Mitochondrial Disease
|
|
|
- Rosamund Rich
- 6 years ago
- Views:
Transcription
1 Mitochondrial Disease Information Booklet for Medical Practitioners Produced by: Dr Karen Crawley MBBS FRACGP Overseen by: Professor Carolyn Sue MBBS PhD FRACP Professor John Cristodoulou MB BS PhD FRACP FRCPA FHGSA Professor David Thorburn BSc(Hons) PhD FHGSA FFS (RCPA) May
2
3 CONTENTS About the Australian Mitochondrial Disease Foundation...2 Could it be Mitochondrial Disease?...3 The Mitochondria...5 Definition of Mitochondrial Disease...9 Genetic Background...12 Mitochondrial Symptoms...15 The Diagnostic Process...18 Common Mitochondrial Disease Presentations...25 Differential Diagnosis...28 Management...29 Prognosis and Further Research...41 Links to Other Diseases...43 Appendix...44 References

4 ABOUT THE AUSTRALIAN MITOCHONDRIAL DISEASE FOUNDATION The Australian Mitochondrial Disease Foundation (AMDF) funds essential research into the diagnosis, treatment and cure of mitochondrial disorders, and supports sufferers and their families. AMDF also works to educate the general public and the medical profession about mitochondrial disease. The AMDF hopes this booklet provides some understanding of mitochondrial or mito disease, an underestimated medical enigma robbing patients of their energy and too often their life! our mission research support educate to fund research into mitochondrial disease to support sufferers of mitochondrial disease & their families to educate the general public & the medical profession about mitochondrial disease 2
5 COULD IT BE MITOCHONDRIAL DISEASE? Most GPs will at some stage cared for a patient that seems to be neurotic or has bad luck. They return repeatedly with symptoms that just don t seem to add up, don t seem to match their appearance, or if they are real, the investigations are negative. The patient ends up with multiple pathological diagnoses that adds to further confusion. The practitioner often feels helpless, to clearly understand them, yet GPs have been taught to listen, believe, show compassion and to look for one pathology. But where is that pathology, where is that link in the variety of presenting complaints? Do we then refer back to the list we so obligingly learnt at university? Have we considered the following possibilities?: Infective Neoplastic Autoimmune Endocrine Nutritional Metabolic Psychological Vascular Neurological... Hopefully this booklet will assist you when the link is not in the organ, nor the hormone, nor the vascular system, nor the mind... but in the cell. More specifically, the problem is in the forgotten essential organelles that produce about 90% of the energy needed by the human body to function, sustain life and support growth. 3

6 This booklet may not be the answer to all your unsolved mysteries, but with recent research 1,2,5,6 demonstrating that mitochondrial DNA mutations are present in one in 200 people with at least one in 5,000 suffering severe disease, it is worth the consideration. That means in a 2,000+ patient general practice approximately 10 patients have a mutation that has the potential to cause mitochondrial disease. So when is it appropriate to consider the mitochondria as the guilty offender, and how do we even begin to investigate it? (Diagram: courtesy of Diagnosing mitochondrial disease can be a nightmare, due to the widespread variety and severity of symptoms in the large number of known sub-groups (>100). Many experts refer to it as the notorious masquerader because it mimics so many different illnesses. It affects both children and adults, many of whom are either undiagnosed or misdiagnosed due to the complexity of the disease. So how do you then summarise the presenting mitochondrial patient? It can be summarised as 4 any symptom, in any organ, at any age

7 THE MITOCHONDRIA Mitochondria help to maintain proper concentration of calcium ions within the various compartments of the cell. Mitochondria store calcium. The major function of the mitochondria is to produce energy. Mitochondria help in the formation of blood components and hormones such as testosterone and oestrogen. Mitochondria in the liver help to detoxify ammonia. Production of heat is another function of mitochondria. Mitochondria help in the regulation of membrane potential, cell proliferation and cell metabolism. Mitochondria cause apoptosis or programmed cell death. Mitochondria help in the biosynthesis of heme and steroids. Functions of the Mitochondria (Diagram: courtesy of Mitochondria perform many functions necessary for cell metabolism (see diagram), but the energy-producing pathways are the most important. These pathways allow us to break down carbohydrate, fat and oxygen to live. This process of burning food to make the energy molecule, adenosine triphosphate (ATP), is called oxidative phosphorylation (OXPHOS). Only mitochondria can do it. This highly efficient manufacturing process requires oxygen and is therefore called aerobic metabolism and produces approximately 90% of the energy needed by the human body. 5

8 Mitochondria also play an intimate role in most of the cell s major metabolic pathways that build, break down, or recycle its molecular building blocks. Cells cannot make the RNA and DNA they need to grow and function without mitochondria 4. However, for the purpose of this booklet, we will discuss in-depth only the mitochondria s main role in energy production. Over 70 different polypeptides or proteins interacting on the inner mitochondrial membrane make up the complex mitochondrial respiratory chain (also known as the electron transport chain) and allow OXPHOS to occur. Energy sources such as glucose are initially metabolized in the cytoplasm then imported via proteins at the beginning of the chain. Other proteins in the chain continue the process of catabolism using metabolic pathways such as the Krebs cycle, fatty acid oxidation, and amino acid oxidation. This produces energy-rich electron donors whose electrons are then passed through the respiratory chain (a series of complex molecules I, II, III, and IV). Cytochrome oxidase or complex IV, passes the electrons to oxygen which is reduced to water. ATP synthase or Complex V then uses the electrochemical proton gradient produced by the respiratory chain to finally make ATP. Although electron transport occurs with great efficiency, a small percentage of electrons are prematurely leaked to oxygen, resulting in the formation of the toxic free-radical superoxide. Energy Production within mitochondria (for more detail see next diagram) (Diagram: courtesy of the Muscular Dystrophy Association and The Mitochondria Research Society) 6

9 Mitochondria synthesise adenosine triphosphate (ATP) from adenosine diphosphate (ADP) and inorganic phosphate in a process called oxidative phosphorylation. Simply put, they burn food in the presence of oxygen to produce ATP. The process, greatly simplified, has three main steps. 1. The citric acid cycle breaks down pyruvate (a product of glucose metabolism) and the beta oxidation spiral breaks down fatty acids. Both use the energy released to reduce (i.e. add electrons to) the electron carriers nicotinamide adenine dinucleotide (NAD+), yielding NADH, and flavin adenine dinucleotide (FAD), yielding FADH 2 2. The electron transport chain (also called the respiratory chain) uses the energy from the electrons to pump hydrogen ions (protons) into the intermembrane space. The electron transport chain comprises five complexes designated I through V. inner membrane intermembrane space outer membrane 3. ATP synthesis takes place at complex V of the electron transport chain, which uses the energy of protons flowing back into the matrix to attach phosphorus atoms to ADP molecules, producing ATP. ATP exits through the adenosine nucleotide translocase (AND) channel, where ATP is exchanged for ADP. ATP (energy) 7

10 Mitochondria take on many different shapes, each being characteristic of the specialised cell in which it resides 7 and tailored to meet the needs of that cell. All told, there are about 250 different cell types in the human body, many having their own distinct mitochondria with its own specialised metabolic function 4. Most of our body s nucleated cells contain 500 to 2,000 mitochondria, though some cell types have only a few mitochondria. For example, platelets have only two to six mitochondria whilst mature red blood cells do not contain any mitochondria, though its cellular precursor, the proerythroblast, is critically dependent on mitochondrial function for differentiation into its mature state. The tissues that require lots of energy have the most mitochondria, and so these highly energy-dependent tissues or organs are the ones most often affected in mitochondrial disease. Therefore, damage is most commonly presenting in the cells of the muscles, brain, heart, liver, gastrointestinal tract, ears and eyes. 8
11 DEFINITION OF MITOCHONDRIAL DISEASE Mitochondrial diseases are a clinically heterogeneous group of disorders that result from a dysfunction in the mitochondrial respiratory chain. The identification of the first genetic cause of mitochondrial diseases was not until Tissues and organs that are highly dependent upon this aerobic metabolism are most likely to be involved in mitochondrial disease. Common clinical features are ptosis, external ophthalmoplegia, proximal myopathy, exercise intolerance, cardiomyopathy, hyperglycaemia, liver failure, sensorineural deafness, optic atrophy, pigmentary retinal changes and central nervous system findings of fluctuating encephalopathy, seizures, dementia, migraine, stroke-like episodes, ataxia, and spasticity. The ability for an organ to function normally depends partly on whether its energy production meets a minimum threshold for that organ, otherwise loss of function occurs. The organs that are more highly energy dependent may show symptoms with even a relatively small drop in energy production. For example, the central nervous system has a lower threshold than other organs at which it will start to show evidence of functional impairment. Some mitochondrial disorders may affect a single organ only, such as the eye in Leber Hereditary Optic Neuropathy (LHON), but many involve multiple organ systems and often present with prominent neurologic and myopathic features. Many affected individuals display a cluster of clinical features that fall into a discrete clinical syndrome, such as mitochondrial encephalopathy with lactic acidosis and strokelike episodes (MELAS), or myoclonic epilepsy with ragged-red fibres (MERRF). However, considerable clinical variability exists and many individuals do not fit neatly into one particular category 3. There is no consistent correlation between the severity of a particular biochemical defect and the severity of that patient s presentation. 9


12 Where does Mitochondrial Disease Hide? Diabetes Clinics Cancer Haematology Clinics Epilepsy Clinics Atypical Learning Disabilities Psychiatric Facilities Cerebal Palsy Clinics Sudden Infant Death Sydnrome SIDS Atypical Leukodystrophy GI Dysmotility Clinics Language Delay Clinics Atypical Autism Rheumatology/ Multiple Sclerosis Clinics Unexplained Liver Failure Unexplained Heart Failure Unexplained Kidney Disease Unexplained Blindness The question often asked, not just by the general public but also by the medical profession, is So why have we not heard of this illness yet? Due to the multi-organ manifestations, its ability to mimic so many other illnesses, the ever-increasing spectrum of recognised phenotypes, the dual genome origins, the varying phenotypes of the same gene even within the same family, an unclear classification system, the relative newness of this illness (of which we have a great deal more to learn), the difficult and non-standardised diagnostic process (clinically, biochemically and genetically), coupled with what can be a very slow, deceptive and insidious onset in the majority (especially in adults), makes mitochondrial disease possibly the ultimate in the world of diagnostic challenges. 10

13 When a cell contains defective mitochondria, it not only becomes deprived of ATP, it also accumulates unused energy molecules and oxygen. The mitochondrial function worsens as these molecules are then used to make ATP by inefficient means, producing potentially harmful by-products such as lactic acid. This lactic acidosis is associated with muscle fatigue and has the potential to damage muscle and nerve tissue. Another harmful by-product, called free radicals or reactive oxygen species may also produce oxidative damage. Therefore, the combined effects of energy deprivation and toxin accumulation in these cells produce the main symptoms of mitochondrial myopathies and encephalomyopathies. 11
 for mitochondrial diseases is complicated by the complexity of the mitochondrion and mitochondrial respiratory chain itself, encoded by both mtdna and ndna (see")
14 GENETIC BACKGROUND Mitochondria are the only cellular organelles known to have their own DNA (mitochondrial DNA or mtdna), distinct from the nuclear DNA (ndna). Genetic testing (mutation analysis) for mitochondrial diseases is complicated by the complexity of the mitochondrion and mitochondrial respiratory chain itself, encoded by both mtdna and ndna (see complexes in diagram). So a dysfunction occurring in complex IV for example, could be due to a mutation in the mtdna or ndna or both. All up it takes about 1,500 genes to make an entire mitochondrion, and mtdna encodes just 37 of those genes; the rest is encoded by the ndna. Less than 10% of these 1,500 genes are actually allocated for making ATP with the remainder involved in the specialised duties of the differentiated cell. Complex I NADH dehydrogenase Complex II Succinate dehydrogenase Complex III Ubiquinol cytochrome C oxidoreductase Complex IV Cytochrome C oxidase Complex V ATP synthase 47 subunits 7 mtdna/40 ndna 4 subunits 0 mtdna/4 ndna 11 subunits 1 mtdna/10 ndna 13 subunits 3 mtdna/10 ndna 17 subunits 2 mtdna/15 ndna The respiratory chain (Diagram: courtesy of 12
15 Defects in ndna can be inherited in a Mendelian pattern from either parent or both, with most showing autosomal recessive inheritance. An estimated 75% of primary paediatric mitochondrial disease results from ndna mutations 32, which tend to present early and are usually fatal 33. Due to a quirk in the process of fertilisation, defects in the genes of the mtdna are usually maternally transmitted. That s because during conception, when the sperm fuses with the egg, the sperm s mitochondria, and its mtdna, are destroyed. Each human cell contains thousands of copies of mtdna which at birth are usually all identical, a state that is called homoplasmy. In contrast, individuals with mitochondrial disorders resulting from mtdna mutations may harbour a mixture of mutant (dysfunctional) and wild-type (normal) mtdna within each cell, and this is called heteroplasmy 8,9. The proportion of mutant mtdna must exceed a critical threshold level, the threshold effect, before a cell expresses a biochemical abnormality of the mitochondrial respiratory chain 10. The percentage level of mutant mtdna may vary among individuals within the same family, and also among organs and tissues within the same individual 11. Therefore, the phenotypic (or symptomatic) expression of the mitochondrial disorder may vary according to the intrinsic pathogenicity of a mutation, its tissue distribution, the variable aerobic energy-demand of different tissues or organs and the individual genetic background 28. Simplistically, a child conceived from a mostly healthy ovum probably won t develop the disease, and a child conceived from a mostly mutant ovum probably will. Please note that in some conditions all the mtdna within a cell is mutant, hence homoplasmy can apply to either wild-type (normal) or mutant mtdna. These mutations usually manifest as single-organ or even single cell-type-failure, like retinal ganglion cells in Leber Hereditary Optic Neuropathy (LHON). 13


