Fatty Acids Synthesis L3
|
|
|
- Mildred Bryan
- 5 years ago
- Views:
Transcription
1 Fatty Acids Synthesis L3 The pathway for fatty acid synthesis occurs in the cytoplasm, whereas, oxidation occurs in the mitochondria. The other major difference is the use of nucleotide co-factors. Oxidation of fats involves the reduction of FAD and NAD +. Synthesis of fats involves the oxidation of NADPH. However, the essential chemistry of the two processes are reversals of each other. Both oxidation and synthesis of fats utilize an activated two carbon intermediate, acetyl-coa. However, the activated form of acetyl-coa in fat synthesis exists temporarily bound to the enzyme complex as malonyl-coa. The synthesis of malonyl-coa is the first committed step of fatty acid synthesis and the enzyme that catalyzes this reaction, acetyl-coa carboxylase (ACC), is the major site of regulation of fatty acid synthesis. Like other enzymes that transfer CO 2 to substrates, ACC requires a biotin co-factor. Acetyl-CoA carboxylase is called an ABC enzyme due to the requirements for ATP,Biotin, and CO 2 for the reaction (ABC enzyme are a family of ATP-dependent transporters that pump amino acids, proteins e.t.c. out of cells against a concentration gradient). The synthesis of fatty acids from acetyl-coa and malonyl-coa is carried out by fatty acid synthase, FAS. Fatty acid synthase is encoded by the FASN gene which is located on chromosome 17q25 and is composed of 43 exons that encode a protein of 2511 amino acids. Lipid Storage disease Lipid storage diseases are a group of inherited metabolic disorders in which harmful amounts of fatty materials (lipids) accumulate in various tissues and cells in the body. Lipids are important parts of the membranes found within and between each cell and in the myelin sheath that coats and protects the nerves. Over time, this excessive storage of fats can cause permanent cellular and tissue damage, particularly in the brain, peripheral nervous system, liver, spleen, and bone marrow. Lipid storage diseases are inherited from one or both parents who carry a defective gene. Symptoms may appear early in life or develop in the teen or even adult years. Neurological complications of the lipid storage diseases may include lack of muscle coordination, brain degeneration, seizures, loss of muscle tone, learning problems, spasticity, feeding and swallowing difficulties, slurred speech, hypersensitivity to touch, pain in the arms and legs, and clouding of the cornea. Inheritance of Lipid Storage disease Lipid storage diseases are inherited from one or both parents who carry a defective gene that regulates a particular protein in a class of the body s cells. They can be inherited in two ways: Autosomal recessive inheritance: this occurs when both parents carry and pass on a copy of the faulty gene, but neither parent is affected by the disorder. 1
2 X-linked inheritance; this occurs when the mother carries the affected gene on the X chromosome that determines the child s gender and passes it to her son. Sons of carriers have a 50 percent chance of inheriting the disorder. Daughters have a 50 percent chance of inheriting the X-linked chromosome but usually are not severely affected by the disorder. Affected men do not pass the disorder to their sons but their daughters will be carriers for the disorder. Some forms of lipid storage diseases 1. Gaucher disease: is caused by a deficiency of the enzyme glucocerebrosidase. Fatty material can collect in the spleen, liver, kidneys, lungs, brain, and bone marrow. Symptoms may include enlarged spleen and liver, liver malfunction, skeletal disorders and bone lesions that may cause pain and fractures, severe neurologic complications, swelling of lymph nodes and adjacent joints, distended abdomen, a brownish tint to the skin, anemia, low blood platelets, and yellow spots in the eyes. Persons affected most seriously may also be more susceptible to infection. The disease affects males and females equally. Gaucher disease has three common clinical subtypes. Type 1 (or non-neuropathic type) is the most common form of the disease in the United States. It occurs most often among persons of Ashkenazi Jewish heritage. Symptoms may begin early in life or in adulthood and include enlarged liver and grossly enlarged spleen, which can rupture and cause additional complications. Skeletal weakness and bone disease may be extensive. The brain is not affected, but there may be lung and, rarely, kidney impairment. Individuals usually bruise easily due to low blood platelets and experience fatigue due to anemia. Depending on disease onset and severity, those with type 1 may live well into adulthood. Many individuals have a mild form of the disease or may not show any symptoms. Type 2 (or acute infantile neuropathic Gaucher disease) typically begins within 3 months of birth. Symptoms include an enlarged liver and spleen, abnormal eye movement, extensive and progressive brain damage, spasticity, seizures, limb rigidity, and a poor ability to suck and swallow. Affected children usually die before age 2. Type 3 (the chronic neuronopathic form) can begin at any time in childhood or even in adulthood. It is characterized by slowly progressive but milder neurological symptoms compared to the acute or Type 2 Gaucher disease. Major symptoms include eye movement disorders, cognitive deficit, poor coordination, an enlarged spleen and/or liver, seizures, skeletal irregularities, blood disorders including anemia, and respiratory problems. Individuals who are successfully treated with enzyme replacement therapy generally live into adulthood. For those with type 1 and most type 3 Gaucher disease, enzyme replacement treatment given intravenously every two weeks can dramatically decrease liver and spleen size, reduce skeletal abnormalities, and reverse other manifestations. Successful bone marrow transplantation cures the non-neurological manifestations of the disease. However, this procedure carries significant risk and is rarely performed in individuals with Gaucher diseases. Surgery to remove the whole or part of the spleen may be required on rare occasions (if the person is anemic or when the enlarged organ affects the person's comfort). Blood transfusion may benefit some 2
3 anemic individuals. Others may require joint replacement surgery to improve mobility and quality of life. There is currently no effective treatment for the brain damage that may occur in types 2 and 3 Gaucher disease. 2. Niemann-Pick disease: is actually a group of autosomal recessive disorders caused by an accumulation of fat and cholesterol in cells of the liver, spleen, bone marrow, lungs, and, in some patients, brain. Neurological complications may include ataxia, eye paralysis, brain degeneration, learning problems, spasticity, feeding and swallowing difficulties, slurred speech, loss of muscle tone, hypersensitivity to touch, and some corneal clouding. A characteristic cherry-red halo develops around the center of the retina in 50 percent of patients. Niemann- Pick disease is currently subdivided into four categories. Onset of type A, the most severe form, is in early infancy. Infants appear normal at birth but develop an enlarged liver and spleen, swollen lymph nodes, nodes under the skin (xanthemas), and profound brain damage by 6 months of age. The spleen may enlarge to as much as 10 times its normal size and can rupture. These children become progressively weaker, lose motor function, may become anemic, and are susceptible to recurring infection. They rarely live beyond 18 months. This form of the disease occurs most often in Jewish families. In the second group, called type B (or juvenile onset), enlargement of the liver and spleen characteristically occurs in the pre-teen years. Most patients also develop ataxia, peripheral neuropathy, and pulmonary difficulties that progress with age, but the brain is generally not affected. Type B patients may live a comparatively long time but many require supplemental oxygen because of lung involvement. Niemann-Pick types A and B result from accumulation of the fatty substance called sphingomyelin, due to deficiency of