures 4A and 4B). As observed in comparisons of naive, effector, and memory CD8 + T cells (Figures S2A S2C), most of these differentially accessible regions were distal to the TSS (Figures S4A and S4B). Similar trends were observed for RNA-seq comparisons between exhausted cells and either effector or memory cells (Figures S4C and S4D), where more genes were differentially expressed between exhausted and memory cells than between exhausted and effector cells. As observed for comparisons of naive, effector, and memory cells, the changes in accessibility were more likely to be positively associated with changes in expression at nearby genes (Figures S4E and S4F). The accessibility profiles of effector and exhausted CD8 + T cells were similar, exemplified by the 7 kb and +19 kb peaks in the Havcr2 (encoding Tim3) locus (Figure 4E). Regions with unique accessibility in exhausted cells were more clearly identifiable in comparison with effector cells (Figures 4B and 4D) than when compared with memory cells (Figures 4A and 4C), such as the 19 kb and 61 kb peaks near the Tox2 locus (Figure 4F) associated with high Tox2 gene expression (Figure 4F). Cells with an exhausted phenotype were also characterized by loss of accessibility compared to effector and memory CD8 + T cells (Figures 4C and 4D), such as intronic peaks in the Satb1 locus, associated with low Satb1 expression (Figure 4G). To identify features that were specific to, or absent from, effector, memory, and exhausted CD8 + T cells, we compared by intersections the differentially accessible regions among these groups. More regions were specific to memory or exhausted cells than to effector cells (Figure 4H). Relatively few regions were less accessible in effector than either memory or exhausted cells (Figure 4I), but there were 762 regions with low accessibility in exhausted cells compared to both effector and memory. 6 Immunity 45, 1 14, December 2, 216
8 Please cite this article in press as: Scott-Browne et al., Dynamic Changes in Chromatin Accessibility Occur in CD8 + T Cells Responding to Viral Infection, Immunity (216), A C E F B D G H I Figure 4. Chronic Activation Profile Identified by Comparison of Viral Antigen-Specific,, and CD8 + T Cells (A and B) Scatterplots of mean ATAC-seq counts per peak comparing the indicated samples. (C and D) Boxplots of ATAC-seq counts per peak from the indicated samples (labeled at bottom) at common or differentially accessible regions from the comparison labeled above. Box indicates interquartile range with whiskers ±1.5 times this range and outlier points. (E G) Mean ATAC-seq coverage at Havcr2 (E), Tox2 (F), and Satb1 (G) loci with a scale of 1,2 (left) or RNA-seq gene expression for the indicated genes (right). (H and I) Venn diagrams illustrating intersection of differentially accessible regions from pairwise comparisons of effector, memory, and exhausted CD8 + T cells characterizing regions specific to a subset (H) or not in a subset (I) with p values and odds ratios from Fisher s test comparisons. ATAC-seq data in (A) (G) are from at least two independent replicates. RNA-seq data in (E) (G) are mean of two independent replicates. See also Figure S4. Despite exhaustion-specific changes, these data indicated that CD8 + T cells with an exhausted phenotype responding to a chronic infection shared a substantial fraction of chromatin accessibility with effector CD8 + T cells responding to acute infection. In particular, the accessibility at key effector-related genes including Ifng, Gzma, Gzmk, Fasl, and Prf1 (Figures 1B, 2I, 2J, and S2J) and inhibitory receptor loci including Havcr2/ Tim3, Lag3, and Ctla4 (Figures 4E and S4G) was similar between effector and exhausted cells. Differentially Accessible Regions Are Associated with Regulators of CD8 + T Cell Differentiation We identified 18,43 regions that were differentially accessible in at least one pairwise comparison of naive cells, effector cells, SLECs, MPECs, memory cells, and exhausted cells and compared the relative location of individual replicates on multidimensional scaling plots computed using ATAC-seq signal at these regions. SLECs, effector cells, and exhausted cells were most distant from naive cells, whereas MPECs and memory cells were closer to naive (Figure 5A). The substantial overlap between groups of differentially accessible regions (Figures 2L, 2M, 4H, and 4I) suggested that there are functionally related groups of accessible regions that may explain the dissimilarity (Figure 5A). Using k-means clustering, we partitioned all 18,43 differentially accessible regions into 12 groups with shared profiles between naive cells, effector cells, SLECs, MPECs, memory cells, and exhausted CD8 + T cells (Figure 5B). In this comparison, relatively few effector- or memory-specific regions were apparent, with slight biases in clusters 11 and 8 in favor of effector and memory, respectively. In contrast, cluster 1 and clusters 3 and 6 identified groups that were largely biased toward naive or exhausted, respectively (Figure 5B). Regions in clusters 1 and 4 that were shared by naive and memory cells were enriched for HMG motifs and a high frequency of TCF1 ChIP-seq peaks from naive CD8 + T cells overlapped these regions (Figures 5C and 5D; Steinke et al., 214). Immunity 45, 1 14, December 2, 216 7
9 Please cite this article in press as: Scott-Browne et al., Dynamic Changes in Chromatin Accessibility Occur in CD8 + T Cells Responding to Viral Infection, Immunity (216), A B C D F E Figure 5. Differentially Accessible Regions in CD8 + T Cells Are Associated with bhlh, bzip, HMG, T-box, NR, and RHD Family TFs (A) Two-dimensional multidimensional scaling plot of ATAC-seq signal for all replicates of naive cells, effector cells, SLECs, MPECs, memory cells, and exhausted cells at 18,43 regions differentially accessible regions identified from comparisons of naive, effector, memory, and exhausted cells. (B) k-means clustered log 2 fold-change from mean ATAC-seq signal for all differentially accessible regions identified from comparisons between naive cells, effector cells, SLECs, MPECs, memory cells, and exhausted CD8 + T cells. (C) Enrichment of all known motifs within each cluster of differentially accessible regions compared to all differentially accessible regions in naive, effector, memory, and exhausted CD8 + T cells. All motifs with an enrichment log p value less than 35 and found in 1% or more regions in at least one cluster are shown. (D) Percent of ChIP-seq peaks overlapping differentially accessible regions in each cluster or percent of ATAC-seq peaks in each cluster (left column). The total number of ChIP-seq peaks for each TF and the fraction of these that overlap any of these differentially accessible regions are shown below the plot. (E) log 2 fold-change from mean RNA-seq counts per transcript are shown for all expressed TFs from families associated with each enriched motif. (F) MeDIP-seq coverage compared to input for naive and effector CD8 + T cells 8 days after LCMV Arm5 infection. The top graph is for all accessible regions in CD8 + T cells, where each graph below is associated with the clusters indicated at left in (B). ATAC-seq data in (A) and (B) are mean of at least two independent replicates and RNA-seq data in (E) are mean of two independent replicates. See also Figure S5. Gene expression data indicated that among the HMG-containing TFs, the expression of Lef1 and Tcf7 was biased toward naive and memory cells (Figure 5E). Altogether, these data agree with the well-characterized contribution of these two TFs to the function of naive and memory CD8 + T cells (Steinke et al., 214; Zhou et al., 21) and suggested that their contribution is related to binding at these differentially accessible regions. The same clusters, along with clusters 9 and 1 (which also shared naive- and memory-biased accessibility), also overlapped E2A ChIP-seq peaks from mouse thymocytes (Leong et al., 216). Another major group including clusters 2, 5, 6, 7, and 11 had higher signal in effector, memory, and exhausted cells than naive (Figure 5B). These groups of accessible regions were enriched for bzip and T-box motifs compared to all differentially accessible regions in CD8 + T cells (Figure 5C) and a substantial fraction of ChIP-seq peaks identified for BATF, IRF4, and T-bet from effector CD8 + T cells overlapped these regions (Figure 5D), supporting the established role for these TFs in effector T cell differentiation (Dominguez et al., 215; Intlekofer et al., 25; Kurachi et al., 214). The regions with accessibility biased most toward exhausted cells (clusters 3 and 6) were enriched for consensus binding motifs characteristic of nuclear hormone receptor (NR)- and rel homology domain (RHD)-containing proteins (Figure 5C). Among NR-family TFs, we found that the expression of Nr4a2 was biased toward the exhausted cells compared to naive, effector, and memory cells (Figures 5E and S5A). Among RHD-family TFs, we did not identify any genes that were primarily expressed in exhausted cells, including those encoding NFAT family members (Figure S5B), although Rel was reduced in exhausted cells compared to the other subsets (Figure 5E). 8 Immunity 45, 1 14, December 2, 216
10 Please cite this article in press as: Scott-Browne et al., Dynamic Changes in Chromatin Accessibility Occur in CD8 + T Cells Responding to Viral Infection, Immunity (216), We observed similar patterns among all 2,466 differentially expressed genes, with prominent clusters of naive- or exhaustedbiased genes, as well as groups of genes with shared expression between naive and memory cells, effector and exhausted cells, or effector, memory, and exhausted cells (Figure S5C). In an effort to associate the groups of differentially accessible regions with global changes in gene expression, we compared the expression of genes within 25 kb of peaks in each cluster from Figure 5B. The changes in accessibility were largely positively correlated with differences in gene expression, although there were examples of some peak-gene pairs with a negatively correlated association (Figure S5D). For example, some differentially accessible peaks near the Tcf7 locus tended to have high signal in naive and memory cells, which was associated with high Tcf7 expression in these cell types (Figure S5D, clusters 4 and 9), but there were other peaks near Tcf7 with high signal in effector cells despite lower expression in these cells (Figure S5D, clusters 11 and 2). Despite these examples, there were many positive associations between changes in accessibility and activation-associated differentially expressed genes; for instance, Ccl5, Tbx21, Gzma, and Batf all showed accessibility and gene expression biased toward effector and exhausted cells (Figure S5D, clusters 2, 5, 6, 7). To associate accessibility with local epigenetic modifications, we compared the MeDIP-seq signal from naive cells and effector cells after LCMV Arm5 infection (Scharer et al., 213) around these groups of differentially accessible peaks; this technique assesses DNA methylation by immunoprecipitation with a methylcytosine-specific antibody. Most enhancers have been found to have moderate to low DNA methylation (Thurman et al., 212) and around the center of all ATAC-seq peaks in both naive and effector cells, we also found low average MeDIPseq coverage compared to input (Figure 5F). Examining the MeDIP-seq signal around the clusters of differentially accessible peaks, we identified several patterns. First, clusters 1, 4, 9, and 1, which had higher ATAC-seq signal in naive and memory T cells than in effector cells, were associated with low MeDIPseq signal in both naive and effector cells, indicating that methylation is not strongly linked with reduced accessibility at these regions in effector cells. Similarly, cluster 2, which had higher accessibility in effector than naive cells, was also associated with low MeDIP-seq signal, suggesting that these regions had low methylation already in naive cells. Clusters 7 and 11 were the only clusters in which increased accessibility from naive to effector was associated with reduced MeDIP-seq signal in effector compared to naive. Finally, clusters 3 and 6, whose accessibility is biased toward exhausted, did not have substantial depletion of MeDIP-seq coverage compared to input; these regions may be demethylated only in exhausted cells, for which MeDIP-seq data are not yet available. NFAT Activity Contributes Directly and Indirectly to the Exhaustion Profile We previously showed that constitutive NFAT activity induces an exhaustion-like profile in T cells (Martinez et al., 215). To assess how NFAT activity influences chromatin accessibility and the exhaustion phenotype, we expressed a constitutively active form of NFAT (NFAT-CA-RIT) with three amino acid substitutions (R468, I469, and T535) that disrupt the interaction between NFAT and AP-1 (Figure 1A). For comparison, cells were transduced with a version of NFAT-CA-RIT unable to bind DNA (NFAT-CA- RIT-DBDmut) or left untransduced (Mock). More than 4, regions were differentially accessible between mock-transduced cells and cells expressing NFAT-CA-RIT (Figure 6A); in contrast, expression of NFAT-CA-RIT-DBDmut did not change accessibility substantially (Figure S6A). Although regions with increased accessibility in NFAT-CA-RIT-expressing cells were also more accessible in effector, memory, and exhausted cells than in naive cells, the greatest increase in accessibility was observed in exhausted cells. Conversely, regions with reduced accessibility in NFAT-CA-RIT-expressing cells were least accessible in exhausted cells (Figure 6B). Moreover, regions with higher accessibility in exhausted than in effector or memory cells had higher ATAC-seq signal in cells expressing NFAT-CA-RIT compared to mock-transduced cells (Figure S6B). Focusing on regions with altered accessibility in exhausted cells in vivo, we asked whether they showed higher enrichment for NFAT-CA-RIT binding based on ChIP-seq analysis. Of the 993 regions highly accessible in exhausted cells (compared to both effector and memory cells, Figure 4H), most showed increased accessibility in NFAT-CA-RIT-expressing compared to mock-transduced cells (Figure 6C, bottom, data points shifted to right of the vertical line), many of which had high NFAT-CA-RIT binding based on ChIP-seq (Figure 6C, bottom; data points shifted above the horizontal line). Conversely, of the 762 regions with low accessibility in exhausted cells (compared to both effector and memory, Figure 4I), most showed reduced accessibility in NFAT-CA-RIT-expressing compared to mock-transduced cells (Figure 6C, top, data points shifted to left of the vertical line) and fewer of these sites showed high ChIP-seq signal for NFAT-CA-RIT (Figure 6C, top, data points shifted above the horizontal line). Together these results extend our prior finding that NFAT activates the program of CD8 + T cell exhaustion and indicate that continuous NFAT activity contributes to the exhaustion profile during chronic infection in vivo (Martinez et al., 215). In contrast, brief restimulation of CD8 + T cells activated and cultured in vitro only partially recapitulated the effect of NFAT- CA-RIT in inducing increased accessibility at exhaustion-related regions. The combination of phorbol myristate acetate (PMA) and ionomycin (Iono), or Iono alone, induced rapid (within 2 hr) changes in accessibility at 3,6 and 7 regions, respectively (Figures S6C and S6D). These changes were partially blocked by addition of the calcineurin inhibitor cyclosporin (CsA), suggesting NFAT involvement. Unlike changes induced by chronic expression of NFAT-CA-RIT (Figure 6B, right), these accessible regions induced by acute stimulation had similar signals in effector, memory, and exhausted cells (Figures S6C and S6D, right). The changes in accessibility observed at regions with high or low accessibility in exhausted cells (Figures 4H and 4I) were more apparent in NFAT-CA-RIT-expressing cells than in PMA-Iono-stimulated cells (Figure S6E). The Pdcd1 locus illustrates several aspects of the contribution of NFAT to changes in accessibility in exhausted-phenotype cells (Figure 6D). Three accessible regions in the Pdcd1 locus had higher signal in exhausted cells compared to naive, memory, or effector cells: two of these were located 1.1 and 2.7 kb 5 of the Pdcd1 TSS whereas the third was located 22 kb 5 of the Immunity 45, 1 14, December 2, 216 9
