Peak-calling for ChIP-seq and ATAC-seq
|
|
|
- Peregrine Atkins
- 6 years ago
- Views:
Transcription


1 Peak-calling for ChIP-seq and ATAC-seq Shamith Samarajiwa CRUK Autumn School in Bioinformatics 2017 University of Cambridge
2 Overview Peak-calling: identify enriched (signal) regions in ChIP-seq or ATAC-seq data Software packages Practical and Statistical aspects (Normalization, IDR, QC measures) MACS peak calling Overview of transcription factor, DNA binding protein, histone mark and nucleosome free region peaks Narrow and Broad peaks A brief look at the MACS2 settings and methodology ATAC-seq signal detection
3 Signal to Noise modified from Carl Herrmann

4 Strand dependent bimodality Wilbanks et al PLOS One
5 Peak Calling Software Comprehensive list is at: MACS2 (MACS1.4) Most widely used peak caller. Can detect narrow and broad peaks. Epic (SICER) Specialised for broad peaks BayesPeak R/Bioconductor Jmosaics Detects enriched regions jointly from replicates T-PIC Shape based EDD Detects megabase domain enrichment GEM Peak calling and motif discovery for ChIP-seq and ChIP-exo SPP Fragment length computation and saturation analysis to determine if read depth is adequate.
 is a measure of library complexity N1= Non redundant, uniquely mapping reads N2= Uniquely mapping")
6 Quality Measures Fraction of reads in peaks (FRiP) is dependant on data type. FRiP can be calculated with deeptools2 PCR Bottleneck Coefficient (PBC) is a measure of library complexity N1= Non redundant, uniquely mapping reads N2= Uniquely mapping reads Preseq and preseqr for determining library complexity Daley et al., 2013, Nat. Methods
7 Quality Measures Relative strand cross-correlation The RSC is the ratio of the fragment-length cross-correlation value minus the background cross-correlation value, divided by the phantom-peak cross-correlation value minus the background cross-correlation value. The minimum possible value is 0 (no signal), highly enriched experiments have values greater than 1, and values much less than 1 may indicate low quality.
8 Quality Measures Normalised strand cross-correlation NSC The ratio of the maximal cross-correlation value (which occurs at strand shift equal to fragment length) divided by the background cross-correlation (minimum cross-correlation value over all possible strand shifts). Higher values indicate more enrichment, values > 1.1 are relatively low NSC scores, and the minimum possible value is 1 (no enrichment). This score is sensitive to technical effects; for example, high-quality antibodies such as H3K4me3 and CTCF score well for all cell types. This score is also sensitive to biological effects; narrow marks score higher than broad marks (H3K4me3 vs H3K36me3, H3K27me3) for all cell types and ENCODE production groups, and features present in some individual cells, but not others, in a population are expected to have lower scores. A measure of enrichment derived without dependence on prior determination of enriched regions.
9 IDR: Irreproducible Discovery Rate If two replicates measure the same underlying biology, the most significant peaks which are likely to be genuine signals, are expected to have high consistency between replicates. Peaks with low significance, which are more likely to be noise, are expected to have low consistency. IDR measures consistency between replicates in high-throughput experiments. The IDR method compare a pair of ranked lists of identifications (such as ChIP-seq peaks). These ranked lists should not be pre-thresholded, i.e they should provide identifications across the entire spectrum of high confidence/enrichment (signal) and low confidence/enrichment (noise). The method uses reproducibility in score rankings between peaks in each replicate to determine an optimal cutoff for significance. The IDR method then fits the bivariate rank distributions over the replicates in order to separate signal from noise based on a defined confidence of rank consistency and reproducibility of identifications. software:
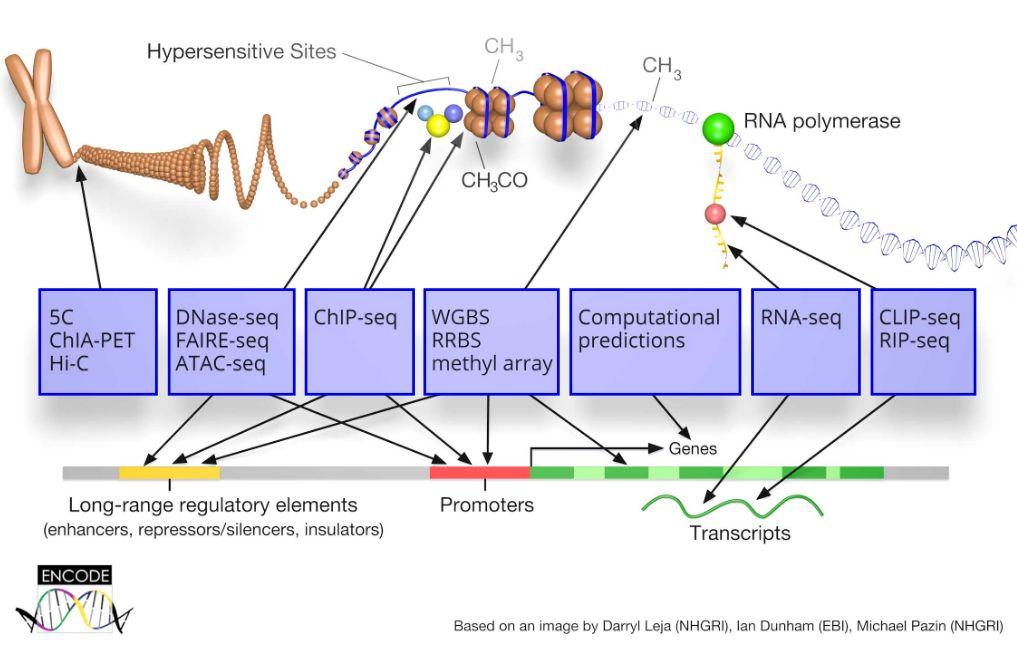
10 ENCODE
11

12 Encode Quality Metrics
13 MACS2 Most widely used peak caller. identifies genome-wide locations of TF binding, histone modification or NFRs from ChIP-seq or ATAC-seq data. Can be used without a control (Input -samples of sonicated chromatin OR IgG - nonspecific antibody) - Not Recommended for ChIP-seq! Controls eliminate bias due to GC content, mappability, DNA repeats or CNVs. Can call narrow and broad peaks. Many settings for optimizing results.
