Inborn Errors of Metabolism (IEM)
|
|
|
- Diane Emily Barker
- 5 years ago
- Views:
Transcription





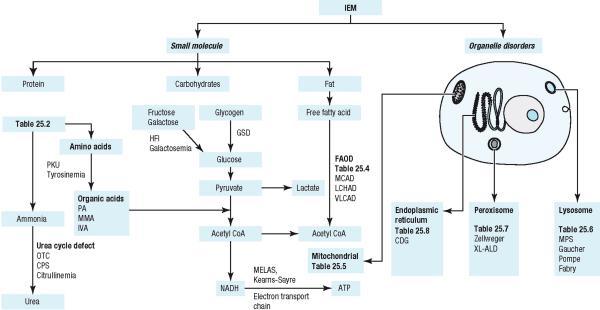
1 Clinical Presentation Inborn Errors of Metabolism (IEM) Click on the following: - Clinical Pearl - link to movie clip - link to picture Investigations Blood Work Urine No Acidosis NH 4 + Metabolic Acidosis No NH 4 + Hypoglycemia General Treatment
2 Inborn Errors of Metabolism Individually rare, collectively common ~1 : Inherent deficiency in a key metabolic pathway: Carbohydrate vs. Fat vs. Protein Pathophysiology : Build up of toxic products cellular intoxication Cellular energy deprivation Combination of the above
3
4 Clinical Presentation Infant Looks like sepsis! Lethargy Hypotonia Decreased level of consciousness (LOC) Seizures Poor feeding Vomiting Sudden deterioration Children Mental retardation Regression Failure to thrive Coarse facial features Skeletal abnormalities Hernias Dietary aversion (protein/carbs) Deterioration after: New foods introduced Fasting
5 Blood Work Blood Work CBC+ Diff Blood Culture Blood gas Rationale/Predicted Result -Rule out sepsis -Pancytopenia (finding in some types of IEM) -Metabolic Acidosis -Alkalosis (seen initially in urea cycle defects from high NH 4 + ) Electrolytes + Ca/Mg/Phos Blood glucose Serum ketones NH + 4 (ammonia) Lactate AST, ALT, AlkPhos, GGT, -Signs of dehydration -Electrolyte derangements can have similar presentation -Hypoglycemia -Hydroxybutyrate: should be present if patient hypoglycemic - *** NO tourniquet, send on ice and process immediately *** NO tourniquet, send on ice and process immediately -Elevated liver enzymes bili, albumin, coags Acyl-carnitine profile, (Pyruvate) *** NO tourniquet, send on ice and process immediately -Pyruvate: only done if lactate high (only needed for pyruvate/ lactate ratio), need Biochemical/genetics lab approval + need special collection tubes. Plasma amino acids Newborn screen Must call to unlock the results
6 Newborn Screen (BC) 22 disorders tested in British Columbia Other provinces test for a spectrum Pertinent to IEM: Amino acid disorders PKU MSUD Citrullinemia Agininosuccinic Aciduria Homocystinuria **Tyrosinemia Type 1 - (not yet included but possibly added soon!) Fatty acid oxidation defects VLCAD LCHAD (Not LCAD- Long-chain 3-hydroxyacyl-CoA dehydrogenase) ****TFP (trifunctional protein deficiency) severe form of LCHAD MCAD Organic Acid Defects PROP (Propionic acidemia) MUT (Methylmalonic Acidemia) Cobalamin Disorders Glutaric Aciduria, Type I Isovaleric Acidemia Galactosemia Galactosemia
7 PKU - Cause: phenylalanine hydroxylase enzyme deficiency - Ddx: rare forms of BH 4 Cofactor deficiency - mousy odour - Light complexion - Eczema - Mental retardation and hyperactivity
8 MSUD - 1 st week of life - Smells sweet - Periods of hypertonicity - Secondary hypoglycemia - Ketosis
9 Tyrosinemia Type 1 - Smells of boiled cabbage
10 MCAD - Encephalopathy - Hypoglycemia
11 Isovaleric Acidemia - Smells like sweaty feet
12 Glutaric Aciduria, Type 1 Subdural bleeds Needs to be included in the non-accidental injury work up
13 Galactosemia Mechanism: Deficiency of galactose-1-phosphate uridyl transferase Leads to galactose-1-phosphate Clinical presentation: ~end of 1 st, start of 2 nd week of life Recurrent Escherichia coli sepsis Galactose-1-phosphate is toxic to the liver: **Jaundice Vomiting Diarrhea Poor weight gain Cataracts Investigations: Urine for reducing substances: looking for non-glucose reducing substances Treatment: Stop galactose containing feeds Only time an infant needs SOY formula Only ISOMIL will work!
14 Homocystinuria ***Increased risk of stroke Clinical Presentation: Marfanoid appearance Dislocated lens etc. Mental retardation Treatment: pyridoxine
15 Urine Urine Tests Urinalysis Rationale/Predicted Result - Rule out sepsis - Ketones - In infants, should only be present if hypoglycemic! Urine Culture - Rule out sepsis Urine organic acids Urine reducing substances -Useful in galactosemia (non-glucose reducing substances), fructose intolerance, tyrosinemia
16 General Treatment Treatment STOP protein intake Rationale -Urea cycle defects/organic acid/amino acid defect -Prophree formula (has no protein) IV glucose infusion rate of 8-10 mmol/kg/min) - Treats hypoglycemia - Provides calories to prevent protein catabolism - May need insulin to control glucose levels **may add IV lipids if no signs of fatty acid oxidation defect IV arginine (6ml/kg 10% arginine HCl over 90 min) IV sodium benzoate -Essential amino acid -Helps to excrete the excess nitrogen waste products in Argininosuccinic Aciduria and Citrullinemia -Increases excretion of ammonia by activating alternate pathways IV phenylacetate IV carnitine -Fatty acid oxidation defects: treats possible carnitine deficiency -Organic acidurias: binds toxic organic acids for excretion in urine Cofactors/vitamin Specific treatments Hemodialysis -Organic acidemia: hydroxycobalamin, biotin -Please see individual disorders for specific treatments -Arrange with ICU/Neprhology -Recall reasons for dialysis (AEIOU) -Acid/Base disorders, Electrolyte disorders (including Ammonia!), Intoxication, Overload, Uremic encephalopathy
")



17 NH 4 + Presentation <24 hours old ph N - (possible 1 o respiratory alkalosis) Normal lactate NH + 4 Acidosis Anion gap ± Lactate ±Ketosis Hypoglycemia No Ketosis LFTs NH + 4 ± Hypoglycemia NH 4 + Transient Hyperammonemia of the Newborn Urea Cycle Defect Organic Acidemia Fatty Acid Oxidation Defect (Most likely )
18 Transient Hyperammonemia Clinical Presentation: Extremely sick of the Newborn Usually large premature neonate (~36wks) Symptomatic pulmonary disease (respiratory distress) Caused by compensatory respiratory alkalosis Does not recur! Treatment: Follow treatment for NH 4 +
19 Urea Cycle Defect Ornithine Transcarbamylase Deficiency The only X-linked urea cycle defect All other urea cycle defects are autosomal recessive
20 Fatty Acid Oxidation Defect Two mechanisms: Impaired capacity to use stored fat as fuel in periods of fasting with depleted glycogen stores Defect in carnitine which transports fatty acids during oxidation Recall Defects lead to a decrease in Acetyl CoA and therefore ketone production Thus presentation is most often a non ketotic hypoglycemia Screening: Urine Organic Acids Plasma acyl-carnitine profile Examples Treatment: IV glucose Carnitine supplementation (except in LCHAD where carnitine can lead to accumulation of toxins that cause severe arrhythmias and cardiomyopathy)
21 Examples VLCAD = Very Long Chain Acyl-CoA Dehydrogenase Deficiency LCHAD = Long-chain 3-hydroxyacyl-CoA Dehydrogenase Deficiency MCAD = Medium Chain Acyl-CoA Dehydrogenase Deficiency Clinical presentation: Non ketotic hypoglycemia Reye like reaction ALTE/Sudden death May have family history of SIDS Fat accumulation in liver or muscle
22 Organic Acidemia 4 key examples: Organic Acid Defect Treatment (cofactor/vitamin to residual enzyme activity) Methylmalonic Acidemia Hydroxocobalamin(B12) Proprionic Acidemia Multiple carboxylase PO/NG Biotin PO/NG Biotin Glutaric acidemia type 1 Riboflavin

 (No ketosis, Normal")


23 Metabolic Acidosis Normal Anion Gap NOT IEM!!! Increased Anion Gap (MUDPILES) (No ketosis, Normal Lactate) - RTA - Diarrhea Acidosis Anion gap ± Lactate ±Ketosis ± Hypoglycemia NH + 4 Lactate Normal Glucose Organic Acidemia Mitochondrial Disorders
24 Organic Acidemia 4 key examples: Organic Acid Defect Treatment (cofactor/vitamin to residual enzyme activity) Methylmalonic Acidemia Hydroxocobalamin(B12) Proprionic Acidemia Multiple carboxylase PO/NG Biotin PO/NG Biotin Glutaric acidemia type 1 Riboflavin
25 (MUDPILES) anion gap In IEM, hidden anions are lactate or organic acids M Methanol U D P I L E S Uremia DKA Peraldehyde Isoniazid, Iron Lactate Ethylene Glycol Salicylates
26 No Acidosis No NH 4 + Non Ketotic Hyperglycinemia Homocystinuria
27 Non-Ketotic Hyperglycinemia Clinical Presentation Severe and progressive encephalopathy Uncontrolled hiccups and apnea Myoclonic seizures Hypotonia Investigations: No urine/serum ketones Increased glycine in blood and CSF
28 Homocystinuria ***Increased risk of stroke Clinical Presentation: Marfanoid appearance Dislocated lens etc. Mental retardation Treatment: pyridoxine





