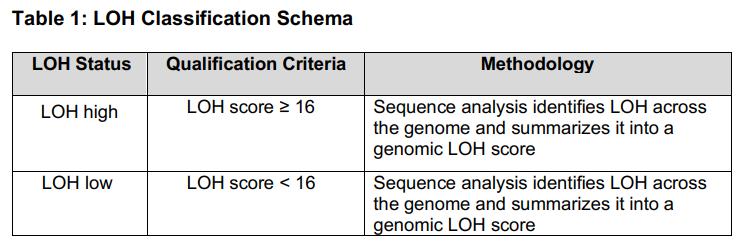
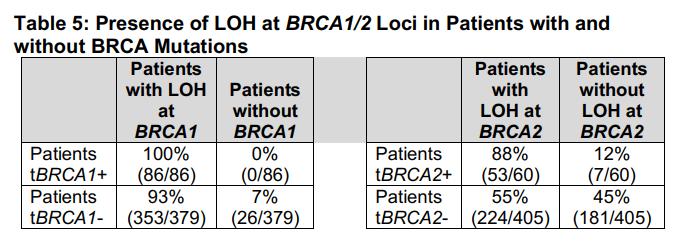
Global Regulatory Landscape of Drug - Companion diagnostics and Revision Proposal of Domestic Guideline
|
|
|
- Bryan Cannon
- 5 years ago
- Views:
Transcription



1 Global Regulatory Landscape of Drug - Companion diagnostics and Revision Proposal of Domestic Guideline 동반진단의약품의해외규제현황및국내가이드라인제안 Young Kee Shin, M.D., Ph.D. College of Pharmacy, Seoul National University N-BIO (Institutes of Entrepreneurial BioConvergence), LOGONE Bio Convergence Research Foundation












2 Paradigm shift : Precision Medicine Precision Diagnostics Patient-first medical care Essential tool Patient:Subject of Healthcare Information Patient InformationDB Healthcare Democratization Regulation Scientification New developed diagnosis test Scientific evidence Patient Safety APPROVED REJECTED Payer-first medical care Risk / Efficacy Cost / Utility
3 Contents 1. Definition of Companion diagnostics (CDx) 2. Global Regulatory trend 3. Korea Regulatory trend 4. Current Regulatory issues in Korea 5. Suggestion
4 1. Definition of Companion Diagnostics



 Joint CBER/ CDRH Mtg.")

 sbla & spma Filed FISH")
5 A Real World Example of Biomarker Application Biomarker HERCEPTIN IHC/FISH CISH Gleevec (Memb. strong) IHC IUO (Herceptest) State Rituxan Phase I IND for rhumab HER2 +2 (<10% memb. strong) Joint CBER/ CDRH Mtg. BLA & PMA Filed +1 (Cytoplasmic) 0 Herceptin & HercepTest Approved Simultaneously FISH (Gene Amplification) sbla & spma Filed FISH spma Approved Herceptin Chromosomal Treatment Analysis FISH sbla Approved IHC 5
6 Approved CDx (U.S) for Herceptin Approved CDx (FDA) Company Biomarker Drug Platform HER2 FISH PharmDx Kit Dako Denmark A/S Her2/neu Herceptin, Perjeta, Kadcyla Fluorescence in situ hybridization (FISH) HERCEPTEST Dako Denmark A/S Her2/neu Herceptin, Perjeta, Kadcyla Immunohistochemical assay (IHC) HER2 CISH PharmDx Kit Dako Denmark A/S Her2/neu Herceptin Chromogenic In Situ Hybridization (CISH) INFORM HER2 DUAL ISH DN Chromogenic In Situ Hybridization Ventana Medical Systems Her2/neu Herceptin A Probe Cocktail (CISH) PATHWAY ANTI-HER-2/NEU (4 B5) Rabbit Monoclonal Primary Ventana Medical Systems Her2/neu Herceptin immunohistochemical assay (IHC) Antibody INFORM HER-2/NEU Ventana Medical Systems Her2/neu Herceptin Fluorescence in situ hybridization (FISH ) Bond Oracle Her2 IHC System Leica Biosystems Her2/neu Herceptin Immunohistochemical assay (IHC) SPOT-LIGHT HER2 CISH Kit Life Technologies Her2/neu Herceptin Chromogenic In Situ Hybridization (CISH) and brightfield microscopy INSITE HER-2/NEU KIT Biogenex Laboratories Her2/neu Herceptin immunohistochemical assay (IHC) PATHVYSION HER-2 DNA Pro be Kit FoundationOne CDx Assay Abbott Her2/neu Herceptin Foundation Medicine EGFR EGFR T790M ALK BRAF HER2 KRAS BRCA1/BRCA2 Gilotrif, Irresa, Tarceva Tagrisso Alecensa, Xalkori, Zykadia Tafinlar, Mekinist, Zelboraf, Cotellic Herceptin, Kadcyla, Perjeta Erbitux, Vectibix Rubraca Fluorescence in situ hybridization (FISH ) NGS

 are eligible for treatment Minor variations in sensitivity between lots, runs, locations may change")
 xxxx xxx xx +400 Positive (above cut-off) Eligible for drug X +300 Borderline (around cut-off) Re-test +200 Negative (below cut-off) Not eligible for drug X 0 Negative (below cut-off)")
7 Definition of Companion diagnostics A companion diagnostics is a medical device, which provides information that is essential for the safe and effective use of a corresponding drug or biological product. The test helps a health care professional determine whether a particular therapeutic product s benefits to patients will outweigh any potential serious side effects or risks. Dx s (IVD) Qualitative yes for no to the presence of the biomarker Minor variation in sensitivity between lots, runs, locations do not change the overall test outcome CDx s (IVD Class III) Generally quantitative by nature (only those patients with expression above a threshold (cut-off) are eligible for treatment Minor variations in sensitivity between lots, runs, locations may change the test outcome IVD (IVD class I) Histology CDx-IVD (IVD class III) YES ( Yes it s a carcinoma ) YES ( Yes it s a carcinoma ) YES ( Yes it s a carcinoma ) No ( Its NOT a carcinoma, but a different tumor type ) xxxx xxx xx +400 Positive (above cut-off) Eligible for drug X +300 Borderline (around cut-off) Re-test +200 Negative (below cut-off) Not eligible for drug X 0 Negative (below cut-off) Not eligible for drug X 7



8 Unfavorable discordance - Increase of Drug development for Targeted therapies - Test increase of detection / measure of biomarker for investigation of Intention to Treat population Discordant result -> accurate, reproducible and clinically useful companion diagnostic test
9 What is Precision Medicine? U.S. FDA Guidance Personalized medicine (also referred to as Precision medicine ) relies on the use of in vitro diagnostic (IVD) devices to detect and measure biomarkers and other individual characteristics of disease or other conditions with the goal of better directing patient treatment. Document issued on: Dec.18,
10 What is Precision Medicine? U.S. FDA Guidance - When accurate testing for molecular alterations is essential for the safe and effective use of the drug, an FDA-cleared or -approved assay should be commercially available at the time of drug approval to identify patients in the clinical setting. Document issued on: Dec.18, The FDA may grant exceptions when the drug is intended to treat a serious or life-threatening condition for which no satisfactory alternative treatment exists, and FDA determines that the benefits from the use of the drug outweigh the risks from the lack of an approved or cleared in vitro companion diagnostic device. 10
11 Diagnostics utility (CDx vs. IVD, LDT) - Laboratory Developed Test - Homebrew Test - IVD - Complementary Dx without Predictive power - Complementary Dx with Predictive power LDT IVD CDx Analytical Validity Minimal Yes Yes Clinical Validity No Yes Reagent Quality Management S ystem with Clinical cut-off No Yes Yes On-Going Proficiency Testing Yes No No Pre-Market Data Review No Yes Yes - Companion Diagnostics - Single Lab PMA - Follow-on CDx Product Labeling Requirements No Yes Yes Post Market Adverse Event rep orting No Yes Yes Lab Personnel Qualifications Yes No No Transparency of Clinical Test performance No Yes Yes



 GMP")

 LDT Evidence & Evidence Level")



12 LDT vs. IVD-CDx LDT Reagents Manufacturing & Quality Management system * Intended use Regulator IVD-CDx FFPE Block Instruments - Who? Will be tested? - What? Are the appropriate specimens? FFPE Block - How? Test results? DNA extraction System (including S/W) GMP (ISO13485) Facilities * Device Performance - Analytical performance Reliability and accuracy DNA extraction IVD Mutation detection (by qpcr) LDT Evidence & Evidence Level considered * Analytical Validity or Performance * Cost-effective * Clinical Validity or Performance * Clinical Utility IVD - Clinical performance Clinical sensitivity and specificity positive and negative predictive values - Labeling Intended use, device design, directions for use, warnings/limitations, result interpretation, performance Tx Mutation detection (by qpcr) Software algorithm No Tx CDx
13 2. Global Regulatory Trend
 is focused on the co-development of the drug diagnostic approach.")
14 U.S regulatory Overview Currently, about 36 companion diagnostic tests for valid genomic biomarkers are approved in the United States. The Food and Drug Administration (FDA) is focused on the co-development of the drug diagnostic approach. Companion diagnostic products are classified as Class III high risk and are subject to approval by the FDA. Source: Frost & Sullivan




15 Drug and companion diagnostic co-development process (U.S) IVD-CDx development process Regulatory concept : Co-approval (exceptions will exist) Co-development Benefits Evaluate drug and device in one trial Regulatory Pathway for companion Diagnostics: Drug vs CDx approval process US Source: The center for anti companion diagnostics modification
16 Comparison of regulatory pathway for CDx US EU China Japan Korea Risk Classification Device Classification Class I Class A Class I Class I Class I Little or no Risk Class II Class III Class B Class II Class II Class II Class C Class III Class III Class III Class D Class IV Class IV Medium Risk High Risk
17 Definition of CDx in Japan A CDx is essential for using the pertinent therapeutic product, and corresponds to either of the following (except in vitro diagnostic agents or medical devices intended simply for disease diagnosis, etc.) : CDx is used to identify patients who are expected to respond better to a specific therapeutic product CDx is used to identify patients who are likely to be at high risk of developing adverse events associated with a particular therapeutic product CDx is necessary for optimizing the treatment including dose, schedule, and discontinuation of a particular therapeutic product Notification on Approval Application for In-vitro companion diagnostics and corresponding therapeutic products Source: T. Suzuki, National institute of Health science
18 Comparison of regulatory pathway for CDx Definition of CDx Labeling Requirments Biomarkernegative patients US EU Japan (i) to identify patients most likely to benefit from a therapeutic product, and (ii) to identify patients likely to be at an increased risk for serious adverse reactions as a result of treatment with the therapeutic product, and (iii) to monitor response to treatment with the therapeutic product for the purpose of adjusting treatment to achieve improved safety or effectiveness. To identify patients in the population for whom the therapeutic product X has been adequately studied, and found safe and effective. In addition to the drug labeling inform ation about sets of companion diagnosti None of the products contained a reference to a specific companion diagnostic cs placed on the corresponding therapeuproduct in the therapeutic indication. tic product. Inclusion of biomarker-negative patients in early-phase clinical trials is mentioned more clearly in the Japanese guidance than in the US guidance or EU draft guidance. Retrospective vs. prospective clinical trials The Japanese guidance goes further, uniquely highlighting prospectively designed retrospective three exceptional cases in which it may be difficult to clinical utility studies may be possible. conduct prospective randomized controlled trials. Nature biotechnology, vol34, no.2, Feb 2016
19 Comparison of regulatory pathway for CDx (continued) The Japanese guidance goes further, uniquely highlighting three exceptional cases in which it may be difficult to conduct prospective randomized controlled trials. First, cases in which running a trial would be questionable on ethical grounds because patients who have a biomarker are subject to extremely serious adverse events when the corresponding therapeutic is administered. Second, cases in which running a trial would be questionable on practical grounds because too few patients in a population carry the biomarker of interest and thus the patient cohort in the trial would be too small. Third, cases in which evaluation of the biomarker on the basis of retrospective analyses is valid even after considering potential biases that arise from such post factum studies.
 Pathway to CE marking of companion diagnostics under the current legal framework and possible scenario under proposed")
20 Pathway to CE marking of companion diagnostics (E.U) Pathway to CE marking of companion diagnostics under the current legal framework and possible scenario under proposed new legal framework under discussion. Clin Cancer Res 2014;20:

