Protocol. This trial protocol has been provided by the authors to give readers additional information about their work.
|
|
|
- Thomasine Barrett
- 5 years ago
- Views:
Transcription
1 Protocol This trial protocol has been provided by the authors to give readers additional information about their work. Protocol for: Taylor PC, Keystone EC, van der Heijde D, et al. Baricitinib versus placebo or adalimumab in rheumatoid arthritis. N Engl J Med 2017;376: DOI: /NEJMoa
2 This supplement contains the following items: 1. Original protocol Pages Final protocol Pages Summary of changes Pages Original statistical analysis plan Pages Final statistical analysis plan Pages a. Summary of changes (revision history) Pages
3 I4V-MC-JADV Clinical Protocol Page 1 1. Protocol I4V-MC-JADV A Randomized, Double-Blind, Placebo- and Active-Controlled, Phase 3 Study Evaluating the Efficacy and Safety of Baricitinib in Patients with Moderately to Severely Active Rheumatoid Arthritis Who Have Had an Inadequate Response to Methotrexate Therapy Baricitinib () Study I4V-MC-JADV is a Phase 3, randomized, double-blind, placebo- and activecontrolled, parallel-group study of baricitinib for the treatment of moderately to severely active rheumatoid arthritis during a 52-week treatment period in patients with inadequate response to methotrexate therapy. Eli Lilly and Company Indianapolis, Indiana USA Protocol Electronically Signed and Approved by Lilly on date provided below. Approval Date: 27-Jul-2012 GMT
4 I4V-MC-JADV Clinical Protocol Page 2 Study Rationale 2. Synopsis Baricitinib () is an oral Janus kinase 1 (JAK1)/Janus kinase 2 (JAK2) selective inhibitor representing a potentially effective therapy for treatment of patients with moderately to severely active rheumatoid arthritis (RA). The rationale for the current study is to confirm the efficacy and to continue to define the safety profile of 4 mg baricitinib when administered once daily (QD) to patients with RA who have had an inadequate response to methotrexate (MTX) therapy. The safety and tolerability data from this study are intended to inform the current understanding of the benefit-risk relationship for baricitinib in patients with RA.
5 I4V-MC-JADV Clinical Protocol Page 3 Clinical Protocol Synopsis: Study I4V-MC-JADV Name of Investigational Product: Baricitinib () Title of Study: A Randomized, Double-Blind, Placebo- and Active-Controlled, Phase 3 Study Evaluating the Efficacy and Safety of Baricitinib in Patients with Moderately to Severely Active Rheumatoid Arthritis Who Have Had an Inadequate Response to Methotrexate Therapy Number of Planned Patients/Subjects: Phase of Development: 3 Entered: 1900 Enrolled/Randomized: 1280 (480 on baricitinib, 480 on placebo, 320 on Humira [adalimumab]) Completed: 1088 Length of Study: 2 years and 3 months Planned first patient visit: Oct 2012 Planned last patient visit: Jan 2015 Objectives: The primary objective of the study is to determine whether baricitinib is superior to placebo in the treatment of patients with moderately to severely active rheumatoid arthritis (RA) despite methotrexate treatment (ie, inadequate response to methotrexate [MTX-IR]), as assessed by the proportion of patients achieving a 20% improvement in American College of Rheumatology criteria (ACR20) at Week 12. The major secondary objectives of the study are to evaluate the efficacy of baricitinib versus placebo or adalimumab as assessed by: change from baseline to Week 24 in structural joint damage as measured by modified Total Sharp Score (mtss [van der Heijde method]) compared to placebo change from baseline to Week 12 in Health Assessment Questionnaire-Disability Index (HAQ-DI) score compared to placebo change from baseline to Week 12 in Disease Activity Score modified to include the 28 diarthroidal joint count (DAS28)-high-sensitivity C-reactive protein (hscrp) compared to placebo proportion of patients achieving ACR20 response at Week 12 compared to adalimumab change from baseline to Week 12 in duration of morning joint stiffness compared to placebo as collected in electronic diaries change from baseline to Week 12 in severity of morning joint stiffness compared to placebo as collected in electronic diaries change from baseline to Week 12 in worst tiredness compared to placebo as collected in electronic diaries change from baseline to Week 12 in worst pain compared to placebo as collected in electronic diaries The other secondary objectives of the study are as follows: to evaluate the efficacy of baricitinib as assessed by: o proportion of patients achieving DAS28-hsCRP 3.2 and DAS28-hsCRP <2.6 at Week 12, Week 24, and Week 52 o proportion of patients achieving HAQ-DI improvement 0.22 and 0.3 at Week 12, Week 24, and Week 52 o change from baseline to Week 16 and Week 52 in structural joint damage as measured by mtss o proportion of patients with mtss change 0 from baseline at Week 16, Week 24, and Week 52 o change from baseline to Week 16, Week 24, and Week 52 in joint space narrowing and bone erosion scores o proportion of patients achieving ACR20 at Week 24 and Week 52 o proportion of patients achieving 50% improvement in American College of Rheumatology criteria (ACR50) at Week 12, Week 24, and Week 52 o proportion of patients achieving 70% improvement in American College of Rheumatology criteria (ACR70) at Week 12, Week 24, and Week 52 o percentage change from baseline to Week 12, Week 24, and Week 52 in individual components
6 I4V-MC-JADV Clinical Protocol Page 4 of the ACR Core Set o change from baseline through Week 52 in DAS28-hsCRP o change from baseline through Week 52 in DAS28-erythrocyte sedimentation rate (ESR) o proportion of patients achieving DAS28-ESR 3.2 and DAS28-ESR <2.6 at Week 12, Week 24, and Week 52 o change from baseline to Week 24 and Week 52 in HAQ-DI score o change from baseline to Week 12, Week 24, and Week 52 in Clinical Disease Activity Index (CDAI) score o change from baseline to Week 12, Week 24, and Week 52 in Simplified Disease Activity Index (SDAI) score o proportion of patients achieving a CDAI score 2.8 at Week 12, Week 24, and Week 52 o proportion of patients achieving an SDAI score 3.3 at Week 12, Week 24, and Week 52 o proportion of patients achieving European League Against Rheumatism (EULAR) Responder Index based on the 28-joint count (EULAR28) at Week 12, Week 24, and Week 52 o change from baseline to Week 12, Week 24, and Week 52 in Functional Assessment of Chronic Illness Therapy Fatigue (FACIT-F) scale scores o change from baseline through Week 52 in each scale and the summary scores for the Mental Component Score (MCS) and Physical Component Score (PCS) of the Medical Outcomes Study 36-Item Short Form Health Survey Version 2 Acute (SF-36v2 Acute) o change from baseline through Week 52 in European Quality of Life-5 Dimensions-5 Level (EQ-5D-5L) scores o change from baseline through Week 52 in Work Productivity and Activity Impairment- Rheumatoid Arthritis (WPAI-RA) scores o change from baseline through Week 52 in healthcare resource utilization to characterize the pharmacokinetics (PK) of baricitinib, determine the magnitude of within- and between-patient variability, and identify the potential factors that may have an impact on the PK of baricitinib to characterize the baricitinib exposure-response time course for key efficacy and safety endpoints and to identify potential factors that may have an impact on these efficacy or safety endpoints of baricitinib The exploratory objective of the study is: to compare baricitinib and adalimumab as regards to various efficacy measures Study Design: Study I4V-MC-JADV (JADV) will be a 52-week, Phase 3, multicenter, randomized, double-blind, double-dummy, placebo- and active-controlled, parallel-group, outpatient study comparing the efficacy of baricitinib versus placebo on signs and symptoms, function, remission, and structural progression in patients with moderately to severely active RA who have had an insufficient response to methotrexate (MTX) and who have never been treated with a biologic disease-modifying antirheumatic drug (DMARD) (ie, MTX-IR patients). The study will include a noninferiority comparison of baricitinib versus adalimumab on signs and symptoms. Baricitinib will be administered as a 4-mg tablet once daily (QD). The dose for patients with renal impairment, defined as estimated glomerular filtration rate (egfr) <60 ml/min/1.73 m2, will be 2 mg baricitinib QD. Patients not assigned to the baricitinib treatment arm will receive either a 4-mg or 2-mg matching placebo tablet. Adalimumab (40 mg) will be administered by subcutaneous (SC) injection biweekly (ie, every 2 weeks). Patients not assigned to adalimumab will receive a matching placebo SC injection biweekly. A total of 1280 patients are planned for enrollment in this study (480 to receive baricitinib, 480 to receive placebo, and 320 to receive adalimumab). After the Week 52 study visit, eligible patients may proceed to a separate extension study (Study I4V-MC-JADY) lasting for up to 2 years or to the posttreatment follow-up period of this study (Part C). Study JADV will consist of 4 parts: Screening: Screening period lasting from 3 to 42 days prior to Visit 2 (Week 0) Part A: double-blind, placebo- and active-controlled period from Week 0 through Week 24
7 I4V-MC-JADV Clinical Protocol Page 5 Part B: double-blind, active-controlled period from Week 24 through Week 52 Part C: posttreatment follow-up period Diagnosis and Main Criteria for Inclusion and Exclusions: The proposed patient population for Study JADV will be ambulatory adult patients with moderately to severely active RA who have had an insufficient response to MTX and who have never been treated with a biologic DMARD. Patients will be eligible for participation only if they have an inadequate response to MTX as evidenced by the presence of at least 6/68 tender joints and 6/66 swollen joints and an hscrp measurement 1.2 times the upper limit of normal (ULN). Investigational Product, Dosage, and Mode of Administration or Intervention: Baricitinib 4 mg administered orally QD Planned Duration of Treatment: 52 weeks. Screening period: 3 to 42 days Treatment period: 52 weeks Follow-up period: 28 days Reference Therapy, Dose, and Mode of Administration or Comparative Intervention: Placebo tablets matching baricitinib will be administered orally QD. As an active comparator, adalimumab 40 mg will be administered by SC injection biweekly (ie, once every 2 weeks). Placebo matching adalimumab for injection will also be administered by SC injection biweekly, as assigned. Patients will continue to take their background MTX therapy during the course of the study. Criteria for Evaluation: Efficacy The following efficacy measures will be assessed in this study: ACR20, ACR50, and ACR70 indices HAQ-DI mtss (includes joint space narrowing score and bone erosion score) DAS28-hsCRP and DAS28-ESR Hybrid ACR (bounded) response measure SDAI CDAI EULAR28 Health Outcomes The following health outcome measures will be administered in this study: duration and severity of morning joint stiffness recurrence of joint stiffness during the day tiredness severity numeric rating scale (Worst Tiredness Numeric Rating Scale [NRS]) pain severity numeric rating scale (Worst Pain NRS) FACIT-F SF-36v2 Acute WPAI-RA EQ-5D-5L Quick Inventory of Depressive Symptomatology Self-Rated-16 (QIDS-SR 16 ) healthcare resource utilization Safety The following safety measures will be assessed in this study: adverse events (AEs) adverse events of special interest (AESIs) serious adverse events (SAEs) suspected unexpected serious adverse reactions (SUSARs)
8 I4V-MC-JADV Clinical Protocol Page 6 physical examinations Electrocardiograms (ECGs) chest x-ray and tuberculosis (TB) testing vital signs (blood pressure and heart rate) and physical characteristics standard laboratory tests (including hematology, clinical chemistry, urinalysis, lipid profile, egfr, iron studies, hscrp, and ESR) concomitant medications AESIs will include: severe or opportunistic infections myelosuppressive events of anemia, leukopenia, neutropenia, lymphopenia, and thrombocytopenia thrombocytosis elevations in alanine aminotransferase (ALT) or aspartate aminotransferase (AST) (>3 times ULN) with total bilirubin (>2 times ULN) Patients with these laboratory value specified events will be identified using the same criteria for the interruption of investigational product with the exception of anemia, which will be identified using the same criteria for the discontinuation of investigational product, and thrombocytosis, which will be defined as a platelet count >600,000/μL. Bioanalytical Concentrations of baricitinib in human plasma will be determined by a validated liquid chromatography tandem mass spectrometry (LC/MS/MS) method. Pharmacokinetics/Pharmacodynamics Plasma concentrations of baricitinib, associated with time from dose, will be obtained at selected visits from all patients. Stored serum, plasma, and messenger ribonucleic acid (mrna) samples will be collected for possible exploratory assessments of pharmacodynamic (PD) markers of RA disease activity or mechanism of action of baricitinib. Pharmacogenetics Samples will be stored, and analysis may be performed on genetic variants thought to play a role in active RA and the Janus kinase (JAK)/signal transducers and activators of transcription (STAT) signaling pathways. Statistical Methods: Sample size: Approximately 1280 patients (480 in the baricitinib group, 480 in the placebo group, and 320 in the adalimumab group) will provide >95% power to detect a difference between baricitinib and placebo groups in ACR20 response rate (60% vs 35%) at Week 12 based on a 2-sided significance level of 0.05; approximately 94% power to detect a difference in mtss between baricitinib and placebo groups for an effect size of 0.25, based on a 2-sided t-test at a significance level of 0.04 (this assumes 90% of randomized patients will have x-ray data available for the mtss analysis at Week 24); and approximately 91% power for the noninferiority analysis of ACR20 response rate at Week 12 between baricitinib and adalimumab groups, where a response rate of 60% for baricitinib and adalimumab, a noninferiority margin of 12%, and a 1-sided significance level of 0.02 are assumed. Analysis Population: The efficacy and health outcome analyses will be conducted on an intention-to-treat (ITT) basis. The ITT analysis set will include all data from all randomized patients treated with at least 1 dose of the study drug. Analysis of structural progression (van der Heijde mtss) will be conducted on the ITT population using patients with available baseline and at least 1 postbaseline value. The primary and major secondary analyses will be repeated using the per-protocol set (PPS), which excludes patients with significant protocol violations. Safety analyses will be performed on the safety population, defined as all randomized patients who received at least 1 dose of the study drug. Missing Data Imputation: 1. Nonresponder imputation (NRI): All patients who discontinue the study or the study treatment at any time for any reason will be defined as nonresponders for the NRI analysis for categorical variables, such as ACR20/50/70, from the time of discontinuation and onward. Patients who are eligible for rescue therapy at Week 16 will be analyzed as nonresponders after Week 16 and onward. Randomized patients without available
9 I4V-MC-JADV Clinical Protocol Page 7 data at a postbaseline visit will be defined as nonresponders for the NRI analysis at that visit. 2. Linear extrapolation method: The linear extrapolation method will be used for analysis of the structural progression endpoint (van der Heijde mtss) to impute missing data at time points when analyses are conducted (including the analyses at Week 24 and Week 52). For patients who discontinue the study or the study treatment or for patients who miss a radiograph for any reason, baseline data and the most recent radiographic data prior to discontinuation or the missed radiograph, adjusted for time, will be used for linear extrapolation to impute missing data at subsequent time points. For patients who receive rescue therapy starting at Week 16 or at any time point thereafter, baseline data and the most recent radiographic data prior to initiation of rescue therapy, adjusted for time, will be used for linear extrapolation. All patients originally randomized to placebo will be switched to active treatment at Week 24; thus, there will be no observed data for the placebo arm for the structural comparison at subsequent time points. Therefore, baseline data and the most recent radiographic data prior to initiation of the new therapy, adjusted for time, will be used for linear extrapolation at Week 52. The linear extrapolation method has been established as an appropriate missing data imputation method in other Phase 3 RA trials. Missing postbaseline values will be imputed only if both a baseline value and a postbaseline value from another time point are available, as long as the patient was on the same treatment at each applicable time point. 3. Multiple imputation: Other methods than linear extrapolation that include multiple imputation will be employed for analysis of the structural progression endpoint (van der Heijde mtss) to impute missing data at Week 24. Details will be provided in the statistical analysis plan (SAP). 4. Modified baseline observation carried forward (mbocf): The mbocf method will be used for the analysis of major secondary continuous endpoints (unless otherwise stated). For patients who discontinue the study or the study treatment because of an AE, including death, the baseline observation will be carried forward to the corresponding endpoint for evaluation. For patients who discontinue the study for reason(s) other than an AE, the last nonmissing postbaseline observation prior to discontinuation will be carried forward to the corresponding endpoint for evaluation. For patients who are eligible for rescue therapy starting at Week 16, the last nonmissing observation at or before rescue will be carried forward to subsequent time points for evaluation. 5. Modified last observation carried forward (mlocf): For all continuous measures including safety analyses, the mlocf will be a general approach to impute missing data unless otherwise specified. For patients who are eligible for rescue therapy starting at Week 16, the last nonmissing observation at or before rescue will be carried forward to subsequent time points for evaluation. For all other patients discontinuing from the study or the study treatment for any reason, the last nonmissing postbaseline observation before discontinuation will be carried forward to subsequent time points for evaluation. If the postbaseline observation is missing, then it will not be included in the analysis even if the baseline observation is not missing. The mlocf will be used as a sensitivity method to the mbocf method with respect to the major secondary endpoints. 6. Other methods for data imputation for analysis of key endpoints other than mtss may be used and will be described in the SAP Adjustment for Multiple Comparisons: A multiple testing procedure will be used to control the family-wise error rate for the primary and key secondary hypotheses for the endpoints listed above as objectives. Each step in the sequential testing procedure uses a local level procedure for the null hypotheses being tested. Testing of hypotheses at each step of the procedure commences only if all null hypotheses of prior steps were rejected. As such, the multiple testing procedure provides strong control of the family-wise error rate at the 1-sided level. Primary and Major Secondary Analyses: A logistic regression model with treatment, region, and joint erosion status in the model will be used to test the treatment difference between baricitinib and placebo in the proportion of patients achieving ACR20 response at Week 12. The NRI method will be used to impute missing data. An ANCOVA model with baseline score, region, joint erosion status, and treatment group in the model will be used to test the treatment difference between baricitinib and placebo in change from baseline to Week 24 of mtss scores. The linear extrapolation method will be used to impute missing data. An analysis of covariance (ANCOVA) model with treatment, region, joint erosion status, and baseline score in the model will be used to test the treatment difference between baricitinib and placebo in change from baseline to Week 12 in HAQ-DI. The mbocf
10 I4V-MC-JADV Clinical Protocol Page 8 approach will be used to impute missing data. An ANCOVA model with treatment, region, joint erosion status, and baseline score in the model will be used to test the treatment difference between baricitinib and placebo in change from baseline to Week 12 in DAS28-hsCRP. The mbocf approach will be used to impute missing data. A logistic regression model with treatment, region, and joint erosion status in the model will be used to test the treatment difference between baricitinib and adalimumab in the percentage of patients achieving ACR20 response at Week 12. A (100-alpha)% confidence interval (CI) for the treatment difference will be constructed using the method described by Newcombe-Wilson without continuity correction. If the lower bound of the (100-alpha)% CI is >-12%, Lilly will conclude that baricitinib is noninferior to adalimumab with respect to ACR20. If the lower bound of the (100-alpha)% CI >0%, Lilly will conclude that baricitinib is superior to adalimumab with respect to ACR20. The NRI method will be used to impute missing data. For duration of morning joint stiffness, severity of morning joint stiffness, worst tiredness, and worst pain, an ANCOVA analysis with treatment, region, joint erosion status, and baseline score in the model will be used to test the treatment difference between baricitinib and placebo in change from baseline to Week 12. The mbocf approach will be used to impute missing data. Safety: All safety data will be descriptively summarized by treatment groups and analyzed using the safety population. Treatment-emergent AEs (TEAEs) are defined as AEs that first occurred or worsened in severity on or after the date of first dose of study treatment. The number and percentage of TEAEs will be summarized using MedDRA (Medical Dictionary for Regulatory Activities) for each system organ class and each preferred term by treatment group. TEAEs will also be summarized by relationship to treatment and by severity within each group. SAEs and AEs that lead to study drug discontinuation will also be summarized by group. Fisher s exact test will be used to perform pairwise comparisons among the treatment groups. Change from baseline in quantitative clinical chemistry will be analyzed using ANCOVA with treatment and baseline value in the model. Categorical variables, including the incidence of abnormal values and incidence of AESIs, will be summarized by frequency and percentage of patients in corresponding categories. Shift tables will be presented for selected measures. Change from baseline to postbaseline in vital signs, QIDS-SR16, and body weight will be analyzed using ANCOVA with treatment and baseline value in the model. Health Outcomes: Health outcome measures will be analyzed using methods described for continuous or categorical data as described for efficacy measures. More details will be provided in the SAP. Bioanalytical: Concentrations of baricitinib in human plasma will be determined by a validated LC/MS/MS method. Pharmacokinetics/Pharmacodynamics: PK analysis will be conducted using all evaluable PK data. PK data for baricitinib will be analyzed using a population modeling approach via a nonlinear mixed-effects modeling (NONMEM) program. Data from this study may be combined with an existing PK model or data from other studies, if appropriate. Intrinsic factors such as age; body weight; gender; ethnic origin; renal function; transaminases/albumin/bilirubin, which are representative of hepatic function; and extrinsic factors such as comedications will be investigated to assess their influence on PK parameters such as clearance and volume of distribution, where applicable. Time-course of response for key efficacy data such as, but not necessarily limited to, ACR20/50/70, DAS28-ESR, DAS28-hsCRP, and safety data (eg, neutropenia, anemia) will be explored using graphical methods. Further investigations using a population-modeling approach via NONMEM to identify potential factors that may influence PD parameters may be warranted. Covariates such as body weight, disease duration, disease severity at baseline, responder status, cytokines concentrations, dose, and baricitinib exposures may be investigated to assess their impact on efficacy and safety endpoints. Data from this study may be combined with an existing PK/PD model or data from other studies, if appropriate. Planned Analyses: The analysis of primary and major secondary endpoints will occur when all patients complete the Week 24 study visit. A final analysis will occur when all patients complete the study. Interim Analyses: A data monitoring committee (DMC), external to Lilly, will oversee the conduct of all the Phase 3 clinical trials evaluating baricitinib in patients with RA. This DMC will follow the rules defined in the DMC charter, focusing on potential and identified risks. The DMC will review and evaluate up to 3 planned interim analyses on an approximately semiannual basis. Access to the unblinded interim data will be limited to the statisticians who conduct the interim analyses and the DMC. The statisticians conducting the interim analyses will
11 I4V-MC-JADV Clinical Protocol Page 9 be independent from the study team who will not have access to the unblinded data. The DMC will review study discontinuations, AEs, clinical laboratory data, vital signs, etc. The DMC may recommend continuation of the study, as designed, temporary suspension of enrollment, or the discontinuation of a particular dose regimen or the entire study. The DMC may request to review efficacy data to investigate the benefit/risk relationship in the context of safety observations for ongoing patients in the study. However, the study will not be stopped for positive efficacy results, and there is no planned futility assessment. Details of the DMC and interim safety analyses will be documented in a DMC charter and DMC analysis plan. Besides DMC members, a limited number of pre-identified individuals may gain access to the unblinded PK and safety data, as specified in the unblinding plan, prior to the final database lock, to initiate the exploration and/or final population PK/PD model development processes for safety data (eg, neutropenia, anemia). Information that may unblind the study during the analyses will not be reported to study sites or the blinded study team until the database lock for the primary and major secondary efficacy analyses.
12 I4V-MC-JADV Clinical Protocol Page Table of Contents A Randomized, Double-Blind, Placebo- and Active-Controlled, Phase 3 Study Evaluating the Efficacy and Safety of Baricitinib in Patients with Moderately to Severely Active Rheumatoid Arthritis Who Have Had an Inadequate Response to Methotrexate Therapy Section Page 1. Protocol I4V-MC-JADV A Randomized, Double-Blind, Placeboand Active-Controlled, Phase 3 Study Evaluating the Efficacy and Safety of Baricitinib in Patients with Moderately to Severely Active Rheumatoid Arthritis Who Have Had an Inadequate Response to Methotrexate Therapy Synopsis Table of Contents Abbreviations and Definitions Introduction Objectives Primary Objective Secondary Objectives Exploratory Objective Investigational Plan Summary of Study Design Screening Period Part A and Part B: 24-Week and 28-Week, Double-Blind, Placebo- and Active-Controlled Periods Part C: Posttreatment Follow-Up Period Study Extensions Discussion of Design and Control Study Population Entry Criteria Inclusion Criteria Exclusion Criteria Enrollment Criteria Inclusion Criteria Exclusion Criteria Rationale for Exclusion of Certain Study Candidates...39
13 I4V-MC-JADV Clinical Protocol Page Discontinuations Discontinuation of Patients Interruption of Study Drug Discontinuation of Study Drug Discontinuation from the Study Discontinuation of Study Sites Discontinuation of the Study Treatment Treatments Administered Materials and Supplies Method of Assignment to Treatment Rationale for Selection of Doses in the Study Selection and Timing of Doses Special Treatment Considerations Continued Access to Investigational Product Blinding Concomitant Therapy Treatment Compliance Efficacy, Health Outcome Measures, Safety Evaluations, Sample Collection and Testing, and Appropriateness of Measurements Efficacy Measures Primary Efficacy Measure Secondary Efficacy Measures Van der Heijde Modified Total Sharp Score Disease Activity Score-Erythrocyte Sedimentation Rate and Disease Activity Score High Sensitivity C-Reactive Protein ACR50 and ACR70 Indices Hybrid American College of Rheumatology Response Measure Simplified Disease Activity Index Clinical Disease Activity Index European League Against Rheumatism Responder Index Health Outcome Measures Safety Evaluations Adverse Events Adverse Events of Special Interest Serious Adverse Events Suspected Unexpected Serious Adverse Reactions...58
14 I4V-MC-JADV Clinical Protocol Page Other Safety Measures Measures Evaluated During Screening Period Measures Evaluated During Screening and Treatment Periods Safety Monitoring Complaint Handling Sample Collection and Testing Samples for Standard Laboratory Testing Samples for Drug Concentration Measurements Pharmacokinetics/Pharmacodynamics Exploratory Stored Samples Pharmacogenetics Nonpharmacogenetic/Biomarker Stored Samples Appropriateness of Measurements Data Quality Assurance Data Capture System Sample Size and Statistical Methods Determination of Sample Size Statistical and Analytical Plans General Considerations Patient Disposition Patient Characteristics Concomitant Therapy Treatment Compliance Primary Outcome and Methodology Efficacy Analyses Pharmacokinetic/Pharmacodynamic Analyses Health Outcome Analyses Safety Analyses Subgroup Analyses Planned Analyses Interim Analyses Informed Consent, Ethical Review, and Regulatory Considerations Informed Consent Ethical Review Regulatory Considerations Investigator Information Protocol Signatures...80
15 I4V-MC-JADV Clinical Protocol Page Final Report Signature References...81
16 I4V-MC-JADV Clinical Protocol Page 14 Table List of Tables Page Table JADV.1. Criteria for Temporary Discontinuation of Investigational Product...40 Table JADV.2. Criteria for Permanent Discontinuation of Investigational Product...41 Table JADV.3. Table JADV.4. Table JADV.5. Treatment Regimens...43 Scoring Method for the Hybrid American College of Rheumatology Response Measure...53 Categorization of Patients as Nonresponders, Moderate Responders, or Good Responders...54
17 I4V-MC-JADV Clinical Protocol Page 15 Figure List of Figures Page Figure JADV.1. Illustration of study design for Clinical Protocol I4V-MC-JADV...29 Figure JADV.2. Illustration of the sequential testing approach within the overall gatekeeping strategy for Study JADV....71
18 I4V-MC-JADV Clinical Protocol Page 16 Attachment List of Attachments Page Attachment 1. Protocol JADV Study Schedule...85 Attachment 2. Attachment 3. Protocol JADV Clinical Laboratory Tests...92 Protocol JADV Hepatic Monitoring Tests for Treatment-Emergent Abnormality...95
19 I4V-MC-JADV Clinical Protocol Page Abbreviations and Definitions Term ACR ACR20 ACR50 ACR70 adverse event (AE) AESI ALT ANCOVA anti-ccp AST audit blinding case report form (CRF) CDAI cdmard CI CL clinical research Definition American College of Rheumatology 20% improvement in American College of Rheumatology criteria 50% improvement in American College of Rheumatology criteria 70% improvement in American College of Rheumatology criteria Any untoward medical occurrence in a patient or clinical investigation subject administered a pharmaceutical product and that does not necessarily have a causal relationship with this treatment. An adverse event can therefore be any unfavorable and unintended sign (including an abnormal laboratory finding), symptom, or disease temporally associated with the use of a medicinal (investigational) product, whether or not related to the medicinal (investigational) product. adverse event of special interest alanine aminotransferase analysis of covariance anticyclic citrullinated peptide aspartate aminotransferase a systematic and independent examination of the trial-related activities and documents to determine whether the evaluated trial-related activities were conducted, and the data were recorded, analyzed, and accurately reported according to the protocol, applicable standard operating procedures (SOPs), good clinical practice (GCP), and the applicable regulatory requirement(s) A procedure in which one or more parties to the trial are kept unaware of the treatment assignment(s). Single-blinding usually refers to the patient(s) being unaware, and double-blinding usually refers to the patient(s), investigator(s), monitor(s), and in some cases, select sponsor personnel being unaware of the treatment assignment(s). sometimes referred to as clinical report form: a printed form for recording study participants data during a clinical study, as required by the protocol Clinical Disease Activity Index conventional disease-modifying antirheumatic drugs confidence interval central compartment clearance Individual responsible for the medical conduct of the study. Responsibilities of
20 I4V-MC-JADV Clinical Protocol Page 18 physician complaint compliance CRP DAS DAS28 DMARD DMC DNA ECG efficacy/ effectiveness egfr end of study (trial) enroll/randomize enter epro EQ-5D-5L ESR ethical review board (ERB) EULAR the clinical research physician may be performed by a physician, clinical research scientist, global safety physician or other medical officer. A complaint is any written, electronic, or oral communication that alleges deficiencies related to the identity, quality, purity, durability, reliability, safety or effectiveness, or performance of a drug or drug delivery system. adherence to all the trial-related requirements, good clinical practice (GCP) requirements, and the applicable regulatory requirements C-reactive protein Disease Activity Score Disease Activity Score modified to include the 28 diarthroidal joint count disease-modifying antirheumatic drug data monitoring committee deoxyribonucleic acid electrocardiogram Efficacy is the ability of a treatment to achieve a beneficial intended result. Effectiveness is the measure of the produced effect of an intervention when carried out in a clinical environment. estimated glomerular filtration rate the date of the last visit or last scheduled procedure shown in the study schedule for the last active patient in the study The act of assigning a patient to a treatment. Patients who are enrolled/randomized in the trial are those who have been assigned to a treatment. The act of obtaining informed consent for participation in a clinical trial from patients deemed eligible or potentially eligible to participate in the clinical trial. Patients entered into a trial are those who sign the informed consent document directly or through their legally acceptable representatives. electronic patient-reported outcome European Quality of Life-5 Dimensions-5 Level erythrocyte sedimentation rate a board or committee (institutional, regional, or national) composed of medical and nonmedical members whose responsibility is to verify that the safety, welfare, and human rights of the patients participating in a clinical trial are protected European League Against Rheumatism
21 I4V-MC-JADV Clinical Protocol Page 19 EULAR28 FACIT-F FSH GCP HAQ-DI HBsAb HBV HCV HDL HIV hscrp IB European League Against Rheumatism Responder Index based on the 28-joint count Functional Assessment of Chronic Illness Therapy Fatigue follicle-stimulating hormone good clinical practice Health Assessment Questionnaire Disability Index hepatitis B surface antibody hepatitis B virus hepatitis C virus high-density lipoprotein human immunodeficiency virus high-sensitivity C-reactive protein Investigator s Brochure IC 50 inhibitory concentration of 50% ICF ICH IL-6 intention to treat (ITT) interim analysis investigator IVRS JAK JAK1 JAK2 informed consent form International Conference on Harmonisation interleukin-6 The principle that asserts that the effect of a treatment policy can be best assessed by evaluating on the basis of the intention to treat a patient (that is, the planned treatment regimen) rather than the actual treatment given. It has the consequence that patients allocated to a treatment group should be followed up, assessed, and analyzed as members of that group irrespective of their compliance to the planned course of treatment. an analysis of clinical study data, separated into treatment groups, that is conducted before the final reporting database is created/locked A person responsible for the conduct of the clinical study at a study site. If a study is conducted by a team of individuals at a study site, the investigator is the responsible leader of the team and may be called the principal investigator. interactive voice-response system Janus kinase 1 of 4 identified members of the family of Janus kinases 1 of 4 identified members of the family of Janus kinases
22 I4V-MC-JADV Clinical Protocol Page 20 JAK3 Ka LC/MS/MS LDL legal representative mbocf MCS MDRD MedDRA mlocf MMRM MRI mrna mtss MTX MTX-IR NONMEM NRI NRS NSAID patient PCS PD per-protocol set (PPS) PK 1 of 4 identified members of the family of Janus kinases absorption rate constant liquid chromatography tandem mass spectrometry low-density lipoprotein an individual, judicial, or other body authorized under applicable law to consent on behalf of a prospective patient, to the patient s participation in the clinical trial modified baseline observation carried forward Mental Component Score Modification of Diet in Renal Disease Medical Dictionary for Regulatory Activities modified last observation carried forward mixed model for repeated measures magnetic resonance imaging messenger ribonucleic acid modified Total Sharp Score methotrexate patients who have had an inadequate response to methotrexate nonlinear mixed-effects modeling nonresponder imputation numeric rating scale nonsteroidal anti-inflammatory drug a study participant who has the disease or condition for which the investigational product is targeted Physical Component Score pharmacodynamic(s) the set of data generated by the subset of patients who sufficiently complied with the protocol to ensure that these data would be likely to exhibit the effects of treatment, according to the underlying scientific model pharmacokinetic(s)
23 I4V-MC-JADV Clinical Protocol Page 21 PPD Q QD QIDS-SR 16 RA SAE SAP SC screening SD SDAI SF-36v2 Acute SJC STAT subject SUSARs TB TJC TNF TNF-α treatmentemergent adverse event (TEAE) TSH purified protein derivative intercompartmental clearance once daily Quick Inventory of Depressive Symptomatology Self-Rated-16 rheumatoid arthritis serious adverse event statistical analysis plan subcutaneous the act of determining if an individual meets minimum requirements to become part of a pool of potential candidates for participation in a clinical study standard deviation Simplified Disease Activity Index Medical Outcomes Study 36-Item Short Form Health Survey Version 2 Acute swollen joint count signal transducers and activators of transcription An individual who is or becomes a participant in clinical research, either as a recipient of the investigational product(s) or as a control. A subject may be either a healthy human or a patient. suspected unexpected serious adverse reactions tuberculosis tender joint count tumor necrosis factor tumor necrosis factor-alpha any untoward medical occurrence that either occurs or worsens at any time after treatment baseline and which does not necessarily have to have a causal relationship with this treatment (also called treatment-emergent signs and symptoms [TESS]) thyroid-stimulating hormone TYK2 tyrosine kinase 2 ULN V1 V2 upper limit of normal central compartment volume of distribution peripheral volume of distribution
24 I4V-MC-JADV Clinical Protocol Page 22 VAS WPAI-RA visual analog scale Work Productivity and Activity Impairment-Rheumatoid Arthritis
25 I4V-MC-JADV Clinical Protocol Page 23 A Randomized, Double-Blind, Placebo- and Active-Controlled, Phase 3 Study Evaluating the Efficacy and Safety of Baricitinib in Patients with Moderately to Severely Active Rheumatoid Arthritis Who Have Had an Inadequate Response to Methotrexate Therapy 5. Introduction Rheumatoid arthritis (RA) is a systemic inflammatory autoimmune disease. The disease has variable expression and outcome ranging from mild, limited disease to severe disease associated with progressive joint destruction, significantly compromised quality of life, and reduced survival (Smolen and Steiner 2003; Colmegna et al. 2012). Substantial comorbidity can be seen outside of the musculoskeletal system, with excess cardiovascular risk, dyslipidemia, and propensity for infection partially related to the need for treatment with immunomodulatory agents (Curtis et al. 2007; Kitas and Gabriel 2011). Management of RA has improved substantially in recent years. In addition to reduction of signs and symptoms, improvement of physical function, and inhibition of structural damage, better patient outcomes and clinical remission are now considered achievable goals. Therefore, the current recommended primary target for treatment of RA should be a state of clinical remission (Smolen et al. 2010; Felson et al. 2011). There are several definitions for clinical remission based on composite scores (commonly including: tender joint counts [TJCs]/swollen joint counts [SJCs], level of acute phase reactants, and assessment of a patient or physician global response). These scores include the Disease Activity Score (DAS), DAS modified to include the 28 diarthroidal joint count (DAS28), Simplified Disease Activity Index (SDAI), Clinical Disease Activity Index (CDAI), and a Boolean definition as recently proposed jointly by American College of Rheumatology (ACR)/European League Against Rheumatism (EULAR) (Felson et al. 2011). The current focus on arresting disease activity results from an understanding that persistent joint inflammation leads to progressive joint destruction manifested by cartilage loss, erosive damage to juxta-articular bone, and resultant functional impairment; that erosive bone changes occur within months of disease onset; and that early and aggressive treatment increases the likelihood of disease control (Klareskog et al. 2009; Colmegna et al. 2012). Despite a variety of approved agents for RA, complete or sustained disease remission is unusual. Conventional disease-modifying anti-rheumatic drugs (cdmards) have been used with some success. Patients often receive 1 or more of these medications (for example, methotrexate [MTX], sulfasalazine, hydroxychloroquine, leflunomide, azathioprine, gold salts, and cyclosporine), typically in combination with low-dose oral or intra-articular glucocorticoids. In addition to cdmards, biological agents that block or antagonize critical inflammatory mediators, T cells, or B cells can reduce pain and swelling and provide joint protection against structural damage. The efficacy of these biologics, particularly in combination with MTX, has been shown to have a clinically important effect on the signs and symptoms of RA (Fleischmann 2005); however, a majority of patients do not go into remission or achieve a 50%
26 I4V-MC-JADV Clinical Protocol Page 24 improvement in ACR criteria (ACR50) in a clinical trial setting. During treatment with tumor necrosis factor-alpha (TNF-α) antagonists, approximately 30% of patients fail to achieve a 20% improvement in ACR criteria (ACR20; primary failure), and more patients lose efficacy during therapy (secondary failure; Rubbert-Roth and Finckh 2009). Disease progression can still occur even for patients who achieve apparent adequate control of their signs and symptoms with cdmards and/or biologic therapies (Klareskog et al. 2009; Rubbert-Roth and Finckh 2009). Accordingly, a significant unmet need remains for more effective and better tolerated treatments for RA. Members of the Janus kinase (JAK) family of protein tyrosine kinases (JAK1, JAK2, JAK3, and tyrosine kinase 2 [TYK2]) play an important role in signal transduction following cytokine and growth factor binding to their receptors. Aberrant production of cytokines and growth factors has been associated with a number of chronic inflammatory conditions, including RA. Many of the pro-inflammatory cytokines implicated in the pathogenesis of RA, including interleukin-6 (IL-6) and interferon-gamma, use cell signaling that involves the JAK/signal transducers and activators of transcription (STAT) pathways. Inhibition of JAK-STAT signaling can target multiple RA-associated cytokine pathways and thereby reduce inflammation, cellular activation, and proliferation of key immune cells as demonstrated with other JAK inhibitors (Williams et al. 2008; Kremer et al. 2009). Baricitinib is an orally available, selective JAK inhibitor with excellent potency and selectivity for JAK1 (inhibitory concentration of 50% [IC 50 ] = 5.9 nm) and JAK2 (IC 50 = 5.7 nm) and less potency for JAK3 (IC nm) or TYK2 (IC 50 = 53 nm) (Fridman et al. 2010). Baricitinib is being developed for treatment of patients with moderately to severely active RA who are either intolerant to MTX treatment or who have had an inadequate response to disease-modifying antirheumatic drugs (DMARDs), either conventional or biologic. One completed and 2 currently ongoing Phase 2 studies have evaluated the clinical utility of baricitinib as treatment for patients with active RA. In the completed study (I4V-MC-JADC [JADC], conducted by Incyte Corporation), administration of baricitinib at doses of 4, 7, or 10 mg daily for up to 24 weeks resulted in improved signs and symptoms in patients with an inadequate response to DMARDs, including biologics. A relatively flat dose response was observed for the efficacy parameters, with the 4-mg dose performing as well as the 7- and 10-mg doses. The proportions of patients who were ACR responders generally decreased in the off-treatment follow-up period (from Week 24 to Week 28). In an ongoing Phase 2 study (I4V-MC-JADA [JADA]), administration of baricitinib at doses of 1, 2, 4, and 8 mg daily for up to 12 weeks with continued administration of the 2-, 4-, and 8-mg doses for up to 24 weeks resulted in improved signs and symptoms in patients with an inadequate response to MTX. The observed treatment effect in the 4- and 8-mg dose groups was similar, confirming the flat dose response observed in Study JADC. The treatment effect in these dose groups was larger than that observed in the 1- and 2-mg dose groups. The 1- and 2-mg dose groups offered some clinical effectiveness relative to placebo treatment. Patients completing 24 weeks of treatment could enter an extension study that is currently ongoing. A third Phase 2
27 I4V-MC-JADV Clinical Protocol Page 25 study (I4V-JE-JADN) is ongoing in Japan, and the sponsor remains blinded to patient treatment allocation. The safety profile for baricitinib has been informed by results from nonclinical and clinical studies evaluating a wide range of doses (up to 20 mg once daily [QD]). There are a number of identified and potential risks recognized for baricitinib that will be followed carefully in the Phase 3 development program. Important identified risks for baricitinib include decreased hemoglobin; increased total cholesterol, high-density lipoprotein (HDL), low-density lipoprotein (LDL), and triglycerides; and decreased neutrophils and other phagocytic white cell lines (within the normal reference range). More information about the known and expected benefits, risks, and reasonably anticipated adverse events (AEs) may be found in the investigator s brochure (IB). Information on AEs expected to be related to the study drug may be found in Section 7 (Development Core Safety Information) of the IB. Information on serious AEs (SAEs) expected in the study population independent of drug exposure will be assessed by the sponsor in aggregate periodically during the course of the study and may be found in Section 6 (Effects in Humans) of the IB. Study I4V-MC-JADV (JADV) is one of 5 Phase 3 clinical trials investigating the efficacy and safety of baricitinib in patients with active RA. Study JADV is a double-blind, placebo- and active-controlled study investigating the efficacy and safety of baricitinib (4 mg QD) in patients with active RA who have had an inadequate response to MTX (MTX-IR). The study is 52 weeks in duration and includes assessment of structural joint damage. Because tumor necrosis factor (TNF) inhibitors are the most commonly used biologic class of therapies for the treatment of moderately to severely active RA, Humira (adalimumab) was selected as an appropriate active comparator to baricitinib. The remaining 4 Phase 3 studies are described below. Study I4V-MC-JADZ is a double-blind, active-controlled study investigating the efficacy and safety of baricitinib (4 mg QD), administered as monotherapy or in combination with MTX, in patients with early RA who have had limited or no treatment with DMARDs. MTX, administered as monotherapy and titrated to the highest tolerated dose, serves as the active comparator. Study I4V-MC-JADX is a doubleblind, placebo-controlled study investigating the efficacy and safety of baricitinib (2 mg QD or 4 mg QD) administered with background cdmard(s) in patients with active RA who have had an inadequate response to conventional DMARDs. Study I4V-MC-JADW is a double-blind, placebo-controlled study investigating the efficacy and safety of baricitinib (2 mg QD or 4 mg QD) administered with background cdmard(s) in patients with active RA who have had an inadequate response to biologic DMARDs. Patients completing any of the above studies are eligible for enrollment in Study I4V-MC-JADY (JADY), a long-term extension study investigating the safety of baricitinib with prolonged administration (up to 2 additional years of treatment). Given the continuing unmet medical need in patients with moderately to severely active RA, the efficacy of baricitinib demonstrated in Phase 2 studies, and the acceptable safety profile for baricitinib observed through the current stage of development, initiation of Phase 3 testing is appropriate.
28 I4V-MC-JADV Clinical Protocol Page Objectives 6.1. Primary Objective The primary objective of the study is to determine whether baricitinib is superior to placebo in the treatment of patients with moderately to severely active RA despite MTX treatment (ie, MTX-IR), as assessed by the proportion of patients achieving ACR20 at Week Secondary Objectives The major secondary objectives of the study are to evaluate the efficacy of baricitinib versus placebo or adalimumab as assessed by: change from baseline to Week 24 in structural joint damage as measured by modified Total Sharp Score (mtss [van der Heijde method]) compared to placebo change from baseline to Week 12 in Health Assessment Questionnaire-Disability Index (HAQ-DI) score compared to placebo change from baseline to Week 12 in DAS28-high-sensitivity C-reactive protein (hscrp) compared to placebo proportion of patients achieving ACR20 response at Week 12 compared to adalimumab change from baseline to Week 12 in duration of morning joint stiffness compared to placebo as collected in electronic diaries change from baseline to Week 12 in severity of morning joint stiffness compared to placebo as collected in electronic diaries change from baseline to Week 12 in worst tiredness compared to placebo as collected in electronic diaries change from baseline to Week 12 in worst pain compared to placebo as collected in electronic diaries The other secondary objectives of the study are as follows: to evaluate the efficacy of baricitinib as assessed by: o proportion of patients achieving DAS28-hsCRP 3.2 and DAS28-hsCRP <2.6 at Week 12, Week 24, and Week 52 o proportion of patients achieving HAQ-DI improvement 0.22 and 0.3 at Week 12, Week 24, and Week 52 o change from baseline to Week 16 and Week 52 in structural joint damage as measured by mtss o proportion of patients with mtss change 0 from baseline at Week 16, Week 24, and Week 52 o change from baseline to Week 16, Week 24, and Week 52 in joint space narrowing and bone erosion scores o proportion of patients achieving ACR20 at Week 24 and Week 52 o proportion of patients achieving ACR50 at Week 12, Week 24, and Week 52 o proportion of patients achieving 70% improvement in ACR criteria (ACR70) at Week 12, Week 24, and Week 52
29 I4V-MC-JADV Clinical Protocol Page 27 o percentage change from baseline to Week 12, Week 24, and Week 52 in individual components of the ACR Core Set o change from baseline through Week 52 in DAS28-hsCRP o change from baseline through Week 52 in DAS28-erythrocyte sedimentation rate (ESR) o proportion of patients achieving DAS28-ESR 3.2 and DAS28-ESR <2.6 at Week 12, Week 24, and Week 52 o change from baseline to Week 24 and Week 52 in HAQ-DI score o change from baseline to Week 12, Week 24, and Week 52 in CDAI score o change from baseline to Week 12, Week 24, and Week 52 in SDAI score o proportion of patients achieving a CDAI score 2.8 at Week 12, Week 24, and Week 52 o proportion of patients achieving an SDAI score 3.3 at Week 12, Week 24, and Week 52 o proportion of patients achieving EULAR Responder Index based on the 28-joint count (EULAR28) at Week 12, Week 24, and Week 52 o change from baseline to Week 12, Week 24, and Week 52 in Functional Assessment of Chronic Illness Therapy Fatigue (FACIT-F) scale scores o change from baseline through Week 52 in each scale and the summary scores for the Mental Component Score (MCS) and Physical Component Score (PCS) of the Medical Outcomes Study 36-Item Short Form Health Survey Version 2 Acute (SF-36v2 Acute) o change from baseline through Week 52 in European Quality of Life- 5 Dimensions-5 Level (EQ-5D-5L) scores o change from baseline through Week 52 in Work Productivity and Activity Impairment-Rheumatoid Arthritis (WPAI-RA) scores o change from baseline through Week 52 in healthcare resource utilization to characterize the pharmacokinetics (PK) of baricitinib, determine the magnitude of within- and between-patient variability, and identify the potential factors that may have an impact on the PK of baricitinib to characterize the baricitinib exposure-response time course for key efficacy and safety endpoints and to identify potential factors that may have an impact on these efficacy or safety endpoints of baricitinib 6.3. Exploratory Objective The exploratory objective of the study is: to compare baricitinib to adalimumab as regards to various efficacy measures
30 I4V-MC-JADV Clinical Protocol Page Investigational Plan 7.1. Summary of Study Design Study JADV will be a 52-week, Phase 3, multicenter, randomized, double-blind, double-dummy, placebo- and active-controlled, parallel-group, outpatient study comparing the efficacy of baricitinib versus placebo on signs and symptoms, function, remission, and structural progression in patients with moderately to severely active RA who have had an insufficient response to MTX and who have never been treated with a biologic DMARD (ie, MTX-IR patients). The study will include a noninferiority comparison of baricitinib versus adalimumab on signs and symptoms. Baricitinib will be administered as a 4-mg tablet QD. The dose for patients with renal impairment, defined as estimated glomerular filtration rate (egfr) <60 ml/min/1.73 m2, will be 2 mg baricitinib QD. Patients not assigned to the baricitinib treatment arm will receive either a 4-mg or 2-mg matching placebo tablet. Adalimumab (40 mg) will be administered by subcutaneous (SC) injection biweekly (ie, every 2 weeks). Patients not assigned to adalimumab will receive a matching placebo SC injection biweekly. Planned enrollment is approximately 1280 randomized patients: 480 to receive baricitinib, 480 to receive placebo, and 320 to receive adalimumab. After the Week 52 study visit, eligible patients may proceed to a separate extension study (Study JADY) lasting for up to 2 years or to the posttreatment follow-up period of this study (Part C). Study JADV will consist of 4 parts: Screening: Screening period lasting from 3 to 42 days prior to Visit 2 (Week 0) Part A: double-blind, placebo- and active-controlled period from Week 0 through Week 24 Part B: double-blind, active-controlled period from the end of Week 24 through Week 52 Part C: posttreatment follow-up period Section outlines information regarding the data monitoring committee (DMC) and interim analyses. Figure JADV.1 illustrates the study design.
31
32 I4V-MC-JADV Clinical Protocol Page 30 Patients randomized to placebo will be administered a placebo tablet QD starting at Week 0 (Visit 2) through Week 24 (the morning of Visit 11). At Week 24, patients will be administered baricitinib 4 mg QD (or 2 mg baricitinib QD if egfr <60 ml/min/1.73 m2) through Week 52 (Visit 15). Patients in this treatment arm will also receive a placebo SC injection biweekly starting at Week 0 (Visit 2) through Week 50. Patients randomized to adalimumab will be administered adalimumab (40 mg) by SC injection biweekly starting at Week 0 (Visit 2) through Week 50 and a placebo tablet QD through Week 52 (the morning of Visit 15). The last SC injection will be administered during Week 50 because patients receiving adalimumab have the option to switch to baricitinib 4 mg QD (or 2 mg QD depending on egfr) if they choose to participate in the extension study. Scheduling the last SC injection at Week 50 ensures approximately a 2-week interval from the last SC injection to the start of baricitinib dosing. Patients will be eligible for rescue treatment beginning at Week 16 (Visit 9). Details of the rescue are listed in Section Patients will remain on background MTX, nonsteroidal anti-inflammatory drugs (NSAIDs), analgesics, and/or corticosteroids (see Section 9.8 for guidelines on adjustments to concomitant therapy). Patients will take their assigned doses of study drugs as directed. The primary efficacy endpoint will be at Week 12, and the final visit during Part A will be at Week 24. The final visit during Part B will be at Week 52 (Visit 15). The structural joint damage endpoints will be at Week 16, Week 24, and Week 52. All patients who complete the Week 52 study visit will be eligible for inclusion in extension Study JADY Part C: Posttreatment Follow-Up Period Patients who are not enrolled in the extension Study JADY will have a Follow-Up Visit (Visit 801) approximately 28 days after the last dose of study drug. Patients who discontinue from the study prior to Week 52 for any reason will have an Early Termination Visit performed, including x-rays. The Early Termination Visit should be performed as soon as logistically possible because this visit includes assessment of safety and PK data. Infrequently, patient and investigator availability may be such that the Early Termination Visit and the Follow-up Visit may occur on or about the same date. In this instance, the visits may be combined and should occur approximately 28 days after the last dose of study drug. All activities required for the Early Termination Visit should be completed according to the schedule of assessments (Attachment 1) Study Extensions Patients who complete this study through Visit 15 will be eligible to participate in Study JADY, if enrollment criteria for Study JADY are met.
33 I4V-MC-JADV Clinical Protocol Page Discussion of Design and Control Based on efficacy, safety, and PK data from the Phase 2 studies (JADC and JADA), Lilly has selected a single dose of 4 mg QD baricitinib (with dose adjustment for renal dysfunction) for evaluation in this study (see Section 9.4 for rationale). The Week 12 primary efficacy endpoint was chosen based on the results of Studies JADC and JADA. In these studies, efficacy as measured by ACR20 response was observed as early as 2 weeks after initial administration of baricitinib followed by near-maximum efficacy response achieved by Week 12. The 52-week treatment period will allow evaluation of the long-term effect of baricitinib on structural joint damage. All patients will be required to be on background MTX treatment at baseline. Other background therapies, including NSAIDs and low dose oral corticosteroids, are permitted during the study for patients who are on stable doses of these treatments at baseline. Patients should remain on the same dose of MTX throughout the study; however, the MTX dose may be adjusted for safety reasons. All patients in Study JADV will be offered rescue therapy starting at Week 16 in line with precedent from recent clinical trials. Patients who are determined to be nonresponders to their originally assigned treatment will be reassigned to treatment with baricitinib at Week 16. Patients originally assigned to baricitinib will be rescued to the same dose of baricitinib to maintain the study blind. Patients not rescued at Week 16 may be rescued to treatment with baricitinib at the discretion of the investigator anytime thereafter. Patients initially randomized to placebo and not rescued will be reassigned to receive baricitinib at Week 24 to limit exposure to inactive treatment. Patients not experiencing improvement in signs and symptoms following approximately 4 weeks of rescue treatment should be discontinued from the study. Patients may be rescued only once. See Section for details of rescue treatment.
34 I4V-MC-JADV Clinical Protocol Page Study Population The study population will be comprised of patients diagnosed with RA with active disease who have failed to adequately respond to MTX. Study investigator(s) will review patient records and screening test results to determine that the patient meets all inclusion and exclusion criteria to qualify for participation in the study Entry Criteria Patients may enter into the study once they have signed the informed consent form (ICF). Patients may be assessed for study entry on the basis of readily available information (ie, information that does not require informed consent from the subject) Inclusion Criteria Patients are eligible for entry into the study (ie, eligible to sign consent) only if they are expected to meet all of the following criteria: [1] are at least 18 years of age [2] have a diagnosis of adult-onset RA as defined by the ACR/EULAR 2010 Criteria for the Classification of RA (Aletaha et al. 2010) [3] have moderately to severely active RA defined as the presence of at least 6/68 tender joints and at least 6/66 swollen joints a. If surgical treatment of a joint has been performed, that joint cannot be counted in the TJC and SJC for entry or enrollment purposes. [4] have a C-reactive protein (CRP) (or hscrp) measurement 1.2 times the upper limit of normal (ULN) based on the most recent data (if available) [5] have had regular use of MTX for at least the 12 weeks prior to study entry at a dose that, in accordance with local clinical practice, is considered acceptable to adequately assess clinical response. The dose of MTX must have been a stable, unchanging oral dose of 7.5 to 25 mg/week (or the equivalent injectable dose) for at least the 8 weeks prior to study entry. The dose of MTX is expected to remain stable throughout the study and may be adjusted only for safety reasons a. For patients entering the trial on MTX doses <15 mg/week, there must be clear documentation in the medical record that higher doses of MTX were not tolerated or that the dose of MTX is the highest acceptable dose based on local clinical practice guidelines. b. Local standard of care should be followed for concomitant administration of folic acid. [6] are able to read, understand, and give written informed consent
35 I4V-MC-JADV Clinical Protocol Page Exclusion Criteria Patients will be excluded from participating in the study if they meet or are expected to meet any of the following criteria: [7] are currently receiving corticosteroids at doses >10 mg of prednisone per day (or equivalent) or have been receiving an unstable dosing regimen of corticosteroids within 2 weeks of study entry or within 6 weeks of planned randomization [8] have started treatment with NSAIDs (for which the NSAID use is intended for treatment of signs and symptoms of RA) within 2 weeks of study entry or within 6 weeks of planned randomization or have been receiving an unstable dosing regimen of NSAIDs within 2 weeks of study entry or within 6 weeks of planned randomization [9] are currently receiving concomitant treatment with MTX, hydroxychloroquine, and sulfasalazine [10] are currently receiving or have received cdmards (eg, gold salts, cyclosporine, azathioprine, or any other immunosuppressives) other than MTX, hydroxychloroquine (up to 400 mg/day), or sulfasalazine (up to 3000 mg/day) within 8 weeks prior to study entry a. Doses of hydroxychloroquine or sulfasalazine should be stable for at least 8 weeks prior to study entry. b. Immunosuppression related to organ transplantation is not permitted. [11] have received leflunomide in the 12 weeks prior to study entry (or within 4 weeks prior to study entry if the standard 11 days of cholestyramine is used to washout leflunomide) [12] have started a new physiotherapy treatment for RA in the 2 weeks prior to study entry [13] have ever received any biologic DMARD (such as TNF, interleukin-1, IL-6, or T-cell- or B-cell-targeted therapies) [14] have received interferon therapy (such as Roferon-A, Intron-A, Rebetron, Alferon-N, Peg-Intron, Avonex, Betaseron, Infergen, Actimmune, Pegasys) within 4 weeks prior to study entry or are anticipated to require interferon therapy during the study [15] have received any parenteral corticosteroid administered by intramuscular or intravenous injection within 2 weeks prior to study entry or within 6 weeks prior to planned randomization or are anticipated to require parenteral injection of corticosteroids during the study [16] have had 3 or more joints injected with intraarticular corticosteroids within 2 weeks prior to study entry or within 6 weeks prior to planned randomization
36 I4V-MC-JADV Clinical Protocol Page 34 a. Joints injected with intraarticular corticosteroids within 2 weeks prior to study entry or within 6 weeks prior to planned randomization cannot be counted in the TJC and SJC for entry or enrollment purposes. [17] have any condition or contraindication as addressed in the local labeling or local clinical practice for adalimumab that would preclude the patient from participating in this protocol [18] have active fibromyalgia that, in the investigator s opinion, would make it difficult to appropriately assess RA activity for the purposes of this study [19] have a diagnosis of any systemic inflammatory condition other than RA such as, but not limited to, juvenile chronic arthritis, spondyloarthropathy, Crohn s disease, ulcerative colitis, psoriatic arthritis, or active vasculitis a. Patients with secondary Sjögren s syndrome are not excluded. [20] have a diagnosis of Felty s syndrome [21] have had any major surgery within 8 weeks prior to study entry or will require major surgery during the study that, in the opinion of the investigator in consultation with Lilly or its designee, would pose an unacceptable risk to the patient [22] have experienced any of the following within 12 weeks of study entry: myocardial infarction, unstable ischemic heart disease, stroke, or New York Heart Association Stage IV heart failure [23] have a history or presence of cardiovascular, respiratory, hepatic, gastrointestinal, endocrine, hematological, neurological, or neuropsychiatric disorders or any other serious and/or unstable illness that, in the opinion of the investigator, could constitute a risk when taking investigational product or could interfere with the interpretation of data [24] are largely or wholly incapacitated permitting little or no self-care, such as being bedridden or confined to a wheelchair [25] have an egfr based on the most recent available serum creatinine using the Modification of Diet in Renal Disease (MDRD) method of <40 ml/min/1.73 m2 [26] have a history of chronic liver disease with the most recent available aspartate aminotransferase (AST) or alanine aminotransferase (ALT) >1.5 times the ULN or the most recent available total bilirubin 1.5 times the ULN [27] have, or have a history of, lymphoproliferative disease; or signs or symptoms suggestive of possible lymphoproliferative disease, including lymphadenopathy or splenomegaly; or active primary or recurrent malignant disease; or been in remission from clinically significant malignancy for <5 years
37 I4V-MC-JADV Clinical Protocol Page 35 a. Patients with cervical carcinoma in situ that has been resected with no evidence of recurrence or metastatic disease for at least 3 years may participate in the study. b. Patients with basal cell or squamous epithelial skin cancers that have been completely resected with no evidence of recurrence for at least 3 years may participate in the study. [28] have been exposed to a live vaccine within 12 weeks prior to planned randomization or are expected to need/receive a live vaccine during the course of the study (with the exception of herpes zoster vaccination) a. All patients who have not received the herpes zoster vaccine at study entry will be encouraged to do so prior to randomization; vaccination must occur >30 days prior to randomization and start of study drug. Patients will be excluded if they were exposed to herpes zoster vaccination within 30 days of planned randomization. b. Investigators should review the vaccination status of their patients and follow the local guidelines for adult vaccination with nonlive vaccines intended to prevent infectious disease prior to entering patients into the study. [29] have a current or recent (<30 days prior to study entry) clinically serious viral, bacterial, fungal, or parasitic infection [30] have had symptomatic herpes zoster infection within 12 weeks prior to study entry [31] have a history of disseminated/complicated herpes zoster (eg, multidermatomal involvement, ophthalmic zoster, central nervous system involvement, or postherpetic neuralgia) [32] are immunocompromised and, in the opinion of the investigator, are at an unacceptable risk for participating in the study [33] have a history of active hepatitis B virus (HBV), hepatitis C virus (HCV), or human immunodeficiency virus (HIV) [34] have had household contact with a person with active tuberculosis (TB) and did not receive appropriate and documented prophylaxis for TB [35] have evidence of active TB or have previously had evidence of active TB and did not receive appropriate and documented treatment [36] are pregnant or nursing at the time of study entry [37] are females of childbearing potential who do not agree to use 2 forms of highly effective birth control when engaging in intercourse while enrolled in the study and for at least 28 days following the last dose of investigational product
38 I4V-MC-JADV Clinical Protocol Page 36 a. Females of nonchildbearing potential are defined as women 60 years of age, women 40 and <60 years of age who have had a cessation of menses for at least 12 months, or women who are congenitally or surgically sterile (that is, have had a hysterectomy or bilateral oophorectomy or tubal ligation). b. The following birth control methods are considered highly effective (the patient should choose 2 to be used with their partner): oral, injectable, or implanted hormonal contraceptives condom with a spermicidal foam, gel, film, cream, or suppository occlusive cap (diaphragm or cervical/vault caps) with a spermicidal foam, gel, film, cream, or suppository intrauterine device intrauterine system (for example, progestin-releasing coil) vasectomized male (with appropriate postvasectomy documentation of the absence of sperm in the ejaculate) [38] are males who do not agree to use 2 forms of highly effective birth control (see above) while engaging in sexual intercourse with female partners of childbearing potential while enrolled in the study and for at least 28 days following the last dose of investigational product [39] have donated >500 ml of blood within 30 days prior to study entry or intend to donate blood during the course of the study [40] have a history of chronic alcohol abuse, intravenous drug abuse, or other illicit drug abuse within the 2 years prior to study entry [41] have previously been randomized in this study or any other study investigating baricitinib [42] are unable or unwilling to make themselves available for the duration of the study and/or are unwilling to follow study restrictions and procedures [43] have received prior treatment with an oral JAK inhibitor [44] are investigator site personnel directly affiliated with this study and/or their immediate families. Immediate family is defined as a spouse, parent, child, or sibling, whether biological or legally adopted [45] are Lilly or Incyte employees or either s designee [46] are currently enrolled in or have discontinued within 30 days of study entry from a clinical trial involving an investigational product or nonapproved use of a drug or device or are concurrently enrolled in any other type of medical research judged not to be scientifically or medically compatible with this study
39 I4V-MC-JADV Clinical Protocol Page Enrollment Criteria Inclusion Criteria Entered patients are eligible for enrollment into the study (ie, eligible for randomization) only if they continue to meet all of the inclusion criteria for entry (see above) at the time of randomization including the following criteria: [47] have moderately to severely active RA defined as the presence of at least 6/68 tender joints and at least 6/66 swollen joints assessed at Visit 1 and Visit 2 at the time of randomization [48] have an hscrp measurement 1.2 times the ULN based on Visit 1 laboratory results by central laboratory testing [49] have at least 1 joint erosion in hand, wrist, or foot joints based on radiographic interpretation by the central reader and be rheumatoid factor or anticyclic citrullinated peptide (anti-ccp) antibody positive; or have at least 3 joint erosions in hand, wrist, or foot joints based on radiographic interpretation by the central reader regardless of rheumatoid factor or anti-ccp antibody status (radiographic images acquired within 4 weeks of study entry may be submitted to the central reader to confirm eligibility) Exclusion Criteria Entered patients are ineligible for enrollment (ie, ineligible for randomization) and should be discontinued from the study if they meet any of the following criteria: [50] have screening laboratory test values, including thyroid-stimulating hormone (TSH), outside the reference range for the population or investigative site that, in the opinion of the investigator, pose an unacceptable risk for the patient s participation in the study a. Patients who are receiving thyroxine as replacement therapy may participate in the study provided stable therapy has been administered for 12 weeks and TSH is within the laboratory s reference range. [51] have any of the following specific abnormalities on screening laboratory tests: AST or ALT >1.5 times the ULN total bilirubin 1.5 times the ULN hemoglobin <10.0 g/dl (100.0 g/l) total white blood cell count <2500 cells/l neutropenia (absolute neutrophil count <1200 cells/l) lymphopenia (lymphocyte count <750 cells/l) thrombocytopenia (platelets <100,000/L)
40 I4V-MC-JADV Clinical Protocol Page 38 egfr <40 ml/min/1.73 m2 In the case of any of the aforementioned laboratory abnormalities, the tests may be repeated once within 2 weeks of the initial values, and values resulting from repeat testing may be accepted for enrollment eligibility if they meet the eligibility criterion. [52] have screening electrocardiogram (ECG) abnormalities that, in the opinion of the investigator or the sponsor, are clinically significant and indicate an unacceptable risk for the patient s participation in the study (eg, Fridericia s corrected QT interval >500 msec for men and >520 msec for women) [53] have symptomatic herpes simplex at the time of study enrollment [54] have evidence of active TB as documented by a positive purified protein derivative (PPD) test ( 5-mm induration between approximately 2 and 3 days after application, regardless of vaccination history), medical history, clinical symptoms, and abnormal chest x-ray at screening a. The QuantiFERON -TB Gold test (if available) may be used instead of the PPD test. Patients are excluded from the study if the test is not negative and there is clinical evidence of active TB. [55] have evidence of latent TB (as documented by a positive PPD, no clinical symptoms consistent with active TB, and a normal chest x-ray at screening) unless patients complete at least 4 weeks of appropriate treatment prior to randomization and agree to complete the remainder of treatment while in the trial a. If the PPD test is positive and the patient has no medical history or chest x-ray findings consistent with active TB, the patient may have a QuantiFERON-TB Gold test (if available). If the test results are not negative, the patient will be considered to have latent TB (for purposes of this study). b. The QuantiFERON-TB Gold test (if available) may be used instead of the PPD test. If the test results are positive, the patient will be considered to have latent TB. If the test is not negative, the test may be repeated once within 2 weeks of the initial value. If the repeat test results are again not negative, the patient will be considered to have latent TB (for purposes of this study). c. Exceptions include patients with a history of active or latent TB who have documented evidence of appropriate treatment. [56] have a positive test for HBV defined as: a. positive for hepatitis B surface antigen, or b. positive for anti-hepatitis B core antibody but negative for hepatitis B surface antibody (HBsAb), or
41 I4V-MC-JADV Clinical Protocol Page 39 c. for patients enrolled in Japan, or elsewhere if required, positive for anti-hbsab and positive for HBV deoxyribonucleic acid (DNA). If any of the HBV tests have an indeterminate result, confirmatory testing will be performed by an alternate method. [57] have HCV (positive for anti-hepatitis C antibody with confirmed presence of HCV) [58] have evidence of human HIV infection and/or positive HIV antibodies [59] Are women 40 and <60 years of age who have had a cessation of menses for at least 12 months, have a follicle-stimulating hormone (FSH) value <40 miu/ml, and do not agree to use 2 forms of highly effective methods of birth control when engaging in intercourse while enrolled in the study and for at least 28 days following the last dose of investigational product, unless they are congenitally or surgically sterile (ie, have had a hysterectomy, bilateral oophorectomy, or tubal ligation). If patients are entered into the study while taking medications that require discontinuation before randomization, those patients should only be asked to discontinue these medications after it has been determined that they will be eligible for randomization. Patients who are entered into the study but do not meet enrollment criteria should be discontinued from the study. These patients can be re-entered into the trial (ie, give repeat consent) if the investigator believes that the patient might meet enrollment criteria at a future date. The investigator should wait approximately 1 month before re-entering a patient into the study Rationale for Exclusion of Certain Study Candidates The rationale for the entry exclusion criteria is as follows: Exclusion Criteria [7] to [16] exclude individuals who are taking or who may take RA medications or treatments that interfere with the ability to assess the safety and efficacy of baricitinib. Exclusion Criteria [17] to [27] exclude individuals with previous or concomitant medical conditions that increase the risk for their participation in the study. Exclusion Criteria [28] to [35] exclude individuals who are at an increased risk for infections or infectious complications. Exclusion Criteria [36] to [38] exclude individuals who are pregnant, breastfeeding, at risk for becoming pregnant, or at risk for impregnating their partner during the study. Exclusion Criteria [39] to [46] exclude individuals who may not be compliant with study-related procedures or whose participation in the study may introduce bias. The rationale for the enrollment exclusion criteria is as follows: Exclusion Criteria [50] to [53] exclude individuals with concomitant medical conditions that increase the risk for their participation in the study. Exclusion Criteria [54] to [58] exclude individuals who are at an increased risk for infections or infectious complications. Exclusion Criterion [59] excludes individuals who are at risk for becoming pregnant during the study.
42 I4V-MC-JADV Clinical Protocol Page Discontinuations Discontinuation of Patients Interruption of Study Drug In some circumstances it may be necessary to temporarily interrupt treatment as a result of AEs or abnormal laboratory values as described in Table JADV.1 that may have an unclear relationship to study drug. Except in cases of emergency, it is recommended that the investigator consult with Lilly (or its designee) before temporarily interrupting therapy. The investigator must obtain approval from Lilly (or its designee) before restarting study drug that was temporarily discontinued because of an AE or abnormal laboratory value. Study drug must be held in the following situations involving laboratory abnormalities and may be resumed as noted in Table JADV.1. Table JADV.1. Criteria for Temporary Discontinuation of Investigational Product Hold Investigational Product if the Following Laboratory Test Results Occur: WBC count <2000 cells/µl ANC <1000 cells/l Lymphocyte count <500 cells/µl Platelet count <75,000/µL egfr <40 ml/min/1.73 m2 (from serum creatinine) for patients receiving the baricitinib 4 mg QD dose egfr <30 ml/min/1.73 m2 (from serum creatinine) for patients receiving the baricitinib 2 mg QD dose ALT or AST >5 times ULN Investigational Product May Be Resumed after Approval from Lilly (or its Designee) and When: WBC count 2500 cells/µl ANC >2000 cells/µl Lymphocyte count 750 cells/µl Platelet count 100,000/µL egfr 50 ml/min/1.73 m2 egfr 40 ml/min/1.73 m2 ALT and AST return to <2 times ULN and investigational product is not considered to be the cause of enzyme elevation Hemoglobin 10 g/dl Hemoglobin <8 g/dl Abbreviations: ALT = alanine aminotransferase; ANC = absolute neutrophil count; AST = aspartate aminotransferase; egfr = estimated glomerular filtration rate; min = minute; QD = once daily; ULN = upper limit of normal; WBC = white blood cell Discontinuation of Study Drug Any patient who is permanently discontinued from study drug for an AE or abnormal laboratory result should have the AE or abnormal laboratory value reported as an SAE (reported as Other Reason Serious). If any of the criteria listed in Section above recur after study drug is restarted, the patient should be permanently discontinued from study drug. In addition, patients will be permanently discontinued from study drug if they experience any of the criteria listed in Table JADV.2.
43 I4V-MC-JADV Clinical Protocol Page 41 Table JADV.2. Criteria for Permanent Discontinuation of Investigational Product Permanently Discontinue Investigational Product if any of the Following are Observed: ALT or AST >8 times the ULN ALT or AST >5 times the ULN persisting for more than 2 weeks after temporary interruption of investigational product ALT or AST >3 times the ULN and total bilirubin level >2 times the ULN ALT or AST >3 times the ULN with the appearance of fatigue, nausea, vomiting, right upper quadrant pain or tenderness, fever, rash, and/or eosinophilia (>5%) WBC count <1000 cells/µl Absolute neutrophil count <500 cells/µl Lymphocyte count <200 cells/µl Hemoglobin <6.5 g/dl Symptomatic herpes zoster Pregnancy Malignancy Severe infection that, in the opinion of the investigator, merits the investigational product being discontinued Anaphylactic or other serious allergic reaction Symptoms suggestive of a lupus-like syndrome Abbreviations: ALT = alanine aminotransferase; AST = aspartate aminotransferase; ULN = upper limit of normal; WBC = white blood cell Discontinuation from the Study The criteria for enrollment must be followed explicitly. If a patient who does not meet enrollment criteria is inadvertently enrolled, the investigator should contact Lilly or its designee to discuss whether the patient should be discontinued from study drug. If the patient is discontinued from study drug, the patient should remain in the study to provide the follow-up data needed for the primary analysis (Week 12) of the entire intention-to-treat (ITT) population. Patients may choose to withdraw from the study for any reason at any time, and the reason for early withdrawal will be documented. In addition, patients will be discontinued from the study in the following circumstances: Patient enrolls in any other clinical trial involving an investigational product or enrollment in any other type of medical research judged not to be scientifically or medically compatible with this study. Investigator decides that the patient should be withdrawn. If this decision is made because of an intolerable AE or a clinically significant laboratory value, the study drug is to be discontinued and appropriate measures are to be taken. Lilly (or designee) is to be notified immediately. Lilly (or designee) decides that the patient should be withdrawn. Patient is unwilling to continue in the study.
44 I4V-MC-JADV Clinical Protocol Page 42 Patient is noncompliant with protocol procedures. Patient withdraws consent. Investigator or Lilly (or designee), for any reason, stops the study. Patients who discontinue the study early will have Early Termination procedures and follow-up performed as shown in the study schedule (Attachment 1) Discontinuation of Study Sites Study site participation may be discontinued if Lilly, the investigator, or the ethical review board (ERB) of the study site judges it necessary for medical, safety, regulatory, or other reasons consistent with applicable laws, regulations, and good clinical practice (GCP) Discontinuation of the Study The study will be discontinued if Lilly judges it necessary for medical, safety, regulatory, or other reasons consistent with applicable laws, regulations, and GCP.
45 I4V-MC-JADV Clinical Protocol Page Treatment 9.1. Treatments Administered This study involves a comparison of baricitinib 4 mg administered orally QD, placebo administered orally QD, and adalimumab 40 mg administered by SC injection biweekly. Patients will continue to take background MTX therapy during the course of the study. Table JADV.3 shows the treatment regimens. Table JADV.3. Treatment Regimens Treatment Group Baricitinib treatment Placebo comparator Active comparator (adalimumab) Treatments Administered during Part A Baricitinib 4 mgb oral once-daily tablet Adalimumab placebo by SC injection biweekly Baricitinib placebo oral once-daily tablet Adalimumab placebo by SC injection biweekly Baricitinib placebo oral once-daily tablet Adalimumab 40 mg by SC injection biweekly Treatments Administered during Part B (Patients Not Rescued) Baricitinib 4 mgb oral once-daily tablet Adalimumab placebo by SC injection biweekly Baricitinib 4 mgb oral once-daily tablet Adalimumab placebo by SC injection biweekly Baricitinib placebo oral once-daily tablet Adalimumab 40 mg by SC injection biweekly Treatments Administered during Part B (after Rescuea) Baricitinib 4 mgb oral once-daily tablet Baricitinib 4 mgb oral once-daily tablet Baricitinib 4 mgb oral once-daily tablet Abbreviation: SC = subcutaneous. a See Section for details of rescue medication. b The dose for patients with renal impairment, defined as estimated glomerular filtration rate <60 ml/min/1.73 m2, will be 2 mg once daily. The investigator or his/her designee will be responsible for explaining the correct use of both the oral and injectable investigational agent(s) to the patient, verifying that instructions are followed properly, maintaining accurate records of investigational product dispensing and collection, and returning all unused medication to Lilly or its designee at the end of the study. Injections of investigational product should be administered by the site personnel, with instructions to the patient, at Visit 2, and the patient should be supervised while self-administering injections at Visits 4 and 5. In some cases, sites may destroy the material if, during the investigator site selection, the evaluator has verified and documented that the site has appropriate facilities and written procedures to dispose of clinical trial materials. Patients will be instructed to contact the investigator as soon as possible if they have a complaint or problem with the investigational product so that the situation can be assessed.
46 I4V-MC-JADV Clinical Protocol Page Materials and Supplies Lilly (or designee) will provide the following primary study materials: tablets containing 4 mg of baricitinib tablets containing 2 mg of baricitinib tablets containing placebo to match 4 mg baricitinib tablets containing placebo to match 2 mg baricitinib single-use syringes containing 40 mg adalimumab for SC injection single-use syringes containing placebo to match 40 mg adalimumab for SC injection Investigational product will be dispensed to the patient at the principal investigator s study site. Investigational product packaging will contain enough tablets and syringes for the longest possible interval between visits. Investigational product will be labeled according to the country s regulatory requirements. All investigational products will be stored, inventoried, reconciled, and destroyed according to applicable regulations. Investigational products will be supplied by Lilly or its representative in accordance with current Good Manufacturing Practices, and will be supplied with lot numbers, expiry dates, and certificates of analysis (as applicable). Adalimumab and placebo to match adalimumab should be stored at 2C to 8C (35F to 46F) in the original carton to protect from light. Sites will be required to monitor the temperature of the on-site storage conditions of the syringes Method of Assignment to Treatment Patients who meet all criteria for enrollment will be randomized in a 3:3:2 ratio (baricitinib:placebo:adalimumab) to double-blind treatment at Week 0 (Visit 2). Randomization will be stratified by region and joint erosion status (1 joint erosion plus seropositivity versus 3 joint erosions). Assignment to treatment groups will be determined by a computer-generated random sequence using an interactive voice-response system (IVRS). The IVRS will be used to assign packages of baricitinib/placebo and packages of prefilled syringes of adalimumab/placebo for SC injection containing double-blind investigational product to each patient. Each package of clinical trial material will supply sufficient medication for the number of weeks between visits. Site personnel will confirm that they have located the correct package by entering a confirmation number found on the packages of investigational product into the IVRS before dispensing the package to the patient Rationale for Selection of Doses in the Study Based on efficacy, safety, and PK data from the Phase 2 studies (JADC and JADA), Lilly has selected a single starting dose of 4 mg QD baricitinib for evaluation in this Phase 3 study. Clinical Efficacy Considerations In Study JADC, baricitinib doses of 4, 7, and 10 mg QD were studied and found to produce similar degrees of efficacy across the measured parameters at both the Week 12 and Week 24
47 I4V-MC-JADV Clinical Protocol Page 45 time points. Study JADA included lower doses of baricitinib (1 and 2 mg QD) to find the minimum clinically effective dose and to confirm the flat dose-response curve in the dose range of 4 to 8 mg QD. In Study JADA, a relatively flat dose-response curve was observed in the 4-and 8-mg QD dose groups at Week 12 across almost all measured efficacy parameters. The 1- and 2-mg QD dose groups are biologically active but did not produce adequate response at Week 12 as assessed by ACR50, ACR70, and low disease activity and remission rates when compared to placebo and the 4- and 8-mg dose groups. All 4 doses were associated with reductions in hscrp and ESR values over time. Assessment of the Week 12 magnetic resonance imaging (MRI) data suggests the potential for the 4- and 8-mg QD baricitinib doses to have a favorable impact on reducing structural damage given the observed significant improvement in osteitis. Week 24 data from Study JADA demonstrated persistence of the flat dose response between the 4- and 8-mg QD doses. Over the second 12-week period, continued administration of the 2-mg QD dose did not result in further improvement in the clinically important endpoints of ACR50, ACR70, DAS28-hsCRP 3.2, DAS28-hsCRP <2.6, and CDAI and SDAI remission rates. Week 24 MRI data further support the potential for the 4- and 8-mg QD doses to have a beneficial effect on prevention of structural damage. Clinical Safety Considerations Baricitinib was generally safe and well tolerated in single doses ranging from 1 to 20 mg and in repeat oral doses ranging from 2 to 20 mg. The most commonly reported treatment-emergent AEs (TEAEs) were in the infections and infestations system organ class. The most common alterations in laboratory values involved decreases in hemoglobin, hematocrit, total red blood cells, and white blood cells (neutrophils and other white cell lines), and increases in platelet counts, HDL, LDL, total cholesterol, and triglycerides. Patients with RA experienced a mean increase in ALT/AST compared to placebo, with the mean values remaining within the normal reference range and clinically significant increases in ALT/AST occurring uncommonly. Patients with impaired renal function have increased exposure to baricitinib. Dose Adjustment for Renal Impairment The therapeutic dose range for baricitinib was identified from concentration-efficacy relationships for ACR20 and DAS28-hsCRP as well as concentration-safety relationships identified for absolute neutrophil counts and hemoglobin. The starting dose for this Phase 3 study will be 4 mg QD. As demonstrated in a study of subjects with normal renal function versus those with mild to end-stage renal disease (Study I4V-MC-JADL), baricitinib exposure increases with decreased renal function. Based on PK simulations of baricitinib exposures for the mild and moderate categories of renal function (stratified as egfr 60 to <90 ml/min/1.73 m2 and egfr 30 to <60 ml/min/1.73 m2, respectively) dose adjustment is not required for patients with egfr 60 ml/min/1.73 m2. Patients with egfr <60 ml/min/1.73 m2 will receive a dose of 2 mg QD, which will ensure that exposures are comparable to, but do not exceed, those of the 4-mg QD dose. Lilly intends to enroll patients with egfr no lower than 40 ml/min/1.73 m2 in the Phase 3 trials. Baricitinib will be discontinued if egfr falls below
48 I4V-MC-JADV Clinical Protocol Page ml/min/1.73 m2. Baricitinib administration to patients with severe renal impairment (egfr <30 ml/min/1.73 m2) is not intended. Summary The clinical data known to date suggest a dosing strategy using a 4-mg QD dose as having the potential for providing clinically relevant efficacy response while minimizing potential risks of AEs Selection and Timing of Doses Each patient will receive blinded investigational product (baricitinib 4 mg or baricitinib placebo tablets orally QD and adalimumab 40 mg or adalimumab placebo biweekly by SC injection) beginning on the day of Visit 2 (Week 0). Baricitinib 4-mg oral dosing will continue daily through Visit 15 (Week 52) and biweekly injections of adalimumab 40 mg or adalimumab placebo will continue biweekly through Week 50. Baricitinib placebo oral dosing will continue daily through Visit 11 (Week 24); at Visit 11 (Week 24), all patients randomized to placebo will be reassigned to baricitinib. Injectable investigational product should be administered at approximately the same time of day, as much as possible. For injections not administered on the scheduled day of the week, the missed dose should be administered within 3 days of the scheduled day during study Weeks 1 to 12 and within 5 days of the scheduled day during study Weeks 13 to 52. Dates of subsequent study visits should not be modified according to this delay. As noted in Section 9.1, injections of investigational product should be administered by site personnel at Visit 2, and the patient should be supervised while self-administering injections at Visits 4 and 5. Thereafter, the patient will self-administer SC injections at home biweekly when no study visit is scheduled (ie, Weeks 6, 10, 18, 22, 26, 30, 34, 36, 38, 42, 44, 46, 48, and 50). On weeks when study visits are scheduled (ie, Weeks 8, 12, 14, 16, 20, 24, 28, 32, and 40), the patient should not administer his/her SC injection at home but should bring the study medication to the clinic for administration during the study visit. This will allow sites to administer rescue dosing when required. Oral investigational product should be taken at home at approximately the same time each day, as much as possible, with the exception of the following visits, to allow for the proper timing of PK sample collection and to ensure that efficacy assessments are made at trough PK levels: At Visit 2 (Week 0), patients will take their investigational product in the clinic, to allow for PK samples to be taken at 15 and 60 minutes postdose. For Visits 7 (Week 12), Visit 9 (Week 16), 11 (Week 24), 13 (Week 32), and 15 (Week 52), patients will be asked not to take their investigational product prior to visiting the clinic, to allow efficacy assessments to be made prior to investigational product dosing and a predose PK sample to be taken at some of these selected visits (Attachment 1).
49 I4V-MC-JADV Clinical Protocol Page Special Treatment Considerations In consideration of the disease severity, all patients in Study JADV will be offered rescue therapy starting at Week 16 if they are determined to be nonresponders. At Week 16, patients who are nonresponders and who were randomized to placebo will be rescued with baricitinib. To maintain study integrity through investigational product blinding, patients who were originally randomized to baricitinib will continue to receive baricitinib, with appropriate ongoing assessment of response and use of concomitant medication as needed. Patients who are nonresponders and who were randomized to adalimumab will be reassigned to baricitinib as rescue therapy. Nonresponse at Week 16 is defined as lack of improvement of at least 20% in both TJC and SJC at both Week 14 and Week 16 compared to baseline. At Week 16, the IVRS will assign rescue treatment based on TJC and SJC values. After Week 16, rescue therapy will be offered to patients at the discretion of the investigator based on TJCs and SJCs. Once a patient has been rescued, new NSAIDS, corticosteroids, and/or analgesics may be added or doses of ongoing concomitant NSAIDs, corticosteroids, and/or analgesics may be increased at the discretion of the investigator. A patient may only be rescued once. If a patient continues to meet nonresponse criteria for 4 weeks after rescue or at any time point thereafter, that patient should be discontinued from the study. Nonresponders who are rescued will receive an oral tablet daily, but no biweekly SC injection, for the remainder of the study. Rescue therapy will be administered through Week Continued Access to Investigational Product After the Week 52 study visit, eligible patients may proceed to a separate extension study (Study JADY) lasting for up to 2 years Blinding This is a double-blind, double-dummy study. To preserve the blinding of the study, a minimum number of Lilly personnel will see the randomization table and treatment assignments before the study is complete. Emergency unblinding for AEs may be performed through an IVRS, which may supplement or take the place of emergency codes generated by a computer drug-labeling system. This option may be used ONLY if the patient s well-being requires knowledge of the patient s treatment assignment. All calls resulting in an unblinding event are recorded and reported by the IVRS. The investigator should make every effort to contact the Lilly clinical research physician prior to unblinding a patient s treatment assignment. If a patient s treatment assignment is unblinded, Lilly must be notified immediately by telephone. If an investigator, site personnel performing assessments, or patient is unblinded, the patient must be discontinued from the study. In cases for which there are ethical reasons to have the
50 I4V-MC-JADV Clinical Protocol Page 48 patient remain in the study, the investigator must obtain specific approval from a Lilly clinical research physician for the patient to continue in the study. Processes to maintain blinding during the interim analysis conducted by the DMC are described in Section Concomitant Therapy Patients will be instructed to consult the investigator or other appropriate study personnel at the site before taking any new medications or supplements during the study. As listed below, all patients will continue to take the background MTX therapy at a stable dose throughout the study. Additional drugs are to be avoided during the study unless required to treat an AE or for the treatment of an ongoing medical condition. Investigators should follow local guidelines for the management of lipid disorders. If the need for other concomitant medications arises, discontinuation of the patient from study drug or the study will be at the discretion of the investigator in consultation with Lilly (or designee). Patients will not be permitted to have received a live vaccination up to 12 weeks prior to baseline or at any time during the study (with the exception of herpes zoster vaccination, which is not permitted within 30 days of planned randomization or at any time during the study). Treatment with concomitant DMARDs during the study is permitted as outlined below and in the inclusion/exclusion criteria: Use of concomitant oral MTX for at least 12 weeks with treatment at a stable dose of 7.5 to 25 mg/week (or the equivalent injectable dose) for at least 8 weeks prior to entry into the study is required for study participation. Patients should remain on the same dose of MTX throughout Part A and Part B of the study; however, the MTX dose may be adjusted for safety reasons. Patients may also be on concomitant hydroxychloroquine or sulfasalazine. If receiving hydroxychloroquine (up to 400 mg/day) or sulfasalazine (up to 3000 mg/day), the patient must be receiving a stable dose for at least 8 weeks prior to entry into the study, and the dose must remain stable throughout the study. Concomitant use of NSAIDs is permitted during Part A of the study only if the patient was on a stable dose for at least 6 weeks before planned randomization. Increase of NSAID dose and/or introduction of new NSAIDs are not permitted during Part A of the study unless a patient receives rescue therapy. After rescue, new NSAIDs or increases in doses of ongoing concomitant NSAIDs are permitted. Dose reductions and/or termination of NSAIDs are permitted at any time. During the study, concomitant use of analgesics is permitted. Increase of an analgesic dose and/or introduction of a new analgesic is not permitted during Part A of the study unless a patient receives rescue therapy. After rescue, new analgesics or increases in doses of ongoing concomitant analgesics are permitted. Dose reductions and/or termination of analgesics are permitted at any time.
51 I4V-MC-JADV Clinical Protocol Page 49 If an unforeseen intra-articular glucocorticoid injection is required during the study, it will be noted as a protocol deviation, and such joints will be excluded from future evaluation for the remainder of the study. Prednisone (or equivalent) at doses up to 10 mg per day is allowed during this study but must be maintained at stable levels from 6 weeks prior to randomization through the treatment phase of the study, unless a patient receives rescue therapy. After rescue, new corticosteroids or increases in doses of ongoing concomitant corticosteroids are permitted. Patients who were not previously on prednisone (or equivalent) prior to randomization should not initiate corticosteroid therapy during the study. Patients should not receive other systemic corticosteroids during the study including intra-muscular or intra-articular corticosteroids. Topical, intranasal, intra-ocular, and inhaled corticosteroids are permitted. Local standard of care should be followed for concomitant administration of folic acid. The following concomitant medications are prohibited after randomization: Live vaccines, including herpes zoster vaccination. Nonlive seasonal vaccinations and/or emergency vaccination, such as rabies or tetanus vaccinations, are allowed. Any DMARDs, other than stable treatment with hydroxychloroquine (up to 400 mg/day), sulfasalazine (up to 3000 mg/day), or MTX Any biologic therapy (such as TNF, interleukin-1, IL-6, T-cell-, or B-cell-targeted therapies) for any indication Any interferon therapy (such as Roferon-A, Intron-A, Rebetron, Alferon-N, Peg-Intron, Avonex, Betaseron, Infergen, Actimmune, Pegasys) Any parenteral corticosteroid administered by intramuscular or IV injection In addition to the concomitant medications guidelines outlined above, all patients who have not already received the herpes zoster vaccine at study entry will be encouraged to do so. Lilly will provide this vaccine where available. After receiving the herpes zoster vaccine, all patients must wait at least 30 days before randomization to study treatment. Any joints that had been injected with intraarticular corticosteroids within 6 weeks prior to randomization will be censored from the TJCs and SJCs for the duration of the study. All concomitant medications taken during the study must be recorded in the Concomitant Medication sections of the case report form (CRF) Treatment Compliance Patient compliance with study medication will be assessed at Visits 5 through 7 and Visits 9 through 15 (and, if necessary, at Early Termination) during the treatment period by counting returned tablets and returned syringes. Deviations from the prescribed dosage regimen should be recorded in the CRF. For baricitinib or oral placebo, a patient will be considered significantly noncompliant if he or she misses >20% of the prescribed doses during the study unless the patient s investigational product was withheld by the investigator for safety reasons. For adalimumab or injectable
52 I4V-MC-JADV Clinical Protocol Page 50 placebo, a patient will be considered significantly noncompliant if his/her study medication compliance rate is <80% or if there are any consecutive missed injections, unless the patient s investigational product was withheld by the investigator for safety reasons. Similarly, a patient will be considered significantly noncompliant if he or she is judged by the investigator to have intentionally or repeatedly taken more than the prescribed amount of medication. Patients found to be noncompliant with the investigational product should be assessed to determine the reason for noncompliance and educated and/or managed as deemed appropriate by the investigator to improve compliance.