11 Please cite this article in press as: Scott-Browne et al., Dynamic Changes in Chromatin Accessibility Occur in CD8 + T Cells Responding to Viral Infection, Immunity (216), A B C D E Figure 6. Constitutively Active NFAT Partially Recapitulates the Chronic Activation Profile In Vitro (A) Scatterplot of ATAC-seq counts per peak comparing in vitro cultured CD8 + T cells after transduction with retroviruses expressing the NFAT-CA-RIT mutant or left untransduced (Mock). (B) Boxplots of ATAC-seq counts per peak in naive, effector, memory, and exhausted CD8 + T cells at differentially accessible regions between mock and NFAT- CA-RIT mutant expressing cells. Box indicates interquartile range with whiskers ±1.5 times this range and outlier points. (C) Scatterplot of NFAT-CA-RIT ChIP-seq coverage with log 2 fold-change ATAC-seq signal between mock and NFAT-CA-RIT mutant expressing cells at regions with lower (top) or higher (bottom) ATAC-seq signal in exhausted compared to effector and memory CD8 + T cells. (D) Mean ATAC-seq and NFAT ChIP-seq coverage at the Pdcd1 locus with a scale of 1,2 for ATAC-seq tracks. (E) Nr4 family member gene expression in CD8 + T cells overexpressing the NFAT-CA-RIT mutant or left untransduced (Mock) showing mean plus range. ATAC-seq data in (A) (D) are from at least two independent replicates. See also Figure S6. TSS (Figure 6D). All three peaks were observed in NFAT-CA-RITexpressing cells, but only the two peaks proximal to the TSS were induced by stimulation with PMA and ionomycin and bound both NFAT and NFAT-CA-RIT. In contrast, the accessible region at +22 kb showed no NFAT binding and minimal change in accessibility after brief stimulation in vitro. Taken together, these data suggest that NFAT activity can directly and indirectly influence the chromatin accessibility profile associated with T cell exhaustion. We compared the consensus TF binding motifs enriched in differentially accessible regions after NFAT-CA-RIT expression or after restimulation of in vitro-cultured cells. Accessible regions associated with NFAT-CA-RIT expression and stimulation with either Iono alone or PMA and Iono together were enriched, as 1 Immunity 45, 1 14, December 2, 216
12 Please cite this article in press as: Scott-Browne et al., Dynamic Changes in Chromatin Accessibility Occur in CD8 + T Cells Responding to Viral Infection, Immunity (216), expected, in NFAT and NFAT-AP1 motifs (Figure S6H). In contrast, the NR-family binding motif that was found in exhaustion-biased regions, including the peak located 22 kb 5 of the TSS in the Pdcd1 locus (Figure 6D), was enriched only in accessible regions induced by NFAT-CA-RIT (Figure S6H). Activation of the NR family under these conditions may be a direct effect of NFAT activity, because NFAT-CA-RIT-expressing cells showed increased expression of Nr4a2 and Nr4a3 (Figure 6E) and NFAT-CA-RIT-occupied accessible regions within the Nr4a2 locus (Figure S6I). Altogether, these data indicated that prolonged stimulation during chronic infection was associated with changes in accessibility that were directly and indirectly induced by NFAT activity. DISCUSSION In this study, we compared chromatin accessibility in CD8 + T cells under different conditions of stimulation and differentiation: in effector, memory, and exhausted cells arising in response to in vivo viral infection; in cells pre-activated in culture and assessed before or after brief restimulation in vitro; and in cells subjected to retroviral transduction with constitutively active versions of NFAT. We identified more than 7, regions that were accessible in at least one subset of activated or differentiated cells. At least half of these accessible regions had similar signals in most cell types examined, demonstrating that there is a core pattern of chromatin accessibility that is relatively stable in CD8 + T cells regardless of their activation or differentiation state. Many commonly accessible regions were located near the TSS, but most were located within introns or at intergenic regions and could potentially function as enhancers (ENCODE Project Consortium, 212). Although these regions remain similarly accessible among CD8 + T cell subsets, their function may be regulated by subsequent DNA or histone modifications on flanking nucleosomes (Kouzarides, 27). Indeed, variable enrichment of the trimethylation of H3K4 and H3K27 at promoters has been associated with differential transcription in naive, effector, and memory CD8 + T cells after influenza infection (Russ et al., 214). At least 3% of all accessible regions were differentially accessible between any pair of cell types. Most of these regions were located far from TSSs but were linked to differentially expressed genes and may represent enhancers that control CD8 + T cell differentiation and function. Among the populations we have compared here, the largest number of differentially accessible regions were observed between naive and effector T cells: approximately 25% of all accessible regions were differentially accessible between these two cell types. Regions that were differentially accessible between naive, effector, and memory CD8 + T cells were enriched for binding sites of several TFs known to contribute to the state of the relevant cell types. Binding of TCF family members, in particular TCF1, was substantially enriched at regions accessible in naive and memory CD8 + T cells. Conversely, binding sites for bzip, IRF, and T-box TFs were enriched within accessible regions specific to effector and memory CD8 + T cells. Our analyses did not identify a substantial number of accessible regions specific to memory CD8 + T cells, which were characterized by a mixture of the accessibility profiles observed in effector and naive cells. Many peaks induced by viral responses at day 8 remained accessible at day 35 in memory cells, providing a potential mechanism for their more rapid responses to antigen compared to naive cells (Williams and Bevan, 27). However, when compared to effector T cells, memory CD8 + T cells also shared certain aspects of their accessibility profile with naive T cells, accompanied by a corresponding similarity in gene expression, as described previously (Best et al., 213). Comparison of MPEC and SLEC subsets from day 8 after acute infection revealed that MPECs are substantially similar to SLECs and total effector populations, with a slight bias toward the memory accessibility profile. Our data do not address whether the shift from MPECs to memory cells reflects the selective survival of cells with a memory-like chromatin accessibility profile after viral infection, or whether cells that originally had an effectorlike pattern of chromatin accessibility revert to accessibility profiles that more closely resemble those of memory cells arising later in the infection. Based on comparisons to DNA methylation data from naive and effector T cells after LCMV infection (Scharer et al., 213), we found low enrichment of DNA methylation around accessible regions in any given cell type, as generally observed in other cells (Thurman et al., 212). Among regions that were accessible in effector cells after activation, some had a corresponding reduction in DNA methylation from naive to effector, whereas other regions were already demethylated in naive cells. Together, these data support the hypothesis that concurrent regulation of DNA methylation and chromatin accessibility controls the activity of regulatory elements in activated T cells to alter gene expression and influence differentiation. In a chronic infection, virus is not completely cleared; antigenspecific cells are continually subjected to chronic stimulation at the site of infection and they acquire a hyporesponsive or exhausted state (Speiser et al., 214; Wherry and Kurachi, 215). Based on the chromatin accessibility described here, exhausted cells were most similar to effector cells: for instance, both functional subsets had similar accessibility profiles at the Ifng locus, which is strongly activated in effector cells but poorly induced upon antigen stimulation of exhausted CD8 + T cells (Barber et al., 26; Kao et al., 211). These observations may explain the effectiveness of antibody therapies, targeting PD-1, PD-L1, and CTLA4 where blockade of the inhibitory signals transmitted by these receptors may enable exhausted T cells to reactivate enhancers that remained as accessible as in effector cells (Barber et al., 26; Pardoll, 212). Regions with similar accessibility between effector and exhausted cells were enriched for T-box motifs and often bound by T-bet in effector cells, consistent with the suggestion that variable expression of T-bet and EOMES among exhausted CD8 + T cells could alter cell function in cells that are similar in their overall accessibility profiles (Doering et al., 212; Intlekofer et al., 25; Wherry and Kurachi, 215). Regions that were uniquely accessible in exhausted cells were enriched for two motifs (NFAT and Nr4 family) that were not substantially enriched in any other viral-specific T cell subset. NFAT can directly induce both Nr4a2 and Nr4a3, whereas only Nr4a2 was induced in exhausted T cells (this study and Martinez et al., 215). Nr4a family members have been associated with the development and function of regulatory T (Treg) cells (Sekiya Immunity 45, 1 14, December 2,
13 Please cite this article in press as: Scott-Browne et al., Dynamic Changes in Chromatin Accessibility Occur in CD8 + T Cells Responding to Viral Infection, Immunity (216), et al., 213); Treg cells also fail to express cytokines characteristic of activated cells after antigen receptor stimulation, and thus Nr4a proteins may mediate similar cell-intrinsic inhibitory programs in Treg cells and exhausted CD8 + cells. We hypothesize that the uniquely exhaustion-specific regions may be induced by continued antigen stimulation during chronic infection, as their accessibility profiles overlap substantially with the profiles of cells activated in vitro and cells expressing constitutively active NFAT, even though only some of the accessible regions bound NFAT in ChIP-seq experiments. Some regions (for instance in the Satb1 and Il7r loci) were less accessible in exhausted cells than in effector and memory cell types, suggesting that at least some aspect of T cell-specific chromatin accessibility is lost in exhausted cells. Loss of chromatin accessibility relative to effector and memory T cells may explain the reported incomplete restoration of function in exhausted cells after checkpoint blockade or after transfer to mice in the absence of antigen (Barber et al., 26; Schietinger et al., 212; Utzschneider et al., 213). Additionally, exhaustion-biased accessible regions showed higher enrichment for DNA methylation in both naive and effector cells, compared to sites accessible in naive and effector cells. These sites may be demethylated specifically in exhausted cells, as shown for two conserved regions in the Pdcd1 locus during chronic infection in mice and humans (Youngblood et al., 211). Thus DNA methylation is an additional factor that may govern the difference between effector and exhausted phenotypes. The changes in chromatin accessibility observed after activation of naive CD8 + T cells, whether in vitro or in vivo, form a continuum reflecting the diverse ways in which TFs expressed in different cellular subsets can cooperate to influence gene expression and cellular function. Based on their patterns of chromatin accessibility, in vitro activated cells, which have been exposed to stimulatory antibodies with limited cytokine exposure and without antigen-presenting cells, are most similar to naive cells that have never encountered antigen, whereas memory, effector, and exhausted cells, which have been exposed to diverse stimulation in a native environment, are progressively less similar. Future experiments will determine whether continuing antigen stimulation or inflammation induces stable changes in chromatin accessibility across an entire population, or whether stochastic changes in accessibility and TF binding and expression give rise to a heterogeneous cell population from which cells with particular phenotypes survive. EXPERIMENTAL PROCEDURES Mice CD8 + T cells were purified from splenocytes of 6- to 1-week-old C57BL/6J mice after infection with LCMV or left uninfected. and memory CD8 + T cell subsets were collected after infection with LCMV Arm5, based on staining with tetramer and antibodies to CD44, IL7Ra, and KLRG1 (Figures S1A, S1C, and S1D). cells were collected after infection LCMV clone 13 based on staining with tetramer and antibodies to PD-1 and Tim3 (Figure S1E). For in vitro stimulation, CD8 + T cells were purified from P14 T cell receptor transgenic mice on a TCRa constant region-deficient background, before stimulation with CD3ε- and CD28-specific antibodies for 2 days and then expanded 1 U/mL or 1 U/mL IL-2 (Pipkin et al., 21). On day 6, cells were restimulated for 2 hr under various conditions (Supplemental Experimental Procedures). For retroviral transduction experiments, CD8 + T cells were isolated by negative selection from C57BL/6J mice. These cells were stimulated for 1 day with antibodies specific for CD3ε and CD28 before infection with retroviruses followed by expansion for three days in 1 U/mL IL-2. All experiments were performed according to protocols approved by the La Jolla Institute animal care and use committee. ATAC-Seq ATAC-seq library preparations were performed as described (Buenrostro et al., 213). Cells were washed with PBS before treatment with lysis buffer followed by labeling with Nextera enzyme (Illumina), before PCR amplification with 1 12 cycles with barcoded primers and 23 5 cycle paired-end sequencing (Illumina). Reads were mapped to mouse genome (mm9) using bowtie (v.1..) (Langmead et al., 29). Unmapped reads were processed with trim_galore, re-mapped with bowtie, and merged with previous mapping output. Duplicate reads identified by picard MarkDuplicates and reads mapping to chrm were excluded. Peak summits were identified with MACS2 (Zhang et al., 28) from individual replicates using short DNA fragments (<1 bp) and expanded to 5 bp regions. We merged overlapping regions from all replicates and excluded regions on chry or those that intersected ENCODE blacklisted regions. The number of transposase insertions within each region was computed for each replicate and differentially accessible regions were identified with DESeq2 (v.1.6) based on an fdr adjusted p value of less than and an estimated fold change of at least 3. Full details are available in Supplemental Experimental Procedures. E2A (Leong et al., 216) (GEO: GSE84974), TCF1 (Steinke et al., 214) (GEO: GSE527), T-bet (Dominguez et al., 215) (SRA: SRR , SRR275585), BATF, and IRF4 (Kurachi et al., 214) (GEO: GSE54191) ChIP-seq and naive and effector MeDIP-seq (Scharer et al., 213) (GEO: GSE44638), including input controls where available, were downloaded from NCBI SRA database and mapped to the mm9 genome (bowtie v.1..). ChIP-seq peaks were called with MACS2. RNA-Seq SMARTseq2 RNA-seq libraries were prepared from naive, effector, memory, and exhausted cells (Picelli et al., 214) sequenced with an Illumina HiSeq25 and mapped to the mm9 genome (TopHat 1.4.1) (Trapnell et al., 29). Lowquality reads were excluded and feature counts were computed with htseqcount (.6. using the union option). Differentially expressed genes were identified with DESeq2 with an fdr adjusted p value of less than and an estimated fold-change of at least 2 and a mean of least 1 transcripts per million transcripts. Full details are available in Supplemental Experimental Procedures. ACCESSION NUMBERS The accession number for the ATAC-seq and RNA-seq data reported in this paper is GEO: GSE SUPPLEMENTAL INFORMATION Supplemental Information includes six figures, one table, and Supplemental Experimental Procedures and can be found with this article online at dx.doi.org/1.116/j.immuni AUTHOR CONTRIBUTIONS J.P.S.-B., R.M.P., S.T., V.W., and A.R. designed experiments; J.P.S.-B., R.M.P., V.W., and S.T. performed experiments; J.P.S.-B., I.F.L.-M., and L.C. analyzed data; and J.P.S.-B., R.M.P., and A.R. wrote the manuscript. ACKNOWLEDGMENTS We thank C. Kim, K. Gunst, and L. Nosworthy for cell sorting, J. Greenbaum, Z. Fu, and J. Day for next-generation sequencing, S. Crotty for LCMV virus (all at La Jolla Institute), the NIH Tetramer Facility for tetramers, and L. Gapin (University of Colorado), P. Marrack (National Jewish Health), and M. Prlic (FHCRC) for providing critical comments. This work was funded by NIH grants R1 12 Immunity 45, 1 14, December 2, 216