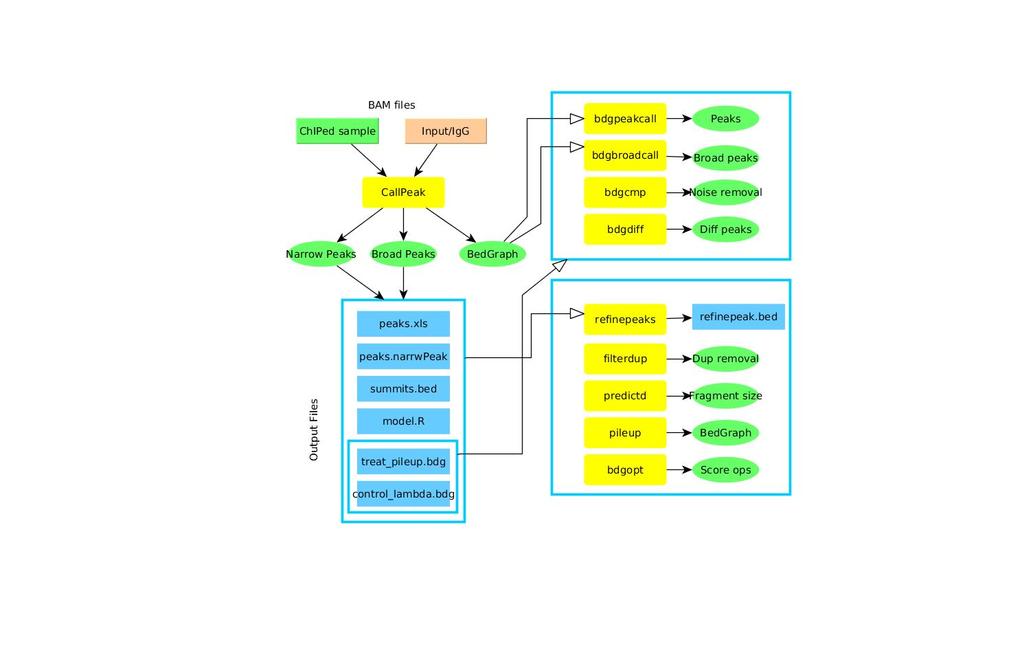
14 MACS2
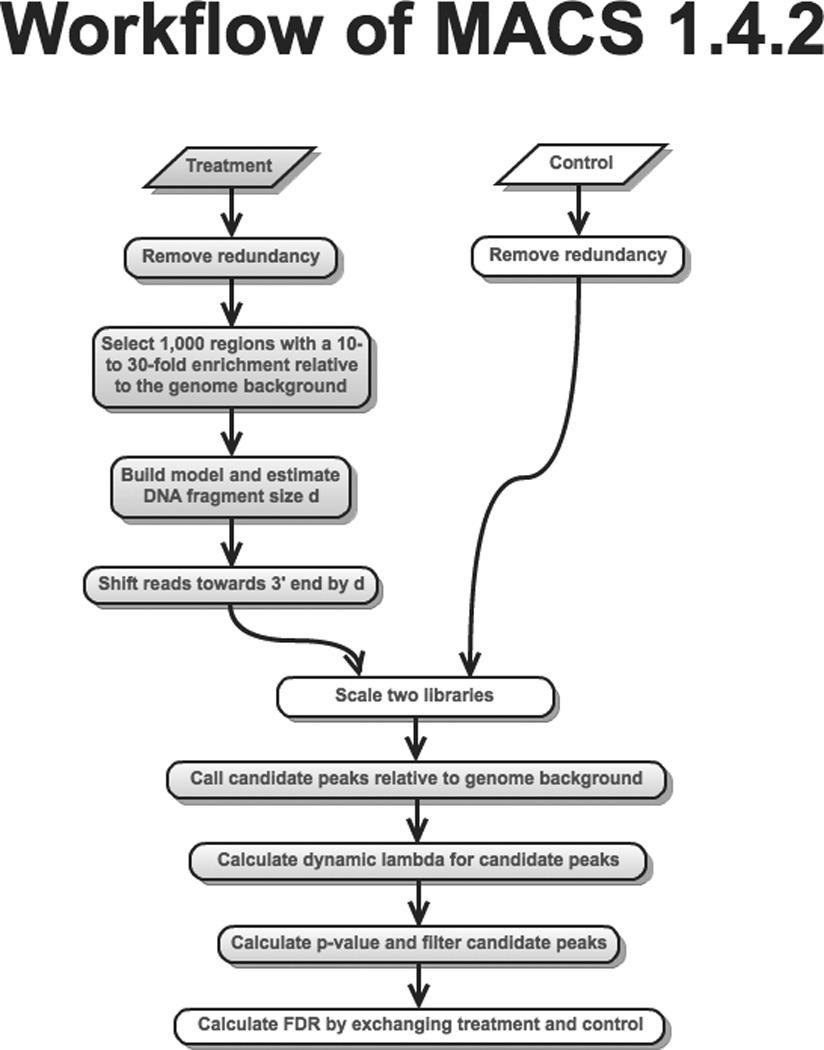
15 MACS1.4

")
16 Peak calling with MACS2 Step 1: Estimate fragment length d and adjust read position Slide a window of length 2 x bw bandwidth (half of estimated sonication size) across genome. Retain windows with > MFOLD ( fold-enrichment of treatment / background) Compute the average +/- strand specific read-densities for these bins.
17 MACS2 Step 2: Identify local noise slide a window of size 2*d across treatment and input estimate local parameter of Poisson distribution modified from Carl Herrmann
18 MACS2 Step 3: Identify enriched (peak) regions determine regions with p-value < PVALUE determine summit position within enriched regions as max density modified from Carl Herrmann
 modified from Carl")
19 MACS2 Step 4: Estimate FDR positive peaks (P-values) swap treatment and input, call negative peaks (P-value) modified from Carl Herrmann
20 Reads to Peaks +ive and -ive strand reads do not represent true binding sites Fragment length d can be detected experimentally or estimated from strand asymmetry in data Reads from both strands can be extended to the length of d OR Reads can be shifted towards 3 by d/2
21 Narrow, Broad and Mixed Peaks Different data types have different peak shapes. Use appropriate peak callers or domain detectors. Same TF may have different peak shapes reflecting differences in biological conditions. Replicates should have similar binding patterns. Most TF peaks are narrow, with particularly sharp peaks from ChIP-exo data. ChIP-seq peaks from epigenomic data can be narrow, broad or gapped. Histone marks such as H3K9me3 or H3K27me3 are broad while others such as H3K4me3 and proteins such as CTCF are narrow. Other DNA binding proteins such as HP1, Lamins (Lamin A or B), HMGA etc. form broad peaks or domains. PolII peaks can be narrow or broad depending on whether its detecting transcription initiation at the TSS or propagation along the gene body. ATAC-seq data representing nucleosome free regions (NFRs) can be narrow or broad depending on the properties of regulatory regions underlying them.

22 Peak Shapes Sims et al., 2014 Nat Rev Genet.
23 Broad peak and Domain callers MACS2: macs2 callpeak --broad Epic: Useful for finding medium or diffusely enriched domains in chip-seq data. Epic is an improvement over the original SICER, by being faster, more memory efficient, multi core, and significantly easier to install and use. Others: Enriched Domain Detector (EDD), RSEG, BroadPeak, PeakRanger (CCAT)

24 ATAC-seq settings If using paired end reads use --format BAMPE to let MACS2 pileup the whole fragments in general. If you want to focus on looking for where the 'cutting sites' are, then --nomodel --shift extsize 200 should work. Since the DNA wrapped on a nucleosome is about 147bp, for single nucleosome detection use --nomodel --shift extsize 73. BASED ON EPIGENETICS CHROMATIN, 7:33, Scientist, Volume 30 Issue 1 January 2016
25 References Landt et al., ChIP-seq guidelines and practices of the ENCODE and modencode consortia. Genome Res. 2012, 22: PMID: Wilbanks et al., Evaluation of algorithm performance in ChIP-seq peak detection. PLoS One. 2010, Jul 8;5(7):e PMID: Nakato et al., Recent advances in ChIP-seq analysis: from quality management to whole-genome annotation. Brief Bioinform Mar 1;18(2): PMID: Buenrostro et al., ATAC-seq: A Method for Assaying Chromatin Accessibility Genome-Wide. Curr Protoc Mol Biol Jan 5;109: PMID: Sims
ChIP-seq analysis. J. van Helden, M. Defrance, C. Herrmann, D. Puthier, N. Servant, M. Thomas-Chollier, O.Sand
 ChIP-seq analysis J. van Helden, M. Defrance, C. Herrmann, D. Puthier, N. Servant, M. Thomas-Chollier, O.Sand Tuesday : quick introduction to ChIP-seq and peak-calling (Presentation + Practical session)
ChIP-seq analysis J. van Helden, M. Defrance, C. Herrmann, D. Puthier, N. Servant, M. Thomas-Chollier, O.Sand Tuesday : quick introduction to ChIP-seq and peak-calling (Presentation + Practical session)
ChIP-seq data analysis
 ChIP-seq data analysis Harri Lähdesmäki Department of Computer Science Aalto University November 24, 2017 Contents Background ChIP-seq protocol ChIP-seq data analysis Transcriptional regulation Transcriptional
ChIP-seq data analysis Harri Lähdesmäki Department of Computer Science Aalto University November 24, 2017 Contents Background ChIP-seq protocol ChIP-seq data analysis Transcriptional regulation Transcriptional
Processing, integrating and analysing chromatin immunoprecipitation followed by sequencing (ChIP-seq) data
 data") Processing, integrating and analysing chromatin immunoprecipitation followed by sequencing (ChIP-seq) data Bioinformatics methods, models and applications to disease Alex Essebier ChIP-seq experiment To
Processing, integrating and analysing chromatin immunoprecipitation followed by sequencing (ChIP-seq) data Bioinformatics methods, models and applications to disease Alex Essebier ChIP-seq experiment To
The Epigenome Tools 2: ChIP-Seq and Data Analysis
 The Epigenome Tools 2: ChIP-Seq and Data Analysis Chongzhi Zang zang@virginia.edu http://zanglab.com PHS5705: Public Health Genomics March 20, 2017 1 Outline Epigenome: basics review ChIP-seq overview
The Epigenome Tools 2: ChIP-Seq and Data Analysis Chongzhi Zang zang@virginia.edu http://zanglab.com PHS5705: Public Health Genomics March 20, 2017 1 Outline Epigenome: basics review ChIP-seq overview
Computational aspects of ChIP-seq. John Marioni Research Group Leader European Bioinformatics Institute European Molecular Biology Laboratory
 Computational aspects of ChIP-seq John Marioni Research Group Leader European Bioinformatics Institute European Molecular Biology Laboratory ChIP-seq Using highthroughput sequencing to investigate DNA
Computational aspects of ChIP-seq John Marioni Research Group Leader European Bioinformatics Institute European Molecular Biology Laboratory ChIP-seq Using highthroughput sequencing to investigate DNA
Computational Analysis of UHT Sequences Histone modifications, CAGE, RNA-Seq
 Computational Analysis of UHT Sequences Histone modifications, CAGE, RNA-Seq Philipp Bucher Wednesday January 21, 2009 SIB graduate school course EPFL, Lausanne ChIP-seq against histone variants: Biological
Computational Analysis of UHT Sequences Histone modifications, CAGE, RNA-Seq Philipp Bucher Wednesday January 21, 2009 SIB graduate school course EPFL, Lausanne ChIP-seq against histone variants: Biological