53 I4V-MC-JADV Clinical Protocol Page Efficacy, Health Outcome Measures, Safety Evaluations, Sample Collection and Testing, and Appropriateness of Measurements Study procedures and their timing (including tolerance limits for timing) are summarized in the study schedule (Attachment 1) Efficacy Measures Primary Efficacy Measure The primary efficacy measure will be the proportion of patients achieving ACR20 at Week 12. ACR20 is defined as at least 20% improvement in the following ACR Core Set values: TJC (68 joint count) SJC (66 joint count) An improvement of 20% in at least 3 of the following 5 assessments: 1. Patient s Assessment of Pain (visual analog scale [VAS]) 2. Patient s Global Assessment of Disease Activity (VAS) 3. Physician s Global Assessment of Disease Activity (VAS) 4. Patient s Assessment of Physical Function as measured by the HAQ-DI 5. Acute phase reactant as measured by hscrp Secondary Efficacy Measures The secondary efficacy measures described below will be collected at the times shown in the study schedule (Attachment 1) Van der Heijde Modified Total Sharp Score Structural progression will be measured using the mtss. During screening, all patients meeting entry criteria will have a single posteroanterior radiographic assessment of the left and right hand/wrist and a single dorsoplantar radiographic image taken of the left and right foot. Alternatively, images taken within 4 weeks prior to baseline may be used. Images taken at screening (or within 4 weeks of screening) will be reviewed centrally by qualified readers for evidence of erosive bony change(s). These initial radiographs will serve as the baseline radiographs for comparison throughout the study. Additional radiographs (ie, a single posteroanterior view of each hand and a single dorsoplantar view of each foot) will be obtained at Week 16, Week 24 (the primary evaluation), and Week 52 (for the purposes of demonstrating durability of structural preservation as requested by regulatory authorities), as shown in the study schedule (Attachment 1). For patients who discontinue prematurely from the study, x-rays will be performed at the early termination visit only if the previous x-rays were taken more than 12 weeks earlier. X-rays of the hands/wrists and feet will be scored using the structural progression as measured using a modification of the mtss (van der Heijde 2000). This methodology quantifies the extent
54 I4V-MC-JADV Clinical Protocol Page 52 of bone erosions and joint space narrowing for 44 and 42 joints, respectively, with higher scores representing greater damage. X-rays will also be assessed for joint space narrowing and bone erosions. Quality control will be performed for proper imaging examination technique prior to allocation of the images to the central readers. The independent read of x-ray images will be performed by 2 primary readers, and 1 adjudicator, when necessary, based on predefined criteria. The 2 primary readers will each read 100% of the study patients. All time points will be displayed in random order for a given patient. The reader will have no knowledge of the true chronologic order, patient identity, or treatment group. To ensure a consistent read by the independent readers, an inter-/intra-reader variability assessment will be included in the central imaging core laboratory independent read. Repeat x-rays will be requested for all images obtained that are of poor quality as defined in the Image Acquisition Guidelines Disease Activity Score-Erythrocyte Sedimentation Rate and Disease Activity Score High Sensitivity C-Reactive Protein The DAS28 is a measure of disease activity in 28 joints that consists of a composite numeric score of the following variables: TJC, SJC, hscrp or ESR, and Patient s Global Assessment of Disease Activity (Vander Cruyssen et al. 2005). The 28 joints to be examined and assessed as tender or not tender for TJC and as swollen or not swollen for SJC include 14 joints on each side of the patient s body: the 2 shoulders, the 2 elbows, the 2 wrists, the 10 metacarpophalangeal joints, the 2 interphalangeal joints of the thumb, the 8 proximal interphalangeal joints, and the 2 knees (Smolen et al. 1995) ACR50 and ACR70 Indices ACR50 and ACR70 responses are secondary efficacy measures that are calculated as improvements of at least 50% and of at least 70%, respectively, in the ACR Core Set values identified in Section Hybrid American College of Rheumatology Response Measure The hybrid ACR (bounded) response measure will be obtained as described by the ACR Committee to Reevaluate Improvement Criteria (2007); see Table JADV.4.
55 I4V-MC-JADV Clinical Protocol Page 53 Table JADV.4. Scoring Method for the Hybrid American College of Rheumatology Response Measure Mean % Change in Core Set Measuresa ACR Status <20 20, <50 50, <70 70 Not ACR20 Mean % change ACR20 but not ACR50 20 Mean % change ACR50 but not ACR Mean % change ACR Mean % change Abbreviations: ACR = American College of Rheumatology; ACR20 = 20% improvement in ACR criteria; ACR50 = 50% improvement in ACR criteria; ACR70 = 70% improvement in ACR criteria. a 1) Calculate the average percentage change in core set measures. For each core set measure, subtract score after treatment from baseline score and determine percentage improvement in each measure. Next, if a core set measure worsened by >100%, limit that percentage change to 100% (set equal to -100% bound). Then, average the percentage changes for all core set measures. 2) Determine whether the patient has achieved ACR20, ACR50, or ACR70. 3) Using the table above, obtain the hybrid ACR response measure. To use the table, take the ACR20, ACR50, or ACR70 status of the patient (left column) and the mean percentage improvement in core set items; the hybrid ACR score is where they intersect in the table Simplified Disease Activity Index The SDAI is a tool for measurement of disease activity in RA that integrates measures of physical examination, acute phase response, patient self-assessment, and evaluator assessment. The SDAI is calculated by adding together scores from the following assessments: number of swollen joints (0 to 28), number of tender joints (0 to 28), CRP in mg/dl (0.1 to 10.0), patient global DAS on VAS (0 to 10.0), and evaluator global health score on VAS (0 to 10.0) (Aletaha and Smolen 2005) Disease remission is defined as an SDAI score of 3.3 (Felson et al. 2011) Clinical Disease Activity Index The CDAI is similar to the SDAI, but it allows for immediate scoring because it does not use a laboratory result. The CDAI is calculated by adding together scores from the following assessments: number of swollen joints (0 to 28), number of tender joints (0 to 28), patient global DAS on VAS (0 to 10.0), and evaluator global health score on VAS (0 to 10.0) (Aletaha and Smolen 2005) Remission is defined as a CDAI score of 2.8 (Felson et al. 2011).
56 I4V-MC-JADV Clinical Protocol Page European League Against Rheumatism Responder Index Assessments of patients with RA by EULAR28 will be used to categorize patients as nonresponders, moderate responders, good responders, or responders (moderate + good responders) according to Table JADV.5 (van Gestel et al. 1998). Table JADV.5. Postbaseline Level of DAS28 DAS Categorization of Patients as Nonresponders, Moderate Responders, or Good Responders Improvement Since Baseline in DAS28 > and > Good response 3.2 < DAS Moderate response DAS28 >5.1 No response Abbreviation: DAS28 = Disease Activity Score modified to include the 28 diarthroidal joint count Health Outcome Measures The health outcome measures listed below will be administered. Country-specific translations of each measure will be available in this study. Morning Joint Stiffness Severity Numeric Rating Scale (NRS): The Morning Joint Stiffness NRS is a patient-administered, single-item, 11-point horizontal scale anchored at 0 and 10, with 0 representing no joint stiffness and 10 representing joint stiffness as bad as you can imagine. Patients rate their morning joint stiffness each day (using an electronic handheld diary) by selecting the 1 number that describes their overall level of joint stiffness at the time they woke up. Duration of morning joint stiffness: The duration of morning joint stiffness is a patient-administered item that allows for the patients to enter the length of time in minutes (using 15-minute increments) that their morning joint stiffness lasted each day (using an electronic handheld diary). Recurrence of joint stiffness: The recurrence of joint stiffness throughout the day is a patient-administered single-item question assessed each day (using an electronic handheld diary) that asks Did you have any joint stiffness throughout today, other than when you woke up? (Yes/No) Tiredness Severity Numeric Rating Scale (Worst Tiredness NRS): The Worst Tiredness NRS is a patient-administered, single-item, 11-point horizontal scale anchored at 0 and 10, with 0 representing no tiredness and 10 representing as bad as you can imagine. Patients rate their tiredness each day (using an electronic handheld diary) by selecting the 1 number that describes their worst level of tiredness during the past 24 hours. Pain Severity Numeric Rating Scale (Worst Pain NRS): The Worst Pain NRS is a patient-administered, single-item, 11-point horizontal scale anchored at 0 and 10, with 0 representing no pain and 10 representing joint pain as bad as you can imagine.
57 I4V-MC-JADV Clinical Protocol Page 55 Patients rate their joint pain each day (using an electronic handheld diary) by selecting the 1 number that describes their worst level of joint pain during the past 24 hours. HAQ-DI: The HAQ-DI is a patient-reported questionnaire that is commonly used in RA to measure disease-associated disability (assessment of physical function). It consists of 24 questions referring to 8 domains: dressing/grooming, arising, eating, walking, hygiene, reach, grip, and activities (Fries et al. 1980, 1982; Ramey et al. 1996). The disability section of the questionnaire scores the patient s self-perception on the degree of difficulty (0 = without any difficulty, 1 = with some difficulty, 2 = with much difficulty, and 3 = unable to do) when dressing and grooming, arising, eating, walking, hygiene, reach, grip, and performing other daily activities. The reported use of special aids or devices and/or the need for assistance of another person to perform these activities is also assessed. The scores for each of the functional domains will be averaged to calculate the functional disability index. FACIT-F scale: The FACIT-F scale (Cella and Webster 1997) is a brief, 13-item, symptom-specific questionnaire that specifically assesses the self-reported severity of fatigue and its impact upon daily activities and functioning. The FACIT-F uses 0 ( not at all ) to 4 ( very much ) numeric rating scales to assess fatigue and its impact in the past 7 days. Scores range from 0 to 52 with higher scores indicating less fatigue. SF-36v2 Acute: The SF-36v2 Acute measure is a subjective, generic, health-related quality of life instrument that is patient-reported and consists of 36 questions covering 8 health domains: physical functioning, bodily pain, role limitations due to physical problems, role limitations due to emotional problems, general health perceptions, mental health, social function, and vitality. Each domain is scored by summing the individual items and transforming the scores into a 0 to 100 scale with higher scores indicating better health status or functioning. In addition, 2 summary scores, the PCS (physical component score) and the MCS (mental component score), will be evaluated based on the 8 SF-36v2 Acute domains (Brazier et al. 1992; Ware and Sherbourne 1992). WPAI-RA: The WPAI-RA questionnaire was developed to measure the effect of general health and symptom severity on work productivity and regular activities. It contains 6 items covering overall work productivity (health), overall work productivity (symptom), impairment of regular activities (health), and impairment of regular activities (symptom). Scores are calculated as impairment percentages (Reilly et al. 1993). EQ-5D-5L: The EQ-5D-5L is a standardized measure of health status that provides a simple, generic measure of health for clinical and economic appraisal. The EQ-5D-5L consists of 2 components: a descriptive system of the respondent s health and a rating of his or her current health state using a 0- to 100-mm VAS. The descriptive system comprises the following 5 dimensions: mobility, self-care, usual activities, pain/discomfort, and anxiety/depression. Each dimension has 5 levels: no problems, slight problems, moderate problems, severe problems, and extreme problems. The respondent is asked to indicate his/her health state by ticking (or placing a cross) in the box associated with the most appropriate statement in each of the 5 dimensions. It should be noted that the numerals 1 to 5 have no arithmetic properties and should not be used as a cardinal score. The VAS records the respondent s self-rated health on a vertical VAS
58 I4V-MC-JADV Clinical Protocol Page 56 in which the endpoints are labeled best imaginable health state and worst imaginable health state. This information can be used as a quantitative measure of health outcome. The EQ-5D-5L health states, defined by the EQ-5D-5L descriptive system, may be converted into a single summary index by applying a formula that essentially attaches a value (also called weights) to each of the levels in each dimension (Brooks 1996; EuroQol Group 2011 [WWW]; Herdman et al. 2011). Quick Inventory of Depressive Symptomatology Self-Rated-16 (QIDS-SR 16 ): The QIDS-SR 16 is a 16-item, self-report instrument intended to assess the existence and severity of symptoms of depression as listed in the American Psychiatric Association's Diagnostic and Statistical Manual of Mental Disorders, 4th Edition (APA 1994). Patients are asked to consider each statement as it relates to the way they have felt for the past 7 days. There is a 4-point scale for each item ranging from 0 to 3. The 16 items corresponding to 9 depression domains are summed to give a single score ranging from 0 to 27. Additional information and the QIDS-SR 16 questions may be found on the University of Pittsburgh Epidemiology Data Center web site (Inventory of Depressive Symptomatology/Quick Inventory of Depressive Symptomatology [WWW]) (Rush et al. 2003; Trivedi et al. 2004). Healthcare resource utilization: The healthcare resource utilization data will be collected by site staff regarding the number of visits to medical care providers such as general practitioners, specialists, physical or occupational therapists, and other non-physical care providers for services outside of the clinical study; emergency room admissions; hospital admissions; and concomitant medications related to the treatment of RA. These data will be collected to support economic evaluations of treatment Safety Evaluations Investigators will be responsible for monitoring the safety of patients who enter this study and for alerting Lilly or its designee to any event that seems unusual, even if this event may be considered an unanticipated benefit to the patient. The investigator will be responsible for the appropriate medical care of patients during the study. The investigator remains responsible for following, through an appropriate health care option, AEs that are serious or that cause the patient to discontinue before completing the study. The patient should be followed until the event is resolved or explained. Frequency of follow-up evaluation will be left to the discretion of the investigator Adverse Events Lilly has standards for reporting AEs that are to be followed regardless of applicable regulatory requirements that may be less stringent. Lack of drug effect is not an AE in clinical studies because the purpose of the clinical study is to establish drug effect.
59 I4V-MC-JADV Clinical Protocol Page 57 Cases of pregnancy that occur during maternal or paternal exposures to investigational product or drug delivery system should be reported. Data on fetal outcome and breastfeeding are collected for regulatory reporting and drug safety evaluation. Study site personnel will record the occurrence and nature of each patient s preexisting conditions, including clinically significant signs and symptoms of the disease under treatment in the study. After the ICF is signed, site personnel will record any change in the condition(s) and the occurrence and nature of any AEs. All AEs related to protocol procedures will be reported to Lilly or its designee. In addition, all AEs occurring after the patient receives the first dose of investigational product must be reported to Lilly or its designee via electronic data entry. Any clinically significant findings from ECGs, laboratory tests, vital sign measurements, or other procedures that result in a diagnosis should be reported to Lilly or its designee. Investigators will be instructed to report to Lilly or its designee their assessment of the potential relatedness of each AE to protocol procedure, studied disease state, investigational product, and/or drug delivery system via electronic data entry. Study site personnel must alert Lilly or its designee within 24 hours of the investigator unblinding a patient s treatment group assignment for any reason. If a patient s dosage is reduced or treatment is discontinued as a result of an AE, study site personnel must clearly report to Lilly or its designee via electronic data entry the circumstances and data leading to any such dosage reduction or discontinuation of treatment Adverse Events of Special Interest Adverse events of special interest (AESIs) will include: severe or opportunistic infections myelosuppressive events of anemia, leukopenia, neutropenia, lymphopenia, and thrombocytopenia thrombocytosis elevations in ALT or AST (>3 times ULN) with total bilirubin (>2 times ULN) Patients with these laboratory value specified events will be identified using the same criteria for the interruption of investigational product with the exception of anemia, which will be identified using the same criteria for the discontinuation of investigational product, and thrombocytosis, which will be defined as a platelet count >600,000/μL Serious Adverse Events SAE collection will begin after the patient has signed informed consent and has received investigational product. If a patient experiences an SAE after signing informed consent but prior to receiving investigational product, the event will NOT be collected unless the investigator feels the event may have been caused by a protocol procedure.
60 I4V-MC-JADV Clinical Protocol Page 58 Previously planned (prior to signing the ICF) surgeries should not be reported as SAEs unless the underlying medical condition has worsened during the course of the study. Study site personnel must alert Lilly or its designee of any serious adverse event (SAE) within 24 hours of investigator awareness of the event via a sponsor-approved method. Alerts issued via telephone are to be immediately followed with official notification on study-specific SAE forms. An SAE is any AE from this study that results in one of the following outcomes: death initial or prolonged inpatient hospitalization a life-threatening experience (that is, immediate risk of dying) persistent or significant disability/incapacity congenital anomaly/birth defect considered significant by the investigator for any other reason Important medical events that may not result in death, be life-threatening, or require hospitalization may be considered serious adverse drug events when, based on appropriate medical judgment, they may jeopardize the patient and may require medical or surgical intervention to prevent one of the outcomes listed in this definition. SAEs occurring after a patient has taken the last dose of investigational product will be collected in the pharmacovigilance system and the clinical data collection database for 28 days after the last dose of investigational product, regardless of the investigator s opinion of causation. Thereafter, SAEs are not required to be reported unless the investigator feels the events were related to investigational product, drug delivery system, or protocol procedure. Information on SAEs expected in the study population independent of drug exposure and that will be assessed by the sponsor in aggregate periodically during the course of the trial may be found in the IB. In the RA population, the occurrence of malignancies, major cerebrocardiovascular events (including death, myocardial infarction, hospitalization for unstable angina, hospitalization for heart failure, serious arrhythmia, resuscitated sudden death, cardiogenic shock due to myocardial infarction, coronary revascularization procedure; neurologic-stroke; and peripheral vascular events), and serious infections is reasonably anticipated because of the age of the population, comorbid conditions, disease state, and concomitant medications Suspected Unexpected Serious Adverse Reactions Suspected unexpected serious adverse reactions (SUSARs) are serious events that are not listed in the IB and that the investigator identifies as related to investigational product or procedure. Lilly has procedures, which are consistent with global regulations and the associated detailed guidances that will be followed for the recording and expedited reporting of SUSARs.
61 I4V-MC-JADV Clinical Protocol Page Other Safety Measures Measures Evaluated During Screening Period Physical examination. One complete physical examination (excluding pelvic and rectal examinations) will be performed at screening. This examination will determine whether the patient meets the criteria required to participate in the study and will also serve as a monitor for preexisting conditions and as a baseline for TEAE assessment. Body weight and height will also be recorded. Electrocardiograms. A single 12-lead ECG will be obtained at screening to determine whether the patient meets entry criteria for the study. ECGs will be locally (machine) read and interpreted by a qualified physician (the investigator or qualified designee) at the site. Chest x-ray and tuberculosis testing. A posterior-anterior view chest x-ray will be obtained locally, unless results from a chest x-ray obtained within 6 months prior to the study are available. The chest x-ray will be reviewed by the investigator or his/her designee to exclude patients with active TB infection. In addition, patients will be tested at screening and entry for evidence of active or latent TB as described in exclusion criteria, Section Measures Evaluated During Screening and Treatment Periods Vital signs and physical characteristics. Vital signs (sitting blood pressure and heart rate) will be measured at times indicated in the study schedule (Attachment 1). Subjects should be seated and relaxed with both feet on the floor for at least 5 minutes prior to taking measurements. Simultaneous blood pressure and pulse measurements should be made using either automated or manual equipment. Three replicate readings should be made at each time point at approximately 30- to 60-second intervals. If measurements are machine averaged, the average reading should be recorded on the CRF. If measurements are manual or the machine does not provide an average reading, then each individual reading should be recorded on the CRF. Measurements should be made before any scheduled blood draws. Any clinically significant findings that result in a diagnosis should be captured on the CRF and reported as an AE. Additional measurements of vital signs may be performed at the discretion of the investigator. Physical characteristics including weight and waist circumference will be measured at times indicated in the study schedule (Attachment 1). Standard laboratory tests. Hematology, clinical chemistry, urinalysis, lipid profile, egfr, iron studies, hscrp, and ESR will be measured at times indicated in the study schedule (Attachment 1) Safety Monitoring The Lilly clinical research physician will monitor safety data throughout the course of the study. Lilly will review SAEs within time frames mandated by company procedures. The Lilly clinical research physician will, as is appropriate, consult with the functionally independent Global Patient Safety therapeutic area physician or clinical scientist and periodically review trends in
62 I4V-MC-JADV Clinical Protocol Page 60 safety data and laboratory analytes. Any concerning trends in frequency or severity noted by an investigator and/or Lilly (or designee) may require further evaluation. All deaths and SAE reports will be reviewed in a blinded manner by Lilly during the clinical trial. These reports will be reviewed to ensure completeness and accuracy but will not be unblinded to Lilly during the clinical trial. If a death or clinical AE is deemed serious, unexpected, and possibly related to investigational product, only Lilly Global Patient Safety will be unblinded for regulatory reporting and safety monitoring purposes. These measures will preserve the integrity of the data collected during this trial and minimize any potential for bias while providing for appropriate safety monitoring. Liver function monitoring will occur frequently throughout the study. If a study patient experiences elevated ALT or AST 3 ULN or elevated total bilirubin 2 ULN, clinical and laboratory monitoring should be initiated by the investigator. Details for hepatic monitoring depend on the severity and persistence of observed laboratory test abnormalities. To ensure patient safety and comply with regulatory guidance, the investigator is to consult with the Lilly designated medical monitor regarding collection of specific recommended clinical information and follow-up laboratory tests (see Attachment 3). Investigators will monitor vital signs and carefully review findings that may be associated with cardiovascular events. AE reports and vital signs will be collected at each study visit. The cardiovascular monitoring plan includes, in addition, the following: regular monitoring of lipid levels adjudication of major adverse cardiovascular events (all deaths and nonfatal myocardial infarctions, hospitalization for unstable angina, hospitalization for heart failure, coronary interventions [such as coronary artery bypass graft or percutaneous coronary intervention], stroke, and transient ischemic attack) by a Cardiovascular Safety Committee at regular intervals estimation of relative rates of occurrence of select AEs using observations obtained from a matched, observational, comparator cohort of RA patients using existing RA registry/registries. A DMC (an advisory group for this study formed to protect the patients; refer to the Interim Analysis in Section ) will also oversee the conduct of all of the baricitinib Phase 3 clinical trials in patients with RA. In the event that safety monitoring uncovers an issue that needs to be addressed by unblinding at the group level, only members of the DMC will be able to conduct additional analyses of the safety data Complaint Handling Lilly collects product complaints on investigational products and drug delivery systems used in clinical studies to ensure the safety of study participants, monitor quality, and facilitate process and product improvements. Complaints related to unblinded concomitant drugs or drug delivery systems are reported directly to the manufacturers of those drugs/devices in accordance with the package inserts. For blinded studies, all product complaints associated with material packaged, labeled, and released by Lilly or its delegate will be reported.
63 I4V-MC-JADV Clinical Protocol Page 61 The investigator or his/her designee is responsible for handling the following aspects of the product complaint process in accordance with the instructions provided for this study: recording a complete description of the product complaint reported and any associated AEs using the study-specific complaint forms provided for this purpose faxing the completed product complaint form within 24 hours to Lilly or its designee If the investigator is asked to return the product for investigation, he/she will return a copy of the product complaint form with the product Sample Collection and Testing Attachment 1 lists the schedule for sample collections in this study, and Attachment 2 lists the specific tests that will be performed for this study Samples for Standard Laboratory Testing Standard laboratory tests, including chemistry, hematology, and urinalysis panels, will be performed. Serum and/or urine pregnancy tests will be performed at each visit as described in the study schedule (Attachment 1). Blood and urine samples will be collected for other laboratory tests at the times specified in the study schedule (Attachment 1). Blood will be collected by venipuncture. Attachment 2 lists the specific tests that will be performed for this study. Routine and other clinical laboratory tests will be analyzed by a central laboratory selected by Lilly. Investigators must document their review of each laboratory safety report. Samples collected for specified laboratory tests will be destroyed within 60 days of receipt of confirmed test results. Tests are run and confirmed promptly whenever scientifically appropriate. When scientific circumstances warrant, however, it is acceptable to retain samples to batch the tests run or to retain the samples until the end of the study to confirm that the results are valid. Certain samples may be retained for a longer period, if necessary, to comply with applicable laws, regulations, or laboratory certification standards Samples for Drug Concentration Measurements Pharmacokinetics/Pharmacodynamics A single venous blood sample will be drawn at the times indicated in the study schedule (Attachment 1). These blood samples will be used to determine the plasma concentrations of baricitinib. Concentrations of baricitinib in human plasma will be determined by a validated liquid chromatography tandem mass spectrometry (LC/MS/MS) method. Blood samples that will be used for other laboratory assessments (chemistry, hematology, etc) should be drawn at approximately the same time as the samples used for plasma concentrations of baricitinib. The timing will be as follows: At Visit 2 (Week 0), patients will take their oral investigational product in the clinic, and PK samples will be drawn at 15 minutes and 1 hour postdose.
64 I4V-MC-JADV Clinical Protocol Page 62 At Visit 5 (Week 4), patients will be asked to take their oral investigational product at home prior to visiting the clinic. The clinic visit should be scheduled so that the sample taken during this visit is 2 to 4 hours after the oral dose taken at home. At Visit 6 (Week 8), patients will be asked to take their oral investigational product at home prior to visiting the clinic. The clinic visit should be scheduled so that the sample taken during this visit is 4 to 6 hours after the oral dose taken at home. For Visits 7 (Week 12), 11 (Week 24), and 13 (Week 32), patients will be asked not to take their oral investigational product prior to visiting the clinic, and a sample will be taken at any time predose on the day of the clinic visits. If the patient has taken his/her oral dose prior to the visit, the sample may be drawn anytime postdose, and the inability to collect a predose sample will not be considered a protocol violation. For the early termination visit, a sample may be drawn anytime if the last dose of oral investigational drug was taken within the last 48 hours. For visits where PK samples will be taken predose, the actual date and exact timing (24-hour clock) of sample collection and the date and time of the last 2 doses prior to the sample being drawn should be recorded. For visits where PK samples are taken postdose, the actual date and exact timing (24-hour clock) of sample collection and the date and time of the last 3 doses prior to the sample being drawn should be recorded. This sampling schedule should be followed as closely as possible; however, failure to take PK samples at these specified times will not be considered a protocol violation. If the patient fails to follow the directions for a particular visit, the sample should still be collected at that visit, and the date and exact timing (24-hour clock) of sample collection and the date and time of the last 2 doses prior to the sample being drawn should be recorded. In the event of an SAE, up to 2 additional blood samples may be taken at the investigator s discretion. Only samples from patients receiving baricitinib will be assayed; samples from patients receiving placebo or adalimumab will not be assayed. PK samples will be kept in storage at a laboratory facility designated by the sponsor. PK samples may also be assayed for additional exploratory analyses. PK results will not be provided to investigative sites until the completion of the study or to the blinded study team until the study has been unblinded. Bioanalytical samples collected to measure investigational product concentration will be retained for a maximum of 1 year following last patient visit for the study Exploratory Stored Samples Pharmacogenetics There is growing evidence that genetic variation may impact a patient s response to therapy. Variable response to therapy may be due to genetic determinants that affect drug absorption, distribution, metabolism, and excretion; the mechanism of action of the drug; the disease etiology; and/or the molecular subtype of the disease being treated. Therefore, where local
65 I4V-MC-JADV Clinical Protocol Page 63 regulations allow, blood samples will be collected for pharmacogenetic analysis, as noted in the study schedule (Attachment 1). Samples will be stored, and analysis may be performed on genetic variants thought to play a role in active RA including, but not limited to, the cluster of immune response genes on chromosome 6, such as HLA-DRB1, HLA-DQA1, HLA-DPA1, HLA-DPB1, TNF-α, and TNF receptors; RANK-Ligand; the JAK signaling pathways; MTX metabolizing genes such as methylene tetrahydrofolate reductase and other folate reductases; PTPN22 and related signaling molecules; IL-6 pathway members; and the STAT signaling family members, including STAT4, to evaluate their association with observed response to baricitinib. In the event of an unexpected AE or the observation of unusual response, the samples may be genotyped and analysis may be performed to evaluate a genetic association with response to baricitinib. These investigations may be limited to a focused candidate gene study or, if appropriate, genome-wide association studies may be performed to identify regions of the genome associated with the variability observed in drug response. Samples will only be used for investigations related to disease and drug or class of drugs under study in the context of this clinical program. They will not be used for broad exploratory unspecified disease or population genetic analysis. The sample will be identified by the patient number (coded) and stored for up to a maximum of 15 years after the last patient visit for the study at a facility selected by the sponsor. The sample and any data generated from it can be linked back to the patient only by investigator site personnel. The duration allows the Sponsor to respond to regulatory requests related to the study drug Nonpharmacogenetic/Biomarker Stored Samples Collection of samples for nonpharmacogenetic biomarker research is a required part of this study. Serum, plasma, whole blood RNA, and urine samples will be collected at the times specified in the study schedule (Attachment 1). Samples may be used for research on the drug target, disease process, pathways associated with disease state, mechanism of action of baricitinib, and/or research method or in validating diagnostic tools or assay(s) related to RA. Samples will be identified by the patient number (coded) and stored for up for a maximum of 15 years after the last patient visit for the study at a facility selected by the sponsor Appropriateness of Measurements All of the clinical and safety assessments made in this study are standard, widely used, and generally recognized as reliable, accurate, and relevant.
66 I4V-MC-JADV Clinical Protocol Page Data Quality Assurance To ensure accurate, complete, and reliable data, Lilly or its representatives will do the following: provide instructional material to the study sites, as appropriate sponsor a start-up training session to instruct the investigators and study coordinators. This session will give instruction on the protocol, the completion of the CRFs, and study procedures make periodic visits to the study site be available for consultation and stay in contact with the study site personnel by mail, telephone, and/or fax review and evaluate CRF data and use standard computer edits to detect errors in data collection In addition, Lilly or its representatives may periodically check a sample of the patient data recorded against source documents at the study site. The study may be audited by Lilly or its representatives and/or regulatory agencies at any time. Investigators will be given notice before an audit occurs. To ensure the safety of participants in the study and to ensure accurate, complete, and reliable data, the investigator will keep records of laboratory tests, clinical notes, and patient medical records in the patient files as original source documents for the study. If requested, the investigator will provide the sponsor, applicable regulatory agencies, and applicable ERBs with direct access to original source documents Data Capture System An electronic data capture system will be used in this study. The site will maintain a separate source for the data entered by the site into the sponsor-provided electronic data capture system. Electronic patient-reported outcome (epro) measures or other data reported directly by the subject will be entered into an epro instrument at the time that the information is obtained. Electronic physician-reported outcome measures will be directly entered by the physician or designee (Physician global assessment of disease activity) or by the blinded joint assessor (TJC/SJC) into the electronic site tablet. In these instances for which there is no prior written or electronic source data at the site, the epro instrument record will serve as the source. The epro records will be stored by a third party, and investigator sites will have continuous access to the source documents during the study and will receive an archival copy at the end of the study for retention. Any data for which the epro instrument record will serve to collect source data will be identified and documented by each site in that site s study file. CRF data will be encoded and stored in a clinical trial database. Data managed by a central vendor, such as laboratory test data or x-ray data, will be stored electronically in the central
67 I4V-MC-JADV Clinical Protocol Page 65 vendor s database system. Data will subsequently be transferred from the central vendor to the Lilly generic laboratories system. Data from complaint forms submitted to Lilly will be encoded and stored in the global product complaint management system.
68 I4V-MC-JADV Clinical Protocol Page Sample Size and Statistical Methods Determination of Sample Size Approximately 1280 patients (480 in the baricitinib group, 480 in the placebo group, and 320 in the adalimumab group) will provide: >95% power to detect a difference between the baricitinib and placebo treatment groups in ACR20 response rate (60% versus 35%) at Week 12 based on a 2-sided chi-square test at a significance level of 0.05 approximately 94% power to detect a difference in mtss between the baricitinib and placebo treatment groups for an effect size of 0.25 (ie, difference in means divided by common standard deviation [SD]), based on a 2-sided t-test at a significance level of This assumes 90% of randomized patients will have x-ray data available for the mtss analysis at Week 24 approximately 91% power for the noninferiority analysis of ACR20 response rate at Week 12 between the baricitinib and adalimumab treatment groups, where a response rate of 60% for baricitinib and adalimumab and a 1-sided significance level of 0.02 are assumed. This power calculation adopted a fixed noninferiority margin of 12%. There is growing support for this margin in recent clinical trials conducted in the MTX-IR patient population (Schiff et al. 2012). The above sample size and power estimates were obtained from nquery Advisor Statistical and Analytical Plans General Considerations Statistical analysis of this study will be the responsibility of Eli Lilly and Company, although implementation of the statistical analysis plan (SAP) may be delegated to a third party. A detailed SAP describing the statistical methodologies will be developed by Lilly or its designee. All statistical tests of treatment effects, besides noninferiority tests specified in Section , will be performed at 2-sided significance levels of 0.05, unless otherwise stated. Treatment comparisons of discrete efficacy variables will be made using a logistic regression analysis with region, joint erosion status (1 joint erosion plus seropositivity vs 3 joint erosions), and treatment group in the model. The percentages, difference in percentages, and (100-alpha)% confidence interval (CI) of the difference in percentages will be reported. Treatment-by-region interaction will be added to the logistic regression model of the primary and key secondary categorical variables as a sensitivity analysis. If this interaction is significant at a 2-sided 0.1 level, further inspection will be used to assess whether the interaction is quantitative (ie, the treatment effect is consistent in direction but not size of effect) or qualitative (the treatment is beneficial for some but not all regions). Treatment comparisons of continuous efficacy variables will be made using analysis of covariance (ANCOVA) with region, joint erosion status, treatment group, and baseline value in the model. Type III sums of squares for the least-squares means will be used for the statistical
69 I4V-MC-JADV Clinical Protocol Page 67 comparison; (100-alpha)% CI will also be reported. Sensitivity analysis will be performed for key secondary continuous variables by adding the treatment-by-baseline interaction to the ANCOVA model. If this interaction is significant at a 2-sided 0.1 level, then additional analyses will be performed. Treatment-by-region interaction will also be added to the model for sensitivity purposes. When a restricted maximum likelihood-based mixed model for repeated measures (MMRM) is used, the model will include treatment, region, joint erosion status, visit, treatment-by-visit-interaction as fixed categorical effects; baseline score and baseline score-byvisit-interaction as fixed continuous effects. An unstructured (co)variance structure will be used to model the between- and within-patient errors. If this analysis fails to converge, other structures will be tested. The Kenward-Roger method will be used to estimate the degrees of freedom. Type III sums of squares for the least-squares means will be used for the statistical comparison; CI will also be reported. Treatment group comparisons versus placebo at Week 12 and other visits will be tested. Further details on the use of MMRM will be described in the SAP. Fisher s exact test will be used for AEs, discontinuations, and other categorical safety data between treatment groups. Continuous vital signs, body weight, and other continuous safety variables, including laboratory variables will be analyzed using ANCOVA with treatment and baseline value in the model. Analysis Population: The efficacy and health outcome analyses will be conducted on an ITT basis unless otherwise specified. The ITT analysis set will include all data from all randomized patients treated with at least 1 dose of the study drug. Analysis of structural progression (van der Heijde mtss) will be conducted on the ITT population using patients with available baseline and at least 1 postbaseline value. In addition, the primary and major secondary analyses will be repeated using the per-protocol set (PPS), which is defined as all randomized patients who are compliant with treatment, who do not have significant protocol violations, and whose investigator site does not have significant GCP issues that require a report to the regulatory agencies. Safety analyses will be performed on the safety population, defined as all randomized patients who received at least 1 dose of the study drug. Patients will be analyzed for efficacy, health outcomes, and safety according to the treatment to which they were assigned. PK analysis will be conducted using all evaluable PK data. Further details will be described in the SAP. Missing Data Imputation: In accordance with precedent set with other Phase 3 RA trials (Keystone et al. 2004, 2008, 2009; Cohen et al. 2006; Smolen et al. 2008, 2009), the following methods for imputation of missing data will be used: 1. Nonresponder imputation (NRI): All patients who discontinue the study or the study treatment at any time for any reason will be defined as nonresponders for the NRI analysis for categorical variables, such as ACR20/50/70, from the time of discontinuation
70 I4V-MC-JADV Clinical Protocol Page 68 and onward. Patients who are eligible for rescue therapy at Week 16 will be analyzed as nonresponders after Week 16 and onward. Randomized patients without available data at a postbaseline visit will be defined as nonresponders for the NRI analysis at that visit. 2. Linear extrapolation method: The linear extrapolation method will be used for analysis of the structural progression endpoint (van der Heijde mtss) to impute missing data at time points when analyses are conducted (including the analyses at Week 24 and Week 52). For patients who discontinue the study or the study treatment or for patients who miss a radiograph for any reason, baseline data and the most recent radiographic data prior to discontinuation or the missed radiograph, adjusted for time, will be used for linear extrapolation to impute missing data at subsequent time points. For patients who receive rescue therapy starting at Week 16 or at any time point thereafter, baseline data and the most recent radiographic data prior to initiation of rescue therapy, adjusted for time, will be used for linear extrapolation. All patients originally randomized to placebo will be switched to active treatment at Week 24; thus, there will be no observed data for the placebo arm for the structural comparison at subsequent time points. Therefore, baseline data and the most recent radiographic data prior to initiation of the new therapy, adjusted for time, will be used for linear extrapolation at Week 52. The linear extrapolation method has been established as an appropriate missing data imputation method in other Phase 3 RA trials (Keystone et al. 2004, 2008, 2009; Cohen et al. 2006; Smolen et al. 2008, 2009). Missing postbaseline values will be imputed only if both a baseline value and a postbaseline value from another time point are available, as long as the patient was on the same treatment at each applicable time point. 3. Multiple imputation: Other methods than linear extrapolation that include multiple imputation will be employed for analysis of the structural progression endpoint (van der Heijde mtss) to impute missing data at Week 24. Details will be provided in the SAP. 4. Modified baseline observation carried forward (mbocf): The mbocf method will be used for the analysis of major secondary continuous endpoints (unless otherwise stated). For patients who discontinue the study or the study treatment because of an AE, including death, the baseline observation will be carried forward to the corresponding endpoint for evaluation. For patients who discontinue the study for reason(s) other than an AE, the last nonmissing postbaseline observation prior to discontinuation will be carried forward to the corresponding endpoint for evaluation. For patients who are eligible for rescue therapy starting at Week 16, the last nonmissing observation at or before rescue will be carried forward to subsequent time points for evaluation. 5. Modified last observation carried forward (mlocf): For all continuous measures including safety analyses, the mlocf will be a general approach to impute missing data unless otherwise specified. For patients who are eligible for rescue therapy starting at Week 16, the last nonmissing observation at or before rescue will be carried forward to subsequent time points for evaluation. For all other patients discontinuing from the study or the study treatment for any reason, the last nonmissing postbaseline observation before discontinuation will be carried forward to subsequent time points for evaluation. If the postbaseline observation is missing, then it will not be included in the analysis even if the
71 I4V-MC-JADV Clinical Protocol Page 69 baseline observation is not missing. The mlocf will be used as a sensitivity method to the mbocf method with respect to the major secondary endpoints. 6. Other methods for data imputation for analysis of key endpoints other than mtss may be used and will be described in the SAP. Adjustment for Multiple Comparisons: The following multiple testing procedure will be used to control the family-wise error rate for the primary and key secondary hypotheses for the endpoints listed in Sections 6.1 and 6.2. Each step in the sequential testing procedure uses a local level procedure for the null hypotheses being tested. Testing of hypotheses at each step of the procedure commences only if all null hypotheses of prior steps were rejected. As such, the multiple testing procedure provides strong control of the family-wise error rate at the 1-sided level. The overall strategy is illustrated (Bretz et al. 2009) in Figure JADV.2. Step 1: Test ACR20 at Week 12 for baricitinib versus placebo at 1-sided alpha = If the null hypothesis is rejected, proceed to Step 2. Otherwise, discontinue testing. Step 2: Test the change from baseline in HAQ-DI at Week 12 and the change from baseline in mtss at Week 24, both tests of baricitinib versus placebo, at 1-sided alpha = via a weighted Holm procedure (Holm 1979) with weights of 0.2 and 0.8, respectively. If both null hypotheses are rejected, proceed to Step 3. Otherwise, discontinue testing. Details of the Holm procedure at this step: Let p2 denote the unadjusted 1-sided p-value resulting from testing the hypothesis regarding HAQ-DI and let p3 denote the unadjusted 1-sided p-value resulting from testing the hypothesis regarding mtss. Let p2* and p3* denote the corresponding weight-adjusted p-values (ie, p2* = p2/0.2 and p3*=p3/0.8). The following decision rules apply: If min(p2*, p3*) >0.025, then Lilly will fail to reject both null hypotheses and discontinue testing. Otherwise, if p2* 0.025, then Lilly will reject the null hypothesis for HAQ-DI (declaring statistical significance) and test mtss using the unadjusted p-value. If p , Lilly will also reject the null hypothesis for mtss (declaring statistical significance); however if p3 >0.025, then Lilly will fail to reject the null hypothesis for mtss and discontinue testing. Otherwise, if p3* 0.025, then Lilly will reject the null hypothesis for mtss (declaring statistical significance) and test HAQ-DI using the unadjusted p-value. If p , Lilly will also reject the null hypothesis for HAQ-DI (declaring statistical significance); however if p2 >0.025, then Lilly will fail to reject the null hypothesis for HAQ-DI and discontinue testing.
72 I4V-MC-JADV Clinical Protocol Page 70 Step 3: Test the change from baseline in DAS28-hsCRP at Week 12 for baricitinib versus placebo and ACR20 at Week 12 for noninferiority of baricitinib to adalimumab at 1-sided alpha=0.025 via a weighted Holm procedure with weights of 0.2 and 0.8, respectively, and following similar details as outlined in Step 2. (Details concerning the noninferiority margin and statistical conclusions are found in Section ) If both null hypotheses are rejected, proceed to Step 4. Otherwise, discontinue testing. Step 4: Test the change from baseline in the duration of morning joint stiffness at Week 12 and the change from baseline in the severity of morning joint stiffness at Week 12, both tests of baricitinib versus placebo, at 1-sided alpha=0.025 via a Hochberg procedure (Hochberg 1988). If both null hypotheses are rejected, proceed to Step 5. Otherwise, discontinue testing. Details of the Hochberg procedure at this step: Let p6 and p7 denote the unadjusted 1-sided p-values resulting from testing the hypotheses regarding duration and severity of morning joint stiffness, respectively. If p6 and p , then Lilly will reject both null hypotheses (declaring statistical significance for both duration and severity). Otherwise, if p and p7 >0.025, then Lilly will reject only the null hypothesis (declaring statistical significance) related to duration of morning joint stiffness. Otherwise if p and p6 >0.025, then Lilly will reject only the null hypothesis (declaring statistical significance) related to severity of morning joint stiffness. Otherwise Lilly will fail to reject both null hypotheses and discontinue testing. Step 5: Test the change from baseline in worst tiredness at Week 12 and the change from baseline in worst pain at Week 12, both tests of baricitinib versus placebo, at 1-sided alpha=0.025 via a Hochberg procedure and following similar details as outlined in Step 4.
73
74 I4V-MC-JADV Clinical Protocol Page 72 Any change to the data analysis methods described in the protocol will require an amendment ONLY if it changes a principal feature of the protocol. Any other change to the data analysis methods described in the protocol, and the justification for making the change, will be described in the SAP and the clinical study report. Additional exploratory analyses of the data will be conducted as deemed appropriate Patient Disposition The number of randomized patients along with ITT and PPS populations will be summarized by treatment group. Frequency counts and percentages in different study parts will be presented for each treatment group. All patients who discontinue from the study or the study treatment will be identified, and the extent of their participation in the study will be reported along with their reason for discontinuation. Reasons for discontinuation from the study will be summarized by treatment group Patient Characteristics Demographic and baseline characteristics will be summarized descriptively by treatment group. Descriptive statistics including number of patients, mean, SD, median, minimum, and maximum will be provided for continuous measures, and frequency counts and percentages will be tabulated for categorical measures. No formal statistical comparisons will be made among treatment groups unless otherwise stated Concomitant Therapy Concomitant medications will be descriptively summarized by treatment group in terms of frequencies and percentages using the safety population. The medications will be coded accordingly Treatment Compliance Compliance to study treatment for baricitinib and oral placebo will be assessed through counts of returned study drug tablets. For baricitinib or oral placebo, a patient will be considered significantly noncompliant if he or she misses more than 20% of the prescribed doses during the study, unless the patient s study drug is withheld by the investigator for safety reasons. For adalimumab or injectable placebo, a patient will be considered significantly noncompliant he or she misses more than 20% of the prescribed doses during the study or if there are any consecutive missed injections, unless the patient s study drug is withheld by the investigator for safety reasons. Similarly, a patient will be considered significantly noncompliant if he or she is judged by the investigator to have intentionally or repeatedly taken more than the prescribed amount of study drug. Patients who are significantly noncompliant will be excluded from the PPS. Patient compliance will be further defined in the SAP.
75 I4V-MC-JADV Clinical Protocol Page Primary Outcome and Methodology A logistic regression model with treatment, region, and joint erosion status in the model will be used to test the treatment difference between baricitinib and placebo in the proportion of patients achieving ACR20 response at Week 12. The NRI method as described above will be used to impute missing data Efficacy Analyses Major secondary analyses: 1. Comparison between baricitinib and placebo in change from baseline to Week 24 in mtss: An ANCOVA model with baseline score, region, joint erosion status, and treatment group in the model will be used to test the treatment difference between baricitinib and placebo in change from baseline to Week 24 of mtss scores. The linear extrapolation method as described above will be used to impute missing data. A permutation-based method will be used as a nonparametric sensitivity analysis to the ANCOVA method. 2. Comparison between baricitinib and placebo in change from baseline to Week 12 in HAQ-DI score: An ANCOVA model with treatment, region, joint erosion status, and baseline score in the model will be used to test the treatment difference between baricitinib and placebo in change from baseline to Week 12 in HAQ-DI. The mbocf approach as described above will be used to impute missing data. 3. Comparison between baricitinib and placebo in change from baseline to Week 12 in DAS28-hsCRP: An ANCOVA model with treatment, region, joint erosion status, and baseline score in the model will be used to test the treatment difference between baricitinib and placebo in change from baseline to Week 12 in DAS28-hsCRP. The mbocf approach as described above will be used to impute missing data. 4. Comparison between baricitinib and adalimumab in ACR20 response at Week 12: A logistic regression model with treatment, region, and joint erosion status in the model will be used to test the treatment difference between baricitinib and adalimumab in the percentage of patients achieving ACR20 response at Week 12. A (100-alpha)% CI for the treatment difference will be constructed using the method described by Newcombe- Wilson (Newcombe 1998) without continuity correction. If a quantitative treatment by region interaction exists, a supportive method to the Newcombe-Wilson method will be used. If the lower bound of the (100-alpha)% CI is >-12%, Lilly will conclude that baricitinib is noninferior to adalimumab with respect to ACR20. Similarly, if the lower bound of the (100-alpha)% CI is >0%, Lilly will conclude that baricitinib is superior to adalimumab with respect to ACR20. The NRI method as described above will be used to impute missing data. 5. Comparison between baricitinib and placebo in change from baseline to Week 12 in duration of morning joint stiffness: An ANCOVA model with treatment, region, joint erosion status, and baseline score in the model will be used to test the treatment difference between baricitinib and placebo in change from baseline to Week 12 in
76 I4V-MC-JADV Clinical Protocol Page 74 duration of morning joint stiffness. The mbocf approach as described above will be used to impute missing data. 6. Comparison between baricitinib and placebo in change from baseline to Week 12 in severity of morning joint stiffness: An ANCOVA model with treatment, region, joint erosion status, and baseline score in the model will be used to test the treatment difference between baricitinib and placebo in change from baseline to Week 12 in severity of morning joint stiffness. The mbocf approach as described above will be used to impute missing data. 7. Comparison between baricitinib and placebo in change from baseline to Week 12 in worst tiredness: An ANCOVA model with treatment, region, joint erosion status, and baseline score in the model will be used to test the treatment difference between baricitinib and placebo in change from baseline to Week 12 in worst tiredness. The mbocf approach as described above will be used to impute missing data. 8. Comparison between baricitinib and placebo in change from baseline to Week 12 in worst pain: An ANCOVA model with treatment, region, joint erosion status, and baseline score in the model will be used to test the treatment difference between baricitinib and placebo in change from baseline to Week 12 in worst pain. The mbocf approach as described above will be used to impute missing data. Other secondary efficacy and health outcome analyses: Besides the analyses listed in the objective section, the following analyses will be performed: change from baseline to Week 16 and Week 52 in structural joint damage as measured by mtss proportion of patients with mtss change 0 change from baseline in joint space narrowing and bone erosion scores hybrid ACR (bounded) response measure proportions of patients achieving ACR20 (other than primary), ACR50, and ACR70 percentage change from baseline in individual components of the ACR Core Set change from baseline in DAS28-ESR change from baseline in DAS28-hsCRP proportion of patients achieving DAS28-ESR 3.2 and <2.6 proportion of patients achieving DAS28-hsCRP 3.2 and <2.6 change from baseline in HAQ-DI proportion of patients achieving HAQ-DI improvement 0.22 and 0.3 change from baseline in CDAI score change from baseline in SDAI score proportion of patients achieving CDAI score 2.8 proportion of patients achieving SDAI score 3.3 proportion of patients achieving EULAR28 response proportion of patients achieving ACR/EULAR remission using both Boolean-based definition and Index-based definition change from baseline in fatigue score in the FACIT-F
77 I4V-MC-JADV Clinical Protocol Page 75 change from baseline in PCS, MCS, and total score in the SF-36v2 Acute change from baseline in health states, VAS score, and index score of EQ-5D-5L change from baseline in absenteeism, presenteeism, work productivity loss, and activity impairment scores of WPAI-RA change from baseline in individual items and total score in healthcare resource utilization characterization of the response time-course for key efficacy endpoints and identification of potential factors that may have an impact on the efficacy endpoints of baricitinib Approaches similar to the primary and major secondary analyses will be used to analyze other secondary efficacy and health outcome measures. In addition, comparisons between baricitinib and adalimumab in other secondary measures will be performed. Analysis methods will consist of ANCOVA and logistic regression, as described above, using mbocf, linear extrapolation, NRI, and mlocf missing data imputation methods, where applicable. In addition, key continuous secondary parameters that are collected at repeated visits throughout the double-blind period may be analyzed using a MMRM approach as described in Section The MMRM will be considered as a sensitivity approach to ANCOVA results. Data collected after initiation of rescue therapy will be summarized as appropriate. Further details will be provided in the SAP Pharmacokinetic/Pharmacodynamic Analyses PK data for baricitinib will be analyzed using a population modeling approach via a nonlinear mixed-effects modeling (NONMEM) program. Data from this study may be combined with an existing PK model or data from other studies, if appropriate. One- and 2-compartment structural models with or without absorption lag-time will be tested. A first-order absorption model whereby a 1-compartment model will be parameterized in terms of absorption rate constant (Ka), central compartment clearance (CL), and central compartment volume of distribution (V1). The 2-compartment model will be parameterized in terms of Ka, V1, CL, intercompartmental clearance (Q), peripheral volume of distribution (V2). In the case of a zero absorption model, the absorption model will be parameterized by absorption duration (D1). Previous modeling results and known predominant renal elimination characteristics of baricitinib indicated close correlation of baricitinib exposures with renal function defined by MDRD-eGFR; hence, partitioning of total clearance into renal and nonrenal clearance guided by mass-balance study results may also be incorporated into the model. Interpatient variability will be assessed separately on each of the PK parameters using an exponential error structure. Residual error will be assessed as proportional, additive, and combined proportional and additive error structures. Intrinsic factors such as age; body weight; gender; ethnic origin; renal function; transaminases/albumin/bilirubin, which are representative of hepatic function; and extrinsic factors such as comedications will be investigated to assess their influence on PK parameters such as clearance and volume of distribution, where applicable. Time-course of response for key efficacy data such as, but not necessarily limited to, ACR20/50/70, DAS28-ESR, DAS28-hsCRP, and safety data (eg, neutropenia, anemia) will be explored using graphical methods. Further investigations using a population-modeling approach
78 I4V-MC-JADV Clinical Protocol Page 76 via NONMEM to identify potential factors that may influence pharmacodynamic (PD) parameters may be warranted. Interpatient variability will then be assessed separately on each of the PD parameters using additive or exponential error structure. Residual error will be assessed as proportional, additive, and combined proportional and additive error structures. Covariates such as body weight, disease duration, disease severity at baseline, responder status, cytokines concentrations, dose, and baricitinib exposures may be investigated to assess their impact on efficacy and safety endpoints. Data from this study may be combined with an existing PK/PD model or data from other studies, if appropriate. Model evaluation will include using sensitivity analysis and visual predictive check methods Health Outcome Analyses The health outcomes will be analyzed using methods described for continuous or categorical data as described for efficacy measures in Section More detailed analytical methods will be described in the SAP Safety Analyses All safety data will be descriptively summarized by treatment groups and analyzed using the safety population. For the purposes of calculating changes from baseline during Part A and Part B, treatment baseline may be considered as the original baseline for Part A prior to any study drug received or at the visits that mark the transition between treatments received, depending on the purpose of the summary or comparison being made. Summaries within Parts A and B will be conducted by treatment group used within each respective part. Additional summaries by treatment group that continue unchanged across multiple parts of the study will be conducted, as appropriate. TEAEs are defined as AEs that first occurred or worsened in severity on or after the date of first dose of study treatment. AEs will be considered treatment emergent for nonresponders assigned to baricitinib if the AEs first occurred or worsened in severity (compared to the maximum severity prior to or on the date of rescue) after the visit at which rescue therapy is assigned. The number of TEAEs as well as the number and percentage of patients who experienced at least 1 TEAE will be summarized using MedDRA (Medical Dictionary for Regulatory Activities) for each system organ class (or a body system) and each preferred term by treatment group. TEAEs will also be summarized by relationship to treatment and by severity within each treatment group. SAEs and AEs that lead to study drug discontinuation will also be summarized by treatment group. Fisher s exact test will be used to perform pairwise comparisons among the treatment groups. All clinical laboratory results will be descriptively summarized by treatment group. Individual results that are outside of normal range will be flagged in data listings. Quantitative clinical hematology, chemistry, and urinalysis variables obtained at the baseline to postbaseline visits will be summarized as changes from baseline by treatment group and analyzed using ANCOVA with treatment and baseline value in the model. Categorical variables, including the incidence of
79 I4V-MC-JADV Clinical Protocol Page 77 abnormal values and incidence of AESIs, will be summarized by frequency and percentage of patients in corresponding categories. Shift tables will be presented for selected measures. Observed values and changes from baseline (predose or screening if missing) for vital signs and physical characteristics will be descriptively summarized by treatment group and time point. Change from baseline to postbaseline in vital signs, QIDS-SR 16, and body weight will be analyzed using ANCOVA with treatment and baseline value in the model. The incidence and average duration of study drug interruptions will be summarized and compared descriptively among treatment groups. Various techniques may be used to estimate the effects of study drug interruptions on safety measures. Further analyses may be performed and will be planned in the SAP. Data collected after initiation of rescue therapy will be summarized as appropriate Subgroup Analyses Subgroups analyses comparing baricitinib to placebo will be performed using ACR20, ACR50, HAQ-DI, DAS28-hsCRP, and DAS28 remission (DAS28-hsCRP <2.6) at Week 12 and mtss at Week 24. Subgroups to be evaluated will include region, renal function, background therapy, joint erosion status, country, gender, age, race, etc. Any other additional subgroups will be defined in the SAP. For ACR20, ACR50, and DAS28 remission analyses, a logistic regression model will be used with treatment, subgroup, and treatment by subgroup interaction included as factors. For the change from baseline to Week 12 in HAQ-DI score and DAS28-hsCRP and to Week 24 in mtss, an ANCOVA model will be used with treatment, baseline score, subgroup, and treatment by subgroup interaction included as factors. Further definitions for the levels of the subgroup variables, the analysis methodology, and any additional subgroup analyses will be defined in the SAP. Because this study is not powered for subgroup analyses, all subgroup analyses will be treated as exploratory Planned Analyses The analysis of primary and major secondary endpoints will occur when all patients complete the Week 24 study visit. This analysis will not be considered an interim analysis, and the study team will become unblinded at this time. A final analysis will occur when all patients complete the study. All investigators, study site personnel, and patients will remain blinded to treatment assignments until the final database lock. Members of the study team not in direct contact with the clinical sites may have access to the data from this study after all patients complete the primary and major secondary endpoints through Week 24 (Visit 11). Members of the study team who become unblinded to the primary and major secondary analyses through Week 24 will have no further contact with study sites until after the final database lock. Unblinding details will be specified in the unblinding plan section of the SAP.
80 I4V-MC-JADV Clinical Protocol Page Interim Analyses A DMC will oversee the conduct of all the Phase 3 clinical trials evaluating baricitinib in patients with RA. The DMC will consist of members external to Lilly. This DMC will follow the rules defined in the DMC charter, focusing on potential and identified risks for this molecule and for this class of compounds. DMC membership will include, at a minimum, specialists with expertise in rheumatology, statistics, and other appropriate specialties. The DMC will review and evaluate up to 3 planned interim analyses on an approximate semiannual basis. This DMC for studies of patients with RA will be coordinated with the DMC(s) for other ongoing studies of baricitinib in other indications, and this coordination may alter the number and timing of the interim analyses. Access to the unblinded interim data will be limited to the statisticians who conduct the interim analyses and the DMC. The statisticians conducting the interim analyses will be independent from the study team. The study team will not have access to the unblinded data. Study sites will receive information about interim results ONLY if they need to know for the safety of their patients. The DMC will review study discontinuation data, AEs including SAEs, clinical laboratory data, vital sign data, etc. The DMC may recommend continuation of the study, as designed, temporary suspension of enrollment, or the discontinuation of a particular dose regimen or the entire study. The DMC may request to review efficacy data to investigate the benefit/risk relationship in the context of safety observations for ongoing patients in the study. However, the study will not be stopped for positive efficacy results, and there is no planned futility assessment. Hence, there will be no alpha adjustment for these interim analyses. Details of the DMC and interim safety analyses will be documented in a DMC charter and DMC analysis plan. Besides DMC members, a limited number of pre-identified individuals may gain access to the unblinded PK and safety data, as specified in the unblinding plan, prior to the final database lock, to initiate the exploration and/or final population PK/PD model development processes for safety data (eg, neutropenia, anemia). Information that may unblind the study during the analyses will not be reported to study sites or the blinded study team until the database lock for the primary and major secondary efficacy analyses.
81 I4V-MC-JADV Clinical Protocol Page Informed Consent, Ethical Review, and Regulatory Considerations Informed Consent The investigator is responsible for ensuring that the patient understands the potential risks and benefits of participating in the study, including answering any questions the patient may have throughout the study and sharing in a timely manner any new information that may be relevant to the patient s willingness to continue his or her participation in the trial. The ICF will be used to explain the potential risks and benefits of study participation to the patient in simple terms before the patient is entered into the study and to document that the patient is satisfied with his or her understanding of the risks and benefits of participating in the study and desires to participate in the study. The investigator is responsible for ensuring that informed consent is given by each patient. This includes obtaining the appropriate signatures and dates on the ICF prior to the performance of any protocol procedures and prior to the administration of investigational product Ethical Review Lilly or its representatives must approve all ICFs before they are submitted to the ERB and are used at investigative sites(s). All ICFs must be compliant with the International Conference on Harmonisation (ICH) guideline on GCP. Documentation of ERB approval of the protocol and the ICF must be provided to Lilly before the study may begin at the investigative site(s). The ERB(s) will review the protocol as required. Any member of the ERB who is directly affiliated with this study as an investigator or as site personnel must abstain from the ERB s vote on the approval of the protocol. The study site s ERB(s) should be provided with the following: the current IB or package labeling and updates during the course of the study ICF relevant curricula vitae Regulatory Considerations This study will be conducted in accordance with: 1) consensus ethics principles derived from international ethics guidelines, including the Declaration of Helsinki and Council for International Organizations of Medical Sciences International Ethical Guidelines 2) the ICH GCP Guideline [E6] 3) applicable laws and regulations The investigator or designee will promptly submit the protocol to the applicable ERB(s).
82 I4V-MC-JADV Clinical Protocol Page 80 An identification code assigned by the investigator to each patient will be used in lieu of the patient s name to protect the patient s identity when reporting AEs and/or other trial-related data Investigator Information Physicians with a specialty in RA will participate as investigators in this clinical trial Protocol Signatures The sponsor s responsible medical officer will approve the protocol, confirming that, to the best of his or her knowledge, the protocol accurately describes the planned design and conduct of the study. After reading the protocol, each principal investigator will sign the protocol signature page and send a copy of the signed page to a Lilly representative Final Report Signature The clinical study report coordinating investigator will sign the final clinical study report for this study, indicating agreement that, to the best of his or her knowledge, the report accurately describes the conduct and results of the study. Lilly will select a qualified investigator from among investigators participating in the design, conduct, and/or analysis of the study to serve as the clinical study report coordinating investigator. The sponsor s responsible medical officer will approve the final clinical study report for this study, confirming that, to the best of his or her knowledge, the report accurately describes the conduct and results of the study.
83 I4V-MC-JADV Clinical Protocol Page References Aletaha D, Neogi T, Silman AJ, Funovits J, Felson DT, Bingham CO 3rd, Birnbaum NS, Burmester GR, Bykerk VP, Cohen MD, Combe B, Costenbader KH, Dougados M, Emery P, Ferraccioli G, Hazes JM, Hobbs K, Huizinga TW, Kavanaugh A, Kay J, Kvien TK, Laing T, Mease P, Ménard HA, Moreland LW, Naden RL, Pincus T, Smolen JS, Stanislawska-Biernat E, Symmons D, Tak PP, Upchurch KS, Vencovskỳ J, Wolfe F, Hawker G Rheumatoid arthritis classification criteria: an American College of Rheumatology/European League Against Rheumatism collaborative initiative. Arthritis Rheum. 2010;62(9): Aletaha D, Smolen J. The Simplified Disease Activity Index (SDAI) and the Clinical Disease Activity Index (CDAI): a review of their usefulness and validity in rheumatoid arthritis. Clin Exp Rheumatol. 2005;23(5 suppl 39):S100-S108. American College of Rheumatology Committee to Reevaluate Improvement Criteria. A proposed revision to the ACR20: the hybrid measure of American College of Rheumatology response. Arthritis Rheum. 2007;57(2): [APA] American Psychiatric Association's Diagnostic and Statistical Manual of Mental Disorders, 4th Edition. Washington, DC; American Psychiatric Association Brazier JE, Harper R, Jones NM, O Cathain A, Thomas KJ, Usherwood T, Westlake L. Validating the SF-36 health survey questionnaire: new outcome measure for primary care. BMJ. 1992;305(6846): Bretz F, Maurer W, Brannath W, Posch M. A graphical approach to sequentially rejective multiple test procedures. Stat Med. 2009;28(4): Brooks R. EuroQol: The current state of play. Health Policy. 1996;37: Cella D, Webster K. Linking outcomes management to quality-of-life measurement. Oncology (Willeston Park). 1997;11(11A): Cohen SB, Emery P, Greenwald MW, Dougados M, Furie RA, Genovese MC, Keystone EC, Loveless JE, Burmester GR, Cravets MW, Hessey EW, Shaw T, Totoritis MC. Rituximab for rheumatoid arthritis refractory to anti-tumor necrosis factor therapy. Results of a multicenter, randomized, double-blind, placebo-controlled phase III trial evaluating primary efficacy and safety at twenty-four weeks. Arthritis Rheum. 2006;54(9): Colmegna I, Ohata BR, Menard HA. Current understanding of rheumatoid arthritis therapy. Clin Pharmacol Ther. 2012;91(4): Curtis JR, Patkar N, Xie A, Martin C, Allison JJ, Saaq M, Shatin D, Saaq KG. Risk of serious bacterial infections among rheumatoid arthritis patients exposed to tumor necrosis factor alpha antagonists. Arthritis Rheum. 2007;56(4): The EuroQol Group. EQ-5D-5L User Guide. Version 1.0. April Available at: EQ-5D-5L.pdf. Accessed July
84 I4V-MC-JADV Clinical Protocol Page 82 Felson DT, Smolen JS, Wells G, Zhang B, van Tuyl LH, Funovits J, Aletaha D, Allaart CF, Bathon J, Bombardieri S, Brooks P, Brown A, Matucci-Cerinic M, Choi H, Combe B, de Wit M, Dougados M, Emery P, Furst D, Gomez-Teino J, Hawker G, Keystone E, Khanna D, Kirwan J, Kvien TK, Landewé R, Listing J, Michaud K, Martin-Mola E, Montie P, Pincus T, Richards P, Siegel JN, Simon LS, Sokka T, Strand V, Tugwell P, Tyndall A, van der Heijde D, Verstappen S, White B, Wolfe F, Zink A, Boers M. American College of Rheumatology/European League against Rheumatism provisional definition of remission in rheumatoid arthritis for clinical trials. Ann Rheum Dis. 2011;70: Fleischmann RM. Is there a need for new therapies for rheumatoid arthritis? J Rheum. 2005;32(suppl 73):3S-7S. Fridman JS, Scherle PA, Collins R, Burn TC, Li Y, Li J, Covington MB, Thomas B, Collier P, Favata MF, Wen X, Shi J, McGee R, Haley PJ, Shepard S, Rodgers JD, Yeleswaram S, Hollis G, Newton RC, Metcalf B, Friedman SM, Vaddi K. Selective inhibition of JAK1 and JAK2 is efficacious in rodent models of arthritis: preclinical characterization of INCB J Immunol. 2010;184(9): Fries JF, Spitz P, Kraines RG, Holman HR. Measurement of patient outcome in arthritis. Arthritis Rheum. 1980;23(2): Fries JF, Spitz PW, Young DY. The dimensions of health outcomes: the health assessment questionnaire, disability and pain scales. J Rheumatol. 1982;9(5): Herdman M, Gudex C, Lloyd A, Janssen MF, Kind P, Parkin D, Bonsel G, Badia X. Development and preliminary testing of the new five-level version of EQ-5D (EQ-5D-5L). Qual Life Res. 2011;20(10): Hochberg Y. A sharper bonferroni procedure for multiple tests of significance. Biometrika. 1988;75(4): Holm S. A simple sequentially rejective multiple test procedure. Scand J Statist. 1979;6: Keystone EC, Kavanaugh AF, Sharp JT, Tannenbaum H, Hua Y, Teoh LS, Fischkoff SA, Chartash EK. Radiographic, clinical, and functional outcomes of treatment with adalimumab (a human anti tumor necrosis factor monoclonal antibody) in patients with active rheumatoid arthritis receiving concomitant methotrexate therapy: a randomized, placebo-controlled, 52-week trial. Arthritis Rheum. 2004;50(5): Keystone E, Heijde D, Mason D Jr, Landewé R, Vollenhoven RV, Combe B, Emery P, Strand V, Mease P, Desai C, Pavelka K. Certolizumab pegol plus methotrexate is significantly more effective than placebo plus methotrexate in active rheumatoid arthritis: findings of a fifty-twoweek, phase III, multicenter, randomized, double-blind, placebo-controlled, parallel-group study. Arthritis Rheum. 2008;58(11): Keystone E, Emery P, Peterfy CG, Tak PP, Cohen S, Genovese MC, Dougados M, Burmester GR, Greenwald M, Kvien TK, Williams S, Hagerty D, Cravets MW, Shaw T. Rituximab inhibits structural joint damage in patients with rheumatoid arthritis with an inadequate response to tumour necrosis factor inhibitor therapies. Ann Rheum Dis. 2009;68(2): Kitas GD, Gabriel SE. Cardiovascular disease in rheumatoid arthritis: state of the art and future perspectives. Ann Rheum Dis. 2011;70(1):8-14. Erratum in Ann Rheum Dis. 2011;70(8):1520.
85 I4V-MC-JADV Clinical Protocol Page 83 Klareskog L, Catrina AI, Paget S. Rheumatoid arthritis. Lancet. 2009;373(9664): Kremer JM, Bloom BJ, Breedveld FC, Coombs JH, Fletcher MP, Gruben D, Krishnaswami S, Burgos-Vargas R, Wilkinson B, Zerbini CA, Zwillich SH. The safety and efficacy of a JAK inhibitor in patients with active rheumatoid arthritis: Results of a double-blind, placebocontrolled phase IIa trial of three dosage levels of CP-690,550 versus placebo. Arthritis Rheum. 2009;60(7): Newcombe RG. Interval estimation for the difference between independent proportions: comparison of eleven methods. Stat Med. 1998;17: Erratum in Stat Med. 1999;18:1293. Ramey DR, Fries JF, Singh G. The Health Assessment Questionnaire 1995: status and review. In: Spiker B, editor. Quality of life and pharmacoeconomics in clinical trials. 2nd ed. Philadelphia: Lippincott-Raven, 1996: p Reilly MC, Zbrozek AS, Dukes EM. The validity and reproducibility of a work productivity and activity impairment instrument. Pharmacoeconomics. 1993;4(5): Rubbert-Roth A, Finckh A. Treatment options in patients with rheumatoid arthritis failing initial TNF inhibitor therapy: a critical review. Arthritis Res Ther. 2009;11(suppl 1):S1. Rush AJ, Trivedi MH, Ibrahim HM, Carmody TJ, Arnow B, Klein DN, Markowitz JC, Ninan PT, Kornstein S, Manber R, Thase ME, Kocsis JH, Keller MB. The 16-Item quick inventory of depressive symptomatology (QIDS), clinician rating (QIDS-C), and self-report (QIDS-SR): a psychometric evaluation in patients with chronic major depression. Biological Psychiatry. 2003;54(50): Erratum in Biol Psychiatry. 2003;54(5);585. Schiff M, Fleischmann R, Weinblatt M, Valente R, van der Heijde D, Citera G, Zhao C, Maldonado M. Abatacept sc versus adalimumab on background methotrexate in RA: one year results from the AMPLE study. Ann Rheum Dis. 2012;71(suppl 3):60. Smolen J, Landewé RB, Mease P, Brzezicki J, Mason D, Luijtens K, van Vollenhoven RF, Kavanaugh A, Schiff M, Burmester GR, Strand V, Vencovskỳ J, van der Heijde D. Efficacy and safety of certolizumab pegol plus methotrexate in active rheumatoid arthritis: the RAPID 2 study. A randomised controlled trial. Ann Rheum Dis. 2009;68(6): Smolen JS, Aletaha D, Bijlsma JW, Breedveld FC, Boumpas D, Burmester G, Combe B, Cutolo M, de Wit M, Dougados M, Emery P, Gibofsky A, Gomez-Reino JJ, Haraoui B, Keystone EC, Kvien TK, McInnes I, Martin-Mola E, Montecucco C, Schoels M, van der Heijde D, T2T Expert Committee. Treating rheumatoid arthritis to target: recommendations of an international task force. Ann Rheum Dis. 2010;69(4): Smolen JS, Beaulieu A, Rubbert-Roth A, Ramos-Remus C, Rovensky J, Alecock E, Woodworth T, Alten R, OPTION investigators. Effect of interleukin-6 receptor inhibition with tocilizumab in patients with rheumatoid arthritis (OPTION study): a double-blind, placebocontrolled, randomised trial. Lancet. 2008;371(9617): Smolen JS, Breedveld FC, Eberl G, Jones I, Leeming M, Wylie GL, Kirkpatrick J. Validity and reliability of the twenty-eight-joint count for the assessment of rheumatoid arthritis activity. Arthritis Rheum. 1995;38(1):38-43.
86 I4V-MC-JADV Clinical Protocol Page 84 Smolen J, Steiner G. Therapeutic strategies for rheumatoid arthritis. Nat Rev Drug Discov. 2003;2(6): Trivedi MH, Rush AJ, Ibrahim HM, Carmody TJ, Biggs MM, Suppes T, Crismon ML, Shores-Wilson K, Toprac MG, Dennehy EB, Witte B, Kashner TM. The Inventory of Depressive Symptomatology, Clinician Rating (IDS-C) and Self-Report (IDS-SR), and the Quick Inventory of Depressive Symptomatology, Clinician Rating (QIDS-C) and Self-Report (QIDS-SR) in public sector patients with mood disorders: a psychometric evaluation. Psychol Med. 2004;34(1): University of Pittsburgh Epidemiology Data Center; IDS/QIDS web site. Available at: Accessed June 13, Vander Cruyssen B, Van Looy S, Wyns B, Westhovens R, Durez P, Van den Bosch F, Veys EM, Mielants H, De Clerck L, Peretz A, Malaise M, Verbruggen L, Vastesaeger N, Geldhof A, Boullart L, De Keyser F. DAS28 best reflects the physician's clinical judgment of response to infliximab therapy in rheumatoid arthritis patients: validation of the DAS28 score in patients under infliximab treatment. Arthritis Res Ther. 2005;7(5):R van der Heijde D. How to read radiographs according to the Sharp/van der Heijde method. J Rheumatol. 2000; 27(1): van Gestel AM, Haagsma CJ, van Riel PL. Validation of rheumatoid arthritis improvement criteria that include simplified joint counts. Arthritis Rheum. 1998;41(10): Ware JE Jr, Sherbourne CD. The MOS 36-item short-form health survey (SF-36), I: Conceptual framework and item selection. Med Care. 1992;30(6): Williams WV, Scherle P, Shi J, Newton R, McKeever E, Fridman J, Vaddi K, Levy R, Moreland L. Initial efficacy of INCB018424, a selective Janus kinase 1& 2 (JAK 1&2) inhibitor in rheumatoid arthritis (RA). Ann Rheum Dis. 2008;67(suppl II):62.
87 I4V-MC-JADV Clinical Protocol Page 85 Attachment 1. Protocol JADV Study Schedule

88

89

90

91

92
93 e If the PPD test is positive and the patient has no medical history or chest x-ray findings consistent with active TB, the patient may have a QuantiFERON-TB Gold test (if available). If the QuantiFERON-TB Gold test results are not negative, the patient will be considered to have latent TB. If the QuantiFERON-TB Gold test is available and in the judgment of the investigator, preferred as an alternative to the PPD skin test for the evaluation of TB infection, it may be used instead of the PPD TB test. If the QuantiFERON-TB Gold test is not negative, the test may be repeated once within 2 weeks of the initial value. If the repeat test results are again not negative, the patient will be considered to have latent TB. f A chest x-ray will be taken locally at screening unless one has been obtained within the past 6 months (provided the x-ray and/or report are available for review). g At Visit 2, the patient will take his/her oral investigational product at the clinic under the supervision of study staff. Injectable investigational product will be administered by the study staff with instructions given to the patient. h At Visits 3, 4, and 8, patients should bring their investigational product with them to the study visit, but no tablet counts need to be performed. i At screening, radiographic images of the hands and feet acquired within 4 weeks prior to study entry may be submitted to the central reader to confirm eligibility. j X-rays will only be taken at early termination if the most recent x-ray was more than 12 weeks earlier. k Morning joint stiffness assessment will be collected using a battery of questions and a daily patient diary to assess duration and severity of morning joint stiffness and recurrence of stiffness during the day. l This item to be collected using patient daily diaries through Week 12. m For all women of childbearing potential, a serum pregnancy test (central laboratory) will be performed at Visit 1 and Visit 2. Urine pregnancy tests (local laboratory) will also be performed at Visit 2 and at all subsequent study visits. n To confirm postmenopausal status for women 40 and <60 years of age who have had a cessation of menses, an FSH test will be performed. Nonchildbearing potential will be defined as an FSH 40 miu/ml and a cessation of menses for at least 12 months. o Clinical chemistry will include the following values calculated from serum creatinine: calculated creatinine clearance (Cockcroft-Gault equation) and egfr (calculated using the Modification of Diet in Renal Disease isotope dilution mass spectrometry traceable method). p Fasting lipid profile. Patients should not eat or drink anything except water for 12 hours prior to sample collection. If a patient attends these visits in a nonfasting state, this will not be considered a protocol violation. q Erythrocyte sedimentation rate will be performed locally using a kit provided by the sponsor. r At Visit 2 (Week 0), patients will take their oral investigational product in the clinic, and PK samples will be drawn at 15 minutes and 1 hour postdose. At Visit 5 (Week 4), patients will be asked to take their oral investigational product at home prior to visiting the clinic. The clinic visit should be scheduled so that the sample taken during this visit is 2 to 4 hours after the oral dose taken at home. At Visit 6 (Week 8), patients will be asked to take their oral investigational product at home prior to visiting the clinic. The clinic visit should be scheduled so that the sample taken during this visit is 4 to 6 hours after the oral dose taken at home. For Visits 7 (Week 12), 11 (Week 24), and 13 (Week 32), patients will be asked not to take their oral investigational product prior to visiting the clinic, and a sample will be taken at any time predose on the day of the clinic visits. If the patient has taken his/her oral dose prior to the visit, the sample may be drawn anytime postdose, and the inability to collect a predose sample will not be considered a protocol violation. For the early termination visit, a sample may be drawn anytime if the last dose of oral investigational drug was taken within the last 48 hours. I4V-MC-JADV Clinical Protocol Page 91
94 I4V-MC-JADV Clinical Protocol Page 92 Attachment 2. Protocol JADV Clinical Laboratory Tests
95 I4V-MC-JADV Clinical Protocol Page 93 Clinical Laboratory Tests Hematologya,b Clinical Chemistrya,b Hemoglobin Serum concentrations of Hematocrit Sodium Erythrocyte count (RBCs) Absolute reticulocyte count Potassium Total bilirubin Mean cell volume Direct bilirubin Mean cell hemoglobin Alkaline phosphatase Mean cell hemoglobin concentration Alanine aminotransaminase/serum glutamic Leukocytes (WBCs) pyruvic transaminase (ALT/SGPT) Absolute count of Neutrophils, segmented Aspartate aminotransferase/serum glutamic oxaloacetic transaminase (AST/SGOT) Neutrophils, juvenile (bands) Lymphocytes Monocytes Eosinophils Basophils Blood urea nitrogen Creatinine Estimated glomerular filtration ratec Calculated creatinine clearance d Uric acid Platelets Calcium Cell morphology CBC, including differential and blood smear Glucosee Albumin Creatine kinase Urinalysisa,b Total protein Color Specific gravity ph Lipid profile includinge: total cholesterol, Protein HDL-C, LDL-C, and triglycerides Glucose ApoA1, ApoB Ketones Blood Bilirubin Urobilinogen Leukocyte esterase Nitrite Microscopic examination of sedimenti Lymphocyte subsets (T, B, NK, and T-cell subsets) Stored serum, plasma, urine, mrna and DNA samples for possible exploratory biomarker analyses Footnotes appear on the following page. Lipoprotein subfractions (NMR) Pregnancy Test (females only)b,f FSH (females only)g Other Tests Hepatitis B surface antigen Hepatitis B surface antibody Hepatitis B core antibody Hepatitis C antibodyh Human immunodeficiency virus Iron studies (iron, TIBC, and ferritin) Thyroid-stimulating hormone serum concentration (PK sample) PPD or QuantiFERON j Rheumatoid factor High-sensitivity C-reactive protein ESRk Anticyclic citrullinated peptide Immunoglobulins (IgG, IgA, IgM)
96 I4V-MC-JADV Clinical Protocol Page 94 Abbreviations: ApoA1 = apolipoprotein A1; ApoB = apolipoprotein B; CBC = complete blood count; CPK = creatine phosphokinase; DNA = deoxyribonucleic acid; ESR = erythrocyte sedimentation rate; FSH = folliclestimulating hormone; HDL-C = high-density lipoprotein cholesterol; Ig = immunoglobulin; LDL-C = lowdensity lipoprotein cholesterol; mrna = messenger ribonucleic acid; NK = natural killer; NMR = nuclear magnetic resonance; PK = pharmacokinetic; PPD = purified protein derivative; RBC = red blood cell; TIBC = total iron binding capacity; WBC = white blood cell. a Assayed/calculated by a sponsor-designated laboratory. b Unscheduled blood chemistry (including CPK), hematology, and urinalysis panels may be performed at the discretion of the investigator. If tests are done to evaluate laboratory results to resume study drug, samples must be assayed centrally. c Estimated glomerular filtration rate for serum creatinine calculated by the central laboratory using the Modification of Diet in Renal Disease isotope dilution mass spectrometry traceable method. d The calculated creatinine clearance will be determined by the central laboratory each time a serum creatinine sample is collected using the Cockcroft-Gault equation: Men: ([140 age] x [weight in kg]) / (72 x [serum creatinine in mg/dl]) or ([140 age] x [weight in kg]) / (0.814 x [serum creatinine in μmol/l]). Women: (above formula) x e Fasting laboratory values for glucose and lipids will be required at baseline, Week 12, Week 24, and Week 52. If a patient attends these visits in a nonfasting state, this will not be considered a protocol violation. These tests may be performed nonfasting at all other visits. f For all women of childbearing potential, a serum pregnancy test (central laboratory) will be performed at Visit 1 and both urine (local laboratory) and serum pregnancy (central laboratory) tests will be performed at Visit 2 to determine study eligibility. Urine pregnancy tests (local laboratory) will also be performed at each subsequent study visit per study schedule. g To confirm postmenopausal status for women 40 and <60 years of age, an FSH test will be performed. Nonchildbearing potential is defined as an FSH 40 miu/ml and a cessation of menses for at least 12 months. h A positive hepatitis C antibody result will be confirmed with a positive hepatitis C virus result. i Microscopic examination of sediment will be performed only if abnormalities are noted on the routine urinalysis. j Test will be required at Visit 1 only to determine eligibility of patient for the study. In countries where the QuantiFERON-TB Gold test is available and, in the judgment of the investigator, medically preferred as an alternative to the PPD test for the evaluation of mycobacterium tuberculosis (MTB) infection, it may be used instead of the PPD test (positive tests excluded) and may be read locally. k ESR analysis will be performed by the local laboratory.
97 I4V-MC-JADV Clinical Protocol Page 95 Attachment 3. Protocol JADV Hepatic Monitoring Tests for Treatment-Emergent Abnormality Selected tests may be obtained in the event of a treatment-emergent hepatic abnormality and may be required in follow up with patients in consultation with the Lilly designated medical monitor. Hepatic Monitoring Tests Hepatic hematologya Hemoglobin Hematocrit RBC WBC Neutrophils, segmented Lymphocytes Monocytes Eosinophils Basophils Platelets Haptoglobina Hepatic coagulationa Prothrombin time Prothrombin time, INR Hepatic serologiesa,b Hepatitis A antibody, total Hepatitis A antibody, IgM Hepatitis B surface antigen Hepatitis B surface antibody Hepatitis B core antibody Hepatitis C antibody Hepatitis E antibody, IgG Hepatitis E antibody, IgM Hepatic chemistrya Total bilirubin Direct bilirubin Alkaline phosphatase ALT Anti-nuclear antibodya AST GGT Anti-smooth muscle antibodya CPK Abbreviations: ALT = alanine aminotransferase; AST = aspartate aminotransferase; CPK = creatine phosphokinase; GGT = gamma glutamyl transferase; Ig = immunoglobulin; INR = international normalized ratio; RBC = red blood cell; WBC = white blood cell. a b Assayed by Lilly-designated or local laboratory. Reflex/confirmation dependent on regulatory requirements and/or testing availability.
98 Leo Document ID = d03b75c1-7a8b f25-c863d311aa43 Approver: Approval Date & Time: 27-Jul :35:40 GMT Signature meaning: Approved Approver: Approval Date & Time: 27-Jul :30:19 GMT Signature meaning: Approved
99 I4V-MC-JADV(c) Clinical Protocol Page 1 1. Protocol I4V-MC-JADV(c) A Randomized, Double-Blind, Placebo- and Active-Controlled, Phase 3 Study Evaluating the Efficacy and Safety of Baricitinib in Patients with Moderately to Severely Active Rheumatoid Arthritis Who Have Had an Inadequate Response to Methotrexate Therapy Baricitinib () Study I4V-MC-JADV is a Phase 3, randomized, double-blind, placebo- and activecontrolled, parallel-group study of baricitinib for the treatment of moderately to severely active rheumatoid arthritis during a 52-week treatment period in patients with inadequate response to methotrexate therapy. Eli Lilly and Company Indianapolis, Indiana USA Protocol Electronically Signed and Approved by Lilly: 27 July 2012 Amendment (a) Electronically Signed and Approved by Lilly: 07 October 2012 Amendment (b) Electronically Signed and Approved by Lilly: 28 November 2012 Amendment (c) electronically signed and approved by Lilly on approval date provided below Approval Date: 19-Jul-2013 GMT
100 I4V-MC-JADV(c) Clinical Protocol Page 2 Study Rationale 2. Synopsis Baricitinib () is an oral Janus kinase 1 (JAK1)/Janus kinase 2 (JAK2) selective inhibitor representing a potentially effective therapy for treatment of patients with moderately to severely active rheumatoid arthritis (RA). The rationale for the current study is to confirm the efficacy and to continue to define the safety profile of 4 mg baricitinib when administered once daily (QD) to patients with RA who have had an inadequate response to methotrexate (MTX) therapy. The safety and tolerability data from this study are intended to inform the current understanding of the benefit-risk relationship for baricitinib in patients with RA.
101 I4V-MC-JADV(c) Clinical Protocol Page 3 Clinical Protocol Synopsis: Study I4V-MC-JADV Name of Investigational Product: Baricitinib () Title of Study: A Randomized, Double-Blind, Placebo- and Active-Controlled, Phase 3 Study Evaluating the Efficacy and Safety of Baricitinib in Patients with Moderately to Severely Active Rheumatoid Arthritis Who Have Had an Inadequate Response to Methotrexate Therapy Number of Planned Patients/Subjects: Phase of Development: 3 Entered: 1900 Enrolled/Randomized: 1280 (480 on baricitinib, 480 on placebo, 320 on Humira [adalimumab]) Completed: 1088 Length of Study: 2 years and 3 months Planned first patient visit: Oct 2012 Planned last patient visit: Jan 2015 Objectives: The primary objective of the study is to determine whether baricitinib is superior to placebo in the treatment of patients with moderately to severely active rheumatoid arthritis (RA) despite methotrexate treatment (ie, inadequate response to methotrexate [MTX-IR]), as assessed by the proportion of patients achieving a 20% improvement in American College of Rheumatology criteria (ACR20) at Week 12. The major secondary objectives of the study are to evaluate the efficacy of baricitinib versus placebo or adalimumab as assessed by: change from baseline to Week 24 in structural joint damage as measured by modified Total Sharp Score (mtss [van der Heijde method]) compared to placebo change from baseline to Week 12 in Health Assessment Questionnaire-Disability Index (HAQ-DI) score compared to placebo change from baseline to Week 12 in DAS28-high-sensitivity C-reactive protein (hscrp) compared to placebo proportion of patients achieving an SDAI score 3.3 at Week 12 compared to placebo proportion of patients achieving ACR20 response at Week 12 compared to adalimumab mean duration of morning joint stiffness in the 7 days prior to Week 12 compared to placebo as collected in electronic diaries change from baseline to Week 12 in DAS28-hsCRP compared to adalimumab mean severity of morning joint stiffness in the 7 days prior to Week 12 compared to placebo as collected in electronic diaries mean Worst Tiredness numeric rating scale (NRS) in the 7 days prior to Week 12 compared to placebo as collected in electronic diaries mean Worst Pain NRS in the 7 days prior to Week 12 compared to placebo as collected in electronic diaries The other secondary objectives of the study are as follows: to evaluate the efficacy of baricitinib as assessed by: o proportion of patients achieving DAS28-hsCRP 3.2 and DAS28-hsCRP <2.6 at Week 12, Week 24, and Week 52 o proportion of patients achieving HAQ-DI improvement 0.22 and 0.3 at Week 12, Week 24, and Week 52 o change from baseline to Week 16 and Week 52 in structural joint damage as measured by mtss o proportion of patients with mtss change 0 from baseline at Week 16, Week 24, and Week 52 o change from baseline to Week 16, Week 24, and Week 52 in joint space narrowing and bone erosion scores o proportion of patients achieving ACR20 at Week 24 and Week 52 o proportion of patients achieving ACR50 at Week 12, Week 24, and Week 52 o proportion of patients achieving 70% improvement in ACR criteria (ACR70) at Week 12, Week
102 I4V-MC-JADV(c) Clinical Protocol Page 4 24, and Week 52 o percentage change from baseline to Week 12, Week 24, and Week 52 in individual components of the ACR Core Set o change from baseline through Week 52 in DAS28-hsCRP o change from baseline through Week 52 in DAS28-erythrocyte sedimentation rate (ESR) o proportion of patients achieving DAS28-ESR 3.2 and DAS28-ESR <2.6 at Week 12, Week 24, and Week 52 o change from baseline to Week 24 and Week 52 in HAQ-DI score o change from baseline to Week 12, Week 24, and Week 52 in CDAI score o change from baseline to Week 12, Week 24, and Week 52 in SDAI score o proportion of patients achieving a CDAI score 2.8 at Week 12, Week 24, and Week 52 o proportion of patients achieving an SDAI score 3.3 at Week 24, and Week 52 o proportion of patients achieving ACR/EULAR remission according to the Boolean-based definition at Week 12, Week 24 and Week 52 o proportion of patients achieving moderate and good EULAR responses based on the 28-joint DAS at Week 12, Week 24, and Week 52 o evaluation of hybrid ACR (bounded) response at Week 12, Week 24 and Week 52 o change from baseline to Week 12, Week 24, and Week 52 in Functional Assessment of Chronic Illness Therapy Fatigue (FACIT-F) scale scores o change from baseline to Week 12, Week 24, and Week 52 in each scale and the summary scores for the Mental Component Score (MCS) and Physical Component Score (PCS) of the Medical Outcomes Study 36-Item Short Form Health Survey Version 2 Acute (SF-36v2 Acute) o change from baseline to Week 12, Week 24, and Week 52 in European Quality of Life- 5 Dimensions-5 Level (EQ-5D-5L) scores o change from baseline to Week 12, Week 24, and Week 52 in Work Productivity and Activity Impairment-Rheumatoid Arthritis (WPAI-RA) scores o evaluation of healthcare resource utilization through Week 52 o change from baseline to Week 12, Week 24, and Week 52 in duration of morning joint stiffness assessed at the visit using an electronic patient-reported outcomes (epro) tablet o change from baseline to Week 12, Week 24, and Week 52 in Worst Tiredness NRS assessed at the visit using an epro tablet o change from baseline to Week 12, Week 24, and Week 52 in Worst Pain NRS assessed at the visit using an epro tablet to characterize the pharmacokinetics (PK) of baricitinib, determine the magnitude of within- and betweenpatient variability, and identify the potential factors that may have an impact on the PK of baricitinib to characterize the baricitinib exposure-response time course for key efficacy and safety endpoints and to identify potential factors that may have an impact on these efficacy or safety endpoints of baricitinib The exploratory objective of the study is: to compare baricitinib and adalimumab as regards to various efficacy measures Study Design: Study I4V-MC-JADV (JADV) will be a 52-week, Phase 3, multicenter, randomized, double-blind, double-dummy, placebo- and active-controlled, parallel-group, outpatient study comparing the efficacy of baricitinib versus placebo on signs and symptoms, function, remission, and structural progression in patients with moderately to severely active RA who have had an insufficient response to methotrexate (MTX) and who have never been treated with a biologic disease-modifying antirheumatic drug (DMARD) (ie, MTX-IR patients). The study will include a noninferiority comparison of baricitinib versus adalimumab on signs and symptoms. Baricitinib will be administered as a 4-mg tablet once daily (QD). The dose for patients with renal impairment, defined as estimated glomerular filtration rate (egfr) <60 ml/min/1.73 m2, will be 2 mg baricitinib QD. Patients not assigned to the baricitinib treatment arm will receive either a 4-mg or 2-mg matching placebo tablet. Adalimumab (40 mg) will be administered by subcutaneous (SC) injection biweekly (ie, every 2 weeks). Patients
103 I4V-MC-JADV(c) Clinical Protocol Page 5 not assigned to adalimumab will receive a matching placebo SC injection biweekly. A total of 1280 patients are planned for enrollment in this study (480 to receive baricitinib, 480 to receive placebo, and 320 to receive adalimumab). After the Week 52 study visit, eligible patients may proceed to a separate extension study (Study I4V-MC-JADY) lasting for up to 2 years or to the posttreatment follow-up period of this study (Part C). Study JADV will consist of 4 parts: Screening: Screening period lasting from 3 to 42 days prior to Visit 2 (Week 0) Part A: double-blind, placebo- and active-controlled period from Week 0 through Week 24 Part B: double-blind, active-controlled period from Week 24 through Week 52 Part C: posttreatment follow-up period Diagnosis and Main Criteria for Inclusion and Exclusions: The proposed patient population for Study JADV will be ambulatory adult patients with moderately to severely active RA who have had an insufficient response to MTX and who have never been treated with a biologic DMARD. Patients will be eligible for participation only if they have an inadequate response to MTX as evidenced by the presence of at least 6/68 tender joints and 6/66 swollen joints and an hscrp measurement 6 mg/l. Investigational Product, Dosage, and Mode of Administration or Intervention: Baricitinib 4 mg administered orally QD Planned Duration of Treatment: 52 weeks. Screening period: 3 to 42 days Treatment period: 52 weeks Follow-up period: 28 days Reference Therapy, Dose, and Mode of Administration or Comparative Intervention: Placebo tablets matching baricitinib will be administered orally QD. As an active comparator, adalimumab 40 mg will be administered by SC injection biweekly (ie, once every 2 weeks). Placebo matching adalimumab for injection will also be administered by SC injection biweekly, as assigned. Patients will continue to take their background MTX therapy during the course of the study. Criteria for Evaluation: Efficacy The following efficacy measures will be assessed in this study: ACR20, ACR50, and ACR70 indices HAQ-DI mtss (includes joint space narrowing score and bone erosion score) DAS28-hsCRP and DAS28-ESR Hybrid ACR (bounded) response measure SDAI CDAI EULAR responses based on DAS28 Health Outcomes The following health outcome measures will be administered in this study: duration and severity of morning joint stiffness recurrence of joint stiffness during the day tiredness severity numeric rating scale (Worst Tiredness Numeric Rating Scale [NRS]) pain severity numeric rating scale (Worst Pain NRS) FACIT-F SF-36v2 Acute WPAI-RA EQ-5D-5L Quick Inventory of Depressive Symptomatology Self-Rated-16 (QIDS-SR 16 )
104 I4V-MC-JADV(c) Clinical Protocol Page 6 healthcare resource utilization Safety The following safety measures will be assessed in this study: adverse events (AEs) adverse events of special interest (AESIs) serious adverse events (SAEs) suspected unexpected serious adverse reactions (SUSARs) physical examinations electrocardiograms (ECGs) chest x-ray and tuberculosis (TB) testing vital signs (blood pressure and heart rate) and physical characteristics standard laboratory tests (including hematology, clinical chemistry, urinalysis, lipid profile, egfr, iron studies, hscrp, and ESR) concomitant medications AESIs will include: severe or opportunistic infections myelosuppressive events of anemia, leukopenia, neutropenia, lymphopenia, and thrombocytopenia thrombocytosis elevations in alanine aminotransferase (ALT) or aspartate aminotransferase (AST) (>3 times ULN) with total bilirubin (>2 times ULN) Patients with these laboratory value specified events will be identified using the same criteria for the interruption of investigational product with the exception of anemia, which will be identified using the same criteria for the discontinuation of investigational product, and thrombocytosis, which will be defined as a platelet count >600,000/μL. Bioanalytical Concentrations of baricitinib in human plasma will be determined by a validated liquid chromatography tandem mass spectrometry (LC/MS/MS) method. Pharmacokinetics/Pharmacodynamics Plasma concentrations of baricitinib, associated with time from dose, will be obtained at selected visits from all patients. Stored serum, plasma, and messenger ribonucleic acid (mrna) samples will be collected for possible exploratory assessments of pharmacodynamic (PD) markers of RA disease activity or mechanism of action of baricitinib. Pharmacogenetics Samples will be stored, and analysis may be performed on genetic variants thought to play a role in active RA and the Janus kinase (JAK)/signal transducers and activators of transcription (STAT) signaling pathways. Statistical Methods: Sample size: Approximately 1280 patients (480 in the baricitinib group, 480 in the placebo group, and 320 in the adalimumab group) will provide >95% power to detect a difference between baricitinib and placebo groups in ACR20 response rate (60% vs 35%) at Week 12 based on a 2-sided significance level of 0.05; approximately 94% power to detect a difference in mtss between baricitinib and placebo groups for an effect size of 0.25, based on a 2-sided t-test at a significance level of 0.04 (this assumes 90% of randomized patients will have x-ray data available for the mtss analysis at Week 24); and approximately 93% power for the noninferiority analysis of ACR20 response rate at Week 12 between baricitinib and adalimumab groups, where a response rate of 60% for baricitinib and adalimumab, a noninferiority margin of 12%, and a 1-sided significance level of are assumed. Analysis Population: The efficacy and health outcome analyses will be conducted on an intention-to-treat (ITT) basis. The ITT analysis set will include all data from all randomized patients treated with at least 1 dose of the study drug. Analysis of structural progression (van der Heijde mtss) will be conducted on the ITT population using patients with available baseline and at least 1 postbaseline value. The primary and major secondary analyses will be repeated using the per-protocol set (PPS), which excludes patients with significant protocol violations.