29 Hypoglycemia Acidosis ± Lactate Hypoglycemia Anion gap Hepatomegaly Liver Failure ±Ketosis ± Lactate LFTs ±Ketosis NH 4 + ± Hypoglycemia NH 4 + Organic Acidemia Glycogen Storage Disorder (GSD) **Note: some exceptions! Tyrosinemias GSD IV Galactosemia Neimann Pick Fatty Acid Oxidation Defects Hyperinsulinemia (Type C)
30 Organic Acidemia 4 key examples: Organic Acid Defect Treatment (cofactor/vitamin to residual enzyme activity) Methylmalonic Acidemia Hydroxocobalamin(B12) Proprionic Acidemia Multiple carboxylase PO/NG Biotin PO/NG Biotin Glutaric acidemia type 1 Riboflavin

31 Fatty Acid Oxidation Defect Two mechanisms: Impaired capacity to use stored fat as fuel in periods of fasting with depleted glycogen stores Defect in carnitine which transports fatty acids during oxidation Recall Defects lead to a decrease in Acetyl CoA and therefore ketone production Thus presentation is most often a non ketotic hypoglycemia Screening: Urine Organic Acids Plasma acyl-carnitine profile Examples Treatment: IV glucose Carnitine supplementation (except in LCHAD where carnitine can lead to accumulation of toxins that cause severe arrhythmias and cardiomyopathy)
32 Glycogen Storage Disorder (GSD) Mechanism: Unable to release glucose from glycogen Several types assigned #s based on order of discovery Liver types: Type 0, I, III, VI, IX Hypoglycemia is presenting symptom Muscle types: Type II (Pompe), V, VII Presents with rhabdomyolysis and weakness
33 Galactosemia Mechanism: Deficiency of galactose-1-phosphate uridyl transferase Leads to galactose-1-phosphate Clinical presentation: ~end of 1 st, start of 2 nd week of life Recurrent Escherichia coli sepsis Galactose-1-phosphate is toxic to the liver: **Jaundice Vomiting Diarrhea Poor weight gain Cataracts Investigations: Urine for reducing substances: looking for non-glucose reducing substances Treatment: Stop galactose containing feeds Only time an infant needs SOY formula Only ISOMIL will work!
34
INBORN ERRORS OF METABOLISM (IEM) IAP UG Teaching slides
 IAP UG Teaching slides") INBORN ERRORS OF METABOLISM (IEM) 1 OBJECTIVES What are IEMs? Categories When to suspect? History and clinical pointers Metabolic presentation Differential diagnosis Emergency and long term management
INBORN ERRORS OF METABOLISM (IEM) 1 OBJECTIVES What are IEMs? Categories When to suspect? History and clinical pointers Metabolic presentation Differential diagnosis Emergency and long term management
Metabolic Disorders. Chapter Thomson - Wadsworth
 Metabolic Disorders Chapter 28 1 Metabolic Disorders Inborn errors of metabolism group of diseases that affect a wide variety of metabolic processes; defective processing or transport of amino acids, fatty
Metabolic Disorders Chapter 28 1 Metabolic Disorders Inborn errors of metabolism group of diseases that affect a wide variety of metabolic processes; defective processing or transport of amino acids, fatty
Newborn Screen & Development Facts about the genetic diseases new since March 2006 (Excluding Cystic Fibrosis)
") Newborn Screen & Development Facts about the genetic diseases new since March 2006 (Excluding Cystic Fibrosis) 1) Argininosuccinic acidemia (ASA) a) Incidence: ~1 in 70,000 b) Deficiency in an enzyme of
Newborn Screen & Development Facts about the genetic diseases new since March 2006 (Excluding Cystic Fibrosis) 1) Argininosuccinic acidemia (ASA) a) Incidence: ~1 in 70,000 b) Deficiency in an enzyme of
Suspected Metabolic Disease in the Newborn Period Acute Management "What do I do?" Barbara Marriage, PhD RD Abbott Nutrition
 Suspected Metabolic Disease in the Newborn Period Acute Management "What do I do?" Barbara Marriage, PhD RD Abbott Nutrition Introduction Review clinical findings that may be suspicious of a metabolic
Suspected Metabolic Disease in the Newborn Period Acute Management "What do I do?" Barbara Marriage, PhD RD Abbott Nutrition Introduction Review clinical findings that may be suspicious of a metabolic
Metabolic Changes in ASD. Norma J. Arciniegas, MD Simón E. Carlo, MD Instituto Filius
 Metabolic Changes in ASD Norma J. Arciniegas, MD Simón E. Carlo, MD Instituto Filius 12 patients 3 Autism: Ages 3/3/3.7 3 PDD: Ages 3/3/6 3 Asperger: Ages 6/7/15.1 3 Speech delay and Sensory Problems (SHL):
Metabolic Changes in ASD Norma J. Arciniegas, MD Simón E. Carlo, MD Instituto Filius 12 patients 3 Autism: Ages 3/3/3.7 3 PDD: Ages 3/3/6 3 Asperger: Ages 6/7/15.1 3 Speech delay and Sensory Problems (SHL):
Routine Newborn Screening, Testing the Newborn Inherited Metabolic Disorders Update August 2015
 Routine Newborn Screening, Testing the Newborn Inherited Metabolic Disorders Update August 2015 Metabolic birth defects can cause physical problems, mental retardation and, in some cases, death. It is
Routine Newborn Screening, Testing the Newborn Inherited Metabolic Disorders Update August 2015 Metabolic birth defects can cause physical problems, mental retardation and, in some cases, death. It is
Inborn Errors of Metabolism in the Emergency Department. Will Davies June 2014
 Inborn Errors of Metabolism in the Emergency Department Will Davies June 2014 Inborn Errors of Metabolism in the Emergency Department Overview Although individually rare, altogether they are 1:1000-2500
Inborn Errors of Metabolism in the Emergency Department Will Davies June 2014 Inborn Errors of Metabolism in the Emergency Department Overview Although individually rare, altogether they are 1:1000-2500
The spectrum and outcome of the. neonates with inborn errors of. metabolism at a tertiary care hospital
 The spectrum and outcome of the neonates with inborn errors of metabolism at a tertiary care hospital Dr. Sevim Ünal Neonatology Division, Ankara Children s Hematology Oncology Research Hospital, Ankara,
The spectrum and outcome of the neonates with inborn errors of metabolism at a tertiary care hospital Dr. Sevim Ünal Neonatology Division, Ankara Children s Hematology Oncology Research Hospital, Ankara,
Medical Foods for Inborn Errors of Metabolism
 Medical Foods for Inborn Errors of Metabolism Policy Number: Original Effective Date: MM.02.014 02/18/2000 Line(s) of Business: Current Effective Date: HMO; PPO; QUEST 08/23/2013 Section: Medicine Place(s)
Medical Foods for Inborn Errors of Metabolism Policy Number: Original Effective Date: MM.02.014 02/18/2000 Line(s) of Business: Current Effective Date: HMO; PPO; QUEST 08/23/2013 Section: Medicine Place(s)
Acute Management of Sick Infants with Suspected Inborn Errors of Metabolism
 Indian J Pediatr (July 2011) 78(7):854 859 DOI 10.1007/s12098-011-0422-0 SYMPOSIUM ON PICU PROTOCOLS OF AIIMS Acute Management of Sick Infants with Suspected Inborn Errors of Metabolism Neerja Gupta Madhulika
Indian J Pediatr (July 2011) 78(7):854 859 DOI 10.1007/s12098-011-0422-0 SYMPOSIUM ON PICU PROTOCOLS OF AIIMS Acute Management of Sick Infants with Suspected Inborn Errors of Metabolism Neerja Gupta Madhulika
HA Convention 2016 Master course How to Handle Abnormal Newborn Metabolic Screening Results Causes, Management and Follow up
 HA Convention 2016 Master course How to Handle Abnormal Newborn Metabolic Screening Results Causes, Management and Follow up Dr. Josephine Chong Clinical Professional Consultant Centre of Inborn Errors
HA Convention 2016 Master course How to Handle Abnormal Newborn Metabolic Screening Results Causes, Management and Follow up Dr. Josephine Chong Clinical Professional Consultant Centre of Inborn Errors
Work-Up and Initial Management of Common Metabolic Emergencies in the Inpatient Setting
 Work-Up and Initial Management of Common Metabolic Emergencies in the Inpatient Setting Kristin Lindstrom, MD Division of Genetics and Metabolism Phoenix Children s Hospital AzAAP Pediatrics in the Red
Work-Up and Initial Management of Common Metabolic Emergencies in the Inpatient Setting Kristin Lindstrom, MD Division of Genetics and Metabolism Phoenix Children s Hospital AzAAP Pediatrics in the Red
Newborn Screening in Manitoba. Information for Health Care Providers
 Newborn Screening in Manitoba Information for Health Care Providers Newborn screening: a healthy start leads to a healthier life Health care professionals have provided newborn screening for phenylketonuria
Newborn Screening in Manitoba Information for Health Care Providers Newborn screening: a healthy start leads to a healthier life Health care professionals have provided newborn screening for phenylketonuria
Inborn Errors of Metabolism. Metabolic Pathway. Digestion and Fasting. How is Expanded Newborn Screening Different? MS/MS. The body is a factory.