21 E.U regulatory trend Class C Medical device.. CE Marking CE Medical Device IVD-CDx should be classified as Class C Update on safety and clinical implication report / year 21
22 New rules for IVDs, EU The set of rules covering IVDs will take effect in 2022, The new rules, the EC said, will improve "market surveillance and traceability" and ensure that all IVDs and medical devices are designed "to reflect the latest scientific and technology state of the art." Additionally, they will create more transparency for consumers and certainty for manufacturers of such products, while strengthening international competitiveness and innovation. Also, a financial mechanism will be created to ensure that patients who have been harmed by defective medical devices will be financially compensated. 22
23 3. Korea Regulatory Trend

24 Medical device classification in korea Medical devices in Korea are regulated by the Ministry of Food and Drug Safety (MFDS). Manufacturers must follow the requirements of the Medical Devices Act and register with the MFDS before entering the Korean market. Source: MFDS, Korea
25 in Korea Oct MFDS IVD-CDx Guideline (Doc#_B ) Currently, the development and approval of new drug and companion diagnostics have been conducted separately in Korea. The companion diagnostics in Korea have labels to show the safety and effectiveness of drugs. However, the labels on the drug does not specify companion diagnostics but refer to biomarker test including homebrew assay. Because the accuracy of diagnostics has a great influence on patients in the case of high-risk therapeutic drugs, a strict regulation system of diagnostic products is essential for the benefit of patient and payers.
26 Follow-on CDx Analytical validation Same gene/variant level validation as original CDx Published Summary of Safety and Effectiveness Data (SSED) as reference Method comparison with approved CDx No drug trial required Banked samples from intended use population Requirement for preservation of drug efficacy Variability between follow-on CDx and originally approved CDx should be within the variability of originally approved CDx
 MFDS Submission of medical device for approval (80 day) NECA New Health Technology Assessment (360 day) HIRA Assessment of item Tech (New or existing HIRA Health")
 80 Day 280 Day HIRA Health insurance review (150 day) 440 Day 360 Day 150 Day Item approval - MFDS: Ministry of Food and Drug Safety - NECA: National Evidence-based")
 46 mutation sites of EGFR gene through 4-well Possible detection with a small")
27 Follow-on CDx (Approval, Health insurance review process in Korea) Before Current: One Stop service (rev. Aug.2014) MFDS Submission of medical device for approval (80 day) NECA New Health Technology Assessment (360 day) HIRA Assessment of item Tech (New or existing HIRA Health insurance review (150 day) MFDS Submission of medical device for approval HIRA Assessment of item Tech (New or existing Tech.) NECA New Health Technology Assessment Tech.) 80 Day 280 Day HIRA Health insurance review (150 day) 440 Day 360 Day 150 Day Item approval - MFDS: Ministry of Food and Drug Safety - NECA: National Evidence-based Healthcare Collaborating Agency - HIRA: Health Insurance Review & Assessment Service First Follow-on CDx in Korea Item approval GenesWell ddegfr mutation test 150 Day Companion Diagnostics for NSCLC patient with EGFR mutation Companion Drugs: TARCEVA (Erlotinib) 46 mutation sites of EGFR gene through 4-well Possible detection with a small amount of DNA (3.3ng/well) Possible with low-frequency mutation (FFPE, about 0.8% )

28 Follow-on CDx Non-inferiority Test (US) vs Equivalence Test (Korea) Terms: CCD: Comparator companion diagnostic FCD: Follow-on Companion diagnostic To support the identical therapeutic indications as CCD, the safety and effectiveness of FCD and CCD should be comparable. There are two approaches: New clinical trial Retest of patients samples from the original clinical trial While the two approaches above are ideal, they may be difficult or impractical to follow. Suggestion: External concordance study to evaluate the clinical performance of FCD when the direct estimate of the therapeutic efficacy in FCD s intented use population is not feasible. Source: Statistics in Biopharmaceutical Research 8.3 (2016):
 <Sep.")
29 Korean regulation on CDx Asian Harmonization Working Party (AHWP) adopted Korea's companion In-vitro diagnostics Based on Korea s in-vitro companion diagnostics device guideline.. Guidance for Additional Considerations to support Conformity Assessment of Companion In vitro Diagnostic Medical Devices(AHWP/WG2/F001:2017) <Sep. 2017>
 Warning & Precautions EGFR T790M mutation positive NSCLC, as detected by an FDA approved test EGFR mutation status should be")
30 Current Issue.. Drug Label (Tagrisso) Tagrisso US EU Japan Korea Approval Date Indicated Use Non-small cell lung cancer (NSCLC) EGFR T790M mutation-positive NSCLC. EGFR T790M mutation- Positive NSCLC Non-small cell lung cancer (NSCLC) Warning & Precautions EGFR T790M mutation positive NSCLC, as detected by an FDA approved test EGFR mutation status should be determined using a validated test method EGFR T790M mutation tests p erformed by a thoroughly expe rienced pathologist or testing laboratory. Approved IVDs should be us ed for the test. EGFR T790M status should be determined using a validated test method In Australia A validated test should be performed in a clinical laboratory using either tumor tissue DNA or circulating tumor DNA (ctdna) obtained from a blood (plasma) sample. Only robust, reliable and sensitive test(s) with demonstrated utility for the determination of EGFR mutation status should be used (see CLINICAL TRIALS_eg.Cobas EGFR test) Korea : It is not specified on the label of the drug. It is not necessary. U.S, Japan : Always use approved CDx for prescription of drugs. It is specified on the label of the drug.
31 4. Current regulatory issues in Korea - Risk of Homebrew Assay - Unfavorable Discordance - NGS Issue




32 PARP inhibitor license 2011 Foundation Medicine s NGS-based CDx Collaboration license 2012 acquisition 2006 Rucaparib BRACAnalysis CDx mychoice HRD license 2012 Olaparib Niraparib license 2015 collaboration acquisition license Talazoparib Veliparib


33 PARP inhibitor CDx Germline BRCA1/2 mutation Germline BRCA1/2 mutation Somatic BRCA1/2 mutation Germline BRCA1/2 mutation Somatic BRCA1/2 mutation HRD score
 BRACAnalysis CDx BRACAnalysis CDx is an in vitro diagnostic device intended for the qualitative detection a nd classification of")
34 PARPi (olaparib) and CDx (Myriad) U.S Approved Japan E.U Korea Mostly Germline BRCA variant Homebrew test (Laboratory developed test??, LDT in US) Patient Safety?? Single Lab PMA concept Drug CDx Intended use Lynparza (olaparib) BRACAnalysis CDx BRACAnalysis CDx is an in vitro diagnostic device intended for the qualitative detection a nd classification of variants of BRCA1 and BRCA2 genes. Results of the test are used as an aid in identifying ovarian cancer patients treatment with Lynparza (olaparib). This assay is fo r professional use only and is to be performed only at Myriad Genetic Laboratories, a single lab oratory site located at 320 Wakara Way, Salt Lake City, UT
35 Homebrew BRCA assay in Korea - False Positive Issue : Interpretation for Deleterious mutation - False Negative Issue : Detection Primer/Probe, Technology, Interpretation Can BRCAAnalysisCDx be used in Korea? - Can BRCAAnalysisCDx reflect the ethnic difference? - Reasonable test : analytical validity and clinical validity - Who should judge the validity? - Drug Risk = Assay Risk
36 FoundationFocus CDx BRCA LOH Rubraca clinical trial
37 Rubraca issues in korea 1. How check the somatic BRCA alteration? MFDS in Korea 2. If.. Home-brew assay is used in Korea.. Unfavorable Discordance issue

38 High risk of LDT Nov. 2015, FDA pointed out the Risk of Laboratory Developed Test 38
 samples for local IHC, central IHC were analyzed Bae et al.")
 samples were concordant with a central IHC results. 39 E.A. Perez et al.")
39 Why CDx pursues IVD * Poor Concordance Between Local and Central laboratory HER2 testing A total of 1,198 breast carcinoma samples were collected from 6 institutions in Korea. Of the 1,198 samples, 959 (80%) samples for local IHC, central IHC were analyzed Bae et al., 2012 Interlaboratory concordance in HER2 status The results of local and central IHC were consistent for 783 samples, with a concordance rate of 81.6% Of the 211 samples with a local IHC result of positive, 140 (66.4%) samples were concordant with a central IHC results. 39 E.A. Perez et al. / Cancer Treatment Reviews 40 (2014)

40 LDT vs. FDA-approved assay Are there performance differences between laboratory-developed tests (LDTs) and US Food and Drug Administration approved companion diagnostics (FDA-CDx)? 1. Lack of no. of specimens 2. Different proportions of specimens using LDT and FDA-approved kit 3. Through the results of just three mutations, can not represent other complicated test methods The risk of LDT still exists.. JAMA Oncol. doi: /jamaoncol
41 Who Have the responsibility of the risk of homebrew assay in Korea? MFDS vs Ministry of Health and Welfare vs Medical Doctor Patients only - Patients right to select Diagnosis - Can patients use an overseas diagnostic laboratory that provides services with CDx products because their clinical evidence level is higher? - If the government or physician does not inform the patient of this fact, is this not a significant infringement of the patient's rights?


 in human- and machinereadable")


