Next Generation Sequencing as a tool for breakpoint analysis in rearrangements of the globin-gene clusters
|
|
|
- Buck Damon Lane
- 6 years ago
- Views:
Transcription





1 Next Generation Sequencing as a tool for breakpoint analysis in rearrangements of the globin-gene clusters XXXth International Symposium on Technical Innovations in Laboratory Hematology Honolulu, Hawaii - Hawaii Convention Center May 4-6, 2017 Barnaby E. Clark 1,2, Claire Shooter 2, Frances Smith 1, David Brawand 1 and Swee Lay Thein 2,* 1 Viapath at King s College Hospital NHS Foundation Trust, London, UK 2 King s College London, Faculty of Life Sciences and Medicine, London, UK * Present Address: NHLBI / NIH, Sickle Cell Branch, Bethesda, USA

2 Disclosures No Relevant Financial Relationships with Commercial Interests
3 Objectives What do we need to detect for hemoglobinopathies Genotypic and phenotypic diversity of hemoglobin disorders Overview of sequential process followed historically for DNA diagnostics of the hemoglobin disorders What is Next Generation sequencing creating and analysing the data Evaluation of next generation sequencing (NGS) as a comprehensive single methodology in Hemoglobin DNA diagnostics Case examples
4 Hemoglobinopathy Cases Requiring Molecular Diagnosis Ø Antenatal assessment: Part of UK national screening programme Ø Prenatal Diagnosis: Part of UK national screening programme Ø Newborn screening: Part of UK national screening programme Ø Specialist workup of individual cases Unusual phenotype detected Unusual presentation Phenotype more/ less severe than expected

5 Genomic structure of the clusters of α-like and β-like globin genes on chromosomes 16 and by American Society of Hematology Schechter A N Blood 2008;;112:
6 Mutations downregulating beta globin gene Point mutations 5 b - LCR e G g A g g y b d b e G g d b 3' -30kb -20kb -10kb 0 10kb 20k 30kb 40kb 50kb 60kb 70kb 80kb Deletions restricted to b gene Large deletions involving b LCR with and without b gene Trans-acting mutations identified in: GATA-1 TFIIH

7 α + thalassemia caused by deletions of one gene Harteveld CL & Higgs DR Orphanet J of Rare Dis 2012;; 5: 13

8 α 0 thalassemia caused by deletions of both a genes Harteveld CL & Higgs DR Orphanet J of Rare Dis 2012;; 5: 13



9 Genetic Testing for Hemoglobinopathies Clinical features Family history FBC Hemoglobin separation, HbA2, HbF Phenotype Osmotic fragility EMA labelling Sequence HBB gene If negative, Gap-PCR HBA cluster, Sequence HBA genes If negative Diagnosis: MLPA HBB, HBA cluster Genotype / phenotype consistent CGH array Or Found Mutation but not consistent with Phenotype Or No Genetic cause identified Genotype

10 Library preparation






11 Bait Capture Agilent SureSelect DNA fragmentation Library preparation Genomic DNA end repair and A overhang ultrasonic shearing adaptor ligation Fragmented DNA ~400bp Sequencing Capture Target library 300 bp Paired end Sequencing low cycle PCR amplification untargeted regions 16 hour hybridisation biotinylated RNA oligos

12 Illumina method
13 Illumina Sequencing 1 read from 1 cluster Each cluster derived from 1 molecule Reads alleles separately Indexing identifies origin Massive multiplexing Targets Samples

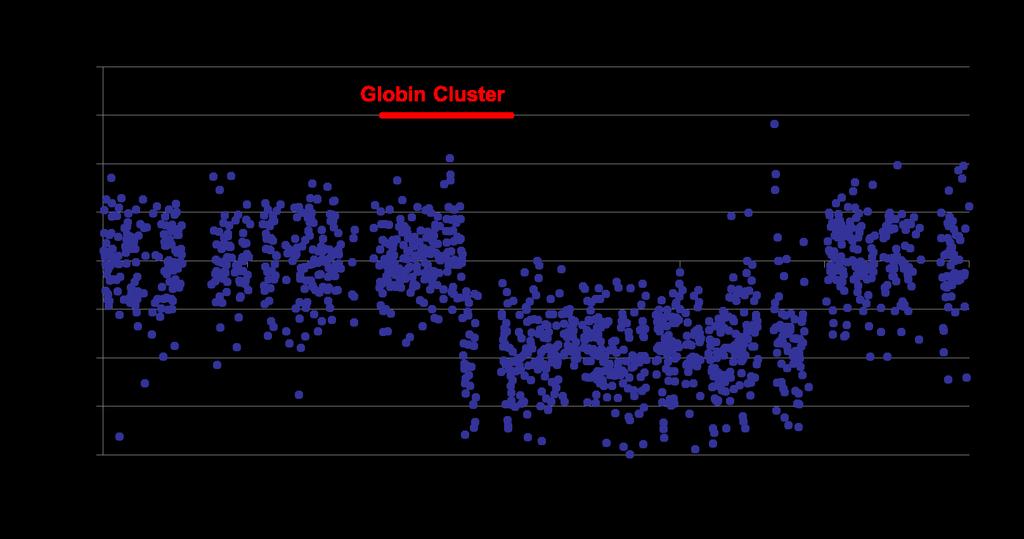
14 Target region Baits designed HBB Globin cluster
15 MiSeq Sequencer M reads/ sample DNA fragment Read 1 Read 2
16 Reference sequence: AGCTGCTCTAGATAGCTCGATAAAAGCTCCGATATAGTGCATCAGCCAGCGCGCGCAGATAGAAAGAGC GCTGCTCTAGATAGCTCGATAAA SNP Statistics G = 5 T = 5 50:50 Heterozygous Deletion = all homozygous Duplication = 66:33 Coverage = 10x

 RPKM Coverage in Control (normalised")
17 Normal control Patient sample Normalisation of coverage within sample by Reads per kb RPKM Then compare patient samples to diploid controls Relative coverage = Coverage in Patient (normalised to total reads) RPKM Coverage in Control (normalised to total reads) RPKM
18 Variation in RPKM values across bait tiled region on chromosome 16 Log2 deviation from control average Bait performance variability Mutation CNV event Variation due to insertions Majority of assay variation Variation due to deletions Chromosomal position
19 Case Example 1
20 PROBAND - 39 year old woman (English Anglo-Saxon) Hypochromic microcytic anemia since infancy, unresponsive to iron supplements Individual Age Hb RBC MCV MCH HbA 2 HbF β/α (gm/dl (x /L) (fl) (μg) (%) (%) Proband 14 yrs yrs < Normal HbA 2 thalassemic RBC indices α thalassemia variant excluded - Globin chain synthesis consistent with phenotype of εγδβ thalassemia Southern blotting, MLPA, qpcr and CGH array confirmed deletion from HBG2 exon 2 to β- LCR BUT not possible to characterise breakpoints
21 PROBAND - 39 year old woman (English Anglo-Saxon) Hypochromic microcytic anemia since infancy, unresponsive to iron supplements Individual Age Hb RBC MCV MCH HbA 2 HbF β/α (gm/dl (x /L) (fl) (μg) (%) (%) Proband 14 yrs yrs < Daughter 3½ yrs Mother 56 yrs Case 2 52 yrs Case 3 24 yrs Daughter of proband received two intra-uterine blood transfusions followed by another blood transfusion at birth when she developed neonatal jaundice and anemia
22 RPKM plot across chromosome 11 in patient Coverage difference: control/patient (Log2 Scale) β δ Ψ γ 1 γ 2 Ɛ LCR Position on Chromosome 11 RPKM Normal variation Deletion
23 Problem Coverage data indicated a deletion Data was consistent with MLPA and CGH array SNP data showed homozygous only SNPs in the deletion and heterozygous SNPs in the region outside of the deletion BUT Can not amplify across the breakpoint with primers to confirm the deletion
24 Alignment information can be used to identify deletions or translocations Fragment size Expected Reference Sequence Opposite direction reads Indicates a deletion? These opposite direction reads did not align in our patient sample near the predicted breakpoints

25 Alignment information can be used to identify inversions Fragment size Reference Sequence Same direction reads? Inversion







