BCM 225 LECTURE ON INBORN ERRORS OF METABOLISM OJEMEKELE O.
|
|
|
- Benjamin Blaise Oliver
- 5 years ago
- Views:
Transcription
1 BCM 225 LECTURE ON INBORN ERRORS OF METABOLISM BY OJEMEKELE O.
2 OUTLINE (week 11 and 12) Inborn errors of carbohydrate metabolism Inborn errors of amino acid metabolism Inborn errors of fatty acid metabolism Organic acidemias Lysosomal storage diseases Mitochondrial disorders Urea cycle disorders Blood chemistry; blood as a tissue
3 INBORN ERRORS OF METABOLISM Virtually every step of metabolic pathway is catalyzed by an enzyme. Defect in enzymes of metabolic pathways that result from abnormalities in their genes are known as inborn errors of metabolism. The cause of enzyme defect are genetic mutations that affect the structure and regulation of the enzyme, or create problems with binding of cofactors. The consequences of an enzyme defect are perturbations of cellular chemistry because of a reduction in the amount of an essential product, the build up of a toxic intermediate or the production of a toxic side product.
4 INBORN ERRORS OF CARBOHYDRATE METABOLISM These are inborn errors that affect the catabolism and anabolism of carbohydrates. Carbohydrates account for a major portion of the human diet and are metabolized into three principal monosaccharides: galactose, fructose and glucose.
5 GALACTOSEMIA Galactose is an aldohexose and is a constituent of lactose of milk. Inability to metabolize galactose results in galactosemia. Classic galactosemia is caused by defect in the enzyme galactose-1- phosphate uridyl transferase ; GAL-1-PUT (which catalyzes the rate limiting step of galatose metabolism pathway). BIOCHEMICAL IMPLICATIONS AND CLINICAL FEATURES Galactose-1-phosphate will accumulate in liver due to the block in the enzyme (GAL-1-PUT). This will inhibit galactokinase as well as glycogen phosphorylase, resulting in hypoglycemia. Bilirubin conjugation is reduced; so unconjugated bilirubin level is increased in blood. There is enlargement of liver, jaundice and severe mental retardation.
6 Free galactose accumulates, leading to galactosemia. It is partly excreted in urine (galactosuria). Galactose is reduced to galactitol. The accumulation of galactitol in the lens results in cataract due to its osmotic effect. This is called congenital cataract and is a very important feature of galactosemia Summary : (hypoglycemia, jaundice and severe mental retardation and cataract)
7 A variant of galactosemia occurs due to the deficiency of galactokinase. But here the symptoms are milder. This is because galactose-1-phosphate is not formed and hence toxic effects of this compound (such as liver insufficiency, hypoglycemia) are not manifested. Cataract is the only manifestations of galactose kinase deficiency.
8 FIG 1: GALACTOSE METABOLISM
9 DIAGNOSIS AND MANAGEMENT Collection of fetal cells by amniocentesis may be useful in prenatal diagnosis. Screening is also performed by measuring GAL- 1-PUT activity Management is by elimination of galactose from the diet. (Elimination of implicating substance; measuring enzyme activity, amnioncentesis)
10 HEREDITARY FRUCTOSE INTOLERANCE (HFI) HFI is caused by a defect in Aldolase B, hence fructose-1- phosphate cannot be metabolized and will accumulate. BIOCHEMICAL IMPLICATIONS AND SYMPTOMS Patients are asymptomatic unless they ingest fructose or sucrose. Accumulation of fructose-1-phosphate will inhibit glycogen phosphorylase. This leads to hypoglycemia. Vomiting and loss of appetite are seen. The infants often fail to thrive. Hepatomegaly and jaundice occur. If liver damage progresses, death will occur. DIAGNOSIS: Genetic testing and Aldolase B assay.
11 MANAGEMENT: Withdrawal of fructose and sucrose from the diet will immediately relieve the symptoms. Fig.2 FRUCTOSE METABOLISM
12 INBORN ERRORS ASSOCIATED WITH GLUCOSE METABOLISM 1. Diabetes mellitus type 1 is a disorder caused by reduced or absent levels of insulin, a hormone that regulates the levels of glucose in blood. It causes hyperglycemia. Due to high osmotic effect of high glucose levels, polyuria (excessive urine output) results, this cause polydipsia (excessive thirst). Diagnosis is by measuring blood glucose concentrations Management can be achieved by insulin replacement therapy
13 2. Glucose 6-Phosphate dehydrogenase deficiency : is an X-linked recessive hereditary disease characterised by abnormally low levels of glucose-6-phosphate dehydrogenase (abbreviated G6PD). G6PD is a metabolic enzyme involved in the pentose phosphate pathway, especially important in red blood cell metabolism. Individuals with the disease may exhibit non immune hemolytic anemia in response to a number of causes, most commonly infection or exposure to certain medications or chemicals. Hemolytic anaemia can lead to paleness, hemolytic jaundice, fatigue, shortness of breath and a rapid heart rate.
14 GLYCOGEN STORAGE DISEASES (GSD) Enzyme deficiencies that leads to impaired synthesis or degradation of glycogen are also considered disorders of carbohydrates metabolism. The two organs most commonly affected are the liver and the skeletal muscle Glycogen storage diseases that affect the liver typically cause hepatomegaly and hypoglycemia, relating to impaired mobilization of glucose for release to the blood during fasting Those that affect skeletal muscle cause exercise intolerance, progressive weakness and cramping resulting from low glucose levels, hence there is inability to increase glucose entry into glycolysis during exercise

15 TABLE 1: GLYCOGEN STORAGE DISEASES
16 DIAGNOSIS AND MANAGEMENT Diagnosis is achieved by evaluating blood glucose levels, measurement of enzyme activity and genetic testing Management; corn starch and sucrose supplementation to increase blood glucose levels have been suggested Gene therapy and enzyme replacement therapy are currently been investigated as possible therapies for GSDs
17 INBORN ERRORS OF AMINO ACID METABOLISM PHENYLKEKONURIA (PKU) This disease is caused by deficiency of phenyl alanine hydroxylase (PAH). The enzyme that converts phenyl alanine to tyrosine. Genetic mutation may be such that either the enzyme is not synthesized, or a non-functional enzyme is synthesized. BIOCHEMICAL ABNORMALITIES Phenylalanine can not be converted to tyrosine. So phenylalanine accumulates. Phenylalanine level in blood is elevated. So, alternate minor pathways are opened (Fig 4) Phenyl ketone (phenyl pyruvate), phenyl lactate and phenyl acetate are produced from pheneylalanine which are are excreted in urine.
18 Clinical Manifestations The classical PKU child is mentally retarded. The mental retardation is caused by the accumulation of phenylalanine (and its toxic metabolities phenylpyruvate, phenyllactate and phenylacetate. Agitation, hyperactivity, tremors and convulsions are often manifested. This may be because high phenylalanine and low tyrosine levels reduces neurotransmitter synthesis. The child often has hypopigmentation (low melanin levels), explained by the decreased level of tyrosine.
19 DIAGNOSIS Blood phenylalanine: Normal level is 1 mg/dl. In PKU, the level is >20 mg/dl. Ferric chloride test: Urine of the patient contains phenyl ketone This could be detected by adding a drop of ferric chloride to the urine. Phenylketone reacts with ferric chloride to produce a transient blue-green color. MANAGEMENT PKU can be managed by providing a diet containing low in phenyl alanine. Food based on tapioca (cassava) have low phenyl alanine content. This special diet is to be continued during the first decade of life; after which the child can have a normal diet. Life-long compliance of special diet is recommended.
20 Fig 3: Tyrosine (a nonessential amino acid) is used not only for protein synthesis, but as shown in the figure above, tyrosine is also the precursor for neurotransmitters; dopamine,epinephrine, norepinephrine, thyroid hormones (T3&T4), as well as, skin pigments (melanin).


21 Fig 4: Alternate pathways for phenylalanine in phenyl ketonuria
22 ALKAPTONURIA This is due to the deficiency of the enzyme homogentisic acid oxidase(an enzyme of tyrosine metabolic pathway) resulting in Homogentisic acid (HGA) accumulation and excretion in urine. Alkaptonuria is an autosomal recessive condition with an incidence of 1 in 250,000 births. It is compatible with fairly normal life. The only abnormality is the blackening of urine on standing. The homogentisic acid is oxidized by polyphenol oxidase to benzoquinone acetate (Fig. 6). It is then polymerized to black coloured alkaptone bodies. By the 3rd or 4th decade of life, patient may develop ochronosis (deposition of alkaptone bodies in intervertebral discs, cartilages of nose, pinna of ear). Black pigments are deposited over the connective tissues including joint cavities to produce arthritis. DIAGNOSIS: 1.Urine becomes black on standing. Blackening is accelerated on exposure to sunlight and oxygen. The urine when kept in a test tube will start to blacken from the top layer. 2. Ferric chloride test will be positive for urine MANAGEMENT: Minimal protein intake with phenylalanine less than 500 mg/day is recommended.

23 FIG 5: PHENYLALANINE AND TYROSINE METABOLISM


24 Black coloured alkaptone bodies FIG 6: ALTERNATIVE OXIDATION OF HOMOGENTISIC ACID IN ALKAPTONURIA Urine becomes black on exposure to air
25 ALBINISM Albinism is an autosomal recessive mutation in the gene for tyrosinase (the rate limiting enzyme of the melanin synthetic pathway), resulting in absent of melanin (the pigment that gives dark colour to the skin, hair and eyes). A deficiency in tyrosinase will result in loss of hair and skin pigments which explains the albino phenotype.
26 There is photophobia, nystagmus and decreased visual acuity. The skin has low pigmentation, and so skin is sensitive to UV rays. Hair is also white. Management includes use of tinted glasses to reduce photophobia, use of sun protection cream on the skin.

