RNA SEQUENCING AND DATA ANALYSIS
|
|
|
- Lester Simmons
- 5 years ago
- Views:
Transcription

1 RNA SEQUENCING AND DATA ANALYSIS
2 Download slides and package
3 Overview Introduction into the topic RNA species Experimental design considerations Analytical approaches Discussion of our analysis pipeline Technical details Application on TCGA data sets Results Hands on
4 All RNA is not the same Types of RNA:
5 All RNA is not the same Types of RNA: Messenger RNA Micro RNA Long non-coding RNA Ribosomal RNA Other
6 Methods for RNA enrichment prior to library construction Poly(A)-RNA selection By hybridization to oligo-dt beads mature mrna highly enriched efficient for quantification of gene expression level and so on limitation: 3 bias correlating with RNA degradation rrna depletion: by hybridization to bead-bound rrna probes rrna sequence-dependent and species-specific all non-rrna retained: premature mrna, long non-coding RNA Small RNA extraction: Specific kits required to retain small RNA Optional fine size-selection by gel or column This lecture focuses on mrna sequencing
7 Length of mrna transcripts in the human genome 5,000 5,000 4,000 3,000 2,000 4,000 1, ,000 2,000 1, ,000 4,000 6,000 8,000 10,000
8 Length of mrna transcripts in the human genome 5,000 4,000 3,000 2,000 5,000 4,000 3,000 2,000 What is the optimal insert 1,000 and read size 0 0 for 200 mrna sequencing? 1, ,000 4,000 6,000 8,000 10,000
9 Alignment versus assembly Assembly Trinity, Cufflinks, ABySS Particularly useful when no reference genome is available, like in bacterial transcriptomes Alignment Bowtie, BWA, Mosaic Maximum sensitivity, fewer false positives
10 Sequencing parameters Read Type, typically 36/51/76/101 bp: Single end read: Paired end read:
11 Sequencing parameters Read Type: Single end read: for efficient counting of transcript copy number and splicing sites Paired end read: longer cdna fragment and read length help to determine transcript structure especially within gene families Applications of RNA-sequencing
12 RNA sequencing applications Quantification of transcript expression levels Detection of splice variation/different isoforms of the same gene Allele specific expression levels Detection of fusion transcripts (such as BCR-ABL in CML) Detection of sequence variation (limited application) Validation of DNA sequence variants
 to normalize for gene length and library size")
13 RNA-seq expression levels are linear where microarrays get saturated or are insensitive Expression is measured as reads per kilobase per million (RPKM) to normalize for gene length and library size
14 Identification of fusion transcripts Popular methods search for Read pairs that map to two different genes Need to correct for gene homology Reads that span fusion junctions Split reads in half and align separate halfs Make a database of all possible fusion junctions and align full reads PRADA, MapSplice, TopHat

15 Variant detection All DNA mutations from TCGA renal cell clear cell carcinoma project Approximately 35% of mutations are covered sufficiently to be detected at a validation rate of ~ 80-90%. Reverse transcriptase step to convert RNA to cdna complicates detection of RNA edits and mutations
16 Sequencing parameters Read Depth Minimum mapped reads: 10 million for quantitative analysis of mammalian transcriptome More reads needed for splicing variant discovery and differential comparison among samples Current output: million raw reads / lane Multiplex level: 4-12 libraries / lane recommended
17 RNA sequencing in The Cancer Genome Atlas mrna: poly-a mrna purified from total RNA using poly-t oligo-attached magnetic beads mirna: Total RNA is mixed with oligo(dt) MicroBeads and loaded into MACS column, which is then placed on a MultiMACS separator. From the flow-through, small RNAs, including mirnas, are recovered by ethanol precipitation.
18 Preprocessing.bam file [PAIRED END].fastq files [END1 & END2] INPUTS Config.txt [location of scripts and reference files] Expression & QC Module Fusion Module GUESS-ft -genea -geneb Processing Module RNA-SeQC Read Alignment Remap alignments Combine two ends Quality Scores Recalibrate d OUTPUTS RPKM & QC metrics Fusion Candidates Supervised search evidence Implementation Results Samples processed >400 KIRC >170 GBM TFG-GPR128 fusion Samples detected 5 KIRC >5 GBM Samples processed 321 normal, 85 tumor (BLCA, BRCA, HNSC, KIRC, KIRP, LIHC, LUAD, LUSC, PRAD, THCA)
19 RNA sequencing read alignment in PRADA Transcripts from same gene Reads are aligned to all possible transcripts Reads are also aligned to genome
20 RNA sequencing read alignment in PRADA Reads are aligned to all possible transcripts Reads are also aligned to genome Final and single placement for each read it determined by re-mapping
21 PRADA alignments advantages versus disadvantages Advantage: Alignment to unannotated transcripts Alignment across exon-exon junctions Disadvantage Alignment approaches such as used by MapSplice, Bowtie/Tophat typically split reads More conservative alignment than split-read
22 Preprocessing.bam file [PAIRED END].fastq files [END1 & END2] INPUTS Config.txt [location of scripts and reference files] Processing Module Expression & QC Module RNA-SeQC Fusion Module GUESS-ft -genea -geneb Read Alignment Remap alignments Combine two ends Quality Scores Recalibrate d OUTPUTS RPKM & QC metrics Fusion Candidates Supervised search evidence PRADA focuses on the analysis of paired-end RNA-sequencing data. Four modules: 1. Processing 2. Expression and Quality Control 3. Gene fusion 4. GUESS-ft: General User defined Supervised Search for fusion transcripts
![Preprocessing.bam file [PAIRED END].fastq files [END1 & END2] Read Alignment Processing Module Remap alignments INPUTS Config.](/docs-images/81/83517087/images/23-6.jpg "txt [location of scripts and reference files] Combine two ends Quality Scores Recalibrate d Expression and QC Module RNA-SeQC Fusion Module GUESS-ft -genea -geneb Samples reads are mapped to:")
23 Preprocessing.bam file [PAIRED END].fastq files [END1 & END2] Read Alignment Processing Module Remap alignments INPUTS Config.txt [location of scripts and reference files] Combine two ends Quality Scores Recalibrate d Expression and QC Module RNA-SeQC Fusion Module GUESS-ft -genea -geneb Samples reads are mapped to: Transcriptome Genome Processing Module Widely use tools by the research community Samtools, BWA, Picard, GATK Enabled References versions hg18 Ensembl52 hg19 Ensembl64 RPKM & QC metrics Fusion Candidates Supervised search evidence
![Preprocessing.bam file [PAIRED END].fastq files [END1 & END2] INPUTS Config.](/docs-images/81/83517087/images/24-0.jpg "txt [location of scripts and reference files] Processing Module Expression & QC Module")


24 Preprocessing.bam file [PAIRED END].fastq files [END1 & END2] INPUTS Config.txt [location of scripts and reference files] Processing Module Expression & QC Module RNA-SeQC Fusion Module GUESS-ft RNAseQC Process (java) -genea -geneb Read Alignment Remap alignments Combine two ends Quality Scores Recalibrate d Expression & QC Module OUTPUTS RNA-SeQC provides three types of quality control metrics: Read Counts Coverage Correlation RPKM Values at transcript level For longest transcript RPKM & QC metrics Fusion Candidates Supervised search evidence
![Preprocessing.bam file [PAIRED END] INPUTS Fusion Module Config.txt.](/docs-images/81/83517087/images/25-5.jpg "fastq files Discordant [location of read scripts and pair: reference files] Each end of the [END1 & END2] read pair maps uniquely to distinct Processing Module protein-coding genes.")