Accessing and Using ENCODE Data Dr. Peggy J. Farnham
 1 William M Keck Professor of Biochemistry Keck School of Medicine University of Southern California How many human genes are encoded in our 3x10 9 bp? C. elegans (worm) 959 cells and 1x10 8 bp 20,000
1 William M Keck Professor of Biochemistry Keck School of Medicine University of Southern California How many human genes are encoded in our 3x10 9 bp? C. elegans (worm) 959 cells and 1x10 8 bp 20,000
ChIP-seq hands-on. Iros Barozzi, Campus IFOM-IEO (Milan) Saverio Minucci, Gioacchino Natoli Labs
 Saverio Minucci, Gioacchino Natoli Labs") ChIP-seq hands-on Iros Barozzi, Campus IFOM-IEO (Milan) Saverio Minucci, Gioacchino Natoli Labs Main goals Becoming familiar with essential tools and formats Visualizing and contextualizing raw data Understand
ChIP-seq hands-on Iros Barozzi, Campus IFOM-IEO (Milan) Saverio Minucci, Gioacchino Natoli Labs Main goals Becoming familiar with essential tools and formats Visualizing and contextualizing raw data Understand
Recent advances in ChIP-seq analysis: from quality management to whole-genome annotation
 Briefings in Bioinformatics Advance Access published March 15, 2016 Briefings in Bioinformatics, 2016, 1 12 doi: 10.1093/bib/bbw023 Paper Recent advances in ChIP-seq analysis: from quality management to
Briefings in Bioinformatics Advance Access published March 15, 2016 Briefings in Bioinformatics, 2016, 1 12 doi: 10.1093/bib/bbw023 Paper Recent advances in ChIP-seq analysis: from quality management to
EPIGENOMICS PROFILING SERVICES
 EPIGENOMICS PROFILING SERVICES Chromatin analysis DNA methylation analysis RNA-seq analysis Diagenode helps you uncover the mysteries of epigenetics PAGE 3 Integrative epigenomics analysis DNA methylation
EPIGENOMICS PROFILING SERVICES Chromatin analysis DNA methylation analysis RNA-seq analysis Diagenode helps you uncover the mysteries of epigenetics PAGE 3 Integrative epigenomics analysis DNA methylation
Session 6: Integration of epigenetic data. Peter J Park Department of Biomedical Informatics Harvard Medical School July 18-19, 2016
 Session 6: Integration of epigenetic data Peter J Park Department of Biomedical Informatics Harvard Medical School July 18-19, 2016 Utilizing complimentary datasets Frequent mutations in chromatin regulators
Session 6: Integration of epigenetic data Peter J Park Department of Biomedical Informatics Harvard Medical School July 18-19, 2016 Utilizing complimentary datasets Frequent mutations in chromatin regulators
Raymond Auerbach PhD Candidate, Yale University Gerstein and Snyder Labs August 30, 2012
 Elucidating Transcriptional Regulation at Multiple Scales Using High-Throughput Sequencing, Data Integration, and Computational Methods Raymond Auerbach PhD Candidate, Yale University Gerstein and Snyder
Elucidating Transcriptional Regulation at Multiple Scales Using High-Throughput Sequencing, Data Integration, and Computational Methods Raymond Auerbach PhD Candidate, Yale University Gerstein and Snyder
Part-II: Statistical analysis of ChIP-seq data
 Part-II: Statistical analysis of ChIP-seq data Outline ChIP-seq data, features, detailed modeling aspects (today). Other ChIP-seq related problems - overview (next lecture). IDR (next lecture) Stat 877
Part-II: Statistical analysis of ChIP-seq data Outline ChIP-seq data, features, detailed modeling aspects (today). Other ChIP-seq related problems - overview (next lecture). IDR (next lecture) Stat 877
Nature Structural & Molecular Biology: doi: /nsmb.2419
 Supplementary Figure 1 Mapped sequence reads and nucleosome occupancies. (a) Distribution of sequencing reads on the mouse reference genome for chromosome 14 as an example. The number of reads in a 1 Mb
Supplementary Figure 1 Mapped sequence reads and nucleosome occupancies. (a) Distribution of sequencing reads on the mouse reference genome for chromosome 14 as an example. The number of reads in a 1 Mb
Exploring chromatin regulation by ChIP-Sequencing
 Exploring chromatin regulation by ChIP-Sequencing From datasets quality assessment, enrichment patterns identification and multi-profiles integration to the reconstitution of gene regulatory wires describing
Exploring chromatin regulation by ChIP-Sequencing From datasets quality assessment, enrichment patterns identification and multi-profiles integration to the reconstitution of gene regulatory wires describing
High Throughput Sequence (HTS) data analysis. Lei Zhou
 data analysis. Lei Zhou") High Throughput Sequence (HTS) data analysis Lei Zhou (leizhou@ufl.edu) High Throughput Sequence (HTS) data analysis 1. Representation of HTS data. 2. Visualization of HTS data. 3. Discovering genomic
High Throughput Sequence (HTS) data analysis Lei Zhou (leizhou@ufl.edu) High Throughput Sequence (HTS) data analysis 1. Representation of HTS data. 2. Visualization of HTS data. 3. Discovering genomic
Comparison of open chromatin regions between dentate granule cells and other tissues and neural cell types.
 Supplementary Figure 1 Comparison of open chromatin regions between dentate granule cells and other tissues and neural cell types. (a) Pearson correlation heatmap among open chromatin profiles of different
Supplementary Figure 1 Comparison of open chromatin regions between dentate granule cells and other tissues and neural cell types. (a) Pearson correlation heatmap among open chromatin profiles of different
RNA-seq Introduction
 RNA-seq Introduction DNA is the same in all cells but which RNAs that is present is different in all cells There is a wide variety of different functional RNAs Which RNAs (and sometimes then translated
RNA-seq Introduction DNA is the same in all cells but which RNAs that is present is different in all cells There is a wide variety of different functional RNAs Which RNAs (and sometimes then translated
Breast cancer. Risk factors you cannot change include: Treatment Plan Selection. Inferring Transcriptional Module from Breast Cancer Profile Data
 Breast cancer Inferring Transcriptional Module from Breast Cancer Profile Data Breast Cancer and Targeted Therapy Microarray Profile Data Inferring Transcriptional Module Methods CSC 177 Data Warehousing
Breast cancer Inferring Transcriptional Module from Breast Cancer Profile Data Breast Cancer and Targeted Therapy Microarray Profile Data Inferring Transcriptional Module Methods CSC 177 Data Warehousing
7SK ChIRP-seq is specifically RNA dependent and conserved between mice and humans.