an enzyme called sphingomyelinase. Niemann-Pick disease also includes two other variant forms called types C and D. These may appear early in life or develop in the teen or even adult years. Niemann-Pick disease types C and D are not caused by a deficiency of sphlingomyelinase but by a lack of the NPC1 or NPC2 proteins. As a result, various lipids and particularly cholesterol accumulate inside nerve cells and cause them to malfunction. Patients with types C and D have only moderate enlargement of their spleens and livers. Brain involvement may be extensive, leading to inability to look up and down, difficulty in walking and swallowing, and progressive loss of vision and hearing. Type D patients typically develop neurologic symptoms later than those with type C and have a progressively slower rate of loss of nerve function. Most type D patients share a common ancestral background in Nova Scotia. The life expectancies of patients with types C and D vary considerably. Some patients die in childhood while others who appear to be less severely affected can live into adulthood. There is currently no cure for Niemann-Pick disease. Treatment is supportive. Children usually die from infection or progressive neurological loss. Bone marrow transplantation has been attempted in a few patients with type B. Patients with types C and D are frequently placed on a low-cholesterol diet and/or cholesterol lowering drugs, although research has not shown these interventions change the abnormal cholesterol metabolism or halt progression of the disease. 3
4 3. Farber s disease: This is also known as Farber s lipogranulomatosis, describes a group of rare autosomal recessive disorders that cause an accumulation of fatty material in the joints, tissues, and central nervous system. The disorder affects both males and females. Disease onset is typically in early infancy but may occur later in life. Children who have the classic form of Farber s disease develop neurological symptoms within the first few weeks of life. These symptoms may include moderately impaired mental ability and problems with swallowing. The liver, heart, and kidneys may also be affected. Other symptoms may include vomiting, arthritis, swollen lymph nodes, swollen joints, joint contractures (chronic shortening of muscles or tendons around joints), hoarseness, and xanthemas which thicken around joints as the disease progresses. Patients with breathing difficulty may require insertion of a breathing tube. Most children with the disease die by age 2, usually from lung disease. In one of the most severe forms of the disease, an enlarged liver and spleen (hepatosplenomegaly) can be diagnosed soon after birth. Children born with this form of the disease usually die within 6 months. Farber's disease is caused by a deficiency of the enzyme called ceramidase. Currently there is no specific treatment for Farber s disease. Corticosteroids may be prescribed to relieve pain. Bone marrow transplants may improve granulomas (small masses of inflamed tissue) on patients with little or no lung or nervous system complications. Older patients may have granulomas surgically reduced or removed. The gangliosidoses are comprised of two distinct groups of genetic diseases. Both are autosomal recessive and affect males and females equally. 1. The GM1 gangliosidoses are caused by a deficiency of the enzyme betagalactosidase, resulting in abnormal storage of acidic lipid materials particularly in the nerve cells in the central and peripheral nervous systems. GM1 gangliosidosis has three clinical presentations: early infantile, late infantile, and adult. Signs of early infantile GM1 (the most severe subtype, with onset shortly after birth) may include neurodegeneration, seizures, liver and spleen enlargement, coarsening of facial features, skeletal irregularities, joint stiffness, distended abdomen, muscle weakness, exaggerated startle response, and problems with gait. About half of affected patients develop cherry-red spots in the eye. Children may be deaf and blind by age 1 and often die by age 3 from cardiac complications or pneumonia. Onset of late infantile GM1 gangliosidosis is typically between ages 1 and 3 years. Neurological signs include ataxia, seizures, dementia, and difficulties with speech. Onset of adult GM1 gangliosidosis is between ages 3 and 30. Symptoms include muscle atrophy, neurological complications that are less severe and progress at a slower rate than in other forms of the disorder, corneal clouding in some patients, and dystonia (sustained muscle contractions that cause twisting and repetitive movements or abnormal postures). Angiokeratomas may develop on the lower part of the trunk of the body. The size of the liver and spleen in most patients is normal.. 2. The GM2 gangliosidoses also cause the body to store excess acidic fatty materials in tissues and cells, most notably in nerve cells. These disorders result from a deficiency of the enzyme beta-hexosaminidase. The GM2 disorders 4
5 include: Tay-Sachs disease (also known as GM2 gangliosidosis-variant B). Tay-Sachs and its variant forms are caused by a deficiency in the enzyme hexosaminidase A. The incidence is particularly high among Eastern European and Ashkenazi Jewish populations, as well as certain French Canadians and Louisianan Cajuns. Affected children appear to develop normally for the first few months of life. Symptoms begin by 6 months of age and include progressive loss of mental ability, dementia, decreased eye contact, increased startle reflex to noise, progressive loss of hearing leading to deafness, difficulty in swallowing, blindness, cherry-red spots in the retinas, and some paralysis. Seizures may begin in the child s second year. Children may eventually need a feeding tube and they often die by age 4 from recurring infection. No specific treatment is available. Anticonvulsant medications may initially control seizures. Other supportive treatment includes proper nutrition and hydration and techniques to keep the airway open. A rarer form of the disorder, called late-onset Tay-Sachs disease, occurs in patients in their twenties and early thirties and is characterized by unsteadiness of gait and progressive neurological deterioration. Sandhoff disease (variant AB). This is a severe form of Tay-Sachs disease. Onset usually occurs at the age of 6 months and is not limited to any ethnic group. Neurological signs may include progressive deterioration of the central nervous system, motor weakness, early blindness, marked startle response to sound, spasticity, myoclonus (shock-like contractions of a muscle), seizures, macrocephaly (an abnormally enlarged head), and cherry-red spots in the eye. Other symptoms may include frequent respiratory infections, murmurs of the heart, doll-like facial features, and an enlarged liver and spleen. There is no specific treatment for Sandhoff disease. As with Tay-Sachs disease, supportive treatment includes keeping the airway open and proper nutrition and hydration. Anticonvulsant medications may initially control seizures. Children generally die by age 3 from respiratory infections. Below table summarizes lipid storage diseases, the deficient enzyme and their symptoms. 5
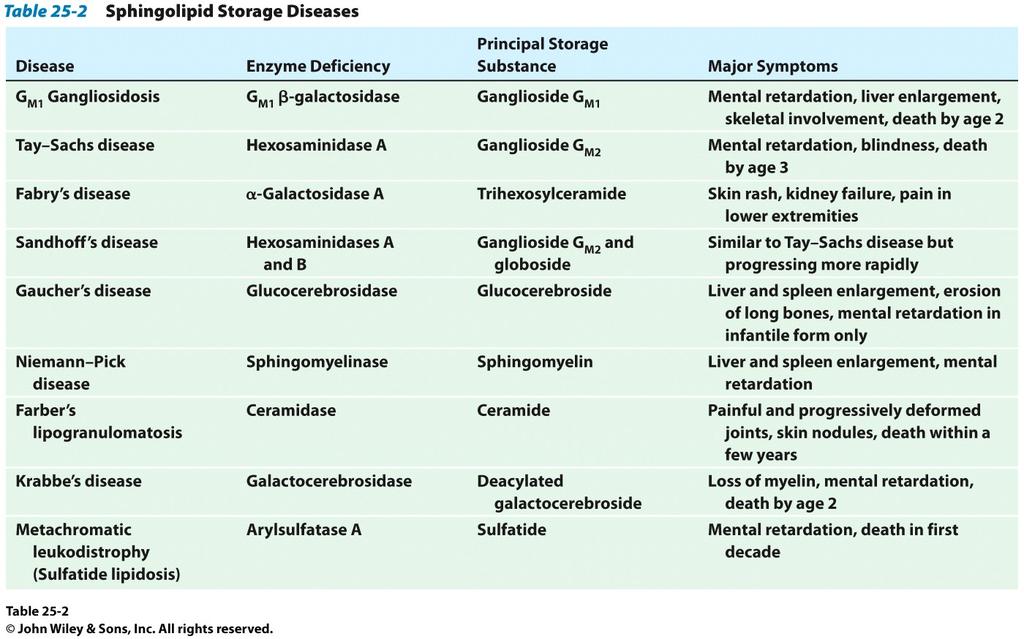
6 6
Lipid storage diseases
 Lipid storage diseases What are lipid storage diseases? Lipid storage diseases, or the lipidoses, are a group of inherited metabolic disorders in which harmful amounts of fatty materials called lipids
Lipid storage diseases What are lipid storage diseases? Lipid storage diseases, or the lipidoses, are a group of inherited metabolic disorders in which harmful amounts of fatty materials called lipids
What s New in Newborn Screening?
 What s New in Newborn Screening? Funded by: Illinois Department of Public Health Information on Newborn Screening Newborn screening in Illinois is administered by the Illinois Department of Public Health.
What s New in Newborn Screening? Funded by: Illinois Department of Public Health Information on Newborn Screening Newborn screening in Illinois is administered by the Illinois Department of Public Health.
What s New in Newborn Screening?