14 Please cite this article in press as: Scott-Browne et al., Dynamic Changes in Chromatin Accessibility Occur in CD8 + T Cells Responding to Viral Infection, Immunity (216), AI19842 and AI4127 (to A.R.). J.P.S.-B. was the Fraternal Order of Eagles Fellow of the Damon Runyon Cancer Research Foundation, DRG R.M.P. was supported by a fellowship from the Pew Latin American Fellows Program in the Biomedical Sciences. I.F.L.-M. was supported by a UC MEXUS-CONACYT fellowship. Received: May 6, 216 Revised: September 8, 216 Accepted: September 23, 216 Published: December 6, 216 REFERENCES Barber, D.L., Wherry, E.J., Masopust, D., Zhu, B., Allison, J.P., Sharpe, A.H., Freeman, G.J., and Ahmed, R. (26). Restoring function in exhausted CD8 T cells during chronic viral infection. Nature 439, Best, J.A., Blair, D.A., Knell, J., Yang, E., Mayya, V., Doedens, A., Dustin, M.L., and Goldrath, A.W.; Immunological Genome Project Consortium (213). Transcriptional insights into the CD8(+) T cell response to infection and memory T cell formation. Nat. Immunol. 14, Blackburn, S.D., Shin, H., Haining, W.N., Zou, T., Workman, C.J., Polley, A., Betts, M.R., Freeman, G.J., Vignali, D.A., and Wherry, E.J. (29). Coregulation of CD8+ T cell exhaustion by multiple inhibitory receptors during chronic viral infection. Nat. Immunol. 1, Buenrostro, J.D., Giresi, P.G., Zaba, L.C., Chang, H.Y., and Greenleaf, W.J. (213). Transposition of native chromatin for fast and sensitive epigenomic profiling of open chromatin, DNA-binding proteins and nucleosome position. Nat. Methods 1, ENCODE Project Consortium (212). An integrated encyclopedia of DNA elements in the human genome. Nature 489, Crespo, J., Sun, H., Welling, T.H., Tian, Z., and Zou, W. (213). T cell anergy, exhaustion, senescence, and stemness in the tumor microenvironment. Curr. Opin. Immunol. 25, Doering, T.A., Crawford, A., Angelosanto, J.M., Paley, M.A., Ziegler, C.G., and Wherry, E.J. (212). Network analysis reveals centrally connected genes and pathways involved in CD8+ T cell exhaustion versus memory. Immunity 37, Dominguez, C.X., Amezquita, R.A., Guan, T., Marshall, H.D., Joshi, N.S., Kleinstein, S.H., and Kaech, S.M. (215). The transcription factors ZEB2 and T-bet cooperate to program cytotoxic T cell terminal differentiation in response to LCMV viral infection. J. Exp. Med. 212, Intlekofer, A.M., Takemoto, N., Wherry, E.J., Longworth, S.A., Northrup, J.T., Palanivel, V.R., Mullen, A.C., Gasink, C.R., Kaech, S.M., Miller, J.D., et al. (25). and memory CD8+ T cell fate coupled by T-bet and eomesodermin. Nat. Immunol. 6, Kao, C., Oestreich, K.J., Paley, M.A., Crawford, A., Angelosanto, J.M., Ali, M.A., Intlekofer, A.M., Boss, J.M., Reiner, S.L., Weinmann, A.S., and Wherry, E.J. (211). Transcription factor T-bet represses expression of the inhibitory receptor PD-1 and sustains virus-specific CD8+ T cell responses during chronic infection. Nat. Immunol. 12, Kouzarides, T. (27). Chromatin modifications and their function. Cell 128, Kurachi, M., Barnitz, R.A., Yosef, N., Odorizzi, P.M., DiIorio, M.A., Lemieux, M.E., Yates, K., Godec, J., Klatt, M.G., Regev, A., et al. (214). The transcription factor BATF operates as an essential differentiation checkpoint in early effector CD8+ T cells. Nat. Immunol. 15, Langmead, B., Trapnell, C., Pop, M., and Salzberg, S.L. (29). Ultrafast and memory-efficient alignment of short DNA sequences to the human genome. Genome Biol. 1, R25. Leong, Y.A., Chen, Y., Ong, H.S., Wu, D., Man, K., Deleage, C., Minnich, M., Meckiff, B.J., Wei, Y., Hou, Z., et al. (216). CXCR5(+) follicular cytotoxic T cells control viral infection in B cell follicles. Nat. Immunol. 17, Love, M.I., Huber, W., and Anders, S. (214). Moderated estimation of fold change and dispersion for RNA-seq data with DESeq2. Genome Biol. 15, 55. Martinez, G.J., Pereira, R.M., Aijo, T., Kim, E.Y., Marangoni, F., Pipkin, M.E., Togher, S., Heissmeyer, V., Zhang, Y.C., Crotty, S., et al. (215). The transcription factor NFAT promotes exhaustion of activated CD8(+) T cells. Immunity 42, Moskophidis, D., Lechner, F., Pircher, H., and Zinkernagel, R.M. (1993). Virus persistence in acutely infected immunocompetent mice by exhaustion of antiviral cytotoxic effector T cells. Nature 362, Pardoll, D.M. (212). The blockade of immune checkpoints in cancer immunotherapy. Nat. Rev. Cancer 12, Picelli, S., Faridani, O.R., Björklund, A.K., Winberg, G., Sagasser, S., and Sandberg, R. (214). Full-length RNA-seq from single cells using Smartseq2. Nat. Protoc. 9, Pipkin, M.E., Sacks, J.A., Cruz-Guilloty, F., Lichtenheld, M.G., Bevan, M.J., and Rao, A. (21). Interleukin-2 and inflammation induce distinct transcriptional programs that promote the differentiation of effector cytolytic T cells. Immunity 32, Russ, B.E., Olshanksy, M., Smallwood, H.S., Li, J., Denton, A.E., Prier, J.E., Stock, A.T., Croom, H.A., Cullen, J.G., Nguyen, M.L., et al. (214). Distinct epigenetic signatures delineate transcriptional programs during virus-specific CD8(+) T cell differentiation. Immunity 41, Scharer, C.D., Barwick, B.G., Youngblood, B.A., Ahmed, R., and Boss, J.M. (213). Global DNA methylation remodeling accompanies CD8 T cell effector function. J. Immunol. 191, Schietinger, A., Delrow, J.J., Basom, R.S., Blattman, J.N., and Greenberg, P.D. (212). Rescued tolerant CD8 T cells are preprogrammed to reestablish the tolerant state. Science 335, Schietinger, A., Philip, M., Krisnawan, V.E., Chiu, E.Y., Delrow, J.J., Basom, R.S., Lauer, P., Brockstedt, D.G., Knoblaugh, S.E., H ammerling, G.J., et al. (216). Tumor-specific T cell dysfunction is a dynamic antigen-driven differentiation program initiated early during tumorigenesis. Immunity 45, Sekiya, T., Kashiwagi, I., Yoshida, R., Fukaya, T., Morita, R., Kimura, A., Ichinose, H., Metzger, D., Chambon, P., and Yoshimura, A. (213). Nr4a receptors are essential for thymic regulatory T cell development and immune homeostasis. Nat. Immunol. 14, Speiser, D.E., Utzschneider, D.T., Oberle, S.G., M unz, C., Romero, P., and Zehn, D. (214). T cell differentiation in chronic infection and cancer: functional adaptation or exhaustion? Nat. Rev. Immunol. 14, Steinke, F.C., Yu, S., Zhou, X., He, B., Yang, W., Zhou, B., Kawamoto, H., Zhu, J., Tan, K., and Xue, H.H. (214). TCF-1 and LEF-1 act upstream of Th-POK to promote the CD4(+) T cell fate and interact with Runx3 to silence Cd4 in CD8(+) T cells. Nat. Immunol. 15, Thurman, R.E., Rynes, E., Humbert, R., Vierstra, J., Maurano, M.T., Haugen, E., Sheffield, N.C., Stergachis, A.B., Wang, H., Vernot, B., et al. (212). The accessible chromatin landscape of the human genome. Nature 489, Trapnell, C., Pachter, L., and Salzberg, S.L. (29). TopHat: discovering splice junctions with RNA-Seq. Bioinformatics 25, Utzschneider, D.T., Legat, A., Fuertes Marraco, S.A., Carrié, L., Luescher, I., Speiser, D.E., and Zehn, D. (213). T cells maintain an exhausted phenotype after antigen withdrawal and population reexpansion. Nat. Immunol. 14, Vierstra, J., Rynes, E., Sandstrom, R., Zhang, M., Canfield, T., Hansen, R.S., Stehling-Sun, S., Sabo, P.J., Byron, R., Humbert, R., et al. (214). Mouse regulatory DNA landscapes reveal global principles of cis-regulatory evolution. Science 346, Wherry, E.J. (211). T cell exhaustion. Nat. Immunol. 12, Wherry, E.J., and Ahmed, R. (24). CD8 T-cell differentiation during viral infection. J. Virol. 78, Wherry, E.J., and Kurachi, M. (215). Molecular and cellular insights into T cell exhaustion. Nat. Rev. Immunol. 15, Wherry, E.J., Teichgr aber, V., Becker, T.C., Masopust, D., Kaech, S.M., Antia, R., von Andrian, U.H., and Ahmed, R. (23). Lineage relationship and protective immunity of memory CD8 T cell subsets. Nat. Immunol. 4, Immunity 45, 1 14, December 2,
15 Please cite this article in press as: Scott-Browne et al., Dynamic Changes in Chromatin Accessibility Occur in CD8 + T Cells Responding to Viral Infection, Immunity (216), Williams, M.A., and Bevan, M.J. (27). and memory CTL differentiation. Annu. Rev. Immunol. 25, Yamamoto, T., Price, D.A., Casazza, J.P., Ferrari, G., Nason, M., Chattopadhyay, P.K., Roederer, M., Gostick, E., Katsikis, P.D., Douek, D.C., et al. (211). Surface expression patterns of negative regulatory molecules identify determinants of virus-specific CD8+ T-cell exhaustion in HIV infection. Blood 117, Youngblood, B., Oestreich, K.J., Ha, S.J., Duraiswamy, J., Akondy, R.S., West, E.E., Wei, Z., Lu, P., Austin, J.W., Riley, J.L., et al. (211). Chronic virus infection enforces demethylation of the locus that encodes PD-1 in antigenspecific CD8(+) T cells. Immunity 35, Zhang, Y., Liu, T., Meyer, C.A., Eeckhoute, J., Johnson, D.S., Bernstein, B.E., Nusbaum, C., Myers, R.M., Brown, M., Li, W., and Liu, X.S. (28). Modelbased analysis of ChIP-Seq (MACS). Genome Biol. 9, R137. Zhou, X., Yu, S., Zhao, D.M., Harty, J.T., Badovinac, V.P., and Xue, H.H. (21). Differentiation and persistence of memory CD8(+) T cells depend on T cell factor 1. Immunity 33, Immunity 45, 1 14, December 2, 216
16 Immunity, Volume 45 Supplemental Information Dynamic Changes in Chromatin Accessibility Occur in CD8 + T Cells Responding to Viral Infection James P. Scott-Browne, Isaac F. López-Moyado, Sara Trifari, Victor Wong, Lukas Chavez, Anjana Rao, and Renata M. Pereira
17 Supplemental Experimental Procedures Experimental Procedures Mice C57BL/6J mice or P14 T cell receptor transgenic (TCR Tg) animals also containing a targeted mutation in the TCRα constant region to eliminate expression of endogenous rearranged TCRα chains (Cα / ) were maintained in specific-pathogen-free barrier facilities and used according to protocols approved by the La Jolla Institute animal care and use committee. CD8 + T cell purification CD8 + T cells were purified from splenocytes of 6-1 week old C57BL/6J mice not infected (for naive cells) or infected with LCMV (for effector, SLEC, MPEC, memory, d35 KLRG1+ or exhausted cells). For effector, SLEC, MPEC, memory and d35 KLRG1+ cell populations, mice were infected with 2 X 1 5 PFU of LCMV Arm5 intraperitoneally. Eight days after infection, spleens were harvested for effector, SLEC, and MPEC populations, while memory and d35 KLRG1+ populations were sorted from mice infected 35 days prior. For exhausted populations, mice were infected intravenously with 3 X 1 6 PFU of LCMV clone 13 for twenty days. For all subsets, splenocytes were enriched for CD8 + T cells using the Dynabeads Untouched CD8 + T Cells Kit (Invitrogen). Cells were then stained with different combinations of antibodies against cell surface markers (CD8, CD44, KLRG1, IL7Rα, PD-1, LAG3 and TIM3) and tetramer (H-2D b -gp33-41, containing the peptide from LCMV glycoprotein amino acids 33-41), and sorted using the gating strategies depicted in Figures S1 A,C-E. For in vitro stimulation, CD8 + T cells were isolated, as above, from P14 TCR Tg Cα / mice. These cells were stimulated for two days with antibodies specific for CD3ε and CD28 followed by expansion with 1 or 1 U/ml recombinant human IL-2, as previously described (Pipkin et al., 21). On day 6, the cells cultured in low (1U) IL-2 were restimulated for 2hrs with Ionomycin (5 nm), PMA (2nM) and Ionomycin, or PMA, Ionomycin and Cyclosporine (2mM, 3 min pre-treatment). For retroviral transduction experiments, CD8 + T cells were isolated by negative selection from C57BL/6J mice. These cells were stimulated for one day with antibodies specific for CD3ε and CD28, at which point they were transduced with retroviruses expressing the indicated genes plus green fluorescent protein as a reporter and expanded for three days. ATAC-seq ATAC-seq library preparations were performed essentially as described (Buenrostro et al., 213), with minor modifications. Briefly, 5, cells were washed once in PBS before incubation for 1 minutes in lysis buffer (1 mm Tris ph 7.5, 1 mm NaCl, 3mM MgCl2,.1% NP-4). For one replicate of memory cells and d35 KLRG1+ cells, 1, cells were used. After lysis, cells were resuspended in 5 ul 1x TD Buffer containing 2.5 ul Nextera enzyme (Illumina, San Diego, CA). Transposition reactions were incubated for 3 min at 37 C before purification with QiaQuick MinElute columns (Qiagen, Valencia, CA). Purified DNA was amplified by PCR using Kapa Hi-Fi real-time library amplification kit (Kapa, Wilmington, Massachusetts) with 1-12 cycles,