16 mother with mild or no symptoms small number of mother s mitochondria, selected randomly, goes into each early egg cell contribution from mother contribution from father possible outcome Bottleneck Effect number of mitochondria increases 80% mutant 50% mutant child with severe disease? child with mild disease? 20% mutant child with no disease? mother s cells may have 20% mutant mitochondria cells that will become egg cells mature egg cells sperm cells (no mitochondria) Heteroplasmy (Diagram: courtesy of The Mitochondria Research Society) Finally, the way that mtdna and ndna mutations interact with each other and with the environment can help determine if disease occurs as well. So, the link between genotype and phenotype in mitochondrial diseases has and will always be recognised as complex 16. When you can t make the diagnosis fit...think of mito! 14
17 MITOCHONDRIAL SYMPTOMS Since mitochondria are present in virtually all cells, symptoms are present in the large majority of organs and as time goes on, we learn also that mitochondrial dysfunction is increasingly being linked to many more pathologies than we could have ever imagined. The first suspicion of a mitochondrial disorder comes from good clinical judgement, not just being aware of the range of its many symptoms but also the quality of those as well. As soon as the word ATYPICAL is used by any specialist to describe an undiagnosed mitochondrial patient, whether it refers to their individual symptoms, their cluster of symptoms, or their previous diagnosis (e.g. atypical MS), or even used in association with a relative s illness (e.g. my aunty died of atypical Parkinsons ), then a mitochondrial disorder needs to be considered as a likely diagnostic candidate. It is the square peg that is too frequently pushed into the round hole. SYMPTOM RANGE Organ / System Brain Nerves Muscles Kidneys Possible Problems Developmental delays, mental retardation/regression, dementia, seizures (especially atypical or refractory), coma, neuro-psychiatric disturbances, atypical cerebral palsy, myoclonus, movement disorders, ataxia, migraines, strokes. Weakness (which may be intermittent), neuropathies, absent reflexes, fainting, absent or excessive sweating resulting in temperature regulation problems. Weakness, hypotonia, cramping, muscle pain, recurrent rhabdomyolysis. Proximal renal tubular wasting resulting in loss of protein, magnesium, phosphorous, calcium and other electrolytes, aminoaciduria, nephrotic syndrome. 15
18 SYMPTOM RANGE (continued) Organ / System Heart Liver Eyes Ears Pancreas Systemic GIT Childhood Skin Endocrine Haematological Possible Problems Conduction defects (e.g., heart blocks, WPW), cardiomyopathy. Hypoglycaemia, unexplained liver failure, Valproateinduced liver failure. Visual loss/blindness, optic atrophy, disorders of extra-ocular muscles, ptosis, retinal degeneration with signs of night blindness, colour-vision deficits, pigmentary retinal changes such as retinitis pigmentosa or salt and pepper retinopathy. Hearing loss and deafness (especially sensorineural). Diabetes and exocrine pancreatic failure (inability to make digestive enzymes). Exercise intolerance not in proportion to weakness, fatigue, short statue, respiratory problems including intermittent air hunger, hypersensitive to general anaesthetics. Gastro-oesophageal reflux, delayed gastric emptying, constipation, pseudo-obstruction, chronic or recurrent vomiting IUGR, unexplained hypotonia, weakness, failure to thrive, or a metabolic acidosis (particularly lactic acidosis), infantile spasms, microcephaly, SIDS. Symmetrical lipomatosis. Diabetes, short stature, hypothyroidism, hypoparathyroidism. Sideroblastic anaemia 16
19 SYMPTOM QUALITY RED FLAGS Clinical Manifestation Sensorineural hearing loss Focal neurological deficits Seizures Ptosis Retinal pigmentary changes Diabetes Features suspicious of a mitochondrial disease Asymmetrical onset Young age of onset History of partial recovery after an insult, i.e., reversible High frequencies affected first Young age of onset Preceded by clinical prodrome Nonvascular territory on neuroimaging Predominantly grey matter affected Associated basal ganglia calcification Good clinical recovery from an event Neuroradiological changes out of proportion to clinical deficit Associated focal seizures or status epilepticus Raised CSF lactate Sudden onset status epilepticus Recurrent physiological trigger Severe episodes of seizures with good interval periods (requiring no ACDs for control) Worsened by sodium valproate Asymmetrical onset Slowly progressive with little diurnal variation Accompanying PEO or retinal pigmentary changes Perimacular distribution No drusen Non-vision threatening No associated diabetic retinopathy/peripheral neuropathy with respect to the length of diabetes onset Easily controlled with OHA with respect to duration of diabetes 17

20 THE DIAGNOSTIC PROCESS As previously mentioned, mitochondrial diseases have varying presentations and the onset may present at any time, from before birth, as IUGR, to late adult life. Even within the same family, the same genetic mutation may affect individuals differently as there is no single identifying feature of mitochondrial disease. So, to begin the diagnostic process, the foremost and utmost diagnostic tool is good clinical judgement. The GP is first required to be aware of the existence and symptomatology of mitochondrial diseases, and to remember it in their differential diagnoses under metabolic. We should begin to consider the possibility of mitochondrial disease when: 1. A common disease has atypical features that set it apart from the pack. 2. Three or more organ systems are involved (or 1-2 of Red Flag symptoms above). 3. Recurrent setbacks/flare ups in a chronic disease occur with infections. 4 When the puzzle is solved, we can now meet the challenge together! 18
21 STEP ONE The diagnostic work-up for suspected mitochondrial disease is a stepwise procedure, which can be likened to constructing a jigsaw puzzle. As the pieces are collected and placed correctly, the picture begins to form. Much of the work however, is done by an initial comprehensive history of the individual s symptoms, the family history, and a full systems review. Clinical investigations are commenced by the GP to complete the systems review and confirm any symptomatology. For example, don t just ask about deafness, but organise a formal audiological assessment as well. STEP TWO In the second step, clinicians need to decide: 1. If the individual s presentation conforms to any of the more common and better understood mitochondrial syndromes such as MELAS or MERRF. These patients may then proceed directly to genetic testing as organised by a mitochondrial specialist, thereby potentially avoiding the long list of other investigations. 2. If the presentation does not conform to a particular syndrome, as is unfortunately the case for the majority, then the next step is direct referral to a mitochondrial specialist as the subsequent investigations include an array of biochemical tests of either serum, CSF and/or urine, additional electrophysiological studies, functional and imaging studies of the brain and other organs such as muscle, and an appropriate tissue biopsy of the muscle, skin, bowel or liver. STEP THREE Based on the patient s presentation and the results thus far, the probability of a mitochondrial disorder should now be approaching a more definitive answer. Although genetic testing is by definition the gold standard to diagnose a genetic disease, the majority of mitochondrial patients currently don t have testing, for reasons explained by the complicated and vastly unknown genetics of 19
22 the illness. So, although the third and final step is genetic testing, diagnosis greatly depends on steps 1 and 2 currently. However, this situation is expected to change quite quickly as exome sequencing or genome sequencing become more widely available in diagnostic rather than just research context within the next 1-2 years. Unfortunately, many patients will have a presentation suspicious of a mitochondrial disorder, but have little evidence to reinforce a diagnosis. In these patients, regular review and assessment for further clinical deterioration and new organ involvement is essential over time, as even the most classical mitochondrial disorders may not present in their full clinical spectrum at the first medical visit. To both aid and unify the diagnosis of mitochondrial disease, there have been a number of attempts to create an internationally accepted standard for its diagnostic criteria, such as those according to the Nijmegen, Bernier or Walker criteria 29,30. Probably the closest so far, which still requires further verification, is the Nijmegen Clinical Criteria for Mitochondrial Disease (see appendix). It uses a point scoring system in the areas of clinical, biochemical and genetic criteria to classify the chance of mitochondrial disease as definite, highly probable, probable, possible or unlikely. So, even though the only way to make a diagnosis of a primary mitochondrial disease with absolute certainty is to identify a mtdna or ndna abnormality that is known to cause disease, it is usually not possible. The best one can do at this point in time is to begin to build a case. Then referral to a mitochondrial specialist would be the next appropriate step to continue to build that case. The gathering of evidence is done by these diagnostic approaches: 20
23 DIAGNOSTIC TESTS IN MITOCHONDRIAL DISEASES Type Test What It Shows Personal & family history Thorough history of patient and family members Can sometimes indicate inheritance pattern by noting soft signs in unaffected relatives. These include deafness, short stature, migraines and PEO (progressive external ophthalmoplegia). Systems review and physical signs Multi-system/organ assessment (dependent upon the individual presentation) Clinical examination, especially neurological Neuroradiological changes out of proportion to clinical deficit Associated focal seizures or status epilepticus Raised CSF lactate 1. Electrophysiology studies, especially EEG, (also NCS, EMG, etc) 2. ECG/Holter/Echo 3. Hearing tests 4. Biochemistry/FBC 5. GIT/transit studies 6. Ophthalmological examination 7. Allied health team assessments (occupational therapist, speech pathologist, physiotherapist) Tests of strength and endurance, and neurological tests including reflexes, vision, speech, basic cognitive skills, and developmental assessment. 1. Monitoring or detection of any seizure activity 2. Detection of cardiac abnormalities 3. Sensorineural hearing loss 4. Renal tubular dysfunction, liver dysfunction, glucose (including HBA1C), TSH, B12, blood film, etc. 5. Gut dysmotility, reflux/cyclical vomiting 6. Ptosis, eye muscles, retinal changes of degeneration, pigmentation, and optic atrophy 7. Initial assessments can be used to look for any abnormalities or used as baselines for further reviews 21
 1. Muscle phosphorus magnetic resonance spectroscopy (MRS) 2. MRI (CT scan if MRI not freely available) 1. Lactate and pyruvate levels 2. Serum creatine kinase 3.")
24 DIAGNOSTIC TESTS IN MITOCHONDRIAL DISEASES Type Test What It Shows Imaging studies (these and the following tests are best done by a mitochondrial specialist) Blood, enzyme and biochemical tests (may include urine and CSF also) 1. Muscle phosphorus magnetic resonance spectroscopy (MRS) 2. MRI (CT scan if MRI not freely available) 1. Lactate and pyruvate levels 2. Serum creatine kinase 3. Serum carnitine levels (including total, acyl, free, and acyl/free ratio), serum ketones 4.Whole blood ammonia 5. Quantitative plasma (fasting) and/or urine amino acids. Urinary screening of amino or organic acids. 1. Measures the levels of lactate, phosphocreatine and ATP (often depleted in muscles affected by mitochondrial disease). 2. Looking for: a) bilateral or symmetric lesions, especially in the basal ganglia or thalamus, brain stem, white matter, or cerebellum b) cerebral and/or cerebellar atrophy c) cortical lesions particularly in non-vascular territories d) diffuse leukoencephalopathy. 1. If elevated, may indicate deficiency in respiratory chain; abnormal ratios of the two may help identify the part of the chain that is blocked. 2. May be slightly elevated in mitochondrial disease but usually only high in cases of mitochondrial DNA depletion Further metabolic evaluations of mitochondrial function, e.g., defects in fatty acid metabolism/ oxidation may demonstrate elevated plasma levels of free fatty acids, hypoketonaemia, hypocarnitinaemia, dicarboxylic aciduria, and the presence of Krebs cycle intermediates. 22

25 DIAGNOSTIC TESTS IN MITOCHONDRIAL DISEASES Type Test What It Shows Tissue biopsy (frequently muscle but not gold standard) Genetic tests (with counselling) NB: Negative test results have a high falsenegative rate Histochemistry 2. Immunohistochemistry 3. Biochemistry 4. Electron microscopy 1. Known mutations 2. Rare or unknown mutations 3. Mitochondrial DNA depletion 1. Detects abnormal proliferation of mitochondria and deficiencies in cytochrome c oxidase (COX, which is complex IV in the respiratory chain). When treated with the modified Gomori trichrome stain, mitochondria become red, so muscle cells with excessive mitochondria appear as ragged red fibres. 2. Detects presence or absence of specific proteins. Can rule out other diseases or confirm loss of respiratory chain proteins. 3. Measures activities of specific enzymes such as complexes I to IV of the respiratory chain. 4. May confirm abnormal size, shape, number and structure of mitochondria. 1. Uses muscle, urine, hair follicle or other tissue sample to screen for known mutations, looking for common mutations first. 2. Can also look for rare or unknown mutations but may require samples from family members; this is more expensive, time-consuming and not freely available in Australia. 3. Uses muscle or liver samples to test whether the amount of mtdna per cell is adequate. mtdna depletion is one of the most common causes of childhood mitochondrial disease. (Original resource: courtesy of the Muscular Dystrophy Association) 23