 Supplementary Figure 1 7SK ChIRP-seq is specifically RNA dependent and conserved between mice and humans. Regions targeted by the Even and Odd ChIRP probes mapped to a secondary structure model 56 of the
Supplementary Figure 1 7SK ChIRP-seq is specifically RNA dependent and conserved between mice and humans. Regions targeted by the Even and Odd ChIRP probes mapped to a secondary structure model 56 of the
Package NarrowPeaks. August 3, Version Date Type Package
 Package NarrowPeaks August 3, 2013 Version 1.5.0 Date 2013-02-13 Type Package Title Analysis of Variation in ChIP-seq using Functional PCA Statistics Author Pedro Madrigal , with contributions
Package NarrowPeaks August 3, 2013 Version 1.5.0 Date 2013-02-13 Type Package Title Analysis of Variation in ChIP-seq using Functional PCA Statistics Author Pedro Madrigal , with contributions
Broad H3K4me3 is associated with increased transcription elongation and enhancer activity at tumor suppressor genes
 Broad H3K4me3 is associated with increased transcription elongation and enhancer activity at tumor suppressor genes Kaifu Chen 1,2,3,4,5,10, Zhong Chen 6,10, Dayong Wu 6, Lili Zhang 7, Xueqiu Lin 1,2,8,
Broad H3K4me3 is associated with increased transcription elongation and enhancer activity at tumor suppressor genes Kaifu Chen 1,2,3,4,5,10, Zhong Chen 6,10, Dayong Wu 6, Lili Zhang 7, Xueqiu Lin 1,2,8,
An epigenetic approach to understanding (and predicting?) environmental effects on gene expression
 environmental effects on gene expression") www.collaslab.com An epigenetic approach to understanding (and predicting?) environmental effects on gene expression Philippe Collas University of Oslo Institute of Basic Medical Sciences Stem Cell Epigenetics
www.collaslab.com An epigenetic approach to understanding (and predicting?) environmental effects on gene expression Philippe Collas University of Oslo Institute of Basic Medical Sciences Stem Cell Epigenetics
Discovery of Novel Human Gene Regulatory Modules from Gene Co-expression and
 Discovery of Novel Human Gene Regulatory Modules from Gene Co-expression and Promoter Motif Analysis Shisong Ma 1,2*, Michael Snyder 3, and Savithramma P Dinesh-Kumar 2* 1 School of Life Sciences, University
Discovery of Novel Human Gene Regulatory Modules from Gene Co-expression and Promoter Motif Analysis Shisong Ma 1,2*, Michael Snyder 3, and Savithramma P Dinesh-Kumar 2* 1 School of Life Sciences, University
Genome-Wide Localization of Protein-DNA Binding and Histone Modification by a Bayesian Change-Point Method with ChIP-seq Data
 Genome-Wide Localization of Protein-DNA Binding and Histone Modification by a Bayesian Change-Point Method with ChIP-seq Data Haipeng Xing, Yifan Mo, Will Liao, Michael Q. Zhang Clayton Davis and Geoffrey
Genome-Wide Localization of Protein-DNA Binding and Histone Modification by a Bayesian Change-Point Method with ChIP-seq Data Haipeng Xing, Yifan Mo, Will Liao, Michael Q. Zhang Clayton Davis and Geoffrey
Heintzman, ND, Stuart, RK, Hon, G, Fu, Y, Ching, CW, Hawkins, RD, Barrera, LO, Van Calcar, S, Qu, C, Ching, KA, Wang, W, Weng, Z, Green, RD,
 Heintzman, ND, Stuart, RK, Hon, G, Fu, Y, Ching, CW, Hawkins, RD, Barrera, LO, Van Calcar, S, Qu, C, Ching, KA, Wang, W, Weng, Z, Green, RD, Crawford, GE, Ren, B (2007) Distinct and predictive chromatin
Heintzman, ND, Stuart, RK, Hon, G, Fu, Y, Ching, CW, Hawkins, RD, Barrera, LO, Van Calcar, S, Qu, C, Ching, KA, Wang, W, Weng, Z, Green, RD, Crawford, GE, Ren, B (2007) Distinct and predictive chromatin
Transcript-indexed ATAC-seq for immune profiling
 Transcript-indexed ATAC-seq for immune profiling Technical Journal Club 22 nd of May 2018 Christina Müller Nature Methods, Vol.10 No.12, 2013 Nature Biotechnology, Vol.32 No.7, 2014 Nature Medicine, Vol.24,
Transcript-indexed ATAC-seq for immune profiling Technical Journal Club 22 nd of May 2018 Christina Müller Nature Methods, Vol.10 No.12, 2013 Nature Biotechnology, Vol.32 No.7, 2014 Nature Medicine, Vol.24,
Yingying Wei George Wu Hongkai Ji
 Stat Biosci (2013) 5:156 178 DOI 10.1007/s12561-012-9066-5 Global Mapping of Transcription Factor Binding Sites by Sequencing Chromatin Surrogates: a Perspective on Experimental Design, Data Analysis,
Stat Biosci (2013) 5:156 178 DOI 10.1007/s12561-012-9066-5 Global Mapping of Transcription Factor Binding Sites by Sequencing Chromatin Surrogates: a Perspective on Experimental Design, Data Analysis,
A Practical Guide to Integrative Genomics by RNA-seq and ChIP-seq Analysis
 A Practical Guide to Integrative Genomics by RNA-seq and ChIP-seq Analysis Jian Xu, Ph.D. Children s Research Institute, UTSW Introduction Outline Overview of genomic and next-gen sequencing technologies
A Practical Guide to Integrative Genomics by RNA-seq and ChIP-seq Analysis Jian Xu, Ph.D. Children s Research Institute, UTSW Introduction Outline Overview of genomic and next-gen sequencing technologies
Supplemental Figure S1. Tertiles of FKBP5 promoter methylation and internal regulatory region
 Supplemental Figure S1. Tertiles of FKBP5 promoter methylation and internal regulatory region methylation in relation to PSS and fetal coupling. A, PSS values for participants whose placentas showed low,
Supplemental Figure S1. Tertiles of FKBP5 promoter methylation and internal regulatory region methylation in relation to PSS and fetal coupling. A, PSS values for participants whose placentas showed low,
Patterns of Histone Methylation and Chromatin Organization in Grapevine Leaf. Rachel Schwope EPIGEN May 24-27, 2016
 Patterns of Histone Methylation and Chromatin Organization in Grapevine Leaf Rachel Schwope EPIGEN May 24-27, 2016 What does H3K4 methylation do? Plant of interest: Vitis vinifera Culturally important
Patterns of Histone Methylation and Chromatin Organization in Grapevine Leaf Rachel Schwope EPIGEN May 24-27, 2016 What does H3K4 methylation do? Plant of interest: Vitis vinifera Culturally important
Genome-wide Association Studies (GWAS) Pasieka, Science Photo Library
 Pasieka, Science Photo Library") Lecture 5 Genome-wide Association Studies (GWAS) Pasieka, Science Photo Library Chi-squared test to evaluate whether the odds ratio is different from 1. Corrected for multiple testing Source: wikipedia.org
Lecture 5 Genome-wide Association Studies (GWAS) Pasieka, Science Photo Library Chi-squared test to evaluate whether the odds ratio is different from 1. Corrected for multiple testing Source: wikipedia.org
Supplementary Figure S1. Gene expression analysis of epidermal marker genes and TP63.
 Supplementary Figure Legends Supplementary Figure S1. Gene expression analysis of epidermal marker genes and TP63. A. Screenshot of the UCSC genome browser from normalized RNAPII and RNA-seq ChIP-seq data
Supplementary Figure Legends Supplementary Figure S1. Gene expression analysis of epidermal marker genes and TP63. A. Screenshot of the UCSC genome browser from normalized RNAPII and RNA-seq ChIP-seq data
Introduction to Systems Biology of Cancer Lecture 2
 Introduction to Systems Biology of Cancer Lecture 2 Gustavo Stolovitzky IBM Research Icahn School of Medicine at Mt Sinai DREAM Challenges High throughput measurements: The age of omics Systems Biology
Introduction to Systems Biology of Cancer Lecture 2 Gustavo Stolovitzky IBM Research Icahn School of Medicine at Mt Sinai DREAM Challenges High throughput measurements: The age of omics Systems Biology
Differential peak calling of ChIP-seq signals with replicates with THOR
 Published online 2 August 2016 Nucleic Acids Research, 2016, Vol. 44, No. 20 e153 doi: 10.1093/nar/gkw680 Differential peak calling of ChIP-seq signals with replicates with THOR Manuel Allhoff 1,2,3, Kristin
Published online 2 August 2016 Nucleic Acids Research, 2016, Vol. 44, No. 20 e153 doi: 10.1093/nar/gkw680 Differential peak calling of ChIP-seq signals with replicates with THOR Manuel Allhoff 1,2,3, Kristin
Nature Genetics: doi: /ng Supplementary Figure 1. Assessment of sample purity and quality.