105 I4V-MC-JADV(c) Clinical Protocol Page 7 Safety analyses will be performed on the safety population, defined as all randomized patients who received at least 1 dose of the study drug. Missing Data Imputation: 1. Nonresponder imputation (NRI): All patients who discontinue the study or the study treatment at any time for any reason will be defined as nonresponders for the NRI analysis for categorical variables, such as ACR20/50/70, from the time of discontinuation and onward. Patients who receive rescue therapy at Week 16 will be analyzed as nonresponders after Week 16 and onward. Randomized patients without available data at a postbaseline visit will be defined as nonresponders for the NRI analysis at that visit. 2. Linear extrapolation method: The linear extrapolation method will be used for analysis of the structural progression endpoint (van der Heijde mtss) to impute missing data at time points when analyses are conducted (including the analyses at Week 24 and Week 52). For patients who discontinue the study or the study treatment or for patients who miss a radiograph for any reason, baseline data and the most recent radiographic data prior to discontinuation or the missed radiograph, adjusted for time, will be used for linear extrapolation to impute missing data at subsequent time points. For patients who receive rescue therapy starting at Week 16 or at any time point thereafter, baseline data and the most recent radiographic data prior to initiation of rescue therapy, adjusted for time, will be used for linear extrapolation. All patients originally randomized to placebo will be switched to active treatment at Week 24; thus, there will be no observed data for the placebo arm for the structural comparison at subsequent time points. Therefore, baseline data and the most recent radiographic data prior to initiation of the new therapy, adjusted for time, will be used for linear extrapolation at Week 52. The linear extrapolation method has been established as an appropriate missing data imputation method in other Phase 3 RA trials. Missing postbaseline values will be imputed only if both a baseline value and a postbaseline value from another time point are available, as long as the patient was on the same treatment at each applicable time point. 3. Multiple imputation: Other methods than linear extrapolation that include multiple imputation will be employed for analysis of the structural progression endpoint (van der Heijde mtss) to impute missing data at Week 24. Details will be provided in the statistical analysis plan (SAP). 4. Modified baseline observation carried forward (mbocf): The mbocf method will be used for the analysis of major secondary continuous endpoints (unless otherwise stated). For patients who discontinue the study or the study treatment because of an AE, including death, the baseline observation will be carried forward to the corresponding endpoint for evaluation. For patients who discontinue the study for reason(s) other than an AE, the last nonmissing postbaseline observation prior to discontinuation will be carried forward to the corresponding endpoint for evaluation. For patients who receive rescue therapy starting at Week 16, the last nonmissing observation at or before rescue will be carried forward to subsequent time points for evaluation. 5. Modified last observation carried forward (mlocf): For all continuous measures including safety analyses, the mlocf will be a general approach to impute missing data unless otherwise specified. For patients who receive rescue therapy starting at Week 16, the last nonmissing observation at or before rescue will be carried forward to subsequent time points for evaluation. For all other patients discontinuing from the study or the study treatment for any reason, the last nonmissing postbaseline observation before discontinuation will be carried forward to subsequent time points for evaluation. After mlocf imputation, data from patients with nonmissing baseline and postbaseline observations will be included in the analyses. The mlocf will be used as a sensitivity method to the mbocf method with respect to the major secondary endpoints. 6. Other methods for data imputation for analysis of key endpoints other than mtss may be used and will be described in the SAP Adjustment for Multiple Comparisons: A multiple testing procedure will be used to control the family-wise error rate for the primary and key secondary hypotheses for the endpoints listed above as objectives. Each step in the sequential testing procedure uses a local level procedure for the null hypotheses being tested. Testing of hypotheses at each step of the procedure commences only if all null hypotheses of prior steps were rejected. As such, the multiple testing procedure provides strong control of the family-wise error rate at the 1-sided level. Primary and Major Secondary Analyses: A logistic regression model with treatment, region, and joint erosion
106 I4V-MC-JADV(c) Clinical Protocol Page 8 status in the model will be used to test the treatment difference between baricitinib and placebo in the proportion of patients achieving ACR20 response at Week 12. The NRI method will be used to impute missing data. An analysis of covariance (ANCOVA) model with baseline score, region, joint erosion status, and treatment group in the model will be used to test the treatment difference between baricitinib and placebo in change from baseline to Week 24 of mtss scores. The linear extrapolation method will be used to impute missing data. An ANCOVA model with treatment, region, joint erosion status, and baseline score in the model will be used to test the treatment difference between baricitinib and placebo in change from baseline to Week 12 in HAQ-DI. The mbocf approach will be used to impute missing data. An ANCOVA model with treatment, region, joint erosion status, and baseline score in the model will be used to test the treatment difference between baricitinib and placebo in change from baseline to Week 12 in DAS28-hsCRP. The mbocf approach will be used to impute missing data. A logistic regression model with treatment, region, and joint erosion status in the model will be used to test the treatment difference between baricitinib and placebo in the proportion of patients achieving an SDAI score 3.3 response at Week 12. The NRI method will be used to impute missing data. A logistic regression model with treatment, region, and joint erosion status in the model will be used to test the treatment difference between baricitinib and adalimumab in the percentage of patients achieving ACR20 response at Week 12. A 95% confidence interval (CI) for the treatment difference will be constructed using the method described by Newcombe-Wilson without continuity correction. If the lower bound of the 95% CI is >-12%, Lilly will conclude that baricitinib is noninferior to adalimumab with respect to ACR20. If the lower bound of the 95% CI >0%, Lilly will conclude that baricitinib is superior to adalimumab with respect to ACR20. The NRI method will be used to impute missing data. For duration of morning joint stiffness, severity of morning joint stiffness, Worst Tiredness NRS, and Worst Pain NRS, an ANCOVA analysis with treatment, region, joint erosion status, and baseline score (Day 1 on the diaries) in the model will be used to test the treatment difference between baricitinib and placebo in mean diary scores in the 7 days prior to Week 12. An ANCOVA model with treatment, region, joint erosion status, and baseline score in the model will be used to test the treatment difference between baricitinib and adalimumab in change from baseline to Week 12 in DAS28-hsCRP. The mbocf approach will be used to impute missing data. Safety: All safety data will be descriptively summarized by treatment groups and analyzed using the safety population. Treatment-emergent AEs (TEAEs) are defined as AEs that first occurred or worsened in severity after the first dose of study treatment. The number and percentage of TEAEs will be summarized using MedDRA (Medical Dictionary for Regulatory Activities) for each system organ class and each preferred term by treatment group. TEAEs will also be summarized by relationship to treatment and by severity within each group. SAEs and AEs that lead to study drug discontinuation will also be summarized by group. Fisher s exact test will be used to perform pairwise comparisons among the treatment groups. Change from baseline in quantitative clinical chemistry will be analyzed using ANCOVA with treatment and baseline value in the model. Categorical variables, including the incidence of abnormal values and incidence of AESIs, will be summarized by frequency and percentage of patients in corresponding categories. Shift tables will be presented for selected measures. Change from baseline to postbaseline in vital signs, QIDS-SR16, and body weight will be analyzed using ANCOVA with treatment and baseline value in the model. Health Outcomes: Health outcome measures will be analyzed using methods described for continuous or categorical data as described for efficacy measures. More details will be provided in the SAP. Bioanalytical: Concentrations of baricitinib in human plasma will be determined by a validated LC/MS/MS method. Pharmacokinetics/Pharmacodynamics: PK analysis will be conducted using all evaluable PK data. PK data for baricitinib will be analyzed using a population modeling approach via a nonlinear mixed-effects modeling (NONMEM) program. Data from this study may be combined with an existing PK model or data from other studies, if appropriate. Intrinsic factors such as age; body weight; gender; ethnic origin; renal function; transaminases/albumin/bilirubin, which are representative of hepatic function; and extrinsic factors such as comedications will be investigated to assess their influence on PK parameters such as clearance and volume of distribution, where applicable. Time-course of response for key efficacy data such as, but not necessarily limited
107 I4V-MC-JADV(c) Clinical Protocol Page 9 to, ACR20/50/70, DAS28-ESR, DAS28-hsCRP, and safety data (eg, neutropenia, anemia) will be explored using graphical methods. Further investigations using a population-modeling approach via NONMEM to identify potential factors that may influence PD parameters may be warranted. Covariates such as body weight, disease duration, disease severity at baseline, responder status, cytokines concentrations, dose, and baricitinib exposures may be investigated to assess their impact on efficacy and safety endpoints. Data from this study may be combined with an existing PK/PD model or data from other studies, if appropriate. Planned Analyses: The analysis of primary and major secondary endpoints (except for x-ray data) will occur when all patients complete the Week 24 study visit. A final analysis will occur when all patients complete the study. Interim Analyses: A data monitoring committee (DMC), external to Lilly, will oversee the conduct of all the Phase 3 clinical trials evaluating baricitinib in patients with RA. This DMC will follow the rules defined in the DMC charter, focusing on potential and identified risks. The DMC will review and evaluate up to 3 planned interim analyses on an approximately semiannual basis. Access to the unblinded interim data will be limited to the statisticians who conduct the interim analyses and the DMC. The statisticians conducting the interim analyses will be independent from the study team who will not have access to the unblinded data. The DMC will review study discontinuations, AEs, clinical laboratory data, vital signs, etc. The DMC may recommend continuation of the study, as designed, temporary suspension of enrollment, or the discontinuation of a particular dose regimen or the entire study. The DMC may request to review efficacy data to investigate the benefit/risk relationship in the context of safety observations for ongoing patients in the study. However, the study will not be stopped for positive efficacy results, and there is no planned futility assessment. Details of the DMC and interim safety analyses will be documented in a DMC charter and DMC analysis plan. Besides DMC members, a limited number of pre-identified individuals may gain access to the unblinded PK and safety data, as specified in the unblinding plan, prior to the final database lock, to initiate the exploration and/or final population PK/PD model development processes for safety data (eg, neutropenia, anemia). Information that may unblind the study during the analyses will not be reported to study sites or the blinded study team until the database lock for the primary and major secondary efficacy analyses.
108 I4V-MC-JADV(c) Clinical Protocol Page Table of Contents A Randomized, Double-Blind, Placebo- and Active-Controlled, Phase 3 Study Evaluating the Efficacy and Safety of Baricitinib in Patients with Moderately to Severely Active Rheumatoid Arthritis Who Have Had an Inadequate Response to Methotrexate Therapy Section Page 1. Protocol I4V-MC-JADV(c) A Randomized, Double-Blind, Placeboand Active-Controlled, Phase 3 Study Evaluating the Efficacy and Safety of Baricitinib in Patients with Moderately to Severely Active Rheumatoid Arthritis Who Have Had an Inadequate Response to Methotrexate Therapy Synopsis Table of Contents Abbreviations and Definitions Introduction Objectives Primary Objective Secondary Objectives Exploratory Objective Investigational Plan Summary of Study Design Screening Period Part A and Part B: 24-Week and 28-Week, Double-Blind, Placebo- and Active-Controlled Periods Part C: Posttreatment Follow-Up Period Study Extensions Discussion of Design and Control Study Population Entry Criteria Inclusion Criteria Exclusion Criteria Enrollment Criteria Inclusion Criteria Exclusion Criteria Rationale for Exclusion of Certain Study Candidates...41
109 I4V-MC-JADV(c) Clinical Protocol Page Discontinuations Discontinuation of Patients Interruption of Study Drug Discontinuation of Study Drug Discontinuation from the Study Discontinuation of Study Sites Discontinuation of the Study Treatment Treatments Administered Materials and Supplies Method of Assignment to Treatment Rationale for Selection of Doses in the Study Selection and Timing of Doses Special Treatment Considerations Continued Access to Investigational Product Blinding Concomitant Therapy Treatment Compliance Efficacy, Health Outcome Measures, Safety Evaluations, Sample Collection and Testing, and Appropriateness of Measurements Efficacy Measures Primary Efficacy Measure Secondary Efficacy Measures Van der Heijde Modified Total Sharp Score Disease Activity Score-Erythrocyte Sedimentation Rate and Disease Activity Score High Sensitivity C-Reactive Protein ACR50 and ACR70 Indices Hybrid American College of Rheumatology Response Measure ACR/EULAR Rheumatoid Arthritis Remission Simplified Disease Activity Index Clinical Disease Activity Index European League Against Rheumatism Responder Index Health Outcome Measures Safety Evaluations Adverse Events Adverse Events of Special Interest Serious Adverse Events...60
110 I4V-MC-JADV(c) Clinical Protocol Page Suspected Unexpected Serious Adverse Reactions Other Safety Measures Measures Evaluated During Screening Period Measures Evaluated During Screening and Treatment Periods Safety Monitoring Complaint Handling Sample Collection and Testing Samples for Standard Laboratory Testing Samples for Drug Concentration Measurements Pharmacokinetics/Pharmacodynamics Exploratory Stored Samples Pharmacogenetics Nonpharmacogenetic/Biomarker Stored Samples Appropriateness of Measurements Data Quality Assurance Data Capture System Sample Size and Statistical Methods Determination of Sample Size Statistical and Analytical Plans General Considerations Patient Disposition Patient Characteristics Concomitant Therapy Treatment Compliance Primary Outcome and Methodology Efficacy Analyses Pharmacokinetic/Pharmacodynamic Analyses Health Outcome Analyses Safety Analyses Subgroup Analyses Planned Analyses Interim Analyses Informed Consent, Ethical Review, and Regulatory Considerations Informed Consent Ethical Review Regulatory Considerations Investigator Information...84
111 I4V-MC-JADV(c) Clinical Protocol Page Protocol Signatures Final Report Signature References...85
112 I4V-MC-JADV(c) Clinical Protocol Page 14 Table List of Tables Page Table JADV.1. Criteria for Temporary Discontinuation of Investigational Product...42 Table JADV.2. Criteria for Permanent Discontinuation of Investigational Product...43 Table JADV.3. Table JADV.4. Table JADV.5. Treatment Regimens...45 Scoring Method for the Hybrid American College of Rheumatology Response Measure...55 Categorization of Patients as Nonresponders, Moderate Responders, or Good Responders...56
113 I4V-MC-JADV(c) Clinical Protocol Page 15 Figure List of Figures Page Figure JADV.1. Illustration of study design for Clinical Protocol I4V-MC-JADV...30 Figure JADV.2. Illustration of the sequential testing approach within the overall gatekeeping strategy for Study JADV....74
114 I4V-MC-JADV(c) Clinical Protocol Page 16 Attachment List of Attachments Page Attachment 1. Protocol JADV Study Schedule...89 Attachment 2. Attachment 3. Attachment 4. Protocol JADV Clinical Laboratory Tests...97 Protocol JADV Hepatic Monitoring Tests for Treatment-Emergent Abnormality Protocol Amendment I4V-MC-JADV(c) Summary...101
115 I4V-MC-JADV(c) Clinical Protocol Page Abbreviations and Definitions Term ACR ACR20 ACR50 ACR70 adverse event (AE) AESI ALT ANCOVA anti-ccp AST audit blinding case report form (CRF) and electronic case report form (ecrf) CDAI cdmard CI CL Definition American College of Rheumatology 20% improvement in American College of Rheumatology criteria 50% improvement in American College of Rheumatology criteria 70% improvement in American College of Rheumatology criteria Any untoward medical occurrence in a patient or clinical investigation subject administered a pharmaceutical product and that does not necessarily have a causal relationship with this treatment. An adverse event can therefore be any unfavorable and unintended sign (including an abnormal laboratory finding), symptom, or disease temporally associated with the use of a medicinal (investigational) product, whether or not related to the medicinal (investigational) product. adverse event of special interest alanine aminotransferase analysis of covariance anticyclic citrullinated peptide aspartate aminotransferase a systematic and independent examination of the trial-related activities and documents to determine whether the evaluated trial-related activities were conducted, and the data were recorded, analyzed, and accurately reported according to the protocol, applicable standard operating procedures (SOPs), good clinical practice (GCP), and the applicable regulatory requirement(s) A procedure in which one or more parties to the trial are kept unaware of the treatment assignment(s). Single-blinding usually refers to the patient(s) being unaware, and double-blinding usually refers to the patient(s), investigator(s), monitor(s), and in some cases, select sponsor personnel being unaware of the treatment assignment(s). sometimes referred to as clinical report form: a printed or electronic form for recording study participants data during a clinical study, as required by the protocol. This study uses an electronic case report form. Clinical Disease Activity Index conventional disease-modifying antirheumatic drugs confidence interval central compartment clearance
116 I4V-MC-JADV(c) Clinical Protocol Page 18 clinical research physician complaint compliance CRP DAS DAS28 DMARD DMC DNA ECG efficacy/ effectiveness egfr end of study (trial) enroll/randomize enter epro EQ-5D-5L ESR ethical review board (ERB) Individual responsible for the medical conduct of the study. Responsibilities of the clinical research physician may be performed by a physician, clinical research scientist, global safety physician or other medical officer. A complaint is any written, electronic, or oral communication that alleges deficiencies related to the identity, quality, purity, durability, reliability, safety or effectiveness, or performance of a drug or drug delivery system. adherence to all the trial-related requirements, good clinical practice (GCP) requirements, and the applicable regulatory requirements C-reactive protein Disease Activity Score Disease Activity Score modified to include the 28 diarthroidal joint count disease-modifying antirheumatic drug data monitoring committee deoxyribonucleic acid electrocardiogram Efficacy is the ability of a treatment to achieve a beneficial intended result. Effectiveness is the measure of the produced effect of an intervention when carried out in a clinical environment. estimated glomerular filtration rate the date of the last visit or last scheduled procedure shown in the study schedule for the last active patient in the study The act of assigning a patient to a treatment. Patients who are enrolled/randomized in the trial are those who have been assigned to a treatment. The act of obtaining informed consent for participation in a clinical trial from patients deemed eligible or potentially eligible to participate in the clinical trial. Patients entered into a trial are those who sign the informed consent document directly or through their legally acceptable representatives. electronic patient-reported outcome European Quality of Life-5 Dimensions-5 Level erythrocyte sedimentation rate a board or committee (institutional, regional, or national) composed of medical and nonmedical members whose responsibility is to verify that the safety, welfare, and human rights of the patients participating in a clinical trial are protected
117 I4V-MC-JADV(c) Clinical Protocol Page 19 EULAR FACIT-F FSH GCP HAQ-DI HBsAb HBV HCV HDL HIV hscrp IB European League Against Rheumatism Functional Assessment of Chronic Illness Therapy Fatigue follicle-stimulating hormone good clinical practice Health Assessment Questionnaire Disability Index hepatitis B surface antibody hepatitis B virus hepatitis C virus high-density lipoprotein human immunodeficiency virus high-sensitivity C-reactive protein Investigator s Brochure IC 50 inhibitory concentration of 50% ICF ICH IL-6 intention to treat (ITT) interim analysis investigator IVRS JAK JAK1 JAK2 informed consent form International Conference on Harmonisation interleukin-6 The principle that asserts that the effect of a treatment policy can be best assessed by evaluating on the basis of the intention to treat a patient (that is, the planned treatment regimen) rather than the actual treatment given. It has the consequence that patients allocated to a treatment group should be followed up, assessed, and analyzed as members of that group irrespective of their compliance to the planned course of treatment. an analysis of clinical study data, separated into treatment groups, that is conducted before the final reporting database is created/locked A person responsible for the conduct of the clinical study at a study site. If a study is conducted by a team of individuals at a study site, the investigator is the responsible leader of the team and may be called the principal investigator. interactive voice-response system Janus kinase 1 of 4 identified members of the family of Janus kinases 1 of 4 identified members of the family of Janus kinases
118 I4V-MC-JADV(c) Clinical Protocol Page 20 JAK3 Ka LC/MS/MS LDL legal representative mbocf MCS MDRD MedDRA mlocf MMRM MRI mrna mtss MTX MTX-IR NONMEM NRI NRS NSAID patient PCS PD per-protocol set (PPS) PK 1 of 4 identified members of the family of Janus kinases absorption rate constant liquid chromatography tandem mass spectrometry low-density lipoprotein an individual, judicial, or other body authorized under applicable law to consent on behalf of a prospective patient, to the patient s participation in the clinical trial modified baseline observation carried forward Mental Component Score Modification of Diet in Renal Disease Medical Dictionary for Regulatory Activities modified last observation carried forward mixed model for repeated measures magnetic resonance imaging messenger ribonucleic acid modified Total Sharp Score methotrexate patients who have had an inadequate response to methotrexate nonlinear mixed-effects modeling nonresponder imputation numeric rating scale nonsteroidal anti-inflammatory drug a study participant who has the disease or condition for which the investigational product is targeted Physical Component Score pharmacodynamic(s) the set of data generated by the subset of patients who sufficiently complied with the protocol to ensure that these data would be likely to exhibit the effects of treatment, according to the underlying scientific model pharmacokinetic(s)
119 I4V-MC-JADV(c) Clinical Protocol Page 21 PPD Q QD QIDS-SR 16 RA SAE SAP SC screening SD SDAI SF-36v2 Acute SJC STAT subject SUSARs TB TJC TNF TNF-α treatmentemergent adverse event (TEAE) TSH purified protein derivative intercompartmental clearance once daily Quick Inventory of Depressive Symptomatology Self-Rated-16 rheumatoid arthritis serious adverse event statistical analysis plan subcutaneous the act of determining if an individual meets minimum requirements to become part of a pool of potential candidates for participation in a clinical study standard deviation Simplified Disease Activity Index Medical Outcomes Study 36-Item Short Form Health Survey Version 2 Acute swollen joint count signal transducers and activators of transcription An individual who is or becomes a participant in clinical research, either as a recipient of the investigational product(s) or as a control. A subject may be either a healthy human or a patient. suspected unexpected serious adverse reactions tuberculosis tender joint count tumor necrosis factor tumor necrosis factor-alpha any untoward medical occurrence that either occurs or worsens at any time after treatment baseline and which does not necessarily have to have a causal relationship with this treatment (also called treatment-emergent signs and symptoms [TESS]) thyroid-stimulating hormone TYK2 tyrosine kinase 2 ULN V1 V2 upper limit of normal central compartment volume of distribution peripheral volume of distribution
120 I4V-MC-JADV(c) Clinical Protocol Page 22 VAS WPAI-RA visual analog scale Work Productivity and Activity Impairment-Rheumatoid Arthritis
121 I4V-MC-JADV(c) Clinical Protocol Page 23 A Randomized, Double-Blind, Placebo- and Active-Controlled, Phase 3 Study Evaluating the Efficacy and Safety of Baricitinib in Patients with Moderately to Severely Active Rheumatoid Arthritis Who Have Had an Inadequate Response to Methotrexate Therapy 5. Introduction Rheumatoid arthritis (RA) is a systemic inflammatory autoimmune disease. The disease has variable expression and outcome ranging from mild, limited disease to severe disease associated with progressive joint destruction, significantly compromised quality of life, and reduced survival (Smolen and Steiner 2003; Colmegna et al. 2012). Substantial comorbidity can be seen outside of the musculoskeletal system, with excess cardiovascular risk, dyslipidemia, and propensity for infection partially related to the need for treatment with immunomodulatory agents (Curtis et al. 2007; Kitas and Gabriel 2011). Management of RA has improved substantially in recent years. In addition to reduction of signs and symptoms, improvement of physical function, and inhibition of structural damage, better patient outcomes and clinical remission are now considered achievable goals. Therefore, the current recommended primary target for treatment of RA should be a state of clinical remission (Smolen et al. 2010; Felson et al. 2011). There are several definitions for clinical remission based on composite scores (commonly including: tender joint counts [TJCs]/swollen joint counts [SJCs], level of acute phase reactants, and assessment of a patient or physician global response). These scores include the Disease Activity Score (DAS), DAS modified to include the 28 diarthroidal joint count (DAS28), Simplified Disease Activity Index (SDAI), Clinical Disease Activity Index (CDAI), and a Boolean definition as recently proposed jointly by American College of Rheumatology (ACR)/European League Against Rheumatism (EULAR) (Felson et al. 2011). The current focus on arresting disease activity results from an understanding that persistent joint inflammation leads to progressive joint destruction manifested by cartilage loss, erosive damage to juxta-articular bone, and resultant functional impairment; that erosive bone changes occur within months of disease onset; and that early and aggressive treatment increases the likelihood of disease control (Klareskog et al. 2009; Colmegna et al. 2012). Despite a variety of approved agents for RA, complete or sustained disease remission is unusual. Conventional disease-modifying anti-rheumatic drugs (cdmards) have been used with some success. Patients often receive 1 or more of these medications (for example, methotrexate [MTX], sulfasalazine, hydroxychloroquine, leflunomide, azathioprine, gold salts, and cyclosporine), typically in combination with low-dose oral or intra-articular glucocorticoids. In addition to cdmards, biological agents that block or antagonize critical inflammatory mediators, T cells, or B cells can reduce pain and swelling and provide joint protection against structural damage. The efficacy of these biologics, particularly in combination with MTX, has been shown to have a clinically important effect on the signs and symptoms of RA (Fleischmann 2005); however, a majority of patients do not go into remission or achieve a 50%
122 I4V-MC-JADV(c) Clinical Protocol Page 24 improvement in ACR criteria (ACR50) in a clinical trial setting. During treatment with tumor necrosis factor-alpha (TNF-α) antagonists, approximately 30% of patients fail to achieve a 20% improvement in ACR criteria (ACR20; primary failure), and more patients lose efficacy during therapy (secondary failure; Rubbert-Roth and Finckh 2009). Disease progression can still occur even for patients who achieve apparent adequate control of their signs and symptoms with cdmards and/or biologic therapies (Klareskog et al. 2009; Rubbert-Roth and Finckh 2009). Accordingly, a significant unmet need remains for more effective and better tolerated treatments for RA. Members of the Janus kinase (JAK) family of protein tyrosine kinases (JAK1, JAK2, JAK3, and tyrosine kinase 2 [TYK2]) play an important role in signal transduction following cytokine and growth factor binding to their receptors. Aberrant production of cytokines and growth factors has been associated with a number of chronic inflammatory conditions, including RA. Many of the pro-inflammatory cytokines implicated in the pathogenesis of RA, including interleukin-6 (IL-6) and interferon-gamma, use cell signaling that involves the JAK/signal transducers and activators of transcription (STAT) pathways. Inhibition of JAK-STAT signaling can target multiple RA-associated cytokine pathways and thereby reduce inflammation, cellular activation, and proliferation of key immune cells as demonstrated with other JAK inhibitors (Williams et al. 2008; Kremer et al. 2009). Baricitinib is an orally available, selective JAK inhibitor with excellent potency and selectivity for JAK1 (inhibitory concentration of 50% [IC 50 ] = 5.9 nm) and JAK2 (IC 50 = 5.7 nm) and less potency for JAK3 (IC nm) or TYK2 (IC 50 = 53 nm) (Fridman et al. 2010). Baricitinib is being developed for treatment of patients with moderately to severely active RA who are either intolerant to MTX treatment or who have had an inadequate response to disease-modifying antirheumatic drugs (DMARDs), either conventional or biologic. One completed and 2 currently ongoing Phase 2 studies have evaluated the clinical utility of baricitinib as treatment for patients with active RA. In the completed study (I4V-MC-JADC [JADC], conducted by Incyte Corporation), administration of baricitinib at doses of 4, 7, or 10 mg daily for up to 24 weeks resulted in improved signs and symptoms in patients with an inadequate response to DMARDs, including biologics. A relatively flat dose response was observed for the efficacy parameters, with the 4-mg dose performing as well as the 7- and 10-mg doses. The proportions of patients who were ACR responders generally decreased in the off-treatment follow-up period (from Week 24 to Week 28). In an ongoing Phase 2 study (I4V-MC-JADA [JADA]), administration of baricitinib at doses of 1, 2, 4, and 8 mg daily for up to 12 weeks with continued administration of the 2-, 4-, and 8-mg doses for up to 24 weeks resulted in improved signs and symptoms in patients with an inadequate response to MTX. The observed treatment effect in the 4- and 8-mg dose groups was similar, confirming the flat dose response observed in Study JADC. The treatment effect in these dose groups was larger than that observed in the 1- and 2-mg dose groups. The 1- and 2-mg dose groups offered some clinical effectiveness relative to placebo treatment. Patients completing 24 weeks of treatment could enter an extension study that is currently ongoing. A third Phase 2
123 I4V-MC-JADV(c) Clinical Protocol Page 25 study (I4V-JE-JADN) is ongoing in Japan, and the sponsor remains blinded to patient treatment allocation. The safety profile for baricitinib has been informed by results from nonclinical and clinical studies evaluating a wide range of doses (up to 20 mg once daily [QD]). There are a number of identified and potential risks recognized for baricitinib that will be followed carefully in the Phase 3 development program. Important identified risks for baricitinib include decreased hemoglobin; increased total cholesterol, high-density lipoprotein (HDL), low-density lipoprotein (LDL), and triglycerides; and decreased neutrophils and other phagocytic white cell lines (within the normal reference range). More information about the known and expected benefits, risks, and reasonably anticipated adverse events (AEs) may be found in the investigator s brochure (IB). Information on AEs expected to be related to the study drug may be found in Section 7 (Development Core Safety Information) of the IB. Information on serious AEs (SAEs) expected in the study population independent of drug exposure will be assessed by the sponsor in aggregate periodically during the course of the study and may be found in Section 6 (Effects in Humans) of the IB. Study I4V-MC-JADV (JADV) is one of 5 Phase 3 clinical trials investigating the efficacy and safety of baricitinib in patients with active RA. Study JADV is a double-blind, placebo- and active-controlled study investigating the efficacy and safety of baricitinib (4 mg QD) in patients with active RA who have had an inadequate response to MTX (MTX-IR). The study is 52 weeks in duration and includes assessment of structural joint damage. Because tumor necrosis factor (TNF) inhibitors are the most commonly used biologic class of therapies for the treatment of moderately to severely active RA, Humira (adalimumab) was selected as an appropriate active comparator to baricitinib. The remaining 4 Phase 3 studies are described below. Study I4V-MC-JADZ is a double-blind, active-controlled study investigating the efficacy and safety of baricitinib (4 mg QD), administered as monotherapy or in combination with MTX, in patients with early RA who have had limited or no treatment with DMARDs. MTX, administered as monotherapy and titrated to the highest tolerated dose, serves as the active comparator. Study I4V-MC-JADX is a doubleblind, placebo-controlled study investigating the efficacy and safety of baricitinib (2 mg QD or 4 mg QD) administered with background cdmard(s) in patients with active RA who have had an inadequate response to conventional DMARDs. Study I4V-MC-JADW is a double-blind, placebo-controlled study investigating the efficacy and safety of baricitinib (2 mg QD or 4 mg QD) administered with background cdmard(s) in patients with active RA who have had an inadequate response to biologic DMARDs. Patients completing any of the above studies are eligible for enrollment in Study I4V-MC-JADY (JADY), a long-term extension study investigating the safety of baricitinib with prolonged administration (up to 2 additional years of treatment). Given the continuing unmet medical need in patients with moderately to severely active RA, the efficacy of baricitinib demonstrated in Phase 2 studies, and the acceptable safety profile for baricitinib observed through the current stage of development, initiation of Phase 3 testing is appropriate.
124 I4V-MC-JADV(c) Clinical Protocol Page Objectives 6.1. Primary Objective The primary objective of the study is to determine whether baricitinib is superior to placebo in the treatment of patients with moderately to severely active RA despite MTX treatment (ie, MTX-IR), as assessed by the proportion of patients achieving ACR20 at Week Secondary Objectives The major secondary objectives of the study are to evaluate the efficacy of baricitinib versus placebo or adalimumab as assessed by: change from baseline to Week 24 in structural joint damage as measured by modified Total Sharp Score (mtss [van der Heijde method]) compared to placebo change from baseline to Week 12 in Health Assessment Questionnaire-Disability Index (HAQ-DI) score compared to placebo change from baseline to Week 12 in DAS28-high-sensitivity C-reactive protein (hscrp) compared to placebo proportion of patients achieving an SDAI score 3.3 at Week 12 compared to placebo proportion of patients achieving ACR20 response at Week 12 compared to adalimumab mean duration of morning joint stiffness in the 7 days prior to Week 12 compared to placebo as collected in electronic diaries change from baseline to Week 12 in DAS28-hsCRP compared to adalimumab mean severity of morning joint stiffness in the 7 days prior to Week 12 compared to placebo as collected in electronic diaries mean Worst Tiredness numeric rating scale (NRS) in the 7 days prior to Week 12 compared to placebo as collected in electronic diaries mean Worst Pain NRS in the 7 days prior to Week 12 compared to placebo as collected in electronic diaries The other secondary objectives of the study are as follows: to evaluate the efficacy of baricitinib as assessed by: o proportion of patients achieving DAS28-hsCRP 3.2 and DAS28-hsCRP <2.6 at Week 12, Week 24, and Week 52 o proportion of patients achieving HAQ-DI improvement 0.22 and 0.3 at Week 12, Week 24, and Week 52 o change from baseline to Week 16 and Week 52 in structural joint damage as measured by mtss o proportion of patients with mtss change 0 from baseline at Week 16, Week 24, and Week 52 o change from baseline to Week 16, Week 24, and Week 52 in joint space narrowing and bone erosion scores o proportion of patients achieving ACR20 at Week 24 and Week 52 o proportion of patients achieving ACR50 at Week 12, Week 24, and Week 52
125 I4V-MC-JADV(c) Clinical Protocol Page 27 o proportion of patients achieving 70% improvement in ACR criteria (ACR70) at Week 12, Week 24, and Week 52 o percentage change from baseline to Week 12, Week 24, and Week 52 in individual components of the ACR Core Set o change from baseline through Week 52 in DAS28-hsCRP o change from baseline through Week 52 in DAS28-erythrocyte sedimentation rate (ESR) o proportion of patients achieving DAS28-ESR 3.2 and DAS28-ESR <2.6 at Week 12, Week 24, and Week 52 o change from baseline to Week 24 and Week 52 in HAQ-DI score o change from baseline to Week 12, Week 24, and Week 52 in CDAI score o change from baseline to Week 12, Week 24, and Week 52 in SDAI score o proportion of patients achieving a CDAI score 2.8 at Week 12, Week 24, and Week 52 o proportion of patients achieving an SDAI score 3.3 at Week 24, and Week 52 o proportion of patients achieving ACR/EULAR remission according to the Boolean-based definition at Week 12, Week 24 and Week 52 o proportion of patients achieving moderate and good EULAR responses based on the 28-joint DAS at Week 12, Week 24, and Week 52 o evaluation of hybrid ACR (bounded) response at Week 12, Week 24 and Week 52 o change from baseline to Week 12, Week 24, and Week 52 in Functional Assessment of Chronic Illness Therapy Fatigue (FACIT-F) scale scores o change from baseline to Week 12, Week 24, and Week 52 in each scale and the summary scores for the Mental Component Score (MCS) and Physical Component Score (PCS) of the Medical Outcomes Study 36-Item Short Form Health Survey Version 2 Acute (SF-36v2 Acute) o change from baseline to Week 12, Week 24, and Week 52 in European Quality of Life-5 Dimensions-5 Level (EQ-5D-5L) scores o change from baseline to Week 12, Week 24, and Week 52 in Work Productivity and Activity Impairment-Rheumatoid Arthritis (WPAI-RA) scores o evaluation of healthcare resource utilization through Week 52 o change from baseline to Week 12, Week 24, and Week 52 in duration of morning joint stiffness assessed at the visit using an electronic patient-reported outcomes (epro) tablet o change from baseline to Week 12, Week 24, and Week 52 in Worst Tiredness NRS assessed at the visit using an epro tablet o change from baseline to Week 12, Week 24, and Week 52 in Worst Pain NRS assessed at the visit using an epro tablet to characterize the pharmacokinetics (PK) of baricitinib, determine the magnitude of within- and between-patient variability, and identify the potential factors that may have an impact on the PK of baricitinib
126 I4V-MC-JADV(c) Clinical Protocol Page 28 to characterize the baricitinib exposure-response time course for key efficacy and safety endpoints and to identify potential factors that may have an impact on these efficacy or safety endpoints of baricitinib 6.3. Exploratory Objective The exploratory objective of the study is: to compare baricitinib to adalimumab as regards to various efficacy measures
127 I4V-MC-JADV(c) Clinical Protocol Page Investigational Plan 7.1. Summary of Study Design Study JADV will be a 52-week, Phase 3, multicenter, randomized, double-blind, double-dummy, placebo- and active-controlled, parallel-group, outpatient study comparing the efficacy of baricitinib versus placebo on signs and symptoms, function, remission, and structural progression in patients with moderately to severely active RA who have had an insufficient response to MTX and who have never been treated with a biologic DMARD (ie, MTX-IR patients). The study will include a noninferiority comparison of baricitinib versus adalimumab on signs and symptoms. Baricitinib will be administered as a 4-mg tablet QD. The dose for patients with renal impairment, defined as estimated glomerular filtration rate (egfr) <60 ml/min/1.73 m2, will be 2 mg baricitinib QD. Patients not assigned to the baricitinib treatment arm will receive either a 4-mg or 2-mg matching placebo tablet. Adalimumab (40 mg) will be administered by subcutaneous (SC) injection biweekly (ie, every 2 weeks). Patients not assigned to adalimumab will receive a matching placebo SC injection biweekly. Planned enrollment is approximately 1280 randomized patients: 480 to receive baricitinib, 480 to receive placebo, and 320 to receive adalimumab. After the Week 52 study visit, eligible patients may proceed to a separate extension study (Study JADY) lasting for up to 2 years or to the posttreatment follow-up period of this study (Part C). Study JADV will consist of 4 parts: Screening: Screening period lasting from 3 to 42 days prior to Visit 2 (Week 0) Part A: double-blind, placebo- and active-controlled period from Week 0 through Week 24 Part B: double-blind, active-controlled period from the end of Week 24 through Week 52 Part C: posttreatment follow-up period Section outlines information regarding the data monitoring committee (DMC) and interim analyses. Figure JADV.1 illustrates the study design.
128
129 I4V-MC-JADV(c) Clinical Protocol Page 31 Week 52 (Visit 15) and a placebo SC injection biweekly starting at Week 0 (Visit 2) through Week 50. Patients randomized to placebo will be administered a placebo tablet QD starting at Week 0 (Visit 2) through Week 24 (the morning of Visit 11). At Week 24, patients will be administered baricitinib 4 mg QD (or 2 mg baricitinib QD if egfr <60 ml/min/1.73 m2) through Week 52 (Visit 15). Patients in this treatment arm will also receive a placebo SC injection biweekly starting at Week 0 (Visit 2) through Week 50. Patients randomized to adalimumab will be administered adalimumab (40 mg) by SC injection biweekly starting at Week 0 (Visit 2) through Week 50 and a placebo tablet QD through Week 52 (the morning of Visit 15). The last SC injection will be administered during Week 50 because patients receiving adalimumab have the option to switch to baricitinib 4 mg QD (or 2 mg QD depending on egfr) if they choose to participate in the extension study. Scheduling the last SC injection at Week 50 ensures approximately a 2-week interval from the last SC injection to the start of baricitinib dosing. Patients will be eligible for rescue treatment beginning at Week 16 (Visit 9). Details of the rescue are listed in Section Patients will remain on background MTX, nonsteroidal anti-inflammatory drugs (NSAIDs), analgesics, and/or corticosteroids (see Section 9.8 for guidelines on adjustments to concomitant therapy). Patients will take their assigned doses of study drugs as directed. The primary efficacy endpoint will be at Week 12, and the final visit during Part A will be at Week 24. The final visit during Part B will be at Week 52 (Visit 15). The structural joint damage endpoints will be at Week 16, Week 24, and Week 52. All patients who complete the Week 52 study visit will be eligible for inclusion in extension Study JADY Part C: Posttreatment Follow-Up Period Patients who are not enrolled in the extension Study JADY will have a Follow-Up Visit (Visit 801) approximately 28 days after the last dose of study drug. Patients who discontinue from the study prior to Week 52 for any reason will have an Early Termination Visit performed, including x-rays. The Early Termination Visit should be performed as soon as logistically possible because this visit includes assessment of safety and PK data. Infrequently, patient and investigator availability may be such that the Early Termination Visit and the Follow-up Visit may occur on or about the same date. In this instance, the visits may be combined and should occur approximately 28 days after the last dose of study drug. All activities required for the Early Termination Visit should be completed according to the schedule of assessments (Attachment 1) Study Extensions Patients who complete this study through Visit 15 will be eligible to participate in Study JADY, if enrollment criteria for Study JADY are met.
130 I4V-MC-JADV(c) Clinical Protocol Page Discussion of Design and Control Based on efficacy, safety, and PK data from the Phase 2 studies (JADC and JADA), Lilly has selected a single dose of 4 mg QD baricitinib (with dose adjustment for renal dysfunction) for evaluation in this study (see Section 9.4 for rationale). The Week 12 primary efficacy endpoint was chosen based on the results of Studies JADC and JADA. In these studies, efficacy as measured by ACR20 response was observed as early as 2 weeks after initial administration of baricitinib followed by near-maximum efficacy response achieved by Week 12. The 52-week treatment period will allow evaluation of the long-term effect of baricitinib on structural joint damage. All patients will be required to be on background MTX treatment at baseline. Other background therapies, including NSAIDs and low dose oral corticosteroids, are permitted during the study for patients who are on stable doses of these treatments at baseline. Patients should remain on the same dose of MTX throughout the study; however, the MTX dose may be adjusted for safety reasons. To prevent potential unblinding due to observed efficacy or laboratory changes, a dual assessor approach will be used to evaluate efficacy and safety. The Joint Assessor (or designee) should be a rheumatologist or skilled arthritis assessor. The Joint Assessor will be responsible for completing the joint counts. To ensure consistent joint evaluation throughout the trial, individual patients should be evaluated by the same Joint Assessor for all study visits. The Joint Assessor must not access or discuss with the patient the patient-reported assessments, Physician s Global Assessment of Disease Activity (visual analog scale [VAS]), and safety assessments. The Safety Assessor (or designee) should be a rheumatologist (or medically qualified physician) and will have access to both safety and efficacy data. The Safety Assessor may be the principal investigator. The Safety Assessor will be responsible for completing the Physician s Global Assessment of Disease Activity (VAS). To ensure consistent Physician s Global Assessment of Disease Activity (VAS) throughout the trial, the instrument should be evaluated by the same physician at all study visits. The Safety Assessor will have access to source documents, laboratory results, and case report forms (CRFs) and will be responsible for making treatment decisions based on a patient s clinical response and laboratory parameters. All patients in Study JADV will be offered rescue therapy starting at Week 16 in line with precedent from recent clinical trials. Patients who are determined to be nonresponders to their originally assigned treatment will be reassigned to treatment with baricitinib at Week 16. Patients originally assigned to baricitinib will be rescued to the same dose of baricitinib to maintain the study blind. Patients not rescued at Week 16 may be rescued to treatment with baricitinib at the discretion of the investigator anytime thereafter. Patients initially randomized to placebo and not rescued will be reassigned to receive baricitinib at Week 24 to limit exposure to inactive treatment. Patients not experiencing improvement in signs and symptoms following approximately 4 weeks of rescue treatment should be discontinued from the study. Patients may be rescued only once. See Section for details of rescue treatment.
131 I4V-MC-JADV(c) Clinical Protocol Page Study Population The study population will be comprised of patients diagnosed with RA with active disease who have failed to adequately respond to MTX. Study investigator(s) will review patient records and screening test results to determine that the patient meets all inclusion and exclusion criteria to qualify for participation in the study Entry Criteria Patients may enter into the study once they have signed the informed consent form (ICF). Patients may be assessed for study entry on the basis of readily available information (ie, information that does not require informed consent from the subject) Inclusion Criteria Patients are eligible for entry into the study (ie, eligible to sign consent) only if they are expected to meet all of the following criteria: [1] are at least 18 years of age [2] have a diagnosis of adult-onset RA as defined by the ACR/EULAR 2010 Criteria for the Classification of RA (Aletaha et al. 2010) [3] have moderately to severely active RA defined as the presence of at least 6/68 tender joints and at least 6/66 swollen joints a. If significant surgical treatment of a joint has been performed, that joint cannot be counted in the TJC and SJC for entry or enrollment purposes. [4] have a C-reactive protein (CRP) (or hscrp) measurement 6 mg/l based on the most recent data (if available) [5] have had regular use of MTX for at least the 12 weeks prior to study entry at a dose that, in accordance with local clinical practice, is considered acceptable to adequately assess clinical response. The dose of MTX must have been a stable, unchanging oral dose of 7.5 to 25 mg/week (or the equivalent injectable dose) for at least the 8 weeks prior to study entry. The dose of MTX is expected to remain stable throughout the study and may be adjusted only for safety reasons a. For patients entering the trial on MTX doses <15 mg/week, there must be clear documentation in the medical record that higher doses of MTX were not tolerated or that the dose of MTX is the highest acceptable dose based on local clinical practice guidelines. b. Local standard of care should be followed for concomitant administration of folic acid. [6] are able to read, understand, and give written informed consent
132 I4V-MC-JADV(c) Clinical Protocol Page Exclusion Criteria Patients will be excluded from participating in the study if they meet or are expected to meet any of the following criteria: [7] are currently receiving corticosteroids at doses >10 mg of prednisone per day (or equivalent) or have been receiving an unstable dosing regimen of corticosteroids within 2 weeks of study entry or within 6 weeks of planned randomization [8] have started treatment with NSAIDs (for which the NSAID use is intended for treatment of signs and symptoms of RA) within 2 weeks of study entry or within 6 weeks of planned randomization or have been receiving an unstable dosing regimen of NSAIDs within 2 weeks of study entry or within 6 weeks of planned randomization [9] are currently receiving concomitant treatment with MTX, hydroxychloroquine, and sulfasalazine or combination of any 3 cdmards [10] are currently receiving or have received cdmards (eg, gold salts, cyclosporine, azathioprine, or any other immunosuppressives) other than MTX, hydroxychloroquine (up to 400 mg/day), or sulfasalazine (up to 3000 mg/day) within 4 weeks prior to study entry a. Doses of hydroxychloroquine or sulfasalazine should be stable for at least 8 weeks prior to study entry; if either has been recently discontinued, the patient must not have taken any dose within 4 weeks prior to study entry. b. Immunosuppression related to organ transplantation is not permitted. [11] have received leflunomide in the 12 weeks prior to study entry (or within 4 weeks prior to study entry if the standard 11 days of cholestyramine is used to washout leflunomide) [12] have started a new physiotherapy treatment for RA in the 2 weeks prior to study entry [13] have ever received any biologic DMARD (such as TNF, interleukin-1, IL-6, or T-cell- or B-cell-targeted therapies) [14] have received interferon therapy (such as Roferon-A, Intron-A, Rebetron, Alferon-N, Peg-Intron, Avonex, Betaseron, Infergen, Actimmune, Pegasys) within 4 weeks prior to study entry or are anticipated to require interferon therapy during the study [15] have received any parenteral corticosteroid administered by intramuscular or intravenous injection within 2 weeks prior to study entry or within 6 weeks prior to planned randomization or are anticipated to require parenteral injection of corticosteroids during the study
133 I4V-MC-JADV(c) Clinical Protocol Page 35 [16] have had 3 or more joints injected with intraarticular corticosteroids or hyaluronic acid within 2 weeks prior to study entry or within 6 weeks prior to planned randomization a. Joints injected with intraarticular corticosteroids or hyaluronic acid within 2 weeks prior to study entry or within 6 weeks prior to planned randomization cannot be counted in the TJC and SJC for entry or enrollment purposes. [17] have any condition or contraindication as addressed in the local labeling or local clinical practice for adalimumab that would preclude the patient from participating in this protocol [18] have active fibromyalgia that, in the investigator s opinion, would make it difficult to appropriately assess RA activity for the purposes of this study [19] have a diagnosis of any systemic inflammatory condition other than RA such as, but not limited to, juvenile chronic arthritis, spondyloarthropathy, Crohn s disease, ulcerative colitis, psoriatic arthritis, active vasculitis or gout a. Patients with secondary Sjögren s syndrome are not excluded. [20] have a diagnosis of Felty s syndrome [21] have had any major surgery within 8 weeks prior to study entry or will require major surgery during the study that, in the opinion of the investigator in consultation with Lilly or its designee, would pose an unacceptable risk to the patient [22] have experienced any of the following within 12 weeks of study entry: myocardial infarction, unstable ischemic heart disease, stroke, or New York Heart Association Stage IV heart failure [23] have a history or presence of cardiovascular, respiratory, hepatic, gastrointestinal, endocrine, hematological, neurological, or neuropsychiatric disorders or any other serious and/or unstable illness that, in the opinion of the investigator, could constitute a risk when taking investigational product or could interfere with the interpretation of data [24] are largely or wholly incapacitated permitting little or no self-care, such as being bedridden or confined to a wheelchair [25] have an egfr based on the most recent available serum creatinine using the Modification of Diet in Renal Disease (MDRD) method of <40 ml/min/1.73 m2 [26] have a history of chronic liver disease with the most recent available aspartate aminotransferase (AST) or alanine aminotransferase (ALT) >1.5 times the ULN or the most recent available total bilirubin 1.5 times the ULN
134 I4V-MC-JADV(c) Clinical Protocol Page 36 [27] have a history of, lymphoproliferative disease; or have signs or symptoms suggestive of possible lymphoproliferative disease, including lymphadenopathy or splenomegaly; or have active primary or recurrent malignant disease; or have been in remission from clinically significant malignancy for <5 years a. Patients with cervical carcinoma in situ that has been resected with no evidence of recurrence or metastatic disease for at least 3 years may participate in the study. b. Patients with basal cell or squamous epithelial skin cancers that have been completely resected with no evidence of recurrence for at least 3 years may participate in the study. [28] have been exposed to a live vaccine within 12 weeks prior to planned randomization or are expected to need/receive a live vaccine during the course of the study (with the exception of herpes zoster vaccination) a. All patients who have not received the herpes zoster vaccine at study entry will be encouraged to do so prior to randomization; vaccination must occur >30 days prior to randomization and start of study drug. Patients will be excluded if they were exposed to herpes zoster vaccination within 30 days of planned randomization. b. Investigators should review the vaccination status of their patients and follow the local guidelines for adult vaccination with nonlive vaccines intended to prevent infectious disease prior to entering patients into the study. [29] have a current or recent (<30 days prior to study entry) clinically serious viral, bacterial, fungal, or parasitic infection [30] have had symptomatic herpes zoster infection within 12 weeks prior to study entry [31] have a history of disseminated/complicated herpes zoster (eg, multidermatomal involvement, ophthalmic zoster, central nervous system involvement, or postherpetic neuralgia) [32] are immunocompromised and, in the opinion of the investigator, are at an unacceptable risk for participating in the study [33] have a history of active hepatitis B virus (HBV), hepatitis C virus (HCV), or human immunodeficiency virus (HIV) [34] have had household contact with a person with active tuberculosis (TB) and did not receive appropriate and documented prophylaxis for TB [35] have evidence of active TB or have previously had evidence of active TB and did not receive appropriate and documented treatment [36] are pregnant or nursing at the time of study entry
135 I4V-MC-JADV(c) Clinical Protocol Page 37 [37] are females of childbearing potential who do not agree to use 2 forms of highly effective birth control when engaging in intercourse while enrolled in the study and for at least 28 days following the last dose of orally administered investigational product and for at least 28 days or the time period specified in the local adalimumab label, whichever is longer, after the last SC injection. a. Females of nonchildbearing potential are defined as women 60 years of age, women 40 and <60 years of age who have had a cessation of menses for at least 12 months, or women who are congenitally or surgically sterile (that is, have had a hysterectomy or bilateral oophorectomy or tubal ligation). b. The following birth control methods are considered highly effective (the patient should choose 2 to be used with their partner): oral, injectable, or implanted hormonal contraceptives condom with a spermicidal foam, gel, film, cream, or suppository occlusive cap (diaphragm or cervical/vault caps) with a spermicidal foam, gel, film, cream, or suppository intrauterine device intrauterine system (for example, progestin-releasing coil) vasectomized male (with appropriate postvasectomy documentation of the absence of sperm in the ejaculate) [38] are males who do not agree to use 2 forms of highly effective birth control (see above) while engaging in sexual intercourse with female partners of childbearing potential while enrolled in the study and for at least 28 days following the last dose of orally administered investigational product and for at least 28 days or the time period specified in the local adalimumab label, whichever is longer, after the last SC injection. [39] have donated >500 ml of blood within 30 days prior to study entry or intend to donate blood during the course of the study [40] have a history of chronic alcohol abuse, intravenous drug abuse, or other illicit drug abuse within the 2 years prior to study entry [41] have previously been randomized in this study or any other study investigating baricitinib [42] are unable or unwilling to make themselves available for the duration of the study and/or are unwilling to follow study restrictions and procedures [43] have received prior treatment with an oral JAK inhibitor
136 I4V-MC-JADV(c) Clinical Protocol Page 38 [44] are investigator site personnel directly affiliated with this study and/or their immediate families. Immediate family is defined as a spouse, parent, child, or sibling, whether biological or legally adopted [45] are Lilly or Incyte employees or either s designee [46] are currently enrolled in or have discontinued within 30 days of study entry from a clinical trial involving an investigational product or nonapproved use of a drug or device or are concurrently enrolled in any other type of medical research judged not to be scientifically or medically compatible with this study 8.2. Enrollment Criteria Inclusion Criteria Entered patients are eligible for enrollment into the study (ie, eligible for randomization) only if they continue to meet all entry criteria (see above) at the time of randomization and meet the following enrollment criteria: [47] have moderately to severely active RA defined as the presence of at least 6/68 tender joints and at least 6/66 swollen joints assessed at Visit 1 and Visit 2 at the time of randomization [48] have an hscrp measurement 6 mg/l based on Visit 1 laboratory results by central laboratory testing The hscrp measurement may be repeated once within approximately 2 weeks of the initial value, and the value resulting from repeat testing may be accepted for enrollment eligibility if it meets the eligibility criterion. [49] have at least 1 joint erosion in hand, wrist, or foot joints based on radiographic interpretation by the central reader and be rheumatoid factor or anticyclic citrullinated peptide (anti-ccp) antibody positive; or have at least 3 joint erosions in hand, wrist, or foot joints based on radiographic interpretation by the central reader regardless of rheumatoid factor or anti-ccp antibody status (radiographic images acquired within 4 weeks of study entry may be submitted to the central reader to confirm eligibility). If a patient undergoes rescreening, radiographic images acquired as part of initial screening and within 10 weeks of randomization may be used Exclusion Criteria Entered patients are ineligible for enrollment (ie, ineligible for randomization) and should be discontinued from the study if they meet any of the following criteria: [50] have screening laboratory test values, including thyroid-stimulating hormone (TSH), outside the reference range for the population or investigative site that, in the opinion of the investigator, pose an unacceptable risk for the patient s participation in the study
137 I4V-MC-JADV(c) Clinical Protocol Page 39 a. Patients who are receiving thyroxine as replacement therapy may participate in the study provided stable therapy has been administered for 12 weeks and TSH is within the laboratory s reference range. The TSH test may be repeated once within approximately 2 weeks of the initial value, and the value resulting from repeat testing may be accepted for enrollment eligibility if it meets the eligibility criterion. [51] have any of the following specific abnormalities on screening laboratory tests: AST or ALT >1.5 times the ULN total bilirubin 1.5 times the ULN hemoglobin <10.0 g/dl (100.0 g/l) total white blood cell count <2500 cells/l neutropenia (absolute neutrophil count <1200 cells/l) lymphopenia (lymphocyte count <750 cells/l) thrombocytopenia (platelets <100,000/L) egfr <40 ml/min/1.73 m2 In the case of any of the aforementioned laboratory abnormalities, the tests may be repeated once within approximately 2 weeks of the initial values, and values resulting from repeat testing may be accepted for enrollment eligibility if they meet the eligibility criterion. [52] have screening electrocardiogram (ECG) abnormalities that, in the opinion of the investigator or the sponsor, are clinically significant and indicate an unacceptable risk for the patient s participation in the study (eg, Fridericia s corrected QT interval >500 msec) [53] have symptomatic herpes simplex at the time of study enrollment [54] have evidence of active TB as documented by a positive purified protein derivative (PPD) test ( 5-mm induration between approximately 2 and 3 days after application, regardless of vaccination history), medical history, clinical symptoms, and abnormal chest x-ray at screening a. The QuantiFERON -TB Gold or the T-SPOT.TB test (if available) may be used instead of the PPD test. Patients are excluded from the study if the test is not negative and there is clinical evidence of active TB.
138 I4V-MC-JADV(c) Clinical Protocol Page 40 [55] have evidence of latent TB (as documented by a positive PPD, no clinical symptoms consistent with active TB, and a normal chest x-ray at screening) unless patients complete at least 4 weeks of appropriate treatment prior to randomization and agree to complete the remainder of treatment while in the trial a. If the PPD test is positive and the patient has no medical history or chest x-ray findings consistent with active TB, the patient may have a QuantiFERON-TB Gold or T-SPOT.TB test (if available). If the test results are not negative, the patient will be considered to have latent TB (for purposes of this study). b. The QuantiFERON-TB Gold or T-SPOT.TB test (if available) may be used instead of the PPD test. If the test results are positive, the patient will be considered to have latent TB. If the test is not negative, the test may be repeated once within approximately 2 weeks of the initial value. If the repeat test results are again not negative, the patient will be considered to have latent TB (for purposes of this study). c. Exceptions include patients with a history of active or latent TB who have documented evidence of appropriate treatment. [56] have a positive test for HBV defined as: a. positive for hepatitis B surface antigen, or b. positive for anti-hepatitis B core antibody but negative for hepatitis B surface antibody (HBsAb), or c. for patients enrolled in Japan, or elsewhere if required, positive for anti-hbsab and positive for HBV deoxyribonucleic acid (DNA). If any of the HBV tests have an indeterminate result, confirmatory testing will be performed by an alternate method. [57] have HCV (positive for anti-hepatitis C antibody with confirmed presence of HCV) [58] have evidence of human HIV infection and/or positive HIV antibodies [59] Are women 40 and <60 years of age who have had a cessation of menses for at least 12 months, have a follicle-stimulating hormone (FSH) value <40 miu/ml, and do not agree to use 2 forms of highly effective methods of birth control when engaging in intercourse while enrolled in the study and for at least 28 days following the last dose of orally administered investigational product and for at least 28 days or the time period specified in the local adalimumab label, whichever is longer, after the last SC injection, unless they are congenitally or surgically sterile (ie, have had a hysterectomy, bilateral oophorectomy, or tubal ligation).
139 I4V-MC-JADV(c) Clinical Protocol Page 41 If patients are entered into the study while taking medications that require discontinuation before randomization, those patients should only be asked to discontinue these medications after it has been determined that they will be eligible for randomization. Patients who are entered into the study but do not meet enrollment criteria should be discontinued from the study. These patients can be re-entered into the trial (ie, give repeat consent) if the investigator believes that the patient might meet enrollment criteria at a future date. The investigator should wait approximately 1 month before re-entering a patient into the study Rationale for Exclusion of Certain Study Candidates The rationale for the entry exclusion criteria is as follows: Exclusion Criteria [7] to [16] exclude individuals who are taking or who may take RA medications or treatments that interfere with the ability to assess the safety and efficacy of baricitinib. Exclusion Criteria [17] to [27] exclude individuals with previous or concomitant medical conditions that increase the risk for their participation in the study. Exclusion Criteria [28] to [35] exclude individuals who are at an increased risk for infections or infectious complications. Exclusion Criteria [36] to [38] exclude individuals who are pregnant, breastfeeding, at risk for becoming pregnant, or at risk for impregnating their partner during the study. Exclusion Criteria [39] to [46] exclude individuals who may not be compliant with study-related procedures or whose participation in the study may introduce bias. The rationale for the enrollment exclusion criteria is as follows: Exclusion Criteria [50] to [53] exclude individuals with concomitant medical conditions that increase the risk for their participation in the study. Exclusion Criteria [54] to [58] exclude individuals who are at an increased risk for infections or infectious complications. Exclusion Criterion [59] excludes individuals who are at risk for becoming pregnant during the study Discontinuations Discontinuation of Patients Interruption of Study Drug In some circumstances it may be necessary to temporarily interrupt treatment as a result of AEs or abnormal laboratory values as described in Table JADV.1 that may have an unclear relationship to study drug. Except in cases of emergency, it is recommended that the investigator consult with Lilly (or its designee) before temporarily interrupting therapy. The investigator must obtain approval from Lilly (or its designee) before restarting study drug that was temporarily discontinued because of an AE or abnormal laboratory value. Study drug must be held in the following situations involving laboratory abnormalities and may be resumed as noted in Table JADV.1.
140 I4V-MC-JADV(c) Clinical Protocol Page 42 Table JADV.1. Criteria for Temporary Discontinuation of Investigational Product Hold Investigational Product if the Following Laboratory Test Results Occur: WBC count <2000 cells/µl ANC <1000 cells/µl Lymphocyte count <500 cells/µl Platelet count <75,000/µL egfr <40 ml/min/1.73 m2 (from serum creatinine) for patients receiving the baricitinib 4 mg QD dose egfr <30 ml/min/1.73 m2 (from serum creatinine) for patients receiving the baricitinib 2 mg QD dose ALT or AST >5 times ULN Investigational Product May Be Resumed after Approval from Lilly (or its Designee) and When: WBC count 2500 cells/µl ANC >2000 cells/µl Lymphocyte count 750 cells/µl Platelet count 100,000/µL egfr 50 ml/min/1.73 m2 egfr 40 ml/min/1.73 m2 ALT and AST return to <2 times ULN and investigational product is not considered to be the cause of enzyme elevation Hemoglobin 10 g/dl Hemoglobin <8 g/dl Abbreviations: ALT = alanine aminotransferase; ANC = absolute neutrophil count; AST = aspartate aminotransferase; egfr = estimated glomerular filtration rate; min = minute; QD = once daily; ULN = upper limit of normal; WBC = white blood cell Discontinuation of Study Drug Any patient who is permanently discontinued from study drug for an AE or abnormal laboratory result should have the AE or abnormal laboratory value reported as an SAE (reported as Other Reason Serious). If any of the criteria listed in Section above recur after study drug is restarted, the patient should be permanently discontinued from study drug. In addition, patients will be permanently discontinued from study drug if they experience any of the criteria listed in Table JADV.2.
141 I4V-MC-JADV(c) Clinical Protocol Page 43 Table JADV.2. Criteria for Permanent Discontinuation of Investigational Product Permanently Discontinue Investigational Product if any of the Following are Observed: ALT or AST >8 times the ULN ALT or AST >5 times the ULN persisting for more than 2 weeks after temporary interruption of investigational product ALT or AST >3 times the ULN and total bilirubin level >2 times the ULN ALT or AST >3 times the ULN with the appearance of fatigue, nausea, vomiting, right upper quadrant pain or tenderness, fever, rash, and/or eosinophilia (>5%) WBC count <1000 cells/µl Absolute neutrophil count <500 cells/µl Lymphocyte count <200 cells/µl Hemoglobin <6.5 g/dl Symptomatic herpes zoster Pregnancy Malignancy Severe infection that, in the opinion of the investigator, merits the investigational product being discontinued Anaphylactic or other serious allergic reaction Symptoms suggestive of a lupus-like syndrome Abbreviations: ALT = alanine aminotransferase; AST = aspartate aminotransferase; ULN = upper limit of normal; WBC = white blood cell Discontinuation from the Study The criteria for enrollment must be followed explicitly. If a patient who does not meet enrollment criteria is inadvertently enrolled, the investigator should contact Lilly or its designee to discuss whether the patient should be discontinued from study drug. If the patient is discontinued from study drug, the patient should remain in the study to provide the follow-up data needed for the primary analysis (Week 12) of the entire intention-to-treat (ITT) population. Patients may choose to withdraw from the study for any reason at any time, and the reason for early withdrawal will be documented. In addition, patients will be discontinued from the study in the following circumstances: Patient enrolls in any other clinical trial involving an investigational product or enrollment in any other type of medical research judged not to be scientifically or medically compatible with this study. Investigator decides that the patient should be withdrawn. If this decision is made because of an intolerable AE or a clinically significant laboratory value, the study drug is to be discontinued and appropriate measures are to be taken. Lilly (or designee) is to be notified immediately. Lilly (or designee) decides that the patient should be withdrawn. Patient is unwilling to continue in the study.
142 I4V-MC-JADV(c) Clinical Protocol Page 44 Patient is noncompliant with protocol procedures. Patient withdraws consent. Investigator or Lilly (or designee), for any reason, stops the study. Patients who discontinue the study early will have Early Termination procedures and follow-up performed as shown in the study schedule (Attachment 1) Discontinuation of Study Sites Study site participation may be discontinued if Lilly, the investigator, or the ethical review board (ERB) of the study site judges it necessary for medical, safety, regulatory, or other reasons consistent with applicable laws, regulations, and good clinical practice (GCP) Discontinuation of the Study The study will be discontinued if Lilly judges it necessary for medical, safety, regulatory, or other reasons consistent with applicable laws, regulations, and GCP.
143 I4V-MC-JADV(c) Clinical Protocol Page Treatment 9.1. Treatments Administered This study involves a comparison of baricitinib 4 mg administered orally QD, placebo administered orally QD, and adalimumab 40 mg administered by SC injection biweekly. Patients will continue to take background MTX therapy during the course of the study. Table JADV.3 shows the treatment regimens. Table JADV.3. Treatment Regimens Treatment Group Baricitinib treatment Placebo comparator Active comparator (adalimumab) Treatments Administered during Part A Baricitinib 4 mgb oral once-daily tablet Adalimumab placebo by SC injection biweekly Baricitinib placebo oral once-daily tablet Adalimumab placebo by SC injection biweekly Baricitinib placebo oral once-daily tablet Adalimumab 40 mg by SC injection biweekly Treatments Administered during Part B (Patients Not Rescued) Baricitinib 4 mgb oral once-daily tablet Adalimumab placebo by SC injection biweekly Baricitinib 4 mgb oral once-daily tablet Adalimumab placebo by SC injection biweekly Baricitinib placebo oral once-daily tablet Adalimumab 40 mg by SC injection biweekly Treatments Administered during Part B (after Rescuea) Baricitinib 4 mgb oral once-daily tablet Baricitinib 4 mgb oral once-daily tablet Baricitinib 4 mgb oral once-daily tablet Abbreviation: SC = subcutaneous. a See Section for details of rescue medication. b The dose for patients with renal impairment, defined as estimated glomerular filtration rate <60 ml/min/1.73 m2, will be 2 mg once daily. The investigator or his/her designee will be responsible for explaining the correct use of both the oral and injectable investigational agent(s) to the patient, verifying that instructions are followed properly, maintaining accurate records of investigational product dispensing and collection, and returning all unused medication to Lilly or its designee at the end of the study. Injections of investigational product should be administered by the site personnel, with instructions to the patient, at Visit 2, and the patient should be supervised while self-administering injections at Visits 4 and 5. In some cases, sites may destroy the material if, during the investigator site selection, the evaluator has verified and documented that the site has appropriate facilities and written procedures to dispose of clinical trial materials. Patients will be instructed to contact the investigator as soon as possible if they have a complaint or problem with the investigational product so that the situation can be assessed.
144 I4V-MC-JADV(c) Clinical Protocol Page Materials and Supplies Lilly (or designee) will provide the following primary study materials: tablets containing 4 mg of baricitinib tablets containing 2 mg of baricitinib tablets containing placebo to match 4 mg baricitinib tablets containing placebo to match 2 mg baricitinib single-use syringes containing 40 mg adalimumab for SC injection single-use syringes containing placebo to match 40 mg adalimumab for SC injection Investigational product will be dispensed to the patient at the principal investigator s study site. Investigational product packaging will contain enough tablets and syringes for the longest possible interval between visits. Investigational product will be labeled according to the country s regulatory requirements. All investigational products will be stored, inventoried, reconciled, and destroyed according to applicable regulations. Investigational products will be supplied by Lilly or its representative in accordance with current Good Manufacturing Practices, and will be supplied with lot numbers, expiry dates, and certificates of analysis (as applicable). Adalimumab and placebo to match adalimumab should be stored at 2C to 8C (35F to 46F) in the original carton to protect from light. Sites will be required to monitor the temperature of the on-site storage conditions of the syringes Method of Assignment to Treatment Patients who meet all criteria for enrollment will be randomized in a 3:3:2 ratio (baricitinib:placebo:adalimumab) to double-blind treatment at Week 0 (Visit 2). Randomization will be stratified by region and joint erosion status (1-2 joint erosions plus seropositivity versus at least 3 joint erosions). Assignment to treatment groups will be determined by a computergenerated random sequence using an interactive voice-response system (IVRS). The IVRS will be used to assign packages of baricitinib/placebo and packages of prefilled syringes of adalimumab/placebo for SC injection containing double-blind investigational product to each patient. Each package of clinical trial material will supply sufficient medication for the number of weeks between visits. Site personnel will confirm that they have located the correct package by entering a confirmation number found on the packages of investigational product into the IVRS before dispensing the package to the patient Rationale for Selection of Doses in the Study Based on efficacy, safety, and PK data from the Phase 2 studies (JADC and JADA), Lilly has selected a single starting dose of 4 mg QD baricitinib for evaluation in this Phase 3 study. Clinical Efficacy Considerations In Study JADC, baricitinib doses of 4, 7, and 10 mg QD were studied and found to produce similar degrees of efficacy across the measured parameters at both the Week 12 and Week 24
145 I4V-MC-JADV(c) Clinical Protocol Page 47 time points. Study JADA included lower doses of baricitinib (1 and 2 mg QD) to find the minimum clinically effective dose and to confirm the flat dose-response curve in the dose range of 4 to 8 mg QD. In Study JADA, a relatively flat dose-response curve was observed in the 4-and 8-mg QD dose groups at Week 12 across almost all measured efficacy parameters. The 1- and 2-mg QD dose groups are biologically active but did not produce adequate response at Week 12 as assessed by ACR50, ACR70, and low disease activity and remission rates when compared to placebo and the 4- and 8-mg dose groups. All 4 doses were associated with reductions in hscrp and ESR values over time. Assessment of the Week 12 magnetic resonance imaging (MRI) data suggests the potential for the 4- and 8-mg QD baricitinib doses to have a favorable impact on reducing structural damage given the observed significant improvement in osteitis. Week 24 data from Study JADA demonstrated persistence of the flat dose response between the 4- and 8-mg QD doses. Over the second 12-week period, continued administration of the 2-mg QD dose did not result in further improvement in the clinically important endpoints of ACR50, ACR70, DAS28-hsCRP 3.2, DAS28-hsCRP <2.6, and CDAI and SDAI remission rates. Week 24 MRI data further support the potential for the 4- and 8-mg QD doses to have a beneficial effect on prevention of structural damage. Clinical Safety Considerations Baricitinib was generally safe and well tolerated in single doses ranging from 1 to 20 mg and in repeat oral doses ranging from 2 to 20 mg. The most commonly reported treatment-emergent AEs (TEAEs) were in the infections and infestations system organ class. The most common alterations in laboratory values involved decreases in hemoglobin, hematocrit, total red blood cells, and white blood cells (neutrophils and other white cell lines), and increases in platelet counts, HDL, LDL, total cholesterol, and triglycerides. Patients with RA experienced a mean increase in ALT/AST compared to placebo, with the mean values remaining within the normal reference range and clinically significant increases in ALT/AST occurring uncommonly. Patients with impaired renal function have increased exposure to baricitinib. Dose Adjustment for Renal Impairment The therapeutic dose range for baricitinib was identified from concentration-efficacy relationships for ACR20 and DAS28-hsCRP as well as concentration-safety relationships identified for absolute neutrophil counts and hemoglobin. The starting dose for this Phase 3 study will be 4 mg QD. As demonstrated in a study of subjects with normal renal function versus those with mild to end-stage renal disease (Study I4V-MC-JADL), baricitinib exposure increases with decreased renal function. Based on PK simulations of baricitinib exposures for the mild and moderate categories of renal function (stratified as egfr 60 to <90 ml/min/1.73 m2 and egfr 30 to <60 ml/min/1.73 m2, respectively) dose adjustment is not required for patients with egfr 60 ml/min/1.73 m2. Patients with egfr <60 ml/min/1.73 m2 will receive a dose of 2 mg QD, which will ensure that exposures are comparable to, but do not exceed, those of the 4-mg QD dose. Lilly intends to enroll patients with egfr no lower than 40 ml/min/1.73 m2 in the Phase 3 trials. Baricitinib will be discontinued if egfr falls below
146 I4V-MC-JADV(c) Clinical Protocol Page ml/min/1.73 m2. Baricitinib administration to patients with severe renal impairment (egfr <30 ml/min/1.73 m2) is not intended. Summary The clinical data known to date suggest a dosing strategy using a 4-mg QD dose as having the potential for providing clinically relevant efficacy response while minimizing potential risks of AEs Selection and Timing of Doses Each patient will receive blinded investigational product (baricitinib 4 mg or baricitinib placebo tablets orally QD and adalimumab 40 mg or adalimumab placebo biweekly by SC injection) beginning on the day of Visit 2 (Week 0). Baricitinib 4-mg oral dosing will continue daily through Visit 15 (Week 52) and biweekly injections of adalimumab 40 mg or adalimumab placebo will continue biweekly through Week 50. Baricitinib placebo oral dosing will continue daily through Visit 11 (Week 24); at Visit 11 (Week 24), all patients randomized to placebo will be reassigned to baricitinib. Injectable investigational product should be administered at approximately the same time of day, as much as possible. For injections not administered on the scheduled day of the week, the missed dose should be administered within 3 days of the scheduled day during study Weeks 1 to 12 and within 5 days of the scheduled day during study Weeks 13 to 52. Dates of subsequent study visits should not be modified according to this delay. As noted in Section 9.1, injections of investigational product should be administered by site personnel at Visit 2, and the patient should be supervised while self-administering injections at Visits 4 and 5. Thereafter, the patient will self-administer SC injections at home biweekly when no study visit is scheduled (ie, Weeks 6, 10, 18, 22, 26, 30, 34, 36, 38, 42, 44, 46, 48, and 50). On weeks when study visits are scheduled (ie, Weeks 8, 12, 14, 16, 20, 24, 28, 32, and 40), the patient should not administer his/her SC injection at home but should bring the study medication to the clinic for administration during the study visit. This will allow sites to administer rescue dosing when required. Oral investigational product should be taken at home at approximately the same time each day, as much as possible, with the exception of the following visits, to allow for the proper timing of PK sample collection and to ensure that efficacy assessments are made at trough PK levels: At Visit 2 (Week 0), patients will take their investigational product in the clinic, to allow for PK samples to be taken at 15 and 60 minutes postdose. For Visits 7 (Week 12), Visit 9 (Week 16), 11 (Week 24), 13 (Week 32), and 15 (Week 52), patients will be asked not to take their investigational product prior to visiting the clinic, to allow efficacy assessments to be made prior to investigational product dosing and a predose PK sample to be taken at some of these selected visits (Attachment 1).
147 I4V-MC-JADV(c) Clinical Protocol Page Special Treatment Considerations In consideration of the disease severity, all patients in Study JADV will be offered rescue therapy starting at Week 16 if they are determined to be nonresponders. At Week 16, patients who are nonresponders and who were randomized to placebo will be rescued with baricitinib. To maintain study integrity through investigational product blinding, patients who were originally randomized to baricitinib will continue to receive baricitinib, with appropriate ongoing assessment of response and use of concomitant medication as needed. Patients who are nonresponders and who were randomized to adalimumab will be reassigned to baricitinib as rescue therapy. Nonresponse at Week 16 is defined as lack of improvement of at least 20% in both TJC and SJC at both Week 14 and Week 16 compared to baseline. At Week 16, the IVRS will assign rescue treatment based on TJC and SJC values. After Week 16, rescue therapy will be offered to patients at the discretion of the investigator based on TJCs and SJCs. Once a patient has been rescued, new NSAIDS, corticosteroids, and/or analgesics may be added or doses of ongoing concomitant NSAIDs, corticosteroids, and/or analgesics may be increased at the discretion of the investigator. A patient may only be rescued once. If a patient continues to meet nonresponse criteria for 4 weeks after rescue or at any time point thereafter, that patient should be discontinued from the study. Nonresponders who are rescued will receive an oral tablet daily, but no biweekly SC injection, for the remainder of the study. Rescue therapy will be administered through Week Continued Access to Investigational Product After the Week 52 study visit, eligible patients may proceed to a separate extension study (Study JADY) lasting for up to 2 years Blinding This is a double-blind, double-dummy study. To preserve the blinding of the study, a minimum number of Lilly personnel will see the randomization table and treatment assignments before the study is complete. Emergency unblinding for AEs may be performed through an IVRS, which may supplement or take the place of emergency codes generated by a computer drug-labeling system. This option may be used ONLY if the patient s well-being requires knowledge of the patient s treatment assignment. All calls resulting in an unblinding event are recorded and reported by the IVRS. The investigator should make every effort to contact the Lilly clinical research physician prior to unblinding a patient s treatment assignment. If a patient s treatment assignment is unblinded, Lilly must be notified immediately by telephone. If an investigator, site personnel performing assessments, or patient is unblinded, the patient must be discontinued from the study. In cases for which there are ethical reasons to have the
148 I4V-MC-JADV(c) Clinical Protocol Page 50 patient remain in the study, the investigator must obtain specific approval from a Lilly clinical research physician for the patient to continue in the study. Processes to maintain blinding during the interim analysis conducted by the DMC are described in Section Concomitant Therapy Patients will be instructed to consult the investigator or other appropriate study personnel at the site before taking any new medications or supplements during the study. As listed below, all patients will continue to take the background MTX therapy at a stable dose throughout the study. Additional drugs are to be avoided during the study unless required to treat an AE or for the treatment of an ongoing medical condition. Investigators should follow local guidelines for the management of lipid disorders. If the need for other concomitant medications arises, discontinuation of the patient from study drug or the study will be at the discretion of the investigator in consultation with Lilly (or designee). Patients will not be permitted to have received a live vaccination up to 12 weeks prior to baseline or at any time during the study (with the exception of herpes zoster vaccination, which is not permitted within 30 days of planned randomization or at any time during the study). Treatment with concomitant DMARDs during the study is permitted as outlined below and in the inclusion/exclusion criteria: Use of concomitant oral MTX for at least 12 weeks with treatment at a stable dose of 7.5 to 25 mg/week (or the equivalent injectable dose) for at least 8 weeks prior to entry into the study is required for study participation. Patients should remain on the same dose of MTX throughout Part A and Part B of the study; however, the MTX dose may be adjusted for safety reasons. Patients may also be on concomitant hydroxychloroquine or sulfasalazine. If receiving hydroxychloroquine (up to 400 mg/day) or sulfasalazine (up to 3000 mg/day), the patient must be receiving a stable dose for at least 8 weeks prior to entry into the study, and the dose must remain stable throughout the study. Concomitant use of NSAIDs is permitted during Part A of the study only if the patient was on a stable dose for at least 6 weeks before planned randomization. Increase of NSAID dose and/or introduction of new NSAIDs are not permitted during Part A of the study unless a patient receives rescue therapy. After rescue, new NSAIDs or increases in doses of ongoing concomitant NSAIDs are permitted. Dose reductions and/or termination of NSAIDs are permitted at any time. During the study, concomitant use of analgesics is permitted. Increase of an analgesic dose and/or introduction of a new analgesic is not permitted during Part A of the study unless a patient receives rescue therapy. After rescue, new analgesics or increases in doses of ongoing concomitant analgesics are permitted. Dose reductions and/or termination of analgesics are permitted at any time.
149 I4V-MC-JADV(c) Clinical Protocol Page 51 Prednisone (or equivalent) at doses up to 10 mg per day is allowed during this study but must be maintained at stable levels from 6 weeks prior to randomization through the treatment phase of the study, unless a patient receives rescue therapy. After rescue, new corticosteroids or increases in doses of ongoing concomitant corticosteroids are permitted. Patients who were not previously on prednisone (or equivalent) prior to randomization should not initiate corticosteroid therapy during the study. Patients should not receive other systemic corticosteroids during the study including intra-muscular or intra-articular corticosteroids. Topical, intranasal, intra-ocular, and inhaled corticosteroids are permitted. If an unforeseen intra-articular glucocorticoid injection is required during the study, it will be noted as a protocol deviation. It is strongly encouraged to avoid hyaluronic acid injections during the course of the study. If required, use should be minimized and discussed with the sponsor. Local standard of care should be followed for concomitant administration of folic acid. The following concomitant medications are prohibited after randomization: Live vaccines, including herpes zoster vaccination. Nonlive seasonal vaccinations and/or emergency vaccination, such as rabies or tetanus vaccinations, are allowed. Any DMARDs, other than stable doses of background DMARDs being used at the time of study entry Any biologic therapy (such as TNF, interleukin-1, IL-6, T-cell-, or B-cell-targeted therapies) for any indication Any interferon therapy (such as Roferon-A, Intron-A, Rebetron, Alferon-N, Peg-Intron, Avonex, Betaseron, Infergen, Actimmune, Pegasys) Any parenteral corticosteroid administered by intramuscular or IV injection In addition to the concomitant medications guidelines outlined above, all patients who have not already received the herpes zoster vaccine at study entry will be encouraged to do so. Lilly will provide this vaccine where available. After receiving the herpes zoster vaccine, all patients must wait at least 30 days before randomization to study treatment. Any joints that had been injected with intraarticular corticosteroids within 6 weeks prior to randomization will be censored from the TJCs and SJCs for the duration of the study. All concomitant medications taken during the study must be recorded in the Concomitant Medication sections of the case report form (CRF) Treatment Compliance Patient compliance with study medication will be assessed at Visits 5 through 7 and Visits 9 through 15 (and, if necessary, at Early Termination) during the treatment period by counting returned tablets and returned syringes. Deviations from the prescribed dosage regimen should be recorded in the CRF. For baricitinib or oral placebo, a patient will be considered significantly noncompliant if he or she misses >20% of the prescribed doses during the study unless the patient s investigational
150 I4V-MC-JADV(c) Clinical Protocol Page 52 product was withheld by the investigator for safety reasons. For adalimumab or injectable placebo, a patient will be considered significantly noncompliant if his/her study medication compliance rate is <80% or if there are any consecutive missed injections, unless the patient s investigational product was withheld by the investigator for safety reasons. Similarly, a patient will be considered significantly noncompliant if he or she is judged by the investigator to have intentionally or repeatedly taken more than the prescribed amount of medication. Patients found to be noncompliant with the investigational product should be assessed to determine the reason for noncompliance and educated and/or managed as deemed appropriate by the investigator to improve compliance.
151 I4V-MC-JADV(c) Clinical Protocol Page Efficacy, Health Outcome Measures, Safety Evaluations, Sample Collection and Testing, and Appropriateness of Measurements Study procedures and their timing (including tolerance limits for timing) are summarized in the study schedule (Attachment 1) Efficacy Measures Primary Efficacy Measure The primary efficacy measure will be the proportion of patients achieving ACR20 at Week 12. ACR20 is defined as at least 20% improvement in the following ACR Core Set values: TJC (68 joint count) SJC (66 joint count) An improvement of 20% in at least 3 of the following 5 assessments: 1. Patient s Assessment of Pain (visual analog scale [VAS]) 2. Patient s Global Assessment of Disease Activity (VAS) 3. Physician s Global Assessment of Disease Activity (VAS) 4. Patient s Assessment of Physical Function as measured by the HAQ-DI 5. Acute phase reactant as measured by hscrp Secondary Efficacy Measures The secondary efficacy measures described below will be collected at the times shown in the study schedule (Attachment 1) Van der Heijde Modified Total Sharp Score Structural progression will be measured using the mtss. During screening, all patients meeting entry criteria will have a single posteroanterior radiographic assessment of the left and right hand/wrist and a single dorsoplantar radiographic image taken of the left and right foot. Alternatively, images taken within 4 weeks prior to baseline may be used. Images taken at screening (or within 4 weeks of screening) will be reviewed centrally by qualified readers for evidence of erosive bony change(s). These initial radiographs will serve as the baseline radiographs for comparison throughout the study. If a patient undergoes rescreening, radiographic images acquired as part of initial screening and within 10 weeks of randomization may be used. Additional radiographs (ie, a single posteroanterior view of each hand and a single dorsoplantar view of each foot) will be obtained at Week 16, Week 24 (the primary evaluation), and Week 52 (for the purposes of demonstrating durability of structural preservation as requested by regulatory authorities), as shown in the study schedule (Attachment 1). For patients who discontinue prematurely from the study, x-rays will be performed at the early termination visit only if the previous x-rays were taken more than 12 weeks earlier.
152 I4V-MC-JADV(c) Clinical Protocol Page 54 X-rays of the hands/wrists and feet will be scored using the structural progression as measured using a modification of the mtss (van der Heijde 2000). This methodology quantifies the extent of bone erosions and joint space narrowing for 44 and 42 joints, respectively, with higher scores representing greater damage. X-rays will also be assessed for joint space narrowing and bone erosions. Quality control will be performed for proper imaging examination technique prior to allocation of the images to the central readers. The independent read of x-ray images will be performed by 2 primary readers, and 1 adjudicator, when necessary, based on predefined criteria. The 2 primary readers will each read 100% of the study patients. All time points will be displayed in random order for a given patient. The reader will have no knowledge of the true chronologic order, patient identity, or treatment group. To ensure a consistent read by the independent readers, an inter-/intra-reader variability assessment will be included in the central imaging core laboratory independent read. Repeat x-rays will be requested for all images obtained that are of poor quality as defined in the Image Acquisition Guidelines Disease Activity Score-Erythrocyte Sedimentation Rate and Disease Activity Score High Sensitivity C-Reactive Protein The DAS28 is a measure of disease activity in 28 joints that consists of a composite numeric score of the following variables: TJC, SJC, hscrp or ESR, and Patient s Global Assessment of Disease Activity (Vander Cruyssen et al. 2005). The 28 joints to be examined and assessed as tender or not tender for TJC and as swollen or not swollen for SJC include 14 joints on each side of the patient s body: the 2 shoulders, the 2 elbows, the 2 wrists, the 10 metacarpophalangeal joints, the 2 interphalangeal joints of the thumb, the 8 proximal interphalangeal joints, and the 2 knees (Smolen et al. 1995) ACR50 and ACR70 Indices ACR50 and ACR70 responses are secondary efficacy measures that are calculated as improvements of at least 50% and of at least 70%, respectively, in the ACR Core Set values identified in Section Hybrid American College of Rheumatology Response Measure The hybrid ACR (bounded) response measure will be obtained as described by the ACR Committee to Reevaluate Improvement Criteria (2007); see Table JADV.4.