26
27 A P1 LTR LTR LINE 1 P2 OR51V1 HBB 5,250,000 HBD HBP1 HBG1 P3 HBG2 HBE LCR 5,300,000 LINE 2 5,400,000 P4 OR51M1 Deletion 5,215, kb Inversion 5,274, ,511 bp 5,397,195 B LTR HBG2 HBG1 HBP1 HBD Inverted sequence HBB OR51V1 LINE 1 LINE 2 OR51M1 P1 P3 P2 P4 1031bp 4499bp Note: not drawn to scale
28 1kb plus ladder A 1kb plus ladder B 12,000 12,000 2,000 1,650 4,000 3,000 1,000 2,000 1, Inversion Gap PCR P1 / P bp 2) Blank 3) Normal Control 1 4) Normal Control 2 5) Case 2 6) Case 3 7) Proband, Case 1 8) Daughter of Proband in Case 1 Deletion Gap PCR P2 /P bp 2) Proband, Case 1 3) Normal Control 4) Daughter of Proband in Case 1 5) Case 2 6) Case 3 7) Blank

29 I1 Family 2 I2 I1 3 Families : Hb 159 g/l Hb 110 g/l 3 unique duplications of the a - globin cluster All β thal carriers who have co- inherited the a - globin MCV MCH 80.1 fl 28.2 pg Hb A2 2.7% Hb F 0.2% HBB sequence normal HBA a a /a a,a a MCV MCH 58.9 fl 19.4 pg Hb A2 4.8% Hb F 0.7% HBB Het c.135delc (Cd 44-C del) HBA a a /a a duplication, have a II1 II2 thalassemia intermedia phenotype Hb MCV 59 g/l 65.6 fl Hb MCV 85 g/l 68.9 fl MCH 19.6 pg MCH 20.7 pg Clark et al, BJH 2016; doi: /bjh Hb A2 3.1% Hb F 13.6% HBB Het c.135delc (Cd 44-C del) HBA a a /a a,a a Hb A2 4.2% Hb F 15.2% HBB Het c.135delc (Cd 44-C del) HBA a a /a a,a a



30 Deviation of RPKM from control average (log2) NGS coverage data Family 2 (proband) Gap PCR Primer 1 Gap PCR Primer 2 Duplication Chromosome 16 Marker size (bp) (L) Sequence at end of balanced region Sequence at start of duplicated region 10, ,000 1,500 1, Ambiguous bases
31 Family 2 I1 I1 I2 Hb 159 g/l Hb 110 g/l MCV 80.1 fl MCV 58.9 fl MCH 28.2 pg MCH 19.4 pg Hb A2 2.7% Hb F 0.2% Hb A2 4.8% Hb F 0.7% HBB sequence normal HBA a a /a a,a a HBB Het c.135delc (Cd 44-C del) HBA a a /a a II1 II2 Hb 59 g/l Hb 85 g/l MCV 65.6 fl MCV 68.9 fl MCH 19.6 pg MCH 20.7 pg Hb A2 3.1% Hb F 13.6% HBB Het c.135delc (Cd 44-C del) HBA a a /a a,a a Hb A2 4.2% Hb F 15.2% HBB Het c.135delc (Cd 44-C del) HBA a a /a a,a a 120,500 bp Duplication WASIR2 MIR6859 POLR3K SNRNP25 RHBDF1 MPG NPRL3 HBZ HBM HBA2 HBA1 HBQ1 LUC7L NPRL3 HBZ HBM HBA2 HBA1 HBQ1 LUC7L FAM234A RGS1 1 ARHGDIG PDIA2 AXIN1

