Protocol. This trial protocol has been provided by the authors to give readers additional information about their work.
|
|
|
- Albert Kelley
- 6 years ago
- Views:
Transcription
1 Protocol This trial protocol has been provided by the authors to give readers additional information about their work. Protocol for: Lok AS, Gardiner DF, Lawitz E, et al. Preliminary study of two antiviral agents for hepatitis C genotype 1. N Engl J Med 2012;366:
2 Page: 1 Protocol Number: IND Number: 79,599 and 100,932 EUDRACT Number Date: 02-Oct-2009 Revised Date 14-Jun-2011 Parallel, Open-Label, Randomized, Multiple-Dose Study to Evaluate the Safety, Pharmacokinetics and Pharmacodynamics of in Combination in Null Responders to Standard of Care Infected with Chronic Hepatitis C Virus Genotype 1 Revised Protocol Number: 07 Incorporates Amendment: 08 Study Director and Medical Monitor David Gardiner, MD Bristol-Myers Squibb Discovery Medicine and Clinical Pharmacology 311 Pennington-Rocky Hill Road Pennington, NJ Telephone (office): (609) Fax: (609) hr Emergency Telephone Number USA: International: Bristol-Myers Squibb Research and Development Avenue de Finlande 8 Building F 1st Floor B-1420 Braine-l Alleud, Belgium and 311 Pennington-Rocky Hill Road Pennington, NJ 08534, USA This document is the confidential and proprietary information of Bristol-Myers Squibb Company and its global affiliates (BMS). By reviewing this document, you agree to keep it confidential and to use and disclose it solely for the purpose of assessing whether your organization will participate in and/or the performance of the proposed BMS-sponsored study. Any permitted disclosures will be made only on a confidential "need to know" basis within your organization or to your
3 independent ethics committee(s). Any other use, copying, disclosure or dissemination of this information is strictly prohibited unless expressly authorized in writing by BMS. Any supplemental information (e.g., amendments) that may be added to this document is also confidential and proprietary to BMS and must be kept in confidence in the same manner as the contents of this document. Any person who receives this document without due authorization from BMS is requested to return it to BMS or promptly destroy it. All other rights reserved. Replace all previous version(s) of the protocol with this revised protocol and please provide a copy of this revised protocol to all study personnel under your supervision, and archive the previous versions. 2
4 DOCUMENT HISTORY Document Date of Issue Summary of Change Revised Protocol Jun-2011 Incorporates Amendment 08 Amendment Jun-2011 In response to a new BMS SOP, a new section was added specific to potential Drug Induced Liver Injury (DILI) providing a program-specific definition and reporting procedures for potential DILI events. The inclusion/exclusion criteria were revised to more accurately reflect the HCV nullresponder patient population the study is targeting. In some places the criteria were also clarified to help the investigators adjudicate subjects eligibility in some common scenarios. Exploratory research on serum/plasma samples including pharmacokinetic samples was added to further investigate the anti-viral immunity in subjects. The analysis plan was clarified to include interim analyses of both the Sentinel and Expansion Cohorts at 48 weeks after end of treatment. Additional prohibitions and restrictions on the concomitant use of some medications that are substrates of CYP2D6 were described. Finally it was clarified that for some post-treatment follow-up visits where the only procedures are AE collection and pregnancy test for women, AE collection can be performed via telephone and pregnancy test can be done in a local lab. This will reduce the burden on sites and patients and enhance protocol adherence. This amendment applies to all subjects. Revised Protocol Dec-2010 Incorporates Amendment 07 Amendment Dec-2010 The purpose of this amendment is to revise the design of the expansion phase of the study. This amendment harmonizes the planned expansion cohorts with the global development plan, lessons learned from the sentinel cohorts, and new data released at the American Liver Meeting held in October There are now five dose cohorts in the expansion. These cohorts will confirm the clinical activity of the four drug therapy (Treatment B) observed in the sentinel cohort using lower doses of BMS The new design will also expand the results seen with two antiviral drugs alone (Treatment A) in genotype 1b-infected subjects with lower doses of BMS These lower doses of BMS will be 200 mg BID and 200 mg QD. Finally, the study will investigate the hypothesis that ribavirin limits the development of resistance to antivirals in antiviral only treatment regimens. Revised Protocol Oct-2010 Incorporates Amendment 06 3
5 Document Date of Issue Summary of Change Amendment Oct-2010 During the 12-week interim analysis of study AI447016, it was found that 600 mg BMS BID or QD was associated with a trend of ALT/AST elevations. The trend is more prominent at the 600 mg doses than the 200 mg BID dose. Importantly, no subjects receiving the 200 mg BID dose discontinued due to AEs related to hepatic laboratory abnormalities, and there were no grade 3-4 AE abnormalities at this dose. Since all three doses (200 mg BID, 600 mg BID, 600 mg QD) explored in study AI as of the week12 analysis demonstrated similar antiviral activity while ALT/AST elevations were primarily limited to the higher doses, the dose of BMS for all subjects in ongoing studies including, will be reduced to 200 mg BID. The dose of BMS was reduced immediately for 5 ongoing subjects, and dosing for additional subjects in this study was halted. The urgent implementation prior to IRB approval is justified by the need to remove subjects from immediate danger. This amendment applies to all subjects. Revised Protocol Jun-2010 Incorporates Amendment 05. Amendment Jun-2010 Preliminary results from the 4-week interim analysis of the Sentinel Cohort indicated that there are fewer virologic breakthroughs in Treatment B than Treatment A in these nullresponder patients. To reduce potential risks and improve potential benefits to patients, it was decided that in Part 2 of the study only treatment B will be expanded, and Treatment A will not be expanded. This amendment added an additional branch to the decision tree to include this option. In Part 2, Treatment B Expanded Cohort 1 with 24 weeks of NS3+NS5A+SOC treatment will be compared to Treatment B Expanded Cohort 2 with 24 weeks of NS3+NS5A+SOC followed by additional 24 weeks of SOC treatment only. A discussion of elevated alanine transaminase (ALT) observed in some subjects in the Sentinel Cohort was added to the Overall Risk/Benefit Assessment section to inform the investigators. Finally four more time points for anti-hcv T- cell sample collection were added to further investigate the immunological changes caused by the study drugs. This amendment applies to all subjects. Revised Protocol May-2010 Incorporates Amendment 04. Amendment May-2010 To further investigate the safety and anti-viral activity in the Sentinel cohort, interim analyses of safety and antiviral activity after all subjects in sentinel cohort reach Treatment Week 12, 24 and Follow-up Week 12 may be performed. Additionally to reduce potential risks to patients, interim 4
6 Document Date of Issue Summary of Change Revised Protocol 02 analyses of safety and antiviral activity after all subjects in both Sentinel and Expanded Cohorts reach Treatment Week 4 and 24 may be performed. This amendment applies to all subjects. 06-Apr-2010 Incorporates Amendment 03 Amendment Apr-2010 The purpose of this amendment is three-fold. First, it will clarify the criteria for adding standard of care (SOC) or discontinuing therapy for individual subject randomized to Treatment A ( only). With this clarification, the decision to add SOC or discontinue therapy will be made by evaluation of HCV viral load data continuously and not just at specific time points. Secondly, the amendment will make it possible for interim analysis of exploratory biomarkers, which will allow us to get data on HCV resistance genotypes earlier. Thirdly, based on data from the Sentinel Cohorts, the amendment will require more frequent measurements of HCV RNA levels in subjects on Treatment A who has not been rescued (from every 4 weeks to every 2 weeks). This amendment applies to all subjects. Revised Protocol Jan-2010 Incorporates Amendment 02 Amendment Jan-2010 Updated background information, provided clarification on meal guidance on Day 14, clarification on inclusion and discontinuation criteria, p-gp substrate guidance, additional interim analysis added and updated SAE reporting procedures. Original Protocol 02-Oct-2009 Not applicable 5
7 SYNOPSIS Title of Study: Protocol : Parallel, Open-Label, Randomized, Multi-Dose Study to Evaluate the Safety, Pharmacokinetics and Pharmacodynamics of in Combination in Null Responders to Standard of Care Infected with Chronic Hepatitis C Virus Genotype 1 Indication: Genotype 1 Chronic Hepatitis C in patients with a null response to prior therapy with pegylated-interferon-α and ribavirin (PegIFN/RBV) which is defined as standard of care (SOC). Estimated Number of Study Centers and Countries/Regions: ~Approximately 19 sites. Study Phase: 2a Research Hypothesis (Part 1): The observed proportion of Hepatitis C Virus (HCV) genotype 1 null responder subjects in the sentinel cohort with successful response to treatment is 70% at Week 2 and rapid virologic response (RVR) is 50% at Week 4 for the combination of (NS5A+NS3) with and without SOC. Successful response to treatment is defined at Week 2 as either undetectable HCV RNA (< 10 IU/mL) or 2 log 10 IU/mL decrease in plasma HCV RNA from baseline without rebound and at Week 4 by RVR defined as undetectable HCV RNA (< 10 IU/mL). Research Hypothesis (Part 2): The observed proportion of null responder subjects achieving 12-week sustained virologic response (SVR 12 ) is 20%. SVR 12 is defined as undetectable HCV RNA (< 10 IU/mL) at follow-up Week 12. Primary Objective (Part 1): To determine the proportion of subjects in the sentinel cohort with successful response to treatment at Week 2 and RVR at Week 4. Primary Objective (Part 2): To determine the proportion of subjects with SVR 12 in each cohort. Secondary Objectives: To assess the safety of co-administration of NS3+NS5A with and without SOC or ribavirin as measured by the frequency of Serious Adverse Events (SAEs) and AEs, discontinuations due to AEs, and abnormalities observed from vital sign and ECG measurements, physical examinations and clinical laboratory results; To assess the decrease in log 10 HCV RNA from baseline to Day 4, Day 7 and Day 14; To assess the Pharmacokinetic (PK) profiles of subjects treated with NS5A+NS3 with and without SOC/RBV; To evaluate the proportion of subjects with RVR; To evaluate the proportion of subjects with extended rapid virologic response (ervr), defined as undetectable HCV RNA at both Week 4 and 12; To evaluate the proportion of subjects with complete early virologic response (cevr), defined as undetectable HCV RNA at Week 12; To evaluate the proportion of subjects with 24-week sustained virologic response (SVR 24 ), defined as undetectable HCV RNA at follow-up Week 24; To describe resistant variants associated with virologic failure. 6
8 Exploratory Objectives: To explore the effect of HCV subtype on antiviral activity. To describe the changes in T-cell immunoregulatory molecules with treatment through analysis of peripheral blood mononuclear cells (PBMCs). To describe T-cell responses (through analysis of PBMCs) to HCV antigens at baseline and their change over time following treatment. To describe the baseline level of IP-10 in serum in all subjects; To explore the relationship between endpoints of safety or antiviral activity and exposure to when co-administered with and without SOC or ribavirin. To explore the relationship between antiviral activity endpoints and single nucleotide polymorphisms (SNPs) in genes encoding proteins of the IFNλ family (IL28A, IL28B, IL29) and potentially the RBV transporter protein ENT1. To describe changes in host gene expression including interferon stimulated genes (ISGs) and immunoregulatory molecules in whole blood following treatment. To potentially explore the relationship between anemia in subjects receiving RBV and SNPs in the gene for inosine triphosphatase (ITPA). Study Design: This is a randomized, open-label, out-patient, multiple-dose study with parallel treatment groups in 2 parts. Approximately 120 HCV genotype 1, null responders to SOC will be randomized to receive either the combination of NS5A+NS3 (treatment group A) or NS5A+NS3 + SOC or ribavirin (treatment group B) for up to 72 weeks. Where indicated, randomization will be stratified by genotype 1a and 1b with the number of lb subjects enrolled capped, allowing for enrollment of more 1a subjects. Other groups will be restricted to genotype 1b only. Subjects who refuse treatment on Day 1 may be replaced. Otherwise, no replacements are allowed. In order to reduce the risk of resistance developing in a large number of subjects, the study will be conducted in two parts. In part 1, a sentinel cohort of approximately 10 subjects per treatment group for a total of approximately 20 subjects will be randomized. The decision to expand Treatment Groups A and B will be made independently and will be based upon results of the pre-planned decision points at Weeks 2 and 4. Part 2 of the study continues as follows: If the decision is made to expand Treatment Group A then approximately 20 additional subjects, all with HCV genotype 1b will be randomized into each Expansion Cohort A1 and A2. The subjects in the sentinel cohorts as well as the subjects in the Expansion Cohorts will continue treatment for up to 24 weeks. See Figure 1. If the decision is made to expand Treatment Group B then approximately 20 additional subjects with HCV genotype 1a or 1b will be randomized into each of Expansion cohorts B1 and B2. Approximately 20 additional subjects might be enrolled into Expansion cohort B3 contingent upon demonstration of adequate antiviral activity in Expansion cohort B1. The subjects in the sentinel cohorts as well as the subjects in the Expansion cohorts will continue treatment for up to 24 weeks. See Figure 2. To further reduce the risk of resistance, group discontinuation criteria are established for Expansion Cohorts.. Part 1: Part 1 is represented by the sentinel cohort with treatment duration up to 28 days and 2 study decisions at Week 2 and 4. In the sentinel cohort, 20 subjects will be randomized in a 1:1 ratio to one of the two treatment groups (A or B). Also, randomization will be stratified by genotype 1a and 1b, with the 7
9 Week 2 number of 1b subjects capped at 2 per treatment group so that each treatment group will have at least 8 subjects with genotype 1a. In treatment group A, approximately 10 subjects will receive 60 mg of BMS QD and 600 mg of BMS BID in combination. In treatment group B, approximately 10 subjects will receive SOC in addition to the combination of the 2 study drugs. During the analysis of HCV RNA levels, subjects in the sentinel cohort will continue treatment for up to 24 weeks as long as all individual criteria for continuation are met (Part 2). Sparse PK samples will be collected up to 24 hours post-dose on Day 1 and serial blood PK samples will be obtained up to 24 hours post-dose on Day 14. At Week 2 and 4, all subjects will have HCV RNA levels measured. The study will continue and expand if criteria are met as outlined in Table 1 below. Otherwise, cohort will not be expanded and only individual subject decision rules will apply to subjects. (See Table 2.) Table 1: Sentinel Cohort Decision Points at Week 2 and 4 Week 4 Treatment A: NS3 + NS5A Part 1 Sentinel cohort decision point at Week 2 a Treatment B: NS3 + NS5A + SOC If 70% of subjects have undetectable HCV RNA or decrease in plasma HCV RNA 2 log 10 IU/mL without rebound continue treatment group. Otherwise, cohort will not be expanded and only individual subject decision rules will apply to subjects (see table 2) Sentinel cohort decision point at Week 4 a If 50% subjects have undetectable HCV RNA (RVR), continue treatment group and expand cohort. Otherwise, cohort will not be expanded and only individual subject decision rules will apply to subjects (see table 2) Study decisions at Weeks 2 and 4 for treatment groups A and B are made independently of each other. Note: On September 21, 2010, the dose of BMS for all on-going subjects was reduced from 600 mg BID to 200 mg BID (Amendment 06). At that time, all subjects on Treatment B and all subjects on Treatment A who had not received rescue with PegIFN/RBV, had completed their treatment. Five subjects in Treatment A who were receiving extended therapy as a result of viral breakthrough and the addition of PegIFN/RBV to their treatment were impacted by the dose change. Part 2: Part 2 is represented by the duration after Week 4 of the sentinel cohort (both Treatments A and B), and the whole study duration of the Expansion Cohorts. Subjects in the sentinel cohorts will continue dosing following individual subject decision rules. (See Table 2). Expansion of a treatment group will occur only after the sentinel cohort satisfies criteria for successful response to treatment at Week 2 and RVR at Week 4. Expansion of each treatment will be made independently. Thus, study expansion may include Treatment A, Treatment B, or both. If the decision is made to expand treatment group A, approximately 40 additional subjects, all with HCV genotype 1b will be randomized to either Expansion Cohort A1 or A2. 8
10 Expansion Cohort A1: Approximately twenty (20) subjects will be randomization to receive NS3 200 mg BID+NS5A 60 mg QD for up to 24 weeks; Expansion Cohort A2: Approximately twenty (20) subjects will be randomized to receive NS3 200 mg QD +NS5A 60 mg QD for up to 24 weeks; Note: Due to viral breakthrough with treatment A in subjects infected with HCV genotype 1a (GT1a) in the Sentinel Cohort, the decision was made to limit the expansion of treatment group A to GT1b subjects only. If the decision is made to expand treatment group B, approximately 40 additional subjects with HCV genotype 1a or 1b will be randomized to either Expansion Cohort B1 or B2: Expansion Cohort B1: Approximately twenty (20) subjects will be randomized to receive NS3 200 mg BID+NS5A 60 mg QD+SOC for up to 24 weeks Expansion Cohort B2: Approximately twenty (20) subjects will be randomized to receive NS3 200 mg QD+NS5A 60 mg QD+SOC for up to 24 weeks. Randomization of Expansion Cohorts B1 and B2 will be stratified by genotype 1a and 1b, with the number of genotype 1b capped at 20% of total subjects in each cohort. Approximately 20 additional subjects with HCV genotype 1a or 1b might be dosed in Expansion Cohort B3: Expansion Cohort B3: Approximately twenty (20) subjects will receive NS3 200 mg BID+NS5A 60 mg QD+Ribavirin for up to 24 weeks The decision to open Expansion Cohort B3 for dosing is contingent upon demonstration of adequate antiviral activity in Expansion Cohort B1. The decision criterion is described in detail in Rule for Initiating Expansion Cohort B3 (NS3 + NS5A + RBV) below. The number of subjects with HCV genotype 1b will be capped at 20% of the total number of subjects in Expansion Cohort B3. For all subjects in the sentinel and Expansion Cohorts: Blood samples for HCV RNA levels and HCV genomic substitution from all subjects will be obtained on Days -1 (baseline), and at specific timepoints during and after treatment (see pharmacodynamic endpoints below). Peripheral blood mononuclear cells (PBMCs) for assessment of antigen-specific Anti-HCV T cell responses will be collected on Days -1, 7 and 14 in the Sentinel Cohorts, and on Days -1, 14, 28, Weeks 8, 12, 16, 24 on treatment and Weeks 4, 12 and 24 post treatment in the Expansion Cohorts. Blood samples for analysis of polymorphisms in genes encoding proteins of the IFNλ family (IL28A, IL28B, IL29) and potentially the RBV transporter protein ENT1will be collected on Day -1 (baseline). Blood samples for concentration will be obtained on Days 1, 2, 7, 14, 15 and Weeks 4, 8, 12 and 16 for assessment. PK samples on Days 1, 2, 14 and 15 will be collected for all subjects in the Sentinel Cohort and the first 12 subjects in each Expansion Cohort. 9
11 All subjects will be closely monitored for adverse events throughout the study. Safety will be assessed by monitoring vital signs, physical examinations, 12-lead electrocardiograms (ECGs), and clinical laboratory tests. Follow-up will be up to 48 weeks post-treatment. Figure 1: Study Design Schematic, Treatment Group A Treatment A, Sentinel Cohort (NS3 + NS5A for 24 Weeks) a N = 10 Follow-up for 48 weeks post-treatment Week 2 Interim Analysis: If 70% of subjects have undetectable HCV RNA or decrease in plasma HCV RNA 2 log 10 IU/mL without rebound continue treatment group Otherwise, cohort will not be expanded and only individual subject decision rules will apply to subjects. Week 4 Interim Analysis: If 50% subjects have undetectable HCV RNA (RVR), continue treatment group and expand cohort. Otherwise, cohort will not be expanded and only individual subject decision rules will apply to subjects. Expand Treatment A? Yes Expansion Cohort A1 NS3 200 mg BID+NS5A 60 mg QD for 24 Weeks a N = 20 (Genotype 1b) b Expansion Cohort A2 NS3 200 mg QD+NS5A 60 mg QD for 24 Weeks a N = 20 (Genotype 1b) b Follow-up for 48 weeks post-treatment Follow-up for 48 weeks post-treatment a If rescue criteria are met for Individual Subject Decision Rules, SOC can be added for up to 48 additional weeks. b Due to viral breakthrough with treatment A in subjects infected with HCV GT1a in the Sentinel Cohort, the decision was made to limit the expansion of treatment group A to GT1b subjects only. 10
12 Figure 2: Study Design Schematic, Treatment Group B Treatment B, Sentinel Cohort (NS3+NS5A+SOC for 24 Weeks) N = 10 Follow-up for 48 weeks post-treatment. Week 2 Interim Analysis: If 70% of subjects have undetectable HCV RNA or decrease in plasma HCV RNA 2 log 10 IU/mL without rebound continue treatment group Otherwise, cohort will not be expanded and only individual subject decision rules will apply to subjects. Week 4 Interim Analysis: If 50% subjects have undetectable HCV RNA (RVR), continue treatment group and expand cohort. Otherwise, cohort will not be expanded and only individual subject decision rules will apply to subjects. Expand Treatment B? Yes Expansion Cohort B1 NS3 200 mg BID+NS5A 60 mg QD + SOC for 24 Weeks N = 20 (Genotype 1a/1b) c Expansion Cohort B2 NS3 200 mg QD+NS5A 60 mg QD + SOC for 24 Weeks N = 20 (Genotype 1a/1b) c Expansion Cohort B3 a NS3 200 mg BID+NS5A 60 mg QD + RBV for 24 Weeks b N = 20 (Genotype 1a/1b) c Follow-up for 48 weeks post-treatment Follow-up for 48 weeks post-treatment Follow-up for 48 weeks post-treatment a The decision to open Expansion Cohort B3 for dosing is contingent upon demonstration of adequate antiviral activity in Expansion Cohort B1. b If rescue criteria are met for Individual Subject Decision Rules, pegylated Interferon can be added for up to 48 additional weeks in addition to continuation BMS , BMS and Ribavirin. c. Randomization will be stratified by subtype 1a and 1b, and the total enrollment of genotype 1b subjects will be capped at 20% in each cohort. 11
13 Individual Subject decision rules: Individual subject decision rules guide the addition of SOC or pegylated Interferon (PegIFN) alone (based upon treatment assignment) as well as discontinuation of therapy during the study. Decisions are based upon an ongoing evaluation of HCV viral loads and are defined by treatment assignment and study week as outlined in Tables 2 and 3 below. These rules are based upon the definitions of HCV viral breakthrough outlined below: Viral Breakthrough Definitions by Treatment: Treatment Group A Sentinel Cohort 1) Any increase in HCV viral load 1 log from nadir (not necessarily from a consecutive sampling). 2) Any quantifiable HCV RNA 25 IU/mL on or after week 4. 3) Any detectable HCV RNA < 25 IU/mL on or after week 4 confirmed by a subsequent consecutive HCV RNA measurement. Expansion Cohorts A1, A2 and B3 1) Any increase in viral load 1 log from nadir 2) Any confirmed detectable HCV RNA < 25 IU/mL on or after Week 8. Confirmation should occur via an immediate unscheduled return visit. 3) Any quantifiable HCV RNA 25 IU/mL on or after Week 8 (no confirmation needed). Expansion Cohorts B1 and B2 1) Any increase in HCV viral load 1 log from nadir (not necessarily from a consecutive sampling). 2) Any confirmed quantifiable HCV RNA 25 IU/mL after confirmed undetectable HCV RNA. Measurements are confirmed at the next scheduled visit. 12
14 Table 2: Individual Subject Decision Rules for the Sentinel Cohorts Weeks 2 and 3 Weeks 4 to 22 Week 24 Treatment A: NS3 + NS5A Sentinel Cohort Undetectable: continue up to 24 weeks. Detectable > 2 log 10 decrease from baseline with no breakthrough: continue up to 24 weeks Detectable 2 log 10 decrease from baseline with breakthrough: add SOC for up to 48 additional weeks Detectable < 2 log 10 decrease from baseline: discontinue All subjects randomized to Treatment A are expected to have undetectable HCV viral load (<10 IU/mL) beyond Week 4. Undetectable: continue up to 24 weeks. Detectable < 25 IU/mL: perform immediate unscheduled visit for repeat HCV VL. If second value is detectable (>10 IU/mL), add SOC for up to 48 additional weeks Quantifiable 25 IU/mL but 2 log10 decrease from baseline: add SOC immediately for up to 48 additional weeks, and draw viral load for pre-soc baseline. Detectable < 2 log10 decrease from baseline: discontinue Undetectable: stop therapy and observe for SVR. Detectable 2 log 10 decrease from baseline: add SOC for up to 48 additional weeks Detectable < 2 log 10 decrease from baseline: discontinue Treatment B: NS3 + NS5A + SOC Sentinel Cohort Undetectable: continue up to 24 weeks Detectable 2 log 10 decrease from baseline: continue up to 24 weeks Detectable < 2 log 10 decrease from baseline: discontinue Undetectable: continue up to 24 weeks Detectable 2 log 10 decrease from baseline: continue up to 24 weeks Detectable < 2 log 10 decrease from baseline: discontinue Undetectable: stop therapy and observe for SVR Detectable: discontinue 13
15 Table 3: Individual Subject Decision Rules for the Expansion Cohorts Weeks 2 and 3 Weeks 4 and 6 Weeks 8 to 22 Expansion Cohorts A1, A2 and B3 NS3 + NS5A +/- RBV Undetectable: continue up to 24 weeks Detectable 2 log 10 decrease from baseline and NOT > 1 log increase in HCV RNA from nadir: continue up to 24 weeks Detectable < 2 log 10 decrease from baseline or 1 log increase in HCV RNA from nadir: add PegIFN or SOC for up to 48 weeks a Undetectable: continue up to 24 weeks. Detectable < 25 IU/mL: recheck HCV RNA level at next visit Quantifiable 25 IU/mL but not 1 log increase from nadir: recheck HCV RNA level at next visit Quantifiable 25 IU/mL and 1 log increase from nadir: add PegIFN or SOC for up to 48 weeks b Undetectable: continue up to 24 weeks. Detectable < 25 IU/mL: perform immediate unscheduled HCV RNA level recheck. If persistently detectable, add PegIFN or SOC for up to 48 weeks c ; if < 10 IU/mL (undetectable) continue therapy Quantifiable 25 IU/mL: add PegIFN or SOC for up to 48 weeks d Week 24 Undetectable: stop therapy and observe for SVR Detectable < 25 IU/mL: stop therapy and perform an immediate unscheduled HCV RNA level recheck. If persistently detectable, start SOC alone for up to 48 weeks; if < 10 IU/mL (undetectable), observe for SVR Quantifiable 25 IU/mL add PegIFN or SOC for up to 48 weeks e a Expansion Cohort B1 and B2 NS3 + NS5A + SOC Undetectable: continue up to 24 weeks. Detectable > 2 log 10 decrease from baseline: continue up to 24 weeks Detectable < 2 log 10 decrease from baseline or 1 log increase in HCV RNA from nadir: consider discontinuation of therapy Undetectable: continue up to 24 weeks. Detectable 2 log10 decrease from baseline: continue up to 24 weeks Detectable < 2 log 10 decrease from baseline or 1 log increase in HCV RNA from nadir: consider discontinuation of therapy Undetectable: continue up to 24 weeks. Detectable 2 log10 decrease from baseline: continue up to 24 weeks Detectable < 2 log 10 decrease from baseline or 1 log increase in HCV RNA from nadir: consider discontinuation of therapy Undetectable: stop therapy and observe for SVR. Detectable: discontinue therapy. For Expansion Cohorts A1 and A2, the rescue is addition of SOC; For Expansion Cohort B3, the rescue is addition o f pegifn. 14
16 Subjects who receive rescue treatment with SOC or PegIFN alone in addition to their assigned treatment regimen must have a decrease in HCV RNA 2 log10 by 12 weeks from the beginning of rescue therapy in order to continue treatment up to Week 72 with SOC + antivirals. Subjects not meeting this criterion will stop all therapy after Week 12. Subjects who experience viral relapse during post treatment follow up periods will not receive rescue therapy as part of the study protocol. Further treatment will be at the discretion of the treating investigator. Group Decision Rules for Suspending An Expansion Cohort Group decision rules provide guidelines for the early suspension ofcohorts that incur unacceptable rates of viral breakthrough. 1) Each Expansion Cohort will be evaluated independently. 2) Antiviral activity will be assessed on an ongoing basis. 3) Should 50% of treated subjects in a cohort with 10 or more subjects experience viral breakthrough, further randomization into the involved cohort will be stopped. 4) A minimum of 5 subjects of the first 10 planned subjects in a cohort must experience viral breakthrough before a cohort will be stopped. 5) Should the decision rule be met in a cohort including the lower dose of NS3 (200mg QD), subjects receiving treatment in that cohort but not experiencing viral breakthrough will have their NS3 dose increased to 200mg BID. 6) Subjects will only receive therapeutic rescue if individual subject decision criteria for addition of rescue therapy are met and not based upon Group decision criteria. 7) Subjects randomized into the failed cohorts but not dosed will be re-randomized into the remaining treatment cohorts. Rule for Initiating Expansion Cohort B3 (NS3 + NS5A + RBV) 1) Dosing in Expansion Cohort B3 is contingent upon demonstration of adequate antiviral activity in Expansion Cohort B1, i.e., at least 16/20 (>75%) treated subjects in Expansion Cohort B1 must reach week 12 on treatment without viral breakthrough for Expansion Cohort B3 to be initiated Duration of Study: Approximately up to 124 weeks (including a 28 day screening period and follow-up visit up to 48 weeks after date of last treatment). Number of Subjects: Approximately 120 treated. Study Population: Eligible subjects are men and women 1) between the ages of with a body mass index (BMI) of kg/m 2, inclusive, 2) who are null responders (defined as subjects who after at least 12 weeks of therapy with the current SOC have never attained 2 log 10 decline in HCV RNA levels), 3) who are chronically infected with HCV genotype 1, 4) who have not received another NS5A replication co-factor inhibitor or NS3 protease inhibitor, 5) who are not co-infected with HIV or HBV. Subjects must have an HCV RNA 100,000 IU/mL and have a documented fibrotest score 0.72 and APRI 2 or negative cirrhosis based on liver biopsy within 24 months. Subjects with prior documented cirrhosis defining equivalent histopathology on liver biopsy are excluded. Female subjects must not be nursing, or pregnant. Women of Child Bearing Potential (WOCBP) and male subjects who have sex with women must agree to practice highly effective contraception methods (two separate forms of contraception, one of which must be an effective barrier method) during study participation and for at least 12 weeks (treatment group A) and 6 months (treatment group B and treatment group A with SOC added) post-treatment. 15
17 Investigational Products, Dose and Mode of Administration, Duration of Treatment with Investigational Products: Treatment A: All subjects in the Sentinel Cohort will receive the investigational products, 60 mg BMS QD and 600 mg BMS BID, orally in tablet form during the treatment period. All subjects in Expansion Cohorts will receive the investigational products, 60 mg BMS QD and 200 mg BMS BID or QD, orally in tablet form during the treatment period. Treatment B: All subjects in the Sentinel Cohort will receive the investigational products, 60 mg BMS QD and 600 mg BMS BID, orally in tablet form with pegylated Interferon injected subcutaneously and ribavirin orally in tablet form during the treatment period. All subjects in the Expansion Cohorts will receive the investigational products, 60 mg BMS QD and 200 mg BMS BID or QD, orally in tablet form with pegylated Interferon injected subcutaneously and ribavirin orally in tablet form, or with ribavirin orally in tablet form alone during the treatment period. Note: For subjects in the Sentinel Cohorts, the dose of BMS was reduced from 600 mg BID to 200 mg BID for 5 rescued Treatment A subjects when they were at Week 32 to 37 of treatment. Other subjects received 600 mg BID and completed treatment before the change. Study Assessments and Endpoints: Safety Outcome Endpoints: Safety assessments will be based on medical review of the frequency of SAEs and AEs, discontinuations due to AEs, and abnormalities observed from vital sign and ECG measurements, physical examinations and clinical laboratory results. Pharmacokinetic Endpoints: Pharmacokinetic parameters [Cmax, Cmin, Tmax, and AUC(TAU)] will be derived from plasma concentration versus time data for up to 24 hours on Day 14. Sparse/trough PK samples will be collected at Days 1, 2, 7 and Weeks 4, 8, 12 and 16. Pharmacodynamic Endpoints: Plasma HCV RNA levels will be measured at screening and on Days -1 (baseline), 1 to 7, 9, 11, 14, Week 3 and every 2 weeks from Week 4 to Week 12 in all subjects. HCV RNA measurements will then proceed as follows: All Treatment group A cohorts and Expansion Cohort B3: Every 2 weeks from Week 12 until EOT unless SOC is added to the regimen, in which case, samples will be collected every 4 weeks from Week 12 until EOT. Subjects who have SOC or pegifn added due to viral breakthrough will have HCV RNA measured immediately prior to administration of the first dose of pegifn and every 4 weeks thereafter until EOT. Treatment group B sentinel cohort and Expansion Cohorts B1 and B2: HCV RNA level measurement every 4 weeks from Week 12 until EOT. All subjects: HCV RNA measured at Week 4, 12, 24, 36, and 48 post-treatment. Characterization of HCV genomic substitutions associated with exposure of BMS and BMS will be determined from blood samples collected from all subjects and for which sufficient HCV RNA permits analysis (generally but not exclusively 1000 IU/mL). 16
18 Part 1 Sentinel Cohorts: Proportion of subjects with successful response to treatment at Week 2. Proportion of subjects with RVR at Week 4. Decrease from baseline in plasma HCV RNA levels at Days 4, 7 and 14. Part 2 Sentinel Cohorts: Proportion of subjects who achieve: ervr; cevr; undetectability at end of treatment; SVR12, SVR24 and SVR48 (defined as undetectable HCV RNA (< 10 IU/mL) at follow-up week 48). Part 2 Expansion Cohorts: Proportion of subjects who achieve: RVR; ervr; cevr; undetectability at end of treatment; SVR12, SVR24 and SVR48. Decrease from baseline in plasma HCV RNA levels at Days 4, 7 and 14. Exploratory Endpoints: Possible exploratory T cell analyses might include IFN-γ production in response to HCV peptide antigens as well as levels of PD-1 expression. Relationship between polymorphisms in genes encoding proteins of the IFNλ family (IL28A, IL28B, IL29) and potentially the RBV transporter protein ENT1with virologic response may be explored. Statistical Methods: Sample Size Determination: In order to reduce the risk of resistance developing in a large number of subjects, the study will be conducted in two parts: In Part 1 approximately 10 subjects will be treated in each of Treatments A or B in the sentinel cohorts. Part 1 includes the sentinel cohorts up to Week 4, after witch the decision to introduce up to 5 expansion cohorts will be made with a target sample size of 20 subjects per treatment group. The subjects in the sentinel cohorts and any subjects in the expansion cohorts will continue treatment for up to 24 weeks (Part 2). The sample size selections for each part of the study and for each treatment group are not based on statistical power calculations. Below are risk assessments calculations based upon the endpoints needed to answer the primary objectives for Part 1 and Part 2 with sample sizes of 10 (sentinel cohorts) or 20 (expansion cohorts). For Part 1 Sentinel Cohorts: Two decision points for each treatment group at Week 2 and 4 will be made independently of each other. For the Week 2 decision, given 10 subjects in each sentinel cohort, the probability of observing 70% or more subjects (at least 7 out of 10) with successful response to treatment is 0.17 if the population rate is 50%. The probability of observing 70% or more subjects (at least 7 out of 10) with successful response to treatment is 0.88 if the population rate is 80%. If the observed rate of success is 70% (7 out of 10), the 90% lower confidence bound is 45%. For the Week 4 decision, given 10 subjects in each sentinel cohort, the probability of observing 50% or more subjects (at least 5 out of 10) with RVR is 0.15 if the population rate is 30%. The probability of observing 50% or more subjects with RVR is 0.83 if the population rate is 60%. If the observed rate of success is 50% (5 out of 10), the 90% lower confidence bound is 27%. For Part 2 Sentinel Cohorts: Given 10 subjects in each cohort, the probability of observing 20% or more subjects (at least 2 of 10) with SVR 12 is 0.26 if the true population rate is 10%. The probability of observing 20% or more subjects with SVR 12 is 0.85 if the true population rate is 30%. In addition, if the true observed rate is 20% (2 out of 10), the 80% CI is (5%, 45%). A target sample size of 10 subjects in each cohort can 17
19 also detect, with 46% or 80% probability a safety event that occurs at incidence rates of 6% or 15%, respectively. For Part 2 Expansion Cohorts: Given 20 subjects in each Expansion Cohort, the probability of observing 20% or more subjects in a cohort (at least 4 of 20) with SVR12 is 0.13 if the population rate is 10%. The probability of observing 20% or more subjects in a cohort with SVR12 is 0.89 if the population rate is 30%. In addition, if the observed rate is 20% (4 out of 20), the 80% CI is (9%, 36%). A target sample size of 20 subjects per Cohort can also detect, with 71% probability, a safety event that occurs at an incident rate of 6% in a cohort. Statistical Analysis: Analyses will be based on treated subjects. Results will be presented by treatment group and cohort separately. Interim analyses of pharmacodynamic, pharmacokinetic, and safety data will be performed for data collected. Analyses may include listings, summaries, and graphs of HCV RNA, genotype subtypes, AEs, ECGs, vital signs, selected laboratory parameters, and exploratory biomarkers. Schedule of Analyses: Sentinel Cohorts Interim analyses may be performed at the following time points: 1) after all subjects in sentinel cohort reach Week 2; 2) after all subjects in sentinel cohort reach Week 4; 3) after all subjects in sentinel cohort reach Week 12; 4) after all subjects in sentinel cohort reach Week 24; 5) after all subjects in sentinel cohort reach Follow-up Week 12; 6) after all subjects in sentinel cohort reach Follow-up Week 24; 7) after all subjects in sentinel cohort reach Follow-up Week 48 Interim analyses will include analysis of safety and antiviral activity by treatment group, and biomarkers including IL28B subject genotype and anti-hcv T-cell. Expansion Cohorts Interim analyses may be performed at the following time points: Expansion cohorts A1, A2, B1 and B2: 1) after all subjects complete 4 weeks of treatment (including analysis of RVR); 2) after all subjects complete 12 weeks of treatment (including analysis of ervr and cevr); 3) after all subjects complete 24 weeks of treatment (including analysis of 4 week sustained virologic response (SVR4)) defined as undetectable HCV RNA at follow-up Week 4; 4) after all subjects complete 12 weeks of follow-up after end of treatment (including SVR12); 5) after all subjects complete 24 weeks of follow-up after end of treatment (including SVR24); 6) after all subjects complete 48 weeks of follow-up after end of treatment (including SVR48). In the event that significant delays in enrollment occur between Expansion Cohorts A1, A2 and B1, B2, Interim analyses may be performed on A1/A2 or B1/B2 separately as needed. Expansion cohort B3: 1) after all subjects complete 4 weeks of treatment (including analysis of RVR); 2) after all subjects complete 12 weeks of treatment (including analysis of ervr and cevr); 3) after all subjects complete 24 weeks of treatment (including analysis of 4 week sustained virologic response (SVR4)) defined as undetectable HCV RNA at follow-up Week 4; 4) after all subjects complete 12 weeks of follow-up after end of treatment (including SVR12); 5) after all subjects complete 24 weeks of follow-up after end of treatment (including SVR24); 6) after all subjects complete 48 weeks of follow-up after end of treatment (including SVR48). In the event one or more group decision criteria are met and a group is stopped, an interim analysis may be performed. These interim analyses all include analyses of antiviral activity by cohort. In addition, analysis of safety and biomarkers including IL28B subject genotype and anti-hcv T-cell might be performed by cohort. 18