153 I4V-MC-JADV(c) Clinical Protocol Page 55 Table JADV.4. Scoring Method for the Hybrid American College of Rheumatology Response Measure Mean % Change in Core Set Measuresa ACR Status <20 20, <50 50, <70 70 Not ACR20 Mean % change ACR20 but not ACR50 20 Mean % change ACR50 but not ACR Mean % change ACR Mean % change Abbreviations: ACR = American College of Rheumatology; ACR20 = 20% improvement in ACR criteria; ACR50 = 50% improvement in ACR criteria; ACR70 = 70% improvement in ACR criteria. a 1) Calculate the average percentage change in core set measures. For each core set measure, subtract score after treatment from baseline score and determine percentage improvement in each measure. Next, if a core set measure worsened by >100%, limit that percentage change to 100% (set equal to -100% bound). Then, average the percentage changes for all core set measures. 2) Determine whether the patient has achieved ACR20, ACR50, or ACR70. 3) Using the table above, obtain the hybrid ACR response measure. To use the table, take the ACR20, ACR50, or ACR70 status of the patient (left column) and the mean percentage improvement in core set items; the hybrid ACR score is where they intersect in the table ACR/EULAR Rheumatoid Arthritis Remission Two ACR/EULAR definitions of RA remission will be evaluated, a Boolean-based definition and an index-based definition (Felson et al. 2011). Boolean-based Definition of Remission: All 4 criteria below must be met: o TJC28 1 o SJC28 1 o hscrp 1 mg/dl (10 mg/l) o patient global DAS on VAS (0 to 10.0) 1 Index-based Definition of Remission (see Section ): o SDAI Simplified Disease Activity Index The SDAI is a tool for measurement of disease activity in RA that integrates measures of physical examination, acute phase response, patient self-assessment, and evaluator assessment. The SDAI is calculated by adding together scores from the following assessments: number of swollen joints (0 to 28), number of tender joints (0 to 28), CRP in mg/dl (0.1 to 10.0), patient global DAS on VAS (0 to 10.0), and evaluator global health score on VAS (0 to 10.0) (Aletaha and Smolen 2005)
154 I4V-MC-JADV(c) Clinical Protocol Page 56 Disease remission according to ACR/EULAR index-based definition of remission is defined as an SDAI score of 3.3 (Felson et al. 2011) Clinical Disease Activity Index The CDAI is similar to the SDAI, but it allows for immediate scoring because it does not use a laboratory result. The CDAI is calculated by adding together scores from the following assessments: number of swollen joints (0 to 28), number of tender joints (0 to 28), patient global DAS on VAS (0 to 10.0), and evaluator global health score on VAS (0 to 10.0) (Aletaha and Smolen 2005) Remission is defined as a CDAI score of 2.8 (Felson et al. 2011) European League Against Rheumatism Responder Index Assessments of patients with RA by EULAR response criteria will be used to categorize patients as nonresponders, moderate responders, good responders, or responders (moderate + good responders) according to Table JADV.5 (van Gestel et al. 1998). Table JADV.5. Postbaseline Level of DAS28 DAS Categorization of Patients as Nonresponders, Moderate Responders, or Good Responders Improvement Since Baseline in DAS28 > and > Good response 3.2 < DAS Moderate response DAS28 >5.1 No response Abbreviation: DAS28 = Disease Activity Score modified to include the 28 diarthroidal joint count Health Outcome Measures The health outcome measures listed below will be administered. Country-specific translations of each measure will be available in this study. Morning Joint Stiffness Severity Numeric Rating Scale (NRS): The Morning Joint Stiffness NRS is a patient-administered, single-item, 11-point horizontal scale anchored at 0 and 10, with 0 representing no joint stiffness and 10 representing joint stiffness as bad as you can imagine. Patients rate their morning joint stiffness each day (using an electronic handheld diary) by selecting the 1 number that describes their overall level of joint stiffness at the time they woke up. Duration of morning joint stiffness: o Electronic patient diary: The duration of morning joint stiffness is a patient-administered item that allows for the patients to enter the length of
155 I4V-MC-JADV(c) Clinical Protocol Page 57 time in minutes (using 15-minute increments) that their morning joint stiffness lasted each day (using an electronic handheld diary). o Electronic PRO (tablet): The duration of morning joint stiffness is a patientadministered item that allows for the patients to enter the length of time in minutes (using 15-minute increments) that their morning joint stiffness lasted on the day prior to that visit (using an electronic patient-reported outcomes [epro] tablet). As the protocol will be approved prior to readiness of some tablet measures, it will not be considered a protocol violation that these measures are not collected until tablet readiness. Recurrence of joint stiffness: The recurrence of joint stiffness throughout the day is a patient-administered single-item question assessed each day (using an electronic handheld diary) that asks Did you have any joint stiffness throughout today, other than when you woke up? (Yes/No). Tiredness Severity Numeric Rating Scale (Worst Tiredness NRS): o Electronic patient diary: The Worst Tiredness NRS is a patient-administered, single-item, 11-point horizontal scale anchored at 0 and 10, with 0 representing no tiredness and 10 representing as bad as you can imagine. Patients rate their tiredness each day (using an electronic handheld diary) by selecting the one number that describes their worst level of tiredness. o Electronic PRO (tablet): The Worst Tiredness NRS is a patient-administered, single-item, 11-point horizontal scale anchored at 0 and 10, with 0 representing no tiredness and 10 representing as bad as you can imagine. Patients rate their tiredness by selecting the one number that describes their worst level of tiredness during the past 24 hours. Severity of joint pain Numeric Rating Scale (Worst Pain NRS): o Electronic patient diary: The Worst Pain NRS is a patient-administered, single-item, 11-point horizontal scale anchored at 0 and 10, with 0 representing no pain and 10 representing joint pain as bad as you can imagine. Patients rate their joint pain each day (using an electronic handheld diary) by selecting the 1 number that describes their worst level of joint pain. o Electronic PRO (tablet): The Worst Pain NRS is a patient-administered, single-item, 11-point horizontal scale anchored at 0 and 10, with 0 representing no pain and 10 representing joint pain as bad as you can imagine. Patients rate their joint pain by selecting the one number that describes their worst level of joint pain during the past 24 hours. HAQ-DI: HAQ-DI is a patient-reported questionnaire that is commonly used in RA to measure disease-associated disability (assessment of physical function). It consists of 24 questions referring to 8 domains: dressing/grooming, arising, eating, walking, hygiene, reach, grip, and activities (Fries et al. 1980, 1982; Ramey et al. 1996). The disability section of the questionnaire scores the patient s self-perception on the degree of difficulty (0 = without any difficulty, 1 = with some difficulty, 2 = with much difficulty, and 3 = unable to do) when dressing and grooming, arising, eating, walking, hygiene, reach, grip, and performing other daily activities. The reported use
156 I4V-MC-JADV(c) Clinical Protocol Page 58 of special aids or devices and/or the need for assistance of another person to perform these activities is also assessed. The scores for each of the functional domains will be averaged to calculate the functional disability index. FACIT-F scale: The FACIT-F scale (Cella and Webster 1997) is a brief, 13-item, symptom-specific questionnaire that specifically assesses the self-reported severity of fatigue and its impact upon daily activities and functioning. The FACIT-F uses 0 ( not at all ) to 4 ( very much ) numeric rating scales to assess fatigue and its impact in the past 7 days. Scores range from 0 to 52 with higher scores indicating less fatigue. SF-36v2 Acute: The SF-36v2 Acute measure is a subjective, generic, health-related quality of life instrument that is patient-reported and consists of 36 questions covering 8 health domains: physical functioning, bodily pain, role limitations due to physical problems, role limitations due to emotional problems, general health perceptions, mental health, social function, and vitality. Each domain is scored by summing the individual items and transforming the scores into a 0 to 100 scale with higher scores indicating better health status or functioning. In addition, 2 summary scores, the PCS (physical component score) and the MCS (mental component score), will be evaluated based on the 8 SF-36v2 Acute domains (Brazier et al. 1992; Ware and Sherbourne 1992). WPAI-RA: The WPAI-RA questionnaire was developed to measure the effect of general health and symptom severity on work productivity and regular activities. It contains 6 items covering overall work productivity (health), overall work productivity (symptom), impairment of regular activities (health), and impairment of regular activities (symptom). Scores are calculated as impairment percentages (Reilly et al. 1993). EQ-5D-5L: The EQ-5D-5L is a standardized measure of health status that provides a simple, generic measure of health for clinical and economic appraisal. The EQ-5D- 5L consists of 2 components: a descriptive system of the respondent s health and a rating of his or her current health state using a 0- to 100-mm VAS. The descriptive system comprises the following 5 dimensions: mobility, self-care, usual activities, pain/discomfort, and anxiety/depression. Each dimension has 5 levels: no problems, slight problems, moderate problems, severe problems, and extreme problems. The respondent is asked to indicate his/her health state by ticking (or placing a cross) in the box associated with the most appropriate statement in each of the 5 dimensions. It should be noted that the numerals 1 to 5 have no arithmetic properties and should not be used as a cardinal score. The VAS records the respondent s self-rated health on a vertical VAS in which the endpoints are labeled best imaginable health state and worst imaginable health state. This information can be used as a quantitative measure of health outcome. The EQ-5D-5L health states, defined by the EQ-5D-5L descriptive system, may be converted into a single summary index by applying a formula that essentially attaches a value (also called weights) to each of the levels in each dimension (Brooks 1996; EuroQol Group 2011 [WWW]; Herdman et al. 2011).
157 I4V-MC-JADV(c) Clinical Protocol Page 59 Quick Inventory of Depressive Symptomatology Self-Rated-16 (QIDS-SR 16 ): The QIDS-SR 16 is a 16-item, self-report instrument intended to assess the existence and severity of symptoms of depression as listed in the American Psychiatric Association's Diagnostic and Statistical Manual of Mental Disorders, 4th Edition (APA 1994). Patients are asked to consider each statement as it relates to the way they have felt for the past 7 days. There is a 4-point scale for each item ranging from 0 to 3. The 16 items corresponding to 9 depression domains are summed to give a single score ranging from 0 to 27. Additional information and the QIDS-SR 16 questions may be found on the University of Pittsburgh Epidemiology Data Center web site (Inventory of Depressive Symptomatology/Quick Inventory of Depressive Symptomatology [WWW]) (Rush et al. 2003; Trivedi et al. 2004). Healthcare resource utilization: The healthcare resource utilization data will be collected by site staff regarding the number of visits to medical care providers such as general practitioners, specialists, physical or occupational therapists, and other nonphysical care providers for services outside of the clinical study; emergency room admissions; hospital admissions; and concomitant medications related to the treatment of RA. These data will be collected to support economic evaluations of treatment Safety Evaluations Investigators will be responsible for monitoring the safety of patients who enter this study and for alerting Lilly or its designee to any event that seems unusual, even if this event may be considered an unanticipated benefit to the patient. The investigator will be responsible for the appropriate medical care of patients during the study. The investigator remains responsible for following, through an appropriate health care option, AEs that are serious or that cause the patient to discontinue before completing the study. The patient should be followed until the event is resolved or explained. Frequency of follow-up evaluation will be left to the discretion of the investigator Adverse Events Lilly has standards for reporting AEs that are to be followed regardless of applicable regulatory requirements that may be less stringent. Lack of drug effect is not an AE in clinical studies because the purpose of the clinical study is to establish drug effect. Cases of pregnancy that occur during maternal or paternal exposures to investigational product or drug delivery system should be reported. Data on fetal outcome and breastfeeding are collected for regulatory reporting and drug safety evaluation. Study site personnel will record the occurrence and nature of each patient s preexisting conditions, including clinically significant signs and symptoms of the disease under treatment in the study.
158 I4V-MC-JADV(c) Clinical Protocol Page 60 After the ICF is signed, site personnel will record any change in the condition(s) and the occurrence and nature of any AEs. All AEs related to protocol procedures will be reported to Lilly or its designee. In addition, all AEs occurring after the patient receives the first dose of investigational product must be reported to Lilly or its designee via electronic data entry. Any clinically significant findings from ECGs, laboratory tests, vital sign measurements, or other procedures that result in a diagnosis should be reported to Lilly or its designee. Investigators will be instructed to report to Lilly or its designee their assessment of the potential relatedness of each AE to protocol procedure, studied disease state, investigational product, and/or drug delivery system via electronic data entry. Study site personnel must alert Lilly or its designee within 24 hours of the investigator unblinding a patient s treatment group assignment for any reason. If a patient s dosage is reduced or treatment is discontinued as a result of an AE, study site personnel must clearly report to Lilly or its designee via electronic data entry the circumstances and data leading to any such dosage reduction or discontinuation of treatment Adverse Events of Special Interest Adverse events of special interest (AESIs) will include: severe or opportunistic infections myelosuppressive events of anemia, leukopenia, neutropenia, lymphopenia, and thrombocytopenia thrombocytosis elevations in ALT or AST (>3 times ULN) with total bilirubin (>2 times ULN) Patients with these laboratory value specified events will be identified using the same criteria for the interruption of investigational product with the exception of anemia, which will be identified using the same criteria for the discontinuation of investigational product, and thrombocytosis, which will be defined as a platelet count >600,000/μL Serious Adverse Events SAE collection will begin after the patient has signed informed consent and has received investigational product. If a patient experiences an SAE after signing informed consent but prior to receiving investigational product, the event will NOT be collected unless the investigator feels the event may have been caused by a protocol procedure. Previously planned (prior to signing the ICF) surgeries should not be reported as SAEs unless the underlying medical condition has worsened during the course of the study. Study site personnel must alert Lilly or its designee of any serious adverse event (SAE) within 24 hours of investigator awareness of the event via a sponsor-approved method. Alerts issued via telephone are to be immediately followed with official notification on study-specific SAE forms. An SAE is any AE from this study that results in one of the following outcomes:
159 I4V-MC-JADV(c) Clinical Protocol Page 61 death initial or prolonged inpatient hospitalization a life-threatening experience (that is, immediate risk of dying) persistent or significant disability/incapacity congenital anomaly/birth defect considered significant by the investigator for any other reason In addition, any patient who is permanently discontinued from study drug for an AE or abnormal laboratory result should have the AE or abnormal laboratory value reported as an SAE (reported as Other Reason Serious). Important medical events that may not result in death, be life-threatening, or require hospitalization may be considered serious adverse drug events when, based on appropriate medical judgment, they may jeopardize the patient and may require medical or surgical intervention to prevent one of the outcomes listed in this definition. SAEs occurring after a patient has taken the last dose of investigational product will be collected in the pharmacovigilance system and the clinical data collection database for 28 days after the last dose of investigational product, regardless of the investigator s opinion of causation. Thereafter, SAEs are not required to be reported unless the investigator feels the events were related to investigational product, drug delivery system, or protocol procedure. Information on SAEs expected in the study population independent of drug exposure and that will be assessed by the sponsor in aggregate periodically during the course of the trial may be found in the IB. In the RA population, the occurrence of malignancies, major cerebrocardiovascular events (including death, myocardial infarction, hospitalization for unstable angina, hospitalization for heart failure, serious arrhythmia, resuscitated sudden death, cardiogenic shock due to myocardial infarction, coronary revascularization procedure; neurologic-stroke; and peripheral vascular events), and serious infections is reasonably anticipated because of the age of the population, comorbid conditions, disease state, and concomitant medications Suspected Unexpected Serious Adverse Reactions Suspected unexpected serious adverse reactions (SUSARs) are serious events that are not listed in the IB and that the investigator identifies as related to investigational product or procedure. Lilly has procedures, which are consistent with global regulations and the associated detailed guidances that will be followed for the recording and expedited reporting of SUSARs Other Safety Measures Measures Evaluated During Screening Period Physical examination. One complete physical examination (excluding pelvic and rectal examinations) will be performed at screening. This examination will determine whether the patient meets the criteria required to participate in the study and will also serve as a monitor for preexisting conditions and as a baseline for TEAE assessment. Body weight and height will also be recorded.
160 I4V-MC-JADV(c) Clinical Protocol Page 62 Electrocardiograms. A single 12-lead ECG will be obtained at screening to determine whether the patient meets entry criteria for the study. ECGs will be locally (machine) read and interpreted by a qualified physician (the investigator or qualified designee) at the site. Chest x-ray and tuberculosis testing. A posterior-anterior view chest x-ray will be obtained locally, unless results from a chest x-ray obtained within 6 months prior to the study are available. The chest x-ray will be reviewed by the investigator or his/her designee to exclude patients with active TB infection. In addition, patients will be tested at screening and entry for evidence of active or latent TB as described in exclusion criteria, Section Measures Evaluated During Screening and Treatment Periods Vital signs and physical characteristics. Vital signs (sitting blood pressure and heart rate) will be measured at times indicated in the study schedule (Attachment 1). Subjects should be seated and relaxed with both feet on the floor for at least 5 minutes prior to taking measurements. Three replicate blood pressure readings should be made at each time point at approximately 30- to 60-second intervals. A single pulse measurement should be taken simultaneously with at least one of the blood pressure readings. Blood pressure and pulse measurements should be made using either automated or manual equipment. If measurements are machine averaged, the average blood pressure reading should be recorded on the CRF. If measurements are manual or the machine does not provide an average reading, then each individual reading should be recorded on the CRF. Measurements should be made before any scheduled blood draws. Any clinically significant findings that result in a diagnosis should be captured on the CRF and reported as an AE. Additional measurements of vital signs may be performed at the discretion of the investigator. Physical characteristics including weight and waist circumference will be measured at times indicated in the study schedule (Attachment 1). Standard laboratory tests. Hematology, clinical chemistry, urinalysis, lipid profile, egfr, iron studies, hscrp, and ESR will be measured at times indicated in the study schedule (Attachment 1) Safety Monitoring The Lilly clinical research physician will monitor safety data throughout the course of the study. Lilly will review SAEs within time frames mandated by company procedures. The Lilly clinical research physician will, as is appropriate, consult with the functionally independent Global Patient Safety therapeutic area physician or clinical scientist and periodically review trends in safety data and laboratory analytes. Any concerning trends in frequency or severity noted by an investigator and/or Lilly (or designee) may require further evaluation. All deaths and SAE reports will be reviewed in a blinded manner by Lilly during the clinical trial. These reports will be reviewed to ensure completeness and accuracy but will not be unblinded to Lilly during the clinical trial. If a death or clinical AE is deemed serious, unexpected, and possibly related to investigational product, only Lilly Global Patient Safety will be unblinded for regulatory reporting and safety monitoring purposes. These measures will
161 I4V-MC-JADV(c) Clinical Protocol Page 63 preserve the integrity of the data collected during this trial and minimize any potential for bias while providing for appropriate safety monitoring. Liver function monitoring will occur frequently throughout the study. If a study patient experiences elevated ALT or AST 3 ULN or elevated total bilirubin 2 ULN, clinical and laboratory monitoring should be initiated by the investigator. Details for hepatic monitoring depend on the severity and persistence of observed laboratory test abnormalities. To ensure patient safety and comply with regulatory guidance, the investigator is to consult with the Lilly designated medical monitor regarding collection of specific recommended clinical information and follow-up laboratory tests (see Attachment 3). Investigators will monitor vital signs and carefully review findings that may be associated with cardiovascular events. AE reports and vital signs will be collected at each study visit. The cardiovascular monitoring plan includes, in addition, the following: regular monitoring of lipid levels adjudication of major adverse cardiovascular events (all deaths and nonfatal myocardial infarctions, hospitalization for unstable angina, hospitalization for heart failure, coronary interventions [such as coronary artery bypass graft or percutaneous coronary intervention], stroke, and transient ischemic attack) by a Cardiovascular Safety Committee at regular intervals estimation of relative rates of occurrence of select AEs using observations obtained from a matched, observational, comparator cohort of RA patients using existing RA registry/registries. A DMC (an advisory group for this study formed to protect the patients; refer to the Interim Analysis in Section ) will also oversee the conduct of all of the baricitinib Phase 3 clinical trials in patients with RA. In the event that safety monitoring uncovers an issue that needs to be addressed by unblinding at the group level, only members of the DMC will be able to conduct additional analyses of the safety data Complaint Handling Lilly collects product complaints on investigational products and drug delivery systems used in clinical studies to ensure the safety of study participants, monitor quality, and facilitate process and product improvements. Complaints related to unblinded concomitant drugs or drug delivery systems are reported directly to the manufacturers of those drugs/devices in accordance with the package inserts. For blinded studies, all product complaints associated with material packaged, labeled, and released by Lilly or its delegate will be reported. The investigator or his/her designee is responsible for handling the following aspects of the product complaint process in accordance with the instructions provided for this study: recording a complete description of the product complaint reported and any associated AEs using the study-specific complaint forms provided for this purpose faxing the completed product complaint form within 24 hours to Lilly or its designee
162 I4V-MC-JADV(c) Clinical Protocol Page 64 If the investigator is asked to return the product for investigation, he/she will return a copy of the product complaint form with the product Sample Collection and Testing Attachment 1 lists the schedule for sample collections in this study, and Attachment 2 lists the specific tests that will be performed for this study Samples for Standard Laboratory Testing Standard laboratory tests, including chemistry, hematology, and urinalysis panels, will be performed. Specifically, hscrp results will be blinded after Visit 2. Serum and/or urine pregnancy tests will be performed at each visit as described in the study schedule (Attachment 1). Blood and urine samples will be collected for other laboratory tests at the times specified in the study schedule (Attachment 1). Blood will be collected by venipuncture. Attachment 2 lists the specific tests that will be performed for this study. Routine and other clinical laboratory tests will be analyzed by a central laboratory selected by Lilly. Investigators must document their review of each laboratory safety report. Samples collected for specified laboratory tests will be destroyed within 60 days of receipt of confirmed test results. Tests are run and confirmed promptly whenever scientifically appropriate. When scientific circumstances warrant, however, it is acceptable to retain samples to batch the tests run or to retain the samples until the end of the study to confirm that the results are valid. Certain samples may be retained for a longer period, if necessary, to comply with applicable laws, regulations, or laboratory certification standards Samples for Drug Concentration Measurements Pharmacokinetics/Pharmacodynamics A single venous blood sample will be drawn at the times indicated in the study schedule (Attachment 1). These blood samples will be used to determine the plasma concentrations of baricitinib. Concentrations of baricitinib in human plasma will be determined by a validated liquid chromatography tandem mass spectrometry (LC/MS/MS) method. Blood samples that will be used for other laboratory assessments (chemistry, hematology, etc) should be drawn at approximately the same time as the samples used for plasma concentrations of baricitinib. The timing will be as follows: At Visit 2 (Week 0), patients will take their oral investigational product in the clinic, and PK samples will be drawn at 15 minutes and 1 hour postdose. At Visit 5 (Week 4), patients will be asked to take their oral investigational product at home prior to visiting the clinic. The clinic visit should be scheduled so that the sample taken during this visit is 2 to 4 hours after the oral dose taken at home.
163 I4V-MC-JADV(c) Clinical Protocol Page 65 At Visit 6 (Week 8), patients will be asked to take their oral investigational product at home prior to visiting the clinic. The clinic visit should be scheduled so that the sample taken during this visit is 4 to 6 hours after the oral dose taken at home. For Visits 7 (Week 12), 11 (Week 24), and 13 (Week 32), patients will be asked not to take their oral investigational product prior to visiting the clinic, and a sample will be taken at any time predose on the day of the clinic visits. If the patient has taken his/her oral dose prior to the visit, the sample may be drawn anytime postdose, and the inability to collect a predose sample will not be considered a protocol violation. For the early termination visit, a sample may be drawn anytime if the last dose of oral investigational drug was taken within the last 48 hours. For visits where PK samples will be taken predose, the actual date and exact timing (24-hour clock) of sample collection and the date and time of the last 2 doses prior to the sample being drawn should be recorded. For visits where PK samples are taken postdose, the actual date and exact timing (24-hour clock) of sample collection and the date and time of the last 3 doses prior to the sample being drawn should be recorded. This sampling schedule should be followed as closely as possible; however, failure to take PK samples at these specified times will not be considered a protocol violation. If the patient fails to follow the directions for a particular visit, the sample should still be collected at that visit, and the date and exact timing (24-hour clock) of sample collection and the date and time of the last 2 doses prior to the sample being drawn should be recorded. In the event of an SAE, up to 2 additional blood samples may be taken at the investigator s discretion. Only samples from patients receiving baricitinib will be assayed; samples from patients receiving placebo or adalimumab will not be assayed. PK samples will be kept in storage at a laboratory facility designated by the sponsor. PK samples may also be assayed for additional exploratory analyses. PK results will not be provided to investigative sites until the completion of the study or to the blinded study team until the study has been unblinded. Bioanalytical samples collected to measure investigational product concentration will be retained for a maximum of 1 year following last patient visit for the study Exploratory Stored Samples Pharmacogenetics There is growing evidence that genetic variation may impact a patient s response to therapy. Variable response to therapy may be due to genetic determinants that affect drug absorption, distribution, metabolism, and excretion; the mechanism of action of the drug; the disease etiology; and/or the molecular subtype of the disease being treated. Therefore, where local regulations allow, blood samples will be collected for pharmacogenetic analysis, as noted in the study schedule (Attachment 1).
164 I4V-MC-JADV(c) Clinical Protocol Page 66 Samples will be stored, and analysis may be performed on genetic variants thought to play a role in active RA including, but not limited to, the cluster of immune response genes on chromosome 6, such as HLA-DRB1, HLA-DQA1, HLA-DPA1, HLA-DPB1, TNF-α, and TNF receptors; RANK-Ligand; the JAK signaling pathways; MTX metabolizing genes such as methylene tetrahydrofolate reductase and other folate reductases; PTPN22 and related signaling molecules; IL-6 pathway members; and the STAT signaling family members, including STAT4, to evaluate their association with observed response to baricitinib. In the event of an unexpected AE or the observation of unusual response, the samples may be genotyped and analysis may be performed to evaluate a genetic association with response to baricitinib. These investigations may be limited to a focused candidate gene study or, if appropriate, genome-wide association studies may be performed to identify regions of the genome associated with the variability observed in drug response. Samples will only be used for investigations related to disease and drug or class of drugs under study in the context of this clinical program. They will not be used for broad exploratory unspecified disease or population genetic analysis. The sample will be identified by the patient number (coded) and stored for up to a maximum of 15 years after the last patient visit for the study at a facility selected by the sponsor. The sample and any data generated from it can be linked back to the patient only by investigator site personnel. The duration allows the Sponsor to respond to regulatory requests related to the study drug Nonpharmacogenetic/Biomarker Stored Samples Collection of samples for nonpharmacogenetic biomarker research is a required part of this study. Serum, plasma, whole blood RNA, and urine samples will be collected at the times specified in the study schedule (Attachment 1). Samples may be used for research on the drug target, disease process, pathways associated with disease state, mechanism of action of baricitinib, and/or research method or in validating diagnostic tools or assay(s) related to RA. Samples will be identified by the patient number (coded) and stored for up for a maximum of 15 years after the last patient visit for the study at a facility selected by the sponsor Appropriateness of Measurements All of the clinical and safety assessments made in this study are standard, widely used, and generally recognized as reliable, accurate, and relevant.
165 I4V-MC-JADV(c) Clinical Protocol Page Data Quality Assurance To ensure accurate, complete, and reliable data, Lilly or its representatives will do the following: provide instructional material to the study sites, as appropriate sponsor a start-up training session to instruct the investigators and study coordinators. This session will give instruction on the protocol, the completion of the CRFs, and study procedures make periodic visits to the study site be available for consultation and stay in contact with the study site personnel by mail, telephone, and/or fax review and evaluate CRF data and use standard computer edits to detect errors in data collection In addition, Lilly or its representatives may periodically check a sample of the patient data recorded against source documents at the study site. The study may be audited by Lilly or its representatives and/or regulatory agencies at any time. Investigators will be given notice before an audit occurs. To ensure the safety of participants in the study and to ensure accurate, complete, and reliable data, the investigator will keep records of laboratory tests, clinical notes, and patient medical records in the patient files as original source documents for the study. If requested, the investigator will provide the sponsor, applicable regulatory agencies, and applicable ERBs with direct access to original source documents Data Capture System An electronic data capture system will be used in this study. The site will maintain a separate source for the data entered by the site into the sponsor-provided electronic data capture system. Electronic patient-reported outcome (epro) measures or other data reported directly by the subject will be entered into an epro instrument at the time that the information is obtained. Electronic physician-reported outcome measures will be directly entered by the physician or designee (Physician global assessment of disease activity) or by the blinded joint assessor (TJC/SJC) into the electronic site tablet. In these instances for which there is no prior written or electronic source data at the site, the epro instrument record will serve as the source. The epro records will be stored by a third party, and investigator sites will have continuous access to the source documents during the study and will receive an archival copy at the end of the study for retention. Any data for which the epro instrument record will serve to collect source data will be identified and documented by each site in that site s study file. CRF data will be encoded and stored in a clinical trial database. Data managed by a central vendor, such as laboratory test data or x-ray data, will be stored electronically in the central
166 I4V-MC-JADV(c) Clinical Protocol Page 68 vendor s database system. Data will subsequently be transferred from the central vendor to the Lilly generic laboratories system. Data from complaint forms submitted to Lilly will be encoded and stored in the global product complaint management system.
167 I4V-MC-JADV(c) Clinical Protocol Page Sample Size and Statistical Methods Determination of Sample Size Approximately 1280 patients (480 in the baricitinib group, 480 in the placebo group, and 320 in the adalimumab group) will provide: >95% power to detect a difference between the baricitinib and placebo treatment groups in ACR20 response rate (60% versus 35%) at Week 12 based on a 2-sided chi-square test at a significance level of 0.05 approximately 94% power to detect a difference in mtss between the baricitinib and placebo treatment groups for an effect size of 0.25 (ie, difference in means divided by common standard deviation [SD]), based on a 2-sided t-test at a significance level of This assumes 90% of randomized patients will have x-ray data available for the mtss analysis at Week 24. Based on a recent publication by van der Heijde et al (2013), the effect size from the comparison of tofacitinib 10 mg BID versus placebo in mtss at Month 6 was approximately If the effect size in Study JADV was at least 0.21, then power of the mtss analysis is at least 85%. approximately 93% power for the noninferiority analysis of ACR20 response rate at Week 12 between the baricitinib and adalimumab treatment groups, where a response rate of 60% for baricitinib and adalimumab and a 1-sided significance level of are assumed. This power calculation adopted a fixed noninferiority margin of 12%. There is growing support for this margin in recent clinical trials conducted in the MTX-IR patient population (Schiff et al. 2012). The above sample size and power estimates were obtained from nquery Advisor Statistical and Analytical Plans General Considerations Statistical analysis of this study will be the responsibility of Eli Lilly and Company, although implementation of the statistical analysis plan (SAP) may be delegated to a third party. A detailed SAP describing the statistical methodologies will be developed by Lilly or its designee. All statistical tests of treatment effects, besides noninferiority tests specified in Section , will be performed at a 2-sided significance level of 0.05, unless otherwise stated. Treatment comparisons of discrete efficacy variables will be made using a logistic regression analysis with region, joint erosion status (1-2 joint erosions plus seropositivity vs at least 3 joint erosions), and treatment group in the model. The percentages, difference in percentages, and 100*(1-alpha)% confidence interval (CI) of the difference in percentages will be reported. Treatment-by-region interaction will be added to the logistic regression model of the primary and key secondary categorical variables as a sensitivity analysis. If this interaction is significant at a 2-sided 0.1 level, further inspection will be used to assess whether the interaction is quantitative (ie, the treatment effect is consistent in direction but not size of effect) or qualitative (the treatment is beneficial for some but not all regions).
168 I4V-MC-JADV(c) Clinical Protocol Page 70 Treatment comparisons of continuous efficacy variables will be made using analysis of covariance (ANCOVA) with region, joint erosion status, treatment group, and baseline value in the model. Type III sums of squares for the least-squares means will be used for the statistical comparison; 100*(1-alpha)% CI will also be reported. Sensitivity analysis will be performed for key secondary continuous variables by adding the treatment-by-baseline interaction to the ANCOVA model. If this interaction is significant at a 2-sided 0.1 level, then additional analyses will be performed. Treatment-by-region interaction will also be added to the model for sensitivity purposes. When a restricted maximum likelihood-based mixed model for repeated measures (MMRM) is used, the model will include treatment, region, joint erosion status, visit, treatment-by-visit-interaction as fixed categorical effects; baseline score and baseline score-byvisit-interaction as fixed continuous effects. An unstructured (co)variance structure will be used to model the between- and within-patient errors. If this analysis fails to converge, other structures will be tested. The Kenward-Roger method will be used to estimate the degrees of freedom. Type III sums of squares for the least-squares means will be used for the statistical comparison; CI will also be reported. Treatment group comparisons versus placebo at Week 12 and other visits will be tested. Further details on the use of MMRM will be described in the SAP. Fisher s exact test will be used for AEs, discontinuations, and other categorical safety data between treatment groups. Continuous vital signs, body weight, and other continuous safety variables, including laboratory variables will be analyzed using ANCOVA with treatment and baseline value in the model. Analysis Population: The efficacy and health outcome analyses will be conducted on an ITT basis unless otherwise specified. The ITT analysis set will include all data from all randomized patients treated with at least 1 dose of the study drug. Analysis of structural progression (van der Heijde mtss) will be conducted on the ITT population using patients with available baseline and at least 1 postbaseline value. In addition, the primary and major secondary analyses will be repeated using the per-protocol set (PPS), which is defined as all randomized patients who are compliant with treatment, who do not have significant protocol violations, and whose investigator site does not have significant GCP issues that require a report to the regulatory agencies. Safety analyses will be performed on the safety population, defined as all randomized patients who received at least 1 dose of the study drug. Patients will be analyzed for efficacy, health outcomes, and safety according to the treatment to which they were assigned. PK analysis will be conducted using all evaluable PK data. Further details will be described in the SAP. Missing Data Imputation: In accordance with precedent set with other Phase 3 RA trials (Keystone et al. 2004, 2008, 2009; Cohen et al. 2006; Smolen et al. 2008, 2009), the following methods for imputation of missing data will be used:
169 I4V-MC-JADV(c) Clinical Protocol Page Nonresponder imputation (NRI): All patients who discontinue the study or the study treatment at any time for any reason will be defined as nonresponders for the NRI analysis for categorical variables, such as ACR20/50/70, from the time of discontinuation and onward. Patients who receive rescue therapy at Week 16 will be analyzed as nonresponders after Week 16 and onward. Randomized patients without available data at a postbaseline visit will be defined as nonresponders for the NRI analysis at that visit. 2. Linear extrapolation method: The linear extrapolation method will be used for analysis of the structural progression endpoint (van der Heijde mtss) to impute missing data at time points when analyses are conducted (including the analyses at Week 24 and Week 52). For patients who discontinue the study or the study treatment or for patients who miss a radiograph for any reason, baseline data and the most recent radiographic data prior to discontinuation or the missed radiograph, adjusted for time, will be used for linear extrapolation to impute missing data at subsequent time points. For patients who receive rescue therapy starting at Week 16 or at any time point thereafter, baseline data and the most recent radiographic data prior to initiation of rescue therapy, adjusted for time, will be used for linear extrapolation. All patients originally randomized to placebo will be switched to active treatment at Week 24; thus, there will be no observed data for the placebo arm for the structural comparison at subsequent time points. Therefore, baseline data and the most recent radiographic data prior to initiation of the new therapy, adjusted for time, will be used for linear extrapolation at Week 52. The linear extrapolation method has been established as an appropriate missing data imputation method in other Phase 3 RA trials (Keystone et al. 2004, 2008, 2009; Cohen et al. 2006; Smolen et al. 2008, 2009). Missing postbaseline values will be imputed only if both a baseline value and a postbaseline value from another time point are available, as long as the patient was on the same treatment at each applicable time point. 3. Multiple imputation: Other methods than linear extrapolation that include multiple imputation will be employed for analysis of the structural progression endpoint (van der Heijde mtss) to impute missing data at Week 24. Details will be provided in the SAP. 4. Modified baseline observation carried forward (mbocf): The mbocf method will be used for the analysis of major secondary continuous endpoints (unless otherwise stated). For patients who discontinue the study or the study treatment because of an AE, including death, the baseline observation will be carried forward to the corresponding endpoint for evaluation. For patients who discontinue the study for reason(s) other than an AE, the last nonmissing postbaseline observation prior to discontinuation will be carried forward to the corresponding endpoint for evaluation. For patients who receive rescue therapy starting at Week 16, the last nonmissing observation at or before rescue will be carried forward to subsequent time points for evaluation. 5. Modified last observation carried forward (mlocf): For all continuous measures including safety analyses, the mlocf will be a general approach to impute missing data unless otherwise specified. For patients who receive rescue therapy starting at Week 16, the last nonmissing observation at or before rescue will be carried forward to subsequent time points for evaluation. For all other patients discontinuing from the study or the study treatment for any reason, the last nonmissing postbaseline observation before
170 I4V-MC-JADV(c) Clinical Protocol Page 72 discontinuation will be carried forward to subsequent time points for evaluation. After mlocf imputation, data from patients with nonmissing baseline and postbaseline observations will be included in the analyses. The mlocf will be used as a sensitivity method to the mbocf method with respect to the major secondary endpoints. 6. Other methods for data imputation for analysis of key endpoints other than mtss may be used and will be described in the SAP. Adjustment for Multiple Comparisons: The following multiple testing procedure will be used to control the family-wise error rate for the primary and key secondary hypotheses for the endpoints listed in Sections 6.1 and 6.2. Each step in the sequential testing procedure uses a local level procedure for the null hypotheses being tested. Testing of hypotheses at each step of the procedure commences only if all null hypotheses of prior steps were rejected. As such, the multiple testing procedure provides strong control of the family-wise error rate at the 1-sided level. The overall strategy is illustrated (Bretz et al. 2009) in Figure JADV.2. Step 1: Test ACR20 at Week 12 for baricitinib versus placebo at 1-sided alpha = If the null hypothesis is rejected, proceed to Step 2. Otherwise, discontinue testing. Step 2: Test the change from baseline in HAQ-DI at Week 12 and the change from baseline in mtss at Week 24, both tests of baricitinib versus placebo, at 1-sided alpha = via a weighted Holm procedure (Holm 1979) with weights of 0.2 and 0.8, respectively. If both null hypotheses are rejected, proceed to Step 3. Otherwise, discontinue testing. Details of the Holm procedure at this step: Let p2 denote the unadjusted 1-sided p-value resulting from testing the hypothesis regarding HAQ-DI and let p3 denote the unadjusted 1-sided p-value resulting from testing the hypothesis regarding mtss. Let p2* and p3* denote the corresponding weight-adjusted p-values (ie, p2* = p2/0.2 and p3*=p3/0.8). The following decision rules apply: If min(p2*, p3*) >0.025, then Lilly will fail to reject both null hypotheses and discontinue testing. Otherwise, if p2* 0.025, then Lilly will reject the null hypothesis for HAQ-DI (declaring statistical significance) and test mtss using the unadjusted p-value. If p , Lilly will also reject the null hypothesis for mtss (declaring statistical significance); however if p3 >0.025, then Lilly will fail to reject the null hypothesis for mtss and discontinue testing. Otherwise, if p3* 0.025, then Lilly will reject the null hypothesis for mtss (declaring statistical significance) and test HAQ-DI using the unadjusted p-value. If p , Lilly will also reject the null hypothesis for HAQ-DI (declaring statistical significance); however if p2 >0.025, then Lilly will fail to reject the null hypothesis for HAQ-DI and discontinue testing.
171 I4V-MC-JADV(c) Clinical Protocol Page 73 Step 3: Test the change from baseline in DAS28-hsCRP at Week 12 and the proportion of patients achieving an SDAI score 3.3 (SDAI remission) at Week 12, both tests of baricitinib versus placebo, at 1-sided alpha=0.025 via a Hochberg procedure (Hochberg 1988). If both null hypotheses are rejected, proceed to Step 4. Otherwise, discontinue testing. Details of the Hochberg procedure at this step: Let p1 and p2 denote the unadjusted 1-sided p-values resulting from testing the hypotheses regarding DAS28-hsCRP and SDAI remission, respectively. If p1and p , then Lilly will reject both null hypotheses (declaring statistical significance for both DAS28-hsCRP and SDAI remission). Otherwise, if p and p2 >0.025, then Lilly will reject only the null hypothesis (declaring statistical significance) related to DAS28-hsCRP. Otherwise if p and p1 >0.025, then Lilly will reject only the null hypothesis (declaring statistical significance) related to SDAI remission. Otherwise Lilly will fail to reject both null hypotheses and discontinue testing. Step 4: Test ACR20 at Week 12 for noninferiority of baricitinib to adalimumab at 1-sided alpha= Details concerning the noninferiority margin and statistical conclusions are found in Section If the null hypothesis is rejected, proceed to Step 5. Otherwise, discontinue testing. Step 5: Test mean duration of morning joint stiffness in the 7 days prior to Week 12 for baricitinib versus placebo at 1-sided alpha= If the null hypothesis is rejected, proceed to Step 6. Otherwise, discontinue testing. Step 6: Test the change from baseline in DAS28-hsCRP at Week 12 for baricitinib versus adalimumab at 1-sided alpha= If the null hypothesis is rejected, proceed to Step 7. Otherwise, discontinue testing. Step 7: Test mean severity of morning joint stiffness in the 7 days prior to Week 12 for baricitinib versus placebo at 1-sided alpha= If the null hypothesis is rejected, proceed to Step 8. Otherwise, discontinue testing. Step 8: Test mean Worst Tiredness NRS in the 7 days prior to Week 12 and mean Worst Pain NRS in the 7 days prior to Week 12, both tests of baricitinib versus placebo, at 1-sided alpha=0.025 via a Hochberg procedure and following similar details as outlined in Step 3.

172 I4V-MC-JADV(c) Clinical Protocol Page 74 Abbreviations: = change from baseline; ACR20 = 20% improvement in American College of Rheumatology criteria; DAS28 = 28 diarthroidal joint count; hscrp = highsensitivity C-reactive protein; HAQ-DI = Health Assessment Questionnaire-Disability Index; SDAI = simplified disease activity index; mtss = modified Total Sharp Score [van der Heijde]; TS = test(s) significant; W = week; BARI = baricitinib; ADA = adalimumab. Figure JADV.2. Illustration of the sequential testing approach within the overall gatekeeping strategy for Study JADV.
173 I4V-MC-JADV(c) Clinical Protocol Page 75 Any change to the data analysis methods described in the protocol will require an amendment ONLY if it changes a principal feature of the protocol. Any other change to the data analysis methods described in the protocol, and the justification for making the change, will be described in the SAP and the clinical study report. Additional exploratory analyses of the data will be conducted as deemed appropriate Patient Disposition The number of randomized patients along with ITT and PPS populations will be summarized by treatment group. Frequency counts and percentages in different study parts will be presented for each treatment group. All patients who discontinue from the study or the study treatment will be identified, and the extent of their participation in the study will be reported along with their reason for discontinuation. Reasons for discontinuation from the study will be summarized by treatment group Patient Characteristics Demographic and baseline characteristics will be summarized descriptively by treatment group. Descriptive statistics including number of patients, mean, SD, median, minimum, and maximum will be provided for continuous measures, and frequency counts and percentages will be tabulated for categorical measures. No formal statistical comparisons will be made among treatment groups unless otherwise stated Concomitant Therapy Concomitant medications will be descriptively summarized by treatment group in terms of frequencies and percentages using the safety population. The medications will be coded accordingly Treatment Compliance Compliance to study treatment for baricitinib and oral placebo will be assessed through counts of returned study drug tablets. For baricitinib or oral placebo, a patient will be considered significantly noncompliant if he or she misses more than 20% of the prescribed doses during the study, unless the patient s study drug is withheld by the investigator for safety reasons. For adalimumab or injectable placebo, a patient will be considered significantly noncompliant he or she misses more than 20% of the prescribed doses during the study or if there are any consecutive missed injections, unless the patient s study drug is withheld by the investigator for safety reasons. Similarly, a patient will be considered significantly noncompliant if he or she is judged by the investigator to have intentionally or repeatedly taken more than the prescribed amount of study drug. Patients who are significantly noncompliant will be excluded from the PPS. Patient compliance will be further defined in the SAP.
174 I4V-MC-JADV(c) Clinical Protocol Page Primary Outcome and Methodology A logistic regression model with treatment, region, and joint erosion status in the model will be used to test the treatment difference between baricitinib and placebo in the proportion of patients achieving ACR20 response at Week 12. The NRI method as described above will be used to impute missing data Efficacy Analyses Major secondary analyses: 1. Comparison between baricitinib and placebo in change from baseline to Week 24 in mtss: An ANCOVA model with baseline score, region, joint erosion status, and treatment group in the model will be used to test the treatment difference between baricitinib and placebo in change from baseline to Week 24 of mtss scores. The linear extrapolation method as described above will be used to impute missing data. A permutation-based method will be used as a nonparametric sensitivity analysis to the ANCOVA method. 2. Comparison between baricitinib and placebo in change from baseline to Week 12 in HAQ-DI score: An ANCOVA model with treatment, region, joint erosion status, and baseline score in the model will be used to test the treatment difference between baricitinib and placebo in change from baseline to Week 12 in HAQ-DI. The mbocf approach as described above will be used to impute missing data. 3. Comparison between baricitinib and placebo in change from baseline to Week 12 in DAS28-hsCRP: An ANCOVA model with treatment, region, joint erosion status, and baseline score in the model will be used to test the treatment difference between baricitinib and placebo in change from baseline to Week 12 in DAS28-hsCRP. The mbocf approach as described above will be used to impute missing data. 4. Comparison between baricitinib and placebo in proportion of patients achieving an SDAI score 3.3 at Week 12: A logistic regression model with treatment, region, and joint erosion status in the model will be used to test the treatment difference between baricitinib and placebo in the proportion of patients achieving an SDAI score 3.3 response at Week 12. The NRI method as described above will be used to impute missing data. 5. Comparison between baricitinib and adalimumab in ACR20 response at Week 12: A logistic regression model with treatment, region, and joint erosion status in the model will be used to test the treatment difference between baricitinib and adalimumab in the percentage of patients achieving ACR20 response at Week 12. A 95% CI for the treatment difference will be constructed using the method described by Newcombe- Wilson (Newcombe 1998) without continuity correction. If a quantitative treatment by region interaction exists, a supportive method to the Newcombe-Wilson method will be used. If the lower bound of the 95% CI is >-12%, Lilly will conclude that baricitinib is noninferior to adalimumab with respect to ACR20. Similarly, if the lower bound of the 95% CI is >0%, Lilly will conclude that baricitinib is superior to adalimumab with
175 I4V-MC-JADV(c) Clinical Protocol Page 77 respect to ACR20. The NRI method as described above will be used to impute missing data. 6. Comparison between baricitinib and placebo in mean duration of morning joint stiffness in the 7 days prior to Week 12: An ANCOVA model with treatment, region, joint erosion status, and baseline score (Day 1 on the diaries) in the model will be used to test the treatment difference between baricitinib and placebo in mean duration of morning joint stiffness in the 7 days prior to Week 12. Refer to the SAP for more details. 7. Comparison between baricitinib and adalimumab in change from baseline to Week 12 in DAS28-hsCRP: An ANCOVA model with treatment, region, joint erosion status, and baseline score in the model will be used to test the treatment difference between baricitinib and adalimumab in change from baseline to Week 12 in DAS28-hsCRP. The mbocf approach as described above will be used to impute missing data 8. Comparison between baricitinib and placebo in mean severity of morning joint stiffness in the 7 days prior to Week 12: An ANCOVA model with treatment, region, joint erosion status, and baseline score (Day 1 on the diaries) in the model will be used to test the treatment difference between baricitinib and placebo in mean severity of morning joint stiffness in the 7 days prior to Week 12. Refer to the SAP for more details. 9. Comparison between baricitinib and placebo in mean Worst Tiredness NRS in the 7 days prior to Week 12: An ANCOVA model with treatment, region, joint erosion status, and baseline score (Day 1 on the diaries) in the model will be used to test the treatment difference between baricitinib and placebo in mean Worst Tiredness NRS in the 7 days prior to Week 12. Refer to the SAP for more details. 10. Comparison between baricitinib and placebo in mean Worst Pain NRS in the 7 days prior to Week 12: An ANCOVA model with treatment, region, joint erosion status, and baseline score (Day 1 on the diaries) in the model will be used to test the treatment difference between baricitinib and placebo in mean Worst Pain NRS in the 7 days prior to Week 12. Refer to the SAP for more details. Other secondary efficacy and health outcome analyses: Besides the analyses listed in the objective section, the following analyses will be performed: change from baseline to Week 16 and Week 52 in structural joint damage as measured by mtss proportion of patients with mtss change 0 change from baseline in joint space narrowing and bone erosion scores hybrid ACR (bounded) response measure proportions of patients achieving ACR20 (other than primary), ACR50, and ACR70 percentage change from baseline in individual components of the ACR Core Set change from baseline in DAS28-ESR change from baseline in DAS28-hsCRP proportion of patients achieving DAS28-ESR 3.2 and <2.6 proportion of patients achieving DAS28-hsCRP 3.2 and <2.6 change from baseline in HAQ-DI
176 I4V-MC-JADV(c) Clinical Protocol Page 78 proportion of patients achieving HAQ-DI improvement 0.22 and 0.3 change from baseline in CDAI score change from baseline in SDAI score proportion of patients achieving CDAI score 2.8 proportion of patients achieving SDAI score 3.3 (ACR/EULAR remission according to the Index-based definition) proportion of patients achieving moderate response and good response based on EULAR response criteria proportion of patients achieving ACR/EULAR remission according to the Boolean-based definition change from baseline in fatigue score in the FACIT-F change from baseline in PCS, MCS, and total score in the SF-36v2 Acute change from baseline in health states, VAS score, and index score of EQ-5D-5L change from baseline in absenteeism, presenteeism, work productivity loss, and activity impairment scores of WPAI-RA individual items and total score in healthcare resource utilization characterization of the response time-course for key efficacy endpoints and identification of potential factors that may have an impact on the efficacy endpoints of baricitinib change from baseline in duration of morning joint stiffness assessed at the visit using an epro tablet change from baseline in Worst Tiredness NRS assessed at the visit using an epro tablet change from baseline in Worst Pain NRS assessed at the visit using an epro tablet Area under the curve (AUC) up to Week 12 in duration of morning joint stiffness collected in electronic diaries AUC up to Week 12 in severity of morning joint stiffness collected in electronic diaries AUC up to Week 12 in Worst Tiredness NRS collected in electronic diaries AUC up to Week 12 in Worst Pain NRS collected in electronic diaries Approaches similar to the primary and major secondary analyses will be used to analyze other secondary efficacy and health outcome measures. In addition, comparisons between baricitinib and adalimumab in other secondary measures will be performed. Analysis methods will consist of ANCOVA and logistic regression, as described above, using mbocf, linear extrapolation, NRI, and mlocf missing data imputation methods, where applicable. In addition, key continuous secondary parameters that are collected at repeated visits throughout the double-blind period may be analyzed using a MMRM approach as described in Section The MMRM will be considered as a sensitivity approach to ANCOVA results. Data collected after initiation of rescue therapy will be summarized as appropriate. Further details will be provided in the SAP Pharmacokinetic/Pharmacodynamic Analyses PK data for baricitinib will be analyzed using a population modeling approach via a nonlinear mixed-effects modeling (NONMEM) program. Data from this study may be combined with an
177 I4V-MC-JADV(c) Clinical Protocol Page 79 existing PK model or data from other studies, if appropriate. One- and 2-compartment structural models with or without absorption lag-time will be tested. A first-order absorption model whereby a 1-compartment model will be parameterized in terms of absorption rate constant (Ka), central compartment clearance (CL), and central compartment volume of distribution (V1). The 2-compartment model will be parameterized in terms of Ka, V1, CL, intercompartmental clearance (Q), peripheral volume of distribution (V2). In the case of a zero absorption model, the absorption model will be parameterized by absorption duration (D1). Previous modeling results and known predominant renal elimination characteristics of baricitinib indicated close correlation of baricitinib exposures with renal function defined by MDRD-eGFR; hence, partitioning of total clearance into renal and nonrenal clearance guided by mass-balance study results may also be incorporated into the model. Interpatient variability will be assessed separately on each of the PK parameters using an exponential error structure. Residual error will be assessed as proportional, additive, and combined proportional and additive error structures. Intrinsic factors such as age; body weight; gender; ethnic origin; renal function; transaminases/albumin/bilirubin, which are representative of hepatic function; and extrinsic factors such as comedications will be investigated to assess their influence on PK parameters such as clearance and volume of distribution, where applicable. Time-course of response for key efficacy data such as, but not necessarily limited to, ACR20/50/70, DAS28-ESR, DAS28-hsCRP, and safety data (eg, neutropenia, anemia) will be explored using graphical methods. Further investigations using a population-modeling approach via NONMEM to identify potential factors that may influence pharmacodynamic (PD) parameters may be warranted. Interpatient variability will then be assessed separately on each of the PD parameters using additive or exponential error structure. Residual error will be assessed as proportional, additive, and combined proportional and additive error structures. Covariates such as body weight, disease duration, disease severity at baseline, responder status, cytokines concentrations, dose, and baricitinib exposures may be investigated to assess their impact on efficacy and safety endpoints. Data from this study may be combined with an existing PK/PD model or data from other studies, if appropriate. Model evaluation will include using sensitivity analysis and visual predictive check methods Health Outcome Analyses The health outcomes will be analyzed using methods described for continuous or categorical data as described for efficacy measures in Section More detailed analytical methods will be described in the SAP Safety Analyses All safety data will be descriptively summarized by treatment groups and analyzed using the safety population. For the purposes of calculating changes from baseline during Part A and Part B, treatment baseline may be considered as the original baseline for Part A prior to any study drug received or at the visits that mark the transition between treatments received, depending on the purpose of the summary or comparison being made. Summaries within Parts A and B will be conducted
178 I4V-MC-JADV(c) Clinical Protocol Page 80 by treatment group used within each respective part. Additional summaries by treatment group that continue unchanged across multiple parts of the study will be conducted, as appropriate. TEAEs are defined as AEs that first occurred or worsened in severity after the first dose of study treatment. AEs will be considered treatment emergent for nonresponders assigned to baricitinib if the AEs first occurred or worsened in severity after the visit at which rescue therapy is assigned. The number of TEAEs as well as the number and percentage of patients who experienced at least 1 TEAE will be summarized using MedDRA (Medical Dictionary for Regulatory Activities) for each system organ class (or a body system) and each preferred term by treatment group. TEAEs will also be summarized by relationship to treatment and by severity within each treatment group. SAEs and AEs that lead to study drug discontinuation will also be summarized by treatment group. Fisher s exact test will be used to perform pairwise comparisons among the treatment groups. All clinical laboratory results will be descriptively summarized by treatment group. Individual results that are outside of normal range will be flagged in data listings. Quantitative clinical hematology, chemistry, and urinalysis variables obtained at the baseline to postbaseline visits will be summarized as changes from baseline by treatment group and analyzed using ANCOVA with treatment and baseline value in the model. Categorical variables, including the incidence of abnormal values and incidence of AESIs, will be summarized by frequency and percentage of patients in corresponding categories. Shift tables will be presented for selected measures. Observed values and changes from baseline (predose or screening if missing) for vital signs and physical characteristics will be descriptively summarized by treatment group and time point. Change from baseline to postbaseline in vital signs, QIDS-SR 16, and body weight will be analyzed using ANCOVA with treatment and baseline value in the model. The incidence and average duration of study drug interruptions will be summarized and compared descriptively among treatment groups. Various techniques may be used to estimate the effects of study drug interruptions on safety measures. Further analyses may be performed and will be planned in the SAP. Data collected after initiation of rescue therapy will be summarized as appropriate Subgroup Analyses Subgroups analyses comparing baricitinib to placebo will be performed using ACR20, ACR50, HAQ-DI and DAS28-hsCRPat Weeks 12 and 24 and mtss at Week 24. Subgroups to be evaluated will include region, renal function, background therapy, joint erosion status, country, gender, age, race, etc. Any other additional subgroups will be defined in the SAP. For ACR20 and ACR50 analyses at Weeks 12 and 24, a logistic regression model will be used with treatment, subgroup, and treatment by subgroup interaction included as factors. For the change from baseline to Weeks 12 and 24 in HAQ-DI score and DAS28-hsCRP and to Week 24 in mtss, an ANCOVA model will be used with treatment, baseline score, subgroup, and treatment by subgroup interaction included as factors.
179 I4V-MC-JADV(c) Clinical Protocol Page 81 Further definitions for the levels of the subgroup variables, the analysis methodology, and any additional subgroup analyses will be defined in the SAP. Because this study is not powered for subgroup analyses, all subgroup analyses will be treated as exploratory Planned Analyses The analysis of primary and major secondary endpoints (except for x-ray data) will occur when all patients complete the Week 24 study visit. This analysis will not be considered an interim analysis, and the study team will become unblinded at this time. A final analysis will occur when all patients complete the study. All investigators, study site personnel, and patients will remain blinded to treatment assignments until the final database lock. Members of the study team not in direct contact with the clinical sites may have access to the data from this study after all patients complete the primary and major secondary endpoints through Week 24 (Visit 11). Members of the study team who become unblinded to the primary and major secondary analyses through Week 24 will have no further contact with study sites until after the final database lock. Unblinding details will be specified in the unblinding plan section of the SAP Interim Analyses A DMC will oversee the conduct of all the Phase 3 clinical trials evaluating baricitinib in patients with RA. The DMC will consist of members external to Lilly. This DMC will follow the rules defined in the DMC charter, focusing on potential and identified risks for this molecule and for this class of compounds. DMC membership will include, at a minimum, specialists with expertise in rheumatology, statistics, and other appropriate specialties. The DMC will review and evaluate up to 3 planned interim analyses on an approximate semiannual basis. This DMC for studies of patients with RA will be coordinated with the DMC(s) for other ongoing studies of baricitinib in other indications, and this coordination may alter the number and timing of the interim analyses. Access to the unblinded interim data will be limited to the statisticians who conduct the interim analyses and the DMC. The statisticians conducting the interim analyses will be independent from the study team. The study team will not have access to the unblinded data. Study sites will receive information about interim results ONLY if they need to know for the safety of their patients. The DMC will review study discontinuation data, AEs including SAEs, clinical laboratory data, vital sign data, etc. The DMC may recommend continuation of the study, as designed, temporary suspension of enrollment, or the discontinuation of a particular dose regimen or the entire study. The DMC may request to review efficacy data to investigate the benefit/risk relationship in the context of safety observations for ongoing patients in the study. However, the study will not be stopped for positive efficacy results, and there is no planned futility assessment. Hence, there will be no alpha adjustment for these interim analyses. Details of the DMC and interim safety analyses will be documented in a DMC charter and DMC analysis plan.
180 I4V-MC-JADV(c) Clinical Protocol Page 82 Besides DMC members, a limited number of pre-identified individuals may gain access to the unblinded PK and safety data, as specified in the unblinding plan, prior to the final database lock, to initiate the exploration and/or final population PK/PD model development processes for safety data (eg, neutropenia, anemia). Information that may unblind the study during the analyses will not be reported to study sites or the blinded study team until the database lock for the primary and major secondary efficacy analyses.
181 I4V-MC-JADV(c) Clinical Protocol Page Informed Consent, Ethical Review, and Regulatory Considerations Informed Consent The investigator is responsible for ensuring that the patient understands the potential risks and benefits of participating in the study, including answering any questions the patient may have throughout the study and sharing in a timely manner any new information that may be relevant to the patient s willingness to continue his or her participation in the trial. The ICF will be used to explain the potential risks and benefits of study participation to the patient in simple terms before the patient is entered into the study and to document that the patient is satisfied with his or her understanding of the risks and benefits of participating in the study and desires to participate in the study. The investigator is responsible for ensuring that informed consent is given by each patient. This includes obtaining the appropriate signatures and dates on the ICF prior to the performance of any protocol procedures and prior to the administration of investigational product Ethical Review Lilly or its representatives must approve all ICFs before they are submitted to the ERB and are used at investigative sites(s). All ICFs must be compliant with the International Conference on Harmonisation (ICH) guideline on GCP. Documentation of ERB approval of the protocol and the ICF must be provided to Lilly before the study may begin at the investigative site(s). The ERB(s) will review the protocol as required. Any member of the ERB who is directly affiliated with this study as an investigator or as site personnel must abstain from the ERB s vote on the approval of the protocol. The study site s ERB(s) should be provided with the following: the current IB or package labeling and updates during the course of the study ICF relevant curricula vitae Regulatory Considerations This study will be conducted in accordance with: 1) consensus ethics principles derived from international ethics guidelines, including the Declaration of Helsinki and Council for International Organizations of Medical Sciences International Ethical Guidelines 2) the ICH GCP Guideline [E6] 3) applicable laws and regulations The investigator or designee will promptly submit the protocol to the applicable ERB(s).
182 I4V-MC-JADV(c) Clinical Protocol Page 84 An identification code assigned by the investigator to each patient will be used in lieu of the patient s name to protect the patient s identity when reporting AEs and/or other trial-related data Investigator Information Physicians with a specialty in RA will participate as investigators in this clinical trial Protocol Signatures The sponsor s responsible medical officer will approve the protocol, confirming that, to the best of his or her knowledge, the protocol accurately describes the planned design and conduct of the study. After reading the protocol, each principal investigator will sign the protocol signature page and send a copy of the signed page to a Lilly representative Final Report Signature The clinical study report coordinating investigator will sign the final clinical study report for this study, indicating agreement that, to the best of his or her knowledge, the report accurately describes the conduct and results of the study. Lilly will select a qualified investigator from among investigators participating in the design, conduct, and/or analysis of the study to serve as the clinical study report coordinating investigator. The sponsor s responsible medical officer will approve the final clinical study report for this study, confirming that, to the best of his or her knowledge, the report accurately describes the conduct and results of the study.
183 I4V-MC-JADV(c) Clinical Protocol Page References Aletaha D, Neogi T, Silman AJ, Funovits J, Felson DT, Bingham CO 3rd, Birnbaum NS, Burmester GR, Bykerk VP, Cohen MD, Combe B, Costenbader KH, Dougados M, Emery P, Ferraccioli G, Hazes JM, Hobbs K, Huizinga TW, Kavanaugh A, Kay J, Kvien TK, Laing T, Mease P, Ménard HA, Moreland LW, Naden RL, Pincus T, Smolen JS, Stanislawska-Biernat E, Symmons D, Tak PP, Upchurch KS, Vencovskỳ J, Wolfe F, Hawker G Rheumatoid arthritis classification criteria: an American College of Rheumatology/European League Against Rheumatism collaborative initiative. Arthritis Rheum. 2010;62(9): Aletaha D, Smolen J. The Simplified Disease Activity Index (SDAI) and the Clinical Disease Activity Index (CDAI): a review of their usefulness and validity in rheumatoid arthritis. Clin Exp Rheumatol. 2005;23(5 suppl 39):S100-S108. American College of Rheumatology Committee to Reevaluate Improvement Criteria. A proposed revision to the ACR20: the hybrid measure of American College of Rheumatology response. Arthritis Rheum. 2007;57(2): [APA] American Psychiatric Association's Diagnostic and Statistical Manual of Mental Disorders, 4th Edition. Washington, DC; American Psychiatric Association Brazier JE, Harper R, Jones NM, O Cathain A, Thomas KJ, Usherwood T, Westlake L. Validating the SF-36 health survey questionnaire: new outcome measure for primary care. BMJ. 1992;305(6846): Bretz F, Maurer W, Brannath W, Posch M. A graphical approach to sequentially rejective multiple test procedures. Stat Med. 2009;28(4): Brooks R. EuroQol: The current state of play. Health Policy. 1996;37: Cella D, Webster K. Linking outcomes management to quality-of-life measurement. Oncology (Willeston Park). 1997;11(11A): Cohen SB, Emery P, Greenwald MW, Dougados M, Furie RA, Genovese MC, Keystone EC, Loveless JE, Burmester GR, Cravets MW, Hessey EW, Shaw T, Totoritis MC. Rituximab for rheumatoid arthritis refractory to anti-tumor necrosis factor therapy. Results of a multicenter, randomized, double-blind, placebo-controlled phase III trial evaluating primary efficacy and safety at twenty-four weeks. Arthritis Rheum. 2006;54(9): Colmegna I, Ohata BR, Menard HA. Current understanding of rheumatoid arthritis therapy. Clin Pharmacol Ther. 2012;91(4): Curtis JR, Patkar N, Xie A, Martin C, Allison JJ, Saaq M, Shatin D, Saaq KG. Risk of serious bacterial infections among rheumatoid arthritis patients exposed to tumor necrosis factor alpha antagonists. Arthritis Rheum. 2007;56(4): The EuroQol Group. EQ-5D-5L User Guide. Version 1.0. April Available at: EQ-5D-5L.pdf. Accessed July
184 I4V-MC-JADV(c) Clinical Protocol Page 86 Felson DT, Smolen JS, Wells G, Zhang B, van Tuyl LH, Funovits J, Aletaha D, Allaart CF, Bathon J, Bombardieri S, Brooks P, Brown A, Matucci-Cerinic M, Choi H, Combe B, de Wit M, Dougados M, Emery P, Furst D, Gomez-Teino J, Hawker G, Keystone E, Khanna D, Kirwan J, Kvien TK, Landewé R, Listing J, Michaud K, Martin-Mola E, Montie P, Pincus T, Richards P, Siegel JN, Simon LS, Sokka T, Strand V, Tugwell P, Tyndall A, van der Heijde D, Verstappen S, White B, Wolfe F, Zink A, Boers M. American College of Rheumatology/European League against Rheumatism provisional definition of remission in rheumatoid arthritis for clinical trials. Ann Rheum Dis. 2011;70: Fleischmann RM. Is there a need for new therapies for rheumatoid arthritis? J Rheum. 2005;32(suppl 73):3S-7S. Fridman JS, Scherle PA, Collins R, Burn TC, Li Y, Li J, Covington MB, Thomas B, Collier P, Favata MF, Wen X, Shi J, McGee R, Haley PJ, Shepard S, Rodgers JD, Yeleswaram S, Hollis G, Newton RC, Metcalf B, Friedman SM, Vaddi K. Selective inhibition of JAK1 and JAK2 is efficacious in rodent models of arthritis: preclinical characterization of INCB J Immunol. 2010;184(9): Fries JF, Spitz P, Kraines RG, Holman HR. Measurement of patient outcome in arthritis. Arthritis Rheum. 1980;23(2): Fries JF, Spitz PW, Young DY. The dimensions of health outcomes: the health assessment questionnaire, disability and pain scales. J Rheumatol. 1982;9(5): Herdman M, Gudex C, Lloyd A, Janssen MF, Kind P, Parkin D, Bonsel G, Badia X. Development and preliminary testing of the new five-level version of EQ-5D (EQ-5D-5L). Qual Life Res. 2011;20(10): Hochberg Y. A sharper bonferroni procedure for multiple tests of significance. Biometrika. 1988;75(4): Holm S. A simple sequentially rejective multiple test procedure. Scand J Statist. 1979;6: Keystone EC, Kavanaugh AF, Sharp JT, Tannenbaum H, Hua Y, Teoh LS, Fischkoff SA, Chartash EK. Radiographic, clinical, and functional outcomes of treatment with adalimumab (a human anti tumor necrosis factor monoclonal antibody) in patients with active rheumatoid arthritis receiving concomitant methotrexate therapy: a randomized, placebo-controlled, 52-week trial. Arthritis Rheum. 2004;50(5): Keystone E, Heijde D, Mason D Jr, Landewé R, Vollenhoven RV, Combe B, Emery P, Strand V, Mease P, Desai C, Pavelka K. Certolizumab pegol plus methotrexate is significantly more effective than placebo plus methotrexate in active rheumatoid arthritis: findings of a fifty-twoweek, phase III, multicenter, randomized, double-blind, placebo-controlled, parallel-group study. Arthritis Rheum. 2008;58(11): Keystone E, Emery P, Peterfy CG, Tak PP, Cohen S, Genovese MC, Dougados M, Burmester GR, Greenwald M, Kvien TK, Williams S, Hagerty D, Cravets MW, Shaw T. Rituximab inhibits structural joint damage in patients with rheumatoid arthritis with an inadequate response to tumour necrosis factor inhibitor therapies. Ann Rheum Dis. 2009;68(2): Kitas GD, Gabriel SE. Cardiovascular disease in rheumatoid arthritis: state of the art and future perspectives. Ann Rheum Dis. 2011;70(1):8-14. Erratum in Ann Rheum Dis. 2011;70(8):1520.
185 I4V-MC-JADV(c) Clinical Protocol Page 87 Klareskog L, Catrina AI, Paget S. Rheumatoid arthritis. Lancet. 2009;373(9664): Kremer JM, Bloom BJ, Breedveld FC, Coombs JH, Fletcher MP, Gruben D, Krishnaswami S, Burgos-Vargas R, Wilkinson B, Zerbini CA, Zwillich SH. The safety and efficacy of a JAK inhibitor in patients with active rheumatoid arthritis: Results of a double-blind, placebocontrolled phase IIa trial of three dosage levels of CP-690,550 versus placebo. Arthritis Rheum. 2009;60(7): Newcombe RG. Interval estimation for the difference between independent proportions: comparison of eleven methods. Stat Med. 1998;17: Erratum in Stat Med. 1999;18:1293. Ramey DR, Fries JF, Singh G. The Health Assessment Questionnaire 1995: status and review. In: Spiker B, editor. Quality of life and pharmacoeconomics in clinical trials. 2nd ed. Philadelphia: Lippincott-Raven, 1996: p Reilly MC, Zbrozek AS, Dukes EM. The validity and reproducibility of a work productivity and activity impairment instrument. Pharmacoeconomics. 1993;4(5): Rubbert-Roth A, Finckh A. Treatment options in patients with rheumatoid arthritis failing initial TNF inhibitor therapy: a critical review. Arthritis Res Ther. 2009;11(suppl 1):S1. Rush AJ, Trivedi MH, Ibrahim HM, Carmody TJ, Arnow B, Klein DN, Markowitz JC, Ninan PT, Kornstein S, Manber R, Thase ME, Kocsis JH, Keller MB. The 16-Item quick inventory of depressive symptomatology (QIDS), clinician rating (QIDS-C), and self-report (QIDS-SR): a psychometric evaluation in patients with chronic major depression. Biological Psychiatry. 2003;54(50): Erratum in Biol Psychiatry. 2003;54(5);585. Schiff M, Fleischmann R, Weinblatt M, Valente R, van der Heijde D, Citera G, Zhao C, Maldonado M. Abatacept sc versus adalimumab on background methotrexate in RA: one year results from the AMPLE study. Ann Rheum Dis. 2012;71(suppl 3):60. Smolen J, Landewé RB, Mease P, Brzezicki J, Mason D, Luijtens K, van Vollenhoven RF, Kavanaugh A, Schiff M, Burmester GR, Strand V, Vencovskỳ J, van der Heijde D. Efficacy and safety of certolizumab pegol plus methotrexate in active rheumatoid arthritis: the RAPID 2 study. A randomised controlled trial. Ann Rheum Dis. 2009;68(6): Smolen JS, Aletaha D, Bijlsma JW, Breedveld FC, Boumpas D, Burmester G, Combe B, Cutolo M, de Wit M, Dougados M, Emery P, Gibofsky A, Gomez-Reino JJ, Haraoui B, Keystone EC, Kvien TK, McInnes I, Martin-Mola E, Montecucco C, Schoels M, van der Heijde D, T2T Expert Committee. Treating rheumatoid arthritis to target: recommendations of an international task force. Ann Rheum Dis. 2010;69(4): Smolen JS, Beaulieu A, Rubbert-Roth A, Ramos-Remus C, Rovensky J, Alecock E, Woodworth T, Alten R, OPTION investigators. Effect of interleukin-6 receptor inhibition with tocilizumab in patients with rheumatoid arthritis (OPTION study): a double-blind, placebocontrolled, randomised trial. Lancet. 2008;371(9617): Smolen JS, Breedveld FC, Eberl G, Jones I, Leeming M, Wylie GL, Kirkpatrick J. Validity and reliability of the twenty-eight-joint count for the assessment of rheumatoid arthritis activity. Arthritis Rheum. 1995;38(1):38-43.
186 I4V-MC-JADV(c) Clinical Protocol Page 88 Smolen J, Steiner G. Therapeutic strategies for rheumatoid arthritis. Nat Rev Drug Discov. 2003;2(6): Trivedi MH, Rush AJ, Ibrahim HM, Carmody TJ, Biggs MM, Suppes T, Crismon ML, Shores-Wilson K, Toprac MG, Dennehy EB, Witte B, Kashner TM. The Inventory of Depressive Symptomatology, Clinician Rating (IDS-C) and Self-Report (IDS-SR), and the Quick Inventory of Depressive Symptomatology, Clinician Rating (QIDS-C) and Self-Report (QIDS-SR) in public sector patients with mood disorders: a psychometric evaluation. Psychol Med. 2004;34(1): University of Pittsburgh Epidemiology Data Center; IDS/QIDS web site. Available at: Accessed June 13, Vander Cruyssen B, Van Looy S, Wyns B, Westhovens R, Durez P, Van den Bosch F, Veys EM, Mielants H, De Clerck L, Peretz A, Malaise M, Verbruggen L, Vastesaeger N, Geldhof A, Boullart L, De Keyser F. DAS28 best reflects the physician's clinical judgment of response to infliximab therapy in rheumatoid arthritis patients: validation of the DAS28 score in patients under infliximab treatment. Arthritis Res Ther. 2005;7(5):R van der Heijde D. How to read radiographs according to the Sharp/van der Heijde method. J Rheumatol. 2000; 27(1): van der Heijde D, Tanaka Y, Fleischmann R, Keystone E, Kremer J, Zerbini C, Cardiel MH, Cohen S, Nash P, Song YW, Tegzová D, Wyman BT, Gruben D, Benda B, Wallenstein G, Krishnaswami S, Zwillich SH, Bradley JD, Connell CA. Tofacitinib (CP-690,550) in patients with rheumatoid arthritis receiving methotrexate: twelve-month data from a twenty-fourmonth phase III randomized radiographic study. Arthritis Rheum. 2013;(3): van Gestel AM, Haagsma CJ, van Riel PL. Validation of rheumatoid arthritis improvement criteria that include simplified joint counts. Arthritis Rheum. 1998;41(10): Ware JE Jr, Sherbourne CD. The MOS 36-item short-form health survey (SF-36), I: Conceptual framework and item selection. Med Care. 1992;30(6): Williams WV, Scherle P, Shi J, Newton R, McKeever E, Fridman J, Vaddi K, Levy R, Moreland L. Initial efficacy of INCB018424, a selective Janus kinase 1& 2 (JAK 1&2) inhibitor in rheumatoid arthritis (RA). Ann Rheum Dis. 2008;67(suppl II):62.
187 I4V-MC-JADV(c) Clinical Protocol Page 89 Attachment 1. Protocol JADV Study Schedule
188
189
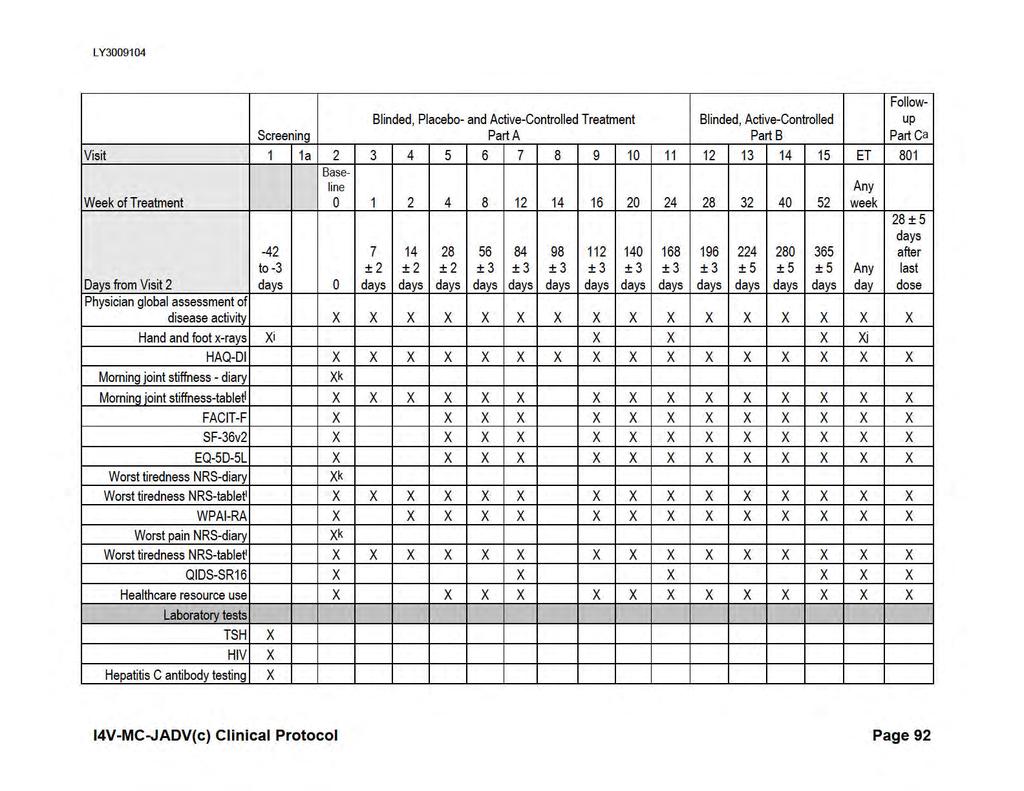
190
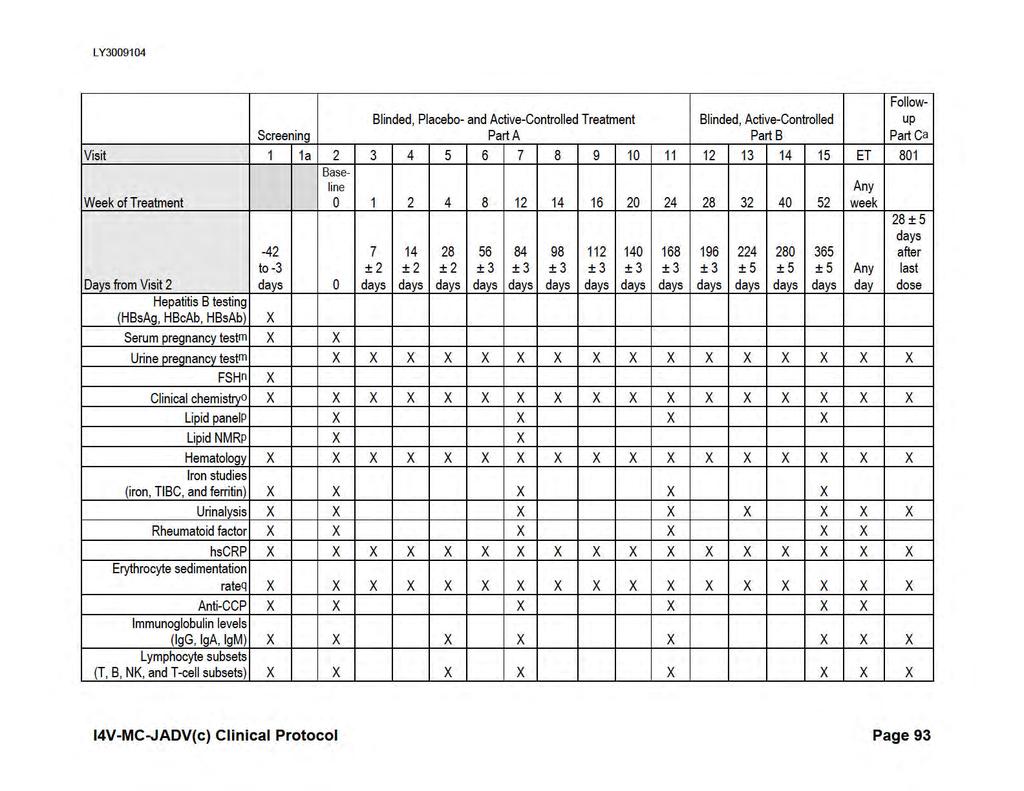
191

192
193 d e f g h i j k l m n o p q r Electrocardiograms will be performed locally and will be locally (machine) read. If the PPD test is positive and the patient has no medical history or chest x-ray findings consistent with active TB, the patient may have a QuantiFERON-TB Gold or T-SPOT.TB test (if available). If the QuantiFERON-TB Gold or T-SPOT.TB test results are not negative, the patient will be considered to have latent TB. If the QuantiFERON-TB Gold or T-SPOT.TB test is available and in the judgment of the investigator, preferred as an alternative to the PPD skin test for the evaluation of TB infection, it may be used instead of the PPD TB test. If the QuantiFERON-TB Gold or T-SPOT.TB test is positive, the patient will be considered to have latent TB. If the test is not negative, the test may be repeated once within 2 approximately weeks of the initial value. If the repeat test results are again not negative, the patient will be considered to have latent TB. A chest x-ray will be taken locally at screening unless one has been obtained within the past 6 months (provided the x-ray and/or report are available for review). At Visit 2, the patient will take his/her oral investigational product at the clinic under the supervision of study staff. Injectable investigational product will be administered by the study staff with instructions given to the patient. At Visits 3, 4, and 8, patients should bring their investigational product with them to the study visit, but no tablet counts need to be performed. At screening, radiographic images of the hands and feet acquired within 4 weeks prior to study entry may be submitted to the central reader to confirm eligibility. If a patient undergoes rescreening, radiographic images acquired as part of initial screening and within 10 weeks of randomization may be used. X-rays will only be taken at early termination if the most recent x-ray was more than 12 weeks earlier. Morning joint stiffness, Worst Tiredness, and Worst Pain assessments will be collected using a battery of questions and a daily patient diary to assess duration, severity of morning joint stiffness, recurrence of stiffness during the day, worst tiredness and worst joint pain. Morning joint stiffness duration, Worst Tiredness, and Worst Pain assessments will be collected using an electronic patient-reported outcomes [epro] tablet at each visit in a subset of patients. For all women of childbearing potential, a serum pregnancy test (central laboratory) will be performed at Visit 1 and Visit 2. Urine pregnancy tests (local laboratory) will also be performed at Visit 2 and at all subsequent study visits. To confirm postmenopausal status for women 40 and <60 years of age who have had a cessation of menses, an FSH test will be performed. Nonchildbearing potential will be defined as an FSH 40 miu/ml and a cessation of menses for at least 12 months. Clinical chemistry will include the following values calculated from serum creatinine: calculated creatinine clearance (Cockcroft-Gault equation) and egfr (calculated using the Modification of Diet in Renal Disease isotope dilution mass spectrometry traceable method). Fasting lipid profile. Patients should not eat or drink anything except water for 12 hours prior to sample collection. If a patient attends these visits in a nonfasting state, this will not be considered a protocol violation. Erythrocyte sedimentation rate will be performed locally using a kit provided by the sponsor. At Visit 2 (Week 0), patients will take their oral investigational product in the clinic, and PK samples will be drawn at 15 minutes and 1 hour postdose. At Visit 5 (Week 4), patients will be asked to take their oral investigational product at home prior to visiting the clinic. The clinic visit should be scheduled so that the sample taken during this visit is 2 to 4 hours after the oral dose taken at home. At Visit 6 (Week 8), patients will be asked to take their oral investigational product at home prior to visiting the clinic. The clinic visit should be scheduled so that the sample taken during this visit is 4 to 6 hours after the oral dose taken at home. For Visits 7 (Week 12), 11 (Week 24), and 13 (Week 32), patients will be asked not to take their oral investigational product prior to visiting the clinic, and a sample will be taken at any time predose on the day of the clinic visits. If the patient has taken his/her oral dose prior to the visit, the sample may be drawn anytime postdose, and the inability to collect a predose sample will not be considered a protocol violation. For the early termination visit, a sample may be drawn anytime if the last dose of oral investigational drug was taken within the last 48 hours. I4V-MC-JADV(c) Clinical Protocol Page 95
194 s At each time point, 3 replicate readings should be made at approximately 30- to 60-second intervals. Blood pressure is recorded as the average of these three readings. A single pulse measurement should be made simultaneously with at least one of the readings at each time point. I4V-MC-JADV(c) Clinical Protocol Page 96
195 I4V-MC-JADV(c) Clinical Protocol Page 97 Attachment 2. Protocol JADV Clinical Laboratory Tests
196 I4V-MC-JADV(c) Clinical Protocol Page 98 Clinical Laboratory Tests Hematologya,b Clinical Chemistrya,b Hemoglobin Serum concentrations of Hematocrit Sodium Erythrocyte count (RBCs) Absolute reticulocyte count Potassium Total bilirubin Mean cell volume Direct bilirubin Mean cell hemoglobin Alkaline phosphatase Mean cell hemoglobin concentration Alanine aminotransaminase/serum glutamic Leukocytes (WBCs) pyruvic transaminase (ALT/SGPT) Absolute count of Neutrophils, segmented Aspartate aminotransferase/serum glutamic oxaloacetic transaminase (AST/SGOT) Neutrophils, juvenile (bands) Lymphocytes Monocytes Eosinophils Basophils Blood urea nitrogen Creatinine Estimated glomerular filtration ratec Calculated creatinine clearance d Uric acid Platelets Calcium Cell morphology CBC, including differential and blood smear Glucosee Albumin Creatine kinase Urinalysisa,b Total protein Color Specific gravity ph Lipid profile includinge: total cholesterol, Protein HDL-C, LDL-C, and triglycerides Glucose ApoA1, ApoB Ketones Blood Bilirubin Urobilinogen Leukocyte esterase Nitrite Microscopic examination of sedimenti Lymphocyte subsets (T, B, NK, and T-cell subsets) Stored serum, plasma, urine, mrna and DNA samples for possible exploratory biomarker analyses Lipoprotein subfractions (NMR) Pregnancy Test (females only)b,f FSH (females only)g Other Tests Hepatitis B surface antigen Hepatitis B surface antibody Hepatitis B core antibody Hepatitis C antibodyh Human immunodeficiency virus Iron studies (iron, TIBC, and ferritin) Thyroid-stimulating hormone plasma concentration (PK sample) PPD, QuantiFERON -TB Goldj, or T-SPOT.TB Rheumatoid factor High-sensitivity C-reactive protein ESRk Anticyclic citrullinated peptide Immunoglobulins (IgG, IgA, IgM)
197 I4V-MC-JADV(c) Clinical Protocol Page 99 Footnotes appear on the following page. Abbreviations: ApoA1 = apolipoprotein A1; ApoB = apolipoprotein B; CBC = complete blood count; CPK = creatine phosphokinase; DNA = deoxyribonucleic acid; ESR = erythrocyte sedimentation rate; FSH = folliclestimulating hormone; HDL-C = high-density lipoprotein cholesterol; Ig = immunoglobulin; LDL-C = lowdensity lipoprotein cholesterol; mrna = messenger ribonucleic acid; NK = natural killer; NMR = nuclear magnetic resonance; PK = pharmacokinetic; PPD = purified protein derivative; RBC = red blood cell; TIBC = total iron binding capacity; WBC = white blood cell. a Assayed/calculated by a sponsor-designated laboratory. b Unscheduled blood chemistry (including CPK), hematology, and urinalysis panels may be performed at the discretion of the investigator. If tests are done to evaluate laboratory results to resume study drug, samples must be assayed centrally. c Estimated glomerular filtration rate for serum creatinine calculated by the central laboratory using the Modification of Diet in Renal Disease isotope dilution mass spectrometry traceable method. d The calculated creatinine clearance will be determined by the central laboratory each time a serum creatinine sample is collected using the Cockcroft-Gault equation: Men: ([140 age] x [weight in kg]) / (72 x [serum creatinine in mg/dl]) or ([140 age] x [weight in kg]) / (0.814 x [serum creatinine in μmol/l]). Women: (above formula) x e Fasting laboratory values for glucose and lipids will be required at baseline, Week 12, Week 24, and Week 52. If a patient attends these visits in a nonfasting state, this will not be considered a protocol violation. These tests may be performed nonfasting at all other visits. f For all women of childbearing potential, a serum pregnancy test (central laboratory) will be performed at Visit 1 and both urine (local laboratory) and serum pregnancy (central laboratory) tests will be performed at Visit 2 to determine study eligibility. Urine pregnancy tests (local laboratory) will also be performed at each subsequent study visit per study schedule. g To confirm postmenopausal status for women 40 and <60 years of age, an FSH test will be performed. Nonchildbearing potential is defined as an FSH 40 miu/ml and a cessation of menses for at least 12 months. h A positive hepatitis C antibody result will be confirmed with a positive hepatitis C virus result. i Microscopic examination of sediment will be performed only if abnormalities are noted on the routine urinalysis. j Test will be required at Visit 1 only to determine eligibility of patient for the study. In countries where the QuantiFERON-TB Gold or T-SPOT.TB test is available and, in the judgment of the investigator, medically preferred as an alternative to the PPD test for the evaluation of mycobacterium tuberculosis (MTB) infection, it may be used instead of the PPD test (positive tests excluded) and may be read locally. k ESR analysis will be performed by the local laboratory.
198 I4V-MC-JADV(c) Clinical Protocol Page 100 Attachment 3. Protocol JADV Hepatic Monitoring Tests for Treatment-Emergent Abnormality Selected tests may be obtained in the event of a treatment-emergent hepatic abnormality and may be required in follow up with patients in consultation with the Lilly designated medical monitor. Hepatic Monitoring Tests Hepatic hematologya Hemoglobin Hematocrit RBC WBC Neutrophils, segmented Lymphocytes Monocytes Eosinophils Basophils Platelets Haptoglobina Hepatic coagulationa Prothrombin time Prothrombin time, INR Hepatic serologiesa,b Hepatitis A antibody, total Hepatitis A antibody, IgM Hepatitis B surface antigen Hepatitis B surface antibody Hepatitis B core antibody Hepatitis C antibody Hepatitis E antibody, IgG Hepatitis E antibody, IgM Hepatic chemistrya Total bilirubin Direct bilirubin Alkaline phosphatase ALT Anti-nuclear antibodya AST GGT Anti-smooth muscle antibodya CPK Abbreviations: ALT = alanine aminotransferase; AST = aspartate aminotransferase; CPK = creatine phosphokinase; GGT = gamma glutamyl transferase; Ig = immunoglobulin; INR = international normalized ratio; RBC = red blood cell; WBC = white blood cell. a b Assayed by Lilly-designated or local laboratory. Reflex/confirmation dependent on regulatory requirements and/or testing availability.
199 I4V-MC-JADV(c) Clinical Protocol Page 101 Attachment 4. Protocol Amendment I4V-MC-JADV(c) Summary Overview Protocol I4V-MC-JADV has been amended. The new protocol is indicated by amendment (c) and will be used to conduct the study in place of any preceding version of the protocol. The overall changes and rationale for the changes made to this protocol are as follows: Selected elements of the gated secondary objectives and analysis have been updated to reflect current endpoints of interest. Change to allow hscrp and TSH retesting during screening to account for intra-patient variability. Changed tuberculosis testing to allow the T.SPOT.TB test as it is approved and validated. Recommended to avoid hyaluronic acid injectsions due to potential to confound efficacy assessments. Specified gout as an exclusion criterion due to the potential to confound efficacy assessments. Selected epro measures to be collected on site tablets to supplement daily diary data Updated duration of period off cdmards prior to study entry to harmonize across protocols Other minor typographical corrections and clarifications not affecting content have been made in the document.
200 I4V-MC-JADV(c) Clinical Protocol Page 102 Revised Protocol Sections Note: Deletions have been identified by strikethroughs. Additions have been identified by the use of underscore.