32 Case example 3 : Shooter et al Brit. J. Haemat. 2015;; 70:
33 Summary We have developed a comprehensive single NGS methodology that can fully characterize all types of variants by analysis of a single data set SNPs, deletions, insertions, inversions and rearrangements of any size Exception: the 3.7 kb a -globin deletion cannot be identified accurately using NGS as currently not possible to uniquely map the reads back to the reference sequence Using this NGS analysis pipeline, we have characterized a novel rearrangement of the HBB cluster, responsible for εγδβ thalassemia in an English family, a globin cluster and b globin cluster duplications. We have automated the library preparation to reduce noise in assay and also automated bioinformatics pipeline analyses. With time, it should be possible to apply NGS to routine diagnostics including newborn and antenatal screening
Haemoglobinopathies case studies 11 th Annual Sickle Cell and Thalassaemia Conference October 2017
 Haemoglobinopathies case studies 11 th Annual Sickle Cell and Thalassaemia Conference 11 13 October 2017 Chris Lambert Haematology Service Delivery Manager Viapath Laboratories Kings College Hospital HUMAN
Haemoglobinopathies case studies 11 th Annual Sickle Cell and Thalassaemia Conference 11 13 October 2017 Chris Lambert Haematology Service Delivery Manager Viapath Laboratories Kings College Hospital HUMAN
Diagnostic difficulties in prevention and control program for thalassemia in Thailand: atypical thalassemia carriers
 Diagnostic difficulties in prevention and control program for thalassemia in Thailand: atypical thalassemia carriers Pranee Winichagoon Fucharoen Thalassemia Research Center Institute of Molecular Biosciences
Diagnostic difficulties in prevention and control program for thalassemia in Thailand: atypical thalassemia carriers Pranee Winichagoon Fucharoen Thalassemia Research Center Institute of Molecular Biosciences
New technologies for the diagnosis of thalassemia:
 New technologies for the diagnosis of thalassemia: Cornelis Harteveld Leiden University Medical Center, The Netherlands ENERCA Ferrara 16 th of November 2013 Introduction to hemoglobinopathies Hb molecule
New technologies for the diagnosis of thalassemia: Cornelis Harteveld Leiden University Medical Center, The Netherlands ENERCA Ferrara 16 th of November 2013 Introduction to hemoglobinopathies Hb molecule
Comprehensive Hemoglobin Analysis HBA1/2 (
 Comprehensive Hemoglobin Analysis HBA1/2 ( α-globin) and HBB (β-globin) mutation and deletion/duplication analysis and HBD (δ-globin) and HBG1/2 (γ-globin) mutation analysis Description: Hemoglobin (Hb)
Comprehensive Hemoglobin Analysis HBA1/2 ( α-globin) and HBB (β-globin) mutation and deletion/duplication analysis and HBD (δ-globin) and HBG1/2 (γ-globin) mutation analysis Description: Hemoglobin (Hb)
Thalassemias. Emanuela Veras, M.D. 01/08/2006
 Thalassemias Emanuela Veras, M.D. 01/08/2006 Structure and Function of normal Hemoglobin molecules: 2/3 1/3 β: increases from 6 th week of fetal life to 12 months of age At birth: HbF: 75-90% HbA: 10-25%
Thalassemias Emanuela Veras, M.D. 01/08/2006 Structure and Function of normal Hemoglobin molecules: 2/3 1/3 β: increases from 6 th week of fetal life to 12 months of age At birth: HbF: 75-90% HbA: 10-25%
6.1 Extended family screening
 CHAPTER 6 CONCLUSION Cost benefit analysis of thalassemia screening programs have shown that the single years treatment for a β-thalassemia major patient was much higher than a total cost per case prevented.
CHAPTER 6 CONCLUSION Cost benefit analysis of thalassemia screening programs have shown that the single years treatment for a β-thalassemia major patient was much higher than a total cost per case prevented.
Dr Banu Kaya Consultant Haematologist Barts Health NHS Trust Royal London Hospital, London, UK SICKLE CELL AND THALASSAEMIA OVERVIEW
 Dr Banu Kaya Consultant Haematologist Barts Health NHS Trust Royal London Hospital, London, UK SICKLE CELL AND THALASSAEMIA OVERVIEW Objectives Gain awareness of haemoglobinopathy inheritance, pathophysiology
Dr Banu Kaya Consultant Haematologist Barts Health NHS Trust Royal London Hospital, London, UK SICKLE CELL AND THALASSAEMIA OVERVIEW Objectives Gain awareness of haemoglobinopathy inheritance, pathophysiology
Thalassemias:general aspects and molecular pathology
 Thalassemias:general aspects and molecular pathology Prof. Renzo Galanello Pediatric Clinic 2 University of Cagliari Ospedale Regionale Microcitemie-ASL8 HEMOGLOBINOPATHIES CLASSIFICATION Structurally
Thalassemias:general aspects and molecular pathology Prof. Renzo Galanello Pediatric Clinic 2 University of Cagliari Ospedale Regionale Microcitemie-ASL8 HEMOGLOBINOPATHIES CLASSIFICATION Structurally
In adults, the predominant Hb (HbA) molecule has four chains: two α and two β chains. In thalassemias, the synthesis of either the α or the β chains
 molecule has four chains: two α and two β chains. In thalassemias, the synthesis of either the α or the β chains") Thalassaemias Thalassemia Thalassemia is an inherited autosomal recessive blood disease. Associated with absence or reduction in a or b globin chains. Reduced synthesis of one of the globin chains can
Thalassaemias Thalassemia Thalassemia is an inherited autosomal recessive blood disease. Associated with absence or reduction in a or b globin chains. Reduced synthesis of one of the globin chains can
When do you have to perform the molecular biology in the hemoglobinopathies diagnosis
 When do you have to perform the molecular biology in the hemoglobinopathies diagnosis Maria Domenica Cappellini MD, FRCP;FACP Fondazione Ca Granda Policlinico IRCCS University of Milan Disclosure Member
When do you have to perform the molecular biology in the hemoglobinopathies diagnosis Maria Domenica Cappellini MD, FRCP;FACP Fondazione Ca Granda Policlinico IRCCS University of Milan Disclosure Member
Line Probe Assay for Detection of Alpha Thalassemia: A Pilot Study
 Line Probe Assay for Detection of Alpha Thalassemia: A Pilot Study Menon PK *, Nimmakayalu M, Bylappa SK, Kumar M, Abdalhaleem HM Center for Advanced Biomedical Research and Innovation, Gulf Medical University,
Line Probe Assay for Detection of Alpha Thalassemia: A Pilot Study Menon PK *, Nimmakayalu M, Bylappa SK, Kumar M, Abdalhaleem HM Center for Advanced Biomedical Research and Innovation, Gulf Medical University,
MOLECULAR BASIS OF THALASSEMIA IN SLOVENIA
 MOLECULAR BASIS OF THALASSEMIA IN SLOVENIA Dijana Plaseska-Karanfilska, MD, PhD Research Centre for Genetic Engineering and Biotechnology Georgi D. Efremov, Macedonian Academy of Sciences and Arts, Skopje,
MOLECULAR BASIS OF THALASSEMIA IN SLOVENIA Dijana Plaseska-Karanfilska, MD, PhD Research Centre for Genetic Engineering and Biotechnology Georgi D. Efremov, Macedonian Academy of Sciences and Arts, Skopje,
Dr. Ayman Mohsen Mashi, MBBS Consultant Hematology & Blood Transfusion Department Head, Laboratory & Blood Bank King Fahad Central Hospital, Gazan,
 Dr. Ayman Mohsen Mashi, MBBS Consultant Hematology & Blood Transfusion Department Head, Laboratory & Blood Bank King Fahad Central Hospital, Gazan, KSA amashi@moh.gov.sa 24/02/2018 β-thalassemia syndromes
Dr. Ayman Mohsen Mashi, MBBS Consultant Hematology & Blood Transfusion Department Head, Laboratory & Blood Bank King Fahad Central Hospital, Gazan, KSA amashi@moh.gov.sa 24/02/2018 β-thalassemia syndromes
Screening for haemoglobinopathies in pregnancy
 Policy Statement All Southern Health patients will receive clinical care that reflects best practice and is based on the best available evidence. Index of chapters within background 1. Prevalence of haemoglobinopathies
Policy Statement All Southern Health patients will receive clinical care that reflects best practice and is based on the best available evidence. Index of chapters within background 1. Prevalence of haemoglobinopathies
Report of Beta Thalassemia in Newar Ethnicity
 Report of Beta Thalassemia in Newar Ethnicity Rajendra Dev Bhatt 1*, Surendra Koju 2, Prabodh Risal 1 Affiliations: 1 Department of Clinical Biochemistry, Dhulikhel Hospital, Kathmandu University Hospital