25 Preprocessing.bam file [PAIRED END] INPUTS Fusion Module Config.txt.fastq files Discordant [location of read scripts and pair: reference files] Each end of the [END1 & END2] read pair maps uniquely to distinct Processing Module protein-coding genes. Expression & QC Module RNA-SeQC Fusion Module GUESS-ft -genea -geneb Read Alignment Remap alignments Combine two ends Quality Scores Recalibrate d OUTPUTS Fusion spanning reads: Chimeric read that maps a putative junction and the mate read maps to either GENE A or GENE B. RPKM & QC metrics Fusion Candidates Supervised search evidence Gene A Gene B


26 Fusion Module Cont d Filters Gene homology using blastn (bitscore 50) Ratio of fusion spanning and discordant reads 49 bp 49 bp 50 bp 50 bp 80 bp 180 bp Number of gene partners within a sample Remove promiscuous fusion pairs, i.e. with large number of partners (e.g. >25) Number of distinct junctions Filtered Candidates: Up to 1 mismatch Unique sequences Unique start positions r t =
27 Fusion Module Cont d SampleID GeneA GeneB TCGA-BP A-01R SFPQ TFE3 Discordant_Pairs 350 Fusion_Reads 220 Fusion_Junctions 1 HomologyScore 26.5 FusionDiscordant_Ratio Positions_Consistent GeneA_Chr GeneB_Chr Fusion_Type Breakpoint_Distance Breakpoint(s) PARTIALLY chr1 chrx Unique reads: gadiffpos 110 Unique reads: gbdiffpos 119 Unique reads: fusdiffseq 35 ga_withinsamplecount 1 gb_withinsamplecount 1 Interchromosomal 1.00E+46 ExonJunction in-frame classification* in-frame chr1.i.e7.e _chr23.e Outputs List all annotated fusions SampleID.annotated.candidates.txt List filtered annotated fusion SampleID.filtered.candidates.txt TAAGACGCATGGAAGAACTTCACAATCAAGAAATGCAGAAACGTAAAGAAATGCAATTGAG * CCTGAACTCTTTGCTTCCGGAATCCGGGATTG TTGCTGACATAGAATTAGAAAACGTCCTT




28 Fusion Module Cont d The identification of in-frame fusion transcripts and their predicted protein sequences. Image Source: Asmann Y W et al. Nucl. Acids Res. 2011;nar.gkr362 The Author(s) Published by Oxford University Press. Out of all the combinations, we consider only those fusion classification which found in primary transcripts. CDR-CDR Non CDR-CDR In-frame Out-of-frame 5 UTR to CDR 5 UTR to 3 UTR 3 UTR to 3 UTR 5 UTR to 5 UTR 3 UTR to 5 UTR CDR to 5 UTR CDR to 3 UTR
29 Preprocessing.bam file [PAIRED END].fastq files [END1 & END2] INPUTS Config.txt [location of scripts and reference files] Expression & QC Module Fusion Module GUESS-ft -genea -geneb Processing Module RNA-SeQC Read Alignment Remap alignments Combine two ends Quality Scores Recalibrate d OUTPUTS RPKM & QC metrics Fusion Candidates Supervised search evidence Implementation Results Samples processed >400 KIRC >170 GBM Works well in MDACC HPC* system PRADA-fusion module validation rate ~85 % (11 out of 13)
30 KIRC fusion results We analyzed 416 RNA-seq samples from clear cell renal carcinoma (ccrcc), available through TCGA. We identified 80 bona-fide fusion transcripts, 57 intrachromosomal 33 interchromosomal in 62 individual samples Recurrent fusions SFPQ-TFE3 (n=5, chr1-chrx) DHX33-NLRP1 (n=2, chr2) TRIP12-SLC16A14 (n=2, chr17) TFG-GRP128 (n=4, chr3)
31 KIRC fusion results Cont d SFPQ-TFE3 TFE3 translocations have been linked to a rare subtype of renal cancer. The five samples harboring a TFE3 fusion did not contain mutations in the ten most frequently mutated genes in ccrcc (PBRM1, PTEN, VHL, SETD2, BAP1, KDM5C, MTOR, ZNF800, PIK3CA, and TP53), except one (in VHL). This suggests that SFPQ-TFE3 fusion plays a unique role in the cancer genomics of these patients.
32 KIRC fusion validation PRADA-fusion module validation rate (11 out of 13) ~85% RT-PCR and FISH assays TFE3-SFPQ was validated in three individual samples Sample ID 5 Gene 3 Gene Discordant Read Pairs Fusion Span Reads Fusion Junction (s) 5 Gene Chr 3 Gene Chr Validated? TCGA-AK A-02R TFE3 SFPQ chrx chr1 Yes TCGA-AK A-02R SFPQ TFE chr1 chrx Yes TCGA-A A-02R C6orf106 LRRC chr6 chr6 Yes TCGA-A A-02R CYP39A1 LEMD chr6 chr6 Yes TCGA-B A-02R FAM172A FHIT chr5 chr3 Yes TCGA-AK A-02R KIAA0802 LRRC chr18 chr1 Yes TCGA-B A-01R GORASP2 WIPF chr2 chr2 Yes TCGA-A A-02R ZNF193 MRPS18A chr6 chr6 Yes TCGA-A A-02R FTSJD2 GPX chr6 chr6 Yes TCGA-B A-01R KIAA0427 GRM chr18 chr6 No TCGA-B A-01R SLC36A1 TTC chr5 chr5 No

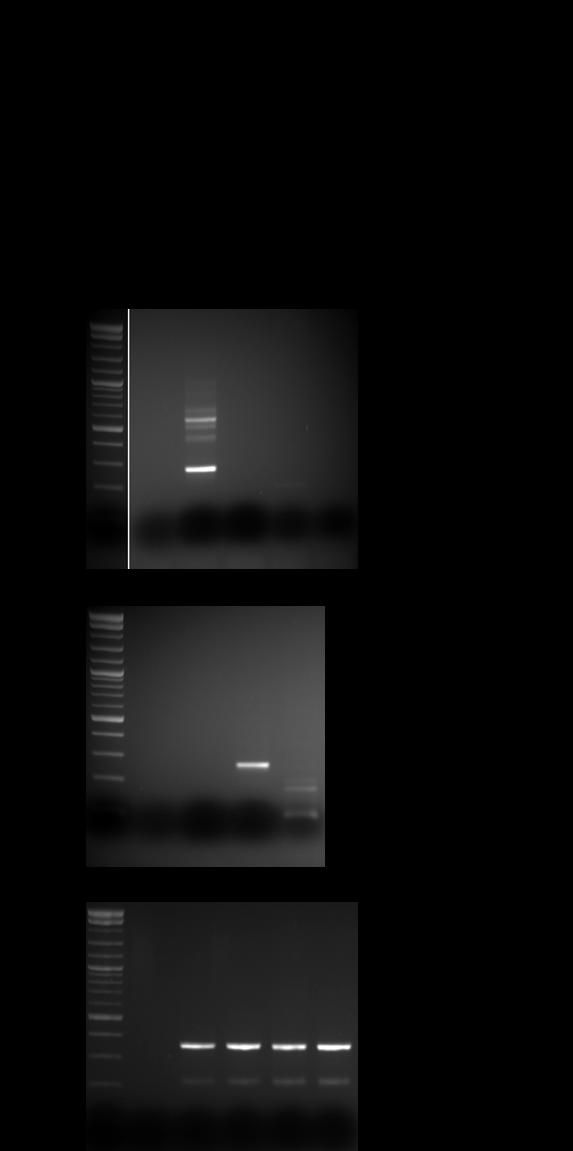
33 KIRC fusion validation: RT-PCR SFPQ-TFE3 TFE3-SFPQ
34 KIRC fusion results We analyzed 416 RNA-seq samples from clear cell renal carcinoma (ccrcc), available through TCGA. We identified 80 bona-fide fusion transcripts, 57 intrachromosomal 33 interchromosomal in 62 individual samples Recurrent fusions SFPQ-TFE3 (n=5, chr1-chrx) DHX33-NLRP1 (n=2, chr2) TRIP12-SLC16A14 (n=2, chr17) TFG-GRP128 (n=4, chr3)
35 TFG-GRP128 has been reported in other cancers
36 TFG-GRP128 has been reported in other cancers

37 TFG-GRP128 has been reported in other cancers TCGA has 1,000s of RNA seq samples - how can we quickly scan many samples for the presence of this fusion?
38 Preprocessing.bam file [PAIRED END] INPUTS Supervised Search Module.fastq files Read Alignment Search Processing for fusion Module transcripts Remap alignments Config.txt [location of scripts and reference files] [END1 & END2] GUESS-ft: General User defined Supervised Use high quality mapping reads only, Checks read orientation fulfills fusion schema, allow up to one mismatch. Two read ends map to A and B respectively Summary report BAM Combine two ends GUESS-ft OUTPUTS Mapped to A or B Discordant reads A-B Quality Scores Recalibrate d Unmapped reads Junction DB Junction spanning reads Expression & QC Module RNA-SeQC Time consuming step Fusion Module RPKM & Fusion Parse QC metrics Candidates Unmapped reads with the other end mapping to A or B Map parsed reads to DB of all possible exon junctions List reads with one end map to junction, the other map to A or B GUESS-ft -genea -geneb Supervised search evidence
39 Tumors with the fusion have higher GPR128 expression levels RPKM expression pattern seen in KIRC tumors Fusion sample(s) Higher expression of GPR128 (activation) TCGA-B w/ 1 discordant read pair in tumor sample w/ 33 discordant read pair in matched normal
40 Identification of TFG-GRP128 fusion All available normal samples in cghub Subset of tumor samples selected based on RPKM expression pattern Table. Samples across cancer types Cancer Type # of normal samples # of tumor samples Bladder Urothelial Carcinoma [BLCA] 11 4 Breast invasive carcinoma [BRCA] Head and Neck squamous cell carcinoma [HNSC] Kidney renal clear cell carcinoma [KIRC] * Kidney renal papillary cell carcinoma [KIRP] 15 4 Liver hepatocellular carcinoma [LIHC] 9 2 Lung adenocarcinoma [LUAD] 51 4 Lung squamous cell carcinoma [LUSC] Prostate adenocarcinoma [PRAD] 7 7 Thyroid carcinoma [THCA] 12 4 * All performed by PRADA fusion module.