201 I4V-MC-JADV(c) Clinical Protocol Page 103 Section 2 Synopsis Objectives: The primary objective of the study is to determine whether baricitinib is superior to placebo in the treatment of patients with moderately to severely active rheumatoid arthritis (RA) despite methotrexate treatment (ie, inadequate response to methotrexate [MTX-IR]), as assessed by the proportion of patients achieving a 20% improvement in American College of Rheumatology criteria (ACR20) at Week 12. The major secondary objectives of the study are to evaluate the efficacy of baricitinib versus placebo or adalimumab as assessed by: change from baseline to Week 24 in structural joint damage as measured by modified Total Sharp Score (mtss [van der Heijde method]) compared to placebo change from baseline to Week 12 in Health Assessment Questionnaire-Disability Index (HAQ-DI) score compared to placebo change from baseline to Week 12 in Disease Activity Score modified to include the 28 diarthroidal joint count (DAS28) high-sensitivity C-reactive protein (hscrp) compared to placebo proportion of patients achieving an SDAI score 3.3 at Week 12 compared to placebo proportion of patients achieving ACR20 response at Week 12 compared to adalimumab change from baseline to Week 12 inmean duration of morning joint stiffness in the 7 days prior to Week 12 compared to placebo as collected in electronic diaries change from baseline to Week 12 in DAS28-hsCRP compared to adalimumab mean severity of morning joint stiffness compared to placebo as collected in electronic diarieschange from baseline in the 7 days prior to Week 12 in worst tiredness compared to placebo as collected in electronic diaries mean Worst Tiredness numeric rating scale (NRS) in the 7 days prior to Week 12 compared to placebo as collected in electronic diaries change from baselinemean Worst Pain NRS in the 7 days prior to Week 12 in worst pain compared to placebo as collected in electronic diaries The other secondary objectives of the study are as follows: to evaluate the efficacy of baricitinib as assessed by: o proportion of patients achieving DAS28-hsCRP 3.2 and DAS28-hsCRP <2.6 at Week 12, Week 24, and Week 52 o proportion of patients achieving HAQ-DI improvement 0.22 and 0.3 at Week 12, Week 24, and Week 52 o change from baseline to Week 16 and Week 52 in structural joint damage as measured by mtss o proportion of patients with mtss change 0 from baseline at Week 16, Week 24, and Week 52 o change from baseline to Week 16, Week 24, and Week 52 in joint space narrowing and bone erosion scores o proportion of patients achieving ACR20 at Week 24 and Week 52 o proportion of patients achieving 50% improvement in American College of Rheumatology criteria (ACR50) at Week 12, Week 24, and Week 52 o proportion of patients achieving 70% improvement in American College of RheumatologyACR criteria (ACR70) at Week 12, Week 24, and Week 52 o percentage change from baseline to Week 12, Week 24, and Week 52 in individual components of the ACR Core Set o change from baseline through Week 52 in DAS28-hsCRP o change from baseline through Week 52 in DAS28-erythrocyte sedimentation rate (ESR) o proportion of patients achieving DAS28-ESR 3.2 and DAS28-ESR <2.6 at Week 12, Week 24, and Week 52 o change from baseline to Week 24 and Week 52 in HAQ-DI score o change from baseline to Week 12, Week 24, and Week 52 in Clinical Disease Activity Index (CDAI)CDAI score
202 I4V-MC-JADV(c) Clinical Protocol Page 104 o change from baseline to Week 12, Week 24, and Week 52 in Simplified Disease Activity Index (SDAI) score o proportion of patients achieving a CDAI score 2.8 at Week 12, Week 24, and Week 52 o proportion of patients achieving an SDAI score 3.3 at Week 12, Week 24, and Week 52 o proportion of patients achieving European League Against Rheumatism (ACR/EULAR) Responder Index based on the 28 joint count (EULAR28) remission according to the Booleanbased definition at Week 12, Week 24 and Week 52 o proportion of patients achieving moderate and good EULAR responses based on the 28-joint DAS at Week 12, Week 24, and Week 52 o evaluation of hybrid ACR (bounded) response at Week 12, Week 24 and Week 52 o change from baseline to Week 12, Week 24, and Week 52 in Functional Assessment of Chronic Illness Therapy Fatigue (FACIT-F) scale scores o change from baseline throughto Week 12, Week 24, and Week 52 in each scale and the summary scores for the Mental Component Score (MCS) and Physical Component Score (PCS) of the Medical Outcomes Study 36-Item Short Form Health Survey Version 2 Acute (SF-36v2 Acute) o change from baseline throughto Week 12, Week 24, and Week 52 in European Quality of Life-5 Dimensions-5 Level (EQ-5D-5L) scores o change from baseline throughto Week 12, Week 24, and Week 52 in Work Productivity and Activity Impairment-Rheumatoid Arthritis (WPAI-RA) scores o change from baseline through Week 52 in evaluation of healthcare resource utilization through Week 52 o change from baseline to Week 12, Week 24, and Week 52 in duration of morning joint stiffness assessed at the visit using an electronic patient-reported outcomes (epro) tablet o change from baseline to Week 12, Week 24, and Week 52 in Worst Tiredness NRS assessed at the visit using an epro tablet o change from baseline to Week 12, Week 24, and Week 52 in Worst Pain NRS assessed at the visit using an epro tablet to characterize the pharmacokinetics (PK) of baricitinib, determine the magnitude of within- and between-patient variability, and identify the potential factors that may have an impact on the PK of baricitinib to characterize the baricitinib exposure-response time course for key efficacy and safety endpoints and to identify potential factors that may have an impact on these efficacy or safety endpoints of baricitinib The exploratory objective of the study is: to compare baricitinib and adalimumab as regards to various efficacy measures Criteria for Evaluation: Efficacy The following efficacy measures will be assessed in this study: ACR20, ACR50, and ACR70 indices HAQ-DI mtss (includes joint space narrowing score and bone erosion score) DAS28-hsCRP and DAS28-ESR Hybrid ACR (bounded) response measure SDAI CDAI EULAR28 EULAR responses based on DAS28
203 I4V-MC-JADV(c) Clinical Protocol Page 105 Statistical Methods: Sample size: Approximately 1280 patients (480 in the baricitinib group, 480 in the placebo group, and 320 in the adalimumab group) will provide >95% power to detect a difference between baricitinib and placebo groups in ACR20 response rate (60% vs 35%) at Week 12 based on a 2-sided significance level of 0.05; approximately 94% power to detect a difference in mtss between baricitinib and placebo groups for an effect size of 0.25, based on a 2-sided t-test at a significance level of 0.04 (this assumes 90% of randomized patients will have x-ray data available for the mtss analysis at Week 24); and approximately 9193% power for the noninferiority analysis of ACR20 response rate at Week 12 between baricitinib and adalimumab groups, where a response rate of 60% for baricitinib and adalimumab, a noninferiority margin of 12%, and a 1-sided significance level of are assumed. Analysis Population: The efficacy and health outcome analyses will be conducted on an intention-to-treat (ITT) basis. The ITT analysis set will include all data from all randomized patients treated with at least 1 dose of the study drug. Analysis of structural progression (van der Heijde mtss) will be conducted on the ITT population using patients with available baseline and at least 1 postbaseline value. The primary and major secondary analyses will be repeated using the per-protocol set (PPS), which excludes patients with significant protocol violations. Safety analyses will be performed on the safety population, defined as all randomized patients who received at least 1 dose of the study drug. Missing Data Imputation: 1. Nonresponder imputation (NRI): All patients who discontinue the study or the study treatment at any time for any reason will be defined as nonresponders for the NRI analysis for categorical variables, such as ACR20/50/70, from the time of discontinuation and onward. Patients who are eligible forreceive rescue therapy at Week 16 will be analyzed as nonresponders after Week 16 and onward. Randomized patients without available data at a postbaseline visit will be defined as nonresponders for the NRI analysis at that visit. 2. Linear extrapolation method: The linear extrapolation method will be used for analysis of the structural progression endpoint (van der Heijde mtss) to impute missing data at time points when analyses are conducted (including the analyses at Week 24 and Week 52). For patients who discontinue the study or the study treatment or for patients who miss a radiograph for any reason, baseline data and the most recent radiographic data prior to discontinuation or the missed radiograph, adjusted for time, will be used for linear extrapolation to impute missing data at subsequent time points. For patients who receive rescue therapy starting at Week 16 or at any time point thereafter, baseline data and the most recent radiographic data prior to initiation of rescue therapy, adjusted for time, will be used for linear extrapolation. All patients originally randomized to placebo will be switched to active treatment at Week 24; thus, there will be no observed data for the placebo arm for the structural comparison at subsequent time points. Therefore, baseline data and the most recent radiographic data prior to initiation of the new therapy, adjusted for time, will be used for linear extrapolation at Week 52. The linear extrapolation method has been established as an appropriate missing data imputation method in other Phase 3 RA trials. Missing postbaseline values will be imputed only if both a baseline value and a postbaseline value from another time point are available, as long as the patient was on the same treatment at each applicable time point. 3. Multiple imputation: Other methods than linear extrapolation that include multiple imputation will be employed for analysis of the structural progression endpoint (van der Heijde mtss) to impute missing data at Week 24. Details will be provided in the statistical analysis plan (SAP). 4. Modified baseline observation carried forward (mbocf): The mbocf method will be used for the analysis of major secondary continuous endpoints (unless otherwise stated). For patients who discontinue the study or the study treatment because of an AE, including death, the baseline observation will be carried forward to the corresponding endpoint for evaluation. For patients who discontinue the study for reason(s) other than an AE, the last nonmissing postbaseline observation prior to discontinuation will be carried forward to the corresponding endpoint for evaluation. For patients who are eligible forreceive rescue therapy starting at Week 16, the last nonmissing observation at or before rescue will be carried forward to subsequent time points for evaluation.
204 I4V-MC-JADV(c) Clinical Protocol Page Modified last observation carried forward (mlocf): For all continuous measures including safety analyses, the mlocf will be a general approach to impute missing data unless otherwise specified. For patients who are eligible forreceive rescue therapy starting at Week 16, the last nonmissing observation at or before rescue will be carried forward to subsequent time points for evaluation. For all other patients discontinuing from the study or the study treatment for any reason, the last nonmissing postbaseline observation before discontinuation will be carried forward to subsequent time points for evaluation. If theafter mlocf imputation, data from patients with nonmissing baseline and postbaseline observation is missing, then itobservations will not be included in the analysis even if the baseline observation is not missing. analyses. The mlocf will be used as a sensitivity method to the mbocf method with respect to the major secondary endpoints. 6. Other methods for data imputation for analysis of key endpoints other than mtss may be used and will be described in the SAP Primary and Major Secondary Analyses: A logistic regression model with treatment, region, and joint erosion status in the model will be used to test the treatment difference between baricitinib and placebo in the proportion of patients achieving ACR20 response at Week 12. The NRI method will be used to impute missing data. An analysis of covariance (ANCOVA) model with baseline score, region, joint erosion status, and treatment group in the model will be used to test the treatment difference between baricitinib and placebo in change from baseline to Week 24 of mtss scores. The linear extrapolation method will be used to impute missing data. An analysis of covariance (ANCOVA) model with treatment, region, joint erosion status, and baseline score in the model will be used to test the treatment difference between baricitinib and placebo in change from baseline to Week 12 in HAQ-DI. The mbocf approach will be used to impute missing data. An ANCOVA model with treatment, region, joint erosion status, and baseline score in the model will be used to test the treatment difference between baricitinib and placebo in change from baseline to Week 12 in DAS28-hsCRP. The mbocf approach will be used to impute missing data. A logistic regression model with treatment, region, and joint erosion status in the model will be used to test the treatment difference between baricitinib and placebo in the proportion of patients achieving an SDAI score 3.3 response at Week 12. The NRI method will be used to impute missing data. A logistic regression model with treatment, region, and joint erosion status in the model will be used to test the treatment difference between baricitinib and adalimumab in the percentage of patients achieving ACR20 response at Week 12. A (100 alpha)%95% confidence interval (CI) for the treatment difference will be constructed using the method described by Newcombe-Wilson without continuity correction. If the lower bound of the (100 alpha)%95% CI is >-12%, Lilly will conclude that baricitinib is noninferior to adalimumab with respect to ACR20. If the lower bound of the (100 alpha)%95% CI >0%, Lilly will conclude that baricitinib is superior to adalimumab with respect to ACR20. The NRI method will be used to impute missing data. For duration of morning joint stiffness, severity of morning joint stiffness, worst tirednessworst Tiredness NRS, and worst painworst Pain NRS, an ANCOVA analysis with treatment, region, joint erosion status, and baseline score (Day 1 on the diaries) in the model will be used to test the treatment difference between baricitinib and placebo in mean diary scores in the 7 days prior to Week 12. An ANCOVA model with treatment, region, joint erosion status, and baseline score in the model will be used to test the treatment difference between baricitinib and adalimumab in change from baseline to Week 12 in DAS28-hsCRP. The mbocf approach will be used to impute missing data. Safety: All safety data will be descriptively summarized by treatment groups and analyzed using the safety population. Treatment-emergent AEs (TEAEs) are defined as AEs that first occurred or worsened in severity on or after the date of first dose of study treatment. The number and percentage of TEAEs will be summarized using MedDRA (Medical Dictionary for Regulatory Activities) for each system organ class and each preferred term by treatment group. TEAEs will also be summarized by relationship to treatment and by severity within each group. SAEs and AEs that lead to study drug discontinuation will also be summarized by group. Fisher s exact test will be used to perform pairwise comparisons among the treatment groups. Change from baseline in quantitative clinical chemistry will be analyzed using ANCOVA with treatment and baseline value in the model. Categorical variables, including the incidence of abnormal values and incidence of AESIs, will be summarized by frequency and percentage of patients in corresponding categories. Shift tables will be presented for selected measures. Change from baseline to postbaseline in vital signs, QIDS-SR16, and body weight will be analyzed using
205 I4V-MC-JADV(c) Clinical Protocol Page 107 ANCOVA with treatment and baseline value in the model. Planned Analyses: The analysis of primary and major secondary endpoints (except for x-ray data) will occur when all patients complete the Week 24 study visit. A final analysis will occur when all patients complete the study. Section 4: Abbreviations EULAR28 European League Against Rheumatism Responder Index based on the 28 joint count Section 6.2: Secondary Objectives The major secondary objectives of the study are to evaluate the efficacy of baricitinib versus placebo or adalimumab as assessed by: change from baseline to Week 24 in structural joint damage as measured by modified Total Sharp Score (mtss [van der Heijde method]) compared to placebo change from baseline to Week 12 in Health Assessment Questionnaire-Disability Index (HAQ-DI) score compared to placebo change from baseline to Week 12 in DAS28-high-sensitivity C-reactive protein (hscrp) compared to placebo proportion of patients achieving an SDAI score 3.3 at Week 12 compared to placebo proportion of patients achieving ACR20 response at Week 12 compared to adalimumab change from baseline to Week 12 inmean duration of morning joint stiffness in the 7 days prior to Week 12 compared to placebo as collected in electronic diaries change from baseline to Week 12 in DAS28-hsCRP compared to adalimumab mean severity of morning joint stiffness compared to placebo as collected in electronic diarieschange from baseline in the 7 days prior to Week 12 in worst tiredness compared to placebo as collected in electronic diaries mean Worst Tiredness numeric rating scale (NRS) in the 7 days prior to Week 12 compared to placebo as collected in electronic diaries change from baselinemean Worst Pain NRS in the 7 days prior to Week 12 in worst pain compared to placebo as collected in electronic diaries The other secondary objectives of the study are as follows: to evaluate the efficacy of baricitinib as assessed by: o proportion of patients achieving DAS28-hsCRP 3.2 and DAS28-hsCRP <2.6 at Week 12, Week 24, and Week 52 o proportion of patients achieving HAQ-DI improvement 0.22 and 0.3 at Week 12, Week 24, and Week 52 o change from baseline to Week 16 and Week 52 in structural joint damage as measured by mtss o proportion of patients with mtss change 0 from baseline at Week 16, Week 24, and Week 52
206 I4V-MC-JADV(c) Clinical Protocol Page 108 o change from baseline to Week 16, Week 24, and Week 52 in joint space narrowing and bone erosion scores o proportion of patients achieving ACR20 at Week 24 and Week 52 o proportion of patients achieving ACR50 at Week 12, Week 24, and Week 52 o proportion of patients achieving 70% improvement in ACR criteria (ACR70) at Week 12, Week 24, and Week 52 o percentage change from baseline to Week 12, Week 24, and Week 52 in individual components of the ACR Core Set o change from baseline through Week 52 in DAS28-hsCRP o change from baseline through Week 52 in DAS28-erythrocyte sedimentation rate (ESR) o proportion of patients achieving DAS28-ESR 3.2 and DAS28-ESR <2.6 at Week 12, Week 24, and Week 52 o change from baseline to Week 24 and Week 52 in HAQ-DI score o change from baseline to Week 12, Week 24, and Week 52 in CDAI score o change from baseline to Week 12, Week 24, and Week 52 in SDAI score o proportion of patients achieving a CDAI score 2.8 at Week 12, Week 24, and o Week 52 proportion of patients achieving an SDAI score 3.3 at Week 12, Week 24, and Week 52 o proportion of patients achieving ACR/EULAR Responder Index based on the 28 joint count (EULAR28)remission according to the Boolean-based definition at Week 12, Week 24, and Week 52 o proportion of patients achieving moderate and good EULAR responses based on the 28-joint DAS at Week 12, Week 24, and Week 52 o evaluation of hybrid ACR (bounded) response at Week 12, Week 24 and Week 52 o o o o o o o change from baseline to Week 12, Week 24, and Week 52 in Functional Assessment of Chronic Illness Therapy Fatigue (FACIT-F) scale scores change from baselinethrough to Week 12, Week 24, and Week 52 in each scale and the summary scores for the Mental Component Score (MCS) and Physical Component Score (PCS) of the Medical Outcomes Study 36-Item Short Form Health Survey Version 2 Acute (SF-36v2 Acute) change from baseline throughto Week 12, Week 24, and Week 52 in European Quality of Life-5 Dimensions-5 Level (EQ-5D-5L) scores change from baseline throughto Week 12, Week 24, and Week 52 in Work Productivity and Activity Impairment-Rheumatoid Arthritis (WPAI-RA) scores change from baseline through Week 52 in evaluation of healthcare resource utilization through Week 52 change from baseline to Week 12, Week 24, and Week 52 in duration of morning joint stiffness assessed at the visit using an electronic patient-reported outcomes (epro) tablet change from baseline to Week 12, Week 24, and Week 52 in Worst Tiredness NRS assessed at the visit using an epro tablet
207 I4V-MC-JADV(c) Clinical Protocol Page 109 o change from baseline to Week 12, Week 24, and Week 52 in Worst Pain NRS assessed at the visit using an epro tablet to characterize the pharmacokinetics (PK) of baricitinib, determine the magnitude of within- and between-patient variability, and identify the potential factors that may have an impact on the PK of baricitinib to characterize the baricitinib exposure-response time course for key efficacy and safety endpoints and to identify potential factors that may have an impact on these efficacy or safety endpoints of baricitinib Section 7.1.1: Screening Period This is a screening period of not less than 3 days and not more than 42 days. In exceptional circumstances, the screening window can be extended after consultation with the Sponsor. Patients will be screened for study eligibility at Visit 1 and will be randomized at Visit 2 to continue in Part A. Section 7.2: Discussion of Design and Control To prevent potential unblinding due to observed efficacy or laboratory changes, a dual assessor approach will be used to evaluate efficacy and safety. The Joint Assessor (or designee) should be a rheumatologist or skilled arthritis assessor. The Joint Assessor will be responsible for completing the joint counts. To ensure consistent joint evaluation throughout the trial, individual patients should be evaluated by the same Joint Assessor for all study visits. The Joint Assessor must not have access to or discuss with the patient the patient-reported assessments, Physician s Global Assessment of Disease Activity (visual analog scale [VAS]), and safety assessments. Section 8.1.1: Inclusion Criteria [3] have moderately to severely active RA defined as the presence of at least 6/68 tender joints and at least 6/66 swollen joints a. If significant surgical treatment of a joint has been performed, that joint cannot be counted in the TJC and SJC for entry or enrollment purposes. Section 8.1.2: Exclusion Criteria [9] are currently receiving concomitant treatment with MTX, hydroxychloroquine, and sulfasalazine or combination of any 3 cdmards [10] are currently receiving or have received cdmards (eg, gold salts, cyclosporine, azathioprine, or any other immunosuppressives) other than MTX, hydroxychloroquine (up to 400 mg/day), or sulfasalazine (up to 3000 mg/day) within 84 weeks prior to study entry a. Doses of hydroxychloroquine or sulfasalazine should be stable for at least 8 weeks prior to study entry.; if either has been recently discontinued, the patient must not have taken any dose within 4 weeks prior to study entry.
208 I4V-MC-JADV(c) Clinical Protocol Page 110 b. Immunosuppression related to organ transplantation is not permitted. [16] have had 3 or more joints injected with intraarticular corticosteroids or hyaluronic acid within 2 weeks prior to study entry or within 6 weeks prior to planned randomization a. Joints injected with intraarticular corticosteroids or hyaluronic acid within 2 weeks prior to study entry or within 6 weeks prior to planned randomization cannot be counted in the TJC and SJC for entry or enrollment purposes. [19] have a diagnosis of any systemic inflammatory condition other than RA such as, but not limited to, juvenile chronic arthritis, spondyloarthropathy, Crohn s disease, ulcerative colitis, psoriatic arthritis, or active vasculitis or gout a. Patients with secondary Sjögren s syndrome are not excluded. [27] have, or have a history of, lymphoproliferative disease; or have signs or symptoms suggestive of possible lymphoproliferative disease, including lymphadenopathy or splenomegaly; or have active primary or recurrent malignant disease; or have been in remission from clinically significant malignancy for <5 years a. Patients with cervical carcinoma in situ that has been resected with no evidence of recurrence or metastatic disease for at least 3 years may participate in the study. b. Patients with basal cell or squamous epithelial skin cancers that have been completely resected with no evidence of recurrence for at least 3 years may participate in the study. Section 8.2.1: Inclusion Criteria Entered patients are eligible for enrollment into the study (ie, eligible for randomization) only if they continue to meet all of the inclusion entry criteria for entry (see above) at the time of randomization includingand meet the following enrollment criteria: [48] have an hscrp measurement 6 mg/l based on Visit 1 laboratory results by central laboratory testing The hscrp measurement may be repeated once within approximately 2 weeks of the initial value, and the value resulting from repeat testing may be accepted for enrollment eligibility if it meets the eligibility criterion.
209 I4V-MC-JADV(c) Clinical Protocol Page 111 [49] have at least 1 joint erosion in hand, wrist, or foot joints based on radiographic interpretation by the central reader and be rheumatoid factor or anticyclic citrullinated peptide (anti-ccp) antibody positive; or have at least 3 joint erosions in hand, wrist, or foot joints based on radiographic interpretation by the central reader regardless of rheumatoid factor or anti-ccp antibody status (radiographic images acquired within 4 weeks of study entry may be submitted to the central reader to confirm eligibility). If a patient undergoes rescreening, radiographic images acquired as part of initial screening and within 10 weeks of randomization may be used. Section 8.2.2: Exclusion Criteria [50] have screening laboratory test values, including thyroid-stimulating hormone (TSH), outside the reference range for the population or investigative site that, in the opinion of the investigator, pose an unacceptable risk for the patient s participation in the study a. Patients who are receiving thyroxine as replacement therapy may participate in the study provided stable therapy has been administered for 12 weeks and TSH is within the laboratory s reference range. The TSH test may be repeated once within approximately 2 weeks of the initial value, and the value resulting from repeat testing may be accepted for enrollment eligibility if it meets the eligibility criterion. [51] have any of the following specific abnormalities on screening laboratory tests: AST or ALT >1.5 times the ULN total bilirubin 1.5 times the ULN hemoglobin <10.0 g/dl (100.0 g/l) total white blood cell count <2500 cells/l neutropenia (absolute neutrophil count <1200 cells/l) lymphopenia (lymphocyte count <750 cells/l) thrombocytopenia (platelets <100,000/L) egfr <40 ml/min/1.73 m2 In the case of any of the aforementioned laboratory abnormalities, the tests may be repeated once within approximately 2 weeks of the initial values, and values resulting from repeat testing may be accepted for enrollment eligibility if they meet the eligibility criterion. [54] have evidence of active TB as documented by a positive purified protein derivative (PPD) test ( 5-mm induration between approximately 2 and 3 days after application, regardless of vaccination history), medical history, clinical symptoms, and abnormal chest x-ray at screening
210 I4V-MC-JADV(c) Clinical Protocol Page 112 a. The QuantiFERON -TB Gold or the T-SPOT.TB test (if available) may be used instead of the PPD test. Patients are excluded from the study if the test is not negative and there is clinical evidence of active TB. [55] have evidence of latent TB (as documented by a positive PPD, no clinical symptoms consistent with active TB, and a normal chest x-ray at screening) unless patients complete at least 4 weeks of appropriate treatment prior to randomization and agree to complete the remainder of treatment while in the trial a. If the PPD test is positive and the patient has no medical history or chest x-ray findings consistent with active TB, the patient may have a QuantiFERON-TB Gold or T-SPOT.TB test (if available). If the test results are not negative, the patient will be considered to have latent TB (for purposes of this study). b. The QuantiFERON-TB Gold or T-SPOT.TB test (if available) may be used instead of the PPD test. If the test results are positive, the patient will be considered to have latent TB. If the test is not negative, the test may be repeated once within approximately 2 weeks of the initial value. If the repeat test results are again not negative, the patient will be considered to have latent TB (for purposes of this study). c. Exceptions include patients with a history of active or latent TB who have documented evidence of appropriate treatment. Section : Interruption of Study Drug
211 I4V-MC-JADV(c) Clinical Protocol Page 113 Table JADV.1. Criteria for Temporary Discontinuation of Investigational Product Hold Investigational Product if the Following Laboratory Test Results Occur: WBC count <2000 cells/µl ANC <1000 cells/lµl Lymphocyte count <500 cells/µl Platelet count <75,000/µL egfr <40 ml/min/1.73 m2 (from serum creatinine) for patients receiving the baricitinib 4 mg QD dose egfr <30 ml/min/1.73 m2 (from serum creatinine) for patients receiving the baricitinib 2 mg QD dose ALT or AST >5 times ULN Investigational Product May Be Resumed after Approval from Lilly (or its Designee) and When: WBC count 2500 cells/µl ANC >2000 cells/µl Lymphocyte count 750 cells/µl Platelet count 100,000/µL egfr 50 ml/min/1.73 m2 egfr 40 ml/min/1.73 m2 ALT and AST return to <2 times ULN and investigational product is not considered to be the cause of enzyme elevation Hemoglobin 10 g/dl Hemoglobin <8 g/dl Abbreviations: ALT = alanine aminotransferase; ANC = absolute neutrophil count; AST = aspartate aminotransferase; egfr = estimated glomerular filtration rate; min = minute; QD = once daily; ULN = upper limit of normal; WBC = white blood cell. Section 9.3: Method of Assignment to Treatment Patients who meet all criteria for enrollment will be randomized in a 3:3:2 ratio (baricitinib:placebo:adalimumab) to double-blind treatment at Week 0 (Visit 2). Randomization will be stratified by region and joint erosion status (1-2 joint erosionerosions plus seropositivity versus at least 3 joint erosions). Assignment to treatment groups will be determined by a computer-generated random sequence using an interactive voice-response system (IVRS). Section 9.8: Concomitant Therapy Treatment with concomitant DMARDs during the study is permitted as outlined below and in the inclusion/exclusion criteria: Use of concomitant oral MTX for at least 12 weeks with treatment at a stable dose of 7.5 to 25 mg/week (or the equivalent injectable dose) for at least 8 weeks prior to entry into the study is required for study participation. Patients should remain on the same dose of MTX throughout Part A and Part B of the study; however, the MTX dose may be adjusted for safety reasons. Patients may also be on concomitant hydroxychloroquine or sulfasalazine. If receiving hydroxychloroquine (up to 400 mg/day) or sulfasalazine (up to 3000 mg/day), the patient must be receiving a stable dose for at least 8 weeks prior to entry into the study, and the dose must remain stable throughout the study. Concomitant use of NSAIDs is permitted during Part A of the study only if the patient was on a stable dose for at least 6 weeks before planned randomization. Increase of NSAID dose and/or introduction of new NSAIDs are not permitted during Part A of the study unless a patient receives rescue therapy. After rescue, new NSAIDs or increases in doses of ongoing concomitant NSAIDs are permitted. Dose reductions and/or termination of NSAIDs are permitted at any time.
212 I4V-MC-JADV(c) Clinical Protocol Page 114 During the study, concomitant use of analgesics is permitted. Increase of an analgesic dose and/or introduction of a new analgesic is not permitted during Part A of the study unless a patient receives rescue therapy. After rescue, new analgesics or increases in doses of ongoing concomitant analgesics are permitted. Dose reductions and/or termination of analgesics are permitted at any time. If an unforeseen intra articular glucocorticoid injection is required during the study, it will be noted as a protocol deviation, and such joints will be excluded from future evaluation for the remainder of the study. Prednisone (or equivalent) at doses up to 10 mg per day is allowed during this study but must be maintained at stable levels from 6 weeks prior to randomization through the treatment phase of the study, unless a patient receives rescue therapy. After rescue, new corticosteroids or increases in doses of ongoing concomitant corticosteroids are permitted. Patients who were not previously on prednisone (or equivalent) prior to randomization should not initiate corticosteroid therapy during the study. Patients should not receive other systemic corticosteroids during the study including intra-muscular or intra-articular corticosteroids. Topical, intranasal, intra-ocular, and inhaled corticosteroids are permitted. If an unforeseen intra-articular glucocorticoid injection is required during the study, it will be noted as a protocol deviation. It is strongly encouraged to avoid hyaluronic acid injections during the course of the study. If required, use should be minimized and discussed with the sponsor. Local standard of care should be followed for concomitant administration of folic acid. The following concomitant medications are prohibited after randomization: Live vaccines, including herpes zoster vaccination. Nonlive seasonal vaccinations and/or emergency vaccination, such as rabies or tetanus vaccinations, are allowed. Any DMARDs, other than stable treatment withdoses of background DMARDs being used at the time of study entry hydroxychloroquine (up to 400 mg/day), sulfasalazine (up to 3000 mg/day), or MTX Any biologic therapy (such as TNF, interleukin-1, IL-6, T-cell-, or B-cell-targeted therapies) for any indication Any interferon therapy (such as Roferon-A, Intron-A, Rebetron, Alferon-N, Peg-Intron, Avonex, Betaseron, Infergen, Actimmune, Pegasys) Any parenteral corticosteroid administered by intramuscular or IV injection Section : Van der Heijde Modified Total Sharp Score Structural progression will be measured using the mtss. During screening, all patients meeting entry criteria will have a single posteroanterior radiographic assessment of the left and right hand/wrist and a single dorsoplantar radiographic image taken of the left and right foot. Alternatively, images taken within 4 weeks prior to baseline may be used. Images taken at screening (or within 4 weeks of screening) will be reviewed centrally by qualified readers for evidence of erosive bony change(s). These initial radiographs will serve as the baseline radiographs for comparison throughout the study. If a patient undergoes rescreening,
213 I4V-MC-JADV(c) Clinical Protocol Page 115 radiographic images acquired as part of initial screening and within 10 weeks of randomization may be used. Section : ACR/EULAR Rheumatoid Arthritis Remission Two ACR/EULAR definitions of RA remission will be evaluated, a Boolean-based definition and an index-based definition (Felson et al. 2011). Boolean-based Definition of Remission: All 4 criteria below must be met: o TJC28 1 o SJC28 1 o hscrp 1 mg/dl (10 mg/l) o patient global DAS on VAS (0 to 10.0) 1 Index-based Definition of Remission (see Section ): o SDAI 3.3 Section : Simplified Disease Activity Index The SDAI is a tool for measurement of disease activity in RA that integrates measures of physical examination, acute phase response, patient self-assessment, and evaluator assessment. The SDAI is calculated by adding together scores from the following assessments: number of swollen joints (0 to 28), number of tender joints (0 to 28), CRP in mg/dl (0.1 to 10.0), patient global DAS on VAS (0 to 10.0), and evaluator global health score on VAS (0 to 10.0) (Aletaha and Smolen 2005) DiseaseDisease remission according to ACR/EULAR index-based definition of remission is defined as an SDAI score of 3.3 (Felson et al. 2011). Section : European League Against Rheumatism Responder Index Assessments of patients with RA by EULAR28EULAR response criteria will be used to categorize patients as nonresponders, moderate responders, good responders, or responders (moderate + good responders) according to Table JADV.5 (van Gestel et al. 1998). Section 10.2: Health Outcome Measures The health outcome measures listed below will be administered. Country-specific translations of each measure will be available in this study. Morning Joint Stiffness Severity Numeric Rating Scale (NRS): The Morning Joint Stiffness NRS is a patient-administered, single-item, 11-point horizontal scale anchored at 0 and 10, with 0 representing no joint stiffness and 10 representing joint stiffness as bad as you can imagine. Patients rate their morning joint stiffness
214 I4V-MC-JADV(c) Clinical Protocol Page 116 each day (using an electronic handheld diary) by selecting the 1 number that describes their overall level of joint stiffness at the time they woke up. Duration of morning joint stiffness: o Electronic patient diary: The duration of morning joint stiffness is a patient-administered item that allows for the patients to enter the length of time in minutes (using 15-minute increments) that their morning joint stiffness lasted each day (using an electronic handheld diary). o Electronic PRO (tablet): The duration of morning joint stiffness is a patientadministered item that allows for the patients to enter the length of time in minutes (using 15-minute increments) that their morning joint stiffness lasted on the day prior to that visit (using an electronic patient-reported outcomes [epro] tablet). As the protocol will be approved prior to readiness of some tablet measures, it will not be considered a protocol violation that these measures are not collected until tablet readiness. Recurrence of joint stiffness: The recurrence of joint stiffness throughout the day is a patient-administered single-item question assessed each day (using an electronic handheld diary) that asks Did you have any joint stiffness throughout today, other than when you woke up? (Yes/No)). Tiredness Severity Numeric Rating Scale (Worst Tiredness NRS): o Electronic patient diary: The Worst Tiredness NRS is a patient-administered, single-item, 11-point horizontal scale anchored at 0 and 10, with 0 representing no tiredness and 10 representing as bad as you can imagine. Patients rate their tiredness each day (using an electronic handheld diary) by selecting the one number that describes their worst level of tiredness. o Electronic PRO (tablet): The Worst Tiredness NRS is a patient-administered, single-item, 11-point horizontal scale anchored at 0 and 10, with 0 representing no tiredness and 10 representing as bad as you can imagine. Patients rate their tiredness each day (using an electronic handheld diary) by selecting the 1 one number that describes their worst level of tiredness during the past 24 hours. Pain Severity of joint pain Numeric Rating Scale (Worst Pain NRS): o Electronic patient diary: The Worst Pain NRS is a patient-administered, single-item, 11-point horizontal scale anchored at 0 and 10, with 0 representing no pain and 10 representing joint pain as bad as you can imagine. Patients rate their joint pain each day (using an electronic handheld diary) by selecting the 1 number that describes their worst level of joint pain during the past 24 hours. o Electronic PRO (tablet): The Worst Pain NRS is a patient-administered, single-item, 11-point horizontal scale anchored at 0 and 10, with 0 representing no pain and 10 representing joint pain as bad as you can imagine. Patients rate their joint pain by selecting the one number that describes their worst level of joint pain during the past 24 hours.
215 I4V-MC-JADV(c) Clinical Protocol Page 117 Section : Serious Adverse Events In addition, any patient who is permanently discontinued from study drug for an AE or abnormal laboratory result should have the AE or abnormal laboratory value reported as an SAE (reported as Other Reason Serious). Section : Measures Evaluated During Screening and Treatment Periods Vital signs and physical characteristics. Vital signs (sitting blood pressure and heart rate) will be measured at times indicated in the study schedule (Attachment 1). Subjects should be seated and relaxed with both feet on the floor for at least 5 minutes prior to taking measurements. Simultaneous blood pressure and pulse measurements should be made using either automated or manual equipment. Three replicate blood pressure readings should be made at each time point at approximately 30- to 60-second intervals. A single pulse measurement should be taken simultaneously with at least one of the blood pressure readings. Blood pressure and pulse measurements should be made using either automated or manual equipment. If measurements are machine averaged, the average blood pressure reading should be recorded on the CRF. If measurements are manual or the machine does not provide an average reading, then each individual reading should be recorded on the CRF. Measurements should be made before any scheduled blood draws. Any clinically significant findings that result in a diagnosis should be captured on the CRF and reported as an AE. Additional measurements of vital signs may be performed at the discretion of the investigator. Physical characteristics including weight and waist circumference will be measured at times indicated in the study schedule (Attachment 1). Section : Samples for Standard Laboratory Testing Standard laboratory tests, including chemistry, hematology, and urinalysis panels, will be performed. Specifically, hscrp results will be blinded after Visit 2. Serum and/or urine pregnancy tests will be performed at each visit as described in the study schedule (Attachment 1). Blood and urine samples will be collected for other laboratory tests at the times specified in the study schedule (Attachment 1). Blood will be collected by venipuncture. Attachment 2 lists the specific tests that will be performed for this study. Section 12.1: Determination of Sample Size Approximately 1280 patients (480 in the baricitinib group, 480 in the placebo group, and 320 in the adalimumab group) will provide: >95% power to detect a difference between the baricitinib and placebo treatment groups in ACR20 response rate (60% versus 35%) at Week 12 based on a 2-sided chi-square test at a significance level of 0.05 approximately 94% power to detect a difference in mtss between the baricitinib and placebo treatment groups for an effect size of 0.25 (ie, difference in means divided by common standard deviation [SD]), based on a 2-sided t-test at a significance level of This assumes 90% of randomized patients will have x-ray data available for the mtss analysis at Week 24. Based on a recent publication by van der Heijde et al (2013), the effect size from the comparison of tofacitinib 10 mg BID versus placebo in mtss at
216 I4V-MC-JADV(c) Clinical Protocol Page 118 Month 6 was approximately If the effect size in Study JADV was at least 0.21, then power of the mtss analysis is at least 85%. approximately 9193% power for the noninferiority analysis of ACR20 response rate at Week 12 between the baricitinib and adalimumab treatment groups, where a response rate of 60% for baricitinib and adalimumab and a 1-sided significance level of are assumed. This power calculation adopted a fixed noninferiority margin of 12%. There is growing support for this margin in recent clinical trials conducted in the MTX- IR patient population (Schiff et al. 2012). Section : General Considerations Statistical analysis of this study will be the responsibility of Eli Lilly and Company, although implementation of the statistical analysis plan (SAP) may be delegated to a third party. A detailed SAP describing the statistical methodologies will be developed by Lilly or its designee. All statistical tests of treatment effects, besides noninferiority tests specified in Section , will be performed at a 2-sided significance level of 0.05, unless otherwise stated. Treatment comparisons of discrete efficacy variables will be made using a logistic regression analysis with region, joint erosion status (1-2 joint erosionerosions plus seropositivity vs at least 3 joint erosions), and treatment group in the model. The percentages, difference in percentages, and (100*(1-alpha)% confidence interval (CI) of the difference in percentages will be reported. Treatment-by-region interaction will be added to the logistic regression model of the primary and key secondary categorical variables as a sensitivity analysis. If this interaction is significant at a 2-sided 0.1 level, further inspection will be used to assess whether the interaction is quantitative (ie, the treatment effect is consistent in direction but not size of effect) or qualitative (the treatment is beneficial for some but not all regions). Treatment comparisons of continuous efficacy variables will be made using analysis of covariance (ANCOVA) with region, joint erosion status, treatment group, and baseline value in the model. Type III sums of squares for the least-squares means will be used for the statistical comparison; (100*(1-alpha)% CI will also be reported. Sensitivity analysis will be performed for key secondary continuous variables by adding the treatment-by-baseline interaction to the ANCOVA model. If this interaction is significant at a 2-sided 0.1 level, then additional analyses will be performed. Treatment-by-region interaction will also be added to the model for sensitivity purposes. When a restricted maximum likelihood-based mixed model for repeated measures (MMRM) is used, the model will include treatment, region, joint erosion status, visit, treatment-by-visit-interaction as fixed categorical effects; baseline score and baseline score-byvisit-interaction as fixed continuous effects. An unstructured (co)variance structure will be used to model the between- and within-patient errors. If this analysis fails to converge, other structures will be tested. The Kenward-Roger method will be used to estimate the degrees of freedom. Type III sums of squares for the least-squares means will be used for the statistical comparison; CI will also be reported. Treatment group comparisons versus placebo at Week 12
217 I4V-MC-JADV(c) Clinical Protocol Page 119 and other visits will be tested. Further details on the use of MMRM will be described in the SAP. Fisher s exact test will be used for AEs, discontinuations, and other categorical safety data between treatment groups. Continuous vital signs, body weight, and other continuous safety variables, including laboratory variables will be analyzed using ANCOVA with treatment and baseline value in the model. Missing Data Imputation: In accordance with precedent set with other Phase 3 RA trials (Keystone et al. 2004, 2008, 2009; Cohen et al. 2006; Smolen et al. 2008, 2009), the following methods for imputation of missing data will be used: 1. Nonresponder imputation (NRI): All patients who discontinue the study or the study treatment at any time for any reason will be defined as nonresponders for the NRI analysis for categorical variables, such as ACR20/50/70, from the time of discontinuation and onward. Patients who are eligible forreceive rescue therapy at Week 16 will be analyzed as nonresponders after Week 16 and onward. Randomized patients without available data at a postbaseline visit will be defined as nonresponders for the NRI analysis at that visit. 2. Linear extrapolation method: The linear extrapolation method will be used for analysis of the structural progression endpoint (van der Heijde mtss) to impute missing data at time points when analyses are conducted (including the analyses at Week 24 and Week 52). For patients who discontinue the study or the study treatment or for patients who miss a radiograph for any reason, baseline data and the most recent radiographic data prior to discontinuation or the missed radiograph, adjusted for time, will be used for linear extrapolation to impute missing data at subsequent time points. For patients who receive rescue therapy starting at Week 16 or at any time point thereafter, baseline data and the most recent radiographic data prior to initiation of rescue therapy, adjusted for time, will be used for linear extrapolation. All patients originally randomized to placebo will be switched to active treatment at Week 24; thus, there will be no observed data for the placebo arm for the structural comparison at subsequent time points. Therefore, baseline data and the most recent radiographic data prior to initiation of the new therapy, adjusted for time, will be used for linear extrapolation at Week 52. The linear extrapolation method has been established as an appropriate missing data imputation method in other Phase 3 RA trials (Keystone et al. 2004, 2008, 2009; Cohen et al. 2006; Smolen et al. 2008, 2009). Missing postbaseline values will be imputed only if both a baseline value and a postbaseline value from another time point are available, as long as the patient was on the same treatment at each applicable time point. 3. Multiple imputation: Other methods than linear extrapolation that include multiple imputation will be employed for analysis of the structural progression endpoint (van der Heijde mtss) to impute missing data at Week 24. Details will be provided in the SAP. 4. Modified baseline observation carried forward (mbocf): The mbocf method will be used for the analysis of major secondary continuous endpoints (unless otherwise stated).
218 I4V-MC-JADV(c) Clinical Protocol Page 120 For patients who discontinue the study or the study treatment because of an AE, including death, the baseline observation will be carried forward to the corresponding endpoint for evaluation. For patients who discontinue the study for reason(s) other than an AE, the last nonmissing postbaseline observation prior to discontinuation will be carried forward to the corresponding endpoint for evaluation. For patients who are eligible forreceive rescue therapy starting at Week 16, the last nonmissing observation at or before rescue will be carried forward to subsequent time points for evaluation. 5. Modified last observation carried forward (mlocf): For all continuous measures including safety analyses, the mlocf will be a general approach to impute missing data unless otherwise specified. For patients who are eligible forreceive rescue therapy starting at Week 16, the last nonmissing observation at or before rescue will be carried forward to subsequent time points for evaluation. For all other patients discontinuing from the study or the study treatment for any reason, the last nonmissing postbaseline observation before discontinuation will be carried forward to subsequent time points for evaluation. If theafter mlocf imputation, data from patients with nonmissing baseline and postbaseline observation is missing, then itobservations will not be included in the analysis even if the baseline observation is not missing. analyses. The mlocf will be used as a sensitivity method to the mbocf method with respect to the major secondary endpoints. 6. Other methods for data imputation for analysis of key endpoints other than mtss may be used and will be described in the SAP. Adjustment for Multiple Comparisons: The following multiple testing procedure will be used to control the family-wise error rate for the primary and key secondary hypotheses for the endpoints listed in Sections 6.1 and 6.2. Each step in the sequential testing procedure uses a local level procedure for the null hypotheses being tested. Testing of hypotheses at each step of the procedure commences only if all null hypotheses of prior steps were rejected. As such, the multiple testing procedure provides strong control of the family-wise error rate at the 1-sided level. The overall strategy is illustrated (Bretz et al. 2009) in Figure JADV.2. Step 1: Test ACR20 at Week 12 for baricitinib versus placebo at 1-sided alpha = If the null hypothesis is rejected, proceed to Step 2. Otherwise, discontinue testing. Step 2: Test the change from baseline in HAQ-DI at Week 12 and the change from baseline in mtss at Week 24, both tests of baricitinib versus placebo, at 1-sided alpha = via a weighted Holm procedure (Holm 1979) with weights of 0.2 and 0.8, respectively. If both null hypotheses are rejected, proceed to Step 3. Otherwise, discontinue testing. Details of the Holm procedure at this step:
219 I4V-MC-JADV(c) Clinical Protocol Page 121 Let p2 denote the unadjusted 1-sided p-value resulting from testing the hypothesis regarding HAQ-DI and let p3 denote the unadjusted 1-sided p- value resulting from testing the hypothesis regarding mtss. Let p2* and p3* denote the corresponding weight-adjusted p-values (ie, p2* = p2/0.2 and p3*=p3/0.8). The following decision rules apply: If min(p2*, p3*) >0.025, then Lilly will fail to reject both null hypotheses and discontinue testing. Otherwise, if p2* 0.025, then Lilly will reject the null hypothesis for HAQ- DI (declaring statistical significance) and test mtss using the unadjusted p- value. If p , Lilly will also reject the null hypothesis for mtss (declaring statistical significance); however if p3 >0.025, then Lilly will fail to reject the null hypothesis for mtss and discontinue testing. Otherwise, if p3* 0.025, then Lilly will reject the null hypothesis for mtss (declaring statistical significance) and test HAQ-DI using the unadjusted p- value. If p , Lilly will also reject the null hypothesis for HAQ-DI (declaring statistical significance); however if p2 >0.025, then Lilly will fail to reject the null hypothesis for HAQ-DI and discontinue testing. Step 3: Test the change from baseline in DAS28-hsCRP at Week 12 for baricitinib versus placebo and ACR20 at Week 12 for noninferiority of baricitinib to adalimumab at 1 sided alpha=0.025 via a weighted Holm procedure with weights of 0.2 and 0.8, respectively, and following similar details as outlined in Step 2. (Details concerning the noninferiority margin and statistical conclusions are found in Section ) If both null hypotheses are rejected, proceed to Step 4. and the proportion of patients achieving an SDAI score 3.3 (SDAI remission)otherwise, discontinue testing.step 4: Test the change from baseline in the duration of morning joint stiffness at Week 12 and the change from baseline in the severity of morning joint stiffness at Week 12, both tests of baricitinib versus placebo, at 1-sided alpha=0.025 via a Hochberg procedure (Hochberg 1988). If both null hypotheses are rejected, proceed to Step 54. Otherwise, discontinue testing. Details of the Hochberg procedure at this step: Let p6p1 and p7p2 denote the unadjusted 1-sided p-values resulting from testing the hypotheses regarding durationdas28-hscrp and severity of morning joint stiffnesssdai remission, respectively.
220 I4V-MC-JADV(c) Clinical Protocol Page 122 If p6 and p7p1and p , then Lilly will reject both null hypotheses (declaring statistical significance for both durationdas28-hscrp and severitysdai remission). Otherwise, if p6p and p7p2 >0.025, then Lilly will reject only the null hypothesis (declaring statistical significance) related to duration of morning joint stiffnessdas28-hscrp. Otherwise if p7p and p6p1 >0.025, then Lilly will reject only the null hypothesis (declaring statistical significance) related to severity of morning joint stiffnesssdai remission. Otherwise Lilly will fail to reject both null hypotheses and discontinue testing. Step 4: Test ACR20 at Week 12 for noninferiority of baricitinib to adalimumab at 1- sided alpha= Details concerning the noninferiority margin and statistical conclusions are found in Section If the null hypothesis is rejected, proceed to Step 5. Otherwise, discontinue testing. Step 5: Test the change from baseline in worst tiredness at Week 12 and the change from baseline in worst pain atstep 5: Test mean duration of morning joint stiffness in the 7 days prior to Week 12 for baricitinib versus placebo at 1-sided alpha= If the null hypothesis is rejected, proceed to Step 6. Otherwise, discontinue testing. Step 6: Test the change from baseline in DAS28-hsCRP at Week 12 for baricitinib versus adalimumab at 1-sided alpha= If the null hypothesis is rejected, proceed to Step 7. Otherwise, discontinue testing. Step 7: Test mean severity of morning joint stiffness in the 7 days prior to Week 12 for baricitinib versus placebo at 1-sided alpha= If the null hypothesis is rejected, proceed to Step 8. Otherwise, discontinue testing. Step 8: Test mean Worst Tiredness NRS in the 7 days prior to Week 12 and mean Worst Pain NRS in the 7 days prior to Week 12, both tests of baricitinib versus placebo, at 1- sided alpha=0.025 via a Hochberg procedure and following similar details as outlined in Step 43.

221 I4V-MC-JADV(c) Clinical Protocol Page 123 Abbreviations: = change from baseline; ACR20 = 20% improvement in American College of Rheumatology criteria; DAS28 = 28 diarthroidal joint count; hscrp = highsensitivity C-reactive protein; HAQ-DI = Health Assessment Questionnaire-Disability Index; SDAI = simplified disease activity index; mtss = modified Total Sharp Score [van der Heijde]; TS = test(s) significant; W = week; BARI = baricitinib; ADA = adalimumab. Figure JADV.3. Illustration of the sequential testing approach within the overall gatekeeping strategy for Study JADV. This new figure replaces figure JADV.2.
222 I4V-MC-JADV(c) Clinical Protocol Page 124 Section : Efficacy Analyses Major secondary analyses: 1. Comparison between baricitinib and placebo in change from baseline to Week 24 in mtss: An ANCOVA model with baseline score, region, joint erosion status, and treatment group in the model will be used to test the treatment difference between baricitinib and placebo in change from baseline to Week 24 of mtss scores. The linear extrapolation method as described above will be used to impute missing data. A permutation-based method will be used as a nonparametric sensitivity analysis to the ANCOVA method. 2. Comparison between baricitinib and placebo in change from baseline to Week 12 in HAQ-DI score: An ANCOVA model with treatment, region, joint erosion status, and baseline score in the model will be used to test the treatment difference between baricitinib and placebo in change from baseline to Week 12 in HAQ-DI. The mbocf approach as described above will be used to impute missing data. 3. Comparison between baricitinib and placebo in change from baseline to Week 12 in DAS28-hsCRP: An ANCOVA model with treatment, region, joint erosion status, and baseline score in the model will be used to test the treatment difference between baricitinib and placebo in change from baseline to Week 12 in DAS28-hsCRP. The mbocf approach as described above will be used to impute missing data. 4. Comparison between baricitinib and placebo in proportion of patients achieving an SDAI score 3.3 at Week 12: A logistic regression model with treatment, region, and joint erosion status in the model will be used to test the treatment difference between baricitinib and placebo in the proportion of patients achieving an SDAI score 3.3 response at Week 12. The NRI method as described above will be used to impute missing data. 5. Comparison between baricitinib and adalimumab in ACR20 response at Week 12: A logistic regression model with treatment, region, and joint erosion status in the model will be used to test the treatment difference between baricitinib and adalimumab in the percentage of patients achieving ACR20 response at Week 12. A (100 alpha)%95% CI for the treatment difference will be constructed using the method described by Newcombe-Wilson (Newcombe 1998) without continuity correction. If a quantitative treatment by region interaction exists, a supportive method to the Newcombe-Wilson method will be used. If the lower bound of the (100 alpha)%95% CI is >-12%, Lilly will conclude that baricitinib is noninferior to adalimumab with respect to ACR20. Similarly, if the lower bound of the (100 alpha)%95% CI is >0%, Lilly will conclude that baricitinib is superior to adalimumab with respect to ACR20. The NRI method as described above will be used to impute missing data. 6. Comparison between baricitinib and placebo in change from baseline to Week 12 in mean duration of morning joint stiffness in the 7 days prior to Week 12: An ANCOVA model with treatment, region, joint erosion status, and baseline score in the model will be
223 I4V-MC-JADV(c) Clinical Protocol Page 125 used to test(day 1 on the treatment difference between baricitinib and placebo in change from baseline to Week 12 in duration of morning joint stiffness. The mbocf approach as described above will be used to impute missing data. 7. Comparison between baricitinib and placebo in change from baseline to Week 12 in severity of morning joint stiffness: An ANCOVA model with treatment, region, joint erosion status, and baseline scorediaries) in the model will be used to test the treatment difference between baricitinib and placebo in change from baseline to Week 12 in severitymean duration of morning joint stiffness. The mbocf approach as described above will be used to impute missing data. 8. Comparison between baricitinib and placebo in change from baseline to Week 12 in worst tiredness: An ANCOVA model with treatment, region, joint erosion status, and baseline score in the model will be used7 days prior to testweek 12. Refer to the treatment difference between baricitinib and placebo in change from baseline to Week 12 in worst tiredness. The mbocf approach as described above will be used to impute missing datasap for more details. 7. Comparison between baricitinib and placeboadalimumab in change from baseline to Week 12 in worst paindas28-hscrp: An ANCOVA model with treatment, region, joint erosion status, and baseline score in the model will be used to test the treatment difference between baricitinib and placeboadalimumab in change from baseline to Week 12 in worst paindas28-hscrp. The mbocf approach as described above will be used to impute missing data. 8. Comparison between baricitinib and placebo in mean severity of morning joint stiffness in the 7 days prior to Week 12: An ANCOVA model with treatment, region, joint erosion status, and baseline score (Day 1 on the diaries) in the model will be used to test the treatment difference between baricitinib and placebo in mean severity of morning joint stiffness in the 7 days prior to Week 12. Refer to the SAP for more details. 9. Comparison between baricitinib and placebo in mean Worst Tiredness NRS in the 7 days prior to Week 12: An ANCOVA model with treatment, region, joint erosion status, and baseline score (Day 1 on the diaries) in the model will be used to test the treatment difference between baricitinib and placebo in mean Worst Tiredness NRS in the 7 days prior to Week 12. Refer to the SAP for more details. 10. Comparison between baricitinib and placebo in mean Worst Pain NRS in the 7 days prior to Week 12: An ANCOVA model with treatment, region, joint erosion status, and baseline score (Day 1 on the diaries) in the model will be used to test the treatment difference between baricitinib and placebo in mean Worst Pain NRS in the 7 days prior to Week 12. Refer to the SAP for more details. Other secondary efficacy and health outcome analyses: Besides the analyses listed in the objective section, the following analyses will be performed: change from baseline to Week 16 and Week 52 in structural joint damage as measured by mtss
224 I4V-MC-JADV(c) Clinical Protocol Page 126 proportion of patients with mtss change 0 change from baseline in joint space narrowing and bone erosion scores hybrid ACR (bounded) response measure proportions of patients achieving ACR20 (other than primary), ACR50, and ACR70 percentage change from baseline in individual components of the ACR Core Set change from baseline in DAS28-ESR change from baseline in DAS28-hsCRP proportion of patients achieving DAS28-ESR 3.2 and <2.6 proportion of patients achieving DAS28-hsCRP 3.2 and <2.6 change from baseline in HAQ-DI proportion of patients achieving HAQ-DI improvement 0.22 and 0.3 change from baseline in CDAI score change from baseline in SDAI score proportion of patients achieving CDAI score 2.8 proportion of patients achieving SDAI score 3.3 (ACR/EULAR remission according to the Index-based definition) proportion of patients achieving EULAR28moderate response and good response based on EULAR response criteria proportion of patients achieving ACR/EULAR remission using bothaccording to the Boolean-based definition and Index based definition change from baseline in fatigue score in the FACIT-F change from baseline in PCS, MCS, and total score in the SF-36v2 Acute change from baseline in health states, VAS score, and index score of EQ-5D-5L change from baseline in absenteeism, presenteeism, work productivity loss, and activity impairment scores of WPAI-RA change from baseline in individual items and total score in healthcare resource utilization characterization of the response time-course for key efficacy endpoints and identification of potential factors that may have an impact on the efficacy endpoints of baricitinib change from baseline in duration of morning joint stiffness assessed at the visit using an epro tablet change from baseline in Worst Tiredness NRS assessed at the visit using an epro tablet change from baseline in Worst Pain NRS assessed at the visit using an epro tablet Area under the curve (AUC) up to Week 12 in duration of morning joint stiffness collected in electronic diaries AUC up to Week 12 in severity of morning joint stiffness collected in electronic diaries AUC up to Week 12 in Worst Tiredness NRS collected in electronic diaries AUC up to Week 12 in Worst Pain NRS collected in electronic diaries Approaches similar to the primary and major secondary analyses will be used to analyze other secondary efficacy and health outcome measures. In addition, comparisons between baricitinib and adalimumab in other secondary measures will be performed. Analysis methods will consist of ANCOVA and logistic regression, as described above, using mbocf, linear extrapolation, NRI, and mlocf missing data imputation methods, where applicable.
225 I4V-MC-JADV(c) Clinical Protocol Page 127 In addition, key continuous secondary parameters that are collected at repeated visits throughout the double-blind period may be analyzed using a MMRM approach as described in Section The MMRM will be considered as a sensitivity approach to ANCOVA results. Data collected after initiation of rescue therapy will be summarized as appropriate. Further details will be provided in the SAP. Section : Safety Analyses TEAEs are defined as AEs that first occurred or worsened in severity on or after the date of first dose of study treatment. AEs will be considered treatment emergent for nonresponders assigned to baricitinib if the AEs first occurred or worsened in severity (compared to the maximum severity prior to or on the date of rescue)after the visit at which rescue therapy is assigned. The number of TEAEs as well as the number and percentage of patients who experienced at least 1 TEAE will be summarized using MedDRA (Medical Dictionary for Regulatory Activities) for each system organ class (or a body system) and each preferred term by treatment group. TEAEs will also be summarized by relationship to treatment and by severity within each treatment group. SAEs and AEs that lead to study drug discontinuation will also be summarized by treatment group. Fisher s exact test will be used to perform pairwise comparisons among the treatment groups. Section : Subgroup Analyses Subgroups analyses comparing baricitinib to placebo will be performed using ACR20, ACR50, HAQ-DI, DAS28 hscrp, and DAS28 remission (DAS28 hscrp <2.6) at Week-hsCRPat Weeks 12 and 24 and mtss at Week 24. Subgroups to be evaluated will include region, renal function, background therapy, joint erosion status, country, gender, age, race, etc. Any other additional subgroups will be defined in the SAP. For ACR20, and ACR50, and DAS28 remission analyses at Weeks 12 and 24, a logistic regression model will be used with treatment, subgroup, and treatment by subgroup interaction included as factors. For the change from baseline to WeekWeeks 12 and 24 in HAQ-DI score and DAS28-hsCRP and to Week 24 in mtss, an ANCOVA model will be used with treatment, baseline score, subgroup, and treatment by subgroup interaction included as factors. Further definitions for the levels of the subgroup variables, the analysis methodology, and any additional subgroup analyses will be defined in the SAP. Because this study is not powered for subgroup analyses, all subgroup analyses will be treated as exploratory. Section : Planned Analyses The analysis of primary and major secondary endpoints (except for x-ray data) will occur when all patients complete the Week 24 study visit. This analysis will not be considered an interim analysis, and the study team will become unblinded at this time. A final analysis will occur when all patients complete the study.
226 I4V-MC-JADV(c) Clinical Protocol Page 128 All investigators, study site personnel, and patients will remain blinded to treatment assignments until the final database lock. Members of the study team not in direct contact with the clinical sites may have access to the data from this study after all patients complete the primary and major secondary endpoints through Week 24 (Visit 11). Members of the study team who become unblinded to the primary and major secondary analyses through Week 24 will have no further contact with study sites until after the final database lock. Unblinding details will be specified in the unblinding plan section of the SAP.

227
228 e i k l s Worst pain NRSltiredness NRS-tabletl X X X X X X X X X X X X X X X If the PPD test is positive and the patient has no medical history or chest x-ray findings consistent with active TB, the patient may have a QuantiFERON-TB Gold or T-SPOT.TB test (if available). If the QuantiFERON-TB Gold or T-SPOT.TB test results are not negative, the patient will be considered to have latent TB. If the QuantiFERON-TB Gold or T-SPOT.TB test is available and in the judgment of the investigator, preferred as an alternative to the PPD skin test for the evaluation of TB infection, it may be used instead of the PPD TB test. If the QuantiFERON-TB Gold or T-SPOT.TB test is positive, the patient will be considered to have latent TB. If the test is not negative, the test may be repeated once within 2 approximately weeks of the initial value. If the repeat test results are again not negative, the patient will be considered to have latent TB. At screening, radiographic images of the hands and feet acquired within 4 weeks prior to study entry may be submitted to the central reader to confirm eligibility. If a patient undergoes rescreening, radiographic images acquired as part of initial screening and within 10 weeks of randomization may be used. Morning joint stiffness assessment, Worst Tiredness, and Worst Pain assessments will be collected using a battery of questions and a daily patient diary to assess duration and, severity of morning joint stiffness and, recurrence of stiffness during the day., worst tiredness and worst joint pain. This item tomorning joint stiffness duration, Worst Tiredness, and Worst Pain assessments will be collected using an electronic patient daily diaries through Week 12.-reported outcomes [epro] tablet at each visit in a subset of patients. At each time point, 3 replicate readings should be made at 30 to 60 second intervals. Approximately 30- to 60- second intervals. Blood pressure is recorded as the average of these three readings. A single pulse measurement should be made simultaneously with at least one of the readings at each time point. I4V-MC-JADV(c) Clinical Protocol Page 130
229 I4V-MC-JADV(c) Clinical Protocol Page 131 Attachment 2: Clinical Laboratory Tests Other Tests Hepatitis B surface antigen Hepatitis B surface antibody Hepatitis B core antibody Hepatitis C antibodyh Human immunodeficiency virus Iron studies (iron, TIBC, and ferritin) Thyroid-stimulating hormone plasma concentration (PK sample) PPD or QuantiFERON j -TB Goldj, or T-SPOT.TB Rheumatoid factor High-sensitivity C-reactive protein ESRk Anticyclic citrullinated peptide Immunoglobulins (IgG, IgA, IgM) j Test will be required at Visit 1 only to determine eligibility of patient for the study. In countries where the QuantiFERON-TB Gold or T-SPOT.TB test is available and, in the judgment of the investigator, medically preferred as an alternative to the PPD test for the evaluation of mycobacterium tuberculosis (MTB) infection, it may be used instead of the PPD test (positive tests excluded) and may be read locally.
230 Leo Document ID = 0a85d402-e423-4e1c-9971-ed435bf1710f Approver: Approval Date & Time: 18-Jul :26:54 GMT Signature meaning: Approved Approver: Approval Date & Time: 19-Jul :11:17 GMT Signature meaning: Approved
231 I4V-MC-JADV Statistical Analysis Plan Version 1.0 Page 1 1. Statistical Analysis Plan: [I4V-MC-JADV]:A Randomized, Double-Blind, Placeboand Active-Controlled, Phase 3 Study Evaluating the Efficacy and Safety of Baricitinib in Patients with Moderately to Severely Active Rheumatoid Arthritis Who Have Had an Inadequate Response to Methotrexate Therapy Baricitinib () Rheumatoid Arthritis Study I4V-MC-JADV is a Phase 3, randomized, double-blind, placebo and activecontrolled, parallel group study of baricitinib for the treatment of moderately to severely active rheumatoid arthritis (RA) during a 52-week treatment period in patients with inadequate response to methotrexate therapy. Eli Lilly and Company Indianapolis, Indiana USA [Protocol I4V-MC-JADV] [Phase 3] Statistical Analysis Plan electronically signed and approved by Lilly on date provided below. Approval Date: 10-Aug-2013 GMT
232 I4V-MC-JADV Statistical Analysis Plan Version 1.0 Page 2 Section 2. Table of Contents Page 1. Statistical Analysis Plan: [I4V-MC-JADV]:A Randomized, Double- Blind, Placebo- and Active-Controlled, Phase 3 Study Evaluating the Efficacy and Safety of Baricitinib in Patients with Moderately to Severely Active Rheumatoid Arthritis Who Have Had an Inadequate Response to Methotrexate Therapy Table of Contents Revision History Study Objectives Primary Objective Secondary Objectives Exploratory Objectives Study Design Summary of Study Design Screening Period Part A and B: 24-week and 28-week, Double-Blind, Placebo- and Active-Controlled Periods Part C: Posttreatment Follow-Up Period Study Extension Determination of Sample Size Method of Treatment Assignment Treatment Administered Selection and Time of Doses Rescue Therapy A Priori Statistical Methods General Considerations Definition of Baseline Derived Data Analysis Populations Adjustment for Covariates Handling of Dropouts or Missing Data Missing Data Imputations Missing Data Imputation for Efficacy and Health Outcome Endpoints Missing Data Imputation for the Structural Progression Endpoint (van der Heijde mtss) Multiple Comparison...28
233 I4V-MC-JADV Statistical Analysis Plan Version 1.0 Page Patient Disposition Protocol Deviation Patient Characteristics Demographics Baseline Disease Characteristics Previous and Concomitant Medication Historical Illnesses and Pre-Existing Conditions Treatment Compliance Treatment Exposure Efficacy Analyses Primary Efficacy Analysis Major Secondary Efficacy Analyses Analysis for Other Secondary Efficacy Endpoints Testing Strategy to Control the Type I Family-Wise Error Rate Pharmacokinetic/Pharmacodynamic Analysis (PK/PD) Health Outcome Analyses Diary Data Data Handling and Missing Data Imputation Analysis of Diary Data Analysis of Non-Diary Data Exploratory Analysis Safety Analysis Adverse Events (AEs) Common Adverse Events Serious Adverse Events (SAEs) Other Significant Adverse Events Vital Signs and Physical Characteristics Laboratory Measurements Safety in Special Groups and Circumstances, including Adverse Events of Special Interest (AESI) Abnormal Hepatic Tests Myelosuppressive Events Lymphocyte Subset Cell Counts Lipids Effects Renal Function Effects Infections, Including Opportunistic Infections Major Adverse Cardiovascular Events (MACE) Malignancies...76
234 I4V-MC-JADV Statistical Analysis Plan Version 1.0 Page Allergic Reactions/Hypersensitivities Symptoms of Depression (QIDS-SR16) Other Safety Analyses Subgroup Analysis Interim Analysis and Data Monitoring Unblinding Plan References Appendices...86
235 I4V-MC-JADV Statistical Analysis Plan Version 1.0 Page 5 Table Table of Contents Page Table JADV.5.1. Countries and their Geographical Regions...16 Table JADV.5.2. Treatment Regimens (on a background of MTX therapy)...16 Table JADV.6.1. Imputation Techniques for Various Efficacy and Safety Variables...28 Table JADV.6.2. Categorical Criteria for Abnormal Treatment-Emergent Blood Pressure and Heart Rate Measurement, and Categorical Criteria for Weight Changes for Adults...59 Table JADV.6.3. Common Terminology Criteria for Adverse Events (CTCAE) Related to Hepatic Tests...65 Table JADV.6.4. CTCAE Related to Myelosuppressive Events...67 Table JADV.6.5. Criteria for Discontinuation of Investigational Product due to Myelosuppression...68 Table JADV.6.6. National Cholesterol Education Program (NCEP) ATP III Criteria...70 Table JADV.6.7. Common Terminology Criteria for Adverse Events (CTCAE) Related to Lipids Effects...71 Table JADV.6.8. Common Terminology Criteria for Adverse Events (CTCAE) Related to Renal Effects...73 Table JADV.6.9. Quick Inventory of Depressive Symptomatology Self-Rated 16 (QIDS-SR 16 ) Severity of Depressive Symptoms Categories based on the QIDS-SR 16 Total Score...79
236 I4V-MC-JADV Statistical Analysis Plan Version 1.0 Page 6 Table of Contents Figure Page Figure JADV.5.1. Illustration of study design for Clinical Protocol I4V-MC-JADV Figure JADV.6.1. Illustration of the sequential testing approach within the overall gatekeeping strategy for Study JADV....44
237 I4V-MC-JADV Statistical Analysis Plan Version 1.0 Page 7 Table of Contents Appendix Page Appendix 1. Algorithm for determining ACR response...87 Appendix 2. Details of Efficacy and Health Outcome Measures...90 Appendix 3. Framingham 10 Year Risk of Cardiovascular Disease Events...106
238 I4V-MC-JADV Statistical Analysis Plan Version 1.0 Page 8 3. Revision History This statistical analysis plan (SAP) Version 1 was approved prior to unblinding.
239 I4V-MC-JADV Statistical Analysis Plan Version 1.0 Page 9 4. Study Objectives The following objectives are replicated from Amendment (c) of Study I4V-MC-JADV (JADV) protocol, dated 19 July Primary Objective The primary objective of this study is to determine whether baricitinib is superior to placebo in the treatment of patients with moderately to severely active rheumatoid arthritis (RA) despite MTX treatment (i.e., MTX-IR), as assessed by the proportion of patients achieving ACR20 at Week Secondary Objectives The major secondary objectives of the study are to evaluate the efficacy of baricitinib versus placebo or adalimumab as assessed by: Change from baseline to Week 24 in structural joint damage as measured by modified Total Sharp Score (mtss; van der Heijde (2000) method) compared to placebo Change from baseline to Week 12 in Health Assessment Questionnaire-Disability Index (HAQ-DI) score compared to placebo Change from baseline to Week 12 in Disease Activity Score modified to include the 28 diarthrodial joint count (DAS28)-high-sensitivity C-reactive protein (hscrp) compared to placebo Proportion of patients achieving an Simplified Disease Activity Index (SDAI) score 3.3 (American College of Rheumatology/ European League Against Rheumatism [ACR/EULAR] remission Index-based definition) at Week 12 compared to placebo Proportion of patients achieving 20% improvement in American College of Rheumatology criteria (ACR20) response at Week 12 compared to adalimumab Mean duration of morning joint stiffness in the 7 days prior to Week 12 compared to placebo as collected in electronic diaries Change from baseline to Week 12 in DAS28-hsCRP compared to adalimumab Mean severity of morning joint stiffness in the 7 days prior to Week 12 compared to placebo as collected in electronic diaries Mean Worst Tiredness numeric rating scale (NRS) in the 7 days prior to Week 12 compared to placebo as collected in electronic diaries Mean Worst Pain NRS in the 7 days prior to Week 12 compared to placebo as collected in electronic diaries The other secondary objectives of the study are as follows: To evaluate the efficacy of baricitinib as assessed by: o proportion of patients achieving DAS28-hsCRP 3.2 and DAS28-hsCRP <2.6 at Week 12, Week 24, and Week 52 o proportion of patients achieving HAQ-DI improvement from baseline 0.22 and 0.3 at Week 12, Week 24, and Week 52
240 I4V-MC-JADV Statistical Analysis Plan Version 1.0 Page 10 o change from baseline to Week 16 and Week 52 in structural joint damage as measured by mtss o proportion of patients with mtss change 0 from baseline at Week 16, Week 24, o and Week 52 change from baseline to Week 16, Week 24, and Week 52 in joint space narrowing and bone erosion scores o proportion of patients achieving ACR20 at Week 24 and Week 52 o proportion of patients achieving ACR50 at Week 12, Week 24, and Week 52 o proportion of patients achieving ACR70 at Week 12, Week 24, and Week 52 o percentage change from baseline to Week 12, Week 24, and Week 52 in individual components of the ACR Core Set o change from baseline through Week 52 in DAS28-hsCRP o change from baseline through Week 52 in DAS28-erythrocyte sedimentation rate (ESR) o proportion of patients achieving DAS28-ESR 3.2 and DAS28-ESR <2.6 at Week 12, Week 24, and Week 52 o change from baseline to Week 24 and Week 52 in HAQ-DI score o change from baseline to Week 12, Week 24, and Week 52 in CDAI score o change from baseline to Week 12, Week 24, and Week 52 in SDAI score o proportion of patients achieving a CDAI score 2.8 at Week 12, Week 24, and Week 52 o proportion of patients achieving an SDAI score 3.3 at Week 24, and Week 52 o proportion of patients achieving ACR/EULAR remission according to the Boolean-based definition at Week 12, Week 24 and Week 52 o proportion of patients achieving moderate and good EULAR responses based on the 28-joint DAS at Week 12, Week 24, and Week 52 o evaluation of hybrid ACR (bounded) response at Week 12, Week 24 and Week 52 o o change from baseline to Week 12, Week 24, and Week 52 in Functional Assessment of Chronic Illness Therapy Fatigue (FACIT-F) scale scores change from baseline to Week 12, Week24, and Week 52 (at Weeks 12, 24, and 52) in each scale and the summary scores for the Mental Component Score (MCS) and Physical Component Score (PCS) of the Medical Outcomes Study 36- Item Short Form Health Survey Version 2 Acute (SF-36v2 Acute) o change from baseline to Week 12, Week 24, and Week 52 in European Quality of Life-5 Dimensions-5 Level (EQ-5D-5L) scores o change from baseline to Week12, Week 24, and Week 52 in Work Productivity and Activity Impairment-Rheumatoid Arthritis (WPAI-RA) scores o evaluation of healthcare resource utilization through Week 52 o o change from baseline to Week 12, Week 24, and Week 52 in duration of morning joint stiffness assessed at the visit using an electronic patient-reported outcomes (epro) tablet change from baseline to Week 12, Week 24, and Week 52 in Worst Tiredness NRS assessed at the visit using an epro tablet
241 I4V-MC-JADV Statistical Analysis Plan Version 1.0 Page 11 o change from baseline to Week 12, Week 24, and Week 52 in Worst Pain NRS assessed at the visit using an epro tablet To characterize the pharmacokinetics (PK) of baricitinib, determine the magnitude of within- and between-patient variability, and identify the potential factors that may have an impact on the PK of baricitinib. To characterize the baricitinib exposure-response time course for key efficacy and safety endpoints and to identify potential factors that may have an impact on these efficacy or safety endpoints of baricitinib Exploratory Objectives The exploratory objective of this study is to Compare baricitinib to adalimumab as regards to various efficacy measures.
242 I4V-MC-JADV Statistical Analysis Plan Version 1.0 Page Study Design 5.1. Summary of Study Design Study JADV is a 52-week, Phase 3, multicenter, randomized, double-blind, double-dummy, placebo- and active-controlled, parallel-group, outpatient study comparing the efficacy of baricitinib versus placebo on signs and symptoms, function, remission, and structural progression in patients with moderately to severely active RA who have had an insufficient response to MTX and who have never been treated with a biologic disease-modifying antirheumatic drugs (DMARD) (ie, MTX-IR patients). The study will include a noninferiority comparison of baricitinib versus adalimumab on signs and symptoms. Baricitinib will be administered as a 4-mg tablet QD. The dose for patients with renal impairment, defined as estimated glomerular filtration rate (egfr) <60 ml/min/1.73 m2, will be 2 mg baricitinib QD. Patients not assigned to the baricitinib treatment arm will receive either a 4-mg or 2-mg matching placebo tablet. Adalimumab (40 mg) will be administered by subcutaneous (SC) injection biweekly (ie, every 2 weeks). Patients not assigned to adalimumab will receive a matching placebo SC injection biweekly. Planned enrollment is approximately 1280 randomized patients: 480 to receive baricitinib, 480 to receive placebo, and 320 to receive adalimumab. After the Week 52 study visit, eligible patients may proceed to a separate extension study (Study I4V-MC-JADY) lasting for up to 2 years or to the posttreatment follow-up period of this study (Part C). Study JADV will consist of 4 parts: Screening: Screening period lasting from 3 to 42 days prior to Visit 2 (Week 0) Part A: double-blind, placebo- and active-controlled period from Week 0 through Week 24 Part B: double-blind, active-controlled period from the end of Week 24 through Week 52 Part C: posttreatment follow-up period Figure JADV.5.1. illustrates the study design.
243
244 I4V-MC-JADV Statistical Analysis Plan Version 1.0 Page 14 <60 ml/min/1.73 m2) through Week 52 (Visit 15). Patients in this treatment arm will also receive a placebo SC injection biweekly starting at Week 0 (Visit 2) through Week 50. Patients randomized to adalimumab will be administered adalimumab (40 mg) by SC injection biweekly starting at Week 0 (Visit 2) through Week 50 and a placebo tablet QD through Week 52 (the morning of Visit 15). The last SC injection will be administered during Week 50 because patients receiving adalimumab have the option to switch to baricitinib 4 mg QD (or 2 mg QD depending on egfr) if they choose to participate in the extension study. Scheduling the last SC injection at Week 50 ensures approximately a 2-week interval from the last SC injection to the start of baricitinib dosing. Patients will be eligible for rescue treatment beginning at Week 16 (Visit 9). Details of the rescue are listed in Section Patients will remain on background MTX, nonsteroidal anti-inflammatory drugs (NSAIDs), analgesics, and/or corticosteroids (see Protocol Section 9.8 for guidelines on adjustments to concomitant therapy). Patients will take their assigned doses of study drugs as directed. The primary efficacy endpoint will be at Week 12, and the final visit during Part A will be at Week 24. The final visit during Part B will be at Week 52 (Visit 15). The structural joint damage endpoints will be assessed at Week 16, Week 24, and Week 52. Patients who complete the Week 52 study visit may be eligible for inclusion in extension Study JADY Part C: Posttreatment Follow-Up Period Patients who discontinue the study or patients who complete Parts A and B of the study and do not enroll in the extension Study JADY will have a Follow-Up Visit (Visit 801) approximately 28 days after the last dose of study drug. Patients who discontinue from the study prior to Week 52 for any reason will have an Early Termination Visit performed, including x-rays. The Early Termination Visit should be performed as soon as logistically possible because this visit includes assessment of safety and PK data. Infrequently, patient and investigator availability may be such that the Early Termination Visit and the Follow-up Visit may occur on or about the same date. In this instance, the visits may be combined and should occur approximately 28 days after the last dose of study drug. All activities required for the Early Termination Visit should be completed according to the schedule of assessments (Protocol Attachment 1) Study Extension Patients who complete this study through Visit 15 will be eligible to participate in Study JADY, if enrollment criteria for Study JADY are met. These patients will not participate in Part C Determination of Sample Size Approximately 1280 patients (480 in the baricitinib group, 480 in the placebo group, and 320 in the adalimumab group) will provide:
245 I4V-MC-JADV Statistical Analysis Plan Version 1.0 Page 15 >95% power to detect a difference between the baricitinib and placebo treatment groups in ACR20 response rate (assuming 60% versus 35%) at Week 12 based on a 2-sided chisquare test at a significance level of approximately 94% power to detect a difference in mtss between the baricitinib and placebo treatment groups for an effect size of 0.25 (ie, difference in means divided by common standard deviation [SD]), based on a 2-sided t-test at a significance level of This assumes 90% of randomized patients will have x-ray data available for the mtss analysis at Week 24. If the effect size in Study JADV was at least 0.21 ( van der Heijde et al. 2013), then power of the mtss analysis is at least 85%. approximately 93% power for the noninferiority analysis of ACR20 response rate at Week 12 between the baricitinib and adalimumab treatment groups, where a response rate of 60% for baricitinib and adalimumab and a 1-sided significance level of are assumed. This power calculation adopted a fixed noninferiority margin of 12%. There is growing support for this margin in recent clinical trials conducted in the MTX-IR patient population (Schiff et al. 2012). These sample size and power estimates were obtained from nquery Advisor Method of Treatment Assignment Patients who meet all criteria for enrollment will be randomized in a 3:3:2 ratio (baricitinib:placebo:adalimumab) to double-blind treatment at Week 0 (Visit 2). Randomization will be stratified by (geographical) region and joint erosion status (1-2 joint erosions plus seropositivity versus at least 3 joint erosions). Assignment to treatment groups will be determined by a computer-generated random sequence using an interactive voice-response system (IVRS). The IVRS will be used to assign packages of baricitinib/placebo and packages of prefilled syringes of adalimumab/placebo for SC injection containing double-blind investigational product to each patient. Each package of clinical trial material will supply sufficient medication for the number of weeks between visits. Site personnel will confirm that they have located the correct package by entering a confirmation number found on the packages of investigational product into the IVRS before dispensing the package to the patient. This study will be conducted internationally in many sites. The following table (Table JADV.5.1) is how region will be defined for the statistical analyses and summaries:
246 I4V-MC-JADV Statistical Analysis Plan Version 1.0 Page 16 Table JADV.5.1. Countries and their Geographical Regions Geographical Region Country or Countries United States and Canada United States, Canada Central and South America and Mexico Argentina, Mexico Eastern Europe Croatia, Czech Republic, Hungary, Latvia, Lithuania, Poland, Romania, Slovakia, Slovenia Western Europe Belgium, France, Germany, Greece, Netherlands, Portugal, Spain, Switzerland, United Kingdom Asia ex-japan China, South Korea, Taiwan Japan Japan Rest of World Australia, Israel, Russia, South Africa Treatment Administered This study involves a comparison of baricitinib 4 mg administered orally QD, placebo administered orally QD and SC biweekly, and adalimumab 40 mg administered by SC injection biweekly. Patients will continue to take background MTX therapy during the course of the study. Table JADV.5.2 shows the treatment regimens. Table JADV.5.2. Treatment Regimens (on a background of MTX therapy) Treatment Group Treatments Administered during Part A Treatments Administered during Part B (Patients Not Rescued) Treatments Administered during Part B (after Rescued a ) Baricitinib 4 mgb oral Baricitinib 4 mgb oral Baricitinib 4 mgb oral Baricitinib once-daily tablet once-daily tablet once-daily tablet treatment Adalimumab placebo by SC injection biweekly Adalimumab placebo by SC injection biweekly Baricitinib placebo oral Baricitinib 4 mgb oral Baricitinib 4 mgb oral Placebo once-daily tablet once-daily tablet once-daily tablet comparator Adalimumab placebo by Adalimumab placebo by SC injection biweekly SC injection biweekly Baricitinib placebo oral Baricitinib placebo oral Baricitinib 4 mgb oral Active once-daily tablet once-daily tablet once-daily tablet comparator Adalimumab 40 mg by SC Adalimumab 40 mg by SC (adalimumab) injection biweekly injection biweekly Abbreviation: SC = subcutaneous. a See Section for details of rescue medication. b The dose for patients with renal impairment, defined as estimated glomerular filtration rate <60 ml/min/1.73 m2, will be 2 mg once daily. The investigator or his/her designee will be responsible for explaining the correct use of both the oral and injectable investigational agent(s) to the patient, verifying that instructions are followed properly, maintaining accurate records of investigational product dispensing and collection, and returning all unused medication to Lilly or its designee at the end of the study.
247 I4V-MC-JADV Statistical Analysis Plan Version 1.0 Page 17 Injections of investigational product should be administered by the site personnel, with instructions to the patient, at Visit 2, and the patient should be supervised while self-administering injections at Visits 4 and 5. In some cases, sites may destroy the material if, during the investigator site selection, the evaluator has verified and documented that the site has appropriate facilities and written procedures to dispose of clinical trial materials. Patients will be instructed to contact the investigator as soon as possible if they have a complaint or problem with the investigational product so that the situation can be assessed. All patients will be required to be on background MTX treatment at baseline. Other background therapies, including NSAIDs and low dose oral corticosteroids, are permitted during the study for patients who are on stable doses (as defined in protocol) of these treatments at baseline. Patients should remain on the same dose of MTX throughout the study; however, the MTX dose may be adjusted for safety reasons Selection and Time of Doses Each patient will receive blinded investigational product (baricitinib 4 mg or baricitinib placebo tablets orally QD and adalimumab 40 mg or adalimumab placebo biweekly by SC injection) beginning on the day of Visit 2 (Week 0). Baricitinib 4-mg oral dosing will continue daily through Visit 15 (Week 52) and biweekly injections of adalimumab 40 mg or adalimumab placebo will continue biweekly through Week 50. Baricitinib placebo oral dosing will continue daily through Visit 11 (Week 24); at Visit 11 (Week 24), all patients randomized to placebo will be reassigned to baricitinib. Injectable investigational product should be administered at approximately the same time of day, as much as possible. For injections not administered on the scheduled day of the week, the missed dose should be administered within 3 days of the scheduled day during study Weeks 1 to 12 and within 5 days of the scheduled day during study Weeks 13 to 52. Dates of subsequent study visits should not be modified according to this delay. As noted in Section 5.3.1, injections of investigational product should be administered by site personnel at Visit 2, and the patient should be supervised while self-administering injections at Visits 4 and 5. Thereafter, the patient will self-administer SC injections at home biweekly when no study visit is scheduled (ie, Weeks 6, 10, 18, 22, 26, 30, 34, 36, 38, 42, 44, 46, 48, and 50). On weeks when study visits are scheduled (ie, Weeks 8, 12, 14, 16, 20, 24, 28, 32, and 40), the patient should not administer his/her SC injection at home but should bring the study medication to the clinic for administration during the study visit. This will allow sites to administer rescue dosing when required. Oral investigational product should be taken at home at approximately the same time each day, as much as possible, with the exception of the following visits, to allow for the proper timing of PK sample collection and to ensure that efficacy assessments are made at trough PK levels:
248 I4V-MC-JADV Statistical Analysis Plan Version 1.0 Page 18 At Visit 2 (Week 0), patients will take their investigational product in the clinic, to allow for PK samples to be taken at 15 and 60 minutes postdose. For Visits 7 (Week 12), Visit 9 (Week 16), 11 (Week 24), 13 (Week 32), and 15 (Week 52), patients will be asked not to take their investigational product prior to visiting the clinic, to allow efficacy assessments to be made prior to investigational product dosing and a predose PK sample to be taken at some of these selected visits (See Protocol Attachment 1) Rescue Therapy In consideration of the disease severity, all patients in Study JADV will be given rescue therapy starting at Week 16 if they are determined to be nonresponders. Starting at Week 16, patients who are nonresponders and who were randomized to placebo will be rescued with baricitinib. To maintain study integrity through investigational product blinding, patients who were originally randomized to baricitinib will continue to receive baricitinib, with appropriate ongoing assessment of response and use of concomitant medication as needed. Patients who are nonresponders and who were randomized to adalimumab will be reassigned to baricitinib as rescue therapy. Nonresponse at Week 16 is defined as lack of improvement of at least 20% in both tender joint counts (TJC) and swollen joint counts (SJC) at both Week 14 and Week 16 compared to baseline. At Week 16, the IVRS will assign rescue treatment based on TJC and SJC values. After Week 16, rescue therapy will be given to patients at the discretion of the investigator based on TJCs and SJCs. Once a patient has been rescued, new NSAIDS, corticosteroids, and/or analgesics may be added or doses of ongoing concomitant NSAIDs, corticosteroids, and/or analgesics may be increased at the discretion of the investigator. A patient may only be rescued once. If a patient continues to meet nonresponse criteria for 4 weeks after rescue or at any time point thereafter, that patient should be discontinued from the study. Patients initially randomized to placebo and not rescued will be reassigned to receive baricitinib at Week 24 in order to limit exposure to inactive treatment. Nonresponders who are rescued will receive an oral tablet daily, but no biweekly SC injection, for the remainder of the study. Rescue therapy will be administered through Week 52.
249 I4V-MC-JADV Statistical Analysis Plan Version 1.0 Page A Priori Statistical Methods 6.1. General Considerations This plan describes a priori statistical analyses for efficacy and safety that will be performed. Statistical analysis of this study will be the responsibility of Eli Lilly and Company (Lilly). For continuous measures, summary statistics will include sample size, means, standard deviations and/or standard errors, medians, minimums, and maximums. For categorical measures, summary statistics will include sample size, frequency counts, and percentages. Statistical tests of treatment effects will be performed at a 2-sided significance level of 0.05, unless otherwise stated (e.g., gated analyses; Section ). All p-values will be rounded up to 3 decimal places. For example, any p-value strictly greater than and less than or equal to 0.05 will be displayed as This guarantees that on any printed statistical output, the unrounded p-value will always be less than or equal to the displayed p-value. A displayed p-value of will always be understood to mean Likewise, any p-value displayed as will be understood to mean >0.999 and 1. Treatment comparisons of categorical efficacy variables (including proportions of subjects achieving ACR20) will be made using a logistic regression analysis with region, baseline joint erosion status (1-2 joint erosions plus seropositivity vs at least 3 joint erosions), and treatment group in the model. The p-value from the logistic model, percentages, difference in percentages, and 100*(1-alpha)% confidence interval (CI) of the difference in percentages using the Newcombe-Wilson method (Newcombe 1998) without continuity correction will be reported (where alpha denotes the significance level for the specific variable of interest). Treatment-byregion interaction will be added to the logistic regression model of the primary and key secondary categorical variables as a sensitivity analysis. If this interaction is significant at a 2- sided 0.1 level, further inspection will be used to assess whether the interaction is quantitative (ie, the treatment effect is consistent in direction across all regions but not size of treatment effect) or qualitative (the treatment is beneficial for some but not all regions). Treatment comparisons of continuous efficacy variables will be made using analysis of covariance (ANCOVA) with region, baseline joint erosion status, treatment group, and baseline value in the model. Type III sums of squares for the least-squares (LS) means will be used for the statistical comparison between treatment groups, the LS mean difference, standard error, p- value and 100*(1-alpha)% CI will also be reported. Sensitivity analysis will be performed for key secondary continuous variables by adding the treatment-by-baseline interaction to the ANCOVA model. If this interaction is significant at a 2-sided 0.1 level, then additional analyses will be performed by subgroups defined by the median of the baseline values using the same terms in the model (with the exception of baseline value). Additional sensitivity analyses will be performed by adding treatment-by-region interaction to the ANCOVA model. If this interaction is significant at a 2-sided 0.1 level, further inspection will be used to assess whether the interaction is quantitative or qualitative.