26 Findings in any one of these categories are not sufficient to make a diagnosis as abnormalities can occur in other illnesses, or may be a secondary phenomenon reflecting mitochondrial dysfunction in another non-mitochondrial disease. Furthermore, some patients with proven disease may not show any biochemical, histological or imaging abnormalities 18. For example, normal EMG findings can still be helpful since patients with most other forms of clinical myopathy (such as inflammatory myopathies) usually have diagnostic abnormalities on EMG testing 31. SYMPTOMS OF MITOCHONDRIAL DISEASE? NERVOUS SYSTEM: Seizures, movement disorders, developmental delays, deafness, dementia, stroke (often at an early age), visual defects, poor balance/coordination, problems with peripheral nerves, migraines/headaches. SKELETAL MUSCLE: Muscle weakness, exercise intolerance, pain, fatigue, cramps, low muscle tone. LIVER: Unexplained liver failure. KIDNEYS: Unexplained renal failure, Fanconi s syndrome (loss of essential metabolites in urine), nephrotic syndrome. EARS: Sensorineural deafness (may be intermittent). EYES: Drooping eyelids (ptosis), inability to move eyes (external ophthalmoplegia), blindness (retinitis pigmentosa, and optic atrophy). HEART: Cardiomyopathy (large heart), conduction abnormalities. DIGESTIVE TRACT: Difficulty swallowing, reflux, vomiting, feeling of being full, chronic constipation, diarrhoea, unexplained intestinal obstruction, irritable bowel type symptoms. ENDOCRINE: Diabetes hypothyroidism, and excess body hair. 24
27 COMMON MITOCHONDRIAL DISEASE PRESENTATIONS The terminology used to describe mitochondrial disease can be confusing and it is still a struggle for many patients to be fitted into the groups we know. A single syndrome, with a combination of symptoms, may have many different genotypes, while more than one syndrome may have the same genotype. A diagnosis may be named for the cause, such as COX deficiency, or it may be based on the symptoms of the disease such as the following examples: MELAS: Mitochondrial Encephalopathy, Lactic Acidosis and Stroke-like episodes. This is the most common type of mitochondrial encephalopathy. Onset: Usually between 2 and 40 years of age, mean age is 10 years, but can be any age. Disease characteristics: Hallmark sign is the MELAS attack. General characteristics are exercise intolerance, seizures, dementia, muscle weakness, hearing loss, blindness, migraine-type headaches, myopathy, gastric dysmotility, polyneuropathy, ptosis, cardiomyopathy, diabetes, renal failure and short stature. Inheritance: Maternal MERRF: Myoclonic Epilepsy with Ragged-Red Fibres Onset: Usually late adolescence to adulthood; variable progression. Disease characteristics: Myoclonic epilepsy, proximal myopathy, sensorineural deafness, ataxia, pigmentary retinopathy, coordination loss, dementia, distal sensory loss and optic atrophy. Inheritance: Sporadic or maternal 25
28 KSS: Kearns-Sayre Syndrome Onset: Before age 20 Disease characteristics: Progressive External Ophthalmoplegia (PEO), ptosis, pigmentary degeneration of retina, heart block, myopathy, dysphagia most commonly associated with cricopharyngeal achalasia, hearing loss, ataxia, and dementia. Inheritance: Sporadic Leigh syndrome: Subacute necrotizing encephalomyopathy Onset: Infancy and progression can be fast or slow. Death often occurs within two years of onset. Disease characteristics: Vomiting, ataxia, hypotonia or spasticity, seizures, feeding and speech difficulties, hearing loss, nystagmus, visual loss, choreoathetosis, peripheral neuropathy, hyperventilation, motor and intellectual regression. Inheritance: Mendelian or maternal MNGIE: Mitochondrial Neuro-GastroIntestinal Encephalopathy Onset: Often before age 20, range five months to 55 years Disease characteristics: External ophthalmoplegia, ptosis, digestive tract disorders due to visceral neuropathy with weight loss, retinal degeneration, neuropathy, short stature, myopathy, loss of coordination, leukoencephalopathy and hearing loss. Inheritance: Mendelian NARP: Neuropathy, Ataxia and Retinitis Pigmentosa Onset: Infancy or childhood Disease characteristics: Retinitis Pigmentosa causing visual loss, lack of coordination, ataxia, weakness, dementia, seizures and developmental delay. This syndrome may represent a less severe form of MILS (Maternally Inherited Leigh Syndrome). Inheritance: Maternal 26

29 PEO: Progressive External Ophthalmoplegia Onset: Usually in adolescence or early adulthood; slow progression. Disease characteristics: Gaze limited in all directions, slow eye movements, bilateral, associated with ptosis, slowly progressive, and usually associated with muscle weakness and fatigue. Inheritance: Maternal, Sporadic, and Mendelian. Often occurs in conjunction with other mitochondrial syndromes. LHON: Leber Hereditary Optic Neuropathy Onset: Male predominance, usually by 30 years of age, range one to 70 years Disease characteristics: Visual loss, pre-excitation cardiac conduction syndromes, spasticity, dystonia, multiple sclerosis-like disorder and encephalopathy. Inheritance: Maternal 27
30 DIFFERENTIAL DIAGNOSIS Paediatric Organic acidaemias: MSUD, propionic, isovaleric, methylmalonic, others Urea cycle defects: carbamyl phosphate synthetase deficiency, OTC, citrullinaemia, argininosuccinic aciduria Carbohydrate disorders: congenital disorders of glycosylation, galactosaemia, hereditary fructose intolerance Aminoacidopathies: homocystinuria, tyrosinemia, nonketotic hyperglycinemia Endocrinopathies: CAR, congenital diabetes OXPHOS disorders Prader-Willi, Angelman syndrome, Rett syndrome Adult Primary endocrine disease Vitamin deficiency: B12 Homocystinuria and associated disorders Primary muscle disease: polymyositis, dystrophin associated glycoprotein muscular dystrophies Chronic fatigue syndrome Autoimmune disorders Glycogen storage disorders Depression and related psychosomatic disorders Other neurodegenerative disorders (MS, Parkinson s, combined systems degeneration) 28
31 MANAGEMENT Due to the complexity and chronicity of mitochondrial disease, the role of the GP is as team leader, coordinator and supervisor. Also, since our medical understanding and general awareness of mitochondrial disease is still in its infancy, the GP must be willing to accommodate these needs for the benefit of the patient: 1. That the physician is interested in learning about a complicated new disease in order to play a more effective role in the patient s care 2. That the physician feels comfortable asking questions and when necessary calling whoever is overseeing the patient s mitochondrial disease management 3. That the physician feels comfortable acting as an advocate for the patient as most medical providers and other services are not familiar with the disease and as a result patients meet many challenges to their care 18. As there is currently no cure, the initial approach and attitude by the medical profession that developed toward mitochondrial disorders in the 1990s and early 2000s, was basically one of despair and helplessness. So management of a patient was limited to diagnosis only with very little attention to other aspects. Unfortunately, this attitude still forms the basis of most management strategies by the medical profession today! Also, too often a formal diagnosis is avoided or delayed due to the belief it won t alter the prognosis. However, before the physician decides to take this pathway, strong consideration needs to be given to the benefits of a diagnosis to the patient, i.e., the dignity of having a diagnosis, access to services/resources, better quality of life by the improved management, understanding and monitoring of the illness, and prevention of unnecessary events. A formal genetic diagnosis if achievable, helps the family in regards to genetic counselling and 29
32 family planning as well. Overall, the data accumulated during the process of a diagnosis also greatly helps research into mitochondrial disease and will therefore increase our knowledge and understanding of the illness for the future. Today, in the larger mitochondrial clinics, the approach is more encouraging with the focus shifted to not just diagnosis, but also full assessment of their disorder, symptomatic treatment, optimisation of function, and prophylaxis through healthy living. Even though these disorders are chronic and incurable, correct and aggressive early management of many issues will optimise the patient s quality of life. For example, at times of stress, early detection or even prevention of a metabolic crisis may limit morbidity and the onset of further handicap. GOALS OF TREATMENT Brain reduces seizures improve attention and concentration improve intellectual functioning prevent headaches prevent strokes improve motor control Muscle improve strength lessen pain lessen fatigue reverse cardiomyopathy Liver improve function, avoid toxins Nerve improve autonomic function lessen pain improve nerve conduction Ears prevent further hearing loss GI improve gastric and intestinal motility Systemic encourage growth and so prevent failure to thrive Eyes prevent further retinitis or optic atrophy The management of the mitochondrial disease itself is largely supportive 14, as there is no way of simply increasing the capacity of the cell to generate energy 21. Treatment therefore involves optimising energy production, reducing energy losses, meeting lifestyle needs such as education, and monitoring for complications 21. Fundamentally, it s about keeping life s energy equation of ENERGY IN = ENERGY OUT balanced. 30
33 OPTIMISING ENERGY PRODUCTION 1. Adequate nutrition Adequate calories and nutrition can dramatically improve a patient s overall clinical state and slow the progression of the illness 21. Eating smaller meals more regularly and avoidance of fasting, particularly prolonged fasting is also extremely important in optimising mitochondrial function. Special diets may benefit some patients, such as high fat diets with restriction of simple carbohydrates, fructose restriction, and/or high complex carbohydrate intake 4. A ketogenic diet, for example, may be used for intractable seizures and this is not contraindicated in mitochondrial disease, particularly with complex I deficiency. However, ketogenic or other high fat diets are not recommended for long-term consumption due to the potential for cardiovascular risks, such as ischaemic heart disease and other atherosclerotic issues Adequate sleep Improving sleep is a correctible component of fatigue. A sleep study may be advisable to identify treatable conditions such as apnoea, hypoxemia, restless legs syndrome, nocturnal myoclonus or seizures 34. Central sleep apnoea can occur in more advanced disease, whereas obstructive sleep apnoea (due to muscle hypotonia or weakness) is more common Promoting activity Regular exercise not only improves stamina it also improves mitochondrial function 25,26. To maximise energy production, mitochondrial patients must remain active, though exhaustion should be avoided and achieving a normal level of endurance is unrealistic

34 4. Supplementation with certain vitamins and cofactors It is considered standard care to trial a cocktail of vitamin and cofactor supplementation for patients with mitochondrial disorders, although there are a large variety of studies with even a larger variety of answers regarding their efficacy. At present, there are no cures for these disorders except in very rare and specific disorders such as primary carnitine deficiency or primary coenzyme Q10 deficiency 4. The goals of supplementation are to improve symptoms and to halt the progression of the illness. The effectiveness of treatment varies with each patient but it will not reverse any damage that has already occurred 23.There is no standard vitamin cocktail, commonly known as the mito cocktail. Ideally, vitamins should be started one at a time to allow observation of any benefit or adverse reaction. Frequently, the patient may not notice much benefit when first commencing various components of the mito cocktail ; however, the difference can often be observed when the vitamin or co-factor is ceased
35 The following table lists substances often suggested as clinically effective in the literature or for which the literature contains anecdotal reports of potential positive treatment effects for mitochondriopathies (17,22,23). However, please note that a Cochrane Review concluded there was no proven efficacy for any of these substances 27. Vitamin/Cofactor Purpose/Effect Clinical Application Quinones (Co-enzyme Q10=Ubiquinone, Synthetic ubiquinone= Idebenone) Vitamin E Thiamine (vitamin B1) Riboflavin (vitamin B2) Nicotinamide (vitamin B3) Antioxidant, efficient electron transport in the respiratory chain depends upon high levels of CoQ10; stabilises the respiratory chain complexes; CoQ10 appears to improve stamina and reduce fatigue. Antioxidants (help to clear free radicals, believed to accumulate in the respiratory chain and contribute to the pathogenesis of mitochondrial disease). Cofactor for decarboxylases, possibly boosts enzyme function, antioxidant, slow disease progression. Cofactor for respiratory chain, possibly boosts enzyme function, antioxidant, slow disease progression May boost respiratory chain activity Co-enzyme Q10 defect. All mitochondrial diseases. Friedreich Ataxia +/- cardiomyopathy (especially using Idebenone) 24, LHON Cardiomyopathy in Friedreich Ataxia. Possibly all mitochondriopathies. Unknown efficacy PDHC E1 defect Possibly all respiratory chain defects (antioxidant) Mitochondrial myopathy All respiratory chain defects (antioxidant) Complex I and II deficiency Anti-migraine agent in some mitochondrial disorders 23 Unknown efficacy 33
36 Vitamin/Cofactor Purpose/Effect Clinical Application Creatine Ascorbic acid (vitamin C) Beneficial to mitochondrial function; supplementation can help with stamina and improve muscle pain, weak antioxidant. Antioxidant (helps to clear free radicals believed to accumulate in the respiratory chain and contribute to the pathogenesis of mitochondrial disease) Mitochondriopathies regardless of the biochemical defect All respiratory chain defects (antioxidant) Vitamin K3 Antioxidant Complex III deficiency Dichloroacetate Alpha lipoic acid Succinate Biotin (vitamin B7) Reduces serum lactate levels through activation of pyruvate dehydrogenase complex, and has been shown to decrease cerebral lactic acidosis in patients. This agent was potentially very toxic and trials were stopped. Antioxidant, co-enzyme for pyruvate dehydrogenase and alpha ketoglutarate dehydrogenase Boosts respiratory chain and citric acid cycle Cofactor for carboxylases, possibly boosts enzyme function, antioxidant, slow disease progression. Severe / chronic lactic acidosis, particularly for pyruvate dehydrogenase (PDHC) deficiency PDHC E3 defect Leigh Syndrome Possibly all respiratory chain defects (antioxidant) Complex 1 deficiency Possibly all respiratory chain defects (antioxidant) Unknown efficacy 34