27 FIG 7: Melanin biosynthesis (reactions 1 and 2 are catalyzed by tyrosinase)
28 HOMOCYSTINURIA Defect in cystathionine synthase (an enzyme involved in conversion of methionine into cysteine) leads to homocystinuria. It causes high concentration of homocysteine and methionine in the blood. There is increased excretion of methionine and homocystine in urine and blood cysteine is markedly reduced. Homocysteine is highly reactive molecule hence the disease is often associated with mental retardation, multi systemic disorder of connective tissue, muscle, CNS, and cardiovascular system.
29 General symptoms are mental retardation, Skeletal deformities. In eyes, ectopia lentis (subluxation of lens), myopia and glaucoma may be observed. Homocysteine causes activation of Hageman's factor. This may lead to increased platelet adhesiveness and life-threatening intra-vascular thrombosis. Diagnosis : Cyanide-nitroprusside test will be positive in urine. Urinary excretion of homocystine is more than 300 mg/24 h. Management : is a diet low in methionine and rich in cysteine. Sometimes the affinity of apo-enzyme to the co-enzyme is reduced. In such cases, pyridoxal phosphate, the co-enzyme given in large quantities (500 mg) will be useful.
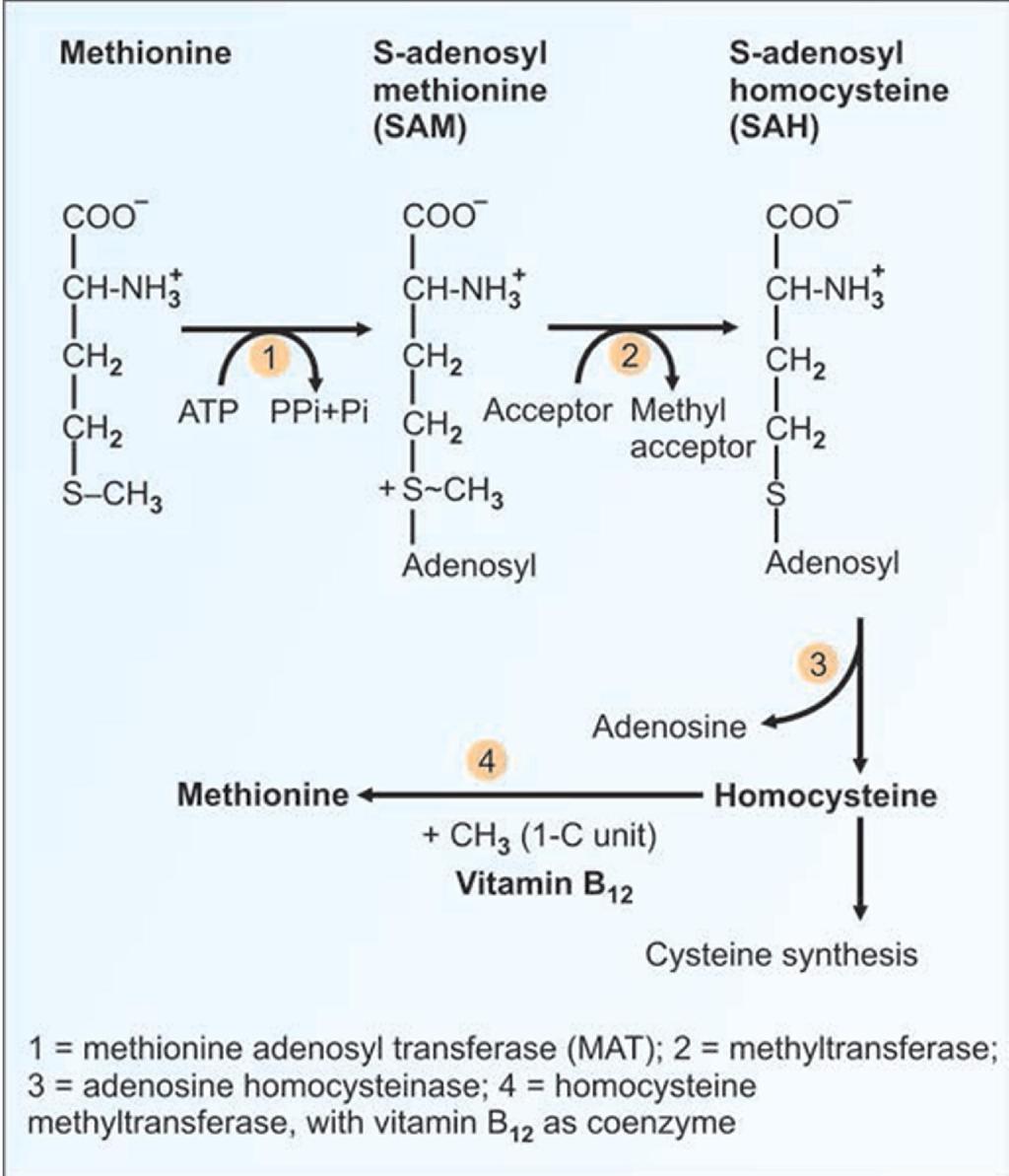
30

31 Fig: Methionine metabolism
32 MAPLE SYRUP URINE DISEASE It is also called branched chain ketonuria. The name originates from the characteristic smell of urine (similar to burnt sugar or maple sugar) due to excretion of branched chain keto acids. BIOCHEMICAL DEFECT: Deficiency of branched chain alpha ketoacid (BKA ) dehydrogenase complex SYMPTOMS AND LABORATORY FINDINGS: Symptoms start in the first week of life. It is characterized by convulsions, severe mental retardation, vomiting, acidosis, coma and death within the first year of life if not properly managed. Urine contains branched chain keto acids derived from valine, leucine and isoleucine. DIAGNOSIS: Diagnosis is based on enzyme analysis in cells. MANAGEMENT: Giving a diet low in branched chain amino acids (valine, leucine and isoleucine).
33 FATTY ACID OXIDATION DISORDERS (FAOD) Fatty acid oxidation disorders are genetic conditions caused by mutations in genes that code for enzymes responsible for oxidation of fatty acids. When there is an alteration in these genes, enzyme levels go down and fatty acids build up in the blood. In the case of fatty acid oxidation disorders, the inability to break down fats for energy and the build up of fatty acids cause serious health problems.
34 DIAGNOSIS: Babies can be tested (newborn screening) for fatty acid oxidation disorders before they leave the hospital. The baby s heel is pricked and a few drops of blood are taken. The blood is sent to the state laboratory to find out if it has more than a normal amount of fatty acids. There are various types of fatty acid oxidation disorders. The following is a list of fatty acid oxidation disorders that can be screened for: Carnitine/Acylcarnitine Translocase deficiency (TRANS) Carnithine transporter deficiency (CTD) Long/Very Long Chain Acyl CoA Dehydrogenase deficiency (LCAD/VLACD) Medium Chain Acyl CoA Dehydrogenase deficiency (MCAD) Short Chain Acyl CoA Dehydrogenase deficiency (SCAD) Short-chain 3-Hydroxyacyl-CoA Dehydrogenase (SCHAD) Deficiency. Long-chain 3-Hydroxyacyl-CoA Dehydrogenase (LCHAD)
35 Carnitine Transporter Deficiency (CTD). Although most of the fatty acid oxidation disorders affect the heart, skeletal muscle and liver, cardiac failure is seen as the major presentation in CTD. Over half of the known cases of CTD first present with progressive heart failure and generalized muscle weakness. During the first years of life, extended fasting stress may provoke an attack of hypoketotic hypoglycemic and coma. This may lead to sudden unexpected infant death. Management: The outcome is usually very good with carnitine therapy. Without carnitine therapy, the cardiac failure can progress rapidly to death.
36 Medium-chain Acyl-CoA Dehydrogenase (MCAD) Deficiency. This is the most common fatty acid oxidation disorder. It is also one of the least severe, with no evidence of chronic muscle or cardiac involvement. Affected individuals appear to be entirely normal until an episode of illness is provoked by an excessive period of fasting. Hypoglycemia develops because of excessive glucose utilization due to the inability to switch to fat as a source of energy. Severe symptoms of lethargy and nausea develop in association with the marked increase in plasma fatty acids.
37 Carnitine/Acylcarnitine Translocase (TRANS) Deficiency Symptoms include : fasting hypoketotic hypoglycemia Coma cardiopulmonary arrest ventricular arrhythmias.
38 Very-long-chain Acyl-CoA Dehydrogenase (VLCAD) Deficiency. Many of the patients with VLCAD deficiency have severe clinical manifestations, including : chronic cardiomyopathy weakness fasting coma.
39 ORGANIC ACIDURIAS They are disorders associated with metabolism of fatty acids, branched chain amino acids, aromatic amino acids and citric acid cycle. They are all characterised by the accumulation of organic acids in body tissues and their excretion in urine. The patients present with acidosis, vomiting, convulsions and coma. The children often die in infancy; in case they survive, there is severe mental and growth retardation. Diagnosis is confirmed by showing the presence of organic acids in urine by chromatography. Management : Dietary restriction, co- enzyme therapy and substrate removal are the general lines of management.

40 SOME IMPORTANT ORGANIC ACIDURIAS Marple syrup Branched chain ketoacid convulsions, mental Urine disease dehydrogenase retardation, acidosis, vomiting, coma

41 Fig: position of some defective enzymes in organic acidurias Several other products
42 MITOCHONDRIAL DISEASES Mitochondrial diseases are group of disorders caused by mutations in mitochondrial DNA (mtdna) or in nuclear genes that code for mitochondrial proteins. SYMPTOMS Poor growth, muscle weakness, heart problems, liver disease, kidney disease, respiratory disorders, neurological problems. Mitochondria diseases are worse when the defective mitochondria are present in the cells of muscles, cerebrum or nerves, because these cells use more energy than most other cells in the body. Recall that mitochondria are double membrane cellular organelles, responsible for producing cellular energy (ATP) by oxidative phosphorylation.