42 Risk of Homebrew assay Laboratory Developed Test In U.S.. Ongoing FDA has established and continues to implement a unique device identification system to adequately identify medical devices through their distribution and use. When fully implemented, the label of most devices will include a unique device identifier (UDI) in human- and machinereadable form. - Adverse effects to patients caused by diagnostic errors - Who take responsibility?? Doctor? Lab? Regulatory Body? - It is impossible to trace the diagnostic errors. - Can you trust the results? - In the case of NGS, different SW &DB are used, so the results may differ.
 U.S.")
 when KRAS Codon 12 or 13 mutation is detected.")
43 CDx for CRC patients U.S Approved CDx Korea Apporved only IVD in Korea IVD vs Homebrew test (Sanger)?? High Risk False negative issue due to Low sensitivity (15%) U.S. (CDx) The test is intended to be used as an aid in the identification of CRC patients who should not be treated with Erbitux (cetuximab) or with Vectibix (panitumumab) when KRAS Codon 12 or 13 mutation is detected. Korea (IVD) The cobas KRAS Mutation Test, for use with the cobas 4800 System, is a real-time PCR test intended for the identification of mutations in codons 12, 13 and 61 of the KRAS gene in DNA derived from FFPE human colorectal cancer tissues.
44 Approved CDx (U.S) in Erbitux an Vectibix Korean IVD vs RUO in Korea but approved CDx in US Approved CDx (FDA) Company Biomarker Drug Platform DAKO EGFR PharmDx KIT Dako, North America EGFR Erbitux therascreen KRAS RGQ PCR Kit Qiagen KRAS The cobas KRAS Mutation Test Roche KRAS Erbitux Vectibix Erbitux Vectibix Praxis Extended RAS Panel Illumina KRAS, NRAS Vectibix immunohistochemical assa y (IHC) Real-time PCR Real-time PCR Targeted high throughput parallel sequencing (NGS) FoundationOne CDx Assay Foundation Medicine EGFR Gilotrif, Irresa, Tarceva NGS EGFR T790M ALK BRAF HER2 KRAS BRCA1/BRCA2 Tagrisso Alecensa, Xalkori, Zykadia Tafinlar, Mekinist, Zelboraf, Cotellic Herceptin, Kadcyla, Perjeta Erbitux, Vectibix Rubraca
 Tecentriq (Atezolizumab) Opdivo")
 PD-L1 IHC 28-8 pharmdx (Dako) Approval condition Intended use CDx PD-L1 IHC 22C3")
.")
 Assay in NSCLC tissue may be associated with enhanced overall survival")
45 CDx vs. Complementary Dx Drug name Keytruda (Pembrolizumab) Tecentriq (Atezolizumab) Opdivo (Nivolumab) Clinical Design Dx Product PD-L1 IHC 22C3 pharmdx (Dako) Ventana PD-L1 IHC SP142 (Roche) PD-L1 IHC 28-8 pharmdx (Dako) Approval condition Intended use CDx PD-L1 IHC 22C3 pharmdx is indicated as an aid in identifying NSCLC patients for treatment with KEYTRUDA (pembrolizumab).. Complementary Dx w/ Predictive power PD-L1 expression in 50% TC or 10% IC determined by VENTANA PD-L1 (SP142) Assay in NSCLC tissue may be associated with enhanced overall survival from TECENTRIQ (atezolizumab). Complementary Dx w/o Predictive power PD-L1 IHC 28-8 pharmdx in nonsquamous NSCLC may be associated with enhanced survival from OPDIVO (nivolumab).



46 Blueprint PD-L1 IHC Assay Comparison Project Different Antibody

47 Blueprint PD-L1 IHC Assay Comparison Project PD-L1 expression was evaluable with all assays in 493 samples. The three assays showed similar patterns of tumor membrane staining, with high correlation between percent PD-L1 staining. An overall percentage agreement of >90% was achieved between assays at multiple expression cutoffs, including 1%, 10%, 25%, and 50% tumor membrane staining. Clin Cancer Res Jul 15;23(14):
48 Current issue (PD-L1 assay) 28-8 Intended Use Korea U.K U.S Non-squamous non-small cell lung cancer Non-squamous NSCLC / SCCHN / Melanoma / Urothelial carcinoma (UC) Non-squamous non-small cell lung cancer (NSCLC) / Melanoma tissues Drug OPDIVO (nivolumab) OPDIVO (nivolumab) OPDIVO (nivolumab) SP263 Intended Use Non-small cell lung cancer/keytruda Non-squamous NSCLC/OPDIVO NSCLC (KEYTRUDA)/ Non-squamous NSCLC patients (OPDIVO)/ Urothelial carcinoma (IMFINZI) Urothelial carcinoma (UC) Drug KEYTRUDA (pembrolizumab) OPDIVO (nivolumab) KEYTRUDA (pembrolizumab) OPDIVO (nivolumab) IMFINZI (durvalumab) IMFINZI (durvalumab) Intended used of Ventana SP263 kit (MFDS) May





49 Drug and companion diagnostic co-development May, 2017 KEYTRUDA is indicated for the treatment of adult and pediatric patients with unresectable or metastatic microsatellite instabilityhigh (MSI-H) or mismatch repair deficient (dmmr) solid tumors that have progressed following prior treatment and who have no satisfactory alternative treatment options, or colorectal cancer that has progressed following treatment with fluoropyrimidine, oxaliplatin, and irinotecan. This indication is approved under accelerated approval based on tumor response rate and durability of response. Continued approval for this indication may be contingent upon verification and description of clinical benefit in the confirmatory trials. The safety and effectiveness of KEYTRUDA in pediatric patients with MSI-H central nervous system cancers have not been established. 49

50 FDA approves first cancer treatment for any solid tumor with a specific genetic feature
 is the total number of mutations per coding area of a tumor genome.")

51 Tumor Mutation Burden (TMB): cut-off issue Source: - Tumor Mutation Burden (TMB) is the total number of mutations per coding area of a tumor genome. - Higher TMB levels are correlated with higher levels of neo-antigens which help our immune system to recognize tumors. - TMB can be measured by tumor genomic profiling assay, as predictor of response to Immune checkpoint therapy.
52 NGS for Insurance beneficiaries with diseases (in Korea) Germline Variants Somatic Variants Hereditary Diseases Solid tumor Hematology malignancy Coverage (Diseases) Hereditary retinitis pigmentosa Hereditary bradyecoia CharcotMarie Tooth syndrome Hereditary disorders excluding the above three diseases Gastric Cancer, Lung Cancer, Colorectal Cancer, Breast Cancer, Ovarian Cancer, Melanoma, Gastrointesrinal stromal tumor, Celebro-spinal cord malignant tumor, Pediatric neuroblastoma, Cancer with unknown primary tumor Plasmacytoma Acute myeloid leukemia Acute lymphocytic leukemia Myelodysplastic disorder, Myeloproliferative tumor Malignant lymphoma Gene (must be included in NGS panel) PRPF31, RHO, RP1, RP2, USH2A, PRPH2, RPGR GJB2, POU3F4, SLC26A4, TECTA GJB1, MFN2, MPZ, PMP22 * RNA fusion gene test: Only Acute myeloid leukemia - ABL1, BCR, CBFB, ETV6, KMT2A, PML, RARA - Specimens should be determined using a validated test method performed by Ministry of Health and Welfare (Korea) validated laboratory - For hereditary genetic testing, only supported once/per disease - For non-hereditary genetic testing, only supported once/per disease, but, in case of recurrence, one addition will be accepted. None HER2, EGFR, ALK, KRAS, NRAS, BRAF, BRCA1, BRCA2, KIT, PDGFRA, IDH1, IDH2, MYC(C-myc), N-myc(MYCN) NRAS, KRAS, TP53 CEBPA, FLT2, JAK2, KIT, NPM1, RUNX1, TP53, IDH1, IDH2 TP53, RB1, JAK2, NRAS, IKZF1 ASXL1, CALR, CSF3R, DNMT3A, JAK2, MPL, RUNX1, SETBP1, SF3B1, SRSF2, TET2 MYD88, BRAF, TP53
53 NGS for Insurance beneficiaries with diseases (in Korea) Applying for Clinical laboratory for NGS test (Insurance coverage) Main management agencies Ministry of Health and Welfare Reagent/Panel Hardware (H/W) Analysis Software (S/W) RUO (IVD is not necessary) MFDS* approved Dx If.. RUO? -> Clinical laboratory RUO devices approved by MFDS* for NGS testing are permitted. RUO (IVD is not necessary) * MFDS : Ministry of Foodand Drug Safety, Korea
54 NGS for Medicare beneficiaries with advanced cancer Proposed Decision Memo for Next Generation Sequencing (NGS) for Medicare Beneficiaries with Advanced Cancer (CAG-00450N) 1. Patient has: 1 either recurrent, relapsed, refractory, metastatic, or advanced stages III or IV cancer 2 either not been previously tested using the same NGS test for the same primary diagnosis of cancer or repeat testing using the same NGS test only when a new primary cancer diagnosis is made by the treating physician 3 decided to seek further cancer treatment (e.g., therapeutic chemotherapy) 2. The diagnostic laboratory test using NGS must have: 1 FDA approval or clearance as a companion in vitro diagnostic 2 an FDA approved or cleared indication for use in that patient s cancer 3 results provided to the treating physician for management of the patient using a report template to specify treatment options. 54
55 NGS based test and Medicare coverage CMS has proposed three pathways through which NGS cancer panels may garner Medicare coverage. - The agency said it will grant full coverage for genetic markers on the panel that have premarket approval (PMA) from the FDA as a companion diagnostic where testing is required for the safe and effective use of a drug. - For markers without CDx status, but that the FDA has cleared or approved for use in a patient's treatment plan with other information (such as MSKCC 468-gene oncopanel MSKIMPACT), CMS is willing to grant coverage with evidence development (CED), when the lab collects patient outcomes in a prospective registry. - For lab-developed tests without the FDA's blessing that are performed in a CLIA-certified lab the majority of tests on the market CMS is proposing CED when tests are included in a National Cancer Institute National Clinical Trial Network study. 55


56 Companion diagnostic development : News_NGS-CDx FoundationOne CDx (F1CDx), Foundation Medicine
 FoundationACT is a liquid biopsy assay for solid tumors that analyzes circulating tumor DNA (ctdna) in")
57 Companion diagnostic development : News_NGS-CDx FDA Grants Breakthrough Device Designation to Foundation Medicine Liquid Biopsy Assay (Apr 26, 2018) FoundationACT is a liquid biopsy assay for solid tumors that analyzes circulating tumor DNA (ctdna) in blood.