 Inborn Errors of Metabolism The body is a factory. Inborn errors of metabolism are rare genetic disorders in which the body cannot properly turn food into energy. The disorders are usually caused by defects
Inborn Errors of Metabolism The body is a factory. Inborn errors of metabolism are rare genetic disorders in which the body cannot properly turn food into energy. The disorders are usually caused by defects
Newborn Screening & Methods for Diagnosing Inborn Errors of Metabolism
 Newborn Screening & Methods for Diagnosing Inborn Errors of Metabolism Patricia Jones, PhD DABCC FACB UT Southwestern Medical Center Children s Medical Center Dallas, Texas Learning Objectives Justify
Newborn Screening & Methods for Diagnosing Inborn Errors of Metabolism Patricia Jones, PhD DABCC FACB UT Southwestern Medical Center Children s Medical Center Dallas, Texas Learning Objectives Justify
3 HYDROXY 3 METHYLGLUTARYL CoA (3 HMG CoA) LYASE DEFICIENCY RECOMMENDATIONS ON EMERGENCY MANAGEMENT OF METABOLIC DISEASES
 LYASE DEFICIENCY RECOMMENDATIONS ON EMERGENCY MANAGEMENT OF METABOLIC DISEASES") 3 HYDROXY 3 METHYLGLUTARYL CoA (3 HMG CoA) LYASE DEFICIENCY RECOMMENDATIONS ON EMERGENCY MANAGEMENT OF METABOLIC DISEASES Patient s name: Date of birth: Please read carefully. Meticulous and prompt treatment
3 HYDROXY 3 METHYLGLUTARYL CoA (3 HMG CoA) LYASE DEFICIENCY RECOMMENDATIONS ON EMERGENCY MANAGEMENT OF METABOLIC DISEASES Patient s name: Date of birth: Please read carefully. Meticulous and prompt treatment
The laboratory investigation of lactic acidaemia. J Bonham/T Laing
 The laboratory investigation of lactic acidaemia J Bonham/T Laing Reference range Typical ranges for blood lactate are: Newborn 0.3-2.2 mmol/l Nielsen J et al1 1994 1-12mo 0.9-1.8 mmol/l Bonnefont et al
The laboratory investigation of lactic acidaemia J Bonham/T Laing Reference range Typical ranges for blood lactate are: Newborn 0.3-2.2 mmol/l Nielsen J et al1 1994 1-12mo 0.9-1.8 mmol/l Bonnefont et al
Introduction to Organic Acidemias. Hilary Vernon, MD PhD Assistant Professor of Genetic Medicine Johns Hopkins University 7.25.
 Introduction to Organic Acidemias Hilary Vernon, MD PhD Assistant Professor of Genetic Medicine Johns Hopkins University 7.25.2014 A Brief Historical Overview Garrod, Archibald E. 1902. The Incidence of
Introduction to Organic Acidemias Hilary Vernon, MD PhD Assistant Professor of Genetic Medicine Johns Hopkins University 7.25.2014 A Brief Historical Overview Garrod, Archibald E. 1902. The Incidence of
Inborn Errors of Metabolism Clinical Approach to Diagnosis and Treatment
 Case Scenario: 15 year old girl presented in ED with aggressive behaviour and hallucinations. No associated fever, vomiting, seizures or developmental concerns Previously well Inborn Errors of Metabolism
Case Scenario: 15 year old girl presented in ED with aggressive behaviour and hallucinations. No associated fever, vomiting, seizures or developmental concerns Previously well Inborn Errors of Metabolism
Genética e Hígado: Cómo contribuye la genética en el algoritmo diagnóstico de la enfermedad hepática pediátrica? Nicholas Ah Mew, MD
 Genética e Hígado: Cómo contribuye la genética en el algoritmo diagnóstico de la enfermedad hepática pediátrica? Nicholas Ah Mew, MD April 24, 2017 SAP 2017 Buenos Aires Genetic disorders are rare why
Genética e Hígado: Cómo contribuye la genética en el algoritmo diagnóstico de la enfermedad hepática pediátrica? Nicholas Ah Mew, MD April 24, 2017 SAP 2017 Buenos Aires Genetic disorders are rare why
Guideline for the diagnosis and management of isovaleryl-coa-dehydrogenase deficiency (isovaleric acidemia) - a systematic review -
 - a systematic review -") Guideline for the diagnosis and management of isovaleryl-coa-dehydrogenase deficiency (isovaleric acidemia) - a systematic review - Guideline development group International interdisciplinary guideline
Guideline for the diagnosis and management of isovaleryl-coa-dehydrogenase deficiency (isovaleric acidemia) - a systematic review - Guideline development group International interdisciplinary guideline
Most common is the congenital adrenogenital syndrome (AGS) or congenital adrenal hyperplasia (CAH) due to 21-hydroxylase deficiency.
 or congenital adrenal hyperplasia (CAH) due to 21-hydroxylase deficiency.") Newborn Screening Examination parameters: TSH-neonatal (hypothyreosis), 17-OH progesterone (AGS), galactose (galactosemia), galactose-uridyl transferase (galacto semia), biotinidase (biotinidase ), phenylalanine
Newborn Screening Examination parameters: TSH-neonatal (hypothyreosis), 17-OH progesterone (AGS), galactose (galactosemia), galactose-uridyl transferase (galacto semia), biotinidase (biotinidase ), phenylalanine
HEREDITARY METABOLIC DISEASES
 HEREDITARY METABOLIC DISEASES Particular risk factors are: Advanced maternal age (e.g. Down's syndrome) Family history of inherited diseases (e.g. fragile X syndrome, Huntington's chorea) Previous child
HEREDITARY METABOLIC DISEASES Particular risk factors are: Advanced maternal age (e.g. Down's syndrome) Family history of inherited diseases (e.g. fragile X syndrome, Huntington's chorea) Previous child
Positive Newborn Screens: What do you do next?