Report of Beta Thalassemia in Newar Ethnicity Rajendra Dev Bhatt 1*, Surendra Koju 2, Prabodh Risal 1 Affiliations: 1 Department of Clinical Biochemistry, Dhulikhel Hospital, Kathmandu University Hospital
HEMOGLOBIN ELECTROPHORESIS DR ARASH ALGHASI SHAFA HOSPITAL-AHWAZ
 HEMOGLOBIN ELECTROPHORESIS DR ARASH ALGHASI SHAFA HOSPITAL-AHWAZ Hemoglobin Hemoglobin (Hb), protein constituting 1/3 of the red blood cells Each red cell has 640 million molecules of Hb sites in the cells:
HEMOGLOBIN ELECTROPHORESIS DR ARASH ALGHASI SHAFA HOSPITAL-AHWAZ Hemoglobin Hemoglobin (Hb), protein constituting 1/3 of the red blood cells Each red cell has 640 million molecules of Hb sites in the cells:
National Haemoglobinopathy Reference Laboratory. Information for Users
 National Haemoglobinopathy Reference Laboratory Information for Users Summary The NHRL offers a service for the identification of haemoglobinopathy genotypes by the molecular analysis of DNA and haematological
National Haemoglobinopathy Reference Laboratory Information for Users Summary The NHRL offers a service for the identification of haemoglobinopathy genotypes by the molecular analysis of DNA and haematological
Unraveling Hemoglobinopathies with Capillary Electrophoresis
 Session Number 2002 Unraveling Hemoglobinopathies with Capillary Electrophoresis David F. Keren, M.D. Professor of Pathology Division Director, Clinical Pathology The University of Michigan dkeren@med.umich.edu
Session Number 2002 Unraveling Hemoglobinopathies with Capillary Electrophoresis David F. Keren, M.D. Professor of Pathology Division Director, Clinical Pathology The University of Michigan dkeren@med.umich.edu
Thalassemia intermedia in HbH-CS disease with compound heterozygosity for β-thalassemia: Challenges in hemoglobin analysis and clinical diagnosis
 Genes Genet. Syst. (2009) 84, p. 67 71 Thalassemia intermedia in HbH-CS disease with compound heterozygosity for β-thalassemia: Challenges in hemoglobin analysis and clinical diagnosis Jin Ai Mary Anne
Genes Genet. Syst. (2009) 84, p. 67 71 Thalassemia intermedia in HbH-CS disease with compound heterozygosity for β-thalassemia: Challenges in hemoglobin analysis and clinical diagnosis Jin Ai Mary Anne
Clinical Characteristics of Pediatric Thalassemia in Korea: A Single Institute Experience
 ORIGINAL ARTICLE Pediatrics http://dx.doi.org/10.3346/jkms.2013.28.11.1645 J Korean Med Sci 2013; 28: 1645-1649 Clinical Characteristics of Pediatric Thalassemia in Korea: A Single Institute Experience
ORIGINAL ARTICLE Pediatrics http://dx.doi.org/10.3346/jkms.2013.28.11.1645 J Korean Med Sci 2013; 28: 1645-1649 Clinical Characteristics of Pediatric Thalassemia in Korea: A Single Institute Experience
Haemoglobinopathy Case Studies. Dr Jill Finlayson Department of Haematology Pathwest Laboratory Medicine
 Haemoglobinopathy Case Studies Dr Jill Finlayson Department of Haematology Pathwest Laboratory Medicine Case 1 KB, 36y M Refugee Afghanistan Screening bloods Hb 101 g/l RCC 3.75 x10 12 /L MCV 90 fl MCH
Haemoglobinopathy Case Studies Dr Jill Finlayson Department of Haematology Pathwest Laboratory Medicine Case 1 KB, 36y M Refugee Afghanistan Screening bloods Hb 101 g/l RCC 3.75 x10 12 /L MCV 90 fl MCH
Gamma gene expression in haemoglobin disorders
 Gamma gene expression in haemoglobin disorders Innovative therapies for Red Cell and Iron related disorders EHA / ESH : April 16-18, Cascais, Portugal Swee Lay Thein King s College London School of Medicine
Gamma gene expression in haemoglobin disorders Innovative therapies for Red Cell and Iron related disorders EHA / ESH : April 16-18, Cascais, Portugal Swee Lay Thein King s College London School of Medicine
Using the Bravo Liquid-Handling System for Next Generation Sequencing Sample Prep
 Using the Bravo Liquid-Handling System for Next Generation Sequencing Sample Prep Tom Walsh, PhD Division of Medical Genetics University of Washington Next generation sequencing Sanger sequencing gold
Using the Bravo Liquid-Handling System for Next Generation Sequencing Sample Prep Tom Walsh, PhD Division of Medical Genetics University of Washington Next generation sequencing Sanger sequencing gold
The diagnosis of Hemoglobinopathies
 1ST IFCC, EFLM, AFCB CONFERENCE "LABORATORY MEDICINE: MEETING THE NEEDS OF MEDITERRANEAN NATIONS" Rome, Italy 02/07/2018 The diagnosis of Hemoglobinopathies Dr Antonino Giambona Unit of Hematology of Rare
1ST IFCC, EFLM, AFCB CONFERENCE "LABORATORY MEDICINE: MEETING THE NEEDS OF MEDITERRANEAN NATIONS" Rome, Italy 02/07/2018 The diagnosis of Hemoglobinopathies Dr Antonino Giambona Unit of Hematology of Rare
Cover Page. The handle holds various files of this Leiden University dissertation.
 Cover Page The handle http://hdl.handle.net/1887/35456 holds various files of this Leiden University dissertation. Author: Hassan, Suha Mustafa Title: Toward prevention of Hemoglobinopathies in Oman Issue
Cover Page The handle http://hdl.handle.net/1887/35456 holds various files of this Leiden University dissertation. Author: Hassan, Suha Mustafa Title: Toward prevention of Hemoglobinopathies in Oman Issue
Detecting and Reporting Alpha Thalassemia In Newborns
 Detecting and Reporting Alpha Thalassemia In Newborns T. Davis, C. Moore, L. Nayak, M.C. Dorley, M. del Pilar Aguinaga, M. Chan, J. Ubaike, C. Yusuf Alpha Thalassemia Screening Status in the US Clinical
Detecting and Reporting Alpha Thalassemia In Newborns T. Davis, C. Moore, L. Nayak, M.C. Dorley, M. del Pilar Aguinaga, M. Chan, J. Ubaike, C. Yusuf Alpha Thalassemia Screening Status in the US Clinical
Haemoglobin BY: MUHAMMAD RADWAN WISSAM MUHAMMAD
 Haemoglobin BY: MUHAMMAD RADWAN WISSAM MUHAMMAD Introduction is the iron-containing oxygen transport metalloprotein in the red blood cells Hemoglobin in the blood carries oxygen from the respiratory organs
Haemoglobin BY: MUHAMMAD RADWAN WISSAM MUHAMMAD Introduction is the iron-containing oxygen transport metalloprotein in the red blood cells Hemoglobin in the blood carries oxygen from the respiratory organs
The Thalassemias in Clinical Practice. Ashutosh Lal, MD Director Comprehensive Thalassemia Program UCSF Benioff Children s Hospital Oakland
 The Thalassemias in Clinical Practice Ashutosh Lal, MD Director Comprehensive Thalassemia Program UCSF Benioff Children s Hospital Oakland Outline Thalassemia: definitions and pathophysiology Epidemiology
The Thalassemias in Clinical Practice Ashutosh Lal, MD Director Comprehensive Thalassemia Program UCSF Benioff Children s Hospital Oakland Outline Thalassemia: definitions and pathophysiology Epidemiology
High Hemoglobin F in a Saudi Child Presenting with Pancytopenia
 Case Report imedpub Journals http://www.imedpub.com Journal of Pediatric Care ISSN 2471-805X DOI: 10.21767/2471-805X.100002 High Hemoglobin F in a Saudi Child Presenting with Pancytopenia Abstract Saudi
Case Report imedpub Journals http://www.imedpub.com Journal of Pediatric Care ISSN 2471-805X DOI: 10.21767/2471-805X.100002 High Hemoglobin F in a Saudi Child Presenting with Pancytopenia Abstract Saudi
Genetics of Thalassemia
 Genetics of Thalassemia Submitted by : Raya Samir Al- Hayaly Sura Zuhair Salih Saad Ghassan Al- Dulaimy Saad Farouq Kassir Sama Naal Salouha Zahraa Jasim Al- Aarajy Supervised by : Dr. Kawkab Adris Mahmod
Genetics of Thalassemia Submitted by : Raya Samir Al- Hayaly Sura Zuhair Salih Saad Ghassan Al- Dulaimy Saad Farouq Kassir Sama Naal Salouha Zahraa Jasim Al- Aarajy Supervised by : Dr. Kawkab Adris Mahmod
EXTERNAL QUALITY ASSESSMENT FOR HAEMOGLOBIN A 2
 EXTERNAL QUALITY ASSESSMENT FOR HAEMOGLOBIN A 2 Dr Barbara abaawild The importance of Hb A 2 measurement Accurate and reliable measurement of Hb A 2 is essential for the diagnosis of beta thalassaemia
EXTERNAL QUALITY ASSESSMENT FOR HAEMOGLOBIN A 2 Dr Barbara abaawild The importance of Hb A 2 measurement Accurate and reliable measurement of Hb A 2 is essential for the diagnosis of beta thalassaemia
SICKLE CELL DISEASE. Dr. MUBARAK ABDELRAHMAN MD PEDIATRICS AND CHILD HEALTH. Assistant Professor FACULTY OF MEDICINE -JAZAN
 SICKLE CELL DISEASE Dr. MUBARAK ABDELRAHMAN MD PEDIATRICS AND CHILD HEALTH Assistant Professor FACULTY OF MEDICINE -JAZAN Objective: The student should be able: To identify the presentation, diagnosis,