41 Identification of TFG-GRP128 fusion All available normal samples in cghub Subset of tumor samples selected based on RPKM expression pattern Table. Samples across cancer types Cancer Type # of normal samples # of tumor samples Bladder Urothelial Carcinoma [BLCA] 0 (0%) 2 (3.6%) Breast invasive carcinoma [BRCA] 1 (0.94%) 13 (1.6%) Head and Neck squamous cell carcinoma [HNSC] 0 (0%) 6 (2.3%) Kidney renal clear cell carcinoma [KIRC] 1 (1.5%) 5 (1.2%) Kidney renal papillary cell carcinoma [KIRP] 0 (0%) 1 (5.9%) Liver hepatocellular carcinoma [LIHC] 0 (0%) 1 (5.9%) Lung adenocarcinoma [LUAD] 0 (0%) 1 (0.79%) Lung squamous cell carcinoma [LUSC] 0 (0%) 9 (4%) Prostate adenocarcinoma [PRAD] 1 (14.3) 2 (1.9%) Thyroid carcinoma [THCA] 0 (0%) 2 (0.89%) * All performed by PRADA fusion module.

42 GUESS-ft module: TFG-GPR128 fusion Cont d Raw Copy Number for KIRC Focal amplification in chr3 (TFG-GPR128)


43 GUESS-ft module: TFG-GPR128 fusion Cont d GWAS
44 In GBM, the gene EGFR is frequently targeted by intragenic deletions Figure. GBM Alterations in EGFR
45 Preprocessing.bam file [PAIRED END] INPUTS Supervised Search Module.fastq files Config.txt [location of scripts and reference files] [END1 & END2] GUESS-ig: GUESS for intragenic rearrangements Processing Module BAM A-A Expression & QC Module RNA-SeQC Fusion Module GUESS-ft -genea -geneb Read Alignment Remap alignments Combine two ends GUESS-IG Quality Scores Recalibrate d Mapped to A OUTPUTS Unmapped reads RPKM & QC metrics Parse Unmapped reads with the other end map to A Fusion Candidates Supervised search evidence Discordant reads Junction DB Map parsed reads to DB of undefined junctions* Summary report Junction spanning reads List reads with one end map to undefined junction, the other maps to A

46 Applying GUESS-ig in GBM identifies intragenic deletion variants Figure. GBM Alterations in EGFR

47 Thanks.
RNA SEQUENCING AND DATA ANALYSIS
 RNA SEQUENCING AND DATA ANALYSIS Length of mrna transcripts in the human genome 5,000 5,000 4,000 3,000 2,000 4,000 1,000 0 0 200 400 600 800 3,000 2,000 1,000 0 0 2,000 4,000 6,000 8,000 10,000 Length
RNA SEQUENCING AND DATA ANALYSIS Length of mrna transcripts in the human genome 5,000 5,000 4,000 3,000 2,000 4,000 1,000 0 0 200 400 600 800 3,000 2,000 1,000 0 0 2,000 4,000 6,000 8,000 10,000 Length
Analysis of Massively Parallel Sequencing Data Application of Illumina Sequencing to the Genetics of Human Cancers
 Analysis of Massively Parallel Sequencing Data Application of Illumina Sequencing to the Genetics of Human Cancers Gordon Blackshields Senior Bioinformatician Source BioScience 1 To Cancer Genetics Studies
Analysis of Massively Parallel Sequencing Data Application of Illumina Sequencing to the Genetics of Human Cancers Gordon Blackshields Senior Bioinformatician Source BioScience 1 To Cancer Genetics Studies
genomics for systems biology / ISB2020 RNA sequencing (RNA-seq)
") RNA sequencing (RNA-seq) Module Outline MO 13-Mar-2017 RNA sequencing: Introduction 1 WE 15-Mar-2017 RNA sequencing: Introduction 2 MO 20-Mar-2017 Paper: PMID 25954002: Human genomics. The human transcriptome
RNA sequencing (RNA-seq) Module Outline MO 13-Mar-2017 RNA sequencing: Introduction 1 WE 15-Mar-2017 RNA sequencing: Introduction 2 MO 20-Mar-2017 Paper: PMID 25954002: Human genomics. The human transcriptome
Machine-Learning on Prediction of Inherited Genomic Susceptibility for 20 Major Cancers
 Machine-Learning on Prediction of Inherited Genomic Susceptibility for 20 Major Cancers Sung-Hou Kim University of California Berkeley, CA Global Bio Conference 2017 MFDS, Seoul, Korea June 28, 2017 Cancer
Machine-Learning on Prediction of Inherited Genomic Susceptibility for 20 Major Cancers Sung-Hou Kim University of California Berkeley, CA Global Bio Conference 2017 MFDS, Seoul, Korea June 28, 2017 Cancer
BWA alignment to reference transcriptome and genome. Convert transcriptome mappings back to genome space
 Whole genome sequencing Whole exome sequencing BWA alignment to reference transcriptome and genome Convert transcriptome mappings back to genome space genomes Filter on MQ, distance, Cigar string Annotate
Whole genome sequencing Whole exome sequencing BWA alignment to reference transcriptome and genome Convert transcriptome mappings back to genome space genomes Filter on MQ, distance, Cigar string Annotate
Iso-Seq Method Updates and Target Enrichment Without Amplification for SMRT Sequencing
 Iso-Seq Method Updates and Target Enrichment Without Amplification for SMRT Sequencing PacBio Americas User Group Meeting Sample Prep Workshop June.27.2017 Tyson Clark, Ph.D. For Research Use Only. Not
Iso-Seq Method Updates and Target Enrichment Without Amplification for SMRT Sequencing PacBio Americas User Group Meeting Sample Prep Workshop June.27.2017 Tyson Clark, Ph.D. For Research Use Only. Not
Exploring TCGA Pan-Cancer Data at the UCSC Cancer Genomics Browser
 Exploring TCGA Pan-Cancer Data at the UCSC Cancer Genomics Browser Melissa S. Cline 1*, Brian Craft 1, Teresa Swatloski 1, Mary Goldman 1, Singer Ma 1, David Haussler 1, Jingchun Zhu 1 1 Center for Biomolecular
Exploring TCGA Pan-Cancer Data at the UCSC Cancer Genomics Browser Melissa S. Cline 1*, Brian Craft 1, Teresa Swatloski 1, Mary Goldman 1, Singer Ma 1, David Haussler 1, Jingchun Zhu 1 1 Center for Biomolecular
Nature Genetics: doi: /ng Supplementary Figure 1. Workflow of CDR3 sequence assembly from RNA-seq data.
 Supplementary Figure 1 Workflow of CDR3 sequence assembly from RNA-seq data. Paired-end short-read RNA-seq data were mapped to human reference genome hg19, and unmapped reads in the TCR regions were extracted
Supplementary Figure 1 Workflow of CDR3 sequence assembly from RNA-seq data. Paired-end short-read RNA-seq data were mapped to human reference genome hg19, and unmapped reads in the TCR regions were extracted
Transcriptome Analysis
 Transcriptome Analysis Data Preprocessing Sample Preparation Illumina Sequencing Demultiplexing Raw FastQ Reference Genome (fasta) Reference Annotation (GTF) Reference Genome Analysis Tophat Accepted hits
Transcriptome Analysis Data Preprocessing Sample Preparation Illumina Sequencing Demultiplexing Raw FastQ Reference Genome (fasta) Reference Annotation (GTF) Reference Genome Analysis Tophat Accepted hits
Selective depletion of abundant RNAs to enable transcriptome analysis of lowinput and highly-degraded RNA from FFPE breast cancer samples
 DNA CLONING DNA AMPLIFICATION & PCR EPIGENETICS RNA ANALYSIS Selective depletion of abundant RNAs to enable transcriptome analysis of lowinput and highly-degraded RNA from FFPE breast cancer samples LIBRARY
DNA CLONING DNA AMPLIFICATION & PCR EPIGENETICS RNA ANALYSIS Selective depletion of abundant RNAs to enable transcriptome analysis of lowinput and highly-degraded RNA from FFPE breast cancer samples LIBRARY
Transcript reconstruction
 Transcript reconstruction Summary I Data types, file formats and utilities Annotation: Genomic regions Genes Peaks bedtools Alignment: Map reads BAM/SAM Samtools Aggregation: Summary files Wig (UCSC) TDF
Transcript reconstruction Summary I Data types, file formats and utilities Annotation: Genomic regions Genes Peaks bedtools Alignment: Map reads BAM/SAM Samtools Aggregation: Summary files Wig (UCSC) TDF
ncounter Assay Automated Process Immobilize and align reporter for image collecting and barcode counting ncounter Prep Station
 ncounter Assay ncounter Prep Station Automated Process Hybridize Reporter to RNA Remove excess reporters Bind reporter to surface Immobilize and align reporter Image surface Count codes Immobilize and
ncounter Assay ncounter Prep Station Automated Process Hybridize Reporter to RNA Remove excess reporters Bind reporter to surface Immobilize and align reporter Image surface Count codes Immobilize and
File Name: Supplementary Information Description: Supplementary Figures and Supplementary Tables. File Name: Peer Review File Description:
 File Name: Supplementary Information Description: Supplementary Figures and Supplementary Tables File Name: Peer Review File Description: Primer Name Sequence (5'-3') AT ( C) RT-PCR USP21 F 5'-TTCCCATGGCTCCTTCCACATGAT-3'
File Name: Supplementary Information Description: Supplementary Figures and Supplementary Tables File Name: Peer Review File Description: Primer Name Sequence (5'-3') AT ( C) RT-PCR USP21 F 5'-TTCCCATGGCTCCTTCCACATGAT-3'
TCGA. The Cancer Genome Atlas