 Supplementary Figure 1 Assessment of sample purity and quality. (a) Hematoxylin and eosin staining of formaldehyde-fixed, paraffin-embedded sections from a human testis biopsy collected concurrently with
Supplementary Figure 1 Assessment of sample purity and quality. (a) Hematoxylin and eosin staining of formaldehyde-fixed, paraffin-embedded sections from a human testis biopsy collected concurrently with
Nature Genetics: doi: /ng Supplementary Figure 1
 Supplementary Figure 1 Expression deviation of the genes mapped to gene-wise recurrent mutations in the TCGA breast cancer cohort (top) and the TCGA lung cancer cohort (bottom). For each gene (each pair
Supplementary Figure 1 Expression deviation of the genes mapped to gene-wise recurrent mutations in the TCGA breast cancer cohort (top) and the TCGA lung cancer cohort (bottom). For each gene (each pair
Tutorial. ChIP Sequencing. Sample to Insight. September 15, 2016
 ChIP Sequencing September 15, 2016 Sample to Insight CLC bio, a QIAGEN Company Silkeborgvej 2 Prismet 8000 Aarhus C Denmark Telephone: +45 70 22 32 44 www.clcbio.com support-clcbio@qiagen.com ChIP Sequencing
ChIP Sequencing September 15, 2016 Sample to Insight CLC bio, a QIAGEN Company Silkeborgvej 2 Prismet 8000 Aarhus C Denmark Telephone: +45 70 22 32 44 www.clcbio.com support-clcbio@qiagen.com ChIP Sequencing
CTCF-Mediated Functional Chromatin Interactome in Pluripotent Cells
 SUPPLEMENTARY INFORMATION CTCF-Mediated Functional Chromatin Interactome in Pluripotent Cells Lusy Handoko 1,*, Han Xu 1,*, Guoliang Li 1,*, Chew Yee Ngan 1, Elaine Chew 1, Marie Schnapp 1, Charlie Wah
SUPPLEMENTARY INFORMATION CTCF-Mediated Functional Chromatin Interactome in Pluripotent Cells Lusy Handoko 1,*, Han Xu 1,*, Guoliang Li 1,*, Chew Yee Ngan 1, Elaine Chew 1, Marie Schnapp 1, Charlie Wah
ChromHMM Tutorial. Jason Ernst Assistant Professor University of California, Los Angeles
 ChromHMM Tutorial Jason Ernst Assistant Professor University of California, Los Angeles Talk Outline Chromatin states analysis and ChromHMM Accessing chromatin state annotations for ENCODE2 and Roadmap
ChromHMM Tutorial Jason Ernst Assistant Professor University of California, Los Angeles Talk Outline Chromatin states analysis and ChromHMM Accessing chromatin state annotations for ENCODE2 and Roadmap
Supplemental Figure 1: Asymmetric chromatin maturation leads to epigenetic asymmetries on sister chromatids.
 Supplemental Material: Annu. Rev. Cell Dev. Biol. 2017. 33:291 318 https://doi.org/10.1146/annurev-cellbio-100616-060447 The Inherent Asymmetry of DNA Replication Snedeker, Wooten, and Chen Supplemental
Supplemental Material: Annu. Rev. Cell Dev. Biol. 2017. 33:291 318 https://doi.org/10.1146/annurev-cellbio-100616-060447 The Inherent Asymmetry of DNA Replication Snedeker, Wooten, and Chen Supplemental
Supervised Learner for the Prediction of Hi-C Interaction Counts and Determination of Influential Features. Tyler Yue Lab
 Supervised Learner for the Prediction of Hi-C Interaction Counts and Determination of Influential Features Tyler Derr @ Yue Lab tsd5037@psu.edu Background Hi-C is a chromosome conformation capture (3C)
Supervised Learner for the Prediction of Hi-C Interaction Counts and Determination of Influential Features Tyler Derr @ Yue Lab tsd5037@psu.edu Background Hi-C is a chromosome conformation capture (3C)
Supplementary Figures
 Supplementary Figures Supplementary Figure 1. Heatmap of GO terms for differentially expressed genes. The terms were hierarchically clustered using the GO term enrichment beta. Darker red, higher positive
Supplementary Figures Supplementary Figure 1. Heatmap of GO terms for differentially expressed genes. The terms were hierarchically clustered using the GO term enrichment beta. Darker red, higher positive
Statistical analysis of ChIP-seq data
 Statistical analysis of ChIP-seq data Sündüz Keleş Department of Statistics Department of Biostatistics and Medical Informatics University of Wisconsin, Madison Feb 9, 2012 BMI 776 (Spring 12) 02/09/2012
Statistical analysis of ChIP-seq data Sündüz Keleş Department of Statistics Department of Biostatistics and Medical Informatics University of Wisconsin, Madison Feb 9, 2012 BMI 776 (Spring 12) 02/09/2012
The Insulator Binding Protein CTCF Positions 20 Nucleosomes around Its Binding Sites across the Human Genome
 The Insulator Binding Protein CTCF Positions 20 Nucleosomes around Its Binding Sites across the Human Genome Yutao Fu 1, Manisha Sinha 2,3, Craig L. Peterson 3, Zhiping Weng 1,4,5 * 1 Bioinformatics Program,
The Insulator Binding Protein CTCF Positions 20 Nucleosomes around Its Binding Sites across the Human Genome Yutao Fu 1, Manisha Sinha 2,3, Craig L. Peterson 3, Zhiping Weng 1,4,5 * 1 Bioinformatics Program,
A novel ATAC-seq approach reveals lineage-specific reinforcement of the open chromatin landscape via cooperation between BAF and p63
 Bao et al. Genome Biology (2015) 16:284 DOI 10.1186/s13059-015-0840-9 RESEARCH A novel ATAC-seq approach reveals lineage-specific reinforcement of the open chromatin landscape via cooperation between BAF
Bao et al. Genome Biology (2015) 16:284 DOI 10.1186/s13059-015-0840-9 RESEARCH A novel ATAC-seq approach reveals lineage-specific reinforcement of the open chromatin landscape via cooperation between BAF
Functional annotation of farm animal genomes: ChIP-seq.
 Functional annotation of farm animal genomes: ChIP-seq Richard Crooijmans 2018, PAGXXVI Richard.Crooijmans@wur.nl Why FAANG is important Understanding the genotype to phenotype link This needs: - genomic
Functional annotation of farm animal genomes: ChIP-seq Richard Crooijmans 2018, PAGXXVI Richard.Crooijmans@wur.nl Why FAANG is important Understanding the genotype to phenotype link This needs: - genomic
Nature Immunology: doi: /ni Supplementary Figure 1. Transcriptional program of the TE and MP CD8 + T cell subsets.
 Supplementary Figure 1 Transcriptional program of the TE and MP CD8 + T cell subsets. (a) Comparison of gene expression of TE and MP CD8 + T cell subsets by microarray. Genes that are 1.5-fold upregulated
Supplementary Figure 1 Transcriptional program of the TE and MP CD8 + T cell subsets. (a) Comparison of gene expression of TE and MP CD8 + T cell subsets by microarray. Genes that are 1.5-fold upregulated
Table S1. Total and mapped reads produced for each ChIP-seq sample
 Tale S1. Total and mapped reads produced for each ChIP-seq sample Sample Total Reads Mapped Reads Col- H3K27me3 rep1 125662 1334323 (85.76%) Col- H3K27me3 rep2 9176437 7986731 (87.4%) atmi1a//c H3K27m3
Tale S1. Total and mapped reads produced for each ChIP-seq sample Sample Total Reads Mapped Reads Col- H3K27me3 rep1 125662 1334323 (85.76%) Col- H3K27me3 rep2 9176437 7986731 (87.4%) atmi1a//c H3K27m3
MIR retrotransposon sequences provide insulators to the human genome
 Supplementary Information: MIR retrotransposon sequences provide insulators to the human genome Jianrong Wang, Cristina Vicente-García, Davide Seruggia, Eduardo Moltó, Ana Fernandez- Miñán, Ana Neto, Elbert
Supplementary Information: MIR retrotransposon sequences provide insulators to the human genome Jianrong Wang, Cristina Vicente-García, Davide Seruggia, Eduardo Moltó, Ana Fernandez- Miñán, Ana Neto, Elbert
Analysis of Massively Parallel Sequencing Data Application of Illumina Sequencing to the Genetics of Human Cancers
 Analysis of Massively Parallel Sequencing Data Application of Illumina Sequencing to the Genetics of Human Cancers Gordon Blackshields Senior Bioinformatician Source BioScience 1 To Cancer Genetics Studies
Analysis of Massively Parallel Sequencing Data Application of Illumina Sequencing to the Genetics of Human Cancers Gordon Blackshields Senior Bioinformatician Source BioScience 1 To Cancer Genetics Studies