SICKLE CELL DISEASE Dr. MUBARAK ABDELRAHMAN MD PEDIATRICS AND CHILD HEALTH Assistant Professor FACULTY OF MEDICINE -JAZAN Objective: The student should be able: To identify the presentation, diagnosis,
 HbF
HbF Corporate Medical Policy
 Corporate Medical Policy Genetic Testing for Alpha Thalassemia File Name: Origination: Last CAP Review: Next CAP Review: Last Review: genetic_testing_for_alpha_thalassemia 9/2013 7/2017 7/2018 7/2017 Description
Corporate Medical Policy Genetic Testing for Alpha Thalassemia File Name: Origination: Last CAP Review: Next CAP Review: Last Review: genetic_testing_for_alpha_thalassemia 9/2013 7/2017 7/2018 7/2017 Description
Genomic structural variation
 Genomic structural variation Mario Cáceres The new genomic variation DNA sequence differs across individuals much more than researchers had suspected through structural changes A huge amount of structural
Genomic structural variation Mario Cáceres The new genomic variation DNA sequence differs across individuals much more than researchers had suspected through structural changes A huge amount of structural
Hematologic Features of Alpha Thalassemia Carriers
 IJMCM Summer 2012, Vol 1, No 3 Original Article Hematologic Features of Alpha Thalassemia Carriers Haleh Akhavan-Niaki 1,2, Reza Youssefi Kamangari 2, Ali Banihashemi 2, Vahid Kholghi Oskooei 1, Mandana
IJMCM Summer 2012, Vol 1, No 3 Original Article Hematologic Features of Alpha Thalassemia Carriers Haleh Akhavan-Niaki 1,2, Reza Youssefi Kamangari 2, Ali Banihashemi 2, Vahid Kholghi Oskooei 1, Mandana
Thalassemia Maria Luz Uy del Rosario, M.D.
 Thalassemia Maria Luz Uy del Rosario, M.D. Philippine Society of Hematology and Blood Transfusion Philippine Society of Pediatric Oncology What is Thalassemia Hereditary Hemoglobin disorder Hemolytic anemia
Thalassemia Maria Luz Uy del Rosario, M.D. Philippine Society of Hematology and Blood Transfusion Philippine Society of Pediatric Oncology What is Thalassemia Hereditary Hemoglobin disorder Hemolytic anemia
Genetic Modulation on the Phenotypic Diversity of Sickle Cell Disease
 Genetic Modulation on the Phenotypic Diversity of Sickle Cell Disease Malay B. Mukherjee Abstract Sickle cell hemoglobin is a β chain structural variant where valine is substituted for glutamic acid in
Genetic Modulation on the Phenotypic Diversity of Sickle Cell Disease Malay B. Mukherjee Abstract Sickle cell hemoglobin is a β chain structural variant where valine is substituted for glutamic acid in
Anemia s. Troy Lund MSMS PhD MD
 Anemia s Troy Lund MSMS PhD MD lundx072@umn.edu Hemoglobinopathy/Anemia IOM take home points. 1. How do we identify the condtion? Smear, CBC Solubility Test (SCD) 2. How does it present clincally? 3. How
Anemia s Troy Lund MSMS PhD MD lundx072@umn.edu Hemoglobinopathy/Anemia IOM take home points. 1. How do we identify the condtion? Smear, CBC Solubility Test (SCD) 2. How does it present clincally? 3. How
Dr.Abdolreza Afrasiabi
 Dr.Abdolreza Afrasiabi Thalassemia & Heamophilia Genetic Reaserch Center Shiraz Medical University Hemoglobin tetramer Hemoglobin Structure % A 1 α 2 β 2 94-97% A 2 α 2 δ 2 2.5% A 1C α 2 (β-n-glucose)
Dr.Abdolreza Afrasiabi Thalassemia & Heamophilia Genetic Reaserch Center Shiraz Medical University Hemoglobin tetramer Hemoglobin Structure % A 1 α 2 β 2 94-97% A 2 α 2 δ 2 2.5% A 1C α 2 (β-n-glucose)
Educational Items Section
 Atlas of Genetics and Cytogenetics in Oncology and Haematology OPEN ACCESS JOURNAL AT INIST-CNRS Educational Items Section Hemoglobin genes; Sickle-cell anemia - Thalassemias Jean-Loup Huret, Xavier Troussard
Atlas of Genetics and Cytogenetics in Oncology and Haematology OPEN ACCESS JOURNAL AT INIST-CNRS Educational Items Section Hemoglobin genes; Sickle-cell anemia - Thalassemias Jean-Loup Huret, Xavier Troussard
Mutation Detection and CNV Analysis for Illumina Sequencing data from HaloPlex Target Enrichment Panels using NextGENe Software for Clinical Research
 Mutation Detection and CNV Analysis for Illumina Sequencing data from HaloPlex Target Enrichment Panels using NextGENe Software for Clinical Research Application Note Authors John McGuigan, Megan Manion,
Mutation Detection and CNV Analysis for Illumina Sequencing data from HaloPlex Target Enrichment Panels using NextGENe Software for Clinical Research Application Note Authors John McGuigan, Megan Manion,
Current Topics in Hemoglobinopathies
 Current Topics in Hemoglobinopathies Bruce R Haas, MS, LCGC 28-29 September 2015 bruce.r.haas@kp.org 1 How malaria escapes effective immunological responses P falciparum exports PfEMP1 proteins and concentrate
Current Topics in Hemoglobinopathies Bruce R Haas, MS, LCGC 28-29 September 2015 bruce.r.haas@kp.org 1 How malaria escapes effective immunological responses P falciparum exports PfEMP1 proteins and concentrate
Anaemia in Pregnancy
 Anaemia in Pregnancy Definition :anaemia is a pathological condition in which the oxygen-carrying capacity of red blood cells is insufficient to meet the body needs. The WHO : haemoglobin concentration
Anaemia in Pregnancy Definition :anaemia is a pathological condition in which the oxygen-carrying capacity of red blood cells is insufficient to meet the body needs. The WHO : haemoglobin concentration
Journal of Medical Science & Technology
 Page82 Original Article Journal of Medical Science & Technology Open Access Screening for hemoglobinopathies among patients in a government hospital and health clinics in Perlis, Malaysia Chin Yuet Meng
Page82 Original Article Journal of Medical Science & Technology Open Access Screening for hemoglobinopathies among patients in a government hospital and health clinics in Perlis, Malaysia Chin Yuet Meng
DNA-seq Bioinformatics Analysis: Copy Number Variation
 DNA-seq Bioinformatics Analysis: Copy Number Variation Elodie Girard elodie.girard@curie.fr U900 institut Curie, INSERM, Mines ParisTech, PSL Research University Paris, France NGS Applications 5C HiC DNA-seq
DNA-seq Bioinformatics Analysis: Copy Number Variation Elodie Girard elodie.girard@curie.fr U900 institut Curie, INSERM, Mines ParisTech, PSL Research University Paris, France NGS Applications 5C HiC DNA-seq
Paolo Moi, Italy. Università di Cagliari Ospedale Pediatrico Microcitemico - A. Cao - CAGLIARI
 T H E O R E T I C A L A N D P R A C T I C A L T R A I N I N G I N HAEMATOLOGICAL RARE DISEASES: from genetic counselling - through bench - to bed Gene Therapy for Hemoglobinopathies Paolo Moi, Italy Università
T H E O R E T I C A L A N D P R A C T I C A L T R A I N I N G I N HAEMATOLOGICAL RARE DISEASES: from genetic counselling - through bench - to bed Gene Therapy for Hemoglobinopathies Paolo Moi, Italy Università
HEMOLYTIC ANEMIA DUE TO ABNORMAL HEMOGLOBIN SYNTHESIS
 Hemolytic Anemia Due to Abnormal Hemoglobin Synthesis MODULE 19 HEMOLYTIC ANEMIA DUE TO ABNORMAL HEMOGLOBIN SYNTHESIS 19.1 INTRODUCTION There are two main mechanisms by which anaemia is produced (a) Thalassemia:
Hemolytic Anemia Due to Abnormal Hemoglobin Synthesis MODULE 19 HEMOLYTIC ANEMIA DUE TO ABNORMAL HEMOGLOBIN SYNTHESIS 19.1 INTRODUCTION There are two main mechanisms by which anaemia is produced (a) Thalassemia:
Characterisation of structural variation in breast. cancer genomes using paired-end sequencing on. the Illumina Genome Analyser
 Characterisation of structural variation in breast cancer genomes using paired-end sequencing on the Illumina Genome Analyser Phil Stephens Cancer Genome Project Why is it important to study cancer? Why
Characterisation of structural variation in breast cancer genomes using paired-end sequencing on the Illumina Genome Analyser Phil Stephens Cancer Genome Project Why is it important to study cancer? Why
Advance Your Genomic Research Using Targeted Resequencing with SeqCap EZ Library
 Advance Your Genomic Research Using Targeted Resequencing with SeqCap EZ Library Marilou Wijdicks International Product Manager Research For Life Science Research Only. Not for Use in Diagnostic Procedures.
Advance Your Genomic Research Using Targeted Resequencing with SeqCap EZ Library Marilou Wijdicks International Product Manager Research For Life Science Research Only. Not for Use in Diagnostic Procedures.