18 according to the manufacturer s instructions with barcoded primers, as described previously (Buenrostro et al., 213). Amplified libraries were purified with QiaQuick MinElute columns (Qiagen, Valencia, CA) and quantitated with Kapa real-time library quantification kit (Kapa, Wilmington, Massachusetts). Paired-end sequencing was performed with the rapid run protocol with an Illumina HiSeq 25 (Illumina, San Diego, CA) with 5 cycles in each direction. Raw data from the sequencer was uploaded to Illumina basespace (Illumina, San Diego, CA) to separate based on barcode sequences and generate individual FASTQ files for each read. Sequencing reads were mapped to mouse genome (mm9) using bowtie (version 1.., (Langmead et al., 29)) with parameters "-p 8 -m 1 --best --strata -X 2 -S --fr --chunkmbs 124." Unmapped reads were processed with trim_galore using parameters --paired --nextera --length 37 --stringency 3 -- three_prime_clip_r1 1 --three_prime_clip_r2 1" before attempting to map again using the above parameters. These two bam files were merged and processed with picard MarkDuplicates to remove duplicate reads and then filtered to remove reads mapping to the mitochondrial genome (total numbers of unique, non-chrm reads for each replicate are reported in Figure S1F). Wiggle files of coverage for individual replicates were computed with MEDIPS (Chavez et al., 21) using full fragments captured by ATAC-seq on 1 bp windows and used to generate average coverage with the Java Genomics Toolkit (Palpant, 216) for each group. To identify peaks, the bam files containing unique, non-chrm reads were processed with samtools and awk using "'{if(sqrt(\$9*\$9)<1){print \$}} " to identify DNA fragments that were less than 1 nt in length and thus could not contain a nucleosome. These subnucleosomal fragments were used to call peak summits for each replicate with MACS2 using parameters "--nomodel -q.1 --keep-dup all --call-summits." The summits for each peak from all replicates were expanded to regions with a uniform size of 5 bp. These regions from each replicates were merged into one global set of peaks. These peaks were filtered to remove peaks that overlapped blacklisted regions with high non-specific background signal in all sequencing experiments from ENCODE (Consortium, 212; Kundaje, 213) and those that were on the Y chromosome. We identified the Tn5 insertion site by isolating the first 9bp of each read (Buenrostro et al., 213) and computed the number of transposase insertions per peak for each replicate with bedtools coverage (version ). For differential coverage, raw ATAC-seq counts in each peak for all replicates of all samples were normalized between replicates with size factors computed with DESeq2 (version 1.6). Size factors were refined using only those regions with a variance across all replicates that was less than 5% of the mean for all replicates. Pairwise contrasts were performed with DESeq2 and differentially accessible regions were filtered based on an fdr adjusted p-value of less than 1x1 3 and an estimated fold-change of at least 3. Commonly accessible regions were selected based on an estimated fold-change of less than 1.5. For downstream visualizations, the ATAC-seq density per peak was normalized to the median peak length of 56 base pairs. Accessible regions were defined as those with a group mean of least 25 length normalized counts per peak. E2A ((Leong et al., 216), GSE84974), TCF1 (Steinke et al., 214), GSE527), T-bet (Dominguez et al., 215), SRR ,SRR275585), BATF and IRF4 (Kurachi et al., 214), GSE54191) ChIP-seq, including input controls where available, were downloaded from NCBI SRA database before processing with fastq-dump, trimming reads to 36 nucleotides with Trimmomatic, and mapping to the mouse genome (mm9) using bowtie
19 (version 1..) with parameters -p 8 -m 1 -q --best --strata --chunkmbs 124. Peaks were called with MACS2 using parameters -f BAM -g mm -B -q.5, including input datasets where available, and filtered against the ENCODE blacklist. The intersection between ChIP-seq and ATAC-seq peaks were computed using bedtools intersect (v2.16.2). MeDIP-seq data for naive and effector T cells and input ((Scharer et al., 213), GSE44638) were downloaded and mapped to the mouse genome (mm9) using bowtie with parameters -p 8 -m 1 -q --best --strata --chunkmbs 124 and coverage was computed around the indicated peaks with HOMER. For comparisons with NFAT binding sites, the NFAT1 ChIP-seq signal at ATAC-seq peaks was computed with HOMER using previously published data for endogenous NFAT1 and over-expressed NFAT-CA-RIT (Martinez et al., 215). Genomic annotations for peaks and motif enrichment were computed with HOMER (Heinz et al., 21) and the identification of peaks near genes was compared using BETA (Wang et al., 213), using default parameters without including CTCF boundary files and manually filtering for peaks within 5 bp of a TSS and within 25kb of a TSS and not at another TSS. The distribution of RNA-seq log 2 fold changes for genes within 25kb was compared using an empirical distribution function and a two-sample Kolmogorov Smirnov test computed in R. To assess the intersection between groups of differentially accessible peaks, we performed Fisher s exact test comparisons using contingency tables using the unique and shared differentially accessible peaks compared to the total accessible peaks. RNA-seq Total RNA was isolated from sorted naive, effector, memory, and exhausted cells (as described above) using Qiagen RNeasy Mini columns (Qiagen, Valencia, CA) according to the manufacturer s instructions. RNA-seq libraries were prepared using the SMARTseq2 protocol, as previously described (Picelli et al., 214). Briefly, purified RNA was evaluated with Bioanalyzer RNA pico kit (Agilent Technologies Inc, Santa Clara, CA) and reverse transcribed using Oligo dt, Template switching Oligo (TSO) and Superscript II, as per manufacturer's recommendation. The cdna was pre-amplified between cycles using IS oligo and Kapa HiFi Hotstart kit. The PCR product was purified using AmpureXP beads (Beckman Coulter, Danvers, MA) and 1 ng was submitted to library prep using Nextera XT Library Prep kit. Tagmented DNA was amplified in a 12 cycles PCR reaction and purified with AmpureXP beads. Size distribution was evaluated on TS Agilent high-sensitivity DNA chip, quantification was performed using Picogreen (ThermoFisher, Waltham, MA) and the median size obtained on the Tapestation (Agilent Technologies Inc) was obtained to calculate the molarity of the final library. RNA-seq libraries were sequenced with the rapid run protocol with an Illumina HiSeq 25. Reads that passed Illumina filters were filtered for reads aligning to trna, rrna, adapter sequences, and spike-in controls. The reads were then aligned to UCSC mm9 reference genome using TopHat (1.4.1, (Trapnell et al., 29)). DUST scores were calculated with PRINSEQ Lite (v.2.3) and low-complexity reads (DUST > 4) were removed from the BAM files. Read counts to each genomic feature were obtained with htseq-count (.6. using the
20 union option. After removing absent features (zero counts in all samples), the raw counts were normalized with DESeq2, followed by differentially expression analysis with DESeq2. Differentially expressed genes were filtered based on an fdr adjusted p-value of less than 1x1 3 and an estimated fold-change of at least 2 and a group mean of least 1 transcripts per million transcripts. Transcript abundance data from CD8 + T cells over-expressing NFAT-CA-RIT or left untransduced (Mock) were collected from processed RNA-seq data (Martinez et al., 215). Supplemental References Buenrostro, J.D., Giresi, P.G., Zaba, L.C., Chang, H.Y., and Greenleaf, W.J. (213). Transposition of native chromatin for fast and sensitive epigenomic profiling of open chromatin, DNA-binding proteins and nucleosome position. Nat Methods 1, Chavez, L., Jozefczuk, J., Grimm, C., Dietrich, J., Timmermann, B., Lehrach, H., Herwig, R., and Adjaye, J. (21). Computational analysis of genome-wide DNA methylation during the differentiation of human embryonic stem cells along the endodermal lineage. Genome Res 2, Consortium, E.P. (212). An integrated encyclopedia of DNA elements in the human genome. Nature 489, Dominguez, C.X., Amezquita, R.A., Guan, T., Marshall, H.D., Joshi, N.S., Kleinstein, S.H., and Kaech, S.M. (215). The transcription factors ZEB2 and T-bet cooperate to program cytotoxic T cell terminal differentiation in response to LCMV viral infection. J Exp Med 212, Heinz, S., Benner, C., Spann, N., Bertolino, E., Lin, Y.C., Laslo, P., Cheng, J.X., Murre, C., Singh, H., and Glass, C.K. (21). Simple combinations of lineage-determining transcription factors prime cis-regulatory elements required for macrophage and B cell identities. Mol Cell 38, Kundaje, A. (213). Blacklisted genomic regions for functional genomics analysis. Kurachi, M., Barnitz, R.A., Yosef, N., Odorizzi, P.M., DiIorio, M.A., Lemieux, M.E., Yates, K., Godec, J., Klatt, M.G., Regev, A., et al. (214). The transcription factor BATF operates as an essential differentiation checkpoint in early effector CD8+ T cells. Nat Immunol 15, Langmead, B., Trapnell, C., Pop, M., and Salzberg, S.L. (29). Ultrafast and memory-efficient alignment of short DNA sequences to the human genome. Genome Biol 1, R25. Martinez, G.J., Pereira, R.M., Aijo, T., Kim, E.Y., Marangoni, F., Pipkin, M.E., Togher, S., Heissmeyer, V., Zhang, Y.C., Crotty, S., et al. (215). The transcription factor NFAT promotes exhaustion of activated CD8(+) T cells. Immunity 42, Palpant, T. (216). Java Genomics Toolkit. Picelli, S., Faridani, O.R., Bjorklund, A.K., Winberg, G., Sagasser, S., and Sandberg, R. (214). Full-length RNA-seq from single cells using Smart-seq2. Nat Protoc 9, Pipkin, M.E., Sacks, J.A., Cruz-Guilloty, F., Lichtenheld, M.G., Bevan, M.J., and Rao, A. (21). Interleukin-2 and inflammation induce distinct transcriptional programs that promote the differentiation of effector cytolytic T cells. Immunity 32, Steinke, F.C., Yu, S., Zhou, X., He, B., Yang, W., Zhou, B., Kawamoto, H., Zhu, J., Tan, K., and Xue, H.H. (214). TCF-1 and LEF-1 act upstream of Th-POK to promote the CD4(+) T cell fate and interact with Runx3 to silence Cd4 in CD8(+) T cells. Nat Immunol 15, Trapnell, C., Pachter, L., and Salzberg, S.L. (29). TopHat: discovering splice junctions with RNA-Seq. Bioinformatics 25, Wang, S., Sun, H., Ma, J., Zang, C., Wang, C., Wang, J., Tang, Q., Meyer, C.A., Zhang, Y., and Liu, X.S. (213). Target analysis by integration of transcriptome and ChIP-seq data with BETA. Nat Protoc 8,
 17.1 (KLRG1+) () 49.")




 rep2 1.16 RV (NFAT-CA-RIT-DBDmut) rep1 1.16 RV (NFAT-CA-RIT-DBDmut) rep2 1.6 RV (NFAT-CA-RIT) rep1.82 RV (NFAT-CA-RIT) rep2 1.")
 Mammalian Conservation L % of Regions log 2 fold enrichment.6.4.2. 1..5.")
21 + A H-2D b -GP33-41 C H-2D b -GP33-41 D H-2D b -GP33-41 E H-2D b -GP33-41 M in vitro RV Mock d8 LCMV Arm5 live CD CD44 d8 LMCV Arm5 SLEC and MPEC live CD8 7.8 CD44 d35 LCMV Arm5 and KLRG1+ live CD live CD KLRG1 PD-1 B 57.2 (SLEC) 17.1 (KLRG1+) () 49.8 ATAC-seq counts/peak 6963 KLRG IL7Rα (MPEC) CD44 IL7Rα d2 LCMV Clone CD % of H-2D b /gp CD8 T cells KLRG1+ KLRG1 IL7Rα IL7Rα N F ex vivo Isolated Cells reads (1x1 7 ) rep1* 2.44 rep1* 2.53 rep rep rep rep1* 2.94 rep1* 2.34 rep SLEC rep SLEC rep2 3.4 SLEC rep MPEC rep MPEC rep MPEC rep rep rep d35 KLRG1+.78 rep rep rep distance (ATAC-seq) rep2 rep1* rep1* SLEC rep2 SLEC rep1 SLEC rep3 MPEC rep2 MPEC rep1 MPEC rep3 rep4 rep3 rep1 rep2 rep1 d35 KLRG1+ rep1* rep1* rep4 rep3 rep2 1U IL-2 rep2 1U IL-2 rep1 1U IL-2 resting rep2 1U IL-2 resting rep1 RV NFAT-CA-RIT-DBDmut rep2 RV NFAT-CA-RIT-DBDmut rep1 RV Mock rep1 RV Mock rep2 1U IL-2 Iono rep2 1U IL-2 Iono rep1 1U IL-2 PMAIonoCsA rep2 1U IL-2 PMAIonoCsA rep1 1U IL-2 PMAIono rep2 1U IL-2 PMAIono rep1 RV NFAT-CA-RIT rep2 RV NFAT-CA-RIT rep1 in vitro Cultured Cells reads (1x1 7 ) 1U IL-2, resting rep U IL-2, resting rep U IL-2, resting rep U IL-2, resting rep U IL-2, Iono rep U IL-2, Iono rep U IL-2, PMA/Iono rep1.91 1U IL-2, PMA/Iono rep U IL-2, PMA/Iono/CsA rep1.91 1U IL-2, PMA/Iono/CsA rep2 1.9 in vitro retrovirus expressing Cells reads (1x1 7 ) RV (Mock) rep RV (Mock) rep RV (NFAT-CA-RIT-DBDmut) rep RV (NFAT-CA-RIT-DBDmut) rep2 1.6 RV (NFAT-CA-RIT) rep1.82 RV (NFAT-CA-RIT) rep TIM3 ATAC-seq counts/peak I in vitro, RV Mock > Common > in vitro, RV Mock O in vitro 1U IL-2 resting 1 1 G H J K 1 density log 1 ( replicate rmse) Mammalian Conservation L % of Regions log 2 fold enrichment counts/peak counts/peak distance (RNA-seq) rep2 rep1 rep2 rep1 rep2 rep1 rep1 rep2 Promoter 5UTR Exon Intron 4 3UTR 2 TTS ATAC-seq counts/peak Distance from center (bp) Intergenic in vitro 1U IL-2 resting 8 Figure S1
22 Figure S1, related to Figure 1) Individual replicate comparison of CD8 + T cell samples collected for ATAC-seq. A,C,D,E) Representative FACS plots indicating sorting strategies for ex vivo isolated LCMV specific effector, memory, and exhausted CD8 + T cells. Data are representative from at least 2 independent experiments. B) Frequency of KLRG1+/IL7R and KLRG1 /IL7R+ expressing cells among H-2D b /gp33-41 tetramer positive events from mice infected 8 days earlier with LCMV Am5. Data are mean plus s.e.m. for three independent experiments. F) Total numbers of unique non-mitochondrial reads for each replicate ATAC-seq sample. G) Histogram of mean ATAC-seq counts per peak in naive CD8 + T cells for all peaks. H) Scatterplot comparing inter-replicate root mean square error (rmse) and the mean ATAC-seq counts per peak between naive CD8 + T cell replicates. I) Pairwise euclidian distance comparison of asinh transformed ATAC-seq signal between individual replicates of all samples at peaks accessible in at least one cell type. Samples labeled with an asterisk (*) were technical replicates of ATAC-seq samples prepared separately from the same pool of ex vivo collected cells and only one of which was included in the statistical comparisons. J) Pairwise euclidian distance comparison of asinh transformed RNA-seq signal (tpm) between individual replicates of all samples at genes expressed in at least one cell type. K) Average placental mammal conservation in the 2kb region around the center of peaks accessible in at least one cell type. L) Genomic annotation of all peaks accessible in at least one cell type. M,O) Scatterplots of mean ATAC-seq counts per peak comparing the indicated samples. N) Boxplots of ATAC-seq counts per peak from the indicated samples (labeled at bottom) at common or differentially accessible regions from the comparison labeled above. Box indicates interquartile range with whiskers +/-1.5 times this range and outlier points. ATAC-seq data in M-O are from at least 2 independent replicates.

23 A B C Density Density Density Common, & > > Distance to nearest TSS (bp) Common, & > > Distance to nearest TSS (bp) Common, & > > Distance to nearest TSS (bp) D 1 4 E F RNA-seq TPM RNA-seq TPM RNA-seq TPM RNA-seq TPM RNA-seq TPM RNA-seq TPM > > > > > > G H I ECDF(x) ECDF(x) ECDF(x) naive > effector 1274 genes p <2e RNA-seq log 2 fold change naive > memory 565 genes p <2e RNA-seq log 2 fold change effector > memory 78 genes p = 1.11e 9 effector > naive 1873 genes p <2e 16 memory > naive 789 genes p <2e 16 memory > effector 389 genes p = 3.22e RNA-seq log 2 fold change J 2 kb >1 Mammalian Conservation Fasl TPM 1 1 <.5 2 kb Fasl Fasl Fasl Fasl +3kb >1 Mammalian Conservation Adamts14 Prf1 Pald1 Adamts14 Prf1 X99384 Adamts14 Prf1 X99384 Prf1 TPM 1 1 <.5 Figure S2
24 Figure S2, related to Figure 2) Gene associations for differentially accessible regions between naive and viral antigen specific effector and memory CD8 + T cells. A-C) Distance to nearest annotated TSS for common or differentially accessible peaks from each indicated comparison. D-F) Scatterplots of mean RNAseq transcripts per million (tpm) comparing the indicated samples (left), with points for differentially expressed genes colored, from two independent replicates for each group. Boxplots of RNA-seq counts per transcript from the indicated samples (bottom labels) for differentially expressed genes in the comparison labeled at the top (right). For boxplots in D-F, boxes indicate interquartile range with whiskers +/-1.5 times this range and outlier points from 2 independent replicates for each group. G-I) ECDF for log 2 fold-change between indicated RNA-seq samples for genes within 25kb of indicated accessible regions. For G,H,I, the numbers of genes within 25kb of indicated peaks are shown. P-values are for one-sided Kolmogorov Smirnov tests against genes within 25kb of commonly accessible peaks (grey). J) Genome browser view of mean ATAC-seq coverage at the Fasl and Prf1 loci with a scale of -12 (left) or RNA-seq gene expression for the indicated genes (right). RNA-seq data in D-F & J are mean of two independent replicates, ATAC-seq data in J are mean of 4 replicates for naïve, 2 replicates for effector, 2 replicates for memory and 3 replicates for exhausted.