20 Final Analyses The final analyses will be performed after all subjects complete 48 weeks of follow-up after end of treatment. Antiviral Activity In general, the proportion of subjects with antiviral activity endpoints will be summarized using modified intent-to-treat (ITT): the numerator is based on treated subjects meeting the response criteria (regardless of add-on SOC); the denominator is based on all treated subjects. Response rates and 80% exact binomial CIs will be presented by treatment group. For treatment group A and Expansion Cohort B3, an additional analysis will classify subjects who added on SOC or pegifn before the endpoint assessment as failures. Select antiviral activity endpoints will also be summarized by HCV subtype (1a, 1b). No multiple testing adjustments will be made. Part 1: Analyses will be based on the sentinel cohort. A co-primary early antiviral activity endpoint is the proportion of subjects with either undetectable HCV RNA at Week 2 or 2 log10 IU/mL decrease in plasma HCV RNA from baseline without rebound during the first 2 weeks. Rebound is defined as 1 log10 IU/mL increase in HCV RNA from nadir either at more than 1 time point (not necessarily consecutive) or at last value through Week 2 or detectable RNA after achieving undetectable RNA. Another co-primary early antiviral activity endpoint is the proportion of subjects with RVR. A secondary antiviral activity endpoint is the change from baseline at Day 4, Day 7 and Day 14 in log10 HCV RNA. The magnitude of the change in log10 HCV RNA levels will be assessed by summarizing changes from baseline, including mean, standard deviation, 90% CIs, median and range by study day and treatment group. Each individual s maximum decrease from baseline in log10 HCV RNA will be summarized by treatment group, and frequency distributions for maximum decrease in log10 HCV RNA will be provided by treatment group. The day of maximum decrease in log10 HCV RNA observed will be estimated and summarized by treatment group. The correlations between exposure to BMS and BMS with or without SOC [Cmax, AUC(TAU) and Cmin] and the magnitude and day of maximum change in HCV RNA levels from baseline will be explored by scatter plots. Log10 HCV RNA levels and changes from baseline will also be summarized by HCV subtype (1a, 1b). Part 2: Analyses will be based on the sentinel and Expansion Cohorts. The following primary and secondary antiviral activity endpoints will be summarized: proportions of subjects with RVR, ervr, cevr, SVR12, SVR24 and SVR48; frequency of genotypic substitutions associated with virologic failure. For treatment group A and Expansion Cohort B3, SVR endpoints will also be summarized by the duration subjects stay on NS3+NS5A only, or NS3+NS5A+RBV (eg, Weeks 2, 4, 12, 24 and 48). For the Expansion Cohorts only: A secondary antiviral activity endpoint is the change from baseline at Days 4, 7 and 14 in log10 HCV RNA. The magnitude of the change in log10 HCV RNA levels will be assessed by summarizing changes from baseline, including mean, standard deviation, 90% CIs, median and range by study day and cohort. Each individual s maximum decrease from baseline in log10 HCV RNA will be summarized by cohort, and frequency distributions. The day of maximum decrease in log10 HCV RNA observed will be estimated and summarized by cohort. 19
21 The relationship between antiviral activity endpoints and exposure to with and without SOC/RBV will be explored graphically. Safety Analysis: All recorded adverse events will be listed and tabulated by system organ class and intensity, preferred term and cohort. Vital signs, clinical laboratory tests will be listed and summarized by cohort. Any physical exam findings and clinical laboratory results will be listed. ECG readings will be evaluated by the investigator and abnormalities, if present, will be listed. Endpoints of safety may also include selected AEs, changes from baseline in laboratory parameters and lab abnormalities by toxicity grades. The relationship between safety endpoints and exposure to with and without SOC/RBV will be explored graphically. PK analysis: The multiple-dose pharmacokinetic parameters of BMS and BMS with and without SOC/RBV will be summarized by treatment and cohort. Biomarker analysis: The relationship between BMS and BMS and the exploratory biomarkers (genotype subtypes, resistant variants, and anti-hcv T cell responses) may be explored graphically and, if appropriate, estimated. 20
22 TABLE OF CONTENTS TITLE PAGE... DOCUMENT HISTORY... SYNOPSIS... TABLE OF CONTENTS... 1 INTRODUCTION AND STUDY RATIONALE Research Hypothesis Investigational Product Development Rationale Summary of Results of BMS and BMS Investigational Program BMS Summary of Non-Clinical Programs for BMS Summary of Clinical Investigational Program for BMS Pharmacokinetic and Pharmacodynamic Results for BMS Studies of Clinical Efficacy of BMS Summary of Clinical Safety of BMS BMS Summary of Non-Clinical Programs for BMS Summary of Clinical Investigational Program for BMS Pharmacokinetic and Pharmacodynamic results for BMS Summary of Clinical Efficacy Summary of Clinical Safety of BMS Combination of BMS and BMS Combination Non-Clinical Toxicology Combination Clinical Data in Humans Study Rationale Rationale for Combining Multiple Direct-acting Antivirals (DAA) Rationale for Dose Selection of BMS
23 1.4.3 Rationale for Dose Selection of BMS Approaches to Reduce Drug Resistance Rationale for Treatment with 24 Weeks of BMS and BMS alone Rationale Supporting Addition of Ribavirin to BMS and BMS Other Rationale Overall Risk/Benefit Assessment BMS BMS Risks of Treatment Including Pegylated Interferon and/or Ribavirin... 2 STUDY OBJECTIVES Primary Objective Secondary Objectives Exploratory Objectives... 3 ETHICAL CONSIDERATIONS Good Clinical Practice Institutional Review Board/Independent Ethics Committee Informed Consent... 4 INVESTIGATIONAL PLAN Study Design and Duration Overview Part Part Individual Subject Decision Rules Viral Breakthrough Definitions by Treatment: Group Decision Rules for Suspending an Expansion Cohort Rule for Initiating Expansion Cohort B3 (NS3 + NS5A + RBV) Study Population Inclusion Criteria Exclusion Criteria Discontinuation of Subjects from Treatment... 5 TREATMENTS Study Treatment
24 5.1.1 Investigational Product Noninvestigational Product Identification Identification of BMS Identification of BMS Identification of Peg-interferon alfa-2a(pegasys ) Identification of RBV (Copegus ) Packaging and Labeling Packaging and Labeling- BMS Packaging and Labeling- BMS Packaging and Labeling - peginterferon alfa-2a Packaging and Labeling - ribavirin (Copegus ) Handling and Dispensing Handling and Dispensing BMS Handling and Dispensing BMS Handling and Dispensing Peg-interferon alfa- 2a(Pegasys ) Handling and Dispensing Ribavirin (Copegus ) Method of Assigning Subjects to a Treatment Selection and Timing of Dose for Each Subject Dose modification Pegylated Interferon and Ribavirin BMS and BMS Blinding/Unblinding Concomitant Treatments Prohibited and/or Restricted Treatments Treatment Compliance... 6 STUDY ASSESSMENTS AND PROCEDURES Flow Chart/Time and Events Schedule Study Materials Safety Assessments Medical History Vital Signs Physical Examinations Physical Measurements Electrocardiograms Other
25 6.3.7 Adverse Event Monitoring Laboratory Test Assessments Efficacy Assessments Primary Efficacy Assessment Secondary Efficacy Assessments Pharmacokinetic Assessments Pharmacokinetics: Collection and Processing Pharmacokinetic Sample Analyses Labeling and Shipping of Biological Samples Pharmacodynamic Assessments Pharmacogenomic/Pharmacogenetic Assessments Outcomes Research Assessments Other Assessments Exploratory Research... 7 ADVERSE EVENTS Definitions Serious Adverse Events Nonserious Adverse Events Assignment of Adverse Event Intensity and Relationship to Study Drug Collection and Reporting Serious Adverse Event Collection and Reporting Handling of Expedited Safety Reports Nonserious Adverse Events Laboratory Test Abnormalities Overdose Potential Drug Induced Liver Injury (DILI) Pregnancy Requirements for Pregnancy Testing Reporting of Pregnancy Other Safety Considerations... 8 STATISTICAL CONSIDERATIONS Sample Size Determination Sample Size for Part 1 Sentinel Cohorts Sample Size for Part 2 Sentinel Cohorts Sample Size for Part 2 Expansion Cohorts Populations for Analyses
26 8.3 Endpoint Definitions Safety Endpoint Pharmacokinetic Endpoint(s) Pharmacodynamic (PD) Endpoints PD Endpoints for Part PD Endpoints for Part Other Endpoint(s) Analyses Demographics and Baseline Characteristics Safety Analyses Efficacy Analyses Efficacy Analyses for Part Efficacy Analyses for Part Pharmacokinetic Analyses Pharmacokinetic/Pharmacodynamic Analyses Pharmacogenomic Analyses Outcomes Research Analyses Other Analyses Interim Analyses Interim Analyses of the Sentinel Cohorts Interim Analyses of the Expansion Cohorts Final Analyses... 9 ADMINISTRATIVE SECTION Compliance Compliance with the Protocol and Protocol Revisions Monitoring Investigational Site Training Records Retention Case Report Forms Study Drug Records Return and Destruction of Study Drug Return of Study Drug Destruction of Study Drug Publications GLOSSARY OF TERMS LIST OF ABBREVIATIONS REFERENCES
27 APPENDIX 1 ADDITIONAL ETHICAL CONSIDERATIONS... APPENDIX 2 DIAGNOSTIC CRITERIA FOR DRUG AND ALCOHOL ABUSE... APPENDIX 3 DIVISION OF AIDS TABLE FOR GRADING THE SEVERITY OF ADULT AND PEDIATRIC ADVERSE EVENTS PUBLISH DATE: DECEMBER,
28 1 INTRODUCTION AND STUDY RATIONALE 1.1 Research Hypothesis Part 1: The observed proportion of Hepatitis C Virus (HCV) genotype 1 null responder subjects in the sentinel cohort with successful response to treatment is 70% at Week 2 and rapid virologic response (RVR) is 50% at Week 4 for the combination of (NS5A+NS3) with and without SOC. Successful response to treatment is defined at Week 2 as either undetectable HCV RNA (< 10 IU/mL) or 2 log 10 IU/mL decrease in plasma HCV RNA from baseline without rebound and at Week 4 by a RVR defined as undetectable HCV RNA (< 10 IU/mL). Part 2: The observed proportion of null responder subjects achieving 12-week sustained virologic response (SVR 12 ) is 20%. SVR 12 is defined as undetectable HCV RNA (< 10 IU/mL) at follow-up Week Investigational Product Development Rationale Approximately 170 million people worldwide are chronically infected with Hepatitis C virus (HCV), including approximately 4 million in the United States. 1 The majority of infected individuals progress to chronic hepatitis, which can lead to cirrhosis, liver failure, and hepatocellular carcinoma (HCC). Currently, 10,000 to 12,000 deaths annually in the United States are due to HCV infection. 1 There are 6 major HCV genotypes with many subtypes based on sequence heterogeneity of the genome. 2 Genotypes 1-3 have a worldwide distribution (with genotype 1 being the major genotype in the United States, Europe, and Japan), genotypes 4 and 5 are found principally in Africa, and genotype 6 is distributed primarily in Asia. Although genotype does not predict the outcome of infection, different genotypes are associated with differential responses to treatment, and allow dosage of current interferon-based treatment to be tailored to the genotype being treated. 3,4,5 27
29 The SOC for treating most patients with chronic HCV infection is a regimen of pegifnα and RBV. In two (2) pivotal clinical trials in treatment-naive patients receiving pegifnα-2b or pegifnα-2a combined with ribavirin, treatment failure, defined as persistent HCV replication up to 24 weeks after the end of treatment (EOT), occurred in 18% and 24% of patients infected by genotype 2 or 3 and in 58% and 54% of patients infected by genotype 1, respectively. 4,5 Treatment failure is more frequent for some groups, including African Americans and individuals coinfected with human immunodeficiency virus (HIV). 6,7,8,9,10,11 In addition, current therapies are unpleasant to administer and are associated with significant side effects resulting in high rates of noncompliance and apprehension about starting treatment. This highlights the unmet medical need for new therapeutic regimens that are more effective (especially in patients infected by genotype 1), less toxic and easier to administer. HCV contains a single-stranded ribonucleic acid (RNA) genome of positive polarity which is translated into a precursor polyprotein and cleaved secondarily into individual proteins. The N-terminus of the non-structural protein NS3 is a serine protease (NS3 protease) which interacts with the non-structural protein NS4A to form an active proteolytic complex. The NS3/4A protease complex is responsible for processing four cleavage events in the HCV polyprotein to yield mature viral replication proteins. This activity is essential for viral replication in vivo. Since efficient propagation of genotype 1 isolates has not been achieved in vitro, surrogate assays were developed to assess the potential antiviral activity of protease inhibitors. 12 BMS has specific activity against the HCV NS3 protease. Proof-of-principle of the HCV NS3 protease as a viable target in humans has been achieved. Schering-Plough and Vertex have demonstrated antiviral activity in man during 2 weeks of TID dosing of similar HCV protease inhibitors, although resistance to monotherapy emerged rapidly during this time. 13,14 Combination studies of the Vertex NS3 protease inhibitor with current standard of care appears to suppress the emergence of resistance. 15 BMS is an inhibitor of HCV non-structural protein 5A (NS5A). HCV NS5A is a multifunctional protein with key functions in HCV replication, modulation of cellular signaling pathways and the interferon response. 16 It contains a putative interferon sensitivity-determining region and may play a role in resistance to IFNα. 17 The essential 28
30 nature of NS5A has been demonstrated in vitro in the cell based replicon assay 18 and in vivo in the chimpanzee model of infection. 19 As a multifunctional protein required for in vivo and in vitro replication with no known human homologues, NS5A represents an attractive target for therapeutic intervention. Proof-of-Principle for NS5A as a viable target has been demonstrated with BMS in early clinical studies conducted in HCV infected subjects and is discussed in Section 1.3. One strategy for competitor compounds under current development is to add the direct acting antiviral (DAA) to SOC with the goal of achieving higher SVR rates in genotype 1 patients. Another strategy is to replace SOC with a combination of direct acting antivirals. In November 2008, Roche initiated the INFORM-1 trial, a potentially ground-breaking study to investigate the activity of a combination of two oral antiviral molecules in the absence of interferon. 20 The study investigated the combination of Pharmasset's R7128, a nucleoside inhibitor, with InterMune's R7227, a protease inhibitor. are being developed to address some of the shortcomings of current SOC therapy and will be developed as add-on to SOC. Finally, at the 2010 AASLD meeting held in Boston Oct 29 to Nov 3rd 2010, several companies (Gilead, Boheringer Ingelhim, Vertex) announced studies combining multiple direct acting antivirals alone, with ribavirin, or in combination with pegylated interferon and ribavirin. 1.3 Summary of Results of Investigational Program Section 1.3 summarizes key non-clinical and clinical study findings that may have potential relevance to. Section 1.3 also includes data that may not yet be incorporated in the Investigator Brochures (IB) at the time the protocol was written. A complete summary of the nonclinical and clinical investigational programs can be found in the IB. 21, BMS BMS is being developed for use in combination with pegifnα/rbv as well as for pegifnα/rbv sparing combinations studies with other DAAs for the treatment of chronic HCV. Details of the pre-clinical and early Phase 1 clinical studies are available in 29
31 the Investigator Brochure (IB). This section will emphasize important data that may be relevant to the management of subjects enrolled in Summary of Non-Clinical Programs for BMS Drug Interactions and Nonclinical Metabolism BMS has the potential to inhibit the transport of compounds that are P-glycoprotein (P-gp) or OATP1B1 substrates. In addition, the disposition of BMS by P-gp and CYP3A4 might be altered by compounds that affect the activities of these molecules. BMS was a substrate of P-gp and both passive permeability and the action of 1 or more transporters appear to be involved in the liver uptake of BMS Systemic clearance of BMS was low to moderate in mouse, rat, dog and monkey. CYP3A4 was the primary enzyme involved in the metabolism of BMS BMS was a weak to moderate in vitro inhibitor of CYP3A4, P-gp, and OATP1B1 but did not induce metabolic enzymes. The elimination of BMS was primarily the result of metabolic clearance, biliary excretion and intestinal secretion. Reproductive Risk Potential In studies of embryo-fetal development in rats and rabbits, preliminary data indicates that BMS is not a selective developmental toxicant in either species. At higher doses however, maternal and developmental (teratogenicity) toxicities were evident in both species. A more detailed summary of these study results can be found in the IB. Carcinogenesis, Mutagenesis, and Impairment of Fertility Nonclinical genetic toxicology studies have shown that BMS was not genotoxic in in vitro or in in vivo assays. A 2-year study in rats and a 6-month study in Tg.rasH2 transgenic mice are planned Summary of Clinical Investigational Program for BMS Twenty-one (21) studies with BMS in healthy volunteers, HCV-infected subjects, and hepatic impaired subjects have been completed or are ongoing. Please see Table for details. 30
32 Table : Completed or On-going Clinical Trials of BMS Clinical Trial Study Number Dosing Duration Test Agent SAD/HV AI Single Dose BMS SAD/HCV AI Single Dose BMS MAD/HV AI days BMS MAD/HCV AI days BMS DDI/HV AI Single Sequence BMS mg ketoconazole ADME AI Single Dose 14 C-BMS SAD/MAD (Japanese subjects) AI Single-Dose/Multipledose 14 days BMS DDI AI days BMS with 5 mg midazolam BA/FE/H2 AI Single Dose BMS and 40 mg famotidine and high fat meal DDI/HV AI Single Sequence BMS mg rifampin Hepatic Impairment AI Single Dose BMS DDI/WOCBP AI Multiple-dose 10 days BMS Ortho-Tricyclen HCV AI Weeks with P/R BMS RBV + pegifnα-2a DDI/HV AI /14 days BMS BMS HCV AI or 24 Weeks with P/R BMS RBV + pegifnα-2a HCV AI Weeks with P/R BMS RBV + pegifnα-2a HCV AI Weeks with P/R BMS RBV + pegifnα-2b HCV AI Weeks with P/R BMS RBV + pegifnα-2a HCV 24 or 48 Weeks BMS BMS /- pegifnα-2a/rbv HCV AI Weeks BMS BMS Abbreviations in the table: ADME - absorption, distribution, metabolism and excretion, BA - bioavailability, DDI - drug-drug interaction, FE - food effect, HCV - HCV genotype 1 infected subjects, HV - healthy volunteers, H2 - H2 receptor antagonist, MAD - 31
33 multiple-ascending dose study, SAD - single-ascending dose study, P/R - pegifnα plus RBV, WOCBP - women of childbearing potential Pharmacokinetic and Pharmacodynamic Results for BMS Single-Ascending Dose Study in Normal Healthy Volunteers (AI444001) BMS was readily absorbed following oral administration of a solution formulation with Tmax reached at ~ 1 to 2 hours after dosing. Exposure to BMS (AUC and Cmax) appeared to increase proportionally with respect to dose in the range of 1 to 200 mg. The mean terminal T-HALF of BMS was ~ 10 to 13 hours, which supports the potential for once-daily administration. Single-Ascending Dose Study in Subjects with Chronic Hepatitis C (AI444002) BMS was readily absorbed following oral administration in HCV-infected subjects with a T-HALF of approximately 10 to 14 hours. The PK in HCV infected subjects in AI and in HV in AI were comparable after single oral doses of 1 to 100 mg of BMS solution formulation; however, increased variability was observed in HCV-infected subjects. Subjects infected with HCV genotype 1a experienced a median decline in HCV RNA (log 10 ) 24 hours after dosing that ranged from 1.9 with BMS (1 mg) to 3.57 with BMS (100 mg) (Table A and Figure A). Subjects infected with HCV genotype 1b experienced median decline in HCV RNA (log 10 ) 24 hours after dosing that ranged from 3.14 with BMS (100 mg) to 3.55 with BMS (10 mg). Table A: Median Decreases in HCV RNA in HCV-1 Infected Subjects by Genotype Subtype Median Decrease in log 10 HCV RNA from Baseline to 24 Hours after Dosing BMS Genotype 1a Genotype 1b Dose (mg) N Median (Range) n Median (Range) (0.18, 3.00) (2.88, 2.98) (3.12, 3.99) (3.53, 3.60) (2.73, 3.40) Source: Study AI
34 Figure A: Median Change in log 10 HCV RNA from Baseline (AI444002) Placebo 1 mg 10 mg 100 mg A 1B log10 HCV RNA Time (Hours) HCV RNA is set to 25 IU/mL if HCV RNA < LLOQ and to 10 IU/mL if HCV RNA is undetectable Placebo n=2 (1A: n=2, 1B: n=0) BMS mg n=6 (1A: n=6, 1B: n=0) BMS mg n=5 (1A: n=3, 1B: n=2) BMS mg n=5 (1A: n=2, 1B: n=3) Note: HCV RNA only available for up to 8 hours for one subject with genotype subtype 1A receiving BMS mg Source: Study AI Multiple-ascending Dose Study in Healthy Volunteers (AI444003) Thirty-three (33) healthy subjects were dosed at 1, 10, 30 and 60 mg once-daily (QD) for 14 days in study AI BMS was readily absorbed following multiple oral doses of the capsule formulation in HV at doses ranging from 1 to 60 mg daily for 14 days. Median Tmax ranged from 1.0 to 2.0 hours and plasma concentrations declined in a multi-phasic manner with mean terminal T-HALF values ranging from 13 to 15 hours. Steady-state was generally achieved between Days 3 and 5. Exposure (Cmax, Cmin, AUC(TAU)) increased with increasing dose in a modestly greater than dose-proportional manner across the dose range studied. Geometric means of accumulation indices for Cmax and AUC(TAU) ranged from 0.88 to 1.22 and 1.04 to 1.37, respectively, which are consistent with the T-HALF of BMS and a QD dosing regimen. 33
35 Multiple-ascending Dose Study in Subjects with Chronic Hepatitis C (AI444004) BMS was readily absorbed following multiple oral doses of the capsule formulation in HCV-infected subjects at doses ranging from 1 to 100 mg daily for 14 days. Similar to HV, steady state appeared to be achieved by ~ 48 to 72 hours (Days 3-4) based on trough levels of BMS The mean accumulation index observed following 1 to 100 mg QD dosing for 14 days was ~ 0.85 to Tmax on Day 14 of mg dosing was reached at around 1 to 2 h post dose. The mean terminal T-HALF was about ~ 13 to 15 h following the last 30 mg or 100 mg dose on Day 14. The overall exposures to BMS in HCV-infected subjects appeared to be lower than those observed in HV at doses from 1 to 30 mg, but similar at 60 mg. Subjects infected with HCV genotype 1a experienced a median decline in HCV RNA (log 10) on Day 4 after dosing that ranged from 1.46 with BMS (1 mg) to 3.64 with BMS (60 mg) (preliminary data as of 02-Mar-2009). Subjects infected with HCV genotype 1b experienced median decline in HCV RNA (log 10) on Day 4 after dosing that ranged from 2.85 with BMS (1 mg) to 4.52 with BMS (100 mg) (Table B and Figure B). HCV RNA values rebounded in most HCVinfected subjects during administration of BMS monotherapy suggesting that resistant viral variants may become clinically relevant in subjects receiving monotherapy. Table B: Median Decreases in HCV RNA in HCV-1 Infected Subjects by Genotype Subtype BMS Dose (mg) Median Decrease in log 10 HCV RNA from Baseline to Day 4 Genotype 1a Genotype 1b N Median (Range) n Median (Range) (1.34, 1.58) (2.36, 3.34) (2.18, 3.12) (3.58, 4.25) (2.70, 3.23) 30 BID (0.00, 3.36) (3.67, 4.44) (2.91, 4.27) (2.37, 3.74) (4.52, 4.52) BID - twice daily Source: Study AI preliminary data 34
36 Figure B: Median Changes from Baseline in log 10 HCV RNA (AI444004) Placebo 1mg 10mg 30mg 30mgBID 60mg 100mg 1 1A B log10 HCV RNA Time (Days) HCV RNA is set to 25 IU/mL if HCV RNA < LLOQ and to 10 IU/mL if HCV RNA is undetectable Placebo n=6 (1A: n=1, 1B: n=5) BMS mg n=4 (1A: n=2, 1B: n=2) BMS mg n=4 (1A: n=2, 1B: n=2) BMS mg n=4 (1A: n=4, 1B: n=0) BMS mg BID n=4 (1A: n=2, 1B: n=2) BMS mg n=4 (1A: n=4, 1B: n=0) BMS mg n=4 (1A: n=3, 1B: n=1) Source: Study AI preliminary data Drug-Drug Interaction Study with Ketoconazole in Healthy Volunteers (AI444005) AI444005, a DDI study in HV, assessed the effect of a potent CYP3A4 inhibitor, ketoconazole, on the PK of BMS Fourteen (14) HV received a single oral dose of BMS mg on Day 1, ketoconazole 400 mg QD on Days 5 to 8, concomitant administration of BMS mg and ketoconazole 400 mg on Day 9 followed by ketoconazole 400 mg QD on Days 10 to 13. PK assessment demonstrated that coadministration of BMS mg and ketoconazole 400 mg resulted in 1.6 fold, 2.9-fold and 3.0-fold increase in geometric mean Cmax, AUC(0-T) and AUC(INF) of BMS , respectively 35
37 ADME Study of 14C-labeled BMS in Healthy Male Volunteers (AI444006) Six (6) subjects were randomized and treated with 25 mg of [14C]-BMS Preliminary biotransformation data suggests that there is minimal metabolism of BMS Of the total dose administered, ~ 87% was recovered in feces, primarily as parent drug, and some as BMS (M2); ~ 6.7% was recovered in urine predominantly as parent drug. In addition, minimal metabolites were present in plasma (< 5%). SAD/MAD Study in Healthy Japanese Subjects (AI444007) and Comparative Pharmacokinetics of BMS in Japanese and Non-Japanese Subjects In Japanese HV, exposure (AUC and Cmax) to BMS appeared to be dose proportional across the range of single doses (oral solution) from 1 to 200 mg and multiple doses (capsule formulation) from 1 to 100 mg administered for 14 days. Exposures (AUC and Cmax) were similar between the Japanese and non-japanese HV after single and multiple-doses of BMS Drug-drug Interaction Study with Midazolam in Healthy Volunteers (AI444008) Eighteen (18) HV were randomized and treated as follows: Day 1, single dose of midazolam (MDZ; 5 mg), Days 2 to 5, BMS (60 mg) QD, and Day 6, single dose of MDZ (5 mg) and BMS (60 mg). The geometric mean ratios for MDZ Cmax were unaffected and AUC(INF) and AUC(0-T) were slightly reduced by ~ 13%. The 90% CIs were estimated to be (0.83, 0.92), and (0.83, 0.93), respectively. Relative Bioavailability/Food Effect/Famotidine Study in Healthy Volunteers (AI444009) Eighteen (18) HV were randomized and treated in this 5-period sequential study which evaluated the relative bioavailability of a dry granulated and a direct compression tablet relative to the capsule formulation previously used in early Phase 1 studies. In addition, the effect of a high fat meal and the effect of an acid modifier (famotidine 40 mg) were assessed. Increased bioavailability measured by geometric mean Cmax (32% to 37%), AUC(0-T) [17% to 22%], and AUC(INF) [17% to 22%] was observed with both dry granulation and direct compression tablets compared with the capsule formulation. 36
38 Administration of a high-fat meal had the effect of reducing the bioavailability of the dry granulation tablet as measured by geometric mean Cmax (43%), AUC(0-T) (26%), and AUC(INF) (26%). Hepatic Impairment Study (AI444013) Study AI evaluated the single-dose PK of BMS in subjects with hepatic impairment relative to healthy subjects. A total of 18 hepatic subjects were evaluated, 6 subjects each in Child-Pugh A (mild), B (moderate) and C (severe), in addition to 12 subjects with normal hepatic function. Total Cmax and AUC were lower in subjects with mild, moderate and severe hepatic impairment relative to subjects with normal hepatic function; however, exposures to unbound BMS suggested that dose adjustment in hepatically impaired subjects is not warranted. Drug-drug Interaction Study with Rifampin (AI444012) Study AI evaluated the effect of the strong CYP3A4 inducer rifampin on the PK of BMS in fourteen healthy subjects. Following 7 days of rifampin 600 mg administration, co-administration of rifampin 600 mg and BMS mg resulted in a 56% and 79% reduction in BMS geometric mean Cmax and AUC(INF), respectively, relative to BMS mg alone. Drug-drug Interaction Study with Ortho Tri-Cyclen (AI444020) Study AI evaluated the multiple-dose PK of Ortho Tri-Cyclen when coadministered with BMS mg in 20 healthy female subjects. Preliminary results suggest that BMS does not have a clinically meaningful impact on the PK of the components of Ortho Tri-Cyclen as assessed by ethinyl estradiol, norgestrel, and norelgestromin Studies of Clinical Efficacy of BMS AI is an ongoing, randomized study of BMS combined with SOC (pegifnα + RBV), administered as triple therapy for 48 weeks. Forty-eight (48) subjects were randomized to 3, 10, or 60 mg BMS (N = 36) once daily or placebo (N = 12). An unblinded interim Week 12 analysis (using a data cutoff of 11-Jan-2010) was 37
39 performed. By Week 2, the mean decrease from baseline in HCV RNA in the 3, 10 and 60 mg BMS dose groups was 4.3, 4.7 and 4.9 (log10 IU/mL), respectively, compared with 1.7 for placebo (Figure ). Figure : Mean Change from Baseline to Week 12 in HCV RNA (AI444014) B/L Mean HCV RNA Change from B/L [80% CI] (log10 IU/mL) B/L Weeks Number of Subjects with Measurements BMS 3 mg BMS 10 mg BMS 60 mg PBO BMS 3 mg (N = 12) BMS 10 mg (N = 12) BMS 60 mg (N = 12) PBO (N = 12) 38
40 Based on modified intent-to-treat (ITT) analyses: Extended rapid virologic response (ervr: undetectable HCV RNA at Weeks 4 and 12) rates of 42% (5/12), 83% (10/12) and 75% (9/12) were achieved for the 3, 10 and 60 mg BMS groups compared with 8% (1/12) for placebo. Complete early virologic response (cevr: undetectable HCV RNA at Week 12) rates of 58% (7/12), 83% (10/12) and 83% (10/12) were achieved for the 3, 10 and 60 mg BMS groups compared with 42% (5/12) for placebo. Rapid virologic response (RVR: undetectable HCV RNA at Week 4) rates of 42% (5/12), 92% (11/12) and 83% (10/12) were achieved for the 3, 10 and 60 mg BMS groups compared with 8% (1/12) for placebo Summary of Clinical Safety of BMS Integrated safety summaries were created based upon studies completed as of December No additional safety signals have been characterized in studies completed since that time. Studies in Healthy Volunteers (Non-HCV Infected, Non-Hepatically Impaired) BMS was administered to HV (non-hcv infected, non-hepatically impaired subjects) in studies AI444001, AI444003, AI444005, AI444006, AI444007, AI444008, AI and AI In these studies, the mean age for all subjects was 33 years and all subjects were < 65 years. Most subjects were male (84%). A large proportion of subjects were white (40%); 28% of subjects were black/african American and 28% were Japanese. In general BMS had a favorable safety profile in these trials. There have been no deaths. There has been 1 serious adverse event (SAE): an episode of increased blood creatine phosphokinase (CK) of IU/L (normal range IU/L) in a subject in AI The CK returned to normal 12 days after discontinuation of dosing. The investigator classified the SAE as not related to study drug. Discontinuations due to AEs have been infrequent. Two (2) subjects in AI treated with BMS (30 mg) discontinued study drug: 1 due to cellulitis below the knee and 1 due to a mild drop in absolute neutrophil count (ANC), a decrease to 1.15 x 10 9 cells/l from a baseline of 2.85 x 10 9 cells/l. The ANC returned to within the normal range prior to discharge. One (1) 39
41 subject in AI discontinued due to ALT increase from baseline 36 U/L to 150 U/L on Day 13 (normal range 5-45 U/L). No concomitant increases in bilirubin were observed and ALT returned to within the normal range prior to discharge. Two (2) subjects in AI discontinued due to abnormal dreams. The most commonly reported AEs ( 3%) in subjects who received any BMS were headache (6%), abdominal pain (3%) and diarrhea (3%). The majority of AEs have been mild. In addition, no clinically relevant trends in vital sign changes, ECG changes, physical examinations or laboratory values have been identified in any of these trials. Studies of BMS Monotherapy in HCV Infected Subjects BMS was administered as monotherapy in HCV infected subjects in studies AI and AI In these studies, the mean age for all subjects was 43 years and all subjects were < 65 years. Most subjects were male (73%). The majority of subjects were white (81%) and 17% were black/african American. In general BMS had a favorable safety profile in these trials. There have been no deaths, SAEs, or discontinuations due to AEs. The most commonly reported AEs in HCV infected subjects were headache (23%) and diarrhea (8%). Back pain, insomnia, abdominal pain, flatulence, and fatigue were reported by 2 HCV infected subjects each (5%). The majority of AEs were mild. In addition, no clinically relevant trends in vital sign changes, ECG changes, physical examinations or laboratory values have been identified in these trials. Study in Hepatic Impaired Subjects (AI444013) Study AI assessed the single-dose safety of BMS in subjects with hepatic impairment. Thirty (30) subjects were dosed and completed the study: 6 subjects in Child-Pugh A, 6 subjects in Child-Pugh B, 6 subjects in Child-Pugh C, and 12 healthy control subjects. Based on preliminary data, BMS had a favorable safety profile with no clinically relevant trends in vital sign changes, ECG changes, physical examinations or laboratory values outside that which would be considered typical for subjects with hepatic impairment. 40
42 BMS in Combination With Standard of Care in HCV-Infected Subjects (AI444014) In this study, 48 HCV-infected subjects received either BMS (36 total: 12 subjects per group received 3, 10, or 60 mg) or placebo (12 subjects) plus SOC. The mean age for all subjects was 51 years and 90% of subjects were < 65 years. Most subjects were male (67%). A large proportion of subjects were white (73%) and 19% were black/african American. As of 11-Jan-2010 (date of the unblinded interim Week 12 analysis), there were no deaths. SAEs were reported by 3 subjects, 1 in each of the BMS dosing groups: 3 mg BMS SOC: A 51-year-old female subject with prior history of epistaxis was hospitalized at Week 4 with severe epistaxis requiring transfusion. The subject also had a brief syncopal episode as well as substernal chest pain. ECG and serial cardiac enzymes were reported as normal. The epistaxis and anemia were considered related to study drugs since it was unclear whether the investigational product was associated with the severity of the event; the chest pain and syncope were considered unrelated to study drug. All symptoms resolved and subject was discharged 2 days after hospital admission. RBV and BMS were interrupted for 5 days and restarted (with a lower dose of RBV). 10 mg BMS SOC: A 67-year-old female subject was hospitalized at Week 20 in Mexico with acute gastroenteritis (abdominal distention, nausea, diarrhea, profuse sweating) and a 15 to 20 second syncopal episode. ECG was unchanged from baseline. The syncopal episode was not considered related to study drug. Subject was discharged the next day with treatment for acute gastroenteritis. No interruption of study medication occurred. 60 mg BMS SOC: A 44-year-old female subject was hospitalized at Week 6 with acute bronchitis, considered unrelated to study drug, and discharged after 2 days. Treatment with BMS and RBV were interrupted for 4 days. This subject discontinued therapy at Week 8 due to worsening baseline anxiety (mentioned below). This subject was hospitalized post-treatment Week 4 for affective disorder considered not related to study drug. Four (4) subjects had AEs leading to discontinuation of study therapy: 1 in the 3 mg group, 2 in the 60 mg group, and 1 in the placebo group. 41
43 3 mg BMS SOC: A 59-year-old male reported fatigue, irritability, aguesia, loss of concentration, headache, auditory hallucinations in Week 1 that led to discontinuation. These AEs were considered related to all study drugs. 60 mg BMS SOC: A 44-year-old female had worsening baseline anxiety (mentioned above) at Week 8 leading to discontinuation. This AE was not considered related to study drugs. 60 mg BMS SOC: A 47-year-old female developed a rash leading to discontinuation at Week 12. The rash resolved after discontinuation of all study drugs. The rash was considered related to all study drugs. Placebo + SOC: A 38-year-old male had mood swings at Week 4 that led to discontinuation. The AE was considered study drug related. Note that investigators were not instructed to identify which of the 3 drugs was associated with the AEs leading to discontinuation, and a determination of relatedness indicates that at least 1 of the 3 drugs (BMS , pegifnα, or RBV) was considered related to the event. The most commonly reported AEs in all treatment groups were fatigue (42% - 58% in the BMS groups vs 75% in the placebo group) and nausea (33% - 42% in the BMS groups vs 42% in the placebo group). There were no clinically relevant trends in AEs through Week 12, with the exception of an increase in skin and subcutaneous (dermatologic) disorders with BMS compared with placebo. Skin and subcutaneous (dermatologic) disorders (all grades) were reported in 5/12, 6/12, 7/12 and 3/12 subjects in the 3, 10, and 60 mg BMS and placebo groups, respectively. In general, the rashes were mild to moderate in intensity not requiring dose modification. Only 1 subject in the 60 mg BMS dose group (mentioned above) discontinued from study due to generalized rash that resolved after stopping all study therapy. The most common (> 10%) dermatologic AEs reported were pruritis, alopecia, rash and hyperhidrosis. The nature and the frequencies of the events were consistent with what has previously been reported in clinical studies for SOC. 42
44 1.3.2 BMS BMS is being developed for use in combination with pegifnα/rbv as well as for pegifnα/rbv sparing combinations studies with other DAAs for the treatment of chronic HCV. Details of the pre-clinical and early Phase 1 clinical studies are available in the Investigator Brochure (IB). This section will emphasize important data that may be relevant to the management of subjects enrolled in Summary of Non-Clinical Programs for BMS Drug Interactions and Nonclinical Metabolism Key information on BMS pharmacology is available in the IB. 22 In vitro studies showed that BMS was an inhibitor of P-gp (IC50 = 11 µm) and OATP1B1 (IC50 ~0.3 µm), a weak inhibitor of CYP3A4 (IC50 = 27.2 µm) and not an inhibitor of other human CYP enzymes. BMS did not transactivate the human pregnane-x receptor (hpxr), nor did it induce CYP3A4 mrna in human immortalized hepatocytes at concentrations up to 10 µm, suggesting that potential induction of CYP3A4 by BMS in humans is not anticipated. Overall, BMS is likely to inhibit the transport of compounds that are P-gp or OATP1B1 substrates and has low potential to increase the exposure of coadministered drugs that are substrates of CYP3A4, but is unlikely to inhibit the metabolism of compounds that are metabolized by the other CYP enzymes. The elimination of BMS involved metabolic clearance, direct biliary excretion, and direct intestinal secretion but very little renal clearance. Reproductive Risk Potential Embryofetal development studies in mice and rabbits have been completed. BMS is not a selective developmental toxicant. The in-life phase of a fertility and early embryonic development study has been completed. Based on preliminary data, no BMS related reproductive effects have been identified. 43
45 Genotoxicity/Phototoxicity BMS was not genotoxic in vitro or in vivo. An in vivo phototoxicity study indicated that BMS was not phototoxic in rats following single oral doses 600 mg/kg (AUC 1440 µg h/ml) Summary of Clinical Investigational Program for BMS Fifteen (15) studies with BMS in HV, HCV-infected subjects, and hepatic impaired subjects have been completed or are ongoing. Please see Table below for details. Table : Phase 1 and 2 Clinical Trials for BMS Clinical Trial Study Number Dosing Duration Test Agent Completed Trials SAD/HV AI Single Dose BMS SAD/HCV-1 AI Single Dose BMS MAD/HV AI days BMS MAD/HCV-1 AI days BMS SAD/MAD/HV (Japan) AI Single Dose / 14 days BMS DDI/HV AI days BMS MDZ BA/FE/HV AI Single Dose BMS DDI/HV AI /14 days BMS BMS ADME/HV AI Single Dose BMS DDI/HV/ WOCBP AI days BMS Ortho-Tricyclen Ongoing Trials HCV Multiple Dose 24 Weeks with SOC BMS SOC Hepatic Impairment AI Multiple-dose / 7 days BMS HCV AI Weeks with SOC BMS SOC 44
46 Table : Phase 1 and 2 Clinical Trials for BMS Clinical Trial Study Number Dosing Duration Test Agent HCV/Japanese subjects Completed Trials AI Weeks with SOC / 48 Weeks with SOC DDI/HV AI treatment sequence / 23 days BMS BMS BMS mg Rifampin Pharmacokinetic and Pharmacodynamic results for BMS Single-ascending Dose (SAD) Study in Normal Healthy Volunteers (AI447001) BMS and matching placebo have been administered in single oral doses to a total of 56 healthy subjects over a dose range of 10 to 1200 mg in Study AI447001; forty-two (42) subjects received BMS and 14 subjects received placebo. The pharmacokinetic results from Study AI indicate that AUC values of BMS following oral administration as a suspension increase more than proportionally from 10 mg to 100 mg and less than proportionally from 100 mg to 1200 mg. Variability in the 600 and 1200 mg doses was high, relative to the other dosing cohorts. Tmax was reached at approximately 2-4 hours post-dose. Concentrations of BMS declined in a biphasic manner with a terminal elimination half-life of ~ 14 to 20 hours. However, apparent total body clearance (CLT/F) was high, ranging from 440 to 1400 L/h (~7,300 to 23,000 ml/min). Renal clearance accounted for less than 1% of the elimination of BMS Single-ascending Dose (SAD) Study in Subjects with Chronic Hepatitis C (AI447002) AI447002, a SAD study in subjects infected with HCV genotype 1, is complete. Twentyfour (24) HCV-1 infected subjects were randomized and treated: 20 (n = 4/dose group) with a single dose of BMS (10, 50, 200, or 600 mg) and 4 with placebo. Following oral administration of single doses of 10 to 600 mg of BMS suspension to HCV-infected subjects, peak plasma concentrations of BMS
47 (Tmax) were achieved in approximately 2-4 hours, with a T-HALF of about hours. Apparent total body clearance (CLT/F) was high and ranged from 300 to 490 L/h (5,000 to 8,200 ml/min). The pharmacokinetic results from Study AI suggest that, following single doses, exposures to BMS in HCV-infected subjects are similar to or slightly higher relative to healthy volunteers. The mean log 10 HCV RNA decline for subjects who received a single dose of BMS mg (2.26; range 1.64, 3.16) and 600 mg (2.87; range 2.31, 3.75) were similar up to 16 hours after dosing, and subjects who received BMS mg (0.28; range 0.05, 0.72) and 50 mg (0.64; range 0.40, 1.12) had lower mean decline in log 10 HCV RNA and lower average rate of decline comparing to subjects treated by BMS mg and 600 mg (Table A and Figure A) Table A: Mean Decreases in HCV RNA in HCV-1 Infected Subjects (AI447002) BMS Dose (mg) N Mean Decrease in log 10 HCV RNA from Baseline to Day 1 Mean (Range) (0.05, 0.72) (0.40, 1.12) (1.64, 3.16) (2.31, 3.75) 46
48 Figure A: Median Change in log 10 HCV RNA from Baseline (AI447002) 10 mg 50 mg mg 600 mg Viral load decline in Log Time (h) Multiple-ascending Dose Study in Normal Healthy Volunteers (AI447003) Forty-six (46) healthy subjects have been randomized and completed treatment in Study AI BMS capsules or matching placebo were administered twice-daily over a dose range of 10 to 600 mg for 14 days. Pharmacokinetic data from the 10, 50, 100, 200, 400 and 600 mg Q12h cohorts in study AI suggest that Tmax (median ~2.5 h post-dose) and terminal elimination half-life (mean ~17-24 h) of BMS following multiple doses was similar to single dose. As with single dose administration, mean apparent oral clearance was high and ranged from L/h. Increases in steady state exposure with dose were greater than dose-proportional throughout the dosing range, but were more pronounced at doses 200 mg. Steady-state accumulation was ~ 4-fold at doses 200 mg, vs. ~2-fold at 400 and 600 mg Q12h. At doses 200 mg, pre-dose BMS concentrations declined 47