58 Development : News_NGS-IVD MSK-IMPACT, MSK Cancer Center A Hybridization Capture-Based Next-Generation Sequencing Clinical Assay for Solid Tumor Molecular Oncology. MSK-IMPACT is a single-site assay performed at Memorial Sloan Kettering Cancer Center. Special instrument requirement: Illumina HiSeq 2500 Sequencer (qualified by MSK)



59 Risk of NGS-based test.. CMS-Proposed Coverage of NGS Cancer Tests Could Lead to Off-Label Scripts, Oncologists Worry (27 th Feb, 2018) CMS: Centers for Medicare & Medicaid Services Solid tumor patient Positive Negative A perspective that hasn't gotten as much attention is that the CMS draft coverage decision is perhaps not restrictive enough, and that the policy could be bad for patients by increasing off-label drug use based on the results of NGS panels. Off-Label prescription (Increase risk of Patient)
 1) In April 2018, the FDA issued two final guidances that recommend approaches to streamline the submission and review of data supporting the clinical and analytical validity of NGS-based tests.")
60 Streamlining FDA s Regulatory Oversight of NGS tests The FDA's Role in Advancing Precision Medicine (April. 2018) 1) In April 2018, the FDA issued two final guidances that recommend approaches to streamline the submission and review of data supporting the clinical and analytical validity of NGS-based tests. 2) These recommendations are intended to provide an efficient and flexible regulatory oversight approach: as technology advances, standards can rapidly evolve and be used to set appropriate metrics for fast growing fields such as NGS. 3) Similarly, as clinical evidence improves, new assertions could be supported. 4) This adaptive approach would ultimately foster innovation among test developers and improve patients' access to these new technologies.
61 5. Suggestion
62 Suggestion 1. Unfavorable discordance of Biomarker test 2. Increase the Single Lab PMA concept test Global Central Lab 3. Patients and Payers First 4. Companion diagnostics and new drugs should be simultaneously developed and applied for approval. 5. The labels of a drug and its companion diagnostics must specifically indicate their association and effectiveness.
 target patients Companion diagnostics (CDx) <Complementary Companion Diagnostics> May be associated with enhanced survival from specific drug (ex.")
63 Suggestion Intended use <Companion Diagnostics> Selection of specific drug (ex.tki) target patients Companion diagnostics (CDx) <Complementary Companion Diagnostics> May be associated with enhanced survival from specific drug (ex. TKI) No Complementary CDx Yes Meets the purpose of assay No Yes <In-vitro Diagnostics> Detection and identification of mutations in specific gene Market surveillance Traceability <Laboratory Developed Test> X No Laboratory developed test In-vitro diagnostics Yes Permission of the assay (Off-label only) Meets the purpose of assay Source : 동반진단의가치반영을위한정책개선제안, 의료기기뉴스라인 ( ) Permission of the assay (Research Use only) 63