37 Vitamin/Cofactor Purpose/Effect Clinical Application L-carnitine (levo-carnitine) Magnesium Orotate L-Arginine Improves stamina and reduces muscle weakness and cramping, reduces headaches, transports longchain fatty acids, binds unused metabolic products In theory it improves CoQ10 function by being converted to an electron donor. Also a potent antioxidant and helps the body synthesize more DNA precursors (purines). Nitric oxide is formed from arginine via the enzyme nitric oxide synthase, which catalyses the conversion of arginine to citrulline, therefore it acts as a nitric oxide precursor 39. Mitochondriopathies regardless of the biochemical defect, with/without secondary carnitine deficiency Animal studies have shown varying benefits in decreasing cardiac injury in times of stress and increasing exercise tolerance in animals with an injured heart muscle The administration of L-arginine during the acute and interictal periods may represent a potential new therapy for MELAS. It reduces the brain damage due to impaired vasodilation in intracerebral arteries owing to nitric oxide depletion 37,38. 35

38 REDUCING ENERGY LOSSES 1. Prevention of infections Patients with mitochondrial disease frequently do not tolerate infections well and they may cause prolonged, debilitating fatigue and weakness, and possibly even death. As a result, their vaccinations should be kept up-to-date including the seasonal vaccinations (e.g. influenza), and antibiotics should be considered early in any infective illness. 2. Avoiding excessive physical activity Overdoing it produces no benefit and can leave a patient exhausted, in pain, nauseous, and miserable. 3. Treating emotional distress Frequent or persistent anxiety, depression, or obsessive-compulsive behaviours are very energy-demanding. 4. Maintaining a suitable ambient temperature. Patients with mitochondrial disease often don t tolerate extremes of temperature. 36
39 AVOIDANCE OF TOXINS Alcohol and smoking have been known to hasten the progression of some conditions so should be avoided. MSG (monosodium glutamate) can trigger migraine headaches in healthy people, so has the potential is to do the same in those susceptible with mitochondrial disease 17. Iron can increase free radical production so although excessive amounts should be avoided, a normal diet containing iron is encouraged 17,23. Antiretroviral medications are toxic to mitochondria, so AZT and other nucleoside drugs such as FIAU (Fialuridine) should be avoided. Similarly, doxorubicin, a chemotherapy medication can cause a cardiomyopathy through mitochondrial damage so it should also be avoided. Sodium valproate, statins, erythromycin, aspirin and other NSAIDS, amphotericin and propofol are best avoided but are not absolute contraindications. Other amino-glycoside antibiotics such as gentamicin, streptomycin, and tobramycin can induce hearing loss by damaging mitochondria so alternatives should be sought. In the anaesthetic situation, IV solutions that contain lactic acid (e.g. Ringers Lactate solution) should be avoided whilst some patients are more sensitive to volatile anaesthetics so lower doses are required. MAJOR EXAMPLES OF MEDICATIONS TO AVOID IN PATIENTS WITH MITOCHONDRIAL DISORDERS 34. Statins HIV Medications Metformin Alcohol Smoking and Nicotine Gum/ Patches Aspirin May deplete CoQ10 Inhibits polymerase gamma (mtdna depletion) Lactic acidosis due to mitochondrial impairment Increases oxidative stress, mitochondrial toxin Inhibits complex IV, damages mitochondria Inhibits mitochondrial function, causes Reye disease in children 37
40 ALLEVIATING SYMPTOMS Close monitoring, a good understanding and therefore early detection of symptoms, allows for optimal management for the patient. These may be in relation to the further deterioration of a currently affected organ or system, the onset of a dysfunction in a new one, or the early awareness of a prodromal episode that could be alleviated with swift action, such as that in a MELAS episode. Some common and current examples of mitochondrial symptom management are given here: Cardiac abnormalities medications, pacemakers, ablation therapy Hearing loss hearing aids, cochlear implants, prophylaxis Eye changes silicone slings, surgery on muscles, monitor intraocular pressures and treat early Acute seizures IV clonazepam, +/-thiopentone induced coma (severe cases) Seizure prophylaxis Good results with levopiracetam and clonazepam (myoclonus), avoid sodium valproate and possibly phenobarbitone. Consider L-Arginine for MELAS Delayed gastric emptying use Motilium, and /or Cisapride Constipation osmotic laxatives such as Movicol, Osmolax, Duphalac, Sorbilax, Epsom salts, and remember surgery is best avoided, especially in cases of pseudo-obstruction EDUCATIONAL NEEDS The majority of children with mitochondrial disease show learning and/ or behavioural problems that are typically unique to each child, so an educational plan should be tailored to each child, not based on his/ her diagnosis. However, a medical plan is frequently needed as well to create an optimal learning environment. Points to consider include: Pacing the child to match their fatigability Never forget they have good and bad days 38
41 A comfortable classroom temperature Avoidance of infective illnesses Avoidance of unnecessary emotional distress 21 MONITORING COMPLICATIONS The caring physician must have a thorough knowledge of the potential complications of mitochondrial disorders in order to prevent unnecessary morbidity and mortality. This includes the early diagnosis and/or management of diabetes mellitus, seizures, cardiac pacing, ptosis correction, and intraocular lens replacement for cataracts. Unless a patient has a very specific disease with a predictable phenotype, routine monitoring of the following organs is generally on a 1-2 year schedule: Blood and urine testing Bone marrow involvement FBC, WBC differential, platelets Liver involvement AST, ALT, bilirubin Kidney involvement Blood urea nitrogen (BUN), creatinine (blood), urinalysis, urine amino acids (quantitative) Muscle involvement CK Endocrine involvement thyroid functions, calcium, phosphorus. Adrenal insufficiency is also a possibility. Metabolic status lactate and pyruvate, carnitine and acylcarnitines, leukocyte coenzyme Q10, urine organic acid analysis. Other testing Ophthalmological evaluation and especially screening for visual function if concerned Audiology testing Cardiac evaluation, including ECG, echocardiogram Developmental or neuropsychological testing according to the patient s needs 21 39
42 PALLIATION Symptom Control including seizures (esp. uncontrolled), autonomic instability, gut dysmotility, constipation, pain/cramps, anxiety/dementia, aspiration, sepsis, weakness, end organ failures (e.g. cardiac, renal), blindness, deafness Physical Support home modification, beds, suction, oxygen, feeding supplies, lifts, bath, home nursing and/or respite care Psychosocial Support treatment, advanced care planning, discussing anticipated course, patient/family values, end-of-life preferences, realistic goals for life, maintenance of functionality reframe/rethink Emotional Support caregivers, siblings of affected children, parents, grandparents and spouses Spiritual Support spiritual care provider Family Considerations genetic counselling, coordination of care and case management, finances (power of attorney) End-of-Life Care early referral to palliative care, helping family with memory making (e.g. scrapbooking) 40
43 PROGNOSIS AND FURTHER RESEARCH At present there is no known cure and it is not possible to predict the future of a person with mitochondrial disease as the expression of the illness in each individual is extremely variable and difficult to assess. The disease might progress quickly or slowly over decades, or it might appear stable for years. Research so far has helped some of the affected children and adults to live fairly normal lives but at the opposite end of the spectrum, many are severely affected, and many children do not survive to their teenage years. Currently, the direction of research in terms of a cure, is more along the lines of preventing a known mitochondrial genetic defect from continuing through a family line. This is available now through prenatal diagnosis and pre-implantation genetic diagnosis for families in whom a molecular diagnosis has been achieved. Although research into mitochondrial genetic manipulation is well on the way around the world, particularly in the UK, there are many ethical and medical obstacles to overcome before it can truly be considered a viable form of mitochondrial disease prevention. The proposed procedure in the UK (see diagram over) requires more research to gather further information on the efficiency and safety before these forms of techniques are considered for introduction into clinical practice. 41
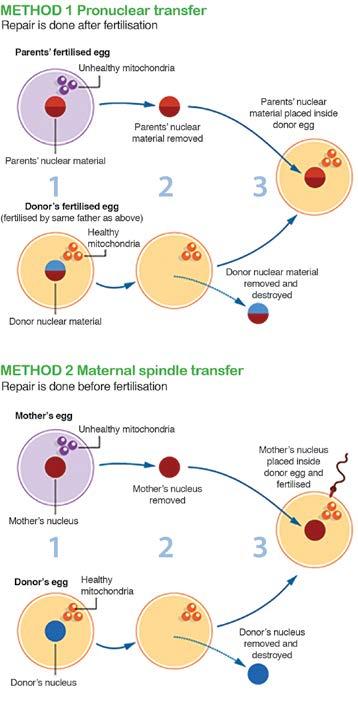
44 (Source: 42
45 LINKS TO OTHER DISEASES Even when mitochondrial dysfunction is confirmed by sophisticated biochemical testing, it can be difficult to know whether the cause is primarily genetic (and directly impacting the mitochondrial respiratory chain), or if it is secondary to another unrelated genetic or environmental cause. For instance, mitochondrial dysfunction may be seen when the primary defect occurs in another energyrelated metabolic pathway, such as fatty acid oxidation or amino acid metabolism. Mitochondrial dysfunction has also been shown to occur in vitro in disorders such as copper-metabolism disorders (Wilson disease and Menkes disease), some lysosomal disorders, and neonatal haemochromatosis, to name a few. In addition, decreased activities of respiratory chain complexes in skeletal muscle may be seen in malnourished children, with correction to normal levels after improved nutrition 13. Researchers are also studying mitochondrial diseases looking for clues to other conditions such as cancer, diabetes, Parkinson s disease, Alzheimer s, and heart disease. Damage to the mitochondria is thought to be involved with all of these conditions, and a lifetime of mitochondrial damage may also be part of the aging process. 43
46 APPENDIX Nijmegen Clinical Criteria for Mitochondrial Disease: Clinical scoring for diagnosis 36 CLINICAL CRITERIA SCORE Neuromuscular manifestations (Maximum of 2 points) Central nervous system and other organ involvement (Maximum of 2 points) Metabolic and imaging studies (Maximum of 4 points) Tissue morphology (Maximum of 4 points) a. Progressive external ophthalmoplegia (2 points) b. Ptosis (1 point) c. Exercise intolerance (1 point) d. Muscle weakness (1 point) e. Rhabdomyolysis (1 point) f. Abnormal electromyogram (1 point) g. Isolated central nervous system involvement (1 point) h. Any other isolated organ system (1 point) i. Two or more organ systems (2 points) j. Elevated blood lactate on three occasions (2 points) k. Elevated cerebrospinal fluid lactate (2 points) l. Elevated blood alanine (2 points) m. Elevated cerebrospinal fluid alanine (2 points) n. Elevated urine tricarboxylic acid (Kreb) cycle intermediates (2 points) o. Elevated urine ethylmalonic, 3-methylglutcaonic, ordicarboxylic acids (1 point) p. Abnormal 31P-MRS (magnetic resonance spectroscopy) inmuscle with reduced Phosphocreatine/Pi ratio (2 points) q. Abnormal T2 signal in basal ganglia on brain MRI (2 points) r. Decreased resting metabolic rate or abnormal exercise studies (cycle ergometry protocol) (2 points) s. Ragged red fibers on muscle biopsy (2 points if present, 4 points if >2%) t. Diffuse reduction in cytochrome c oxidase histochemical reaction or scattered COX deficient fibres (4 points) u. Strongly succinate dehydrogenase positive vessels by histochemistry (1 point) 44 Scoring for evaluation of Clinical Criteria Mitochondrial Diagnostic Criteria Definite: 8-12 points Probable: 5-7 points Possible: 2-4 points Unlikely: 1 point