 What s New in Newborn Screening? Funded by: Illinois Department of Public Health Information on Newborn Screening Newborn screening in Illinois is mandated and administered by the Illinois Department of
What s New in Newborn Screening? Funded by: Illinois Department of Public Health Information on Newborn Screening Newborn screening in Illinois is mandated and administered by the Illinois Department of
A Lawyer s Perspective on Genetic Screening Performed by Cryobanks
 A Lawyer s Perspective on Genetic Screening Performed by Cryobanks As a lawyer practicing in the area of sperm bank litigation, I have, unfortunately, represented too many couples that conceived a child
A Lawyer s Perspective on Genetic Screening Performed by Cryobanks As a lawyer practicing in the area of sperm bank litigation, I have, unfortunately, represented too many couples that conceived a child
Clinical Summaries. CLN1 Disease, infantile onset and others
 Clinical Summaries CLN1 Disease, infantile onset and others The gene called CLN1 lies on chromosome 1. CLN1 disease is inherited as an autosomal recessive disorder, which means that both chromosomes carry
Clinical Summaries CLN1 Disease, infantile onset and others The gene called CLN1 lies on chromosome 1. CLN1 disease is inherited as an autosomal recessive disorder, which means that both chromosomes carry
METABOLISM OF ACYLGLYCEROLS AND SPHINGOLIPDS. Ben S. Ashok MSc.,FAGE.,PhD., Dept. of Biochemistry
 METABOLISM OF ACYLGLYCEROLS AND SPHINGOLIPDS Ben S. Ashok MSc.,FAGE.,PhD., Dept. of Biochemistry STORAGE AND MEMBRANE LIPIDS STORAGE LIPIDS Mainly as triacylglycerols (triglycerides) in adipose cells Constitute
METABOLISM OF ACYLGLYCEROLS AND SPHINGOLIPDS Ben S. Ashok MSc.,FAGE.,PhD., Dept. of Biochemistry STORAGE AND MEMBRANE LIPIDS STORAGE LIPIDS Mainly as triacylglycerols (triglycerides) in adipose cells Constitute
Newborn Screening for Lysosomal Storage Diseases in Missouri. Outline
 Newborn Screening for Lysosomal Storage Diseases in Missouri Dr. Kathy Grange Division of Genetics and Genomic Medicine Department of Pediatrics Washington University Outline Brief overview of clinical
Newborn Screening for Lysosomal Storage Diseases in Missouri Dr. Kathy Grange Division of Genetics and Genomic Medicine Department of Pediatrics Washington University Outline Brief overview of clinical
Pompe. Lysosomal Disorders. Introduction
 Introduction disease is an inherited, genetic disorder which results in the lack of an enzyme 'acid alpha-glucosidase. disease is also known as acid maltase deficiency or glycogen storage disease type
Introduction disease is an inherited, genetic disorder which results in the lack of an enzyme 'acid alpha-glucosidase. disease is also known as acid maltase deficiency or glycogen storage disease type
Genetic Testing FOR DISEASES OF INCREASED FREQUENCY IN THE ASHKENAZI JEWISH POPULATION
 Carrier Screening and Diagnostic Testing for the Ashkenazi Jewish Population Genetic Testing FOR DISEASES OF INCREASED FREQUENCY IN THE ASHKENAZI JEWISH POPULATION Our Science. Your Care. An extensive
Carrier Screening and Diagnostic Testing for the Ashkenazi Jewish Population Genetic Testing FOR DISEASES OF INCREASED FREQUENCY IN THE ASHKENAZI JEWISH POPULATION Our Science. Your Care. An extensive
Inborn Error Of Metabolism :
 Inborn Error Of Metabolism : Inborn Error Of Metabolism inborn error of metabolism are a large group of hereditary biochemical diseases in which specific gene mutation cause abnormal or missing proteins
Inborn Error Of Metabolism : Inborn Error Of Metabolism inborn error of metabolism are a large group of hereditary biochemical diseases in which specific gene mutation cause abnormal or missing proteins
SELECTIVE VULNERABILITY (HYPOXIA AND HYPOGLYCEMIA)
") DEFICIENCY OF METABOLITE -HYPOXIA AND HYPOGLYCEMIA -HYPOVITAMINOSIS SELECTIVE VULNERABILITY (HYPOXIA AND HYPOGLYCEMIA) -SPECIFIC CELL TYPE NEURONS>OLIGODENDROCYTES>ASTROCYTES -SPECIFIC BRAIN REGION PYRAMIDAL
DEFICIENCY OF METABOLITE -HYPOXIA AND HYPOGLYCEMIA -HYPOVITAMINOSIS SELECTIVE VULNERABILITY (HYPOXIA AND HYPOGLYCEMIA) -SPECIFIC CELL TYPE NEURONS>OLIGODENDROCYTES>ASTROCYTES -SPECIFIC BRAIN REGION PYRAMIDAL
Macular Cherry-Red Spots: Causes and Consequences
 Macular Cherry-Red Spots: Causes and Consequences 2018 Victoria Conference James Kundart OD MEd FAAO FCOVD-A Pacific University College of Optometry Financial Disclosures: Nothing to Disclose Learning
Macular Cherry-Red Spots: Causes and Consequences 2018 Victoria Conference James Kundart OD MEd FAAO FCOVD-A Pacific University College of Optometry Financial Disclosures: Nothing to Disclose Learning
Understanding Late-Onset Pompe Disease
 Understanding Late-Onset Pompe Disease What Is a Lysosome? Millions of tiny units called cells make up the human body. Each cell has its own job to keep the body running. Within each cell, there are organelles,
Understanding Late-Onset Pompe Disease What Is a Lysosome? Millions of tiny units called cells make up the human body. Each cell has its own job to keep the body running. Within each cell, there are organelles,
INTRODUCTION. 1.
 KRABBE DISEASE INTRODUCTION Krabbe disease is a genetic defect that affects the nervous system. It is caused by the shortage (deficiency) of an enzyme called galactosylceramidase. This enzyme deficiency
KRABBE DISEASE INTRODUCTION Krabbe disease is a genetic defect that affects the nervous system. It is caused by the shortage (deficiency) of an enzyme called galactosylceramidase. This enzyme deficiency
A Case Refort of Sandhoff Disease
 Korean J Ophthalmol Vol. 19:68-72, 2005 A Case Refort of Sandhoff Disease Yie-Min Yun, MD, Su-Na Lee, MD Department of Ophthalmology, College of Medicine, Chungnam National University, Daejeon, Korea Sandhoff
Korean J Ophthalmol Vol. 19:68-72, 2005 A Case Refort of Sandhoff Disease Yie-Min Yun, MD, Su-Na Lee, MD Department of Ophthalmology, College of Medicine, Chungnam National University, Daejeon, Korea Sandhoff
Inheritance of Gaucher Disease
 Sarah Mother of a child with Gaucher Working toward a healthy future Helping her son achieve his own dreams, too Straight Talk For Patients and Families Inheritance of Gaucher Disease Genzyme Corporation
Sarah Mother of a child with Gaucher Working toward a healthy future Helping her son achieve his own dreams, too Straight Talk For Patients and Families Inheritance of Gaucher Disease Genzyme Corporation
Should Universal Carrier Screening be Universal?