64 Thank you for your attention! 64
Regulatory Landscape for Precision Medicine
 Regulatory Landscape for Precision Medicine Adam C. Berger, Ph.D. Office of In Vitro Diagnostics and Radiological Health, FDA FOCR-Alexandria A Blueprint for Breakthrough Meeting September 13, 2017 1 What
Regulatory Landscape for Precision Medicine Adam C. Berger, Ph.D. Office of In Vitro Diagnostics and Radiological Health, FDA FOCR-Alexandria A Blueprint for Breakthrough Meeting September 13, 2017 1 What
Transform genomic data into real-life results
 CLINICAL SUMMARY Transform genomic data into real-life results Biomarker testing and targeted therapies can drive improved outcomes in clinical practice New FDA-Approved Broad Companion Diagnostic for
CLINICAL SUMMARY Transform genomic data into real-life results Biomarker testing and targeted therapies can drive improved outcomes in clinical practice New FDA-Approved Broad Companion Diagnostic for
NGS ONCOPANELS: FDA S PERSPECTIVE
 NGS ONCOPANELS: FDA S PERSPECTIVE CBA Workshop: Biomarker and Application in Drug Development August 11, 2018 Rockville, MD You Li, Ph.D. Division of Molecular Genetics and Pathology Food and Drug Administration
NGS ONCOPANELS: FDA S PERSPECTIVE CBA Workshop: Biomarker and Application in Drug Development August 11, 2018 Rockville, MD You Li, Ph.D. Division of Molecular Genetics and Pathology Food and Drug Administration
NGS IN ONCOLOGY: FDA S PERSPECTIVE
 NGS IN ONCOLOGY: FDA S PERSPECTIVE ASQ Biomed/Biotech SIG Event April 26, 2018 Gaithersburg, MD You Li, Ph.D. Division of Molecular Genetics and Pathology Food and Drug Administration (FDA) Center for
NGS IN ONCOLOGY: FDA S PERSPECTIVE ASQ Biomed/Biotech SIG Event April 26, 2018 Gaithersburg, MD You Li, Ph.D. Division of Molecular Genetics and Pathology Food and Drug Administration (FDA) Center for
Keytruda. Keytruda (pembrolizumab) Description
 Description") Federal Employee Program 1310 G Street, N.W. Washington, D.C. 20005 202.942.1000 Fax 202.942.1125 5.21.50 Subject: Keytruda Page: 1 of 7 Last Review Date: December 8, 2017 Keytruda Description Keytruda
Federal Employee Program 1310 G Street, N.W. Washington, D.C. 20005 202.942.1000 Fax 202.942.1125 5.21.50 Subject: Keytruda Page: 1 of 7 Last Review Date: December 8, 2017 Keytruda Description Keytruda
Patient Leader Education Summit. Precision Medicine: Today and Tomorrow March 31, 2017
 Patient Leader Education Summit Precision Medicine: Today and Tomorrow March 31, 2017 Precision Medicine: Presentation Outline Agenda What is a Precision Medicine What is its clinical value Overview of
Patient Leader Education Summit Precision Medicine: Today and Tomorrow March 31, 2017 Precision Medicine: Presentation Outline Agenda What is a Precision Medicine What is its clinical value Overview of
Innovation, Uncertainty and Reimbursement Processes in Precision Medicine: The Case of PD-L1
 Innovation, Uncertainty and Reimbursement Processes in Precision Medicine: The Case of PD-L1 Monday, October 17, 2016 MaRS Discovery District, Toronto This session was generously sponsored by Merck Canada
Innovation, Uncertainty and Reimbursement Processes in Precision Medicine: The Case of PD-L1 Monday, October 17, 2016 MaRS Discovery District, Toronto This session was generously sponsored by Merck Canada
Keytruda. Keytruda (pembrolizumab) Description
 Description") Federal Employee Program 1310 G Street, N.W. Washington, D.C. 20005 202.942.1000 Fax 202.942.1125 5.21.50 Subject: Keytruda Page: 1 of 9 Last Review Date: September 20, 2018 Keytruda Description Keytruda
Federal Employee Program 1310 G Street, N.W. Washington, D.C. 20005 202.942.1000 Fax 202.942.1125 5.21.50 Subject: Keytruda Page: 1 of 9 Last Review Date: September 20, 2018 Keytruda Description Keytruda
Keytruda. Keytruda (pembrolizumab) Description
 Description") Federal Employee Program 1310 G Street, N.W. Washington, D.C. 20005 202.942.1000 Fax 202.942.1125 5.21.50 Subject: Keytruda Page: 1 of 6 Last Review Date: September 15, 2017 Keytruda Description Keytruda
Federal Employee Program 1310 G Street, N.W. Washington, D.C. 20005 202.942.1000 Fax 202.942.1125 5.21.50 Subject: Keytruda Page: 1 of 6 Last Review Date: September 15, 2017 Keytruda Description Keytruda
Keytruda. Keytruda (pembrolizumab) Description
 Description") Federal Employee Program 1310 G Street, N.W. Washington, D.C. 20005 202.942.1000 Fax 202.942.1125 5.21.50 Subject: Keytruda Page: 1 of 9 Last Review Date: November 30, 2018 Keytruda Description Keytruda
Federal Employee Program 1310 G Street, N.W. Washington, D.C. 20005 202.942.1000 Fax 202.942.1125 5.21.50 Subject: Keytruda Page: 1 of 9 Last Review Date: November 30, 2018 Keytruda Description Keytruda
PLENARY SESSION 1: CLINICAL TRIAL DESIGN IN AN ERA OF HORIZONTAL DRUG DEVELOPMENT Industry Perspective
 PLENARY SESSION 1: CLINICAL TRIAL DESIGN IN AN ERA OF HORIZONTAL DRUG DEVELOPMENT Industry Perspective Davy Chiodin, VP - Regulatory Science, QA and Compliance, Acerta Pharma (A Member of the AstraZeneca
PLENARY SESSION 1: CLINICAL TRIAL DESIGN IN AN ERA OF HORIZONTAL DRUG DEVELOPMENT Industry Perspective Davy Chiodin, VP - Regulatory Science, QA and Compliance, Acerta Pharma (A Member of the AstraZeneca
GLOBAL REGISTRATION STRATEGIES:
 GLOBAL REGISTRATION STRATEGIES: Therapeutic Product (TP) and the Companion Diagnostic (CDx) Erin Pedalino Regulatory Affairs International MSD Unique Role as CDx Regulatory Liaison at a Therapeutic Product
GLOBAL REGISTRATION STRATEGIES: Therapeutic Product (TP) and the Companion Diagnostic (CDx) Erin Pedalino Regulatory Affairs International MSD Unique Role as CDx Regulatory Liaison at a Therapeutic Product
Comprehensive genomic profiling for various solid tumors
 Content Highlight Test Specification Test Content Performance Validation Test Report I II III IV V The NovoPM TM comprehensive cancer genomic profiling test Comprehensive genomic profiling for various
Content Highlight Test Specification Test Content Performance Validation Test Report I II III IV V The NovoPM TM comprehensive cancer genomic profiling test Comprehensive genomic profiling for various
Opdivo. Opdivo (nivolumab) Description
 Description") Federal Employee Program 1310 G Street, N.W. Washington, D.C. 20005 202.942.1000 Fax 202.942.1125 5.21.53 Subsection: Antineoplastic nts Original Policy Date: January 16, 2015 Subject: Opdivo Page: 1 of
Federal Employee Program 1310 G Street, N.W. Washington, D.C. 20005 202.942.1000 Fax 202.942.1125 5.21.53 Subsection: Antineoplastic nts Original Policy Date: January 16, 2015 Subject: Opdivo Page: 1 of
Out-Patient Billing CPT Codes
 Out-Patient Billing CPT Codes Updated Date: August 3, 08 Client Billed Molecular Tests HPV DNA Tissue Testing 8764 No Medicare Billed - Molecular Tests NeoARRAY NeoARRAY SNP/Cytogenetic No 89 NeoLAB NeoLAB
Out-Patient Billing CPT Codes Updated Date: August 3, 08 Client Billed Molecular Tests HPV DNA Tissue Testing 8764 No Medicare Billed - Molecular Tests NeoARRAY NeoARRAY SNP/Cytogenetic No 89 NeoLAB NeoLAB
Media Contact: Ron Rogers Investor Contact: Scott Gleason (801) (801)
 (801)") News Release Media Contact: Ron Rogers Investor Contact: Scott Gleason (801) 584-3065 (801) 584-1143 rrogers@myriad.com sgleason@myriad.com Myriad Receives FDA Approval of BRACAnalysis CDx as Companion
News Release Media Contact: Ron Rogers Investor Contact: Scott Gleason (801) 584-3065 (801) 584-1143 rrogers@myriad.com sgleason@myriad.com Myriad Receives FDA Approval of BRACAnalysis CDx as Companion
Molecular Testing Updates. Karen Rasmussen, PhD, FACMG Clinical Molecular Genetics Spectrum Medical Group, Pathology Division Portland, Maine
 Molecular Testing Updates Karen Rasmussen, PhD, FACMG Clinical Molecular Genetics Spectrum Medical Group, Pathology Division Portland, Maine Keeping Up with Predictive Molecular Testing in Oncology: Technical
Molecular Testing Updates Karen Rasmussen, PhD, FACMG Clinical Molecular Genetics Spectrum Medical Group, Pathology Division Portland, Maine Keeping Up with Predictive Molecular Testing in Oncology: Technical
Molecular. Oncology & Pathology. Diagnostic, Prognostic, Therapeutic, and Predisposition Tests in Precision Medicine. Liquid Biopsy.
 Molecular Oncology & Pathology Hereditary Cancer Somatic Cancer Liquid Biopsy Next-Gen Sequencing qpcr Sanger Sequencing Diagnostic, Prognostic, Therapeutic, and Predisposition Tests in Precision Medicine
Molecular Oncology & Pathology Hereditary Cancer Somatic Cancer Liquid Biopsy Next-Gen Sequencing qpcr Sanger Sequencing Diagnostic, Prognostic, Therapeutic, and Predisposition Tests in Precision Medicine
GENETIC TESTING FOR TARGETED THERAPY FOR NON-SMALL CELL LUNG CANCER (NSCLC)
") CANCER (NSCLC) Non-Discrimination Statement and Multi-Language Interpreter Services information are located at the end of this document. Coverage for services, procedures, medical devices and drugs are
CANCER (NSCLC) Non-Discrimination Statement and Multi-Language Interpreter Services information are located at the end of this document. Coverage for services, procedures, medical devices and drugs are
Review of NEO Testing Platforms. Lawrence M. Weiss, MD Medical Director, Aliso Viejo
 Review of NEO Testing Platforms Lawrence M. Weiss, MD Medical Director, Aliso Viejo Lawrence Weiss, M.D. Medical Director, Aliso Viejo Dr. Weiss currently serves as NeoGenomics Medical Director, Aliso
Review of NEO Testing Platforms Lawrence M. Weiss, MD Medical Director, Aliso Viejo Lawrence Weiss, M.D. Medical Director, Aliso Viejo Dr. Weiss currently serves as NeoGenomics Medical Director, Aliso
MEDICAL POLICY. SUBJECT: MOLECULAR PANEL TESTING OF CANCERS TO IDENTIFY TARGETED THERAPIES (Excluding NSCLC and CRC) EFFECTIVE DATE: 12/21/17
 EFFECTIVE DATE: 12/21/17") MEDICAL POLICY SUBJECT: MOLECULAR PANEL TESTING OF PAGE: 1 OF: 5 If a product excludes coverage for a service, it is not covered, and medical policy criteria do not apply. If a commercial product, including
MEDICAL POLICY SUBJECT: MOLECULAR PANEL TESTING OF PAGE: 1 OF: 5 If a product excludes coverage for a service, it is not covered, and medical policy criteria do not apply. If a commercial product, including
Predictive markers for treatment with Immune checkpoint inhibitors - PD-L1 et al -
 Predictive markers for treatment with Immune checkpoint inhibitors - PD-L1 et al - Lukas Bubendorf Pathology Improved overall survival as a result of combination therapy Predictive biomarkers for the treatment
Predictive markers for treatment with Immune checkpoint inhibitors - PD-L1 et al - Lukas Bubendorf Pathology Improved overall survival as a result of combination therapy Predictive biomarkers for the treatment
Overview of Biomarker Development for Immune PD-1/L1 Checkpoint Blockade
 Overview of Biomarker Development for Immune PD-1/L1 Checkpoint Blockade David L. Rimm MD-PhD Professor Departments of Pathology and Medicine (Oncology) Director, Yale Pathology Tissue Services Disclosures
Overview of Biomarker Development for Immune PD-1/L1 Checkpoint Blockade David L. Rimm MD-PhD Professor Departments of Pathology and Medicine (Oncology) Director, Yale Pathology Tissue Services Disclosures
Disruptive innovation in molecular diagnostics. Hilde Windels CEO Biocartis 25 March 2017
 Disruptive innovation in molecular diagnostics Hilde Windels CEO Biocartis 25 March 2017 1 One of the key innovations in healthcare in the last decade PERSONALISED MEDICINE or HIGH PRECISION MEDICINE From
Disruptive innovation in molecular diagnostics Hilde Windels CEO Biocartis 25 March 2017 1 One of the key innovations in healthcare in the last decade PERSONALISED MEDICINE or HIGH PRECISION MEDICINE From
Approval of pembrolizumab (MSI- H/dMMR) and considerations for site-agnostic development of drugs in oncology
 and considerations for site-agnostic development of drugs in oncology") Approval of pembrolizumab (MSI- H/dMMR) and considerations for site-agnostic development of drugs in oncology Steven Lemery, MD, MHS Associate Director, DOP2 Traditional development paradigm Based on tumor