49 beginning on Day 3, suggesting possible auto-induction. Steady state appeared to be achieved by Day 7 in all dose cohorts. Multiple-ascending Dose Study in Subjects with Chronic Hepatitis C (AI447004) Based on the data from AI447002, three doses were selected for a 3-day multiple dose study in HCV-infected subjects: 200 mg, 400 mg and 600 mg twice daily. AI has completed drug administration. Fifteen (15) subjects have been randomized and have received twice daily for 3 days: 12 (n = 4/dose group) with BMS (200, 400 or 600 mg) and 3 with placebo. Pharmacokinetic data from the 3-day 200 mg, 400 mg and 600 mg Q12h cohorts from study AI suggest that Tmax of BMS following multiple doses in HCVinfected subjects is similar to that in healthy subjects. Exposures on Day 3 were not expected to be at steady state, and were higher than those in healthy volunteers on Day 14 of dosing; this outcome was anticipated based on the patterns observed in pre-dose concentrations in AI447003, suggesting auto-induction at doses 200 mg Q12h. Variability at 400 mg and 600 mg was high, with considerable overlap in exposures between the 400 mg dose cohort and the other two dose cohorts. Terminal half-life was not assessed in this study and neither time to steady state nor corresponding accumulation index could be reliably estimated after 3 days of dosing. HCV-infected subjects administered BMS mg twice daily showed an HCV RNA mean change from baseline at Day 4 of ~ 3.1 log 10 (preliminary data). At 400 mg twice daily, the mean change at Day 4 was ~3.3 log 10 and at 600 mg twice daily the mean change at Day 4 was ~2.8 log 10. Viral RNA rebounds upon discontinuation of BMS , and returned to baseline at around Day 10 (Table B and Figure B). Table B: Mean Decreases in HCV RNA in HCV-1 Infected Subjects (AI447004) BMS Dose (mg) N Mean Decrease in log 10 HCV RNA from Baseline to Day 4 Mean (Range) (2.1, 4.1) (2.7, 3.8) (2.2, 3.5) 48
50 Figure B: Median Changes from Baseline in log 10 HCV RNA (AI447004) 0.5 HCV RNA (log10 IU/mL Change from Baseline Expa nsion -4.0 Treatment Placebo BID 200 mg BID 400 mg BID 600 mg BID -1 or Screening Time (Days) Clinical Pharmacokinetics of BMS in Japanese Subjects (AI447005) Preliminary pharmacokinetic data are available in Japanese subjects at single doses from 200 to 1200 mg in the SAD phase of AI Following single doses of suspension, Tmax was reached within 3 to 4 hours post-dose, and T-HALF was approximately 15 to 21 hours. Exposures (i.e. Cmax and AUC) increased in a dose-dependent manner from 200 to 400 mg. Exposures at 600 mg were lower than those observed at 400 mg and while geometric mean AUC(INF) values were higher than 400 mg at 900 and 1200 mg, Cmax values were lower. Exposures at 900 and 1200 mg were generally similar. Variability was high at doses 600 mg. Analysis of the pharmacokinetic samples from the MAD phase of the study is ongoing. Drug-Drug Interaction Study with Midazolam in Healthy Volunteers (AI447007) AI447007, a drug-drug interaction (DDI) study in healthy subjects, has been completed. Eighteen (18) subjects received a single oral dose of midazolam (MDZ) 5 mg on Day 1, BMS mg BID on Days 2 to 7, and a single oral dose of MDZ 5 mg 49
51 concomitantly with BMS mg on Day 8. BMS caused an approximate 34% and 44% reduction in MDZ geometric mean Cmax and AUC(INF), respectively. Based on these findings, administration of sensitive CYP3A4 substrates and those CYP3A4 substrates with narrow therapeutic indices may need to be adjusted when given with BMS Relative Bioavailability and Food Effect Study in Healthy Volunteers (AI447008) AI447008, a relative bioavailability and food effect study in eighteen (18) healthy subjects, has been completed. Subjects received four separate single oral 600 mg doses of BMS , either using a capsule (6 100mg) or two tablet formulations (3 200 mg; one with micronized active pharmaceutical ingredient (API), one with unmicronized API fasted in a randomized fashion or the unmicronized API tablet with a high-fat meal. Seventeen (17) subjects completed the study. The PK results indicate that both tablet formulations performed similarly and were ~50% bioavailable relative to the capsule when fasted. When the unmicronized tablet was administered following a high fat meal, it resulted in a ~30-fold increase in Cmax and ~11.5-fold increase in AUC relative to the tablet fasted. Compared to the capsule fasted, the increased Cmax and AUC following tablet with food were ~9-fold and ~5.5-fold, respectively. The food effect appears to be caused by multiple factors, including enhanced solubility and saturation of some process upon absorption and hepatic first pass. The increase in bioavailability of the tablet when given with food suggests that the absolute bioavailability of the tablet when fasted is <10%. Data from AI further suggest that the food effect is dose dependent but independent of the dosage form itself. Due to robust exposure margins, administering BMS with a high fat meal does not produce exposures greater than the lowest NOAEL in animal toxicology studies. ADME Study of 14C-labeled BMS in Healthy Male Volunteers (AI447010) AI447010, an ADME study in healthy subjects, has been completed. Nine (9) subjects were randomized and dosed with 200 mg of [14C]BMS as a solution. Over several days, all subjects provided samples of feces, urine and blood/plasma for mass balance assessment of total radioactivity (TRA) and exploration of routes of elimination and the metabolic profile of BMS in humans. TRA data suggest that fecal excretion of parent and metabolites is the major route of excretion for BMS Of 50
52 the total dose administered, approximately 84% of TRA was recovered in feces, and less than 3% recovered in urine. Preliminary metabolite profiling data indicate BMS comprised the majority of the circulating radioactivity in human plasma. A few minor metabolites were detected in human plasma, with the AUC values generally below 5% of total plasma radioactivity. All metabolites in human plasma were also present in plasma from at least one animal species. BMS was a prominent drug-related component in human feces, representing 7.5% of the dose. Two (2) metabolites, M8 (amide hydrolysis product) and M12 (oxidative metabolite), were abundant in human feces, representing 14.6 and 8.4% of the dose, respectively. Phase 2a/2b Study of BMS in Combination with PegInterferon Alfa-2a (Pegasys) and Ribavirin (Copegus) in Treatment-Naive Subjects with Genotypes 1 and 4 Chronic Hepatitis C (AI447016) The Phase 2a phase of the study has fully enrolled and reached 12 weeks on study. There are 3 BMS doses being evaluated in Phase 2a: 200 mg BID, 600 mg BID, and 600 mg QD. Intensive PK samples for BMS were collected at the Week 12 visit from all subjects, steady state PK parameters were derived and they are summarized in Table C. All subjects with valid Week 12 PK are summarized. Exposure-response analyses for AI are ongoing. Table C: Summary Statistics for BMS Pharmacokinetic Parameters in Study AI Study Visit Week 12 Cmax (ng/ml) - Geo. Mean (%CV) Tmax (h) - Median (min, Max) AUC(TAU) (ng/ml*h) - Geo. Mean (%CV) Cmin (ng/ml) - Geo. Mean (%CV) BMS mg BID N= 12 BMS mg BID N = 10 BMS mg QD N = (86) 1440 (78) 1840 (97) 2 (1, 4) 4 (2, 12) 4 (2, 8) 1845 (77) 5676 (87) 9113 (82) 43.6 (161) 127 (169) 21.8 (113) 51
53 Drug-Drug Interaction Study with an Oral Contraceptive in Healthy Female Volunteers (AI447019) AI447019, a drug-drug interaction (DDI) study in healthy female volunteers has been completed. Preliminary PK data indicate that BMS at 600 mg twice daily causes decreases in the plasma AUC(TAU) of ethinyl estradiol and norelgestromin (the major circulating active metabolite of norgestimate) of ~28% and ~34%, respectively. Based on these results, women of child bearing potential (WOCBP) enrolled in Phase 2 and Phase 3 studies with BMS would need to use two reliable methods of birth control other than oral contraceptives to ensure against pregnancy during treatment Summary of Clinical Efficacy Protocol AI is a comparative, randomized, multi-center, combined Phase 2a/2b study that is intended to establish the safety and efficacy of BMS in combination with standard of care pegifnα/ RBV in Phase 2a (Stage 1), and to select doses being evaluated in Phase 2a for inclusion in Phase 2b (Stage 2). In Stage 1, 47 treatment-naive, genotype 1 HCV-infected patients were randomized (1:1:1:1) to receive 200 mg BID, 600 mg BID and 600 mg QD of BMS plus pegifnα/rbv versus placebo daily plus pegifnα/rbv. The unblinded Week 12 primary analysis was performed after the last subject was treated for 12 weeks to evaluate safety and antiviral activity to support dose selection for Stage 2. The primary endpoint for this study was the proportion of subjects with extended rapid virologic response (ervr), defined as undetectable HCV RNA at both Weeks 4 and 12. Other antiviral activity endpoints of interest were the proportion of subjects with rapid virologic response (RVR: undetectable HCV RNA at Week 4), proportion with early virologic response (EVR: 2 log10 decrease in HCV RNA from baseline or HCV RNA < limit of quantitation [LOQ] at Week 12), proportion with complete early virologic response (cevr: undetectable HCV RNA at Week 12), proportion with HCV RNA < LOQ, and log10 HCV RNA levels and changes from baseline. Based on preliminary data as of 01-Sep-2010, BMS demonstrated a rapid and persistent antiviral activity during the first 12 weeks of administration. The antiviral activity of BMS , as demonstrated by the proportion of subjects with ervr 52
54 (undetectable HCV RNA at Weeks 4 and 12) and mean change in HCV RNA from baseline at Week 12, were comparable across all BMS regimens (200 mg BID, 600 mg BID, 600 mg QD), and significantly higher than placebo (PBO). By the Cobas TaqMan assay, for regimens containing BMS , ervr rates ranged from 75.0% to 91.7%. All three BMS regimens met the protocol-specified efficacy dose selection criteria for ervr Summary of Clinical Safety of BMS Studies in Healthy Volunteers (Non-HCV Infected, Non-Hepatically Impaired) BMS was administered to HV (non-hcv infected, non-hepatically impaired subjects) in single doses (up to 1200 mg) and multiple doses (up to 600 mg BID for up to 14 days) in studies AI447001, AI447003, AI447005, AI447007, AI447008, AI447009, AI and AI Study AI was also performed in HV with dosing just completed but data are not available for inclusion in this analysis. In these studies, the mean age for all subjects was 31 years and all subjects were < 65 years. Most subjects were male (85.9%). A large proportion were white (43.1%); 26.0% of subjects were black/african American and 24.8% were Japanese. In general, BMS had a favorable safety profile in these trials. There have been no deaths. One (1) serious adverse event (SAE) occurred during studies of HVs: an episode of increased blood creatine phosphokinase (CPK) of IU/L (normal range IU/L) in a subject in AI The CPK measurements were performed by the investigator to investigate the occurrence of AST increase, both which were determined likely the result of physical exercise by the subject, a prohibited activity during this study. No clinical symptoms were reported by the subject during the study, and both laboratory AEs were resolved by Study Day 23. The CPK returned to normal 12 days after discontinuation of dosing. The investigator classified the SAEs as not related to study drug. Discontinuations due to AEs have been infrequent. One (1) subject in AI who was administered BMS (600 mg) discontinued study drug due to nephrolithiasis. The subject also had an AE of mild diarrhea that lasted approximately 20 minutes. Both AEs resolved without treatment and were considered by the investigator as unrelated to study 53
55 medication. Based on preliminary data, 2 subjects in AI discontinued study drug: 1 due to streptococcal pharyngitis and 1 due to gastroenteritis. Both AEs resolved and were considered by the investigator as not related to study drug. The most commonly reported AEs ( 3%) in subjects who received any BMS were: diarrhea (12.1%), headache (12.1%), dizziness (4.7%), nausea (3.3%), flatulence (3.3%), and pollakiuria (3.3%). The reported AEs of pollakiuria (mild to moderate in intensity) resolved spontaneously in 4 subjects while still on treatment and in 3 subjects at 1, 2 and 6 days after treatment cessation. All but 1 AE of pollakiuria were considered related to study drug. Based on urinalysis and serum creatinine measurements, there was no evidence of altered kidney function. Increased urinary frequency was not observed in the higher dose (400 and 600 mg) groups. In addition, no clinically relevant trends in vital sign changes, ECG changes, physical examinations or laboratory values reproducible across the studies have been identified in any of these trials. Results from AI are not available for inclusion in this version of the Investigator Brochure. Studies of BMS Monotherapy in HCV-Infected Subjects BMS was administered as monotherapy in HCV-infected subjects in single doses (up to 600 mg) and multiple doses (up to 600 mg BID for up to 3 days) in studies AI and AI447004, respectively. In these studies, the mean age for all subjects was 47 years and all subjects were < 65 years. Most subjects were male (79.5%). The majority of subjects were white (71.8%) and 25.6% were black/african American. In general, BMS had a favorable safety profile in these trials. There was 1 death due to cardiac arrest and shock secondary to amphetamine toxicity 18 days after BMS (600mg, Q12h) dosing was completed for a subject in AI Other SAEs for this subject included renal failure, rhabdomyolysis, respiratory failure, and hyperkalemia also due to amphetamine toxicity. No other subjects discontinued due to AEs in these trials. There were 2 SAEs reported among subjects in AI447002: 1 subject had a seizure 21 hours following administration of placebo and 1 subject had moderate chest pain requiring hospitalization, beginning 29 days after administration of BMS (50 mg). A Thallium Stress Test was negative. Results of subsequent evaluation were 54
56 consistent with amyotrophic lateral sclerosis (ALS).Both events were related to pre existing conditions and were considered by the investigator as unrelated to study drug. The most commonly reported AEs in HCV infected subjects who received BMS were: headache (18.8%) and nausea (6.3%). All other AEs were reported by 1 HCVinfected subject each (3.1%). The majority of AEs were mild to moderate. In addition, no consistent clinically relevant trends in vital sign changes, ECG changes, physical examinations or laboratory values have been identified across these trials. Transaminitis in subjects treated with BMS Please see section for a detailed description of the hepatic effects of BMS observed to date Combination of BMS and BMS Combination Non-Clinical Toxicology : One-month Oral Combination Toxicity Study in Rats (DS08126) In a 1-month oral toxicity study in rats, vehicle alone or in combination at low and high doses were administered to 3 groups of 10 rats/sex/group. Doses were: 0 (vehicle control), BMS at 10 or 60 mg/kg/day and BMS at 30 or 60 mg/kg/day. Criteria for evaluation included survival, toxicokinetics, clinical signs, body weights, food consumption, physical examinations, clinical-pathology assessments, and gross- and microscopic-pathology evaluations. BMS (doses 60 mg/kg/day, Day 28 AUC 35.6 µg h/ml) and BMS (doses 60 mg/kg/day, Day 28 AUCs 54.9 µg h/ml) were clinically well tolerated when administered in combination to rats for 1 month. Both BMS and BMS combinations were associated with minor clinical-pathology changes and target-organ effects limited to adrenal cortical-cell vacuolation. Based on earlier toxicity studies in rats using either BMS or BMS alone, there was no evidence of a toxicologic interaction between. 55
57 : One-month Oral Combination Toxicity Study in Monkey (DS08143) In a 1-month oral combination toxicity study in monkeys, vehicle alone or BMS and BMS in combination at low and high doses were administered to 3 groups of 4/sex/group. Doses were: 0 (vehicle control), BMS at 15 or 50 mg/kg/day and BMS at 72 or mg/kg/day. Criteria for evaluation included survival, toxicokinetics, clinical signs, body weights, physical examinations, electrocardiographic examinations, arterial oxygen saturation, clinical-pathology assessments, and gross- and microscopic-pathology evaluations. BMS (doses 50 mg/kg/day, mean Day 28 AUC 35.2 µg h/ml) and BMS (doses mg/kg/day, mean Day 28 AUC 60.2 µg h/ml) were clinically well tolerated when administered to monkeys for 1 month. BMS and BMS combinations were associated with minor clinical findings, and microscopic changes of inflammation in the intestines, minimal lymphoid depletion of the thymus, and adrenal gland decreased vacuolation and/or hypertrophy (high dose combination only). Based on earlier toxicity studies using either BMS or BMS alone, there was no evidence of a toxicologic interaction between BMS and BMS However, the data indicate a trend toward increased AUCs when administered in combination in monkeys Combination Clinical Data in Humans DDI study AI A multiple dose, drug-drug interaction study was conducted in healthy subjects to assess the pharmacokinetics and safety of when coadministered (AI447009). BMS mg QD and BMS mg BID were administered alone for 7 days followed by coadministration for 14 days at 30 mg QD and 200 mg BID, respectively. Pharmacokinetic sampling was performed on Days 7 and 21. The rationale for the reduced doses during the combination treatment was based on the nonclinical monkey and rat toxicokinetic findings where a modest to large increase in BMS exposure was observed when coadministered with BMS The exposures on Day 56
58 21 were dose normalized (assuming dose proportionality) and compared to exposures on Day 7. In addition, Day 21 exposures were compared to Day 14 historical data obtained from the multiple dose studies AI and AI conducted in healthy subjects. The dose normalized and historical comparisons for BMS are summarized in Tables A and B, respectively. Table A: Results of Statistical Analysis of BMS mg after Day 21 A.M. Coadministration with BMS mg to BMS mg on Day 7 A.M. PK Parameter BMS mg BID Geo Mean (CV) BMS mg Normalized to 600 mg BID + BMS mg QD Geo Mean (CV) GMR (90% CI) AUC(TAU) AM 1019 (58) 926 (44) 0.91 (0.69, 1.19) Cmax AM 420 (48) 247 (64) 0.59 (0.41, 0.83) Cmin AM 10.9 (55) 19.3 (28) 1.78 (1.43, 2.21) Table B: Results of Statistical Analysis of BMS mg after Day 21 A.M. Coadministration with BMS mg to Historical Data from AI PK Parameter BMS mg BID + BMS mg BMS mg BID AI GMR (90% CI) AUC(TAU) - AM 309 (44) 300 (39) 1.02 (0.74, 1.44) Cmax - AM 82 (64) 88 (62) 0.94 (0.57, 1.53) C (28) 8.44 (60) 0.76 (0.58, 1.01) As noted in Tables A and B overall exposures were similar on Days 7 and 21 indicating a minimal pharmacokinetic interaction when BMS is coadministered with BMS The AUC(TAU) point estimates were 0.91 and 1.02 for the dose normalized and historical comparisons, respectively. The point estimates for Cmax and C12 ranged from 0.59 to 1.78 which suggests that a PK interaction, if observed, would most likely be minimal. 57
59 A similar comparison was performed for BMS as depicted in Tables C and D. As noted in Tables C and D, overall exposures were similar on Days 7 and 21 indicating a lack of a PK interaction when BMS is coadministered with BMS The AUC(TAU) point estimates were 1.12 and 1.16 for the dose normalized and historical comparisons, respectively. Furthermore, the point estimates for Cmax and Cmin were close to 1.0 indicating minimal if any impact on these PK parameters. Table C: Results of Statistical Analysis of BMS after Day 21 A.M. Coadministration with 200 mg BMS to BMS mg on Day 7 PK Parameter BMS mg Geo Mean (CV) BMS mg Normalized to 60 mg + BMS mg BID Geo Mean (CV) GMR (90% CI) AUC(TAU) AM (25) (28) 1.12 (0.95, 1.32) Cmax AM 1567 (24) 1598 (26) 1.02 (0.87, 1.19) C24 AM 186 (39) 215 (35) 1.16 (0.93, 1.45) Table D: Results of Statistical Analysis of BMS after Day 21 A.M. Coadministration with 200 mg BMS to BMS Historical Data from AI PK Parameter BMS mg Geo Mean (CV) BMS mg AI Geo Mean (CV) GMR (90% CI) AUC(TAU) AM 7259 (28) 6275 (39) 1.16 (0.90, 1.49) Cmax AM 799 (26) 734 (30) 1.09 (0.86, 1.37) C24 AM 108 (35) 92 (54) 1.17 (0.85, 1.62) Overall, no PK interaction was observed when were coadministered. were well tolerated when coadministered for 14 days. 58
60 DDI study Preliminary results from Sentinel cohort Preliminary analysis of trough PK samples on Day 14 of dosing from the 21 subjects enrolled in the sentinel subjects of this study has been performed. Intensive PK was collected after the morning dose at Week 2 during the study. All doses of study drug followed a meal in the morning and were ~2-2.5 hours after a meal in the evening. The preliminary PK data suggest that the PK of the BMS compounds are similar across the cohorts, suggesting that Pegylated Interferon alpha/ribavirin have no clinically relevant effect on the PK of either compound. Furthermore, the PK data for BMS were the first steady state PK data in HCV-infected subjects and were similar to the evening PK in HV in AI suggesting that the steady state PK following meals is similar in HV and HCV-infected subjects. Table E: Summary Statistics for BMS and BMS Pharmacokinetic Parameters in Study BMS BMS Study Day Day 14 Treatment A Treatment B Treatment A Treatment B Cmax (ng/ml) - Geo. Mean (%CV) 1820 (83.4) 1640 (103) 1020 (31.9) 1430 (28.4) Tmax (h) - Median (min, Max) AUC(TAU) (ng/ml*h) - Geo. Mean (%CV) Cmin (ng/ml) - Geo. Mean (%CV) 2 (2, 4) 2 (1, 4) 2 (1, 24) 1 (1, 4) 6590 (75.8) 6150 (102) (30.7) (17.7) 86 (87.1) 76.4 (234) 202 (91.2) 255 (65.8) 59
61 1.4 Study Rationale Rationale for Combining Multiple Direct-acting Antivirals (DAA) An important goal in HCV therapy is to develop a more effective, well-tolerated, simple treatment regimen than the current SOC, PEG-IFN-α and RBV. To date, the novel NS5A cofactor inhibitor BMS has demonstrated potent antiviral activity in chronically HCV-infected subjects when administered as either a single dose or multiple QD doses for up to 14 days of dosing. Similar results have been obtained with the NS3 protease inhibitor BMS , when administered as either a single dose or multiple BID doses for 3 days. Since drug-resistant substitutions rapidly emerge against HCV-specific antivirals when given in monotherapy, combination of 2 or 3 DAA agents may be required in a successful therapy. In a pre-clinical study employing the HCV replicon system, additive to synergistic effects were observed when BMS was combined with BMS These results support the concept that their combined use in patients could enhance the treatment efficacy and possibly overcome the need for SOC. The aim of this Phase II proof-of-concept study (Study ) is to combine both a NS5A inhibitor BMS and a protease inhibitor BMS with or without SOC or ribavirin for up to 48 weeks Rationale for Dose Selection of BMS The choice of the 60 mg QD dose of BMS was driven by the available antiviral and safety and tolerability data which are presented above. Importantly, the 60 mg QD dose was deemed safe and tolerable and demonstrated robust antiviral activity. Doses higher than 60 mg (up to 200 mg as single doses and up to 100 mg for 14 days) were safe and tolerable, but did not show increased antiviral effect. The 60 mg QD dose is the highest dose selected to move forward into the ongoing FDA approved phase IIa study of BMS SOC (AI444014). A safety analysis of the blinded treatment cohorts was performed for AI at Week 4, and this demonstrated no safety signals in any of the treatment arms, and no observed safety differences between any of the 3 treatment arms and the placebo/control arm. When the two molecules were co-administered in the DDI study AI for 14 days at doses of 30 mg QD (BMS ) and 200 mg BID (BMS ), the agents were 60
62 safe and well tolerated and there did not appear to be a clinically significant pharmacokinetic interaction between BMS and BMS Based on these results, there appears to be a low probability of a clinically significant interaction even at the highest projected therapeutic doses. Due to the similarity in AM PK at the doses in the combination period compared to historical data and the dose-normalized analysis within AI between periods, there does not appear to be a clinically significant interaction between BMS and BMS Based on these results, there appears to be a low probability of a clinically significant interaction even at the highest projected therapeutic doses of BMS (60 mg QD) up to and including the previous high dose of BMS (600 mg BID) previously selected for this study. This remains true for use of BMS at 200mg BID. Since long-term response with only small molecule combinations has not been demonstrated to date, this study employs the maximal tolerable doses of the tablet formulations available which are thought to offer the best chance of success. As single agents, these compounds have each been effective in reducing HCV viral load of at least 3 logs over 1 to 3 days of treatment in naive patients Rationale for Dose Selection of BMS The 600 mg BID dose of BMS was initially chosen based upon antiviral, safety and tolerability data. As discussed below, the 600 mg BID dose was the highest dose selected to move forward into the ongoing phase IIa study of BMS SOC (AI ) and was the dose administered in the sentinel cohorts of this study (). The results from a Week 12 interim analysis of study AI447016, A Phase 2a/2b study of BMS in Combination With Peginterferon Alfa-2a (Pegasys) and Ribavirin (Copegus) in Treatment-Naive Subjects with Genotypes 1 and 4 Chronic Hepatitis C Infection. have resulted in an adjustment to 200mg BID in the Expansion Cohorts of this study. In AI447016, treatment-naive, genotype 1 HCV-infected patients were randomized (1:1:1:1) to receive 200 mg BID, 600 mg BID and 600 mg QD of BMS plus pegifnα/rbv versus placebo daily plus pegifnα/rbv. A review of the safety at the time of the 12 week analysis revealed that one grade 3-4 elevation in ALT and AST in each of the 600 mg regimens (BID and QD) and one grade 3-4 elevation in total bilirubin in the 600 mg QD dose group occurred. An SAE of cytolytic hepatitis was also observed in the 600 mg QD dose group. In addition, there was an 61
63 overall trend in ALT/AST values that differed between dose groups. While there was a decrease in median ALT values from baseline over time with the placebo and 200 mg BID dose groups (-44 and -8), the median ALT values in the 600 mg BID and 600 mg QD dose group either remained constant or increased by treatment week 12 (0.0 and +10.5). Elevations in total bilirubin were observed, but much less frequently and inconsistently. Therefore, although subjects generally tolerated all BMS doses well and the majority of adverse events were consistent with the AE profile of peginterferon alfa-2a and ribavirin, there appears to be a trend in AST/ALT elevations in the treatment arms relative to the placebo arm. The trend is more prominent at the two 600 mg dose levels than the 200 mg BID dose. Importantly, while transient changes in ALT/AST occurred in subjects receiving 200mg BID, no subjects in this arm discontinued due to AEs related to hepatic laboratory abnormalities, and there were no grade 3-4 AE abnormalities at this dose. It is also important to note that although ALT/AST levels increased in a number of subjects at approximately 8 to 16 weeks of treatment, most subjects began to return to within normal limits despite continued dosing of BMS In addition, in three subjects who were in the 600 mg BID arm and discontinued BMS secondary to elevated AST/ALT levels, these laboratory abnormalities rapidly corrected while continuing to administer peginterferon alfa-2a and ribavirin. Since all three doses (200 mg BID, 600 mg BID, 600 mg QD) explored in study AI as of the week12 analysis demonstrated similar antiviral activity and the laboratory abnormalities observed with ALT and AST were primarily limited to the higher doses, the dose for all subjects in ongoing Phase 2a studies, including, will be reduced to no more than 200 mg BID. Note that in when the dose reduction decision was made, all subjects on Treatment B and all subjects on Treatment A who had not been rescued, had completed their treatment. One of the six rescued subjects discontinued treatment before the dose reduction. Thus only 5 rescued subjects were impacted by the dose change. The dose for subjects in the Expansion Cohorts of study is limited to 200 mg BID. The selection of 200 mg QD as an alternate dose of BMS is based on the observed antiviral activity at all doses in AI that showed similar antiviral effects and weak exposure-response relationships suggesting that the doses selected for Phase 62
64 2A were at an effect plateau and that a lower dose could have similar antiviral activity. The disproportional plasma PK and the emergence of auto-induction at the 200 mg BID dose range suggest that the 200 mg QD AUC(24h) will overlap with 200 mg BID but with a lower Cmin value. Of note, the single dose antiviral activity of BMS was tested under fasting conditions. Additional data has demonstrated a positive food effect that enhances the bioavailability of BMS and as in AI and this study, dosing with meals may potentiate the antiviral effect of BMS Especially in combination with BMS and Pegylated interferon/ribavirin, a dose of 200 mg QD should be sufficiently active to produce a robust antiviral response Approaches to Reduce Drug Resistance Since the emergence of drug-resistant viral variants is a major concern, the study will start with 2 parallel sentinel cohorts of 10 patients monitored closely for viral load. The first sentinel cohort decision point will take place at Week 2, where the desired threshold is the achievement of undetectable HCV RNA or decreased plasma HCV RNA 2 log 10 IU/ml in 70% or more of the patients. If this threshold is achieved, the study will continue. If this threshold is not achieved, the cohort will not be expanded and only individual subject decision rules will apply to subjects. The second sentinel cohort decision point will take place at Week 4, where the desired endpoint is undetectable HCV RNA in 50% patients. If this is achieved, the study will continue enrolling additional subjects. If this is not achieved, will not be expanded and only individual subject decision rules will apply to subjects. These are the only two decision points affecting the entire cohorts. The fate of the individual patients will follow the individual decision rules illustrated in Tables A and B. To reduce the number of subjects exposed to regimens with reduced antiviral activity in Expansion Cohorts, group specific decisions will be employed. These rules will halt further randomization into specific cohorts in the event that reduced efficacy is observed (see Group Decision Rules). 63
65 1.4.5 Rationale for Treatment with 24 Weeks of BMS and BMS alone A similar study has been conducted by Roche and Pharmasset (INFORM-1), combining a protease inhibitor (R-7227) with a polymerase inhibitor (R-7128) in naive patients. Initial viral load data (presented at the April 09 annual EASL meeting) after 14 days of dosing looked quite encouraging. All subjects achieved consistent and sustained viral load reduction with no apparent rebounds or selection of variants. Four (4) of thirty-one (31) patients became HCV RNA undetectable at the end of the first two weeks of treatment. No SAEs were reported and AEs were moderate. AEs included an increase of LDL/cholesterol and glucose, and decrease of total neutrophils. As of Amendment 7 in this study, data from the Treatment A sentinel cohort demonstrated that treatment with BMS and BMS provided robust early antiviral activity with a mean HCV RNA decline at week 2 of -5.1 log 10. However, 6/9 subjects infected with GT1a experienced viral breakthrough between weeks 3 and 12, indicating the need of additional antiviral potency to this DAA combination in subjects infected with GT1a. Alternatively, 2/2 subjects infected with GT1b have maintained viral control. Data from a similar study (BMS study AI447017) being conducted in Japan, a region with a predominance of GT1b subjects, has shown similarly promising data in GT1b infected subjects receiving BMS and BMS alone. These data are consistent with known in vitro differences between the resistance profiles of GT1a and GT1b viruses to BMS and BMS Thus, a growing body of data supports the use of these two drugs for treatment of GT1b infected subjects Rationale Supporting Addition of Ribavirin to BMS and BMS The utility of adding ribavirin alone to direct acting antivirals has been subject of considerable debate. Generation of in vitro data documenting potential benefit is hindered by the cytotoxicity of ribavirin in the replicon model system. Two recent clinical studies have highlighted the individual contribution of ribavirin to HCV antiviral regimens. First, the Prove 2 study (NEJM 360:18, p ) examined the protease inhibitor telaprevir and pegylated interferon with or without ribavirin in HCV infected treatment naive subjects. The removal of ribavirin from the treatment regimen resulted in reduced SVR at 64
66 24 weeks post treatment and higher rates of viral breakthrough on treatment. The second and most direct support is found in a recently published interim analysis from Gilead. This study examined two HCV antivirals, GS-9256 and tegobuvir in combination alone, with ribavirin, or with pegylated interferon and ribavirin for 4 weeks. This was followed by completion of 44 weeks of pegylated Interferon and ribavirin. At the end of the 4-week initial therapy, addition of ribavirin provided significantly improved antiviral activity compared to the 2 DAAs alone (Zeuzem et.al. Presentation #LB-1, AASLD Nov 1, 2010) Other Rationale Exploratory endpoints for host anti-hcv T cell responses may include IFN-γ production to measure host responses to HCV peptide antigens as well as changes in levels of PD-1 expression on T cells. 1.5 Overall Risk/Benefit Assessment A summary of the nonclinical toxicology findings for are discussed in detail in each respective IB. A summary of the preliminary combination toxicology of is discussed in Section One month oral toxicity combination studies in rats and monkeys with BMS and BMS have not identified any clinical findings or unique toxicity. The toxicology profile has been similar to the findings observed in the single toxicity studies with no overlapping toxicity observed. Toxicokinetic evaluation in the monkeys demonstrated ~2 fold increase in the exposure of BMS when co-administered with BMS As outlined in Section , the mechanism of this interaction is not unexpected and may be due to multiple pathways. Overall, as described in Section 1.3, the clinical safety data for both compounds is favorable BMS In clinical studies, BMS has been generally well tolerated at single oral doses up to 200 mg and at multiple oral doses up to 100 mg when administered for 14 days. In 65
67 clinical studies noteworthy findings include: one subject was discontinued after experiencing a decrease in ANC in study AI (MAD in healthy subjects) (Section ), 2 subjects discontinued due to reported AEs of abnormal dreams in study AI and one subject discontinued due to elevated AST with accompanying CK elevation. Otherwise, there have been no other significant hematologic or clinical laboratory findings BMS There have been no drug-related deaths in clinical studies of BMS To date, 3 SAEs considered related to BMS have been reported: 1 event of cytolytic hepatitis, 1 event of increased bilirubin, and 1 event of pyrexia. A detailed description of the integrated safety summary is available above and in the IB. Diarrhea has been the most common AE encountered in clinical studies of BMS This effect is generally mild to moderate in intensity and does not lead to treatment discontinuation. As discussed in the details above, transaminitis has been the only clinical safety signal requiring intervention. In spite of this finding, it is believed that there is a positive risk/benefit for subjects on BMS and the study overall moving forward with BMS dosed at 200 mg BID. This is based upon the following: The changes in transaminases seen in AI are monitorable with resolution of the most serious cases following removal of BMS These trends are of lower magnitude and frequency in subjects receiving 200 mg. With the exception of subjects who met criteria for discontinuation in AI447016, most elevations improve or resolve despite continued dosing. Antiviral activity endpoints in study AI at 12 weeks of therapy were very similar among groups receiving 200 mg versus 600 mg of BMS Thus, antiviral activity at this dose does not appear to be compromised. Thus far, the four drug therapy of results has provided unprecedented antiviral activity in the difficult-to-treat population of HCV infected null responders. Group specific criteria will stop randomization of subjects into dose panels which meet pre-specified criteria. 66
68 1.5.3 Risks of Treatment Including Pegylated Interferon and/or Ribavirin Subjects in treatment group B will receive 24 weeks of treatment with ribavirin with or without pegifnα, in addition to. Additionally, subjects in treatment group A may be switched to antivirals plus SOC depending on their response to antiviral treatment alone. Subjects participating in this study might achieve RVR, EVR, and potentially SVR if the treatment with BMS and BMS is successful. Null patients retreated with SOC alone typically have a response rate between 5 and 10%. As of this writing, 100% of subjects in treatment group B sentinel cohort (10/10) have maintained viral control through 12 weeks of treatment with BMS and BMS with SOC. Should this predict SVR, this would be an unprecedented achievement in this patient group (HCV null responders). The risks associated with pegifnα and RBV are well described in the package inserts. 43, 44 The risks and benefits of erythropoiesis-stimulating agents, (ESAs) for the management of HCV treatment related anemia has not been standardized or established in well controlled clinical trials. The primary indication for the use of an ESA is to decrease the chance that a red blood cell transfusion will be needed. The use of ESAs has been associated with thrombo-embolic disorders such as deep venous thrombosis, myocardial infarction, stroke and pulmonary embolism. Investigators should exercise clinical judgment and follow the treatment guidelines provided in section when considering the use of ESAs. In addition to these findings, the possibility of drug induced liver injury (DILI) may be increased with direct anti-hcv agents for HCV, such as BMS and BMS , which are concentrated and metabolized by the liver, and administered to a population (chronic hepatitis C patients) at increased risk for DILI. The type of liver injury that leads to severe DILI is predominantly hepatocellular, and is therefore associated with a rise in ALT and AST, but is extensive enough to affect the liver s functional ability to clear bilirubin or to synthesize coagulation factors. Therefore, increased total bilirubin and/or INR associated with marked ALT/AST elevations should warrant consideration of DILI, 67
69 and patient interventions should include discontinuation of study drugs (see Section 4.2.3). Finally, it is possible that subjects treated with may develop NS5A and/or NS3 drug-resistant HCV which may decrease the effectiveness of future treatment with HCV antivirals of the same class. In the MAD study (AI444004) involving HCV-infected subjects, HCV RNA levels rebounded in most individuals dosed with BMS monotherapy. The experience from other DAA molecules indicates that most resistant variants are suppressed when these drugs are used in combination with pegifnα/rbv. Therefore, resistant variants which emerged on monotherapy may be fully or partially suppressed by alone or in combination with pegifnα/rbv. Characterization of drug resistance will be performed as part of this study (Section 6.9). 2 STUDY OBJECTIVES 2.1 Primary Objective Primary Objectives: Part 1: To determine the proportion of subjects in the sentinel cohort with successful response to treatment at Week 2 and RVR at Week 4. Part 2: To determine the proportion of subjects with SVR 12 in each cohort. 2.2 Secondary Objectives Secondary Objectives: To assess the safety of co-administration of NS3+NS5A with and without SOC or ribavirin as measured by the frequency of Serious Adverse Events (SAEs) and AEs, discontinuations due to AEs, and abnormalities observed from vital sign and ECG measurements, physical examinations and clinical laboratory results; To assess the decrease in log 10 HCV RNA from baseline to Day 4, Day 7 and Day 14; To assess the Pharmacokinetic (PK) profiles of subjects treated with NS5A+NS3 with and without SOC/RBV; To evaluate the proportion of subjects with RVR; 68
70 To evaluate the proportion of subjects with extended rapid virologic response (ervr), defined as undetectable HCV RNA at both Week 4 and 12; To evaluate the proportion of subjects with complete early virologic response (cevr), defined as undetectable HCV RNA at Week 12; To evaluate the proportion of subjects with 24-week sustained virologic response (SVR 24 ), defined as undetectable HCV RNA at follow-up Week 24; To describe resistant variants associated with virologic failure. 2.3 Exploratory Objectives Exploratory Objectives: To explore the effect of HCV subtype on antiviral activity; To describe the changes in T-cell immunoregulatory molecules with treatment through analysis of peripheral blood mononuclear cells (PBMCs). To describe T-cell responses (through analysis of PBMCs) to HCV antigens at baseline and their change over time following treatment. To describe the baseline level of IP-10 in serum in all subjects; To explore the relationship between endpoints of safety or antiviral activity and exposure to when co-administered with and without SOC or ribavirin. To explore the relationship between antiviral activity endpoints and single nucleotide polymorphisms (SNPs) in genes encoding proteins of the IFNλ family (IL28A, IL28B, IL29) and potentially the RBV transporter protein ENT1. To describe changes in host gene expression including interferon stimulated genes (ISGs) and immunoregulatory molecules in whole blood following treatment. To potentially explore the relationship between anemia in subjects receiving RBV and SNPs in the gene for inosine triphosphatase (ITPA). 3 ETHICAL CONSIDERATIONS 3.1 Good Clinical Practice This study will be conducted in accordance with Good Clinical Practice (GCP), as defined by the International Conference on Harmonisation (ICH) and in accordance with 69