 TCGA The Cancer Genome Atlas TCGA: History and Goal History: Started in 2005 by the National Cancer Institute (NCI) and the National Human Genome Research Institute (NHGRI) with $110 Million to catalogue
TCGA The Cancer Genome Atlas TCGA: History and Goal History: Started in 2005 by the National Cancer Institute (NCI) and the National Human Genome Research Institute (NHGRI) with $110 Million to catalogue
ncounter Assay Automated Process Capture & Reporter Probes Bind reporter to surface Remove excess reporters Hybridize CodeSet to RNA
 ncounter Assay Automated Process Hybridize CodeSet to RNA Remove excess reporters Bind reporter to surface Immobilize and align reporter Image surface Count codes mrna Capture & Reporter Probes slides
ncounter Assay Automated Process Hybridize CodeSet to RNA Remove excess reporters Bind reporter to surface Immobilize and align reporter Image surface Count codes mrna Capture & Reporter Probes slides
Supplementary Figures
 Supplementary Figures Supplementary Figure 1. Pan-cancer analysis of global and local DNA methylation variation a) Variations in global DNA methylation are shown as measured by averaging the genome-wide
Supplementary Figures Supplementary Figure 1. Pan-cancer analysis of global and local DNA methylation variation a) Variations in global DNA methylation are shown as measured by averaging the genome-wide
The Cancer Genome Atlas Pan-cancer analysis Katherine A. Hoadley
 The Cancer Genome Atlas Pan-cancer analysis Katherine A. Hoadley Department of Genetics Lineberger Comprehensive Cancer Center The University of North Carolina at Chapel Hill What is TCGA? The Cancer Genome
The Cancer Genome Atlas Pan-cancer analysis Katherine A. Hoadley Department of Genetics Lineberger Comprehensive Cancer Center The University of North Carolina at Chapel Hill What is TCGA? The Cancer Genome
AVENIO family of NGS oncology assays ctdna and Tumor Tissue Analysis Kits
 AVENIO family of NGS oncology assays ctdna and Tumor Tissue Analysis Kits Accelerating clinical research Next-generation sequencing (NGS) has the ability to interrogate many different genes and detect
AVENIO family of NGS oncology assays ctdna and Tumor Tissue Analysis Kits Accelerating clinical research Next-generation sequencing (NGS) has the ability to interrogate many different genes and detect
SUPPLEMENTARY INFORMATION
 doi: 1.138/nature8645 Physical coverage (x haploid genomes) 11 6.4 4.9 6.9 6.7 4.4 5.9 9.1 7.6 125 Neither end mapped One end mapped Chimaeras Correct Reads (million ns) 1 75 5 25 HCC1187 HCC1395 HCC1599
doi: 1.138/nature8645 Physical coverage (x haploid genomes) 11 6.4 4.9 6.9 6.7 4.4 5.9 9.1 7.6 125 Neither end mapped One end mapped Chimaeras Correct Reads (million ns) 1 75 5 25 HCC1187 HCC1395 HCC1599
BIMM 143. RNA sequencing overview. Genome Informatics II. Barry Grant. Lecture In vivo. In vitro.
 RNA sequencing overview BIMM 143 Genome Informatics II Lecture 14 Barry Grant http://thegrantlab.org/bimm143 In vivo In vitro In silico ( control) Goal: RNA quantification, transcript discovery, variant
RNA sequencing overview BIMM 143 Genome Informatics II Lecture 14 Barry Grant http://thegrantlab.org/bimm143 In vivo In vitro In silico ( control) Goal: RNA quantification, transcript discovery, variant
Breast and ovarian cancer in Serbia: the importance of mutation detection in hereditary predisposition genes using NGS
 Breast and ovarian cancer in Serbia: the importance of mutation detection in hereditary predisposition genes using NGS dr sc. Ana Krivokuća Laboratory for molecular genetics Institute for Oncology and
Breast and ovarian cancer in Serbia: the importance of mutation detection in hereditary predisposition genes using NGS dr sc. Ana Krivokuća Laboratory for molecular genetics Institute for Oncology and
Session 4 Rebecca Poulos
 The Cancer Genome Atlas (TCGA) & International Cancer Genome Consortium (ICGC) Session 4 Rebecca Poulos Prince of Wales Clinical School Introductory bioinformatics for human genomics workshop, UNSW 28
The Cancer Genome Atlas (TCGA) & International Cancer Genome Consortium (ICGC) Session 4 Rebecca Poulos Prince of Wales Clinical School Introductory bioinformatics for human genomics workshop, UNSW 28
Introduction to Systems Biology of Cancer Lecture 2
 Introduction to Systems Biology of Cancer Lecture 2 Gustavo Stolovitzky IBM Research Icahn School of Medicine at Mt Sinai DREAM Challenges High throughput measurements: The age of omics Systems Biology
Introduction to Systems Biology of Cancer Lecture 2 Gustavo Stolovitzky IBM Research Icahn School of Medicine at Mt Sinai DREAM Challenges High throughput measurements: The age of omics Systems Biology
Supplementary Figure 1: LUMP Leukocytes unmethylabon to infer tumor purity
 Supplementary Figure 1: LUMP Leukocytes unmethylabon to infer tumor purity A Consistently unmethylated sites (30%) in 21 cancer types 174,696
Supplementary Figure 1: LUMP Leukocytes unmethylabon to infer tumor purity A Consistently unmethylated sites (30%) in 21 cancer types 174,696
Session 4 Rebecca Poulos
 The Cancer Genome Atlas (TCGA) & International Cancer Genome Consortium (ICGC) Session 4 Rebecca Poulos Prince of Wales Clinical School Introductory bioinformatics for human genomics workshop, UNSW 20
The Cancer Genome Atlas (TCGA) & International Cancer Genome Consortium (ICGC) Session 4 Rebecca Poulos Prince of Wales Clinical School Introductory bioinformatics for human genomics workshop, UNSW 20
RASA: Robust Alternative Splicing Analysis for Human Transcriptome Arrays
 Supplementary Materials RASA: Robust Alternative Splicing Analysis for Human Transcriptome Arrays Junhee Seok 1*, Weihong Xu 2, Ronald W. Davis 2, Wenzhong Xiao 2,3* 1 School of Electrical Engineering,
Supplementary Materials RASA: Robust Alternative Splicing Analysis for Human Transcriptome Arrays Junhee Seok 1*, Weihong Xu 2, Ronald W. Davis 2, Wenzhong Xiao 2,3* 1 School of Electrical Engineering,
Variant Classification. Author: Mike Thiesen, Golden Helix, Inc.
 Variant Classification Author: Mike Thiesen, Golden Helix, Inc. Overview Sequencing pipelines are able to identify rare variants not found in catalogs such as dbsnp. As a result, variants in these datasets
Variant Classification Author: Mike Thiesen, Golden Helix, Inc. Overview Sequencing pipelines are able to identify rare variants not found in catalogs such as dbsnp. As a result, variants in these datasets
PSSV User Manual (V2.1)
") PSSV User Manual (V2.1) 1. Introduction A novel pattern-based probabilistic approach, PSSV, is developed to identify somatic structural variations from WGS data. Specifically, discordant and concordant
PSSV User Manual (V2.1) 1. Introduction A novel pattern-based probabilistic approach, PSSV, is developed to identify somatic structural variations from WGS data. Specifically, discordant and concordant
Whole Genome and Transcriptome Analysis of Anaplastic Meningioma. Patrick Tarpey Cancer Genome Project Wellcome Trust Sanger Institute
 Whole Genome and Transcriptome Analysis of Anaplastic Meningioma Patrick Tarpey Cancer Genome Project Wellcome Trust Sanger Institute Outline Anaplastic meningioma compared to other cancers Whole genomes
Whole Genome and Transcriptome Analysis of Anaplastic Meningioma Patrick Tarpey Cancer Genome Project Wellcome Trust Sanger Institute Outline Anaplastic meningioma compared to other cancers Whole genomes
RNA-seq Introduction
 RNA-seq Introduction DNA is the same in all cells but which RNAs that is present is different in all cells There is a wide variety of different functional RNAs Which RNAs (and sometimes then translated
RNA-seq Introduction DNA is the same in all cells but which RNAs that is present is different in all cells There is a wide variety of different functional RNAs Which RNAs (and sometimes then translated
CRISPR/Cas9 Enrichment and Long-read WGS for Structural Variant Discovery
 CRISPR/Cas9 Enrichment and Long-read WGS for Structural Variant Discovery PacBio CoLab Session October 20, 2017 For Research Use Only. Not for use in diagnostics procedures. Copyright 2017 by Pacific Biosciences
CRISPR/Cas9 Enrichment and Long-read WGS for Structural Variant Discovery PacBio CoLab Session October 20, 2017 For Research Use Only. Not for use in diagnostics procedures. Copyright 2017 by Pacific Biosciences
Supplementary Figure 1. Copy Number Alterations TP53 Mutation Type. C-class TP53 WT. TP53 mut. Nature Genetics: doi: /ng.