43 EXAMPLES OF MITOCHONDRIAL DISEASES Mitochondrial myopathy (disease of muscles) Leber s hereditary optic neuropathy (LHON) usually characterized by deterioration in vision Leigh syndrome: clinical features include encephalopathy (disorder that affects the brain), seizures, dementia. Mitochondrial encephalomyopathy with lactic acidosis and stroke-like episodes (MELAS)
44 DIAGNOSIS Genetic testing: Although high levels of mutations can be detected in blood, it is always advisable to use muscle for detecting mtdna mutations, because it contains much mitochondria. MANAGEMENT Coenzyme Q and creatine supplementation. Coenzyme Q plays important role in oxidative phosphorylation. Creatine enhances ATP production in muscle cells.
45 UREA CYCLE DISORDERS The urea cycle disorders (UCD) result from mutations in genes that code for enzymes of urea cycle. This leads to defects in the metabolism of the extra nitrogen produced by the breakdown of protein and other nitrogen-containing molecules. Severe deficiency or total absence of activity of any of the first four enzymes (CPSI, OTC, ASS, ASL) in the urea cycle or the cofactor producer (NAGS) results in the accumulation of ammonia and other precursor metabolites during the first few days of life.

46 Fig: urea cycle
47 Symptoms of Newborns with Urea Cycle Defects Normal appearance at birth Irritability progressing to lethargy and later coma Loss of thermoregulation (hypothermia) Neurologic posturing (from cerebral edema) Seizures Hyperventilation and then hypoventilation
48 Examples are: CPS1 (Carbamoylphosphate Synthetase I) Deficiency ASS (Argininosuccinate Synthetase) Deficiency, ASL (Argininosuccinate Lyase) Deficiency, ARG (Arginase) Deficiency NAGS (N-Acetylglutamate Synthetase) Deficiency OTC (Ornithine Transcarbamylase) Deficiency
49 DIAGNOSIS The most important step in diagnosing urea cycle disorders is clinical suspicion of hyperammonemia. A blood ammonia level is the first laboratory test in evaluating a patient with UCD Enzymatic and genetic diagnosis MANAGEMENT Fluids, dextrose to mitigate dehydration Remove protein from intake Dialysis is very effective for the removal of ammonia from blood.
50 LSDs frequently involve the central nervous system (CNS), where neuronal dysfunction or loss results in progressive neurodegeneration and premature death. LYSOSOMAL STORAGE DISEASES (LSD) Lysosomes are involved in the degradation and recycling of extracellular material (via endocytosis) and intracellular material (via autophagy). Lysosomal storage diseases (LSDs) describe a group of rare inherited metabolic disorders that result from the defect in lysosomal hydrolases or transporters, resulting in the progressive accumulation of undigested material in lysosomes. The accumulation of these substances affects the function of lysosomes and other organelles, resulting in secondary alterations such as impairment of autophagy, mitochondrial dysfunction, inflammation and apoptosis.
51 Gaucher s disease Gaucher's disease is a sphingolipidosis resulting from glucocerebrosidase deficiency, causing deposition of glucocerebroside and related compounds. Type I (non-neuronopathic) is the adult form of the disease. Onset ranges from age 2 yr to late adulthood. Type I Gaucher's disease cause anemia, fatigue, lung impairment and kidney impairment.
52 Type II (acute neuronopathic) is very rare and residual enzyme activity in this type is lowest. Onset occurs during infancy. Type II Gaucher's disease causes severe neurological disorder, liver enlargement and spleen enlargement. Type III (subacute neuronopathic) falls between types I and II in incidence, enzyme activity and clinical severity. Type III Gaucher's disease cause progressive brain damage and seizures. Diagnosis : is by enzyme analysis of WBCs. Treatment: Enzyme replacement with placental or recombinant glucocerebrosidase is effective in types I and III; there is no treatment for type II disease. An oral treatment drug for Gaucher's disease (Zavesca/Miglustat) was licensed by the European authorities in April 2003 for those patients deemed unsuitable for enzyme replacement[
53 Tay-sachs disease Gangliosides are complex sphingolipids present in the brain. There are 2 major forms, GM1 and GM2. Taysachs disease is caused by the deficiency of hexosaminidase A (an enzyme that breaks down GM2) results in accumulation of GM2 in the brain. Children develop progressive cognitive and motor deterioration resulting in seizures, mental retardation and paralysis. Diagnosis is clinical and can be confirmed by enzyme assay. Treatment : Hexosaminidase A from human urine infused intravenously in patients with Tay-Sachs disease results in 43% reduction in the elevated quantity of globoside in the circulation.
54 Niemann-pick disease Niemann-Pick disease is a rare inherited autosomal recessive disease cause by defect in acid sphingomyelinase, this results in accumulation of sphingomyelin in lysosomes. Type A Niemann-Pick disease is a severe neurodegenerative disorder of infancy that leads to death by age 3 years. Patients have enlarged liver and spleen. Type B disease has a later age at onset, little or no neurologic involvement, and survival of most patients into adulthood. Patients also have enlarged liver and spleen.. Diagnosis: Prenatal diagnosis of Niemann-Pick disease types A and B is routinely accomplished by sphingomyelinase assay. Treatment: At present no specific treatment is available for patients with any Niemann-Pick disease subtypes and treatment is symptomatic. Bone marrow transplantation in a patient with Niemann-Pick disease type B was successful in reducing spleen and liver volumes
55 Sandhoff s disease Sandhoff s disease is caused by the combined deficiency of hexosaminidase A and B. Clinical manifestations include progressive cerebral degeneration beginning at 6 months accompanied by seizures, blindness, cherry-red macular spot and hyperacusis (a hearing disorder in which patients cannot tolerate normal environmental sound). Diagnosis involves a test to measure enzyme activity of the two hexosaminidase. If the enzyme activity results indicate that there is no hexosaminidase activity, it means that the patient has Sandhoff disease. However, if there is still B subunit activity, then this indicates that the patient might have Tay-Sachs disease.
56 BLOOD AS A TISSUE Blood is a liquid connective tissue The total blood volume makes up about 6-8 percent of the body s weight A 70kg person has approximately 5 to 6 litres of blood. Normal ph of blood is 7.35(venous blood)-7.45 (arterial blood)
57 BLOOD COMPOSITION Blood consists of: Liquid plasma (volume 55-60%) Formed elements or cells (volume 40-45) The formed elements include red blood cells, white blood cells and platelets
58 ERYTHROCYTES Red blood cells or erythrocytes are the most abundant type of blood cell They are involved in the transport of oxygen and carbon dioxide. They lack nucleus and mitochondria in order to accommodate maximum space for hemoglobin
59 LEUKOCYTES Leukocytes or white blood cells have nuclei The major function of leukocytes is protective function, they provide immunity and thus defends the body Lymphocytes, neutrophils, monocytes, eosinophils and basophils are types of lymphocytes
60 PLATELETS These are tiny cell fragments They play critical roles in blood clotting They are produced in the bone marrow by large cells called megakarocytes
61 SERUM AND PLASMA Serum is the liquid portion of blood obtained after centrifugation of coagulated blood. Serum therefore lacks clotting factors. Plasma on the other hand is the liquid portion of blood obtained by centrifuging blood sample that has been treated with anticoagulant (such as EDTA, heparin). Plasma therefore has clotting factors.
62
63 FUNCTIONS OF BLOOD TRANSPORTATION of dissolved gases (Carbon dioxide and oxygen), nutrients, hormones and metabolic wastes. PROTECTION Platelets in the blood minimize blood loss when a blood vessel is damaged White blood cells protect the body against infectious diseases
64 REGULATION Blood regulates ph and electrolyte composition of the body blood regulates body temperature
INBORN ERRORS OF METABOLISM (IEM) IAP UG Teaching slides
 IAP UG Teaching slides") INBORN ERRORS OF METABOLISM (IEM) 1 OBJECTIVES What are IEMs? Categories When to suspect? History and clinical pointers Metabolic presentation Differential diagnosis Emergency and long term management
INBORN ERRORS OF METABOLISM (IEM) 1 OBJECTIVES What are IEMs? Categories When to suspect? History and clinical pointers Metabolic presentation Differential diagnosis Emergency and long term management
Metabolic Disorders. Chapter Thomson - Wadsworth
 Metabolic Disorders Chapter 28 1 Metabolic Disorders Inborn errors of metabolism group of diseases that affect a wide variety of metabolic processes; defective processing or transport of amino acids, fatty
Metabolic Disorders Chapter 28 1 Metabolic Disorders Inborn errors of metabolism group of diseases that affect a wide variety of metabolic processes; defective processing or transport of amino acids, fatty
PROTEIN METABOLISM: SPECIFIC WAYS OF AMINO ACIDS CATABOLISM AND SYNTHESIS
 PROTEIN METABOLISM: SPECIFIC WAYS OF AMINO ACIDS CATABOLISM AND SYNTHESIS SPECIFIC WAYS OF AMINO ACID CATABOLISM After removing of amino group the carbon skeletons of amino acids are transformed into metabolic
PROTEIN METABOLISM: SPECIFIC WAYS OF AMINO ACIDS CATABOLISM AND SYNTHESIS SPECIFIC WAYS OF AMINO ACID CATABOLISM After removing of amino group the carbon skeletons of amino acids are transformed into metabolic
7 Medical Genetics. Hemoglobinopathies. Hemoglobinopathies. Protein and Gene Structure. and Biochemical Genetics
 SESSION 7 Medical Genetics Hemoglobinopathies and Biochemical Genetics J a v a d F a s a J a m s h i d i U n i v e r s i t y o f M e d i c a l S c i e n c e s, N o v e m b e r 2 0 1 7 Hemoglobinopathies
SESSION 7 Medical Genetics Hemoglobinopathies and Biochemical Genetics J a v a d F a s a J a m s h i d i U n i v e r s i t y o f M e d i c a l S c i e n c e s, N o v e m b e r 2 0 1 7 Hemoglobinopathies
Newborn Screen & Development Facts about the genetic diseases new since March 2006 (Excluding Cystic Fibrosis)
") Newborn Screen & Development Facts about the genetic diseases new since March 2006 (Excluding Cystic Fibrosis) 1) Argininosuccinic acidemia (ASA) a) Incidence: ~1 in 70,000 b) Deficiency in an enzyme of
Newborn Screen & Development Facts about the genetic diseases new since March 2006 (Excluding Cystic Fibrosis) 1) Argininosuccinic acidemia (ASA) a) Incidence: ~1 in 70,000 b) Deficiency in an enzyme of
HEREDITARY METABOLIC DISEASES
 HEREDITARY METABOLIC DISEASES Particular risk factors are: Advanced maternal age (e.g. Down's syndrome) Family history of inherited diseases (e.g. fragile X syndrome, Huntington's chorea) Previous child
HEREDITARY METABOLIC DISEASES Particular risk factors are: Advanced maternal age (e.g. Down's syndrome) Family history of inherited diseases (e.g. fragile X syndrome, Huntington's chorea) Previous child
Inborn Error Of Metabolism :
 Inborn Error Of Metabolism : Inborn Error Of Metabolism inborn error of metabolism are a large group of hereditary biochemical diseases in which specific gene mutation cause abnormal or missing proteins
Inborn Error Of Metabolism : Inborn Error Of Metabolism inborn error of metabolism are a large group of hereditary biochemical diseases in which specific gene mutation cause abnormal or missing proteins
Amino Acid Oxidation and the Urea Cycle
 Amino Acid Oxidation and the Urea Cycle Amino Acids: Final class of biomolecules whose oxidation contributes significantly to the generation of energy Undergo oxidation in three metabolic circumstances
Amino Acid Oxidation and the Urea Cycle Amino Acids: Final class of biomolecules whose oxidation contributes significantly to the generation of energy Undergo oxidation in three metabolic circumstances
Jana Novotná, Bruno Sopko. Department of the Medical Chemistry and Clinical Biochemistry The 2nd Faculty of Medicine, Charles Univ.