71 the ethical principles underlying European Union Directive 2001/20/EC and the United States Code of Federal Regulations, Title 21, Part 50 (21CFR50). The study will be conducted in compliance with the protocol. The protocol and any amendments and the subject informed consent will receive Institutional Review Board/Independent Ethics Committee (IRB/IEC) approval/favorable opinion prior to initiation of the study. All potential serious breaches must be reported to BMS immediately. A serious breach is a breach of the conditions and principles of GCP in connection with the study or the protocol, which is likely to affect, to a significant degree, the safety or physical or mental integrity of the subjects of the study or the scientific value of the study. Study personnel involved in conducting this study will be qualified by education, training, and experience to perform their respective task(s). This study will not use the services of study personnel where sanctions have been invoked or where there has been scientific misconduct or fraud (eg, loss of medical licensure, debarment). Systems with procedures that assure the quality of every aspect of the study will be implemented. 3.2 Institutional Review Board/Independent Ethics Committee Before study initiation, the investigator must have written and dated approval/favorable opinion from the IRB/IEC for the protocol, consent form, subject recruitment materials/process (eg, advertisements), and any other written information to be provided to subjects. The investigator or sponsor should also provide the IRB/IEC with a copy of the Investigator Brochure or product labeling, information to be provided to subjects and any updates. The investigator or sponsor should provide the IRB/IEC with reports, updates and other information (eg, expedited safety reports, amendments, and administrative letters) according to regulatory requirements or institution procedures. 70
72 3.3 Informed Consent Investigators must ensure that subjects, or, in those situations where consent cannot be given by subjects, their legally acceptable representatives, are clearly and fully informed about the purpose, potential risks, and other critical issues regarding clinical studies in which they volunteer to participate. Freely given written informed consent must be obtained from every subject or, in those situations where consent cannot be given by subjects, their legally acceptable representative, and prior to clinical study participation, including informed consent for any screening procedures conducted to establish subject eligibility for the study. The rights, safety, and well-being of the study subjects are the most important considerations and should prevail over interests of science and society. Appendix 1 contains BMS procedures on obtaining informed consent from subjects, or, in those situations where consent cannot be given by subjects, their legally acceptable representative prior to participating in a clinical study. Procedures are described for all subjects, including those who are unable to give informed consent. The relevant procedures must be used whenever they are applicable (see subject selection criteria in Sections and 4.2.2). 4 INVESTIGATIONAL PLAN 4.1 Study Design and Duration Overview This is a randomized, open-label, out-patient, multiple-dose study with parallel treatment groups in 2 parts. Approximately 120 HCV genotype 1, null responders to SOC will be randomized to receive either the combination of NS5A+NS3 (treatment group A) or NS5A+NS3 + SOC or ribavirin (treatment group B) for up to 72 weeks. Where indicated, randomization will be stratified by genotype 1a and 1b with the number of lb subjects enrolled capped, allowing for enrollment of more 1a subjects. Other groups will be restricted to genotype 1b only. Subjects who refuse treatment on Day 1 may be replaced. Otherwise, no replacements are allowed. 71
73 In order to reduce the risk of resistance developing in a large number of subjects, the study will be conducted in two parts. In part 1, a sentinel cohort of approximately 10 subjects per treatment group for a total of approximately 20 subjects will be randomized. The decision to expand Treatment Groups A and B will be made independently and will be based upon results of the pre-planned decision points at Weeks 2 and 4. Part 2 of the study continues as follows: If the decision is made to expand Treatment Group A then approximately 20 additional subjects, all with HCV genotype 1b will be randomized into each Expansion Cohort A1 and A2. The subjects in the sentinel cohorts as well as the subjects in the Expansion Cohorts will continue treatment for up to 24 weeks. See Figure 4.1.1A. If the decision is made to expand Treatment Group B then approximately 20 additional subjects with HCV genotype 1a or 1b will be randomized into each of Expansion cohorts B1 and B2. Approximately 20 additional subjects might be enrolled into Expansion cohort B3 contingent upon demonstration of adequate antiviral activity in Expansion cohort B1. The subjects in the sentinel cohorts as well as the subjects in the Expansion cohorts will continue treatment for up to 24 weeks. See Figure 4.1.1B. To further reduce the risk of resistance, group discontinuation criteria are established for Expansion Cohorts. Subjects (for sentinel and Expansion Cohorts) will undergo screening evaluations to determine eligibility within 28 days prior to study enrollment. A subject is considered enrolled when the protocol-specific informed consent is signed. Subjects will report to the clinical facility the morning prior to dosing (Day 1). On Day -1 or Day 1, subjects will be randomized to receive NS5A+NS3 or NS5A+NS3+SOC or ribavirin. Subjects will self-administer a daily oral dose of NS5A+NS3 or NS5A+NS3 + SOC or ribavirin for up to 72 weeks and report to the study site for scheduled visits as outlined in Tables 6.1A, B, C and D. 72
74 Figure 4.1.1A: Study Design Schematic for Treatment A Treatment A, Sentinel Cohort (NS3 + NS5A for 24 Weeks) a N = 10 Follow-up for 48 weeks post-treatment Week 2 Interim Analysis: If 70% of subjects have undetectable HCV RNA or decrease in plasma HCV RNA 2 log 10 IU/mL without rebound continue treatment group Otherwise, cohort will not be expanded and only individual subject decision rules will apply to subjects. Week 4 Interim Analysis: If 50% subjects have undetectable HCV RNA (RVR), continue treatment group and expand cohort. Otherwise, cohort will not be expanded and only individual subject decision rules will apply to subjects. Expand Treatment A? Yes Expansion Cohort A1 NS3 200 mg BID+NS5A 60 mg QD for 24 Weeks a N = 20 (Genotype 1b) b Expansion Cohort A2 NS3 200 mg QD+NS5A 60 mg QD for 24 Weeks a N = 20 (Genotype 1b) b Follow-up for 48 weeks post-treatment Follow-up for 48 weeks post-treatment a If rescue criteria are met for Individual Subject Decision Rules, SOC can be added for up to 48 additional weeks. b Due to viral breakthrough with treatment A in subjects infected with HCV GT1a in the Sentinel Cohort, the decision was made to limit the expansion of treatment group A to GT1b subjects only. 73
75 Figure 4.1.1B: Study Design Schematic for Treatment B Treatment B, Sentinel Cohort (NS3+NS5A+SOC for 24 Weeks) N = 10 Follow-up for 48 weeks post-treatment. Week 2 Interim Analysis: If 70% of subjects have undetectable HCV RNA or decrease in plasma HCV RNA 2 log 10 IU/mL without rebound continue treatment group Otherwise, cohort will not be expanded and only individual subject decision rules will apply to subjects. Week 4 Interim Analysis: If 50% subjects have undetectable HCV RNA (RVR), continue treatment group and expand cohort. Otherwise, cohort will not be expanded and only individual subject decision rules will apply to subjects. Expand Treatment B? Yes Expansion Cohort B1 NS3 200 mg BID+NS5A 60 mg QD + SOC for 24 Weeks N = 20 (Genotype 1a/1b) c Expansion Cohort B2 NS3 200 mg QD+NS5A 60 mg QD + SOC for 24 Weeks N = 20 (Genotype 1a/1b) c Expansion Cohort B3 a NS3 200 mg BID+NS5A 60 mg QD + RBV for 24 Weeks b N = 20 (Genotype 1a/1b) c Follow-up for 48 weeks post-treatment Follow-up for 48 weeks post-treatment Follow-up for 48 weeks post-treatment a The decision to open Expansion Cohort B3 for dosing is contingent upon demonstration of adequate antiviral activity in Expansion Cohort B1. b If rescue criteria are met for Individual Subject Decision Rules, pegylated Interferon can be added for up to 48 additional weeks. c. Randomization will be stratified by subtype 1a and 1b, and the total enrollment of genotype 1b subjects will be capped at 20% in each cohort. 74
76 Plasma HCV RNA levels will be measured at screening and on Days -1 (baseline), 1 to 7, 9, 11, 14, Week 3 and every 2 weeks from Week 4 to Week 12 in all subjects. HCV RNA measurements will then proceed as follows: All Treatment group A cohorts and Expansion Cohort B3: Every 2 weeks from Week 12 until EOT unless SOC is added to the regimen, in which case, samples will be collected every 4 weeks from Week 12 until EOT. Subjects who have SOC or pegifn added due to viral breakthrough will have HCV RNA measured immediately prior to administration of the first dose of pegifn and every 4 weeks thereafter until EOT. Treatment group B sentinel cohort and Expansion Cohorts B1 and B2: HCV RNA level measurement every 4 weeks from Week 12 until EOT. All subjects: HCV RNA measured at Week 4, 12, 24, 36, and 48 post-treatment. Characterization of HCV genomic substitutions associated with exposure of will be determined from blood samples collected from all subjects and for which sufficient HCV RNA permits analysis (generally but not exclusively 1000 IU/mL). Peripheral blood mononuclear cells (PBMCs) for assessment of antigen-specific anti-hcv T cell responses will be collected on Days -1, 7 and 14 in the Sentinel Cohorts. In the Expansion Cohorts, PBMCs will be collected at Days -1, 14 and 28, Weeks 8, 12, 16, 24 on treatment and Weeks 4, 12 and 24 post treatment. Blood samples for analysis of polymorphisms in genes encoding proteins of the IFNλ family (IL28A, IL28B, IL29) and potentially the RBV transporter protein ENT1 will be collected on Day -1 (baseline). For all subjects in the sentinel cohorts and the first 12 subjects in each Expansion Cohort: Sparse PK samples will be collected up to 24 hours post-dose on Day 1. Serial blood PK samples will be obtained up to 24 hours post-dose on Day 14. For all subjects: Blood samples for trough concentration will be obtained on Day 7 and Weeks 4, 8, and 12 for assessment. Sparse PK samples will be collected 2 hours post-am dose on Weeks 4, 8 and
77 Physical examinations, vital sign measurements, 12-lead electrocardiograms (ECG), and clinical laboratory evaluations will be performed at selected times throughout the dosing interval. Subjects will be closely monitored for adverse events throughout the study. Blood will be collected for PK and Pharmacodynamic (PD) analyses at selected times throughout the study. Approximately 500 ml of blood will be drawn from each subject during the study. End of study is defined as date of the last visit of the last subject undergoing the study. Last visit is defined as the date last follow-up visit. The approximate duration of the study is up to 124 weeks (including a 28 day screening period, a treatment period of up to 72 week and a follow-up visit up 48 weeks after date of last treatment Part 1 Part 1 is represented by the sentinel cohort with treatment duration up to 28 days and 2 study decisions at Week 2 and 4. In the sentinel cohort, 20 subjects will be randomized in a 1:1 ratio to one of the two treatment groups (A or B). Also, randomization will be stratified by genotype 1a and 1b, with the number of 1b subjects capped at 2 per treatment group so that each treatment group will have at least 8 subjects with genotype 1a. In treatment group A, approximately 10 subjects will receive 60 mg of BMS QD and 600 mg of BMS BID in combination. In treatment group B, approximately 10 subjects will receive SOC in addition to the combination of the 2 study drugs. During the analysis of HCV RNA levels, subjects in the sentinel cohort will continue treatment for up to 24 weeks as long as all individual criteria for continuation are met (Part 2). Sparse PK samples will be collected up to 24 hours post-dose on Day 1 and serial blood PK samples will be obtained up to 24 hours post dose on Day 14. At Week 2 and 4, all subjects will have HCV RNA levels measured. The study will continue and expand if criteria are met as outlined in Table below. Otherwise, 76
78 2 cohort will not be expanded and only individual subject decision rules will apply to subjects. (See Table 4.1.4A). Table 4.1.2: Sentinel Cohort Study Decision Points at Weeks 2 and 4 a Week Week 4 Treatment A: NS3 + NS5A Treatment B: NS3 + NS5A + SOC Part 1 Sentinel cohort decision point at Week 2 a If 70% of subjects have undetectable HCV RNA or decrease in plasma HCV RNA 2 log 10 IU/mL without rebound continue treatment group. Otherwise, cohort will not be expanded and only individual subject decision rules will apply to subjects Sentinel cohort decision point at Week 4 a If 50% subjects have undetectable HCV RNA (RVR), continue treatment group and expand cohort. Otherwise, cohort will not be expanded and only individual subject decision rules will apply to subjects Study decisions at Weeks 2 and 4 for treatment groups A and B are made independently of each other. Note: On September 21, 2010, the dose of BMS for all on-going subjects was reduced from 600 mg BID to 200 mg BID (Amendment 06). At that time, all subjects on Treatment B and all subjects on Treatment A who had not received rescue with PegIFN/RBV, had completed their treatment. Five subjects in Treatment A who were receiving extended therapy as a result of viral breakthrough and the addition of PegIFN/RBV to their treatment were impacted by the dose change Part 2 Part 2 is represented by the duration after Week 4 of the Sentinel Cohorts (both Treatments A and B), and the whole study duration of the Expansion Cohorts. Subjects in the sentinel cohorts will continue dosing following individual subject decision rules (See Table 4.1.4A). Expansion of a treatment group will occur only after the sentinel cohort satisfies criteria for successful response to treatment at Week 2 and RVR at Week 4. Expansion of each 77
79 treatment will be made independently. Thus, study expansion may include Treatment A, Treatment B, or both. If the decision is made to expand treatment group A, approximately 40 additional subjects, all with HCV genotype 1b will be randomized to either Expansion Cohort A1 or A2. Expansion Cohort A1: Approximately twenty (20) subjects will be randomization to receive NS3 200 mg BID+NS5A 60 mg QD for up to 24 weeks; Expansion Cohort A2: Approximately twenty (20) subjects will be randomized to receive NS3 200 mg QD +NS5A 60 mg QD for up to 24 weeks; Note: Due to viral breakthrough with treatment A in subjects infected with HCV genotype 1a (GT1a) in the Sentinel Cohort, the decision was made to limit the expansion of treatment group A to GT1b subjects only. If the decision is made to expand treatment group B, approximately 40 additional subjects with HCV genotype 1a or 1b will be randomized to either Expansion Cohort B1 or B2: Expansion Cohort B1: Approximately twenty (20) subjects will be randomized to receive NS3 200 mg BID+NS5A 60 mg QD+SOC for up to 24 weeks Expansion Cohort B2: Approximately twenty (20) subjects will be randomized to receive NS3 200 mg QD+NS5A 60 mg QD+SOC for up to 24 weeks. Randomization of Expansion Cohorts B1 and B2 will be stratified by genotype 1a and 1b, with the number of genotype 1b capped at 20% of total subjects in each cohort. Approximately 20 additional subjects with HCV genotype 1a or 1b might be dosed in Expansion Cohort B3: Expansion Cohort B3: Approximately twenty (20) subjects will receive NS3 200 mg BID+NS5A 60 mg QD+Ribavirin for up to 24 weeks The decision to open Expansion Cohort B3 for dosing is contingent upon demonstration of adequate antiviral activity in Expansion Cohort B1. The decision criterion is described in detail in Section below. The number of subjects with HCV genotype 1b will be capped at 20% of the total number of subjects in Expansion Cohort B3. 78
80 4.1.4 Individual Subject Decision Rules Individual subject decision rules guide the addition of SOC or pegylated Interferon (PegIFN) alone (based upon treatment assignment) as well as discontinuation of therapy during the study. Decisions are based upon an ongoing evaluation of HCV viral loads and are defined by treatment assignment and study week as outlined in Tables 4.1.4A and B below. These rules are based upon the definitions of HCV viral breakthrough outlined below: Viral Breakthrough Definitions by Treatment: Treatment Group A Sentinel Cohort 1) Any increase in HCV viral load 1 log from nadir (not necessarily from a consecutive sampling). 2) Any quantifiable HCV RNA 25 IU/mL on or after week 4. 3) Any detectable HCV RNA < 25 IU/mL on or after week 4 confirmed by a subsequent consecutive HCV RNA measurement. Expansion Cohorts A1, A2 and B3 1) Any increase in viral load 1 log from nadir 2) Any confirmed detectable HCV RNA < 25 IU/mL on or after Week 8. Confirmation should occur via an immediate unscheduled return visit. 3) Any quantifiable HCV RNA 25 IU/mL on or after Week 8 (no confirmation needed). Expansion Cohorts B1 and B2 1) Any increase in HCV viral load 1 log from nadir (not necessarily from a consecutive sampling). 2) Any confirmed quantifiable HCV RNA 25 IU/mL after confirmed undetectable HCV RNA. Measurements are confirmed at the next scheduled visit. 79
81 Table 4.1.4A: Individual Subject Decision Rules for the Sentinel Cohorts Weeks 2 and 3 Weeks 4 to 22 Week 24 Treatment A: NS3 + NS5A Sentinel Cohort Undetectable: continue up to 24 weeks. Detectable > 2 log 10 decrease from baseline with no breakthrough: continue up to 24 weeks Detectable 2 log 10 decrease from baseline with breakthrough: add SOC for up to 48 additional weeks Detectable < 2 log 10 decrease from baseline: discontinue All subjects randomized to Treatment A are expected to have undetectable HCV viral load (<10 IU/mL) beyond Week 4. Undetectable: continue up to 24 weeks. Detectable < 25 IU/mL: perform immediate unscheduled visit for repeat HCV VL. If second value is detectable (>10 IU/mL), add SOC for up to 48 additional weeks Quantifiable 25 IU/mL but 2 log10 decrease from baseline: add SOC immediately for up to 48 additional weeks, and draw viral load for pre-soc baseline. Detectable < 2 log10 decrease from baseline: discontinue Undetectable: stop therapy and observe for SVR. Detectable 2 log 10 decrease from baseline: add SOC for up to 48 additional weeks Detectable < 2 log 10 decrease from baseline: discontinue Treatment B: NS3 + NS5A + SOC Sentinel Cohort Undetectable: continue up to 24 weeks Detectable 2 log 10 decrease from baseline: continue up to 24 weeks Detectable < 2 log 10 decrease from baseline: discontinue Undetectable: continue up to 24 weeks Detectable 2 log 10 decrease from baseline: continue up to 24 weeks Detectable < 2 log 10 decrease from baseline: discontinue Undetectable: stop therapy and observe for SVR Detectable: discontinue 80
82 Table 4.1.4B: Individual Subject Decision Rules for the Expansion Cohorts Weeks 2 and 3 Weeks 4 and 6 Weeks 8 to 22 Expansion Cohorts A1, A2 and B3 NS3 + NS5A +/- RBV Undetectable: continue up to 24 weeks Detectable 2 log 10 decrease from baseline and NOT 1 log increase in HCV RNA from nadir: continue up to 24 weeks Detectable < 2 log 10 decrease from baseline or 1 log increase in HCV RNA from nadir: add PegIFN or SOC for up to 48 weeks a Undetectable: continue up to 24 weeks. Detectable < 25 IU/mL: recheck HCV RNA level at next visit Quantifiable 25 IU/mL but not 1 log increase from nadir: recheck HCV RNA level at next visit Quantifiable 25 IU/mL and 1 log increase from nadir: add PegIFN or SOC for up to 48 weeks b Undetectable: continue up to 24 weeks. Detectable < 25 IU/mL: perform immediate unscheduled HCV RNA level recheck. If persistently detectable, add PegIFN or SOC for up to 48 weeks c ; if < 10 IU/mL (undetectable) continue therapy Quantifiable 25 IU/mL: add PegIFN or SOC for up to 48 weeks d Week 24 Undetectable: stop therapy and observe for SVR Detectable < 25 IU/mL: stop therapy and perform an immediate unscheduled HCV RNA level recheck. If persistently detectable, start SOC alone for up to 48 weeks; if < 10 IU/mL (undetectable), observe for SVR Quantifiable 25 IU/mL add PegIFN or SOC for up to 48 weeks e a Expansion Cohort B1 and B2 NS3 + NS5A + SOC Undetectable: continue up to 24 weeks. Detectable > 2 log 10 decrease from baseline: continue up to 24 weeks Detectable < 2 log 10 decrease from baseline or 1 log increase in HCV RNA from nadir: consider discontinuation of therapy Undetectable: continue up to 24 weeks. Detectable 2 log10 decrease from baseline: continue up to 24 weeks Detectable < 2 log 10 decrease from baseline or 1 log increase in HCV RNA from nadir: consider discontinuation of therapy Undetectable: continue up to 24 weeks. Detectable 2 log10 decrease from baseline: continue up to 24 weeks Detectable < 2 log 10 decrease from baseline or 1 log increase in HCV RNA from nadir: consider discontinuation of therapy Undetectable: stop therapy and observe for SVR. Detectable: discontinue therapy. For Expansion Cohorts A1 and A2, the rescue is addition of SOC; For Expansion Cohort B3, the rescue is addition o f pegifn. 81
83 Subjects who receive rescue treatment with SOC or PegIFN must have a decrease in HCV RNA 2 log10 by 12 weeks from the beginning of rescue therapy in order to continue treatment up to Week 72. Subjects not meeting this criterion will stop all therapy after Week 12. Subjects who experience viral relapse during post treatment follow up periods will not receive rescue therapy as part of the study protocol. Further treatment will be at the discretion of the treating investigator Group Decision Rules for Suspending an Expansion Cohort Group decision rules provide guidelines for the early suspension of cohorts that incur unacceptable rates of viral breakthrough. 1) Each Expansion Cohort will be evaluated independently. 2) Antiviral activity will be assessed on an ongoing basis. 3) Should 50% of treated subjects in a cohort with 10 or more subjects experience viral breakthrough, further randomization into the involved cohort will be stopped. 4) A minimum of 5 subjects of the first 10 planned subjects in a cohort must experience viral breakthrough before a cohort will be stopped. 5) Should the decision rule be met in a cohort including the lower dose of NS3 (200mg QD), subjects receiving treatment in that cohort but not experiencing viral breakthrough will have their NS3 dose increased to 200mg BID. 6) Subjects will only receive therapeutic rescue if individual subject decision criteria for addition of rescue therapy are met and not based upon Group decision criteria. 7) Subjects randomized into the failed cohorts but not dosed will be re-randomized into the remaining treatment cohorts Rule for Initiating Expansion Cohort B3 (NS3 + NS5A + RBV) Two antivirals alone resulted in an unacceptable rate of treatment failure when administered to subjects infected with genotype 1a while four medications (2 antivirals plus SOC) provided 100% virologic control. Given this was achieved with a regimen including a higher dose of BMS (600 mg BID), it is prudent to require a similar antiviral activity of the four drugs with the lower dose of BMS (200mg BID) before investigating the antiviral activity of 3 drugs in subjects infected with genotype 1a. 82
84 1) Dosing in Expansion cohort B3 is contingent upon demonstration of adequate antiviral activity in Expansion cohort B1, i.e., at least 16/20 (>75%) treated subjects in Expansion cohort B1 must reach week 12 on treatment without viral breakthrough for Expansion cohort B3 to be initiated. 4.2 Study Population For entry into the study, the following criteria MUST be met prior to dosing on Day 1. No exceptions will be granted Inclusion Criteria 1) Signed Written Informed Consent a) The signed informed consent form. 2) Target Population a) Subjects chronically infected with HCV Genotype 1 who are null-responders defined as subjects who after at least 12 weeks of therapy with the current standard of care (pegifnα and RBV) have never attained 2 log 10 decline in HCV RNA level. Investigator will verify null-responder status. In the event of multiple prior treatment courses, the most recent response may be used to qualify the subject as a null responder. b) Subjects should have chronic hepatitis C (CHC) as documented by: i) Positive for anti-hcv antibody, HCV RNA, or a positive HCV genotype test at least 6 months prior to screening, and positive for HCV RNA and Anti- HCV antibody at the time of screening, or ii) Positive for anti-hcv antibody and HCV RNA at the time of screening with a liver biopsy consistent with chronic HCV infection (or a liver biopsy performed prior to enrollment with evidence of chronic hepatitis C disease, such as the presence of fibrosis) c) HCV RNA viral load of 10 5 IU/mL (100,000 IU/mL) at screening and a history of either a positive HCV antibody or a positive HCV RNA test at least 6 months prior to screening. d) Have not received another NS5A replication co-factor inhibitor and/or NS3 protease inhibitor. e) Have one of the following: 83
85 i) Documented Fibrotest score of 0.72 and AST to platelet ratio index (APRI) 2; or ii) Documented liver biopsy within 24 months preceding Day 1 showing absence of cirrhosis. Please note that Liver biopsy results may be used to supersede a Fibrotest score for study entry f) Not co-infected with HIV or HBV. g) Subjects with chronic medical disorders that are commonly seen in advancing age groups may participate in the study if the investigator determines: i) That the volunteer is medically stable; ii) It is unlikely that the study will be interrupted because of the need for treatment or significant change of medications; iii) The medical disorder will not cause any undue physical discomfort for the volunteer; iv) The medical disorder will not necessitate any special accommodations that are deemed by the investigator to be excessive or problematic; v) The medical disorder shall not be disruptive to the medical staff or other volunteers. h) Body Mass Index (BMI) of 18 to 35 kg/m 2, inclusive. BMI = weight (kg)/ [height (m)] 2. 3) Age and Sex a) Men and women, ages 18 to 70, Women of childbearing potential (WOCBP), unless non-heterosexually active or have a vasectomized partner and sexually active men (unless vasectomized) with partners who are WOCBP must be using highly effective contraception methods (two separate forms of contraception, one of which must be an effective barrier method) to avoid pregnancy. Subjects in treatment group A are required to follow these directions throughout the study and for up to 12 weeks after the last dose has been administered, and subjects in treatment group B (includes subjects in treatment group A that have SOC added on) will be required to do the same for 6 months following the last dose of ribavirin. Acceptable methods of barrier contraception include: Condom with spermicide Diaphragm and spermicide 84
86 Cervical cap and spermicide Use of hormonal contraceptives are allowed in the study, but may not be counted as one of the methods of contraception. Drug interaction studies examining concomitant use of oral contraceptives with BMS or BMS have not been completed. The use of intrauterine devises, (IUDs) shall be at the discretion of the investigator. The increased risk of anemia from associated heavy menses and possibility of infection while on ribavirin shall be considered. WOCBP include any female who has experienced menarche and who has not undergone successful surgical sterilization (hysterectomy, bilateral tubal ligation, or bilateral oophorectomy) or is not postmenopausal. Post menopause is defined as: Amenorrhea 12 consecutive months without another cause or For women with irregular menstrual periods and on hormone replacement therapy (HRT), a documented serum follicle stimulating hormone (FSH) level > 40 miu/ml Women who are using oral contraceptives, other hormonal contraceptives (vaginal products, skin patches, or implanted or injectable products), or mechanical products such as an intrauterine device or barrier methods (diaphragm, condoms, spermicides) to prevent pregnancy, or are practicing abstinence or where their partner is sterile (eg, vasectomy) should be considered to be of childbearing potential. WOCBP must have a negative serum or urine pregnancy test (minimum sensitivity 25 IU/L or equivalent units of HCG) within 24 hours prior to the start of investigational product and must agree to the pregnancy testing specified in this protocol. (See Tables 6.1A, B, C and D) Men (unless vasectomized) with female partners who are WOCBP must agree to inform their female partners of the protocol-specified contraception requirements and pregnancy testing recommendations during treatment and post-treatment (for subjects on antivirals only, monthly pregnancy testing will occur on-treatment, as well as for 12 weeks after discontinuation of treatment; for subjects who take 85
87 RBV in this study, monthly pregnancy testing should occur on-treatment, as well as 6 months after the discontinuation of RBV) and agree to adhere to these recommendations for both the on-treatment and post-dosing follow-up period Exclusion Criteria 1) Sex and Reproductive Status a) WOCBP who are unwilling or unable to use highly effective contraception methods (two separate forms of contraception, one of which must be an effective barrier method) to avoid pregnancy for the entire study period and for up to 12 weeks for subjects in treatment group A and 6 months for subjects in treatment group B (includes subjects in treatment group A that have SOC added on) after the last dose of investigational product. b) Women who are pregnant or breastfeeding c) Women with a positive pregnancy test on enrollment or prior to investigational product administration. d) Sexually active fertile men not using effective birth control methods as described above. Men who are not willing to refrain from sperm donation during study participation and for at least 12 weeks for subjects in treatment group A and 6 months for subjects in treatment group B after dosing has been concluded. 2) Medical History and Concurrent Diseases a) Evidence of a medical condition associated with chronic liver disease other than HCV (such as but not limited to: hemochromatosis, autoimmune hepatitis, metabolic liver disease, alcoholic liver disease, toxin exposures); b) History of variceal bleeding, hepatic encephalopathy, or ascites requiring management with diuretics or paracentesis; c) Current or known history of cancer (except in situ carcinoma of the cervix or adequately treated basal or squamous cell carcinoma of the skin) within 5 years prior to enrollment; d) Any gastrointestinal disease or surgical procedure (cholecystectomy is allowed) that may impact the absorption of study drug; e) Donation of blood or plasma to a blood bank or in a clinical study (except a screening visit or follow up visit of more than 50 ml) within 4 weeks of study drug administration. f) Blood transfusion within 4 weeks of study drug administration. g) Inability to tolerate oral medication. h) Poor venous access. i) A history of clinically significant cardiac disease. 86
88 j) Recent (within 6 months) drug or alcohol abuse as defined in DSM IV, Diagnostic Criteria for Drug and Alcohol Abuse (Appendix 2). k) Any other medical, psychiatric and/or social reason which, in the opinion of the Investigator, would make the candidate inappropriate for participation in this study. l) HCV infected subjects who are treatment naive. m) HCV infected subjects who are treatment non-responder defined as a subject who received at least 12 weeks of SOC and continue to have a detectable HCV RNA level or subjects who may have attained a 2 log 10 decline in HCV RNA levels but did not sustain it at 12 weeks and stopped treatment. Non-responders are different from null-responders in that non-responders have at some point achieved a 2 log 10 decline in HCV RNA. n) HCV infected subjects who are treatment intolerant (defined as subject who are unable to receive at least 12 weeks of SOC due to toxicities associated with IFN and/or RBV). 3) Physical and Laboratory Test Findings a) Evidence of organ dysfunction that is not consistent with age or a condition that will likely progress to a clinically unstable state. b) Documented cirrhosis on histopathology from liver biopsy done within 24 months prior to dosing. c) QTcF value 450 msec at Screening or Day 1 (prior to dosing), confirmed by repeat ECG. d) Evidence of second or third degree heart block at screening or Day 1 (prior to dosing) confirmed by repeat ECG. e) Positive blood screen hepatitis B surface antigen. f) Positive blood screen for HIV-1 and/or -2 antibodies. g) History of G6PD deficiency. h) Positive urine or serum test for βhcg (females only) at Screening or prior to dosing. i) Any of the following laboratory results at Screening or prior to dosing outside of the ranges specified below as defined by the central laboratory, confirmed by repeat analysis. i) Hemoglobin 12 g/dl (120 g/l) for women and 13 g/dl (130 g/l) for men; ii) Neutrophils (absolute) 1500/mL ( 1200/mL for subjects of black race) iii) Platelet count 90,000/mL iv) ALT > 5xupper limit of normal (ULN) v) Direct bilirubin > 1.5xULN 87
89 vi) Albumin < 3.2 g/dl vii) Creatinine clearance (as estimated by method of Cockcroft and Gault 45 ) < 50 ml/min. 4) Medical History or Laboratory Findings that Exclude Subject from pegifnα or RBV Therapy The following exclusion criteria are based on guidelines or recommendations from the pegifnα 43 and Ribavirin 44 package inserts. a) Severe psychiatric disease, especially untreated or unstable depression, that would prohibit use of pegifnα as judged by investigator; b) History of hemoglobinopathies (i.e., thalassemia major or sickle cell anemia), diagnoses associated with an increased baseline risk for anemia (eg spherocytosis), hemolytic anemia, or diseases in which anemia would be medically problematic; c) History of thyroid dysfunction not adequately controlled. (Subjects with a medically controlled thyroid condition should be monitored with regular thyroid function tests including baseline clinical laboratory testing prior to randomization.) d) History of chronic pulmonary disease associated with functional limitation; e) History of cardiomyopathy, coronary artery disease (including angina), interventive procedure for coronary artery disease (including angioplasty, stent procedure, or cardiac bypass surgery), ventricular arrhythmia, or other clinically significant cardiac disease; f) Pre-existing ophthalmologic disorders considered clinically significant on eye or retinal exam (all subjects with history of diabetes or hypertension must have a documented eye exam within 12 months prior to randomization); 5) Allergies and Adverse Drug Reactions a) History of allergy to BMS , BMS , peg-ifnα, RBV or related compounds. b) History of any significant drug allergy (such as anaphylaxis or hepatotoxicity). 6) Prohibited Treatments and/or Therapies a) Prior exposure to any investigational agent or drug with potential anti-hcv activity unless otherwise reviewed and approved by the BMS medical monitor. Prior treatments with direct acting antivirals with activity against HCV are specifically excluded. b) Therapies including interferon, peg-interferon and/or RBV within the previous 12 weeks. Subjects may be screened following a 12 week "washout" period. 88
90 c) Exposure to any investigational drug or placebo within 4 weeks of study drug administration; d) Use of any prescription drugs within 4 weeks prior to study drug administration. However, certain drugs may be allowed if approved by the BMS medical monitor. e) Use of any other drugs, including over-the-counter medications, vitamins and/or herbal preparations (e.g. St. John s wort, milk thistle), within 1 week prior to study drug administration. However, certain drugs may be allowed if approved by the BMS medical monitor. f) Use of alcohol-containing beverages within 3 days prior to study drug administration. g) Use of grapefruit or grapefruit-containing products or Seville oranges within 3 days prior to study drug administration. h) Long-term treatment with immunosuppressive agents or with drugs that are associated with a high risk for nephrotoxicity or hepatotoxicity; i) Strong or moderate inhibitors or inducers of CYP3A4 (refer to Section for a detailed listing). j) Use of proton pump inhibitors. Restricted use of H2 receptor antagonists or other acid modifying agents such as antacids are allowed (see Section 5.5.1). 7) Other Exclusion Criteria a) Prisoners or subjects who are involuntarily incarcerated b) Subjects who are compulsorily detained for treatment of either a psychiatric or physical (eg, infectious disease) illness Eligibility criteria for this study have been carefully considered to ensure the safety of the study subjects and to ensure that the results of the study can be used. It is imperative that subjects fully meet all eligibility criteria Discontinuation of Subjects from Treatment Subjects MUST discontinue investigational product (and noninvestigational product at the discretion of the investigator) for any of the following reasons: Withdrawal of informed consent (subject s decision to withdraw for any reason) Any clinical adverse event (AE), laboratory abnormality or intercurrent illness which, in the opinion of the investigator, indicates that continued participation in the study is not in the best interest of the subject Any Grade 4 AE considered study drug related (as indicated in Appendix 3) 89
91 Pregnancy (see Section 7.6.2) Termination of the study by Bristol-Myers Squibb (BMS) Loss of ability to freely provide consent through imprisonment or involuntarily incarceration for treatment of either a psychiatric or physical (eg, infectious disease) illness Inability to comply with protocol Discretion of the investigator QTcF > 500 msec (confirmed by repeat ECG) Confirmed second (Mobitz type II) or third-degree heart block. Evidence of confirmed hepatic decompensation (Child-Pugh Class B or C, Score > 6) ALT > 2xbaseline and 5xULN and either total bilirubin > 2xULN or INR > 2xULN Platelets < 25x10 9 cells/l If criteria to continue study outlined in Tables 4.1.4A and B. (Individual Subject Decision Rules) are not met. Dosing within any cohort (sentinel or Expansion Cohorts) must be stopped and the study halted until safety information can be reviewed in the event of the following situations: Three or more subjects within any given cohort of 10 (or 6 per cohort of 20) subjects experience onset of QTcF values > 500 msec following dosing and confirmed by repeat ECG. Three or more subjects within a cohort of 10 (or 6 per cohort of 20) subjects experience confirmed second (Mobitz type II) or third-degree heart block following dosing. Three or more subjects within a cohort of 10 (or 6 per cohort of 20) subjects with confirmed elevation in ALT > 10xbaseline (Day -1) result. Life threatening ventricular arrhythmia, including torsades de pointes, occurs in any subject in any given cohort of subjects. All subjects who discontinue should comply with protocol specified follow-up procedures as outlined in Section 6. The only exception to this requirement is when a subject withdraws consent for all study procedures or loses the ability to consent freely (i.e., is imprisoned or involuntarily incarcerated for the treatment of either a psychiatric or physical illness). 90
92 If a subject was withdrawn before completing the study, the reason for withdrawal must be entered on the appropriate case report form (CRF) page. 5 TREATMENTS 5.1 Study Treatment All protocol-specified investigational and noninvestigational products are considered study drug. Restrictions related to food intake are described in Section Investigational Product An investigational product, also known as investigational medicinal product in some regions, is defined as follows: A pharmaceutical form of an active substance or placebo being tested or used as a reference in a clinical study, including products already with a marketing authorization but used or assembled (formulated or packaged) in a way different from the authorized form, or used for an unauthorized indication, or when used to gain further information about the authorized form. In this protocol, investigational products are: BMS mg tablets, BMS mg tablets and ribavirin (Copegus ) Noninvestigational Product Other medications used in the study as support or escape medication for preventative, diagnostic, or therapeutic reasons, as components of the standard of care for a given diagnosis, are considered noninvestigational products. In this protocol, noninvestigational product is: Peg-interferon alfa-2a (Pegasys ). 91
93 5.1.3 Identification Identification of BMS BMS will be supplied by Bristol-Myers Squibb Research and Development as shown below: PRODUCT POTENCY APPEARANCE BMS Film-Coated Tablet 30 mg (as the free base) A plain, round, biconvex, beveledged, film-coated, white tablet Identification of BMS BMS will be supplied by Bristol-Myers Squibb Research and Development as shown below: PRODUCT POTENCY APPEARANCE BMS Film Coated Tablet 200 mg Plain, white to off-white, convex, round, film coated tablet Identification of Peg-interferon alfa-2a(pegasys ) Peg-interferon alfa-2a (Pegasys ) will be supplied by Bristol-Myers Squibb Research and Development as shown below: PRODUCT POTENCY APPEARANCE Peg-interferon alfa-2a(pegasys ) solution for injection 180 mcg /vial 0.5mL prefilled syringe Solution is clear and colorless to light yellow 92
94 5.1.4 Identification of RBV (Copegus ) Ribavirin (Copegus ) will be supplied by Bristol-Myers Squibb Research and Development as shown below: PRODUCT POTENCY APPEARANCE Ribavirin (Copegus ) 200mg Light pink, flat oval-shaped film-coated tablet. Marked with RIB 200 on one side and Roche on the opposite side Packaging and Labeling Packaging and Labeling- BMS The BMS study medication will be packaged in HDPE bottles. Each bottle of BMS mg (as the free base) will contain 35 tablets, and will be labeled with a single panel open label. The label will contain the protocol stem, number of tablets/bottle, container number, batch number, storage conditions, directions of use and caution statements required by country regulations Packaging and Labeling- BMS The BMS study medication will be packaged in HDPE bottles. Each bottle of BMS mg will contain 70 tablets, and will be labeled with a single panel open label. The label will contain the protocol stem, number of tablets/bottle, container number, batch number, storage conditions, directions of use and caution statements required by country regulations Packaging and Labeling peginterferon alfa-2a Each peg-interferon alfa-2a 180 mcg will be packaged in a single use, graduated, clear glass pre-filled syringe. Each pre-filled syringe provides 0.5 ml containing 180 mcg peg-interferon alfa-2a for subcutaneous injection. Each package contains one pre-filled syringe. 93