 Supplementary Figure a Copy Number Alterations in M-class b TP53 Mutation Type Recurrent Copy Number Alterations 8 6 4 2 TP53 WT TP53 mut TP53-mutated samples (%) 7 6 5 4 3 2 Missense Truncating M-class
Supplementary Figure a Copy Number Alterations in M-class b TP53 Mutation Type Recurrent Copy Number Alterations 8 6 4 2 TP53 WT TP53 mut TP53-mutated samples (%) 7 6 5 4 3 2 Missense Truncating M-class
Deploying the full transcriptome using RNA sequencing. Jo Vandesompele, CSO and co-founder The Non-Coding Genome May 12, 2016, Leuven
 Deploying the full transcriptome using RNA sequencing Jo Vandesompele, CSO and co-founder The Non-Coding Genome May 12, 2016, Leuven Roadmap Biogazelle the power of RNA reasons to study non-coding RNA
Deploying the full transcriptome using RNA sequencing Jo Vandesompele, CSO and co-founder The Non-Coding Genome May 12, 2016, Leuven Roadmap Biogazelle the power of RNA reasons to study non-coding RNA
Pan-cancer analysis of expressed somatic nucleotide variants in long intergenic non-coding RNA
 Pan-cancer analysis of expressed somatic nucleotide variants in long intergenic non-coding RNA Travers Ching 1,2, Lana X. Garmire 1,2 1 Molecular Biosciences and Bioengineering Graduate Program, University
Pan-cancer analysis of expressed somatic nucleotide variants in long intergenic non-coding RNA Travers Ching 1,2, Lana X. Garmire 1,2 1 Molecular Biosciences and Bioengineering Graduate Program, University
Fluxion Biosciences and Swift Biosciences Somatic variant detection from liquid biopsy samples using targeted NGS
 APPLICATION NOTE Fluxion Biosciences and Swift Biosciences OVERVIEW This application note describes a robust method for detecting somatic mutations from liquid biopsy samples by combining circulating tumor
APPLICATION NOTE Fluxion Biosciences and Swift Biosciences OVERVIEW This application note describes a robust method for detecting somatic mutations from liquid biopsy samples by combining circulating tumor
The Cancer Genome Atlas & International Cancer Genome Consortium
 The Cancer Genome Atlas & International Cancer Genome Consortium Session 3 Dr Jason Wong Prince of Wales Clinical School Introductory bioinformatics for human genomics workshop, UNSW 31 st July 2014 1
The Cancer Genome Atlas & International Cancer Genome Consortium Session 3 Dr Jason Wong Prince of Wales Clinical School Introductory bioinformatics for human genomics workshop, UNSW 31 st July 2014 1
ChIP-seq hands-on. Iros Barozzi, Campus IFOM-IEO (Milan) Saverio Minucci, Gioacchino Natoli Labs
 Saverio Minucci, Gioacchino Natoli Labs") ChIP-seq hands-on Iros Barozzi, Campus IFOM-IEO (Milan) Saverio Minucci, Gioacchino Natoli Labs Main goals Becoming familiar with essential tools and formats Visualizing and contextualizing raw data Understand
ChIP-seq hands-on Iros Barozzi, Campus IFOM-IEO (Milan) Saverio Minucci, Gioacchino Natoli Labs Main goals Becoming familiar with essential tools and formats Visualizing and contextualizing raw data Understand
Characterisation of structural variation in breast. cancer genomes using paired-end sequencing on. the Illumina Genome Analyser
 Characterisation of structural variation in breast cancer genomes using paired-end sequencing on the Illumina Genome Analyser Phil Stephens Cancer Genome Project Why is it important to study cancer? Why
Characterisation of structural variation in breast cancer genomes using paired-end sequencing on the Illumina Genome Analyser Phil Stephens Cancer Genome Project Why is it important to study cancer? Why
Advance Your Genomic Research Using Targeted Resequencing with SeqCap EZ Library
 Advance Your Genomic Research Using Targeted Resequencing with SeqCap EZ Library Marilou Wijdicks International Product Manager Research For Life Science Research Only. Not for Use in Diagnostic Procedures.
Advance Your Genomic Research Using Targeted Resequencing with SeqCap EZ Library Marilou Wijdicks International Product Manager Research For Life Science Research Only. Not for Use in Diagnostic Procedures.
MODULE 4: SPLICING. Removal of introns from messenger RNA by splicing
 Last update: 05/10/2017 MODULE 4: SPLICING Lesson Plan: Title MEG LAAKSO Removal of introns from messenger RNA by splicing Objectives Identify splice donor and acceptor sites that are best supported by
Last update: 05/10/2017 MODULE 4: SPLICING Lesson Plan: Title MEG LAAKSO Removal of introns from messenger RNA by splicing Objectives Identify splice donor and acceptor sites that are best supported by
PSSV User Manual (V1.0)
") PSSV User Manual (V1.0) 1. Introduction A novel pattern-based probabilistic approach, PSSV, is developed to identify somatic structural variations from WGS data. Specifically, discordant and concordant
PSSV User Manual (V1.0) 1. Introduction A novel pattern-based probabilistic approach, PSSV, is developed to identify somatic structural variations from WGS data. Specifically, discordant and concordant
Computational Analysis of UHT Sequences Histone modifications, CAGE, RNA-Seq
 Computational Analysis of UHT Sequences Histone modifications, CAGE, RNA-Seq Philipp Bucher Wednesday January 21, 2009 SIB graduate school course EPFL, Lausanne ChIP-seq against histone variants: Biological
Computational Analysis of UHT Sequences Histone modifications, CAGE, RNA-Seq Philipp Bucher Wednesday January 21, 2009 SIB graduate school course EPFL, Lausanne ChIP-seq against histone variants: Biological
Ambient temperature regulated flowering time
 Ambient temperature regulated flowering time Applications of RNAseq RNA- seq course: The power of RNA-seq June 7 th, 2013; Richard Immink Overview Introduction: Biological research question/hypothesis
Ambient temperature regulated flowering time Applications of RNAseq RNA- seq course: The power of RNA-seq June 7 th, 2013; Richard Immink Overview Introduction: Biological research question/hypothesis
Supplementary Tables. Supplementary Figures
 Supplementary Files for Zehir, Benayed et al. Mutational Landscape of Metastatic Cancer Revealed from Prospective Clinical Sequencing of 10,000 Patients Supplementary Tables Supplementary Table 1: Sample
Supplementary Files for Zehir, Benayed et al. Mutational Landscape of Metastatic Cancer Revealed from Prospective Clinical Sequencing of 10,000 Patients Supplementary Tables Supplementary Table 1: Sample
Trinity: Transcriptome Assembly for Genetic and Functional Analysis of Cancer [U24]
![Trinity: Transcriptome Assembly for Genetic and Functional Analysis of Cancer [U24]](/thumbs/87/95271099.jpg "Trinity: Transcriptome Assembly for Genetic and Functional Analysis of Cancer [U24]") Trinity: Transcriptome Assembly for Genetic and Functional Analysis of Cancer [U24] ITCR meeting, June 2016 The Cancer Transcriptome A window into the (expressed) genetic and epigenetic state of a tumor
Trinity: Transcriptome Assembly for Genetic and Functional Analysis of Cancer [U24] ITCR meeting, June 2016 The Cancer Transcriptome A window into the (expressed) genetic and epigenetic state of a tumor
MODULE 3: TRANSCRIPTION PART II
 MODULE 3: TRANSCRIPTION PART II Lesson Plan: Title S. CATHERINE SILVER KEY, CHIYEDZA SMALL Transcription Part II: What happens to the initial (premrna) transcript made by RNA pol II? Objectives Explain
MODULE 3: TRANSCRIPTION PART II Lesson Plan: Title S. CATHERINE SILVER KEY, CHIYEDZA SMALL Transcription Part II: What happens to the initial (premrna) transcript made by RNA pol II? Objectives Explain
A Statistical Framework for Classification of Tumor Type from microrna Data
 DEGREE PROJECT IN MATHEMATICS, SECOND CYCLE, 30 CREDITS STOCKHOLM, SWEDEN 2016 A Statistical Framework for Classification of Tumor Type from microrna Data JOSEFINE RÖHSS KTH ROYAL INSTITUTE OF TECHNOLOGY
DEGREE PROJECT IN MATHEMATICS, SECOND CYCLE, 30 CREDITS STOCKHOLM, SWEDEN 2016 A Statistical Framework for Classification of Tumor Type from microrna Data JOSEFINE RÖHSS KTH ROYAL INSTITUTE OF TECHNOLOGY
Simple, rapid, and reliable RNA sequencing
 Simple, rapid, and reliable RNA sequencing RNA sequencing applications RNA sequencing provides fundamental insights into how genomes are organized and regulated, giving us valuable information about the
Simple, rapid, and reliable RNA sequencing RNA sequencing applications RNA sequencing provides fundamental insights into how genomes are organized and regulated, giving us valuable information about the
Circular RNAs (circrnas) act a stable mirna sponges