 Positive Newborn Screens: What do you do next? James B. Gibson, MD, Ph.D. Biochemical Geneticist at Specially for Children Clinical Associate Professor of Pediatrics UTHSCSA Carla R. Scott, MD Pediatric
Positive Newborn Screens: What do you do next? James B. Gibson, MD, Ph.D. Biochemical Geneticist at Specially for Children Clinical Associate Professor of Pediatrics UTHSCSA Carla R. Scott, MD Pediatric
Newborn Screening: Focus on Treatment
 Newborn Screening: Focus on Treatment Alan R. Fleischman, M.D. Senior Vice President and Medical Director March of Dimes National Conference of State Legislatures July 21, 2008 Newborn Screening: A Public
Newborn Screening: Focus on Treatment Alan R. Fleischman, M.D. Senior Vice President and Medical Director March of Dimes National Conference of State Legislatures July 21, 2008 Newborn Screening: A Public
ANATOMY OF A METABOLIC CRISIS: FAOD-style. Mark S. Korson, MD Tufts Medical Center Boston, MA
 ANATOMY OF A METABOLIC CRISIS: FAOD-style Mark S. Korson, MD Tufts Medical Center Boston, MA NORMAL PHYSIOLOGY Anabolic Eating well Calories eaten > body s needs BRAIN uses GLUCOSE MUSCLE uses GLUCOSE
ANATOMY OF A METABOLIC CRISIS: FAOD-style Mark S. Korson, MD Tufts Medical Center Boston, MA NORMAL PHYSIOLOGY Anabolic Eating well Calories eaten > body s needs BRAIN uses GLUCOSE MUSCLE uses GLUCOSE
Overview of Newborn Screening, Potential Uses of Residual Dried Blood Spots, and Protection of Privacy
 Overview of Newborn Screening, Potential Uses of Residual Dried Blood Spots, and Protection of Privacy Alan R. Fleischman, M.D. Senior Vice President and Medical Director Chair, Federal Advisory Committee,
Overview of Newborn Screening, Potential Uses of Residual Dried Blood Spots, and Protection of Privacy Alan R. Fleischman, M.D. Senior Vice President and Medical Director Chair, Federal Advisory Committee,
7 Medical Genetics. Hemoglobinopathies. Hemoglobinopathies. Protein and Gene Structure. and Biochemical Genetics
 SESSION 7 Medical Genetics Hemoglobinopathies and Biochemical Genetics J a v a d F a s a J a m s h i d i U n i v e r s i t y o f M e d i c a l S c i e n c e s, N o v e m b e r 2 0 1 7 Hemoglobinopathies
SESSION 7 Medical Genetics Hemoglobinopathies and Biochemical Genetics J a v a d F a s a J a m s h i d i U n i v e r s i t y o f M e d i c a l S c i e n c e s, N o v e m b e r 2 0 1 7 Hemoglobinopathies
ESPEN Congress Madrid 2018
 ESPEN Congress Madrid 2018 Inborn Errors Of Metabolism Urea Cycle Disorders Diagnosis And Care F. Feillet (FR) Urea cycle disorders, diagnosis and care F Feillet National reference centre for Inborn errors
ESPEN Congress Madrid 2018 Inborn Errors Of Metabolism Urea Cycle Disorders Diagnosis And Care F. Feillet (FR) Urea cycle disorders, diagnosis and care F Feillet National reference centre for Inborn errors
THE ED APPROACH OF THE CHILD WITH SUSPECTED METABOLIC DISEASE
 THE ED APPROACH OF THE CHILD WITH SUSPECTED METABOLIC DISEASE Dr. Nadeem Qureshi M.D, FAAP, FCCM Associate Professor Pediatrics School of Medicine. St Louis University Attending Physician Pediatric Emergency
THE ED APPROACH OF THE CHILD WITH SUSPECTED METABOLIC DISEASE Dr. Nadeem Qureshi M.D, FAAP, FCCM Associate Professor Pediatrics School of Medicine. St Louis University Attending Physician Pediatric Emergency
ARGININOSUCCINIC ACIDEMIA (or ARGININOSUCCINATE LYASE DEFICIENCY) RECOMMENDATIONS ON EMERGENCY MANAGEMENT OF METABOLIC DISEASES
 RECOMMENDATIONS ON EMERGENCY MANAGEMENT OF METABOLIC DISEASES") ARGININOSUCCINIC ACIDEMIA (or ARGININOSUCCINATE LYASE DEFICIENCY) RECOMMENDATIONS ON EMERGENCY MANAGEMENT OF METABOLIC DISEASES Patient s name: Date of birth: Please read carefully. Meticulous and prompt
ARGININOSUCCINIC ACIDEMIA (or ARGININOSUCCINATE LYASE DEFICIENCY) RECOMMENDATIONS ON EMERGENCY MANAGEMENT OF METABOLIC DISEASES Patient s name: Date of birth: Please read carefully. Meticulous and prompt
Clinical Management of Organic Acidemias and OAA Natural History Registry. Kim Chapman MD PhD Children s National Rare Disease Institute
 Clinical Management of Organic Acidemias and OAA Natural History Registry Kim Chapman MD PhD Children s National Rare Disease Institute Disclosure Nothing to disclose concerning this lecture Organic acid?
Clinical Management of Organic Acidemias and OAA Natural History Registry Kim Chapman MD PhD Children s National Rare Disease Institute Disclosure Nothing to disclose concerning this lecture Organic acid?
e-learning Fatty Acid Oxidation Defects Camilla Reed and Dr Simon Olpin Sheffield Children s Hospital
 e-learning Fatty Acid Oxidation Defects Camilla Reed and Dr Simon Olpin Sheffield Children s Hospital Fatty Acids Fatty acids are a major source of energy and body fat is an energy dense material. They
e-learning Fatty Acid Oxidation Defects Camilla Reed and Dr Simon Olpin Sheffield Children s Hospital Fatty Acids Fatty acids are a major source of energy and body fat is an energy dense material. They
Newborn Screening: Blood Spot Disorders
 Newborn Screening: Blood Spot Disorders Arizona s Newborn Screening Program Program Overview Panel of Disorders Disorder Descriptions Program Components Hospitals ADHS Lab ADHS Follow-up ADHS Billing Medical
Newborn Screening: Blood Spot Disorders Arizona s Newborn Screening Program Program Overview Panel of Disorders Disorder Descriptions Program Components Hospitals ADHS Lab ADHS Follow-up ADHS Billing Medical
Urea Cycle Defects. Dr Mick Henderson. Biochemical Genetics Leeds Teaching Hospitals Trust. MetBioNet IEM Introductory Training
 Urea Cycle Defects Dr Mick Henderson Biochemical Genetics Leeds Teaching Hospitals Trust The Urea Cycle The urea cycle enables toxic ammonia molecules to be converted to the readily excreted and non toxic
Urea Cycle Defects Dr Mick Henderson Biochemical Genetics Leeds Teaching Hospitals Trust The Urea Cycle The urea cycle enables toxic ammonia molecules to be converted to the readily excreted and non toxic
Title: Assessing Recommendations Related To Timeliness of Newborn Screening
 Title: Assessing Recommendations Related To Timeliness of Newborn Screening Purpose: In January 2014, the Secretary s Discretionary Advisory Committee on Heritable Diseases on Newborns and Children (Committee)
Title: Assessing Recommendations Related To Timeliness of Newborn Screening Purpose: In January 2014, the Secretary s Discretionary Advisory Committee on Heritable Diseases on Newborns and Children (Committee)
Beyond the case for NBS in South Africa. Chris Vorster 28/05/2016
 Beyond the case for NBS in South Africa Chris Vorster 28/05/2016 The case for NBS in SA Economic justification Cost Utility analysis = Cost/QALY GDP/Capita Immediate implementation (WHO) Political justification
Beyond the case for NBS in South Africa Chris Vorster 28/05/2016 The case for NBS in SA Economic justification Cost Utility analysis = Cost/QALY GDP/Capita Immediate implementation (WHO) Political justification
For Your Baby s Health Department of Health
 Newborn Screening For Your Baby s Health Department of Health Why is my baby tested? To help make sure your baby will be as healthy as possible. The blood test provides important information about your
Newborn Screening For Your Baby s Health Department of Health Why is my baby tested? To help make sure your baby will be as healthy as possible. The blood test provides important information about your
Diagnosis of IEM. And Emergency Management
 Diagnosis of IEM And Emergency Management Susan Sklower Brooks, M.D., F.A.C.M.G. Professor of Pediatrics Professor of Obstetrics, Gynecology and Reproductive Sciences Robert Wood Johnson Medical School
Diagnosis of IEM And Emergency Management Susan Sklower Brooks, M.D., F.A.C.M.G. Professor of Pediatrics Professor of Obstetrics, Gynecology and Reproductive Sciences Robert Wood Johnson Medical School
Metabolic diseases of the liver
 Metabolic diseases of the liver Central role in metabolism Causes and mechanisms of dysfunction Clinical patterns of metabolic disease Clinical approach to problem-solving Specific disorders Liver s central
Metabolic diseases of the liver Central role in metabolism Causes and mechanisms of dysfunction Clinical patterns of metabolic disease Clinical approach to problem-solving Specific disorders Liver s central
Organic acidaemias (OAs) & Urea cycle disorders (UCDs) PRESENTATION & MANAGEMENT
 & Urea cycle disorders (UCDs) PRESENTATION & MANAGEMENT") Great Ormond Street Hospital London 20/04/2018 Organic acidaemias (OAs) & Urea cycle disorders (UCDs) PRESENTATION & MANAGEMENT Spyros P. Batzios, MD, MSc, PhD OAs & UCDs How do they present? neonatal
Great Ormond Street Hospital London 20/04/2018 Organic acidaemias (OAs) & Urea cycle disorders (UCDs) PRESENTATION & MANAGEMENT Spyros P. Batzios, MD, MSc, PhD OAs & UCDs How do they present? neonatal
TRANSAMINATION AND UREA CYCLE
 TRANSAMINATION AND UREA CYCLE USMAN SUMO FRIEND TAMBUNAN ARLI ADITYA PARIKESIT SEPTIANA BIOINFORMATICS GROUP DEPARTEMENT OF CHEMISTRY FACULTY OF MATHEMATIC AND SCIENCE UNIVERSITY OF INDONESIA What is transamination?