Not IN Our Genes - A Different Kind of Inheritance.! Christopher Phiel, Ph.D. University of Colorado Denver Mini-STEM School February 4, 2014
 Not IN Our Genes - A Different Kind of Inheritance! Christopher Phiel, Ph.D. University of Colorado Denver Mini-STEM School February 4, 2014 Epigenetics in Mainstream Media Epigenetics *Current definition:
Not IN Our Genes - A Different Kind of Inheritance! Christopher Phiel, Ph.D. University of Colorado Denver Mini-STEM School February 4, 2014 Epigenetics in Mainstream Media Epigenetics *Current definition:
Single-strand DNA library preparation improves sequencing of formalin-fixed and paraffin-embedded (FFPE) cancer DNA
 cancer DNA") www.impactjournals.com/oncotarget/ Oncotarget, Supplementary Materials 2016 Single-strand DNA library preparation improves sequencing of formalin-fixed and paraffin-embedded (FFPE) DNA Supplementary Materials
www.impactjournals.com/oncotarget/ Oncotarget, Supplementary Materials 2016 Single-strand DNA library preparation improves sequencing of formalin-fixed and paraffin-embedded (FFPE) DNA Supplementary Materials
MBios 478: Systems Biology and Bayesian Networks, 27 [Dr. Wyrick] Slide #1. Lecture 27: Systems Biology and Bayesian Networks
![MBios 478: Systems Biology and Bayesian Networks, 27 [Dr. Wyrick] Slide #1. Lecture 27: Systems Biology and Bayesian Networks](/thumbs/80/82116384.jpg "MBios 478: Systems Biology and Bayesian Networks, 27 [Dr. Wyrick] Slide #1. Lecture 27: Systems Biology and Bayesian Networks") MBios 478: Systems Biology and Bayesian Networks, 27 [Dr. Wyrick] Slide #1 Lecture 27: Systems Biology and Bayesian Networks Systems Biology and Regulatory Networks o Definitions o Network motifs o Examples
MBios 478: Systems Biology and Bayesian Networks, 27 [Dr. Wyrick] Slide #1 Lecture 27: Systems Biology and Bayesian Networks Systems Biology and Regulatory Networks o Definitions o Network motifs o Examples
Sudin Bhattacharya Institute for Integrative Toxicology
 Beyond the AHRE: the Role of Epigenomics in Gene Regulation by the AHR (or, Varied Applications of Computational Modeling in Toxicology and Ingredient Safety) Sudin Bhattacharya Institute for Integrative
Beyond the AHRE: the Role of Epigenomics in Gene Regulation by the AHR (or, Varied Applications of Computational Modeling in Toxicology and Ingredient Safety) Sudin Bhattacharya Institute for Integrative
SUPPLEMENTARY INFORMATION
 doi:10.1038/nature16931 Contents 1 Nomenclature 3 2 Fertility of homozygous B6 H/H and heterozygous B6 B6/H mice 3 3 DSB hotspot maps 3 3.1 DSB hotspot caller............................... 3 3.2 Calling
doi:10.1038/nature16931 Contents 1 Nomenclature 3 2 Fertility of homozygous B6 H/H and heterozygous B6 B6/H mice 3 3 DSB hotspot maps 3 3.1 DSB hotspot caller............................... 3 3.2 Calling
cis-regulatory enrichment analysis in human, mouse and fly
 cis-regulatory enrichment analysis in human, mouse and fly Zeynep Kalender Atak, PhD Laboratory of Computational Biology VIB-KU Leuven Center for Brain & Disease Research Laboratory of Computational Biology
cis-regulatory enrichment analysis in human, mouse and fly Zeynep Kalender Atak, PhD Laboratory of Computational Biology VIB-KU Leuven Center for Brain & Disease Research Laboratory of Computational Biology
Transcriptional control in Eukaryotes: (chapter 13 pp276) Chromatin structure affects gene expression. Chromatin Array of nuc
 Chromatin structure affects gene expression. Chromatin Array of nuc") Transcriptional control in Eukaryotes: (chapter 13 pp276) Chromatin structure affects gene expression Chromatin Array of nuc 1 Transcriptional control in Eukaryotes: Chromatin undergoes structural changes
Transcriptional control in Eukaryotes: (chapter 13 pp276) Chromatin structure affects gene expression Chromatin Array of nuc 1 Transcriptional control in Eukaryotes: Chromatin undergoes structural changes
Yue Wei 1, Rui Chen 2, Carlos E. Bueso-Ramos 3, Hui Yang 1, and Guillermo Garcia-Manero 1
 Genome-wide CHIP-Seq Analysis of Histone Methylation Reveals Modulators of NF- B Signaling And the Histone Demethylase JMJD3 Implicated in Myelodysplastic Syndrome Yue Wei 1, Rui Chen 2, Carlos E. Bueso-Ramos
Genome-wide CHIP-Seq Analysis of Histone Methylation Reveals Modulators of NF- B Signaling And the Histone Demethylase JMJD3 Implicated in Myelodysplastic Syndrome Yue Wei 1, Rui Chen 2, Carlos E. Bueso-Ramos
Discovery of two identities of neuroblastoma cells via the analysis of super-enhancer landscapes
 Discovery of two identities of neuroblastoma cells via the analysis of super-enhancer landscapes Valentina BOEVA Computational (Epi-)Genetics of Cancer Institut Cochin, Inserm U1016 / CNRS UMR 8104 / Université
Discovery of two identities of neuroblastoma cells via the analysis of super-enhancer landscapes Valentina BOEVA Computational (Epi-)Genetics of Cancer Institut Cochin, Inserm U1016 / CNRS UMR 8104 / Université
genomics for systems biology / ISB2020 RNA sequencing (RNA-seq)
") RNA sequencing (RNA-seq) Module Outline MO 13-Mar-2017 RNA sequencing: Introduction 1 WE 15-Mar-2017 RNA sequencing: Introduction 2 MO 20-Mar-2017 Paper: PMID 25954002: Human genomics. The human transcriptome
RNA sequencing (RNA-seq) Module Outline MO 13-Mar-2017 RNA sequencing: Introduction 1 WE 15-Mar-2017 RNA sequencing: Introduction 2 MO 20-Mar-2017 Paper: PMID 25954002: Human genomics. The human transcriptome
Epigenetics. Jenny van Dongen Vrije Universiteit (VU) Amsterdam Boulder, Friday march 10, 2017
 Amsterdam Boulder, Friday march 10, 2017") Epigenetics Jenny van Dongen Vrije Universiteit (VU) Amsterdam j.van.dongen@vu.nl Boulder, Friday march 10, 2017 Epigenetics Epigenetics= The study of molecular mechanisms that influence the activity of
Epigenetics Jenny van Dongen Vrije Universiteit (VU) Amsterdam j.van.dongen@vu.nl Boulder, Friday march 10, 2017 Epigenetics Epigenetics= The study of molecular mechanisms that influence the activity of
DNA-seq Bioinformatics Analysis: Copy Number Variation
 DNA-seq Bioinformatics Analysis: Copy Number Variation Elodie Girard elodie.girard@curie.fr U900 institut Curie, INSERM, Mines ParisTech, PSL Research University Paris, France NGS Applications 5C HiC DNA-seq
DNA-seq Bioinformatics Analysis: Copy Number Variation Elodie Girard elodie.girard@curie.fr U900 institut Curie, INSERM, Mines ParisTech, PSL Research University Paris, France NGS Applications 5C HiC DNA-seq
Supplemental Materials
 Supplemental Materials Sequence Features and Chromatin Structure around the Genomic Regions Bound by 119 Human Transcription Factors Running title: Analysis of Sites of 119 TFs in the Human Genome Jie
Supplemental Materials Sequence Features and Chromatin Structure around the Genomic Regions Bound by 119 Human Transcription Factors Running title: Analysis of Sites of 119 TFs in the Human Genome Jie
A fully Bayesian approach for the analysis of Whole-Genome Bisulfite Sequencing Data
 A fully Bayesian approach for the analysis of Whole-Genome Bisulfite Sequencing Data Leonardo Bottolo 1,2,3 1 Department of Medical Genetics, University of Cambridge, UK 2 The Alan Turing Institute, London,
A fully Bayesian approach for the analysis of Whole-Genome Bisulfite Sequencing Data Leonardo Bottolo 1,2,3 1 Department of Medical Genetics, University of Cambridge, UK 2 The Alan Turing Institute, London,
RNA- seq Introduc1on. Promises and pi7alls
 RNA- seq Introduc1on Promises and pi7alls DNA is the same in all cells but which RNAs that is present is different in all cells There is a wide variety of different func1onal RNAs Which RNAs (and some1mes
RNA- seq Introduc1on Promises and pi7alls DNA is the same in all cells but which RNAs that is present is different in all cells There is a wide variety of different func1onal RNAs Which RNAs (and some1mes