95 Each peg-interferon alfa-2a (Pegasys ) 180 mcg 0.5mL pre-filled syringe will be labeled with an open label. The syringe label will contain the batch number, storage conditions, route of administration, directions of use, and IND caution statement. Additionally, the outer carton will have a label containing the protocol prefix, number of syringes per box, container number, batch number, storage conditions, directions of use, expiry date and caution statements required by country regulations. For additional information on peginterferon alfa-2a, please refer to the package insert Packaging and Labeling - ribavirin (Copegus ) Ribavirin (Copegus ) tablets will be supplied in a bottle containing 180 tablets. The label will contain the protocol prefix, number of tablets per bottle, container number, batch number, storage conditions, directions of use, expiry date and caution statements required by country regulations. For additional information on ribavirin, please refer to the package insert Handling and Dispensing Study drug supplied by the sponsor or sourced by the investigator should be stored in a secure area according to local regulations. It is the responsibility of the investigator to ensure that study drug is only dispensed to study subjects. The study drug must be dispensed only from official study sites by authorized personnel according to local regulations. The investigator should ensure that the study drug is stored in accordance with the environmental conditions (temperature, light, and humidity) as determined by the sponsor. If concerns regarding the quality or appearance of the study drug arise, do not dispense the study drug and contact the sponsor immediately. Please refer to Section for information on study drug record retention and 9.3 for return and destruction instructions. 94
96 Handling and Dispensing BMS Storage: BMS mg tablets should be stored in a tightly closed container at temperatures between 15 C to 25 C (59 F to 77 F) and protected from light. Maximum allowable time for exposure to ambient light is 12 hours. Administration: The investigator (or assigned designee, ie, study pharmacist) will dispense the proper number of bottles (as assigned by the IVRS) to the subject to satisfy dosing requirements until the subject s next visit Handling and Dispensing BMS Storage: BMS mg tablets should be stored in a tightly closed container at temperatures between 15 C to 25 C (59 F to 77 F). Administration: The investigator (or assigned designee, ie, study pharmacist) will dispense the proper number of bottles (as assigned by the IVRS) to the subject to satisfy dosing requirements until the subject s next visit Handling and Dispensing Peg-interferon alfa-2a(pegasys ) Storage: Unopened pre-filled syringes of Peg-interferon alfa-2a are stable until the expiration date indicated on the package when stored between 2 C - 8 C (36 F - 46 F), in the original package. Keep the pre-filled syringes in the outer carton to protect from light. Do not freeze or shake. Administration: The investigator (or assigned designee, i.e., study pharmacist) will dispense the proper number of pre-filled syringes (as assigned by the IVRS) to the subject to satisfy dosing requirements until the subject s next visit. The solution for injection is for single use only. It should be inspected visually for particulate matter and discoloration before administration. Pre-filled syringes that contain particulate matter or discoloration should not be used, and should be returned to the investigator. 95
97 Handling and Dispensing Ribavirin (Copegus ) Storage: Ribavirin (Copegus ) tablets should be stored in a tightly closed container at 25 C (77 F). Excursions permitted between 15 C to 30 C (59 F to 86 F). Administration: The investigator (or assigned designee, i.e., study pharmacist) will dispense the proper number of bottles (as assigned by the IVRS) to the subject to satisfy dosing requirements until the subject s next visit. 5.2 Method of Assigning Subjects to a Treatment Subjects will be randomized to receive either Treatment A (BMS and BMS ) or Treatment B ( with SOC/RBV) according to computer generated randomization schemes prepared by a Randomization Coordinator within the Drug Supply Management Department of the Bristol-Myers Squibb Research and Development. Approximately twenty subjects will be randomized in a ratio of 1:1 in the sentinel cohort to treatment group A and B. If Treatment A is expanded, approximately 40 additional subjects will be randomized to Expansion cohorts A1 and A2. If Treatment B is expanded, approximately 40 additional subjects will be randomized to Expansion cohorts B1 and B2, and approximately 20 additional subjects will be assigned to Expansion Cohort B3, where assignment to Expansion cohort B3 is dependent upon observed antiviral activity in Expansion cohort B1. Randomization is conditional on HCV genotype, with genotype 1b capped at 2 subjects per 10 (or 4 per 20) in the sentinel cohorts and in expansion cohorts B1, B2, and B3. Subjects with HCV genotype subtype 1b will be randomized in a ratio of 5:5:1:1 to expansion cohorts A1, A2, B1, and B2 if both Treatments A and B are expanded, or in a ratio 1:1 to expansion cohorts A1 and A2, or B1 and B2 if only Treatment A or Treatment B is expanded, respectively. Subjects with HCV genotype subtype 1a will be randomized in a ratio of 1:1 to expansion cohorts B1 and B2 if Treatment B is expanded. Randomization in Expansion cohort A1 and A2 will be restricted to genotype 1b subjects only. Upon randomization, subjects will be assigned to a treatment group. All subjects enrolled (whether dosed or not) will be assigned a subject number starting with
98 Subjects who refuse treatment on Day 1 may be replaced. Otherwise, no replacements are allowed. If a subject is replaced, the replacement subject will receive the same treatment as the original subject but a new randomization number will assigned to him or her. The replacement subject will be assigned the original subject s randomization number plus 200. For example, randomization number 1004 for Subject would be replaced by randomization number 1204 for replacement Subject More detailed information regarding IVRS will be provided in a separate document to the sites. 5.3 Selection and Timing of Dose for Each Subject On Day 1, after all Day 1 procedures have been performed, eligible subjects will start study drugs. The screening period for this study is 28 days. Eligible subjects must be dosed within 28 days from the day of screening. The Day 1 and 14 dosing must occur in the office/clinic in order to comply with PK assessments (See Tables 6.5.1A and B). Selection and timing of dose for each subject are as follows (with the exceptions described below): All Cohorts: BMS tablets should be taken once daily. BMS should be taken with a meal, and may be taken with Ribavirin (RBV). All subjects, regardless of which treatment regimen they are randomized to, will take two (2) BMS mg once a day. BMS should be taken with a meal, and may be taken with RBV and BMS All subjects in the Sentinel Cohorts will take three (3) BMS mg tablets twice a day. All subjects in Expansion Cohorts A1, B1 and B3 will take one (1) BMS mg tablets twice a day. All subjects in Expansion Cohorts A2 and B2 will take one (1) BMS mg tablets once a day. Note: For subjects in the Sentinel Cohorts, the dose of BMS was reduced from 600 mg BID to 200 mg BID for 5 rescued Treatment A subjects when they 97
99 were at Week 32 to 37 of treatment. Other subjects received 600 mg BMS BID and completed treatment before the change. For subjects participating in Day 14 intensive PK study, ie, all subjects in the Sentinel Cohorts, the first 12 subjects in each Expansion Cohort, a standard (moderate fat) breakfast should be consumed prior to dosing on the morning of Day 14. An example of a standard fat breakfast is provided (Table 5.3). It is preferred that the contents of the meal be as described; however, substitutions are permitted so long as the overall caloric content and the calories derived from protein, fat and carbohydrate are very similar to (within 10%) the example provided. Table 5.3: Representative Standard Breakfast Meal Food Item Calories (kcal) Fat (g) Carbohydrates (g) Protein (g) 1 egg fried slices white bread toasted tablespoon jam 56 trace 13.8 trace 8 fluid ounces (237 ml) of whole milk Total Grams (g) Total Calories (kcal) % of Total Calories Source: US Department of Agriculture Nutrient Database for Standard Reference, Release 18 (August 2005) 46 Cohorts with Regimen Including Pegylated Interferon (Treatment B Sentinel cohort, Expansion Cohorts B1 and B2, and All Subjects Receiving Rescue): Peg-interferon alfa-2a (pegifnα): Subjects will self-administer 180 µg/0.5 ml pegifnα injection subcutaneously once weekly throughout the entire treatment period. 98
100 Cohorts with Regimen Including oral Ribavirin (Treatment B Sentinel Cohort, Expansion Cohort B1, B2 and B3, and All Subjects Receiving Rescue): RBV: For subjects < 75 kg, the total dose is 1000 mg per day. Subjects should take 400 mg (2 tablets) in the morning with a meal and 600 mg (3 tablets) in the evening with a meal. For subjects 75 kg, the dose is 1200 mg per day. Subjects should take 600 mg (3 tablets) in the morning and evening with a meal. Subjects will continue therapy up to Week 72 (for subjects who receive rescue therapy after 24 weeks of standard therapy), and will continue to be followed up to 48 weeks post-treatment Dose modification Pegylated Interferon and Ribavirin Dose modifications and/or study drug discontinuations of pegifn or RBV are permitted for the management of adverse events or clinical laboratory findings believed to be related to administration RBV or pegifn. Management of the abnormality should be undertaken in consultation with guidelines suggested in the package inserts for each medication (Copegus,44 and Pegasys,43 from Roche, Inc.). Investigators are encouraged to discuss the clinical plan with the Study Sponsor (BMS Medical Monitor) prior to any dose modifications or drug discontinuation BMS and BMS Dose modifications of BMS and BMS are not permitted. Parameters for discontinuation are described in section Blinding/Unblinding Not applicable. This is an open-label study. 99
101 5.5 Concomitant Treatments Restrictions on medications taken prior to enrollment in the study are described in Section Medications taken within 4 weeks prior to administration of study medication must be recorded on the CRF. No concomitant medications (prescription, over-the-counter or herbal) are to be administered during study unless they are prescribed by the investigator for treatment of specific clinical events. Any concomitant therapies must be recorded on the CRF. Guidelines for the use of drugs with established or other potentially significant drug interactions listed in the package inserts of the marketed agents used by subjects participating in this study should be followed. Medications listed in the package inserts as contra-indicated with the marketed agents used by subjects participating in this study are not permitted Prohibited and/or Restricted Treatments Strong or moderate inhibitors of CYP3A4, including: ketoconazole, troleandomycin, itraconazole, voriconazole, mibefradil, clarithromycin, telithromycin, grapefruit juice and grapefruit-containing products, Seville oranges, juices and products that contain Seville oranges, conivaptan, nefazodone, fluconazole, erythromycin, diltiazem, aprepitant, imatinib, verapamil, tofisopam; CYP3A4 inducers, including: rifampin, rifabutin, rifapentin, dexamethasone, phenytoin, carbamazepine, phenobarbital, and St John's wort; Drugs Metabolized by CYP2D6: BMS is a moderate inhibitor of CYP2D6. CYP2D6 substrates with a narrow therapeutic index (eg, Flecainide, propafenone) are prohibited. Additionally, due to the risk of serious ventricular arrhythmias and sudden death potentially associated with elevated plasma levels of thioridazine, thioridazine is prohibited. Co-administration of BMS with other drugs that are metabolized by CYP2D6 (eg, metoprolol, arapiprazole, tricyclic antidepressants such as amitryptyline, nortriptyline, imipramine and desipramine) should be approached with caution. Close clinical monitoring for adverse events, use of therapeutic drug monitoring when available, and decreased doses of medications should be considered. Additionally, medications with an alternate elimination pathway should be considered. Medications with known or potential anti-hcv activity other than the assigned study treatment; 100
102 Any prescription or herbal product which is not prescribed by the investigator or licensed physician for treatment of a specific clinical condition; Oral estrogen (e.g., ethinylestradiol) and progestin (e.g., norethindrone) hormonal contraceptives as a sole method of contraception. Two methods of contraception other than, or in addition to, oral hormonal contraceptives must be used; Long-term treatment ( 2 weeks) with agents that are immunosuppressive, or have a high risk for nephrotoxicity or hepatotoxicity, should be discussed with the BMS central medical monitor. Proton pump inhibitors are prohibited. H2 receptor antagonists will be allowed but must be administered at least 2 hours post dosing of BMS and at least 10 hours prior to administration of BMS Other acid modifying agents such as antacids will be allowed but must be taken at least 2 hours prior and 2 hours post administration of BMS ; P-gp substrates with a narrow therapeutic index (e.g. digoxin) are prohibited and sensitive P-gp substrates (e.g. fexofenadine, vincristine, colchicine, topotecan, and paclitaxel) should be used with caution. Erythropoiesis-stimulating agents (ESAs): The risks and benefits of the use of ESAs for the management of HCV treatment-related anemia has not been standardized nor established in well controlled clinical trials. Although the use of ESA for treatment of HCV treatment-related anemia is at the investigator s discretion, the following guidelines are recommended (please refer to the ESAs package inserts for additional information): ESAs should not be initiated until the hemoglobin falls below 10 g/dl Iron studies should be obtained prior to and during treatment with ESA Iron supplementation should be initiated for deficient subjects and to maintain transferrin saturation at a level that will support erythropoiesis Once an ESA is initiated, hemoglobin levels and blood pressure must be monitored weekly until the hemoglobin level stabilizes Treatment should target a hemoglobin level sufficient to avoid transfusion ESA dose should be titrated to treatment response ESA dose should be reduced if the hemoglobin increases by more than 1g/dL in a 2 week period ESA dose should not exceed those recommended for currently approved indications ESAs should not be used in subjects at increased risk for thromboembolic events, cardiovascular events, including those with inadequately controlled hypertension, and in subjects diagnosed with malignancies 101
103 Precautionary therapies: Drugs that are highly dependent on CYP3A4 for elimination, especially those with a narrow therapeutic window, should be used with caution. BMS may cause decreases in plasma concentrations and in some cases a corresponding decrease in effectiveness; therefore, dose adjustments may be necessary and therapeutic drug monitoring may be considered, if available. Such medications include, but are not limited to: alfentanil, cyclosporine, fentanyl, midazolam, quinidine, sirolimus, tacrolimus, and triazolam. Drugs that are highly dependent on OATP1B1 for liver uptake should be used with caution. BMS may cause increases in plasma concentrations and in some cases a corresponding decrease in effectiveness or an increase in adverse events; therefore dose adjustments may be necessary and careful monitoring for adverse events is warranted. Such medications include, but are not limited to: rosuvastatin, pravastatin, atorvastatin, methotrexate, and thyroxine. 5.6 Treatment Compliance Study drug will be administered via out-patient care. Site staff will be required to conduct courtesy calls to remind subjects about dosing procedures and timing. In addition, assessment of study medication use will be performed at each clinical evaluation visit. Each subject will be required to maintain a drug diary. The study coordinator shall track the following information that will be noted by each subject in a diary: compliance with study medications onset and resolution of any unexpected symptoms or illness This information shall be reviewed by the investigator regularly or sooner if symptoms greater than grade I. The subject should be instructed to bring all used and unused study medication in the original bottle to each visit. The dates and number of tablets (or volume of oral solution) dispensed and returned must be recorded on the Master Clinical Supply inventory form 102
104 maintained on-site. Opened bottles of study drug will be returned to the subject and dosing should continue from the in-use container until it has been emptied. 103
105 6 STUDY ASSESSMENTS AND PROCEDURES 6.1 Flow Chart/Time and Events Schedule Table 6.1A: Procedure Screening and On-treatment Procedure Outlines for the Sentinel Cohorts Screening Treatment EOT a Protocol Section Days - 28 to -2 Sign Protocol Specific Informed Consent Form/ Enrollment d X Day -1 Day 1 Days 2 to 6 Day 7 Day 9 Day 11 Day 14 Day 15 Day 21 Day 28 Wks 6, 8, 10, 12 Wks 16, b Wk 24c 20 Medical History X 4.2 Vital Signs X X X X X X Wks 8, 12 X X Physical Measurements e X X X X X Physical X Examination X f X X X 12-lead ECG X X X X X X Wks 8, 12 Wks 8, 12 Wks 8, and X X X X X X
106 Table 6.1A: Screening and On-treatment Procedure Outlines for the Sentinel Cohorts Procedure Clinical Laboratory Tests Serology, HCV Genotyping Screening Treatment EOT a Protocol Section Days - 28 to -2 Day -1 Day 1 Days 2 to 6 Day 7 Day 9 Day 11 Day 14 Day 15 Day 21 X X X X X Day 28 Wks 6, 8, 10, 12 Wks 8, 12 Wks 16, b Wk 24c 20 X X X Fibrotest and APRI X Urine Drug Screen X X FSH Testing for women on HRT X Pregnancy Test X X X X X Wks 8, 12 X X Randomization X g 5.2 Study Drug Administration Dispense Study Drug Daily for BMS , BMS and ribavirin. Weekly for pegifnα until end of treatment. Every 4 weeks through an Interactive Voice Response System (IVRS) until end of treatment. Sparse/Trough PK X Day 2 X X Wks 8,
107 Table 6.1A: Screening and On-treatment Procedure Outlines for the Sentinel Cohorts Procedure Screening Treatment EOT a Protocol Section Days - 28 to -2 Day -1 Day 1 Days 2 to 6 Day 7 Day 9 Day 11 Day 14 Day 15 Day 21 Day 28 Wks 6, 8, 10, 12 Wks 16, b Wk 24c 20 Serial PK X X 6.5 HCV RNA Level X X X X X X X X X X X X h X 6.6 HCV Genomic Substitution X X X X X X X X X h X 6.9 SNP X Anti-HCV T-cell Response X X X Monitor for SAEs All SAEs must be collected from the date of subject s written consent until 30 days post discontinuation of dosing or subject s participation in the study if the last scheduled visit occurs at a later time Monitor for Non- SAEs (NSAEs) All NSAEs will be collected from Day 1 until the subject s last scheduled visit a These evaluations should also be performed prior to discharge for subjects who are discontinued. Subjects who are discontinued should be followed up according to Table 6.1D unless subjects withdraw consent. b For subjects in treatment group A who are rescued, the treatment duration is longer than 24 weeks and the exact duration depends on when the subject is rescued. 106
108 c For subjects in treatment group A who are rescued, the end of treatment is later than Week 24 and the exact EOT date depends on when the subject is rescued. d A subject is considered enrolled only when a protocol specified inform consent is signed. e Weight only after screening. f Within 24 hours of study drug administration. g Randomization can occur on Day -1 or Day 1. h For subjects on Treatment B, follow the schedule in the table. For subjects on Treatment A, every 2 weeks from Week 12 until EOT unless SOC is added to the regimen (ie, rescued), in which case, samples will be collected every 4 weeks from Week 12 until EOT. HCV RNA levels/genomic substitution will also be studied on the day when SOC is added. 107
109 Table 6.1B: Screening and On-treatment Procedure Outlines for the Expansion Cohorts Screening Treatment EOT a Protocol Section Procedure Days - 28 to - 2 Sign Protocol Specific Informed Consent Form/ Enrollment e X Day -1 Day 1 Days 2 to 6 Day 7 Day 9 Day 11 Medical History X 4.2 Vital Signs X X X X X X Physical Measurements f X X X X X Physical Examination X X g X X X 12-lead ECG X X X X X X Clinical Laboratory Tests Serology, HCV Genotyping Day 14 Day 15 Day 21 X X X X X Day 28 Wks 6, 8, 10, 12 Wks 8, 12 Wks 8, 12 Wks 8, 12 Wks 8, 12 Wks 8, 12 Wks 14, 18, 22 b Wks 16, 20 c Wk 24 d 3.3 and X X X X X X X X X X X
110 Table 6.1B: Screening and On-treatment Procedure Outlines for the Expansion Cohorts Screening Treatment EOT a Protocol Section Procedure Days - 28 to - 2 Day -1 Day 1 Days 2 to 6 Day 7 Day 9 Day 11 Fibrotest and APRI X Urine Drug Screen X X FSH Testing for women on HRT Day 14 Day 15 Day 21 Day 28 Wks 6, 8, 10, 12 X Pregnancy Test X X X X X Wks 8, 12 Wks 14, 18, 22 b Wks 16, 20 c Wk 24 d X X Randomization X h 5.2 Study Drug Administration Dispense Study Drug Daily for BMS , BMS and ribavirin. Weekly for pegifnα until end of treatment. Every 4 weeks through an Interactive Voice Response System (IVRS) until end of treatment. Sparse/Trough PK X i Day 2 i X X Wks 8, 12 Wk Serial PK i X X 6.5 HCV RNA Level X X X X X X X X X X X X X X
111 Table 6.1B: Screening and On-treatment Procedure Outlines for the Expansion Cohorts Screening Treatment EOT a Protocol Section Procedure HCV Genomic Substitution Days - 28 to - 2 Day -1 Day 1 Days 2 to 6 Day 7 Day 9 Day 11 Day 14 Day 15 Day 21 Day 28 Wks 6, 8, 10, 12 X X X X X X X X X X X X X 6.9 SNP X T cell Immunoregulatory Molecules via PBMCs X X X Anti-HCV T-cell Response X X X Wks 8, 12 Wks 8, 12 Wks 14, 18, 22 b Wks 16, 20 c Wk 16 Wk 16 Wk 24 d X X IP-10 X X X Wk 12 X Host Gene Expression X X X Wk 12 X
112 Table 6.1B: Screening and On-treatment Procedure Outlines for the Expansion Cohorts Screening Treatment EOT a Protocol Section Procedure Monitor for SAEs Monitor for Non-SAEs (NSAEs) Days - 28 to - 2 Day -1 Day 1 Days 2 to 6 Day 7 Day 9 Day 11 Day 14 Day 15 Day 21 Day 28 Wks 6, 8, 10, 12 All SAEs must be collected from the date of subject s written consent until 30 days post discontinuation of dosing or subject s participation in the study if the last scheduled visit occurs at a later time. All NSAEs will be collected from Day 1 until the subject s last scheduled visit a These evaluations should also be performed prior to discharge for subjects who are discontinued. Subjects who are discontinued should be followed up according to Table 6.1D unless subjects withdraw consent. b Subjects in Expansion Cohort A1, A2 and B3 only, before they are rescued. c Treatment duration may be longer than 24 weeks for some subjects who need to have SOC or pegifn added. d End of treatment may be later than Week 24 for some subjects who need to have SOC or pegifn added. Wks 14, 18, 22 b Wks 16, 20 c Wk 24 d e A subject is considered enrolled only when a protocol specified inform consent is signed. f Weight only after screening. g Within 24 hours of study drug administration. h Randomization can occur on Day -1 or Day 1 i Sparse PK samples on Days 1 and 2, and serial PK samples will be collected from the first 12 subjects in each Expansion Cohort. 111
113 Table 6.1C: On-Treatment Procedure Outlines for Subjects Who Are Rescued Day of Rescue (SOC or PegIFN added) Every 4 Weeks for up to 48 Weeks End of Treatment Protocol Section Vital Signs X X X Body Weight X X X Physical Examination X X X lead ECG X X X Clinical Laboratory Tests X X X Pregnancy Test X X X Rescue IVRS Call X 5.2 Study Drug Administration Daily for BMS , BMS and ribavirin. Weekly for pegifnα until end of treatment Dispense Study Drug Every 4 weeks through an Interactive Voice Response System (IVRS) until end of treatment Sparse/Trough PK Weeks 4, 8, 12, and 16 only counting from randomization 6.5 HCV RNA Level X X X 6.6 HCV Genomic Substitution X X X 6.9 T cell Immunoregulatory Molecules via PBMCs a Anti-HCV T-cell Response a X X Weeks 4, 12, 24 and 48 or EOT counting from Rescue b X Weeks 4, 12, 24 and 48 or EOT counting from Rescue b X
114 Table 6.1C: On-Treatment Procedure Outlines for Subjects Who Are Rescued IP-10 a Host Gene Expression a Day of Rescue (SOC or PegIFN added) X X Every 4 Weeks for up to 48 Weeks End of Treatment Protocol Section Weeks 4, 12, 24 and 48 or EOT counting from Rescue b X Weeks 4, 12, 24 and 48 or EOT counting from Rescue b X Monitor for SAEs All SAEs must be collected from the date of subject s written consent until 30 days post discontinuation of dosing or subject s participation in the study if the last scheduled visit occurs at a later time. Monitor for Non-SAEs (NSAEs) All NSAEs will be collected from Day 1 until the subject s last scheduled visit a Rescued subjects in the Expansion Cohorts only. b A +/- 2 weeks window is allowed for these biomarker timepoints. 113
115 Table 6.1D: Post-treatment Procedure Outlines for All Cohorts Procedure PTWk 4 PTWk 8 Post-Treatment (PT) Follow-up Discharge Protocol Section PTWk 12 PTWks 16, 20 PTWk 24 PTWks 28, 32 PTWk 36 PTWks 40, 44 PTWk 48 Pregnancy Test X X X X a X a HCV RNA Level X X X X X 6.6 HCV Genomic Substitution X X X X X 6.9 T cell Immunoregulatory Molecules via PBMCs b X X X Anti-HCV T-cell Response b X X X Host Gene Expression b X IP-10 b X Monitor for SAEs All SAEs must be collected from the date of subject s written consent until 30 days post discontinuation of dosing or subject s participation in the study if the last scheduled visit occurs at a later time. Monitor for Non-SAEs (NSAEs) All NSAEs will be collected from Day 1 until the subject s last scheduled visit a Subjects who are women of child-bearing potential (WOCBP) should have a pregnancy test every 4 weeks for 12 weeks if they are not exposed to ribavirin (Treatment A not rescued). WOCBP subjects should have a pregnancy test every 4 weeks for 24 weeks if they are exposed to ribavirin (Treatment A rescued, Treatment B Sentinel and Expansion Cohorts). b Subjects in Expansion Cohorts only
116 In general, no visit window is allowed for the first 2 weeks of the study. From Week 3 to Week 12, a 3-day visit window is allowed. After Week 12, a 5-day visit window is allowed. 6.2 Study Materials The site will provide all required materials for the tests performed locally (i.e., relevant clinical laboratory tests and urine drug screens). The site will have available a well-calibrated scale for recording body weight, a 12-lead ECG machine, and a calibrated sphygmomanometer and thermometer for vital signs assessments. A current and fully stocked advanced cardiac life support (ACLS) cart will be immediately available on the premises. The site will have a refrigerated centrifuge (or approved alternative), a monitored and alarmed refrigerator, and freezer (-20 C or below), as well as containers and dry ice for shipment and storage of blood samples. The site will provide all materials required for accurate source documentation of study activities and for housing the subjects during the study. BMS will provide a BMS-approved protocol and any amendments or administrative letters (if required). Case report forms (electronic) will be provided by BMS. The Central Laboratory will provide labels and tubes for the collection of blood samples for safety, PK/PD and for genotyping analysis. 6.3 Safety Assessments Data for the procedures and assessments specified in this protocol should be submitted to BMS on a case report form. Additional procedures and assessments should remain in the subject s medical record and should not be provided to BMS, unless specifically requested from the sponsor Medical History A detailed medical history will be obtained at screening. Please include in the history any toxicities or allergy related to previous treatments. 115
117 6.3.2 Vital Signs Vital signs (body temperature, respiratory rate, seated blood pressure and heart rate) will be recorded during the screening visit, Days -1, 1, 7, 14, and every four weeks beginning on Week 4 through EOT (up to Week 72). Blood pressure and heart rate should be measured after the subject has been seated quietly for at least 5 minutes Physical Examinations A physical examination will be performed at the screening visit, on Days 1, 7, 14 and every four weeks beginning on Week 4 through EOT (up to Week 72). If the screening physical examination is performed within 24 hours of dosing on Day 1 then a single examination may count as both the screening and predose evaluation Physical Measurements Height and weight will be measured and body mass index (BMI) calculated at screening as part of the physical exam. Only body weight measurements will be recorded on Days 1, 7, 14, every four weeks beginning on Week 4 through EOT (up to Week 72). The recommendations for weighing subjects are outlined below: Scales should be calibrated in accordance with the manufacturer s recommendations. Body weights shall be recorded at the same time point ± 30 minutes. Prior to weighing, subjects should remove shoes and any supplementary clothing (such as sweaters, jackets or hats) and any electronic or mechanical device(s) (i.e. ipods, headphones cell phone gear) Subject s pockets should be empty, verified by asking them to turn pockets inside out Electrocardiograms A 12-lead ECG will be recorded at screening, Days -1, 1, 7, 14, and every four weeks beginning on Week 4 through EOT (up to Week 72). ECGs should be recorded after the subject has been supine for at least 5 minutes. 116
118 6.3.6 Other Not applicable Adverse Event Monitoring Subjects will be closely monitored throughout the study for adverse events and will not be discharged from the study until the investigator has determined that adverse events have either completely resolved or are not of clinical significance (refer to Section 7). For post-treatment visits that only include AE monitoring, monitoring may be conducted by phone if preferred by the site and the subject. Details of the phone visits must be documented in subjects study records Laboratory Test Assessments Blood samples will be obtained at selected times: screening, Days -1, 7, 14, and every four weeks beginning on Week 4 through EOT (up to Week 72) for clinical laboratory evaluations. Subjects are required to fast for at least 8 hours prior to the collection of specimens for clinical laboratory tests. A central laboratory will perform the analyses and will provide reference ranges for these tests. Results of clinical laboratory tests performed on Day -1 must be available prior to dosing. The following clinical laboratory tests will be performed: Hematology Hemoglobin Hematocrit Total leukocyte count, including differential Platelet count Serum Chemistry Aspartate aminotransferase (AST) Alanine aminotransferase (ALT) γ-glutamyl transferase (GGT) Total bilirubin Direct bilirubin Alkaline phosphatase Fasting glucose Total Protein Albumin Sodium Potassium Chloride 117
119 Lactate dehydrogenase (LDH) Creatinine Blood Urea Nitrogen (BUN) Uric acid Calcium Phosphorus Bicarbonate Cholesterol Urinalysis Protein Glucose Blood Leukocyte esterase Microscopic examination of the sediment if blood, protein or leukocytes esterase are positive on the dipstick Serology Serum for hepatitis C antibody, hepatitis B surface antigen, HIV-1, -2 antibody (screening only) Other Analyses Urine for drugs of abuse (screening and on Day -1) Pregnancy test (all women) FSH testing for women on HRT (screening) Blood Sampling for: HCV RNA Fibrotest APRI (AST to platelet ratio index) HCV Genotype Assay HCV Genomic Substitution Anti-HCV T-cell Response T cell immuno-regulatory Molecules IFN, ENT1 and ITPA SNP IP-10 Host Gene Expression Results of all laboratory tests required by this protocol must be provided to BMS, either recorded on the laboratory pages of the CRF or by another mechanism as agreed upon between the investigator and BMS (eg, provided electronically). If the units of a test result differ from those printed on the CRF, the recorded laboratory values must specify the correct units. Any abnormal laboratory test result considered clinically significant by 118
120 the investigator must be recorded on the appropriate AE page of the CRF (see Section 7.4 Laboratory Test Abnormalities). For post treatment follow-up visits that only include pregnancy testing and adverse event monitoring, the pregnancy test may be performed by a certified local lab, and AE monitoring may be conducted by phone. A specific site visit is may not be needed. Reports from all local lab testing, such as Day -1 clinical labs, repeat lab or posttreatment visit pregnancy tests, etc, must be sent to Local Lab Data Services promptly to be included in the study database. 6.4 Efficacy Assessments Not applicable Primary Efficacy Assessment Not applicable Secondary Efficacy Assessments Not applicable. 6.5 Pharmacokinetic Assessments Pharmacokinetics: Collection and Processing Tables 6.5.1A and B list the sampling schedule to be followed for the assessment of pharmacokinetics. Further details of blood collection and processing will be provided to the site in the procedure manual. The Day 1 and 14 dosing must occur in the office/clinic in order to comply with PK assessments. 119
121 Table 6.5.1A: Pharmacokinetic Sampling Schedule for the Sentinel Cohorts Study Day 1 Time (Event) Hour Time (Relative to Dosing) Hour:Min PK Blood Sample for BMS and BMS (pre AM dose) 00:00 X 4 04:00 X 8 08:00 X 2 0 (pre AM dose) 24:00 X 14 0 (pre AM dose) 00:00 X 1 01:00 X 2 02:00 X 4 04:00 X 8 08:00 X 12 12:00 X 15 0 (pre AM dose) 24:00 X Day 7 and Weeks 4, 8, and 12 0 (pre AM dose) 00:00 X Weeks 4, 8, and :00 X Table 6.5.1B: Study Day 1 a Pharmacokinetic Sampling Schedule for the Expansion Cohorts Time (Event) Hour Time (Relative to Dosing) Hour:Min PK Blood Sample for BMS and BMS :00 X 2 02:00 X 4 04:00 X 8 08:00 X 2 a 0 (pre AM dose) 24:00 X 120
122 Table 6.5.1B: Study Day 14 a Pharmacokinetic Sampling Schedule for the Expansion Cohorts Time (Event) Hour Time (Relative to Dosing) Hour:Min PK Blood Sample for BMS and BMS (pre AM dose) 00:00 X 1 01:00 X 2 02:00 X 4 04:00 X 8 08:00 X 12 12:00 X 15 a 0 (pre AM dose) 24:00 X Day 7 and Weeks 4, 8, and 12 0 (pre AM dose) 00:00 X Week :30 X Week :00 X Week 16 0 (pre AM dose) 00:00 X a The first 12 subjects in each Expansion Cohort only Pharmacokinetic Sample Analyses The plasma samples will be analyzed for by a validated LC/MS/MS assay. In addition, remaining plasma samples will be archived for exploratory metabolite analysis, if the need arises and to the extent possible Labeling and Shipping of Biological Samples Detailed instructions for the pharmacokinetic blood collection, labeling, processing, storage, and shipping will be provided to the site in the procedure manual. 121
123 6.6 Pharmacodynamic Assessments Blood will be drawn at the times indicated in Tables 6.6A and B for the measurement of HCV RNA, HCV genomic substitution, Anti-HCV T Cell and IFN SNP. Further details of blood collection and processing will be provided to the site in the procedure manual. The COBAS TaqMan HCV Test will be used to measure HCV RNA levels in subjects serum samples. The assay uses the High Pure System Viral Nucleic Acid Kit for manual sample preparation and the COBAS TaqMan 48 Analyzer for automated amplification and detection. The assay will be performed by ICON Central Laboratories at two different locations: Samples from the subjects in France will be analyzed at Dublin Ireland, samples from the US will be analyzed at Farmingdale, New York, the US. The same assay is used at both locations. The lower limit of quantitation (LLOQ) of the assay is 25 IU/mL. 47 Table 6.6A: Screening Treatment Phase PD Sampling Schedule for the Sentinel Cohorts Study Day Time (Relative To Dosing) Hour HCV RNA HCV Genomic Substitution Anti- HCV T Cell IFN SNP Screening --- X X X X X 0 (pre AM dose) X X X X X X (pre AM dose) X X X X X X X X X X X X X X X X X X (week 3) --- X X
124 Table 6.6A: PD Sampling Schedule for the Sentinel Cohorts Study Day Time (Relative To Dosing) Hour HCV RNA HCV Genomic Substitution Anti- HCV T Cell IFN SNP 28 (Week 4) --- X X Every 2 weeks from Week 4 to Week X X Post-Treatment Every 4 weeks from Week 12 to EOT Weeks 4, 12, 24, 36, 48 posttreatment --- X a X a X X a For subjects on Treatment B, every 4 weeks from Week 12 until EOT as shown in the table. For subjects on Treatment A, every 2 weeks from Week 12 until SOC is added (rescued) or EOT, whichever is earlier; after rescue, every 4 weeks until EOT. For rescued subjects, blood samples for HCV RNA levels and HCV genomic substitution will be obtained on the day SOC is added. 123
125 Table 6.6B: Screening Treatment Phase Study Day PD Sampling Schedule for the Expansion Cohorts Time (Relative To Dosing) Hour HCV RNA HCV Genomic Substitution T cell Immuno- Regulatory Molecules via PBMCs T cell Responses to HCV Antigens via PBMCs Host Gene Expression IP-10 IFN, ENT1 and ITPA SNP Screening --- X X X X X X X X (pre AM dose) X X X X X X (pre AM dose) X X X X X X X X X X X X X X X X X X X X X X
126 Table 6.6B: PD Sampling Schedule for the Expansion Cohorts Post- Treatment Study Day Time (Relative To Dosing) Hour HCV RNA HCV Genomic Substitution T cell Immuno- Regulatory Molecules via PBMCs T cell Responses to HCV Antigens via PBMCs Host Gene Expression IP-10 IFN, ENT1 and ITPA SNP 21 (week 3) --- X X (Week 4) --- X X X X X X --- Every 2 weeks from Week 4 to Week 12 Every 4 weeks from Week 12 to EOT Weeks 4, 12, 24, 36, 48 post-treatment --- X X Wks 8, 12 Wks 8, 12 Wk 12 Wk X a X a Wks 16, 24 Wks 16, 24 Wk 24 Wk X X PT Wks 4, 12, 24 PT Wks 4, 12, 24 PT Wk 24 PT Wk a For subjects in Expansion Cohorts B1and B2, every 4 weeks from Week 12 until EOT as shown in the table. For subjects in Expansion Cohorts A1, A2 and B3, every 2 weeks from Week 12 until SOC or pegifn is added (rescued) or EOT, whichever is earlier; after rescue, every 4 weeks until EOT. For rescued subjects, blood samples for HCV RNA levels and HCV genomic substitution will be obtained on the day SOC or pegifn is added. 125
127 6.7 Pharmacogenomic/Pharmacogenetic Assessments See Amendment 01 for pharmacogenomics blood sampling. 6.8 Outcomes Research Assessments Not applicable. 6.9 Other Assessments To characterize HCV genomic substitutions associated with exposure of BMS and BMS , blood samples from all subjects will be obtained on Days -1 (baseline), 1 to 7, 14, Week 3, every two weeks from Week 4 to Week 12. After Week 12, HCV genomic substitutions samples collection will proceed as follows: All Treatment group A cohorts and Expansion Cohort B3: Every 2 weeks from Week 12 until EOT unless SOC is added to the regimen, in which case, samples will be collected every 4 weeks from Week 12 until EOT. Subjects who have SOC or pegifn added due to viral breakthrough will have HCV RNA measured immediately prior to administration of the first dose of pegifn and every 4 weeks thereafter until EOT. Treatment group B sentinel cohort and Expansion Cohorts B1 and B2: HCV RNA level measurement every 4 weeks from Week 12 until EOT. All subjects: HCV RNA measured at Week 4, 12, 24, 36, and 48 post-treatment. All baseline samples from randomized subjects will be analyzed for resistance genotypes using population sequencing. Resistance testing will also be performed on samples in all randomized subjects with HCV RNA 1000 IU/mL who have virologic breakthrough as defined previously in Section Testing will include the HCV NS3 and NS5A genes. Resistance testing will be performed on samples with HCV RNA 1000 IU/mL obtained at time points representing virologic breakthrough. Analysis of additional time points representing virologic breakthrough with HCV RNA < 1000 IU/mL will be performed at the discretion of the virology, statistical and medical teams. 126
128 Study data relevant to the performance and interpretation of the resistance testing will be shared between the medical, statistical and virology teams under the direction of the medical monitor. Exploratory resistance testing to evaluate for low level variants may be performed on subsets of samples depending on patterns of viral load response at time points determined by the medical, statistical and virology teams. The pre-existence of variants at baseline, the emergence of drug-resistant variants or any sequence changes in the gene target of interest (NS3 protease domain and nucleotides encoding the first ~100 amino acids of NS5A, followed by the complete NS5A gene if necessary) will be monitored and compared with the respective baseline sample sequences and reference sequences 1a(H77) and 1b(Con1). For patients who have detectable, well characterized resistance substitutions at the EOT that are known to persist, the Week 48 post-treatment time-point will be evaluated. If the resistance substitution has reverted to the baseline sequence, genotypic analysis will be carried out at earlier time-points collected post-treatment. For less characterized or novel resistance substitutions, resistance testing will be carried out every 12 weeks post-treatment to determine the pattern of decay up to Week 48 post-treatment. If these less characterized or novel substitutions have reverted to baseline sequence, genotypic analysis of the Week 4 post-treatment time-point will be evaluated. For patients who have undetectable viral RNA at the EOT, genotypic analysis of sample time-points post-treatment will be evaluated if viral relapse is detected. If the virologic failure of a particular patient sample cannot be explained genotypically, the patient sample quasi-species encoding the respective target regions will be phenotyped. The following possibilities of linked NS3 and NS5A resistance substitutions causing virological failure are anticipated: (i) previously uncharacterized NS5A resistance substitution(s) paired with a known NS3 resistance substitution, (ii) previously uncharacterized NS3 resistance substitution(s) paired with a known NS5A resistance substitution, or (iii) previously uncharacterized NS5A resistance substitution(s) paired with a previously uncharacterized NS3 resistance substitution. In each case the NS3 plus NS5A substitutions will be introduced into a wild type replicon background. For possibility (i), a replicon construct containing specific substitutions or the first 100 amino acids or the entire coding region of NS5A from a patient sample containing uncharacterized NS5A substitution(s) plus a specific substitution from a previously 127
129 characterized NS3 resistance variant will be analyzed. The resistance profile and replication ability (fitness) will be determined in the presence of BMS , BMS and the inhibitor combination in the transient replication assay. In addition, a replicon construct containing the NS5A substitutions as described above in combination with wild type NS3 will be analyzed for the resistance profile to BMS For possibility (ii) an analogous approach will be applied. In the case of possibility (iii), subject sequences from both the NS3 protease and NS5A gene (either the first 100 amino acids or the entire gene) will be introduced into the replicon system and inhibitor potency evaluated. If the resistant phenotype is not observed, genotypic analysis will be expanded to include all the nonstructural genes. Alternatively, if the replication capacity of this replicon is severely impaired, the NS3 protease domain will be introduced into a bacterial expression construct. NS3 protease will be expressed, purified, and sensitivity to BMS determined Exploratory Research PBMC Samples will be taken on Days -1 (baseline), 7 and 14 for analysis of host anti HCV T cell responses in the Sentinel Cohort. In the Expansion Cohort, PBMCs will be collected at Days -1, 14 and 28, Weeks 8, 12, 16, 24 on treatment and Weeks 4, 12 and 24 post treatment. Exploratory analysis to describe T cell responses to HCV antigens at baseline and their change over time following treatment may include measurement of IFN-γ production in response to HCV peptide antigens. PBMCs will also be analyzed to describe the changes in T cell immunoregulatory molecules with treatment by measurement of the levels of PD-1 expression on T cells. All exploratory samples will be analyzed as deemed relevant. Serum IP-10 will be measured at baseline and during treatment to explore the relationship between IP-10 levels and virologic response. Blood samples will be collected on Day -1 (baseline) to explore the relationship between polymorphisms in genes encoding proteins of the IFN lambda family (IL28A, IL28B, and IL29) and virologic response. Recent studies have identified single nucleotide polymorphisms in the IL28B region associated with increased rates of SVR with PEG-IFNα/RBV treatment in HCV-infected individuals. 48,49,50 IL28B, along with IL28A and IL29, encode proteins of the IFN lambda family. IFN lambda proteins have been 128