 act a stable mirna sponges") Circular RNAs (circrnas) act a stable mirna sponges cernas compete for mirnas Ancestal mrna (+3 UTR) Pseudogene RNA (+3 UTR homolgy region) The model holds true for all RNAs that share a mirna binding
Circular RNAs (circrnas) act a stable mirna sponges cernas compete for mirnas Ancestal mrna (+3 UTR) Pseudogene RNA (+3 UTR homolgy region) The model holds true for all RNAs that share a mirna binding
Solving Problems of Clustering and Classification of Cancer Diseases Based on DNA Methylation Data 1,2
 APPLIED PROBLEMS Solving Problems of Clustering and Classification of Cancer Diseases Based on DNA Methylation Data 1,2 A. N. Polovinkin a, I. B. Krylov a, P. N. Druzhkov a, M. V. Ivanchenko a, I. B. Meyerov
APPLIED PROBLEMS Solving Problems of Clustering and Classification of Cancer Diseases Based on DNA Methylation Data 1,2 A. N. Polovinkin a, I. B. Krylov a, P. N. Druzhkov a, M. V. Ivanchenko a, I. B. Meyerov
Module 3: Pathway and Drug Development
 Module 3: Pathway and Drug Development Table of Contents 1.1 Getting Started... 6 1.2 Identifying a Dasatinib sensitive cancer signature... 7 1.2.1 Identifying and validating a Dasatinib Signature... 7
Module 3: Pathway and Drug Development Table of Contents 1.1 Getting Started... 6 1.2 Identifying a Dasatinib sensitive cancer signature... 7 1.2.1 Identifying and validating a Dasatinib Signature... 7
Abstract. Optimization strategy of Copy Number Variant calling using Multiplicom solutions APPLICATION NOTE. Introduction
 Optimization strategy of Copy Number Variant calling using Multiplicom solutions Michael Vyverman, PhD; Laura Standaert, PhD and Wouter Bossuyt, PhD Abstract Copy number variations (CNVs) represent a significant
Optimization strategy of Copy Number Variant calling using Multiplicom solutions Michael Vyverman, PhD; Laura Standaert, PhD and Wouter Bossuyt, PhD Abstract Copy number variations (CNVs) represent a significant
Nature Genetics: doi: /ng Supplementary Figure 1. SEER data for male and female cancer incidence from
 Supplementary Figure 1 SEER data for male and female cancer incidence from 1975 2013. (a,b) Incidence rates of oral cavity and pharynx cancer (a) and leukemia (b) are plotted, grouped by males (blue),
Supplementary Figure 1 SEER data for male and female cancer incidence from 1975 2013. (a,b) Incidence rates of oral cavity and pharynx cancer (a) and leukemia (b) are plotted, grouped by males (blue),
Supplemental Methods RNA sequencing experiment
 Supplemental Methods RNA sequencing experiment Mice were euthanized as described in the Methods and the right lung was removed, placed in a sterile eppendorf tube, and snap frozen in liquid nitrogen. RNA
Supplemental Methods RNA sequencing experiment Mice were euthanized as described in the Methods and the right lung was removed, placed in a sterile eppendorf tube, and snap frozen in liquid nitrogen. RNA
Hands-On Ten The BRCA1 Gene and Protein
 Hands-On Ten The BRCA1 Gene and Protein Objective: To review transcription, translation, reading frames, mutations, and reading files from GenBank, and to review some of the bioinformatics tools, such
Hands-On Ten The BRCA1 Gene and Protein Objective: To review transcription, translation, reading frames, mutations, and reading files from GenBank, and to review some of the bioinformatics tools, such
Aliccia Bollig-Fischer, PhD Department of Oncology, Wayne State University Associate Director Genomics Core Molecular Therapeutics Program Karmanos
 Aliccia Bollig-Fischer, PhD Department of Oncology, Wayne State University Associate Director Genomics Core Molecular Therapeutics Program Karmanos Cancer Institute Development of a multiplexed assay to
Aliccia Bollig-Fischer, PhD Department of Oncology, Wayne State University Associate Director Genomics Core Molecular Therapeutics Program Karmanos Cancer Institute Development of a multiplexed assay to
Genomic structural variation
 Genomic structural variation Mario Cáceres The new genomic variation DNA sequence differs across individuals much more than researchers had suspected through structural changes A huge amount of structural
Genomic structural variation Mario Cáceres The new genomic variation DNA sequence differs across individuals much more than researchers had suspected through structural changes A huge amount of structural
Computer Science, Biology, and Biomedical Informatics (CoSBBI) Outline. Molecular Biology of Cancer AND. Goals/Expectations. David Boone 7/1/2015
 Outline. Molecular Biology of Cancer AND. Goals/Expectations. David Boone 7/1/2015") Goals/Expectations Computer Science, Biology, and Biomedical (CoSBBI) We want to excite you about the world of computer science, biology, and biomedical informatics. Experience what it is like to be a
Goals/Expectations Computer Science, Biology, and Biomedical (CoSBBI) We want to excite you about the world of computer science, biology, and biomedical informatics. Experience what it is like to be a
Lecture 8 Understanding Transcription RNA-seq analysis. Foundations of Computational Systems Biology David K. Gifford
 Lecture 8 Understanding Transcription RNA-seq analysis Foundations of Computational Systems Biology David K. Gifford 1 Lecture 8 RNA-seq Analysis RNA-seq principles How can we characterize mrna isoform
Lecture 8 Understanding Transcription RNA-seq analysis Foundations of Computational Systems Biology David K. Gifford 1 Lecture 8 RNA-seq Analysis RNA-seq principles How can we characterize mrna isoform
AVENIO ctdna Analysis Kits The complete NGS liquid biopsy solution EMPOWER YOUR LAB
 Analysis Kits The complete NGS liquid biopsy solution EMPOWER YOUR LAB Analysis Kits Next-generation performance in liquid biopsies 2 Accelerating clinical research From liquid biopsy to next-generation
Analysis Kits The complete NGS liquid biopsy solution EMPOWER YOUR LAB Analysis Kits Next-generation performance in liquid biopsies 2 Accelerating clinical research From liquid biopsy to next-generation
ACE ImmunoID Biomarker Discovery Solutions ACE ImmunoID Platform for Tumor Immunogenomics
 ACE ImmunoID Biomarker Discovery Solutions ACE ImmunoID Platform for Tumor Immunogenomics Precision Genomics for Immuno-Oncology Personalis, Inc. ACE ImmunoID When one biomarker doesn t tell the whole
ACE ImmunoID Biomarker Discovery Solutions ACE ImmunoID Platform for Tumor Immunogenomics Precision Genomics for Immuno-Oncology Personalis, Inc. ACE ImmunoID When one biomarker doesn t tell the whole
TCGA-Assembler: Pipeline for TCGA Data Downloading, Assembling, and Processing. (Supplementary Methods)
") TCGA-Assembler: Pipeline for TCGA Data Downloading, Assembling, and Processing (Supplementary Methods) Yitan Zhu 1, Peng Qiu 2, Yuan Ji 1,3 * 1. Center for Biomedical Research Informatics, NorthShore University
TCGA-Assembler: Pipeline for TCGA Data Downloading, Assembling, and Processing (Supplementary Methods) Yitan Zhu 1, Peng Qiu 2, Yuan Ji 1,3 * 1. Center for Biomedical Research Informatics, NorthShore University
NGS in tissue and liquid biopsy
 NGS in tissue and liquid biopsy Ana Vivancos, PhD Referencias So, why NGS in the clinics? 2000 Sanger Sequencing (1977-) 2016 NGS (2006-) ABIPrism (Applied Biosystems) Up to 2304 per day (96 sequences
NGS in tissue and liquid biopsy Ana Vivancos, PhD Referencias So, why NGS in the clinics? 2000 Sanger Sequencing (1977-) 2016 NGS (2006-) ABIPrism (Applied Biosystems) Up to 2304 per day (96 sequences
A Practical Guide to Integrative Genomics by RNA-seq and ChIP-seq Analysis
 A Practical Guide to Integrative Genomics by RNA-seq and ChIP-seq Analysis Jian Xu, Ph.D. Children s Research Institute, UTSW Introduction Outline Overview of genomic and next-gen sequencing technologies
A Practical Guide to Integrative Genomics by RNA-seq and ChIP-seq Analysis Jian Xu, Ph.D. Children s Research Institute, UTSW Introduction Outline Overview of genomic and next-gen sequencing technologies
Mutation Detection and CNV Analysis for Illumina Sequencing data from HaloPlex Target Enrichment Panels using NextGENe Software for Clinical Research
 Mutation Detection and CNV Analysis for Illumina Sequencing data from HaloPlex Target Enrichment Panels using NextGENe Software for Clinical Research Application Note Authors John McGuigan, Megan Manion,
Mutation Detection and CNV Analysis for Illumina Sequencing data from HaloPlex Target Enrichment Panels using NextGENe Software for Clinical Research Application Note Authors John McGuigan, Megan Manion,