Approval of pembrolizumab (MSI- H/dMMR) and considerations for site-agnostic development of drugs in oncology Steven Lemery, MD, MHS Associate Director, DOP2 Traditional development paradigm Based on tumor
Learning from the Impact of the Drug-Diagnostics Strategy in Oncology
 Learning from the Impact of the Drug-Diagnostics Strategy in Oncology Critical Path to TB Drug Regimens 2017 Workshop Washington DC March 22, 2017 Jan Trøst Jørgensen, M.Sc.Pharm., Ph.D. Dx-Rx Institute
Learning from the Impact of the Drug-Diagnostics Strategy in Oncology Critical Path to TB Drug Regimens 2017 Workshop Washington DC March 22, 2017 Jan Trøst Jørgensen, M.Sc.Pharm., Ph.D. Dx-Rx Institute
August 17, Dear Valued Client:
 August 7, 08 Re: CMS Announces 6-Month Period of Enforcement Discretion for Laboratory Date of Service Exception Policy Under the Medicare Clinical Laboratory Fee Schedule (the 4 Day Rule ) Dear Valued
August 7, 08 Re: CMS Announces 6-Month Period of Enforcement Discretion for Laboratory Date of Service Exception Policy Under the Medicare Clinical Laboratory Fee Schedule (the 4 Day Rule ) Dear Valued
QIAGEN's Growing Immuno-Oncology Testing Portfolio
 QIAGEN's Growing Immuno-Oncology Testing Portfolio QIAGEN Your Partner in Translational Medicine Current Biomarkers of Immuno-Oncology Focus: Immuno-Oncology Testing Portfolio Tumor mutation burden (TMB)
QIAGEN's Growing Immuno-Oncology Testing Portfolio QIAGEN Your Partner in Translational Medicine Current Biomarkers of Immuno-Oncology Focus: Immuno-Oncology Testing Portfolio Tumor mutation burden (TMB)
Matthew Smolkin, MD HCLD Medical Director Molecular Pathology Diagnostic Laboratory
 Molecular Profiling Matthew Smolkin, MD HCLD Medical Director Molecular Pathology Diagnostic Laboratory Objectives Defining molecular profiling Technologies Why do we profile tumors? Current testing &
Molecular Profiling Matthew Smolkin, MD HCLD Medical Director Molecular Pathology Diagnostic Laboratory Objectives Defining molecular profiling Technologies Why do we profile tumors? Current testing &
Implementation of nation-wide molecular testing in oncology in the French Health care system : quality assurance issues & challenges
 Implementation of nation-wide molecular testing in oncology in the French Health care system : quality assurance issues & challenges Frédérique Nowak - 21 october 2015 "Putting Science into Standards event:
Implementation of nation-wide molecular testing in oncology in the French Health care system : quality assurance issues & challenges Frédérique Nowak - 21 october 2015 "Putting Science into Standards event:
Companion & Complementary Diagnostics: Clinical and Regulatory Perspectives
 Companion & Complementary Diagnostics: Clinical and Regulatory Perspectives Workshop on Companion Diagnostics January 31, 2017 Jan Trøst Jørgensen, M.Sc.Pharm., Ph.D. Dx-Rx Institute Fredensborg, Denmark
Companion & Complementary Diagnostics: Clinical and Regulatory Perspectives Workshop on Companion Diagnostics January 31, 2017 Jan Trøst Jørgensen, M.Sc.Pharm., Ph.D. Dx-Rx Institute Fredensborg, Denmark
Evolution of Pathology
 1 Traditional pathology Molecular pathology 2 Evolution of Pathology Gross Pathology Cellular Pathology Morphologic Pathology Molecular/Predictive Pathology Antonio Benivieni (1443-1502): First autopsy
1 Traditional pathology Molecular pathology 2 Evolution of Pathology Gross Pathology Cellular Pathology Morphologic Pathology Molecular/Predictive Pathology Antonio Benivieni (1443-1502): First autopsy
Advances in Pathology and molecular biology of lung cancer. Lukas Bubendorf Pathologie
 Advances in Pathology and molecular biology of lung cancer Lukas Bubendorf Pathologie Agenda The revolution of predictive markers Liquid biopsies PD-L1 Molecular subtypes (non-squamous NSCLC) Tsao AS et
Advances in Pathology and molecular biology of lung cancer Lukas Bubendorf Pathologie Agenda The revolution of predictive markers Liquid biopsies PD-L1 Molecular subtypes (non-squamous NSCLC) Tsao AS et
September 23, The Role of In Vitro Diagnostic Tests in Pediatric Master Protocol Development
 The Role of In Vitro Diagnostic Tests in Pediatric Master Protocol Development September 23, 2016 Anand Pathak, MD, PhD, MPH Medical Officer Molecular Genetics Branch Division of Molecular Genetics and
The Role of In Vitro Diagnostic Tests in Pediatric Master Protocol Development September 23, 2016 Anand Pathak, MD, PhD, MPH Medical Officer Molecular Genetics Branch Division of Molecular Genetics and
How Personalized Medicine is Changing the Biopharmaceutical Marketplace
 How Personalized Medicine is Changing the Biopharmaceutical Marketplace Marc Chioda, PharmD Associate Medical Director, Pfizer Oncology. Presentation to the Cancer Action Coalition of Virginia January
How Personalized Medicine is Changing the Biopharmaceutical Marketplace Marc Chioda, PharmD Associate Medical Director, Pfizer Oncology. Presentation to the Cancer Action Coalition of Virginia January
Agilent companion diagnostics for cancer immunotherapy
 Agilent companion diagnostics for cancer immunotherapy Annika Eklund, PhD Global Product Manager Companion Diagnostics Agilent Technologies Aalborg 1 Agilent Trusted Answers. Together OUR FOCUS life sciences,
Agilent companion diagnostics for cancer immunotherapy Annika Eklund, PhD Global Product Manager Companion Diagnostics Agilent Technologies Aalborg 1 Agilent Trusted Answers. Together OUR FOCUS life sciences,
QIAGEN Complete Solutions for Liquid Biopsy Molecular Testing
 QIAGEN Complete Solutions for Liquid Biopsy Molecular Testing Christopher Swagell, PhD Market Development Manager, Advanced Molecular Pathology QIAGEN 1 Agenda QIAGEN Solid Tumor Testing and Liquid Biopsy
QIAGEN Complete Solutions for Liquid Biopsy Molecular Testing Christopher Swagell, PhD Market Development Manager, Advanced Molecular Pathology QIAGEN 1 Agenda QIAGEN Solid Tumor Testing and Liquid Biopsy
Updated Molecular Testing Guideline for the Selection of Lung Cancer Patients for Treatment with Targeted Tyrosine Kinase Inhibitors
 Q: How is the strength of recommendation determined in the new molecular testing guideline? A: The strength of recommendation is determined by the strength of the available data (evidence). Strong Recommendation:
Q: How is the strength of recommendation determined in the new molecular testing guideline? A: The strength of recommendation is determined by the strength of the available data (evidence). Strong Recommendation:
Corporate Medical Policy
 Corporate Medical Policy Molecular Analysis for Targeted Therapy for Non-Small Cell Lung File Name: Origination: Last CAP Review: Next CAP Review: Last Review: molecular_analysis_for_targeted_therapy_for_non_small_cell_lung_cancer
Corporate Medical Policy Molecular Analysis for Targeted Therapy for Non-Small Cell Lung File Name: Origination: Last CAP Review: Next CAP Review: Last Review: molecular_analysis_for_targeted_therapy_for_non_small_cell_lung_cancer
Companion Diagnostics: Technologies and Markets. Robert Hunter. March Report Code: BIO077C
 Companion Diagnostics: Technologies and Markets March 2017 Robert Hunter Report Code: BIO077C Table of Contents Chapter 1: Introduction... 1 Study Goals and Objectives... 1 Reasons for Doing the Study...
Companion Diagnostics: Technologies and Markets March 2017 Robert Hunter Report Code: BIO077C Table of Contents Chapter 1: Introduction... 1 Study Goals and Objectives... 1 Reasons for Doing the Study...
FDA Companion Diagnostic Testing and Implications for Pharmacy and Medical Directors
 FDA Companion Diagnostic Testing and Implications for Pharmacy and Medical Directors Diana Brixner, PhD, RPh, FAMCP Professor, University of Utah Health Sciences Executive Director, Pharmacotherapy Outcomes
FDA Companion Diagnostic Testing and Implications for Pharmacy and Medical Directors Diana Brixner, PhD, RPh, FAMCP Professor, University of Utah Health Sciences Executive Director, Pharmacotherapy Outcomes
MEDICAL POLICY. Proprietary Information of YourCare Health Plan
 MEDICAL POLICY SUBJECT: HER-2 TESTING IN INVASIVE BREAST OR PAGE: 1 OF: 7 If the member's subscriber contract excludes coverage for a specific service it is not covered under that contract. In such cases,
MEDICAL POLICY SUBJECT: HER-2 TESTING IN INVASIVE BREAST OR PAGE: 1 OF: 7 If the member's subscriber contract excludes coverage for a specific service it is not covered under that contract. In such cases,
Keytruda (pembrolizumab)
") Keytruda (pembrolizumab) Line(s) of Business: HMO; PPO; QUEST Integration Akamai Advantage Original Effective Date: 10/01/2015 Current Effective Date: 07/24/2017TBD03/01/2018 POLICY A. INDICATIONS The
Keytruda (pembrolizumab) Line(s) of Business: HMO; PPO; QUEST Integration Akamai Advantage Original Effective Date: 10/01/2015 Current Effective Date: 07/24/2017TBD03/01/2018 POLICY A. INDICATIONS The
NeoTYPE Cancer Profiles
 NeoTYPE Cancer Profiles 30+ Multimethod Assays for Hematologic Diseases and Solid Tumors Molecular FISH Anatomic Pathology The next generation of diagnostic, prognostic, and therapeutic assessment What
NeoTYPE Cancer Profiles 30+ Multimethod Assays for Hematologic Diseases and Solid Tumors Molecular FISH Anatomic Pathology The next generation of diagnostic, prognostic, and therapeutic assessment What
Keytruda. Keytruda (pembrolizumab) Description
 Description") Federal Employee Program 1310 G Street, N.W. Washington, D.C. 20005 202.942.1000 Fax 202.942.1125 5.21.50 Subject: Keytruda Page: 1 of 5 Last Review Date: June 24, 2016 Keytruda Description Keytruda (pembrolizumab)
Federal Employee Program 1310 G Street, N.W. Washington, D.C. 20005 202.942.1000 Fax 202.942.1125 5.21.50 Subject: Keytruda Page: 1 of 5 Last Review Date: June 24, 2016 Keytruda Description Keytruda (pembrolizumab)
Next Generation Sequencing in Haematological Malignancy: A European Perspective. Wolfgang Kern, Munich Leukemia Laboratory
 Next Generation Sequencing in Haematological Malignancy: A European Perspective Wolfgang Kern, Munich Leukemia Laboratory Diagnostic Methods Cytomorphology Cytogenetics Immunophenotype Histology FISH Molecular
Next Generation Sequencing in Haematological Malignancy: A European Perspective Wolfgang Kern, Munich Leukemia Laboratory Diagnostic Methods Cytomorphology Cytogenetics Immunophenotype Histology FISH Molecular
Immunotherapy in NSCLC Pathologist role
 Immunotherapy in NSCLC Pathologist role Pimpin Incharoen, M.D. Assistant Professor, Thoracic Pathology Department of Pathology, Ramathibodi Hospital Genetic alterations in NSCLC Khono et al, Trans Lung
Immunotherapy in NSCLC Pathologist role Pimpin Incharoen, M.D. Assistant Professor, Thoracic Pathology Department of Pathology, Ramathibodi Hospital Genetic alterations in NSCLC Khono et al, Trans Lung
Precision Genetic Testing in Cancer Treatment and Prognosis
 Precision Genetic Testing in Cancer Treatment and Prognosis Deborah Cragun, PhD, MS, CGC Genetic Counseling Graduate Program Director University of South Florida Case #1 Diana is a 47 year old cancer patient
Precision Genetic Testing in Cancer Treatment and Prognosis Deborah Cragun, PhD, MS, CGC Genetic Counseling Graduate Program Director University of South Florida Case #1 Diana is a 47 year old cancer patient
Opportunities and Challenges in the Development of Companion Diagnostics
 Opportunities and Challenges in the Development of Companion Diagnostics E. Patrick Groody, Ph.D. Divisional Vice President Research and Development Abbott Molecular Agenda Value of Personalized Medicine
Opportunities and Challenges in the Development of Companion Diagnostics E. Patrick Groody, Ph.D. Divisional Vice President Research and Development Abbott Molecular Agenda Value of Personalized Medicine
Clinical Policy: Nivolumab (Opdivo) Reference Number: CP.PHAR.121 Effective Date: Last Review Date: Line of Business: Medicaid