Inferring Biological Meaning from Cap Analysis Gene Expression Data
 Inferring Biological Meaning from Cap Analysis Gene Expression Data HRYSOULA PAPADAKIS 1. Introduction This project is inspired by the recent development of the Cap analysis gene expression (CAGE) method,
Inferring Biological Meaning from Cap Analysis Gene Expression Data HRYSOULA PAPADAKIS 1. Introduction This project is inspired by the recent development of the Cap analysis gene expression (CAGE) method,
BST227: Introduction to Statistical Genetics
 BST227: Introduction to Statistical Genetics Lecture 11: Heritability from summary statistics & epigenetic enrichments Guest Lecturer: Caleb Lareau Success of GWAS EBI Human GWAS Catalog As of this morning
BST227: Introduction to Statistical Genetics Lecture 11: Heritability from summary statistics & epigenetic enrichments Guest Lecturer: Caleb Lareau Success of GWAS EBI Human GWAS Catalog As of this morning
Assignment 5: Integrative epigenomics analysis
 Assignment 5: Integrative epigenomics analysis Due date: Friday, 2/24 10am. Note: no late assignments will be accepted. Introduction CpG islands (CGIs) are important regulatory regions in the genome. What
Assignment 5: Integrative epigenomics analysis Due date: Friday, 2/24 10am. Note: no late assignments will be accepted. Introduction CpG islands (CGIs) are important regulatory regions in the genome. What
STAT1 regulates microrna transcription in interferon γ stimulated HeLa cells
 CAMDA 2009 October 5, 2009 STAT1 regulates microrna transcription in interferon γ stimulated HeLa cells Guohua Wang 1, Yadong Wang 1, Denan Zhang 1, Mingxiang Teng 1,2, Lang Li 2, and Yunlong Liu 2 Harbin
CAMDA 2009 October 5, 2009 STAT1 regulates microrna transcription in interferon γ stimulated HeLa cells Guohua Wang 1, Yadong Wang 1, Denan Zhang 1, Mingxiang Teng 1,2, Lang Li 2, and Yunlong Liu 2 Harbin
High-resolution characterization of sequence signatures due to non-random cleavage of cell-free DNA
 Chandrananda et al. BMC Medical Genomics (2015) 8:29 DOI 10.1186/s12920-015-0107-z RESEARCH ARTICLE High-resolution characterization of sequence signatures due to non-random cleavage of cell-free DNA Dineika
Chandrananda et al. BMC Medical Genomics (2015) 8:29 DOI 10.1186/s12920-015-0107-z RESEARCH ARTICLE High-resolution characterization of sequence signatures due to non-random cleavage of cell-free DNA Dineika
DNA Sequence Bioinformatics Analysis with the Galaxy Platform
 DNA Sequence Bioinformatics Analysis with the Galaxy Platform University of São Paulo, Brazil 28 July - 1 August 2014 Dave Clements Johns Hopkins University Robson Francisco de Souza University of São
DNA Sequence Bioinformatics Analysis with the Galaxy Platform University of São Paulo, Brazil 28 July - 1 August 2014 Dave Clements Johns Hopkins University Robson Francisco de Souza University of São
Chapter 1. Introduction
 Chapter 1 Introduction 1.1 Motivation and Goals The increasing availability and decreasing cost of high-throughput (HT) technologies coupled with the availability of computational tools and data form a
Chapter 1 Introduction 1.1 Motivation and Goals The increasing availability and decreasing cost of high-throughput (HT) technologies coupled with the availability of computational tools and data form a
Supplementary Information
 Supplementary Information for Ho et al., modencode and ENCODE resources for analysis of metazoan chromatin organization Supplementary Content Supplementary Methods ---------------------------------------------------------
Supplementary Information for Ho et al., modencode and ENCODE resources for analysis of metazoan chromatin organization Supplementary Content Supplementary Methods ---------------------------------------------------------
Allelic reprogramming of the histone modification H3K4me3 in early mammalian development
 Allelic reprogramming of the histone modification H3K4me3 in early mammalian development 张戈 Method and material STAR ChIP seq (small-scale TELP-assisted rapid ChIP seq) 200 mouse embryonic stem cells PWK/PhJ
Allelic reprogramming of the histone modification H3K4me3 in early mammalian development 张戈 Method and material STAR ChIP seq (small-scale TELP-assisted rapid ChIP seq) 200 mouse embryonic stem cells PWK/PhJ
Supplementary Figure 1 IL-27 IL
 Tim-3 Supplementary Figure 1 Tc0 49.5 0.6 Tc1 63.5 0.84 Un 49.8 0.16 35.5 0.16 10 4 61.2 5.53 10 3 64.5 5.66 10 2 10 1 10 0 31 2.22 10 0 10 1 10 2 10 3 10 4 IL-10 28.2 1.69 IL-27 Supplementary Figure 1.
Tim-3 Supplementary Figure 1 Tc0 49.5 0.6 Tc1 63.5 0.84 Un 49.8 0.16 35.5 0.16 10 4 61.2 5.53 10 3 64.5 5.66 10 2 10 1 10 0 31 2.22 10 0 10 1 10 2 10 3 10 4 IL-10 28.2 1.69 IL-27 Supplementary Figure 1.
Histone Modifications Are Associated with Transcript Isoform Diversity in Normal and Cancer Cells
 Histone Modifications Are Associated with Transcript Isoform Diversity in Normal and Cancer Cells Ondrej Podlaha 1, Subhajyoti De 2,3,4, Mithat Gonen 5, Franziska Michor 1 * 1 Department of Biostatistics
Histone Modifications Are Associated with Transcript Isoform Diversity in Normal and Cancer Cells Ondrej Podlaha 1, Subhajyoti De 2,3,4, Mithat Gonen 5, Franziska Michor 1 * 1 Department of Biostatistics
Sequence and chromatin determinants of cell-type specific transcription factor binding
 Method Sequence and chromatin determinants of cell-type specific transcription factor binding Aaron Arvey, 1 Phaedra Agius, 1 William Stafford Noble, 2 and Christina Leslie 1,3 1 Computational Biology
Method Sequence and chromatin determinants of cell-type specific transcription factor binding Aaron Arvey, 1 Phaedra Agius, 1 William Stafford Noble, 2 and Christina Leslie 1,3 1 Computational Biology
TrainMALTA Summer School on Epigenomics
 TrainMALTA Summer School on Epigenomics 23-27 th July 2018, University of Malta TrainMALTA Partners: University of Malta University of Cambridge Katholieke Universiteit Leuven This project has received
TrainMALTA Summer School on Epigenomics 23-27 th July 2018, University of Malta TrainMALTA Partners: University of Malta University of Cambridge Katholieke Universiteit Leuven This project has received
AVENIO ctdna Analysis Kits The complete NGS liquid biopsy solution EMPOWER YOUR LAB
 Analysis Kits The complete NGS liquid biopsy solution EMPOWER YOUR LAB Analysis Kits Next-generation performance in liquid biopsies 2 Accelerating clinical research From liquid biopsy to next-generation
Analysis Kits The complete NGS liquid biopsy solution EMPOWER YOUR LAB Analysis Kits Next-generation performance in liquid biopsies 2 Accelerating clinical research From liquid biopsy to next-generation
PRC2 inhibition counteracts the culture-associated loss of engraftment potential of human cord blood-derived hematopoietic stem and progenitor cells
 Supplementary Informations PRC2 inhibition counteracts the culture-associated loss of engraftment potential of human cord blood-derived hematopoietic stem and progenitor cells Linda Varagnolo 1, Qiong
Supplementary Informations PRC2 inhibition counteracts the culture-associated loss of engraftment potential of human cord blood-derived hematopoietic stem and progenitor cells Linda Varagnolo 1, Qiong
Lecture 8 Understanding Transcription RNA-seq analysis. Foundations of Computational Systems Biology David K. Gifford
 Lecture 8 Understanding Transcription RNA-seq analysis Foundations of Computational Systems Biology David K. Gifford 1 Lecture 8 RNA-seq Analysis RNA-seq principles How can we characterize mrna isoform
Lecture 8 Understanding Transcription RNA-seq analysis Foundations of Computational Systems Biology David K. Gifford 1 Lecture 8 RNA-seq Analysis RNA-seq principles How can we characterize mrna isoform
Nature Immunology: doi: /ni Supplementary Figure 1. Characteristics of SEs in T reg and T conv cells.