130 shown to inhibit HCV replication in vitro. 51 Exploratory analysis will be performed in this study to determine if SNPs in genes encoding IL28A, IL28B, or IL29 are associated with virologic response when individuals are treated with BMS and BMS plus RBV with or without PEG-IFNα. A SNP in the RBV transporter gene ENT1 has been identified to be associated with RVR to PEG IFNα/RBV therapy. This SNP may modulate intracellular RBV exposure in hepatocytes. 52 Exploratory analysis may be performed in this study to determine if this SNP is associated with virologic response when individuals are treated with BMS and BMS plus RBV with or without PEG IFNα. Several SNPs have been identified in the gene for inosine triphosphatase (ITPA) which are associated with hemolytic anemia in HCV infected patients receiving RBV. These SNPs will only be analyzed if an imbalance in the number of subjects experiencing anemia in different study arms is observed. Whole blood will be collected at baseline and during the study to explore changes in host gene expression. Genes to be measured may include, but are not limited to: Interferon stimulated genes (MX1, IFIT1, ISG15, OAS1, OAS2, OAS3, OASL), IFNα (IFNA1), inhibitory receptors (PDCD1, LAG3, CTLA4) and signaling genes (STAT1, STAT2, SOCS1, SOCS3). Additional exploratory research on viral RNA, serum/plasma samples including pharmacokinetic samples, and/or PBMC samples may be conducted on an exploratory basis limited to anti-viral immunity or viral dynamics. 7 ADVERSE EVENTS 7.1 Definitions An Adverse Event (AE) is defined as any new untoward medical occurrence or worsening of a pre-existing medical condition in a patient or clinical investigation subject administered an investigational (medicinal) product and that does not necessarily have a causal relationship with this treatment. An AE can therefore be any unfavorable and unintended sign (including an abnormal laboratory finding, for example), symptom, or 129
131 disease temporally associated with the use of investigational product, whether or not considered related to the investigational product Serious Adverse Events A serious AE (SAE) is any untoward medical occurrence that at any dose: results in death is life-threatening (defined as an event in which the subject was at risk of death at the time of the event; it does not refer to an event which hypothetically might have caused death if it were more severe) requires inpatient hospitalization or causes prolongation of existing hospitalization (see note below for exceptions) results in persistent or significant disability/incapacity is a congenital anomaly/birth defect is an important medical event (defined as a medical event(s) that may not be immediately life-threatening or result in death or hospitalization but, based upon appropriate medical and scientific judgment, may jeopardize the subject or may require intervention [eg, medical, surgical] to prevent one of the other serious outcomes listed in the definition above.) Examples of such events include, but are not limited to, intensive treatment in an emergency room or at home for allergic bronchospasm; blood dyscrasias or convulsions that do not result in hospitalization.) Suspected transmission of an infectious agent (eg, any organism, virus or infectious particle, pathogenic or non-pathogenic) via the study drug is an SAE and must be reported accordingly. Although overdose and cancer are not always serious by regulatory definition, these events should be reported on an SAE form and sent to BMS in an expedited manner. All pregnancies, regardless of outcome, must be reported to the sponsor on a Pregnancy Surveillance Form, not an SAE form (see Section 7.6). 130
132 NOTE: The following hospitalizations are not considered SAEs in BMS clinical studies: a visit to the emergency room or other hospital department < 24 hours, that does not result in admission (unless considered "important medical event" or event life threatening) elective surgery, planned prior to signing consent admissions as per protocol for a planned medical/surgical procedure routine health assessment requiring admission for baseline/trending of health status (eg, routine colonoscopy) medical/surgical admission for purpose other than remedying ill health state and was planned prior to entry into the study. Appropriate documentation is required in these cases admission encountered for another life circumstance that carries no bearing on health status and requires no medical/surgical intervention (eg, lack of housing, economic inadequacy, care-giver respite, family circumstances, administrative) Nonserious Adverse Events All AEs that are not classified as serious. 7.2 Assignment of Adverse Event Intensity and Relationship to Study Drug The following categories and definitions of intensity as determined by a physician should be used for all BMS clinical study AEs: Mild (Grade 1) - Awareness of event but easily tolerated Moderate (Grade 2) - Discomfort enough to cause some interference with usual activity Severe (Grade 3) - Inability to carry out usual activity Very Severe (Grade 4) - Debilitating, significantly incapacitates subject despite symptomatic therapy The following categories and definitions of causal relationship to study drug as determined by a physician should be used for all BMS clinical study AEs: 131
133 Related: There is a reasonable causal relationship to study drug administration and the AE Not related: There is not a reasonable causal relationship to study drug administration and the AE The expression "reasonable causal relationship" is meant to convey in general that there are facts (eg, evidence such as de-challenge/re-challenge) or other arguments to suggest a positive causal relationship. 7.3 Collection and Reporting Adverse events can be spontaneously reported or elicited during open-ended questioning, examination, or evaluation of a subject. (In order to prevent reporting bias, subjects should not be questioned regarding the specific occurrence of one or more AEs.) If known, the diagnosis of the underlying illness or disorder should be recorded, rather than its individual symptoms. The following information should be captured for all AEs: onset, duration, intensity, seriousness, relationship to study drug, action taken, and treatment required. If treatment for the AE was administered, it should be recorded on the appropriate CRF page. The investigator shall supply the sponsor and Ethics Committee with any additional requested information, notably for reported deaths of subjects. Completion of supplemental CRFs may be requested for AEs and/or laboratory abnormalities that are reported/identified during the course of the study Serious Adverse Event Collection and Reporting Following the subject s written consent to participate in the study, all SAEs, whether related or not related to study drug, must be collected, including those thought to be associated with protocol-specified procedures. All SAEs must be collected that occur within 30 days of discontinuation of dosing or within 30 days of the last visit for screen failures. If applicable, SAEs must be collected that relate to any later protocol-specified procedure (eg, a follow-up skin biopsy). The investigator should collect any SAE occurring after these time periods that is believed to be related to study drug or protocol-specified procedure. 132
134 An SAE report should be completed for any event where doubt exists regarding its status of seriousness. If the investigator believes that an SAE is not related to study drug, but is potentially related to the conditions of the study (such as withdrawal of previous therapy, or a complication of a study procedure), the relationship should be specified in the narrative section of the SAE Report Form. SAEs must be recorded on the BMS SAE Report Form; pregnancies on a BMS Pregnancy Surveillance Form. These original BMS Forms are to remain on site. SAEs, whether related or not related to study drug, and pregnancies, must be reported to BMS within 24 hours via confirmed facsimile (fax) transmission, or scanned and reported via electronic mail to: SAE Address: Worldwide.Safety@BMS.com SAE Facsimile Number: See Contact Information list. SAE Telephone Contact (required for pregnancy reporting): See Contact Information list. If only limited information is initially available, follow-up reports are required. (Note: Follow-up SAE reports should include the same investigator term(s) initially reported.) If an ongoing SAE changes in its intensity or relationship to study drug or if new information becomes available, a follow-up SAE report should be sent within 24 hours to the BMS using the same procedure used for transmitting the initial SAE report. All SAEs should be followed to resolution or stabilization Handling of Expedited Safety Reports In accordance with local regulations, BMS will notify investigators of all SAEs that are suspected (related to the investigational product) and unexpected (i.e., not previously described in the Investigator Brochure). In the European Union (EU), an event meeting these criteria is termed a Suspected, Unexpected Serious Adverse Reaction (SUSAR). Investigator notification of these events will be in the form of an expedited safety report (ESR). 133
135 Other important findings which may be reported by the sponsor as an ESR include: increased frequency of a clinically significant expected SAE, an SAE considered associated with study procedures that could modify the conduct of the study, lack of efficacy that poses significant hazard to study subjects, clinically significant safety finding from a nonclinical (eg, animal) study, important safety recommendations from a study data monitoring committee, or sponsor decision to end or temporarily halt a clinical study for safety reasons. Upon receiving an ESR from BMS, the investigator must review and retain the ESR with the Investigator Brochure. Where required by local regulations or when there is a central IRB/IEC for the study, the sponsor will submit the ESR to the appropriate IRB/IEC. The investigator and IRB/IEC will determine if the informed consent requires revision. The investigator should also comply with the IRB/IEC procedures for reporting any other safety information. In addition, suspected serious adverse reactions (whether expected or unexpected) shall be reported by BMS to the relevant competent health authorities in all concerned countries according to local regulations (either as expedited and/or in aggregate reports) Nonserious Adverse Events The collection of nonserious AE information should begin at initiation of study drug. Nonserious AE information should also be collected from the start of a placebo lead-in period or other observational period intended to establish a baseline status for the subjects. If an ongoing nonserious AE worsens in its intensity or its relationship to the study drug changes, a new nonserious AE entry for the event should be completed. Nonserious AEs should be followed to resolution or stabilization, or reported as SAEs if they become serious (see Section 7.3.1). Follow-up is also required for nonserious AEs that cause interruption or discontinuation of study drug, or those that are present at the end of study treatment as appropriate. All identified nonserious AEs must be recorded and described on the appropriate non serious AE page of the CRF (paper or electronic). 134
136 7.4 Laboratory Test Abnormalities All laboratory test values captured as part of the study should be recorded on the appropriate laboratory test results pages of the CRF, or be submitted electronically from a central laboratory. In addition, the following laboratory abnormalities should also be captured on the nonserious AE CRF page (paper or electronic) or SAE paper CRF page as appropriate: Any laboratory test result that is clinically significant or meets the definition of an SAE Any laboratory abnormality that required the subject to have study drug discontinued or interrupted Any laboratory abnormality that required the subject to receive specific corrective therapy It is expected that wherever possible, the clinical, rather than the laboratory term would be used by the reporting investigator (eg, anemia versus low hemoglobin value). 7.5 Overdose An overdose is defined as the accidental or intentional ingestion or infusion of any dose of a product that is considered both excessive and medically important. All occurrences of overdose must be reported as an SAE (see Section for reporting details.) 7.6 Potential Drug Induced Liver Injury (DILI) Initial liver-related laboratory abnormalities should be confirmed in 3-5 days prior to the reporting of a potential DILI event and discussed with the sponsor. All confirmed occurrences of potential DILIs meeting the defined criteria must be reported as SAEs (see Section for reporting details). Potential drug induced liver injury is defined as concurrent: 135
137 1) ALT 5 X baseline value AND 10 X ULN AND 2) Total bilirubin 2 X ULN, AND 3) No other immediately apparent possible causes of ALT elevation and hyperbilirubinemia, including, but not limited to, acute viral hepatitis, cholestasis, pre-existing hepatic disease excluding HCV or the administration of other drug(s), herbal medications and substances known to be hepatotoxic. 7.7 Pregnancy Requirements for Pregnancy Testing Pregnancy testing must be performed throughout the study as specified in Section 6.1 and the results of all pregnancy tests (positive or negative) recorded on the CRF or transferred electronically. The minimum sensitivity of the pregnancy test must be 25 IU/L or equivalent units of HCG. If the pregnancy test is positive, the subject must not receive the investigational product and must not continue in the study. In addition, all WOCBP should be instructed to contact the investigator immediately if they suspect they might be pregnant (eg, missed or late menstrual period) at any time during study participation Reporting of Pregnancy If following initiation of the investigational product, it is subsequently discovered that a study subject is pregnant or may have been pregnant at the time of investigational product exposure, including during at least 6 half-lives after product administration, the investigational product will be permanently discontinued in an appropriate manner (eg, dose tapering if necessary for subject safety). The investigator must immediately notify the BMS medical monitor of this event, record the pregnancy on the Pregnancy Surveillance Form (not an SAE form). Initial information on a pregnancy must be reported immediately to BMS and the outcome information provided once the outcome is known. Completed Pregnancy Surveillance Forms must be forwarded to BMS according to SAE reporting procedures described in Section
138 Protocol-required procedures for study discontinuation and follow-up must be performed on the subject unless contraindicated by pregnancy (eg, x-ray studies). Other appropriate pregnancy follow-up procedures should be considered if indicated. Follow-up information regarding the course of the pregnancy, including perinatal and neonatal outcome must be reported on the Pregnancy Surveillance Form. Any pregnancy that occurs in a female partner of a male study participant should be reported to the sponsor. Information on this pregnancy will be collected on the Pregnancy Surveillance Form. 7.8 Other Safety Considerations Any significant worsening noted during interim or final physical examinations, electrocardiograms, x-rays, and any other potential safety assessments, whether or not these procedures are required by the protocol, should also be recorded on the appropriate nonserious AE page of the CRF (paper or electronic) or SAE paper CRF page. 8 STATISTICAL CONSIDERATIONS Part 1 of the study represents the first 28 days (4 weeks) of treatment for the Sentinel Cohorts (both Treatments A and B). Part 2 of the study represents post-week 4 treatment for the Sentinel Cohorts and all study duration for the Expansion Cohorts. 8.1 Sample Size Determination In order to reduce the risk of resistance developing in a large number of subjects, the study will be conducted in two parts: In Part 1 approximately 10 subjects will be treated in each of Treatments A or B in the sentinel cohorts. Part 1 includes the sentinel cohorts up to Week 4, after witch the decision to introduce up to 5 expansion cohorts will be made with a target sample size of 20 subjects per treatment group. The subjects in the sentinel cohorts and any subjects in the expansion cohorts will continue treatment for up to 24 weeks (Part 2). The sample size selections for each part of the study and for each treatment group are not based on statistical power calculations. Below are risk assessments calculations based upon the endpoints needed to answer the primary 137
139 objectives for Part 1 and Part 2 with sample sizes of 10 (sentinel cohorts) or 20 (expansion cohorts) Sample Size for Part 1 Sentinel Cohorts Two decision points for each treatment group at Week 2 and 4 will be made independently of each other. For the Week 2 decision, given 10 subjects in each sentinel cohort, the probability of observing 70% or more subjects (at least 7 out of 10) with successful response to treatment is 0.17 if the population rate is 50%. The probability of observing 70% or more subjects (at least 7 out of 10) with successful response to treatment is 0.88 if the population rate is 80%. If the observed rate of success is 70% (7 out of 10), the 90% lower confidence bound is 45%. For the Week 4 decision, given 10 subjects in each sentinel cohort, the probability of observing 50% or more subjects (at least 5 out of 10) with RVR is 0.15 if the population rate is 30%. The probability of observing 50% or more subjects with RVR is 0.83 if the population rate is 60%. If the observed rate of success is 50% (5 out of 10), the 90% lower confidence bound is 27% Sample Size for Part 2 Sentinel Cohorts Given 10 subjects in each cohort, the probability of observing 20% or more subjects (at least 2 of 10) with SVR12 is 0.26 if the true population rate is 10%. The probability of observing 20% or more subjects with SVR12 is 0.85 if the true population rate is 30%. In addition, if the true observed rate is 20% (2 out of 10), the 80% CI is (5%, 45%). A target sample size of 10 subjects in each cohort can also detect, with 46% or 80% probability a safety event that occurs at incidence rates of 6% or 15%, respectively Sample Size for Part 2 Expansion Cohorts Given 20 subjects in each Expansion cohort, the probability of observing 20% or more subjects in a cohort (at least 4 of 20) with SVR12 is 0.13 if the population rate is 10%. The probability of observing 20% or more subjects in a cohort with SVR12 is 0.89 if the population rate is 30%. In addition, if the observed rate is 20% (4 out of 20), the 80% CI 138
140 is (9%, 36%). A target sample size of 20 subjects per cohort can also detect, with 71% probability, a safety event that occurs at an incident rate of 6% in a cohort. No multiple testing adjustments will be made. 8.2 Populations for Analyses All subjects who receive study medication will be included in the safety data set. All available concentration-time data from subjects who receive BMS and BMS will be reported. All available derived PK parameter values will be included in the PK data set and reported, but only subjects with adequate pharmacokinetic profiles will be included in the summary statistics and statistical analysis. All available data from subjects for whom pharmacodynamic measurements are available at baseline and at least one other time will be included in the pharmacodynamic data set. 8.3 Endpoint Definitions Safety Endpoint Safety endpoints will be based on medical review of the frequency of SAEs and AEs, discontinuations due to AEs, and abnormalities observed from vital sign and ECGs measurements, physical examinations and clinical laboratory results Pharmacokinetic Endpoint(s) Pharmacokinetics of will be derived from plasma concentration versus time data. The pharmacokinetic parameters to be assessed include: Cmax Cmin Tmax AUC(TAU) Maximum observed plasma concentration Trough observed plasma concentration Time of maximum observed plasma concentration Area under the concentration-time curve in one dosing interval 139
141 Individual subject pharmacokinetic parameter values will be derived by non-compartmental methods by a validated pharmacokinetic analysis program Pharmacodynamic (PD) Endpoints For all subjects, Plasma HCV RNA levels will be measured at screening and on Days -1 (baseline), 1 to 7, 9, 11, 14, Week 3 and every 2 weeks from Week 4 to Week 12 in all subjects. HCV RNA measurements will then proceed as follows: All Treatment group A cohorts and Expansion Cohort B3: Every 2 weeks from Week 12 until EOT unless SOC is added to the regimen, in which case, samples will be collected every 4 weeks from Week 12 until EOT. Subjects who have SOC or pegifn added due to viral breakthrough will have HCV RNA measured immediately prior to administration of the first dose of pegifn and every 4 weeks thereafter until EOT. Treatment group B sentinel cohort and Expansion Cohorts B1 and B2: HCV RNA level measurement every 4 weeks from Week 12 until EOT. All subjects: HCV RNA measured at Week 4, 12, 24, 36, and 48 post-treatment. Characterization of HCV genomic substitutions associated with exposure of, will be determined from blood samples collected from all subjects and for which sufficient HCV RNA permits analysis (generally but not exclusively 1000 IU/mL) PD Endpoints for Part 1 Proportion of subjects with successful response to treatment at Week 2 and 4. Proportion of subjects with RVR at Week 4. Decrease from baseline in plasma HCV RNA levels at Days 4, 7 and 14. Additional endpoints used for describing the magnitude of the change in log 10 HCV RNA are: absolute changes from baseline, maximum changes from baseline, and the proportion of subjects who reach undetectable viral load. The rate of the change in log 10 HCV RNA levels will be assessed by the time point for which a subject s maximum decrease is observed. Baseline HCV RNA is defined as the observed level on Day -1. The screening value will serve as baseline if the Day -1 value is not available. 140
142 PD Endpoints for Part 2 Proportion of subjects by cohort (Sentinel and Expansion cohorts) who achieve: RVR; ervr; cevr; undetectable at end of treatment; SVR12; SVR24 and SVR48. For the Expansion Cohorts additional endpoints used for describing the magnitude of the change in log10 HCV RNA are: absolute changes from baseline, maximum changes from baseline, and the proportion of subjects who reach undetectable viral load. The rate of the change in log10 HCV RNA levels will be assessed by the time point for which a subject s maximum decrease is observed. Baseline HCV RNA is defined as the observed level on Day -1. The screening value will serve as baseline if the Day -1 value is not available Other Endpoint(s) Other endpoints include characterization of HCV genomic substitutions, anti-hcv T cell response analysis and analysis of changes in host gene expression. Possible exploratory analysis of host anti-hcv T cell responses might include IFN-γ production in response to HCV peptide antigens as well as levels of PD-1 expression. Anti-HCV T cell responses and changes in host gene expression will be analyzed as deemed relevant. The relationship between antiviral activity endpoints and SNPs in genes encoding proteins of the IFNλ family (IL28A, IL28B, IL29) and potentially the RBV transporter protein ENT1 will be explored. The relationship between hemolytic anemia and SNPs in ITPA may be explored. 8.4 Analyses Analyses will be based on treated subjects. Results will be presented by treatment group and cohort separately. Categorical variables will be summarized with counts and percents. Continuous variables will be summarized with univariate statistics (eg, mean, median, standard error). Longitudinal summaries of safety and efficacy endpoints will use pre-defined visit week windows. Windows around planned measurement times will be constructed based on the 141
143 midpoint between planned study visits. Laboratory measures will be summarized using US standard values and units. On-treatment endpoints will be assessed with measurements through the last dose of study therapy plus 7 days (based on the half-lives of BMS and BMS ). Follow-up endpoints will be assessed with measurements after the last dose of study therapy plus 7 days Demographics and Baseline Characteristics The following will be summarized by treatment group and overall: Demographics: age, race, gender, geographic region; Disease characteristics at baseline: HCV RNA level, HCV subtype; Physical measurements at baseline: height, weight, body mass index (BMI); Laboratory tests at baseline; Prior medications Safety Analyses All recorded adverse events will be listed and tabulated by system organ class and intensity, preferred term, treatment and cohort. Vital signs, clinical laboratory tests will be listed and summarized by treatment and cohort. Any physical exam findings and clinical laboratory results will be listed. ECG readings will be evaluated by the investigator and abnormalities, if present, will be listed. Endpoints of safety may also include changes in selected AEs, changes from baseline in laboratory parameters and lab abnormalities by toxicity grades. For the effect of BMS and BMS on key safety endpoints, please see section Efficacy Analyses In general, the proportion of subjects with antiviral activity endpoints will be assessed using modified intent to treat (ITT): the numerator is based on treated subjects meeting the response criteria (regardless of add-on SOC); the denominator is based on all treated 142
144 subjects. Response rates and 80% exact binomial CIs will be presented by treatment group. For treatment group A and Expansion Cohort B3, an additional analysis will classify subjects who added on SOC or pegifn before the endpoint assessment as failures. Primary and secondary antiviral activity endpoints will also be summarized by HCV subtype (1a, 1b). For the effect of BMS and BMS on key antiviral activity endpoints, please see section Efficacy Analyses for Part 1 Analyses will be based on the sentinel cohort. Primary antiviral activity endpoints A co-primary early antiviral activity endpoint is the proportion of subjects with either undetectable HCV RNA at Week 2 or 2 log 10 IU/mL decrease in plasma HCV RNA from baseline without rebound during the first 2 weeks. Rebound is defined as 1 log 10 IU/mL increase in HCV RNA from nadir either at more than 1 time point (not necessarily consecutive) or at last value through Week 2 or detectable RNA after achieving undetectable RNA. Another co-primary early antiviral activity endpoint is the proportion of subjects with RVR. Secondary antiviral activity endpoint Secondary antiviral activity endpoint is the change from baseline at Day 4, Day 7 and Day 14 in log 10 HCV RNA. The magnitude of the change in log 10 HCV RNA levels will be assessed by summarizing changes from baseline, including mean, standard deviation, 90% CIs, median and range by study day and treatment group. Each individual s maximum decrease from baseline in log 10 HCV RNA will be summarized by treatment group, frequency distributions for maximum decrease in log 10 HCV RNA will be provided by treatment group, e.g., the proportion of subjects with a decrease of <1.0, 143
145 1.0-<2.0, 2.0-<3.0 and 3.0 log10 HCV RNA. The proportion of subjects with undetectable HCV RNA will be summarized by study day and treatment group. The day of maximum viral load drop in log 10 HCV RNA observed will be estimated and summarized by treatment group. The slope of the decrease from baseline to the day with maximum decrease in log 10 HCV RNA may be estimated using mixed effect model and summarized, if appropriate Efficacy Analyses for Part 2 Analyses will be based on the sentinel and Expansion Cohorts. Primary antiviral activity endpoint The proportion of subjects with SVR 12, defined as undetectable HCV RNA at follow-up Week 12, will be summarized. Secondary antiviral activity endpoints The following antiviral activity endpoints will be summarized: Proportion of subjects with RVR, i.e., undetectable HCV RNA at Week 4 on treatment; Proportion of subjects with ervr, i.e., undetectable HCV RNA at both Weeks 4 and 12 on treatment; Proportion of subjects with cevr, i.e., undetectable HCV RNA at Week 12 on treatment; Proportion of subjects with SVR24, i.e., undetectable HCV RNA at follow-up Week 24; Frequency of genotypic substitutions associated with virologic failure. Primary and secondary antiviral activity endpoints will also be summarized by HCV subtype (1a, 1b). The proportion of subjects with SVR48, i.e., undetectable HCV RNA at follow-up Week 48 will be similarly summarized. In addition, if applicable, for treatment group A (sentinel cohort and Expansion Cohorts A1 and A2) and Expansion Cohort B3, SVR endpoints will be summarized by the 144
146 duration subjects stay on BMS BMS only, or BMS BMS RBV, respectively (e.g., Weeks 2, 4, 12, 24 and 48). Secondary antiviral activity endpoint for the Expansion Cohorts is the change from baseline at Day 4, Day 7 and Day 14 in log10 HCV RNA. The magnitude of the change in log10 HCV RNA levels will be assessed by summarizing changes from baseline, including mean, standard deviation, 90% CIs, median and range by study day and cohort. Each individual s maximum decrease from baseline in log10 HCV RNA will be summarized by cohort, frequency distributions for maximum decrease in log10 HCV RNA will be provided by cohort, e.g., the proportion of subjects with a decrease of <1.0, 1.0-<2.0, 2.0-<3.0 and 3.0 log10 HCV RNA. The proportion of subjects with undetectable HCV RNA will be summarized by study day and cohort. The day of maximum viral load drop in log10 HCV RNA observed will be estimated and summarized by cohort. The slope of the decrease from baseline to the day with maximum decrease in log10 HCV RNA may be estimated using mixed effect model and summarized, if appropriate Pharmacokinetic Analyses Pharmacokinetic parameters listed in Section will be summarized by treatment group and cohort. Trough concentrations will be summarized by treatment group, cohort and study day Pharmacokinetic/Pharmacodynamic Analyses In addition to the efficacy and safety analyses described in Section and 8.4.3, the relationship between exposures to BMS and BMS and measures of safety/antiviral activity will be explored graphically. Measures of antiviral activity may include the proportions of subjects with ervr, SVR 24, and the change from baseline in HCV RNA. Measures of safety may include SAEs, AEs leading to discontinuation of study therapy, selected AEs and changes from baseline in laboratory tests. 145
147 8.4.6 Pharmacogenomic Analyses Analyses will focus on SNPs in genes encoding proteins of the IFNλ family (IL28A, IL28B, IL29), and potentially the RBV transporter protein ENT1 and inosine triphosphatase (ITPA) for treated subjects by treatment group and cohort. For each SNP in each candidate gene, genotypes will be summarized by treatment group and cohort. Minor allele frequencies and departures from Hardy-Weinberg Equilibrium will be summarized for each SNP pooled across treatment groups and cohorts. Antiviral activity endpoints (eg. ervr, SVR24 and other measures) will be summarized using modified ITT by treatment group, cohort and genotype for each SNP Outcomes Research Analyses Not applicable Other Analyses Summary statistics for genomic substitution and anti-hcv T cell measurements will be tabulated by treatment group, cohort/cohort, study day and time point. Possible associations between the biomarkers of interest and viral RNA may be explored graphically and by suitable statistical models, if appropriate. 8.5 Interim Analyses Analyses will be based on treated subjects. Results will be presented by treatment group and cohort. Interim analyses of pharmacodynamic, pharmacokinetic, and safety data will be performed for data collected. Analyses may include listings, summaries, and graphs of HCV RNA, genotype subtypes, AEs, ECGs, vital signs, selected laboratory parameters, and exploratory biomarkers Interim Analyses of the Sentinel Cohorts Interim analyses may be performed at the following time points. Interim analyses time points are determined by subjects not requiring rescue treatment. 146
148 1) after all subjects in sentinel cohort reach Week 2; 2) after all subjects in sentinel cohort reach Week 4; 3) after all subjects in sentinel cohort reach Week 12; 4) after all subjects in sentinel cohort reach Week 24; 5) after all subjects in sentinel cohort reach Follow-up Week 12; 6) after all subjects in sentinel cohort reach Follow-up Week 24; 7) after all subjects in sentinel cohort reach Follow-up Week 48. Interim analyses will include analysis of safety and antiviral activity by treatment group, and biomarkers including IL28B subject genotype and anti-hcv T-cell Interim Analyses of the Expansion Cohorts Interim analyses may be performed at the following time points. Interim analyses time points are determined by subjects not requiring rescue treatment. Expansion Cohort A1, A2, B1 and B2: 1) after all subjects complete 4 weeks of treatment (including analysis of RVR); 2) after all subjects complete 12 weeks of treatment (including analysis of ervr and cevr); 3) after all subjects complete 24 weeks of treatment (including analysis of 4 week sustained virologic response (SVR4)) defined as undetectable HCV RNA at followup Week 4; 4) after all subjects complete 12 weeks of follow-up after end of treatment (including SVR12); 5) after all subjects complete 24 weeks of follow-up after end of treatment (including SVR24); 6) after all subjects complete 48 weeks of follow-up after end of treatment (including SVR48). In the event that significant delays in enrollment occur between Expansion cohorts A1, A2 and B1, B2, Interim analyses may be performed on A1/A2 or B1/B2 separately as needed. 147
149 Expansion Cohort B3: 1) after all subjects complete 4 weeks of treatment (including analysis of RVR); 2) after all subjects complete 12 weeks of treatment (including analysis of ervr and cevr); 3) after all subjects complete 24 weeks of treatment (including analysis of 4 week sustained virologic response (SVR4)) defined as undetectable HCV RNA at followup Week 4; 4) after all subjects complete 12 weeks of follow-up after end of treatment (including SVR12) 5) after all subjects complete 24 weeks of follow-up after end of treatment (including SVR24); 6) after all subjects complete 48 weeks of follow-up after end of treatment (including SVR48). In the event one or more group decision criteria are met and a group is stopped, an interim analysis may be performed. These interim analyses all include analyses of antiviral activity by cohort. In addition, analysis of safety and biomarkers including IL28B subject genotype and anti-hcv T-cell might be included Final Analyses The final analyses will be performed after all subjects complete 48 weeks of follow-up after end of treatment. Analyses will be based on treated subjects. 9 ADMINISTRATIVE SECTION 9.1 Compliance Compliance with the Protocol and Protocol Revisions The study shall be conducted as described in this approved protocol. All revisions to the protocol must be discussed with, and be prepared by, BMS. The investigator should not implement any deviation or change to the protocol without prior review and documented approval/favorable opinion from the IRB/IEC of an amendment, except where necessary 148
150 to eliminate an immediate hazard(s) to study subjects. Any significant deviation must be documented in the CRF. If a deviation or change to a protocol is implemented to eliminate an immediate hazard(s) prior to obtaining IRB/IEC approval/favorable opinion, as soon as possible the deviation or change will be submitted to: IRB/IEC for review and approval/favorable opinion Bristol-Myers Squibb Regulatory Authority(ies), if required by local regulations Documentation of approval signed by the chairperson or designee of the IRB(s)/IEC(s) must be sent to BMS. If an amendment substantially alters the study design or increases the potential risk to the subject: (1) the consent form must be revised and submitted to the IRB(s)/IEC(s) for review and approval/favorable opinion; (2) the revised form must be used to obtain consent from subjects currently enrolled in the study if they are affected by the amendment; and (3) the new form must be used to obtain consent from new subjects prior to enrollment. If the revision is an administrative letter, investigators must inform their IRB(s)/IEC(s) Monitoring Representatives of BMS must be allowed to visit all study site locations periodically to assess the data quality and study integrity. On site they will review study records and directly compare them with source documents, discuss the conduct of the study with the investigator, and verify that the facilities remain acceptable. In addition, the study may be evaluated by BMS internal auditors and government inspectors who must be allowed access to CRFs, source documents, other study files, and study facilities. BMS audit reports will be kept confidential. 149
151 THE INVESTIGATOR MUST NOTIFY BMS PROMPTLY OF ANY INSPECTIONS SCHEDULED BY REGULATORY AUTHORITIES, AND PROMPTLY FORWARD COPIES OF INSPECTION REPORTS TO BMS Investigational Site Training Bristol-Myers Squibb will provide quality investigational staff training prior to study initiation. Training topics will include but are not limited to: GCP, AE reporting, study details and procedure, study documentation, informed consent, and enrollment of WOCBP. For sites using the BMS electronic data capture tool, each individual making entries and/or corrections on electronic CRFs must meet BMS training requirements and must only access the BMS electronic data capture tool using the unique user account provided by the sponsor. User accounts are not to be shared or reassigned to other individuals. For electronic CRFs, corrections are made through the BMS electronic data capture tool that generates an automated audit trail including date and timestamp, full name of the person making the correction and original entry. The system also prompts the user to document reason for change that is also maintained in the audit trail. Each individual electronically signing electronic CRFs must meet BMS training requirements and must only access the BMS electronic data capture tool using the unique user account provided by the sponsor. User accounts are not to be shared or reassigned to other individuals. 9.2 Records Retention The investigator must retain study drug (those supplied by the sponsor or sourced by the investigator) disposition records, copies of CRFs (or electronic files), and source documents for the maximum period required by applicable regulations and guidelines, or institution procedures, or for the period specified by the sponsor, whichever is longer. The investigator must contact BMS prior to destroying any records associated with the study. BMS will notify the investigator when the study records are no longer needed. 150
152 If the investigator withdraws from the study (eg, relocation, retirement), the records shall be transferred to a mutually agreed upon designee (eg, another investigator, IRB). Notice of such transfer will be given in writing to BMS Case Report Forms An investigator is required to prepare and maintain adequate and accurate case histories designed to record all observations and other data pertinent to the investigation on each individual treated or entered as a control in the investigation. Data reported on the CRF that are derived from source documents must be consistent with the source documents or the discrepancies must be explained. For sites using the BMS electronic data capture tool, electronic CRFs will be prepared for all data collection fields except for fields specific to SAEs and pregnancy, which will be reported on the Pregnancy Surveillance Form. Spaces may be left blank only in those circumstances permitted by study-specific CRF completion guidelines provided by the sponsor. Paper CRFs must be completed legibly in ink. Subjects are to be identified by birth date and subject number, if applicable. All requested information must be entered on the CRF in the spaces provided. If an item is not available or is not applicable, it must be documented as such; do not leave a space blank. Electronic data transfer is acceptable. The confidentiality of records that could identify subjects must be protected, respecting the privacy and confidentiality rules in accordance with the applicable regulatory requirement(s). The investigator will maintain a signature sheet to document signatures and initials of all persons authorized to make entries and/or corrections on CRFs. For paper CRFs, a correction must be made by striking through the incorrect entry with a single line and entering the correct information adjacent to the incorrect entry. The correction must be dated, initialed and explained (if necessary) by the person making the correction and must not obscure the original entry. 151
153 The completed CRF, including any paper SAE/pregnancy CRFs, must be promptly reviewed, signed, and dated by a qualified physician who is an investigator or subinvestigator. For electronic CRFs, review and approval/signature is completed electronically through the BMS electronic data capture tool. The investigator must retain a copy of the CRFs including records of the changes and corrections Study Drug Records It is the responsibility of the investigator to ensure that a current disposition record of investigational product (those supplied by the sponsor) is maintained at each study site where study drug is inventoried and disposed. Records or logs must comply with applicable regulations and guidelines and should include: amount received and placed in storage area amount currently in storage area container number or batch number and use date or expiry date dates and initials of person responsible for the inventory /entry/ movement of each study drug amount dispensed to and returned by each subject, including unique subject identifiers amount transferred to another area/site for dispensing or storage non-study disposition (eg, lost, wasted, broken) amount returned to the sponsor amount destroyed at study site, if applicable retain samples sent to third party for bioavailability/bioequivalence, if applicable The sponsor will provide forms to facilitate inventory control if the staff at the investigational site does not have an established system that meets these requirements. 9.3 Return and Destruction of Study Drug Return of Study Drug Upon completion or termination of the study, all unused and/or partially used study drug, which was supplied by the sponsor, must be returned to BMS, if not authorized by BMS to be destroyed at the site. 152