 Reference Number: CP.PHAR.121 Effective Date: Last Review Date: Line of Business: Medicaid") Clinical Policy: (Opdivo) Reference Number: CP.PHAR.121 Effective Date: 07.15 Last Review Date: 01.18 Line of Business: Medicaid Revision Log See Important Reminder at the end of this policy for important
Clinical Policy: (Opdivo) Reference Number: CP.PHAR.121 Effective Date: 07.15 Last Review Date: 01.18 Line of Business: Medicaid Revision Log See Important Reminder at the end of this policy for important
Big data vs. the individual liver from a regulatory perspective
 Big data vs. the individual liver from a regulatory perspective Robert Schuck, Pharm.D., Ph.D. Genomics and Targeted Therapy Office of Clinical Pharmacology Center for Drug Evaluation and Research Food
Big data vs. the individual liver from a regulatory perspective Robert Schuck, Pharm.D., Ph.D. Genomics and Targeted Therapy Office of Clinical Pharmacology Center for Drug Evaluation and Research Food
NCCN Non-Small Cell Lung Cancer V Meeting June 15, 2018
 Guideline Page and Request Illumina Inc. requesting to replace Testing should be conducted as part of broad molecular profiling with Consider NGS-based assays that include EGFR, ALK, ROS1, and BRAF as
Guideline Page and Request Illumina Inc. requesting to replace Testing should be conducted as part of broad molecular profiling with Consider NGS-based assays that include EGFR, ALK, ROS1, and BRAF as
HER2 status assessment in breast cancer. Marc van de Vijver Academic Medical Centre (AMC), Amsterdam
, Amsterdam") HER2 status assessment in breast cancer Marc van de Vijver Academic Medical Centre (AMC), Amsterdam 13e Bossche Mamma Congres 17 th June 2015 Modern cancer therapies are based on sophisticated molecular
HER2 status assessment in breast cancer Marc van de Vijver Academic Medical Centre (AMC), Amsterdam 13e Bossche Mamma Congres 17 th June 2015 Modern cancer therapies are based on sophisticated molecular
Role of the Pathologist in Guiding Immuno-oncological Therapies. Scott Rodig MD, PhD
 Role of the Pathologist in Guiding Immuno-oncological Therapies Scott Rodig MD, PhD Department of Pathology, Brigham & Women s Hospital Center for Immuno-Oncology, Dana-Farber Cancer Institute Associate
Role of the Pathologist in Guiding Immuno-oncological Therapies Scott Rodig MD, PhD Department of Pathology, Brigham & Women s Hospital Center for Immuno-Oncology, Dana-Farber Cancer Institute Associate
Opdivo. Opdivo (nivolumab) Description
 Description") Federal Employee Program 1310 G Street, N.W. Washington, D.C. 20005 202.942.1000 Fax 202.942.1125 5.21.53 Subsection: Antineoplastic Agents Original Policy Date: January 16, 2015 Subject: Opdivo Page:
Federal Employee Program 1310 G Street, N.W. Washington, D.C. 20005 202.942.1000 Fax 202.942.1125 5.21.53 Subsection: Antineoplastic Agents Original Policy Date: January 16, 2015 Subject: Opdivo Page:
MICROSCOPY PREDICTIVE PROFILING
 Immunomodulatory therapy in NSCLC: a year into clinical practice Professor J R Gosney Consultant Thoracic Pathologist Royal Liverpool University Hospital Disclosure JRG is a paid advisor to and speaker
Immunomodulatory therapy in NSCLC: a year into clinical practice Professor J R Gosney Consultant Thoracic Pathologist Royal Liverpool University Hospital Disclosure JRG is a paid advisor to and speaker
May 31, NCCN Guidelines: T-Cell Lymphomas
 May 31, 2017 Maria Rivas, MD Senior Vice-President Global Medical Affairs Merck & Co. Kenilworth - Galloping Hill, +1 908 740 6533 K6-1624G maria.rivas1@merck.com NCCN Guidelines: T-Cell Lymphomas On behalf
May 31, 2017 Maria Rivas, MD Senior Vice-President Global Medical Affairs Merck & Co. Kenilworth - Galloping Hill, +1 908 740 6533 K6-1624G maria.rivas1@merck.com NCCN Guidelines: T-Cell Lymphomas On behalf
Policy. Medical Policy Manual Approved Revised: Do Not Implement until 6/30/2019. Nivolumab
 Medical Manual Approved Revised: Do Not Implement until 6/30/2019 Nivolumab NDC CODE(S) 00003-3772-XX Opdivo 40 MG/4ML SOLN (B-M SQUIBB U.S. (PRIMARY CARE)) 00003-3774-XX Opdivo 100 MG/10ML SOLN (B-M SQUIBB
Medical Manual Approved Revised: Do Not Implement until 6/30/2019 Nivolumab NDC CODE(S) 00003-3772-XX Opdivo 40 MG/4ML SOLN (B-M SQUIBB U.S. (PRIMARY CARE)) 00003-3774-XX Opdivo 100 MG/10ML SOLN (B-M SQUIBB
Introduction of an NGS gene panel into the Haemato-Oncology MPN service
 Introduction of an NGS gene panel into the Haemato-Oncology MPN service Dr. Anna Skowronska, Dr Jane Bryon, Dr Samuel Clokie, Dr Yvonne Wallis and Professor Mike Griffiths West Midlands Regional Genetics
Introduction of an NGS gene panel into the Haemato-Oncology MPN service Dr. Anna Skowronska, Dr Jane Bryon, Dr Samuel Clokie, Dr Yvonne Wallis and Professor Mike Griffiths West Midlands Regional Genetics
1. Q: What has changed from the draft recommendations posted for public comment in November/December 2011?
 Frequently Asked Questions (FAQs) in regard to Molecular Testing Guideline for Selection of Lung Cancer Patients for EGFR and ALK Tyrosine Kinase Inhibitors 1. Q: What has changed from the draft recommendations
Frequently Asked Questions (FAQs) in regard to Molecular Testing Guideline for Selection of Lung Cancer Patients for EGFR and ALK Tyrosine Kinase Inhibitors 1. Q: What has changed from the draft recommendations
Product Introduction
 Product Introduction Product Codes: HCL026, HCL027 and HCL028 Contents Introduction to HER2 2 HER2 immunohistochemistry 3 Cell lines as controls 5 HER2 Analyte Control DR IHC 7 HER2 Analyte Control DR
Product Introduction Product Codes: HCL026, HCL027 and HCL028 Contents Introduction to HER2 2 HER2 immunohistochemistry 3 Cell lines as controls 5 HER2 Analyte Control DR IHC 7 HER2 Analyte Control DR
FDA Regulation of Diagnostic Tests Jeffrey N. Gibbs Hyman, Phelps & McNamara, P.C. Washington, DC
 AIPLA Annual Meeting Joint Biotechnology Committee/ Special Committee on FDA Law Program October 21, 2010 Marriott Wardman Park Hotel Washington, DC FDA Regulation of Diagnostic Tests Jeffrey N. Gibbs
AIPLA Annual Meeting Joint Biotechnology Committee/ Special Committee on FDA Law Program October 21, 2010 Marriott Wardman Park Hotel Washington, DC FDA Regulation of Diagnostic Tests Jeffrey N. Gibbs
MEDICAL POLICY Genetic Testing for Breast and Ovarian Cancers
 POLICY: PG0067 ORIGINAL EFFECTIVE: 07/30/02 LAST REVIEW: 01/25/18 MEDICAL POLICY Genetic Testing for Breast and Ovarian Cancers GUIDELINES This policy does not certify benefits or authorization of benefits,
POLICY: PG0067 ORIGINAL EFFECTIVE: 07/30/02 LAST REVIEW: 01/25/18 MEDICAL POLICY Genetic Testing for Breast and Ovarian Cancers GUIDELINES This policy does not certify benefits or authorization of benefits,
The Center for PERSONALIZED DIAGNOSTICS
 The Center for PERSONALIZED DIAGNOSTICS Precision Diagnostics for Personalized Medicine A joint initiative between The Department of Pathology and Laboratory Medicine & The Abramson Cancer Center The (CPD)
The Center for PERSONALIZED DIAGNOSTICS Precision Diagnostics for Personalized Medicine A joint initiative between The Department of Pathology and Laboratory Medicine & The Abramson Cancer Center The (CPD)
Novel Test for Improving Patient Outcomes in Hematologic Cancers. predictive test that determines sensitivity to therapeutic options
 Novel Test for Improving Patient Outcomes in Hematologic Cancers predictive test that determines sensitivity to therapeutic options Implementing A Surrogate Functional Biomarker Practical application of
Novel Test for Improving Patient Outcomes in Hematologic Cancers predictive test that determines sensitivity to therapeutic options Implementing A Surrogate Functional Biomarker Practical application of
Molecular Diagnostics Overview JAN A. NOWAK, PHD, MD PATHOLOGY AND LABORATORY MEDICINE MOLECULAR DIAGNOSTICS LABORATORY FEBRUARY 15, 2018
 Molecular Diagnostics Overview JAN A. NOWAK, PHD, MD PATHOLOGY AND LABORATORY MEDICINE MOLECULAR DIAGNOSTICS LABORATORY FEBRUARY 15, 2018 Some Key Points Molecular Testing has applications in every section
Molecular Diagnostics Overview JAN A. NOWAK, PHD, MD PATHOLOGY AND LABORATORY MEDICINE MOLECULAR DIAGNOSTICS LABORATORY FEBRUARY 15, 2018 Some Key Points Molecular Testing has applications in every section
Assessment Run C1 2017
 Assessment Run C1 2017 PD-L1 The first assessment in this new NordiQC Companion module C1 focused on the accuracy of the PD-L1 IHC assays performed by the participating laboratories to identify patients
Assessment Run C1 2017 PD-L1 The first assessment in this new NordiQC Companion module C1 focused on the accuracy of the PD-L1 IHC assays performed by the participating laboratories to identify patients
Clinical Policy: Atezolizumab (Tecentriq) Reference Number: CP.PHAR.235 Effective Date: 06/16 Last Review Date: 05/17
 Reference Number: CP.PHAR.235 Effective Date: 06/16 Last Review Date: 05/17") Clinical Policy: (Tecentriq) Reference Number: CP.PHAR.235 Effective Date: 06/16 Last Review Date: 05/17 Coding Implications Revision Log See Important Reminder at the end of this policy for important
Clinical Policy: (Tecentriq) Reference Number: CP.PHAR.235 Effective Date: 06/16 Last Review Date: 05/17 Coding Implications Revision Log See Important Reminder at the end of this policy for important
Vernieuwing en diagnostiek bij NSCLC: Immunotherapy: PD-L1 analyse: waar staan we
 9e avondsymposium: "Nieuwe ontwikkelingen in de behandeling van NSCLC" 9 november 2016, UMCG Vernieuwing en diagnostiek bij NSCLC: Immunotherapy: PD-L1 analyse: waar staan we Wim Timens Professor and Chair
9e avondsymposium: "Nieuwe ontwikkelingen in de behandeling van NSCLC" 9 november 2016, UMCG Vernieuwing en diagnostiek bij NSCLC: Immunotherapy: PD-L1 analyse: waar staan we Wim Timens Professor and Chair
Clinical Policy: Nivolumab (Opdivo) Reference Number: ERX.SPA.302 Effective Date:
 Reference Number: ERX.SPA.302 Effective Date:") Clinical Policy: (Opdivo) Reference Number: ERX.SPA.302 Effective Date: 03.01.19 Last Review Date: 02.19 Revision Log See Important Reminder at the end of this policy for important regulatory and legal
Clinical Policy: (Opdivo) Reference Number: ERX.SPA.302 Effective Date: 03.01.19 Last Review Date: 02.19 Revision Log See Important Reminder at the end of this policy for important regulatory and legal
Cancer Immunotherapy Survey
 CHAPTER 8: Cancer Immunotherapy Survey All (N=100) Please classify your organization. Academic lab or center Small biopharmaceutical company Top 20 Pharma Mid-size pharma Diagnostics company Other (please
CHAPTER 8: Cancer Immunotherapy Survey All (N=100) Please classify your organization. Academic lab or center Small biopharmaceutical company Top 20 Pharma Mid-size pharma Diagnostics company Other (please
Role of the pathologist in the diagnosis and mutational analysis of lung cancer Professor J R Gosney
 Role of the pathologist in the diagnosis and mutational analysis of lung cancer Professor J R Gosney Consultant Thoracic Pathologist Royal Liverpool University Hospital Disclosure JRG is a paid advisor
Role of the pathologist in the diagnosis and mutational analysis of lung cancer Professor J R Gosney Consultant Thoracic Pathologist Royal Liverpool University Hospital Disclosure JRG is a paid advisor
Clovis Oncology Announces First Quarter 2017 Operating Results. May 3, :06 PM ET
 Clovis Oncology Announces First Quarter 2017 Operating Results May 3, 2017 4:06 PM ET Strong Q1 launch quarter for Rubraca (rucaparib) in U.S. with $7M reported in net sales Clovis notified that ARIEL3
Clovis Oncology Announces First Quarter 2017 Operating Results May 3, 2017 4:06 PM ET Strong Q1 launch quarter for Rubraca (rucaparib) in U.S. with $7M reported in net sales Clovis notified that ARIEL3
Personalised cancer care Information for Medical Specialists. A new way to unlock treatment options for your patients