250 I4V-MC-JADV Statistical Analysis Plan Version 1.0 Page 20 When a restricted maximum likelihood-based mixed model for repeated measures (MMRM) is used (as sensitivity model for applicable major or key secondary variables [see Section ]), the model will include treatment, region, joint erosion status, visit, treatment-by-visit-interaction as fixed categorical effects; baseline score and baseline score-by-visit-interaction as fixed continuous effects to estimate change from baseline across post-baseline visits. An unstructured covariance structure will be used to model the between- and within-patient errors. If this analysis fails to converge, other structures will be tested such as heterogeneous autoregressive [ARH(1)], heterogeneous compound symmetry (CSH) or heterogeneous Toeplitz (TOEPH). The variance-covariance structure that results in the smallest AIC will be used. The Kenward-Roger method will be used to estimate the degrees of freedom. Type III sums of squares for the LS means will be used for the statistical comparison. The LS mean difference, standard error, p- value and CIs will also be reported. Treatment group comparisons versus placebo at Week 12 and other visits will be tested using the t-test obtained from the MMRM results. The Fisher exact test will be used to perform pairwise comparisons for AEs, discontinuations, and other categorical safety data among treatment groups. Change from baseline in continuous vital signs, body weight, and other continuous safety variables, including laboratory variables will be analyzed using an ANCOVA model with explanatory terms for treatment and the baseline value as a covariate. Type 3 sums of squares will be used. The significance of withintreatment group changes from baseline will be evaluated by testing whether the treatment group LS mean changes from baseline are different from zero using a t-statistic; the standard error for the LS mean change will also be displayed. Differences in LS means will be displayed with the p-value associated with the LS mean comparison to placebo and a 95% CI on the LS mean difference. In addition to the LS means for each group, the within-group p-value, the standard deviation, minimum, Q1, median, Q3, and maximum will be displayed. Data collected at early termination visits will be mapped to the next planned visit number for that patient. There will be two planned data locks for this study, one when all patients have completed Part A and a final one at end of the study. Refer to Section 7 for a description of the unblinding plan for this study. The following is a description of the analysis groups planned for this study. Refer to Section for the definitions of analysis populations used for the efficacy and safety analyses. Analysis Groups for the Efficacy, and Health Outcome Data Summaries of efficacy and health outcome data will be presented according to the following treatment/ cohort groups: Baricitinib: patients randomized to baricitinib excluding data obtained after the receipt of rescue therapy, Placebo: patients randomized to placebo excluding data obtained after the receipt of baricitinib, either as rescue therapy or according to the protocol-designated switch to baricitinib,
251 I4V-MC-JADV Statistical Analysis Plan Version 1.0 Page 21 Adalimumab: patients randomized to adalimumab excluding data obtained after the receipt of rescue therapy, Placebo to baricitinib: patients switched to baricitinib from placebo excluding data obtained after the receipt of rescue therapy (using data collected after the switch), Baricitinib (rescued): patients in baricitinib group who were rescued (using data collected after rescue), Placebo (rescued): patients in the placebo group who were rescued before or after switch to baricitinib (using data collected after rescue), Adalimumab (rescued): patients in adalimumab group who were rescued (using data collected after rescue). Analysis Groups for the Safety Data Summaries of safety data will be presented according to the following treatment/ cohort groups: Baricitinib: patients randomized to baricitinib excluding data obtained after the receipt of rescue therapy, Placebo: patients randomized to placebo using data obtained prior to the receipt of baricitinib, either as rescue therapy or according to the protocol-designated switch to baricitinib, Adalimumab: patients randomized to adalimumab excluding data obtained after the receipt of rescue therapy, Placebo (rescued): patients in the placebo group who were rescued before or after switch to baricitinib (using data collected after rescue), Placebo (switched): patients in the placebo group who were switched to baricitinib excluding data obtained after the receipt of rescue therapy (using data collected after switch), Adalimumab (rescued): patients in adalimumab group who were rescued (using data collected after rescue), All baricitinib exposures across all periods, regardless of initial treatment or use of rescue, ie, pooled across all cohorts who received baricitinib (using data collected after initiation of baricitinib). Patients who have impaired renal function who are randomized to baricitinib 4 mg will be given the 2 mg dose but analyzed in the baricitinib 4 mg group. Additional analyses for selected efficacy and safety endpoints will use this subgroup of patients [see Section 6.16 (Safety Analyses) and Section 6.17 (Subgroup Analyses)]. Summaries of safety data collected during the Post-Treatment Follow-up Period (Part C) will be presented separately from the summaries for Parts A and B and according to the following cohorts:
252 I4V-MC-JADV Statistical Analysis Plan Version 1.0 Page 22 Baricitinib: patients who were randomized to baricitinib, were receiving baricitinib at the time of discontinuing from the study or completing Part B, Adalimumab: patients who were randomized to adalimumab, were receiving adalimumab at the time of discontinuing from the study or completing Part B, Placebo: patients who were randomized to placebo, were receiving placebo at the time of discontinuation (during Part A), Other: all other patients who were not included in any of the 3 cohorts described previously Definition of Baseline The baseline period is defined as Visit 1 through pre-dose Visit 2. Treatment period for the blinded placebo and active controlled period (Part A) starts after the first study drug administration at Visit 2 (Week 0) and ends on the date of Visit 11 (Week 24) or the early discontinuation visit. Treatment period for the blinded active controlled period (Part B) starts after the first study drug administration at Visit 11 (Week 24) and ends on the date of Visit 15 (Week 52) or the early discontinuation visit. The Post-Treatment Follow-up Period (Part C) starts after the date of Visit 15 (Week 52) or the early discontinuation visit and lasts until the follow-up visit, which occurs approximately 28 days following the last dose of the study drug, for patients who do not enter long-term extension Study JADY. Baseline for the period up to rescue/ switch or end of study (if patients are not rescued/ switched to baricitinib) is defined as the last non-missing measurement on or prior to the date of first study drug administration (Visit 2, Week 0). Postbaseline measurements are collected after study drug administration (Week 0, Visit 2) through Week 52 (Visit 15) or early discontinuation visit. Baseline for the post-rescue/ switch period for patients who are rescued/ switched is defined as the last non-missing measurement obtained prior to initiation of the new treatment (baricitinib). Postbaseline measurements for rescued/ switched patients are collected after the rescue/ switch visit through Week 52 (Visit 15) or early discontinuation visit. Baseline for the Post-Treatment Follow-Up Period is defined as the last non-missing assessment on or prior to Week 52 (Visit 15) or early discontinuation visit. These baseline and postbaseline definitions will be considered for both safety and efficacy analyses and detailed information is found in Sections 6.12 to Derived Data Age (year) = (Date of informed consent (year of birth, July, 1) +1) / , and then truncated to a whole-year (integer) age Age group (<65 years old, 65 years old) Age group (<65 years old, 65 <75 years old, 75 <85 years old, 85 years old) BMI (kg/m 2 ) = Weight (kg)/((height (cm)/100)**2)
253 I4V-MC-JADV Statistical Analysis Plan Version 1.0 Page 23 Duration of RA (years) = [(Date of informed consent Date of RA onset) +1]/ Date of RA onset is defined as the date when the patient had symptoms of RA. If year of onset is missing, duration of RA will be set as missing. Otherwise, unknown month will be taken as January, and unknown day will be taken as 01. The duration of RA will be rounded to one decimal place. Duration of RA from diagnosis (years) = [(Date of informed consent Date of RA diagnosis+1]/ Date of RA diagnosis is defined as the date when the patient was diagnosed with RA. If year of diagnosis is missing, duration of RA will be set as missing. Otherwise, unknown month will be taken as January, and unknown day will be taken as 01. The duration of RA will be rounded to one decimal place. Duration from onset of RA (years): 0 to < 1; 1 to < 5; 5 Baseline and postbaseline Framingham 10-year risk of cardiovascular disease events category for men and women [Framingham percentage risk < 10% (low risk), 10% to 20% (intermediate risk), and >20% (high risk)] will be derived. The Framingham score is calculated from gender, age, diabetes diagnosis, smoking status, blood pressure medications, cholesterol, HDL, and systolic blood pressure. See Appendix 3 for the algorithm to calculate the Framingham risk assessment absolute score. The list of blood pressure medications used in the calculation will be identified via medical review prior to data lock. Baseline and postbaseline Reynolds Risk Score (RRS) 10-year risk of cardiovascular disease events category for non-diabetic men and women aged 45 years and hscrp 20mg/L [< 5% (low risk), 5% to < 10% (low to moderate), 10% to < 20% (moderate to high) and 20% (high risk)] will be derived (Ridker et al. 2007, 2008). The RRS is calculated from gender, age, smoking status, hscrp, total cholesterol, HDL, systolic blood pressure and whether either parent had a heart attack prior to age of 60 years old. Refer to Ridker et al for the calculation of the RRS for females and to Ridker et al for the calculation of the RRS for males. Change from baseline = postbaseline measurement at Visit x baseline measurement Percentage change from baseline at Visit x: o ((Postbaseline measurement at Visit x baseline measurement)/ baseline measurement)*100 Weight (kg) = weight (lbs) * Height (cm) = height (in) * 2.54 Temperature ( C) = [temperature ( F) 32]*5/9
254 I4V-MC-JADV Statistical Analysis Plan Version 1.0 Page 24 Baseline disease activity based on the DAS28 (calculated separately for DAS28-ESR and DAS28-hsCRP) o Low disease activity: DAS28 3.2, o Moderate disease activity: DAS28 >3.2 to 5.1, o High disease activity: DAS28 >5.1 SDAI, CDAI, EULAR Responder Index categories along with other efficacy and health outcome questionnaires scoring are included in Appendix Analysis Populations Modified Intent-to-treat (mitt) analysis set: The mitt analysis set will include all randomized patients who received at least one dose of the study drug. Patients will be analyzed according to the study drug to which they were randomized or assigned per protocol. The efficacy and health outcome analyses will be conducted on a mitt basis unless otherwise specified. If a patient is randomized but not treated with any dose of the study drug, he/she will not be included in any of the efficacy and health outcome analyses. Analysis of structural progression (van der Heijde mtss) will be conducted on a subset of the mitt that includes all randomized patients who received at least one dose of the study drug, have non-missing baseline and at least one non-missing postbaseline measurement. Per-protocol (PP) analysis set: The PP analysis set will include all mitt patients who are not deemed noncompliant with treatment in Part A, who do not have significant protocol deviations (refer to Section 6.6 for more details) in Part A, and whose investigator site does not have significant GCP issues that require a report to regulatory agencies (regardless of study part). Qualifications and identification of the specific significant protocol deviations that result in exclusion from the PP subsets will be determined while the study remains blinded, prior to first data lock. The primary and major secondary analyses will be repeated using the PP analysis set. Therefore, the PP will only be used for Part A. Safety analysis set: The safety set is defined as all randomized patients who received at least one dose of the study drug and who did not discontinue from the study for the reason Lost to Follow-up at the first post-baseline visit. Patients will be analyzed according to the study drug to which they were randomized or assigned per protocol. Safety analyses up to end of Part B will be conducted on this safety analysis set. Post-Treatment Follow-up analysis set: Safety analyses for Part C will be conducted on the follow-up population, defined as patients valid for the safety analysis set who completed the Post-Treatment Follow-up Period (Part C). All details involving the PK analysis will be provided in a separate PK analysis plan.
255 I4V-MC-JADV Statistical Analysis Plan Version 1.0 Page Adjustment for Covariates The covariates used in the parametric statistical model (ANCOVA) of continuous data generally will include the parameter value at baseline. Inclusion of baseline in the ANCOVA model provides estimated adjusted (least squares) means for each treatment reflecting averages for the average baseline patient. Other covariates that will be included in the models to adjust estimates of treatment effect include the stratifying variables region and joint erosion status. The covariates used in the logistic regression models will include the stratifying variables region and joint erosion status (refer to Section 6.1). The stratification variables used in the analyses will be derived based on ecrf data and x-ray data collected by Synarc Handling of Dropouts or Missing Data Missing Data Imputations Missing Data Imputation for Efficacy and Health Outcome Endpoints In accordance with precedent set with other Phase 3 RA trials (Keystone et al. 2004, 2008, 2009; Cohen et al. 2006; Smolen et al. 2008, 2009), the following methods for imputation of missing data will be used: 1. Non-responder imputation (NRI) All patients who discontinue the study or permanently discontinued the study treatment at any time for any reason will be defined as nonresponders for the NRI analysis for categorical variables, such as ACR20/ ACR50/ ACR70, from the time of discontinuation and onward. Patients who receive rescue therapy starting at Week 16 will be analyzed as nonresponders after the rescue visit and onward. Randomized patients without available data at a postbaseline visit will be defined as nonresponders for the NRI analysis at that visit. 2. Modified baseline observation carried forward (mbocf) The mbocf method will be used for the analysis of major secondary continuous endpoints (unless otherwise stated). For patients who discontinue the study or permanently discontinue the study treatment because of an AE, including death, the baseline observation will be carried forward to subsequent time points for evaluation. For patients who discontinue the study or permanently discontinue the study treatment for reason(s) other than an AE, the last nonmissing postbaseline observation prior to discontinuation will be carried forward to the subsequent time points for evaluation. For patients who receive rescue therapy starting at Week 16, the last nonmissing observation at or before rescue will be carried forward to subsequent time points for evaluation. For patients whose baseline value is missing or who have no change from baseline values after the aforementioned imputation, then the change from baseline will take a value of zero indicating no improvement from baseline. 3. Modified last observation carried forward (mlocf)
256 I4V-MC-JADV Statistical Analysis Plan Version 1.0 Page 26 For all continuous measures including safety analyses, the mlocf will be a general approach to impute missing data unless otherwise specified. For patients who receive rescue therapy starting at Week 16, the last nonmissing observation at or before rescue will be carried forward to subsequent time points for evaluation. For all other patients who discontinue from the study or permanently discontinue the study treatment for any reason, the last nonmissing postbaseline observation before discontinuation will be carried forward to subsequent time points for evaluation. After mlocf imputation, data from patients with nonmissing baseline and postbaseline observations will be included in the analyses. The mlocf will be used as a sensitivity method to the mbocf method with respect to the major secondary endpoints Missing Data Imputation for the Structural Progression Endpoint (van der Heijde mtss) 1. Primary method: linear extrapolation method The primary approach to the analysis of the structural progression data will be the linear extrapolation method, which assumes that individual patients would continue to accrue structural damage in their joints at the same rate that was observed at their time of last observation. The linear extrapolation method has been established as an appropriate missing data imputation method in other Phase 3 RA trials (Keystone et al. 2004, 2008, 2009; Cohen et al. 2006; Smolen et al. 2008, 2009). The linear extrapolation method will be used for analysis of the structural progression endpoint (van der Heijde mtss), including bone erosion scores and joint space narrowing (JSN), to impute missing data at time points when analyses are conducted (including the analyses at Week 24 and Week 52). For patients who discontinue the study or permanently discontinue the study treatment or for patients who miss a radiograph for any reason, baseline data and the most recent radiographic data prior to discontinuation or prior to the missed radiograph, adjusted for time, will be used for linear extrapolation to impute missing data at subsequent scheduled time points, including the primary analysis at Week 24. For patients who receive rescue therapy starting at Week 16 or at any time point thereafter, baseline data and the most recent radiographic data prior to initiation of rescue therapy, adjusted for time, will be used for linear extrapolation. For patients who discontinue from the study or permanently discontinue from the study treatment after receiving rescue therapy, the radiographic data collected prior to initiation of rescue therapy should supercede the data collected before discontinuation. All patients originally randomized to placebo will be switched to active treatment at Week 24; thus, there will be no observed data for the placebo arm for the structural comparison at subsequent time points. Therefore, baseline data and the most recent radiographic data prior to initiation of the new therapy, adjusted for time, will be used for linear extrapolation at Week 52. Missing postbaseline values will be imputed only if both a baseline value and a postbaseline value from another time point are available, as long as the patient was receiving the same treatment at each applicable time point. Missing post-baseline values will not be imputed using post-baseline data from later time points. For example: (1) For a baricitinib- or adalimumab-treated patient who did not require rescue therapy:
257 I4V-MC-JADV Statistical Analysis Plan Version 1.0 Page 27 Missing data at Week 16 will be extrapolated from baseline and an early discontinuation visit prior to Week 16, if available. Missing data at Week 24 will be extrapolated from baseline and the last available postbaseline value prior to Week 24. Missing data at Week 52 will be extrapolated from baseline and the last available postbaseline value prior to Week 52, if available or Week 24. (2) For a baricitinib- or adalimumab- or placebo-treated patient who was rescued at Week 16: Missing data at Weeks 24 or 52 will be extrapolated from baseline and Week 16. (3) For a placebo-treated patient who did not require rescue therapy: Missing data at Week 16 will be extrapolated from baseline and an early discontinuation visit prior to Week 16, if available. Missing data at Week 24 will be extrapolated from baseline and the last available postbaseline value prior to Week 24. Missing data at Week 52 will be extrapolated from baseline and the last available postbaseline value at or prior to Week Sensitivity method: multiple imputation (MI) A MI approach will also be used for the analyses of mtss, bone erosion scores, and JSN for the Week 24 analysis. A set of Bayesian regressions using SAS Proc MI or equivalent computational procedures will be utilized for the imputation of missing values due to dropout, rescue or treatment re-assignment, unevaluable images, or otherwise missing data such as from bones/joints that were surgically modified (e.g., joint replacement). The regression models will be utilized in order to impute missing values sequentially through study time. In all cases, observations obtained on patients after rescue, even to the same treatment, or re-assignment will not be utilized in the analysis. For each randomized treatment, the following steps will be used: Step 1. Impute missing Week 16 using data obtained at the early termination visit or the treatment rescue visit, depending on which case applies to the individual patient, using only patients with data at these early visits. A. Fit change from baseline = α weeks + β baseline*weeks interaction + error [no intercept], with the variance of the error term defined by ε. B. From the estimated joint distribution of (α,β,ε), randomly sample α and β for the regression equation and ε for each individual patient. C. Assign first scheduled score = baseline + 16*α + 16*β *baseline + ε for patients with missing data.
258 I4V-MC-JADV Statistical Analysis Plan Version 1.0 Page 28 Step 2. Impute missing Week 24 observations using standard imputation methodology in Proc MI to predict the Week 24 scores from baseline and first scheduled observation scores (both actual and imputed). Additional demographic or baseline disease severity covariates that are generally regarded or suspected to be predictive of progression of joint structural damage may also be included in the regression models. These Bayesian regression models utilize the variability inherent in the estimated model parameters to create random observations for patients with missing data at the specific time point. In order to properly account for the variability present in such predictions, multiple imputed data sets will be created and standard statistical analyses, such as ANCOVA, averaged over the sets of data using SAS PROC MIANALYZE. The multiple imputation method will be considered as a supportive approach to the linear extrapolation method for handling missing structural progression data. Details of the missing data imputation rules, methods for between-reader adjudication, score derivations, and other details for the bone erosion scores, joint space narrowing scores, and mtss are provided in Appendix 2 (Van der Heijde Modified Total Sharp Score). Table JADV.6.1. Imputation Techniques for Various Efficacy and Safety Variables Endpoint Imputation Method ACR 20/50/70 responses, DAS28-hsCRP and DAS28-ESR Low Disease NRI Activity (LDA) and remission, EULAR response criteria based on the 28-joint DAS, ACR/EULAR remission according to the Boolean-based definition, HAQ-DI improvement from baseline 0.22 and 0.3, CDAI LDA and remission and SDAI LDA and remission JSN, bone erosion scores, and mtss at Weeks 24 and 52 Linear extrapolation method JSN, bone erosion scores, and mtss at Week 24 MI HAQ-DI, DAS28-hsCRP mbocf Duration and severity of morning joint stiffness, worst tiredness and worst See Section 6.14 joint pain assessed in the diaries Continuous efficacy, health outcome, and safety measures (labs, vital signs, etc.) mlocf Abbreviations: ACR = American College of Rheumatology; CDAI = Clinical Disease Activity Index; DAS28 = Disease Activity Score modified to include the 28 diarthrodial joint count; EULAR = European League Against Rheumatism; HAQ-DI = Health Assessment Questionnaire Disability Index; hscrp = high-sensitivity C-reactive protein; JSN = joint space narrowing; LDA = low disease activity; mlocf = modified last observation carried forward; mtss = modified Total Sharp Score; NRI = nonresponder imputation. SDAI = Simplified Disease Activity Index Multiple Comparison Adjustments to multiple comparisons among the treatment groups using the primary and major secondary endpoints will use the methods described in Section These multiple testing procedures will be used to control the family-wise error rate for the primary and key secondary hypotheses for the endpoints. Each step in the sequential testing procedure uses a local 1-sided level procedure for the null hypotheses being tested. Testing of hypotheses at each step of the procedure commences only if all null hypotheses of prior steps were rejected. As such, the
259 I4V-MC-JADV Statistical Analysis Plan Version 1.0 Page 29 multiple testing procedure provides strong control of the family-wise error rate at the 1-sided level Patient Disposition Frequency counts and percentages of patients excluded prior to randomization by primary reason for exclusion will be provided. Patient disposition will be summarized using the mitt and PP (only Part A) analysis sets. The number of randomized patients will be summarized by treatment group and primary reason for discontinuation from the study and discontinuations to the study drug. Frequency counts and percentages of patients who are randomized, who complete Parts A or B or the study, along with their reason for discontinuation from the study and reason for study drug discontinuation, will be presented by treatment group for the non-rescued and rescued/ switched patients. A listing of all patients who discontinue from the study will be provided, with the extent of their participation in the study and the reason for discontinuation Protocol Deviation Protocol deviations will be tracked by the clinical team, and the importance will be assessed by key team members during the protocol deviation review meetings. Out of all important protocol deviations identified, a subset occurring during Part A with the potential to affect efficacy analyses will result in exclusion from the PP analysis set. Potential examples of deviations might include patients who receive excluded concomitant antirheumatic therapy, significant non-compliance (< 80%) with study medication (including failure to take study medication and taking incorrect study medication), patients incorrectly enrolled in the study, patients whose data are questionable due to significant site quality or compliance issues, etc. A summary of the number and percentage of patients with an important protocol deviation by treatment group, overall, and by type of deviation will be provided. Although the PP Set is for Part A only, important protocol deviations will be summarized for Parts A and B. Individual patient listings of important protocol deviations will be provided Patient Characteristics Patient characteristics including demographics will be summarized using the mitt analysis set by treatment group. The summary will include descriptive statistics such as the number of patients (n), mean, standard deviation (SD), median, minimum (min), and maximum (max) for continuous measures and frequency counts and percentages for categorical measures. No formal statistical comparisons will be made among treatment groups, unless otherwise stated Demographics Patient demographics will be summarized by treatment group using the mitt analysis set. The following demographic information will be presented for this study.
260 I4V-MC-JADV Statistical Analysis Plan Version 1.0 Page 30 o o o o o o o o o o Age and Age group Gender Race Ethnicity Region Country Weight Height BMI Waist circumference Baseline Disease Characteristics Baseline disease characteristics will be summarized by treatment group using the mitt analysis set. The following baseline disease information (but not limited to only these), will be presented for this study. o o o o o o o o o o o o o o o o o o o o o o o Habits (Alcohol, smoking) Baseline Framingham 10-year risk of cardiovascular disease events score Baseline RRS 10-year risk of cardiovascular disease events score Duration from onset of Rheumatoid Arthritis (years), Duration from onset of RA category (<1 years, 1 to < 5, 5 years) Years since RA Diagnosis Patient s assessment of pain (by VAS in mm) Patient s global assessment of disease activity (by VAS in mm) Physician s global assessment of disease activity (by VAS in mm) HAQ-DI total score Erythrocyte Sedimentation Rate (ESR) (mm/hr) hscrp (mg/l) DAS28-ESR and DAS28-hsCRP Anti-cyclic citrullinated Peptide (Anti-CCP) positivity and Rheumatoid factor (RF) positivity status Number of tender joints out of 28 (TJC28) Number of swollen joints out of 28 (SJC28) Number of tender joints out of 68 (TJC68) Number of swollen joints out of 66 (SJC66) Estimated Glomerular Filtration Rate (egfr) egfr < 60 ml/min/1.73 m2 egfr 60 ml/min/1.73 m2 MTX dose (mg/week) Use of cdmards Number of cdmards use (1, >1) Use of MTX only Use of MTX + other cdmards Use of oral corticosteroid and dose (mg/day) expressed as prednisone-equivalent dose Use of statins
261 I4V-MC-JADV Statistical Analysis Plan Version 1.0 Page 31 In addition to the summary table mentioned above, a listing of patient baseline characteristics will be produced Previous and Concomitant Medication Summaries of previous and concomitant medications used for RA including cdmards (MTX only, MTX and other cdmard; see Section 6.7) will be based on the mitt analysis set. At screening, medications used for RA that had been previously discontinued are recorded for each subject. A summary of previous medications used for RA will be prepared using frequency counts and percentages by World Health Organization (WHO) drug anatomical therapeutic chemical (ATC) classification and treatment group. Concomitant therapy will be recorded at each visit and will be classified according to the WHO drug dictionary and summarized using frequency counts and percentages by WHO drug ATC classification sorted by decreasing frequency and treatment group. Concomitant medications are defined as: Concomitant medications with a stop date on or after the date of the first dose of the study drug and that precedes Week 24 (Visit 11) or early discontinuation visit will be included in the concomitant medications summaries for the analysis at the end of Part A. Should there be insufficient data to make this comparison (for example the concomitant therapy stop year is the same as the treatment start year, but the concomitant therapy stop month and day are missing), the medication will be considered as concomitant for the Treatment Period. For patients who continue in Part B, concomitant medications with a stop date that is after Week 24 or ongoing will be included in the concomitant medications summaries at end of Parts A and B. For patients who complete Part C, concomitant medications with a stop date that is after Week 52 (Visit 15) or ongoing will be included in the concomitant medications summaries for Part C. Summaries of concomitant medications will also be summarized for the all baricitinib exposures group including the rescued and switched placebo patients. An individual patient listing will be provided for previous and concomitant medications. Also, an individual patient listing will be provided for the patients using prohibited concomitant medications Historical Illnesses and Pre-Existing Conditions The number and percentage of patients with significant historical illnesses, as listed on the ecrf, will be summarized by system organ class (SOC) and preferred term (PT) and by treatment group using the mitt analysis set. Historical illnesses will be coded using the Medical Dictionary for Regulatory Activities (MedDRA, most current available version). Pre-existing conditions are defined as those conditions recorded on the Pre-existing Conditions and AEs ecrf page with a start date prior to the date of informed consent. Note that conditions
262 I4V-MC-JADV Statistical Analysis Plan Version 1.0 Page 32 with a partial or missing start date will be assumed to be not pre-existing unless there is evidence, through comparison of partial dates, to suggest otherwise. Pre-existing conditions will be coded using the MedDRA dictionary. Frequency counts and percentages of patients with preexisting conditions will be summarized by treatment group using the mitt analysis set by SOC and PT Treatment Compliance Patient compliance with study medication will be assessed from randomization visit (Week 0) to Visit 15 (Week 52) during the treatment period. Compliance will be summarized in different periods of the study such as, from randomization to rescue, from rescue to Week 24, etc. Compliance for baricitinib and placebo treatment group: For patients receiving baricitinib or placebo where one tablet is expected to be taken daily in Parts A and B regardless of rescue, a patient is considered noncompliant if he or she misses >20% of the prescribed doses during the study. For patients who have their dose interrupted due to safety reasons, the period of dose withholding will be taken into account in the compliance calculation as outlined below. The compliance for the placebo treatment group will be calculated based on the placebo tablets instead of placebo syringes. Compliance to baricitinib and placebo treatment in the period of interest will be calculated as follows: actual total # of tablets used Expected total # of tablets used 100 Where: the expected total # tablets used = the number of days in the period of interest; the actual total # of tablets used = total number dispensed total number returned in the period. If a patient has dose interrupted for safety reasons during the period, the total number of days drug was withheld will be deducted from the expected total # of tablets used. Compliance for adalimumab treatment group: For patients receiving adalimumab, a patient is considered noncompliant if >20% of prescribed doses are missing or if there are any consecutive missed injections. For patients who have dose interrupted due to safety reasons, the period of dose withholding will be taken into account in the compliance calculation as outlined below. Compliance to adalimumab treatment in the period of interest will be calculated as follows:
263 I4V-MC-JADV Statistical Analysis Plan Version 1.0 Page 33 Where: actual total # of syringes used Expected total # of syringes used 100 Expected total number of syringes used = integer of [(total number of days in the period of interest)/14] + 1. Actual total # of syringes used = total # of syringes dispensed total # of syringes returned. For patients with drug interruptions, the expected number of syringes used will be calculated for the period before and after interruption in the period of interest separately, then the sum of the two will be the expected total for the period of interest. Patients who are significantly noncompliant in Part A will be excluded from the PP analysis set. The number of tablets dispensed/returned, number of syringes dispensed/returned, and percent compliance will be listed by treatment group Treatment Exposure Duration of exposure to each study drug will be summarized for the safety analysis set using descriptive statistics (n, mean, standard deviation, minimum, median, maximum). Cumulative exposure and duration of exposure will be summarized in terms of frequency counts and percentages by category and treatment group. Overall exposure will be summarized in total patient-years which are calculated according to the following: Exposure in patient-years = sum of duration of exposure in days (for all patients in treatment group) / The summary for the first data lock (end of Part A) will include exposure to each treatment plus exposure for the subset of patients who are rescued during Part A from each randomized therapy. For the baricitinib treatment group, exposure will be calculated including the subset of patients who are rescued during Part A. The summary for the final lock (end of Parts A and B) will include all exposures for patients randomized to baricitinib, placebo and adalimumab, plus all exposures subsequent to rescue or switch to baricitinib. For the baricitinib treatment group, exposure will be calculated including the subset of patients who are rescued. Duration of exposure will be calculated as follows: 1. Baricitinib group: a. Non-rescued: duration of exposure to baricitinib will be calculated as: (date of last baricitinib treatment date of first dose (Visit 2) + 1). b. Rescued: duration of exposure to baricitinib will be calculated as: (date of last baricitinib treatment prior to rescue date of first dose (Visit 2) + 1).
264 I4V-MC-JADV Statistical Analysis Plan Version 1.0 Page Adalimumab group: a. Non-rescued: duration of exposure to adalimumab will be calculated as (date of last visit in the study Visit 2 date + 1). b. Rescued: duration of exposure to baricitinib 4 mg will be calculated as: (date of last baricitinib treatment date of first baricitinib dose + 1). 3. Placebo group: a. Non-rescued: duration of exposure to placebo will be calculated as: (date of last placebo treatment date of first dose (Visit 2) + 1). b. Rescued: duration of exposure to baricitinib will be calculated as: (date of last baricitinib treatment date of first baricitinib dose after rescue + 1). c. Switched: duration of exposure to baricitinib will be calculated as: (date of last baricitinib treatment (before rescue if applicable) date of first baricitinib dose + 1). Summaries of treatment exposure will also include all baricitinib exposures, covering groups 1a, 1b, 2b, 3b and 3c above Efficacy Analyses Primary Efficacy Analysis The primary efficacy measure will be the proportion of patients achieving ACR20 at Week 12 based on the change in ACR response values from baseline. ACR20 is defined as at least 20% improvement from baseline in the following ACR Core Set variables: TJC (68 joint count) SJC (66 joint count) An improvement of 20% from baseline in at least 3 of the following 5 assessments: o Patient s Assessment of Pain (visual analog scale [VAS]) o Patient s Global Assessment of Disease Activity (VAS) o Physician s Global Assessment of Disease Activity (VAS) o Patient s Assessment of Physical Function as measured by the HAQ-DI o Acute phase reactant as measured by hscrp Refer to the algorithm to calculate ACR response in Appendix 3. The primary efficacy analysis compares ACR20 at Week 12 between baricitinib and placebo using the mitt analysis set. The PP analysis set results will be used as supportive to the mitt results. A logistic regression model with treatment, region, and joint erosion status (1-2 joint erosions plus seropositivity vs. at least 3 joint erosions) in the model will be used to test the treatment difference between baricitinib and placebo in the proportion of patients achieving ACR20 response at Week 12 using the Wald test at 2-sided significance level of The NRI approach will be used to impute response status for patients with completely missing data. The percentages, difference in percentages, p-value (from the logistic model) and 95% CI of the difference in percentages will be reported. The 95% CI will be calculated using the Newcombe- Wilson method (Newcombe 1998) without continuity correction. Treatment-by-region interaction will be added to the logistic regression model as a sensitivity analysis and results
265 I4V-MC-JADV Statistical Analysis Plan Version 1.0 Page 35 from this model will be compared to the primary fixed effects model (without the interaction effect). If the treatment-by-region interaction is significant at a 2-sided 0.1-alpha level, the nature of this interaction will be inspected as to whether it is quantitative (ie, the treatment effect is consistent in direction but not size of effect) or qualitative (the treatment is beneficial in some but not all regions). If the treatment-by-region interaction effect is found to be quantitative, results from the primary fixed effect model will be presented. If the treatment-by-region interaction effect is found to be qualitative, further inspection will be used to identify in which regions baricitinib was found to be more beneficial Major Secondary Efficacy Analyses Unless otherwise stated, the following analyses will be performed to test superiority of baricitinib comparing to the placebo group. The major (key) secondary efficacy measures are: Change from baseline to Week 24 in mtss Change from baseline to Week 12 in HAQ-DI Change from baseline to Week 12 in DAS28-hsCRP Proportion of patients achieving an SDAI score 3.3 at Week 12 Proportion of patients achieving ACR20 at Week 12 with respect to noninferiority of baricitinib to adalimumab Mean duration of morning joint stiffness (7 days prior to Week 12) Change from baseline to Week 12 in DAS28-hsCRP with respect to superiority of baricitinib to adalimumab Mean severity of morning joint stiffness (7 days prior to Week 12) Mean Worst Tiredness NRS (7 days prior to Week 12) Mean Worst Pain NRS (7 days prior to Week 12) The major (key) secondary efficacy measures will be analyzed using the mitt analysis set. The PP analysis set results will be used as supportive to the mitt results. These analyses are described as follows: Comparison between baricitinib and placebo in change from baseline to Week 24 in mtss: an ANCOVA model with baseline score, region, joint erosion status and treatment group in the model will be used to test the treatment difference between baricitinib and placebo in change from baseline to Week 24 in mtss scores. Type III sums of squares for the LS means will be used for the statistical comparison; 100*(1- alpha)% CI will also be reported. The alpha level will be defined according to the gatekeeping analyses described in Section The LS means, SE, CI, and p-values
266 I4V-MC-JADV Statistical Analysis Plan Version 1.0 Page 36 will be presented. The linear extrapolation method described in Section 6.3 will be used. A MI approach described in Section 6.3 will be used as supportive to the linear extrapolation method to impute missing data. A permutation-based method will be used as a nonparametric sensitivity (supportive) analysis to the ANCOVA method. Comparison between baricitinib and placebo in change from baseline to Week 12 in HAQ-DI score: an ANCOVA model with baseline score, region, joint erosion status and treatment group in the model will be used to test the treatment difference between baricitinib and placebo in change from baseline to Week 12 in HAQ-DI. Type III sums of squares for the LS means will be used for the statistical comparison; 100*(1-alpha)% CI will also be reported. The alpha level will be defined according to the gatekeeping analyses described in Section The LS means, SE, CI, and p-values will be presented. The mbocf approach described in Section 6.3 will be used to impute missing data. The mlocf approach described in Section 6.3 will also be used as supportive to the mbocf approach. In addition, supportive MMRM analysis will be used (refer to Section 6.1 for more details) to present the results of treatment effect at Week 12. Comparison between baricitinib and placebo in change from baseline to Week 12 in DAS28-hsCRP: an ANCOVA model with baseline score, region, joint erosion status and treatment group in the model will be used to test the treatment difference between baricitinib and placebo in change from baseline to Week 12 in DAS28-hsCRP. Type III sums of squares for the LS means will be used for the statistical comparison; 100*(1- alpha)% CI will also be reported. The alpha level will be defined according to the gatekeeping analyses described in Section The LS means, SE, CI, and p-values will be presented. The mbocf approach described in Section 6.3 will be used to impute missing data. The mlocf approach described in Section 6.3 will also be used as supportive to the mbocf approach. In addition, supportive MMRM analysis will be used (refer to Section 6.1 for more details) to present the results of treatment effect at Week 12. Comparison between baricitinib and placebo in proportion of patients achieving an SDAI score 3.3 at Week 12: A logistic regression model with treatment, region, and joint erosion status in the model will be used to test the treatment difference between baricitinib and placebo in the proportion of patients achieving an SDAI score 3.3 response at Week 12. The NRI method as described in Section 6.3 will be used to impute missing data. The percentages, difference in percentages, p-value (from the logistic model) and 100*(1-alpha)% CI of the difference in percentages will be reported. The 100*(1-alpha)% CI will be calculated using the Newcombe-Wilson method (Newcombe 1998) without continuity correction. The alpha level will be defined according to the gatekeeping analyses described in Section Comparison between baricitinib and placebo in mean duration of morning joint
267 I4V-MC-JADV Statistical Analysis Plan Version 1.0 Page 37 stiffness in the 7 days prior to Week 12: refer to Section 6.14 for the statistical methods used for the analysis of mean duration of morning joint stiffness. Noninferiority comparison of baricitinib to adalimumab in ACR20 response at Week 12: a logistic regression model with region, joint erosion status and treatment group in the model will be used to test the treatment difference between baricitinib and placebo in the percentage of patients achieving ACR20 response at Week 12. The NRI approach will be used to impute response status for patients with completely missing data. The percentages, difference in percentages, and 95% CI of the difference in percentages will be reported. The 95% CI will be calculated using the Newcombe- Wilson method (Newcombe 1998) without continuity correction. If adalimumab is found to be superior to placebo as assessed by the ACR20 response rate at Week 12 using same model specification as for the primary analysis, and if the lower bound of the 95% CI (1- sided 97.5% CI) between baricitinib and adalimumab is > -12%, Lilly will conclude that baricitinib is noninferior to adalimumab with respect to ACR20. If the lower bound of the 95% CI is > 0%, Lilly will conclude that baricitinib is superior to adalimumab with respect to ACR20. Treatment-by-region interaction will be inspected. If this interaction is significant at a 2-sided 0.1-alpha level, and the interaction is quantitative, a supportive method to the Newcombe-Wilson method will be used such as the extended Mantel- Haenszel method to construct a stratified 95% CI. However, conclusions will be based on the Newcombe-Wilson method without continuity correction. Comparison between baricitinib and adalimumab in change from baseline to Week 12 in DAS28-hsCRP: An ANCOVA model with treatment, region, joint erosion status, and baseline score in the model will be used to test the treatment difference between baricitinib and adalimumab in change from baseline to Week 12 in DAS28-hsCRP. Type III sums of squares for the LS means will be used for the statistical comparison; 95% CI will also be reported. The LS means, SE, CI, and p-values will be presented. The mbocf approach described in Section 6.3 will be used to impute missing data. The mlocf approach described in Section 6.3 will also be used as supportive to the mbocf approach. In addition, supportive MMRM analysis will be used (refer to Section 6.1 for more details) to present the results of treatment effect at Week 12. Comparison between baricitinib and placebo in mean severity of morning joint stiffness in the 7 days prior to Week 12: refer to Section 6.14 for the statistical methods used for the analysis of severity of morning joint stiffness. Comparison between baricitinib and placebo in mean Worst Tiredness NRS in the 7 days prior to Week 12: refer to Section 6.14 for the statistical methods used for the analysis of worst tiredness. Comparison between baricitinib and placebo in mean Worst Pain NRS in the 7 days prior to Week 12: refer to Section 6.14 for the statistical methods used for the
268 I4V-MC-JADV Statistical Analysis Plan Version 1.0 Page 38 analysis of worst joint pain. Sensitivity analyses will be performed by adding the treatment-by-baseline interaction to the ANCOVA model. If this interaction is significant at a 2-sided 0.1-alpha level, then additional analyses will be performed to determine if the interaction is qualitative or quantitative. Such analyses may require defining category levels to the continuous baseline score. Treatment-byregion interaction will also be added to the fixed effects model for sensitivity purposes. If the treatment-by-region interaction is significant at a 2-sided 0.1-alpha level, the nature of this interaction will be inspected as to whether it is quantitative or qualitative. If the interaction is quantitative, results from the primary fixed effect model will be presented. If the interaction effect is qualitative, further inspection will be used to identify in which regions baricitinib was found to be more beneficial Analysis for Other Secondary Efficacy Endpoints All statistical comparisons described in this section will compare baricitinib to placebo. Summary statistics will be provided for the proportion of patients achieving DAS28-ESR 3.2 and <2.6 and for DAS28-hsCRP 3.2 and <2.6 at post-baseline visits. A logistic regression analysis will be conducted to analyze the patients with DAS28-ESR 3.2 and <2.6 and for DAS28-hsCRP 3.2 and <2.6 at Weeks 12 up to Week 24. The model for the logistic regression will consist of region, joint erosion, and treatment as fixed effects to compare baricitinib to placebo. The proportion of patients achieving HAQ-DI improvement from baseline 0.22 and 0.3 will be summarized at post-baseline visits. A logistic regression analysis with a model consisting of region, joint erosion, and treatment will be applied to compare baricitinib to placebo. The actual score and change from baseline scores at Week 16 and Week 52 in mtss will be summarized and ANCOVA will be performed for the change from baseline score at Weeks 16 and 52 using the same model specification for the change from baseline to Week 24 in mtss mentioned above. Summary statistics for the proportion of patients with mtss change 0 by treatment group will be provided. A logistic regression analysis will be conducted to analyze the patients with mtss change 0 at Weeks 16, 24, and 52 between baricitinib and placebo. The model for the logistic regression will consist of region, joint erosion, and treatment as fixed effects. Summary statistics by treatment group will be provided for the proportion of patients who improved (mtss <0), no change (mtss=0), or worsened (mtss >0). In addition, the proportion of patients with change in mtss SDC at Week 24 will be analyzed in a similar manner as described for mtss change 0. SDC is defined as the smallest detectable change which is computed from the variability in Week 0 to Week 24 change in scores assigned by the blinded imaging assessors (Bruynesteyn 2005).
269 I4V-MC-JADV Statistical Analysis Plan Version 1.0 Page 39 The actual score and change from baseline scores at Week 16, Week 24, and Week 52 in joint space narrowing and in bone erosion scores will be summarized. In addition, ANCOVA analysis will be performed for the change from baseline scores at Weeks 16, 24, and 52 using the same model specified for mtss. Furthermore, as an exploratory analysis, the change from baseline in mtss to Week 24 (Visit 11) will be compared to the change from Week 24 (Visit 11) to Week 52 (Visit 15) for each treatment group using a paired t-test to explore changes in the rate of structural damage progression over time or as a result of treatment switch. Note that patients randomized to placebo will be switched to baricitinib after Week 24 (Visit 11). The proportion of patients achieving ACR20, ACR50, and ACR70 at each post-baseline visit up to Visit 15 (Week 52) will be summarized. A logistic regression with the same model specification as the primary analysis will be used for the treatment comparisons up to Week 24 for ACR20, ACR50, and ACR70. The observed ( as-is ) values of the ACR20, ACR50, and ACR70 will be summarized as a sensitivity approach to the primary method of NRI. Using the as-is approach, the ACR values are calculated without applying any imputation to the components. The percent change from baseline in individual components of the ACR core set will be summarized at post-baseline visits. An ANCOVA model with region, joint erosion status, treatment group, and baseline value of each individual component of the ACR core set in the model will be used to compare baricitinib to placebo in the percent change in individual components of the ACR core set at Weeks 12 up to Week 24. Summary statistics of the actual score and change in DAS28-hsCRP and in DAS28-ESR from baseline to post-baseline visits will be provided. Change from baseline to postbaseline visits up to Week 24 in DAS28-hsCRP and in DAS28-ESR will be analyzed using an ANCOVA model with treatment, region, joint erosion status, and baseline score in the model to test the treatment difference between baricitinib and placebo. Summary statistics of the actual score and change in HAQ-DI from baseline to postbaseline visits will be provided. Change from baseline to post-baseline visits up to Week 24 in HAQ-DI score will be analyzed using an ANCOVA model with treatment, region, joint erosion status, and baseline score in the model to test the treatment difference between baricitinib and placebo. Summary statistics of actual score and change in CDAI and in SDAI from baseline to post-baseline visits will be provided. Change from baseline in CDAI and in SDAI up to Week 24 will be analyzed using ANCOVA. The model for the ANCOVA analysis will include treatment, region, and joint erosion status as independent fixed effects and the baseline score as a covariate with change from baseline SDAI or CDAI as the dependent variable. The proportion of patients achieving SDAI score 3.3 (ACR/EULAR remission indexbased definition) and CDAI score 2.8 up to Week 52 will be summarized. Remission based upon SDAI score 3.3 (Felson et al. 2011) up to Week 24 and remission based
270 I4V-MC-JADV Statistical Analysis Plan Version 1.0 Page 40 upon CDAI score 2.8 up to Week 24 will be analyzed using a logistic regression model with region, joint erosion, and treatment as fixed effects. The proportion of patients achieving CDAI score 10 and proportion of patients achieving SDAI score 11 will also be analyzed up to Week 24 and summarized up to Week 52. The proportion of patients achieving European League Against Rheumatism (EULAR) Responder Index based on the 28-joint count at each post-baseline visit will be summarized. EULAR nonresponders and responders (moderate + good responders) will be summarized. The NRI method will be used as the missing data imputation technique. A logistic regression analysis will be performed to analyze EULAR responders between baricitinib and placebo up to Week 24. The model consists of region, joint erosion, and treatment as fixed effects. Apart from the aforementioned categories, a summary table with frequency and percentages will be presented for the patients categorized as good responder, moderate responder, and nonresponder, and the treatment comparison between baricitinib and placebo will be conducted using a Cochran-Mantel-Haenszel (CMH) test controlling for the region effect. A summary table will be provided for the proportion of patients achieving ACR/EULAR remission for the Boolean-based definition by treatment group. A logistic regression analysis will be used and defined similar to what was described for SDAI or CDAI. Hybrid ACR (bounded) response will be summarized at all post-baseline visits and using an ANOVA model with region, joint erosion status, and treatment group as fixed effects and the post-baseline hybrid ACR response as the dependent variable to compare baricitinib to placebo up to Week 24. Actual measures and change from baseline measures will be summarized by visit and treatment group for TJC68, SJC66, patient s assessment of arthritis pain, patient s global assessment of disease activity and physician s global assessment of disease activity. Change from baseline measures in these endpoints up to Week 24 will be analyzed between baricitinib and placebo using ANCOVA. The model for the ANCOVA will include treatment, region, and joint erosion status as fixed effects and the baseline measurement as a covariate with the change from baseline as the dependent variable. Summary statistics of the actual and change in duration of morning joint stiffness, severity of tiredness using the Worst Tiredness NRS, and severity of pain using the Worst Pain NRS from baseline to post-baseline visits will be provided. The change from baseline to post-baseline visits up to Week 24 will be analyzed using data obtained from the epro tablet in ANCOVA to test the treatment difference between baricitinib and placebo Testing Strategy to Control the Type I Family-Wise Error Rate The following multiple testing procedure will be used to control the family-wise error rate for the primary and key secondary hypotheses for the endpoints listed in Section 4.2. Each step in the sequential testing procedure uses a local level procedure for the null hypotheses being
271 I4V-MC-JADV Statistical Analysis Plan Version 1.0 Page 41 tested. Testing of hypotheses at each step of the procedure commences only if all null hypotheses of prior steps were rejected. As such, the multiple testing procedure provides strong control of the family-wise error rate at the 1-sided level. The overall strategy is illustrated (Bretz et al. 2009) below. This strategy is appropriate for inclusion of significant results in product labeling. If any of the secondary hypotheses in the list are not rejected, the subsequent test(s) will still be performed but the results will be treated as exploratory. Hypotheses to be tested: 1. Baricitinib vs. placebo for superiority in ACR20 at Week Baricitinib vs. placebo for superiority in change from baseline in HAQ-DI at Week Baricitinib vs. placebo for superiority in change from baseline in mtss at Week Baricitinib vs. placebo for superiority in change from baseline in DAS28-hsCRP at Week Baricitinib vs. placebo for superiority in proportion of patients achieving an SDAI score 3.3 at Week Baricitinib vs. adalimumab for noninferiority in ACR20 at Week Baricitinib vs. placebo for superiority in mean duration of morning joint stiffness in the 7 days prior to Week Baricitinib vs. Adalimumab for superiority in change from baseline in DAS28-hsCRP at Week Baricitinib vs. placebo for superiority in mean severity of morning joint stiffness in the 7 days prior to Week Baricitinib vs. placebo for superiority in mean Worst Tiredness NRS in the 7 days prior to Week Baricitinib vs. placebo for superiority in Worst Pain NRS in the 7 days prior to Week 12. The following steps will be followed to test the aforementioned hypotheses. Step 1: Test ACR20 at Week 12 for baricitinib versus placebo at 1-sided alpha = If the null hypothesis is rejected, proceed to Step 2. Otherwise, discontinue testing. Step 2: Test the change from baseline in HAQ-DI at Week 12 and the change from baseline in mtss at Week 24, both tests of baricitinib versus placebo, at 1-sided alpha = via a weighted Holm procedure (Holm 1979) with weights of 0.2 and 0.8, respectively. If both null hypotheses are rejected, proceed to Step 3. Otherwise, discontinue testing.
272 I4V-MC-JADV Statistical Analysis Plan Version 1.0 Page 42 Details of the Holm procedure at this step: Let p2 denote the unadjusted 1-sided p-value resulting from testing the hypothesis regarding HAQ-DI and let p3 denote the unadjusted 1-sided p-value resulting from testing the hypothesis regarding mtss. Let p2* and p3* denote the corresponding weight-adjusted p-values (ie, p2* = p2/0.2 and p3*=p3/0.8). The following decision rules apply: If min (p2*, p3*) >0.025, then Lilly will fail to reject both null hypotheses and discontinue testing. Otherwise, if p2* 0.025, then Lilly will reject the null hypothesis for HAQ-DI (declaring statistical significance) and test mtss using the unadjusted p-value. If p , Lilly will also reject the null hypothesis for mtss (declaring statistical significance); however if p3 >0.025, then Lilly will fail to reject the null hypothesis for mtss and discontinue testing. Otherwise, if p3* 0.025, then Lilly will reject the null hypothesis for mtss (declaring statistical significance) and test HAQ-DI using the unadjusted p-value. If p , Lilly will also reject the null hypothesis for HAQ-DI (declaring statistical significance); however if p2 >0.025, then Lilly will fail to reject the null hypothesis for HAQ-DI and discontinue testing. Step 3: Test the change from baseline in DAS28-hsCRP at Week 12 and the proportion of patients achieving an SDAI score 3.3 (SDAI remission) at Week 12, both tests of baricitinib versus placebo, at 1-sided alpha=0.025 via a Hochberg procedure (Hochberg 1988). If both null hypotheses are rejected, proceed to Step 4. Otherwise, discontinue testing. Details of the Hochberg procedure at this step: Let p1 and p2 denote the unadjusted 1-sided p-values resulting from testing the hypotheses regarding DAS28-hsCRP and SDAI remission, respectively. If p1and p , then Lilly will reject both null hypotheses (declaring statistical significance for both DAS28-hsCRP and SDAI remission). Otherwise, if p and p2 >0.025, then Lilly will reject only the null hypothesis (declaring statistical significance) related to DAS28-hsCRP. Otherwise if p and p1 >0.025, then Lilly will reject only the null hypothesis (declaring statistical significance) related to SDAI remission. Otherwise Lilly will fail to reject both null hypotheses and discontinue testing. Step 4: Test ACR20 at Week 12 for noninferiority of baricitinib to adalimumab at 1-sided alpha= Details concerning the noninferiority margin and statistical conclusions are found in Section If the null hypothesis is rejected, proceed to Step 5. Otherwise, discontinue testing.
273 I4V-MC-JADV Statistical Analysis Plan Version 1.0 Page 43 Step 5: Test mean duration of morning joint stiffness in the 7 days prior to Week 12 for baricitinib versus placebo at 1-sided alpha= If the null hypothesis is rejected, proceed to Step 6. Otherwise, discontinue testing. Step 6: Test the change from baseline in DAS28-hsCRP at Week 12 for baricitinib versus adalimumab at 1-sided alpha= If the null hypothesis is rejected, proceed to Step 7. Otherwise, discontinue testing. Step 7: Test mean severity of morning joint stiffness in the 7 days prior to Week 12 for baricitinib versus placebo at 1-sided alpha= If the null hypothesis is rejected, proceed to Step 8. Otherwise, discontinue testing. Step 8: Test mean Worst Tiredness NRS in the 7 days prior to Week 12 and mean Worst Pain NRS in the 7 days prior to Week 12, both tests of baricitinib versus placebo, at 1-sided alpha=0.025 via a Hochberg procedure and following similar details as outlined in Step 3.

 significant; W = week; BARI = baricitinib; ADA = adalimumab. Figure JADV.6.1.")
274 I4V-MC-JADV Statistical Analysis Plan Version 1.0 Page 44 Abbreviations: = change from baseline; ACR20 = 20% improvement in American College of Rheumatology criteria; DAS28 = 28 diarthroidal joint count; hscrp = high-sensitivity C-reactive protein; HAQ-DI = Health Assessment Questionnaire-Disability Index; SDAI = simplified disease activity index; mtss = modified Total Sharp Score [van der Heijde]; TS = test(s) significant; W = week; BARI = baricitinib; ADA = adalimumab. Figure JADV.6.1. Illustration of the sequential testing approach within the overall gatekeeping strategy for Study JADV.
275 I4V-MC-JADV Statistical Analysis Plan Version 1.0 Page Pharmacokinetic/Pharmacodynamic Analysis (PK/PD) All PK/PD analyses will be performed by Eli Lilly and Company and documented in a separate SAP Health Outcome Analyses Psychometric analyses will be performed by Global Health Outcomes at Eli Lilly and Company and documented in a separate SAP Diary Data Data Handling and Missing Data Imputation Diary data will be assessed daily from Week 0 (Visit 2) to Week 12 (Visit 7). Two analyses will be performed for the diary data. The primary analysis is based on the scores collected in the 7 days prior to the Week 12 visit date, which addresses one of the major secondary objectives included in the gatekeeping strategy. The supportive analysis is based on daily data modeled by weekly means. The weeks are defined as follows: Day 1 on diary (baseline), Days 2-8 (Week1), Days 9-15 (Week2), and so on up to Days (Week 12). For the primary analysis, the average of the scores collected in the 7 days prior to Week 12 will be used. If there are fewer than 4 nonmissing scores, the 7-day window will be shifted back in time (toward baseline) one day at a time until there are 4 nonmissing scores available in the 7- day window. Then the average of the 4 scores will be used in the Week 12 analysis. For patients who discontinue the study or permanently discontinue the study treatment, the scores collected in the last 7 days prior to discontinuation will be used in the analysis. If there are fewer than 4 nonmissing scores, a similar approach will be used to find 4 nonmissing scores in a 7-day window and the average score will be used in the Week 12 analysis. Patients who do not have at least 4 scores in any postbaseline 7-day window up to Week 12 will have their baseline score carried forward to the endpoint for analysis. For patients without a baseline score, the first available postbaseline score will be used to impute the baseline score so that the patient can be included in the analysis. For the supportive analysis, if more than 4 scores during a defined week are missing, the daily scores collected in the week will not be used in the analysis as a protective measure against potentially unreliable or non-representative data Analysis of Diary Data The impact of baricitinib compared to placebo up to Week 12 with regard to patient-reported outcomes (PROs) will be evaluated by the following measures: Duration of morning joint stiffness Duration of morning joint stiffness is a patient-administered item that allows for the patients to enter the length of time in minutes (using 15-minute increments) that their morning joint stiffness lasted each day (using an electronic handheld diary) through Visit 7 (Week 12).
276 I4V-MC-JADV Statistical Analysis Plan Version 1.0 Page 46 Duration of morning stiffness will be recorded in minutes. Durations recorded as longer than 12 hours (720 minutes) will be censored at 720 minutes. For the analysis used in the gatekeeping strategy (Section 6.12), an ANCOVA model will be used to test the treatment differences in the average durations (of morning joint stiffness) collected in the 7 days prior to Week 12, with treatment, region and joint erosion status as fixed effects and baseline (Day 1 on the diary) as a covariate. Missing data will be handled as described for the main analysis in Section Type III sums of squares for the LS means will be used for the statistical comparison; 100*(1-alpha)% CI will also be reported. The alpha level will be defined according to the gatekeeping analyses described in Section The LS means, SE, CI, and p-values will be presented for the treatment differences between baricitinib and placebo. In addition, a supportive analysis using a MMRM model will be performed to compare the treatment differences in weekly mean durations (of morning joint stiffness). The missing data will be handled as described for the supportive analysis in Section A MMRM model will be fit on the daily individual-patient durations with fixed effects and covariates for treatment, region, joint erosion status, baseline (Day 1 on the diary), study week, and the interactions of baseline-by-week and treatment-by-week. The choice of variance-covariance matrix to model the between-day covariance structure for observations from the same patient may allow for different variances at different weeks, different covariances between observations from different pairs of weeks, and different (across weeks) within-week covariances for observations within the same week. However, the final selection may require simplification to aid in model convergence and may be guided by model fit criteria. Summary data of the treatment comparisons from this model will be presented by week and treatment group through Week 12. For the protocol-specified analysis of AUC up to Week 12, the test of difference between treatment groups will be equivalent to the test using the MMRM model with only treatment included as the fixed factor. Therefore, the p-value will be generated from the MMRM main model. If data are skewed and the nomal assumption is violated, a nonparametric analysis such as a permutation method may be used as a sensitivity analysis. Severity of morning joint stiffness numeric rating scale (NRS) Severity of morning joint stiffness NRS is a patient-administered, single-item, 11-point horizontal scale anchored at 0 and 10, with 0 representing no joint stiffness and 10 representing joint stiffness as bad as you can imagine. Patients rate their morning joint stiffness each day (using an electronic handheld diary) up to Visit 7 (Week 12) by selecting one number that describes their overall level of joint stiffness at the time they woke up. Severity of morning joint stiffness will be analyzed in a similar manner as described for duration of morning joint stiffness.
277 I4V-MC-JADV Statistical Analysis Plan Version 1.0 Page 47 Recurrence of joint stiffness during the day Recurrence of joint stiffness throughout the day is a patient-administered single-item question assessed each day (using an electronic handheld diary) through Visit 7 (Week 12) that asks Did you have any joint stiffness throughout today, other than when you woke up? (Yes/No). Recurrence of stiffness during the day will be summarized as a percentage of days with the event at weekly intervals (as defined in Section ) up to Visit 7 (Week 12). Percentage of days will be analyzed by week using ANCOVA with the same model specification described for duration of morning joint stiffness. Missing data imputation will follow what was described for the supportive analysis in Section Percentage of days will also be analyzed using MMRM with the same model specification as described for duration of morning joint stiffness. Tiredness severity NRS ( Worst Tiredness ) Worst tiredness NRS is a patient-administered, single-item, 11-point horizontal scale anchored at 0 and 10, with 0 representing no tiredness and 10 representing as bad as you can imagine. Patients rate their tiredness each day (using an electronic handheld diary) through Visit 7 (Week 12) by selecting the 1 number that describes their worst level of tiredness during the past 24 hours. Worst tiredness will be analyzed in a similar manner as described for duration of morning joint stiffness. Pain severity NRS ( Worst Joint Pain ) Worst joint pain NRS is a patient-administered, single-item, 11-point horizontal scale anchored at 0 and 10, with 0 representing no pain and 10 representing joint pain as bad as you can imagine. Patients rate their joint pain each day (using an electronic handheld diary) through Visit 7 (Week 12) by selecting the 1 number that describes their worst level of joint pain during the past 24 hours. Worst joint pain will be analyzed in a similar manner as described for duration of morning joint stiffness. These analyses may be repeated for the subset of patients who enroll within the study after the epro tablets are introduced in amendment (c) of Study JADV protocol Analysis of Non-Diary Data The impact of baricitinib compared to placebo from baseline up to Week 24 and to Week 52 in terms of descriptive summaries with regard to patient-reported outcomes (PROs) will be evaluated by the following measures:
278 I4V-MC-JADV Statistical Analysis Plan Version 1.0 Page 48 Duration of Morning Joint Stiffness The duration of morning joint stiffness is a patient-administered item that allows for the patients to enter the length of time in minutes (using 15-minute increments) that their morning joint stiffness lasted on the day prior to that visit using an electronic patient-reported outcomes (epro) tablet. Duration of morning joint stiffness will be measured at baseline and at postbaseline visits. Summary statistics of the actual score and change from baseline will be displayed by treatment and protocol-specified visit. In addition, an individual patient listing will be provided. The change from baseline in duration of morning joint stiffness from baseline to Week 24 will be analyzed using ANCOVA. The ANCOVA model will consider treatment, region, and joint erosion status as fixed effects and baseline score as a covariate with change from baseline being the dependent variable. To test the treatment difference between baricitinib and placebo, LS means, 95% CI, and p-values will be presented. The mlocf approach will be used to impute missing data. Tiredness Severity Numeric Rating Scale (Worst Tiredness NRS) The Worst Tiredness NRS is a patient-administered, single-item, 11-point horizontal scale anchored at 0 and 10, with 0 representing no tiredness and 10 representing as bad as you can imagine. Patients rate their tiredness by selecting the one number that describes their worst level of tiredness during the past 24 hours using an electronic patient-reported outcomes (epro) tablet. Worst Tiredness NRS will be measured at baseline and at postbaseline visits. Summary statistics of the actual score and change from baseline will be displayed by treatment and protocol-specified visit. In addition, an individual patient listing will be provided. The change from baseline in Worst Tiredness NRS from baseline to Week 24 will be analyzed using ANCOVA. The ANCOVA model will consider treatment, region, and joint erosion status as fixed effects and baseline score as a covariate with change from baseline being the dependent variable. To test the treatment difference between baricitinib and placebo, LS means, 95% CI, and p-values will be presented. The mlocf approach will be used to impute missing data. Severity of Joint Pain Numeric Rating Scale (Worst Pain NRS) The Worst Pain NRS is a patient-administered, single-item, 11-point horizontal scale anchored at 0 and 10, with 0 representing no pain and 10 representing as bad as you can imagine. Patients rate their pain by selecting the one number that describes their worst level of pain during the past 24 hours using an electronic patient-reported outcomes (epro) tablet. Worst Pain NRS will be measured at baseline and at postbaseline visits. Summary statistics of the actual score and change from baseline will be displayed by treatment and protocolspecified visit. In addition, an individual patient listing will be provided.
279 I4V-MC-JADV Statistical Analysis Plan Version 1.0 Page 49 The change from baseline in Worst Pain NRS from baseline to Week 24 will be analyzed using ANCOVA. The ANCOVA model will consider treatment, region, and joint erosion status as fixed effects and baseline score as a covariate with change from baseline being the dependent variable. To test the treatment difference between baricitinib and placebo, LS means, 95% CI, and p-values will be presented. The mlocf approach will be used to impute missing data. FACIT-F The FACIT-F scale (Cella and Webster 1997) is a 13-item, symptom-specific questionnaire that specifically assesses the self-reported severity of fatigue and its impact upon daily activities and functioning. The FACIT-F uses 0 ( not at all ) to 4 ( very much ) numeric rating scales to assess fatigue and its impact in the past 7 days. Scores range from 0 to 52 with higher scores indicating less fatigue. FACIT-F scores will be measured at baseline and at postbaseline visits. Summary statistics of the actual score and change from baseline will be displayed by treatment and protocol-specified visit. In addition, an individual patient listing will be provided. The change from baseline in FACIT-F scale score from baseline to Week 24 will be analyzed using ANCOVA. The ANCOVA model will consider treatment, region, and joint erosion status as fixed effects and baseline score as a covariate with change from baseline being the dependent variable. To test the treatment difference between baricitinib and placebo, LS means, 95% CI, and p-values will be presented. The mlocf approach will be used to impute missing data. The Minimum Clinically Important Difference (MCID) FACIT-F Score is defined as 3.56 in the change from baseline value of the Fatigue Subscale Score. The summary and analysis of the FACIT-F Score will include the number and percent of patients achieving MCID. SF-36v2 Acute Summaries of domain and summary scores (scoring details in Appendix 2) will be provided by visit and change from baseline to post-baseline visits. An ANCOVA model will be used for the change from baseline in the physical component score (PCS), the mental component score (MCS), and total SF-36v2 acute scores to Week 24. The ANCOVA model will use treatment, region, and joint erosion status as fixed effects and baseline score as a covariate with change from baseline as the dependent variable. To test the treatment difference between baricitinib and placebo, LS means, 95% CI, and p-values will be presented. The mlocf approach will be used to impute missing data. Analyses will be repeated for the eight health domains (physical functioning, bodily pain, role limitations due to physical problems, role limitations due to emotional problems, general health perceptions, mental health, social function, and vitality).
280 I4V-MC-JADV Statistical Analysis Plan Version 1.0 Page 50 Summary statistics per treatment group will be tabulated for the transformed scale scores (physical functioning, role limitations / physical health, bodily pain, general health perceptions, vitality, social functioning, role limitations / emotional, mental health) and the two summary scores (PCS and MCS). Minimum Clinically Important Differences: The summary and analyses of the SF-36v2 Acute Score will include the number of patients achieving a MCID, which is defined for each summary score, physical component score (PCS) and the mental component score (MCS), as: MCID at least 5 point improvement in PCS MCID at least 5 point improvement in MCS In addition, an individual patient listing will be provided. WPAI-RA The WPAI-RA questionnaire was developed to measure the effect of general health and symptom severity on work productivity and regular activities in the last 7 days of the visit. It contains 6 items covering overall work productivity (health), overall work productivity (symptom), impairment of regular activities (health), and impairment of regular activities (symptom). Scores are calculated as impairment percentages (Reilly et al. 1993). The WPAI-RA yields four types of scores: o o o o Absenteeism (work time missed) Presenteeism (impairment at work / reduced on-the-job effectiveness) Work productivity loss (overall work impairment / absenteeism plus presenteeism) Activity Impairment Six questions will be answered: o o o o o o Q1 = currently employed Q2 = hours missed due to specified problem Q3 = hours missed due to other reasons Q4 = hours actually worked Q5 = degree that RA affected productivity while working Q6 = degree that RA affected regular activities WPAI-RA outcomes are expressed as impairment percentages, with higher numbers indicating greater impairment and less productivity, ie, worse outcomes, as follows: o Percentage work time missed due to RA absenteeism : Q2/(Q2+Q4)
281 I4V-MC-JADV Statistical Analysis Plan Version 1.0 Page 51 o Percentage impairment while working due to RA presenteeism : Q5/10 o Percentage overall work impairment due to RA work productivity loss : Q2/(Q2+Q4)+[(1-Q2/(Q2+Q4))x(Q5/10)] o Percentage activity impairment due to RA activity impairment : Q6/10 o Multiply scores by 100 to express in percents. WPAI-RA scores will be measured at baseline and at postbaseline visits. Summary statistics of the actual and percent change from baseline scores based on absenteeism, presenteeism, work productivity loss, and activity impairment will be displayed by treatment and protocol-specified visit. In addition, an individual patient listing will be provided. The change from baseline in each of the four types of scores of WPAI-RA (absenteeism, presenteeism, work productivity loss, and activity impairment) from baseline to Week 24, will be analyzed using ANCOVA. The ANCOVA model will consider treatment, region, and joint erosion status as independent fixed effects and the baseline score as a covariate with change from baseline being the dependent variable. To test the treatment difference between baricitinib and placebo, the treatment difference LS mean, 95% CI, and p-value will be presented. The mlocf approach will be used to impute missing data. European Quality of Life 5 Dimensions 5 Level (EQ-5D-5L) The EQ-5D-5L is a standardized measure of health status of the patient at the visit (same day) that provides a simple, generic measure of health for clinical and economic appraisal. The EQ-5D-5L consists of 2 components: a descriptive system of the respondent s health and a rating of his or her current health state using a 0- to 100-mm VAS. The descriptive system comprises the following 5 dimensions: mobility, self-care, usual activities, pain/discomfort, and anxiety/depression. Each dimension has 5 levels: no problems, slight problems, moderate problems, severe problems, and extreme problems. The respondent is asked to indicate his/her health state by ticking (or placing a cross) in the box associated with the most appropriate statement in each of the 5 dimensions. It should be noted that the numerals 1 to 5 have no arithmetic properties and should not be used as a cardinal score. The VAS records the respondent s self-rated health on a vertical VAS in which the endpoints are labeled best imaginable health state and worst imaginable health state. This information can be used as a quantitative measure of health outcome. The EQ-5D-5L health states, defined by the EQ-5D-5L descriptive system, may be converted into a single summary index by applying a formula that essentially attaches a value (also called weights) to each of the levels in each dimension (Brooks 1996; EuroQol Group 2011 [WWW]; Herdman et al. 2011). The EQ-5D-5L questionnaire is self-completed by the patient in this study at baseline and post-baseline visits up to Week 52 (Visit 15). The EQ-5D-5L will be scored according to the developer s instructions (scoring guidelines). Three outcomes are described for each treatment group by visit according to the following:
282 I4V-MC-JADV Statistical Analysis Plan Version 1.0 Page 52 A health profile: summary statistics will be provided, including number of patients and proportions of categorical responses for the 5 dimensions (mobility, self-care, usual activity, pain/discomfort, and anxiety/depression) A health state index score: the UK algorithm (Szende et al. 2006) will be used to produce a patient-level index score between and 1.0 (continuous variable); the same will be done using the US algorithm which produces a patient-level index score between and 1.0. The mean, standard deviation (SD), minimum, median and maximum scores will be provided by visit and by treatment. A self-perceived current health score: calculated from patient level EQ VAS responses (continuous variable). The mean, SD, minimum, median and maximum scores will be provided by visit and by treatment. The change in health state index, in each of the 5 dimensions (mobility, self-care, usual activities, pain/ discomfort, and anxiety/ depression) and EQ VAS score from baseline up to Week 24 (Visit 11) will be analyzed using an ANCOVA model with the following terms: treatment, region, joint erosion status, and baseline score. Within each treatment arm, the LS mean, SE of the LS mean, and p-value will be reported. The LS mean difference between baricitinib and placebo, 95% CI for LS mean difference and p-value will be reported. ANCOVA analysis will be computed after imputing the post-baseline missing values using mlocf. Healthcare Resource Utilization The healthcare resource utilization data will be collected by site staff regarding the number of visits to medical care providers such as general practitioners, specialists, physical or occupational therapists, and other non-physical care providers for services outside of the clinical study; emergency room admissions; hospital admissions; and concomitant medications related to the treatment of RA. These data will be collected to support economic evaluations of treatment. A summary table including the following will be presented for the period of the past two months prior to study treatment as baseline and summarized at baseline and at post-baseline intervals (Week 0 to Week 24, Week 24 to Week 52 and Week 0 to Week 52). Healthcare resource utilization Healthcare providers: A summary of the mean number of consultations of medical care providers for care/treatment of RA will be provided for the period of the past 2 months prior to the study treatment. It will also be summarized at the post-baseline intervals described earlier. The number of consultations will be totaled by days and patients for the categories: Healthcare Provider Consultation Emergency Room Consultation The Healthcare Provider Consultation comprises the following: General Practitioner (Such as family doctor or primary care physician)
283 I4V-MC-JADV Statistical Analysis Plan Version 1.0 Page 53 Specialist Rheumatologist Occupational Therapist Physical therapist Non-physician care providers For the Emergency Room Consultation every necessary Emergency Room (ER) or equivalent facility consultation will be counted. Healthcare resource utilization Inpatient Hospitalization: A summary table including the following will be presented from the period of the past two months prior to study treatment and at post-baseline intervals for the following details: Frequency of patient s hospitalization and need for staying overnight because of their rheumatoid arthritis. Frequency of hospitalization type (general ward / intensive care ward). Descriptive statistics for the total number of days in hospital. Healthcare resource utilization Work Status: A frequency table for the non-missing statements on work status for the period of the past 2 months prior to study treatment and at post-baseline intervals will be provided for the following details: Working for pay full-time (> 36 hours per week) Working for pay part-time ( 36 hours per week) Unemployed, unrelated to study disease disability Unemployed due to study disease disability Other [Keeping house (full-time), Student (full-time), Student (part-time), Retired, Volunteer work] All the aforementioned health outcome analyses will be performed on the mitt analysis set. Descriptive statistics will be provided for all the parameters by visit and treatment group Exploratory Analysis Comparisons between adalimumab and baricitinib will be performed as exploratory for all primary and major secondary endpoints with the exception of the ACR20 and DAS28-hsCRP comparison at Week 12. Other endpoints to be explored are as follows: change from baseline to Week 52 in mtss, proportion of patients with change in mtss 0, ACR20 (other than Week 12), ACR50/ACR70, CDAI score 2.8, CDAI score 10, SDAI score 3.3, SDAI score 11, change from baseline in DAS28 (ESR and hscrp), proportion of patients with DAS28 (ESR and hscrp) 3.2 and <2.6, change from baseline in HAQ-DI, and proportion of patients with HAQ- DI improvement from baseline of 0.22 and 0.3.
284 I4V-MC-JADV Statistical Analysis Plan Version 1.0 Page 54 In addition, the comparison between baricitinib and adalimumab in diary data (morning joint stiffness severity NRS, duration of morning joint stiffness, recurrence of joint stiffness, worst tiredness NRS, and worst pain NRS) as calculated by averaging the scores collected in the 3 days prior to each injection at Week 2, 4, 6, 8, 10, and 12 may be explored. The 3-day time period will be defined based on the adalimumab injection date for adalimumab patients and placebo injection date for baricitinib patients. Other health outcome measures such as FACIT-F, EQ-5D, SF-36, and WPAI may be analyzed to compare differences between baricitinib and adalimumab treatment Safety Analysis Safety analyses for this study follow a hierarchy of importance: Of primary importance is the analysis of safety parameters during Part A wherein the comparison between baricitinib and placebo are of primary interest, informing risks to treatment that can be juxtaposed against the potential efficacy advantages shown over the same acute treatment time course. The baricitinib-placebo differences will be formally statistically compared, where applicable. Secondarily, a summary of the safety parameters associated with the patients treated with adalimumab will be provided and compared to placebo to complete the summary of safety for all randomized and treated patients. Of secondary importance is the analysis of safety parameters which includes data collected during Part B. The Part B portion of the trial is weakly-controlled for the baricitinib-toadalimumab comparison since the baricitinib group contains rescued (but not randomized) patients and is subject to attrition bias in each treatment relative to part A. Thus, analysis of part B provides descriptive summaries of the safety parameters for the patients over a longer course of treatment with baricitinib. Summaries will be provided for all patients exposed to baricitinib and according to the cohorts described in Section 6.1. No formal statistical comparisons are planned between cohorts. Of tertiary importance will be summarization of safety parameters after switching from treatment with either placebo or adalimumab to baricitinib from the point of rescue or reassignment in comparison to treatment with baricitinib. As these comparisons do not result from some randomization, no formal statistical comparisons will be performed. Thus, safety results will generally be prepared according to 5 formats: Part A, Week 0 (Visit 2) to Week 16 (Visit 9) covering exposures to randomized treatments prior to the possibility of rescue, Part A, Week 0 (Visit 2) to Week 24 (Visit 11) covering all of Part A but with censoring for patients after the point of rescue, Parts A and B combined from Week 0 (Visit 2) to Week 52 (Visit 15) for patients randomized to adalimumab or baricitinib but with censoring after the point of rescue,
285 I4V-MC-JADV Statistical Analysis Plan Version 1.0 Page 55 Part B, Week 24 (Visit 11) to Week 52 (Visit 15) for patients randomized to adalimumab or baricitinib who did not rescue prior to Week 24 (Visit 11), but with censoring after the point of rescue during Part B, and Parts A and B combined from Week 0 (Visit 2) to Week 52 (Visit 15) for patients randomized to baricitinib and for patients who change to baricitinib from either placebo or adalimumab covering all exposure to baricitinib. Safety analyses will take place using the Safety Analysis Set defined in Section Safety topics that will be addressed include the following: adverse events (AEs), clinical laboratory evaluations, vital signs and weight, safety in special groups and circumstances, including Adverse Events of Special Interest (AESI) (see Section ), and investigational product interruptions Adverse Events (AEs) Adverse events are recorded in the ecrfs. Each AE will be coded to system organ class and preferred term using the Medical Dictionary for Regulatory Activities (MedDRA) using the MedDRA version that is current at the time of each data lock. Severity of AEs is recorded as mild, moderate, severe or more severe than baseline; the more severe than baseline and severe categories will be combined for analysis and presentation. A treatment-emergent adverse event (TEAE) is defined as an event that either first occurred or worsened in severity after the first dose of study treatment and on or prior to the date of the last visit within the treatment period. Adverse events are classified based upon the MedDRA term. The MedDRA Lowest Level Term (LLT) will be used in the treatment-emergent computation. The maximum severity for each LLT during the baseline period will be used as baseline. The treatment period will be included as post-baseline for the analysis. If severity is missing during the baseline period, then any AE severity that is reported after baseline will be considered as treatment emergent. Events with a missing severity during the treatment period will be considered treatment-emergent. Should there be insufficient data for adverse event start date to make this comparison (for example the adverse event start year is the same as the treatment start year, but the adverse event start month and day are missing), the adverse event will be considered treatment-emergent. For events occurring on the day of the first dose of study treatment, a case report form (CRF)-collected indicator may be used to determine whether the event was pre- versus post-treatment. For patients who were randomized to placebo and required rescue therapy or who were switched to baricitinib at the end of Part A, the AE severity just prior to rescue or switch to baricitinib will be used as baseline for TEAE during baricitinib treatment. Similarly, for patients who were randomized to adalimumab and required rescue therapy, the AE severity just prior to rescue to baricitinib will be used as baseline for TEAE during baricitinib treatment. For some AEs of interest, time to onset of event analyses may be performed using the Kaplan- Meier technique. The time to onset of a TEAE will be defined as the start date of the first occurrence or first worsening of the TEAE minus the date of initiation of treatment plus 1; if a
286 I4V-MC-JADV Statistical Analysis Plan Version 1.0 Page 56 specific onset date is not available, then the start date of the visit when the treatment-emergence of the event was recorded will be used as a proxy. If a patient does not have the TEAE, they will be censored at the date of study drug discontinuation or the date of discontinuation from the treatment period/study, as appropriate. Kaplan-Meier estimates of the proportion of patients experiencing the TEAE at 28, 168 and 365 days (or other reasonable time points) and the estimated median time to onset will be provided for each treatment group/cohort, if calculable. A Kaplan-Meier plot of the time to onset may also be presented by treatment group/cohort. The log-rank test may be used to compare treatments with respect to time-to-event. Further, for some AEs of interest, duration of the TEAE may be summarized. The duration of each event will be defined as the end date of AE minus the start date of treatment-emergence of the AE plus 1. If an AE has not ended by the date of study drug discontinuation, early discontinuation of the study/period or completion of the study, it will be censored as of that appropriate final date. Descriptive statistics for the duration of treatment-emergent and followup emergent AEs will be summarized by treatment group/cohort. Duration of AEs may also be expressed conversely as the time to resolution of the AE. In general, TEAEs, serious adverse events (SAEs), AEs that lead to discontinuation from the study and AEs that lead to temporary or permanent discontinuation of study drug will be expressed with the frequency and relative frequency (or incidence) of occurrence. For common AEs, AEs in special groups and circumstances and in certain other cases, parallel presentation of incidence rates adjusted for patient exposure will be presented. These exposure-adjusted incidence rates (EAIR) will be expressed as the number of patients experiencing an adverse event per 100 patient years (PY) of exposure to treatment and are derived as 100 times the number of patients with the event divided by the sum of all patient exposure times (in years) to the treatment for the study/period. In an overview table, the number and percentage of patients in the safety population who experienced a death, an SAE, discontinuation from the study due to an AE, a TEAE, or an AE possibly related to study drug will be summarized by treatment. In addition, other notable events including discontinuation from study drug due to AE, interruption from study drug due to AE, and interruption from study drug due to lab abnormality will be summarized by treatment. Exposure-adjusted incidence rates will also be displayed for these outcomes in the overview table. The number of unevaluable patients, that is, those who were randomized but did not receive any study medication or those lost to follow-up at the first post-baseline visit, will be summarized in this table, but not included in any subsequent AE analysis. The number and percentage of patients with TEAEs will be summarized by treatment group for each treatment period using MedDRA PT nested within SOC. System Organ Class will be ordered alphabetically, and events will be ordered within each SOC by decreasing frequency in the baricitinib group (or combined baricitinib group in the event of multiple cohorts/doses). As an additional table, the number and percentage of patients with TEAEs will be summarized by treatment using MedDRA PT (without regard to SOC). Events will be ordered by decreasing frequency in the baricitinib group or combined baricitinib group, as appropriate.
287 I4V-MC-JADV Statistical Analysis Plan Version 1.0 Page 57 Adverse events considered possibly related to study drug by the investigator will be summarized by treatment group for each treatment period using MedDRA PT nested within SOC. A possibly-related event is defined as an event recorded on the ecrf as Yes to the question Is the event possibly related to study drug? Events will be ordered by decreasing frequency in the baricitinib group or combined baricitinib group, as appropriate, within alphabetical SOC. If the investigator s determination of possible relationship to study drug is missing, then the event will be considered as related. Follow-up emergent adverse events (FEAEs) will be established for the Post-Treatment Follow- Up Period. An FEAE is an event that either first occurred or worsened in severity after the last visit during the treatment period. The baseline severity for FEAE is the severity level at the last visit from the treatment period; otherwise, FEAE processing is similar to TEAE processing. Follow-up AEs will be summarized in a similar manner as TEAEs Common Adverse Events The number and percentage of patients with TEAEs will be summarized by treatment using MedDRA PT nested within SOC for common TEAEs (ie, TEAEs that occurred in 2% (before rounding) of all treated patients regardless of treatment. Events will be ordered by decreasing frequency in the baricitinib group (or combined baricitinib groups when appropriate) within alphabetical SOC. In addition, parallel summary tables providing EAIR using MedDRA PT nested within SOC will be generated for common TEAEs. The number and percentage of patients with TEAEs will be summarized by maximum severity by treatment using MedDRA PT nested within SOC for the common TEAEs. For each patient and TEAE, the maximum severity for the MedDRA level being displayed (PT, HLT, or SOC) is the maximum post-baseline severity observed from all associated LLTs mapping to that MedDRA level Serious Adverse Events (SAEs) Consistent with the ICH E2A guideline, a serious adverse event (SAE) is any AE that results in one of the following outcomes: Death Initial or prolonged inpatient hospitalization A life-threating experience (ie, immediate risk of dying) Persistent or significant disability/incapacity Congenital anomaly/birth defect Considered significant by the investigator for any other reason. In addition, a protocol-specific SAE was defined as an AE or laboratory value that resulted in permanent study drug discontinuation. The number and percentage of patients who experienced any SAE, separately, only an ICHdefined SAE and, separately, only a protocol-defined SAE, will be summarized by treatment using MedDRA PT nested within SOC. Events will be ordered by decreasing frequency in the baricitinib group (or combined baricitinib group in the event of multiple cohorts/doses) within
288 I4V-MC-JADV Statistical Analysis Plan Version 1.0 Page 58 alphabetical SOC. Serious AEs overall and of each type will be summarized by treatment period, and during the Post-Treatment Follow-up period. Summaries of SAEs by treatment period will include EAIR Other Significant Adverse Events The number and percentage of patients who discontinued from the study due to an AE or death during the treatment period will be summarized by treatment using MedDRA PT nested within SOC. Events will be ordered by decreasing frequency in the baricitinib group (or combined baricitinib group in the event of multiple cohorts/doses) within alphabetical SOC. The number and percentage of patients who temporarily interrupted or who permanently discontinued study drug during the treatment period will each be summarized by treatment overall and by reason for interruption or discontinuation. For AEs leading to interruptions or discontinuation of study drug, additional detail using MedDRA PT nested within SOC will be provided, with events ordered by decreasing frequency in the baricitinib group (or combined baricitinib group in the event of multiple cohorts/doses) within alphabetical SOC. For the duration of temporary interruptions of study drug, in days, basic descriptive statistics (mean, standard deviation, minimum, Q1, median, Q3, and maximum) will be displayed Vital Signs and Physical Characteristics Vital signs and physical characteristics include systolic blood pressure (SBP), diastolic blood pressure (DBP), pulse, weight, BMI, and waist circumference. Triplicate assessments of SBP and DBP are collected at each visit. When each triplicate is recorded on the ecrf, the average will be derived; otherwise, the average which is recorded on the ecrf will be used for the summaries. Only the average across the triplicates will be analyzed. When these parameters are analyzed as continuous numerical variables, unscheduled visits will be excluded. When these parameters are analyzed as categorical outcomes and/or treatment-emergent abnormalities, unscheduled visits will be included. In general, vital signs and physical characteristics will be summarized with descriptive statistics (n, mean, standard deviation median, minimum, and maximum) at baseline and at each postbaseline visit by treatment group, including change from baseline for post-baseline visits. These summaries will be based on patients who have both baseline and at least one post-baseline assessment. Three approaches to the evaluation of change from baseline will be taken: Change from baseline to last observation during the treatment period considering baseline as the last non-missing observation from the baseline period; missing data will be imputed using mlocf; Change from baseline to the minimum value during the treatment period considering baseline as the minimum value of the non-missing observations in the baseline period; Change from baseline to the maximum value during the treatment considering baseline as the maximum value of the non-missing observations in the baseline period.
289 I4V-MC-JADV Statistical Analysis Plan Version 1.0 Page 59 Change from baseline in vital sign parameters (each approach above) will be summarized by visit up to Week 52 and analyzed by visit up to Week 24 using an ANCOVA model with explanatory terms for treatment and the baseline value as a covariate. Treatment comparisons will be provided showing the LS mean for each treatment, the pairwise differences in LS means, 95% CI for pairwise treatment differences, within-treatment p-values, and p-values for the treatment differences obtained from the model. Missing data will be imputed using mlocf imputation. Vital signs and physical characteristics parameters will be summarized across time points using box plots which include accompanying summary statistics. The number and percentage of patients with treatment-emergent high or low vital signs and weight will be summarized by visit and at anytime during study periods of interest by treatment group or treatment cohort. Comparisons between treatments up to Week 24 will be made using Fisher exact test. The treatment-emergent high or low vital signs and weight outcomes area treatmentemergent high result is defined as a change from a value less than or equal to the high limit at all baseline visits to a value greater than the high limit at any time that meets the specified change criteria in Table JADV.3 during the treatment period. A treatment-emergent low result is defined as a change from a value greater than or equal to the low limit at all baseline visits to a value less than the low limit at any time that meets the specified change criteria in Table JADV.3 during the treatment period. Treatment-emergent weight gain/loss is based on percent increase/decrease from baseline. Table JADV.6.2 defines the low and high baseline values as well as the criteria used to define treatment-emergence based on post-baseline values. Table JADV.6.2. Categorical Criteria for Abnormal Treatment-Emergent Blood Pressure and Heart Rate Measurement, and Categorical Criteria for Weight Changes for Adults Parameter Low High SBP (mm Hg) (sitting) 90 (low limit) and decrease from lowest value during baseline 20 if > 90 at each baseline visit 140 (high limit) and increase from highest value during baseline 20 if < 140 at each baseline visit DBP (mm Hg) (sitting) Heart rate (bpm) (sitting) Weight (kg) 50(low limit) and decrease from lowest value during baseline 10 if > 50 at each baseline visit < 50 (low limit) and decrease from lowest value during baseline 15 if > 50 at each baseline visit (Loss) decrease 7% from lowest value during baseline 90 (high limit) and increase from highest value during baseline 10 if < 90 at each baseline visit > 100 (high limit) and increase from highest value during baseline 15 if 100 at each baseline visit (Gain) increase 7% from highest value during baseline
290 I4V-MC-JADV Statistical Analysis Plan Version 1.0 Page 60 Individual vital sign parameter values for each patient will be listed Laboratory Measurements Two sets of tables for laboratory tests will be presented for standard clinical laboratory evaluations. All laboratory tests will be presented using units conventional to the United States and also using the International System (SI) of units. For topics of safety in special groups and circumstances, laboratory test units will be specified for each analysis. Large clinical trial population based reference limits (Lilly-defined) will be used to define the low and high limits since it is generally desirable for limits used for analyses to have greater specificity (identify fewer false positive cases) than reference limits used for individual patient management. For the hepatic laboratory assessments (alanine aminotransferase [ALT], aspartate aminotransferase [AST], total bilirubin, and alkaline phosphatase [ALP]), central laboratory reference ranges (Quintiles) will be used and all results pertaining to these assessments will be included as a separate analysis to address the risk of liver injury as a special safety topic (see Section ). Central laboratory reference ranges (Quintiles) will also be used to evaluate lymphocyte cell subsets (see Section ). Change from baseline to last observation for laboratory tests will be summarized for patients who have both baseline and at least one post-baseline result. Baseline will be the last nonmissing observation in the baseline period. The last non-missing observation in the treatment period will be analyzed. Untransformed data will be analyzed. Unplanned visits and unplanned measurements will be excluded. Change from the minimum value during the baseline period to the minimum value during the treatment period for selected laboratory tests will be summarized for patients who have both baseline and at least one post-baseline result. Baseline will be the minimum of non-missing observations in the baseline period. The minimum value in the treatment period will be analyzed. Similarly, change from the maximum value during the baseline period to the maximum value during the treatment period for selected laboratory tests will be summarized for patients who have both baseline and at least one post-baseline result. Baseline will be the maximum of non-missing observations in the baseline period. The maximum value in the treatment period will be analyzed. Untransformed data will be analyzed. Planned visits, unplanned visits, and unplanned measurements will be included. The number and percentage of patients with treatment-emergent abnormal, high, or low laboratory results at any time will be summarized by treatment group. A treatment-emergent abnormal result is defined as a change from normal at all baseline visits to abnormal at any time during the treatment period. A treatment-emergent high result is defined as a change from a value less than or equal to the high limit at all baseline visits to a value greater than the high limit at any time during the treatment period.
291 I4V-MC-JADV Statistical Analysis Plan Version 1.0 Page 61 A treatment-emergent low result is defined as a change from a value greater than or equal to the low limit at all baseline visits to a value less than the low limit at any time during the treatment period. Planned visits, unplanned visits, and unplanned measurements will be included in analysis of treatment-emergent abnormal, high, or low laboratory results. Treatment differences in mean change for laboratory tests will be analyzed using an ANCOVA model with explanatory terms for treatment and the baseline value as a covariate. When appropriate, treatment comparisons will be provided showing the LS mean for each treatment, the pairwise differences in LS means, 95% CI for pairwise treatment differences, withintreatment p-values, and p-values for the treatment differences obtained from the model. Missing data will be imputed using mlocf imputation. Differences between treatments (when appropriate) in incidence of treatment-emergent abnormal, high or low laboratory results will be assessed using the Fisher exact test. Additionally, values at each visit (starting at randomization) and change from baseline to each visit for laboratory tests will be displayed in box plots (with outliers displayed) for subjects who have both a baseline and a result for the specified visit. Baseline will be the last non-missing observation in the baseline period. Original-scale data will be used for the display. Unplanned measurements will be excluded. In each box-plot, lines indicating the appropriate reference limits will be added; in cases where limits vary across age, race and/or gender, the lowest of the high limits and the highest of the low limits will be used. Displays using both SI and conventional units will be provided (when different). The following summary statistics will be included in a table below the box plot: N, mean, standard deviation, minimum, Q1, median, Q3, and maximum. P-values and confidence limits will not be included in the summary statistics at the bottom of the box plot. Box plots will be used to evaluate trends over time and to assess a potential impact of outliers on central tendency summaries Safety in Special Groups and Circumstances, including Adverse Events of Special Interest (AESI) In addition to general safety parameters, safety information on specific topics of special interest will also be presented. Topics will be identified by laboratory results, standardized MedDRA queries (SMQs), Lilly-defined MedDRA listings, or a combination of these methods. The primary focus of analyses will be on laboratory test values, where applicable, with a secondary focus on reported adverse events related to laboratory values. Additional special safety topics may be added as warranted. The topics outlined in this section include the protocol-specified AESI: severe or opportunistic infections myelosuppressive events of anemia, leukopenia, neutropenia, lymphopenia, and thrombocytopenia thrombocytosis elevations in ALT or AST (>3 times ULN) with total bilirubin (>2 times ULN).
292 I4V-MC-JADV Statistical Analysis Plan Version 1.0 Page 62 In general, for topics regarding safety in special groups and circumstances, patient profiles and/or patient listings will be provided when needed to allow medical review of the time course of cases/events, related parameters, patient demographics, study drug treatment and meaningful concomitant medication use. In addition to the safety topics for which provision or review of patient data is specified below we anticipate that these will be provided when summary data are insufficient to permit adequate understanding of the safety topic Abnormal Hepatic Tests Analyses for change from baseline to last observation, change from the minimum value during the baseline period to the minimum value during the treatment period, change from the maximum value during the baseline period to the maximum value during the treatment period, and treatment-emergent high or low laboratory results at any time are described in Section This section describes additional analyses for the topic. Recall also that central laboratory reference ranges (Quintiles) will be used for the some hepatic laboratory assessments (Alanine transaminase [ALT], aspartate aminotransferase [AST], total bilirubin, and alkaline phosphatase [ALP]). The number and percentage of patients with the following elevations in hepatic laboratory tests at any time will be summarized between treatment groups: The percentages of patients with an ALT measurement greater than or equal to 3 times (3X), 5 times (5X), and 10 times (10X) the central laboratory upper limit of normal (ULN) during the treatment period will be summarized for all patients with a postbaseline value and for subsets based on various levels of baseline. o o o The analysis of 3X ULN will contain four subsets: patients whose non-missing maximum baseline value is less than or equal to 1X ULN, patients whose maximum baseline is greater than 1X ULN but less than 3X ULN, patients whose maximum baseline value is greater than or equal 3X ULN, and patients whose baseline values are missing. The analysis of 5X ULN will be contain five subsets: patients whose non-missing maximum baseline value is less than or equal to 1X ULN, patients whose maximum baseline is greater than 1X ULN but less than 3X ULN, patients whose maximum baseline is greater than or equal to 3X ULN but less than 5X ULN, patients whose maximum baseline value is greater than or equal to 5X ULN, and patients whose baseline values are missing. The analysis of 10X ULN will contain six subsets: patients whose non-missing maximum baseline value is less than or equal to 1X ULN, patients whose maximum baseline is greater than 1X ULN but less than 3X ULN, patients whose maximum baseline is greater than or equal to 3X ULN but less than 5X ULN, patients whose maximum baseline is greater than or equal to 5X ULN but less than 10X ULN, patients whose maximum baseline value is greater than or equal to 10X ULN, and patients whose baseline values are missing.