47 The Nijmegen Biochemical Criteria are modified for incorporation of contemporary technologies. BIOCHEMICAL CRITERIA SCORE 1. Abnormal high resolution respirometry in muscle or fibroblasts (measurements <5% reference level). (Live or fresh tissue assessment of Complex V function, coupling and protein leak across the mitochondrial membrane) 2. Abnormal OXPHOS subunit immunohistochemistry or immunofluorescence in skeletal muscle tissue sections (Qualitative assessment of OXPHOS enzyme assembly within tissue sections. Defects in OXPHOS enzyme assembly are readily recognized by this testing.) 3. Abnormal OXPHOS enzymology (single or multiple enzyme defects) (activity measurements <5% reference level) 4. Abnormal quantitative Western Blot of Selected OXPHOS subunits from Complexes I-V (levels <5% reference level for subunit) (Western blot can detect defects that are not evident by other techniques). 5. Abnormal muscle CoQ10 level (<50% of control mean) (Allows assessment for primary defects in Coenzyme Q10 synthesis). 6. Supercomplex evaluation: Optimal OXPHOS function requires aggregation of individual OXPHOS enzymes into supercomplexes which allows efficient and rapid transport of electrons. Supercomplexes allow efficient formation of an electrochemical (proton) gradient created by Complexes I, III, and IV that is then used by Complex V to synthesize ATP. Supercomplex formation is impaired in a variety of OXPHOS diseases. a. Abnormal supercomplex formation (Score 0.5 if present only in Blue Native OR OXPHOS Clear Native Immunoblot. Score 1 if present in BOTH tests.) b. Abnormal monomeric OXPHOS enzyme complex formation (Score 0.5 if present only in Blue Native OR OXPHOS Clear Native Immunoblot. Score 1 if present in BOTH tests.) 7. Abnormal in-gel OXPHOS enzyme activity. (Qualitative in-gel assessment of OXPHOS enzyme activity in intact enzymes. This is particularly important in assessment of the ATPase activity of Complex V and Complex V assembly). Scoring for evaluation of Biochemical Criteria Mitochondrial Diagnostic Criteria Unlikely: Criteria Score = 0 Possible: Criteria Score is 1 and <2 Probable: Criteria Score = 2 Highly Probable: Criteria Score > 2 45
 4.")
48 The Nijmegen Genetic Criteria GENETIC CRITERIA SCORE 1. mtdna depletion (mtdna copy number <5% reference interval) 2. Identification of confirmed pathogenic mtdna or nuclear DNA mutation 3. Identification of provisional pathogenic mtdna or nuclear DNA mutation (i.e., mutation requires additional data supporting pathogenicity) 4. No mutation identified Scoring for evaluation of Genetic Criteria Definite: Criteria 1 or 2 are abnormal Probable: Criteria for 3 is abnormal Undetermined: Criteria 4 is present Note: Criteria 4 The failure to find a mutation does not exclude mitochondrial disease due to the large number of genes associated with mitochondrial disease and the large number of undiscovered genetic associations. 46
Nutritional Interventions in Primary Mitochondrial Disorders
 Nutritional Interventions in Primary Mitochondrial Disorders Carolyn J Ellaway MBBS PhD FRACP CGHGSA Genetic Metabolic Disorders Service Sydney Children s Hospital Network Disciplines of Child and Adolescent
Nutritional Interventions in Primary Mitochondrial Disorders Carolyn J Ellaway MBBS PhD FRACP CGHGSA Genetic Metabolic Disorders Service Sydney Children s Hospital Network Disciplines of Child and Adolescent
MITOCHONDRIAL DISEASE. Amel Karaa, MD Mitochondrial Disease Program Massachusetts General Hospital
 MITOCHONDRIAL DISEASE Amel Karaa, MD Mitochondrial Disease Program Massachusetts General Hospital Disclosures & Disclaimers United Mitochondrial Disease Foundation Research Grant North American Mitochondrial
MITOCHONDRIAL DISEASE Amel Karaa, MD Mitochondrial Disease Program Massachusetts General Hospital Disclosures & Disclaimers United Mitochondrial Disease Foundation Research Grant North American Mitochondrial
REQUISITION FORM NOTE: ALL FORMS MUST BE FILLED OUT COMPLETELY FOR SAMPLE TO BE PROCESSED. Last First Last First
 #: DEPARTMENT OF NEUROLOGY COLUMBIA COLLEGE OF PHYSICIANS & SURGEONS Room 4-420 630 West 168th Street, New York, NY 10032 Telephone #: 212-305-3947 Fax#: 212-305-3986 REQUISITION FORM NOTE: ALL FORMS MUST
#: DEPARTMENT OF NEUROLOGY COLUMBIA COLLEGE OF PHYSICIANS & SURGEONS Room 4-420 630 West 168th Street, New York, NY 10032 Telephone #: 212-305-3947 Fax#: 212-305-3986 REQUISITION FORM NOTE: ALL FORMS MUST
Presentation and investigation of mitochondrial disease in children
 Presentation and investigation of mitochondrial disease in children Andrew Morris Willink Unit, Manchester Mitochondrial function Carbohydrate Fat Respiratory chain Energy Mitochondria are the product
Presentation and investigation of mitochondrial disease in children Andrew Morris Willink Unit, Manchester Mitochondrial function Carbohydrate Fat Respiratory chain Energy Mitochondria are the product
THIAMINE TRANSPORTER TYPE 2 DEFICIENCY
 THIAMINE TRANSPORTER TYPE 2 DEFICIENCY WHAT IS THE THIAMINE TRANSPORTER TYPE 2 DEFICIENCY (hthtr2)? The thiamine transporter type 2 deficiency (hthtr2) is a inborn error of thiamine metabolism caused by
THIAMINE TRANSPORTER TYPE 2 DEFICIENCY WHAT IS THE THIAMINE TRANSPORTER TYPE 2 DEFICIENCY (hthtr2)? The thiamine transporter type 2 deficiency (hthtr2) is a inborn error of thiamine metabolism caused by
Marah Bitar. Faisal Nimri ... Nafeth Abu Tarboosh
 8 Marah Bitar Faisal Nimri... Nafeth Abu Tarboosh Summary of the 8 steps of citric acid cycle Step 1. Acetyl CoA joins with a four-carbon molecule, oxaloacetate, releasing the CoA group and forming a six-carbon
8 Marah Bitar Faisal Nimri... Nafeth Abu Tarboosh Summary of the 8 steps of citric acid cycle Step 1. Acetyl CoA joins with a four-carbon molecule, oxaloacetate, releasing the CoA group and forming a six-carbon
Urea Cycle Defects. Dr Mick Henderson. Biochemical Genetics Leeds Teaching Hospitals Trust. MetBioNet IEM Introductory Training
 Urea Cycle Defects Dr Mick Henderson Biochemical Genetics Leeds Teaching Hospitals Trust The Urea Cycle The urea cycle enables toxic ammonia molecules to be converted to the readily excreted and non toxic
Urea Cycle Defects Dr Mick Henderson Biochemical Genetics Leeds Teaching Hospitals Trust The Urea Cycle The urea cycle enables toxic ammonia molecules to be converted to the readily excreted and non toxic
Energy Production In A Cell (Chapter 25 Metabolism)
") Energy Production In A Cell (Chapter 25 Metabolism) Large food molecules contain a lot of potential energy in the form of chemical bonds but it requires a lot of work to liberate the energy. Cells need
Energy Production In A Cell (Chapter 25 Metabolism) Large food molecules contain a lot of potential energy in the form of chemical bonds but it requires a lot of work to liberate the energy. Cells need
Newborn Screen & Development Facts about the genetic diseases new since March 2006 (Excluding Cystic Fibrosis)
") Newborn Screen & Development Facts about the genetic diseases new since March 2006 (Excluding Cystic Fibrosis) 1) Argininosuccinic acidemia (ASA) a) Incidence: ~1 in 70,000 b) Deficiency in an enzyme of
Newborn Screen & Development Facts about the genetic diseases new since March 2006 (Excluding Cystic Fibrosis) 1) Argininosuccinic acidemia (ASA) a) Incidence: ~1 in 70,000 b) Deficiency in an enzyme of
Higher Biology. Unit 2: Metabolism and Survival Topic 2: Respiration. Page 1 of 25
 Higher Biology Unit 2: Metabolism and Survival Topic 2: Respiration Page 1 of 25 Sub Topic: Respiration I can state that: All living cells carry out respiration. ATP is the energy currency of the cell
Higher Biology Unit 2: Metabolism and Survival Topic 2: Respiration Page 1 of 25 Sub Topic: Respiration I can state that: All living cells carry out respiration. ATP is the energy currency of the cell
Cellular Respiration
 Cellular Respiration 1. To perform cell work, cells require energy. a. A cell does three main kinds of work: i. Mechanical work, such as the beating of cilia, contraction of muscle cells, and movement
Cellular Respiration 1. To perform cell work, cells require energy. a. A cell does three main kinds of work: i. Mechanical work, such as the beating of cilia, contraction of muscle cells, and movement
An Introduction to mitochondrial disease.
 9 th September 2017 An Introduction to mitochondrial disease. Dr Andy Schaefer Consultant Neurologist and Clinical Lead NHS Highly Specialised Rare Mitochondrial Disease Service and Wellcome Trust Centre
9 th September 2017 An Introduction to mitochondrial disease. Dr Andy Schaefer Consultant Neurologist and Clinical Lead NHS Highly Specialised Rare Mitochondrial Disease Service and Wellcome Trust Centre
Citric Acid Cycle and Oxidative Phosphorylation
 Citric Acid Cycle and Oxidative Phosphorylation Page by: OpenStax Summary The Citric Acid Cycle In eukaryotic cells, the pyruvate molecules produced at the end of glycolysis are transported into mitochondria,
Citric Acid Cycle and Oxidative Phosphorylation Page by: OpenStax Summary The Citric Acid Cycle In eukaryotic cells, the pyruvate molecules produced at the end of glycolysis are transported into mitochondria,
Electron Transport Chain and Oxidative phosphorylation
 Electron Transport Chain and Oxidative phosphorylation So far we have discussed the catabolism involving oxidation of 6 carbons of glucose to CO 2 via glycolysis and CAC without any oxygen molecule directly
Electron Transport Chain and Oxidative phosphorylation So far we have discussed the catabolism involving oxidation of 6 carbons of glucose to CO 2 via glycolysis and CAC without any oxygen molecule directly
Mitochondrial Cytopathies
 Mitochondrial Cytopathies Bruce H. Cohen, M.D. Neuroscience Institute Brain Tumor and Neuro-Oncology Center Pediatric Neurology Center Cleveland Clinic 2008 What Are Mitochondria? Engines within the cell
Mitochondrial Cytopathies Bruce H. Cohen, M.D. Neuroscience Institute Brain Tumor and Neuro-Oncology Center Pediatric Neurology Center Cleveland Clinic 2008 What Are Mitochondria? Engines within the cell
Robert Barski. Biochemical Genetics St James s University Hospital, Leeds. MetBioNet IEM Introductory Training
 Robert Barski Biochemical Genetics St James s University Hospital, Leeds Lactate is produced as the fate of anaerobic metabolism of pyruvate. It is an important intermediary metabolite especially with
Robert Barski Biochemical Genetics St James s University Hospital, Leeds Lactate is produced as the fate of anaerobic metabolism of pyruvate. It is an important intermediary metabolite especially with
Ch. 9 Cell Respiration. Title: Oct 15 3:24 PM (1 of 53)
") Ch. 9 Cell Respiration Title: Oct 15 3:24 PM (1 of 53) Essential question: How do cells use stored chemical energy in organic molecules and to generate ATP? Title: Oct 15 3:28 PM (2 of 53) Title: Oct 19
Ch. 9 Cell Respiration Title: Oct 15 3:24 PM (1 of 53) Essential question: How do cells use stored chemical energy in organic molecules and to generate ATP? Title: Oct 15 3:28 PM (2 of 53) Title: Oct 19
Chapter 8 Mitochondria and Cellular Respiration
 Chapter 8 Mitochondria and Cellular Respiration Cellular respiration is the process of oxidizing food molecules, like glucose, to carbon dioxide and water. The energy released is trapped in the form of
Chapter 8 Mitochondria and Cellular Respiration Cellular respiration is the process of oxidizing food molecules, like glucose, to carbon dioxide and water. The energy released is trapped in the form of
MIDDLETOWN HIGH SCHOOL SOUTH BIOLOGY
 MIDDLETOWN HIGH SCHOOL SOUTH BIOLOGY BOOKLET 10 NAME: CLASS: 1 S.Tagore Middletown South High School March 2013 LEARNING OUTCOMES The role and production of ATP (a) Importance, role and structure of ATP
MIDDLETOWN HIGH SCHOOL SOUTH BIOLOGY BOOKLET 10 NAME: CLASS: 1 S.Tagore Middletown South High School March 2013 LEARNING OUTCOMES The role and production of ATP (a) Importance, role and structure of ATP
LESSON 2.2 WORKBOOK. Metabolism: Glucose is the middleman for ATP
 DEFINITIONS OF TERMS Homeostasis The tendency toward a relatively stable equilibrium that is maintained by physiological processes. For a complete list of defined terms, see the Glossary. LESSON 2.2 WORKBOOK
DEFINITIONS OF TERMS Homeostasis The tendency toward a relatively stable equilibrium that is maintained by physiological processes. For a complete list of defined terms, see the Glossary. LESSON 2.2 WORKBOOK
What s the point? The point is to make ATP! ATP
 2006-2007 What s the point? The point is to make ATP! ATP Glycolysis 2 ATP Kreb s cycle 2 ATP Life takes a lot of energy to run, need to extract more energy than 4 ATP! There s got to be a better way!
2006-2007 What s the point? The point is to make ATP! ATP Glycolysis 2 ATP Kreb s cycle 2 ATP Life takes a lot of energy to run, need to extract more energy than 4 ATP! There s got to be a better way!
Mitochondria and ATP Synthesis
 Mitochondria and ATP Synthesis Mitochondria and ATP Synthesis 1. Mitochondria are sites of ATP synthesis in cells. 2. ATP is used to do work; i.e. ATP is an energy source. 3. ATP hydrolysis releases energy
Mitochondria and ATP Synthesis Mitochondria and ATP Synthesis 1. Mitochondria are sites of ATP synthesis in cells. 2. ATP is used to do work; i.e. ATP is an energy source. 3. ATP hydrolysis releases energy