25 A SLEC ATAC-seq counts/peak ATAC-seq counts/peak13 SLEC > > SLEC B MPEC ATAC-seq counts/peak ATAC-seq counts/peak13 MPEC > > MPEC C 2 kb 5 kb Peaks (M) (M) SLEC (F) MPEC (F) (M) d35 KLRG1+ (M) (M) 1U IL-2,resting (F) 1U IL-2,resting (F) 1U IL-2,Iono (F) 1U IL-2,PMA/Iono (F) 1U IL-2,PMA/Iono/CsA (F) RV (Mock) (M) RV (NFAT-CA-RIT-DBDmut) (M) RV (NFAT-CA-RIT) (M) Mammalian Conservation Tsix Xist Xist Firre Figure S3
26 Figure S3, related to Figure 3) Minimal differences between male effector CD8 + T cell and female SLEC T cells after response to acute viral infection. A,B) Scatterplots of ATAC-seq counts per peak comparing the indicated samples (left) and boxplots (right) of ATAC-seq counts per peak from the indicated samples (labeled at bottom) at common or differentially accessible regions from the comparison labeled above. For boxplots, box indicates interquartile range with whiskers +/-1.5 times this range and outlier points. C) Genome browser view of mean ATAC-seq coverage at Xist/Tsix and Firre loci with a scale of -12. Within track names at left, male (M) or female (F) are indicated. ATAC-seq data are mean of at least 2 independent replicates for all cell types, except for a single replicate for d35 KLRG1+.
27 A Density B Density Common, & > > Distance to nearest TSS (bp) Common, & > > Distance to nearest TSS (bp) C D RNA-seq TPM RNA-seq TPM RNA-seq TPM RNA-seq TPM > > > > E F ECDF(x) ECDF(x) memory > exhausted 82 genes p = 4.36e RNA-seq log 2 fold change effector > exhausted 617 genes p = 1.31e 13 exhausted > memory 873 genes p = 3.6e 14 exhausted > effector 414 genes p = 2.3e RNA-seq log 2 fold change G 2 kb >1 Mammalian Conservation Ctla4 Ctla4 Ctla4 ctla-4 Ctla4 TPM 1 1 <.5 2 kb >1 Mammalian Conservation Cd4 Cd4 Cd4 Cd4 Lag3 Ptms Lag3 Ptms A2383G16Rik Mlf2 Mlf2 Mlf2 Mlf2 Mlf2 Lag3 TPM 1 1 <.5 Figure S4
28 Figure S4, related to Figure 4) Gene associations for differentially accessible regions between viral antigen specific effector, memory, and exhausted CD8 + T cells. A,B) Distance to nearest annotated TSS for common or differentially accessible peaks from each indicated comparison. C,D) Scatterplots of mean RNA-seq transcripts per million (tpm) comparing the indicated samples (left), with points for differentially expressed genes colored. Boxplots of RNA-seq counts per transcript from the indicated samples (bottom labels) for differentially expressed genes in the comparison labeled at the top (right). For boxplots, box indicates interquartile range with whiskers +/-1.5 times this range and outlier points. E,F) ECDF for log 2 foldchange between indicated RNA-seq samples for genes within 25kb of indicated accessible regions. For E and F the numbers of genes within 25kb of indicated peaks are shown. P-values are for one-sided Kolmogorov Smirnov tests against genes within 25kb of commonly accessible peaks (grey). G) Genome browser view of mean ATAC-seq coverage at the Ctla4 and Lag3 loci with a scale of -12 (left) or RNA-seq gene expression for the indicated genes (right). RNA-seq data in C,D & G are from 2 independent replicates, ATAC-seq data in G are from at least 2 indpendent replicates.




















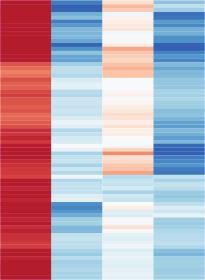
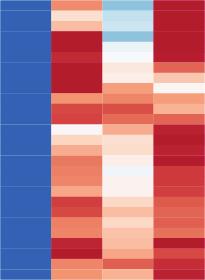
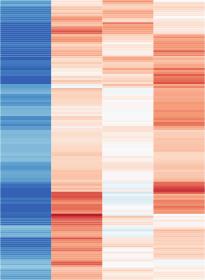
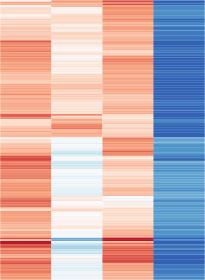
29 A C RNA-seq all diff. genes D ATAC-seq all diff. peaks within 25kb of TSS RNA-seq for genes within 25kb of peaks Nr4a1 TPM Nr4a2 TPM Nr4a3 TPM B Nfatc1 TPM >1 1 1 <.5 >1 1 1 <.5 >1 1 1 <.5 >1 1 1 <.5 >1 Sell Ifngr2 Ccr7 Tcf7 Bach2 Dmrta1 S1pr1 Btg1 Bcl2 Trib2 Ly6c2 Ifitm3 Aqp9 Klrk1 S1pr5 Cd28 Tcf3 Prf1 Bcl2l1 Rel Satb1 Gpr183 Il7r Jun Cd2r1 Nr4a2 Nfil3 Cd2r4 Ctla4 Cd244 Tox2 Nr4a1 Tox Cluster Peak-gene pairs (#) SLEC MPEC Peak-gene pair examples Ccr Sell +7 Bach Tox 1365 Tox 286 Ifngr Usp Ccr Lef Lag Foxp Ccr Tcf Il7r Sell +183 Irf4 663 Irf Sell Tcf7 99 Cd Rpa Dmrta Dmrta Bach Ctla Satb Satb Lef Il7r 1621 Ccr Ccr7 754 Pdcd Tox Lag Cd2r4 382 Pdcd Fasl 2334 Ctla Batf Cd Nr4a Genes (#) within 25kb of peaks Nfatc2 TPM 1 1 <.5 >1 Il2rb Hells Dnmt1 Smarca4 Irf4 Uhrf1 Havcr Klrg Gzma Rora Tbx Ccl Ifitm Il18rap Gzmk Klrg Nfatc3 TPM 1 1 <.5 RNA-seq log 2 FC from mean Hmgb2 Cxcr3 Tbx21 Ikzf3 Id2 Lag3 Pdcd1 Gzma Gzmb Klrg1 Ifng Bhlhe4 Gzmk Fasl ATAC-seq or RNA-seq log 2 FC from mean Tcf Zbtb Slamf Il2rb Tbx Tcf Bhlhe Runx Pdcd Gzma Usp Figure S
Transcript-indexed ATAC-seq for immune profiling
 Transcript-indexed ATAC-seq for immune profiling Technical Journal Club 22 nd of May 2018 Christina Müller Nature Methods, Vol.10 No.12, 2013 Nature Biotechnology, Vol.32 No.7, 2014 Nature Medicine, Vol.24,
Transcript-indexed ATAC-seq for immune profiling Technical Journal Club 22 nd of May 2018 Christina Müller Nature Methods, Vol.10 No.12, 2013 Nature Biotechnology, Vol.32 No.7, 2014 Nature Medicine, Vol.24,
Supplementary Figure 1 IL-27 IL
 Tim-3 Supplementary Figure 1 Tc0 49.5 0.6 Tc1 63.5 0.84 Un 49.8 0.16 35.5 0.16 10 4 61.2 5.53 10 3 64.5 5.66 10 2 10 1 10 0 31 2.22 10 0 10 1 10 2 10 3 10 4 IL-10 28.2 1.69 IL-27 Supplementary Figure 1.
Tim-3 Supplementary Figure 1 Tc0 49.5 0.6 Tc1 63.5 0.84 Un 49.8 0.16 35.5 0.16 10 4 61.2 5.53 10 3 64.5 5.66 10 2 10 1 10 0 31 2.22 10 0 10 1 10 2 10 3 10 4 IL-10 28.2 1.69 IL-27 Supplementary Figure 1.
Nature Immunology: doi: /ni Supplementary Figure 1. Transcriptional program of the TE and MP CD8 + T cell subsets.
 Supplementary Figure 1 Transcriptional program of the TE and MP CD8 + T cell subsets. (a) Comparison of gene expression of TE and MP CD8 + T cell subsets by microarray. Genes that are 1.5-fold upregulated
Supplementary Figure 1 Transcriptional program of the TE and MP CD8 + T cell subsets. (a) Comparison of gene expression of TE and MP CD8 + T cell subsets by microarray. Genes that are 1.5-fold upregulated
Nature Immunology: doi: /ni Supplementary Figure 1. Characteristics of SEs in T reg and T conv cells.
 Supplementary Figure 1 Characteristics of SEs in T reg and T conv cells. (a) Patterns of indicated transcription factor-binding at SEs and surrounding regions in T reg and T conv cells. Average normalized
Supplementary Figure 1 Characteristics of SEs in T reg and T conv cells. (a) Patterns of indicated transcription factor-binding at SEs and surrounding regions in T reg and T conv cells. Average normalized
Exposure of virus-specific or tumor-reactive CD8 + T cells to
 Exhaustion-associated regulatory regions in CD8 + tumor-infiltrating T cells Giuliana P. Mognol a,1, Roberto Spreafico b,1, Victor Wong a,1,2, James P. Scott-Browne a, Susan Togher a, Alexander Hoffmann
Exhaustion-associated regulatory regions in CD8 + tumor-infiltrating T cells Giuliana P. Mognol a,1, Roberto Spreafico b,1, Victor Wong a,1,2, James P. Scott-Browne a, Susan Togher a, Alexander Hoffmann
Distinct Epigenetic Signatures Delineate Transcriptional Programs during Virus-Specific CD8 + T Cell Differentiation
 Resource Distinct Epigenetic Signatures Delineate Transcriptional Programs during Virus-Specific CD8 + T Cell Differentiation Brendan E. Russ, 1 Moshe Olshanksy, 2 Heather S. Smallwood, 3 Jasmine Li, 1
Resource Distinct Epigenetic Signatures Delineate Transcriptional Programs during Virus-Specific CD8 + T Cell Differentiation Brendan E. Russ, 1 Moshe Olshanksy, 2 Heather S. Smallwood, 3 Jasmine Li, 1
Comparison of open chromatin regions between dentate granule cells and other tissues and neural cell types.
 Supplementary Figure 1 Comparison of open chromatin regions between dentate granule cells and other tissues and neural cell types. (a) Pearson correlation heatmap among open chromatin profiles of different
Supplementary Figure 1 Comparison of open chromatin regions between dentate granule cells and other tissues and neural cell types. (a) Pearson correlation heatmap among open chromatin profiles of different
Session 6: Integration of epigenetic data. Peter J Park Department of Biomedical Informatics Harvard Medical School July 18-19, 2016
 Session 6: Integration of epigenetic data Peter J Park Department of Biomedical Informatics Harvard Medical School July 18-19, 2016 Utilizing complimentary datasets Frequent mutations in chromatin regulators
Session 6: Integration of epigenetic data Peter J Park Department of Biomedical Informatics Harvard Medical School July 18-19, 2016 Utilizing complimentary datasets Frequent mutations in chromatin regulators
7SK ChIRP-seq is specifically RNA dependent and conserved between mice and humans.
 Supplementary Figure 1 7SK ChIRP-seq is specifically RNA dependent and conserved between mice and humans. Regions targeted by the Even and Odd ChIRP probes mapped to a secondary structure model 56 of the
Supplementary Figure 1 7SK ChIRP-seq is specifically RNA dependent and conserved between mice and humans. Regions targeted by the Even and Odd ChIRP probes mapped to a secondary structure model 56 of the
Nature Genetics: doi: /ng Supplementary Figure 1. Assessment of sample purity and quality.
 Supplementary Figure 1 Assessment of sample purity and quality. (a) Hematoxylin and eosin staining of formaldehyde-fixed, paraffin-embedded sections from a human testis biopsy collected concurrently with
Supplementary Figure 1 Assessment of sample purity and quality. (a) Hematoxylin and eosin staining of formaldehyde-fixed, paraffin-embedded sections from a human testis biopsy collected concurrently with
DEVELOPMENT AND REVERSAL
 DEVELOPMENT AND REVERSAL OF T CELL EXHAUSTION E. John Wherry Institute for Immunology University of Pennsylvania IAS 2016 Towards an HIV Cure Symposium Durban, South Africa www.iasociety.org T cell exhaustion
DEVELOPMENT AND REVERSAL OF T CELL EXHAUSTION E. John Wherry Institute for Immunology University of Pennsylvania IAS 2016 Towards an HIV Cure Symposium Durban, South Africa www.iasociety.org T cell exhaustion
Nature Structural & Molecular Biology: doi: /nsmb.2419
 Supplementary Figure 1 Mapped sequence reads and nucleosome occupancies. (a) Distribution of sequencing reads on the mouse reference genome for chromosome 14 as an example. The number of reads in a 1 Mb
Supplementary Figure 1 Mapped sequence reads and nucleosome occupancies. (a) Distribution of sequencing reads on the mouse reference genome for chromosome 14 as an example. The number of reads in a 1 Mb
The epigenetic landscape of T cell subsets in SLE identifies known and potential novel drivers of the autoimmune response
 Abstract # 319030 Poster # F.9 The epigenetic landscape of T cell subsets in SLE identifies known and potential novel drivers of the autoimmune response Jozsef Karman, Brian Johnston, Sofija Miljovska,
Abstract # 319030 Poster # F.9 The epigenetic landscape of T cell subsets in SLE identifies known and potential novel drivers of the autoimmune response Jozsef Karman, Brian Johnston, Sofija Miljovska,
Accessing and Using ENCODE Data Dr. Peggy J. Farnham
 1 William M Keck Professor of Biochemistry Keck School of Medicine University of Southern California How many human genes are encoded in our 3x10 9 bp? C. elegans (worm) 959 cells and 1x10 8 bp 20,000
1 William M Keck Professor of Biochemistry Keck School of Medicine University of Southern California How many human genes are encoded in our 3x10 9 bp? C. elegans (worm) 959 cells and 1x10 8 bp 20,000
Nature Immunology: doi: /ni Supplementary Figure 1. DNA-methylation machinery is essential for silencing of Cd4 in cytotoxic T cells.