 Should Universal Carrier Screening be Universal? Disclosures Research funding from Natera Mary E Norton MD University of California, San Francisco Antepartum and Intrapartum Management June 15, 2017 Burden
Should Universal Carrier Screening be Universal? Disclosures Research funding from Natera Mary E Norton MD University of California, San Francisco Antepartum and Intrapartum Management June 15, 2017 Burden
Increased Density of the Thalamus on CT Scans in Patients with GM2 Gangliosidoses
 125 Increased Density of the Thalamus on CT Scans in Patients with GM2 Gangliosidoses Jan Brismar 1 Gudrun Brismar 2 Robert Coates 1 Generoso Gascon 3 Pinar Ozand 3 In 13 patients, the GM2 gangliosidoses,
125 Increased Density of the Thalamus on CT Scans in Patients with GM2 Gangliosidoses Jan Brismar 1 Gudrun Brismar 2 Robert Coates 1 Generoso Gascon 3 Pinar Ozand 3 In 13 patients, the GM2 gangliosidoses,
The role of the laboratory in diagnosing lysosomal disorders
 The role of the laboratory in diagnosing lysosomal disorders Dr Guy Besley, formerly Willink Biochemical Genetics Unit, Manchester Children s Hospital, Manchester M27 4HA, UK. Lysosomal disorders What
The role of the laboratory in diagnosing lysosomal disorders Dr Guy Besley, formerly Willink Biochemical Genetics Unit, Manchester Children s Hospital, Manchester M27 4HA, UK. Lysosomal disorders What
Abdallah Q& Razi. Faisal
 27 & Ahmad Attari م ح م د ي وس ف Abdallah Q& Razi Faisal Sphingophospolipids - The backbone of sphingophospholipids is sphingosine, unlike glycerophospholipids with a glycerol as the backbone. Which contains
27 & Ahmad Attari م ح م د ي وس ف Abdallah Q& Razi Faisal Sphingophospolipids - The backbone of sphingophospholipids is sphingosine, unlike glycerophospholipids with a glycerol as the backbone. Which contains
TEST INFORMATION Test: CarrierMap GEN (Genotyping) Panel: CarrierMap Expanded Diseases Tested: 311 Genes Tested: 299 Mutations Tested: 2647
 Panel: CarrierMap Expanded Diseases Tested: 311 Genes Tested: 299 Mutations Tested: 2647") Ordering Practice Jane Smith John Smith Practice Code: 675 Miller MD 374 Broadway New York, NY 10000 Physician: Dr. Frank Miller Report Generated: 2016-02-03 DOB: 1973-02-19 Gender: Female Ethnicity: European
Ordering Practice Jane Smith John Smith Practice Code: 675 Miller MD 374 Broadway New York, NY 10000 Physician: Dr. Frank Miller Report Generated: 2016-02-03 DOB: 1973-02-19 Gender: Female Ethnicity: European
Lysosomal Acid Lipase Deficiency (LAL-D)
") Lysosomal Acid Lipase Deficiency (LAL-D) What is LAL-D? Lysosomal acid lipase deficiency (LAL-D) is a rare, chronic, progressive inherited disorder. It affects the body s ability to produce an enzyme called
Lysosomal Acid Lipase Deficiency (LAL-D) What is LAL-D? Lysosomal acid lipase deficiency (LAL-D) is a rare, chronic, progressive inherited disorder. It affects the body s ability to produce an enzyme called
Peroxisomal Disorders
 Peroxisomal Disorders George Gray Birmingham Childrens Hospital Peroxisomal Disorders Peroxisomes are large single membrane bound organelles that are present in the cytoplasm of all cells. They are formed
Peroxisomal Disorders George Gray Birmingham Childrens Hospital Peroxisomal Disorders Peroxisomes are large single membrane bound organelles that are present in the cytoplasm of all cells. They are formed
Tay Sachs, Cystic Fibrosis, Sickle Cell Anemia and PKU. Tay Sachs Disease (also called Hexosaminidase deficiency)
") Tay Sachs, Cystic Fibrosis, Sickle Cell Anemia and PKU Tay Sachs Disease (also called Hexosaminidase deficiency) Introduction 1. Tay Sachs is a rare condition named after 2 physicians, Tay and Sachs, who
Tay Sachs, Cystic Fibrosis, Sickle Cell Anemia and PKU Tay Sachs Disease (also called Hexosaminidase deficiency) Introduction 1. Tay Sachs is a rare condition named after 2 physicians, Tay and Sachs, who
Genetic screening. Martin Delatycki
 7 Genetic screening Martin Delatycki Case study 1 Vanessa and John are planning a family. They see their general practitioner and ask whether they should have any tests prior to falling pregnant to maximise
7 Genetic screening Martin Delatycki Case study 1 Vanessa and John are planning a family. They see their general practitioner and ask whether they should have any tests prior to falling pregnant to maximise
Cardiovascular Genetics Clinic Vascular Questionnaire
 Name: Address: Home Phone: Cell Phone: Email Address: Date of Birth: Primary Care Physician: Why have you been referred for a Cardiovascular Genetics Appointment? Have you had a genetics evaluation? If
Name: Address: Home Phone: Cell Phone: Email Address: Date of Birth: Primary Care Physician: Why have you been referred for a Cardiovascular Genetics Appointment? Have you had a genetics evaluation? If
Citric Acid Cycle and Oxidative Phosphorylation
 Citric Acid Cycle and Oxidative Phosphorylation Page by: OpenStax Summary The Citric Acid Cycle In eukaryotic cells, the pyruvate molecules produced at the end of glycolysis are transported into mitochondria,
Citric Acid Cycle and Oxidative Phosphorylation Page by: OpenStax Summary The Citric Acid Cycle In eukaryotic cells, the pyruvate molecules produced at the end of glycolysis are transported into mitochondria,
DNA Day Illinois 2013 Webinar: Newborn Screening and Family Health History. Tuesday, April 16, 2013
 DNA Day Illinois 2013 Webinar: Newborn Screening and Family Health History Tuesday, April 16, 2013 Objectives Recognize the importance & impact of newborn screening Describe the process of newborn screening
DNA Day Illinois 2013 Webinar: Newborn Screening and Family Health History Tuesday, April 16, 2013 Objectives Recognize the importance & impact of newborn screening Describe the process of newborn screening
Human Genetic Diseases (non mutation)
") mutation) Pedigrees mutation) 1. Autosomal recessive inheritance: this is the inheritance of a disease through a recessive allele. In order for the person to have the condition they would have to be homozygous
mutation) Pedigrees mutation) 1. Autosomal recessive inheritance: this is the inheritance of a disease through a recessive allele. In order for the person to have the condition they would have to be homozygous
Cardiovascular Genetics Clinic Arrhythmia Questionnaire
 Name: Address: Home Phone: Cell Phone: Email Address: Date of Birth: Primary Care Physician: Why have you been referred for a Cardiovascular Genetics Appointment? Have you had a genetics evaluation? If
Name: Address: Home Phone: Cell Phone: Email Address: Date of Birth: Primary Care Physician: Why have you been referred for a Cardiovascular Genetics Appointment? Have you had a genetics evaluation? If
PATIENT EDUCATION. carrier screening INFORMATION
 PATIENT EDUCATION carrier screening INFORMATION carrier screening AT A GLANCE Why is carrier screening recommended? Carrier screening is one of many tests that can help provide information to you and your
PATIENT EDUCATION carrier screening INFORMATION carrier screening AT A GLANCE Why is carrier screening recommended? Carrier screening is one of many tests that can help provide information to you and your
Citric Acid Cycle and Oxidative Phosphorylation
 Citric Acid Cycle and Oxidative Phosphorylation Bởi: OpenStaxCollege The Citric Acid Cycle In eukaryotic cells, the pyruvate molecules produced at the end of glycolysis are transported into mitochondria,
Citric Acid Cycle and Oxidative Phosphorylation Bởi: OpenStaxCollege The Citric Acid Cycle In eukaryotic cells, the pyruvate molecules produced at the end of glycolysis are transported into mitochondria,
Newborn screening for congenital metabolic diseases Optional out-of-pocket tests Information Sheet
 Newborn screening for congenital metabolic diseases Optional out-of-pocket tests Information Sheet Website:www.tipn.org.tw Telephone:(02)85962050 Ext. 401-403 Service line:(02)85962065 Fax:(02)85962067
Newborn screening for congenital metabolic diseases Optional out-of-pocket tests Information Sheet Website:www.tipn.org.tw Telephone:(02)85962050 Ext. 401-403 Service line:(02)85962065 Fax:(02)85962067
Orofacial function of persons having. Report from questionnaires. Salla disease
 7-- Orofacial function of persons having Salla disease Report from questionnaires The survey comprises questionnaires. Synonyms: Sialic acid storage disorder, SIASD, Sialuria Finnish type. Estimated occurrence:
7-- Orofacial function of persons having Salla disease Report from questionnaires The survey comprises questionnaires. Synonyms: Sialic acid storage disorder, SIASD, Sialuria Finnish type. Estimated occurrence:
Genetic Disorders. n A genetic disorder is an abnormality
 + GENETIC DISORDERS + Genetic Disorders n A genetic disorder is an abnormality in an individual's DNA. Abnormalities can range from a small mutation in a single gene to the addition or subtraction of an
+ GENETIC DISORDERS + Genetic Disorders n A genetic disorder is an abnormality in an individual's DNA. Abnormalities can range from a small mutation in a single gene to the addition or subtraction of an
Pedigree. Tracking Genetic Traits: How it s done!