 Amino acid metabolism II. Urea cycle Jana Novotná, Bruno Sopko Department of the Medical Chemistry and Clinical Biochemistry The 2nd Faculty of Medicine, Charles Univ. Nitrogen balance Tissue proteins
Amino acid metabolism II. Urea cycle Jana Novotná, Bruno Sopko Department of the Medical Chemistry and Clinical Biochemistry The 2nd Faculty of Medicine, Charles Univ. Nitrogen balance Tissue proteins
Urea Cycle Defects. Dr Mick Henderson. Biochemical Genetics Leeds Teaching Hospitals Trust. MetBioNet IEM Introductory Training
 Urea Cycle Defects Dr Mick Henderson Biochemical Genetics Leeds Teaching Hospitals Trust The Urea Cycle The urea cycle enables toxic ammonia molecules to be converted to the readily excreted and non toxic
Urea Cycle Defects Dr Mick Henderson Biochemical Genetics Leeds Teaching Hospitals Trust The Urea Cycle The urea cycle enables toxic ammonia molecules to be converted to the readily excreted and non toxic
TRANSAMINATION AND UREA CYCLE
 TRANSAMINATION AND UREA CYCLE USMAN SUMO FRIEND TAMBUNAN ARLI ADITYA PARIKESIT SEPTIANA BIOINFORMATICS GROUP DEPARTEMENT OF CHEMISTRY FACULTY OF MATHEMATIC AND SCIENCE UNIVERSITY OF INDONESIA What is transamination?
TRANSAMINATION AND UREA CYCLE USMAN SUMO FRIEND TAMBUNAN ARLI ADITYA PARIKESIT SEPTIANA BIOINFORMATICS GROUP DEPARTEMENT OF CHEMISTRY FACULTY OF MATHEMATIC AND SCIENCE UNIVERSITY OF INDONESIA What is transamination?
SYNTHESIS OF NON-ESSENTIAL AMINO ACIDS [LIPPINCOTT S ] Deeba S. Jairajpuri
![SYNTHESIS OF NON-ESSENTIAL AMINO ACIDS [LIPPINCOTT S ] Deeba S. Jairajpuri](/thumbs/80/80689583.jpg "SYNTHESIS OF NON-ESSENTIAL AMINO ACIDS [LIPPINCOTT S ] Deeba S. Jairajpuri") SYNTHESIS OF NON-ESSENTIAL AMINO ACIDS [LIPPINCOTT S 267-274] Deeba S. Jairajpuri TYPES OF AMINO ACIDS Amino acids that cannot be synthesized by the human body and are required to be taken in the diet
SYNTHESIS OF NON-ESSENTIAL AMINO ACIDS [LIPPINCOTT S 267-274] Deeba S. Jairajpuri TYPES OF AMINO ACIDS Amino acids that cannot be synthesized by the human body and are required to be taken in the diet
Inborn Errors of Metabolism (IEM)
") Clinical Presentation Inborn Errors of Metabolism (IEM) Click on the following: - Clinical Pearl - link to movie clip - link to picture Investigations Blood Work Urine No Acidosis NH 4 + Metabolic Acidosis
Clinical Presentation Inborn Errors of Metabolism (IEM) Click on the following: - Clinical Pearl - link to movie clip - link to picture Investigations Blood Work Urine No Acidosis NH 4 + Metabolic Acidosis
Module : Clinical correlates of disorders of metabolism Block 3, Week 2
 Module : Clinical correlates of disorders of metabolism Block 3, Week 2 Department of Paediatrics and Child Health University of Pretoria Tutor : Prof DF Wittenberg : dwittenb@medic.up.ac.za Aim of this
Module : Clinical correlates of disorders of metabolism Block 3, Week 2 Department of Paediatrics and Child Health University of Pretoria Tutor : Prof DF Wittenberg : dwittenb@medic.up.ac.za Aim of this
The breakdown of fats to provide energy occurs in segregated membrane-bound compartments
 CPT1a deficiency The breakdown of fats to provide energy occurs in segregated membrane-bound compartments of the cell known as mitochondria. Carnitine palmitoyltransferase Ia (CPT1a) is a protein that
CPT1a deficiency The breakdown of fats to provide energy occurs in segregated membrane-bound compartments of the cell known as mitochondria. Carnitine palmitoyltransferase Ia (CPT1a) is a protein that
Very-long-chain acyl-coa dehydrogenase deficiency
 Very-long-chain acyl-coa dehydrogenase deficiency Introductory information Written by: V. Prietsch & P. Burgard Reviewed & Revised for North America by: S. van Calcar Very-long-chain acyl-coa dehydrogenase
Very-long-chain acyl-coa dehydrogenase deficiency Introductory information Written by: V. Prietsch & P. Burgard Reviewed & Revised for North America by: S. van Calcar Very-long-chain acyl-coa dehydrogenase
Routine Newborn Screening, Testing the Newborn Inherited Metabolic Disorders Update August 2015
 Routine Newborn Screening, Testing the Newborn Inherited Metabolic Disorders Update August 2015 Metabolic birth defects can cause physical problems, mental retardation and, in some cases, death. It is
Routine Newborn Screening, Testing the Newborn Inherited Metabolic Disorders Update August 2015 Metabolic birth defects can cause physical problems, mental retardation and, in some cases, death. It is
BCM 317 LECTURE OJEMEKELE O.
 BCM 317 LECTURE BY OJEMEKELE O. JAUNDICE Jaundice is yellowish discoloration of the skin, sclera and mucous membrane, resulting from an increased bilirubin concentration in the body fluid. It is usually
BCM 317 LECTURE BY OJEMEKELE O. JAUNDICE Jaundice is yellowish discoloration of the skin, sclera and mucous membrane, resulting from an increased bilirubin concentration in the body fluid. It is usually
Metabolism of pentoses, glycogen, fructose and galactose. Jana Novotna
 Metabolism of pentoses, glycogen, fructose and galactose Jana Novotna 1. The Pentose Phosphate Pathway The pentose phosphate pathway (PPP): (hexose monophosphate or 6-phosphogluconate patway) Process that
Metabolism of pentoses, glycogen, fructose and galactose Jana Novotna 1. The Pentose Phosphate Pathway The pentose phosphate pathway (PPP): (hexose monophosphate or 6-phosphogluconate patway) Process that
Amino acid Catabolism
 Enzymatic digestion of dietary proteins in gastrointestinal-tract. Amino acid Catabolism Amino acids: 1. There are 20 different amino acid, they are monomeric constituents of proteins 2. They act as precursors
Enzymatic digestion of dietary proteins in gastrointestinal-tract. Amino acid Catabolism Amino acids: 1. There are 20 different amino acid, they are monomeric constituents of proteins 2. They act as precursors
Medical Foods for Inborn Errors of Metabolism
 Medical Foods for Inborn Errors of Metabolism Policy Number: Original Effective Date: MM.02.014 02/18/2000 Line(s) of Business: Current Effective Date: HMO; PPO; QUEST 08/23/2013 Section: Medicine Place(s)
Medical Foods for Inborn Errors of Metabolism Policy Number: Original Effective Date: MM.02.014 02/18/2000 Line(s) of Business: Current Effective Date: HMO; PPO; QUEST 08/23/2013 Section: Medicine Place(s)
Carnitine palmitoyl transferase 2 deficiency (CPT2) is a rare inherited disorder that occurs when
 is a rare inherited disorder that occurs when") CPT2 Deficiency Carnitine palmitoyl transferase 2 deficiency (CPT2) is a rare inherited disorder that occurs when the last step in the entry of fats into sac-like bodies called mitochondria is blocked.
CPT2 Deficiency Carnitine palmitoyl transferase 2 deficiency (CPT2) is a rare inherited disorder that occurs when the last step in the entry of fats into sac-like bodies called mitochondria is blocked.