4. Which step shows a split of one molecule into two smaller molecules? a. 2. d. 5
 1. Which of the following statements about NAD + is false? a. NAD + is reduced to NADH during both glycolysis and the citric acid cycle. b. NAD + has more chemical energy than NADH. c. NAD + is reduced
1. Which of the following statements about NAD + is false? a. NAD + is reduced to NADH during both glycolysis and the citric acid cycle. b. NAD + has more chemical energy than NADH. c. NAD + is reduced
Enzymes and Metabolism
 PowerPoint Lecture Slides prepared by Vince Austin, University of Kentucky Enzymes and Metabolism Human Anatomy & Physiology, Sixth Edition Elaine N. Marieb 1 Protein Macromolecules composed of combinations
PowerPoint Lecture Slides prepared by Vince Austin, University of Kentucky Enzymes and Metabolism Human Anatomy & Physiology, Sixth Edition Elaine N. Marieb 1 Protein Macromolecules composed of combinations
III. 6. Test. Respiració cel lular
 III. 6. Test. Respiració cel lular Chapter Questions 1) What is the term for metabolic pathways that release stored energy by breaking down complex molecules? A) anabolic pathways B) catabolic pathways
III. 6. Test. Respiració cel lular Chapter Questions 1) What is the term for metabolic pathways that release stored energy by breaking down complex molecules? A) anabolic pathways B) catabolic pathways
Cellular Respiration Harvesting Chemical Energy ATP
 Cellular Respiration Harvesting Chemical Energy ATP 2006-2007 What s the point? The point is to make ATP! ATP 2006-2007 Harvesting stored energy Energy is stored in organic molecules carbohydrates, fats,
Cellular Respiration Harvesting Chemical Energy ATP 2006-2007 What s the point? The point is to make ATP! ATP 2006-2007 Harvesting stored energy Energy is stored in organic molecules carbohydrates, fats,
Cell Respiration. Anaerobic & Aerobic Respiration
 Cell Respiration Anaerobic & Aerobic Respiration Understandings/Objectives 2.8.U1: Cell respiration is the controlled release of energy from organic compounds to produce ATP. Define cell respiration State
Cell Respiration Anaerobic & Aerobic Respiration Understandings/Objectives 2.8.U1: Cell respiration is the controlled release of energy from organic compounds to produce ATP. Define cell respiration State
Oxidative Phosphorylation
 Oxidative Phosphorylation Energy from Reduced Fuels Is Used to Synthesize ATP in Animals Carbohydrates, lipids, and amino acids are the main reduced fuels for the cell. Electrons from reduced fuels are
Oxidative Phosphorylation Energy from Reduced Fuels Is Used to Synthesize ATP in Animals Carbohydrates, lipids, and amino acids are the main reduced fuels for the cell. Electrons from reduced fuels are
g) Cellular Respiration Higher Human Biology
 Cellular Respiration Higher Human Biology") g) Cellular Respiration Higher Human Biology What can you remember about respiration? 1. What is respiration? 2. What are the raw materials? 3. What are the products? 4. Where does it occur? 5. Why does
g) Cellular Respiration Higher Human Biology What can you remember about respiration? 1. What is respiration? 2. What are the raw materials? 3. What are the products? 4. Where does it occur? 5. Why does
Citric Acid Cycle and Oxidative Phosphorylation
 Citric Acid Cycle and Oxidative Phosphorylation Bởi: OpenStaxCollege The Citric Acid Cycle In eukaryotic cells, the pyruvate molecules produced at the end of glycolysis are transported into mitochondria,
Citric Acid Cycle and Oxidative Phosphorylation Bởi: OpenStaxCollege The Citric Acid Cycle In eukaryotic cells, the pyruvate molecules produced at the end of glycolysis are transported into mitochondria,
How Cells Harvest Chemical Energy
 How Cells Harvest Chemical Energy Global Athlete Outreach Program US CytoThesis Systems Medicine Center www.cytothesis.us US OncoTherapy Systems BioMedicine Group CytoThesis Bioengineering Research Group
How Cells Harvest Chemical Energy Global Athlete Outreach Program US CytoThesis Systems Medicine Center www.cytothesis.us US OncoTherapy Systems BioMedicine Group CytoThesis Bioengineering Research Group
3.7.1 Define cell respiration [Cell respiration is the controlled release of energy from organic compounds in cells to form ATP]
![3.7.1 Define cell respiration [Cell respiration is the controlled release of energy from organic compounds in cells to form ATP]](/thumbs/72/66350985.jpg "3.7.1 Define cell respiration [Cell respiration is the controlled release of energy from organic compounds in cells to form ATP]") 3.7 Cell respiration ( Chapter 9 in Campbell's book) 3.7.1 Define cell respiration [Cell respiration is the controlled release of energy from organic compounds in cells to form ATP] Organic compounds store
3.7 Cell respiration ( Chapter 9 in Campbell's book) 3.7.1 Define cell respiration [Cell respiration is the controlled release of energy from organic compounds in cells to form ATP] Organic compounds store
CHY2026: General Biochemistry UNIT 7& 8: CARBOHYDRATE METABOLISM
 CHY2026: General Biochemistry UNIT 7& 8: CARBOHYDRATE METABOLISM Metabolism Bioenergetics is the transfer and utilization of energy in biological systems The direction and extent to which a chemical reaction
CHY2026: General Biochemistry UNIT 7& 8: CARBOHYDRATE METABOLISM Metabolism Bioenergetics is the transfer and utilization of energy in biological systems The direction and extent to which a chemical reaction
Hompes Method Lesson 29 Organic Acids Part Three
 Hompes Method Lesson 29 Organic Acids Part Three Health for the People Ltd not for reuse without expressed permission Organic Acids - Review Fats, carbohydrates, and amino acids are converted into carboxylic
Hompes Method Lesson 29 Organic Acids Part Three Health for the People Ltd not for reuse without expressed permission Organic Acids - Review Fats, carbohydrates, and amino acids are converted into carboxylic
Lecture 5: Cell Metabolism. Biology 219 Dr. Adam Ross
 Lecture 5: Cell Metabolism Biology 219 Dr. Adam Ross Cellular Respiration Set of reactions that take place during the conversion of nutrients into ATP Intricate regulatory relationship between several
Lecture 5: Cell Metabolism Biology 219 Dr. Adam Ross Cellular Respiration Set of reactions that take place during the conversion of nutrients into ATP Intricate regulatory relationship between several
Page 2 of 51 WJEC/CBAC 2016 pdfcrowd.com
 1. Page 2 of 51 WJEC/CBAC 2016 Page 3 of 51 WJEC/CBAC 2016 2. Page 4 of 51 WJEC/CBAC 2016 Page 5 of 51 WJEC/CBAC 2016 3. Page 6 of 51 WJEC/CBAC 2016 Page 7 of 51 WJEC/CBAC 2016 4. Page 8 of 51 WJEC/CBAC
1. Page 2 of 51 WJEC/CBAC 2016 Page 3 of 51 WJEC/CBAC 2016 2. Page 4 of 51 WJEC/CBAC 2016 Page 5 of 51 WJEC/CBAC 2016 3. Page 6 of 51 WJEC/CBAC 2016 Page 7 of 51 WJEC/CBAC 2016 4. Page 8 of 51 WJEC/CBAC
Training Syllabus CLINICAL SYLLABUS
 Training Syllabus CLINICAL SYLLABUS SYLLABUS FOR TRAINING IN CLINICAL PAEDIATRIC METABOLIC MEDICINE Updated July 2006 This syllabus is intended as a guide. Whilst the training should be comprehensive,
Training Syllabus CLINICAL SYLLABUS SYLLABUS FOR TRAINING IN CLINICAL PAEDIATRIC METABOLIC MEDICINE Updated July 2006 This syllabus is intended as a guide. Whilst the training should be comprehensive,
Chapter 7 Cellular Respiration and Fermentation*
 Chapter 7 Cellular Respiration and Fermentation* *Lecture notes are to be used as a study guide only and do not represent the comprehensive information you will need to know for the exams. Life Is Work
Chapter 7 Cellular Respiration and Fermentation* *Lecture notes are to be used as a study guide only and do not represent the comprehensive information you will need to know for the exams. Life Is Work
Plant Respiration. Exchange of Gases in Plants:
 Plant Respiration Exchange of Gases in Plants: Plants do not have great demands for gaseous exchange. The rate of respiration in plants is much lower than in animals. Large amounts of gases are exchanged
Plant Respiration Exchange of Gases in Plants: Plants do not have great demands for gaseous exchange. The rate of respiration in plants is much lower than in animals. Large amounts of gases are exchanged
National Metabolic Biochemistry Network Guidelines for the investigation of hypoglycaemia in infants and children
 National Metabolic Biochemistry Network Guidelines for the investigation of hypoglycaemia in infants and children Aim To provide guidance on the biochemical investigation of hypoglycaemia in infants and
National Metabolic Biochemistry Network Guidelines for the investigation of hypoglycaemia in infants and children Aim To provide guidance on the biochemical investigation of hypoglycaemia in infants and
Cellular Respiration. Biochemistry Part II 4/28/2014 1
 Cellular Respiration Biochemistry Part II 4/28/2014 1 4/28/2014 2 The Mitochondria The mitochondria is a double membrane organelle Two membranes Outer membrane Inter membrane space Inner membrane Location
Cellular Respiration Biochemistry Part II 4/28/2014 1 4/28/2014 2 The Mitochondria The mitochondria is a double membrane organelle Two membranes Outer membrane Inter membrane space Inner membrane Location
Structure of the Mitochondrion. Cell Respiration. Cellular Respiration. Catabolic Pathways. Photosynthesis vs. Cell Respiration ATP 10/14/2014
 Structure of the Mitochondrion Cellular Respiration Chapter 9 Pgs. 163 183 Enclosed by a double membrane Outer membrane is smooth Inner, or cristae, membrane is folded - this divides the mitochondrion
Structure of the Mitochondrion Cellular Respiration Chapter 9 Pgs. 163 183 Enclosed by a double membrane Outer membrane is smooth Inner, or cristae, membrane is folded - this divides the mitochondrion
AP BIOLOGY Chapter 7 Cellular Respiration =
 1 AP BIOLOGY Chapter 7 Cellular Respiration = Day 1 p. I. Overview A. Cellular Respiration 1. Respiration breathing, exchange of O 2 for CO 2 2. Cellular respiration aerobic harvesting of energy from food
1 AP BIOLOGY Chapter 7 Cellular Respiration = Day 1 p. I. Overview A. Cellular Respiration 1. Respiration breathing, exchange of O 2 for CO 2 2. Cellular respiration aerobic harvesting of energy from food
Chemical Energy. Valencia College
 9 Pathways that Harvest Chemical Energy Valencia College 9 Pathways that Harvest Chemical Energy Chapter objectives: How Does Glucose Oxidation Release Chemical Energy? What Are the Aerobic Pathways of
9 Pathways that Harvest Chemical Energy Valencia College 9 Pathways that Harvest Chemical Energy Chapter objectives: How Does Glucose Oxidation Release Chemical Energy? What Are the Aerobic Pathways of
Cellular Pathways That Harvest Chemical Energy. Cellular Pathways That Harvest Chemical Energy. Cellular Pathways In General
 Cellular Pathways That Harvest Chemical Energy A. Obtaining Energy and Electrons from Glucose Lecture Series 12 Cellular Pathways That Harvest Chemical Energy B. An Overview: Releasing Energy from Glucose
Cellular Pathways That Harvest Chemical Energy A. Obtaining Energy and Electrons from Glucose Lecture Series 12 Cellular Pathways That Harvest Chemical Energy B. An Overview: Releasing Energy from Glucose
Metabolic Disorders. Chapter Thomson - Wadsworth
 Metabolic Disorders Chapter 28 1 Metabolic Disorders Inborn errors of metabolism group of diseases that affect a wide variety of metabolic processes; defective processing or transport of amino acids, fatty
Metabolic Disorders Chapter 28 1 Metabolic Disorders Inborn errors of metabolism group of diseases that affect a wide variety of metabolic processes; defective processing or transport of amino acids, fatty
Electron Transport and oxidative phosphorylation (ATP Synthesis) Dr. Howaida Nounou Biochemistry department Sciences college
 Dr. Howaida Nounou Biochemistry department Sciences college") Electron Transport and oxidative phosphorylation (ATP Synthesis) Dr. Howaida Nounou Biochemistry department Sciences college The Metabolic Pathway of Cellular Respiration All of the reactions involved
Electron Transport and oxidative phosphorylation (ATP Synthesis) Dr. Howaida Nounou Biochemistry department Sciences college The Metabolic Pathway of Cellular Respiration All of the reactions involved
Cellular Respiration Harvesting Chemical Energy ATP
 Cellular Respiration Harvesting Chemical Energy ATP 2006-2007 What s the point? The point is to make ATP! ATP 2006-2007 Harvesting stored energy Energy is stored in organic molecules carbohydrates, fats,
Cellular Respiration Harvesting Chemical Energy ATP 2006-2007 What s the point? The point is to make ATP! ATP 2006-2007 Harvesting stored energy Energy is stored in organic molecules carbohydrates, fats,