TRANSAMINATION AND UREA CYCLE USMAN SUMO FRIEND TAMBUNAN ARLI ADITYA PARIKESIT SEPTIANA BIOINFORMATICS GROUP DEPARTEMENT OF CHEMISTRY FACULTY OF MATHEMATIC AND SCIENCE UNIVERSITY OF INDONESIA What is transamination?
UREA CYCLE DISORDERS - The What, Why, How and When
 UREA CYCLE DISORDERS - The What, Why, How and When George A. Diaz, MD, PhD Program for Inherited Metabolic Diseases Department of Genetics and Genomic Sciences Department of Pediatrics Icahn School of
UREA CYCLE DISORDERS - The What, Why, How and When George A. Diaz, MD, PhD Program for Inherited Metabolic Diseases Department of Genetics and Genomic Sciences Department of Pediatrics Icahn School of
Training Syllabus CLINICAL SYLLABUS
 Training Syllabus CLINICAL SYLLABUS SYLLABUS FOR TRAINING IN CLINICAL PAEDIATRIC METABOLIC MEDICINE Updated July 2006 This syllabus is intended as a guide. Whilst the training should be comprehensive,
Training Syllabus CLINICAL SYLLABUS SYLLABUS FOR TRAINING IN CLINICAL PAEDIATRIC METABOLIC MEDICINE Updated July 2006 This syllabus is intended as a guide. Whilst the training should be comprehensive,
Newborn screening for congenital metabolic diseases Optional out-of-pocket tests Information Sheet
 Newborn screening for congenital metabolic diseases Optional out-of-pocket tests Information Sheet Website:www.tipn.org.tw Telephone:(02)85962050 Ext. 401-403 Service line:(02)85962065 Fax:(02)85962067
Newborn screening for congenital metabolic diseases Optional out-of-pocket tests Information Sheet Website:www.tipn.org.tw Telephone:(02)85962050 Ext. 401-403 Service line:(02)85962065 Fax:(02)85962067
UK NATIONAL METABOLIC BIOCHEMISTRY NETWORK GUIDELINES FOR THE INVESTIGATION OF HYPERAMMONAEMIA
 UK NATIONAL METABOLIC BIOCHEMISTRY NETWORK GUIDELINES FOR THE INVESTIGATION OF HYPERAMMONAEMIA Hyperammonaemia results from defective catabolism of amino acids to urea. Recognition and treatment of hyperammonaemia,
UK NATIONAL METABOLIC BIOCHEMISTRY NETWORK GUIDELINES FOR THE INVESTIGATION OF HYPERAMMONAEMIA Hyperammonaemia results from defective catabolism of amino acids to urea. Recognition and treatment of hyperammonaemia,
TIMELINESS OF NEWBORN SCREENING: RECOMMENDATIONS FROM ADVISORY COMMITTEE ON HERITABLE DISORDERS IN NEWBORNS AND CHILDREN
 TIMELINESS OF NEWBORN SCREENING: RECOMMENDATIONS FROM ADVISORY COMMITTEE ON HERITABLE DISORDERS IN NEWBORNS AND CHILDREN Susan Tanksley, PhD May 19, 2015 TIMELINESS - BACKGROUND In order to effectively
TIMELINESS OF NEWBORN SCREENING: RECOMMENDATIONS FROM ADVISORY COMMITTEE ON HERITABLE DISORDERS IN NEWBORNS AND CHILDREN Susan Tanksley, PhD May 19, 2015 TIMELINESS - BACKGROUND In order to effectively
Module : Clinical correlates of disorders of metabolism Block 3, Week 2
 Module : Clinical correlates of disorders of metabolism Block 3, Week 2 Department of Paediatrics and Child Health University of Pretoria Tutor : Prof DF Wittenberg : dwittenb@medic.up.ac.za Aim of this
Module : Clinical correlates of disorders of metabolism Block 3, Week 2 Department of Paediatrics and Child Health University of Pretoria Tutor : Prof DF Wittenberg : dwittenb@medic.up.ac.za Aim of this
Isovaleric Acidemia: Quick reference guide
 Isovaleric Acidemia: Quick reference guide Introduction Isovaleric acidemia (IVA) is an inborn error of the leucine pathway caused by defects of the isovaleryl-oadehydrogenase (IV). The clinical presentation
Isovaleric Acidemia: Quick reference guide Introduction Isovaleric acidemia (IVA) is an inborn error of the leucine pathway caused by defects of the isovaleryl-oadehydrogenase (IV). The clinical presentation
Lynne A. Wolfe, MS, ACNP, PNP, BC Department of Genetics Yale School of Medicine
 Lynne A. Wolfe, MS, ACNP, PNP, BC Department of Genetics Yale School of Medicine Harvey Levy, MD Mark Korson, MD Piero Rinaldo, MD, PhD Larry Sweetman, PhD K. Michael Gibson, PhD Charlie Roe, MD Jerry
Lynne A. Wolfe, MS, ACNP, PNP, BC Department of Genetics Yale School of Medicine Harvey Levy, MD Mark Korson, MD Piero Rinaldo, MD, PhD Larry Sweetman, PhD K. Michael Gibson, PhD Charlie Roe, MD Jerry
Jana Novotná, Bruno Sopko. Department of the Medical Chemistry and Clinical Biochemistry The 2nd Faculty of Medicine, Charles Univ.