Lecture 10 - Protein Turnover and Amino Acid Catabolism
 Lecture 10 - Protein Turnover and Amino Acid Catabolism Chem 454: Regulatory Mechanisms in Biochemistry University of Wisconsin-Eau Claire 1 Introduction 2 Proteins are degraded into amino acids. Protein
Lecture 10 - Protein Turnover and Amino Acid Catabolism Chem 454: Regulatory Mechanisms in Biochemistry University of Wisconsin-Eau Claire 1 Introduction 2 Proteins are degraded into amino acids. Protein
CHY2026: General Biochemistry UNIT 7& 8: CARBOHYDRATE METABOLISM
 CHY2026: General Biochemistry UNIT 7& 8: CARBOHYDRATE METABOLISM Metabolism Bioenergetics is the transfer and utilization of energy in biological systems The direction and extent to which a chemical reaction
CHY2026: General Biochemistry UNIT 7& 8: CARBOHYDRATE METABOLISM Metabolism Bioenergetics is the transfer and utilization of energy in biological systems The direction and extent to which a chemical reaction
number Done by Corrected by Doctor Nayef Karadsheh
 number 16 Done Huda shaheen by Corrected by حسام أبو عوض Doctor Nayef Karadsheh 1 In the previous lecture, we talked about glycogen metabolism and regulation. In this sheet we will talk about the metabolism
number 16 Done Huda shaheen by Corrected by حسام أبو عوض Doctor Nayef Karadsheh 1 In the previous lecture, we talked about glycogen metabolism and regulation. In this sheet we will talk about the metabolism
Fatty Acids Synthesis L3
 Fatty Acids Synthesis L3 The pathway for fatty acid synthesis occurs in the cytoplasm, whereas, oxidation occurs in the mitochondria. The other major difference is the use of nucleotide co-factors. Oxidation
Fatty Acids Synthesis L3 The pathway for fatty acid synthesis occurs in the cytoplasm, whereas, oxidation occurs in the mitochondria. The other major difference is the use of nucleotide co-factors. Oxidation
Urea Cycle Disorders Prior Authorization Program Summary
 Urea Cycle Disorders Prior Authorization Program Summary FDA APPROVED INDICATIONS DOSAGE 1,2 Agent(s) Indication(s) Dosing -Adjunctive therapy in the chronic management of patients with urea cycle disorders
Urea Cycle Disorders Prior Authorization Program Summary FDA APPROVED INDICATIONS DOSAGE 1,2 Agent(s) Indication(s) Dosing -Adjunctive therapy in the chronic management of patients with urea cycle disorders
AMINOACID METABOLISM FATE OF AMINOACIDS & UREA CYCLE
 AMINOACID METABOLISM FATE OF AMINOACIDS & UREA CYCLE SOURCE & FATE OF AA The aminoacids obtained from DIETARY SOURCE or BODY PROTEIN TURNOVER are utilized for protein biosynthesis and the production of
AMINOACID METABOLISM FATE OF AMINOACIDS & UREA CYCLE SOURCE & FATE OF AA The aminoacids obtained from DIETARY SOURCE or BODY PROTEIN TURNOVER are utilized for protein biosynthesis and the production of
The molecule that serves as the major source of readily available body fuel is: a. fat. b. glucose. c. acetyl CoA. d. cellulose.
 The molecule that serves as the major source of readily available body fuel is: a. fat. b. glucose. c. acetyl CoA. d. cellulose. Dietary fats are important because: a. they keep blood pressure normal.
The molecule that serves as the major source of readily available body fuel is: a. fat. b. glucose. c. acetyl CoA. d. cellulose. Dietary fats are important because: a. they keep blood pressure normal.
Urea is the major end product of nitrogen catabolism in humans One nitrogen free NH3 other nitrogen aspartate. carbon oxygen CO2 liver,
 Urea is the major end product of nitrogen catabolism in humans Urea is the major disposal form of amino groups derived from amino acids, and accounts about 90% percent of the nitrogencontaining components
Urea is the major end product of nitrogen catabolism in humans Urea is the major disposal form of amino groups derived from amino acids, and accounts about 90% percent of the nitrogencontaining components
Integration Of Metabolism
 Integration Of Metabolism Metabolism Consist of Highly Interconnected Pathways The basic strategy of catabolic metabolism is to form ATP, NADPH, and building blocks for biosyntheses. 1. ATP is the universal
Integration Of Metabolism Metabolism Consist of Highly Interconnected Pathways The basic strategy of catabolic metabolism is to form ATP, NADPH, and building blocks for biosyntheses. 1. ATP is the universal
number Done by Corrected by Doctor
 number 33 Done by Omar Sami Corrected by Waseem abu obeida Doctor Diala Too late for second guessing, too late to go back to sleep 1 P age Just try defying gravity This sheet will be divided to two parts,
number 33 Done by Omar Sami Corrected by Waseem abu obeida Doctor Diala Too late for second guessing, too late to go back to sleep 1 P age Just try defying gravity This sheet will be divided to two parts,
Newborn Screening & Methods for Diagnosing Inborn Errors of Metabolism
 Newborn Screening & Methods for Diagnosing Inborn Errors of Metabolism Patricia Jones, PhD DABCC FACB UT Southwestern Medical Center Children s Medical Center Dallas, Texas Learning Objectives Justify
Newborn Screening & Methods for Diagnosing Inborn Errors of Metabolism Patricia Jones, PhD DABCC FACB UT Southwestern Medical Center Children s Medical Center Dallas, Texas Learning Objectives Justify
AMINO ACID METABOLISM
 AMINO ACID METABOLISM Synthesis of Urea in Liver The series of reactions that form urea is known as the Urea Cycle or the Krebs-Henseleit Cycle. The urea cycle operates only to eliminate excess nitrogen.
AMINO ACID METABOLISM Synthesis of Urea in Liver The series of reactions that form urea is known as the Urea Cycle or the Krebs-Henseleit Cycle. The urea cycle operates only to eliminate excess nitrogen.
Metabolic Changes in ASD. Norma J. Arciniegas, MD Simón E. Carlo, MD Instituto Filius
 Metabolic Changes in ASD Norma J. Arciniegas, MD Simón E. Carlo, MD Instituto Filius 12 patients 3 Autism: Ages 3/3/3.7 3 PDD: Ages 3/3/6 3 Asperger: Ages 6/7/15.1 3 Speech delay and Sensory Problems (SHL):
Metabolic Changes in ASD Norma J. Arciniegas, MD Simón E. Carlo, MD Instituto Filius 12 patients 3 Autism: Ages 3/3/3.7 3 PDD: Ages 3/3/6 3 Asperger: Ages 6/7/15.1 3 Speech delay and Sensory Problems (SHL):
Biochemistry: A Short Course
 Tymoczko Berg Stryer Biochemistry: A Short Course Second Edition CHAPTER 16 Glycolysis 2013 W. H. Freeman and Company Chapter 16 Outline Why is glucose such a prominent fuel in all life forms? 1. Glucose
Tymoczko Berg Stryer Biochemistry: A Short Course Second Edition CHAPTER 16 Glycolysis 2013 W. H. Freeman and Company Chapter 16 Outline Why is glucose such a prominent fuel in all life forms? 1. Glucose
e-learning Fatty Acid Oxidation Defects Camilla Reed and Dr Simon Olpin Sheffield Children s Hospital
 e-learning Fatty Acid Oxidation Defects Camilla Reed and Dr Simon Olpin Sheffield Children s Hospital Fatty Acids Fatty acids are a major source of energy and body fat is an energy dense material. They
e-learning Fatty Acid Oxidation Defects Camilla Reed and Dr Simon Olpin Sheffield Children s Hospital Fatty Acids Fatty acids are a major source of energy and body fat is an energy dense material. They
Genética e Hígado: Cómo contribuye la genética en el algoritmo diagnóstico de la enfermedad hepática pediátrica? Nicholas Ah Mew, MD
 Genética e Hígado: Cómo contribuye la genética en el algoritmo diagnóstico de la enfermedad hepática pediátrica? Nicholas Ah Mew, MD April 24, 2017 SAP 2017 Buenos Aires Genetic disorders are rare why
Genética e Hígado: Cómo contribuye la genética en el algoritmo diagnóstico de la enfermedad hepática pediátrica? Nicholas Ah Mew, MD April 24, 2017 SAP 2017 Buenos Aires Genetic disorders are rare why
Medium-chain acyl-coa dehydrogenase deficiency
 Medium-chain acyl-coa dehydrogenase deficiency Introductory information Written by: V. Prietsch & P. Burgard Reviewed & Revised for North America by: S. van Calcar Medium-chain acyl-coa dehydrogenase MCAD
Medium-chain acyl-coa dehydrogenase deficiency Introductory information Written by: V. Prietsch & P. Burgard Reviewed & Revised for North America by: S. van Calcar Medium-chain acyl-coa dehydrogenase MCAD
HA Convention 2016 Master course How to Handle Abnormal Newborn Metabolic Screening Results Causes, Management and Follow up
 HA Convention 2016 Master course How to Handle Abnormal Newborn Metabolic Screening Results Causes, Management and Follow up Dr. Josephine Chong Clinical Professional Consultant Centre of Inborn Errors
HA Convention 2016 Master course How to Handle Abnormal Newborn Metabolic Screening Results Causes, Management and Follow up Dr. Josephine Chong Clinical Professional Consultant Centre of Inborn Errors
Glycogen Storage Disease
 Glycogen Storage Disease 1 Introduction The food we eat is usually used for growth, tissue repair and energy. The body stores what it does not use. Excess sugar, or glucose, is stored as glycogen in the
Glycogen Storage Disease 1 Introduction The food we eat is usually used for growth, tissue repair and energy. The body stores what it does not use. Excess sugar, or glucose, is stored as glycogen in the
Conversion of amino acids الفريق الطبي األكاديمي
 الفريق الطبي األكاديمي لكية الطب البرشي البلقاء التطبيقية / املركز 2022 2016/ -the ones with * are the essential amino acids- - Histidine decarboxylation -> histamine Page 1 - Glutamine is a nitrogen carrier