 Supplementary Figure 1 DNA-methylation machinery is essential for silencing of Cd4 in cytotoxic T cells. (a) Scheme for the retroviral shrna screen. (b) Histogram showing CD4 expression (MFI) in WT cytotoxic
Supplementary Figure 1 DNA-methylation machinery is essential for silencing of Cd4 in cytotoxic T cells. (a) Scheme for the retroviral shrna screen. (b) Histogram showing CD4 expression (MFI) in WT cytotoxic
Computational Analysis of UHT Sequences Histone modifications, CAGE, RNA-Seq
 Computational Analysis of UHT Sequences Histone modifications, CAGE, RNA-Seq Philipp Bucher Wednesday January 21, 2009 SIB graduate school course EPFL, Lausanne ChIP-seq against histone variants: Biological
Computational Analysis of UHT Sequences Histone modifications, CAGE, RNA-Seq Philipp Bucher Wednesday January 21, 2009 SIB graduate school course EPFL, Lausanne ChIP-seq against histone variants: Biological
Processing, integrating and analysing chromatin immunoprecipitation followed by sequencing (ChIP-seq) data
 data") Processing, integrating and analysing chromatin immunoprecipitation followed by sequencing (ChIP-seq) data Bioinformatics methods, models and applications to disease Alex Essebier ChIP-seq experiment To
Processing, integrating and analysing chromatin immunoprecipitation followed by sequencing (ChIP-seq) data Bioinformatics methods, models and applications to disease Alex Essebier ChIP-seq experiment To
Cancer immunity and immunotherapy. General principles
 1 Cancer immunity and immunotherapy Abul K. Abbas UCSF General principles 2 The immune system recognizes and reacts against cancers The immune response against tumors is often dominated by regulation or
1 Cancer immunity and immunotherapy Abul K. Abbas UCSF General principles 2 The immune system recognizes and reacts against cancers The immune response against tumors is often dominated by regulation or
Supplemental Figure 1
 Supplemental Figure 1 1a 1c PD-1 MFI fold change 6 5 4 3 2 1 IL-1α IL-2 IL-4 IL-6 IL-1 IL-12 IL-13 IL-15 IL-17 IL-18 IL-21 IL-23 IFN-α Mut Human PD-1 promoter SBE-D 5 -GTCTG- -1.2kb SBE-P -CAGAC- -1.kb
Supplemental Figure 1 1a 1c PD-1 MFI fold change 6 5 4 3 2 1 IL-1α IL-2 IL-4 IL-6 IL-1 IL-12 IL-13 IL-15 IL-17 IL-18 IL-21 IL-23 IFN-α Mut Human PD-1 promoter SBE-D 5 -GTCTG- -1.2kb SBE-P -CAGAC- -1.kb
Computational aspects of ChIP-seq. John Marioni Research Group Leader European Bioinformatics Institute European Molecular Biology Laboratory
 Computational aspects of ChIP-seq John Marioni Research Group Leader European Bioinformatics Institute European Molecular Biology Laboratory ChIP-seq Using highthroughput sequencing to investigate DNA
Computational aspects of ChIP-seq John Marioni Research Group Leader European Bioinformatics Institute European Molecular Biology Laboratory ChIP-seq Using highthroughput sequencing to investigate DNA
Nature Structural & Molecular Biology: doi: /nsmb Supplementary Figure 1
 Supplementary Figure 1 Effect of HSP90 inhibition on expression of endogenous retroviruses. (a) Inducible shrna-mediated Hsp90 silencing in mouse ESCs. Immunoblots of total cell extract expressing the
Supplementary Figure 1 Effect of HSP90 inhibition on expression of endogenous retroviruses. (a) Inducible shrna-mediated Hsp90 silencing in mouse ESCs. Immunoblots of total cell extract expressing the
Supplementary Figure 1. Efficiency of Mll4 deletion and its effect on T cell populations in the periphery. Nature Immunology: doi: /ni.
 Supplementary Figure 1 Efficiency of Mll4 deletion and its effect on T cell populations in the periphery. Expression of Mll4 floxed alleles (16-19) in naive CD4 + T cells isolated from lymph nodes and
Supplementary Figure 1 Efficiency of Mll4 deletion and its effect on T cell populations in the periphery. Expression of Mll4 floxed alleles (16-19) in naive CD4 + T cells isolated from lymph nodes and
Therapeutic PD L1 and LAG 3 blockade rapidly clears established blood stage Plasmodium infection
 Supplementary Information Therapeutic PD L1 and LAG 3 blockade rapidly clears established blood stage Plasmodium infection Noah S. Butler, Jacqueline Moebius, Lecia L. Pewe, Boubacar Traore, Ogobara K.
Supplementary Information Therapeutic PD L1 and LAG 3 blockade rapidly clears established blood stage Plasmodium infection Noah S. Butler, Jacqueline Moebius, Lecia L. Pewe, Boubacar Traore, Ogobara K.
In Vivo RNA Interference Screens Identify Regulators of Antiviral CD4 + and CD8 + T Cell Differentiation
 Resource In Vivo RNA Interference Screens Identify Regulators of Antiviral CD + and CD8 + T Cell Differentiation Runqiang Chen,,,,5 Simon Bélanger,,5 Megan A. Frederick, Bin Li, Robert J. Johnston, Nengming
Resource In Vivo RNA Interference Screens Identify Regulators of Antiviral CD + and CD8 + T Cell Differentiation Runqiang Chen,,,,5 Simon Bélanger,,5 Megan A. Frederick, Bin Li, Robert J. Johnston, Nengming
Supplementary Figures
 Supplementary Figures Supplementary Figure 1. Heatmap of GO terms for differentially expressed genes. The terms were hierarchically clustered using the GO term enrichment beta. Darker red, higher positive
Supplementary Figures Supplementary Figure 1. Heatmap of GO terms for differentially expressed genes. The terms were hierarchically clustered using the GO term enrichment beta. Darker red, higher positive
Nature Immunology: doi: /ni Supplementary Figure 1. Gene expression profile of CD4 + T cells and CTL responses in Bcl6-deficient mice.
 Supplementary Figure 1 Gene expression profile of CD4 + T cells and CTL responses in Bcl6-deficient mice. (a) Gene expression profile in the resting CD4 + T cells were analyzed by an Affymetrix microarray
Supplementary Figure 1 Gene expression profile of CD4 + T cells and CTL responses in Bcl6-deficient mice. (a) Gene expression profile in the resting CD4 + T cells were analyzed by an Affymetrix microarray
Table S1. Total and mapped reads produced for each ChIP-seq sample
 Tale S1. Total and mapped reads produced for each ChIP-seq sample Sample Total Reads Mapped Reads Col- H3K27me3 rep1 125662 1334323 (85.76%) Col- H3K27me3 rep2 9176437 7986731 (87.4%) atmi1a//c H3K27m3
Tale S1. Total and mapped reads produced for each ChIP-seq sample Sample Total Reads Mapped Reads Col- H3K27me3 rep1 125662 1334323 (85.76%) Col- H3K27me3 rep2 9176437 7986731 (87.4%) atmi1a//c H3K27m3
Broad H3K4me3 is associated with increased transcription elongation and enhancer activity at tumor suppressor genes
 Broad H3K4me3 is associated with increased transcription elongation and enhancer activity at tumor suppressor genes Kaifu Chen 1,2,3,4,5,10, Zhong Chen 6,10, Dayong Wu 6, Lili Zhang 7, Xueqiu Lin 1,2,8,
Broad H3K4me3 is associated with increased transcription elongation and enhancer activity at tumor suppressor genes Kaifu Chen 1,2,3,4,5,10, Zhong Chen 6,10, Dayong Wu 6, Lili Zhang 7, Xueqiu Lin 1,2,8,
T Cell Activation, Costimulation and Regulation
 1 T Cell Activation, Costimulation and Regulation Abul K. Abbas, MD University of California San Francisco 2 Lecture outline T cell antigen recognition and activation Costimulation, the B7:CD28 family
1 T Cell Activation, Costimulation and Regulation Abul K. Abbas, MD University of California San Francisco 2 Lecture outline T cell antigen recognition and activation Costimulation, the B7:CD28 family
T cell maturation. T-cell Maturation. What allows T cell maturation?
 T-cell Maturation What allows T cell maturation? Direct contact with thymic epithelial cells Influence of thymic hormones Growth factors (cytokines, CSF) T cell maturation T cell progenitor DN DP SP 2ry
T-cell Maturation What allows T cell maturation? Direct contact with thymic epithelial cells Influence of thymic hormones Growth factors (cytokines, CSF) T cell maturation T cell progenitor DN DP SP 2ry
Allelic reprogramming of the histone modification H3K4me3 in early mammalian development
 Allelic reprogramming of the histone modification H3K4me3 in early mammalian development 张戈 Method and material STAR ChIP seq (small-scale TELP-assisted rapid ChIP seq) 200 mouse embryonic stem cells PWK/PhJ
Allelic reprogramming of the histone modification H3K4me3 in early mammalian development 张戈 Method and material STAR ChIP seq (small-scale TELP-assisted rapid ChIP seq) 200 mouse embryonic stem cells PWK/PhJ
fl/+ KRas;Atg5 fl/+ KRas;Atg5 fl/fl KRas;Atg5 fl/fl KRas;Atg5 Supplementary Figure 1. Gene set enrichment analyses. (a) (b)
 (b)") KRas;At KRas;At KRas;At KRas;At a b Supplementary Figure 1. Gene set enrichment analyses. (a) GO gene sets (MSigDB v3. c5) enriched in KRas;Atg5 fl/+ as compared to KRas;Atg5 fl/fl tumors using gene set
KRas;At KRas;At KRas;At KRas;At a b Supplementary Figure 1. Gene set enrichment analyses. (a) GO gene sets (MSigDB v3. c5) enriched in KRas;Atg5 fl/+ as compared to KRas;Atg5 fl/fl tumors using gene set
Sirt1 Hmg20b Gm (0.17) 24 (17.3) 877 (857)
 24 (17.3) 877 (857)") 3 (0.17) 24 (17.3) Sirt1 Hmg20 Gm4763 877 (857) c d Suppl. Figure 1. Screen validation for top candidate antagonists of Dot1L (a) Numer of genes with one (gray), two (cyan) or three (red) shrna scored
3 (0.17) 24 (17.3) Sirt1 Hmg20 Gm4763 877 (857) c d Suppl. Figure 1. Screen validation for top candidate antagonists of Dot1L (a) Numer of genes with one (gray), two (cyan) or three (red) shrna scored
Heintzman, ND, Stuart, RK, Hon, G, Fu, Y, Ching, CW, Hawkins, RD, Barrera, LO, Van Calcar, S, Qu, C, Ching, KA, Wang, W, Weng, Z, Green, RD,
 Heintzman, ND, Stuart, RK, Hon, G, Fu, Y, Ching, CW, Hawkins, RD, Barrera, LO, Van Calcar, S, Qu, C, Ching, KA, Wang, W, Weng, Z, Green, RD, Crawford, GE, Ren, B (2007) Distinct and predictive chromatin
Heintzman, ND, Stuart, RK, Hon, G, Fu, Y, Ching, CW, Hawkins, RD, Barrera, LO, Van Calcar, S, Qu, C, Ching, KA, Wang, W, Weng, Z, Green, RD, Crawford, GE, Ren, B (2007) Distinct and predictive chromatin
FOCiS. Lecture outline. The immunological equilibrium: balancing lymphocyte activation and control. Immunological tolerance and immune regulation -- 1
 1 Immunological tolerance and immune regulation -- 1 Abul K. Abbas UCSF FOCiS 2 Lecture outline Principles of immune regulation Self-tolerance; mechanisms of central and peripheral tolerance Inhibitory
1 Immunological tolerance and immune regulation -- 1 Abul K. Abbas UCSF FOCiS 2 Lecture outline Principles of immune regulation Self-tolerance; mechanisms of central and peripheral tolerance Inhibitory
Epigenetic Instability of Cytokine and Transcription Factor Gene Loci Underlies Plasticity of the T Helper 17 Cell Lineage
 Article Epigenetic Instability of Cytokine and Transcription Factor Gene Loci Underlies Plasticity of the T Helper 17 Cell Lineage Ryuta Mukasa, 1,3 Anand Balasubramani, 1,2,5 Yun Kyung Lee, 1,2,5 Sarah
Article Epigenetic Instability of Cytokine and Transcription Factor Gene Loci Underlies Plasticity of the T Helper 17 Cell Lineage Ryuta Mukasa, 1,3 Anand Balasubramani, 1,2,5 Yun Kyung Lee, 1,2,5 Sarah
Chronic Virus Infection Enforces Demethylation of the Locus that Encodes PD-1 in Antigen-Specific CD8 + T Cells
 Article Chronic Virus Infection Enforces Demethylation of the Locus that Encodes PD-1 in Antigen-Specific CD8 + T Cells Ben Youngblood, 1,2,6 Kenneth J. Oestreich, 2,3,6 Sang-Jun Ha, 1,2,4 Jaikumar Duraiswamy,
Article Chronic Virus Infection Enforces Demethylation of the Locus that Encodes PD-1 in Antigen-Specific CD8 + T Cells Ben Youngblood, 1,2,6 Kenneth J. Oestreich, 2,3,6 Sang-Jun Ha, 1,2,4 Jaikumar Duraiswamy,
Peak-calling for ChIP-seq and ATAC-seq
 Peak-calling for ChIP-seq and ATAC-seq Shamith Samarajiwa CRUK Autumn School in Bioinformatics 2017 University of Cambridge Overview Peak-calling: identify enriched (signal) regions in ChIP-seq or ATAC-seq
Peak-calling for ChIP-seq and ATAC-seq Shamith Samarajiwa CRUK Autumn School in Bioinformatics 2017 University of Cambridge Overview Peak-calling: identify enriched (signal) regions in ChIP-seq or ATAC-seq
Supplementary Figure S1. Gene expression analysis of epidermal marker genes and TP63.
 Supplementary Figure Legends Supplementary Figure S1. Gene expression analysis of epidermal marker genes and TP63. A. Screenshot of the UCSC genome browser from normalized RNAPII and RNA-seq ChIP-seq data
Supplementary Figure Legends Supplementary Figure S1. Gene expression analysis of epidermal marker genes and TP63. A. Screenshot of the UCSC genome browser from normalized RNAPII and RNA-seq ChIP-seq data
SUPPLEMENTARY FIGURES: Supplementary Figure 1
 SUPPLEMENTARY FIGURES: Supplementary Figure 1 Supplementary Figure 1. Glioblastoma 5hmC quantified by paired BS and oxbs treated DNA hybridized to Infinium DNA methylation arrays. Workflow depicts analytic
SUPPLEMENTARY FIGURES: Supplementary Figure 1 Supplementary Figure 1. Glioblastoma 5hmC quantified by paired BS and oxbs treated DNA hybridized to Infinium DNA methylation arrays. Workflow depicts analytic
Transduction of lentivirus to human primary CD4+ T cells
 Transduction of lentivirus to human primary CD4 + T cells Human primary CD4 T cells were stimulated with anti-cd3/cd28 antibodies (10 µl/2 5 10^6 cells of Dynabeads CD3/CD28 T cell expander, Invitrogen)
Transduction of lentivirus to human primary CD4 + T cells Human primary CD4 T cells were stimulated with anti-cd3/cd28 antibodies (10 µl/2 5 10^6 cells of Dynabeads CD3/CD28 T cell expander, Invitrogen)
Supplemental Figure 1. Genes showing ectopic H3K9 dimethylation in this study are DNA hypermethylated in Lister et al. study.
 mc mc mc mc SUP mc mc Supplemental Figure. Genes showing ectopic HK9 dimethylation in this study are DNA hypermethylated in Lister et al. study. Representative views of genes that gain HK9m marks in their
mc mc mc mc SUP mc mc Supplemental Figure. Genes showing ectopic HK9 dimethylation in this study are DNA hypermethylated in Lister et al. study. Representative views of genes that gain HK9m marks in their
Nature Immunology: doi: /ni Supplementary Figure 1. RNA-Seq analysis of CD8 + TILs and N-TILs.