293 I4V-MC-JADV Statistical Analysis Plan Version 1.0 Page 63 The percentages of patients with an AST measurement greater than or equal to 3 times (3X), 5 times (5X), and 10 times (10X) the central laboratory upper limit of normal (ULN) during the treatment period will be summarized for all patients with a postbaseline value and for subsets based on various levels of baseline. Analyses will be constructed as described above for ALT. The percentages of patients with a total bilirubin measurement greater than or equal to 2 times (2X) the central laboratory upper limit of normal (ULN) during the treatment period will be summarized for all patients with a post-baseline value, and subset into four subsets: patients whose non-missing maximum baseline value is less than or equal to 1X ULN, patients whose maximum baseline is greater than 1X ULN but less than 2X ULN, patients whose maximum baseline value is greater than or equal to 2X ULN, and patients whose baseline values are missing. The percentages of patients with an ALP measurement greater than or equal to 1.5 times (1.5X) the central laboratory upper limit of normal (ULN) during the treatment period will be summarized for all patients with a post-baseline value, and subset into four subsets: patients whose non-missing maximum baseline value is less than or equal to 1X ULN, patients whose maximum baseline is greater than 1X ULN but less than 1.5X ULN, patients whose maximum baseline value is greater than or equal to 1.5X ULN, and patients whose baseline values are missing. The percentages of patients potentially meeting Hy s rule (greater than or equal to 3X ULN for ALT or AST, less than 2X ULN for alkaline phosphatase, and greater than or equal to 2X ULN for total bilirubin all at the same time) during the treatment period will be summarized for patients with at least one post-baseline value for ALT or AST, alkaline phosphatase, and total bilirubin measured at the same time. Narratives for subjects with an ALT or AST measurement greater than or equal to 3X ULN or with an ALP measurement greater than or equal to 2X ULN will be prepared. The review for these subjects includes an assessment of the proximity of any ALT or AST elevation to any total bilirubin elevation, alkaline phosphatase levels, other potential causes, and the temporal association with events such as nausea, vomiting, anorexia, abdominal pain, or fatigue. To facilitate this review, a patient profile listing will be created. The listing will include patient demographics, concomitant medications, ALT, AST, alkaline phosphatase, and total bilirubin by visit, treatment start and stop dates, and reason for discontinuation. A patient profile graph may be created instead of or in addition to the listing to facilitate this review. To further evaluate potential hepatotoxicity, an edish (Evaluation of Drug-Induced Serious Hepatotoxicity) plot may be created. Each patient with at least one post-baseline ALT and total bilirubin contributes one point to the plot. The points correspond to maximum total bilirubin and maximum ALT, even if not obtained from the same blood draw. Criteria for AE grading from the Common Terminology Criteria for Adverse Events (CTCAE) will be applied for the hepatic laboratory tests AST, ALT, ALP, total bilirubin and albumin (Table JADV.5.1). These CTCAE grading schemes are consistent with both Version 3.0 and
294 I4V-MC-JADV Statistical Analysis Plan Version 1.0 Page 64 Version 4.03 of the CTCAE guidelines. Treatment-emergent lab abnormalities related to hepatic abnormalities occurring at any time during the treatment period and shift tables of baseline to maximum during the treatment period will be tabulated. Treatment-emergence will be characterized using three criteria (as appropriate to the grading scheme): any increase in post-baseline CTCAE grade from baseline grade an increase from baseline less than Grade 3 to Grade 3 or 4 at worst post-baseline an increase from baseline less than Grade 4 to Grade 4 at worst post-baseline. Shift tables will show the number and percentage of patients based on baseline to maximum CTCAE grade during the treatment period, with baseline depicted by the most extreme grade during the baseline period. With each shift table, a shift table summary displaying the number and percentage of patients with maximum post-baseline results will be presented overall and by treatment group for each treatment period within the following categories: Decreased; post-baseline category < baseline category, Increased; post-baseline category > baseline category, Same; post-baseline category = baseline category. Treatment differences for the shift table summary categorizations will be assessed with the Likelihood Ratio Chi-Square test. Planned visits, unplanned visits, and unplanned measurements will be included.
295 I4V-MC-JADV Statistical Analysis Plan Version 1.0 Page 65 Table JADV.6.3. Common Terminology Criteria for Adverse Events (CTCAE) Related to Hepatic Tests Lab Test Grade Criteria Albumin 0 (nomal) LLN < LLN and 30 g/l < 30 g/l and 20 g/l < 20 g/l ALT AST ALP Total bilirubin 0 (nomal) (nomal) (nomal) (nomal) ULN > ULN and 2.5X ULN > 2.5X ULN and 5X ULN > 5X ULN and 20X ULN > 20X ULN ULN > ULN and 2.5X ULN > 2.5X ULN and 5X ULN > 5X ULN and 20X ULN > 20X ULN ULN > ULN and 2.5X ULN > 2.5X ULN and < =5X ULN > 5X ULN and < =20X ULN > 20X ULN ULN > ULN and 1.5X ULN > 1.5X ULN and < =3X ULN > 3X ULN and 10X ULN > 10X ULN Abbreviations: ALP = alkaline phosphatase; ALT = alanine transaminase; AST = aspartate aminotransferase; LLN=lower limit of normal; ULN=upper limit of normal. Treatment-emergent potentially drug-related hepatic disorders will be summarized by treatment using MedDRA PTs contained in each of the following SMQs: Broad and narrow terms in the Liver related investigations, signs and symptoms SMQ (SMQ ) Broad and narrow terms in the Cholestasis and jaundice of hepatic origin SMQ (SMQ ) Broad and narrow terms in the Hepatitis non-infections SMQ (SMQ ) Broad and narrow terms in the Hepatic failure, fibrosis and cirrhosis and other liver damage SMQ (SMQ ) Narrow terms in the Liver-related coagulation and bleeding disturbances SMQ (SMQ ) Frequency and relative frequency for each PT will be provided, first pooling narrow and broad terms together, and then for narrow terms only, ordered by decreasing frequency in the
296 I4V-MC-JADV Statistical Analysis Plan Version 1.0 Page 66 baricitinib group (or combined baricitinib group in the event of multiple cohorts/doses). The percentages of subjects with potentially drug-related hepatic disorders that led to study drug discontinuation will be summarized similarly. A by-patient listing will be provided for all patients having a treatment-emergent drug related hepatic adverse event Myelosuppressive Events Myelosuppressive events will be defined based on TEAEs and clinical laboratory assessments. Myelosuppressive TEAEs will be identified using target surveillance terms which are either standardized MedDRA queries (SMQs) or Lilly-defined lists of MedDRA PTs, involving both narrow and broad search terms, as appropriate. The specific target surveillance terms are: Agranulocytosis (SMQ ) Haematopoietic cytopenias affecting more than one type of blood cell (SMQ ) Haematopoietic erythropenia (SMQ ) Haematopoietic leukopenia (SMQ ) o with a Lilly-defined subset of specific terms for Neutropenia o with a Lilly-defined subset of specific terms for Lymphopenia o with a Lilly-defined subset of specific terms for Eosinophils decreased Haematopoietic thrombocytopenia (SMQ ) Haemorrhage terms excluding laboratory terms (SMQ ). Frequency and relative frequency for each PT will be provided, first pooling narrow and broad terms together, and then for narrow terms only nested within target surveillance terms, ordered by decreasing frequency in the baricitinib group (or combined baricitinib group in the event of multiple cohorts/doses). In addition, parallel summary tables comparing EAIR will be generated. The specific search terms associated with neutropenia, lymphopenia and decreased eosinophils from MedDRA SMQ are shown in the appendices of the current version of the Baricitinib Program Safety Analysis Plan (PSAP). A by-patient listing will be provided for all patients having a myelosuppressive event within individual clinical study reports. Using clinical laboratory assessments, the myelosuppressive events of anemia, leukopenia, neutropenia, lymphopenia, and thrombocytopenia will be assessed through analysis of decreased hemoglobin, decreased white cell count, decreased neutrophils absolute count, decreased lymphocytes absolute count, and decreased platelet count, respectively. Laboratory tests for these measures will be summarized across time points using box plots which include accompanying summary statistics. Criteria for AE grading from the CTCAEs will be applied for laboratory tests related to myelosuppressive events (Table JADV.6.4). These CTCAE grading schemes are consistent with both Version 3.0 and Version 4.03 of the CTCAE guidelines. Treatment-emergent lab
297 I4V-MC-JADV Statistical Analysis Plan Version 1.0 Page 67 abnormalities related to myelosuppression occurring at any time during the treatment period and shift tables of baseline to maximum during the treatment period will be tabulated. Treatment-emergence will be characterized using three criteria: any increase in post-baseline CTCAE grade from baseline grade an increase from baseline less than Grade 3 to Grade 3 or Grade 4 at worst post-baseline an increase from baseline less than Grade 4 to Grade 4 at worst post-baseline. Table JADV.6.4. CTCAE Related to Myelosuppressive Events Event Lab Test Grade Criteria Anemia Hemoglobin 0 (nomal) LLN < LLN and 10.0 g/dl < 10.0 g/dl and 8.0 g/dl < 8.0 g/dl and 6.5 g/dl < 6.5 g/dl Leukopenia White blood cell (WBC) count 0 (nomal) LLN < LLN and 3000 cells/μl < 3000 cells/μl and 2000 cells/μl < 2000 cells/μl and 1000 cells/μl < 1000 cells/μl Neutropenia Absolute neutrophil count (ANC) 0 (nomal) LLN < LLN and 1500 cells/μl < 1500 cells/μl and 1000 cells/μl < 1000 cells/μl and 500 cells/μl < 500 cells/μl Lymphopenia Lymphocyte count 0 (nomal) LLN < LLN and 800 cells/μl < 800 cells/μl and 500 cells/μl < 500 cells/μl and 200 cells/μl < 200 cells/μl Thrombocytopenia Platelet count 0 (nomal) LLN < LLN and cells/μl < cells/μl and cells/μl < cells/μl and cells/μl < cells/μl Abbreviations: LLN=lower limit of normal based on Lilly-defined large clinical trial population based reference limits. Shift tables will show the number and percentage of patients based on each of baseline to last CTCAE grade and baseline to maximum during the treatment period, with baseline depicted by the most extreme grade during the baseline period. With each shift table, a shift table summary displaying the number and percentage of patients with last and maximum post-baseline results,
298 I4V-MC-JADV Statistical Analysis Plan Version 1.0 Page 68 respectively, overall and by treatment group for each treatment period within the following categories: Decreased; post-baseline category < baseline category, Increased; post-baseline category > baseline category, Same; post-baseline category = baseline category. Treatment differences for the shift table summary categorizations will be assessed with the Likelihood Ratio Chi-Square test. A laboratory-based treatment emergent outcome related to increased platelet count will be summarized in similar fashion. Treatment-emergent thrombocytosis as a laboratory-based abnormality will be defined as an increase in platelet count from a maximum baseline value 600,000/μL to any post-baseline value > 600,000 cells/μl. A frequency table overall and by treatment group for each treatment period showing the number and percentage of patients meeting criteria for thrombocytosis will be provided. A frequency table overall and by treatment group for each treatment period showing the number and percentage of patients meeting criteria for temporary or permanent discontinuation of investigational product will be provided. Criteria are shown in Table JADV Table JADV.6.5. Criteria for Discontinuation of Investigational Product due to Myelosuppression Type of Discontinuation Myelosuppressive Event Criteria Temporary Anemia Hemoglobin <8 g/dl Leukopenia WBC count <2000 cells/μl Neutropenia ANC <1000 cells/μl Lymphopenia Lymphocyte count <500 cells/μl Thrombocytopenia Platelet count <75,000 cells/μl Permanent Anemia Hemoglobin <6.5 g/dl Leukopenia WBC count <1000 cells/μl Neutropenia ANC <500 cells/μl Lymphopenia Lymphocyte count <200 cells/μl Thrombocytopenia None Abbreviations: ANC = absolute neutrophil count; WBC=white blood cell Lymphocyte Subset Cell Counts The following lymphocyte subsets will be analyzed; those subsets marked with a ( ) will be evaluated only in patients from United Kingdom: CD3+ T cells CD3+CD4+ T cells (CD4) CD3+CD8+ T cells (CD8) CD4+CD45RA-CCR7- effector memory T cells ( ) CD4+CD45RA+CCR7- effector memory T cells ( )
299 I4V-MC-JADV Statistical Analysis Plan Version 1.0 Page 69 CD4+CD45RA+CCR7+ naïve T cells ( ) CD4+CD45RA-CCR7+ central memory T cells ( ) CD4+CXCR3+CCR6- Th1 cells CD4+CXCR3-CCR6+ Th17 cells CD3+CD4+CD127-/loCD25+FoxP3+ (CD4+) T regulatory cells CD3+CD4+CD127-/loCD25+ (CD4+)IL2-producing naïve and central memory Helper T cells CD8+CD45RA+CCR7+ naïve T cells ( ) CD8+CD45RA-CCR7+ central memory T cells ( ) CD8+CD45RA-CCR7- effector memory T cells( ) CD8+CD45RA+CCR7- effector memory T cells ( ) CD56+/CD16+ NK cells CD19+ B cells CD20+ B cells CD19+CD27-IgD+ mature naïve B cells CD19+CD27+IgD- switched memory B cells CD19+CD27+IgD+ non-switched memory B cells CD19+CD27-IgD- Immature/transitional B cells. For each type of cells, both the absolute count and the relative count (i.e. as a percentage of the total lymphocyte population) will be analyzed. In addition, the CD4:CD8 ratio will be analyzed. For these parameters, the analyses of: change from baseline to endpoint, change from minimum at baseline to minimum post-baseline, change from maximum at baseline to maximum post-baseline, and the number and percentage of patients with treatment-emergent abnormal, high, or low cell counts at any time, will be performed using the same approaches as described for analysis of clinical laboratory measurements in Section For determining treatment-emergent abnormal, high, or low lymphocyte subset cell counts, central laboratory reference ranges (Quintiles) will be used when available Lipids Effects Lipids effects will be assessed through analysis of elevated total cholesterol, elevated LDL cholesterol, decreased HDL cholesterol, and elevated triglycerides and with selected treatmentemergent adverse events. National Cholesterol Education Program (NCEP) ATP III and CTCAE criteria (Version 3.0) will be applied for laboratory tests related to lipids effects (Table JADV.6.6 and Table JADV.6.7). Shift tables will show the number and percentage of patients based on baseline to maximum grade/category during the treatment period, with baseline depicted by the most extreme grade/category during the baseline period. With each shift table, a shift table summary
300 I4V-MC-JADV Statistical Analysis Plan Version 1.0 Page 70 displaying the number and percentage of patients with last and maximum post-baseline results, respectively, overall and by treatment group for each treatment period within the following categories: Decreased; post-baseline category < baseline category, Increased; post-baseline category > baseline category, Same; post-baseline category = baseline category. Pairwise treatment differences for the shift table summary categorizations will be assessed with the Likelihood Ratio Chi-Square test. Table JADV.6.6. National Cholesterol Education Program (NCEP) ATP III Criteria Lab Test Category Criteria Total Cholesterol Desirable Borderline high High < 200 mg/dl 200 mg/dl and < 240 mg/dl 240 mg/dl LDL Cholesterol HDL Cholesterol Optimal Near optimal Borderline high High Very high Low Normal High < 100 mg/dl 100 mg/dl and < 130 mg/dl 130 mg/dl and < 160 mg/dl 160 mg/dl and < 190 mg/dl 190 mg/dl < 40 mg/dl 40 mg/dl and < 60 mg/dl 60 mg/dl Triglycerides Normal Borderline high High Very high < 150 mg/dl 150 mg/dl and < 200 mg/dl 200 mg/dl and < 500 mg/dl 500 mg/dl Abbreviations: HDL = high-density lipoproteins; LDL = low-density lipoproteins.
301 I4V-MC-JADV Statistical Analysis Plan Version 1.0 Page 71 Table JADV.6.7. Common Terminology Criteria for Adverse Events (CTCAE) Related to Lipids Effects Event Lab Test Grade Criteria Hypercholesterolemia Total Cholesterol 0 (nomal) ULN > ULN and 300 mg/dl > 300 mg/dl and 400 mg/dl > 400 mg/dl and 500 mg/dl > 500 mg/dl Hypertriglyceridemia Triglycerides 0 (nomal) ULN > ULN and 2.5X ULN > 2.5X ULN and 5X ULN > 5X ULN and 10X ULN > 10X ULN Abbreviations: ULN=upper limit of normal based on Lilly-defined large clinical trial population based reference limits. Treatment-emergent lab abnormalities related to elevated total cholesterol and elevated triglycerides occurring at endpoint and at any time during the treatment period will be tabulated using the CTCAE grades shown in Table JADV.6.7. Treatment-emergence will be characterized using three criteria: any increase in post-baseline CTCAE grade from baseline grade an increase from baseline less than Grade 3 to Grade 3 or Grade 4 at worst post-baseline an increase from baseline less than Grade 4 to Grade 4 at worst post-baseline. Treatment-emergent lab abnormalities related to elevated LDL cholesterol and decreased HDL cholesterol occurring at endpoint and at any time during the treatment period will be tabulated using the NCEP categories shown in Table JADV.6.6. Treatment-emergent elevated LDL cholesterol will be characterized as movement from categories Optimal, Near optimal or Borderline high to categories High or Very high considering patients who are at risk for the abnormality, ie, neither High nor Very high based on the maximum baseline LDL cholesterol category. Treatment-emergent decreased HDL cholesterol will be characterized as movement from categories High or Normal to category Low considering patients who are at risk for the abnormality, ie, not Low based on the minimum baseline HDL cholesterol category. Treatment-emergent hyperlipidemia events will also be analyzed using reported adverse events. The target surveillance term Hyperlipidemia is a Lilly-defined MedDRA listing which is a subset of the SMQ Dyslipidemia related to elevated or increased lipids. MedDRA PTs for broad and narrow search categories for the target surveillance term are shown in Appendix 2. Frequency and relative frequency for each PT will be provided, first pooling narrow and broad terms together, and then for narrow terms only, ordered by decreasing frequency in the baricitinib group (or combined baricitinib group in the event of multiple cohorts/doses).
302 I4V-MC-JADV Statistical Analysis Plan Version 1.0 Page 72 A by-patient listing will be provided for all patients having a treatment-emergent hypercholesterolemia or hypertriglyceridemia adverse event based on an increase from baseline less than Grade 3 to Grade 3 or Grade 4 at worst post-baseline using CTCAE grades Renal Function Effects Effects on renal function will be assessed through analysis of elevated creatinine and decreased estimated glomerular filtration rate (egfr). Laboratory tests for creatinine and egfr will be summarized across time points using box plots which include accompanying summary statistics. Common Terminology Criteria for Adverse Events will be applied for laboratory tests related to renal effects (Table JADV.6.8). The creatinine grades come from CTCAE Version 3.0 guidelines whereas the egfr grades come from Version The updated grading for egfr is used to facilitate implementation as it is based exclusively on the laboratory measurement; Version 3.0 grading for egfr provides no value-based breakpoints and requires knowledge of the patient s chronic use of dialysis to implement Grade 3 and Grade 4. Shift tables will show the number and percentage of patients based on baseline to maximum during the treatment period, with baseline depicted by the most extreme grade during the baseline period. With each shift table, a shift table summary displaying the number and percentage of patients with last and maximum post-baseline results, respectively, overall and by treatment group for each treatment period within the following categories: Decreased; post-baseline category < baseline category, Increased; post-baseline category > baseline category, Same; post-baseline category = baseline category. Pairwise treatment differences for the shift table summary categorizations will be assessed with the Likelihood Ratio Chi-Square test. Treatment-emergent lab abnormalities related to elevated creatinine and decreased estimated glomerular filtration rate (egfr) occurring at endpoint and at any time during the treatment period will be tabulated using the CTCAE grades shown in Table JADV.6.8. Treatment-emergence will be characterized using three characterizations: any increase in post-baseline CTCAE grade from baseline grade an increase from baseline less than Grade 3 to Grade 3 or Grade 4 at worst post-baseline an increase from baseline less than Grade 4 to Grade 4 at worst post-baseline.
303 I4V-MC-JADV Statistical Analysis Plan Version 1.0 Page 73 Table JADV.6.8. Common Terminology Criteria for Adverse Events (CTCAE) Related to Renal Effects Lab Test CTCAE Version Grade Criteria Elevated creatinine (nomal) ULN > ULN and 1.5*ULN > 1.5*ULN and 3*ULN > 3*ULN and 6*ULN > 6*ULN Decreased estimated glomerular filtration rate (egfr) (nomal) LLN 60 ml/min/1.73m**2 and < LLN 30 ml/min/1.73m**2 and < 60 ml/min/1.73m**2 15 ml/min/1.73m**2 and < 30 ml/min/1.73m**2 < 15 ml/min/1.73m**2 Abbreviations: LLN=lower limit of normal based on Lilly-defined large clinical trial population based reference limits, ULN=upper limit of normal based on Lilly-defined large clinical trial population based reference limits. A by-patient listing will be provided for all patients having a treatment-emergent Grade 3 or Grade 4 post-baseline elevated creatinine or decreased egfr Infections, Including Opportunistic Infections Infections will be defined using the PTs from the MedDRA Infections and Infestations SOC, with additional terms from the Investigations SOC being used in selected instances, as described below. Treatment-emergent infections will be analyzed according to various groups of infectious events including: all infections o all PTs in the Infections and Infestations SOC, serious infections o all PTs in the Infections and Infestations SOC that are SAEs, infections that require therapeutic intervention (antibiotics, antivirals, antifungals, etc.) o all PTs in the Infections and Infestations System Organ Class for which there is an antimicrobial concomitant medication associated with that event for that patient, herpes zoster and simplex o specific Lilly-defined PTs from the Herpes Viral Infections HLT in the Infections and Infestations SOC, shown in the appendices of the current version of the Baricitinib PSAP tuberculosis o specific Lilly-defined PTs from the Tuberculous Infections HLT and the Atypical Mycobacterial infections HLT of the Infections and Infestations SOC and the Investigations SOC, shown in the appendices of the current version of the Baricitinib PSAP
304 I4V-MC-JADV Statistical Analysis Plan Version 1.0 Page 74 viral hepatitis o all PTs from the Hepatitis Viral Infections HLT (HLT code ) in the Infections and Infestations SOC, opportunistic infections, as described in detail below. For each of all infections, serious infections, infections that require therapeutic intervention, herpes zoster infections, herpes simplex infections, tuberculosis and viral hepatitis, the frequency and relative frequency for each PT will be provided, first pooling narrow and broad terms together, and then for narrow terms only, ordered by decreasing frequency in the baricitinib group (or combined baricitinib group in the event of multiple cohorts/doses). In addition to incidence of infectious AEs by MedDRA Preferred Term and SOC as described above, the number and percentage of patients with treatment-emergent infectious AEs by infection diagnosis during the study will be provided by treatment group, using the following diagnostic categories: Gram-positive bacterial infection, Gram-negative bacterial infection, Mixed aerobic-anaerobic bacterial infection, Fungal infections, Viral infections, and Other infections. Frequency counts and percentages of patients experiencing treatment-emergent infectious AEs will also be presented by treatment group and primary site of infection. Opportunistic infections: Infections which may be opportunistic vary with the nature of the immune function impairment that is present. Data will be analyzed: to identify patients with an infection typically considered to be opportunistic in settings of neutropenia, or impaired lymphocyte function or numbers, to assess the impact of known immunocompromising states on the development of infections, and to discern patients with a host defense impairment that is not evident by other data. Infections will be analyzed to discern events that may be opportunistic in a variety of immunocompromised states using a specified list of MedDRA PTs that are typically considered to be opportunistic infections (organized to distinguish events associated with neutropenia, or impaired lymphocyte function or numbers). Three categories of these opportunistic infections will be considered: infections typically considered to be opportunistic (Category 1) infections due to common pathogens in neutropenia (Category 2) additional types of infection possibly associated with other immune-compromised states (Category 3).
305 I4V-MC-JADV Statistical Analysis Plan Version 1.0 Page 75 The list of MedDRA terms used to identify these infections that are typically considered to be opportunistic infections are found in the appendices of the current version of the Baricitinib PSAP. These lists contain PTs as contained within HLT and certain specific PTs from the Infections and Infestations SOC and from the Investigations SOC which can assist in identifying patients of interest. The complete data of some patients identified using this list of MedDRA PTs may require further review to determine if the patient had an opportunistic infection, particularly if the only PT identifying the patient is from the Investigations SOC. For these treatment-emergent infections that are typically considered to be opportunistic infections, the frequency and relative frequency overall, and for each PT will be provided, first pooling narrow and broad terms together, and then for narrow terms only, ordered by decreasing frequency in the baricitinib group (or combined baricitinib group in the event of multiple cohorts/doses). Summarization should include category and the specific opportunistic infection terms shown in the related appendices. The impact of known immunocompromising states will be assessed by analyzing laboratory data and all infections (all PTs in the Infections and Infestations SOC) to identify treatment-emergent infections that were preceded or accompanied by either neutropenia or lymphopenia. For this analysis, neutropenia and lymphopenia are defined as Grade 2 or greater for neutropenia or lymphopenia as defined by CTCAE and as shown in Table JADV.6.4; these criteria need to be met at the visit when onset of the event was noted, or at the visit prior to when onset of the event was noted. For these treatment-emergent infections preceded/accompanied by neutropenia/lymphopenia, the frequency and relative frequency overall, and for each PT will be provided, first pooling narrow and broad terms together, and then for narrow terms only, ordered by decreasing frequency in the baricitinib group (or combined baricitinib group in the event of multiple cohorts/doses). The number and percentage of patients with treatment-emergent infection events meeting this criterion will be summarized further by neutropenia category and leukopenia category and by treatment group using MedDRA PT nested within SOC. The neutropenia category will be assigned in two ways: (1) by lowest ANC at any post-baseline visit 1200 cells/μl versus >1200 cells/μl and (2) by lowest ANC at any post-baseline visit 1000 cells/μl versus >1000 cells/μl. Lymphopenia category will be assigned in two ways: (1) by lowest lymphocyte count at any post-baseline visit 750 cells/μl versus > 750 cells/μl and (2) by lowest lymphocyte count at any post-baseline visit 500 cells/μl versus >500 cells/μl. Events will be ordered by decreasing frequency in the combined baricitinib group within alphabetical SOC. Patients who may have an immunocompromised state that is not manifest as neutropenia or lymphopenia, or who are not identified using the list of MedDRA terms described above, will be identified for further assessment by identifying patients with multiple and/or recurrent infections. This analysis will consider treatment-emergent infections based on use of the PTs within the Infections and Infestations SOC. For this analysis, a recurrent infection is an infection which occurs at one visit, resolves at a later visit and then re-occurs at an even later visit while on the same therapy. A multiple infection requires occurrence of infections with 2 different MedDRA PTs at different times during treatment with baricitinib. A summary of the frequency and relative frequency for each PT associated with recurrent or multiple infections will be provided,
306 I4V-MC-JADV Statistical Analysis Plan Version 1.0 Page 76 first pooling narrow and broad terms together, and then for narrow terms only, nested within the SOC, ordered by decreasing frequency in the baricitinib group (or combined baricitinib group in the event of multiple cohorts/doses). Treatment-emergent infectious events will be reviewed in context of other clinical and laboratory parameters. A listing of patients experiencing treatment-emergent infectious AEs will be provided, which will include patient demographics, treatment group, treatment start and stop dates, infectious event, event start and stop dates, total leukocytes, total lymphocytes, absolute neutrophils, and event seriousness, event outcome, and concomitant medications Major Adverse Cardiovascular Events (MACE) Major adverse cardiovascular events (MACE) requiring adjudication will be summarized. Subcategories analyzed will include the following: Death Myocardial infarction (MI) Hospitalization for unstable angina Hospitalization for heart failure Coronary interventions (such as coronary artery bypass graft or percutaneous coronary intervention) Cardiogenic shock Resuscitated sudden death Serious arrhythmia Stroke Transient ischemic attack The number and percentage of patients with positively adjudicated MACE events will be summarized by treatment group using MedDRA PT nested within the subcategory. Events will be ordered by decreasing frequency in the combined baricitinib group within subcategory. An additional treatment-specific summarization of the total number of patients with MACE which are SAEs, AEs leading to discontinuation from the study or adverse events resulting in study drug discontinuation will be provided. A by-patient listing for all patients having a positively adjudicated major adverse cardiovascular event at any time will be provided Malignancies Malignancies will be identified using terms from the malignant tumours SMQ (SMQ ). The number and percentage of patients with TEAEs associated with malignant tumors will be summarized by treatment group/cohort using MedDRA PT. Events will be ordered by decreasing frequency in the combined baricitinib group within SMQ. Additional summaries will indicate the total number of patients with malignancies which are SAEs, AEs leading to discontinuation from the study or adverse events resulting in study drug discontinuation. A by-patient listing for all patients having a malignancy event at any time will be provided.
307 I4V-MC-JADV Statistical Analysis Plan Version 1.0 Page Allergic Reactions/Hypersensitivities Allergic reactions/hypersensitivities will be examined within the context of anaphylactic reactions. Anaphylaxis has been broadly defined as a serious allergic reaction that is rapid in onset and may cause death (Sampson et al. 2006). Identification of cases of anaphylaxis from the clinical trial data involves two criteria, one designed to specifically identify cases based on narrow terms from the MedDRA SMQ for anaphylactic reaction (SMQ ), and the second to identify possible cases, following criterion 2 as defined in Sampson (2006). Specifically, Criterion 1 for anaphylaxis is defined by the presence of a TEAE based on the following MedDRA PTs from the anaphylactic reaction SMQ: Anaphylactic reaction Anaphylactic shock Anaphylactoid reaction Anaphylactoid shock Kounis Syndrome Narrow PTs from the anaphylactic reaction SMQ not included in this determination are: Circulatory collapse First use syndrome Shock Type I hypersensitivity Criterion 2 for anaphylaxis requires having adverse events from two or more of four categories of adverse events as described by Sampson et al. (2006). Occurrence of these events should be nearly coincident and develop rapidly after exposure to the most recent injection; based on recording of events on CRFs, a window wherein the events occur within 2 days of one another (based on the onset dates of the events) is allowed. The four categories to be considered in Criterion 2 are: Category A: Involvement of the skin-mucosal tissue Category B: Respiratory compromise Category C: Reduced BP or associated symptoms Category D: Persistent gastrointestinal symptoms Summaries of Criterion 2 anaphylactic adverse events will be provided by the specific combination of categories of such events: AB: events based on meeting Category A and Category B (but no other category); AC: events based on meeting Category A and Category C (but no other category); AD: events based on meeting Category A and Category D (but no other category); BC: events based on meeting Category B and Category C (but no other category); BD: events based on meeting Category B and Category D (but no other category); CD: events based on meeting Category C and Category D (but no other category);
308 I4V-MC-JADV Statistical Analysis Plan Version 1.0 Page 78 ABC: events based on meeting Category A, Category B and Category C (but no other category); ABD: events based on meeting Category A, Category B and Category D (but no other category); ACD: events based on meeting Category A, Category C and Category D (but no other category); BCD: events based on meeting Category B, Category C and Category D (but no other category); ABCD: events based on meeting each of the 4 Criterion 2 categories. Summaries of treatment-emergent anaphylactic AEs will be provided for patients meeting each of the two criteria and for patients who meet either criteria overall. Separate tallies will be provided for serious anaphylactic adverse events and for severe treatment-emergent anaphylactic AEs. Severity of treatment-emergent Criterion 2 anaphylactic AEs will be based on the maximum severity of the specific events met by the patient. Maximum severity of an (overall) treatment-emergent anaphylactic AE will be based on the maximum severity within Criterion 1 and/or Criterion 2. The number and percentage of patients with treatment-emergent anaphylactic AEs will be summarized by treatment group using MedDRA PT nested within the SMQ and additional terms. Events will be ordered by decreasing frequency in the combined baricitinib group within the SMQ and additional terms. Additional rows in the table will indicate the total number of patients with allergic reactions/hypersensitivities which are SAEs and adverse events resulting in study drug discontinuation. The specific MedDRA PTs covered by each of these Criterion 2 categories are shown in the appendices of the current version of the Baricitinib PSAP. A by-patient listing for all patients having an allergic/hypersensitivity event at any time will be provided. Please note that the approach to identifying cases of anaphylaxis and/or hypersensitivity reactions may be adjusted to accommodate changes in MedDRA SMQs Symptoms of Depression (QIDS-SR16) The Quick Inventory of Depressive Symptomatology Self-Rated (QIDS-SR 16 ) is a 16-item, selfreport instrument intended to assess the existence and severity of symptoms of depression as listed in the American Psychiatric Association s Diagnostic and Statistical Manual of Mental Disorders, 4th Edition (DSM-IV; APA 1994). Patients are asked to consider each statement as it relates to the way they have felt for the past 7 days. There is a unique 4-point ordinal scale for each item with scores ranging from 0 to 3 reflecting increasing depressive symptoms as the item score increases. Additional information and the QIDS-SR 16 questions may be found on the University of Pittsburgh Epidemiology Data Center web site ( A key reference for this instrument covers its psychometric properties for use in patients with chronic major depression. (Rush et al. 2003).
309 I4V-MC-JADV Statistical Analysis Plan Version 1.0 Page 79 The QIDS-SR 16 total score is derived as the sum of the scores across the 9 scale domains. For scale domains that contain more than one item, the domain score is the highest item rating given across the items within that domain): Sleep: the highest score on any one of the four sleep items (items 1 to 4). Depressed mood: item 5. Weight/appetite change: the highest score on any one of the four weight items (items 6 to 9). Psychomotor changes: the highest score on either of the two psychomotor items (items 15 and 16). Concentration: item 10. Worthlessness/guilt: item 11. Suicidal ideation: item 12. Decreased interest: item 13. Decreased energy: item 14. The QIDS-SR 16 total scores will also be categorized in the following severity classes: Table JADV.6.9. Quick Inventory of Depressive Symptomatology Self-Rated 16 (QIDS-SR 16 ) Severity of Depressive Symptoms Categories based on the QIDS-SR 16 Total Score QIDS-SR 16 Severity of Depressive Symptoms Category QIDS-SR 16 Total Score 0=None 0-5 1=Mild =Moderate =Severe =Very Severe Treatment differences in mean change in the QIDS-SR 16 total score will be analyzed using the methods specified in Section 6.1 for continuous safety measures. Frequency counts, and percentages will be used to summarize the QIDS-SR 16 total score severity categories and the item responses for the QIDS-SR 16 suicidal ideation item at each visit by treatment group. Using the QIDS-SR 16 Severity of Depressive Symptoms Categories shown in Table JADV.6.9, shift tables will show the number and percentage of patients based on each of baseline to last category and baseline to maximum category during the treatment period, with baseline depicted by the most extreme category during the baseline period, with further summarization of change from baseline in severity using categories of any improvement, no change, and any worsening, by treatment. Similarly, shift tables will be created for the QIDS-SR 16 suicidal ideation item (Item 12) responses. Pairwise treatment differences for the shift table summary categorizations will be assessed with the Likelihood Ratio Chi-Square test.
310 I4V-MC-JADV Statistical Analysis Plan Version 1.0 Page 80 In addition, an individual patient listing of the QIDS-SR 16 data will be provided Other Safety Analyses Descriptive summary of common AEs (reported by at least 5% of patients overall) for the subgroup of folic acid intake ( 1mg, > 1mg). Common AEs will be described in terms of frequency counts and percentages by treatment groups. The Framingham risk score represents the likelihood of experiencing a cardiovascular disease (CVD) event in the next 10 years for an individual patient. It will be analyzed as continuous and categorical variables (ordinal endpoint of Framingham risk score). The difference between baricitinib and placebo in change from baseline up to Week 24 will be analyzed using ANCOVA which includes treatment, region, and joint erosion status as fixed effects, the baseline measurement as covariate and change from baseline as the dependent variable. The ordinal endpoint of Framingham risk score is defined as low risk (risk<10%), intermediate risk (risk between 10% and 20%), and high risk (risk>20%). The CMH method controlling for region will be used to analyze the ordinal endpoint of Framingham risk score, comparing baricitinib and placebo up to Week 24. The Reynolds Risk Score (RRS) is an alternative characterization to the Framingham risk score for non-diabetic men and women aged 45 years and hscrp 20mg/L. The RRS will be analyzed similarly to what was described for the Framingham risk score Subgroup Analysis Subgroup analyses comparing baricitinib to placebo will be performed using ACR20, ACR50, HAQ-DI, and DAS28-hsCRP at Week 12 (Visit 7) and Week 24 (Visit 11), and mtss at Week 24 (Visit 11). The following subgroups (but not limited to only these), will be evaluated: Gender: (Male, Female) Age: (<65 years, 65 years) Age: (<65 years, 65 to <75 years, 75 to <85 years, 85 years) Weight: tertiles of baseline weight Ethnicity: (Hispanic/Latino, non-hispanic/non-latino) Race: (Asian, Black/African American, White, Other (American Indian/Alaska Native, Native Hawaiian or other Pacific Islander), Multiple) Smoker: (yes, no) Region (as defined in Section 5.3) Specific region: (Europe, other) Specific country: (USA, other) Specific country: (Japan, other) Baseline renal function status: impaired (egfr <60 ml/min/1.73 m2) or not impaired (egfr 60 ml/min/1.73 m2) Duration of RA: (<1 year, 1 to <5 years, 5 to < 10 years, 10 years) Baseline disease severity: (DAS28-hsCRP 5.1, DAS28-hsCRP >5.1)
311 I4V-MC-JADV Statistical Analysis Plan Version 1.0 Page 81 Joint erosion status: (1-2 joint erosions plus seropositivity, at least 3 joint erosions) Seropositivity: [either rheumatoid factor (RF) positive or anti-citrillunated protein antibody (ACPA) positive, both RF negative and ACPA negative] Seropositivity: (RF positive, RF negative) Seropositivity: (ACPA positive, ACPA negative) Background therapy: (MTX only, MTX + Other cdmards) Baseline mtss: ( median, > median) Corticosteroid use at baseline: (yes, no) The analyses of ACR20 and ACR50 will be performed using logistic regression. The model will include ACR20/50 as the dependent variable and treatment, subgroup, and treatment-bysubgroup interaction as explanatory variables. The treatment-by-subgroup interaction will be tested at the 0.1 significance level. Counts, percentages and 95% CI will be presented. ANCOVA will be used to analyze the change from baseline in HAQ-DI and DAS28-hsCRP at Weeks 12 and 24 and mtss at Week 24 as dependent, and baseline, treatment, subgroup and treatment-by-subgroup as explanatory variables. The treatment-by-subgroup interaction will be tested at the 0.1 significance level. The LS mean difference, SE and 95% CI will be presented Interim Analysis and Data Monitoring A DMC will oversee the conduct of all the Phase 3 clinical trials evaluating baricitinib in patients with RA. The DMC will consist of members external to Lilly. This DMC will follow the rules defined in the DMC charter, focusing on potential and identified risks for this molecule and for this class of compounds. DMC membership will include, at a minimum, specialists with expertise in rheumatology, statistics, and other appropriate specialties. The DMC will review and evaluate up to 3 planned interim analyses on an approximate semiannual basis and the coordination of these analyses will be documented in the DMC Charter. Access to the unblinded interim data will be limited to the statisticians who conduct the interim analyses and the DMC. The statisticians conducting the interim analyses will be independent from the study team. The study team will not have access to the unblinded data. Study sites will receive information about interim results ONLY if they need to know for the safety of their patients. The DMC will review study discontinuation data, AEs including SAEs, clinical laboratory data, vital sign data, etc. The DMC may recommend continuation of the study, as designed, temporary suspension of enrollment, or the discontinuation of a particular treatment regimen or the entire study. The DMC may request to review efficacy data to investigate the benefit/risk relationship in the context of safety observations for ongoing patients in the study. However, the study will not be stopped for positive efficacy results, and there is no planned futility assessment. Hence, there will be no alpha adjustment for these interim analyses. Details of the DMC and interim safety analyses will be documented in a DMC charter and DMC analysis plan. Besides DMC members, a limited number of pre-identified individuals may gain access to the unblinded PK and safety data, as specified in the unblinding plan, prior to the primary data lock, to initiate the exploration and/or final population PK/PD model development processes for the primary analysis and/or to evaluate safety data (e.g., neutropenia, anemia). Information that may
312 I4V-MC-JADV Statistical Analysis Plan Version 1.0 Page 82 unblind the study during the analyses will not be reported to study sites or the blinded study team until the data lock for the primary and major secondary efficacy analyses.
313 I4V-MC-JADV Statistical Analysis Plan Version 1.0 Page Unblinding Plan There will be two planned data locks for this study: a primary data lock after all patients complete Part A and a final data lock at end of the study. A data lock is planned after all patients complete Part A in which by-visit data through Week 24 (Visit 11) will be locked but ongoing log-type entry forms for ongoing patients such as adverse events and concomitant medications will remain unlocked. Analyses will include the primary and major (key) secondary endpoints (included in the gatekeeping strategy except for x-ray data), efficacy, health outcome measures, and safety endpoints that are collected through Week 24 according to the treatment/ cohort groups defined in section 6.1. This analysis will not be considered an interim analysis conducted by the DMC. A limited number of pre-identified individuals at Eli Lilly and Company may gain access to the unblinded PK data prior to the primary data lock in order to initiate the final population PK/PD model development processes for the primary analysis and/or to evaluate safety data. Information that may unblind the study during the analyses will not be shared with study team and study site personnel who still remain blinded at the time. Members of the study team not in direct contact with the study sites may have access to the unblinded data from this study after all patients complete Week 24 (Visit 11) and the database has been locked. Members of the study team who become unblinded will have no further contact with study sites until after the final data lock. A final data lock is planned at the end of the study when all patients complete the study. The final analysis will include all data that are collected during the study. All investigators, study site personnel, and patients will remain blinded to treatment assignments until the final data lock.
314 I4V-MC-JADV Statistical Analysis Plan Version 1.0 Page References [APA] American Psychiatric Association. Diagnostic and Statistical manual of Mental Disorders. 4 th ed. DSM-IV. Washington, DC: American Psychiatric Association; Bretz F, Maurer W, Brannath W, Posch M. A graphical approach to sequentially rejective multiple test procedures. Stat Med. 2009;28(4): Brooks R. EuroQol: The current state of play. Health Policy. 1996;37: Bruynesteyn K, Boers M, Kostense P, van der Linden S, van der Heijde D. Decidingon progression of joint damage in paired films of individual patients: smallest detectable difference or change. Ann Rheum Dis. 2005;64(2): Cancer Therapy Evaluation Program, Common Terminology Criteria for Adverse Events, Version 3.0, DCTD, NCI, NIH, DHHS, March 31, Cohen SB, Emery P, Greenwald MW, Dougados M, Furie RA, Genovese MC, Keystone EC, Loveless JE, Burmester GR, Cravets MW, Hessey EW, Shaw T, Totoritis MC. Rituximab for rheumatoid arthritis refractory to anti-tumor necrosis factor therapy. Results of a multicenter, randomized, double-blind, placebo-controlled phase III trial evaluating primary efficacy and safety at twenty-four weeks. Arthritis Rheum. 2006;54(9): Common Terminology Criteria for Adverse Events, Version 4.03, DHHS NIH NCI, NIH Publication No , June 14, D'Agostino, Vasan, Pencina, Wolf, Cobain, Massaro, Kannel, A General Cardiovascular Risk Profile for Use in Primary Care: The Framingham Heart Study. Circulation Keystone EC, Kavanaugh AF, Sharp JT, Tannenbaum H, Hua Y, Teoh LS, Fischkoff SA, Chartash EK. Radiographic, clinical, and functional outcomes of treatment with adalimumab (a human anti tumor necrosis factor monoclonal antibody) in patients with active rheumatoid arthritis receiving concomitant methotrexate therapy: a randomized, placebo-controlled, 52-week trial. Arthritis Rheum. 2004;50(5): Keystone E, Heijde D, Mason D Jr, Landewé R, Vollenhoven RV, Combe B, Emery P, Strand V, Mease P, Desai C, Pavelka K. Certolizumab pegol plus methotrexate is significantly more effective than placebo plus methotrexate in active rheumatoid arthritis: findings of a fifty-twoweek, phase III, multicenter, randomized, double-blind, placebo-controlled, parallel-group study. Arthritis Rheum. 2008;58(11): Keystone E, Emery P, Peterfy CG, Tak PP, Cohen S, Genovese MC, Dougados M, Burmester GR, Greenwald M, Kvien TK, Williams S, Hagerty D, Cravets MW, Shaw T. Rituximab inhibits structural joint damage in patients with rheumatoid arthritis with an inadequate response to tumour necrosis factor inhibitor therapies. Ann Rheum Dis. 2009;68(2): National Kidney Foundation. K/DOQI Clinical Practice Guidelines for Chronic KidneyDisease: Evaluation, Classification and Stratification. Am J Kidney Dis ; 39(2)(suppl 1):S1-S266. Newcombe RG. Interval estimation for the difference between independent proportions: comparison of eleven methods. Stat Med. 1998;17: Erratum in Stat Med. 1999;18:1293.
315 I4V-MC-JADV Statistical Analysis Plan Version 1.0 Page 85 Protocol I4V-MC-JADV. A Randomized, Double-Blind, Placebo- and Active-Controlled, Phase 3 Study Evaluating the Efficacy and Safety of Baricitinib in Patients with Moderately to Severely Active Rheumatoid Arthritis Who Have Had an Inadequate Response to Methotrexate Therapy. Ridker PM, Buring JE, Rifari N, Cook NF. Development and Validation of Improved Algorithms for the Assessment of Global Cardiovascular Risk in Women: The Reynolds Risk Score. JAMA. 2007;(297):6. Ridker PM, Paynter NP, Rifai N, Gaziano JM, Cook NR. C-Reactive Protein and Parental History Improve Global Cardiovascular Risk Prediction: The Reynolds Risk Score for Men. Circulation. 2008;118: Rush AJ, Trivedi MH, Ibrahim HM, Carmody TJ, Arnow B, Klein DN, Markowitz JC,Ninan PT, Kornstein S, Manber R, Thase ME, Kocsis JH, Keller MB. The 16-item Quick Inventory of Depressive Symptomatology (QIDS) Clinician Rating (QIDS-C) and Self-Report (QIDS-SR): A psychometric evaluation in patients with chronic major depression. Biological Psychiatry. 2003;54: Sampson, HA, Muñoz-Furlong, A, Campbell RL, Adkinson NF Jr, Bock SA, Branum A, Brown SG, Camargo CA Jr, Cydulka R, Galli SJ, Gidudu J, Gruchalla RS, Harlor AD Jr, Hepner DL, Lewis LM, Liberman PL, Metcalfe DD, O Connor R, Muraro A, Rudman A, Schmitt C, Scherrer D, Simons FE, Thomas S, Wood JP, Decker WW. Second symposium on the definition and management of anaphylaxis: summary report - second National Institute of Allergy and Infectious Disease/Food Allergy and Anaphylaxis Network symposium. J Allergy Clin Immunol. 2006;117(2): Smolen J, Landewé RB, Mease P, Brzezicki J, Mason D, Luijtens K, van Vollenhoven RF, Kavanaugh A, Schiff M, Burmester GR, Strand V, Vencovskỳ J, van der Heijde D. Efficacy and safety of certolizumab pegol plus methotrexate in active rheumatoid arthritis: the RAPID 2 study. A randomised controlled trial. Ann Rheum Dis. 2009;68(6): Smolen JS, Beaulieu A, Rubbert-Roth A, Ramos-Remus C, Rovensky J, Alecock E, Woodworth T, Alten R, OPTION investigators. Effect of interleukin-6 receptor inhibition with tocilizumab in patients with rheumatoid arthritis (OPTION study): a double-blind, placebocontrolled, randomised trial. Lancet. 2008;371(9617): Szende A, Oppe M, Devlin N. EQ-5D value sets: Inventory, comparative review and user guide. EuroQol Group Monographs. Volume 2. Springer, Third Report of the National Cholesterol Education Program (NCEP) Expert Panel on Detection, Evaluation, and Treatment of High Blood Cholesterol in Adults (Adult Treatment Panel III) Final Report, National Cholesterol Education Program, National Heart, Lung, and Blood Institute, National Institutes of Health, NIH Publication No , September van der Heijde D, Tanaka Y, Fleischmann R, Keystone E, Kremer J, Zerbini C, Cardiel MH, Cohen S, Nash P, Song YW, Tegzová D, Wyman BT, Gruben D, Benda B, Wallenstein G, Krishnaswami S, Zwillich SH, Bradley JD, Connell CA. Tofacitinib (CP-690,550) in patients with rheumatoid arthritis receiving methotrexate: twelve-month data from a twenty-fourmonth phase III randomized radiographic study. Arthritis Rheum. 2013;65(3):
316 I4V-MC-JADV Statistical Analysis Plan Version 1.0 Page Appendices
317 I4V-MC-JADV Statistical Analysis Plan Version 1.0 Page 87 Appendix 1. Algorithm for determining ACR response Details presented in this appendix will use x as a generic symbol, and the appropriate number (either 20, 50, or 70) is to be filled in when implementing in dataset programming code. ACRx response is defined as x% improvement from baseline in tender joint count (68 joint counts), and x% improvement in swollen joint count (66 joint counts), and x% improvement in at least three of the following five items: Patient s global assessment of arthritis pain; Patient s global assessment of disease activity; Physician s global assessment of disease activity; HAQ-DI; hscrp. The following abbreviations will be used throughout this appendix to refer to the items needed in the algorithm definitions: Parameter Abbreviation for the Parameter % improvement in tender joint count TJC68 % improvement in swollen joint count SJC66 % improvement in patient s assessment pain PATPAIN % improvement in patient s global assessment of disease PATGA activity % improvement in physician s global assessment of disease PHYGA activity % improvement in HAQ-DI HAQ % improvement in hscrp hscrp For all seven parameters mentioned above, % improvement at a visit is calculated as: (baseline value value at visit) * 100 / baseline value. To calculate the observed ACRx response at a visit: Step 1: If the patient discontinued from the study prior to reaching the visit, then STOP assign ACRx response as blank (ie, missing). Otherwise, calculate the % improvement at the visit for all seven parameters as described above. Step 2: o o If TJC68 AND SJC66 are BOTH x%, then proceed to step3. If both are non-missing but one or both is < x%, then STOP assign the patient as a non-responder for ACRx.
318 I4V-MC-JADV Statistical Analysis Plan Version 1.0 Page 88 o If either or both are missing, proceed as follows: a. If both are missing, then STOP assign ACRx response as blank (ie, missing). b. If one of TJC68 or SJC66 is missing and the non-missing value is < x%, then STOP assign the patient as a non-responder for ACRx. c. If one of TJC68 or SJC66 is missing and the non-missing value is x%, then STOP assign ACRx response as blank (ie, missing). Step 3: Consider the following five variables: PATPAIN, PTGA, PHYGA, HAQ, and hscrp. o o If three or more items are missing, then STOP assign ACRx response as blank (ie, missing). If three or more items are non-missing, then proceed with the following order: a. If at least three items are x%, then STOP assign the patient as a responder for ACRx. b. If at least three items are < x%, then STOP assign the patient as a nonresponder for ACRx. c. If less than three items are x%, then STOP assign ACRx response as blank. To calculate the ACRx response at post baseline visits up to Week 52 using NRI: Step 1: Calculate the % improvement at the visit for all seven parameters as described above. Step 2: If all seven parameters are missing at the visit, or if the patient discontinued from the study prior to reaching the visit, then STOP assign the patient as a nonresponder for ACRx. Step 3: If at least one of the seven parameters is non-missing at the visit and the patient is still enrolled in the study, then use LOCF to fill in any missing values. Step 4: If TJC68 AND SJC66 are BOTH x%, then proceed to step 5. If both are nonmissing but one or both is < x%, then STOP assign the patient as a nonresponder for ACRx. If either or both are missing, then STOP assign the patient as a non-responder for ACRx. Step 5: Consider the following five variables: PATPAIN, PATGA, PHYGA, HAQ, hscrp. o If three or more of the five items are non-missing, then: If three or more of the five items are x%, then STOP assign the patient as a responder for ACRx. If less than three of those five items are x%, then STOP assign the patient as a non-responder for ACRx.
319 I4V-MC-JADV Statistical Analysis Plan Version 1.0 Page 89 o If three or more of the five items are missing, then STOP assign the patient as a non-responder for ACRx.
320 I4V-MC-JADV Statistical Analysis Plan Version 1.0 Page 90 Appendix 2. Details of Efficacy and Health Outcome Measures Van der Heijde Modified Total Sharp Score X-rays of the hands/wrists and feet will be scored (according to the Synarc predefined imaging charter) for structural progression as measured using the mtss (van der Heijde, 2000). This methodology quantifies the extent of bone erosions for 44 joints and joint space narrowing for 42 joints with higher scores representing greater damage. Structural progression will be measured using the mtss. During screening, all patients meeting entry criteria will have a single posteroanterior radiographic assessment of the left and right hand/wrist and a single dorsoplantar radiographic image taken of the left and right foot. Alternatively, images taken within 4 weeks prior to baseline may be used. Images taken at screening (or within 4 weeks of screening) will be reviewed centrally by qualified readers for evidence of erosive bony change(s). These initial radiographs will serve as the baseline radiographs for comparison throughout the study. Additional radiographs (ie, a single posteroanterior view of each hand and a single dorsoplantar view of each foot) will be obtained at Week 16, Week 24 (the primary evaluation), and Week 52 (for the purposes of demonstrating durability of structural preservation as requested by regulatory authorities), as shown in the study schedule (See Protocol Appendix). For patients who discontinue prematurely from the study, x-rays will be performed at the early termination visit only if the previous x-rays were taken more than 12 weeks earlier. Bone Joint Erosion Score The joint erosion score is a summary of erosion severity in 32 joints of the hands and 12 joints of the feet. The maximum erosion score for a hand joint is 5 and for a foot joint is 10. Thus, the maximal erosion score is 280 for a timepoint (160 for both hands/ wrists and 120 for both feet). Each joint is scored according to the surface area involved from 0 to 5 for hand joints and 0 to 10 for the foot joints. The highest score (5 for the hand and 10 for the foot) indicates extensive loss of bone from more than one half of the articulating bone. A score of 0 in either the hand or foot joints indicates no erosion. Erosion for each hand joint (16 joints per hand) is graded on the following scale. 0 Normal (no erosion) 1 Discrete small erosion 2 Two discrete erosions or one large erosion not passing the joint middle line 3 One large erosion passing the joint middle line of the bone or combination of the above 4 Combinations of discrete and/or large erosions adding up to 4
321 I4V-MC-JADV Statistical Analysis Plan Version 1.0 Page 91 5 Combinations of discrete and/or large erosions adding up to 5 or more NA - Not assessable due to poor radiographic depiction S - Surgically modified (e.g. joint replacement, amputation or other surgery) Discrete erosions will be graded 1 if small and 2 or 3 if larger, with a score of 3 if the erosion is large and extends across the imaginary middle of the bone. Discrete erosions of each grade will then be summed over both sides of the joint to a maximum of 5 to give a total score for each location in the hands. When erosions in the carpal bones of the wrist are confluent rather than discrete and focal, the percent surface eroded will be scored from 0 to 5 in approximately 20% intervals. A score of NA will be assigned in the case that the location is not evaluable due to advanced subluxation or poor radiographic depiction. A score of S will be assigned in the case had joint replacement, amputation or other surgery. Erosion for each side of a foot joint (6 joints per foot) is graded on the following scale. 0 Normal (no erosion) 1 Discrete small erosion 2 Two discrete erosions or one large erosion not passing the joint middle line 3 One large erosion passing the joint middle line 4-10 Combination of the above conditions. NA - Not assessable due to poor radiographic depiction S - Surgically modified (e.g. joint replacement, amputation or other surgery) Since both sides of each foot joint are graded, the maximum score for a foot joint is 10 (a score of 5 on each side). Joint Space Narrowing (JSN) Score The JSN score summarizes the severity of JSN in 30 joints of both hands and 12 joints of both feet. Assessment of JSN for each hand (15 joints per hand) and foot (6 joints per foot), including subluxation, is scored from 0 to 4, with 0 indicating no (normal) JSN and 4 indicating complete loss of joint space, bony ankylosis or luxation. Thus, the maximum JSN score is 168 (120 for both hands/ wrists and 48 for both feet) at a time point. JSN for each hand and foot joint is graded on the following scale: 0 Normal or no narrowing 1 Asymmetrical and/ or minimal narrowing- up to 25% 2 Definite narrowing with loss of up to 50% of the normal space 3 Definite narrowing with loss of 50-99% of the normal space or subluxation 4 Absence of a joint space, presumptive evidence of ankylosis or complete luxation NA - Not assessable due to poor radiographic depiction
322 I4V-MC-JADV Statistical Analysis Plan Version 1.0 Page 92 S - Surgically modified (e.g. joint replacement, amputation or other surgery) Total Modified Sharp Score (mtss) The total mtss score at a time point is the sum of the erosion (maximum of 280) and JSN (maximum of 168) scores, for a maximum score of 448. Handling of Missing Joint Data Refer to a separate document for a detailed description of imputation of missing joint data. Adjudication Process The read of x-ray images will be performed by 2 independent readers. An adjudication of results will be in place to show any discrepancy between the two independent readers. The adjudication process will involve re-reading by a third independent reader. Cases that require adjudication will be identified once the two primary independent reviews are completed. To ensure a consistent read by the two independent readers, an inter-/intra-reader variability assessment will be evaluated. Refer to the Synarc imaging charter and a separate document describing missing data imputation for more details. ACR Core Set The individual components that make up the ACR Core Set of measures for RA are: Tender Joint Count (TJC) For ACR measures, the number of tender and painful joints will be determined by examination of 68 joints (34 joints on each side of the patient s body). These 68 joints will be assessed and classified as tender, not tender, or not evaluable. The number of tender joints ranges from 0 to 68. The algorithm to calculate TJC is in the Joint Assessment section of this Appendix. Swollen Joint Count (SJC) For ACR measures, the number of swollen joints will be determined by examination of 66 joints (33 joints on each side of the patient s body). These 66 joints will be assessed and classified as swollen, not swollen, or not evaluable. The number of swollen joints ranges from 0 to 66. Refer to the Joint Assessment section of this Appendix for the algorithm to calculate SJC. Patient s Assessment of Pain (VAS) The patient will be asked to assess his or her current level of pain by marking a vertical tick on a 100-mm horizontal VAS with the left end marked as no pain and the right end marked as worst possible pain. Results will be expressed in millimeters measured between the left end of the scale and the crossing point of the vertical line of the tick. This procedure is applicable for all VAS scales used in the trial. Patient s Global Assessment of Disease Activity (VAS) The patients will be asked to give an overall assessment of how their rheumatoid arthritis is affecting them at present by marking a vertical tick on a VAS from very well to very poor.
323 I4V-MC-JADV Statistical Analysis Plan Version 1.0 Page 93 Physician s Global Assessment of Disease Activity (VAS) The physician s assessment of a patient s disease activity assessed at the visit will be recorded using the VAS from none to extremely active arthritis. Patient s Assessment of Physical Function (HAQ-DI) The patient will be asked to complete the HAQ-DI to measure the impact of arthritis on their ability to function in daily life. The HAQ-DI functional disability index score ranges from 0 to 3, with lower scores indicating better physical functioning. See Section HAQ-DI of this appendix for the algorithm to calculate the HAQ-DI functional disability index score. High sensitivity C-Reactive Protein (hscrp) hscrp will be the ACR Core Set measure of acute phase reactant. It will be measured at the central laboratory. Joint Assessment Each of 28 or 68 joints will be evaluated for tenderness and 28 or 66 joints will be evaluated for swelling (hips are excluded for swelling) at the specified visits as shown in the schedule of events of the protocol. The 68/66 joint count will be performed at screening, Week 0 (baseline), Week 1 to Week 24 in Part A, Week 25 to Week 52 in Part B, end of treatment and at the followup visit. For scores using 28 joints, the following subset will be assessed (both right and left side): Shoulder, Elbow, Wrist, Metacarpophalangeal (MCP) I, II, III, IV, and V, Thumb interphalangeal, Proximal interphalangeal (PIP) II, III, IV, and V, Knee. Joints will be assessed for tenderness by pressure and joint manipulation on physical examination. Any positive response on pressure, movement, or both will then be translated into a single tender-versus non-tender dichotomy. Joints will be classified as either swollen or not swollen. Swelling is defined as palpable fluctuating synovitis of the joint. Swelling secondary to osteoarthrosis will be assessed as not swollen, unless there is unmistakable fluctuation. The following joint count imputation rules will be applied. (1) Joints that had any of the following procedures or disease conditions that occur at the screening (Visit 1) up to and including baseline (Visit 2) will be imputed as follows:
324 I4V-MC-JADV Statistical Analysis Plan Version 1.0 Page 94 Arthroplasty, fusion, synovectomy, ankylosis, amputation, injury, fracture and infection: these will be considered non-evaluable from baseline up to end of the study. Joints that include non-evaluable joints will be prorated from baseline up to end of the study. For example, the swollen joint count score for 6 swollen plus 2 non-evaluable joints for DAS28 is calculated as [6/(28-2)]*28 = Injection: joints that have received intra-articular and bursal injections will be collected in the data but set to pain/ tender or swollen after the injection for 6 months (up to Week 24 [Visit 11]). (2) Joints that had any of the following procedures or disease conditions that occur postbaseline (anytime during treatment with the study drugs) will be imputed as follows: Amputation: amputation that occurs postbaseline will be considered non-evaluable from the visit after amputation up to end of the study. Joints that include non-evaluable joints will be prorated from the visit after amputation up to end of the study. For example, the swollen joint count score for 6 swollen plus 2 non-evaluable joints for DAS28 is calculated as [6/(28-2)]*28 = Arthroplasty, fusion, synovectomy and ankylosis: if any of these procedures occur postbaseline, joints will be set to pain/ tender or swollen from the visit after the procedure up to end of the study. Infection, injury and fracture: if any of these conditions occur postbaseline, joints will be set to pain/ tender or swollen from the visit after the condition up to condition is resolved. Injection: joints that have received intra-articular and bursal injections postbaseline will be collected in the data but set to pain/ tender or swollen after the injection for 6 months or end of study (whichever comes first). The number of tender and swollen joints will be calculated by summing all joints. For patients who have an incomplete set of joints evaluated, the joint count will be adjusted to a 28- or 68- joint count for tenderness and a 28- or 66-joint count for swelling by dividing the number of affected joints by the number of evaluated joints and multiplying by 28 or 68 for tenderness and 28 or 66 for swelling. Patient s Assessment of Arthritis Pain The patient s arthritis pain will be assessed with the use of VAS (range, 0 to 100-mm, with higher scores indicating more severe pain). Patient s Global Assessment of Disease Activity The Patient s Global Assessment of Disease Activity will be recorded on a VAS (range, 0 to 100-mm, with higher scores indicating poorer status or more active arthritis). Physician s Global Assessment of Disease Activity The Physician s Global Assessments of Disease Activity will be recorded on a VAS (range, 0 to 100-mm, with higher scores indicating poorer status or more active arthritis).
325 I4V-MC-JADV Statistical Analysis Plan Version 1.0 Page 95 Disease Activity Score-Erythrocyte Sedimentation Rate (DAS28- ESR) and Disease Activity Score-High Sensitivity C-Reactive Protein (DAS28-hsCRP) The DAS28 is a measure of disease activity in 28 joints that consists of a composite numeric score of the following variables: TJC, SJC, hscrp or ESR, and Patient s Global Assessment of Disease Activity (Vander Cruyssen et al. 2005). The 28 joints to be examined and assessed as tender or not tender for TJC and as swollen or not swollen for SJC include 14 joints on each side of the patient s body: the 2 shoulders, the 2 elbows, the 2 wrists, the 10 metacarpophalangeal joints, the 2 interphalangeal joints of the thumb, the 8 proximal interphalangeal joints, and the 2 knees (Smolen et al. 1995). The following equation will be used to calculate the DAS28- hscrp (Vander Cruyssen et al. 2005): DAS28-hsCRP = 0.56(TJC28) 1/ ( SJC28) 1/ (ln(hsCRP +1)) (VAS) The hscrp value in mg/l must be used. In order to calculate DAS28-hsCRP data must be present for all 4 components: TJC28, SJC28, patient s global assessment (VAS) and hscrp. No component-level imputation will be performed for the calculation of DAS28-hsCRP. If any components are missing, the DAS28-hsCRP should be missing. Based on the DAS- hscrp, the following will be calculated: DAS28-hsCRP 3.2 DAS28-hsCRP < 2.6 Similarly, DAS28-ESR=0.56(TJC28) 1/ (SJC28) 1/ ln(esr) (VAS) [If ESR = 0 in analysis data, set ln(esr)=0] The ESR (Erythrocyte sedimentation rate) in mm/first hour must be used. In order to calculate DAS28-ESR data must be present for all 4 components: TJC28, SJC28, patient s global assessment (VAS) and ESR. No component-level imputation will be performed for the calculation of DAS28-ESR. If any components are missing, the DAS28-ESR should be missing. Based on the DAS-ESR, the following will be calculated: DAS28-ESR 3.2 DAS28-ESR < 2.6 A NRI rule for the DAS28 binary endpoints will be applied for each visit as follows: if the DAS28 is missing at the visit or if the patient discontinued treatment or the study prior to the visit, then assign the patient as DAS28 non-responder. Hybrid American College of Rheumatology Response Measure The hybrid ACR (bounded) response measure will be obtained as described by the ACR Committee to Reevaluate Improvement Criteria (2007);
326 I4V-MC-JADV Statistical Analysis Plan Version 1.0 Page 96 Table APP.1. Scoring Method for the Hybrid American College of Rheumatology Response Measure Mean % Change in Core Set Measuresa ACR Status <20 20, <50 50, <70 70 Not ACR20 Mean % change ACR20 but not ACR50 20 Mean % change ACR50 but not ACR Mean % change ACR Mean % change Abbreviations: ACR = American College of Rheumatology; ACR20 = 20% improvement in ACR criteria; ACR50 = 50% improvement in ACR criteria; ACR70 = 70% improvement in ACR criteria. a 1) Calculate the average percentage change in core set measures. For each core set measure, subtract score after treatment from baseline score and determine percentage improvement in each measure. Next, if a core set measure worsened by >100%, limit that percentage change to 100% (set equal to -100% bound). Then, average the percentage changes for all core set measures. 2) Determine whether the patient has achieved ACR20, ACR50, or ACR70. 3) Using the table above, obtain the hybrid ACR response measure. To use the table, take the ACR20, ACR50, or ACR70 status of the patient (left column) and the mean percentage improvement in core set items; the hybrid ACR score is where they intersect in the table. Simplified Disease Activity Index (SDAI) The SDAI is a tool for measurement of disease activity in RA that integrates measures of physical examination, acute phase response, patient self-assessment, and evaluator assessment. The SDAI is calculated by adding together scores from the following assessments: number of swollen joints (0 to 28), number of tender joints (0 to 28), hscrp in mg/dl (0.1 to 10.0), patient global assessment of DAS on VAS (0 to 10.0, measured in cm), and evaluator or physician global assessment of DAS on VAS (0 to 10.0, measured in cm) (Aletaha and Smolen 2005) Disease remission according to ACR/EULAR index-based definition of remission is defined as an SDAI score of 3.3 (Felson et al. 2011). Low disease activity according to SDAI is defined as a SDAI score 11 (Aletaha and Smolen 2005). Clinical Disease Activity Index (CDAI) The CDAI is similar to the SDAI, but it allows for immediate scoring because it does not use a laboratory result. The CDAI is calculated by adding together scores from the following assessments: number of swollen joints (0 to 28), number of tender joints (0 to 28), patient global assessment of DAS on VAS (0 to 10.0, measured in cm), and
327 I4V-MC-JADV Statistical Analysis Plan Version 1.0 Page 97 evaluator or physician global assessment of DAS on VAS (0 to 10.0, measured in cm) (Aletaha and Smolen 2005) Remission according to CDAI is defined as a CDAI score of 2.8 (Felson et al. 2011). Low disease activity according to CDAI is defined as a CDAI score 10 (Aletaha and Smolen 2005). European League Against Rheumatism (EULAR) Responder Index Assessments of patients with RA by the EULAR Responder Index based on the 28-joint count will be used to categorize patients as nonresponders, moderate responders, good responders, or responders (moderate + good responders) according to van Gestel et al Table APP.2. Postbaseline Level of DAS28-hsCRP DAS28-hsCRP 3.2 Categorization of Patients as Nonresponders, Moderate Responders, or Good Responders Improvement Since Baseline in DAS28-hsCRP (Decline in DAS28-hsCRP) > and > Good response 3.2 < DAS28-hsCRP 5.1 Moderate response DAS28-hsCRP >5.1 No response Abbreviation: DAS28-hsCRP = Disease Activity Score modified to include the 28 diarthroidal joint count used for hscrp. There are 3 categories, no improvement (or no response), moderate improvement (or moderate response), and good improvement (highest kind of improvement or good response). The category of no improvement also includes a worsening of RA. Hence, relative to baseline, a patient can only have a 0 change in categories, a change of 1 category or a change of 2 categories. ACR/EULAR Rheumatoid Arthritis Remission Two new ACR/EULAR definitions of RA remission will be evaluated, a boolean-based definition and an index-based definition (Felson et al. 2011). Boolean-based Definition of Remission: All 4 criteria below must be met (at the same visit): o TJC28 1 o SJC28 1 o hscrp 1 mg/dl (10 mg/l) o Patient Global Assessment of Disease Activity (on a mm scale) / 10 1

328
329 I4V-MC-JADV Statistical Analysis Plan Version 1.0 Page 99 The FACIT-F uses a 13-item symptom-specific questionnaire to assess the burden of selfreported fatigue caused by a chronic disease and its impact upon daily activities and function. A 5-point Likert-type scale (0 = not at all; 1 = a little bit; 2 = somewhat; 3 = quite a bit; 4 = very much) is used. As each of the 13 items of the FACIT-Fatigue scale ranges from 0 4, the range of possible scores is 0 52, with 0 being the worst possible score and 52 the best. To obtain the 0 52 score, each negatively worded item response is transformed so that 0 is a bad response and 4 is good response. All responses are added with equal weight to obtain the total score. Lower values of the FACIT-Fatigue score denote higher fatigue. Specifically, all questions with the exception of I have energy and I am able to perform usual activities will have their responses recoded so that 0 = Very much, 1 = Quite a Bit, 2 = Somewhat, 3 = A little bit, 4 = Not at all) prior to calculation of the FACIT-Fatigue scale score. To score FACIT-Fatigue Subscale (FACIT_F), do the following: 1. Perform reversals for 11 out of the 13 individual items, namely HI7, HI12, An1, An2, An3, An4, An8, An12, An14, An15, An16: item score = 4 minus item response 2. For the individual items An5 and An7: items score = item response 3. Sum the individual items to obtain a score 4. Multiply the sum of the items scores by 13 (the number of items in the subscale), then divide by the number of items answered which produces the Fatigue Subscale score. Missing items are acceptable as long as more than 50% of the items are answered (i.e. a minimum of 7 out of 13 items). If less than 7 items are answered, the Fatigue Subscale score will be set to missing. SF-36v2 Acute: The SF-36v2 Acute measure is a subjective, generic, health-related quality of life instrument that is patient-reported and consists of 36 questions covering 8 health domains: physical functioning, bodily pain, role limitations due to physical problems, role limitations due to emotional problems, general health perceptions, mental health, social function, and vitality. Each domain is scored by summing the individual items and transforming the scores into a 0 to 100 scale with higher scores indicating better health status or functioning. In addition, 2 summary scores, the PCS (physical component score) and the MCS (mental component score), will be evaluated based on the 8 SF-36v2 Acute domains (Brazier et al. 1992; Ware and Sherbourne 1992). How to Calculate the SF-36v2 Scores This protocol uses the SF-36 version 2 acute (1-week recall) form. Scoring for the form is as follows, based on the manual How to Score version 2 of the SF-36 Health Survey (Ware JE, et al. 2000):

330

331
332
333 I4V-MC-JADV Statistical Analysis Plan Version 1.0 Page 103 European Quality of Life-5 Dimensions-5 Level (EQ-5D-5L) Scores: The European Quality of Life - 5 dimensions (EQ-5D) questionnaire is a widely used, generic questionnaire that assesses health status (The EuroQol Group 1990/Herdman et al. 2011). The questionnaire consists of 2 parts: The first part assesses 5 dimensions: mobility, self-care, usual activities, pain/discomfort and anxiety/depression. The levels of response for mobility, self-care and usual activities are no problems, slight problems, moderate problems, severe problems or unable to. For pain/ discomfort, the levels of response are no pain/discomfort, slight pain/discomfort, moderate pain/discomfort, severe pain/discomfort and extreme pain/discomfort. Lastly, for anxiety/ depression, the levels of response are not anxious or depressed, slightly anxious or depressed, moderately anxious or depressed, severely anxious or depressed, and extremely anxious or depressed. This part of the EQ-5D can be used to generate a health state index score, which is often used to compute quality-adjusted life years (QALY) for utilization in health economic analyses. The health state index score is calculated based on the responses to the 5 dimensions, providing a single value on a scale from less than 0 (where zero is a health state equivalent to death; negative values are valued as worse than dead) to 1 (perfect health), with higher scores indicating better health utility. The second part of the questionnaire consists of a visual analog scale (VAS) on which the patient rates their perceived health state from 0 (worst imaginable health state/the worst health you can imagine) to 100 (best imaginable health state/the best health you can imagine).
334 I4V-MC-JADV Statistical Analysis Plan Version 1.0 Page 104 Health Assessment Questionnaire Disability Index (HAQ-DI) The HAQ-DI is a patient-reported questionnaire that is commonly used in RA to measure disease-associated disability (assessment of physical function). It consists of 24 questions referring to 8 domains: dressing/grooming, arising, eating, walking, hygiene, reach, grip, and activities (Fries et al. 1980, 1982; Ramey et al. 1996). 1. Dressing and grooming (C1. Dress yourself, including tying shoelaces and doing buttons, C2. Shampooing your Hair) Includes 2 component questions, 1 device checkbox (devices used for dressing), 1 help checkbox. 2. Arising (C1. Stand up from straight chair, C2. Get in and out of bed) Includes 2 component questions, 1 device checkbox (built-up or special chair), 1 help checkbox. 3. Eating (C1. Cut your meat, C2. Lift a cup or glass to your mouth, C3. Open a new carton of milk) Includes 3 component questions, 1 device checkbox (built-up or special utensils), 1 help checkbox. 4. Walking (C1. Walk outdoors on flat ground, C2. Climb up five steps) Includes 2 component questions, 4 device checkboxes (cane, walker, crutches, wheelchair), 1 help checkbox. 5. Hygiene (C1. Wash and dry your body, C2. Take a tub bath, C3. Get on and off the toilet) Includes 3 component questions, 4 device checkboxes (raised toilet seat, bathtub seat, bathtub bar, long-handled appliances in bathroom), 1 help checkbox. 6. Reach (C1. Reach and get down a 5 pound object (such as a bag of sugar) from just above your head, C2. Bend down to pick up clothing from the floor) Includes 2 component questions, 1 device checkbox (long-handled appliances for reach), 1 help checkbox. 7. Grip (C1. Open car doors, C2. Open jars which have been previously opened, C3. Turn faucets on and off) Includes 3 component questions, 1 device checkbox (jar opener), 1 help checkbox. 8. Activities (C1. Run errands and shop, C2. Get in and out of a car, C3. Do chores such as vacuuming or yard work) Includes 3 component questions, 1 help checkbox. In order to compute the HAQ-DI (Standard Disability Index) score, the following scores are assigned to the responses: Without any difficulty = 0 With some difficulty = 1 With much difficulty = 2
335 I4V-MC-JADV Statistical Analysis Plan Version 1.0 Page 105 Unable to do = 3. The disability section of the questionnaire scores the patient s self-perception on the degree of difficulty (0 = without any difficulty, 1 = with some difficulty, 2 = with much difficulty, and 3 = unable to do) when dressing and grooming, arising, eating, walking, hygiene, reach, grip, and performing other daily activities. The reported use of special aids or devices and/or the need for assistance of another person to perform these activities is also assessed. The scores for each of the functional domains will be averaged to calculate the functional disability index. Calculating the HAQ-DI: The patient must have a score for at least 6 of the 8 categories. If there are less than six categories completed, a HAQ-DI cannot be computed. A category score is determined from the highest score of the sub-categories, or components, in that category. (For example, in the category EATNG there are three sub-category items. If a patient responds with a 1, 2, and 0, respectively; the category score is 2.) Adjust for use of aids/devices and/or help from another person when indicated : o When there are no aids or devices or help indicated for a category, the category s score is not modified. o When aids or devices or help ARE indicated by the patient, adjust the score for a category by increasing a zero or a one to a two. If a patient's highest score for that sub-category is a two it remains a two, and if a three, it remains a three. o Sum the eight category scores o Divide the sum by the number of categories answered (range 6-8) The scale is not truly continuous but has 25 possible values (i.e., 0, 0.125, 0.250, ).The mapping of the aids or devices to the categories is the following: HAQ-DI Category Dressing and Grooming Arising Eating Walking Hygiene Reach Grip Companion aids or devices item Devices used for dressing (button hook, zipper pull, long handled shoe horn, etc.) Built up or special chair Built up or special utensils Cane, walker, crutches Long handled appliances in bathroom Long handled appliances for reach Jar opener (for jars previously opened) Minimum Clinically Important Differences: The summary and analyses of the HAQ-DI Score will include the number of patients achieving a MCID, which is defined as: MCID 0.22 reduction in HAQ-DI Score. Also, the number of patients having 0.3 reduction in HAQ-DI Score will be assessed.
336 I4V-MC-JADV Statistical Analysis Plan Version 1.0 Page 106 Appendix 3. Framingham 10 Year Risk of Cardiovascular Disease Events The Framingham risk score is a 10-year prediction of cardiovascular disease (CVD) events (D Agostino et al. 2008). The outcome CVD comprises: coronary death, myocardial infarction, coronary insufficiency, angina, ischemic stroke, hemorrhagic stroke, transient ischemic attack, peripheral artery disease, and heart failure. Following predictors will be assessed in the primary model: Age [years] Diabetes [Yes / No] Smoking [Yes / No] Treated and untreated Systolic Blood Pressure [mmhg] Total cholesterol [mg/dl] HDL cholesterol [mg/dl] The following data will be used to calculate the Framingham score: Age - year of birth recorded in the CRF. Diabetes - coded pre-existing condition will be evaluated. Smoking - current tobacco usage recorded in the CRF. Treated and untreated hypertension - the coded concomitant therapy will be evaluated to distinguish between treated and untreated hypertension. Systolic Blood Pressure - vital signs data. Total cholesterol - LAB data. HDL cholesterol - LAB data. The measurements must be entered into the calculation using the units mentioned above. The 10-year risk for women can be calculated as exp(σßx ) and the risk for men is given as exp(σßx ) where ß is the regression coefficient and X is the level (value) for each risk factor. The estimated regression coefficients (Beta) are shown in the following table.
337
338 Leo Document ID = 42130ade-b9b2-43b6-81b5-d8cb48a6df71 Approver: Approval Date & Time: 10-Aug :32:15 GMT Signature meaning: Approved
339 I4V-MC-JADV Statistical Analysis Plan Version 2 Page 1 1. Statistical Analysis Plan: I4V-MC-JADV:A Randomized, Double-Blind, Placebo- and Active-Controlled, Phase 3 Study Evaluating the Efficacy and Safety of Baricitinib in Patients with Moderately to Severely Active Rheumatoid Arthritis Who Have Had an Inadequate Response to Methotrexate Therapy Baricitinib () Rheumatoid Arthritis Study I4V-MC-JADV is a Phase 3, randomized, double-blind, placebo and activecontrolled, parallel group study of baricitinib for the treatment of moderately to severely active rheumatoid arthritis (RA) during a 52-week treatment period in patients with inadequate response to methotrexate therapy. Eli Lilly and Company Indianapolis, Indiana USA Protocol I4V-MC-JADV Phase 3 Statistical Analysis Plan Version 1 electronically signed and approved by Lilly: 10 August 2013 Statistical Analysis Plan Version 2 electronically signed and approved by Lilly on date provided below. Approval Date: 17-Apr-2015 GMT
340 I4V-MC-JADV Statistical Analysis Plan Version 2 Page 2 Section 2. Table of Contents Page 1. Statistical Analysis Plan: I4V-MC-JADV:A Randomized, Double- Blind, Placebo- and Active-Controlled, Phase 3 Study Evaluating the Efficacy and Safety of Baricitinib in Patients with Moderately to Severely Active Rheumatoid Arthritis Who Have Had an Inadequate Response to Methotrexate Therapy Table of Contents Revision History Study Objectives Primary Objective Secondary Objectives Exploratory Objectives Study Design Summary of Study Design Screening Period Parts A and B: 24-week Double-Blind, Placebo- and Active- Controlled Period (Part A) and 28-week Double-Blind, Active-Controlled Period (Part B) Part C: Post-Treatment Follow-Up Period Study Extension Determination of Sample Size Method of Treatment Assignment Treatment Administered Selection and Time of Doses Rescue Therapy A Priori Statistical Methods General Considerations Definition of Baseline and Postbaseline Measures Derived Data Analysis Populations Handling of X-ray Data Adjustment for Covariates Handling of Dropouts or Missing Data Missing Data Imputations Missing Data Imputation for Efficacy and Health Outcome Endpoints...30
341 I4V-MC-JADV Statistical Analysis Plan Version 2 Page Missing Data Imputation for the Structural Progression Endpoint (van der Heijde mtss) Multiple Comparison Patient Disposition Protocol Deviations Patient Characteristics Demographics Baseline Disease Characteristics Previous and Concomitant Medication Historical Diagnoses and Pre-Existing Conditions Treatment Compliance Treatment Exposure Efficacy Analyses Primary Efficacy Analysis Major Secondary Efficacy Analyses Analysis for Other Secondary Efficacy Endpoints Efficacy Analyses in Follow-up Period Testing Strategy to Control the Type I Family-Wise Error Rate Pharmacokinetic/Pharmacodynamic Analysis (PK/PD) Health Outcomes Analyses Diary Data Data Handling and Missing Data Imputation Multiple Imputation (MI) Analysis of Diary Data Analysis of Non-Diary Data Exploratory Analysis Safety Analyses Adverse Events (AEs) Common Adverse Events (AEs) Serious Adverse Events (SAEs) Other Significant Adverse Events Vital Signs and Physical Characteristics Laboratory Measurements Safety in Special Groups and Circumstances, including Adverse Events of Special Interest (AESI) Abnormal Hepatic Tests Myelosuppressive Events Lymphocyte Subset Cell Counts...87
342 I4V-MC-JADV Statistical Analysis Plan Version 2 Page Lipids Effects Renal Function Effects Elevations in Creatine Phosphokinase (CPK) Infections, Including Potential Opportunistic Infections (POIs) Major Adverse Cardiovascular Events (MACE) Malignancies Allergic Reactions/Hypersensitivities Gastrointestinal (GI) Perforations Symptoms of Depression (QIDS-SR16) Other Safety Analyses Subgroup Analysis Interim Analysis and Data Monitoring Unblinding Plan References Appendices...112
343 I4V-MC-JADV Statistical Analysis Plan Version 2 Page 5 Table Table of Contents Page Table JADV.6.1. Imputation Techniques for Various Efficacy and Safety Variables...34 Table JADV.6.2. Categorical Criteria for Abnormal Treatment-Emergent Blood Pressure and Heart Rate Measurement, and Categorical Criteria for Weight Changes for Adults...74 Table JADV.6.3. Common Terminology Criteria for Adverse Events (CTCAEs) to Hepatic Tests...81 Table JADV.6.4. Common Terminology Criteria for Adverse Events (CTCAEs) Related to Myelosuppressive Events...84 Table JADV.6.5. National Cholesterol Education Adult Treatment Panel III (NCEP ATP III) Criteria...90 Table JADV.6.6. Common Terminology Criteria for Adverse Events (CTCAEs) Related to Renal Effects...92 Table JADV.6.7. Common Terminology Criteria for Adverse Events (CTCAE) Related to Elevations in CPK...94 Table JADV.6.8. Quick Inventory of Depressive Symptomatology Self-Rated 16 (QIDS-SR 16 ) Severity of Depressive Symptoms Categories based on the QIDS-SR 16 Total Score...101
344 I4V-MC-JADV Statistical Analysis Plan Version 2 Page 6 Table of Contents Figure Page Figure JADV.5.1. Illustration of study design for Clinical Protocol I4V-MC-JADV Figure JADV.6.1. Illustration of graphical multiple testing procedure within Family 1 for Study JADV Figure JADV.6.2. Illustration of graphical multiple testing procedure within Family 2 for Study JADV....57
345 I4V-MC-JADV Statistical Analysis Plan Version 2 Page 7 Appendix Appendix 1. Appendix 2. Table of Contents Page Algorithm for Determining ACR Response Details of Efficacy and Health Outcomes Measures Appendix 3. Framingham Risk Assessment and Reynolds Risk Assessment: 10- Year Risk of Cardiovascular Disease Events Appendix 4. Appendix 5. Corticosteroids X-ray Missing Data Imputation Plan...137
346 I4V-MC-JADV Statistical Analysis Plan Version 2 Page 8 3. Revision History Version 1 of this Statistical Analysis Plan was approved on 10 August 2013, prior to the first unblinding of the external Data Monitoring Committee (DMC). Version 2 of this Statistical Analysis Plan was approved on the date shown on the header page, prior to unblinding for the primary analysis. Some of the major changes incorporated in Version 2 are as follows: General o o o Provided more detail on safety and efficacy analysis groups and periods Redefined the follow-up population as any patient who entered the follow-up period; added the safety follow-up population Included the list of protocol deviation categories Imputation Methods o o o o Added baseline-observation-carried-forward (BOCF) method for major primary and secondary analyses as a supportive analysis to meet regulatory requests Included component-level imputation for all efficacy measures comprised of or derived from multiple components Included imputation rules for intermediate missed visits Added nonresponder imputation (NRI) as randomized imputation method for x-ray categorical analyses Efficacy Analyses o o o o o o Updated list of other secondary and exploratory objectives Included windowing method for efficacy electronic patient reported outcomes (epro) assessments Included pooling strategy for subgroup and sensitivity analyses Specified use of the Fisher exact test if logistic regression sample requirements aren t satisfied Morning joint stiffness analyses Truncated values to 720 minutes Specified the use of non-parametric summary statistics Work productivity analyses Included employment status in summary
347 I4V-MC-JADV Statistical Analysis Plan Version 2 Page 9 o o o o e-diary data analyses Included analyses for all scheduled visits prior to Week 12 using the primary analysis method X-ray data analyses: Included additional supportive analyses Updated the Gatekeeping Procedure to control for Multiplicity Added tipping point analyses for the continuous major secondary efficacy outcomes Adverse Events (AEs) o o o o o o Specified that percentages and patient-years should be adjusted for gender-specific events Updated the definition for classifying treatment-emergence Specified how serious adverse events (SAEs) are classified as International Conference on Harmonisation (ICH)-defined or protocoldefined Removed hepatic and myelosuppressive events based on preferred terms analyses Revised the methods and terminology in the allergic reaction, major adverse cardiovascular events (MACE), and opportunistic infections special interest sections Added gastrointestinal perforations analyses Common Terminology Criteria for Adverse Events (CTCAE) Criteria o o o o Updated albumin and analytes related to myelosuppressive events to use specific threshold values instead of the lower limit of normal (LLN) to distinguish normal versus Grade 1 Updated lipids to only use National Cholesterol Education Program (NCEP) criteria Removed estimated glomerular filtration rate (egfr) grading Added creatine phosphokinase (CPK) grading.
348 I4V-MC-JADV Statistical Analysis Plan Version 2 Page Study Objectives The following objectives are replicated from Amendment (c) of Study I4V-MC-JADV (JADV) protocol, dated 19 July Primary Objective The primary objective of this study is to determine whether baricitinib 4 mg once daily (QD) is superior to placebo in the treatment of patients with moderately to severely active rheumatoid arthritis (RA) despite methotrexate (MTX) treatment (i.e., MTX-IR), as assessed by the proportion of patients achieving the American College of Rheumatology (ACR) criteria (ACR20) at Week Secondary Objectives The major secondary objectives of the study which are controlled under graphical testing procedure for multiplicity are to evaluate the efficacy of baricitinib versus placebo or adalimumab as assessed by: change from baseline to Week 24 in structural joint damage as measured by modified Total Sharp Score (mtss; van der Heijde [2000]) method compared to placebo change from baseline to Week 12 in Health Assessment Questionnaire- Disability Index (HAQ-DI) score compared to placebo change from baseline to Week 12 in Disease Activity Score modified to include the 28 diarthrodial joint count (DAS28)-high-sensitivity C-reactive protein (hscrp) compared to placebo proportion of patients achieving an Simplified Disease Activity Index (SDAI) score 3.3 (American College of Rheumatology/ European League Against Rheumatism [ACR/EULAR] remission Index-based definition) at Week 12 compared to placebo proportion of patients achieving ACR20 response at Week 12 compared to adalimumab mean duration of morning joint stiffness in the 7 days prior to Week 12 compared to placebo as collected in electronic diaries change from baseline to Week 12 in DAS28-hsCRP compared to adalimumab mean severity of morning joint stiffness in the 7 days prior to Week 12 compared to placebo as collected in electronic diaries mean Worst Tiredness numeric rating scale (NRS) in the 7 days prior to Week 12 compared to placebo as collected in electronic diaries mean Worst Pain NRS in the 7 days prior to Week 12 compared to placebo as collected in electronic diaries.
349 I4V-MC-JADV Statistical Analysis Plan Version 2 Page 11 The other secondary objectives of the study are to evaluate the efficacy of baricitinib 4 mg compared to placebo as assessed by: proportion of patients achieving DAS28-hsCRP 3.2 and DAS28-hsCRP <2.6 at Week 12, Week 24, and Week 52 proportion of patients achieving HAQ-DI improvement from baseline 0.22 and 0.3 at Week 12, Week 24, and Week 52 change from baseline to Week 16 and Week 52 in structural joint damage as measured by mtss proportion of patients with mtss change 0 from baseline at Week 16, Week 24, and Week 52 change from baseline to Week 16, Week 24, and Week 52 in joint space narrowing and bone erosion scores proportion of patients achieving ACR20 at Week 24 and Week 52 proportion of patients achieving ACR50 at Week 12, Week 24, and Week 52 proportion of patients achieving ACR70 at Week 12, Week 24, and Week 52 percentage of change from baseline to Week 12, Week 24, and Week 52 in individual components of the ACR Core Set including tender joint count based on 68 joint counts (TJC68), swollen joint count based on 66 joint counts (SJC66), patient s assessment of arthritis pain, patient s global assessment of disease activity, physician s global assessment of disease activity, HAQ-DI and hscrp change from baseline through Week 52 in DAS28-hsCRP change from baseline through Week 52 in DAS28-erythrocyte sedimentation rate (ESR) proportion of patients achieving DAS28-ESR 3.2 and DAS28-ESR <2.6 at Week 12, Week 24, and Week 52 change from baseline to Week 24 and Week 52 in HAQ-DI score change from baseline to Week 12, Week 24, and Week 52 in Clinical Disease Activity Index (CDAI) score change from baseline to Week 12, Week 24, and Week 52 in SDAI score proportion of patients achieving a CDAI score 2.8 at Week 12, Week 24, and Week 52 proportion of patients achieving an SDAI score 3.3 at Week 24 and Week 52 proportion of patients achieving ACR/EULAR remission according to the Boolean-based definition at Week 12, Week 24 and Week 52
350 I4V-MC-JADV Statistical Analysis Plan Version 2 Page 12 proportion of patients achieving moderate and good EULAR responses based on the 28-joint DAS at Week 12, Week 24, and Week 52 evaluation of hybrid ACR (bounded) response at Week 12, Week 24 and Week 52 change from baseline to Week 12, Week 24, and Week 52 in Functional Assessment of Chronic Illness Therapy Fatigue (FACIT-F) scale scores change from baseline to Week 12, Week 24, and Week 52 (at Weeks 12, 24, and 52) in each scale and the summary scores for the Mental Component Score (MCS) and Physical Component Score (PCS) of the Medical Outcomes Study 36-Item Short Form Health Survey Version 2 Acute (SF-36v2 Acute) change from baseline to Week 12, Week 24, and Week 52 in European Quality of Life-5 Dimensions-5 Level (EQ-5D-5L) scores change from baseline to Week12, Week 24, and Week 52 in Work Productivity and Activity Impairment-Rheumatoid Arthritis (WPAI-RA) scores evaluation of healthcare resource utilization through Week 52 change from baseline to Week 12, Week 24, and Week 52 in duration of morning joint stiffness assessed at the visit using an electronic patientreported outcomes (epro) tablet change from baseline to Week 12, Week 24, and Week 52 in Worst Tiredness NRS assessed at the visit using an epro tablet change from baseline to Week 12, Week 24, and Week 52 in Worst Pain NRS assessed at the visit using an epro tablet To characterize the pharmacokinetics (PK) of baricitinib, determine the magnitude of within- and between-patient variability, and identify the potential factors that may have an impact on the PK of baricitinib. To characterize the baricitinib exposure-response time course for key efficacy and safety endpoints and to identify potential factors that may have an impact on these efficacy or safety endpoints of baricitinib. In addition to the aforementioned other secondary objectives, the following analyses are also evaluated: the proportion of patients achieving a SDAI score 11 at Week 12, Week 24 and Week 52 the proportion of patients achieving a CDAI score 10 at Week 12, Week 24 and Week 52 the proportion of patients achieving improvement from baseline in FACIT-F score 3.56 at Week 12, Week 24 and Week 52
351 I4V-MC-JADV Statistical Analysis Plan Version 2 Page 13 change from baseline to Week 12, Week 24 and Week 52 and shift analyses in rheumatoid factor (RF) and anti-citrullinated peptide antibody (ACPA) Exploratory Objectives The exploratory objective of this study is to compare baricitinib to adalimumab as regards to various efficacy and health outcomes measures.