 Pedigree Tracking Genetic Traits: How it s done! REVIEW Many traits in humans are controlled by genes. Some of these traits are common features like eye color, straight or curly hair, baldness, attached
Pedigree Tracking Genetic Traits: How it s done! REVIEW Many traits in humans are controlled by genes. Some of these traits are common features like eye color, straight or curly hair, baldness, attached
Newborn Screen & Development Facts about the genetic diseases new since March 2006 (Excluding Cystic Fibrosis)
") Newborn Screen & Development Facts about the genetic diseases new since March 2006 (Excluding Cystic Fibrosis) 1) Argininosuccinic acidemia (ASA) a) Incidence: ~1 in 70,000 b) Deficiency in an enzyme of
Newborn Screen & Development Facts about the genetic diseases new since March 2006 (Excluding Cystic Fibrosis) 1) Argininosuccinic acidemia (ASA) a) Incidence: ~1 in 70,000 b) Deficiency in an enzyme of
Human Genetic Diseases (Ch. 15)
") Human Genetic Diseases (Ch. 15) 1 2 2006-2007 3 4 5 6 Genetic counseling Pedigrees can help us understand the past & predict the future Thousands of genetic disorders are inherited as simple recessive
Human Genetic Diseases (Ch. 15) 1 2 2006-2007 3 4 5 6 Genetic counseling Pedigrees can help us understand the past & predict the future Thousands of genetic disorders are inherited as simple recessive
The exact cause of sarcoidosis is unknown. However, gender, race, and genetics can increase the risk of developing the condition:
 What is sarcoidosis? Sarcoidosis is an inflammatory disease in which granulomas, or clumps of inflammatory cells, form in various organs. This causes organ inflammation. Sarcoidosis may be triggered by
What is sarcoidosis? Sarcoidosis is an inflammatory disease in which granulomas, or clumps of inflammatory cells, form in various organs. This causes organ inflammation. Sarcoidosis may be triggered by
Fatty Acid Oxidation Disorders
 Genetic Fact Sheets for Parents Fatty Acid Oxidation Disorders Screening, Technology, and Research in Genetics is a multi-state project to improve information about the financial, ethical, legal, and social
Genetic Fact Sheets for Parents Fatty Acid Oxidation Disorders Screening, Technology, and Research in Genetics is a multi-state project to improve information about the financial, ethical, legal, and social
Spinal Muscular Atrophy: Case Study. Spinal muscular atrophy (SMA) is a fairly common genetic disorder, affecting
 is a fairly common genetic disorder, affecting") Spinal Muscular Atrophy: Case Study Spinal muscular atrophy (SMA) is a fairly common genetic disorder, affecting approximately one in 6,000 babies. It is estimated that one in every 40 Americans carries
Spinal Muscular Atrophy: Case Study Spinal muscular atrophy (SMA) is a fairly common genetic disorder, affecting approximately one in 6,000 babies. It is estimated that one in every 40 Americans carries
A CASE OF GIANT AXONAL NEUROPATHY HEMANANTH T SECOND YEAR POST GRADUATE IN PAEDIATRICS INSTITUTE OF SOCIAL PAEDIATRICS GOVERNMENT STANLEY HOSPITAL
 A CASE OF GIANT AXONAL NEUROPATHY HEMANANTH T SECOND YEAR POST GRADUATE IN PAEDIATRICS INSTITUTE OF SOCIAL PAEDIATRICS GOVERNMENT STANLEY HOSPITAL CASE HISTORY Nine year old male child Second born Born
A CASE OF GIANT AXONAL NEUROPATHY HEMANANTH T SECOND YEAR POST GRADUATE IN PAEDIATRICS INSTITUTE OF SOCIAL PAEDIATRICS GOVERNMENT STANLEY HOSPITAL CASE HISTORY Nine year old male child Second born Born
Genetic Diseases. Genetic diseases occur when an individual s DNA has one or more abnormalities.
 Genetic Diseases Genetic diseases occur when an individual s DNA has one or more abnormalities. Autosomal dominant genetic disorders refer to diseases in which only one copy, the dominant allele, is needed
Genetic Diseases Genetic diseases occur when an individual s DNA has one or more abnormalities. Autosomal dominant genetic disorders refer to diseases in which only one copy, the dominant allele, is needed
Human Genetic Diseases. AP Biology
 Human Genetic Diseases 1 3 4 2 5 2006-2007 6 Pedigree analysis n Pedigree analysis reveals Mendelian patterns in human inheritance u data mapped on a family tree = male = female = male w/ trait = female
Human Genetic Diseases 1 3 4 2 5 2006-2007 6 Pedigree analysis n Pedigree analysis reveals Mendelian patterns in human inheritance u data mapped on a family tree = male = female = male w/ trait = female
AFL NSW/ACT Exemption & Dispensation Policy
 AFL NSW/ACT Exemption & Dispensation Policy Last reviewed 14 May 2012. AFL NSW/ACT s policy is that age exceptions and/or dispensations will only exist in cases of disability These exemptions require approval
AFL NSW/ACT Exemption & Dispensation Policy Last reviewed 14 May 2012. AFL NSW/ACT s policy is that age exceptions and/or dispensations will only exist in cases of disability These exemptions require approval
Lipid Metabolism. Remember fats?? Triacylglycerols - major form of energy storage in animals
 Remember fats?? Triacylglycerols - major form of energy storage in animals Your energy reserves: ~0.5% carbs (glycogen + glucose) ~15% protein (muscle, last resort) ~85% fat Why use fat for energy? 1 gram
Remember fats?? Triacylglycerols - major form of energy storage in animals Your energy reserves: ~0.5% carbs (glycogen + glucose) ~15% protein (muscle, last resort) ~85% fat Why use fat for energy? 1 gram
The breakdown of fats to provide energy occurs in segregated membrane-bound compartments
 CPT1a deficiency The breakdown of fats to provide energy occurs in segregated membrane-bound compartments of the cell known as mitochondria. Carnitine palmitoyltransferase Ia (CPT1a) is a protein that
CPT1a deficiency The breakdown of fats to provide energy occurs in segregated membrane-bound compartments of the cell known as mitochondria. Carnitine palmitoyltransferase Ia (CPT1a) is a protein that
Human Genetic Diseases. AP Biology
 Human Genetic Diseases 1 2 2006-2007 3 4 5 6 Pedigree analysis Pedigree analysis reveals Mendelian patterns in human inheritance data mapped on a family tree = male = female = male w/ trait = female w/
Human Genetic Diseases 1 2 2006-2007 3 4 5 6 Pedigree analysis Pedigree analysis reveals Mendelian patterns in human inheritance data mapped on a family tree = male = female = male w/ trait = female w/
Genetic Disorders. and. blood vessels the and. How many genes are affected by this deletion? Turner s Syndrome- An incomplete or missing chromosome
 Genetic Disorders A genetic disorder is an abnormality in the. They can range for a deletion of a gene to the deletion of an entire chromosome. List the types of genetic disorders. Williams Syndrome- A
Genetic Disorders A genetic disorder is an abnormality in the. They can range for a deletion of a gene to the deletion of an entire chromosome. List the types of genetic disorders. Williams Syndrome- A
THIAMINE TRANSPORTER TYPE 2 DEFICIENCY
 THIAMINE TRANSPORTER TYPE 2 DEFICIENCY WHAT IS THE THIAMINE TRANSPORTER TYPE 2 DEFICIENCY (hthtr2)? The thiamine transporter type 2 deficiency (hthtr2) is a inborn error of thiamine metabolism caused by
THIAMINE TRANSPORTER TYPE 2 DEFICIENCY WHAT IS THE THIAMINE TRANSPORTER TYPE 2 DEFICIENCY (hthtr2)? The thiamine transporter type 2 deficiency (hthtr2) is a inborn error of thiamine metabolism caused by
Klinefelter syndrome ( 47, XXY )
") Sex Chromosome Abnormalities, Sex Chromosome Aneuploidy It has been estimated that, overall, approximately one in 400 infants have some form of sex chromosome aneuploidy. A thorough discussion of sex chromosomes
Sex Chromosome Abnormalities, Sex Chromosome Aneuploidy It has been estimated that, overall, approximately one in 400 infants have some form of sex chromosome aneuploidy. A thorough discussion of sex chromosomes
MEMBRANE LIPIDS I and II: GLYCEROPHOSPHOLIPIDS AND SPHINGOLIPIDS
 December 6, 2011 Lecturer: Eileen M. Lafer MEMBRANE LIPIDS I and II: GLYCEROPHOSPHOLIPIDS AND SPHINGOLIPIDS Reading: Stryer Edition 6: Chapter 26 Images: All images in these notes were taken from Lehninger,
December 6, 2011 Lecturer: Eileen M. Lafer MEMBRANE LIPIDS I and II: GLYCEROPHOSPHOLIPIDS AND SPHINGOLIPIDS Reading: Stryer Edition 6: Chapter 26 Images: All images in these notes were taken from Lehninger,
1/28/2019. OSF HealthCare INI Care Center Team. Neuromuscular Disease: Muscular Dystrophy. OSF HealthCare INI Care Center Team: Who are we?