154 All study drug, which was supplied by the sponsor, returned to BMS must be accompanied by the appropriate documentation and be clearly identified by protocol number and study site number on the outermost shipping container. Returned supplies should be in the original containers (eg, patient kits that have clinical labels attached). Empty containers should not be returned to BMS. It is the investigator s responsibility to arrange for disposal of all empty containers, provided that procedures for proper disposal have been established according to applicable federal, state, local, and institutional guidelines and procedures, and provided that appropriate records of disposal are kept. The return of unused study drug, those that were supplied by the sponsor, should be arranged by the responsible Study Monitor Destruction of Study Drug If study drugs (those supplied by the sponsor or sourced by the investigator) are to be destroyed on site, it is the investigator s responsibility to ensure that arrangements have been made for the disposal, written authorization has been granted by BMS, procedures for proper disposal have been established according to applicable regulation and guidelines and institutional procedures, and appropriate records of the disposal have been documented. The unused study drug can only be destroyed after being inspected and reconciled by the responsible BMS Study Monitor. 9.4 Publications The data collected during this study are confidential and proprietary to the sponsor. Any publications or abstracts arising from this study require approval by the sponsor prior to publication or presentation and must adhere to the sponsor s publication requirements as set forth in the approved clinical trial agreement (CTA). All draft publications, including abstracts or detailed summaries of any proposed presentations, must be submitted to the sponsor at the earliest practicable time for review, but at any event not less than 30 days before submission or presentation unless otherwise set forth in the CTA. Sponsor shall have the right to delete any confidential or proprietary information contained in any proposed presentation or abstract and may delay publication for up to 60 days for purposes of filing a patent application. 153
155 10 GLOSSARY OF TERMS Term Adverse Reaction Expedited Safety Report Non-responder Null-responder Post-treatment period Rebound RVR Successful response to treatment at Week 2 SUSAR SVR SVR 12 Definition An adverse event that is considered by either the investigator or the sponsor as related to the investigational product Rapid notification to investigators of all SAEs that are suspected (related to the investigational product) and unexpected (i.e., not previously described in the Investigator Brochure), or that could be associated with the study procedures. Subjects who received at least 12 weeks of SOC and continue to have a detectable HCV RNA level or subjects who may have attained a 2 log 10 decline in HCV RNA levels but did not sustain it at 12 weeks and stopped treatment. Subjects who after at least 12 weeks of therapy with the current standard of care (pegifnα and RBV) have never attained 2 log 10 decline in HCV RNA level. Period after date of last study drug administration. Rebound is defined as 1 log 10 IU/mL increase in HCV RNA from nadir either at more than 1 time point (not necessarily consecutive) or last value through Week 2 or detectable RNA after achieving undetectable RNA. RVR defined as undetectable HCV RNA (< 10 IU/mL) at Week 4. Successful response to treatment at Week 2 is defined as either undetectable HCV RNA (< 10 IU/mL) or 2 log 10 IU/mL decrease in plasma HCV RNA from baseline without rebound. Suspected, Unexpected, Serious Adverse Reaction as termed by the European Clinical Trial Directive (2001/20/EC). Sustained Virologic Response (undetectable viral load) HCV RNA < 10 IU/mL at 12 weeks post-treatment 154
156 Term SVR 24 SVR 48 Treatment intolerant Unexpected Adverse Reaction Definition HCV RNA < 10 IU/mL at 24 weeks post-treatment HCV RNA < 10 IU/mL at 48 weeks post-treatment Subjects who are unable to receive at least 12 weeks of SOC due to toxicities associated with IFN and/or RBV. An adverse reaction, the nature or severity of which is not consistent with the applicable product information (eg, Investigator Brochure for an unapproved investigational product) 155
157 11 LIST OF ABBREVIATIONS Term ADME AE ACLS AI ALT ANC ANOVA aptt APRI AST ALT API AUC AUC(INF) AUC(0-T) AUC(TAU) AUMC(INF) AV A-V β-hcg BA/BE %BE BLQ BMI Definition Absorption, Distribution, Metabolism, and Excretion adverse event advanced cardiac life support accumulation index alanine aminotransferase absolute neutrophil count analysis of variance activated partial thromboplastin time AST to platelet ratio index aspartate aminotransferase alanine transaminase active pharmaceutical ingredient area under the concentration-time curve area under the concentration-time curve from time zero extrapolated to infinite time area under the concentration-time curve from time zero to the time of the last quantifiable concentration area under the concentration-time curve in one dosing interval area under the moment concentration time curve extrapolated to infinity Antiviral atrioventricular beta-human chorionic gonadotrophin bioavailability/bioequivalence percent biliary excretion below limit of quantification body mass index 156
158 Term Definition BMS Bristol-Myers Squibb BP blood pressure BUN blood urea nitrogen C Celsius Ca ++ CBC cevr CFR CHC CI C1 - CLcr CLNR CLR CLT CLT/F cm Cmax, CMAX Cmin, CMIN Ctrough COSTART CNS CRC CRF CV calcium complete blood count complete early virologic response Code of Federal Regulations chronic hepatitis C confidence interval chloride creatinine clearance nonrenal clearance renal clearance total body clearance apparent total body clearance centimeter maximum observed concentration trough observed concentration trough concentration Coding Symbols for Thesaurus of Adverse Reaction Terms Central nervous system Clinical Research Center Case Report Form, paper or electronic coefficient of variation CYP cytochrome p-450 D/C discontinue 157
159 Term DAA DEV %DEV DHWE DILI dl DSM IV EA ECG ecrf EDC EEG eg EOT ESR F FDA FSH %FE g GC GCP GGT GFR h HBsAg HBV HCC Definition direct acting antiviral deviation from the nominal value percent deviation departures from Hardy-Weinberg equilibrium Drug induced liver injury deciliter Diagnostic and Statistical Manual of Mental Disorders (4 th Edition) extent of absorption electrocardiogram Electronic Case Report Form Electronic Data Capture electroencephalogram exempli gratia (for example) end of treatment Expedited Safety Report bioavailability Food and Drug Administration follicle stimulating hormone percent fecal excretion gram gas chromatography Good Clinical Practice gamma-glutamyl transferase glomerular filtration rate hour hepatitis B surface antigen hepatitis B virus Hepatocellular Carcinoma 158
160 Term HCV - HCO 3 HIV HPF HPLC HR HRT hpxr IB ICD ie IEC IMSL IND IRB IU IV K K 3 EDTA K + kg L LAS LC LDH LLQ LPF Definition hepatitis C virus bicarbonate Human Immunodeficiency Virus high power field High-performance liquid chromatography heart rate hormone replacement therapy human pregnane X receptor Investigator Brochure International Classification of Diseases id est (that is) Independent Ethics Committee International Mathematical Statistical Library Investigational New Drug Exemption Institutional Review Board International Unit intravenous slope of the terminal phase of the log concentration-time curve potassium ethylenediaminetetraacetic acid potassium kilogram liter Laboratory Acquisition System liquid chromatography lactate dehydrogenase lower limit of quantitation low power field 159
161 Term Definition ln natural logarithm m meter MAD multiple ascending dose mg milligram Mg ++ MIC min ml mmhg MRT MRT(INF) MRT(PO) MRT(SS) MS MTD magnesium minimum inhibitory concentration minute milliliter millimeters of mercury mean residence time mean residence time adjusted for infusion time mean residence time following oral administration mean residence time at steady-state mass spectrometry maximum tolerated dose µg microgram NS3 non-structural protein 3 (BMS ) NS4A non-structural protein 4A NS5A non-structural protein 5A (BMS ) NS5A+NS3 in combination N number of subjects or observations Na + sodium N/A not applicable ng nanogram NSAID nonsteroidal anti-inflammatory drug PBMC Peripheral Blood Mononuclear Cell Pct percentage 160
162 Term PD pegifnα P-gp PK PO PT PTT QC RBC RBV RNA RSD %RSD RVR SAD SAE SD Seq SOC SOP sp. Subj SVR SVR 12 SVR 24 SVR 48 t Definition pharmacodynamics pegylated interferon-alfa permeability glycoprotein pharmacokinetics per os (by mouth route of administration) prothrombin time partial thromboplastin time quality control red blood cell ribavirin ribonucleic acid relative standard deviation percent relative standard deviation Rapid Virologic Response single ascending dose serious adverse event standard deviation sequence Standard of Care Standard Operating Procedures species subject Sustained Virologic Response (undetectable viral load) Sustained Virologic Response at 12 weeks post-treatment Sustained Virologic Response at 24 weeks post-treatment Sustained Virologic Response at 48 weeks post-treatment temperature 161
163 Term T TAO TAUC(TAU) TAUC(0-T) T-HALF TK Tmax, TMAX TID UR %UR ULN ULQ UV VdBeta VdBeta/F Vss WBC WOCBP Definition time Trial Access Online, the BMS implementation of an EDC capability trapezoidal area under the concentration-time curve in one dosing interval trapezoidal area under the concentration-time curve from time zero to the time of the last quantifiable concentration half-life toxicokinetic time of maximum observed concentration thrice daily urinary recovery percent urinary recovery upper limit of normal upper limit of quantitation ultraviolet volume of distribution during the terminal phase apparent volume of distribution during the terminal phase volume of distribution at steady-state white blood cell women of childbearing potential 162
164 12 REFERENCES National Institute of Health Consensus Development Conference Statement: Management of hepatitis C. Hepatology. June 2002; 36 (5 Suppl 1):S3-20 Simmonds P. Viral heterogenicity of the hepatitis C virus. J Hepatol 1999;31 Suppl 1: Manns MP, McHutchinson JG, Gordon SC, et al. Peginterferon alfa-2b plus ribavirin compared with interferon alfa-2b plus ribavirin for initial treatment of chronic hepatitis C; a randomized trial. Lancet. Sept 2001;358(9286): Fried NW, Shiffman ML, Reddy KR, et al. Peginterferon alfa-2a plus ribavirin for chronic hepatitis C virus infection. N Engl J Med. Sept 2002;347(13): Hadziyannis SJ, Sette H, Jr, Norgan TR, et al. Peginterferon-alpha-2a and ribavirin combination therapy in chronic hepatitis C: a randomized study of treatment duration and ribavirin dose. Ann Intern Med. 2004;140(5): Reddy KR, Hoofnagle JH, Tong MJ, et al. Racial differences in responses to therapy with interferon in chronic hepatitis C. Consensus Interferon Study Group. Hepatology. 1999;30(3): Jeffers LJ, Cassidy W, Howell CD, Hu S, Reddy KR. Peginterferon alfa-2a (40kd) and ribavirin for black American patients with chronic HCV genotype 1. Hepatology. 2004;39(6): Muir AJ, Bornstein JD, Killenberg PG. Peginterferon alfa-2b and ribavirin for the treatment of chronic hepatitis C in blacks and non-hispanic whites. N Engl J Med. 2004;350(22):
165 Chung RT, Anderson J, Volberding P, et al. Peginterferon alfa-2a plus ribavirin versus interferon alfa-2a plus ribavirin for chronic hepatitis C in HIV-coinfected persons. N Engl J Med. 2004;351(5): Torriani FJ, Rodriguez-Torres M, Rockstroh JK, et al. Peginterferon alfa-2a plus ribavirin for chronic hepatitis C virus infection in HIV-infected patients. N Engl J Med. 2004;351(5): Perronne C, Carrat F, Bani-Sadr F, et al. Final results of ANRS HC02-RIBAVIC: a randomized controlled trial of pegylated-interferon-alfa-2b plus ribavirin vs. interferon-alfa-2b plus ribavirin for the initial treatment of chronic hepatitis C in HIV coinfected patients. Paper presented at: 11th Conference on Retroviruses and Opportunistic Infections; 2004; San Francisco, California. Abstract 117LB. McPhee F, Yeung K.-S, Good AC, Meanwell NA. Hepatitis C virus NS3 serine protease as a drug discovery target. Drugs of the Future (5): Sarrazin C, Rouzier R, Wagner F, Forestier N, Larrey D, Gupta SK, Hussain M, Shah A, Cutler D, Zhang J, Zeuzem S. SCH , a Novel Hepatitis C Virus Protease Inhibitor, Plus Pegylated Interferon α-2b for Genotype 1 Nonresponders. Gastroenterology. 2007; 132: Reesink HW, Zeuzem S, Weegink C, Forestier N, Van Vliet A, Van de Wetering de Rooij J, McNair L, Purdy S, Kauffman R, Alam J, Jansen PM. Rapid Decline of Viral RNA in Hepatitis C Patients Treated with VX-950: A Phase 1b, Placebo- Controlled, Randomized Study. Gastroenterology. 2006; 131: Kieffer TL, Sarrazin C, Miller JS, Welker MW, Forestier N, Kwong AD, Zeuzem S. Telaprevir and pegylated interferon-alpha-2a inhibit wild-type and resistant genotype 1 hepatitis C virus replication in patients. Hepatology Aug 6; [Epub ahead of print]. 164
166 Macdonald A, Harris M. Hepatitis C virus NS5A: tales of a promiscuous protein. J. Gen. Virol. 2004;85: Tan SL and Katze MG. How HCV counteracts in interferon response: the jury is still out on NS5A. Virology. 2001;284:1-12. Bukh J., Pietschmann. T., Lohmann V., Krieger N., Faulk K., Engle RE., Govindarajan S., Shapiro M., Claire MS., and Bartenschlager R. Mutations that permit efficient replication of HCV RNA in Huh-7 cells prevent productive replication in chimpanzees. PNAS. 2002; 99: Lindenbach BD., Meuleman P., Ploss A., Vanwolleghem T., Syder AJ., McKeating JA., Lanford RE., Feinstone SM., Major ME., Leroux-Roels G., and Rice C. Cell culture-grown HCV is infectious in vivo and can be recultured in vitro. PNAS. 2006;103: National Aids Treatment Advocacy Project. (Accessed 24-Feb, 2009). Roche, InterMune and Pharmasset Announce Initiation of INFORM-1, the First Dual- Combination Clinical Trial with Oral Antivirals in Hepatitis C. BMS Investigator Brochure, version 4, 01-Mar Bristol-Myers Squibb Research and Development; BMS Document Control No BMS Investigator Brochure, version 4, 24-Oct Bristol-Myers Squibb Research and Development; BMS Document Control No AI Final clinical study report: Placebo-controlled, single ascending dose study to evaluate the safety, tolerability and pharmacokinetics of BMS in healthy subjects. Bristol-Myers Squibb Research and Development; Document Control No
167 24 AI Final Clinical Study Report: Placebo-controlled, single ascending dose study to evaluate the safety, tolerability, pharmacokinetics, and antiviral activity of BMS in subjects chronically infected with hepatitis C virus Genotype 1. Bristol-Myers Squibb Research and Development; Document Control No AI Final Clinical Study Report: Placebo-controlled, multiple ascending dose study to evaluate the safety, tolerability, and pharmacokinetics of BMS in healthy subjects. Bristol-Myers Squibb Research and Development; Document Control No AI Final Clinical Study Report: Clinical Study Report: Double-blind, placebo-controlled, multiple ascending dose study to evaluate the antiviral activity and safety, tolerability, pharmacokinetics of BMS in subjects infected with hepatitis C virus genotype 1. Bristol-Myers Squibb Research and Development; Document Control No AI Final Clinical Study Report: Study to Evaluate the Effect of the CYP3A4 Inhibitor Ketoconazole on the Pharmacokinetics of BMS in Healthy Subjects. Bristol-Myers Squibb Research and Development; Document Control No AI Final Clinical Study Report: Pharmacokinetics and Metabolism of [14C]- labeled BMS in Healthy Male Subjects. Bristol-Myers Squibb Research and Development; Document Control No AI Final Clinical Study Report: Study to Evaluate the Effect of BMS on the Pharmacokinetics of the CYP3A4 Probe Midazolam Administered Orally in Healthy Subjects. Bristol-Myers Squibb Research and Development; Document Control No
168 30 AI Final Clinical Study Report: Bioavailability of BMS Dry Granulated Tablet and Direct Compression Tablet Relative to BMS Capsule and Effect of a High Fat Meal and Famotidine on the Pharmacokinetics of BMS in Healthy Subjects. Bristol-Myers Squibb Research and Development; Document Control No AI Final Clinical Study Report: Study to Evaluate the Effect of Rifampin on the Pharmacokinetics of BMS in Healthy Subjects. Bristol-Myers Squibb Research and Development; Document Control No AI Final Clinical Study Report: Single-Dose Pharmacokinetics of BMS in Subjects with Hepatic Impairment Compared to Healthy Subjects. Bristol-Myers Squibb Research and Development; Document Control No AI Final Clinical Study Report: The Effect of the Co-administration of BMS on the Pharmacokinetics of a Combined Oral Contraceptive Containing Ethinyl Estradiol and Norgestimate (Ortho Tri-Cyclen ) in Healthy Female Subjects. Bristol-Myers Squibb Research and Development; Document Control No AI Final Clinical Study Report: A Phase 2a Study of BMS in Combination with Peginterferon Alfa-2a (Pegasys ) and Ribavirin (Copegus ) in Treatment Naive Subjects with Chronic Hepatitis C Virus Genotype 1 Infection. Bristol-Myers Squibb Research and Development; Document Control No AI Clinical Study Report: Placebo-controlled, single ascending dose study to evaluate the safety, tolerability and pharmacokinetics of BMS in healthy subjects. Bristol-Myers Squibb Research and Development; Document Control No
169 AI Clinical Study Report: Placebo-controlled, single ascending dose study to evaluate the safety, tolerability, pharmacokinetics, and antiviral activity of BMS in subjects chronically infected with hepatitis C virus Genotype 1. Bristol- Myers Squibb Research and Development; Document Control No AI Clinical Study Report: Placebo-controlled, multiple ascending dose study to evaluate the safety, tolerability, and pharmacokinetics of BMS in healthy subjects. Bristol-Myers Squibb Research and Development; Document Control No AI Clinical Study Report: Double-blind, placebo-controlled, multiple ascending dose study to evaluate the antiviral activity and safety, tolerability, pharmacokinetics of BMS in subjects infected with hepatitis C virus genotype 1. Bristol-Myers Squibb Research and Development; Document Control No AI Clinical Study Report: Study to evaluate the effect of BMS on the pharamacokinetics of the CYP3A4 probe midazolam given orally in healthy subjects. Bristol-Myers Squibb Research and Development Document Control No AI Clinical Study Report: Bioavailability of two tablet formulations of BMS relative to capsule and effect of a high fat meal on the pharmacokinetics of BMS administered as the non-micronized tablet in healthy subjects. Bristol- Myers Squibb Research and Development; Document Control No AI Clinical Study Report: Open-label, randomized, multiple dose, drug-drug interaction study to assess the pharmacokinetics and safety of BMS and BMS co-administered in healthy subjects. Bristol-Myers Squibb Research and Development; Document Control No
170 42 AI Clinical Study Report: Pharmacokinetics and metabolism of [14C]BMS in healthy male subjects. Bristol-Myers Squibb Research and Development Document Control No Pegaysys (peginterferon alfa-2a) U.S. Package Insert. Roche Pharmaceuticals. October Copegus (ribavirin) U.S. Package Insert. Roche Pharmaceuticals. April Cockroft DW, Gault MH. Prediction of creatinine clearance from serum creatinine. Nephron, 1976; 16: U.S. Department of Agriculture, Agricultural Research Service USDA National Nutrient Database for Standard Reference, Release 18. Nutrient Data Laboratory Home Page, HCV RNA (TAQ/PCR) V2 Assay Summary. ICON Central Laboratories, Inc. March 1, Ge, D., et al. Nature 461, (2009). Suppiah V., et al. Nat Genet. advance online publication, doi: /ng.447 (13 September 2009) Tanaka, Y., et al. Nat Genet. advance online publication, doi: /ng.449 (13 September 2009) 169
171 51 Robek, M.D., Boyd, B.S., Chisari F.V. J Virol 79, (2005). 52 Morello J., Cuenca L., Soriano V., Medrano J., Madejón A., Vispo E., Barreiro P., Labarga P., JiménezNácher I., and RodríguezNóvoa S. Influence of a Single Nucleotide Polymorphism at the Main Ribavirin Transporter Gene on the Rapid Virological Response to Pegylated Interferon Ribavirin Therapy in Patients with Chronic Hepatitis C Virus Infection. J. Infect. Diseas., 2010, 202 (8),
172 APPENDIX 1 ADDITIONAL ETHICAL CONSIDERATIONS 1 INFORMED CONSENT PROCEDURES BMS will provide the investigator with an appropriate (ie, Global or Local) sample informed consent form which will include all elements required by ICH, GCP and applicable regulatory requirements. The sample informed consent form will adhere to the ethical principles that have their origin in the Declaration of Helsinki. The consent form must also include a statement that BMS and regulatory authorities have direct access to subject records. Prior to the beginning of the study, the investigator must have the IRB/IEC s written approval/favorable opinion of the written informed consent form and any other information to be provided to the subjects. The investigator must provide the subject, or, in those situations where consent cannot be given by subjects, their legally acceptable representative with a copy of the consent form and written information about the study in the language in which the subject is most proficient. The language must be non-technical and easily understood. The investigator should allow time necessary for subject or subject's legally acceptable representative to inquire about the details of the study, then informed consent must be signed and personally dated by the subject or the subject's legally acceptable representative and by the person who conducted the informed consent discussion. The subject or legally acceptable representative should receive a copy of the signed informed consent and any other written information provided to study subjects prior to subject's participation in the study. 1.1 Subjects Unable to Give Written Informed Consent Minors For minors, according to local legislation, one or both parents or a legally acceptable representative must be informed of the study procedures and must sign the informed consent form approved for the study prior to clinical study participation. (In the event that the parents or legal guardians are unable to read, then an impartial witness should be present during the entire informed consent discussion). Whenever feasible, minors who 171
173 are judged to be of an age of reason must also give their written assent by signing and dating the completed informed consent. All local laws, rules and regulations regarding informed consent of minors must be followed Subjects Experiencing Acute Events or Emergencies A legally acceptable representative or legal guardian must provide informed consent when consent of the subject is not possible prior to clinical study participation, eg, for subjects experiencing an acute medical event such as myocardial infarction or stroke. Informed consent of the subject must additionally be obtained if they become capable of making and communicating their informed consent during the clinical study. All local laws, rules and regulations regarding informed consent of adult subjects incapable of giving informed consent must be followed Mentally Impaired or Incapacitated Subjects Investigators (or whoever required by local regulations) should determine whether or not a mentally impaired or incapacitated subject is capable of giving informed consent and should sign a statement to that effect. If the subject is deemed mentally competent to give informed consent, the investigator should follow standard procedures. If the subject is deemed not to be mentally competent to give informed consent, a fully informed legal guardian or legally acceptable representative can be asked to give consent for, or on behalf of, the subject. All local laws, rules and regulations regarding informed consent of mentally impaired or incapacitated subjects must be followed. Patients who are involuntarily hospitalized because of mental illness must not be enrolled in clinical studies Other Circumstances Subjects who are imprisoned or involuntarily detained for treatment of either a psychiatric or physical (eg, infectious disease) illness must not be enrolled in clinical studies. In circumstances where a subject s only access to treatment is through enrollment in a clinical study, eg, for subjects in developing countries with limited resources or for 172
174 subjects with no marketed treatment options, the investigator must take special care to explain the potential risks and benefits associated with the study and ensure that the subject is giving informed consent. When a subject may be in a dependent relationship with the investigator, a well-informed physician who is not engaged in the clinical study and is completely independent of the relationship between the subject and investigator should obtain the subject s informed consent Illiterate Subjects If the subject, or, in those situations where consent cannot be given by the subject, their legally acceptable representative is unable to read, a reliable and independent witness should be present during the entire informed consent discussion. The choice of the witness must not breach the subject s rights to confidentiality. A reliable independent witness is defined as one not affiliated with the institution or engaged in the investigation. A family member or acquaintance is an appropriate independent witness. After the subject or legally acceptable representative orally consents and has signed, if capable, the witness should sign and personally date the consent form attesting that the information is accurate and that the subject, or, in those situations where consent cannot be given by subjects, their legally acceptable representative has fully understood the content of the informed consent agreement and is giving true informed consent. 1.2 Update of Informed Consent The informed consent and any other information provided to subjects, or, in those situations where consent cannot be given by subjects, the subject's legally acceptable representative, should be revised whenever important new information becomes available that is relevant to the subject's consent, and should receive IRB/IEC approval/favorable opinion prior to use. The investigator, or a person designated by the investigator should fully inform the subject or the subject's legally acceptable representative of all pertinent aspects of the study and of any new information relevant to the subject's willingness to continue participation in the study. This communication should be documented. 173
175 During a subject's participation in the study, any updates to the consent form and any updates to the written information will be provided to the subject. 174
176 APPENDIX 2 DIAGNOSTIC CRITERIA FOR DRUG AND ALCOHOL ABUSE The following is taken from DSM-IV: Diagnostic Criteria for Psychoactive Substance Dependence A maladaptive pattern of substance use, leading to clinically significant impairment or distress as manifested by three (or more) of the following, occurring at any time in the same 12-month period: 1) Tolerance, as defined by either of the following: a) A need for markedly increased amounts of the substance to achieve intoxication or desired effect, b) Markedly diminished effect with continued use of the same amount of the substance. 2) Withdrawal, as manifested by either of the following: a) The characteristic withdrawal syndrome for the substance, b) The same (or closely related) substance is taken to relieve or avoid withdrawal symptoms. 3) The substance is often taken in larger amounts or over a longer period than was intended. 4) There is a persistent desire or unsuccessful efforts to cut down or control substance use. 5) A great deal of time is spent in activities necessary to obtain the substance (eg visiting multiple doctors or driving long distances), use the substance (eg chain-smoking) or recover from its effects. 6) Important social, occupational or recreational activities are given up or reduced because of substance use. 7) The substance use is continued despite knowledge of having a persistent or recurring physical or psychological problem that is likely to have been caused or exacerbated by the substance (eg current cocaine use despite recognition of cocaine-induce depression, or continued drinking despite recognition that an ulcer was made worse by alcohol consumption.) 175
177 Criteria for Severity of Psychoactive Substance Dependence: Mild: Few, if any, symptoms in excess of those required to make the diagnosis, and the symptoms result in no more than mild impairment in occupational functioning or in usual social activities or relationships with others. Moderate: Symptoms or functional impairment between mild and severe. Severe: Many symptoms in excess of those required to make the diagnosis, and the symptoms markedly interfere with occupational functioning or with usual social activities or relationships with others. In Partial Remission: During the past six months, some use of the substance and some symptoms of dependence. In Full Remission: During the past six months, either no use of the substance, or use of the substance and no symptoms of dependence. Diagnostic Criteria for Psychoactive Substance Abuse A. A maladaptive pattern of psychoactive substance use, leading to clinically significant impairment or distress as manifested by one (or more) of the following, occurring at any time in the same 12-month period: 1) Recurrent substance use resulting in a failure to fulfill major role obligations at work, school, or home (eg repeated absences or poor work performance related to substance use; substance-related absences, suspensions, or expulsions from school, neglect of children or household). 2) Recurrent substance use in situations in which it is physically hazardous (eg driving an automobile or operating a machine when impaired by substance use). 3) Recurrent substance-related legal problems (eg arrests for substance-related disorderly conduct). 4) Continued substance use despite having persistent or recurrent social or interpersonal problems caused or exacerbated by the effects of the substance (eg arguments with spouse about consequences of intoxication, physical fights). B. The symptoms have never met the criteria for substance dependence for this class of substance. 176
178 APPENDIX 3 DIVISION OF AIDS TABLE FOR GRADING THE SEVERITY OF ADULT AND PEDIATRIC ADVERSE EVENTS PUBLISH DATE: DECEMBER,
179 178
180 179
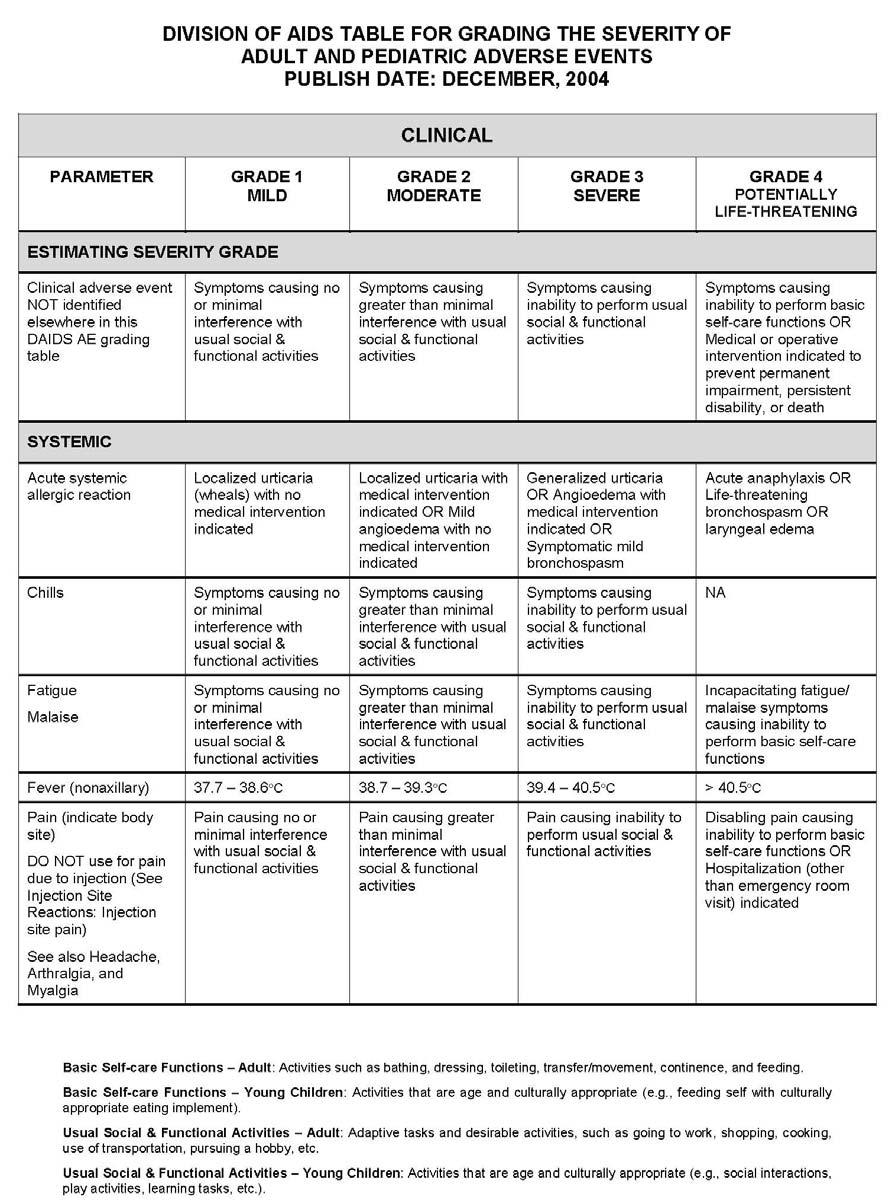
181 180
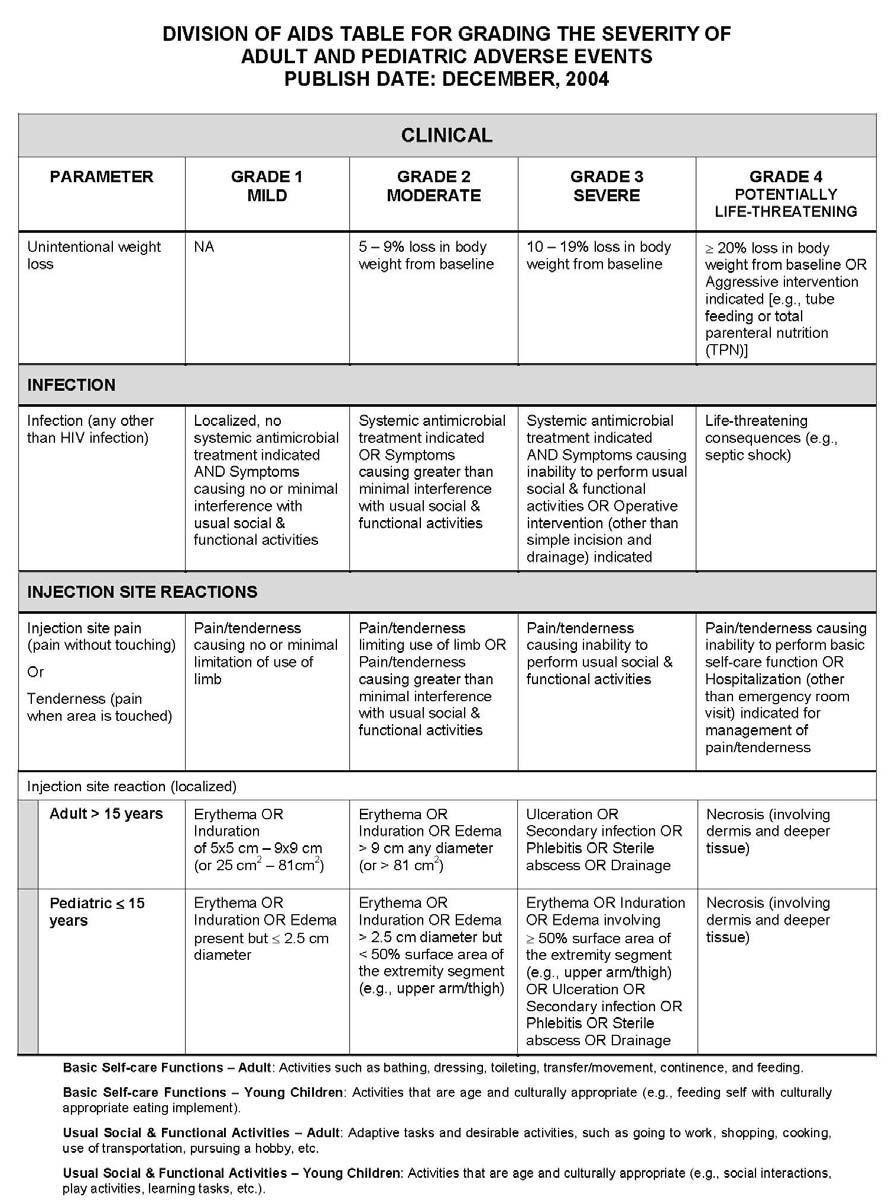
182 181
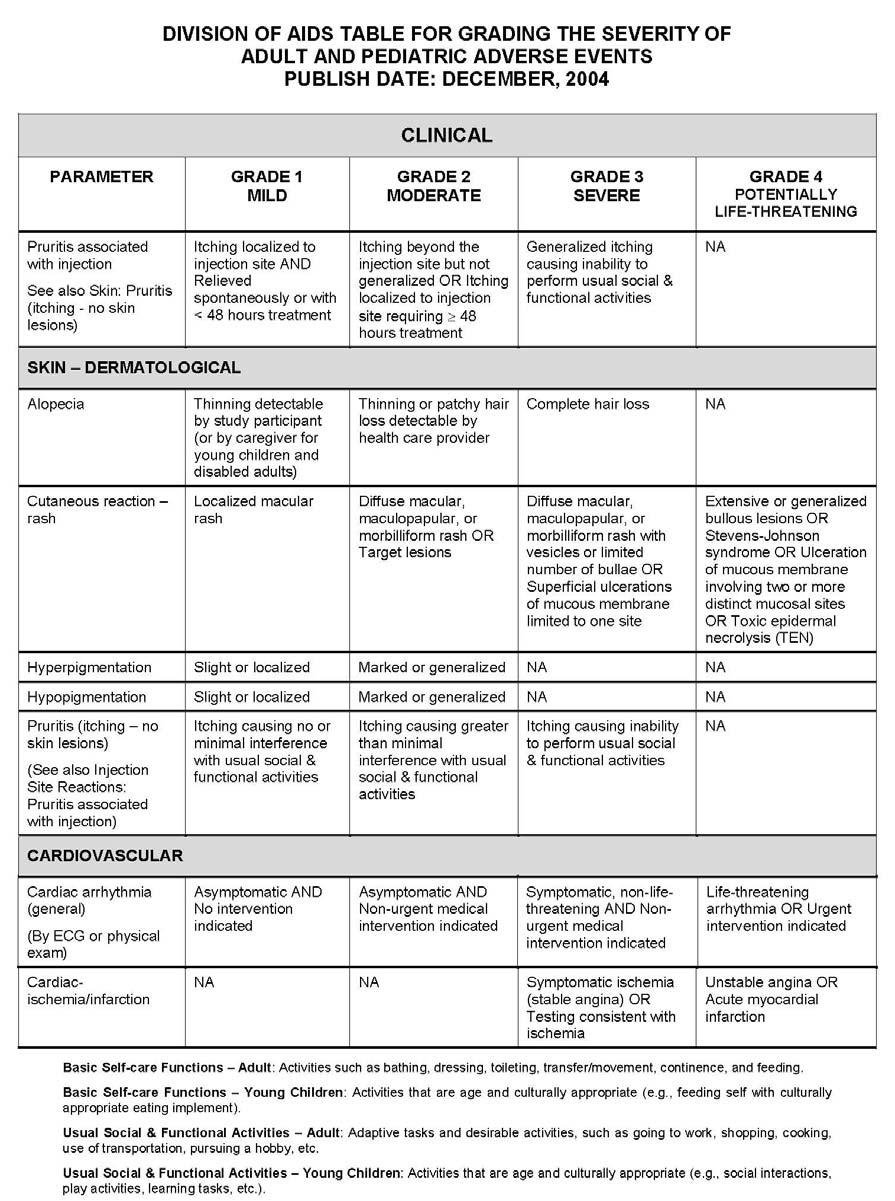
183 182
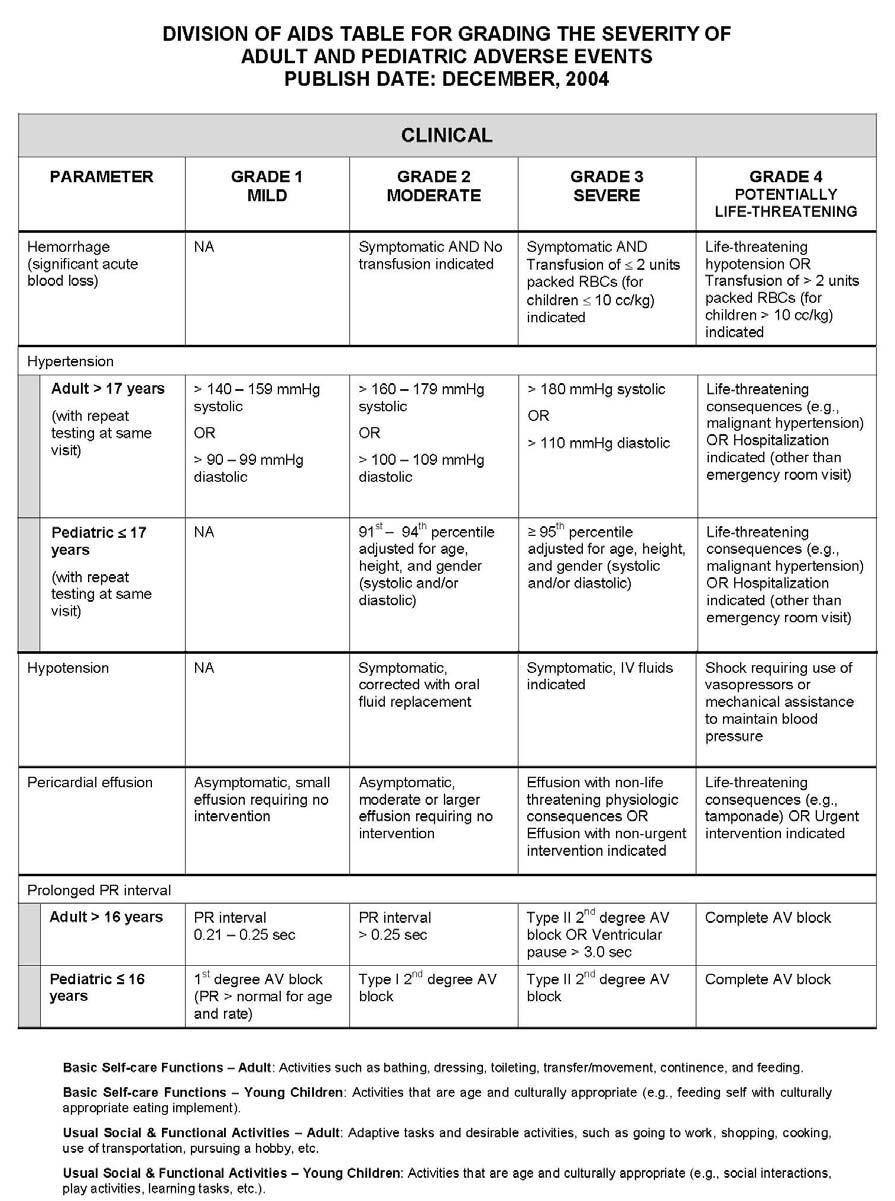
184 183
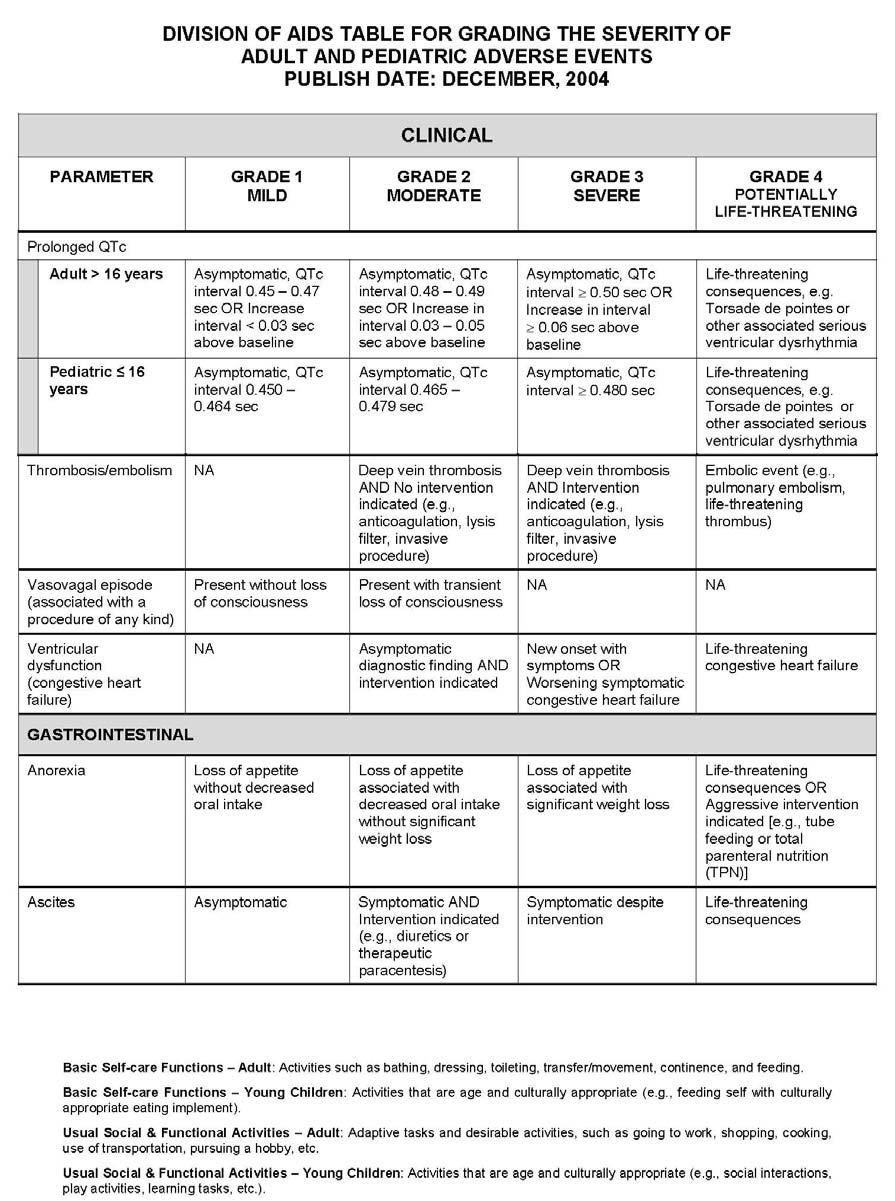
185 184
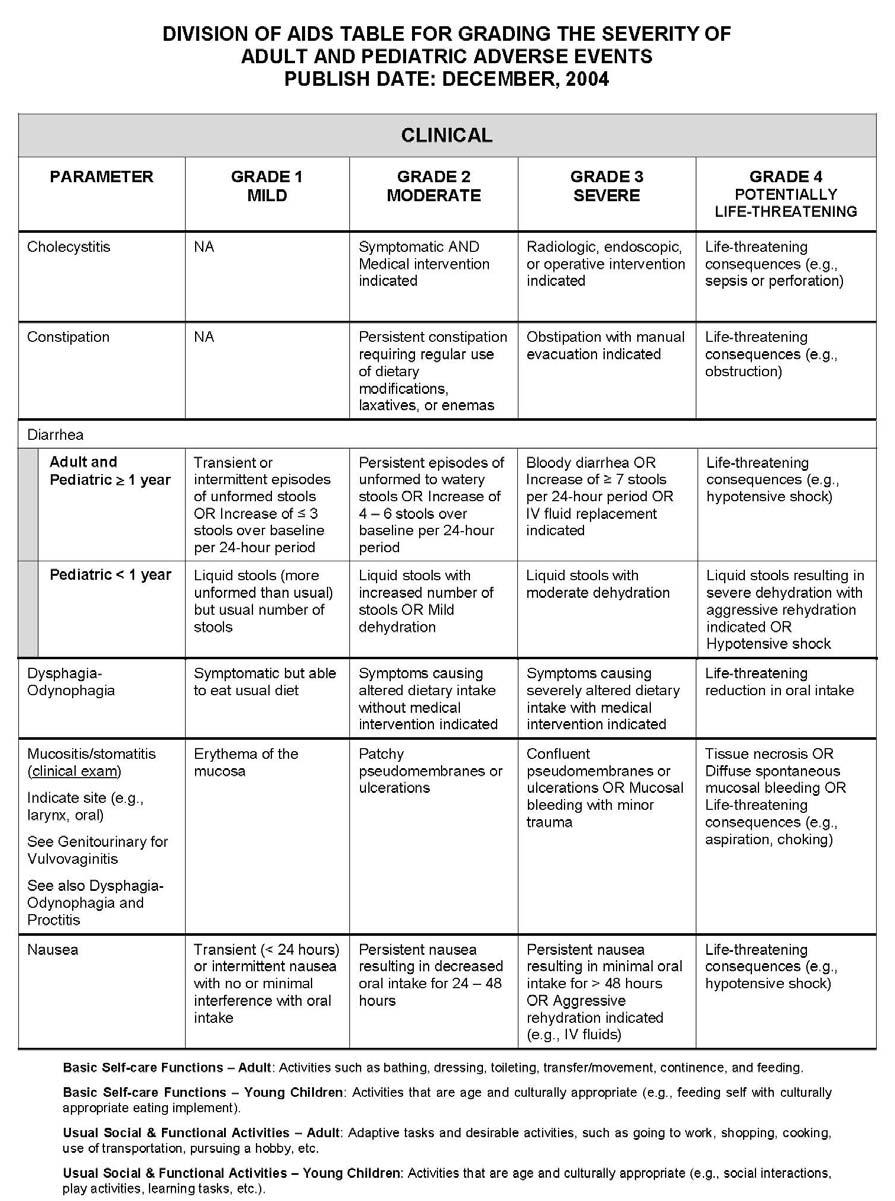
186 185
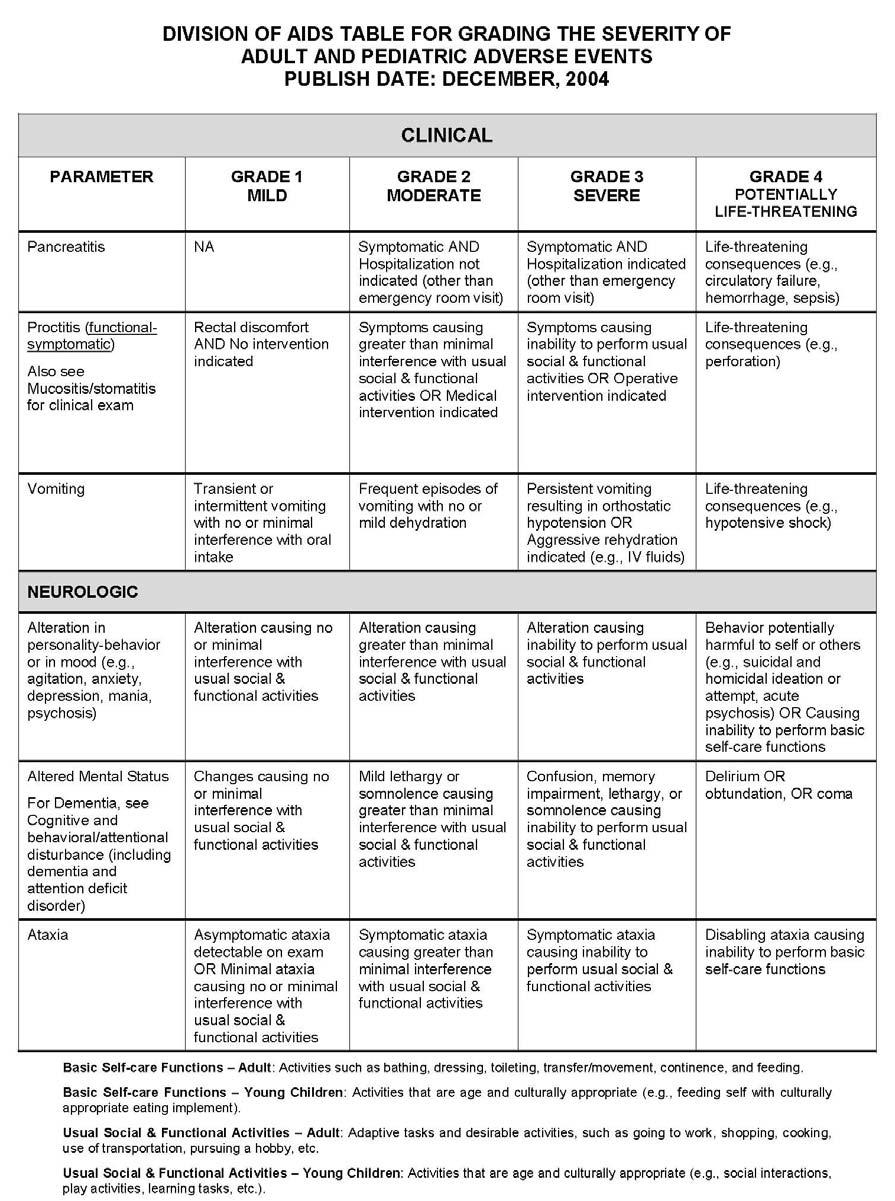
187 186
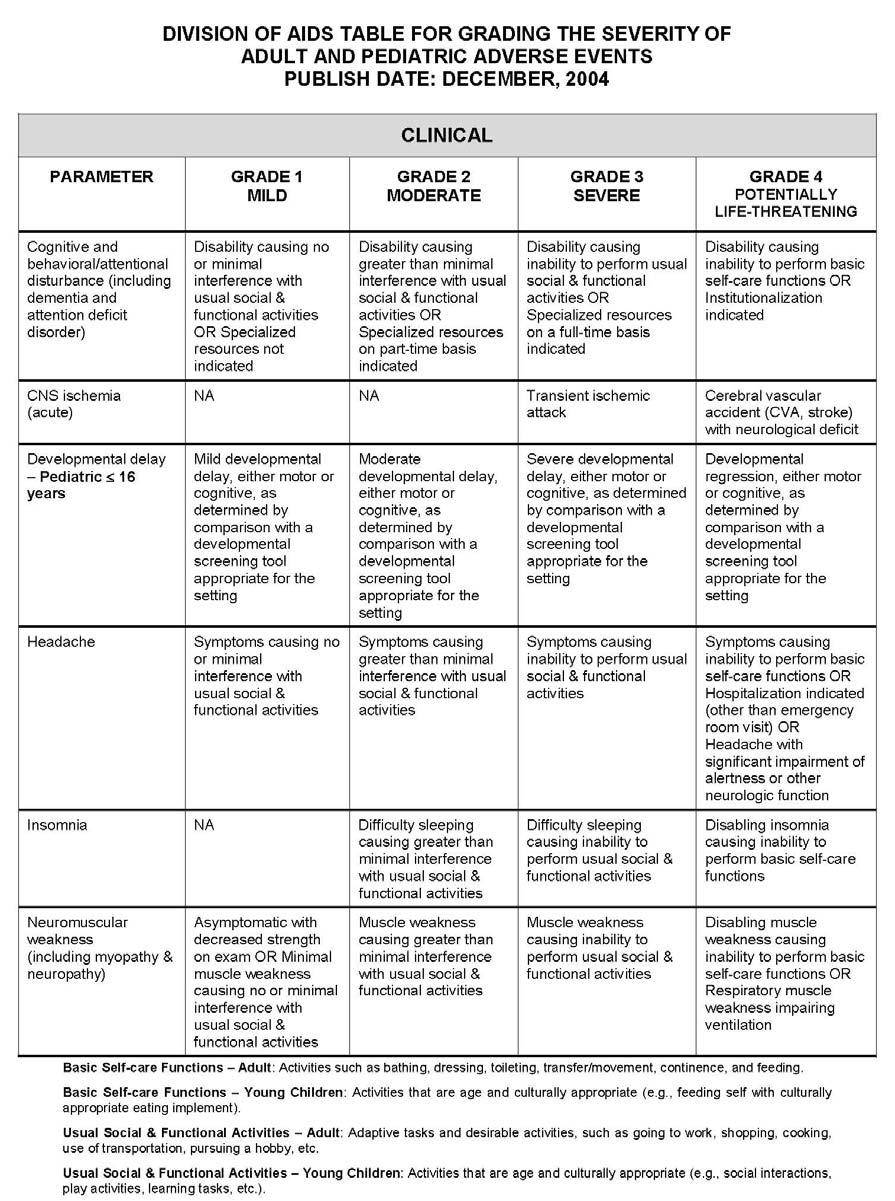
188 187
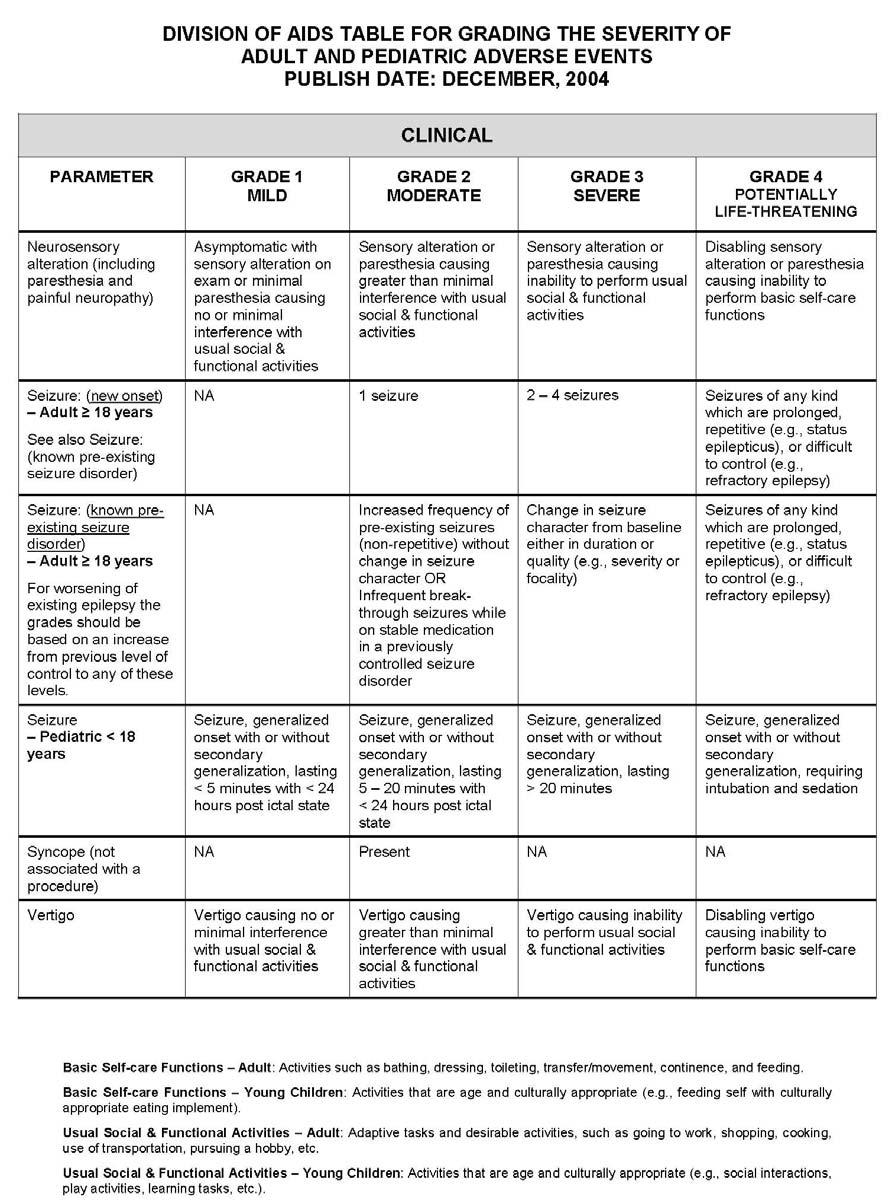
189 188
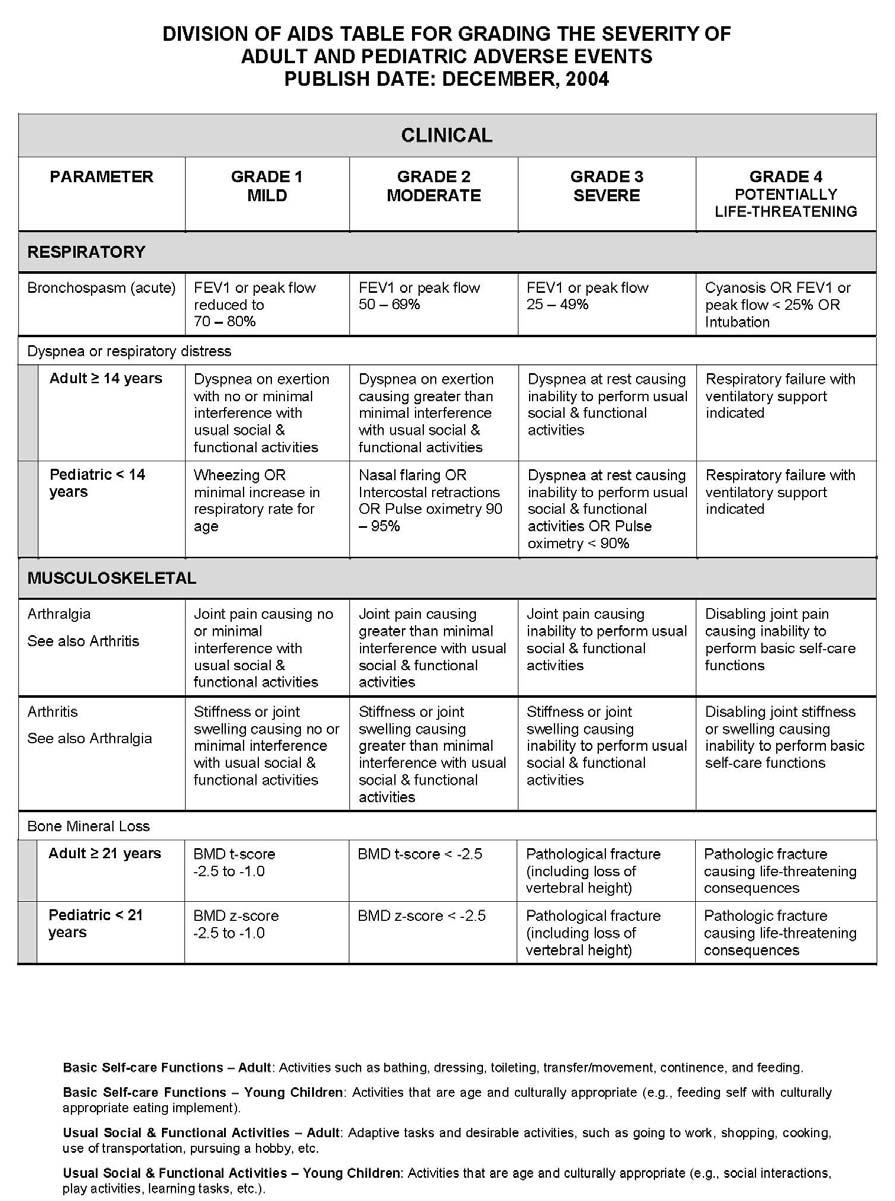
190 189
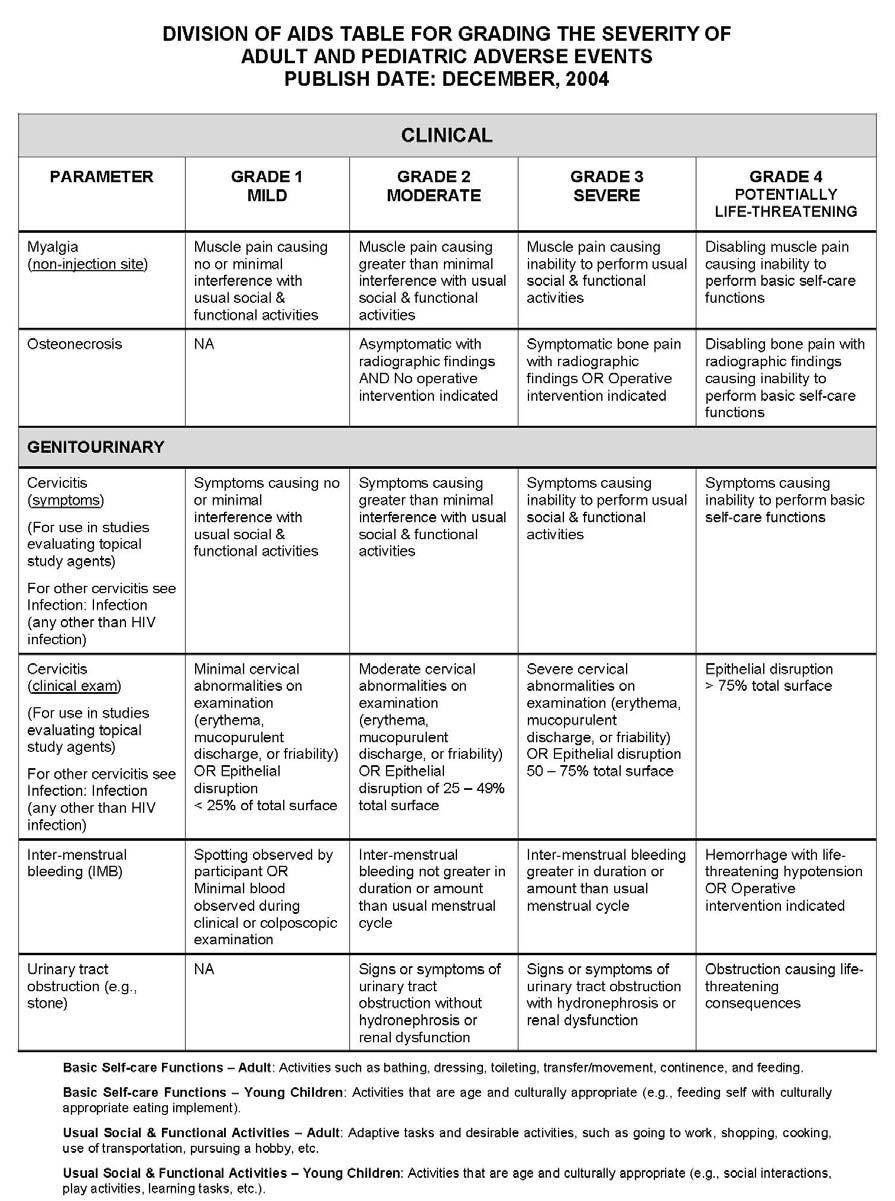
191 190
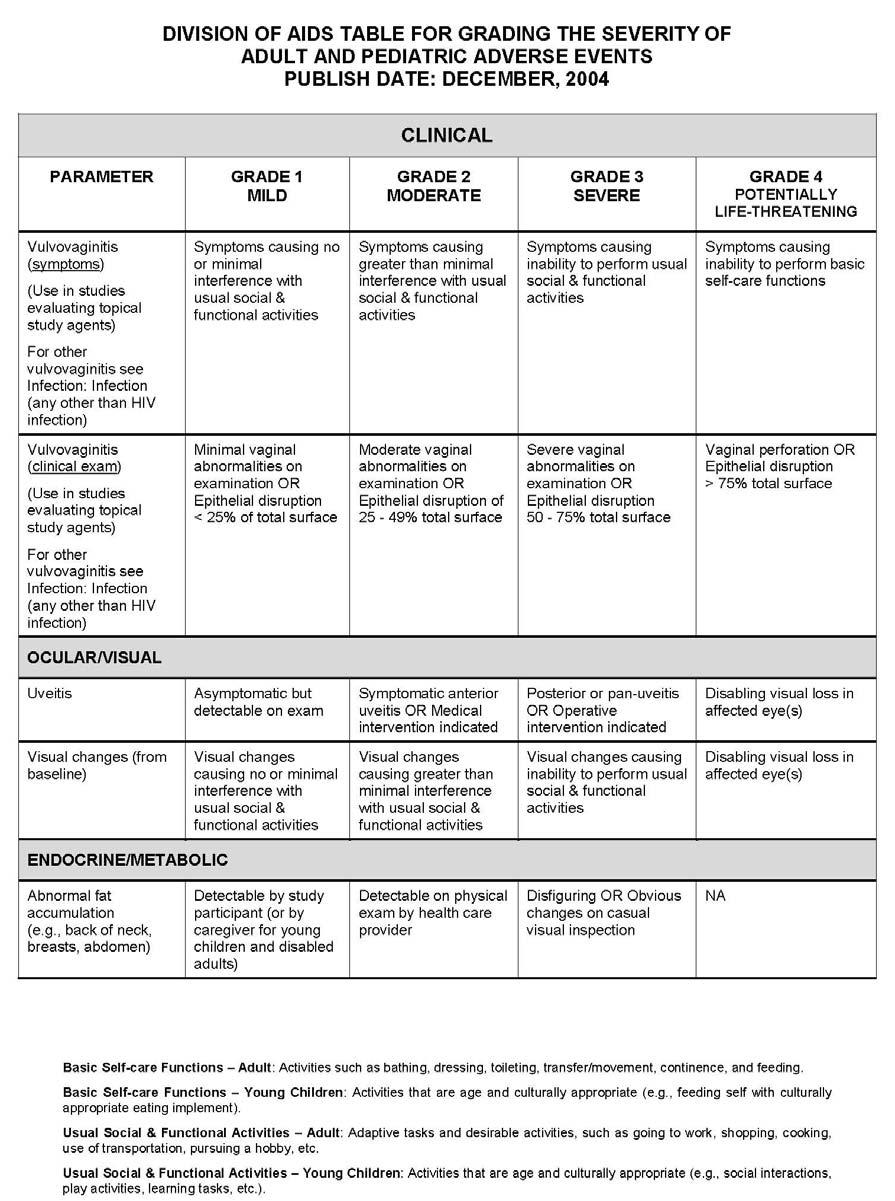
192 191
193 192
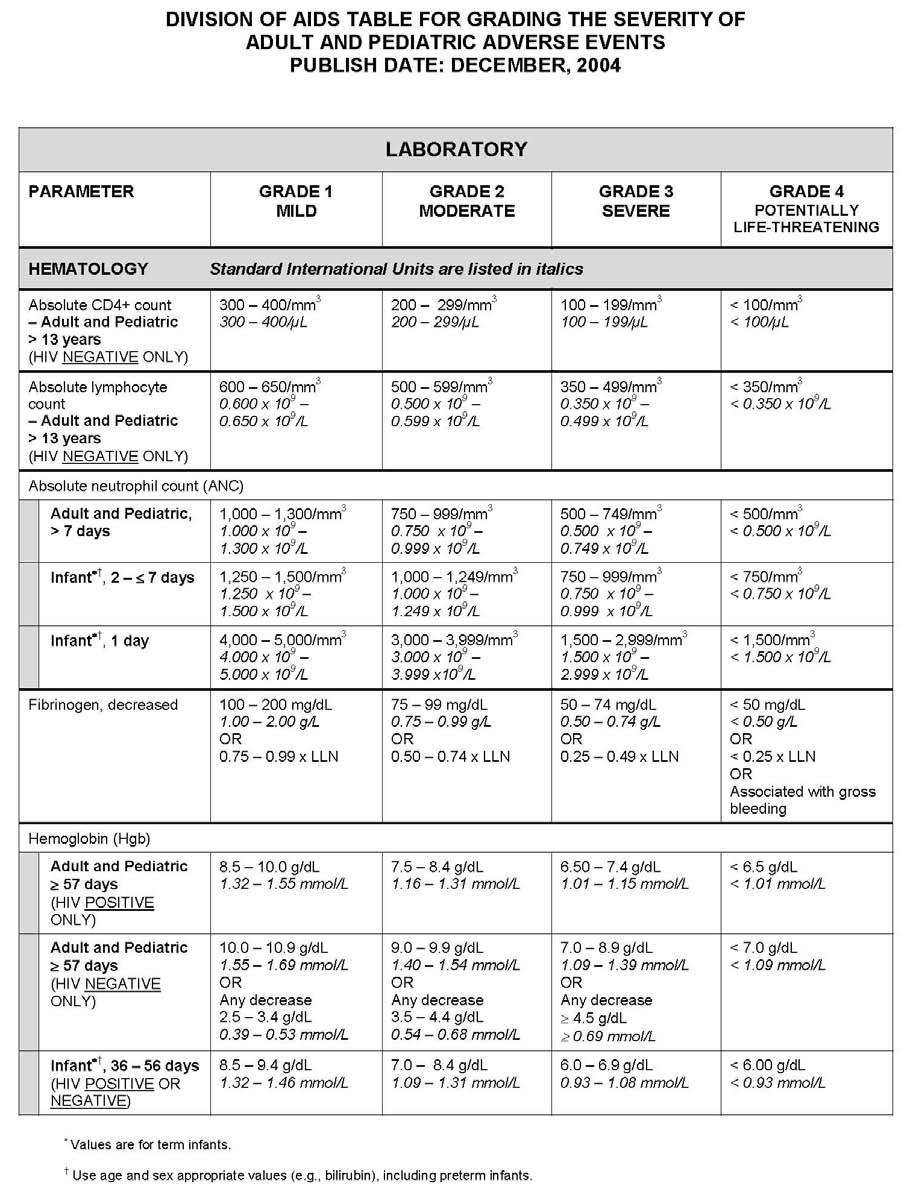
194 193
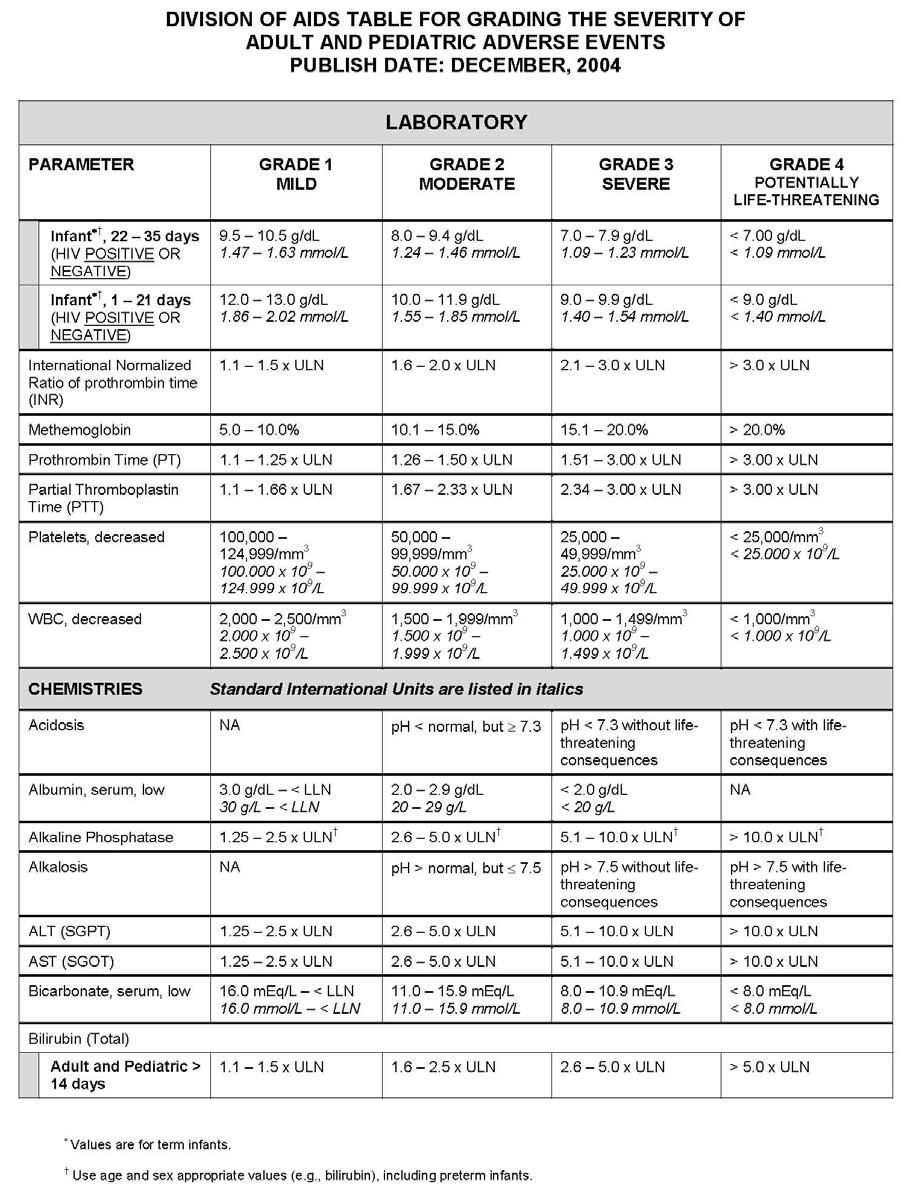
195 194
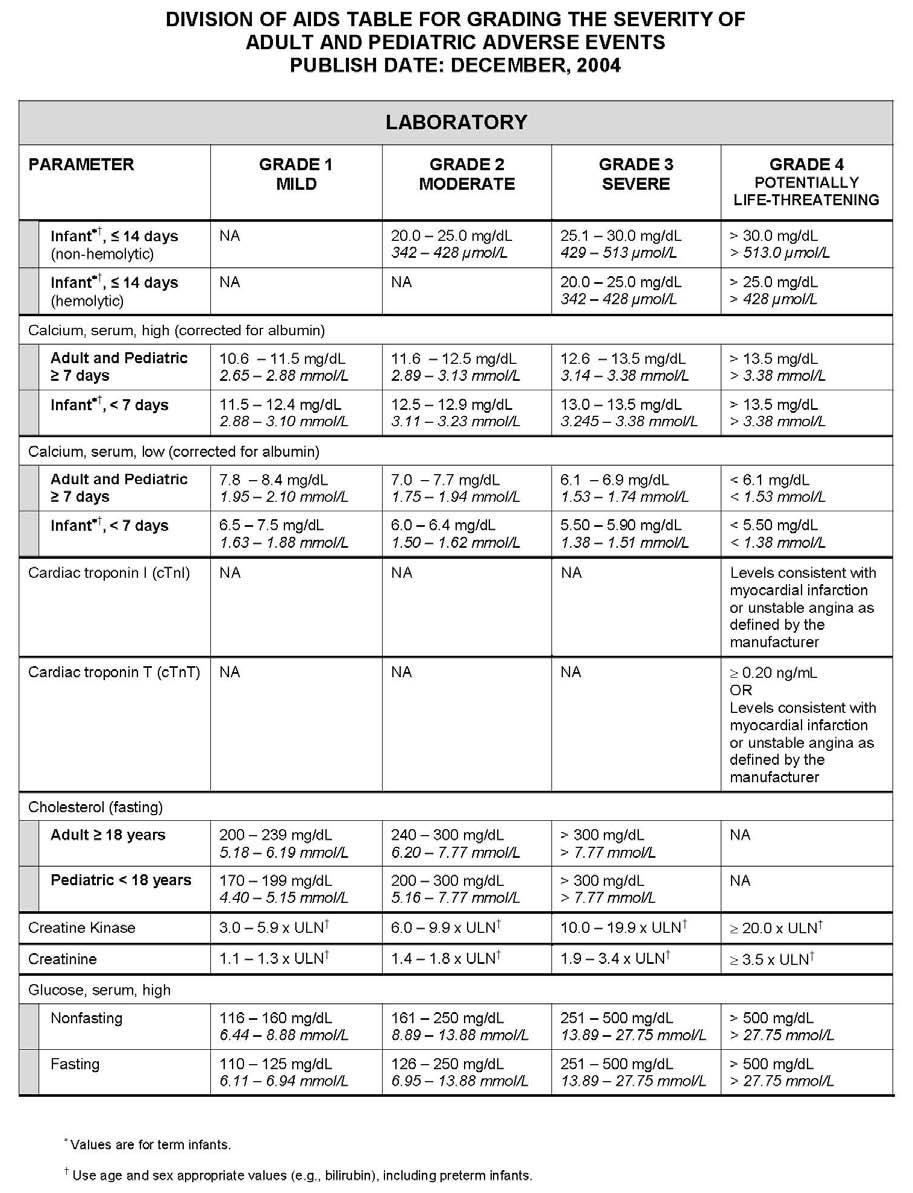
196 195
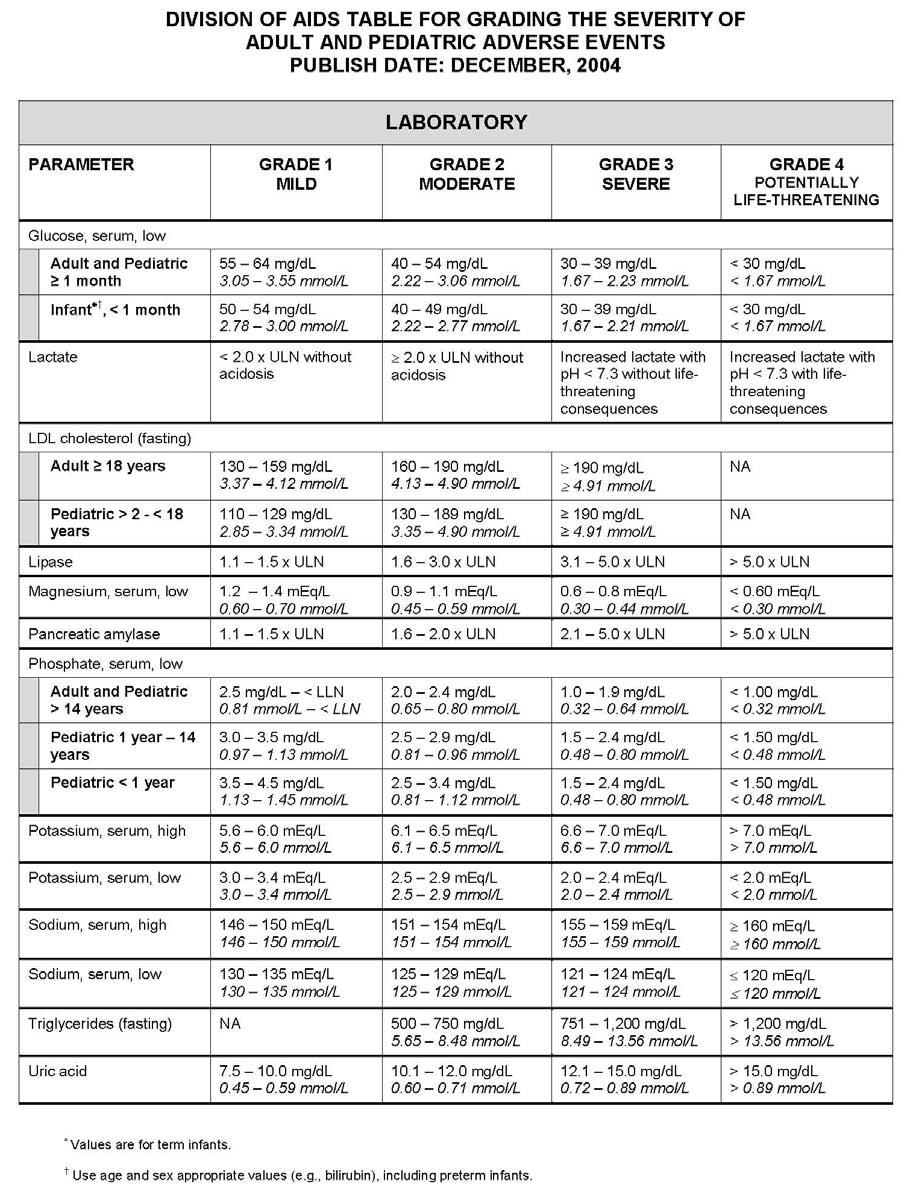
197 196
SYNOPSIS Final Clinical Study Report for Study AI444031
 Name of Sponsor/Company: Bristol-Myers Squibb Name of Finished Product: Name of Active Ingredient: () Individual Study Table Referring to the Dossier (For National Authority Use Only) SYNOPSIS for Study
Name of Sponsor/Company: Bristol-Myers Squibb Name of Finished Product: Name of Active Ingredient: () Individual Study Table Referring to the Dossier (For National Authority Use Only) SYNOPSIS for Study
Bristol-Myers Squibb. HCV Full Development Portfolio Overview. Richard Bertz Int Workshop CP HIV Meeting Amsterdam, Netherlands 24 April 2013
 Bristol-Myers Squibb HCV Full Development Portfolio Overview Richard Bertz Int Workshop CP HIV Meeting Amsterdam, Netherlands 24 April 2013 1 BMS Agents in Clinical Development: DAAs and INF Lambda Lambda
Bristol-Myers Squibb HCV Full Development Portfolio Overview Richard Bertz Int Workshop CP HIV Meeting Amsterdam, Netherlands 24 April 2013 1 BMS Agents in Clinical Development: DAAs and INF Lambda Lambda
2.0 Synopsis. ABT-450/r, ABT-267 M Clinical Study Report R&D/17/0539. (For National Authority Use Only)
") 2.0 Synopsis AbbVie Inc. Name of Study Drug: ABT-450, ritonavir, ABT-267, ribavirin, pegylated interferon Name of Active Ingredient: ABT-450, Ritonavir, ABT-267, Ribavirin, Pegylated interferon Individual
2.0 Synopsis AbbVie Inc. Name of Study Drug: ABT-450, ritonavir, ABT-267, ribavirin, pegylated interferon Name of Active Ingredient: ABT-450, Ritonavir, ABT-267, Ribavirin, Pegylated interferon Individual
ABT-493/ABT-530 M Clinical Study Report Post-Treatment Week 12 Primary Data R&D/16/0145. Referring to Part of Dossier: Volume: Page:
 2.0 Synopsis AbbVie Inc. Name of Study Drug: ABT-493/ABT-530 Name of Active Ingredient: ABT-493: (3aR,7S,10S,12R,21E,24aR)- 7-tert-butyl-N-{(1R,2R)-2- (difluoromethyl)-1-[(1- methylcyclopropane-1- sulfonyl)carbamoyl]cyclopropyl}-20,20-
2.0 Synopsis AbbVie Inc. Name of Study Drug: ABT-493/ABT-530 Name of Active Ingredient: ABT-493: (3aR,7S,10S,12R,21E,24aR)- 7-tert-butyl-N-{(1R,2R)-2- (difluoromethyl)-1-[(1- methylcyclopropane-1- sulfonyl)carbamoyl]cyclopropyl}-20,20-
This clinical study synopsis is provided in line with Boehringer Ingelheim s Policy on Transparency and Publication of Clinical Study Data.
 abcd Clinical Study for Public Disclosure This clinical study synopsis is provided in line with s Policy on Transparency and Publication of Clinical Study Data. The synopsis which is part of the clinical
abcd Clinical Study for Public Disclosure This clinical study synopsis is provided in line with s Policy on Transparency and Publication of Clinical Study Data. The synopsis which is part of the clinical
2.0 Synopsis. ABT-333 M Clinical Study Report R&D/09/956
 2.0 Synopsis Abbott Laboratories Individual Study Table Referring to Part of Dossier: (For National Authority Use Only) Name of Study Drug: ABT-333 Volume: Name of Active Ingredient: Page: Sodium N-{6-[3-tert-butyl-5-(2,4-
2.0 Synopsis Abbott Laboratories Individual Study Table Referring to Part of Dossier: (For National Authority Use Only) Name of Study Drug: ABT-333 Volume: Name of Active Ingredient: Page: Sodium N-{6-[3-tert-butyl-5-(2,4-
Introduction. The ELECTRON Trial
 63rd AASLD November 9-13, 12 Boston, Massachusetts Faculty Douglas T. Dieterich, MD Professor of Medicine and Director of CME Department of Medicine Director of Outpatient Hepatology Division of Liver
63rd AASLD November 9-13, 12 Boston, Massachusetts Faculty Douglas T. Dieterich, MD Professor of Medicine and Director of CME Department of Medicine Director of Outpatient Hepatology Division of Liver
Referring to Part of Dossier: Volume: Page:
 Synopsis AbbVie Inc. Name of Study Drug: ABT-450, ritonavir, ABT-333, ribavirin Name of Active Ingredient: ABT-450: (2R,6S,12Z,13aS,14aR,16aS)-N- (cyclopropylsulfonyl)-6-{[(5- methylpyrazin-2-yl)carbonyl]amino}-
Synopsis AbbVie Inc. Name of Study Drug: ABT-450, ritonavir, ABT-333, ribavirin Name of Active Ingredient: ABT-450: (2R,6S,12Z,13aS,14aR,16aS)-N- (cyclopropylsulfonyl)-6-{[(5- methylpyrazin-2-yl)carbonyl]amino}-
ClinicalTrials.gov Protocol Registration and Results System (PRS) Receipt Release Date: June 25, ClinicalTrials.gov ID: NCT
 Receipt Release Date: June 25, ClinicalTrials.gov ID: NCT") ClinicalTrials.gov Protocol Registration and Results System (PRS) Receipt Release Date: June 25, 2014 ClinicalTrials.gov ID: NCT00372385 Study Identification Unique Protocol ID: VX05-950-104EU Brief Title:
ClinicalTrials.gov Protocol Registration and Results System (PRS) Receipt Release Date: June 25, 2014 ClinicalTrials.gov ID: NCT00372385 Study Identification Unique Protocol ID: VX05-950-104EU Brief Title:
This clinical study synopsis is provided in line with Boehringer Ingelheim s Policy on Transparency and Publication of Clinical Study Data.
 abcd Clinical Study Synopsis for Public Disclosure This clinical study synopsis is provided in line with Boehringer Ingelheim s Policy on Transparency and Publication of Clinical Study Data. The synopsis
abcd Clinical Study Synopsis for Public Disclosure This clinical study synopsis is provided in line with Boehringer Ingelheim s Policy on Transparency and Publication of Clinical Study Data. The synopsis
Glecaprevir-Pibrentasvir in HCV GT 1 or 4 & Prior DAA Treatment MAGELLAN-1 (Part 2)
") Phase 3 Treatment-Experienced in HCV GT 1 or 4 & Prior DAA Treatment MAGELLAN-1 (Part 2) in HCV GT 1 or 4 & Prior DAA Treatment MAGELLAN-1 (Part 2): Study Features MAGELLAN-1 (Part 2) Trial Design: Randomized,
Phase 3 Treatment-Experienced in HCV GT 1 or 4 & Prior DAA Treatment MAGELLAN-1 (Part 2) in HCV GT 1 or 4 & Prior DAA Treatment MAGELLAN-1 (Part 2): Study Features MAGELLAN-1 (Part 2) Trial Design: Randomized,
Clinical Study Report AI Final 28 Feb Volume: Page:
 Study Design, Continued Electrocardiogram (ECG) and vital sign assessments were done at select times during the study. Blood and urine samples for clinical laboratory evaluations were collected at specified
Study Design, Continued Electrocardiogram (ECG) and vital sign assessments were done at select times during the study. Blood and urine samples for clinical laboratory evaluations were collected at specified
This clinical study synopsis is provided in line with Boehringer Ingelheim s Policy on Transparency and Publication of Clinical Study Data.
 abcd Clinical Study for Public Disclosure This clinical study synopsis is provided in line with s Policy on Transparency and Publication of Clinical Study Data. The synopsis which is part of the clinical
abcd Clinical Study for Public Disclosure This clinical study synopsis is provided in line with s Policy on Transparency and Publication of Clinical Study Data. The synopsis which is part of the clinical
VII CURSO AVANCES EN INFECCIÓN VIH Y HEPATITIS VIRALES
 VII CURSO AVANCES EN INFECCIÓN VIH Y HEPATITIS VIRALES REGIMENES TERAPÊUTICOS DE LA HEPATITIS C, INTERFERÓN FREE A Coruña 2 Febrero 2013 Rui Sarmento e Castro Centro Hospitalar do Porto HJU ECS Universidade
VII CURSO AVANCES EN INFECCIÓN VIH Y HEPATITIS VIRALES REGIMENES TERAPÊUTICOS DE LA HEPATITIS C, INTERFERÓN FREE A Coruña 2 Febrero 2013 Rui Sarmento e Castro Centro Hospitalar do Porto HJU ECS Universidade
Direct acting anti-virals: the near future
 Direct acting anti-virals: the near future Heiner Wedemeyer Hannover Medical School Germany Will IFN-free treatment be possible in the near future? Interferon-free regimens to treat hepatitis C What should
Direct acting anti-virals: the near future Heiner Wedemeyer Hannover Medical School Germany Will IFN-free treatment be possible in the near future? Interferon-free regimens to treat hepatitis C What should
New developments in HCV research and their implications for front-line practice
 New developments in HCV research and their implications for front-line practice Dr. Curtis Cooper Associate Professor, University of Ottawa Director, Ottawa Hospital Viral Hepatitis Program June 17, 2013
New developments in HCV research and their implications for front-line practice Dr. Curtis Cooper Associate Professor, University of Ottawa Director, Ottawa Hospital Viral Hepatitis Program June 17, 2013
Phase 3. Treatment Experienced. Ledipasvir-Sofosbuvir +/- Ribavirin in HCV Genotype 1 ION-2. Afdhal N, et al. N Engl J Med. 2014;370:
 Phase 3 Treatment Experienced Ledipasvir-Sofosbuvir +/- Ribavirin in HCV Genotype 1 ION-2 Afdhal N, et al. N Engl J Med. 2014;370:1483-93. Ledipasvir-Sofosbuvir +/- Ribavirin in Treatment-Experienced HCV
Phase 3 Treatment Experienced Ledipasvir-Sofosbuvir +/- Ribavirin in HCV Genotype 1 ION-2 Afdhal N, et al. N Engl J Med. 2014;370:1483-93. Ledipasvir-Sofosbuvir +/- Ribavirin in Treatment-Experienced HCV
ABT-493/ABT-530 M Clinical Study Report Post-Treatment Week 12 Primary Data R&D/16/0162. Referring to Part of Dossier: Volume: Page:
 2.0 Synopsis AbbVie Inc. Name of Study Drug: ABT-493/ABT-530 Name of Active Ingredient: ABT-493: (3aR,7S,10S,12R,21E,24aR)- 7-tert-butyl-N-{(1R,2R)-2- (difluoromethyl)-1-[(1- methylcyclopropane-1- sulfonyl)carbamoyl]cyclopropyl}-20,20-
2.0 Synopsis AbbVie Inc. Name of Study Drug: ABT-493/ABT-530 Name of Active Ingredient: ABT-493: (3aR,7S,10S,12R,21E,24aR)- 7-tert-butyl-N-{(1R,2R)-2- (difluoromethyl)-1-[(1- methylcyclopropane-1- sulfonyl)carbamoyl]cyclopropyl}-20,20-
ABT-493/ABT-530 M Clinical Study Report Post-Treatment Week 12 Primary Data R&D/16/0144. Referring to Part of Dossier: Volume: Page:
 2.0 Synopsis AbbVie Inc. Name of Study Drug: ABT-493/ABT-530 Name of Active Ingredient: ABT-493: (3aR,7S,10S,12R,21E,24aR)- 7-tert-butyl-N-{(1R,2R)-2- (difluoromethyl)-1-[(1- methylcyclopropane-1- sulfonyl)carbamoyl]cyclopropyl}-20,20-
2.0 Synopsis AbbVie Inc. Name of Study Drug: ABT-493/ABT-530 Name of Active Ingredient: ABT-493: (3aR,7S,10S,12R,21E,24aR)- 7-tert-butyl-N-{(1R,2R)-2- (difluoromethyl)-1-[(1- methylcyclopropane-1- sulfonyl)carbamoyl]cyclopropyl}-20,20-
STATISTICAL ANALYSIS PLAN FOR HCV DAA
 STATISTICAL ANALYSIS PLAN FOR HCV DAA A PHASE 3 EVALUATION OF A DACLATASVIR/ASUNAPREVIR/BMS-791325 FIXED DOSE COMBINATION IN SUBJECTS WITH GENOTYPE 1 CHRONIC HEPATITIS C AND COMPENSATED CIRRHOSIS PROTOCOL
STATISTICAL ANALYSIS PLAN FOR HCV DAA A PHASE 3 EVALUATION OF A DACLATASVIR/ASUNAPREVIR/BMS-791325 FIXED DOSE COMBINATION IN SUBJECTS WITH GENOTYPE 1 CHRONIC HEPATITIS C AND COMPENSATED CIRRHOSIS PROTOCOL
ABT-493, ABT-530, ABT-493/ABT-530 M Clinical Study Report Primary Analysis R&D/16/0160. Referring to Part of Dossier: Volume: Page:
 2.0 Synopsis AbbVie Inc. Name of Study Drug: ABT-493, ABT-530, ABT-493/ABT-530 Name of Active Ingredient: ABT-493: (3aR,7S,10S,12R,21E,24aR)- 7-tert-butyl-N-{(1R,2R)-2- (difluoromethyl)-1-[(1- methylcyclopropane-1-
2.0 Synopsis AbbVie Inc. Name of Study Drug: ABT-493, ABT-530, ABT-493/ABT-530 Name of Active Ingredient: ABT-493: (3aR,7S,10S,12R,21E,24aR)- 7-tert-butyl-N-{(1R,2R)-2- (difluoromethyl)-1-[(1- methylcyclopropane-1-
Treatment Options in HCV Relapsers and Nonresponders. Raymond T. Chung, M.D.