 Supplementary Figure 1 Characteristics of SEs in T reg and T conv cells. (a) Patterns of indicated transcription factor-binding at SEs and surrounding regions in T reg and T conv cells. Average normalized
Supplementary Figure 1 Characteristics of SEs in T reg and T conv cells. (a) Patterns of indicated transcription factor-binding at SEs and surrounding regions in T reg and T conv cells. Average normalized
Stem Cell Epigenetics
 Stem Cell Epigenetics Philippe Collas University of Oslo Institute of Basic Medical Sciences Norwegian Center for Stem Cell Research www.collaslab.com Source of stem cells in the body Somatic ( adult )
Stem Cell Epigenetics Philippe Collas University of Oslo Institute of Basic Medical Sciences Norwegian Center for Stem Cell Research www.collaslab.com Source of stem cells in the body Somatic ( adult )
SUPPLEMENTARY FIGURE 1: f-i
 SUPPLEMENTARY FIGURE 1: Comparisons of the biological replicates of ChIP-seq, Input and Bisulfite-seq. (a) Density plot of MeCP2 ChIP-seq genome coverage (calculated using tiled 15 bp windows) shows high
SUPPLEMENTARY FIGURE 1: Comparisons of the biological replicates of ChIP-seq, Input and Bisulfite-seq. (a) Density plot of MeCP2 ChIP-seq genome coverage (calculated using tiled 15 bp windows) shows high
Histones modifications and variants
 Histones modifications and variants Dr. Institute of Molecular Biology, Johannes Gutenberg University, Mainz www.imb.de Lecture Objectives 1. Chromatin structure and function Chromatin and cell state Nucleosome
Histones modifications and variants Dr. Institute of Molecular Biology, Johannes Gutenberg University, Mainz www.imb.de Lecture Objectives 1. Chromatin structure and function Chromatin and cell state Nucleosome
Mechanisms of alternative splicing regulation
 Mechanisms of alternative splicing regulation The number of mechanisms that are known to be involved in splicing regulation approximates the number of splicing decisions that have been analyzed in detail.
Mechanisms of alternative splicing regulation The number of mechanisms that are known to be involved in splicing regulation approximates the number of splicing decisions that have been analyzed in detail.
Genome-wide relationship between histone H3 lysine 4 mono- and tri-methylation and transcription factor binding
 Letter Genome-wide relationship between histone H3 lysine 4 mono- and tri-methylation and transcription factor binding A. Gordon Robertson, 1,4 Mikhail Bilenky, 1,4 Angela Tam, 1 Yongjun Zhao, 1 Thomas
Letter Genome-wide relationship between histone H3 lysine 4 mono- and tri-methylation and transcription factor binding A. Gordon Robertson, 1,4 Mikhail Bilenky, 1,4 Angela Tam, 1 Yongjun Zhao, 1 Thomas
Chromatin marks identify critical cell-types for fine-mapping complex trait variants
 Chromatin marks identify critical cell-types for fine-mapping complex trait variants Gosia Trynka 1-4 *, Cynthia Sandor 1-4 *, Buhm Han 1-4, Han Xu 5, Barbara E Stranger 1,4#, X Shirley Liu 5, and Soumya
Chromatin marks identify critical cell-types for fine-mapping complex trait variants Gosia Trynka 1-4 *, Cynthia Sandor 1-4 *, Buhm Han 1-4, Han Xu 5, Barbara E Stranger 1,4#, X Shirley Liu 5, and Soumya
Modeling gene expression using five histone modifications
 Modeling gene expression using five histone modifications Fifth Annual Primes MIT Conference Lalita Devadas Mentor: Angela Yen May 17, 2015 Lalita Devadas Modeling gene expression using five histone modifications
Modeling gene expression using five histone modifications Fifth Annual Primes MIT Conference Lalita Devadas Mentor: Angela Yen May 17, 2015 Lalita Devadas Modeling gene expression using five histone modifications
Multiplex target enrichment using DNA indexing for ultra-high throughput variant detection
 Multiplex target enrichment using DNA indexing for ultra-high throughput variant detection Dr Elaine Kenny Neuropsychiatric Genetics Research Group Institute of Molecular Medicine Trinity College Dublin
Multiplex target enrichment using DNA indexing for ultra-high throughput variant detection Dr Elaine Kenny Neuropsychiatric Genetics Research Group Institute of Molecular Medicine Trinity College Dublin
ChipSeq. Technique and science. The genome wide dynamics of the binding of ldb1 complexes during erythroid differentiation
 Center for Biomics ChipSeq Technique and science The genome wide dynamics of the binding of ldb1 complexes during erythroid differentiation Wilfred van IJcken Sequencing Seminar Illumina September 8 Scheme
Center for Biomics ChipSeq Technique and science The genome wide dynamics of the binding of ldb1 complexes during erythroid differentiation Wilfred van IJcken Sequencing Seminar Illumina September 8 Scheme
QIAsymphony DSP Circulating DNA Kit
 QIAsymphony DSP Circulating DNA Kit February 2017 Performance Characteristics 937556 Sample to Insight Contents Performance Characteristics... 4 Basic performance... 4 Run precision... 6 Equivalent performance
QIAsymphony DSP Circulating DNA Kit February 2017 Performance Characteristics 937556 Sample to Insight Contents Performance Characteristics... 4 Basic performance... 4 Run precision... 6 Equivalent performance
The corrected Figure S1J is shown below. The text changes are as follows, with additions in bold and deletions in bracketed italics:
 Correction H3K4me3 readth Is Linked to Cell Identity and Transcriptional Consistency érénice A. enayoun, Elizabeth A. Pollina, Duygu Ucar, Salah Mahmoudi, Kalpana Karra, Edith D. Wong, Keerthana Devarajan,
Correction H3K4me3 readth Is Linked to Cell Identity and Transcriptional Consistency érénice A. enayoun, Elizabeth A. Pollina, Duygu Ucar, Salah Mahmoudi, Kalpana Karra, Edith D. Wong, Keerthana Devarajan,
Use Case 9: Coordinated Changes of Epigenomic Marks Across Tissue Types. Epigenome Informatics Workshop Bioinformatics Research Laboratory
 Use Case 9: Coordinated Changes of Epigenomic Marks Across Tissue Types Epigenome Informatics Workshop Bioinformatics Research Laboratory 1 Introduction Active or inactive states of transcription factor
Use Case 9: Coordinated Changes of Epigenomic Marks Across Tissue Types Epigenome Informatics Workshop Bioinformatics Research Laboratory 1 Introduction Active or inactive states of transcription factor
Transcription and chromatin. General Transcription Factors + Promoter-specific factors + Co-activators
 Transcription and chromatin General Transcription Factors + Promoter-specific factors + Co-activators Cofactor or Coactivator 1. work with DNA specific transcription factors to make them more effective
Transcription and chromatin General Transcription Factors + Promoter-specific factors + Co-activators Cofactor or Coactivator 1. work with DNA specific transcription factors to make them more effective
Integrated analysis of sequencing data
 Integrated analysis of sequencing data How to combine *-seq data M. Defrance, M. Thomas-Chollier, C. Herrmann, D. Puthier, J. van Helden *ChIP-seq, RNA-seq, MeDIP-seq, Transcription factor binding ChIP-seq
Integrated analysis of sequencing data How to combine *-seq data M. Defrance, M. Thomas-Chollier, C. Herrmann, D. Puthier, J. van Helden *ChIP-seq, RNA-seq, MeDIP-seq, Transcription factor binding ChIP-seq
Genome-Wide Analysis of Histone Modifications in Human Endometrial Stromal Cells
 ORIGINAL RESEARCH Genome-Wide Analysis of Histone Modifications in Human Endometrial Stromal Cells Isao Tamura, Yasuyuki Ohkawa, Tetsuya Sato, Mikita Suyama, Kosuke Jozaki, Maki Okada, Lifa Lee, Ryo Maekawa,
ORIGINAL RESEARCH Genome-Wide Analysis of Histone Modifications in Human Endometrial Stromal Cells Isao Tamura, Yasuyuki Ohkawa, Tetsuya Sato, Mikita Suyama, Kosuke Jozaki, Maki Okada, Lifa Lee, Ryo Maekawa,
Lecture 10. Eukaryotic gene regulation: chromatin remodelling
 Lecture 10 Eukaryotic gene regulation: chromatin remodelling Recap.. Eukaryotic RNA polymerases Core promoter elements General transcription factors Enhancers and upstream activation sequences Transcriptional
Lecture 10 Eukaryotic gene regulation: chromatin remodelling Recap.. Eukaryotic RNA polymerases Core promoter elements General transcription factors Enhancers and upstream activation sequences Transcriptional
Supplementary Figure 1. Efficiency of Mll4 deletion and its effect on T cell populations in the periphery. Nature Immunology: doi: /ni.
 Supplementary Figure 1 Efficiency of Mll4 deletion and its effect on T cell populations in the periphery. Expression of Mll4 floxed alleles (16-19) in naive CD4 + T cells isolated from lymph nodes and
Supplementary Figure 1 Efficiency of Mll4 deletion and its effect on T cell populations in the periphery. Expression of Mll4 floxed alleles (16-19) in naive CD4 + T cells isolated from lymph nodes and