 Amino acid metabolism II. Urea cycle Jana Novotná, Bruno Sopko Department of the Medical Chemistry and Clinical Biochemistry The 2nd Faculty of Medicine, Charles Univ. Nitrogen balance Tissue proteins
Amino acid metabolism II. Urea cycle Jana Novotná, Bruno Sopko Department of the Medical Chemistry and Clinical Biochemistry The 2nd Faculty of Medicine, Charles Univ. Nitrogen balance Tissue proteins
Metabolic Emergencies and the Pediatrician
 Metabolic Emergencies and the Pediatrician Stephen G. Kahler, MD Professor of Pediatrics Section of Genetics and Metabolism, UAMS and ACH Hot Springs, AR March 8, 2012 INHERITED METABOLIC DISORDERS HOW
Metabolic Emergencies and the Pediatrician Stephen G. Kahler, MD Professor of Pediatrics Section of Genetics and Metabolism, UAMS and ACH Hot Springs, AR March 8, 2012 INHERITED METABOLIC DISORDERS HOW
Salicylate (Aspirin) Ingestion California Poison Control Background 1. The prevalence of aspirin-containing analgesic products makes
 Ingestion California Poison Control Background 1. The prevalence of aspirin-containing analgesic products makes") Salicylate (Aspirin) Ingestion California Poison Control 1-800-876-4766 Background 1. The prevalence of aspirin-containing analgesic products makes these agents, found in virtually every household, common
Salicylate (Aspirin) Ingestion California Poison Control 1-800-876-4766 Background 1. The prevalence of aspirin-containing analgesic products makes these agents, found in virtually every household, common
بنام خدا هیپوگلیسمی درنوزادان و گاالکتوزمی دکتر انتظاری
 بنام خدا هیپوگلیسمی درنوزادان و گاالکتوزمی دکتر انتظاری Serum glucose< 35 mg/dl 1-3 hr of life < 40 mg/dl 3-24 hr < 45 mg/dl after 24 hr 10% NL newborns can t maintain BS>30 if delayed feeding >3-6 hrs
بنام خدا هیپوگلیسمی درنوزادان و گاالکتوزمی دکتر انتظاری Serum glucose< 35 mg/dl 1-3 hr of life < 40 mg/dl 3-24 hr < 45 mg/dl after 24 hr 10% NL newborns can t maintain BS>30 if delayed feeding >3-6 hrs
MEDICAL COVERAGE GUIDELINES ORIGINAL EFFECTIVE DATE: 12/19/17 SECTION: MEDICINE LAST REVIEW DATE: LAST CRITERIA REVISION DATE: ARCHIVE DATE:
 MEDICAL FOODS Non-Discrimination Statement and Multi-Language Interpreter Services information are located at the end of this document. Coverage for services, procedures, medical devices and drugs are
MEDICAL FOODS Non-Discrimination Statement and Multi-Language Interpreter Services information are located at the end of this document. Coverage for services, procedures, medical devices and drugs are
Dr Rodney Itaki Lecturer Anatomical Pathology Discipline. University of Papua New Guinea School of Medicine & Health Sciences Division of Pathology
 Fetal & Neonatal Pathology Dr Rodney Itaki Lecturer Anatomical Pathology Discipline University of Papua New Guinea School of Medicine & Health Sciences Division of Pathology Self-Directed Reading Topics
Fetal & Neonatal Pathology Dr Rodney Itaki Lecturer Anatomical Pathology Discipline University of Papua New Guinea School of Medicine & Health Sciences Division of Pathology Self-Directed Reading Topics
Neonatal Guidelines. Chapter 10: Metabolic V Date Revised : January 2017 Ratified 6 th February Date for Review: 1 st of March 2021
 Neonatal Guidelines Chapter 10: Metabolic V2017.1 Specialty: Neonatal Medicine Revised by: Jean Matthes Date Revised : January 2017 Ratified 6 th February 2017 Approved by: ABMU Joint Perinatal Forum Date
Neonatal Guidelines Chapter 10: Metabolic V2017.1 Specialty: Neonatal Medicine Revised by: Jean Matthes Date Revised : January 2017 Ratified 6 th February 2017 Approved by: ABMU Joint Perinatal Forum Date
Providing the Right Fuels for FOD s. Elaina Jurecki, MS, RD Regional Metabolic Nutritionist Kaiser Permanente Medical Center Oakland, CA
 Providing the Right Fuels for FOD s Elaina Jurecki, MS, RD Regional Metabolic Nutritionist Kaiser Permanente Medical Center Oakland, CA Providing the Right Fuels for FOD s The Body s Use of Energy Fat
Providing the Right Fuels for FOD s Elaina Jurecki, MS, RD Regional Metabolic Nutritionist Kaiser Permanente Medical Center Oakland, CA Providing the Right Fuels for FOD s The Body s Use of Energy Fat
Nutritional Management of Inborn Errors of Metabolism. Kay Davis, RD, CSP Esther Berenhaut, RD, CSP, CSR Aug 28, 2017
 Nutritional Management of Inborn Errors of Metabolism Kay Davis, RD, CSP Esther Berenhaut, RD, CSP, CSR Aug 28, 2017 OBJECTIVES Brief overview of newborn screening of metabolic disorders, inheritance patterns.
Nutritional Management of Inborn Errors of Metabolism Kay Davis, RD, CSP Esther Berenhaut, RD, CSP, CSR Aug 28, 2017 OBJECTIVES Brief overview of newborn screening of metabolic disorders, inheritance patterns.
Carnitine palmitoyl transferase 2 deficiency (CPT2) is a rare inherited disorder that occurs when
 is a rare inherited disorder that occurs when") CPT2 Deficiency Carnitine palmitoyl transferase 2 deficiency (CPT2) is a rare inherited disorder that occurs when the last step in the entry of fats into sac-like bodies called mitochondria is blocked.
CPT2 Deficiency Carnitine palmitoyl transferase 2 deficiency (CPT2) is a rare inherited disorder that occurs when the last step in the entry of fats into sac-like bodies called mitochondria is blocked.
MISSOURI DEPARTMENT OF HEALTH AND SENIOR SERVICES
 NEWBORN SCREENING Protecting your newborn MISSOURI DEPARTMENT OF HEALTH AND SENIOR SERVICES [ 1 ] NEWBORN SCREENING [ 2 ] FREQUENTLY ASKED QUESTIONS [ 4 ] DISORDERS INCLUDED IN NEWBORN SCREENING [ 12 ]
NEWBORN SCREENING Protecting your newborn MISSOURI DEPARTMENT OF HEALTH AND SENIOR SERVICES [ 1 ] NEWBORN SCREENING [ 2 ] FREQUENTLY ASKED QUESTIONS [ 4 ] DISORDERS INCLUDED IN NEWBORN SCREENING [ 12 ]
Ornithine Transcarbamylase Deficiency (OTCD) Recommendations on Emergency Management of Metabolic Disease
 Recommendations on Emergency Management of Metabolic Disease") Ornithine Transcarbamylase Deficiency (OTCD) Recommendations on Emergency Management of Metabolic Disease Patient s Name: Date of Birth: MRN in KFSH&RC: Please read carefully. Meticulous and prompt treatment
Ornithine Transcarbamylase Deficiency (OTCD) Recommendations on Emergency Management of Metabolic Disease Patient s Name: Date of Birth: MRN in KFSH&RC: Please read carefully. Meticulous and prompt treatment
Inborn Error Of Metabolism :
 Inborn Error Of Metabolism : Inborn Error Of Metabolism inborn error of metabolism are a large group of hereditary biochemical diseases in which specific gene mutation cause abnormal or missing proteins
Inborn Error Of Metabolism : Inborn Error Of Metabolism inborn error of metabolism are a large group of hereditary biochemical diseases in which specific gene mutation cause abnormal or missing proteins
Testing Strategy for Inborn Errors of Metabolism in the Neonate Aditi I. Dagli, Roberto T. Zori and Bryce A. Heese. DOI: /neo.
 Testing Strategy for Inborn Errors of Metabolism in the Neonate Aditi I. Dagli, Roberto T. Zori and Bryce A. Heese Neoreviews 2008;9;e291 DOI: 10.1542/neo.9-7-e291 The online version of this article, along
Testing Strategy for Inborn Errors of Metabolism in the Neonate Aditi I. Dagli, Roberto T. Zori and Bryce A. Heese Neoreviews 2008;9;e291 DOI: 10.1542/neo.9-7-e291 The online version of this article, along
NEWBORN SCREENING. health.mo.gov/newbornscreening MISSOURI DEPARTMENT OF HEALTH AND SENIOR SERVICES
 NEWBORN SCREENING health.mo.gov/newbornscreening MISSOURI DEPARTMENT OF HEALTH AND SENIOR SERVICES Table of Contents NEWBORN SCREENING...1 FREQUENTLY ASKED QUESTIONS...2 DISORDERS INCLUDED IN NEWBORN SCREENING...4
NEWBORN SCREENING health.mo.gov/newbornscreening MISSOURI DEPARTMENT OF HEALTH AND SENIOR SERVICES Table of Contents NEWBORN SCREENING...1 FREQUENTLY ASKED QUESTIONS...2 DISORDERS INCLUDED IN NEWBORN SCREENING...4
TITLE: NURSING GUIDELINES FOR THE MANAGEMENT OF CHILDREN WITH METHYLMALONIC ACIDURIA. Eilish O Connell, Clinical Education Facilitator, NCIMD