Review. Respiration. Glycolysis. Glycolysis is the decomposition (lysis) of glucose (glyco) to pyruvate (or pyruvic acid).
 of glucose (glyco) to pyruvate (or pyruvic acid).") Review Photosynthesis is the process of incorporating energy from light into energy-rich molecules like glucose. Respiration is the opposite process extracting that stored energy from glucose to form ATP
Review Photosynthesis is the process of incorporating energy from light into energy-rich molecules like glucose. Respiration is the opposite process extracting that stored energy from glucose to form ATP
MEMBRANE-BOUND ELECTRON TRANSFER AND ATP SYNTHESIS (taken from Chapter 18 of Stryer)
") MEMBRANE-BOUND ELECTRON TRANSFER AND ATP SYNTHESIS (taken from Chapter 18 of Stryer) FREE ENERGY MOST USEFUL THERMODYNAMIC CONCEPT IN BIOCHEMISTRY Living things require an input of free energy for 3 major
MEMBRANE-BOUND ELECTRON TRANSFER AND ATP SYNTHESIS (taken from Chapter 18 of Stryer) FREE ENERGY MOST USEFUL THERMODYNAMIC CONCEPT IN BIOCHEMISTRY Living things require an input of free energy for 3 major
Oxidative Phosphorylation
 Electron Transport Chain (overview) The NADH and FADH 2, formed during glycolysis, β- oxidation and the TCA cycle, give up their electrons to reduce molecular O 2 to H 2 O. Electron transfer occurs through
Electron Transport Chain (overview) The NADH and FADH 2, formed during glycolysis, β- oxidation and the TCA cycle, give up their electrons to reduce molecular O 2 to H 2 O. Electron transfer occurs through
Unit 2: Metabolic Processes
 How is energy obtained biologically? Recall: Red Ox Reactions Unit 2: Metabolic Processes Oxidation Is the chief mechanism by which chemical potential energy is released This energy comes from reduced
How is energy obtained biologically? Recall: Red Ox Reactions Unit 2: Metabolic Processes Oxidation Is the chief mechanism by which chemical potential energy is released This energy comes from reduced
Childhood epilepsy: the biochemical epilepsies. Dr Colin D Ferrie Consultant Paediatric Neurologist Leeds General Infirmary
 Childhood epilepsy: the biochemical epilepsies Dr Colin D Ferrie Consultant Paediatric Neurologist Leeds General Infirmary Definitions Epileptic Seizure Manifestation(s) of epileptic (excessive and/or
Childhood epilepsy: the biochemical epilepsies Dr Colin D Ferrie Consultant Paediatric Neurologist Leeds General Infirmary Definitions Epileptic Seizure Manifestation(s) of epileptic (excessive and/or
Cell Respiration Assignment Score. Name Sec.. Date.
 Cell Respiration Assignment Score. Name Sec.. Date. Working by alone or in a group, answer the following questions about Cell Respiration. This assignment is worth 30 points with the possible points for
Cell Respiration Assignment Score. Name Sec.. Date. Working by alone or in a group, answer the following questions about Cell Respiration. This assignment is worth 30 points with the possible points for
Electron transport chain chapter 6 (page 73) BCH 340 lecture 6
 BCH 340 lecture 6") Electron transport chain chapter 6 (page 73) BCH 340 lecture 6 The Metabolic Pathway of Cellular Respiration All of the reactions involved in cellular respiration can be grouped into three main stages
Electron transport chain chapter 6 (page 73) BCH 340 lecture 6 The Metabolic Pathway of Cellular Respiration All of the reactions involved in cellular respiration can be grouped into three main stages
Chapter 9: Cellular Respiration
 Chapter 9: Cellular Respiration Breaking down glucose a little at a time.. It s like turning a five pound bag of sugar into several tiny sugar packets worth of energy in the form of ATP. Remember the carbon
Chapter 9: Cellular Respiration Breaking down glucose a little at a time.. It s like turning a five pound bag of sugar into several tiny sugar packets worth of energy in the form of ATP. Remember the carbon
Principles of Anatomy and Physiology
 Principles of Anatomy and Physiology 14 th Edition CHAPTER 25 Metabolism and Nutrition Metabolic Reactions Metabolism refers to all of the chemical reactions taking place in the body. Reactions that break
Principles of Anatomy and Physiology 14 th Edition CHAPTER 25 Metabolism and Nutrition Metabolic Reactions Metabolism refers to all of the chemical reactions taking place in the body. Reactions that break
Metabolism. Chapter 5. Catabolism Drives Anabolism 8/29/11. Complete Catabolism of Glucose
 8/29/11 Metabolism Chapter 5 All of the reactions in the body that require energy transfer. Can be divided into: Cell Respiration and Metabolism Anabolism: requires the input of energy to synthesize large
8/29/11 Metabolism Chapter 5 All of the reactions in the body that require energy transfer. Can be divided into: Cell Respiration and Metabolism Anabolism: requires the input of energy to synthesize large
for a bright reaching resources
 resources e Glut1 Deficiency Foundation is a non-profit family organization dedicated to improving the lives of those in the Glut1 Deficiency community through its mission of: increased awareness improved
resources e Glut1 Deficiency Foundation is a non-profit family organization dedicated to improving the lives of those in the Glut1 Deficiency community through its mission of: increased awareness improved
number Done by Corrected by Doctor Nafeth Abu Tarboush
 number 7 Done by حسام أبو عوض Corrected by Shahd Alqudah Doctor Nafeth Abu Tarboush 1 P a g e As we have studied before, in the fourth reaction of the Krebs cycle, α- ketoglutarate is converted into Succinyl-CoA
number 7 Done by حسام أبو عوض Corrected by Shahd Alqudah Doctor Nafeth Abu Tarboush 1 P a g e As we have studied before, in the fourth reaction of the Krebs cycle, α- ketoglutarate is converted into Succinyl-CoA
Biol 219 Lec 7 Fall 2016
 Cellular Respiration: Harvesting Energy to form ATP Cellular Respiration and Metabolism Glucose ATP Pyruvate Lactate Acetyl CoA NAD + Introducing The Players primary substrate for cellular respiration
Cellular Respiration: Harvesting Energy to form ATP Cellular Respiration and Metabolism Glucose ATP Pyruvate Lactate Acetyl CoA NAD + Introducing The Players primary substrate for cellular respiration
ATP. Principles of Energy Harvest. Chapter 9~ The point is to make ATP! Cellular Respiration: Harvesting Chemical Energy. What s the point?
 Chapter 9~ Cellular Respiration: Harvesting Chemical Energy What s the point? The point is to make! 2006-2007 Principles of Energy Harvest Catabolic pathway Fermentation Cellular Respiration C6H126 + 62
Chapter 9~ Cellular Respiration: Harvesting Chemical Energy What s the point? The point is to make! 2006-2007 Principles of Energy Harvest Catabolic pathway Fermentation Cellular Respiration C6H126 + 62
CHE 242 Exam 3 Practice Questions
 CHE 242 Exam 3 Practice Questions Glucose metabolism 1. Below is depicted glucose catabolism. Indicate on the pathways the following: A) which reaction(s) of glycolysis are irreversible B) where energy
CHE 242 Exam 3 Practice Questions Glucose metabolism 1. Below is depicted glucose catabolism. Indicate on the pathways the following: A) which reaction(s) of glycolysis are irreversible B) where energy
Chapter Seven (Cellular Respiration)
") Chapter Seven (Cellular Respiration) 1 SECTION ONE: GLYCOLYSIS AND FERMENTATION HARVESTING CHEMICAL ENERGY Cellular respiration is the process in which cells make adenosine triphosphate (ATP) by breaking
Chapter Seven (Cellular Respiration) 1 SECTION ONE: GLYCOLYSIS AND FERMENTATION HARVESTING CHEMICAL ENERGY Cellular respiration is the process in which cells make adenosine triphosphate (ATP) by breaking
Chapter 5. Microbial Metabolism
 Chapter 5 Microbial Metabolism Metabolism Collection of controlled biochemical reactions that take place within a microbe Ultimate function of metabolism is to reproduce the organism Metabolic Processes
Chapter 5 Microbial Metabolism Metabolism Collection of controlled biochemical reactions that take place within a microbe Ultimate function of metabolism is to reproduce the organism Metabolic Processes
Cellular Respiration Harvesting Chemical Energy ATP
 Cellular Respiration Harvesting Chemical Energy ATP 2009-2010 Ch.8.3 Section Objectives: Compare and contrast cellular respiration and fermentation. Explain how cells obtain energy from cellular respiration.
Cellular Respiration Harvesting Chemical Energy ATP 2009-2010 Ch.8.3 Section Objectives: Compare and contrast cellular respiration and fermentation. Explain how cells obtain energy from cellular respiration.
NBCE Mock Board Questions Biochemistry
 1. Fluid mosaic describes. A. Tertiary structure of proteins B. Ribosomal subunits C. DNA structure D. Plasma membrane structure NBCE Mock Board Questions Biochemistry 2. Where in the cell does beta oxidation
1. Fluid mosaic describes. A. Tertiary structure of proteins B. Ribosomal subunits C. DNA structure D. Plasma membrane structure NBCE Mock Board Questions Biochemistry 2. Where in the cell does beta oxidation
BY: RASAQ NURUDEEN OLAJIDE
 BY: RASAQ NURUDEEN OLAJIDE LECTURE CONTENT INTRODUCTION CITRIC ACID CYCLE (T.C.A) PRODUCTION OF ACETYL CoA REACTIONS OF THE CITIRC ACID CYCLE THE AMPHIBOLIC NATURE OF THE T.C.A CYCLE THE GLYOXYLATE CYCLE
BY: RASAQ NURUDEEN OLAJIDE LECTURE CONTENT INTRODUCTION CITRIC ACID CYCLE (T.C.A) PRODUCTION OF ACETYL CoA REACTIONS OF THE CITIRC ACID CYCLE THE AMPHIBOLIC NATURE OF THE T.C.A CYCLE THE GLYOXYLATE CYCLE
2. What are the products of cellular respiration? Include all forms of energy that are products.
 Name Per Cellular Respiration An Overview Why Respire Anyhoo? Because bucko all cells need usable chemical energy to do work. The methods cells use to convert glucose into ATP vary depending on the availability
Name Per Cellular Respiration An Overview Why Respire Anyhoo? Because bucko all cells need usable chemical energy to do work. The methods cells use to convert glucose into ATP vary depending on the availability
Cellular Metabolism 6/20/2015. Metabolism. Summary of Cellular Respiration. Consists of all the chemical reactions that take place in a cell!
 Cellular Metabolism Biology 105 Lecture 6 Chapter 3 (pages 56-61) Metabolism Consists of all the chemical reactions that take place in a cell! Cellular metabolism: Aerobic cellular respiration requires
Cellular Metabolism Biology 105 Lecture 6 Chapter 3 (pages 56-61) Metabolism Consists of all the chemical reactions that take place in a cell! Cellular metabolism: Aerobic cellular respiration requires
The mitochondrion and its disorders
 100 PRACTICAL NEUROLOGY H O W T O U N D E R S T A N D I T The mitochondrion and its disorders Patrick F. Chinnery Department of Neurology, Regional Neurosciences Centre, Newcastle General Hospital and
100 PRACTICAL NEUROLOGY H O W T O U N D E R S T A N D I T The mitochondrion and its disorders Patrick F. Chinnery Department of Neurology, Regional Neurosciences Centre, Newcastle General Hospital and
These factors should be taken into consideration when addressing fatigue or low energy because each factor will be approached slightly differently.
 1 2 These factors should be taken into consideration when addressing fatigue or low energy because each factor will be approached slightly differently. 3 4 First we must understand the systems of the body
1 2 These factors should be taken into consideration when addressing fatigue or low energy because each factor will be approached slightly differently. 3 4 First we must understand the systems of the body
LAB 6 Fermentation & Cellular Respiration
 LAB 6 Fermentation & Cellular Respiration INTRODUCTION The cells of all living organisms require energy to keep themselves alive and fulfilling their roles. Where does this energy come from? The answer
LAB 6 Fermentation & Cellular Respiration INTRODUCTION The cells of all living organisms require energy to keep themselves alive and fulfilling their roles. Where does this energy come from? The answer
Mitochondrial Diseases
 Mitochondrial Diseases Simon Heales SWIM Conference Taunton, November 29 th 2018 Respiratory Failure Cardiomyopathy Optic Atrophy / Retinitis Pigmentosa Seizures / Developmental delay Liver Failure Deafness
Mitochondrial Diseases Simon Heales SWIM Conference Taunton, November 29 th 2018 Respiratory Failure Cardiomyopathy Optic Atrophy / Retinitis Pigmentosa Seizures / Developmental delay Liver Failure Deafness
2) The molecule that functions as the reducing agent (electron donor) in a redox or oxidationreduction
 The molecule that functions as the reducing agent (electron donor) in a redox or oxidationreduction") Campbell Biology in Focus (Urry) Chapter 7 Cellular Respiration and Fermentation 7.1 Multiple-Choice Questions 1) What is the term for metabolic pathways that release stored energy by breaking down complex