Supplemental Data. Integrating omics and alternative splicing i reveals insights i into grape response to high temperature
 Supplemental Data Integrating omics and alternative splicing i reveals insights i into grape response to high temperature Jianfu Jiang 1, Xinna Liu 1, Guotian Liu, Chonghuih Liu*, Shaohuah Li*, and Lijun
Supplemental Data Integrating omics and alternative splicing i reveals insights i into grape response to high temperature Jianfu Jiang 1, Xinna Liu 1, Guotian Liu, Chonghuih Liu*, Shaohuah Li*, and Lijun
User s Manual Version 1.0
 User s Manual Version 1.0 #639 Longmian Avenue, Jiangning District, Nanjing,211198,P.R.China. http://tcoa.cpu.edu.cn/ Contact us at xiaosheng.wang@cpu.edu.cn for technical issue and questions Catalogue
User s Manual Version 1.0 #639 Longmian Avenue, Jiangning District, Nanjing,211198,P.R.China. http://tcoa.cpu.edu.cn/ Contact us at xiaosheng.wang@cpu.edu.cn for technical issue and questions Catalogue
The Cancer Genome Atlas
 The Cancer Genome Atlas July 14, 2011 Kenna M. Shaw, Ph.D. Deputy Director The Cancer Genome Atlas Program TCGA: Core Objectives Launched in 2006 as a pilot and expanded in 2009, the goals of TCGA are
The Cancer Genome Atlas July 14, 2011 Kenna M. Shaw, Ph.D. Deputy Director The Cancer Genome Atlas Program TCGA: Core Objectives Launched in 2006 as a pilot and expanded in 2009, the goals of TCGA are
Supplementary Information
 Supplementary Information Guided Visual Exploration of Genomic Stratifications in Cancer Marc Streit 1,6, Alexander Lex 2,6, Samuel Gratzl¹, Christian Partl³, Dieter Schmalstieg³, Hanspeter Pfister², Peter
Supplementary Information Guided Visual Exploration of Genomic Stratifications in Cancer Marc Streit 1,6, Alexander Lex 2,6, Samuel Gratzl¹, Christian Partl³, Dieter Schmalstieg³, Hanspeter Pfister², Peter
Patnaik SK, et al. MicroRNAs to accurately histotype NSCLC biopsies
 Patnaik SK, et al. MicroRNAs to accurately histotype NSCLC biopsies. 2014. Supplemental Digital Content 1. Appendix 1. External data-sets used for associating microrna expression with lung squamous cell
Patnaik SK, et al. MicroRNAs to accurately histotype NSCLC biopsies. 2014. Supplemental Digital Content 1. Appendix 1. External data-sets used for associating microrna expression with lung squamous cell
Structural Variation and Medical Genomics
 Structural Variation and Medical Genomics Andrew King Department of Biomedical Informatics July 8, 2014 You already know about small scale genetic mutations Single nucleotide polymorphism (SNPs) Deletions,
Structural Variation and Medical Genomics Andrew King Department of Biomedical Informatics July 8, 2014 You already know about small scale genetic mutations Single nucleotide polymorphism (SNPs) Deletions,
Nature Getetics: doi: /ng.3471
 Supplementary Figure 1 Summary of exome sequencing data. ( a ) Exome tumor normal sample sizes for bladder cancer (BLCA), breast cancer (BRCA), carcinoid (CARC), chronic lymphocytic leukemia (CLLX), colorectal
Supplementary Figure 1 Summary of exome sequencing data. ( a ) Exome tumor normal sample sizes for bladder cancer (BLCA), breast cancer (BRCA), carcinoid (CARC), chronic lymphocytic leukemia (CLLX), colorectal
Using the Bravo Liquid-Handling System for Next Generation Sequencing Sample Prep
 Using the Bravo Liquid-Handling System for Next Generation Sequencing Sample Prep Tom Walsh, PhD Division of Medical Genetics University of Washington Next generation sequencing Sanger sequencing gold
Using the Bravo Liquid-Handling System for Next Generation Sequencing Sample Prep Tom Walsh, PhD Division of Medical Genetics University of Washington Next generation sequencing Sanger sequencing gold
The 16th KJC Bioinformatics Symposium Integrative analysis identifies potential DNA methylation biomarkers for pan-cancer diagnosis and prognosis
 The 16th KJC Bioinformatics Symposium Integrative analysis identifies potential DNA methylation biomarkers for pan-cancer diagnosis and prognosis Tieliu Shi tlshi@bio.ecnu.edu.cn The Center for bioinformatics
The 16th KJC Bioinformatics Symposium Integrative analysis identifies potential DNA methylation biomarkers for pan-cancer diagnosis and prognosis Tieliu Shi tlshi@bio.ecnu.edu.cn The Center for bioinformatics
RNA-Seq Preparation Comparision Summary: Lexogen, Standard, NEB
 RNA-Seq Preparation Comparision Summary: Lexogen, Standard, NEB CSF-NGS January 22, 214 Contents 1 Introduction 1 2 Experimental Details 1 3 Results And Discussion 1 3.1 ERCC spike ins............................................
RNA-Seq Preparation Comparision Summary: Lexogen, Standard, NEB CSF-NGS January 22, 214 Contents 1 Introduction 1 2 Experimental Details 1 3 Results And Discussion 1 3.1 ERCC spike ins............................................
Accessing and Using ENCODE Data Dr. Peggy J. Farnham
 1 William M Keck Professor of Biochemistry Keck School of Medicine University of Southern California How many human genes are encoded in our 3x10 9 bp? C. elegans (worm) 959 cells and 1x10 8 bp 20,000
1 William M Keck Professor of Biochemistry Keck School of Medicine University of Southern California How many human genes are encoded in our 3x10 9 bp? C. elegans (worm) 959 cells and 1x10 8 bp 20,000
Figure S4. 15 Mets Whole Exome. 5 Primary Tumors Cancer Panel and WES. Next Generation Sequencing
 Figure S4 Next Generation Sequencing 15 Mets Whole Exome 5 Primary Tumors Cancer Panel and WES Get coverage of all variant loci for all three Mets Variant Filtering Sequence Alignments Index and align
Figure S4 Next Generation Sequencing 15 Mets Whole Exome 5 Primary Tumors Cancer Panel and WES Get coverage of all variant loci for all three Mets Variant Filtering Sequence Alignments Index and align
Dr Rick Tearle Senior Applications Specialist, EMEA Complete Genomics Complete Genomics, Inc.
 Dr Rick Tearle Senior Applications Specialist, EMEA Complete Genomics Topics Overview of Data Processing Pipeline Overview of Data Files 2 DNA Nano-Ball (DNB) Read Structure Genome : acgtacatgcattcacacatgcttagctatctctcgccag
Dr Rick Tearle Senior Applications Specialist, EMEA Complete Genomics Topics Overview of Data Processing Pipeline Overview of Data Files 2 DNA Nano-Ball (DNB) Read Structure Genome : acgtacatgcattcacacatgcttagctatctctcgccag
OncoPPi Portal A Cancer Protein Interaction Network to Inform Therapeutic Strategies
 OncoPPi Portal A Cancer Protein Interaction Network to Inform Therapeutic Strategies 2017 Contents Datasets... 2 Protein-protein interaction dataset... 2 Set of known PPIs... 3 Domain-domain interactions...
OncoPPi Portal A Cancer Protein Interaction Network to Inform Therapeutic Strategies 2017 Contents Datasets... 2 Protein-protein interaction dataset... 2 Set of known PPIs... 3 Domain-domain interactions...
NGS in Cancer Pathology After the Microscope: From Nucleic Acid to Interpretation
 NGS in Cancer Pathology After the Microscope: From Nucleic Acid to Interpretation Michael R. Rossi, PhD, FACMG Assistant Professor Division of Cancer Biology, Department of Radiation Oncology Department
NGS in Cancer Pathology After the Microscope: From Nucleic Acid to Interpretation Michael R. Rossi, PhD, FACMG Assistant Professor Division of Cancer Biology, Department of Radiation Oncology Department
DNA-seq Bioinformatics Analysis: Copy Number Variation
 DNA-seq Bioinformatics Analysis: Copy Number Variation Elodie Girard elodie.girard@curie.fr U900 institut Curie, INSERM, Mines ParisTech, PSL Research University Paris, France NGS Applications 5C HiC DNA-seq
DNA-seq Bioinformatics Analysis: Copy Number Variation Elodie Girard elodie.girard@curie.fr U900 institut Curie, INSERM, Mines ParisTech, PSL Research University Paris, France NGS Applications 5C HiC DNA-seq
A complete next-generation sequencing workfl ow for circulating cell-free DNA isolation and analysis
 APPLICATION NOTE Cell-Free DNA Isolation Kit A complete next-generation sequencing workfl ow for circulating cell-free DNA isolation and analysis Abstract Circulating cell-free DNA (cfdna) has been shown
APPLICATION NOTE Cell-Free DNA Isolation Kit A complete next-generation sequencing workfl ow for circulating cell-free DNA isolation and analysis Abstract Circulating cell-free DNA (cfdna) has been shown
of TERT, MLL4, CCNE1, SENP5, and ROCK1 on tumor development were discussed.