 Personalised cancer care Information for Medical Specialists A new way to unlock treatment options for your patients Contents Optimised for clinical benefit 4 Development history 4 Full FIND IT panel vs
Personalised cancer care Information for Medical Specialists A new way to unlock treatment options for your patients Contents Optimised for clinical benefit 4 Development history 4 Full FIND IT panel vs
What s New Medical Policy Updates January 2019
 What s New Medical Policy Updates January 2019 Listed below are the recent changes made to policies within the Geisinger Health Plan Medical Policy Portfolio during the month of December that will become
What s New Medical Policy Updates January 2019 Listed below are the recent changes made to policies within the Geisinger Health Plan Medical Policy Portfolio during the month of December that will become
Wells Fargo Healthcare Conference September 6, 2018
 Wells Fargo Healthcare Conference September 6, 2018 Safe Harbor Statement To the extent that statements contained in this presentation are not descriptions of historical facts regarding TESARO, they are
Wells Fargo Healthcare Conference September 6, 2018 Safe Harbor Statement To the extent that statements contained in this presentation are not descriptions of historical facts regarding TESARO, they are
Clinical Utility of Droplet ddpcr, moving to diagnostics. Koen De Gelas, PhD, CRIG ddpcr mini symposium, 15/05/2018
 1 Clinical Utility of Droplet ddpcr, moving to diagnostics 2 Koen De Gelas, PhD, CRIG ddpcr mini symposium, 15/05/2018 Disclaimer Disclaimer: all consumables, instruments, applications and software covered
1 Clinical Utility of Droplet ddpcr, moving to diagnostics 2 Koen De Gelas, PhD, CRIG ddpcr mini symposium, 15/05/2018 Disclaimer Disclaimer: all consumables, instruments, applications and software covered
Policy. Medical Policy Manual Approved Revised: Do Not Implement Until 3/2/19. Nivolumab (Intravenous)
") Nivolumab (Intravenous) NDC CODE(S) 00003-3772-XX Opdivo 40 MG/4ML SOLN (B-M SQUIBB U.S. (PRIMARY CARE)) 00003-3774-XX Opdivo 100 MG/10ML SOLN (B-M SQUIBB U.S. (PRIMARY CARE)) 00003-3734-XX Opdivo 240
Nivolumab (Intravenous) NDC CODE(S) 00003-3772-XX Opdivo 40 MG/4ML SOLN (B-M SQUIBB U.S. (PRIMARY CARE)) 00003-3774-XX Opdivo 100 MG/10ML SOLN (B-M SQUIBB U.S. (PRIMARY CARE)) 00003-3734-XX Opdivo 240
Harnessing Complexity: Companion Diagnostics in Oncology
 Ipsos Healthcare Harnessing Complexity: Companion Diagnostics in Oncology Pieter De Richter, Head of Global Molecular Diagnostics Portfolio, Ipsos Healthcare First Published: October 2015 Updated: February
Ipsos Healthcare Harnessing Complexity: Companion Diagnostics in Oncology Pieter De Richter, Head of Global Molecular Diagnostics Portfolio, Ipsos Healthcare First Published: October 2015 Updated: February
Immunotherapy in Colorectal cancer
 Immunotherapy in Colorectal cancer Ahmed Zakari, MD Associate Professor University of Central Florida, College of Medicine Medical Director, Gastro Intestinal Cancer Program Florida Hospital Cancer Institute
Immunotherapy in Colorectal cancer Ahmed Zakari, MD Associate Professor University of Central Florida, College of Medicine Medical Director, Gastro Intestinal Cancer Program Florida Hospital Cancer Institute
PMDA Perspectives on Oncology Panel. REIKO YANAGIHARA, Ph.D. Office of In Vitro Diagnostics PMDA
 15th DIA Japan Annual Meeting 2018 Promoting Better Collaboration to Drive Global Health and Innovation in an Era of Medical and Scientific Transformation November 11-13, 2018 Tokyo Big Sight PMDA Perspectives
15th DIA Japan Annual Meeting 2018 Promoting Better Collaboration to Drive Global Health and Innovation in an Era of Medical and Scientific Transformation November 11-13, 2018 Tokyo Big Sight PMDA Perspectives
Agenda. What is a Liquid Biopsy? Biocept technology. Concordance With Tissue. Clinical Applications. Billing and Reimbursement.
 Agenda What is a Liquid Biopsy? Biocept technology Concordance With Tissue Clinical Applications Billing and Reimbursement Recap & Questions 1 Targets of Tumor Found in Liquid Biopsy 1 Eric Topol, Professor
Agenda What is a Liquid Biopsy? Biocept technology Concordance With Tissue Clinical Applications Billing and Reimbursement Recap & Questions 1 Targets of Tumor Found in Liquid Biopsy 1 Eric Topol, Professor
See Important Reminder at the end of this policy for important regulatory and legal information.
 Clinical Policy: Pembrolizumab (Keytruda) Reference Number: CP.PHAR.322 Effective Date: 07.01.18 Last Review Date: 11.17 Line of Business: Oregon Health Plan Revision Log See Important Reminder at the
Clinical Policy: Pembrolizumab (Keytruda) Reference Number: CP.PHAR.322 Effective Date: 07.01.18 Last Review Date: 11.17 Line of Business: Oregon Health Plan Revision Log See Important Reminder at the
Personalised Healthcare (PHC) with Foundation Medicine (FMI) Fatma Elçin KINIKLI, FMI Turkey, Science Leader
 with Foundation Medicine (FMI) Fatma Elçin KINIKLI, FMI Turkey, Science Leader") Personalised Healthcare (PHC) with Foundation Medicine (FMI) Fatma Elçin KINIKLI, FMI Turkey, Science Leader Agenda PHC Approach Provides Better Patient Outcome FMI offers Comprehensive Genomic Profiling,
Personalised Healthcare (PHC) with Foundation Medicine (FMI) Fatma Elçin KINIKLI, FMI Turkey, Science Leader Agenda PHC Approach Provides Better Patient Outcome FMI offers Comprehensive Genomic Profiling,
CDx in oncology Prof. Christophe Le Tourneau, MD, PhD FEAM Geneva September 27, 2018
 CDx in oncology Prof. Christophe Le Tourneau, MD, PhD Institut Curie Paris & Saint-Cloud France Head, Department of Drug Development and Innovation (D 3 i) INSERM U900 Research unit Versailles Saint-Quentin-en-Yvelines
CDx in oncology Prof. Christophe Le Tourneau, MD, PhD Institut Curie Paris & Saint-Cloud France Head, Department of Drug Development and Innovation (D 3 i) INSERM U900 Research unit Versailles Saint-Quentin-en-Yvelines
Personalized Healthcare Update
 Dr. Kai - Oliver Wesche Market Development Manager, Personalized Healthcare QIAGEN Personalized Healthcare Update Pioneering Personalized Medicine through Partnering TOMTOVOK BKM120 Zelboraf QIAGEN partners:
Dr. Kai - Oliver Wesche Market Development Manager, Personalized Healthcare QIAGEN Personalized Healthcare Update Pioneering Personalized Medicine through Partnering TOMTOVOK BKM120 Zelboraf QIAGEN partners:
Circulating Tumor DNA in GIST and its Implications on Treatment
 Circulating Tumor DNA in GIST and its Implications on Treatment October 2 nd 2017 Dr. Ciara Kelly Assistant Attending Physician Sarcoma Medical Oncology Service Objectives Background Liquid biopsy & ctdna
Circulating Tumor DNA in GIST and its Implications on Treatment October 2 nd 2017 Dr. Ciara Kelly Assistant Attending Physician Sarcoma Medical Oncology Service Objectives Background Liquid biopsy & ctdna
Results you can trust
 PRODUCT I NF OR MAT ION pharmdx Results you can trust The first and only FDA-approved PD-L1 test to assess the magnitude of treatment effect on progression-free survival in melanoma patients from OPDIVO
PRODUCT I NF OR MAT ION pharmdx Results you can trust The first and only FDA-approved PD-L1 test to assess the magnitude of treatment effect on progression-free survival in melanoma patients from OPDIVO
Molecular Testing in Lung Cancer
 Molecular Testing in Lung Cancer Pimpin Incharoen, M.D. Assistant Professor, Thoracic Pathology Department of Pathology, Ramathibodi Hospital Genetic alterations in lung cancer Source: Khono et al, Trans
Molecular Testing in Lung Cancer Pimpin Incharoen, M.D. Assistant Professor, Thoracic Pathology Department of Pathology, Ramathibodi Hospital Genetic alterations in lung cancer Source: Khono et al, Trans
Novartis Oncology. The Precision Oncology Annual Trend Report. Perspectives From Payers, Oncologists, and Pathologists Third Edition
 Novartis Oncology The Precision Oncology Annual Trend Report Perspectives From Payers, Oncologists, and Pathologists Third Edition Table of Contents Foreword 3 Introduction 4 Executive Summary 5 Methodology
Novartis Oncology The Precision Oncology Annual Trend Report Perspectives From Payers, Oncologists, and Pathologists Third Edition Table of Contents Foreword 3 Introduction 4 Executive Summary 5 Methodology
Illumina Trusight Myeloid Panel validation A R FHAN R A FIQ
 Illumina Trusight Myeloid Panel validation A R FHAN R A FIQ G E NETIC T E CHNOLOGIST MEDICAL G E NETICS, CARDIFF To Cover Background to the project Choice of panel Validation process Genes on panel, Protocol
Illumina Trusight Myeloid Panel validation A R FHAN R A FIQ G E NETIC T E CHNOLOGIST MEDICAL G E NETICS, CARDIFF To Cover Background to the project Choice of panel Validation process Genes on panel, Protocol
Biomarkers for Cancer Immunotherapy Debate
 Biomarkers for Cancer Immunotherapy Debate Moderator: Maria Karasarides, PhD AstraZeneca Pro: Daniel S. Chen, MD, PhD Genentech Con: Steve Averbuch, MD Bristol-Myers Squibb Biomarkers to Select Patients
Biomarkers for Cancer Immunotherapy Debate Moderator: Maria Karasarides, PhD AstraZeneca Pro: Daniel S. Chen, MD, PhD Genentech Con: Steve Averbuch, MD Bristol-Myers Squibb Biomarkers to Select Patients
1.5. Research Areas Treatment Selection
 1.5. Research Areas Cancer biomarker research embraces many areas of study, including tumorigenesis, metastasis, clinical trial and surrogate endpoints, cell isolation, target identification, drug resistance,
1.5. Research Areas Cancer biomarker research embraces many areas of study, including tumorigenesis, metastasis, clinical trial and surrogate endpoints, cell isolation, target identification, drug resistance,
MP Circulating Tumor DNA Management of Non-Small-Cell Lung Cancer (Liquid Biopsy)
") Medical Policy BCBSA Ref. Policy: 2.04.143 Last Review: 10/18/2018 Effective Date: 10/18/2018 Section: Medicine Related Policies 2.04.121 Miscellaneous Genetic and Molecular Diagnostic Tests 2.04.45 Molecular
Medical Policy BCBSA Ref. Policy: 2.04.143 Last Review: 10/18/2018 Effective Date: 10/18/2018 Section: Medicine Related Policies 2.04.121 Miscellaneous Genetic and Molecular Diagnostic Tests 2.04.45 Molecular
Prior Authorization Required: Additional Information:
 Genetic Testing for Somatic Tumor Markers MP9486 Covered Service: Prior Authorization Required: Additional Information: Yes when meets criteria below No A first-degree relative is defined as an individual
Genetic Testing for Somatic Tumor Markers MP9486 Covered Service: Prior Authorization Required: Additional Information: Yes when meets criteria below No A first-degree relative is defined as an individual
NeoTYPE Cancer Profiles
 NeoTYPE Cancer Profiles Multimethod Analysis of 25+ Hematologic Diseases and Solid Tumors Anatomic Pathology FISH Molecular The next generation of diagnostic, prognostic, and therapeutic assessment NeoTYPE
NeoTYPE Cancer Profiles Multimethod Analysis of 25+ Hematologic Diseases and Solid Tumors Anatomic Pathology FISH Molecular The next generation of diagnostic, prognostic, and therapeutic assessment NeoTYPE
A Blueprint for Drug/Diagnostic Development: Facilitating Development and Use of Curated Genetic Databases
 A Blueprint for Drug/Diagnostic Development: Facilitating Development and Use of Curated Genetic Databases TABLE OF CONTENTS Forum Goals Summary Terminology and Considerations Proposals o Proposal #1:
A Blueprint for Drug/Diagnostic Development: Facilitating Development and Use of Curated Genetic Databases TABLE OF CONTENTS Forum Goals Summary Terminology and Considerations Proposals o Proposal #1:
Clinical Utility of Diagnostic Tests
 Clinical Utility of Diagnostic Tests David A. Eberhard MD, PhD Director, Pre-Clinical Genomic Pathology, Lineberger Comprehensive Cancer Center Associate Professor, Depts. of Pathology and Pharmacology
Clinical Utility of Diagnostic Tests David A. Eberhard MD, PhD Director, Pre-Clinical Genomic Pathology, Lineberger Comprehensive Cancer Center Associate Professor, Depts. of Pathology and Pharmacology