George R. Honig Junius G. Adams III. Human Hemoglobin. Genetics. Springer-Verlag Wien New York
 George R. Honig Junius G. Adams III Human Hemoglobin Genetics Springer-Verlag Wien New York George R. Honig, M.D., Ph.D. Professor and Head Department of Pediatrics, College of Medicine University of Illinois
George R. Honig Junius G. Adams III Human Hemoglobin Genetics Springer-Verlag Wien New York George R. Honig, M.D., Ph.D. Professor and Head Department of Pediatrics, College of Medicine University of Illinois
Multiplex target enrichment using DNA indexing for ultra-high throughput variant detection
 Multiplex target enrichment using DNA indexing for ultra-high throughput variant detection Dr Elaine Kenny Neuropsychiatric Genetics Research Group Institute of Molecular Medicine Trinity College Dublin
Multiplex target enrichment using DNA indexing for ultra-high throughput variant detection Dr Elaine Kenny Neuropsychiatric Genetics Research Group Institute of Molecular Medicine Trinity College Dublin
Genetic Modifiers of Sickle Cell Disease Severity. Kunle Adekile, MD, PhD Professor Department of Pediatrics Kuwait University
 Genetic Modifiers of Sickle Cell Disease Severity Kunle Adekile, MD, PhD Professor Department of Pediatrics Kuwait University Outline Hb Molecule and Genetic control of globin synthesis Pathophysiology
Genetic Modifiers of Sickle Cell Disease Severity Kunle Adekile, MD, PhD Professor Department of Pediatrics Kuwait University Outline Hb Molecule and Genetic control of globin synthesis Pathophysiology
An overview of Thalassaemias and Complications
 An overview of Thalassaemias and Complications Haemoglobin Haemoglobin is the most abundant protein in blood, and exists as three main types in normal adults: HbA ( ) - 97% HbA 2 ( ) - 2.5% HbF ( ) - 0.5%
An overview of Thalassaemias and Complications Haemoglobin Haemoglobin is the most abundant protein in blood, and exists as three main types in normal adults: HbA ( ) - 97% HbA 2 ( ) - 2.5% HbF ( ) - 0.5%
CRISPR/Cas9 Enrichment and Long-read WGS for Structural Variant Discovery
 CRISPR/Cas9 Enrichment and Long-read WGS for Structural Variant Discovery PacBio CoLab Session October 20, 2017 For Research Use Only. Not for use in diagnostics procedures. Copyright 2017 by Pacific Biosciences
CRISPR/Cas9 Enrichment and Long-read WGS for Structural Variant Discovery PacBio CoLab Session October 20, 2017 For Research Use Only. Not for use in diagnostics procedures. Copyright 2017 by Pacific Biosciences
Hb S/β + -thalassemia due to Hb sickle and a novel deletion of DNase I hypersensitive sites HS3 and HS4 of the b Locus Control Region
 Published Ahead of Print on February 14, 2015, as doi:10.3324/haematol.2014.117408. Copyright 2015 Ferrata Storti Foundation. Hb S/β + -thalassemia due to Hb sickle and a novel deletion of DNase I hypersensitive
Published Ahead of Print on February 14, 2015, as doi:10.3324/haematol.2014.117408. Copyright 2015 Ferrata Storti Foundation. Hb S/β + -thalassemia due to Hb sickle and a novel deletion of DNase I hypersensitive
Extra Notes 3. Warm. In the core (center) of the body, where the temperature is 37 C.
 of the body, where the temperature is 37 C.") Extra Notes 3 *The numbers of the slides are according to the last year slides. Slide 33 Autoimmune hemolytic anemia : Abnormal circulating antibodies that target normal antigen on the RBC and cause lysis.
Extra Notes 3 *The numbers of the slides are according to the last year slides. Slide 33 Autoimmune hemolytic anemia : Abnormal circulating antibodies that target normal antigen on the RBC and cause lysis.
Molecular Diagnosis of Thalassemias and Hemoglobinopathies. An ACLPS Critical Review CME/SAM. Daniel E. Sabath, MD, PhD ABSTRACT.
 Molecular Diagnosis of Thalassemias and Hemoglobinopathies An ACLPS Critical Review Daniel E. Sabath, MD, PhD From the Department of Laboratory Medicine, University of Washington, Seattle. CME/SAM Key
Molecular Diagnosis of Thalassemias and Hemoglobinopathies An ACLPS Critical Review Daniel E. Sabath, MD, PhD From the Department of Laboratory Medicine, University of Washington, Seattle. CME/SAM Key
DEVELOPMENT OF A HAEMOGLOBINOPATHY GENETIC DIAGNOSTIC SERVICE FOR THE NORTH WEST OF ENGLAND
 DEVELOPMENT OF A HAEMOGLOBINOPATHY GENETIC DIAGNOSTIC SERVICE FOR THE NORTH WEST OF ENGLAND A thesis submitted to the Manchester Metropolitan University for the degree of Master of Philosophy in the Faculty
DEVELOPMENT OF A HAEMOGLOBINOPATHY GENETIC DIAGNOSTIC SERVICE FOR THE NORTH WEST OF ENGLAND A thesis submitted to the Manchester Metropolitan University for the degree of Master of Philosophy in the Faculty
Cytogenetics 101: Clinical Research and Molecular Genetic Technologies
 Cytogenetics 101: Clinical Research and Molecular Genetic Technologies Topics for Today s Presentation 1 Classical vs Molecular Cytogenetics 2 What acgh? 3 What is FISH? 4 What is NGS? 5 How can these
Cytogenetics 101: Clinical Research and Molecular Genetic Technologies Topics for Today s Presentation 1 Classical vs Molecular Cytogenetics 2 What acgh? 3 What is FISH? 4 What is NGS? 5 How can these
Expanded Carrier Screening: What s Best?
 Expanded Carrier Screening: What s Best? James D Goldberg, MD September 17, 2017 Disclosures James D. Goldberg, M.D. Chief Medical Officer, Counsyl 3 Learning Objectives Guidelines Data Design Practice
Expanded Carrier Screening: What s Best? James D Goldberg, MD September 17, 2017 Disclosures James D. Goldberg, M.D. Chief Medical Officer, Counsyl 3 Learning Objectives Guidelines Data Design Practice
New: P077 BRCA2. This new probemix can be used to confirm results obtained with P045 BRCA2 probemix.
 SALSA MLPA KIT P045-B2 BRCA2/CHEK2 Lot 0410, 0609. As compared to version B1, four reference probes have been replaced and extra control fragments at 100 and 105 nt (X/Y specific) have been included. New:
SALSA MLPA KIT P045-B2 BRCA2/CHEK2 Lot 0410, 0609. As compared to version B1, four reference probes have been replaced and extra control fragments at 100 and 105 nt (X/Y specific) have been included. New:
HPLC profile of sickle cell disease in central India
 Original Research Article HPLC profile of sickle cell disease in central India Shweta P. Bijwe * Department of Pathology, IGGMC, Nagpur, Maharashtra, India * Corresponding author email: dr.shwetabijwe@gmail.com
Original Research Article HPLC profile of sickle cell disease in central India Shweta P. Bijwe * Department of Pathology, IGGMC, Nagpur, Maharashtra, India * Corresponding author email: dr.shwetabijwe@gmail.com
HAEMOGLOBINOPATHIES. Editing file. References: 436 girls & boys slides 435 teamwork slides. Color code: Important. Extra.
 HAEMOGLOBINOPATHIES Objectives: normal structure and function of haemoglobin. how the globin components of haemoglobin change during development, and postnatally. the mechanisms by which the thalassaemias
HAEMOGLOBINOPATHIES Objectives: normal structure and function of haemoglobin. how the globin components of haemoglobin change during development, and postnatally. the mechanisms by which the thalassaemias
Investigating rare diseases with Agilent NGS solutions
 Investigating rare diseases with Agilent NGS solutions Chitra Kotwaliwale, Ph.D. 1 Rare diseases affect 350 million people worldwide 7,000 rare diseases 80% are genetic 60 million affected in the US, Europe
Investigating rare diseases with Agilent NGS solutions Chitra Kotwaliwale, Ph.D. 1 Rare diseases affect 350 million people worldwide 7,000 rare diseases 80% are genetic 60 million affected in the US, Europe
SALSA MLPA KIT P050-B2 CAH
 SALSA MLPA KIT P050-B2 CAH Lot 0510, 0909, 0408: Compared to lot 0107, extra control fragments have been added at 88, 96, 100 and 105 nt. The 274 nt probe gives a higher signal in lot 0510 compared to
SALSA MLPA KIT P050-B2 CAH Lot 0510, 0909, 0408: Compared to lot 0107, extra control fragments have been added at 88, 96, 100 and 105 nt. The 274 nt probe gives a higher signal in lot 0510 compared to
Type and frequency of hemoglobinopathies, diagnosed in the area of Karachi, in Pakistan
 HEMATOLOGY RESEARCH ARTICLE Type and frequency of hemoglobinopathies, diagnosed in the area of Karachi, in Pakistan Received: 09 March 2016 Accepted: 09 May 2016 First Published: 13 May 2016 *Corresponding