 Supplementary Note The potential association and implications of HBV integration at known and putative cancer genes of TERT, MLL4, CCNE1, SENP5, and ROCK1 on tumor development were discussed. Human telomerase
Supplementary Note The potential association and implications of HBV integration at known and putative cancer genes of TERT, MLL4, CCNE1, SENP5, and ROCK1 on tumor development were discussed. Human telomerase
Supplementary Material for IPred - Integrating Ab Initio and Evidence Based Gene Predictions to Improve Prediction Accuracy
 1 SYSTEM REQUIREMENTS 1 Supplementary Material for IPred - Integrating Ab Initio and Evidence Based Gene Predictions to Improve Prediction Accuracy Franziska Zickmann and Bernhard Y. Renard Research Group
1 SYSTEM REQUIREMENTS 1 Supplementary Material for IPred - Integrating Ab Initio and Evidence Based Gene Predictions to Improve Prediction Accuracy Franziska Zickmann and Bernhard Y. Renard Research Group
Lectures 13: High throughput sequencing: Beyond the genome. Spring 2017 March 28, 2017
 Lectures 13: High throughput sequencing: Beyond the genome Spring 2017 March 28, 2017 h@p://www.fejes.ca/2009/06/science- cartoons- 5- rna- seq.html Omics Transcriptome - the set of all mrnas present in
Lectures 13: High throughput sequencing: Beyond the genome Spring 2017 March 28, 2017 h@p://www.fejes.ca/2009/06/science- cartoons- 5- rna- seq.html Omics Transcriptome - the set of all mrnas present in
MSI positive MSI negative
 Pritchard et al. 2014 Supplementary Figure 1 MSI positive MSI negative Hypermutated Median: 673 Average: 659.2 Non-Hypermutated Median: 37.5 Average: 43.6 Supplementary Figure 1: Somatic Mutation Burden
Pritchard et al. 2014 Supplementary Figure 1 MSI positive MSI negative Hypermutated Median: 673 Average: 659.2 Non-Hypermutated Median: 37.5 Average: 43.6 Supplementary Figure 1: Somatic Mutation Burden
Elevated RNA Editing Activity Is a Major Contributor to Transcriptomic Diversity in Tumors
 Cell Reports Supplemental Information Elevated RNA Editing Activity Is a Major Contributor to Transcriptomic Diversity in s Nurit Paz-Yaacov, Lily Bazak, Ilana Buchumenski, Hagit T. Porath, Miri Danan-Gotthold,
Cell Reports Supplemental Information Elevated RNA Editing Activity Is a Major Contributor to Transcriptomic Diversity in s Nurit Paz-Yaacov, Lily Bazak, Ilana Buchumenski, Hagit T. Porath, Miri Danan-Gotthold,
Cancer Informatics Lecture
 Cancer Informatics Lecture Mayo-UIUC Computational Genomics Course June 22, 2018 Krishna Rani Kalari Ph.D. Associate Professor 2017 MFMER 3702274-1 Outline The Cancer Genome Atlas (TCGA) Genomic Data Commons
Cancer Informatics Lecture Mayo-UIUC Computational Genomics Course June 22, 2018 Krishna Rani Kalari Ph.D. Associate Professor 2017 MFMER 3702274-1 Outline The Cancer Genome Atlas (TCGA) Genomic Data Commons
Data mining with Ensembl Biomart. Stéphanie Le Gras
 Data mining with Ensembl Biomart Stéphanie Le Gras (slegras@igbmc.fr) Guidelines Genome data Genome browsers Getting access to genomic data: Ensembl/BioMart 2 Genome Sequencing Example: Human genome 2000:
Data mining with Ensembl Biomart Stéphanie Le Gras (slegras@igbmc.fr) Guidelines Genome data Genome browsers Getting access to genomic data: Ensembl/BioMart 2 Genome Sequencing Example: Human genome 2000:
EXAMPLE. - Potentially responsive to PI3K/mTOR and MEK combination therapy or mtor/mek and PKC combination therapy. ratio (%)
") Dr Kate Goodhealth Goodhealth Medical Clinic 123 Address Road SUBURBTOWN NSW 2000 Melanie Citizen Referring Doctor Your ref Address Dr John Medico 123 Main Street, SUBURBTOWN NSW 2000 Phone 02 9999 9999
Dr Kate Goodhealth Goodhealth Medical Clinic 123 Address Road SUBURBTOWN NSW 2000 Melanie Citizen Referring Doctor Your ref Address Dr John Medico 123 Main Street, SUBURBTOWN NSW 2000 Phone 02 9999 9999
Inference of Isoforms from Short Sequence Reads
 Inference of Isoforms from Short Sequence Reads Tao Jiang Department of Computer Science and Engineering University of California, Riverside Tsinghua University Joint work with Jianxing Feng and Wei Li
Inference of Isoforms from Short Sequence Reads Tao Jiang Department of Computer Science and Engineering University of California, Riverside Tsinghua University Joint work with Jianxing Feng and Wei Li
Fusion Analysis of Solid Tumors Reveals Novel Rearrangements in Breast Carcinomas
 Fusion Analysis of Solid Tumors Reveals Novel Rearrangements in Breast Carcinomas Igor Astsaturov Philip Ellis Jeff Swensen Zoran Gatalica David Arguello Sandeep Reddy Wafik El-Deiry Disclaimers Dr. Igor
Fusion Analysis of Solid Tumors Reveals Novel Rearrangements in Breast Carcinomas Igor Astsaturov Philip Ellis Jeff Swensen Zoran Gatalica David Arguello Sandeep Reddy Wafik El-Deiry Disclaimers Dr. Igor
Genomic Medicine: What every pathologist needs to know
 Genomic Medicine: What every pathologist needs to know Stephen P. Ethier, Ph.D. Professor, Department of Pathology and Laboratory Medicine, MUSC Director, MUSC Center for Genomic Medicine Genomics and
Genomic Medicine: What every pathologist needs to know Stephen P. Ethier, Ph.D. Professor, Department of Pathology and Laboratory Medicine, MUSC Director, MUSC Center for Genomic Medicine Genomics and
Supplemental Information. Integrated Genomic Analysis of the Ubiquitin. Pathway across Cancer Types
 Cell Reports, Volume 23 Supplemental Information Integrated Genomic Analysis of the Ubiquitin Pathway across Zhongqi Ge, Jake S. Leighton, Yumeng Wang, Xinxin Peng, Zhongyuan Chen, Hu Chen, Yutong Sun,
Cell Reports, Volume 23 Supplemental Information Integrated Genomic Analysis of the Ubiquitin Pathway across Zhongqi Ge, Jake S. Leighton, Yumeng Wang, Xinxin Peng, Zhongyuan Chen, Hu Chen, Yutong Sun,
Copy Number Varia/on Detec/on. Alex Mawla UCD Genome Center Bioinforma5cs Core Tuesday June 16, 2015
 Copy Number Varia/on Detec/on Alex Mawla UCD Genome Center Bioinforma5cs Core Tuesday June 16, 2015 Today s Goals Understand the applica5on and capabili5es of using targe5ng sequencing and CNV calling
Copy Number Varia/on Detec/on Alex Mawla UCD Genome Center Bioinforma5cs Core Tuesday June 16, 2015 Today s Goals Understand the applica5on and capabili5es of using targe5ng sequencing and CNV calling
Expert-guided Visual Exploration (EVE) for patient stratification. Hamid Bolouri, Lue-Ping Zhao, Eric C. Holland
 for patient stratification. Hamid Bolouri, Lue-Ping Zhao, Eric C. Holland") Expert-guided Visual Exploration (EVE) for patient stratification Hamid Bolouri, Lue-Ping Zhao, Eric C. Holland Oncoscape.sttrcancer.org Paul Lisa Ken Jenny Desert Eric The challenge Given - patient clinical
Expert-guided Visual Exploration (EVE) for patient stratification Hamid Bolouri, Lue-Ping Zhao, Eric C. Holland Oncoscape.sttrcancer.org Paul Lisa Ken Jenny Desert Eric The challenge Given - patient clinical
RNA- seq Introduc1on. Promises and pi7alls
 RNA- seq Introduc1on Promises and pi7alls DNA is the same in all cells but which RNAs that is present is different in all cells There is a wide variety of different func1onal RNAs Which RNAs (and some1mes
RNA- seq Introduc1on Promises and pi7alls DNA is the same in all cells but which RNAs that is present is different in all cells There is a wide variety of different func1onal RNAs Which RNAs (and some1mes
Role of FISH in Hematological Cancers
 Role of FISH in Hematological Cancers Thomas S.K. Wan PhD,FRCPath,FFSc(RCPA) Honorary Professor, Department of Pathology & Clinical Biochemistry, Queen Mary Hospital, University of Hong Kong. e-mail: wantsk@hku.hk
Role of FISH in Hematological Cancers Thomas S.K. Wan PhD,FRCPath,FFSc(RCPA) Honorary Professor, Department of Pathology & Clinical Biochemistry, Queen Mary Hospital, University of Hong Kong. e-mail: wantsk@hku.hk
Breast cancer. Risk factors you cannot change include: Treatment Plan Selection. Inferring Transcriptional Module from Breast Cancer Profile Data
 Breast cancer Inferring Transcriptional Module from Breast Cancer Profile Data Breast Cancer and Targeted Therapy Microarray Profile Data Inferring Transcriptional Module Methods CSC 177 Data Warehousing
Breast cancer Inferring Transcriptional Module from Breast Cancer Profile Data Breast Cancer and Targeted Therapy Microarray Profile Data Inferring Transcriptional Module Methods CSC 177 Data Warehousing
Analysis with SureCall 2.1
 Analysis with SureCall 2.1 Danielle Fletcher Field Application Scientist July 2014 1 Stages of NGS Analysis Primary analysis, base calling Control Software FASTQ file reads + quality 2 Stages of NGS Analysis
Analysis with SureCall 2.1 Danielle Fletcher Field Application Scientist July 2014 1 Stages of NGS Analysis Primary analysis, base calling Control Software FASTQ file reads + quality 2 Stages of NGS Analysis