 Neuromuscular Disease: Muscular Dystrophy Muscular Dystrophy Association (MDA) and OSF HealthCare Illinois Neurological Institute (INI) Care Center Team The Neuromuscular clinic is a designated MDA Care
Neuromuscular Disease: Muscular Dystrophy Muscular Dystrophy Association (MDA) and OSF HealthCare Illinois Neurological Institute (INI) Care Center Team The Neuromuscular clinic is a designated MDA Care
Muscular System. Disorders & Conditions
 Muscular System Disorders & Conditions Fibromyalgia Fibromyalgia is a disorder characterized by widespread musculoskeletal pain accompanied by fatigue, sleep, memory and mood issues. Often is described
Muscular System Disorders & Conditions Fibromyalgia Fibromyalgia is a disorder characterized by widespread musculoskeletal pain accompanied by fatigue, sleep, memory and mood issues. Often is described
Amino Acid Disorders. What is ASAL deficiency? Genetic Fact Sheets for Parents
 Genetic Fact Sheets for Parents Amino Acid Disorders Screening, Technology, and Research in Genetics is a multi-state project to improve information about the financial, ethical, legal, and social issues
Genetic Fact Sheets for Parents Amino Acid Disorders Screening, Technology, and Research in Genetics is a multi-state project to improve information about the financial, ethical, legal, and social issues
Update on the Genetics of Ataxia. Vicki Wheelock MD UC Davis Department of Neurology GHPP Clinic
 Update on the Genetics of Ataxia Vicki Wheelock MD UC Davis Department of Neurology GHPP Clinic Outline Definitions Review of genetics Autosomal Dominant cerebellar ataxias Autosomal Recessive cerebellar
Update on the Genetics of Ataxia Vicki Wheelock MD UC Davis Department of Neurology GHPP Clinic Outline Definitions Review of genetics Autosomal Dominant cerebellar ataxias Autosomal Recessive cerebellar
Amino Acid Disorders. Disorder name: Tyrosinemia, type 1. What is tyrosinemia 1? Genetic Fact Sheets for Parents
 Genetic Fact Sheets for Parents Amino Acid Disorders Screening, Technology, and Research in Genetics is a multi-state project to improve information about the financial, ethical, legal, and social issues
Genetic Fact Sheets for Parents Amino Acid Disorders Screening, Technology, and Research in Genetics is a multi-state project to improve information about the financial, ethical, legal, and social issues
CONSULTATION ADMITTANCE FORM
 CONSULTATION ADMITTANCE FORM Last Name: First Name: Address: City Postal Code: Home Phone: Work Phone: Age: Birth date (dd/mm/yr): Sex: M / F Height Weight Occupation: Alberta Health Care #: PLEASE CHECK
CONSULTATION ADMITTANCE FORM Last Name: First Name: Address: City Postal Code: Home Phone: Work Phone: Age: Birth date (dd/mm/yr): Sex: M / F Height Weight Occupation: Alberta Health Care #: PLEASE CHECK
Admission Medical Information Form
 Return Form to: Admission Medical Information Form Part I: To Be Completed by Family or Staff of Birth: Sex: M F Race: Marital Status: Home Address: Phone Number: Number/Street City State Zip Last Time
Return Form to: Admission Medical Information Form Part I: To Be Completed by Family or Staff of Birth: Sex: M F Race: Marital Status: Home Address: Phone Number: Number/Street City State Zip Last Time
HEREDITARY METABOLIC DISEASES
 HEREDITARY METABOLIC DISEASES Particular risk factors are: Advanced maternal age (e.g. Down's syndrome) Family history of inherited diseases (e.g. fragile X syndrome, Huntington's chorea) Previous child
HEREDITARY METABOLIC DISEASES Particular risk factors are: Advanced maternal age (e.g. Down's syndrome) Family history of inherited diseases (e.g. fragile X syndrome, Huntington's chorea) Previous child
Cerebral Palsy. By:Carrie Siders and Kelsey Hampsey. 3rd hour.
 Cerebral Palsy By:Carrie Siders and Kelsey Hampsey 3rd hour. What is Cerebral Palsy? Cerebral palsy is a physical disability It affects movement and posture It is a permanent life-long condition does not
Cerebral Palsy By:Carrie Siders and Kelsey Hampsey 3rd hour. What is Cerebral Palsy? Cerebral palsy is a physical disability It affects movement and posture It is a permanent life-long condition does not
WOMENCARE. Herpes. Source: PDR.net Page 1 of 8. A Healthy Woman is a Powerful Woman (407)
") WOMENCARE A Healthy Woman is a Powerful Woman (407) 898-1500 Herpes Basics: Herpes is a common viral disease characterized by painful blisters of the mouth or genitals. The herpes simplex virus (HSV) causes
WOMENCARE A Healthy Woman is a Powerful Woman (407) 898-1500 Herpes Basics: Herpes is a common viral disease characterized by painful blisters of the mouth or genitals. The herpes simplex virus (HSV) causes
Familial Mediterranean Fever
 Familial Mediterranean Fever FMF most often occurs in individuals of Mediterranean and Middle Eastern descent, and the first episodes typically begin in childhood Fast Facts FMF causes episodic fevers
Familial Mediterranean Fever FMF most often occurs in individuals of Mediterranean and Middle Eastern descent, and the first episodes typically begin in childhood Fast Facts FMF causes episodic fevers
Movement Disorders. Psychology 372 Physiological Psychology. Background. Myasthenia Gravis. Many Types
 Background Movement Disorders Psychology 372 Physiological Psychology Steven E. Meier, Ph.D. Listen to the audio lecture while viewing these slides Early Studies Found some patients with progressive weakness
Background Movement Disorders Psychology 372 Physiological Psychology Steven E. Meier, Ph.D. Listen to the audio lecture while viewing these slides Early Studies Found some patients with progressive weakness
Original Article:http://www.mayoclinic.com/health/sickle-cell-anemia/DS00324
 1 Original Article:http://www.mayoclinic.com/health/sickle-cell-anemia/DS00324 Sickle Cell Anemia [Mayo Clinic Staff. Sickle cell anemia. MayoClinic.com; Mar 28, 2007: http://www.mayoclinic.com/health/sickle-cell-anemia/ds00324]
1 Original Article:http://www.mayoclinic.com/health/sickle-cell-anemia/DS00324 Sickle Cell Anemia [Mayo Clinic Staff. Sickle cell anemia. MayoClinic.com; Mar 28, 2007: http://www.mayoclinic.com/health/sickle-cell-anemia/ds00324]
There are four different forms of Tarui disease, which are classed by their signs and symptoms and age of presentation.
 Tarui Disease Tarui disease (also known as phosphofructokinase deficiency, or glycogen storage disease type VII) was first described in 1965 by the Japanese physician Seiichiro Tarui and his co-workers.
Tarui Disease Tarui disease (also known as phosphofructokinase deficiency, or glycogen storage disease type VII) was first described in 1965 by the Japanese physician Seiichiro Tarui and his co-workers.