الفريق الطبي األكاديمي لكية الطب البرشي البلقاء التطبيقية / املركز 2022 2016/ -the ones with * are the essential amino acids- - Histidine decarboxylation -> histamine Page 1 - Glutamine is a nitrogen carrier
Newborn screening for congenital metabolic diseases Optional out-of-pocket tests Information Sheet
 Newborn screening for congenital metabolic diseases Optional out-of-pocket tests Information Sheet Website:www.tipn.org.tw Telephone:(02)85962050 Ext. 401-403 Service line:(02)85962065 Fax:(02)85962067
Newborn screening for congenital metabolic diseases Optional out-of-pocket tests Information Sheet Website:www.tipn.org.tw Telephone:(02)85962050 Ext. 401-403 Service line:(02)85962065 Fax:(02)85962067
Connections of Carbohydrate, Protein, and Lipid Metabolic Pathways
 OpenStax-CNX module: m44441 1 Connections of Carbohydrate, Protein, and Lipid Metabolic Pathways OpenStax College This work is produced by OpenStax-CNX and licensed under the Creative Commons Attribution
OpenStax-CNX module: m44441 1 Connections of Carbohydrate, Protein, and Lipid Metabolic Pathways OpenStax College This work is produced by OpenStax-CNX and licensed under the Creative Commons Attribution
Nitrogen Metabolism. Overview
 Nitrogen Metabolism Pratt and Cornely Chapter 18 Overview Nitrogen assimilation Amino acid biosynthesis Nonessential aa Essential aa Nucleotide biosynthesis Amino Acid Catabolism Urea Cycle Juicy Steak
Nitrogen Metabolism Pratt and Cornely Chapter 18 Overview Nitrogen assimilation Amino acid biosynthesis Nonessential aa Essential aa Nucleotide biosynthesis Amino Acid Catabolism Urea Cycle Juicy Steak
METABOLISM OF ACYLGLYCEROLS AND SPHINGOLIPDS. Ben S. Ashok MSc.,FAGE.,PhD., Dept. of Biochemistry
 METABOLISM OF ACYLGLYCEROLS AND SPHINGOLIPDS Ben S. Ashok MSc.,FAGE.,PhD., Dept. of Biochemistry STORAGE AND MEMBRANE LIPIDS STORAGE LIPIDS Mainly as triacylglycerols (triglycerides) in adipose cells Constitute
METABOLISM OF ACYLGLYCEROLS AND SPHINGOLIPDS Ben S. Ashok MSc.,FAGE.,PhD., Dept. of Biochemistry STORAGE AND MEMBRANE LIPIDS STORAGE LIPIDS Mainly as triacylglycerols (triglycerides) in adipose cells Constitute
The spectrum and outcome of the. neonates with inborn errors of. metabolism at a tertiary care hospital
 The spectrum and outcome of the neonates with inborn errors of metabolism at a tertiary care hospital Dr. Sevim Ünal Neonatology Division, Ankara Children s Hematology Oncology Research Hospital, Ankara,
The spectrum and outcome of the neonates with inborn errors of metabolism at a tertiary care hospital Dr. Sevim Ünal Neonatology Division, Ankara Children s Hematology Oncology Research Hospital, Ankara,
Biochemistry: A Short Course
 Tymoczko Berg Stryer Biochemistry: A Short Course Second Edition CHAPTER 30 Amino Acid Degradation and the Urea Cycle 2013 W. H. Freeman and Company Chapter 30 Outline Amino acids are obtained from the
Tymoczko Berg Stryer Biochemistry: A Short Course Second Edition CHAPTER 30 Amino Acid Degradation and the Urea Cycle 2013 W. H. Freeman and Company Chapter 30 Outline Amino acids are obtained from the
Nutritional Management of Inborn Errors of Metabolism. Kay Davis, RD, CSP Esther Berenhaut, RD, CSP, CSR Aug 28, 2017
 Nutritional Management of Inborn Errors of Metabolism Kay Davis, RD, CSP Esther Berenhaut, RD, CSP, CSR Aug 28, 2017 OBJECTIVES Brief overview of newborn screening of metabolic disorders, inheritance patterns.
Nutritional Management of Inborn Errors of Metabolism Kay Davis, RD, CSP Esther Berenhaut, RD, CSP, CSR Aug 28, 2017 OBJECTIVES Brief overview of newborn screening of metabolic disorders, inheritance patterns.
Disorders of Carbohydrates. Disorders of Galactose Metabolism Glycogen Storage Diseases Diabetes Mellitus
 Disorders of Carbohydrates Metabolism Disorders of Galactose Metabolism Glycogen Storage Diseases Diabetes Mellitus Disorders of Galactose Metabolism GALACTOSE Galactose is a sugar that is found mainly
Disorders of Carbohydrates Metabolism Disorders of Galactose Metabolism Glycogen Storage Diseases Diabetes Mellitus Disorders of Galactose Metabolism GALACTOSE Galactose is a sugar that is found mainly
Inborn Errors of Metabolism. Metabolic Pathway. Digestion and Fasting. How is Expanded Newborn Screening Different? MS/MS. The body is a factory.
 Inborn Errors of Metabolism The body is a factory. Inborn errors of metabolism are rare genetic disorders in which the body cannot properly turn food into energy. The disorders are usually caused by defects
Inborn Errors of Metabolism The body is a factory. Inborn errors of metabolism are rare genetic disorders in which the body cannot properly turn food into energy. The disorders are usually caused by defects
Published on Second Faculty of Medicine, Charles University (http://www.lf2.cuni.cz )
") Published on Second Faculty of Medicine, Charles University (http://www.lf2.cuni.cz ) Biochemistry Submitted by Marie Havlová on 8. February 2012-0:00 Syllabus of Biochemistry Mechanisms of enzyme catalysis.
Published on Second Faculty of Medicine, Charles University (http://www.lf2.cuni.cz ) Biochemistry Submitted by Marie Havlová on 8. February 2012-0:00 Syllabus of Biochemistry Mechanisms of enzyme catalysis.
LOW CITRULLINE AS A MARKER FOR THE PROXIMAL UREA CYCLE DEFECTS EXPERIENCE OF THE NEW ENGLAND NEWBORN SCREENING PROGRAM
 LOW CITRULLINE AS A MARKER FOR THE PROXIMAL UREA CYCLE DEFECTS EXPERIENCE OF THE NEW ENGLAND NEWBORN SCREENING PROGRAM Inderneel Sahai, MD, FACMG Newborn Screening and Genetic Testing Symposium Oct 2014
LOW CITRULLINE AS A MARKER FOR THE PROXIMAL UREA CYCLE DEFECTS EXPERIENCE OF THE NEW ENGLAND NEWBORN SCREENING PROGRAM Inderneel Sahai, MD, FACMG Newborn Screening and Genetic Testing Symposium Oct 2014
ESPEN Congress Madrid 2018
 ESPEN Congress Madrid 2018 Inborn Errors Of Metabolism Urea Cycle Disorders Diagnosis And Care F. Feillet (FR) Urea cycle disorders, diagnosis and care F Feillet National reference centre for Inborn errors
ESPEN Congress Madrid 2018 Inborn Errors Of Metabolism Urea Cycle Disorders Diagnosis And Care F. Feillet (FR) Urea cycle disorders, diagnosis and care F Feillet National reference centre for Inborn errors
Chapter 24 Lecture Outline
 Chapter 24 Lecture Outline Carbohydrate Lipid and Protein! Metabolism! In the catabolism of carbohydrates, glycolysis converts glucose into pyruvate, which is then metabolized into acetyl CoA. Prepared
Chapter 24 Lecture Outline Carbohydrate Lipid and Protein! Metabolism! In the catabolism of carbohydrates, glycolysis converts glucose into pyruvate, which is then metabolized into acetyl CoA. Prepared
NITROGEN METABOLISM An Overview
 1 University of Papua New Guinea School of Medicine and Health Sciences Division of Basic Medical Sciences Discipline of Biochemistry and Molecular Biology PBL Seminar & Health Sciences NITROGEN METABOLISM
1 University of Papua New Guinea School of Medicine and Health Sciences Division of Basic Medical Sciences Discipline of Biochemistry and Molecular Biology PBL Seminar & Health Sciences NITROGEN METABOLISM
Newborn Screening in Manitoba. Information for Health Care Providers
 Newborn Screening in Manitoba Information for Health Care Providers Newborn screening: a healthy start leads to a healthier life Health care professionals have provided newborn screening for phenylketonuria
Newborn Screening in Manitoba Information for Health Care Providers Newborn screening: a healthy start leads to a healthier life Health care professionals have provided newborn screening for phenylketonuria
AMINO ACID METABOLISM. Sri Widia A Jusman Dept. of Biochemistry & Molecular Biology FMUI
 AMINO ACID METABOLISM Sri Widia A Jusman Dept. of Biochemistry & Molecular Biology FMUI Amino acids derived from dietary protein absorbed from intestine through blood taken up by tissues used for biosynthesis
AMINO ACID METABOLISM Sri Widia A Jusman Dept. of Biochemistry & Molecular Biology FMUI Amino acids derived from dietary protein absorbed from intestine through blood taken up by tissues used for biosynthesis
Fatty Acid Oxidation Disorders
 Genetic Fact Sheets for Parents Fatty Acid Oxidation Disorders Screening, Technology, and Research in Genetics is a multi-state project to improve information about the financial, ethical, legal, and social
Genetic Fact Sheets for Parents Fatty Acid Oxidation Disorders Screening, Technology, and Research in Genetics is a multi-state project to improve information about the financial, ethical, legal, and social
Nutrients. Chapter 25 Nutrition, Metabolism, Temperature Regulation
 Chapter 25 Nutrition, Metabolism, Temperature Regulation 25-1 Nutrients Chemicals used by body to produce energy, provide building blocks or function in other chemical reactions Classes Carbohydrates,
Chapter 25 Nutrition, Metabolism, Temperature Regulation 25-1 Nutrients Chemicals used by body to produce energy, provide building blocks or function in other chemical reactions Classes Carbohydrates,
Presentation and investigation of mitochondrial disease in children
 Presentation and investigation of mitochondrial disease in children Andrew Morris Willink Unit, Manchester Mitochondrial function Carbohydrate Fat Respiratory chain Energy Mitochondria are the product