 Supplementary Figure 1 RNA-Seq analysis of CD8 + TILs and N-TILs. (a) Schematic representation of the tumor and cell types used for the study. HNSCC, head and neck squamous cell cancer; NSCLC, non-small
Supplementary Figure 1 RNA-Seq analysis of CD8 + TILs and N-TILs. (a) Schematic representation of the tumor and cell types used for the study. HNSCC, head and neck squamous cell cancer; NSCLC, non-small
Supplemental Figure 1. Signature gene expression in in vitro differentiated Th0, Th1, Th2, Th17 and Treg cells. (A) Naïve CD4 + T cells were cultured
 Naïve CD4 + T cells were cultured") Supplemental Figure 1. Signature gene expression in in vitro differentiated Th0, Th1, Th2, Th17 and Treg cells. (A) Naïve CD4 + T cells were cultured under Th0, Th1, Th2, Th17, and Treg conditions. mrna
Supplemental Figure 1. Signature gene expression in in vitro differentiated Th0, Th1, Th2, Th17 and Treg cells. (A) Naïve CD4 + T cells were cultured under Th0, Th1, Th2, Th17, and Treg conditions. mrna
Nature Structural & Molecular Biology: doi: /nsmb Supplementary Figure 1
 Supplementary Figure 1 Frequency of alternative-cassette-exon engagement with the ribosome is consistent across data from multiple human cell types and from mouse stem cells. Box plots showing AS frequency
Supplementary Figure 1 Frequency of alternative-cassette-exon engagement with the ribosome is consistent across data from multiple human cell types and from mouse stem cells. Box plots showing AS frequency
Scott Abrams, Ph.D. Professor of Oncology, x4375 Kuby Immunology SEVENTH EDITION
 Scott Abrams, Ph.D. Professor of Oncology, x4375 scott.abrams@roswellpark.org Kuby Immunology SEVENTH EDITION CHAPTER 11 T-Cell Activation, Differentiation, and Memory Copyright 2013 by W. H. Freeman and
Scott Abrams, Ph.D. Professor of Oncology, x4375 scott.abrams@roswellpark.org Kuby Immunology SEVENTH EDITION CHAPTER 11 T-Cell Activation, Differentiation, and Memory Copyright 2013 by W. H. Freeman and
Patterns of Histone Methylation and Chromatin Organization in Grapevine Leaf. Rachel Schwope EPIGEN May 24-27, 2016
 Patterns of Histone Methylation and Chromatin Organization in Grapevine Leaf Rachel Schwope EPIGEN May 24-27, 2016 What does H3K4 methylation do? Plant of interest: Vitis vinifera Culturally important
Patterns of Histone Methylation and Chromatin Organization in Grapevine Leaf Rachel Schwope EPIGEN May 24-27, 2016 What does H3K4 methylation do? Plant of interest: Vitis vinifera Culturally important
Analysis of the peroxisome proliferator-activated receptor-β/δ (PPARβ/δ) cistrome reveals novel co-regulatory role of ATF4
 cistrome reveals novel co-regulatory role of ATF4") Khozoie et al. BMC Genomics 2012, 13:665 RESEARCH ARTICLE Open Access Analysis of the peroxisome proliferator-activated receptor-β/δ (PPARβ/δ) cistrome reveals novel co-regulatory role of ATF4 Combiz Khozoie
Khozoie et al. BMC Genomics 2012, 13:665 RESEARCH ARTICLE Open Access Analysis of the peroxisome proliferator-activated receptor-β/δ (PPARβ/δ) cistrome reveals novel co-regulatory role of ATF4 Combiz Khozoie
Supplementary Figure 1. Immune profiles of untreated and PD-1 blockade resistant EGFR and Kras mouse lung tumors (a) Total lung weight of untreated
 Total lung weight of untreated") 1 Supplementary Figure 1. Immune profiles of untreated and PD-1 blockade resistant EGFR and Kras mouse lung tumors (a) Total lung weight of untreated (U) EGFR TL mice (n=7), Kras mice (n=7), PD-1 blockade
1 Supplementary Figure 1. Immune profiles of untreated and PD-1 blockade resistant EGFR and Kras mouse lung tumors (a) Total lung weight of untreated (U) EGFR TL mice (n=7), Kras mice (n=7), PD-1 blockade
T Lymphocyte Activation and Costimulation. FOCiS. Lecture outline
 1 T Lymphocyte Activation and Costimulation Abul K. Abbas, MD UCSF FOCiS 2 Lecture outline T cell activation Costimulation, the B7:CD28 family Inhibitory receptors of T cells Targeting costimulators for
1 T Lymphocyte Activation and Costimulation Abul K. Abbas, MD UCSF FOCiS 2 Lecture outline T cell activation Costimulation, the B7:CD28 family Inhibitory receptors of T cells Targeting costimulators for
Supplemental Information. Checkpoint Blockade Immunotherapy. Induces Dynamic Changes. in PD-1 CD8 + Tumor-Infiltrating T Cells
 Immunity, Volume 50 Supplemental Information Checkpoint Blockade Immunotherapy Induces Dynamic Changes in PD-1 CD8 + Tumor-Infiltrating T Cells Sema Kurtulus, Asaf Madi, Giulia Escobar, Max Klapholz, Jackson
Immunity, Volume 50 Supplemental Information Checkpoint Blockade Immunotherapy Induces Dynamic Changes in PD-1 CD8 + Tumor-Infiltrating T Cells Sema Kurtulus, Asaf Madi, Giulia Escobar, Max Klapholz, Jackson
Chapter 7 Conclusions
 VII-1 Chapter 7 Conclusions VII-2 The development of cell-based therapies ranging from well-established practices such as bone marrow transplant to next-generation strategies such as adoptive T-cell therapy
VII-1 Chapter 7 Conclusions VII-2 The development of cell-based therapies ranging from well-established practices such as bone marrow transplant to next-generation strategies such as adoptive T-cell therapy
T-cell activation T cells migrate to secondary lymphoid tissues where they interact with antigen, antigen-presenting cells, and other lymphocytes:
 Interactions between innate immunity & adaptive immunity What happens to T cells after they leave the thymus? Naïve T cells exit the thymus and enter the bloodstream. If they remain in the bloodstream,
Interactions between innate immunity & adaptive immunity What happens to T cells after they leave the thymus? Naïve T cells exit the thymus and enter the bloodstream. If they remain in the bloodstream,
T-cell activation T cells migrate to secondary lymphoid tissues where they interact with antigen, antigen-presenting cells, and other lymphocytes:
 Interactions between innate immunity & adaptive immunity What happens to T cells after they leave the thymus? Naïve T cells exit the thymus and enter the bloodstream. If they remain in the bloodstream,
Interactions between innate immunity & adaptive immunity What happens to T cells after they leave the thymus? Naïve T cells exit the thymus and enter the bloodstream. If they remain in the bloodstream,
ChIP-seq data analysis
 ChIP-seq data analysis Harri Lähdesmäki Department of Computer Science Aalto University November 24, 2017 Contents Background ChIP-seq protocol ChIP-seq data analysis Transcriptional regulation Transcriptional
ChIP-seq data analysis Harri Lähdesmäki Department of Computer Science Aalto University November 24, 2017 Contents Background ChIP-seq protocol ChIP-seq data analysis Transcriptional regulation Transcriptional
SUPPLEMENTARY INFORMATION
 doi: 1.138/nature89 IFN- (ng ml ) 5 4 3 1 Splenocytes NS IFN- (ng ml ) 6 4 Lymph node cells NS Nfkbiz / Nfkbiz / Nfkbiz / Nfkbiz / IL- (ng ml ) 3 1 Splenocytes IL- (ng ml ) 1 8 6 4 *** ** Lymph node cells
doi: 1.138/nature89 IFN- (ng ml ) 5 4 3 1 Splenocytes NS IFN- (ng ml ) 6 4 Lymph node cells NS Nfkbiz / Nfkbiz / Nfkbiz / Nfkbiz / IL- (ng ml ) 3 1 Splenocytes IL- (ng ml ) 1 8 6 4 *** ** Lymph node cells
Immunology Basics Relevant to Cancer Immunotherapy: T Cell Activation, Costimulation, and Effector T Cells
 Immunology Basics Relevant to Cancer Immunotherapy: T Cell Activation, Costimulation, and Effector T Cells Andrew H. Lichtman, M.D. Ph.D. Department of Pathology Brigham and Women s Hospital and Harvard
Immunology Basics Relevant to Cancer Immunotherapy: T Cell Activation, Costimulation, and Effector T Cells Andrew H. Lichtman, M.D. Ph.D. Department of Pathology Brigham and Women s Hospital and Harvard
MHC class I MHC class II Structure of MHC antigens:
 MHC class I MHC class II Structure of MHC antigens: MHC class I antigens consist of a transmembrane heavy chain (α chain) that is non-covalently associated with β2- microglobulin. Membrane proximal domain
MHC class I MHC class II Structure of MHC antigens: MHC class I antigens consist of a transmembrane heavy chain (α chain) that is non-covalently associated with β2- microglobulin. Membrane proximal domain
7.012 Quiz 3 Answers
 MIT Biology Department 7.012: Introductory Biology - Fall 2004 Instructors: Professor Eric Lander, Professor Robert A. Weinberg, Dr. Claudette Gardel Friday 11/12/04 7.012 Quiz 3 Answers A > 85 B 72-84
MIT Biology Department 7.012: Introductory Biology - Fall 2004 Instructors: Professor Eric Lander, Professor Robert A. Weinberg, Dr. Claudette Gardel Friday 11/12/04 7.012 Quiz 3 Answers A > 85 B 72-84
Comprehensive nucleosome mapping of the human genome in cancer progression
 /, Vol. 7, No. 12 Comprehensive nucleosome mapping of the human genome in cancer progression Brooke R. Druliner 1,5, Daniel Vera 1,6, Ruth Johnson 2, Xiaoyang Ruan 3, Lynn M. Apone 4, Eileen T. Dimalanta
/, Vol. 7, No. 12 Comprehensive nucleosome mapping of the human genome in cancer progression Brooke R. Druliner 1,5, Daniel Vera 1,6, Ruth Johnson 2, Xiaoyang Ruan 3, Lynn M. Apone 4, Eileen T. Dimalanta
The Epigenome Tools 2: ChIP-Seq and Data Analysis
 The Epigenome Tools 2: ChIP-Seq and Data Analysis Chongzhi Zang zang@virginia.edu http://zanglab.com PHS5705: Public Health Genomics March 20, 2017 1 Outline Epigenome: basics review ChIP-seq overview
The Epigenome Tools 2: ChIP-Seq and Data Analysis Chongzhi Zang zang@virginia.edu http://zanglab.com PHS5705: Public Health Genomics March 20, 2017 1 Outline Epigenome: basics review ChIP-seq overview
Supplementary Information. Supplementary Figure 1
 Supplementary Information Supplementary Figure 1 1 Supplementary Figure 1. Functional assay of the hcas9-2a-mcherry construct (a) Gene correction of a mutant EGFP reporter cell line mediated by hcas9 or
Supplementary Information Supplementary Figure 1 1 Supplementary Figure 1. Functional assay of the hcas9-2a-mcherry construct (a) Gene correction of a mutant EGFP reporter cell line mediated by hcas9 or
CTCF-Mediated Functional Chromatin Interactome in Pluripotent Cells
 SUPPLEMENTARY INFORMATION CTCF-Mediated Functional Chromatin Interactome in Pluripotent Cells Lusy Handoko 1,*, Han Xu 1,*, Guoliang Li 1,*, Chew Yee Ngan 1, Elaine Chew 1, Marie Schnapp 1, Charlie Wah
SUPPLEMENTARY INFORMATION CTCF-Mediated Functional Chromatin Interactome in Pluripotent Cells Lusy Handoko 1,*, Han Xu 1,*, Guoliang Li 1,*, Chew Yee Ngan 1, Elaine Chew 1, Marie Schnapp 1, Charlie Wah
The Insulator Binding Protein CTCF Positions 20 Nucleosomes around Its Binding Sites across the Human Genome
 The Insulator Binding Protein CTCF Positions 20 Nucleosomes around Its Binding Sites across the Human Genome Yutao Fu 1, Manisha Sinha 2,3, Craig L. Peterson 3, Zhiping Weng 1,4,5 * 1 Bioinformatics Program,
The Insulator Binding Protein CTCF Positions 20 Nucleosomes around Its Binding Sites across the Human Genome Yutao Fu 1, Manisha Sinha 2,3, Craig L. Peterson 3, Zhiping Weng 1,4,5 * 1 Bioinformatics Program,
Supplementary Figure 1: Attenuation of association signals after conditioning for the lead SNP. a) attenuation of association signal at the 9p22.
 attenuation of association signal at the 9p22.") Supplementary Figure 1: Attenuation of association signals after conditioning for the lead SNP. a) attenuation of association signal at the 9p22.32 PCOS locus after conditioning for the lead SNP rs10993397;
Supplementary Figure 1: Attenuation of association signals after conditioning for the lead SNP. a) attenuation of association signal at the 9p22.32 PCOS locus after conditioning for the lead SNP rs10993397;
User Guide. Association analysis. Input
 User Guide TFEA.ChIP is a tool to estimate transcription factor enrichment in a set of differentially expressed genes using data from ChIP-Seq experiments performed in different tissues and conditions.
User Guide TFEA.ChIP is a tool to estimate transcription factor enrichment in a set of differentially expressed genes using data from ChIP-Seq experiments performed in different tissues and conditions.
Breast cancer. Risk factors you cannot change include: Treatment Plan Selection. Inferring Transcriptional Module from Breast Cancer Profile Data
 Breast cancer Inferring Transcriptional Module from Breast Cancer Profile Data Breast Cancer and Targeted Therapy Microarray Profile Data Inferring Transcriptional Module Methods CSC 177 Data Warehousing
Breast cancer Inferring Transcriptional Module from Breast Cancer Profile Data Breast Cancer and Targeted Therapy Microarray Profile Data Inferring Transcriptional Module Methods CSC 177 Data Warehousing
Supplemental Figure S1. Expression of Cirbp mrna in mouse tissues and NIH3T3 cells.
 SUPPLEMENTAL FIGURE AND TABLE LEGENDS Supplemental Figure S1. Expression of Cirbp mrna in mouse tissues and NIH3T3 cells. A) Cirbp mrna expression levels in various mouse tissues collected around the clock
SUPPLEMENTAL FIGURE AND TABLE LEGENDS Supplemental Figure S1. Expression of Cirbp mrna in mouse tissues and NIH3T3 cells. A) Cirbp mrna expression levels in various mouse tissues collected around the clock
Discovery of Novel Human Gene Regulatory Modules from Gene Co-expression and
 Discovery of Novel Human Gene Regulatory Modules from Gene Co-expression and Promoter Motif Analysis Shisong Ma 1,2*, Michael Snyder 3, and Savithramma P Dinesh-Kumar 2* 1 School of Life Sciences, University
Discovery of Novel Human Gene Regulatory Modules from Gene Co-expression and Promoter Motif Analysis Shisong Ma 1,2*, Michael Snyder 3, and Savithramma P Dinesh-Kumar 2* 1 School of Life Sciences, University
Immune Checkpoints. PD Dr med. Alessandra Curioni-Fontecedro Department of Hematology and Oncology Cancer Center Zurich University Hospital Zurich
 Immune Checkpoints PD Dr med. Alessandra Curioni-Fontecedro Department of Hematology and Oncology Cancer Center Zurich University Hospital Zurich Activation of T cells requires co-stimulation Science 3
Immune Checkpoints PD Dr med. Alessandra Curioni-Fontecedro Department of Hematology and Oncology Cancer Center Zurich University Hospital Zurich Activation of T cells requires co-stimulation Science 3
Where a licence is displayed above, please note the terms and conditions of the licence govern your use of this document.