Campbell Biology in Focus (Urry) Chapter 7 Cellular Respiration and Fermentation 7.1 Multiple-Choice Questions 1) What is the term for metabolic pathways that release stored energy by breaking down complex
A cell has enough ATP to last for about three seconds.
 Energy Transformation: Cellular Respiration Outline 1. Energy and carbon sources in living cells 2. Sources of cellular ATP 3. Turning chemical energy of covalent bonds between C-C into energy for cellular
Energy Transformation: Cellular Respiration Outline 1. Energy and carbon sources in living cells 2. Sources of cellular ATP 3. Turning chemical energy of covalent bonds between C-C into energy for cellular
How Cells Harvest Energy. Chapter 7. Respiration
 How Cells Harvest Energy Chapter 7 Respiration Organisms classified on how they obtain energy: autotrophs: produce their own organic molecules through photosynthesis heterotrophs: live on organic compounds
How Cells Harvest Energy Chapter 7 Respiration Organisms classified on how they obtain energy: autotrophs: produce their own organic molecules through photosynthesis heterotrophs: live on organic compounds
Cellular Respiration Stage 2 & 3. Glycolysis is only the start. Cellular respiration. Oxidation of Pyruvate Krebs Cycle.
 Cellular Respiration Stage 2 & 3 Oxidation of Pyruvate Krebs Cycle AP 2006-2007 Biology Glycolysis is only the start Glycolysis glucose pyruvate 6C 2x 3C Pyruvate has more energy to yield 3 more C to strip
Cellular Respiration Stage 2 & 3 Oxidation of Pyruvate Krebs Cycle AP 2006-2007 Biology Glycolysis is only the start Glycolysis glucose pyruvate 6C 2x 3C Pyruvate has more energy to yield 3 more C to strip
There are four different forms of Tarui disease, which are classed by their signs and symptoms and age of presentation.
 Tarui Disease Tarui disease (also known as phosphofructokinase deficiency, or glycogen storage disease type VII) was first described in 1965 by the Japanese physician Seiichiro Tarui and his co-workers.
Tarui Disease Tarui disease (also known as phosphofructokinase deficiency, or glycogen storage disease type VII) was first described in 1965 by the Japanese physician Seiichiro Tarui and his co-workers.
The Organism as a system
 The Organism as a system PATIENT 1: Seven-year old female with a history of normal development until age two. At this point she developed episodic vomiting, acidosis, epilepsy, general weakness, ataxia
The Organism as a system PATIENT 1: Seven-year old female with a history of normal development until age two. At this point she developed episodic vomiting, acidosis, epilepsy, general weakness, ataxia
Cellular Metabolism 9/24/2013. Metabolism. Cellular Metabolism. Consists of all the chemical reactions that take place in a cell!
 Cellular Metabolism Biology 105 Lecture 6 Chapter 3 (pages 56-61) Metabolism Consists of all the chemical reactions that take place in a cell! Cellular Metabolism Aerobic cellular respiration requires
Cellular Metabolism Biology 105 Lecture 6 Chapter 3 (pages 56-61) Metabolism Consists of all the chemical reactions that take place in a cell! Cellular Metabolism Aerobic cellular respiration requires
1- Which of the following statements is TRUE in regards to eukaryotic and prokaryotic cells?
 Name: NetID: Exam 3 - Version 1 October 23, 2017 Dr. A. Pimentel Each question has a value of 4 points and there are a total of 160 points in the exam. However, the maximum score of this exam will be capped
Name: NetID: Exam 3 - Version 1 October 23, 2017 Dr. A. Pimentel Each question has a value of 4 points and there are a total of 160 points in the exam. However, the maximum score of this exam will be capped
CURRENT GENETIC TESTING TOOLS IN NEONATAL MEDICINE. Dr. Bahar Naghavi
 2 CURRENT GENETIC TESTING TOOLS IN NEONATAL MEDICINE Dr. Bahar Naghavi Assistant professor of Basic Science Department, Shahid Beheshti University of Medical Sciences, Tehran,Iran 3 Introduction Over 4000
2 CURRENT GENETIC TESTING TOOLS IN NEONATAL MEDICINE Dr. Bahar Naghavi Assistant professor of Basic Science Department, Shahid Beheshti University of Medical Sciences, Tehran,Iran 3 Introduction Over 4000
19 Oxidative Phosphorylation and Photophosphorylation W. H. Freeman and Company
 19 Oxidative Phosphorylation and Photophosphorylation 2013 W. H. Freeman and Company CHAPTER 19 Oxidative Phosphorylation and Photophosphorylation Key topics: Electron transport chain in mitochondria Capture
19 Oxidative Phosphorylation and Photophosphorylation 2013 W. H. Freeman and Company CHAPTER 19 Oxidative Phosphorylation and Photophosphorylation Key topics: Electron transport chain in mitochondria Capture
Bioenergetics. Finding adequate sources of energy is a constant challenge for all living organisms, including this bear.
 33 Bioenergetics Finding adequate sources of energy is a constant challenge for all living organisms, including this bear. Introduction to General, Organic, and Biochemistry, 10e John Wiley & Sons, Inc
33 Bioenergetics Finding adequate sources of energy is a constant challenge for all living organisms, including this bear. Introduction to General, Organic, and Biochemistry, 10e John Wiley & Sons, Inc
Cellular respiration and fermentation 04/18/2016 BI102
 Cellular respiration and fermentation 04/18/2016 BI102 Announcements Exam 1 after lecture Don t forget to do the online assignments every week! Quiz 2 and lab 2 review Cellular Respiration Cells require
Cellular respiration and fermentation 04/18/2016 BI102 Announcements Exam 1 after lecture Don t forget to do the online assignments every week! Quiz 2 and lab 2 review Cellular Respiration Cells require
Energy and life. Generation of Biochemical Energy Chapter 21. Energy. Energy and biochemical reactions: 4/5/09
 Energy and life Generation of Biochemical Energy Chapter 21 1 Biological systems are powered by oxidation of biomolecules made mainly of C, H and O. The food biomolecules are mainly Lipids (fats) Carbohydrates
Energy and life Generation of Biochemical Energy Chapter 21 1 Biological systems are powered by oxidation of biomolecules made mainly of C, H and O. The food biomolecules are mainly Lipids (fats) Carbohydrates
CLINICAL SIGNS SUGGESTIVE OF A NEUROMETABOLIC DISEASE. Bwee Tien Poll-The Amsterdam UMC The Netherlands
 CLINICAL SIGNS SUGGESTIVE OF A NEUROMETABOLIC DISEASE Bwee Tien Poll-The Amsterdam UMC The Netherlands FRAMEWORK OF PRINCIPALS 1. Problem-oriented clinical approach 2. Biomarkers in plasma, urine, CSF
CLINICAL SIGNS SUGGESTIVE OF A NEUROMETABOLIC DISEASE Bwee Tien Poll-The Amsterdam UMC The Netherlands FRAMEWORK OF PRINCIPALS 1. Problem-oriented clinical approach 2. Biomarkers in plasma, urine, CSF
Biochemistry of cellular organelles
 Kontinkangas, L101A Biochemistry of cellular organelles Lectures: 1. Membrane channels; 2. Membrane transporters; 3. Soluble lipid/metabolite-transfer proteins; 4. Mitochondria as cellular organelles;
Kontinkangas, L101A Biochemistry of cellular organelles Lectures: 1. Membrane channels; 2. Membrane transporters; 3. Soluble lipid/metabolite-transfer proteins; 4. Mitochondria as cellular organelles;
Carnitine palmitoyl transferase 2 deficiency (CPT2) is a rare inherited disorder that occurs when
 is a rare inherited disorder that occurs when") CPT2 Deficiency Carnitine palmitoyl transferase 2 deficiency (CPT2) is a rare inherited disorder that occurs when the last step in the entry of fats into sac-like bodies called mitochondria is blocked.
CPT2 Deficiency Carnitine palmitoyl transferase 2 deficiency (CPT2) is a rare inherited disorder that occurs when the last step in the entry of fats into sac-like bodies called mitochondria is blocked.
Cellular Metabolism. Biology 105 Lecture 6 Chapter 3 (pages 56-61)
") Cellular Metabolism Biology 105 Lecture 6 Chapter 3 (pages 56-61) Metabolism Consists of all the chemical reactions that take place in a cell! Cellular Metabolism Aerobic cellular respiration requires
Cellular Metabolism Biology 105 Lecture 6 Chapter 3 (pages 56-61) Metabolism Consists of all the chemical reactions that take place in a cell! Cellular Metabolism Aerobic cellular respiration requires
Energy Transformation: Cellular Respiration Outline 1. Sources of cellular ATP 2. Turning chemical energy of covalent bonds between C-C into energy
 Energy Transformation: Cellular Respiration Outline 1. Sources of cellular ATP 2. Turning chemical energy of covalent bonds between C-C into energy for cellular work (ATP) 3. Importance of electrons and
Energy Transformation: Cellular Respiration Outline 1. Sources of cellular ATP 2. Turning chemical energy of covalent bonds between C-C into energy for cellular work (ATP) 3. Importance of electrons and
Hompes Method Lesson 29 Organic Acids Part One
 Hompes Method Lesson 29 Organic Acids Part One Health for the People Ltd not for reuse without expressed permission Organic Acids - Introduction The ultimate tool for laboratory evaluations in nutritional
Hompes Method Lesson 29 Organic Acids Part One Health for the People Ltd not for reuse without expressed permission Organic Acids - Introduction The ultimate tool for laboratory evaluations in nutritional
Chapter 10. Cellular Respiration Pearson Education Ltd
 Chapter 10 Cellular Respiration Life Is Work a) Living cells require energy from outside sources b) Some animals, such as the giraffe, obtain energy by eating plants, and some animals feed on other organisms
Chapter 10 Cellular Respiration Life Is Work a) Living cells require energy from outside sources b) Some animals, such as the giraffe, obtain energy by eating plants, and some animals feed on other organisms
Biology Chapter-7 Cellular Respiration
 Biology-1406 Chapter-7 Cellular Respiration Energy is stored in Chemicals Catabolism- the breaking down of complex molecules, such as glucose, to release their stored energy. Catabolism may or may not
Biology-1406 Chapter-7 Cellular Respiration Energy is stored in Chemicals Catabolism- the breaking down of complex molecules, such as glucose, to release their stored energy. Catabolism may or may not
Ch 9: Cellular Respiration
 Ch 9: Cellular Respiration Cellular Respiration An overview Exergonic reactions and catabolic pathway Energy stored in bonds of food molecules is transferred to ATP Cellular respiration provides the energy
Ch 9: Cellular Respiration Cellular Respiration An overview Exergonic reactions and catabolic pathway Energy stored in bonds of food molecules is transferred to ATP Cellular respiration provides the energy
Cellular Respiration- -conversion of stored energy in glucose to usable energy for the cell -energy in cells is stored in the form of ATP
 Cellular Respiration Notes Chapter 7 How Cells Make ATP Energy Releasing Pathways Cellular Respiration- -conversion of stored energy in glucose to usable energy for the cell -energy in cells is stored
Cellular Respiration Notes Chapter 7 How Cells Make ATP Energy Releasing Pathways Cellular Respiration- -conversion of stored energy in glucose to usable energy for the cell -energy in cells is stored
Chapter 14 - Electron Transport and Oxidative Phosphorylation
 Chapter 14 - Electron Transport and Oxidative Phosphorylation The cheetah, whose capacity for aerobic metabolism makes it one of the fastest animals Prentice Hall c2002 Chapter 14 1 14.4 Oxidative Phosphorylation
Chapter 14 - Electron Transport and Oxidative Phosphorylation The cheetah, whose capacity for aerobic metabolism makes it one of the fastest animals Prentice Hall c2002 Chapter 14 1 14.4 Oxidative Phosphorylation
Biochemistry #02. The biochemical basis of skeletal muscle and bone disorders Dr. Nabil Bashir Bara Sami. 0 P a g e
 ]Type text[ ]Type text[ ]Type text[ Biochemistry #02 The biochemical basis of skeletal muscle and bone disorders Dr. Nabil Bashir Bara Sami 0 P a g e Greetings everyone, ladies and gentlemen The biochemical
]Type text[ ]Type text[ ]Type text[ Biochemistry #02 The biochemical basis of skeletal muscle and bone disorders Dr. Nabil Bashir Bara Sami 0 P a g e Greetings everyone, ladies and gentlemen The biochemical
Harvesting Energy: Glycolysis and Cellular Respiration
 Lesson 5 Harvesting Energy: Glycolysis and Cellular Respiration Introduction to Life Processes - SCI 102 1 How Cells Obtain Energy Cells require a constant flow of energy Most cellular energy is stored
Lesson 5 Harvesting Energy: Glycolysis and Cellular Respiration Introduction to Life Processes - SCI 102 1 How Cells Obtain Energy Cells require a constant flow of energy Most cellular energy is stored