Presentation and investigation of mitochondrial disease in children Andrew Morris Willink Unit, Manchester Mitochondrial function Carbohydrate Fat Respiratory chain Energy Mitochondria are the product
BY: RASAQ NURUDEEN OLAJIDE
 BY: RASAQ NURUDEEN OLAJIDE AROMATIC AMINO ACID DEGRADATION The aromatic amino acids are those containing aromatic or benzene ring in their side chains Examples include phenylalanine, tyrosine and tryptophan
BY: RASAQ NURUDEEN OLAJIDE AROMATIC AMINO ACID DEGRADATION The aromatic amino acids are those containing aromatic or benzene ring in their side chains Examples include phenylalanine, tyrosine and tryptophan
Cellular Respiration
 Cellular Respiration 1. To perform cell work, cells require energy. a. A cell does three main kinds of work: i. Mechanical work, such as the beating of cilia, contraction of muscle cells, and movement
Cellular Respiration 1. To perform cell work, cells require energy. a. A cell does three main kinds of work: i. Mechanical work, such as the beating of cilia, contraction of muscle cells, and movement
Carbohydrate Metabolism
 Chapter 34 Carbohydrate Metabolism Carbohydrate metabolism is important for both plants and animals. Introduction to General, Organic, and Biochemistry, 10e John Wiley & Sons, Inc Morris Hein, Scott Pattison,
Chapter 34 Carbohydrate Metabolism Carbohydrate metabolism is important for both plants and animals. Introduction to General, Organic, and Biochemistry, 10e John Wiley & Sons, Inc Morris Hein, Scott Pattison,
Fatty acid oxidation. Naomi Rankin
 Fatty acid oxidation Naomi Rankin Fatty acid oxidation Provides energy to muscles from lipid stores, spares glucose for the brain Lipolysis of triglycerides results in FFA, mainly C16 and C18 FA oxidation
Fatty acid oxidation Naomi Rankin Fatty acid oxidation Provides energy to muscles from lipid stores, spares glucose for the brain Lipolysis of triglycerides results in FFA, mainly C16 and C18 FA oxidation
3 HYDROXY 3 METHYLGLUTARYL CoA (3 HMG CoA) LYASE DEFICIENCY RECOMMENDATIONS ON EMERGENCY MANAGEMENT OF METABOLIC DISEASES
 LYASE DEFICIENCY RECOMMENDATIONS ON EMERGENCY MANAGEMENT OF METABOLIC DISEASES") 3 HYDROXY 3 METHYLGLUTARYL CoA (3 HMG CoA) LYASE DEFICIENCY RECOMMENDATIONS ON EMERGENCY MANAGEMENT OF METABOLIC DISEASES Patient s name: Date of birth: Please read carefully. Meticulous and prompt treatment
3 HYDROXY 3 METHYLGLUTARYL CoA (3 HMG CoA) LYASE DEFICIENCY RECOMMENDATIONS ON EMERGENCY MANAGEMENT OF METABOLIC DISEASES Patient s name: Date of birth: Please read carefully. Meticulous and prompt treatment
Integration of Metabolism 1. made by: Noor M. ALnairat. Sheet No. 18
 Integration of Metabolism 1 made by: Noor M. ALnairat Sheet No. 18 Data :24/11/2016 SLIDE 2: Metabolism Consist of Highly Interconnected Pathways The basic strategy of catabolic metabolism is to form ATP,
Integration of Metabolism 1 made by: Noor M. ALnairat Sheet No. 18 Data :24/11/2016 SLIDE 2: Metabolism Consist of Highly Interconnected Pathways The basic strategy of catabolic metabolism is to form ATP,
ANATOMY OF A METABOLIC CRISIS: FAOD-style. Mark S. Korson, MD Tufts Medical Center Boston, MA
 ANATOMY OF A METABOLIC CRISIS: FAOD-style Mark S. Korson, MD Tufts Medical Center Boston, MA NORMAL PHYSIOLOGY Anabolic Eating well Calories eaten > body s needs BRAIN uses GLUCOSE MUSCLE uses GLUCOSE
ANATOMY OF A METABOLIC CRISIS: FAOD-style Mark S. Korson, MD Tufts Medical Center Boston, MA NORMAL PHYSIOLOGY Anabolic Eating well Calories eaten > body s needs BRAIN uses GLUCOSE MUSCLE uses GLUCOSE
Newborn Screening: Blood Spot Disorders
 Newborn Screening: Blood Spot Disorders Arizona s Newborn Screening Program Program Overview Panel of Disorders Disorder Descriptions Program Components Hospitals ADHS Lab ADHS Follow-up ADHS Billing Medical
Newborn Screening: Blood Spot Disorders Arizona s Newborn Screening Program Program Overview Panel of Disorders Disorder Descriptions Program Components Hospitals ADHS Lab ADHS Follow-up ADHS Billing Medical
Fatty Acid Oxidation Disorders
 Genetic Fact Sheets for Parents Fatty Acid Oxidation Disorders Screening, Technology, and Research in Genetics is a multi-state project to improve information about the financial, ethical, legal, and social
Genetic Fact Sheets for Parents Fatty Acid Oxidation Disorders Screening, Technology, and Research in Genetics is a multi-state project to improve information about the financial, ethical, legal, and social
Chemistry 1120 Exam 4 Study Guide
 Chemistry 1120 Exam 4 Study Guide Chapter 12 12.1 Identify and differentiate between macronutrients (lipids, amino acids and saccharides) and micronutrients (vitamins and minerals). Master Tutor Section
Chemistry 1120 Exam 4 Study Guide Chapter 12 12.1 Identify and differentiate between macronutrients (lipids, amino acids and saccharides) and micronutrients (vitamins and minerals). Master Tutor Section
Amino Acid Metabolism
 Amino Acid Metabolism The continuous degradation and synthesis of cellular proteins occur in all forms of life. Each day humans turn over 1 2% of their total body protein, principally muscle protein. Approximately
Amino Acid Metabolism The continuous degradation and synthesis of cellular proteins occur in all forms of life. Each day humans turn over 1 2% of their total body protein, principally muscle protein. Approximately
Medical Policy An independent licensee of the Blue Cross Blue Shield Association
 Urea Cycle Disorders Page 1 of 7 Medical Policy An independent licensee of the Blue Cross Blue Shield Association Title: Urea Cycle Disorders Prime Therapeutics will review Prior Authorization requests
Urea Cycle Disorders Page 1 of 7 Medical Policy An independent licensee of the Blue Cross Blue Shield Association Title: Urea Cycle Disorders Prime Therapeutics will review Prior Authorization requests
Biochemistry: A Short Course
 Tymoczko Berg Stryer Biochemistry: A Short Course Second Edition CHAPTER 30 Amino Acid Degradation and the Urea Cycle 2013 W. H. Freeman and Company In the cytosol of a cell amino groups from amino acids
Tymoczko Berg Stryer Biochemistry: A Short Course Second Edition CHAPTER 30 Amino Acid Degradation and the Urea Cycle 2013 W. H. Freeman and Company In the cytosol of a cell amino groups from amino acids
Suspected Metabolic Disease in the Newborn Period Acute Management "What do I do?" Barbara Marriage, PhD RD Abbott Nutrition
 Suspected Metabolic Disease in the Newborn Period Acute Management "What do I do?" Barbara Marriage, PhD RD Abbott Nutrition Introduction Review clinical findings that may be suspicious of a metabolic
Suspected Metabolic Disease in the Newborn Period Acute Management "What do I do?" Barbara Marriage, PhD RD Abbott Nutrition Introduction Review clinical findings that may be suspicious of a metabolic
Human inherited diseases
 Human inherited diseases A genetic disorder that is caused by abnormality in an individual's DNA. Abnormalities can range from small mutation in a single gene to the addition or subtraction of a whole
Human inherited diseases A genetic disorder that is caused by abnormality in an individual's DNA. Abnormalities can range from small mutation in a single gene to the addition or subtraction of a whole
Higher Biology. Unit 2: Metabolism and Survival Topic 2: Respiration. Page 1 of 25
 Higher Biology Unit 2: Metabolism and Survival Topic 2: Respiration Page 1 of 25 Sub Topic: Respiration I can state that: All living cells carry out respiration. ATP is the energy currency of the cell
Higher Biology Unit 2: Metabolism and Survival Topic 2: Respiration Page 1 of 25 Sub Topic: Respiration I can state that: All living cells carry out respiration. ATP is the energy currency of the cell
MEMBRANE LIPIDS I and II: GLYCEROPHOSPHOLIPIDS AND SPHINGOLIPIDS
 December 6, 2011 Lecturer: Eileen M. Lafer MEMBRANE LIPIDS I and II: GLYCEROPHOSPHOLIPIDS AND SPHINGOLIPIDS Reading: Stryer Edition 6: Chapter 26 Images: All images in these notes were taken from Lehninger,
December 6, 2011 Lecturer: Eileen M. Lafer MEMBRANE LIPIDS I and II: GLYCEROPHOSPHOLIPIDS AND SPHINGOLIPIDS Reading: Stryer Edition 6: Chapter 26 Images: All images in these notes were taken from Lehninger,
Unit Seven Blood and Immunity
 Unit Seven Blood and Immunity I. Introduction A. Definition Blood is a sticky fluid that is heavier and thicker than water. Blood is a type of, whose cells and suspended in a liquid intercellular material.
Unit Seven Blood and Immunity I. Introduction A. Definition Blood is a sticky fluid that is heavier and thicker than water. Blood is a type of, whose cells and suspended in a liquid intercellular material.
Glycogen Storage Diseases (A group of genetic diseases)
") Glycogen Storage Diseases (A group of genetic diseases) Glycogen Storage Diseases (GSD) Inherited genetic defects related to glycogen metabolism. Glycogenosis. Characterized by deposition of glycogen in
Glycogen Storage Diseases (A group of genetic diseases) Glycogen Storage Diseases (GSD) Inherited genetic defects related to glycogen metabolism. Glycogenosis. Characterized by deposition of glycogen in
1- Which of the following statements is TRUE in regards to eukaryotic and prokaryotic cells?