 NO. OF PAGES: Page 1 of 17 SUPERCEDES: N/A NURSING GUIDELINES FOR THE MANAGEMENT OF CHILDREN WITH METHYLMALONIC ACIDURIA NAME/ Eilish O Connell, Clinical Education Facilitator, NCIMD SIGNATURE: DATE: NAME/
NO. OF PAGES: Page 1 of 17 SUPERCEDES: N/A NURSING GUIDELINES FOR THE MANAGEMENT OF CHILDREN WITH METHYLMALONIC ACIDURIA NAME/ Eilish O Connell, Clinical Education Facilitator, NCIMD SIGNATURE: DATE: NAME/
National Metabolic Biochemistry Network Guidelines for the investigation of hypoglycaemia in infants and children
 National Metabolic Biochemistry Network Guidelines for the investigation of hypoglycaemia in infants and children Aim To provide guidance on the biochemical investigation of hypoglycaemia in infants and
National Metabolic Biochemistry Network Guidelines for the investigation of hypoglycaemia in infants and children Aim To provide guidance on the biochemical investigation of hypoglycaemia in infants and
Very-long-chain acyl-coa dehydrogenase deficiency
 Very-long-chain acyl-coa dehydrogenase deficiency Introductory information Written by: V. Prietsch & P. Burgard Reviewed & Revised for North America by: S. van Calcar Very-long-chain acyl-coa dehydrogenase
Very-long-chain acyl-coa dehydrogenase deficiency Introductory information Written by: V. Prietsch & P. Burgard Reviewed & Revised for North America by: S. van Calcar Very-long-chain acyl-coa dehydrogenase
How MS/MS Revolutionized Newborn Screening
 How MS/MS Revolutionized Newborn Screening David S Millington, PhD Medical Research Professor of Pediatrics Director, Biochemical Genetics Laboratory Duke University Medical Center NEWBORN SCREENING IN
How MS/MS Revolutionized Newborn Screening David S Millington, PhD Medical Research Professor of Pediatrics Director, Biochemical Genetics Laboratory Duke University Medical Center NEWBORN SCREENING IN
UNIVERSITY OF PNG SCHOOL OF MEDICINE AND HEALTH SCIENCES DIVISION OF BASIC MEDICAL SCIENCES Discipline of Biochemistry and Molecular Biology
 UNIVERSITY OF PNG SCHOOL OF MEDICINE AND HEALTH SCIENCES DIVISION OF BASIC MEDICAL SCIENCES Discipline of Biochemistry and Molecular Biology 1 PBL SEMINAR ACUTE & CHRONIC ETHANOL EFFECTS An Overview Sites
UNIVERSITY OF PNG SCHOOL OF MEDICINE AND HEALTH SCIENCES DIVISION OF BASIC MEDICAL SCIENCES Discipline of Biochemistry and Molecular Biology 1 PBL SEMINAR ACUTE & CHRONIC ETHANOL EFFECTS An Overview Sites
Diagnose a broad range of metabolic disorders with a single test, Global MAPS
 PEDIATRIC Assessing or diagnosing a metabolic disorder commonly requires several tests. Global Metabolomic Assisted Pathway Screen, commonly known as Global MAPS, is a unifying test for analyzing hundreds
PEDIATRIC Assessing or diagnosing a metabolic disorder commonly requires several tests. Global Metabolomic Assisted Pathway Screen, commonly known as Global MAPS, is a unifying test for analyzing hundreds
NEWBORN SCREENING. in Massachusetts: Answers for You and Your Baby. University of Massachusetts Medical School
 University of Massachusetts Medical School NEWBORN SCREENING in Massachusetts: Answers for You and Your Baby New England Newborn Screening Program Biotech 4, 2nd Floor UMass Medical School 377 Plantation
University of Massachusetts Medical School NEWBORN SCREENING in Massachusetts: Answers for You and Your Baby New England Newborn Screening Program Biotech 4, 2nd Floor UMass Medical School 377 Plantation
Inborn Errors of Metabolism
 30 Inborn Errors of Metabolism Inborn errors of metabolism (IEM) are disorders in which there is a block in the normal metabolic pathway that is caused by a genetic defect of a specific enzyme. The number
30 Inborn Errors of Metabolism Inborn errors of metabolism (IEM) are disorders in which there is a block in the normal metabolic pathway that is caused by a genetic defect of a specific enzyme. The number
Understanding metabolic disease
 Understanding metabolic disease Let s build a restaurant Chris Hendriksz Birmingham Children s Hospital 2006 The Task Let s build a restaurant! 2 Partners join to draw the plan. How can we change the
Understanding metabolic disease Let s build a restaurant Chris Hendriksz Birmingham Children s Hospital 2006 The Task Let s build a restaurant! 2 Partners join to draw the plan. How can we change the
Fatty Acid Oxidation Disorders- an update. Fiona Carragher Biochemical Sciences, GSTS Pathology St Thomas Hospital, London
 Fatty Acid Oxidation Disorders- an update Fiona Carragher Biochemical Sciences, GSTS Pathology St Thomas Hospital, London An update. Overview of metabolism Clinical presentation and outcome Diagnostic
Fatty Acid Oxidation Disorders- an update Fiona Carragher Biochemical Sciences, GSTS Pathology St Thomas Hospital, London An update. Overview of metabolism Clinical presentation and outcome Diagnostic
Inborn error of metabolism
 Inborn error of metabolism The lecture is given by Dr Kefah Al qa qa, Consultant metabolic pediatrician, in Queen Rania Al Abdulla children hospital, on Thursday 11/12/2014. Let s tart Metabolism: refers
Inborn error of metabolism The lecture is given by Dr Kefah Al qa qa, Consultant metabolic pediatrician, in Queen Rania Al Abdulla children hospital, on Thursday 11/12/2014. Let s tart Metabolism: refers
Urea Cycle Disorders and Hyperammonemia: Diagnosable Treatable Screenable
 Urea Cycle Disorders and Hyperammonemia: Diagnosable Treatable Screenable Marshall L. Summar, M.D. Chief, Division of Genetics and Metabolism Children s National Medical Center Washington, DC, USA Disclosure
Urea Cycle Disorders and Hyperammonemia: Diagnosable Treatable Screenable Marshall L. Summar, M.D. Chief, Division of Genetics and Metabolism Children s National Medical Center Washington, DC, USA Disclosure
Integrative Metabolism: Significance
 Integrative Metabolism: Significance Energy Containing Nutrients Carbohydrates Fats Proteins Catabolism Energy Depleted End Products H 2 O NH 3 ADP + Pi NAD + NADP + FAD + Pi NADH+H + NADPH+H + FADH2 Cell
Integrative Metabolism: Significance Energy Containing Nutrients Carbohydrates Fats Proteins Catabolism Energy Depleted End Products H 2 O NH 3 ADP + Pi NAD + NADP + FAD + Pi NADH+H + NADPH+H + FADH2 Cell
[R23-13-MET/HRG] STATE OF RHODE ISLAND AND PROVIDENCE PLANTATIONS DEPARTMENT OF HEALTH. February October October 1992 (E) September 1995
![[R23-13-MET/HRG] STATE OF RHODE ISLAND AND PROVIDENCE PLANTATIONS DEPARTMENT OF HEALTH. February October October 1992 (E) September 1995](/thumbs/95/122505027.jpg "[R23-13-MET/HRG] STATE OF RHODE ISLAND AND PROVIDENCE PLANTATIONS DEPARTMENT OF HEALTH. February October October 1992 (E) September 1995") RULES AND REGULATIONS PERTAINING TO THE NEWBORN METABOLIC, ENDOCRINE, AND HEMOGLOBINOPATHY SCREENING PROGRAM AND THE NEWBORN HEARING LOSS SCREENING PROGRAM [R23-13-MET/HRG] STATE OF RHODE ISLAND AND PROVIDENCE
RULES AND REGULATIONS PERTAINING TO THE NEWBORN METABOLIC, ENDOCRINE, AND HEMOGLOBINOPATHY SCREENING PROGRAM AND THE NEWBORN HEARING LOSS SCREENING PROGRAM [R23-13-MET/HRG] STATE OF RHODE ISLAND AND PROVIDENCE
Metabolic Precautions & ER Recommendations
 Metabolic Precautions & ER Recommendations * To whom correspondence Sumit Parikh, should MD be addressed Center for Pediatric Neurology Cleveland Clinic Cleveland, OH UMDF 2010 The catabolic state Entering
Metabolic Precautions & ER Recommendations * To whom correspondence Sumit Parikh, should MD be addressed Center for Pediatric Neurology Cleveland Clinic Cleveland, OH UMDF 2010 The catabolic state Entering
Genomics & Modern Health Care Caring for the Special Children & Adults of Isolated Populations. Propionic Acidemia
 Genomics & Modern Health Care Caring for the Special Children & Adults of Isolated Populations Propionic Acidemia D. Holmes Morton MD Pediatrician, Clinic for Special Children Strasburg, Pennsylvania 17579
Genomics & Modern Health Care Caring for the Special Children & Adults of Isolated Populations Propionic Acidemia D. Holmes Morton MD Pediatrician, Clinic for Special Children Strasburg, Pennsylvania 17579
BCM 225 LECTURE ON INBORN ERRORS OF METABOLISM OJEMEKELE O.