Corporate Medical Policy
 Corporate Medical Policy File Name: Origination: Last CAP Review: Next CAP Review: Last Review: nusinersen_spinraza 03/2017 10/2017 10/2018 10/2017 Description of Procedure or Service Spinal muscular atrophy
Corporate Medical Policy File Name: Origination: Last CAP Review: Next CAP Review: Last Review: nusinersen_spinraza 03/2017 10/2017 10/2018 10/2017 Description of Procedure or Service Spinal muscular atrophy
FABRY DISEASE 12/30/2012. Ataxia-Telangiectasia. Ophthalmologic Signs of Genetic Neurological Disease
 Ophthalmologic Signs of Genetic Neurological Disease ES ROACH,MD. Ophthalmologic Signs of Genetic Neurological Disease Conjunctival lesions Corneal lesions Lesions of iris & lens Retinal vascular lesions
Ophthalmologic Signs of Genetic Neurological Disease ES ROACH,MD. Ophthalmologic Signs of Genetic Neurological Disease Conjunctival lesions Corneal lesions Lesions of iris & lens Retinal vascular lesions
City State Zip Code. Ethnic Background: Caucasian African-American Asian Hispanic Native American. Previous. Hobbies/Leisure activities:,,,
 History # UPIN # (Please leave blank) Name: First M.I. Last Address: Street (Apt #) City State Zip Code Phone number: ( ) ( ) Home Business Birth Date: / / Day-Month-Year Gender: M F Marital status: (Maiden
History # UPIN # (Please leave blank) Name: First M.I. Last Address: Street (Apt #) City State Zip Code Phone number: ( ) ( ) Home Business Birth Date: / / Day-Month-Year Gender: M F Marital status: (Maiden
How Cells Release Chemical Energy. Chapter 7
 How Cells Release Chemical Energy Chapter 7 7.1 Overview of Carbohydrate Breakdown Pathways All organisms (including photoautotrophs) convert chemical energy of organic compounds to chemical energy of
How Cells Release Chemical Energy Chapter 7 7.1 Overview of Carbohydrate Breakdown Pathways All organisms (including photoautotrophs) convert chemical energy of organic compounds to chemical energy of
Biology 3201 Nervous System # 7: Nervous System Disorders
 Biology 3201 Nervous System # 7: Nervous System Disorders Alzheimer's Disease first identified by German physician, Alois Alzheimer, in 1906 most common neurodegenerative disease two thirds of cases of
Biology 3201 Nervous System # 7: Nervous System Disorders Alzheimer's Disease first identified by German physician, Alois Alzheimer, in 1906 most common neurodegenerative disease two thirds of cases of
Genetic Disorders. PART ONE: Detecting Genetic Disorders. Amniocentesis Chorionic villus sampling Karyotype Triple Screen Blood Test
 Genetic Disorders PART ONE: Detecting Genetic Disorders Amniocentesis Chorionic villus sampling Karyotype Triple Screen Blood Test Amniocentesis A technique for determining genetic abnormalities in a fetus
Genetic Disorders PART ONE: Detecting Genetic Disorders Amniocentesis Chorionic villus sampling Karyotype Triple Screen Blood Test Amniocentesis A technique for determining genetic abnormalities in a fetus
CHAPTER 3 1/21/2016. Typical Bacteria Cell. The Cell
 CHAPTER 3 The Cell Chapter 3 Learning Objectives Compare and contrast the features of prokaryotic and eukaryotic cells. Explain why surface area-to-volume ratios constrain cell size. Contrast light microscopy
CHAPTER 3 The Cell Chapter 3 Learning Objectives Compare and contrast the features of prokaryotic and eukaryotic cells. Explain why surface area-to-volume ratios constrain cell size. Contrast light microscopy
Types of Muscle. Skeletal striated & voluntary Smooth involuntary Cardiac - heart
 Muscular System Types of Muscle Skeletal striated & voluntary Smooth involuntary Cardiac - heart The word striated means striped. Skeletal muscle appears striped under a microscope. Muscles and Muscle
Muscular System Types of Muscle Skeletal striated & voluntary Smooth involuntary Cardiac - heart The word striated means striped. Skeletal muscle appears striped under a microscope. Muscles and Muscle
Asking questions Misunderstood questions or inappropriate responses Presence of a aid Sign language or
 1 Chapter 45 The Challenged Patient 2 Hearing Impairments 3 Types of Hearing Impairments Deafness: a blockage of the transmission of sound waves through the external ear canal to the middle or inner ear.
1 Chapter 45 The Challenged Patient 2 Hearing Impairments 3 Types of Hearing Impairments Deafness: a blockage of the transmission of sound waves through the external ear canal to the middle or inner ear.
Clinical Approach to Diagnosis of Lysosomal Storage Diseases
 Clinical Approach to Diagnosis of Lysosomal Storage Diseases M. Rohrbach, MD, PhD FMH Pädiatrie und FMH Medizinische Genetik Abteilung Stoffwechsel Universitätskinderklinik Zürich Lysosomal storage disorders
Clinical Approach to Diagnosis of Lysosomal Storage Diseases M. Rohrbach, MD, PhD FMH Pädiatrie und FMH Medizinische Genetik Abteilung Stoffwechsel Universitätskinderklinik Zürich Lysosomal storage disorders
ataxia, head tremors and mild inappentence, was given palliative care
 2013-5-2 Cerebellum, Spleen-Raccoon Ahmed M. Abubakar BOVINE PATHOLOGY CONTRIBUTING INSTITUTION :College of Veterinary Medicine UC Davies Signalment: Wild-caught juvenile male raccoon, ( Procyon lotor)
2013-5-2 Cerebellum, Spleen-Raccoon Ahmed M. Abubakar BOVINE PATHOLOGY CONTRIBUTING INSTITUTION :College of Veterinary Medicine UC Davies Signalment: Wild-caught juvenile male raccoon, ( Procyon lotor)
Fatty Acid Oxidation Disorders
 Genetic Fact Sheets for Parents Fatty Acid Oxidation Disorders Screening, Technology, and Research in Genetics is a multi-state project to improve information about the financial, ethical, legal, and social
Genetic Fact Sheets for Parents Fatty Acid Oxidation Disorders Screening, Technology, and Research in Genetics is a multi-state project to improve information about the financial, ethical, legal, and social
Autoimmune lymphoproliferative syndrome (ALPS)
") ALPS Autoimmune lymphoproliferative syndrome (ALPS) Information for families hello@piduk.org 0800 987 8986 www.piduk.org About this leaflet This leaflet is designed to help answer the questions families
ALPS Autoimmune lymphoproliferative syndrome (ALPS) Information for families hello@piduk.org 0800 987 8986 www.piduk.org About this leaflet This leaflet is designed to help answer the questions families
Chapter Goal. Learning Objectives 9/12/2012. Chapter 36. Geriatrics. Use assessment findings to formulate management plan for geriatric patients
 Chapter 36 Geriatrics Chapter Goal Use assessment findings to formulate management plan for geriatric patients Learning Objectives Describe dependent & independent living environments Identify local resources
Chapter 36 Geriatrics Chapter Goal Use assessment findings to formulate management plan for geriatric patients Learning Objectives Describe dependent & independent living environments Identify local resources
Alpha-1 Antitrypsin Deficiency: Liver Disease
 Alpha-1 Antitrypsin Deficiency: Liver Disease Who is at risk to develop Alpha-1 liver disease? Alpha-1 liver disease may affect children and adults who have abnormal Alpha-1 antitrypsin genes. Keys to
Alpha-1 Antitrypsin Deficiency: Liver Disease Who is at risk to develop Alpha-1 liver disease? Alpha-1 liver disease may affect children and adults who have abnormal Alpha-1 antitrypsin genes. Keys to
 https://www.printo.it/pediatric-rheumatology/gb/intro Majeed Syndrome Version of 2016 1. WHAT IS MAJEED 1.1 What is it? Majeed syndrome is a rare genetic disease. Affected children suffer from Chronic
https://www.printo.it/pediatric-rheumatology/gb/intro Majeed Syndrome Version of 2016 1. WHAT IS MAJEED 1.1 What is it? Majeed syndrome is a rare genetic disease. Affected children suffer from Chronic
MULTIPLE SCLEROSIS INTRODUCTION OBJECTIVES. When the student has finished this module, he/she will be able to:
 MULTIPLE SCLEROSIS INTRODUCTION Multiple sclerosis (MS) is a chronic disease of the nervous system. Multiple sclerosis causes inflammation and damage to the protective coatings in the brain and the nerves.