 Name: NetID: Exam 3 - Version 1 October 23, 2017 Dr. A. Pimentel Each question has a value of 4 points and there are a total of 160 points in the exam. However, the maximum score of this exam will be capped
Name: NetID: Exam 3 - Version 1 October 23, 2017 Dr. A. Pimentel Each question has a value of 4 points and there are a total of 160 points in the exam. However, the maximum score of this exam will be capped
In glycolysis, glucose is converted to pyruvate. If the pyruvate is reduced to lactate, the pathway does not require O 2 and is called anaerobic
 Glycolysis 1 In glycolysis, glucose is converted to pyruvate. If the pyruvate is reduced to lactate, the pathway does not require O 2 and is called anaerobic glycolysis. If this pyruvate is converted instead
Glycolysis 1 In glycolysis, glucose is converted to pyruvate. If the pyruvate is reduced to lactate, the pathway does not require O 2 and is called anaerobic glycolysis. If this pyruvate is converted instead
BIOB111 - Tutorial activity for Session 25
 BIOB111 - Tutorial activity for Session 25 General topics for week 14 Session 25 The metabolism of proteins Students are asked to draw the concept map showing all details of protein metabolism 1 Instructions:
BIOB111 - Tutorial activity for Session 25 General topics for week 14 Session 25 The metabolism of proteins Students are asked to draw the concept map showing all details of protein metabolism 1 Instructions:
Summary. Syndromic versus Etiologic. Definitions. Why does it matter? ASD=autism
 Summary It is becoming clear that multiple genes with complex interactions underlie autism spectrum (ASD). A small subset of people with ASD, however, actually suffer from rare single-gene Important to
Summary It is becoming clear that multiple genes with complex interactions underlie autism spectrum (ASD). A small subset of people with ASD, however, actually suffer from rare single-gene Important to
Lynne A. Wolfe, MS, ACNP, PNP, BC Department of Genetics Yale School of Medicine
 Lynne A. Wolfe, MS, ACNP, PNP, BC Department of Genetics Yale School of Medicine Harvey Levy, MD Mark Korson, MD Piero Rinaldo, MD, PhD Larry Sweetman, PhD K. Michael Gibson, PhD Charlie Roe, MD Jerry
Lynne A. Wolfe, MS, ACNP, PNP, BC Department of Genetics Yale School of Medicine Harvey Levy, MD Mark Korson, MD Piero Rinaldo, MD, PhD Larry Sweetman, PhD K. Michael Gibson, PhD Charlie Roe, MD Jerry
Abdallah Q& Razi. Faisal
 27 & Ahmad Attari م ح م د ي وس ف Abdallah Q& Razi Faisal Sphingophospolipids - The backbone of sphingophospholipids is sphingosine, unlike glycerophospholipids with a glycerol as the backbone. Which contains
27 & Ahmad Attari م ح م د ي وس ف Abdallah Q& Razi Faisal Sphingophospolipids - The backbone of sphingophospholipids is sphingosine, unlike glycerophospholipids with a glycerol as the backbone. Which contains
Overview of AMINO ACIDS
 Overview of AMINO ACIDS Amino Acids are the chemical units or "building blocks" of the body that make up proteins. Protein substances make up the muscles, tendons, organs, glands, nails, and hair. Growth,
Overview of AMINO ACIDS Amino Acids are the chemical units or "building blocks" of the body that make up proteins. Protein substances make up the muscles, tendons, organs, glands, nails, and hair. Growth,
Most common is the congenital adrenogenital syndrome (AGS) or congenital adrenal hyperplasia (CAH) due to 21-hydroxylase deficiency.
 or congenital adrenal hyperplasia (CAH) due to 21-hydroxylase deficiency.") Newborn Screening Examination parameters: TSH-neonatal (hypothyreosis), 17-OH progesterone (AGS), galactose (galactosemia), galactose-uridyl transferase (galacto semia), biotinidase (biotinidase ), phenylalanine
Newborn Screening Examination parameters: TSH-neonatal (hypothyreosis), 17-OH progesterone (AGS), galactose (galactosemia), galactose-uridyl transferase (galacto semia), biotinidase (biotinidase ), phenylalanine
CHEMISTRY OF LIFE 30 JANUARY 2013
 CHEMISTRY OF LIFE 30 JANUARY 2013 Lesson Description In this lesson, we will: Investigate the structure and function of molecules that are essential for life. Key Concepts Terminology A molecule is any
CHEMISTRY OF LIFE 30 JANUARY 2013 Lesson Description In this lesson, we will: Investigate the structure and function of molecules that are essential for life. Key Concepts Terminology A molecule is any
National Metabolic Biochemistry Network Guidelines for the investigation of hypoglycaemia in infants and children
 National Metabolic Biochemistry Network Guidelines for the investigation of hypoglycaemia in infants and children Aim To provide guidance on the biochemical investigation of hypoglycaemia in infants and
National Metabolic Biochemistry Network Guidelines for the investigation of hypoglycaemia in infants and children Aim To provide guidance on the biochemical investigation of hypoglycaemia in infants and
Citric Acid Cycle and Oxidative Phosphorylation
 Citric Acid Cycle and Oxidative Phosphorylation Page by: OpenStax Summary The Citric Acid Cycle In eukaryotic cells, the pyruvate molecules produced at the end of glycolysis are transported into mitochondria,
Citric Acid Cycle and Oxidative Phosphorylation Page by: OpenStax Summary The Citric Acid Cycle In eukaryotic cells, the pyruvate molecules produced at the end of glycolysis are transported into mitochondria,
So Much More Than The PKU Test
 Newborn Metabolic Screening So Much More Than The PKU Test Sarah Viall, MSN, PPCNP BC Newborn Screening Program Coordinator Division of Genetics & Metabolism Conflicts of Interest I have no conflicts of
Newborn Metabolic Screening So Much More Than The PKU Test Sarah Viall, MSN, PPCNP BC Newborn Screening Program Coordinator Division of Genetics & Metabolism Conflicts of Interest I have no conflicts of
Metabolism. Metabolic pathways. BIO 5099: Molecular Biology for Computer Scientists (et al) Lecture 11: Metabolic Pathways
 Lecture 11: Metabolic Pathways") BIO 5099: Molecular Biology for Computer Scientists (et al) Lecture 11: Metabolic Pathways http://compbio.uchsc.edu/hunter/bio5099 Larry.Hunter@uchsc.edu Metabolism Metabolism is the chemical change of
BIO 5099: Molecular Biology for Computer Scientists (et al) Lecture 11: Metabolic Pathways http://compbio.uchsc.edu/hunter/bio5099 Larry.Hunter@uchsc.edu Metabolism Metabolism is the chemical change of
Fatty Acid Oxidation Disorders Organic Acid Disorders
 Genetic Fact Sheets for Parents Fatty Acid Oxidation Disorders Organic Acid Disorders Screening, Technology, and Research in Genetics is a multi-state project to improve information about the financial,
Genetic Fact Sheets for Parents Fatty Acid Oxidation Disorders Organic Acid Disorders Screening, Technology, and Research in Genetics is a multi-state project to improve information about the financial,
Metabolism. Chapter 5. Catabolism Drives Anabolism 8/29/11. Complete Catabolism of Glucose
 8/29/11 Metabolism Chapter 5 All of the reactions in the body that require energy transfer. Can be divided into: Cell Respiration and Metabolism Anabolism: requires the input of energy to synthesize large
8/29/11 Metabolism Chapter 5 All of the reactions in the body that require energy transfer. Can be divided into: Cell Respiration and Metabolism Anabolism: requires the input of energy to synthesize large
AMINO ACID METABOLISM
 AMINO ACID METABOLISM PHL-285 Biochemistry-2 Mahmoud N. Nagi, Ph.D. Professor of Biochemistry Overview of amino acid metabolism. Classification of amino acids. Biosynthesis of nonessential amino acids.
AMINO ACID METABOLISM PHL-285 Biochemistry-2 Mahmoud N. Nagi, Ph.D. Professor of Biochemistry Overview of amino acid metabolism. Classification of amino acids. Biosynthesis of nonessential amino acids.
NBCE Mock Board Questions Biochemistry
 1. Fluid mosaic describes. A. Tertiary structure of proteins B. Ribosomal subunits C. DNA structure D. Plasma membrane structure NBCE Mock Board Questions Biochemistry 2. Where in the cell does beta oxidation
1. Fluid mosaic describes. A. Tertiary structure of proteins B. Ribosomal subunits C. DNA structure D. Plasma membrane structure NBCE Mock Board Questions Biochemistry 2. Where in the cell does beta oxidation
Structure. Lysosomes are membrane-enclosed organelles. Hydrolytic enzymes. Variable in size & shape need
 Lysosomes Structure Lysosomes are membrane-enclosed organelles Hydrolytic enzymes Variable in size & shape need Degrade material taken up from outside and inside the cell Variable in size and shape Lysosomal
Lysosomes Structure Lysosomes are membrane-enclosed organelles Hydrolytic enzymes Variable in size & shape need Degrade material taken up from outside and inside the cell Variable in size and shape Lysosomal
Nitrogen Metabolism. Pratt and Cornely Chapter 18
 Nitrogen Metabolism Pratt and Cornely Chapter 18 Overview Nitrogen assimilation Amino acid biosynthesis Nonessential aa Essential aa Nucleotide biosynthesis Amino Acid Catabolism Urea Cycle Juicy Steak
Nitrogen Metabolism Pratt and Cornely Chapter 18 Overview Nitrogen assimilation Amino acid biosynthesis Nonessential aa Essential aa Nucleotide biosynthesis Amino Acid Catabolism Urea Cycle Juicy Steak
Salicylate (Aspirin) Ingestion California Poison Control Background 1. The prevalence of aspirin-containing analgesic products makes
 Ingestion California Poison Control Background 1. The prevalence of aspirin-containing analgesic products makes") Salicylate (Aspirin) Ingestion California Poison Control 1-800-876-4766 Background 1. The prevalence of aspirin-containing analgesic products makes these agents, found in virtually every household, common
Salicylate (Aspirin) Ingestion California Poison Control 1-800-876-4766 Background 1. The prevalence of aspirin-containing analgesic products makes these agents, found in virtually every household, common