HEMATOLOGY RESEARCH ARTICLE Type and frequency of hemoglobinopathies, diagnosed in the area of Karachi, in Pakistan Received: 09 March 2016 Accepted: 09 May 2016 First Published: 13 May 2016 *Corresponding
HST.161 Molecular Biology and Genetics in Modern Medicine Fall 2007
 MIT OpenCourseWare http://ocw.mit.edu HST.161 Molecular Biology and Genetics in Modern Medicine Fall 2007 For information about citing these materials or our Terms of Use, visit: http://ocw.mit.edu/terms.
MIT OpenCourseWare http://ocw.mit.edu HST.161 Molecular Biology and Genetics in Modern Medicine Fall 2007 For information about citing these materials or our Terms of Use, visit: http://ocw.mit.edu/terms.
Changes in hematological parameters in α-thalassemia individuals co-inherited with erythroid Krüppel-like factor mutations
 Clin Genet 2015: 88: 56 61 Printed in Singapore. All rights reserved Short Report 2014 John Wiley & Sons A/S. Published by John Wiley & Sons Ltd CLINICAL GENETICS doi: 10.1111/cge.12443 Changes in hematological
Clin Genet 2015: 88: 56 61 Printed in Singapore. All rights reserved Short Report 2014 John Wiley & Sons A/S. Published by John Wiley & Sons Ltd CLINICAL GENETICS doi: 10.1111/cge.12443 Changes in hematological
Clinical, haematological, and genetic studies of type 2
 Journal of Medical Genetics 1988, 25, 195-199 Clinical, haematological, and genetic studies of type 2 normal Hb A2 thalassaemia ANNA METAXOTOU-MAVROMATI, CHRISTOS KATTAMIS, LILIAN MATATHIA, MARIA TZETIS,
Journal of Medical Genetics 1988, 25, 195-199 Clinical, haematological, and genetic studies of type 2 normal Hb A2 thalassaemia ANNA METAXOTOU-MAVROMATI, CHRISTOS KATTAMIS, LILIAN MATATHIA, MARIA TZETIS,
Counselling and prenatal diagnosis. Antonis Kattamis, Greece
 Counselling and prenatal diagnosis Antonis Kattamis, Greece Epidemiology of Hemoglobinopathies 7% of world population carriers of hemoglobinopathies 500.000 newborns annually affected 300.000 : Thalassemias
Counselling and prenatal diagnosis Antonis Kattamis, Greece Epidemiology of Hemoglobinopathies 7% of world population carriers of hemoglobinopathies 500.000 newborns annually affected 300.000 : Thalassemias
BRITISH BIOMEDICAL BULLETIN
 Journal Home Page www.bbbulletin.org BRITISH BIOMEDICAL BULLETIN Original A Long Term Screening of Iranian Populations with Thalassemia and Hemoglobinopathies Soudabeh Hosseini 1,2, Ebrahim Kalantar 3,2
Journal Home Page www.bbbulletin.org BRITISH BIOMEDICAL BULLETIN Original A Long Term Screening of Iranian Populations with Thalassemia and Hemoglobinopathies Soudabeh Hosseini 1,2, Ebrahim Kalantar 3,2
Abstract. Optimization strategy of Copy Number Variant calling using Multiplicom solutions APPLICATION NOTE. Introduction
 Optimization strategy of Copy Number Variant calling using Multiplicom solutions Michael Vyverman, PhD; Laura Standaert, PhD and Wouter Bossuyt, PhD Abstract Copy number variations (CNVs) represent a significant
Optimization strategy of Copy Number Variant calling using Multiplicom solutions Michael Vyverman, PhD; Laura Standaert, PhD and Wouter Bossuyt, PhD Abstract Copy number variations (CNVs) represent a significant
Evaluation of the Molecular basis of KLF1 Gene in Iranian Thalassemia individuals with borderline hemoglobin A2
 Advances in Bioresearch Adv. Biores., Vol 7 (5) September 2016: 11-15 2016 Society of Education, India Print ISSN 0976-4585; Online ISSN 2277-1573 Journal s URL:http://www.soeagra.com/abr.html CODEN: ABRDC3
Advances in Bioresearch Adv. Biores., Vol 7 (5) September 2016: 11-15 2016 Society of Education, India Print ISSN 0976-4585; Online ISSN 2277-1573 Journal s URL:http://www.soeagra.com/abr.html CODEN: ABRDC3
No mutations were identified.
 Hereditary High Cholesterol Test ORDERING PHYSICIAN PRIMARY CONTACT SPECIMEN Report date: Aug 1, 2017 Dr. Jenny Jones Sample Medical Group 123 Main St. Sample, CA Kelly Peters Sample Medical Group 123
Hereditary High Cholesterol Test ORDERING PHYSICIAN PRIMARY CONTACT SPECIMEN Report date: Aug 1, 2017 Dr. Jenny Jones Sample Medical Group 123 Main St. Sample, CA Kelly Peters Sample Medical Group 123
Antenatal Detection of Hemoglobinopathies using Red Blood Cells Indices for Screening
 Sukanya Singh et al. ORIGINAL ARTICLE 10.5005/jp-journals-10054-0064 Antenatal Detection of Hemoglobinopathies using Red Blood Cells Indices for Screening 1 Sukanya Singh, 2 Lalna R Takale, 3 Mona Tilak
Sukanya Singh et al. ORIGINAL ARTICLE 10.5005/jp-journals-10054-0064 Antenatal Detection of Hemoglobinopathies using Red Blood Cells Indices for Screening 1 Sukanya Singh, 2 Lalna R Takale, 3 Mona Tilak
Red cell disorder. Dr. Ahmed Hasan
 Red cell disorder Dr. Ahmed Hasan Things to be learned in this lecture Definition and clinical feature of anemia. Classification of anemia. Know some details of microcytic anemia Question of the lecture:
Red cell disorder Dr. Ahmed Hasan Things to be learned in this lecture Definition and clinical feature of anemia. Classification of anemia. Know some details of microcytic anemia Question of the lecture:
Biology 2C03: Genetics What is a Gene?
 Biology 2C03: Genetics What is a Gene? September 9 th, 2013 Model Organisms - E. coli - Yeast - Worms - Arabodopsis - Fruitflie - Mouse What is a Gene? - Define, recognize, describe and apply Mendel s
Biology 2C03: Genetics What is a Gene? September 9 th, 2013 Model Organisms - E. coli - Yeast - Worms - Arabodopsis - Fruitflie - Mouse What is a Gene? - Define, recognize, describe and apply Mendel s
The Beats of Natural Sciences Issue 3-4 (September-December) Vol. 3 (2016)
 Vol. 3 (2016)") Frequency of β (Beta Thalassaemia) Trait and Haemaglobin E (HbE) Trait: Case Study in a Thalassaemia Carrier Detection Camp in Gurudas College, West Bengal, India Mitu De Department of Botany, Gurudas
Frequency of β (Beta Thalassaemia) Trait and Haemaglobin E (HbE) Trait: Case Study in a Thalassaemia Carrier Detection Camp in Gurudas College, West Bengal, India Mitu De Department of Botany, Gurudas
POLICY PRODUCT VARIATIONS DESCRIPTION/BACKGROUND RATIONALE DEFINITIONS BENEFIT VARIATIONS DISCLAIMER CODING INFORMATION REFERENCES POLICY HISTORY
 Original Issue Date (Created): November 26, 2013 Most Recent Review Date (Revised): November 26, 2013 Effective Date: April 1, 2014 POLICY PRODUCT VARIATIONS DESCRIPTION/BACKGROUND RATIONALE DEFINITIONS
Original Issue Date (Created): November 26, 2013 Most Recent Review Date (Revised): November 26, 2013 Effective Date: April 1, 2014 POLICY PRODUCT VARIATIONS DESCRIPTION/BACKGROUND RATIONALE DEFINITIONS
IRON2009_CAP.10( ):EBMT :24 Pagina 250 CHAPTER 10. Molecular basis of thalassaemia syndromes. Bill Wood, Doug Higgs
:EBMT :24 Pagina 250 CHAPTER 10. Molecular basis of thalassaemia syndromes. Bill Wood, Doug Higgs") IRON2009_CAP.10(250-263):EBMT2008 4-12-2009 16:24 Pagina 250 * CHAPTER 10 Molecular basis of thalassaemia syndromes Bill Wood, Doug Higgs IRON2009_CAP.10(250-263):EBMT2008 4-12-2009 16:24 Pagina 251 CHAPTER
IRON2009_CAP.10(250-263):EBMT2008 4-12-2009 16:24 Pagina 250 * CHAPTER 10 Molecular basis of thalassaemia syndromes Bill Wood, Doug Higgs IRON2009_CAP.10(250-263):EBMT2008 4-12-2009 16:24 Pagina 251 CHAPTER
MRC-Holland MLPA. Description version 19;
 SALSA MLPA probemix P6-B2 SMA Lot B2-712, B2-312, B2-111, B2-511: As compared to the previous version B1 (lot B1-11), the 88 and 96 nt DNA Denaturation control fragments have been replaced (QDX2). SPINAL
SALSA MLPA probemix P6-B2 SMA Lot B2-712, B2-312, B2-111, B2-511: As compared to the previous version B1 (lot B1-11), the 88 and 96 nt DNA Denaturation control fragments have been replaced (QDX2). SPINAL
Thalassemia and other hemoglobinopathies among anemic individuals in Metro Manila: Preliminary findings from the National Nutrition Survey
 Thalassemia and other hemoglobinopathies among anemic individuals in Metro Manila: Preliminary findings from the National Nutrition Survey Sofia V. Amarra, R.D., Ph.D ILSI Southeast Asia region Definitions
Thalassemia and other hemoglobinopathies among anemic individuals in Metro Manila: Preliminary findings from the National Nutrition Survey Sofia V. Amarra, R.D., Ph.D ILSI Southeast Asia region Definitions