MULTIPLE SCLEROSIS INTRODUCTION Multiple sclerosis (MS) is a chronic disease of the nervous system. Multiple sclerosis causes inflammation and damage to the protective coatings in the brain and the nerves.
 http://www.ncbi.nlm.nih.gov/pubmedhealth/pmh0002150/ PKU; Phenylketonuria Phenylketonuria (PKU) is a rare condition in which a baby is born without the ability to properly break down an amino acid called
http://www.ncbi.nlm.nih.gov/pubmedhealth/pmh0002150/ PKU; Phenylketonuria Phenylketonuria (PKU) is a rare condition in which a baby is born without the ability to properly break down an amino acid called
Chapter 12. Ataxia-Telangiectasia
 Chapter 12 Ataxia-Telangiectasia Ataxia-Telangiectasia (A-T) is an inherited disease that affects several body systems, including the immune system. People with A-T have an unsteady, wobbly gait (ataxia)
Chapter 12 Ataxia-Telangiectasia Ataxia-Telangiectasia (A-T) is an inherited disease that affects several body systems, including the immune system. People with A-T have an unsteady, wobbly gait (ataxia)
CHRONIC CONDITIONS FYI
 CHRONIC CONDITIONS FYI AIDS More than 2,500 cases of HIV/AIDS have been identified in Nebraska. ALS (Amyotrophic Lateral Sclerosis) Approximately 95 people in Nebraska have ALS. As many as 800 Nebraskans
CHRONIC CONDITIONS FYI AIDS More than 2,500 cases of HIV/AIDS have been identified in Nebraska. ALS (Amyotrophic Lateral Sclerosis) Approximately 95 people in Nebraska have ALS. As many as 800 Nebraskans
SEX-LINKED INHERITANCE. Dr Rasime Kalkan
 SEX-LINKED INHERITANCE Dr Rasime Kalkan Human Karyotype Picture of Human Chromosomes 22 Autosomes and 2 Sex Chromosomes Autosomal vs. Sex-Linked Traits can be either: Autosomal: traits (genes) are located
SEX-LINKED INHERITANCE Dr Rasime Kalkan Human Karyotype Picture of Human Chromosomes 22 Autosomes and 2 Sex Chromosomes Autosomal vs. Sex-Linked Traits can be either: Autosomal: traits (genes) are located
Biol 219 Lec 7 Fall 2016
 Cellular Respiration: Harvesting Energy to form ATP Cellular Respiration and Metabolism Glucose ATP Pyruvate Lactate Acetyl CoA NAD + Introducing The Players primary substrate for cellular respiration
Cellular Respiration: Harvesting Energy to form ATP Cellular Respiration and Metabolism Glucose ATP Pyruvate Lactate Acetyl CoA NAD + Introducing The Players primary substrate for cellular respiration
Mitrochondrial Disease
 Mitrochondrial Disease - Information for patients, parents and families Neuromuscular and Neurometabolic Clinic McMaster University Medical Centre 1200 Main Street West Hamilton ON 905-521-7933 What is
Mitrochondrial Disease - Information for patients, parents and families Neuromuscular and Neurometabolic Clinic McMaster University Medical Centre 1200 Main Street West Hamilton ON 905-521-7933 What is
Sialic Acid Storage Diseases
 Sialic Acid Storage Diseases Class: BIOL 10001 Instructor: Dr. Vivegananthan Submitted by: Lyndsay Grover Date Submitted: Thursday March 24 th, 2011 Introduction to Sialic Acid Storage Diseases Sialic
Sialic Acid Storage Diseases Class: BIOL 10001 Instructor: Dr. Vivegananthan Submitted by: Lyndsay Grover Date Submitted: Thursday March 24 th, 2011 Introduction to Sialic Acid Storage Diseases Sialic
Overview of Neurodegeneration with Brain Iron Accumulation (NBIA) Disorders
 Disorders") Overview of Neurodegeneration with Brain Iron Accumulation (NBIA) Disorders What is NBIA? Neurodegeneration with Brain Iron Accumulation is a group of inherited neurological disorders characterized by
Overview of Neurodegeneration with Brain Iron Accumulation (NBIA) Disorders What is NBIA? Neurodegeneration with Brain Iron Accumulation is a group of inherited neurological disorders characterized by
Natural History of JNCL and other NCLs
 Natural History of JNCL and other NCLs Jonathan W. Mink, MD PhD Departments of Neurology, Neurobiology & Anatomy, Brain & Cognitive Sciences, and Pediatrics University of Rochester Neuronal Ceroid Lipofuscinosis
Natural History of JNCL and other NCLs Jonathan W. Mink, MD PhD Departments of Neurology, Neurobiology & Anatomy, Brain & Cognitive Sciences, and Pediatrics University of Rochester Neuronal Ceroid Lipofuscinosis
Marah Bitar. Faisal Nimri ... Nafeth Abu Tarboosh
 8 Marah Bitar Faisal Nimri... Nafeth Abu Tarboosh Summary of the 8 steps of citric acid cycle Step 1. Acetyl CoA joins with a four-carbon molecule, oxaloacetate, releasing the CoA group and forming a six-carbon
8 Marah Bitar Faisal Nimri... Nafeth Abu Tarboosh Summary of the 8 steps of citric acid cycle Step 1. Acetyl CoA joins with a four-carbon molecule, oxaloacetate, releasing the CoA group and forming a six-carbon
Genetics of Thalassemia
 Genetics of Thalassemia Submitted by : Raya Samir Al- Hayaly Sura Zuhair Salih Saad Ghassan Al- Dulaimy Saad Farouq Kassir Sama Naal Salouha Zahraa Jasim Al- Aarajy Supervised by : Dr. Kawkab Adris Mahmod
Genetics of Thalassemia Submitted by : Raya Samir Al- Hayaly Sura Zuhair Salih Saad Ghassan Al- Dulaimy Saad Farouq Kassir Sama Naal Salouha Zahraa Jasim Al- Aarajy Supervised by : Dr. Kawkab Adris Mahmod
Cellular Pathways That Harvest Chemical Energy. Cellular Pathways That Harvest Chemical Energy. Cellular Pathways In General
 Cellular Pathways That Harvest Chemical Energy A. Obtaining Energy and Electrons from Glucose Lecture Series 12 Cellular Pathways That Harvest Chemical Energy B. An Overview: Releasing Energy from Glucose
Cellular Pathways That Harvest Chemical Energy A. Obtaining Energy and Electrons from Glucose Lecture Series 12 Cellular Pathways That Harvest Chemical Energy B. An Overview: Releasing Energy from Glucose
OBJECTIVES. Unit 7:5 PROPERTIES OR CHARACTERISTICS OF MUSCLES. Introduction. 3 Kinds of Muscles. 3 Kinds of Muscles 4/17/2018 MUSCULAR SYSTEM
 OBJECTIVES Unit 7:5 MUSCULAR SYSTEM Compare the three main kinds of muscles by describing the action of each Differentiate between voluntary and involuntary muscles List at least three functions of muscles
OBJECTIVES Unit 7:5 MUSCULAR SYSTEM Compare the three main kinds of muscles by describing the action of each Differentiate between voluntary and involuntary muscles List at least three functions of muscles
DISORDERS OF THE NERVOUS SYSTEM
 DISORDERS OF THE NERVOUS SYSTEM Bell Work What s your reaction time? Go to this website and check it out: https://www.justpark.com/creative/reaction-timetest/ Read the following brief article and summarize
DISORDERS OF THE NERVOUS SYSTEM Bell Work What s your reaction time? Go to this website and check it out: https://www.justpark.com/creative/reaction-timetest/ Read the following brief article and summarize
CHRONIC CONDITIONS FYI
 CHRONIC CONDITIONS FYI AIDS More than 2,500 cases of HIV/AIDS have been identified in Nebraska. Nationwide, this number is more than 1.2 million. ALS (Amyotrophic Lateral Sclerosis) There are 87 people
CHRONIC CONDITIONS FYI AIDS More than 2,500 cases of HIV/AIDS have been identified in Nebraska. Nationwide, this number is more than 1.2 million. ALS (Amyotrophic Lateral Sclerosis) There are 87 people
Review. Respiration. Glycolysis. Glycolysis is the decomposition (lysis) of glucose (glyco) to pyruvate (or pyruvic acid).
 of glucose (glyco) to pyruvate (or pyruvic acid).") Review Photosynthesis is the process of incorporating energy from light into energy-rich molecules like glucose. Respiration is the opposite process extracting that stored energy from glucose to form ATP
Review Photosynthesis is the process of incorporating energy from light into energy-rich molecules like glucose. Respiration is the opposite process extracting that stored energy from glucose to form ATP
Pompe Disease. Family Education Booklet. Division of Pediatric Genetics Metabolism and Genomic Medicine
 Pompe Disease Family Education Booklet Division of Pediatric Genetics Metabolism and Genomic Medicine Table of Contents What is Pompe disease?... 1 How common is Pompe disease?... 1 Basic genetic concepts...
Pompe Disease Family Education Booklet Division of Pediatric Genetics Metabolism and Genomic Medicine Table of Contents What is Pompe disease?... 1 How common is Pompe disease?... 1 Basic genetic concepts...