 Session IV Treatment Options in HCV Relapsers and Nonresponders Raymond T. Chung, M.D. Director of Hepatology, Massachusetts General Hospital, Associate Professor of Medicine, Harvard Medical School, Boston,
Session IV Treatment Options in HCV Relapsers and Nonresponders Raymond T. Chung, M.D. Director of Hepatology, Massachusetts General Hospital, Associate Professor of Medicine, Harvard Medical School, Boston,
Oral combination therapy: future hepatitis C virus treatment? "Lancet Oct 30;376(9751): Oral combination therapy with a nucleoside
: Oral combination therapy with a nucleoside") Author manuscript, published in "Journal of Hepatology 2011;55(4):933-5" DOI : 10.1016/j.jhep.2011.04.018 Oral combination therapy: future hepatitis C virus treatment? Commentary article on the following
Author manuscript, published in "Journal of Hepatology 2011;55(4):933-5" DOI : 10.1016/j.jhep.2011.04.018 Oral combination therapy: future hepatitis C virus treatment? Commentary article on the following
Clinical Management: Treatment of HCV Mono-infection
 Clinical Management: Treatment of HCV Mono-infection Curtis Cooper, MD, FRCPC Associate Professor-University of Ottawa The Ottawa Hospital- Infections Diseases Viral Hepatitis Program- Director Industry
Clinical Management: Treatment of HCV Mono-infection Curtis Cooper, MD, FRCPC Associate Professor-University of Ottawa The Ottawa Hospital- Infections Diseases Viral Hepatitis Program- Director Industry
Hepatitis C Treatment 2014
 Hepatitis C Treatment 214 Brendan M. McGuire, MD UAB Liver Center Outline Epidemiology/National History Terminology for Treatment Treatment Considerations Current Treatment Options Genotype 1 (GT 1) Genotype
Hepatitis C Treatment 214 Brendan M. McGuire, MD UAB Liver Center Outline Epidemiology/National History Terminology for Treatment Treatment Considerations Current Treatment Options Genotype 1 (GT 1) Genotype
Update in the Management of Hepatitis C: What Does the Future Hold
 Update in the Management of Hepatitis C: What Does the Future Hold Paul Y Kwo, MD, FACG Professor of Medicine Mdi Medical ldirector, Liver Transplantation tti Gastroenterology/Hepatology Division Indiana
Update in the Management of Hepatitis C: What Does the Future Hold Paul Y Kwo, MD, FACG Professor of Medicine Mdi Medical ldirector, Liver Transplantation tti Gastroenterology/Hepatology Division Indiana
Patients must have met all of the following inclusion criteria to be eligible for participation in this study.
 Supplementary Appendix S1: Detailed inclusion/exclusion criteria Patients must have met all of the following inclusion criteria to be eligible for participation in this study. Inclusion Criteria 1) Willing
Supplementary Appendix S1: Detailed inclusion/exclusion criteria Patients must have met all of the following inclusion criteria to be eligible for participation in this study. Inclusion Criteria 1) Willing
Glecaprevir-Pibrentasvir in Non-Cirrhotic Genotype 2 ENDURANCE-2
 Phase 3 Treatment Naïve or Experienced Glecaprevir-Pibrentasvir in Non-Cirrhotic Genotype 2 ENDURANCE-2 *ENDURANCE-2: Study Features ENDURANCE-2 Trial Design: Randomized, double-blind, placebo-controlled
Phase 3 Treatment Naïve or Experienced Glecaprevir-Pibrentasvir in Non-Cirrhotic Genotype 2 ENDURANCE-2 *ENDURANCE-2: Study Features ENDURANCE-2 Trial Design: Randomized, double-blind, placebo-controlled
Treatment of chronic hepatitis C in HIV co-infected patients
 Treatment of chronic hepatitis C in HIV co-infected patients Vicente Soriano Department of Infectious Diseases Hospital Carlos III, Madrid, Spain The most prevalent chronic viral infections in humans HBV
Treatment of chronic hepatitis C in HIV co-infected patients Vicente Soriano Department of Infectious Diseases Hospital Carlos III, Madrid, Spain The most prevalent chronic viral infections in humans HBV
Evolution of Therapy in HCV
 Hepatitis C: Update on New Therapies and AASLD 13 David Bernstein, MD, FACP, AGAF, FACP Professor of Medicine Hofstra North Shore-LIJ School of Medicine Evolution of Therapy in HCV 199 1999 1 13 (%) SVR
Hepatitis C: Update on New Therapies and AASLD 13 David Bernstein, MD, FACP, AGAF, FACP Professor of Medicine Hofstra North Shore-LIJ School of Medicine Evolution of Therapy in HCV 199 1999 1 13 (%) SVR
Strategies towards cure of HCV infection: a personalized approach. Heiner Wedemeyer Hannover Medical School Hannover, Germany
 Strategies towards cure of HCV infection: a personalized approach Heiner Wedemeyer Hannover Medical School Hannover, Germany 1 Disclosures Honoraria for consulting or speaking (last 5 years): Abbott, Biolex,
Strategies towards cure of HCV infection: a personalized approach Heiner Wedemeyer Hannover Medical School Hannover, Germany 1 Disclosures Honoraria for consulting or speaking (last 5 years): Abbott, Biolex,
The HCV pipeline: Will IFN-free treatment be possible? Heiner Wedemeyer. Hannover Medical School Germany
 : Will IFN-free treatment be possible? Heiner Wedemeyer Hannover Medical School Germany Interferon-free regimens to treat hepatitis C What should be the goal of interferon-free treatment regimens: Sustained
: Will IFN-free treatment be possible? Heiner Wedemeyer Hannover Medical School Germany Interferon-free regimens to treat hepatitis C What should be the goal of interferon-free treatment regimens: Sustained
2.0 Synopsis. Ombitasvir/Paritaprevir/Ritonavir, Dasabuvir, RBV M Clinical Study Report R&D/16/1328. (For National Authority Use Only)
") 2.0 Synopsis AbbVie Inc. Name of Study Drug: ombitasvir (OBV), paritaprevir (PTV), ritonavir (r), dasabuvir (DSV), ribavirin (RBV) Name of Active Ingredient: ombitasvir: Dimethyl ([(2S,5S)-1-(4-tert- butylphenyl)pyrrolidine-2,5-
2.0 Synopsis AbbVie Inc. Name of Study Drug: ombitasvir (OBV), paritaprevir (PTV), ritonavir (r), dasabuvir (DSV), ribavirin (RBV) Name of Active Ingredient: ombitasvir: Dimethyl ([(2S,5S)-1-(4-tert- butylphenyl)pyrrolidine-2,5-
Antiviral agents in HCV
 Antiviral agents in HCV : Upcoming Therapeutic Options Su Jong Yu, M.D., Ph.D. Department of Internal Medicine, Liver Research Institute, Seoul National University College of Medicine Estimated 170 Million
Antiviral agents in HCV : Upcoming Therapeutic Options Su Jong Yu, M.D., Ph.D. Department of Internal Medicine, Liver Research Institute, Seoul National University College of Medicine Estimated 170 Million
Express Scripts, Inc. monograph dated 5/25/2011; selected revision 6/1/2011
 BENEFIT DESCRIPTION AND LIMITATIONS OF COVERAGE ITEM: PRODUCT LINES: COVERED UNDER: DESCRIPTION: CPT/HCPCS Code: Company Supplying: Setting: Coverage Criteria: Approval Period: Victrelis (boceprevir capsules)
BENEFIT DESCRIPTION AND LIMITATIONS OF COVERAGE ITEM: PRODUCT LINES: COVERED UNDER: DESCRIPTION: CPT/HCPCS Code: Company Supplying: Setting: Coverage Criteria: Approval Period: Victrelis (boceprevir capsules)
Gane et al. BMC Infectious Diseases (2017) 17:389 DOI /s
 17:389 DOI /s") Gane et al. BMC Infectious Diseases (2017) 17:389 DOI 10.1186/s12879-017-2444-3 RESEARCH ARTICLE Simeprevir with peginterferon α-2a/ribavirin for chronic hepatitis C virus genotype 1 infection in treatment-experienced
Gane et al. BMC Infectious Diseases (2017) 17:389 DOI 10.1186/s12879-017-2444-3 RESEARCH ARTICLE Simeprevir with peginterferon α-2a/ribavirin for chronic hepatitis C virus genotype 1 infection in treatment-experienced
Management of CHC G1 patients who are relapsers or non-responders to Peg IFN and RBV therapy: Wait or Triple Therapy?
 Management of CHC G1 patients who are relapsers or non-responders to Peg IFN and RBV therapy: Wait or Triple Therapy? Prof. Teerha Piratvisuth NKC Institute of Gastroenterology and Hepatology Prince of
Management of CHC G1 patients who are relapsers or non-responders to Peg IFN and RBV therapy: Wait or Triple Therapy? Prof. Teerha Piratvisuth NKC Institute of Gastroenterology and Hepatology Prince of
Future strategies with new DAAs
 Future strategies with new DAAs Ola Weiland professor New direct antiviral drugs Case no 1 male with genotype 2b Male with gt 2b chronic HCV Male with gt 2b relapse afer peg-ifn + RBV during 24 weeks
Future strategies with new DAAs Ola Weiland professor New direct antiviral drugs Case no 1 male with genotype 2b Male with gt 2b chronic HCV Male with gt 2b relapse afer peg-ifn + RBV during 24 weeks
ASSAYS UTILZIED TO MONITOR HCV AND ITS TREATMENT
 ASSAYS UTILZIED TO MONITOR HCV AND ITS TREATMENT Mitchell L Shiffman, MD Liver Institute of Virginia Bon Secours Health System Richmond and Newport News, VA Liver Institute of Virginia Education, Research
ASSAYS UTILZIED TO MONITOR HCV AND ITS TREATMENT Mitchell L Shiffman, MD Liver Institute of Virginia Bon Secours Health System Richmond and Newport News, VA Liver Institute of Virginia Education, Research
ةي : لآا ةرقبلا ةروس
 سورة البقرة: اآلية HCV RELAPSERS AND NONRESPONDERS: How to deal with them? BY Prof. Mohamed Sharaf-Eldin Prof. of Hepatology and Gastroenterology Tanta University Achieving SVR The ability to achieve a
سورة البقرة: اآلية HCV RELAPSERS AND NONRESPONDERS: How to deal with them? BY Prof. Mohamed Sharaf-Eldin Prof. of Hepatology and Gastroenterology Tanta University Achieving SVR The ability to achieve a
Should Elderly CHC Patients (>70 years old) be Treated?
 be Treated?") Should Elderly CHC Patients (>70 years old) be Treated? Deepak Amarapurkar Consultant Gastroenterologist & Hepatologist Bombay Hospital & Medical Research Center, Mumbai & Jagjivanram Western Railway Hospital,
Should Elderly CHC Patients (>70 years old) be Treated? Deepak Amarapurkar Consultant Gastroenterologist & Hepatologist Bombay Hospital & Medical Research Center, Mumbai & Jagjivanram Western Railway Hospital,
Emerging Therapies for HCV: Highlights from AASLD 2012 (Part 2)
") Emerging Therapies for HCV: Highlights from AASLD 2012 (Part 2) Goals for Hepatitis C Therapy Compared to PegIFN α/rbv, new treatment regimens for chronic hepatitis C should offer: Improved efficacy Efficacy
Emerging Therapies for HCV: Highlights from AASLD 2012 (Part 2) Goals for Hepatitis C Therapy Compared to PegIFN α/rbv, new treatment regimens for chronic hepatitis C should offer: Improved efficacy Efficacy
PIB. Next Generation Direct-Acting Antivirals. Collectively: G/P. Pibrentasvir (formerly ABT-530) pangenotypic NS5A inhibitor
 pangenotypic NS5A inhibitor") Surveyor-II, Part 3: Efficacy and Safety of Glecaprevir/Pibrentasvir (Abt-493/Abt-53) in Patients with Hepatitis C Virus Genotype 3 Infection with Prior Treatment Experience and/or Cirrhosis David L. Wyles,
Surveyor-II, Part 3: Efficacy and Safety of Glecaprevir/Pibrentasvir (Abt-493/Abt-53) in Patients with Hepatitis C Virus Genotype 3 Infection with Prior Treatment Experience and/or Cirrhosis David L. Wyles,
Glecaprevir-Pibrentasvir in Cirrhotic Genotype 1, 2, 4, 5, and 6 EXPEDITION-1
 Phase 3 Treatment-Naïve and Treatment-Experienced Glecaprevir-Pibrentasvir in Cirrhotic Genotype 1, 2, 4, 5, and 6 EXPEDITION-1 EXPEDITION-1: Study Features EXPEDITION-1 Trial Design: Open-label, single-arm,
Phase 3 Treatment-Naïve and Treatment-Experienced Glecaprevir-Pibrentasvir in Cirrhotic Genotype 1, 2, 4, 5, and 6 EXPEDITION-1 EXPEDITION-1: Study Features EXPEDITION-1 Trial Design: Open-label, single-arm,
29th Viral Hepatitis Prevention Board Meeting
 29th Viral Hepatitis Prevention Board Meeting Madrid, November 2006 Treatment of chronic hepatitis C José M. Sánchez-Tapias Liver Unit Hospital Clínic University of Barcelona Spain CHRONIC HEPATITIS C
29th Viral Hepatitis Prevention Board Meeting Madrid, November 2006 Treatment of chronic hepatitis C José M. Sánchez-Tapias Liver Unit Hospital Clínic University of Barcelona Spain CHRONIC HEPATITIS C
Protocol. This trial protocol has been provided by the authors to give readers additional information about their work.
 Protocol This trial protocol has been provided by the authors to give readers additional information about their work. Protocol for: Wyles DL, Ruane PJ, Sulkowski MS, et al. Daclatasvir plus sofosbuvir
Protocol This trial protocol has been provided by the authors to give readers additional information about their work. Protocol for: Wyles DL, Ruane PJ, Sulkowski MS, et al. Daclatasvir plus sofosbuvir
Hepatitis C: Management of Previous Non-responders with First Line Protease Inhibitors
 Hepatitis C: Management of Previous Non-responders with First Line Protease Inhibitors Fred Poordad, MD The Texas Liver Institute Clinical Professor of Medicine University of Texas Health Science Center
Hepatitis C: Management of Previous Non-responders with First Line Protease Inhibitors Fred Poordad, MD The Texas Liver Institute Clinical Professor of Medicine University of Texas Health Science Center
Emerging Therapies for HCV: Highlights from AASLD 2012 (Part 2)
") Emerging Therapies for HCV: Highlights from AASLD 2012 (Part 2) PegIFN and RBV remain vital components of HCV therapy-- selected presentations from: Program Disclosure This activity has been planned and
Emerging Therapies for HCV: Highlights from AASLD 2012 (Part 2) PegIFN and RBV remain vital components of HCV therapy-- selected presentations from: Program Disclosure This activity has been planned and
How to optimize current therapy for GT1 patients Shortened therapy with IFNa-based therapy
 How to optimize current therapy for GT1 patients Shortened therapy with IFNa-based therapy Thomas Berg Sektion Hepatologie Klinik und Poliklinik für Gastroenterologie und Rheumatologie Leber- und Studienzentrum
How to optimize current therapy for GT1 patients Shortened therapy with IFNa-based therapy Thomas Berg Sektion Hepatologie Klinik und Poliklinik für Gastroenterologie und Rheumatologie Leber- und Studienzentrum
Emerging Approaches for the Treatment of Hepatitis C Virus
 Emerging Approaches for the Treatment of Hepatitis C Virus Gap Analysis 1 Physicians may not be sufficiently familiar with the latest guidelines for chronic HCV treatment, including the initiation and
Emerging Approaches for the Treatment of Hepatitis C Virus Gap Analysis 1 Physicians may not be sufficiently familiar with the latest guidelines for chronic HCV treatment, including the initiation and
Treatement Experienced patients without cirrhosis. Rafael Esteban Hospital Universitario Valle Hebron Barcelona
 Treatement Experienced patients without cirrhosis Rafael Esteban Hospital Universitario Valle Hebron Barcelona Agenda With IFN PegIFN+ Ribavirin + Simeprevir PegIFN+ Ribavirin+ Sofosbuvir Without IFN Sofosbuvir
Treatement Experienced patients without cirrhosis Rafael Esteban Hospital Universitario Valle Hebron Barcelona Agenda With IFN PegIFN+ Ribavirin + Simeprevir PegIFN+ Ribavirin+ Sofosbuvir Without IFN Sofosbuvir
The Liver Meeting 2013: The 64th Annual Meeting of the AASLD Washington, DC, November 1 5, 2013 Presentation LB-1
 Phase 2b Study of the Interferon-Free and Ribavirin-Free Combination of Daclatasvir, Asunaprevir, and BMS-791325 for 12 Weeks in Treatment-Naive Patients With Chronic HCV Genotype 1 Infection Everson GT,
Phase 2b Study of the Interferon-Free and Ribavirin-Free Combination of Daclatasvir, Asunaprevir, and BMS-791325 for 12 Weeks in Treatment-Naive Patients With Chronic HCV Genotype 1 Infection Everson GT,
Update on Real-World Experience With HARVONI
 Update on Real-World Experience With A RESOURCE FOR PAYERS MAY 217 This information is intended for payers only. The HCV-TARGET study was supported by Gilead Sciences, Inc. Real-world experience data were
Update on Real-World Experience With A RESOURCE FOR PAYERS MAY 217 This information is intended for payers only. The HCV-TARGET study was supported by Gilead Sciences, Inc. Real-world experience data were
Hepatitis C: Management of Treatment Naïve Patients with First Line Protease Inhibitors
 Hepatitis C: Management of Treatment Naïve Patients with First Line Protease Inhibitors Eric Lawitz, MD, AGAF, CPI The Texas Liver Institute Clinical Professor of Medicine University of Texas Health Science
Hepatitis C: Management of Treatment Naïve Patients with First Line Protease Inhibitors Eric Lawitz, MD, AGAF, CPI The Texas Liver Institute Clinical Professor of Medicine University of Texas Health Science
from 29 March 2012 Effect estimates [95% CI] Telaprevir + PegIFN/RBV vs. PegIFN/RBV
![from 29 March 2012 Effect estimates [95% CI] Telaprevir + PegIFN/RBV vs. PegIFN/RBV](/thumbs/78/77297477.jpg "from 29 March 2012 Effect estimates [95% CI] Telaprevir + PegIFN/RBV vs. PegIFN/RBV") Resolution by the Federal Joint Committee on an amendment to the Pharmaceutical Directive (AM-RL): Appendix XII Resolutions on the benefit assessment of pharmaceuticals with new active ingredients, in
Resolution by the Federal Joint Committee on an amendment to the Pharmaceutical Directive (AM-RL): Appendix XII Resolutions on the benefit assessment of pharmaceuticals with new active ingredients, in
CENTENE PHARMACY AND THERAPEUTICS DRUG REVIEW 3Q17 July August
 BRAND NAME Technivie GENERIC NAME Ombitasvir/paritaprevir/ritonavir MANUFACTURER AbbVie, Inc. DATE OF APPROVAL February 27, 2017 PRODUCT LAUNCH DATE Already available on the market REVIEW TYPE Review type
BRAND NAME Technivie GENERIC NAME Ombitasvir/paritaprevir/ritonavir MANUFACTURER AbbVie, Inc. DATE OF APPROVAL February 27, 2017 PRODUCT LAUNCH DATE Already available on the market REVIEW TYPE Review type
EASL 2013 Interferon Free, All Oral Regimens for Hepatitis C. Maria Buti Hospital Universitario Valle Hebron Barcelona Spain
 EASL 2013 Interferon Free, All Oral Regimens for Hepatitis C Maria Buti Hospital Universitario Valle Hebron Barcelona Spain The first Results with Oral therapy: a Protease Inhibitor and NS5A inhibitor
EASL 2013 Interferon Free, All Oral Regimens for Hepatitis C Maria Buti Hospital Universitario Valle Hebron Barcelona Spain The first Results with Oral therapy: a Protease Inhibitor and NS5A inhibitor
Tratamiento de la Hepatitis C Rafael Esteban Hospital General Universitario Valle de Hebrón Barcelona
 Tratamiento de la Hepatitis C Rafael Esteban Hospital General Universitario Valle de Hebrón Barcelona rrent HCV Therapy 8% % sustained response 6% 4% 2% % 54-61% 41% 34% 25% 16% 6% IFN 24w IFN 48w Peg
Tratamiento de la Hepatitis C Rafael Esteban Hospital General Universitario Valle de Hebrón Barcelona rrent HCV Therapy 8% % sustained response 6% 4% 2% % 54-61% 41% 34% 25% 16% 6% IFN 24w IFN 48w Peg
SECTION 1: OLYSIO with (PEGASYS) AND RIBAVIRIN SECTION 2: OLYSIO with (PEGINTRON) AND RIBAVIRIN RATIONALE FOR INCLUSION IN PA PROGRAM
 AND RIBAVIRIN SECTION 2: OLYSIO with (PEGINTRON) AND RIBAVIRIN RATIONALE FOR INCLUSION IN PA PROGRAM") SECTION 1: OLYSIO with (PEGASYS) AND RIBAVIRIN SECTION 2: OLYSIO with (PEGINTRON) AND RIBAVIRIN RATIONALE FOR INCLUSION IN PA PROGRAM SECTION 1: OLYSIO with (PEGASYS) AND RIBAVIRIN Background Hepatitis
SECTION 1: OLYSIO with (PEGASYS) AND RIBAVIRIN SECTION 2: OLYSIO with (PEGINTRON) AND RIBAVIRIN RATIONALE FOR INCLUSION IN PA PROGRAM SECTION 1: OLYSIO with (PEGASYS) AND RIBAVIRIN Background Hepatitis
Pharmacological management of viruses in obese patients
 Cubist Pharmaceuticals The Shape of Cures to Come Pharmacological management of viruses in obese patients Dr. Dimitar Tonev, Medical Director UKINORD 1 Disclosures } The author is a pharmaceutical physician
Cubist Pharmaceuticals The Shape of Cures to Come Pharmacological management of viruses in obese patients Dr. Dimitar Tonev, Medical Director UKINORD 1 Disclosures } The author is a pharmaceutical physician
Highlights of AASLD 2012 CCO Official Conference Coverage of the 2012 Annual Meeting of the American Association for the Study of Liver Diseases
 Highlights of AASLD 12 CCO Official Conference Coverage of the 12 Annual Meeting of the American Association for the Study of Liver Diseases November 9-13, 12 Boston, Massachusetts In partnership with
Highlights of AASLD 12 CCO Official Conference Coverage of the 12 Annual Meeting of the American Association for the Study of Liver Diseases November 9-13, 12 Boston, Massachusetts In partnership with
Individual Study Table Referring to the Dossier SYNOPSIS. Final Clinical Study Report for Study AI424136
 Name of Sponsor/Company: Bristol-Myers Squibb Name of Finished Product: Reyataz Name of Active Ingredient: Individual Study Table Referring to the Dossier (For National Authority Use Only) SYNOPSIS for
Name of Sponsor/Company: Bristol-Myers Squibb Name of Finished Product: Reyataz Name of Active Ingredient: Individual Study Table Referring to the Dossier (For National Authority Use Only) SYNOPSIS for
The role of triple therapy with protease inhibitors in hepatitis C virus genotype 1na «ve patients
 The role of triple therapy with protease inhibitors in hepatitis C virus genotype 1na «ve patients David R. Nelson Clinical and Translational Science Institute, University of Florida, FL, USA Liver International
The role of triple therapy with protease inhibitors in hepatitis C virus genotype 1na «ve patients David R. Nelson Clinical and Translational Science Institute, University of Florida, FL, USA Liver International
Topic: Sovaldi, sofosbuvir Date of Origin: March 14, Committee Approval Date: August 15, 2014 Next Review Date: March 2015
 Medication Policy Manual Policy No: dru332 Topic: Sovaldi, sofosbuvir Date of Origin: March 14, 2014 Committee Approval Date: August 15, 2014 Next Review Date: March 2015 Effective Date: October 1, 2014
Medication Policy Manual Policy No: dru332 Topic: Sovaldi, sofosbuvir Date of Origin: March 14, 2014 Committee Approval Date: August 15, 2014 Next Review Date: March 2015 Effective Date: October 1, 2014
TECHNICAL SUMMARY OF RESULTS
 TECHNICAL SUMMARY OF RESULTS 2008-004605-34 [Debio 025-HCV-205] Sponsor: Debiopharm S.A. Name of Finished Product: Tabulated Study Report (For National Authority Use Only) Name of Active Ingredient: Debio
TECHNICAL SUMMARY OF RESULTS 2008-004605-34 [Debio 025-HCV-205] Sponsor: Debiopharm S.A. Name of Finished Product: Tabulated Study Report (For National Authority Use Only) Name of Active Ingredient: Debio
Background: Narlaprevir (SCH )
") Once Daily Narlaprevir (SCH 9518) in Combination with Peginterferon alfa-2b/ Ribavirin for Treatment-Naive Patients with Genotype-1 Chronic Hepatitis C: Interim Results from the NEXT-1 Study Vierling J,
Once Daily Narlaprevir (SCH 9518) in Combination with Peginterferon alfa-2b/ Ribavirin for Treatment-Naive Patients with Genotype-1 Chronic Hepatitis C: Interim Results from the NEXT-1 Study Vierling J,
GSK Medicine: Study Number: Title: Rationale: Phase: Study Period: Study Design: Centres: Indication: Treatment: Objectives:
 The study listed may include approved and non-approved uses, formulations or treatment regimens. The results reported in any single study may not reflect the overall results obtained on studies of a product.
The study listed may include approved and non-approved uses, formulations or treatment regimens. The results reported in any single study may not reflect the overall results obtained on studies of a product.
Determinants of Response to Pegylated Interferon and Ribavirin for Acute Hepatitis C Infection in Patients with Human Immunodeficiency Virus
 Determinants of Response to Pegylated Interferon and Ribavirin for Acute Hepatitis C Infection in Patients with Human Immunodeficiency Virus Leah Burke, M.D. 1, Daniel Fierer, M.D. 2, David Cassagnol,
Determinants of Response to Pegylated Interferon and Ribavirin for Acute Hepatitis C Infection in Patients with Human Immunodeficiency Virus Leah Burke, M.D. 1, Daniel Fierer, M.D. 2, David Cassagnol,
Ledipasvir-Sofosbuvir (Harvoni)
") HEPATITIS WEB STUDY HEPATITIS C ONLINE Ledipasvir-Sofosbuvir (Harvoni) Robert G. Gish MD Professor, Consultant, Stanford University Medical Center Senior Medical Director, St Josephs Hospital and Medical
HEPATITIS WEB STUDY HEPATITIS C ONLINE Ledipasvir-Sofosbuvir (Harvoni) Robert G. Gish MD Professor, Consultant, Stanford University Medical Center Senior Medical Director, St Josephs Hospital and Medical
Simeprevir + PEG + RBV in Treatment-Naïve Genotype 1 QUEST-1 Trial
 Phase 3 Treatment Naïve Simeprevir + in Treatment-Naïve Genotype 1 QUEST-1 Trial Jacobson IM, et al. Lancet. 2014;384:403-13. Simeprevir + PEG + Ribavirin for Treatment-Naïve HCV GT1 QUEST-1 Trial QUEST-1
Phase 3 Treatment Naïve Simeprevir + in Treatment-Naïve Genotype 1 QUEST-1 Trial Jacobson IM, et al. Lancet. 2014;384:403-13. Simeprevir + PEG + Ribavirin for Treatment-Naïve HCV GT1 QUEST-1 Trial QUEST-1
Clinical Cases Hepatitis C Naïve Patients. Rafael Esteban Liver Unit. Hospital General Universitari Vall Hebron. Barcelona.
 Clinical Cases Hepatitis C Naïve Patients Rafael Esteban Liver Unit. Hospital General Universitari Vall Hebron. Barcelona. Case study 1 27 year old woman, Diagnosed with Chronic Hepatitis C 3 years ago
Clinical Cases Hepatitis C Naïve Patients Rafael Esteban Liver Unit. Hospital General Universitari Vall Hebron. Barcelona. Case study 1 27 year old woman, Diagnosed with Chronic Hepatitis C 3 years ago
COMMITTEE FOR MEDICINAL PRODUCTS FOR HUMAN USE (CHMP) DRAFT
 DRAFT") European Medicines Agency London, 24 April 2008 Doc. Ref. EMEA/CHMP/EWP/30039/2008 COMMITTEE FOR MEDICINAL PRODUCTS FOR HUMAN USE (CHMP) DRAFT GUIDELINE ON THE CLINICAL EVALUATION OF DIRECT ACTING ANTIVIRAL
European Medicines Agency London, 24 April 2008 Doc. Ref. EMEA/CHMP/EWP/30039/2008 COMMITTEE FOR MEDICINAL PRODUCTS FOR HUMAN USE (CHMP) DRAFT GUIDELINE ON THE CLINICAL EVALUATION OF DIRECT ACTING ANTIVIRAL
SYNOPSIS. Clinical Study Report AI Addendum #1. Open-label Dosing Phase
 Name of Sponsor/Company: Bristol-Myers Squibb Name of Finished Product: Individual Study Table Referring to the Dossier (For National Authority Use Only) Name of Active Ingredient: Entecavir SYNOPSIS Clinical
Name of Sponsor/Company: Bristol-Myers Squibb Name of Finished Product: Individual Study Table Referring to the Dossier (For National Authority Use Only) Name of Active Ingredient: Entecavir SYNOPSIS Clinical
ABCs of Hepatitis C: What s New. The Long-Awaited New Era: Protease Inhibitors for HCV Genotype 1
 ABCs of Hepatitis C: What s New ACG Postgraduate Course Washington, DC October 30, 2011 Ira M. Jacobson, M.D. Vincent Astor Professor of Medicine Chief, Division of Gastronterology and Hepatology Medical
ABCs of Hepatitis C: What s New ACG Postgraduate Course Washington, DC October 30, 2011 Ira M. Jacobson, M.D. Vincent Astor Professor of Medicine Chief, Division of Gastronterology and Hepatology Medical
1.0 Abstract. Title. Keywords
 1.0 Abstract Title Real World Evidence of the Effectiveness of Paritaprevir/r Ombitasvir, ± Dasabuvir, ± Ribavirin in Patients with Chronic Hepatitis C - An Observational Study in Austria (REAL) Keywords
1.0 Abstract Title Real World Evidence of the Effectiveness of Paritaprevir/r Ombitasvir, ± Dasabuvir, ± Ribavirin in Patients with Chronic Hepatitis C - An Observational Study in Austria (REAL) Keywords
TREATMENT OF GENOTYPE 2
 Treatment of Genotype 2, 3,and 4 David E. Bernstein, MD, FACG Advisory Committee/Board Member: AbbVie Pharmaceuticals, Gilead, Merck, Janssen Consultant: AbbVie Pharmaceuticals, Bristol-Myers Squibb, Gilead,
Treatment of Genotype 2, 3,and 4 David E. Bernstein, MD, FACG Advisory Committee/Board Member: AbbVie Pharmaceuticals, Gilead, Merck, Janssen Consultant: AbbVie Pharmaceuticals, Bristol-Myers Squibb, Gilead,
Selected Properties of Daclatasvir
 Selected Properties of Daclatasvir Other names Manufacturer Pharmacology / Mechanism of Action Activity Resistance Genotypic Daklinza, BMS-790052 Bristol-Myers Squibb Daclatasvir is a highly potent and
Selected Properties of Daclatasvir Other names Manufacturer Pharmacology / Mechanism of Action Activity Resistance Genotypic Daklinza, BMS-790052 Bristol-Myers Squibb Daclatasvir is a highly potent and
Final Clinical Study Report. to the Dossier SYNOPSIS. Final Clinical Study Report for Study AI463110
 BMS-475 AI463 Name of Sponsor/Company: Bristol-Myers Squibb Individual Study Table Referring to the Dossier For National Authority Use Only) Name of Finished Product: Baraclude Name of Active Ingredient:
BMS-475 AI463 Name of Sponsor/Company: Bristol-Myers Squibb Individual Study Table Referring to the Dossier For National Authority Use Only) Name of Finished Product: Baraclude Name of Active Ingredient:
Latest Treatment Updates for GT 2 and GT 3 Patients
 Latest Treatment Updates for GT 2 and GT 3 Patients Eric Lawitz, MD, AGAF, CPI Vice President, Scientific and Research Development The Texas Liver Institute Clinical Professor of Medicine University of
Latest Treatment Updates for GT 2 and GT 3 Patients Eric Lawitz, MD, AGAF, CPI Vice President, Scientific and Research Development The Texas Liver Institute Clinical Professor of Medicine University of
Handelsbanken November Bertil Samuelsson VP Discovery Research Rein Piir CFO / IR
 Handelsbanken November 18 2009 Bertil Samuelsson VP Discovery Research Rein Piir CFO / IR Pipeline Project Indication(s) Partners/- date of Explorati Optimiz agreement Terms Medivir's markets ve phase
Handelsbanken November 18 2009 Bertil Samuelsson VP Discovery Research Rein Piir CFO / IR Pipeline Project Indication(s) Partners/- date of Explorati Optimiz agreement Terms Medivir's markets ve phase
5/12/2016. Learning Objectives. Management of Hepatitis C Virus Genotype 2 or 3 Infected Treatment-Naive or Experienced Patients
 5/12/216 Management of Hepatitis C Virus Genotype 2 or 3 Infected Treatment-Naive or Experienced Patients Alexander Monto, MD Professor of Clinical Medicine University of California San Francisco San Francisco,
5/12/216 Management of Hepatitis C Virus Genotype 2 or 3 Infected Treatment-Naive or Experienced Patients Alexander Monto, MD Professor of Clinical Medicine University of California San Francisco San Francisco,
Bristol-Myers Squibb
 A Study of the Safety and Efficacy of plus Tenofovir in Adults with Chronic Hepatitis B Virus Infection with Previous Nucleoside/Nucleotide Treatment Failure () FINAL CLINICAL STUDY REPORT EUDRACT Number:
A Study of the Safety and Efficacy of plus Tenofovir in Adults with Chronic Hepatitis B Virus Infection with Previous Nucleoside/Nucleotide Treatment Failure () FINAL CLINICAL STUDY REPORT EUDRACT Number:
Interferon-based and interferon-free new treatment options
 Interferon-based and interferon-free new treatment options White Nights of Hepatology St. Petersburg, 7. June 2013 Christoph Sarrazin Klinikum der J. W. Goethe-Universität Medizinische Klinik I Frankfurt
Interferon-based and interferon-free new treatment options White Nights of Hepatology St. Petersburg, 7. June 2013 Christoph Sarrazin Klinikum der J. W. Goethe-Universität Medizinische Klinik I Frankfurt
Monitoring Patients Who Are Starting HCV Treatment, Are On Treatment, Or Have Completed Therapy
 Monitoring Patients Who Are Starting HCV Treatment, Are On Treatment, Or Have Completed Therapy WV ECHO August 10, 2017 Selection of patients for HCV treatment Despite current guidance to treat everyone,
Monitoring Patients Who Are Starting HCV Treatment, Are On Treatment, Or Have Completed Therapy WV ECHO August 10, 2017 Selection of patients for HCV treatment Despite current guidance to treat everyone,
The legally binding text is the original French version TRANSPARENCY COMMITTEE OPINION. 14 December 2011
 The legally binding text is the original French version TRANSPARENCY COMMITTEE OPINION 14 December 2011 INCIVO 375 mg, film-coated tablet B/4 bottles of 42 tablets (CIP code: 217 378-5) B/1 bottle of 42
The legally binding text is the original French version TRANSPARENCY COMMITTEE OPINION 14 December 2011 INCIVO 375 mg, film-coated tablet B/4 bottles of 42 tablets (CIP code: 217 378-5) B/1 bottle of 42
Virological Tools and Monitoring in the DAA Era
 Virological Tools and Monitoring in the DAA Era Prof. Jean-Michel Pawlotsky, MD, PhD National Reference Center for Viral Hepatitis B, C and delta Department of Virology & INSERM U955 Henri Mondor Hospital
Virological Tools and Monitoring in the DAA Era Prof. Jean-Michel Pawlotsky, MD, PhD National Reference Center for Viral Hepatitis B, C and delta Department of Virology & INSERM U955 Henri Mondor Hospital
Treatments of Genotype 2, 3,and 4: Now and in the future
 Treatments of Genotype 2, 3,and 4: Now and in the future THERAPY FOR THE TREATMENT OF GENOTYPE 2 1 GT 2 and GT 3 Treatment-Naïve: SOF+RBV vs PEG-IFN+RBV FISSION Study Design HCV GT 2 and GT 3 Treatment-naïve
Treatments of Genotype 2, 3,and 4: Now and in the future THERAPY FOR THE TREATMENT OF GENOTYPE 2 1 GT 2 and GT 3 Treatment-Naïve: SOF+RBV vs PEG-IFN+RBV FISSION Study Design HCV GT 2 and GT 3 Treatment-naïve
Abstract 233. Post-treatment follow up. Pawlotsky JM, et al. 63rd AASLD; Boston, MA; November 9-13, 2012:. Abst ALV loading* RVR at W4
 Jean-Michel Pawlotsky, Shiv K. Sarin, Graham R. Foster, Cheng-Yuan Peng, Jens Rasenack, Robert Flisiak, Teerha Piratvisuth, Heiner Wedemeyer, Wan-Long Chuang, Wei Zhang and Nikolai V. Naoumov Abstract
Jean-Michel Pawlotsky, Shiv K. Sarin, Graham R. Foster, Cheng-Yuan Peng, Jens Rasenack, Robert Flisiak, Teerha Piratvisuth, Heiner Wedemeyer, Wan-Long Chuang, Wei Zhang and Nikolai V. Naoumov Abstract
Treatment of Hepatitis C in HIV-Coinfected Patients. Vincent Soriano Department of Infectious Diseases Hospital Carlos III Madrid, Spain
 Treatment of Hepatitis C in HIV-Coinfected Patients Vincent Soriano Department of Infectious Diseases Hospital Carlos III Madrid, Spain Estimated no. of persons infected with HIV and hepatitis viruses
Treatment of Hepatitis C in HIV-Coinfected Patients Vincent Soriano Department of Infectious Diseases Hospital Carlos III Madrid, Spain Estimated no. of persons infected with HIV and hepatitis viruses
SASKATCHEWAN FORMULARY BULLETIN Update to the 62nd Edition of the Saskatchewan Formulary
 April 1, 2017 Bulletin #165 ISSN 1923-0761 SASKATCHEWAN FORMULARY BULLETIN Update to the 62nd Edition of the Saskatchewan Formulary Related Information for Prescribers: Only prescribers who have completed
April 1, 2017 Bulletin #165 ISSN 1923-0761 SASKATCHEWAN FORMULARY BULLETIN Update to the 62nd Edition of the Saskatchewan Formulary Related Information for Prescribers: Only prescribers who have completed
Update on Real-World Experience With HARVONI
 Update on Real-World Experience With A RESOURCE FOR PAYERS This information is intended for payers only. The HCV-TARGET and TRIO studies were supported by Gilead Sciences, Inc. Real-world experience data
Update on Real-World Experience With A RESOURCE FOR PAYERS This information is intended for payers only. The HCV-TARGET and TRIO studies were supported by Gilead Sciences, Inc. Real-world experience data
Olysio Pegasys Ribavirin
 Federal Employee Program 1310 G Street, N.W. Washington, D.C. 20005 202.942.1000 Fax 202.942.1125 5.01.28 Subject: Olysio Pegasys Ribavirin Page: 1 of 7 Last Review Date: March 18, 2016 Olysio Pegasys
Federal Employee Program 1310 G Street, N.W. Washington, D.C. 20005 202.942.1000 Fax 202.942.1125 5.01.28 Subject: Olysio Pegasys Ribavirin Page: 1 of 7 Last Review Date: March 18, 2016 Olysio Pegasys
Clinical Study Synopsis
 Clinical Study Synopsis This Clinical Study Synopsis is provided for patients and healthcare professionals to increase the transparency of Bayer's clinical research. This document is not intended to replace
Clinical Study Synopsis This Clinical Study Synopsis is provided for patients and healthcare professionals to increase the transparency of Bayer's clinical research. This document is not intended to replace
Olysio PegIntron Ribavirin
 Federal Employee Program 1310 G Street, N.W. Washington, D.C. 20005 202.942.1000 Fax 202.942.1125 5.01.27 Subject: Olysio PegIntron Ribavirin Page: 1 of 7 Last Review Date: March 18, 2016 Olysio PegIntron
Federal Employee Program 1310 G Street, N.W. Washington, D.C. 20005 202.942.1000 Fax 202.942.1125 5.01.27 Subject: Olysio PegIntron Ribavirin Page: 1 of 7 Last Review Date: March 18, 2016 Olysio PegIntron
Ribavirin (Medicare Prior Authorization)
") Prior Authorization Form ARKANSAS BLUE CROSS AND BLUE SHIELD Medi-Pak Rx (PDP), Medi-Pak Advantage (PFFS), and Medi-Pak Advantage PPO Ribavirin (Medicare Prior Authorization) This fax machine is located
Prior Authorization Form ARKANSAS BLUE CROSS AND BLUE SHIELD Medi-Pak Rx (PDP), Medi-Pak Advantage (PFFS), and Medi-Pak Advantage PPO Ribavirin (Medicare Prior Authorization) This fax machine is located
Clinical Study Synopsis
 Clinical Study Synopsis This Clinical Study Synopsis is provided for patients and healthcare professionals to increase the transparency of Bayer's clinical research. This document is not intended to replace
Clinical Study Synopsis This Clinical Study Synopsis is provided for patients and healthcare professionals to increase the transparency of Bayer's clinical research. This document is not intended to replace
Olysio Pegasys Ribavirin
 Federal Employee Program 1310 G Street, N.W. Washington, D.C. 20005 202.942.1000 Fax 202.942.1125 5.01.28 Subject: Olysio Pegasys Ribavirin Page: 1 of 8 Last Review Date: December 18, 2017 Olysio Pegasys
Federal Employee Program 1310 G Street, N.W. Washington, D.C. 20005 202.942.1000 Fax 202.942.1125 5.01.28 Subject: Olysio Pegasys Ribavirin Page: 1 of 8 Last Review Date: December 18, 2017 Olysio Pegasys
New Therapies on the Horizon in Hepatitis C Patients Paul Y. Kwo, MD
 Viral Targets for HCV New Therapies on the Horizon in Hepatitis C Patients Paul Y. Kwo, MD Sites for development of inhibitors Metalloproteinase Serine protease (trans) Core E E2 NS2 NS3 NS4a/NS4b NS5a/NS5b
Viral Targets for HCV New Therapies on the Horizon in Hepatitis C Patients Paul Y. Kwo, MD Sites for development of inhibitors Metalloproteinase Serine protease (trans) Core E E2 NS2 NS3 NS4a/NS4b NS5a/NS5b
Interferon Side Effects and The Future of Interferon Sparing Regimens. Todd Wills, MD ETAC Infectious Disease Specialist
 Interferon Side Effects and The Future of Interferon Sparing Regimens Todd Wills, MD ETAC Infectious Disease Specialist HEPATITIS C TREATMENT EXPANSION INITIATIVE MULTISITE CONFERENCE CALL FEBRUARY 15,
Interferon Side Effects and The Future of Interferon Sparing Regimens Todd Wills, MD ETAC Infectious Disease Specialist HEPATITIS C TREATMENT EXPANSION INITIATIVE MULTISITE CONFERENCE CALL FEBRUARY 15,
Experience with pre-transplant antiviral treatment: PEG/RBV and DAA. Xavier Forns, MD Liver Unit Hospital Clínic IDIBAPS and CIBREHD Barcelona
 Experience with pre-transplant antiviral treatment: PEG/RBV and DAA Xavier Forns, MD Liver Unit Hospital Clínic IDIBAPS and CIBREHD Barcelona Interferon-free regimens G1b nulls Asunaprevir (PI) + Daclatasvir
Experience with pre-transplant antiviral treatment: PEG/RBV and DAA Xavier Forns, MD Liver Unit Hospital Clínic IDIBAPS and CIBREHD Barcelona Interferon-free regimens G1b nulls Asunaprevir (PI) + Daclatasvir