Beta Thalassemia Frequency in Bahrain: A Ten Year Study. Shaikha Salim Al-Arrayed, MB,ChB, DHCG, PhD*
 Bahrain Medical Bulletin, Vol. 2, No. 2, June 200 Beta Thalassemia Frequency in Bahrain: A Ten Year Study Shaikha Salim Al-Arrayed, MB,ChB, DHCG, PhD* Background: Sickle-cell disease and Thalassanemia
Bahrain Medical Bulletin, Vol. 2, No. 2, June 200 Beta Thalassemia Frequency in Bahrain: A Ten Year Study Shaikha Salim Al-Arrayed, MB,ChB, DHCG, PhD* Background: Sickle-cell disease and Thalassanemia
Introduction to LOH and Allele Specific Copy Number User Forum
 Introduction to LOH and Allele Specific Copy Number User Forum Jonathan Gerstenhaber Introduction to LOH and ASCN User Forum Contents 1. Loss of heterozygosity Analysis procedure Types of baselines 2.
Introduction to LOH and Allele Specific Copy Number User Forum Jonathan Gerstenhaber Introduction to LOH and ASCN User Forum Contents 1. Loss of heterozygosity Analysis procedure Types of baselines 2.
Joanne Mallon, Nora O Neill, Dr D Hull Antenatal midwife coordinator, Consultant Haematologist
 Title: CLINICAL GUIDELINES ID TAG Guideline for Antenatal Screening for Haemoglobinopathies Author: Designation: Speciality / Division: Directorate: Joanne Mallon, Nora O Neill, Dr D Hull Antenatal midwife
Title: CLINICAL GUIDELINES ID TAG Guideline for Antenatal Screening for Haemoglobinopathies Author: Designation: Speciality / Division: Directorate: Joanne Mallon, Nora O Neill, Dr D Hull Antenatal midwife
Prof Sanath P Lamabadusuriya
 Prof Sanath P Lamabadusuriya What is Thalassaemia? It is the commonest inherited variety of anaemia It is the commonest haemoglobinopathy in Sri Lanka Of all the different types, Beta-Thalassaemia major
Prof Sanath P Lamabadusuriya What is Thalassaemia? It is the commonest inherited variety of anaemia It is the commonest haemoglobinopathy in Sri Lanka Of all the different types, Beta-Thalassaemia major
SALSA MLPA KIT P060-B2 SMA
 SALSA MLPA KIT P6-B2 SMA Lot 111, 511: As compared to the previous version B1 (lot 11), the 88 and 96 nt DNA Denaturation control fragments have been replaced (QDX2). Please note that, in contrast to the
SALSA MLPA KIT P6-B2 SMA Lot 111, 511: As compared to the previous version B1 (lot 11), the 88 and 96 nt DNA Denaturation control fragments have been replaced (QDX2). Please note that, in contrast to the
Hypochromic Anaemias
 Hypochromic Anaemias Dr Mere Kende MBBS, MMED (Path), MAACB, MACTM, MACRRM LECTURER-SMHS Anaemia LOW HEMOGLOBIN Anaemia Definition: Hb
Hypochromic Anaemias Dr Mere Kende MBBS, MMED (Path), MAACB, MACTM, MACRRM LECTURER-SMHS Anaemia LOW HEMOGLOBIN Anaemia Definition: Hb
Chem*3560 Lecture 4: Inherited modifications in hemoglobin
 Chem*3560 Lecture 4: Inherited modifications in hemoglobin Genetic modifications fall into two classes: Thalassemias, which are the result of failure to express globin genes. Thalassa is Greek for the
Chem*3560 Lecture 4: Inherited modifications in hemoglobin Genetic modifications fall into two classes: Thalassemias, which are the result of failure to express globin genes. Thalassa is Greek for the
12.1 X-linked Inheritance in Humans. Units of Heredity: Chromosomes and Inheritance Ch. 12. X-linked Inheritance. X-linked Inheritance
 Units of Heredity: Chromosomes and Inheritance Ch. 12 12.1 in Humans X-chromosomes also have non genderspecific genes Called X-linked genes Vision Blood-clotting X-linked conditions Conditions caused by
Units of Heredity: Chromosomes and Inheritance Ch. 12 12.1 in Humans X-chromosomes also have non genderspecific genes Called X-linked genes Vision Blood-clotting X-linked conditions Conditions caused by
Epidemiological Study among Thalassemia Intermedia Pediatric Patients
 Med. J. Cairo Univ., Vol. 78, No. 2, December 651-655, 2010 www.medicaljournalofcairouniversity.com Epidemiological Study among Thalassemia Intermedia Pediatric Patients NERMEEN KADDAH, M.D.; KHALED SALAMA,
Med. J. Cairo Univ., Vol. 78, No. 2, December 651-655, 2010 www.medicaljournalofcairouniversity.com Epidemiological Study among Thalassemia Intermedia Pediatric Patients NERMEEN KADDAH, M.D.; KHALED SALAMA,
Case Report Iron Depletion: An Ameliorating Factor for Sickle Cell Disease?
 International Scholarly Research Network ISRN Hematology Volume 2011, Article ID 473152, 4 pages doi:10.5402/2011/473152 Case Report Iron Depletion: An Ameliorating Factor for Sickle Cell Disease? P. C.
International Scholarly Research Network ISRN Hematology Volume 2011, Article ID 473152, 4 pages doi:10.5402/2011/473152 Case Report Iron Depletion: An Ameliorating Factor for Sickle Cell Disease? P. C.
Evaluation of MIA FORA NGS HLA test and software. Lisa Creary, PhD Department of Pathology Stanford Blood Center Research & Development Group
 Evaluation of MIA FORA NGS HLA test and software Lisa Creary, PhD Department of Pathology Stanford Blood Center Research & Development Group Disclosure Alpha and Beta Studies Sirona Genomics Reagents,
Evaluation of MIA FORA NGS HLA test and software Lisa Creary, PhD Department of Pathology Stanford Blood Center Research & Development Group Disclosure Alpha and Beta Studies Sirona Genomics Reagents,
Nuovi strumenti diagnostici : NGS era
 Nuovi strumenti diagnostici : NGS era Achille Iolascon, MD, PhD Dept. Molecular Medicine and Medical Biotechnology, University Federico II, Naples, Italy achille.iolascon@unina.it madre padre figlio neonato
Nuovi strumenti diagnostici : NGS era Achille Iolascon, MD, PhD Dept. Molecular Medicine and Medical Biotechnology, University Federico II, Naples, Italy achille.iolascon@unina.it madre padre figlio neonato
SALSA MLPA probemix P169-C2 HIRSCHSPRUNG-1 Lot C As compared to version C1 (lot C1-0612), the length of one probe has been adjusted.
, the length of one probe has been adjusted.") mix P169-C2 HIRSCHSPRUNG-1 Lot C2-0915. As compared to version C1 (lot C1-0612), the length of one has been adjusted. Hirschsprung disease (HSCR), or aganglionic megacolon, is a congenital disorder characterised
mix P169-C2 HIRSCHSPRUNG-1 Lot C2-0915. As compared to version C1 (lot C1-0612), the length of one has been adjusted. Hirschsprung disease (HSCR), or aganglionic megacolon, is a congenital disorder characterised
Laboratory for diagnosis of THALASSEMIA
 SCBM343 CLINICAL PATHOLOGY 2(1-2-3) Laboratory for diagnosis of THALASSEMIA PORNTHIP CHAICHOMPOO pornthip.chh@mahidol.ac.th Acknowledgements Dr. Pranee Winichagoon Fucharoen Ms. Pornnapa Khampan Thalassemia
SCBM343 CLINICAL PATHOLOGY 2(1-2-3) Laboratory for diagnosis of THALASSEMIA PORNTHIP CHAICHOMPOO pornthip.chh@mahidol.ac.th Acknowledgements Dr. Pranee Winichagoon Fucharoen Ms. Pornnapa Khampan Thalassemia
2/10/2016. Evaluation of MIA FORA NGS HLA test and software. Disclosure. NGS-HLA typing requirements for the Stanford Blood Center
 Evaluation of MIA FORA NGS HLA test and software Lisa Creary, PhD Department of Pathology Stanford Blood Center Research & Development Group Disclosure Alpha and Beta Studies Sirona Genomics Reagents,
Evaluation of MIA FORA NGS HLA test and software Lisa Creary, PhD Department of Pathology Stanford Blood Center Research & Development Group Disclosure Alpha and Beta Studies Sirona Genomics Reagents,
Beta thalassaemia traits in Nigerian patients with sickle cell anaemia
 JMBR: A Peer-review Journal of Biomedical Sciences June 2005 Vol. 4 No.1 pp-37-43 Beta thalassaemia traits in Nigerian patients with sickle cell anaemia CE Omoti ABSTRACT Haematological values were determined
JMBR: A Peer-review Journal of Biomedical Sciences June 2005 Vol. 4 No.1 pp-37-43 Beta thalassaemia traits in Nigerian patients with sickle cell anaemia CE Omoti ABSTRACT Haematological values were determined
CRISPR-mediated Editing of Hematopoietic Stem Cells for the Treatment of β-hemoglobinopathies
 CRISPR-mediated Editing of Hematopoietic Stem Cells for the Treatment of β-hemoglobinopathies Jennifer Gori American Society of Gene & Cell Therapy May 11, 2017 editasmedicine.com 1 Highlights Developed
CRISPR-mediated Editing of Hematopoietic Stem Cells for the Treatment of β-hemoglobinopathies Jennifer Gori American Society of Gene & Cell Therapy May 11, 2017 editasmedicine.com 1 Highlights Developed