 BCM 225 LECTURE ON INBORN ERRORS OF METABOLISM BY OJEMEKELE O. OUTLINE (week 11 and 12) Inborn errors of carbohydrate metabolism Inborn errors of amino acid metabolism Inborn errors of fatty acid metabolism
BCM 225 LECTURE ON INBORN ERRORS OF METABOLISM BY OJEMEKELE O. OUTLINE (week 11 and 12) Inborn errors of carbohydrate metabolism Inborn errors of amino acid metabolism Inborn errors of fatty acid metabolism
Newborn Screening in Washington State Saving lives with a simple blood spot
 Newborn Screening in Washington State Saving lives with a simple blood spot Ashleigh Ragsdale, MPH Gauri Gupta, MScPH Objectives Newborn Screening Overview Process and Law Completing Collection Cards Video
Newborn Screening in Washington State Saving lives with a simple blood spot Ashleigh Ragsdale, MPH Gauri Gupta, MScPH Objectives Newborn Screening Overview Process and Law Completing Collection Cards Video
SYLLABUS FOR TRAINING IN CLINICAL PAEDIATRIC METABOLIC MEDICINE
 SYLLABUS FOR TRAINING IN CLINICAL PAEDIATRIC METABOLIC MEDICINE Updated December 2014: Vassili Valayannopoulos and Andrew Morris Paediatrics is an independent medical specialty based on the knowledge and
SYLLABUS FOR TRAINING IN CLINICAL PAEDIATRIC METABOLIC MEDICINE Updated December 2014: Vassili Valayannopoulos and Andrew Morris Paediatrics is an independent medical specialty based on the knowledge and
BIOTIN (BIOTINIDASE) DEFICIENCY Marc E. Tischler, PhD; University of Arizona
 DEFICIENCY Marc E. Tischler, PhD; University of Arizona") BIOTIN (BIOTINIDASE) DEFICIENCY Marc E. Tischler, PhD; University of Arizona BIOTIN (BIOTINIDASE) DEFICIENCY biotin in the body is recycled by its removal from carboxylase enzymes to which it is attached
BIOTIN (BIOTINIDASE) DEFICIENCY Marc E. Tischler, PhD; University of Arizona BIOTIN (BIOTINIDASE) DEFICIENCY biotin in the body is recycled by its removal from carboxylase enzymes to which it is attached
My Experiences and Understanding of VLCAD Deficiency and its Treatment Charles R. Roe, MD June 26, 2011 (Now retired)
") My Experiences and Understanding of VLCAD Deficiency and its Treatment Charles R. Roe, MD June 26, 2011 (Now retired) This disorder is characterized by the deficiency of the VLCAD enzyme that is required
My Experiences and Understanding of VLCAD Deficiency and its Treatment Charles R. Roe, MD June 26, 2011 (Now retired) This disorder is characterized by the deficiency of the VLCAD enzyme that is required
ACUTE & CHRONIC ETHANOL EFFECTS An Overview
 ACUTE & CHRONIC ETHANOL EFFECTS An Overview University of Papua New Guinea School of Medicine & Health Sciences, Division of Basic Medical Sciences Clinical Biochemistry: PBL Seminar MBBS Yr 4 VJ Temple
ACUTE & CHRONIC ETHANOL EFFECTS An Overview University of Papua New Guinea School of Medicine & Health Sciences, Division of Basic Medical Sciences Clinical Biochemistry: PBL Seminar MBBS Yr 4 VJ Temple
A Guide for Prenatal Educators
 A Guide for Prenatal Educators Why Teach Newborn Screening This booklet is designed to make it easy for you, a prenatal educator, to effectively inform expectant parents about newborn screening. All of
A Guide for Prenatal Educators Why Teach Newborn Screening This booklet is designed to make it easy for you, a prenatal educator, to effectively inform expectant parents about newborn screening. All of
Arizona Newborn Screening Program
 ARIZONA DEPARTMENT OF HEALTH SERVICES BUREAU OF STATE LABORATORY SERVICES Arizona Newborn Screening Program GUIDELINES DIVISION OF PUBLIC HEALTH SERVICES AUGUST 2010 with revisions (January 2011) Table
ARIZONA DEPARTMENT OF HEALTH SERVICES BUREAU OF STATE LABORATORY SERVICES Arizona Newborn Screening Program GUIDELINES DIVISION OF PUBLIC HEALTH SERVICES AUGUST 2010 with revisions (January 2011) Table
Metabolic Disorders Screened Overseas but not Screened in Australia Condition Features Inherited Diagnosis Treatment Newborn Screen
 Metabolic Disorders ed Overseas but not ed in Australia Biotinidase Deficiency Severe form causes seizures & delay Biotin can prevent complications NZ, USA Tyrosinaemia Type I Coma & death before age 10
Metabolic Disorders ed Overseas but not ed in Australia Biotinidase Deficiency Severe form causes seizures & delay Biotin can prevent complications NZ, USA Tyrosinaemia Type I Coma & death before age 10
Fatty acid oxidation. Naomi Rankin
 Fatty acid oxidation Naomi Rankin Fatty acid oxidation Provides energy to muscles from lipid stores, spares glucose for the brain Lipolysis of triglycerides results in FFA, mainly C16 and C18 FA oxidation
Fatty acid oxidation Naomi Rankin Fatty acid oxidation Provides energy to muscles from lipid stores, spares glucose for the brain Lipolysis of triglycerides results in FFA, mainly C16 and C18 FA oxidation
Inborn errors of metabolism
 ESPEN Congress Nice 2010 From child to adult nutrition Inborn errors of metabolism Pascal Crenn Inborn errors of metabolism: from child to adult Pascal Crenn Hôpital Raymond Poincaré 92380 Garches. France
ESPEN Congress Nice 2010 From child to adult nutrition Inborn errors of metabolism Pascal Crenn Inborn errors of metabolism: from child to adult Pascal Crenn Hôpital Raymond Poincaré 92380 Garches. France
NEONATAL HYPOGLYCEMIA HEATHER MCKNIGHT-MENCI, MSN, CRNP CHILDREN S HOSPITAL OF PHILADELPHIA
 NEONATAL HYPOGLYCEMIA HEATHER MCKNIGHT-MENCI, MSN, CRNP CHILDREN S HOSPITAL OF PHILADELPHIA WHAT IS NEONATAL HYPOGLYCEMIA? Glucose concentration low enough to cause signs and symptoms of impaired brain
NEONATAL HYPOGLYCEMIA HEATHER MCKNIGHT-MENCI, MSN, CRNP CHILDREN S HOSPITAL OF PHILADELPHIA WHAT IS NEONATAL HYPOGLYCEMIA? Glucose concentration low enough to cause signs and symptoms of impaired brain
Fatty Acid Oxidation Disorders Organic Acid Disorders
 Genetic Fact Sheets for Parents Fatty Acid Oxidation Disorders Organic Acid Disorders Screening, Technology, and Research in Genetics is a multi-state project to improve information about the financial,
Genetic Fact Sheets for Parents Fatty Acid Oxidation Disorders Organic Acid Disorders Screening, Technology, and Research in Genetics is a multi-state project to improve information about the financial,
Good Morning! OCTOBER 2, 2014
 Good Morning! OCTOBER, 04 Inborn Errors of Metabolism Disorders of Amino Acid Metabolism PKU 4 4 My disorder is AR AKA Følling s disease I Appeared normal at birth But now I have an eczematoid rash and
Good Morning! OCTOBER, 04 Inborn Errors of Metabolism Disorders of Amino Acid Metabolism PKU 4 4 My disorder is AR AKA Følling s disease I Appeared normal at birth But now I have an eczematoid rash and
Glycogen Storage Disease
 Glycogen Storage Disease 1 Introduction The food we eat is usually used for growth, tissue repair and energy. The body stores what it does not use. Excess sugar, or glucose, is stored as glycogen in the
Glycogen Storage Disease 1 Introduction The food we eat is usually used for growth, tissue repair and energy. The body stores what it does not use. Excess sugar, or glucose, is stored as glycogen in the
UPDATE ON UREA CYCLE DISORDERS TREATMENT
 UPDATE ON UREA CYCLE DISORDERS TREATMENT George A. Diaz, MD, PhD Program for Inherited Metabolic Diseases Department of Genetics and Genomic Sciences Icahn School of Medicine at Mount Sinai Disclosure
UPDATE ON UREA CYCLE DISORDERS TREATMENT George A. Diaz, MD, PhD Program for Inherited Metabolic Diseases Department of Genetics and Genomic Sciences Icahn School of Medicine at Mount Sinai Disclosure
BROADENING YOUR PATIENT S OPTIONS FOR GENETIC CARRIER SCREENING.
 BROADENING YOUR PATIENT S OPTIONS FOR GENETIC CARRIER SCREENING. The Inheritest SM Carrier Screen provides relevant genetic screening for many inherited diseases found throughout the pan-ethnic US population.
BROADENING YOUR PATIENT S OPTIONS FOR GENETIC CARRIER SCREENING. The Inheritest SM Carrier Screen provides relevant genetic screening for many inherited diseases found throughout the pan-ethnic US population.
OVERVIEW M ET AB OL IS M OF FR EE FA TT Y AC ID S
 LIPOLYSIS LIPOLYSIS OVERVIEW CATABOLISM OF FREE FATTY ACIDS Nonesterified fatty acids Source:- (a) breakdown of TAG in adipose tissue (b) action of Lipoprotein lipase on plasma TAG Combined with Albumin
LIPOLYSIS LIPOLYSIS OVERVIEW CATABOLISM OF FREE FATTY ACIDS Nonesterified fatty acids Source:- (a) breakdown of TAG in adipose tissue (b) action of Lipoprotein lipase on plasma TAG Combined with Albumin