 Inducible chromatin priming is associated with the establishment of immunological memory in T cells Bevington, Sarah; Cauchy, Pierre; Bonifer, Constanze; Ott, S; Cockerill, Peter DOI:.55/embj.59534 License:
Inducible chromatin priming is associated with the establishment of immunological memory in T cells Bevington, Sarah; Cauchy, Pierre; Bonifer, Constanze; Ott, S; Cockerill, Peter DOI:.55/embj.59534 License:
Tumor Immunity and Immunotherapy. Andrew Lichtman M.D., Ph.D. Brigham and Women s Hospital Harvard Medical School
 Tumor Immunity and Immunotherapy Andrew Lichtman M.D., Ph.D. Brigham and Women s Hospital Harvard Medical School Lecture Outline Evidence for tumor immunity Types of tumor antigens Generation of anti-tumor
Tumor Immunity and Immunotherapy Andrew Lichtman M.D., Ph.D. Brigham and Women s Hospital Harvard Medical School Lecture Outline Evidence for tumor immunity Types of tumor antigens Generation of anti-tumor
H3K4 demethylase KDM5B regulates global dynamics of transcription elongation and alternative splicing in embryonic stem cells
 Nucleic Acids Research, 2017 1 doi: 10.1093/nar/gkx251 H3K4 demethylase KDM5B regulates global dynamics of transcription elongation and alternative splicing in embryonic stem cells Runsheng He 1,2 and
Nucleic Acids Research, 2017 1 doi: 10.1093/nar/gkx251 H3K4 demethylase KDM5B regulates global dynamics of transcription elongation and alternative splicing in embryonic stem cells Runsheng He 1,2 and
Page 32 AP Biology: 2013 Exam Review CONCEPT 6 REGULATION
 Page 32 AP Biology: 2013 Exam Review CONCEPT 6 REGULATION 1. Feedback a. Negative feedback mechanisms maintain dynamic homeostasis for a particular condition (variable) by regulating physiological processes,
Page 32 AP Biology: 2013 Exam Review CONCEPT 6 REGULATION 1. Feedback a. Negative feedback mechanisms maintain dynamic homeostasis for a particular condition (variable) by regulating physiological processes,
Lecture 11. Immunology and disease: parasite antigenic diversity
 Lecture 11 Immunology and disease: parasite antigenic diversity RNAi interference video and tutorial (you are responsible for this material, so check it out.) http://www.pbs.org/wgbh/nova/sciencenow/3210/02.html
Lecture 11 Immunology and disease: parasite antigenic diversity RNAi interference video and tutorial (you are responsible for this material, so check it out.) http://www.pbs.org/wgbh/nova/sciencenow/3210/02.html
Polycomb Repressive Complex 2-Mediated Chromatin Repression Guides Effector CD8 + T Cell Terminal Differentiation and Loss of Multipotency
 Article Polycomb Repressive Complex 2-Mediated Chromatin Repression Guides Effector CD8 + T Cell Terminal Differentiation and Loss of Multipotency Graphical Abstract Authors Simon M. Gray, Robert A. Amezquita,
Article Polycomb Repressive Complex 2-Mediated Chromatin Repression Guides Effector CD8 + T Cell Terminal Differentiation and Loss of Multipotency Graphical Abstract Authors Simon M. Gray, Robert A. Amezquita,
Lecture outline. Immunological tolerance and immune regulation. Central and peripheral tolerance. Inhibitory receptors of T cells. Regulatory T cells
 1 Immunological tolerance and immune regulation Abul K. Abbas UCSF 2 Lecture outline Central and peripheral tolerance Inhibitory receptors of T cells Regulatory T cells 1 The immunological equilibrium:
1 Immunological tolerance and immune regulation Abul K. Abbas UCSF 2 Lecture outline Central and peripheral tolerance Inhibitory receptors of T cells Regulatory T cells 1 The immunological equilibrium:
SUPPLEMENTAL INFORMATION
 SUPPLEMENTAL INFORMATION GO term analysis of differentially methylated SUMIs. GO term analysis of the 458 SUMIs with the largest differential methylation between human and chimp shows that they are more
SUPPLEMENTAL INFORMATION GO term analysis of differentially methylated SUMIs. GO term analysis of the 458 SUMIs with the largest differential methylation between human and chimp shows that they are more
Supplementary figure 1
 Supplementary figure 1 Nature Medicine: doi:1.138/nm.275 CLUSTER BY SELF-ORGANIZING MAPS SELECTED PATHWAY ANALISYS TERMS Cluster : up-regulated genes in acute patients Cell cycle/dna repair Fatty acid
Supplementary figure 1 Nature Medicine: doi:1.138/nm.275 CLUSTER BY SELF-ORGANIZING MAPS SELECTED PATHWAY ANALISYS TERMS Cluster : up-regulated genes in acute patients Cell cycle/dna repair Fatty acid
Results. Abstract. Introduc4on. Conclusions. Methods. Funding
 . expression that plays a role in many cellular processes affecting a variety of traits. In this study DNA methylation was assessed in neuronal tissue from three pigs (frontal lobe) and one great tit (whole
. expression that plays a role in many cellular processes affecting a variety of traits. In this study DNA methylation was assessed in neuronal tissue from three pigs (frontal lobe) and one great tit (whole
Supplementary information for: Community detection for networks with. unipartite and bipartite structure. Chang Chang 1, 2, Chao Tang 2
 Supplementary information for: Community detection for networks with unipartite and bipartite structure Chang Chang 1, 2, Chao Tang 2 1 School of Life Sciences, Peking University, Beiing 100871, China
Supplementary information for: Community detection for networks with unipartite and bipartite structure Chang Chang 1, 2, Chao Tang 2 1 School of Life Sciences, Peking University, Beiing 100871, China
Mapping the Antigenic and Genetic Evolution of Influenza Virus
 Mapping the Antigenic and Genetic Evolution of Influenza Virus Derek J. Smith, Alan S. Lapedes, Jan C. de Jong, Theo M. Bestebroer, Guus F. Rimmelzwaan, Albert D. M. E. Osterhaus, Ron A. M. Fouchier Science
Mapping the Antigenic and Genetic Evolution of Influenza Virus Derek J. Smith, Alan S. Lapedes, Jan C. de Jong, Theo M. Bestebroer, Guus F. Rimmelzwaan, Albert D. M. E. Osterhaus, Ron A. M. Fouchier Science
Supplemental Figure S1. Tertiles of FKBP5 promoter methylation and internal regulatory region
 Supplemental Figure S1. Tertiles of FKBP5 promoter methylation and internal regulatory region methylation in relation to PSS and fetal coupling. A, PSS values for participants whose placentas showed low,
Supplemental Figure S1. Tertiles of FKBP5 promoter methylation and internal regulatory region methylation in relation to PSS and fetal coupling. A, PSS values for participants whose placentas showed low,
Supplementary Fig. 1: Ex vivo tetramer enrichment with anti-c-myc beads
 Supplementary Fig. 1: Ex vivo tetramer enrichment with anti-c-myc beads Representative example of comparative ex vivo tetramer enrichment performed in three independent experiments with either conventional
Supplementary Fig. 1: Ex vivo tetramer enrichment with anti-c-myc beads Representative example of comparative ex vivo tetramer enrichment performed in three independent experiments with either conventional
T Cell Activation. Patricia Fitzgerald-Bocarsly March 18, 2009
 T Cell Activation Patricia Fitzgerald-Bocarsly March 18, 2009 Phases of Adaptive Immune Responses Phases of T cell responses IL-2 acts as an autocrine growth factor Fig. 11-11 Clonal Expansion of T cells
T Cell Activation Patricia Fitzgerald-Bocarsly March 18, 2009 Phases of Adaptive Immune Responses Phases of T cell responses IL-2 acts as an autocrine growth factor Fig. 11-11 Clonal Expansion of T cells
Nature Medicine: doi: /nm.2109
 HIV 1 Infects Multipotent Progenitor Cells Causing Cell Death and Establishing Latent Cellular Reservoirs Christoph C. Carter, Adewunmi Onafuwa Nuga, Lucy A. M c Namara, James Riddell IV, Dale Bixby, Michael
HIV 1 Infects Multipotent Progenitor Cells Causing Cell Death and Establishing Latent Cellular Reservoirs Christoph C. Carter, Adewunmi Onafuwa Nuga, Lucy A. M c Namara, James Riddell IV, Dale Bixby, Michael
During acute infections, antigen-specific CD8 T cells are activated
 Progressive Loss of Memory T Cell Potential and Commitment to Exhaustion during Chronic Viral Infection Jill M. Angelosanto, Shawn D. Blackburn, Alison Crawford,* and E. John Wherry Department of Microbiology
Progressive Loss of Memory T Cell Potential and Commitment to Exhaustion during Chronic Viral Infection Jill M. Angelosanto, Shawn D. Blackburn, Alison Crawford,* and E. John Wherry Department of Microbiology
Supplemental Information. Genomic Characterization of Murine. Monocytes Reveals C/EBPb Transcription. Factor Dependence of Ly6C Cells
 Immunity, Volume 46 Supplemental Information Genomic Characterization of Murine Monocytes Reveals C/EBPb Transcription Factor Dependence of Ly6C Cells Alexander Mildner, Jörg Schönheit, Amir Giladi, Eyal
Immunity, Volume 46 Supplemental Information Genomic Characterization of Murine Monocytes Reveals C/EBPb Transcription Factor Dependence of Ly6C Cells Alexander Mildner, Jörg Schönheit, Amir Giladi, Eyal
Raymond Auerbach PhD Candidate, Yale University Gerstein and Snyder Labs August 30, 2012
 Elucidating Transcriptional Regulation at Multiple Scales Using High-Throughput Sequencing, Data Integration, and Computational Methods Raymond Auerbach PhD Candidate, Yale University Gerstein and Snyder
Elucidating Transcriptional Regulation at Multiple Scales Using High-Throughput Sequencing, Data Integration, and Computational Methods Raymond Auerbach PhD Candidate, Yale University Gerstein and Snyder
Lecture 6. Burr BIO 4353/6345 HIV/AIDS. Tetramer staining of T cells (CTL s) Andrew McMichael seminar: Background
 Andrew McMichael seminar: Background") Lecture 6 Burr BIO 4353/6345 HIV/AIDS Andrew McMichael seminar: Background Tetramer staining of T cells (CTL s) 1. Vβ 19: There are 52 T cell receptor (TCR) Vβ gene segments in germ line DNA (See following
Lecture 6 Burr BIO 4353/6345 HIV/AIDS Andrew McMichael seminar: Background Tetramer staining of T cells (CTL s) 1. Vβ 19: There are 52 T cell receptor (TCR) Vβ gene segments in germ line DNA (See following
Autoimmunity. Autoimmunity arises because of defects in central or peripheral tolerance of lymphocytes to selfantigens
 Autoimmunity Autoimmunity arises because of defects in central or peripheral tolerance of lymphocytes to selfantigens Autoimmune disease can be caused to primary defects in B cells, T cells and possibly
Autoimmunity Autoimmunity arises because of defects in central or peripheral tolerance of lymphocytes to selfantigens Autoimmune disease can be caused to primary defects in B cells, T cells and possibly
SUPPLEMENTAL INFORMATIONS
 1 SUPPLEMENTAL INFORMATIONS Figure S1 Cumulative ZIKV production by testis explants over a 9 day-culture period. Viral titer values presented in Figure 1B (viral release over a 3 day-culture period measured
1 SUPPLEMENTAL INFORMATIONS Figure S1 Cumulative ZIKV production by testis explants over a 9 day-culture period. Viral titer values presented in Figure 1B (viral release over a 3 day-culture period measured
Molecular profiling of cytomegalovirus-induced human CD8 + T cell differentiation
 Molecular profiling of cytomegalovirus-induced human CD8 + T cell differentiation Kirsten M.L. Hertoghs,, Ineke J.M. ten Berge, René A.W. van Lier J Clin Invest. 2010;120(11):4077-4090. https://doi.org/10.1172/jci42758.
Molecular profiling of cytomegalovirus-induced human CD8 + T cell differentiation Kirsten M.L. Hertoghs,, Ineke J.M. ten Berge, René A.W. van Lier J Clin Invest. 2010;120(11):4077-4090. https://doi.org/10.1172/jci42758.
Supplemental Information. T Cells Enhance Autoimmunity by Restraining Regulatory T Cell Responses via an Interleukin-23-Dependent Mechanism
 Immunity, Volume 33 Supplemental Information T Cells Enhance Autoimmunity by Restraining Regulatory T Cell Responses via an Interleukin-23-Dependent Mechanism Franziska Petermann, Veit Rothhammer, Malte
Immunity, Volume 33 Supplemental Information T Cells Enhance Autoimmunity by Restraining Regulatory T Cell Responses via an Interleukin-23-Dependent Mechanism Franziska Petermann, Veit Rothhammer, Malte
Nature Immunology: doi: /ni Supplementary Figure 1. Id2 and Id3 define polyclonal T H 1 and T FH cell subsets.
 Supplementary Figure 1 Id2 and Id3 define polyclonal T H 1 and T FH cell subsets. Id2 YFP/+ (a) or Id3 GFP/+ (b) mice were analyzed 7 days after LCMV infection. T H 1 (SLAM + CXCR5 or CXCR5 PD-1 ), T FH
Supplementary Figure 1 Id2 and Id3 define polyclonal T H 1 and T FH cell subsets. Id2 YFP/+ (a) or Id3 GFP/+ (b) mice were analyzed 7 days after LCMV infection. T H 1 (SLAM + CXCR5 or CXCR5 PD-1 ), T FH
Fluorochrome Panel 1 Panel 2 Panel 3 Panel 4 Panel 5 CTLA-4 CTLA-4 CD15 CD3 FITC. Bio) PD-1 (MIH4, BD) ICOS (C398.4A, Biolegend) PD-L1 (MIH1, BD)
 PD-1 (MIH4, BD) ICOS (C398.4A, Biolegend) PD-L1 (MIH1, BD)") Additional file : Table S. Antibodies used for panel stain to identify peripheral immune cell subsets. Panel : PD- signaling; Panel : CD + T cells, CD + T cells, B cells; Panel : Tregs; Panel :, -T, cdc,
Additional file : Table S. Antibodies used for panel stain to identify peripheral immune cell subsets. Panel : PD- signaling; Panel : CD + T cells, CD + T cells, B cells; Panel : Tregs; Panel :, -T, cdc,
T cell memory Friday, February 10, 17
 T cell memory 2-10-2017 expansion contraction memory recall EFFECTORS 10 6 pathogen load secondary challenge MEMORY 10 2 NAIVE 2 7-8 30 >1 year days post-infection In mouse, ~100 naïve, CD8s become ~3x10
T cell memory 2-10-2017 expansion contraction memory recall EFFECTORS 10 6 pathogen load secondary challenge MEMORY 10 2 NAIVE 2 7-8 30 >1 year days post-infection In mouse, ~100 naïve, CD8s become ~3x10
Variant Classification. Author: Mike Thiesen, Golden Helix, Inc.
 Variant Classification Author: Mike Thiesen, Golden Helix, Inc. Overview Sequencing pipelines are able to identify rare variants not found in catalogs such as dbsnp. As a result, variants in these datasets
Variant Classification Author: Mike Thiesen, Golden Helix, Inc. Overview Sequencing pipelines are able to identify rare variants not found in catalogs such as dbsnp. As a result, variants in these datasets