352 I4V-MC-JADV Statistical Analysis Plan Version 2 Page Study Design 5.1. Summary of Study Design Study JADV is a 52-week, Phase 3, multicenter, randomized, double-blind, double-dummy, placebo- and active-controlled, parallel-group, outpatient study comparing the efficacy of baricitinib versus placebo on signs and symptoms, function, remission, and structural progression in patients with moderately to severely active RA who have had an insufficient response to MTX and who have never been treated with a biologic disease-modifying antirheumatic drug (DMARD) (i.e., MTX-IR patients). The study will include a noninferiority comparison of baricitinib versus adalimumab on signs and symptoms. Baricitinib will be administered as a 4-mg tablet QD. The dose for patients with renal impairment, defined as egfr <60 ml/min/1.73 m2, will be 2 mg baricitinib QD. Patients not assigned to the baricitinib treatment arm will receive either a 4-mg or 2-mg matching placebo tablet. Adalimumab (40 mg) will be administered by subcutaneous (SC) injection biweekly (i.e., every 2 weeks). Patients not assigned to adalimumab will receive a matching placebo SC injection biweekly. Planned enrollment is approximately 1280 randomized patients. After the Week 52 study visit, eligible patients may proceed to a separate extension study (Study I4V-MC-JADY [JADY]) lasting for up to 4 years or to the post-treatment follow-up period of this study (Part C). Study JADV will consist of 4 parts: Screening: Screening period lasting from 3 to 42 days prior to Visit 2 (Week 0) Part A: double-blind, placebo- and active-controlled period from Week 0 through Week 24 Part B: double-blind, active-controlled period from the end of Week 24 through Week 52 Part C: post-treatment follow-up period (28 days). Figure JADV.5.1. illustrates the study design.
353
354 I4V-MC-JADV Statistical Analysis Plan Version 2 Page 16 Patients randomized to placebo will be administered a placebo tablet QD starting at Week 0 (Visit 2) through Week 24 (the morning of Visit 11). At Week 24, patients will be administered baricitinib 4 mg QD (or 2 mg baricitinib QD if screening egfr <60 ml/min/1.73 m2) through Week 52 (Visit 15). Patients in this treatment arm will also receive a placebo SC injection biweekly starting at Week 0 (Visit 2) through Week 50. Patients randomized to adalimumab will be administered adalimumab (40 mg) by SC injection biweekly starting at Week 0 (Visit 2) through Week 50 and a placebo tablet QD through Week 52 (the morning of Visit 15). The last SC injection will be administered during Week 50 because patients receiving adalimumab have the option to switch to baricitinib 4 mg QD (or 2 mg QD depending on egfr) if they choose to participate in the extension study. Scheduling the last SC injection at Week 50 ensures approximately a 2-week interval from the last SC injection to the start of baricitinib dosing. Patients will be eligible for rescue treatment beginning at Week 16 (Visit 9). Details of the rescue are listed in Section Patients will remain on background MTX, nonsteroidal anti-inflammatory drugs (NSAIDs), analgesics, and/or corticosteroids (see Protocol Section 9.8 for guidelines on adjustments to concomitant therapy). Patients will be eligible for rescue treatment starting at Week 16 (Visit 9). Patients will take their assigned doses of study drugs as directed. The primary efficacy endpoint will be at Week 12, and the final visit during Part A will be at Week 24. The final visit during Part B will be at Week 52 (Visit 15). The structural joint damage endpoints will be assessed at Week 16, Week 24, and Week 52. Patients who complete the Week 52 study visit may be eligible for inclusion in extension Study JADY. Patients who do not enroll in Study JADY will continue to Part C of this study. Patients who discontinue from the study prior to Week 52 for any reason will have an Early Termination Visit performed, including x-rays. The Early Termination Visit should be performed as soon as logistically possible because this visit includes assessment of safety and PK data. Infrequently, patient and investigator availability may be such that the Early Termination Visit and the Follow-up Visit may occur on or about the same date. In this instance, the visits may be combined and should occur approximately 28 days after the last dose of investigational product. All activities required for the Early Termination Visit should be completed according to the schedule of assessments (Protocol Attachment 1) Part C: Post-Treatment Follow-Up Period Patients who discontinue the study or patients who complete Parts A and B of the study and do not enroll in the extension Study JADY will have a Follow-Up Visit (Visit 801) approximately 28 days after the last dose of study drug Study Extension Patients who complete this study through Visit 15 will be eligible to participate in Study JADY, if enrollment criteria for Study JADY are met. These patients will not participate in Part C.
355 I4V-MC-JADV Statistical Analysis Plan Version 2 Page Determination of Sample Size Approximately 1280 patients (480 in the baricitinib group, 480 in the placebo group, and 320 in the adalimumab group) will provide: >95% power to detect a difference between the baricitinib and placebo treatment groups in ACR20 response rate (assuming 60% versus 35%) at Week 12 based on a 2-sided chi-square test at a significance level of approximately 94% power to detect a difference in mtss between the baricitinib and placebo treatment groups for an effect size of 0.25 (i.e., difference in means divided by common standard deviation [SD]), based on a 2-sided t-test at a significance level of This assumes 90% of randomized patients will have x-ray data available for the mtss analysis at Week 24. If the effect size in Study JADV was at least 0.21 (van der Heijde et al. 2013), then power of the mtss analysis is at least 85%. approximately 93% power for the noninferiority analysis of ACR20 response rate at Week 12 between the baricitinib and adalimumab treatment groups, where a response rate of 60% for baricitinib and adalimumab and a 1-sided significance level of are assumed. This power calculation adopted a fixed noninferiority margin of 12%. There is growing support for this margin in recent clinical trials conducted in the MTX-IR patient population (Schiff et al. 2012). These sample size and power estimates were obtained from nquery Advisor Method of Treatment Assignment Patients who meet all criteria for enrollment will be randomized in a 3:3:2 ratio (baricitinib:placebo:adalimumab) to double-blind treatment at Week 0 (Visit 2). Randomization will be stratified by (geographical) region and joint erosion status (1-2 joint erosions plus seropositivity versus at least 3 joint erosions). Assignment to treatment groups will be determined by a computer-generated random sequence using the interactive voice-response system (IVRS). The IVRS will be used to assign packages of baricitinib/placebo and packages of prefilled syringes of adalimumab/placebo for SC injection containing double-blind investigational product to each patient. Each package of clinical trial material will supply sufficient medication for the number of weeks between visits. Site personnel will confirm that they have located the correct package by entering a confirmation number found on the packages of investigational product into the IVRS before dispensing the package to the patient. This study will be conducted internationally in many sites. Table JADV.5.1 explains how region will be defined for the statistical analyses and summaries.
356 I4V-MC-JADV Statistical Analysis Plan Version 2 Page 18 Table JADV.5.1. Countries and their Geographical Regions Geographical Region United States and Canada Central and South America and Mexico Eastern Europe Western Europe Asia ex-japan Japan Rest of World Country or Countries United States, Canada Argentina, Mexico Croatia, Czech Republic, Hungary, Latvia, Lithuania, Poland, Romania, Slovakia, Slovenia Belgium, France, Germany, Greece, Netherlands, Portugal, Spain, Switzerland, United Kingdom China, South Korea, Taiwan Japan Australia, Israel, Russia, South Africa Treatment Administered This study involves a comparison of baricitinib 4 mg administered orally QD, placebo administered orally QD and SC biweekly, and adalimumab 40 mg administered by SC injection biweekly. Patients will continue to take background MTX therapy during the course of the study. Table JADV.5.2 shows the treatment regimens. Table JADV.5.2. Treatment Regimens (on a background of MTX therapy) Treatment Group Baricitinib treatment Placebo comparator Active comparator (adalimumab) Treatments Administered during Part A (Patients Not Rescued) Baricitinib 4 mgb oral once-daily tablet Adalimumab placebo by SC injection biweekly Baricitinib placebo oral once-daily tablet Adalimumab placebo by SC injection biweekly Baricitinib placebo oral once-daily tablet Adalimumab 40 mg by SC injection biweekly Treatments Administered during Part B (Patients Not Rescued) Baricitinib 4 mgb oral once-daily tablet Adalimumab placebo by SC injection biweekly Baricitinib 4 mgb oral once-daily tablet Adalimumab placebo by SC injection biweekly Baricitinib placebo oral once-daily tablet Adalimumab 40 mg by SC injection biweekly Treatments Administered during Parts A and B (after Rescue a ) Baricitinib 4 mgb oral once-daily tablet Baricitinib 4 mgb oral once-daily tablet Baricitinib 4 mgb oral once-daily tablet Abbreviation: SC = subcutaneous. a See Section for details of rescue medication. b The dose for patients with renal impairment, defined as estimated glomerular filtration rate <60 ml/min/1.73 m2, will be 2 mg once daily. The investigator or his/her designee will be responsible for explaining the correct use of both the oral and injectable investigational agent(s) to the patient, verifying that instructions are followed properly, maintaining accurate records of investigational product dispensing and collection, and returning all unused medication to Eli Lilly and Company (Lilly) or its designee at the end of the study.
357 I4V-MC-JADV Statistical Analysis Plan Version 2 Page 19 Injections of investigational product should be administered by the site personnel, with instructions to the patient, at Visit 2, and the patient should be supervised while self-administering injections at Visits 4 and 5. In some cases, sites may destroy the material if, during the investigator site selection, the evaluator has verified and documented that the site has appropriate facilities and written procedures to dispose of clinical trial materials. Patients will be instructed to contact the investigator as soon as possible if they have a complaint or problem with the investigational product so that the situation can be assessed. All patients will be required to be on background MTX treatment at baseline. Other background therapies, including NSAIDs and low dose oral corticosteroids, are permitted during the study for patients who are on stable doses (as defined in protocol) of these treatments at baseline. Patients should remain on the same dose of MTX throughout the study; however, the MTX dose may be adjusted for safety reasons Selection and Time of Doses Each patient will receive blinded investigational product (baricitinib 4 mg or baricitinib placebo tablets orally QD and adalimumab 40 mg or adalimumab placebo biweekly by SC injection) beginning on the day of Visit 2 (Week 0). Baricitinib 4-mg and baricitinib placebo oral dosing will continue daily through Visit 15 (Week 52) and biweekly injections of adalimumab 40 mg or adalimumab placebo will continue biweekly through Week 50. At Visit 11 (Week 24), all patients randomized to placebo will be reassigned to baricitinib 4 mg while preserving the double-blind nature of the study. Injectable investigational product should be administered at approximately the same time of day, as much as possible. For injections not administered on the scheduled day of the week, the missed dose should be administered within 3 days of the scheduled day during study Weeks 1 to 12 and within 5 days of the scheduled day during study Weeks 13 to 52. Dates of subsequent study visits should not be modified according to this delay. As noted in Section 5.3.1, injections of investigational product should be administered by site personnel at Visit 2, and the patient should be supervised while self-administering injections at Visits 4 and 5. Thereafter, the patient will self-administer SC injections at home biweekly when no study visit is scheduled (i.e., Weeks 6, 10, 18, 22, 26, 30, 34, 36, 38, 42, 44, 46, 48, and 50). On weeks when study visits are scheduled (i.e., Weeks 8, 12, 14, 16, 20, 24, 28, 32, and 40), the patient should not administer his/her SC injection at home but should bring the study medication to the clinic for administration during the study visit. This will allow sites to administer rescue dosing when required. Oral investigational product should be taken at home at approximately the same time each day, as much as possible, with the exception of the following visits, to allow for the proper timing of PK sample collection and to ensure that efficacy assessments are made at trough PK levels:
358 I4V-MC-JADV Statistical Analysis Plan Version 2 Page 20 At Visit 2 (Week 0), patients will take their investigational product in the clinic, to allow for PK samples to be taken at 15 and 60 minutes postdose. For Visits 7 (Week 12), Visit 9 (Week 16), 11 (Week 24), 13 (Week 32), and 15 (Week 52), patients will be asked not to take their investigational product prior to visiting the clinic, to allow efficacy assessments to be made prior to investigational product dosing and a predose PK sample to be taken at some of these selected visits (See Protocol Attachment 1) Rescue Therapy In consideration of the disease severity, all patients in Study JADV will be given rescue therapy starting at Week 16 if they are determined to be nonresponders. Nonresponse at Week 16 is defined as lack of improvement of at least 20% in both tender joint counts (TJC) and swollen joint counts (SJC) at both Week 14 and Week 16 compared to baseline. At Week 16, the IVRS will assign rescue treatment based on TJC and SJC values. Starting at Week 16, patients who are nonresponders and who were randomized to placebo will be rescued with baricitinib 4 mg. To maintain study integrity through investigational product blinding, patients who were originally randomized to baricitinib will continue to receive baricitinib, with appropriate ongoing assessment of response and use of concomitant medication as needed. Patients who are nonresponders and who were randomized to adalimumab will be reassigned to baricitinib as rescue therapy. After Week 16, rescue therapy will be given to patients at the discretion of the investigator based on TJCs and SJCs. Once a patient has been rescued, new NSAIDs, corticosteroids, and/or analgesics may be added or doses of ongoing concomitant NSAIDs, corticosteroids, and/or analgesics may be increased at the discretion of the investigator. A patient may only be rescued once. If a patient continues to meet nonresponse criteria for 4 weeks after rescue or at any time point thereafter, that patient should be discontinued from the study. Patients initially randomized to placebo and not rescued before Week 24 will be reassigned to receive baricitinib 4 mg at Week 24 in order to limit exposure to inactive treatment. Nonresponders who are rescued will receive an oral tablet daily, but no biweekly SC injection, for the remainder of the study. Rescue therapy will be administered through Week 52.
359 I4V-MC-JADV Statistical Analysis Plan Version 2 Page A Priori Statistical Methods 6.1. General Considerations This plan describes a priori statistical analyses for efficacy, health outcomes, and safety that will be performed. Statistical analysis of this study will be the responsibility of Lilly. For continuous measures, summary statistics will include the sample size, means, SDs, medians, minimums, 1st quartiles (Q1), 3rd quartiles (Q3) and maximums. For categorical measures, summary statistics will include the sample size, frequency counts, and percentages. Statistical tests of treatment effects will be performed at a 2-sided significance level of 0.05, unless otherwise stated (e.g., graphical multiple testing analyses; Section ). All p-values will be rounded up to 3 decimal places. For example, any p-value strictly greater than and less than or equal to 0.05 will be displayed as This guarantees that on any printed statistical output, the unrounded p-value will always be less than or equal to the displayed p-value. A displayed p-value of will always be understood to mean Likewise, any p-value displayed as will be understood to mean >0.999 and 1. Lilly will use both an effectiveness estimand (that is, the relative effect attributable to the originally randomized treatments, baricitinib, placebo, and adalimumab, at the primary time point in all treated patients, even if patients change treatment) and an efficacy estimand (that is, the relative effect attributable to the originally randomized treatments, baricitinib, placebo, and adalimumab, at the primary time point in all treated patients if taken as directed) to evaluate efficacy measures at the primary time point of Week 24 for Study JADV. All primary and key (gated) secondary endpoints related to improvements in symptomatic features (that is, RA signs and symptoms, physical function, and patient-reported outcomes) are assessed prior to patients having the option to receive rescue therapy. Therefore, the only practical difference between the efficacy and effectiveness estimands for these endpoints concerns assumptions about patient response after permanent discontinuation of study drug or from the study prior to the analysis time point. For outcome measures defined by response/nonresponse categories, NRI is utilized with both estimands. This approach considers study discontinuation to be a negative outcome that outweighs any potential benefit gained from hypothetical continued treatment or actual treatment and therefore fits in either the efficacy or effectiveness frameworks. For continuous outcome measures that quantify a treatment effect, Lilly utilizes modified baseline observation carried forward (mbocf). At the time of discontinuation, some patients may have positive outcomes (or even a range of effects) and discontinue for reasons unrelated to treatment; it seems reasonable to reflect those outcomes in the expected effects attributable to the originally randomized treatments. Other patients may discontinue because they were not seeing benefit, in which case their outcomes at the time of discontinuation should reflect their level of expected efficacy (or lack thereof) attributable to the originally randomized treatments. Finally,
360 I4V-MC-JADV Statistical Analysis Plan Version 2 Page 22 other patients may see positive outcomes (or even a range of effects) but be unable to tolerate or continue receiving the medicine because of AEs. Because the latter is a negative outcome in spite of potentially positive efficacy data, these patients will be considered to have received no improvement in their symptomatic features; therefore, the patient s baseline observation is carried forward for analysis. These approaches are reasonable for use with both the efficacy and effectiveness estimands when the inference concerns the originally randomized treatments. Therefore, in each of the situations whereby patients may not take a complete course of study drug between the time of randomization and the primary time point, an approach that is appropriate in this setting can be defined. For Study JADV, Lilly plans to use this described approach for evaluating the primary endpoints and all gated secondary endpoints. Additional sensitivity analyses, described in Section and Section 6.12, are planned to investigate the robustness of this approach. Treatment comparisons of categorical efficacy variables (including proportions of patients achieving ACR20) will be made using a logistic regression analysis with region, baseline joint erosion status (1-2 joint erosions plus seropositivity vs at least 3 joint erosions), and treatment group in the model. In general, the p-value and 100*(1-alpha)% confidence interval (CI) for the odds ratio (where alpha denotes the significance level for the specific hypothesis of interest) from the logistic regression model are used for primary statistical inference, except that the noninferiority comparison of baricitinib to adalimumab in ACR20 response at Week 12 is tested using the Newcombe-Wilson method (Newcombe 1998) without continuity correction. The difference in percentages and 100*(1-alpha)% CI of the difference in percentages using the Newcombe-Wilson method without continuity correction are used for descriptive purposes unless otherwise specified. When logistic regression sample size requirements (<5 responders in any category for any factor) are not met, the p-value from the Fisher exact test is produced instead of the odds ratio and 95% CI. Treatment-by-region interaction will be added to the logistic regression model of the primary and key secondary categorical variables as a sensitivity analysis. If this interaction is significant at a 2-sided 0.10 level, further analytical inspection will be used to assess whether the interaction is quantitative (i.e., the treatment effect is consistent in direction across all regions but not size of treatment effect) or qualitative (the treatment is beneficial for some but not all regions). Additional sensitivity analyses will be performed by adding treatment-by-joint erosion status interaction to the logistic regression model. Treatment comparisons of continuous efficacy and health outcomes variables will be made using analysis of covariance (ANCOVA) with region, baseline joint erosion status, treatment group, and baseline value in the model. Type III sums of squares for the least-squares (LS) means will be used for the statistical comparison between treatment groups. The LS mean difference, standard error, p-value and 100*(1-alpha)% CI will also be reported. Sensitivity analysis will be performed for key secondary continuous variables by adding the treatment-by-baseline interaction to the ANCOVA model. If this interaction is significant at a 2-sided 0.1 level, then additional analyses will be performed by subgroups defined by the median of the baseline values using the same terms in the model (with the exception of baseline value). Additional sensitivity analyses will be performed by adding treatment-by-region interaction to the ANCOVA model,
361 I4V-MC-JADV Statistical Analysis Plan Version 2 Page 23 and adding treatment-by-joint erosion status interaction to the ANCOVA model. If this interaction is significant at a 2-sided 0.1 level, further inspection will be used to assess whether the interaction is quantitative or qualitative. When a restricted maximum likelihood-based mixed model for repeated measures (MMRM) is used (as a sensitivity model for applicable key secondary continuous variables [see Section ]), the model will include treatment, region, joint erosion status (1-2 joint erosions plus seropositivity vs at least 3 joint erosions), visit, treatment-by-visit-interaction as fixed categorical effects; baseline score and baseline score-by-visit-interaction as fixed continuous effects to estimate change from baseline across postbaseline visits. An unstructured covariance structure will be used to model the between- and within-patient errors. If this analysis fails to converge, other structures will be tested such as heterogeneous autoregressive [ARH(1)], heterogeneous compound symmetry (CSH) or heterogeneous Toeplitz (TOEPH). The variance-covariance structure that results in the smallest AIC will be used. The Kenward-Roger method will be used to estimate the denominator degrees of freedom. Type III sums of squares for the LS means will be used for the statistical comparison. The LS mean difference, standard error, p-value and CIs will also be reported. Treatment group comparisons versus placebo at Week 12 and other visits will be tested using the t-test obtained from the MMRM results. The Fisher exact test will be used to perform pairwise comparisons for AEs, discontinuations, and other categorical safety data among treatment groups. Change from baseline in continuous vital signs, physical characteristics, and other continuous safety variables, including laboratory variables will be analyzed using an ANCOVA model with explanatory terms for treatment and the baseline value as a covariate. Type 3 sums of squares will be used. The significance of within-treatment group changes from baseline will be evaluated by testing whether the treatment group LS mean changes from baseline are different from zero using a t-statistic; the standard error for the LS mean change will also be displayed. Differences in LS means will be displayed with the p-value associated with the LS mean comparison to placebo and a 95% CI on the LS mean difference. In addition to the LS means for each group, the within-group p-value, the SD, minimum, Q1, median, Q3, and maximum will be displayed. Data collected at early termination visits will be mapped to the next scheduled visit number for that patient. There will be two planned database locks for this study, one when all patients have completed Part A and a final one at end of the study. Refer to Section 7 for a description of the unblinding plan for this study. The following is a description of the analysis groups planned for this study. Refer to Section for the definitions of analysis populations used for the efficacy and safety analyses.
362 I4V-MC-JADV Statistical Analysis Plan Version 2 Page 24 Analysis Groups for the Efficacy and Health Outcomes Data Summaries of efficacy and health outcomes data for the Week 0 to Week 12 period, the Week 0 to Week 24 period with data up to rescue, and the Week 0 to Week 52 period with data up to rescue/switch will be presented according to the following treatment groups: Placebo: patients randomized to placebo excluding data obtained after the receipt of baricitinib 4 mg, either as rescue therapy or according to the protocol-designated switch to baricitinib 4 mg, Baricitinib 4 mg: patients randomized to baricitinib 4 mg excluding data obtained after the receipt of rescue therapy, Adalimumab: patients randomized to adalimumab excluding data obtained after the receipt of rescue therapy. Summaries of efficacy and health outcomes data for patients who are rescued/switched will be presented according to the following treatment groups: Placebo (rescued): patients randomized to placebo who were rescued on or before Week 24, Placebo (switched, not rescued): patients switched to baricitinib from placebo at Week 24 and not rescued after the initiation of baricitinib, Placebo (switched, rescued): patients switched to baricitinib from placebo at Week 24 and rescued after initiation of baricitinib, Baricitinib (rescued): patients randomized to baricitinib who were rescued, Adalimumab (rescued): patients randomized to adalimumab who were rescued. Analysis Groups for the Safety Data Summaries of safety data for the Week 0 to Week 12 period and the Week 0 to Week 24 period with data up to rescue will be presented according to the following treatment groups: Baricitinib 4 mg: patients randomized to baricitinib excluding data obtained after the receipt of rescue therapy, Placebo: patients randomized to placebo using data obtained prior to the receipt of baricitinib, either as rescue therapy or according to the protocoldesignated switch to baricitinib, Adalimumab: patients randomized to adalimumab excluding data obtained after the receipt of rescue therapy. Summaries of safety data for the Week 0 through Week 52 period with data up to rescue for patients as randomized to baricitinib or adalimumab will be presented according to the following treatment groups:
363 I4V-MC-JADV Statistical Analysis Plan Version 2 Page 25 Baricitinib 4 mg: patients randomized to baricitinib excluding data obtained after the receipt of rescue therapy, Adalimumab: patients randomized to adalimumab excluding data obtained after the receipt of rescue therapy. Summaries of safety data for the Week 0 to Week 52 period (including data after rescue/switch), considering all baricitinib exposures, will be presented according to the following groups: Placebo (rescued): patients randomized to placebo who were rescued on or before Week 24 (using data collected after initiation of baricitinib), Placebo (switched, not rescued): patients switched to baricitinib from placebo at Week 24 and not rescued after initiation of baricitinib (using data collected after initiation of baricitinib), Placebo (switched, rescued): patients switched to baricitinib from placebo at Week 24 and rescued after initiation of baricitinib (using data collected after initiation of baricitinib), Baricitinib (rescued): patients randomized to baricitinib who were rescued (using data collected after initiation of baricitinib), Adalimumab (rescued): patients randomized to adalimumab who were rescued (using data collected after initiation of baricitinib), All baricitinib exposures across all periods, regardless of initial treatment, use of rescue treatment, or protocol-defined switch of assigned treatment, i.e., pooled across all cohorts who received baricitinib (using data collected after initiation of baricitinib). Patients who have impaired renal function who are randomized to baricitinib 4 mg will be given a 2 mg dose but analyzed in the baricitinib 4 mg group. Additional analyses for selected efficacy and safety endpoints will use this subgroup of patients (see Section 6.16 [Safety Analyses] and Section 6.17 [Subgroup Analyses]). Summaries of efficacy and safety data collected during the post-treatment follow-up period (Part C) will be presented separately from the summaries for Parts A and B and according to the following cohorts: Placebo (not rescued): patients randomized to placebo who discontinued on or before Week 24 and had never been rescued at the time of discontinuation, Placebo (rescued): patients randomized to placebo who were rescued on or before Week 24, Placebo (switched, not rescued): patients switched to baricitinib from placebo and not rescued after initiation of baricitinib, Placebo (switched, rescued): patients switched to baricitinib from placebo and rescued after initiation of baricitinib,
364 I4V-MC-JADV Statistical Analysis Plan Version 2 Page 26 Baricitinib (not rescued): patients randomized to baricitinib who were never rescued, Baricitinib (rescued): patients randomized to baricitinib who were rescued, Adalimumab (not rescued): patients randomized to adalimumab who were never rescued, Adalimumab (rescued): patients randomized to adalimumab who were rescued, All baricitinib exposures across all periods, regardless of initial treatment, use of rescue treatment, or protocol-defined switch of assigned treatment, i.e., pooled across all cohorts who received baricitinib prior to entering Part C Definition of Baseline and Postbaseline Measures The baseline period is defined as Visit 1 through pre-dose (expected at Visit 2). The treatment period for the blinded placebo- and active-controlled period (Part A) starts after the first study drug administration at Visit 2 (Week 0) and ends on the date of Visit 11 (Week 24) or an early discontinuation visit before Visit 11 (Week 24). The treatment period for the blinded activecontrolled period (Part B) starts after the first study drug administration at Visit 11 (Week 24) and ends on the date of Visit 15 (Week 52) or an early discontinuation visit in the treatment period. The post-treatment follow-up period (Part C) starts after the date of Visit 15 (Week 52) or an early discontinuation visit and lasts until the follow-up visit, which occurs approximately 28 days following the last dose of the study drug, for patients who do not enter long-term extension Study JADY. The baseline value for the efficacy analyses of the treatment period, including postrescue analyses, is defined as the last non-missing measurement on or prior to the date of first study drug administration (expected at Week 0, Visit 2), regardless of rescue status. If the physician s global assessment of disease activity score is recorded 5 days after the date of first study drug administration, it will still be used as the baseline value. Postbaseline measurements are collected after study drug administration (expected at Week 0, Visit 2) through Week 52 (Visit 15) or early discontinuation visit. For data collected in the epro tablet related to efficacy assessments, unscheduled postbaseline visits that fall within the visit windows defined by Lilly will be summarized in the by-visit analyses if there is no scheduled visit available. That is, a +4 day window is applied to Visit 3 (Week 1) and Visit 4 (Week 2), and a +7 day window is applied to Visits 5 through 15 (Weeks 4 through 52) within the same treatment period (and while receiving the same treatment). For example, data should not be pulled across study periods for placebo group, and data cannot be pulled from postrescue visits to previous nonrescued visits. If there is more than one unscheduled visit within the defined visit window and no scheduled visit available, the unscheduled visit closest to the scheduled visit date will be used. The baseline value for the safety analysis of the treatment period until rescue, protocol-defined switch, or end of study (if patients are not rescued to baricitinib 4 mg) is defined as the last non-
365 I4V-MC-JADV Statistical Analysis Plan Version 2 Page 27 missing measurement on or prior to the date of first study drug administration (expected at Week 0, Visit 2). The baseline value for the safety postrescue/switch period for patients randomized to placebo or adalimumab who are rescued/switched is defined as the last nonmissing measurement obtained prior to initiation of the new treatment (baricitinib). The baseline value for the safety analyses of the postrescue period for patients randomized to baricitinib 4 mg who are rescued is the same as their treatment-period baseline. Postbaseline measurements for rescued/switched patients are collected after the rescue/switch visit through Week 52 (Visit 15) or an early discontinuation visit. The baseline value for the efficacy analysis of the post-treatment follow-up period is defined as the last nonmissing assessment at the time of Week 52 (Visit 15) or early discontinuation visit. The baseline value for the safety analysis of the post-treatment follow-up period is defined as the last non-missing assessment on or prior to Week 52 (Visit 15) or early discontinuation visit. These baseline and postbaseline definitions will be considered for safety, health outcomes, and efficacy analyses and detailed information is found in Sections 6.12 to Derived Data Age (year) = (number of months between first dose date and July of birth year) / 12, and then truncated to a whole-year (integer) age Age group (<65 years old, 65 years old) Age group (<75 years old, 75 years old) Age group (<85 years old, 85 years old) Age group (<65 years old, 65 <75 years old, 75 <85 years old, 85 years old) Body mass index (kg/m 2 ) = Weight (kg)/((height (cm)/100)**2) Duration of RA from onset (years) = [(Date of first dose Date of RA onset) +1]/ Date of RA onset is defined as the date when the patient had symptoms of RA. If year of onset is missing, duration of RA from onset will be set as missing. Otherwise, unknown month will be taken as January, and unknown day will be taken as 01. The duration of RA from onset will be rounded to one decimal place. Duration of RA from diagnosis (years) = [(Date of first dose Date of RA diagnosis)+1]/ Date of RA diagnosis is defined as the date when the patient was diagnosed with RA. If year of diagnosis is missing, duration of RA from diagnosis will be set as missing. Otherwise, unknown month will be taken as January, and unknown day will be taken as 01. The duration of RA from diagnosis will be rounded to one decimal place. Duration from onset of RA (years): 0 to <1; 1 to <5; 5
366 I4V-MC-JADV Statistical Analysis Plan Version 2 Page 28 Baseline and postbaseline Framingham 10-year risk of cardiovascular disease (CVD) events category for men and women aged 30 and 74 years with no prior history of CVD will be derived [Framingham percentage risk < 10% (low risk), 10% to 20% (intermediate risk), and >20% (high risk)] The Framingham score is calculated from gender, age, diabetes diagnosis, smoking status, blood pressure medications, cholesterol, HDL, and systolic blood pressure (treated or untreated). See Appendix 3 for the algorithm to calculate the Framingham risk score (FRS) and the algorithm to identify patients using hypertension medications. Baseline and postbaseline Reynolds Risk Score (RRS) 10-year risk of CVD events category for non-diabetic men and women aged 45 and 80 years with no prior history of CVD and/or cerebrovascular events will be derived [<5% (low risk), 5% to <10% (low to moderate), 10% to <20% (moderate to high) and 20% (high risk)] (Ridker et al. 2007, 2008). The RRS is calculated from gender, age, smoking status, hscrp, total cholesterol, HDL, systolic blood pressure and whether either parent had a heart attack prior to age of 60 years old. Refer to Ridker et al for the calculation of the RRS for females and to Ridker et al for the calculation of the RRS for males. See Appendix 3 for the algorithm to calculate the RRS. Change from baseline = postbaseline measurement at Visit x baseline measurement Percentage change from baseline at Visit x: o ((Postbaseline measurement at Visit x baseline measurement)/ baseline measurement)*100 Weight (kg) = weight (lbs) * Height (cm) = height (in) * 2.54 Temperature ( C) = [temperature ( F) 32]*5/9 Baseline disease activity based on the DAS28 (calculated separately for DAS28-ESR and DAS28-hsCRP) o o Low disease activity: DAS28 3.2, Moderate disease activity: DAS28 >3.2 to 5.1, o High disease activity: DAS28 >5.1 Simplified Disease Activity Index, CDAI, EULAR Responder Index categories along with other efficacy and health outcomes questionnaires scoring are included in Appendix 2.
367 I4V-MC-JADV Statistical Analysis Plan Version 2 Page Analysis Populations Modified Intent-to-treat (mitt) Population: The mitt population will be used for the Full Analysis Set and will include all randomized patients who received at least one dose of study drug. The intention-to-treat principle remains preserved since the decision of whether or not to begin treatment could not be influenced by knowledge of the assigned treatment in this doubleblind study. Patients will be analyzed according to the study drug to which they were randomized or assigned per protocol. The efficacy and health outcomes analyses will be conducted on an mitt basis unless otherwise specified. If a patient is randomized but not treated with any dose of the study drug, he/she will not be included in any of the efficacy and health outcomes analyses. Analysis of structural progression (van der Heijde mtss) will be conducted on a subset of the mitt population that includes all randomized patients who received at least one dose of the study drug and have non-missing baseline and at least one non-missing postbaseline measurement. Per-protocol (PP) Population: The PP population will include all mitt patients who are not deemed noncompliant with treatment in Part A, who do not have selected important protocol deviations (refer to Section 6.6 for more details) in Part A, and whose investigator site does not have significant GCP issues that require a report to regulatory agencies (regardless of study part). Qualifications and identification of the specific selected important protocol deviations that result in exclusion from the PP subsets will be determined while the study remains blinded, prior to first database lock. The primary and major secondary analyses will be repeated using the PP population. Therefore, the PP population will only be used for Part A. Safety Population: The safety population is defined as all randomized patients who received at least one dose of study drug and who did not discontinue from the study for the reason Lost to Follow-up at the first postbaseline visit. Patients will be analyzed according to the analysis groups defined in Section 6.1. Safety analyses up to the end of Part B will be conducted on this safety population. Follow-up Population: The follow-up population is defined as patients who entered the followup period. Safety Follow-up Population: The safety follow-up population is defined as patients in the safety population who entered the follow-up period and who did not discontinue in the follow-up period for the reason 'Lost to Follow-up'. All details involving the PK analysis will be provided in a separate PK analysis plan.
368 I4V-MC-JADV Statistical Analysis Plan Version 2 Page Handling of X-ray Data X-rays are scheduled at the following visits: Screening (Visit 1), Week 16 (Visit 9), Week 24 (Visit 11) and Week 52 (Visit 15). X-rays are obtained at an early termination only if the most recent x-ray was more than 12 weeks ago. There may be instances in which the x-rays are obtained on a date different from the scheduled InForm visit date. Also, there may be instances in which the set of x-rays for a visit (2 hands/wrists, 2 feet) are split across dates, resulting from the need to re-acquire some images in the set. To maximize the use of the observed data and avoid imputation, the following rules will be applied in the analysis: 1. Postbaseline x-rays administered within 14 days prior to the scheduled InForm visit date, or within 30 days after the scheduled InForm visit date, will be associated with that respective InForm visit for analysis. 2. An x-ray set split across different dates will have the data combined as long as the images are collected within 30 days of each other. The earlier x-ray date will be considered to be the date of the x-ray set. Any linear extrapolation will be based on the date of the x-ray set instead of the InForm visit date Adjustment for Covariates The covariates used in the parametric statistical model (ANCOVA) of continuous data generally will include the parameter value at baseline. Inclusion of baseline in the ANCOVA model provides estimated adjusted (least squares) means for each treatment reflecting averages for the average baseline patient. Other independent variables that will be included in the models for efficacy and health outcomes analyses to adjust estimates of treatment effect include the stratifying variables region and joint erosion status. The independent variables used in the logistic regression models will include the stratifying variables region and joint erosion status (refer to Section 6.1). The stratification variables used in the analyses will be derived based on ecrf data and x-ray data collected by Synarc Handling of Dropouts or Missing Data Missing Data Imputations Missing Data Imputation for Efficacy and Health Outcome Endpoints In accordance with precedent set with other Phase 3 RA trials (Keystone et al. 2004, 2008, 2009; Cohen et al. 2006; Smolen et al. 2008, 2009), the following methods for imputation of missing data will be used: 1. Non-responder imputation (NRI) All patients who discontinue the study or permanently discontinue the study treatment at any time for any reason will be defined as nonresponders for the NRI analysis for categorical
369 I4V-MC-JADV Statistical Analysis Plan Version 2 Page 31 variables, such as ACR20/ACR50/ACR70, from the time of discontinuation and onward. If, at the time of study database lock, there are patients with unresolved interruptions of study drug (i.e., not yet re-started, to be confirmed as a temporary interruption, and not yet permanently discontinued), these patients will be treated as having been permanently discontinued from study drug for the purposes of data imputation for analysis purposes. Patients who are randomized to baricitinib 4 mg or adalimumab and who receive rescue therapy starting from Week 16 will be analyzed as nonresponders after the rescue visit and onward. Patients who are randomized to placebo and who receive rescue therapy starting from Week 16 will be analyzed as nonresponders after the rescue visit and on or prior to Week 24. Randomized patients without available data at a postbaseline visit will be defined as nonresponders for the NRI analysis at that visit. 2. Modified baseline-observation-carried-forward (mbocf) The mbocf method will be used for the analysis of key secondary continuous endpoints (unless otherwise stated). For patients who discontinue the study or permanently discontinue the study treatment because of an AE, including death and abnormal lab results reported as AEs, the baseline observation will be carried forward to subsequent time points for evaluation. For patients who discontinue the study or permanently discontinue the study treatment for reason(s) other than an AE, the last nonmissing postbaseline observation prior to discontinuation will be carried forward to the subsequent time points for evaluation. If, at the time of study database lock, there are patients with unresolved interruptions of study drug (i.e., not yet re-started, to be confirmed as a temporary interruption, and not yet permanently discontinued), these patients will be treated as having been permanently discontinued from study drug for the purposes of data imputation for analysis purposes. For patients who are randomized to baricitinib 4 mg or adalimumab and who receive rescue therapy starting from Week 16, the last nonmissing observation at or before rescue will be carried forward to subsequent time points for evaluation. For patients who are randomized to placebo and who receive rescue therapy starting from Week 16, the last nonmissing observation at or before rescue will be carried forward to subsequent time points on or prior to Week 24 for evaluation. For patients whose baseline value is missing or who have no change from baseline values after the aforementioned imputation, then the change from baseline will take a value of zero indicating no improvement from baseline. If a patient does not have a nonmissing observed record (or one imputed by other means) for a postbaseline visit after applying mbocf, the last postbaseline record prior to the missed visit will be used for this postbaseline visit. 3. Modified last observation carried forward (mlocf) For all continuous measures including safety analyses, the mlocf method will be a general approach to impute missing data unless otherwise specified. For patients who are randomized to baricitinib 4 mg or adalimumab and who receive rescue therapy starting from Week 16, the last nonmissing observation at or before rescue will be carried forward to subsequent time points for evaluation. For patients who are randomized to placebo and who receive rescue therapy starting from Week 16, the last nonmissing observation at or before rescue will be carried forward to subsequent time points on or prior to Week 24 for evaluation. For all other patients who
370 I4V-MC-JADV Statistical Analysis Plan Version 2 Page 32 discontinue from the study or permanently discontinue the study treatment for any reason, the last nonmissing postbaseline observation before discontinuation will be carried forward to subsequent time points for evaluation. If, at the time of study database lock, there are patients with unresolved interruptions of study drug (i.e., not yet re-started, to be confirmed as a temporary interruption, and not yet permanently discontinued), these patients will be treated as having been permanently discontinued from study drug for the purposes of data imputation for analysis purposes. If a patient does not have a nonmissing observed record (or one imputed by other means) for a postbaseline visit, the last postbaseline record prior to the missed visit will be used for this postbaseline visit. After mlocf imputation, data from patients with nonmissing baseline and postbaseline observations will be included in the analyses. The mlocf method will be used as a sensitivity approach to the mbocf method with respect to the key secondary endpoints. For safety analyses of the Week 0 to Week 52 period which includes data collected after rescue, the last nonmissing assessment prior to a missed visit will be carried forward if the assessment occurred after the first dose of baricitinib (i.e., after first study dose for groups originally randomized to baricitinib 4 mg, after first rescue dose for patients randomized to placebo or adalimumab who are later rescued, and after first baricitinib dose for patients randomized to placebo who switch treatment at Week 24 according to the protocol). 4. Baseline observation carried forward (BOCF) The BOCF method will be used for the analysis of key secondary continuous endpoints (HAQ-DI and DAS28-hsCRP) at Week 12 as a sensitivity analysis to satisfy specific regulatory requests. For patients who discontinue the study or permanently discontinue the study treatment, the baseline observation will be carried forward to subsequent time points for evaluation. If, at the time of study database lock, there are patients with unresolved interruptions of study drug (i.e., not yet re-started, to be confirmed as a temporary interruption, and not yet permanently discontinued), these patients will be treated as having been permanently discontinued from study drug for the purposes of data imputation for analysis purposes. For patients whose baseline value is missing or who have no change from baseline scores after the aforementioned imputation, then the change from baseline will take a value of zero, indicating no improvement from baseline. If a patient does not have a nonmissing observed record (or one imputed by other means) for a postbaseline visit after applying BOCF, the last postbaseline record prior to the missed visit will be used for this postbaseline visit Missing Data Imputation for the Structural Progression Endpoint (van der Heijde mtss) 1. Primary method: Linear extrapolation method The primary approach to the analysis of the structural progression data will be the linear extrapolation method, which assumes that individual patients would continue to accrue structural damage in their joints at the same rate that was observed at their time of last observation. The linear extrapolation method has been established as an appropriate missing data imputation method in other Phase 3 RA trials (Keystone et al. 2004, 2008, 2009; Cohen et al. 2006; Smolen et al. 2008, 2009). The linear extrapolation method will be used for analysis of the structural progression endpoint (van der Heijde mtss), including bone erosion scores and joint space
371 I4V-MC-JADV Statistical Analysis Plan Version 2 Page 33 narrowing (JSN), to impute missing data at time points when analyses are conducted (including the analyses at Week 24 and Week 52). For patients who discontinue from the study or for patients who miss a radiograph for any reason, baseline data and the most recent radiographic data prior to discontinuation or prior to the missed radiograph, adjusted for time, will be used for linear extrapolation to impute missing data at subsequent scheduled time points, including the primary analysis at Week 24. For patients who receive rescue therapy starting at Week 16 or at any time point thereafter, baseline data and the most recent radiographic data prior to initiation of rescue therapy, adjusted for time, will be used for linear extrapolation. For patients who discontinue from the study or permanently discontinue the study treatment after receiving rescue therapy, the radiographic data collected prior to initiation of rescue therapy should supersede the data collected before discontinuation. All patients originally randomized to placebo will be switched to active treatment at Week 24; thus, there will be no observed data for the placebo arm from patients still receiving placebo for the structural comparison at subsequent time points. Therefore, baseline data and the most recent radiographic data prior to initiation of the new therapy, adjusted for time, will be used for linear extrapolation at Week 52. Missing postbaseline values will be imputed only if both a baseline value and a postbaseline value from another time point are available, as long as the patient was receiving the same treatment at each applicable time point. Missing postbaseline values will not be imputed using postbaseline data from later time points. For example: (1) For a baricitinib- or adalimumab-treated patient who did not require rescue therapy: Missing data at Week 16 will be extrapolated from baseline and an early discontinuation visit prior to Week 16, if available. Missing data at Week 24 will be extrapolated from baseline and the last available postbaseline value prior to Week 24. Missing data at Week 52 will be extrapolated from baseline and the last available postbaseline value prior to Week 52, if available. (2) For a baricitinib-, adalimumab-, or placebo-treated patient who was rescued at Week 16: Missing data at Weeks 24 or 52 will be extrapolated from baseline and Week 16. (3) For a placebo-treated patient who did not require rescue therapy: Missing data at Week 16 will be extrapolated from baseline and an early discontinuation visit prior to Week 16, if available. Missing data at Week 24 will be extrapolated from baseline and the last available postbaseline value prior to Week 24. Missing data at Week 52 will be extrapolated from baseline and the last available postbaseline value at or prior to Week Sensitivity method: Last observation carried forward (LOCF)
372 I4V-MC-JADV Statistical Analysis Plan Version 2 Page 34 An LOCF approach will also be used for the Week 24 analyses of mtss, bone erosion scores, and JSN. Data observed at Week 24 will be analyzed according to randomized treatment group regardless of rescue status. Patients with missing data at Week 24, as a result of study discontinuation or unreadable x-rays, will have their data imputed by last postbaseline observation carried forward. 3. Other method: Non-responder imputation (NRI) All patients who discontinue the study early or receive rescue treatment will be defined as nonresponders for the NRI analysis for categorical endpoints, from the time of discontinuation/rescue and onward. Randomized patients without available data at any postbaseline visit will be defined as nonresponders for the NRI analysis at all visits. If a patient does not have a nonmissing observed or imputed record for a postbaseline visit after applying NRI, the patient will be imputed as a nonresponder at that postbaseline visit. Details of the missing data imputation rules, methods for between-reader adjudication, score derivations, and other details for the bone erosion scores, joint space narrowing scores, and mtss are provided in Appendix 2 (van der Heijde Modified Total Sharp Score) and Appendix 5. Table JADV.6.1. Imputation Techniques for Various Efficacy and Safety Variables Endpoint Imputation Method ACR 20/50/70 responses, DAS28-hsCRP and DAS28-ESR Low Disease NRI Activity (LDA) and remission, EULAR response criteria based on the 28-joint DAS, ACR/EULAR remission according to the Boolean-based definition, HAQ-DI improvement from baseline 0.22 and 0.3, CDAI LDA and remission, and SDAI LDA and remission JSN, bone erosion scores, and mtss Linear extrapolation method JSN, bone erosion scores, and mtss LOCF mtss 0 and mtss SDC NRI; see Section HAQ-DI, DAS28-hsCRP mbocf HAQ-DI, DAS28-hsCRP at Week 12 BOCF Duration and severity of morning joint stiffness, worst tiredness and worst See Section 6.14 joint pain assessed in the diaries Continuous efficacy, health outcomes, and safety measures (labs, vital signs, mlocf etc.) Abbreviations: ACR = American College of Rheumatology; BOCF = baseline observation carried forward; CDAI = Clinical Disease Activity Index; DAS28 = Disease Activity Score modified to include the 28 diarthrodial joint count; EULAR = European League Against Rheumatism; HAQ-DI = Health Assessment Questionnaire Disability Index; hscrp = high-sensitivity C-reactive protein; JSN = joint space narrowing; LDA = low disease activity; mbocf = modified BOCF; mlocf = modified last observation carried forward; mtss = modified Total Sharp Score; NRI = nonresponder imputation; SDAI = Simplified Disease Activity Index; SDC = smallest detectable changes Multiple Comparison Adjustments to multiple comparisons among the treatment groups using the primary and major secondary endpoints will use the methods described in Section These multiple testing procedures will be used to control the family-wise error rate for the primary and major secondary
373 I4V-MC-JADV Statistical Analysis Plan Version 2 Page 35 hypotheses for the endpoints. The multiple testing procedure provides strong control of the family-wise error rate at the 1-sided level Patient Disposition Frequency counts and percentages of patients excluded prior to randomization by primary reason for exclusion will be provided for patients who failed to meet study entry requirements at screening. An overview of patient populations will be summarized by treatment group using all entered patients. Patient disposition will be summarized using the mitt population. Frequency counts and percentages of patients who complete through Week 24 or discontinue early from the study on or prior to Week 24 will be summarized separately by treatment group for patients who are not rescued and for patients who are rescued, along with their reason for study discontinuation. Frequency counts and percentages of patients who complete through Week 52 or discontinue early from the study after Week 24 and on or prior to Week 52 will be summarized separately by treatment group for patients who are not rescued/switched and for patients who are rescued/switched, along with their reason for study discontinuation. Among patients who completed the study, frequency counts and percentages of patients who entered Study JADY, completed the post-treatment follow-up visit, or did not complete the follow-up visit will be will be presented by treatment group. Among patients who discontinued early from the study, frequency counts and percentages of patients who completed the post-treatment follow-up period or did not complete the follow-up visit will be presented by treatment group. Reasons for not completing the post-treatment follow-up visit will also be presented. A listing of patient disposition will be provided for all randomized patients, with the extent of their participation in the study and the reason for discontinuation. A listing of all randomized patients will also be provided Protocol Deviations Protocol deviations will be tracked by the clinical team, and their importance will be assessed by key team members during protocol deviation review meetings. Out of all important protocol deviations identified, a subset occurring during Part A prior to the primary endpoint (Week 12) with the potential to affect efficacy analyses will result in exclusion from the PP population. Potential examples of deviations include patients who receive excluded concomitant antirheumatic therapy, significant non-compliance with study medication (<80% of assigned doses taken, failure to take study medication and taking incorrect study medication), patients incorrectly enrolled in the study, and patients whose data are questionable due to significant site quality or compliance issues. Important protocol deviations are the following (those indicated with an asterisk [*] may exclude a patient from the per protocol analysis population): Protocol inclusion or exclusion criteria:
374 I4V-MC-JADV Statistical Analysis Plan Version 2 Page 36 o o o General Are unable to read, understand and give written informed consent Are pregnant or nursing at the time of study entry(*) Prior infection or comorbidity Have a history of lymphoproliferative disease; or have signs or symptoms suggestive of possible lymphoproliferative disease, including lymphadenopathy or splenomegaly; or have active primary or recurrent malignant disease; or have been in remission from clinically significant malignancy for <5 years Have been exposed to a live vaccine within 12 weeks prior to planned randomization Have been exposed to herpes zoster vaccination within 30 days of planned randomization Have a current or recent (30 days prior to study entry) clinically serious viral, bacterial, fungal, or parasitic infection Have had symptomatic herpes zoster infection within 12 weeks prior to study entry Have a history of disseminated/complicated herpes zoster (multi dermatomal involvement, ophthalmic zoster, central nervous system involvement, or post therapeutic neuralgia) Have a history of active Hepatitis B (HBV), Hepatitis C (HCV), or Human Immunodeficiency Virus (HIV) Have had household contact with a person with active Tuberculosis (TB) and did not receive appropriate and documented prophylaxis for TB Have evidence of active TB or have previously had evidence of active TB and did not receive appropriate and documented treatment Have a diagnosis of Felty's syndrome Current infection Have evidence of active TB as documented by a PPD test ( 5-mm induration between approximately 2 and 3 days after application, regardless of vaccination history), medical history, clinical symptoms, and abnormal chest x-ray at screening (*) Have evidence of latent TB (as documented by a positive PPD, no clinical symptoms consistent with active TB and a normal chest x-ray at screening ) unless patients complete at least 4 weeks of appropriate treatment prior to
375 I4V-MC-JADV Statistical Analysis Plan Version 2 Page 37 randomization and agree to complete the remainder of treatment while in the trial Have a positive test for HBV Have HCV (positive for anti-hepatitis C antibody with confirmed presence of HCV) Have evidence of HIV infection or positive HIV antibodies o o Medications Have not had regular use of MTX for at least the 12 weeks prior to study entry. The dose of MTX must have been a stable, unchanging oral dose of 7.5 to 25 mg/week (or the equivalent injectable dose) for at least the 8 weeks prior to study entry. Have received cdmards (e.g., gold salts, cyclosporine, azathioprine, or any other immunosuppressives) other than MTX, hydroxychloroquine (up to 400 mg/day), or sulfasalazine (up to 3000 mg/day) within 4 weeks prior to study entry at a stable dose for at least 8 weeks prior to study entry; if either was recently discontinued, the patient must not have taken any dose within 4 weeks prior to study entry. Immunosuppression related to organ transplantation was not permitted. Have ever received any biologic DMARD (such as TNF, interleukin-1 [IL-1], IL-6, or T-cell- or B-cell-targeted therapies) Have been receiving an unstable dosing regimen of oral, intramuscular (IM) or intravenous (IV) corticosteroids within 6 weeks of randomization Are currently receiving corticosteroids at doses >10 mg per day of prednisone (or equivalent) at study entry Have started NSAID treatment for signs and symptoms of RA within 6 weeks of randomization Have been receiving an unstable dosing regimen of NSAIDs within 6 weeks of randomization Have received interferon therapy (such as Roferon-A, Intron-A, Rebetron, Alferon-N, Peg-Intron, Avonex, Betaseron, Infergen, Actimmune, Pegasys) within 4 weeks prior to study entry or are anticipated to require interferon therapy during the study Have any specific abnormalities on screening laboratory tests Have AST >1.5 times the ULN on screening laboratory testing (1 retest allowed within approximately 2 weeks)
376 I4V-MC-JADV Statistical Analysis Plan Version 2 Page 38 Have ALT >1.5 times the ULN on screening laboratory testing (1 retest allowed within approximately 2 weeks) Have total bilirubin 1.5 times the ULN on screening laboratory testing (1 retest allowed within approximately 2 weeks) Have hemoglobin <10.0 g/dl on screening laboratory testing (1 retest allowed within approximately 2 weeks) Have total white blood cell count <2500 cells/µl on screening laboratory testing (1 retest allowed within approximately 2 weeks) Have absolute neutrophil count < 1200 cells/µl on screening laboratory testing (1 retest allowed within approximately 2 weeks) Have lymphocyte count <750 cells/µl on screening laboratory testing (1 retest allowed within approximately 2 weeks) Have platelets <100,00/µL on screening laboratory testing (1 retest allowed within approximately 2 weeks) Have egfr <40 ml/min/1.73 m 2 on screening laboratory testing (1 retest allowed within approximately 2 weeks) Use of prohibited concomitant medication during study o o o o Use of any new or any change of dose in steroids in the absence of an AE or prior to rescue. Use for treatment of RA disease activity will be considered an important protocol deviation.(*) Use of any new or increased analgesics or NSAIDs in the absence of an AE or prior to rescue Change in cdmard therapy in the absence of an AE. Change for treatment of RA disease activity will be considered an important protocol deviation.(*) Use of any biologic or interferon therapy(*) Study drug administration and compliance o o o o o Drug Dispensing Error (e.g. patient dispensed wrong package and received incorrect dose or drug)(*) Patient received drug that was declared not Fit for Use (*) Use of expired CT Material(*) Significant non-compliance as defined per protocol(*) Interactive voice response system data entry errors that impact patient stratification at randomization Protocol non-compliance
377 I4V-MC-JADV Statistical Analysis Plan Version 2 Page 39 o o o o Patient met temporary or permanent IP discontinuation criteria and IP not discontinued per protocol Inadvertent unblinding(*) Fraud(*) Other significant GCP issues(*). Since the primary and secondary key efficacy endpoint analyses will be repeated using the PP population at Week 12 only (except that mtss will be analyzed at Week 24), time constraints will be applied to the important protocol deviations that exclude a patient from the per protocol analysis population. For example, for treatment non-compliance, only patients with <80% compliance rate in the first 12 week period will be excluded from the per protocol population. A summary of the number and percentage of patients with an important protocol deviation by treatment group, overall, and by type of deviation will be provided. Although the PP population is for the primary endpoint (Week 12) in Part A only, important protocol deviations will be summarized for Parts A and B. Individual patient listings of important protocol deviations will be provided. A summary of reasons patients were excluded from the PP population will be provided by treatment group, and a patient listing will also be generated Patient Characteristics Patient characteristics including demographics will be summarized using the mitt population and PP population by treatment group. The summary will include descriptive statistics such as the number of patients (n), mean, SD, median, minimum (min), and maximum (max) for continuous measures and frequency counts and percentages for categorical measures. No formal statistical comparisons will be made among treatment groups, unless otherwise stated Demographics Patient demographics will be summarized by treatment group using the mitt population. The following demographic information will be presented for this study. Age and age group Gender Race Ethnicity (US patients only) Region Country Weight Height Body mass index
378 I4V-MC-JADV Statistical Analysis Plan Version 2 Page 40 Waist circumference Habits (alcohol, smoking, caffeine) In addition, patient demographics will be summarized by country using the mitt population. A listing of patient demographics will be produced Baseline Disease Characteristics Baseline disease characteristics will be summarized by treatment group using the mitt population. The following baseline disease information (but not limited to only these) will be categorized and presented for baseline cardiovascular and renal comorbidities, baseline RA clinical characteristics, baseline health outcome measures, and other baseline demographic and disease characteristics: Baseline Framingham 10-year risk of CVD events score Baseline RRS 10-year risk of CVD events score Duration from onset of RA (years), Duration from onset of RA category (<1 years, 1 to <5, 5 years) Years since RA Diagnosis Patient s assessment of pain (by VAS in mm) Patient s global assessment of disease activity (by VAS in mm) Physician s global assessment of disease activity (by VAS in mm) HAQ-DI total score Erythrocyte Sedimentation Rate (ESR) (mm/hr) hscrp (mg/l) DAS28-ESR and DAS28-hsCRP DAS28-ESR and DAS28-hsCRP category ( 3.2, >3.2 to 5.1, >5.1) Anti citrullinated peptide antibody (ACPA) positivity and Rheumatoid factor (RF) positivity status Number of tender joints out of 28 (TJC28) Number of swollen joints out of 28 (SJC28) Number of tender joints out of 68 (TJC68) Number of swollen joints out of 66 (SJC66) Estimated Glomerular Filtration Rate (egfr) o o egfr <60 ml/min/1.73 m2 egfr 60 ml/min/1.73 m2
379 I4V-MC-JADV Statistical Analysis Plan Version 2 Page 41 MTX dose (mg/week) Number of cdmards currently used (0, 1, 2, 3) Number of cdmards previously used (0, 1, 2, 3) Type of cdmards currently used o o Use of MTX only Use of MTX + other cdmards Use of oral corticosteroid and dose (mg/day) expressed as prednisoneequivalent dose Use of statin therapy (Yes/No). In addition, the baseline RA clinical characteristics will be summarized by country using the mitt population. A listing of patient demographics and selected baseline characteristics will be produced Previous and Concomitant Medication Summaries of previous and concomitant medications used for RA including cdmards (MTX only, MTX and other cdmards; see Section 6.7) will be based on the mitt population. At screening, medications used for RA that had been previously discontinued are recorded for each patient. A summary of previous medications used for RA will be prepared using frequency counts and percentages by World Health Organization (WHO) drug anatomical therapeutic chemical (ATC) classification and treatment group. Concomitant therapy will be recorded at each visit and will be classified according to the WHO drug dictionary and summarized using frequency counts and percentages by WHO drug ATC classification sorted by decreasing frequency and treatment group. Concomitant medications are defined as: Concomitant medications with a stop date on or after the date of the first dose of the study drug and that precedes Week 24 (Visit 11) or an early discontinuation visit prior to Week 24 will be included in the concomitant medications summaries for the analysis at the end of Part A. Should there be insufficient data to make this comparison (for example, the concomitant therapy stop year is the same as the treatment start year, but the concomitant therapy stop month and day are missing), the medication will be considered as concomitant for Part A. For patients who continue in Part B, concomitant medications with a stop date that is after Week 24 or ongoing will be included in the concomitant medications summaries at the end of Part B. Summaries of concomitant medications will be provided using data up to rescue/switch for the following categories:
380 I4V-MC-JADV Statistical Analysis Plan Version 2 Page 42 concomitant medications used for RA concomitant medications excluding RA and statin therapies concomitant medications - statins and will also be summarized for the all baricitinib exposures group including data after rescue/switch Historical Diagnoses and Pre-Existing Conditions The number and percentage of patients with selected historical diagnoses, as listed on the ecrf, will be summarized by treatment group using the mitt population. Historical diagnoses will be identified using the MedDRA (most current available version) algorithmic standardized MedDRA queries (SMQs) or similar Lilly-defined lists of preferred terms (PTs) of interest. Pre-existing conditions are defined as those conditions recorded on the Pre-existing Conditions and AEs ecrf page with a start date prior to the date of informed consent. Note that conditions with a partial or missing start date will be assumed to be not pre-existing unless there is evidence, through comparison of partial dates, to suggest otherwise. Pre-existing conditions will be identified using the MedDRA SMQs or similar Lilly-defined lists of PTs of interest. Frequency counts and percentages of patients with selected pre-existing conditions will be summarized by treatment group using the mitt population Treatment Compliance Patient compliance with study medication will be assessed from randomization visit (Week 0) to Visit 15 (Week 52) during the treatment period. Compliance will be summarized by forms of study drug (tablets and injections) and by treatment groups from randomization to visit of rescue/switch using the mitt population. Compliance for tablets (baricitinib and baricitinib placebo): All patients are expected to be taken one tablet baricitinib or baricitinib placebo daily in Parts A and B regardless of rescue, a patient is considered noncompliant to tablets if he or she misses >20% of the prescribed doses during the study. For patients who have their dose interrupted because of safety reasons, the period of dose withholding will be taken into account in the compliance calculation as outlined below. Compliance to baricitinib and baricitinib placebo tablet in the period of interest will be calculated as follows: Where: actual total # of tablets used Expected total # of tablets used 100 the expected total # tablets used = the number of days in the period of interest; the actual total # of tablets used = total number dispensed total number returned in the period.
381 I4V-MC-JADV Statistical Analysis Plan Version 2 Page 43 If a patient has dose interrupted for safety reasons during the period, the total number of days drug was withheld will be deducted from the expected total # of tablets used. Compliance for injections (adalimumab or adalimumab placebo): All patients are expected to be taken adalimumab or adalimumab placebo injection biweekly in Parts A and B regardless of rescue, a patient is considered noncompliant to injections if he or she misses >20% of the prescribed doses during the study. For patients who have dose interrupted because of safety reasons, the period of dose withholding will be taken into account in the compliance calculation as outlined below. Compliance to adalimumab or adalimumab placebo injection in the period of interest will be calculated as follows: Where: actual total # of syringes used Expected total # of syringes used 100 Expected total number of syringes used = integer of [(total number of days in the period of interest)/14] + 1. Actual total # of syringes used = total # of syringes dispensed total # of syringes returned. For patients with drug interruptions, the expected number of syringes used will be calculated for the period before and after interruption in the period of interest separately, then the sum of the two will be the expected total for the period of interest. Patients who are significantly noncompliant in Part A will be excluded from the PP population. The summary statistics of percent compliance and non-compliance rate will be summarized by forms of study drug (tablets and injections) and by treatment group. The sub-total percent compliance for Weeks 0 through 12, the sub-total percent compliance for Weeks 0 through 24, and grand-total percent compliance for Weeks 0 through 52 with data up to the time of rescue/switch will be presented, as well, along with the associated non-compliance rates. The number of expected doses, tablets dispensed, tablets returned, and percent compliance will be listed by patient. The number of tablets dispensed/returned, number of syringes dispensed/returned, and percent compliance will be listed by treatment group Treatment Exposure Duration of exposure to each study drug will be summarized for the safety population using descriptive statistics (n, mean, SD, minimum, Q1, median, Q3, maximum). Cumulative exposure and duration of exposure will be summarized in terms of frequency counts and percentages by category and treatment group. Overall exposure will be summarized in total patient-years which are calculated according to the following:
382 I4V-MC-JADV Statistical Analysis Plan Version 2 Page 44 Exposure in patient-years = sum of duration of exposure in days (for all patients in treatment group) / The summary for the first database lock (end of Part A) will include exposure to each treatment plus exposure for the subset of patients who are rescued during Part A from each randomized therapy. For the baricitinib treatment group, exposure will be calculated including the subset of patients who are rescued during Part A. The summary for the final lock (end of Parts A and B) will include all exposures for patients randomized to baricitinib, placebo, and adalimumab, plus all exposures subsequent to rescue or switch to baricitinib. For the baricitinib treatment group, exposure will be calculated including the subset of patients who are rescued. Duration of exposure will be calculated as follows: 1. Baricitinib group: Duration of exposure to baricitinib excluding exposure after the initiation of rescue therapy will be calculated as: [date of last study drug treatment prior to rescue date of first study drug dose (Visit 2) + 1]. Duration of exposure to baricitinib after the initiation of rescue therapy will be calculated as: (date of last study drug treatment date of first baricitinib dose as rescue therapy + 1). 2. Adalimumab group: 3. Placebo group: Duration of exposure to adalimumab excluding exposure after the initiation of rescue therapy will be calculated as: [date of last study drug treatment date of first study drug dose (Visit 2) + 1]. Duration of exposure to baricitinib after the initiation of rescue therapy will be calculated as: (date of last study drug treatment date of first baricitinib dose as rescue therapy + 1). Duration of exposure to placebo excluding exposure after the initiation of rescue therapy will be calculated as: [date of last study drug treatment date of first study drug dose (Visit 2) + 1]. Duration of exposure to baricitinib after the initiation of rescue therapy will be calculated as: [date of last study drug treatment date of first baricitinib dose as rescue therapy + 1]. Duration of exposure to baricitinib after switch will be calculated as: (date of last study drug treatment (before rescue, if applicable) date of first dose of study drug after switch + 1). Summaries of treatment exposure will also include all baricitinib exposures.
383 I4V-MC-JADV Statistical Analysis Plan Version 2 Page Efficacy Analyses Efficacy analyses will use the mitt Population defined in Section and will generally be analyzed according to the following 2 formats: Week 0 (Visit 2) to Week 52 (Visit 15) with data included up to the time of rescue/switch. Formal statistical tests will be used to compare the baricitinib group to the placebo group for these efficacy outcomes through Week 24; Week 0 (Visit 2) to Week 52 (Visit 15) for rescued/switched patients with data included after rescue/switch. These efficacy results will be summarized by treatment group, but no formal statistical comparisons will be performed Primary Efficacy Analysis The primary efficacy measure will be the proportion of patients achieving ACR20 at Week 12 based on the change in ACR response values from baseline. ACR20 is defined as at least 20% improvement from baseline in the following ACR Core Set variables: TJC68 SJC66 An improvement of 20% from baseline in at least 3 of the following 5 assessments: o o o o o Patient s Assessment of Pain (visual analog scale [VAS]) Patient s Global Assessment of Disease Activity (VAS) Physician s Global Assessment of Disease Activity (VAS) Patient s assessment of physical function as measured by the HAQ-DI Acute phase reactant as measured by hscrp. Refer to the algorithm to calculate ACR response in Appendix 1. The primary efficacy analysis compares ACR20 at Week 12 between baricitinib and placebo using the mitt population. The PP population results will be used as supportive to the mitt results. A logistic regression model with treatment, region, and joint erosion status (1-2 joint erosions plus seropositivity versus at least 3 joint erosions) in the model will be used to test the treatment difference between baricitinib and placebo in the proportion of patients achieving ACR20 response at Week 12 using the Wald test at a 2-sided significance level of The NRI approach will be used to impute response status for patients with completely missing data. The p-value and 95% CI of odds ratio from the logistic regression model will be reported. The 95% CI of responder rate difference will be calculated using the Newcombe-Wilson method (Newcombe 1998) without continuity correction. Treatment-by-region interaction will be added to the logistic regression model as a sensitivity analysis and results from this model will be compared to the primary model (without the interaction effect). If the treatment-by-region interaction is significant at a 2-sided 0.1-alpha level, the nature of this interaction will be inspected as to whether it is quantitative (i.e., the treatment effect is consistent in direction but
384 I4V-MC-JADV Statistical Analysis Plan Version 2 Page 46 not size of effect) or qualitative (the treatment is beneficial in some but not all regions). If the treatment-by-region interaction effect is found to be quantitative, results from the primary model will be presented. If the treatment-by-region interaction effect is found to be qualitative, further inspection will be used to identify in which regions baricitinib is found to be more beneficial Major Secondary Efficacy Analyses Unless otherwise stated, the following analyses will be performed to test for superiority of baricitinib relative to the placebo group. The key secondary efficacy measures are: Change from baseline to Week 24 in mtss Change from baseline to Week 12 in HAQ-DI Change from baseline to Week 12 in DAS28-hsCRP Proportion of patients achieving an SDAI score 3.3 at Week 12 Proportion of patients achieving ACR20 at Week 12 with respect to noninferiority of baricitinib to adalimumab Mean duration of morning joint stiffness (epro Diary; 7 days prior to Week 12) Change from baseline to Week 12 in DAS28-hsCRP with respect to superiority of baricitinib to adalimumab Mean severity of morning joint stiffness (epro Diary; 7 days prior to Week 12) Mean Worst Tiredness NRS (epro Diary; 7 days prior to Week 12) Mean Worst Pain NRS (epro Diary; 7 days prior to Week 12). The key secondary efficacy measures will be analyzed using the mitt population. The PP population results will be used as supportive to the mitt results. These analyses are described as follows: Comparison between baricitinib and placebo in change from baseline to Week 24 in mtss: The actual scores and change from baseline scores at Week 24 in mtss will be summarized. An ANCOVA model with baseline score, region, joint erosion status, and treatment group in the model will be used to test the treatment difference between baricitinib and placebo in change from baseline to Week 24 in mtss scores. Type III sums of squares for the LS means will be used for the statistical comparison; 100*(1- alpha)% CI will also be reported. The alpha level will be defined according to the graphical multiple testing procedure described in Section The LS mean difference, SE, CI, and p-value will be presented. The linear extrapolation method described in Section 6.3 will be used. The LOCF approach described in Section 6.3 will
385 I4V-MC-JADV Statistical Analysis Plan Version 2 Page 47 be used as a sensitivity approach to the linear extrapolation method to impute missing data. To address the normality assumption of ANCOVA, rank-based ANCOVA with van der Waerden scores will be used as a sensitivity analysis to ANCOVA. For comparison between baricitinib and placebo, the median score and its 95% CI from the Hodges- Lehmann estimator will be presented. A 95% CI for the median score within each treatment group will be calculated using distribution-free confidence limits. The p-value for the median difference will be calculated using the rank-based ANCOVA with van der Waerden scores. Both linear extrapolation and the LOCF method will be used to impute missing data for the rank-based ANCOVA sensitivity analysis. A second distribution-free method that does not enforce a particular transformation onto the data will be conducted as a sensitivity analysis. Initially, a 2-sample t test statistic comparing baricitinib to placebo (and later comparing adalimumab to placebo) will be calculated using the randomized treatment assignments. Then 50,000 (or more) samples will be obtained whereby the assigned treatments are randomly permuted among the patients included in the analysis and the same test statistic re-computed. An estimated p- value for comparing the data distributions of the 2 treatments groups is obtained by comparing the observed test statistic to the 50,000 randomly generated test statistics, counting the fraction for which the observed test statistic is greater than or equal to in magnitude (for example, in absolute value). An upper bound for the p-value will be obtained using a normal approximation to the binomial distribution. Both linear extrapolation and the LOCF method will be used to impute missing data for the permutation test sensitivity analysis. Supportive analyses using the cumulative responder curves, in which patients who have discontinued their randomized treatment are defined as progressors across the spectrum of choices of analysis thresholds will be provided. Comparison between baricitinib and placebo in change from baseline to Week 12 in HAQ-DI score: an ANCOVA model with baseline score, region, joint erosion status, and treatment group in the model will be used to test the treatment difference between baricitinib and placebo in change from baseline to Week 12 in HAQ-DI. Type III sums of squares for the LS means will be used for the statistical comparison; 100*(1-alpha)% CI will also be reported. The alpha level will be defined according to the graphical multiple testing procedure described in Section The LS mean difference, SE, CI, and p-value will be presented. The mbocf approach described in Section will be used to impute missing data. The mlocf approach described in Section will also be used as supportive to the mbocf approach. The BOCF approach described in Section will be used as an additional supportive analysis to satisfy regulatory
386 I4V-MC-JADV Statistical Analysis Plan Version 2 Page 48 requests. In addition, a supportive MMRM analysis will be used (refer to Section 6.1 for more details) to present the estimates of treatment effect at Week 12. Comparison between baricitinib and placebo in change from baseline to Week 12 in DAS28-hsCRP: an ANCOVA model with baseline score, region, joint erosion status, and treatment group in the model will be used to test the treatment difference between baricitinib and placebo in change from baseline to Week 12 in DAS28-hsCRP. Type III sums of squares for the LS means will be used for the statistical comparison; 100*(1- alpha)% CI will also be reported. The alpha level will be defined according to the graphical multiple testing procedure described in Section The LS mean difference, SE, CI, and p-value will be presented. The mbocf approach described in Section will be used to impute missing data. The mlocf approach described in Section will also be used as supportive to the mbocf approach. The BOCF approach described in Section will be used as an additional supportive analysis to satisfy regulatory requests. In addition, supportive MMRM analysis will be used (refer to Section 6.1 for more details) to present the estimates of treatment effect at Week 12. Comparison between baricitinib and placebo in proportion of patients achieving an SDAI score 3.3 at Week 12: A logistic regression model with treatment, region, and joint erosion status in the model will be used to test the treatment difference between baricitinib and placebo in the proportion of patients achieving an SDAI score 3.3 response at Week 12. The NRI method as described in Section will be used to impute missing data. The p-value and 95% CI of odds ratio from the logistic regression model will be reported for statistical inference. The 100*(1-alpha)% CI of responder rate difference will be calculated using the Newcombe-Wilson method (Newcombe 1998) without continuity correction. The alpha level will be defined according to the graphical multiple testing procedure described in Section Comparison between baricitinib and placebo in mean duration of morning joint stiffness in the 7 days prior to Week 12: refer to Section 6.14 for the statistical methods used for the analysis of mean duration of morning joint stiffness. Noninferiority comparison of baricitinib to adalimumab in ACR20 response at Week 12: The noninferiority test will be conducted using the Newcombe-Wilson method (Newcombe 1998) without continuity correction. The responder rate difference and its 95% CI will be reported. If adalimumab is found to be superior to placebo as assessed by the ACR20 response rate at Week 12 using same model specification as for the primary analysis, and if the lower bound of the 95% CI (1-sided 97.5% CI) for the response rate difference between baricitinib and adalimumab is > -12%, Lilly will conclude that baricitinib is noninferior to adalimumab with respect to ACR20. If the lower bound of the 95% CI is > 0%, Lilly will conclude that baricitinib is superior to adalimumab with respect to ACR20. A logistic regression model with treatment, region, and joint erosion status in the model will also be used to test the treatment difference
387 I4V-MC-JADV Statistical Analysis Plan Version 2 Page 49 between baricitinib and adalimumab in the proportion of patients achieving an ACR20 response at Week 12. The NRI method as described in Section will be used to impute missing data. The p-value and 95% CI of odds ratio from the logistic regression model will be reported for statistical inference. Comparison between baricitinib and adalimumab in change from baseline to Week 12 in DAS28-hsCRP: An ANCOVA model with treatment, region, joint erosion status, and baseline score in the model will be used to test the treatment difference between baricitinib and adalimumab in change from baseline to Week 12 in DAS28-hsCRP. Type III sums of squares for the LS means will be used for the statistical comparison; the 95% CI will also be reported. The LS mean differences, SE, CI, and p- value will be presented. The mbocf approach described in Section will be used to impute missing data. The mlocf approach described in Section will also be used as supportive to the mbocf approach. In addition, supportive MMRM analysis will be used (refer to Section 6.1 for more details) to present the estimates of treatment effect at Week 12. Comparison between baricitinib and placebo in mean severity of morning joint stiffness in the 7 days prior to Week 12: refer to Section 6.14 for the statistical methods used for the analysis of severity of morning joint stiffness. Comparison between baricitinib and placebo in mean Worst Tiredness NRS in the 7 days prior to Week 12: refer to Section 6.14 for the statistical methods used for the analysis of worst tiredness. Comparison between baricitinib and placebo in mean Worst Pain NRS in the 7 days prior to Week 12: refer to Section 6.14 for the statistical methods used for the analysis of worst joint pain Analysis for Other Secondary Efficacy Endpoints Besides the analyses described in Section and , the following analyses will be performed between baricitinib and placebo using data through Week 24, as appropriate. Descriptive statistics for the proportion of patients achieving DAS28-ESR 3.2 and <2.6 and for DAS28-hsCRP 3.2 and <2.6 at postbaseline visits will be provided by visit and treatment group. Logistic regression will be conducted to analyze the proportion of patients with DAS28-ESR 3.2 and <2.6 and with DAS28-hsCRP 3.2 and <2.6 through Week 24. The model for the logistic regression will consist of region, joint erosion, and treatment as fixed effects to compare baricitinib to placebo. The observed values will be analyzed as a sensitivity approach to the primary method that utilizes NRI. Descriptive statistics for the proportion of patients achieving HAQ-DI improvement from baseline 0.22 and 0.3 at postbaseline visits will be provided by visit and treatment group. A logistic regression analysis with a model consisting of region, joint erosion, and
388 I4V-MC-JADV Statistical Analysis Plan Version 2 Page 50 treatment will be applied to compare baricitinib to placebo. The observed values will be analyzed as a sensitivity approach to the primary method that utilizes NRI. Descriptive statistics for the actual score and change from baseline scores at Week 16 and Week 52 in mtss will be provided by visit and treatment group. ANCOVA will be performed for the change from baseline score at Weeks 16 and 52 using the same model specifications for the change from baseline to Week 24 in mtss mentioned above. Except that the supportive analyses using the cumulative responder curves is not performed for Weeks 16 and 52. Descriptive statistics for the proportion of patients with mtss change 0 by treatment group at Weeks 16, 24, and 52 will be provided by visit and treatment group. Logistic regression will be conducted to analyze the proportion of patients with mtss change 0 at Weeks 16, 24, and 52 between baricitinib and placebo. The model for the logistic regression will consist of region, joint erosion, and treatment as fixed effects. Summaries by treatment group will be provided for the proportion of patients who improved or experienced no change (mtss 0), (or worsened (mtss >0). In addition, the proportion of patients with change in mtss SDC at Weeks 16, 24, and 52 will be analyzed in a similar manner as described for mtss change 0. The smallest detectable changes (SDCs) at Weeks 16, 24 and 52 are defined as the smallest detectable change which is computed from the variability in Week 0 to Week 16, Week 24 or Week 52 change in scores assigned by the blinded imaging assessors, respectively (Bruynesteyn 2005). The NRI method described in Section 6.3 will be used to impute missing data. Nonresponder imputation as randomized will also be used to impute missing data as a supportive approach to the primary method, where data observed at Week 52 will be analyzed according to randomized treatment group even if the Week 52 observation is obtained after the patient receives rescue or switch treatment. Patients with missing data at Week 52, as a result of study discontinuation or unreadable x-rays, will have their data imputed by NRI. The categorical analyses described above will be applied to both mitt population and a subset of mitt population that includes all randomized patients who received at least 1 dose of study drug, have non-missing baseline and at least 1 nonmissing postbaseline mtss measurement. For the analysis of the proportion of patients with change in mtss 0 and mtss SDC, additional imputation method will be applied by imputing the underlying continuous response of mtss scores using linear extrapolation or LOCF and then categorizing patients as a responder or non-responder after imputations.. Descriptive statistics for the actual score and change from baseline scores at Week 16, Week 24, and Week 52 in joint space narrowing and in bone erosion scores will be provided by visit and treatment group. In addition, ANCOVA analysis will be performed for the change from baseline scores at Weeks 16, 24, and 52 using the same model specified for mtss. Descriptive statistics for the proportion of patients achieving ACR20, ACR50, and ACR70 at each postbaseline visit through Visit 15 (Week 52) will be provided by visit
389 I4V-MC-JADV Statistical Analysis Plan Version 2 Page 51 and treatment group. A logistic regression with the same model specification as the primary analysis will be used for the treatment comparisons through Week 24 for ACR20, ACR50, and ACR70. The observed ( as-is ) values of the ACR20, ACR50, and ACR70 will be summarized as a sensitivity approach to the primary method of NRI. Using the as-is approach, the ACR values are calculated without applying any imputation to the components. Descriptive statistics for the percent change from baseline in individual components of the ACR core set, including TJC68, SJC66, patient s assessment of arthritis pain, patient s global assessment of disease activity, physician s global assessment of disease activity, and hscrp at postbaseline visits will be provided by visit and treatment group. An ANCOVA model with region, joint erosion status, treatment group, and baseline value of each individual component of the ACR core set in the model will be used to compare baricitinib to placebo in the percent change in individual components of the ACR core set at all postbaseline visits through Week 24. The mlocf approach described in Section will be used to impute missing data. The observed values will be analyzed as a sensitivity approach to the primary method that utilizes mlocf. Descriptive statistics for the actual score and change in DAS28-hsCRP and in DAS28- ESR from baseline to postbaseline visits will be provided by visit and treatment group. Change from baseline to postbaseline visits through Week 24 in DAS28-hsCRP and in DAS28-ESR will be analyzed using an ANCOVA model with treatment, region, joint erosion status, and baseline score in the model to test the treatment difference between baricitinib and placebo. The mlocf approach described in Section will be used to impute missing data. The observed values will be analyzed as a sensitivity approach to the primary method that utilizes mlocf. Descriptive statistics for the actual score and change in HAQ-DI from baseline to postbaseline visits will be provided by visit and treatment group. Change from baseline to postbaseline visits through Week 24 in HAQ-DI score will be analyzed using an ANCOVA model with treatment, region, joint erosion status, and baseline score in the model to test the treatment difference between baricitinib and placebo. The mlocf approach described in Section will be used to impute missing data. The observed values will be analyzed as a sensitivity approach to the primary method that utilizes mlocf. Descriptive statistics for the actual score and change in CDAI and in SDAI from baseline to postbaseline visits will be provided by visit and treatment group. Change from baseline in CDAI and in SDAI through Week 24 will be analyzed using ANCOVA. The model for the ANCOVA analysis will include treatment, region, and joint erosion status as independent fixed effects and the baseline score as a covariate with change from baseline SDAI or CDAI as the dependent variable. The mlocf approach described in Section will be used to impute missing data. The observed values will be analyzed as a sensitivity approach to the primary method that utilizes mlocf.
390 I4V-MC-JADV Statistical Analysis Plan Version 2 Page 52 Descriptive statistics for the proportion of patients achieving SDAI score 3.3 (ACR/EULAR remission index-based definition) and CDAI score 2.8 through Week 52 will be provided by visit and treatment group. Remission based upon SDAI score 3.3 (Felson et al. 2011) through Week 24 and remission based upon CDAI score 2.8 through Week 24 will be analyzed using a logistic regression model with region, joint erosion, and treatment as fixed effects. The proportion of patients achieving CDAI score 10 and proportion of patients achieving SDAI score 11 will also be analyzed through Week 24 and summarized through Week 52. The observed values will be analyzed as a sensitivity approach to the primary method that utilizes NRI. Descriptive statistics for the proportion of patients achieving European League Against Rheumatism (EULAR) Responder Index based on the 28-joint count at each postbaseline visit will be provided by visit and treatment group. European League Against Rheumatism nonresponders and responders (moderate + good responders) will be summarized. The NRI method will be used as the missing data imputation technique. A logistic regression analysis will be performed to compare the proportion of EULAR responders between baricitinib and placebo through Week 24. The model will consist of region, joint erosion, and treatment as fixed effects. The analysis will be repeated to compare the treatment groups in proportion of patients achieving good responses. Apart from the aforementioned categories, a summary table with frequencies and percentages will be presented for the patients categorized as good responder, moderate responder, and nonresponder, and the treatment comparison between baricitinib and placebo will be conducted using a Cochran-Mantel-Haenszel (CMH) test controlling for the region effect. The observed values will be analyzed as a sensitivity approach to the primary method that utilizes NRI. Descriptive statistics for the proportion of patients achieving ACR/EULAR remission according to the Boolean-based definition by treatment group at postbaseline visits will be provided by visit and treatment group. A logistic regression analysis will be used and defined similar to what was described for SDAI or CDAI remission. The observed values will be analyzed as a sensitivity approach to the primary method that utilizes NRI. Descriptive statistics for the Hybrid ACR (bounded) response at all postbaseline visits will be provided by visit and treatment group. An ANOVA model with region, joint erosion status, and treatment group as fixed effects and the postbaseline hybrid ACR response as the dependent variable will be used to compare baricitinib to placebo through Week 24. The mlocf approach described in Section will be used to impute missing data. The observed values will be analyzed as a sensitivity approach to the primary method that utilizes mlocf. Descriptive statistics for actual score and change from baseline will be provided by visit and treatment group for TJC68, SJC66, patient s assessment of arthritis pain, patient s global assessment of disease activity, physician s global assessment of disease activity, and hscrp. The change from baseline through Week 24 will be analyzed using ANCOVA, including treatment, region, and baseline joint erosion status as fixed effects
391 I4V-MC-JADV Statistical Analysis Plan Version 2 Page 53 and the baseline measurement as a covariate with the change from baseline as the dependent variable. The mlocf approach described in Section will be used to impute missing data. The observed values will be analyzed as a sensitivity approach to the primary method that utilizes mlocf. Descriptive statistics of the actual score and change from baseline in duration of morning joint stiffness, severity of tiredness using the Worst Tiredness NRS, and severity of pain using the Worst Pain NRS from baseline to postbaseline visits will be provided by visit and treatment group. The change from baseline to postbaseline visits through Week 24 will be analyzed using data obtained from the epro tablet by ANCOVA to test the treatment difference between baricitinib and placebo. Descriptive statistics for the actual score, changes from baseline, and percent changes from baseline will be provided by visit and treatment group for TJC28 and SJC28. Change from baseline and percent change from baseline measures through Week 24 will be analyzed using ANCOVA. The model for the ANCOVA will include treatment, region, and joint erosion status as fixed effects, baseline as a covariate, and change from baseline or percent change from baseline as the dependent variable. The mlocf approach described in Section will be used to impute missing data. The observed values will be analyzed as a sensitivity approach to the primary method that utilizes mlocf. Descriptive statistics for the actual score, change from baseline, and percent change from baseline will be provided by visit and treatment group for ESR. Change and percent change from baseline through Week 24 will be analyzed using ANCOVA. The model for the ANCOVA will include treatment, region, and joint erosion status as fixed effects, baseline as a covariate, and change from baseline or percent change from baseline as the dependent variable. The mlocf approach described in Section will be used to impute missing data. The observed values will be analyzed as a sensitivity approach to the primary method that utilizes mlocf. Descriptive statistics for the actual score and changes from baseline will be provided by visit and treatment group for ACPA and RF. Change from baseline through Week 24 will be analyzed using ANCOVA. The model for the ANCOVA will include treatment, region, and joint erosion status as fixed effects, baseline as a covariate, and change from baseline from baseline as the dependent variable. The mlocf approach described in Section will be used to impute missing data. The observed values will be analyzed as a sensitivity approach to the primary method that utilizes mlocf. In addition, shift tables of baseline seropositivity to postbaseline seropositivity will be produced for each postbaseline visit. For these key secondary continuous efficacy measures, tipping point analyses will be utilized to further investigate the impact that varying the magnitude of assumptions concerning imputation has on the robustness of the conclusions.
392 I4V-MC-JADV Statistical Analysis Plan Version 2 Page 54 For all the efficacy analyses, formal statistical tests will also be provided for comparison between the adalimumab and the placebo using data through Week 24. The comparison between baricitinib and adalimumab will be discussed in the exploratory analysis section (Section 6.15) Efficacy Analyses in Follow-up Period Summaries of efficacy data collected during the post-treatment follow-up period (Part C) will be presented separately from the main summaries (Parts A and B). The efficacy measures to be summarized include DAS28-hsCRP, DAS28-ESR, ACR component scores (TJC68, SJC66, patient s assessment of arthritis pain, patient s global assessment of disease activity, physician s global assessment of disease activity, HAQ-DI, and hscrp), CDAI and SDAI. The summary statistics of these measures at study baseline, follow-up baseline, at follow-up visit, and for the changes from study baseline and from follow-up baseline to follow-up visit will be presented based on the groups defined in Section 6.1 for the Follow-up population. The follow-up baseline is defined as the data collected at the last visit in Parts A and B prior to entering the follow-up period. Patients who have data for both the follow-up baseline and the follow-up visit are included in the analysis. Some other analyses for efficacy measures will be explored as well Testing Strategy to Control the Type I Family-Wise Error Rate The testing procedure of the primary and key secondary endpoints was reorganized to reduce the risk of order misspecification. The graphical multiple testing procedure in Bretz et al (2011) will be used to test the primary and key secondary endpoints. It is a closed testing procedure, hence it strongly controls the familywise error rate (FWER) across all endpoints. In comparison to the arrangement outlined in the protocol, the primary endpoint did not change and there were no key secondary objectives added or removed within the graphical multiple testing procedure. There are a total of 11 hypotheses to be tested: Family 1 (Figure JADV.6.1): H 1 : Proportion of patients achieving ACR20 at Week 12, baricitinib vs placebo H 2 : Mean change from baseline in mtss at Week 24, baricitinib vs placebo H 3 : Mean change from baseline in HAQ-DI at Week 12, baricitinib vs placebo H 4 : Mean change from baseline in DAS28-hsCRP at Week 12, baricitinib vs placebo H 5 : Proportion of patients achieving SDAI 3.3 at Week 12, baricitinib vs placebo H 6 : Proportion of patients achieving ACR20 at Week 12, baricitinib vs adalimumab (noninferiority) H 7 : Mean change from baseline in DAS28-hsCRP at Week 12, baricitinib vs adalimumab
393 I4V-MC-JADV Statistical Analysis Plan Version 2 Page 55 H 8 : Mean duration of morning joint stiffness (ediary) in the 7 days prior to Week 12, baricitinib vs placebo. Family 2 (Figure JADV.6.2): H 9 : Mean severity of morning joint stiffness (ediary) in the 7 days prior to Week 12, baricitinib vs placebo H 10 : Mean Worst Tiredness NRS (ediary) in the 7 days prior to Week 12, baricitinib vs placebo H 11 : Mean Worst Pain NRS (ediary) in the 7 days prior to Week 12, baricitinib vs placebo The sequentially rejective weighted Bonferonni multiple testing procedure is completely specified by Figure JADV.6.1 and Figure JADV.6.2. Specifically, the testing procedure begins with Family 1 as shown in Figure JADV.6.1. The primary endpoint H1 will first be tested at α=0.05. If successful, α will be propagated to H2 and H3 according to the weights on the corresponding edges (50% for each edge). The testing process continues for the remaining endpoints (H4 through H8) in Figure JADV.6.1 as long as there is at least one hypothesis that can be rejected at its allocated α level. Each time a hypothesis is rejected, the graph is updated to reflect the reallocation of α, which is considered recycled by Alosh et al. (2014). If at least one hypothesis associated with endpoint H7 or H8 is rejected, the remaining α will be used to test the remaining endpoints in Family 2 (H9, H10, and H11) using a similar testing procedure as shown in Figure JADV.6.2. This iterative process of updating the graph and reallocating α is repeated until all hypotheses have been tested, or when no remaining hypotheses can be rejected at their corresponding α levels.

394 I4V-MC-JADV Statistical Analysis Plan Version 2 Page 56 Figure JADV.6.1. Illustration of graphical multiple testing procedure within Family 1 for Study JADV.
 All PK/PD analyses will be performed by Lilly and documented in a separate SAP. 6.14.")
395 I4V-MC-JADV Statistical Analysis Plan Version 2 Page 57 Figure JADV.6.2. Illustration of graphical multiple testing procedure within Family 2 for Study JADV Pharmacokinetic/Pharmacodynamic Analysis (PK/PD) All PK/PD analyses will be performed by Lilly and documented in a separate SAP Health Outcomes Analyses Psychometric analyses will be performed by Lilly Global Health Outcomes and documented in a separate SAP Diary Data Data Handling and Missing Data Imputation The diary data (duration of morning joint stiffness, severity of morning joint stiffness, worst joint pain, worst tiredness, and recurrence of morning joint stiffness during the day) will be assessed daily from Week 0 (Visit 2) to Week 12 (Visit 7). The primary analysis is based on the scores collected in the 7 days prior to the Week 12 visit date, which addresses one of the major secondary objectives included in the graphical multiple testing procedure. Similar analysis will be done for 7 days prior to each scheduled visit (Week 1, Week 2, Week 4, and Week 8) using the primary analysis method. The supportive analysis is based on daily data modeled by weekly means. The weeks are defined as follows: Day 1 on diary, Days 2-8 (Week 1), Days 9-15 (Week 2), and so on up to Days (Week 12).
396 I4V-MC-JADV Statistical Analysis Plan Version 2 Page 58 For the primary analysis, the average of the scores collected in the 7 days prior to Week 12 will be used. If there are fewer than 4 nonmissing scores, the 7-day window will be shifted back in time (toward baseline) one day at a time until there are 4 nonmissing scores available in the 7- day window. Then the average of the 4 scores will be used in the Week 12 analysis. This will be repeated for each scheduled visit (Week 1, Week 2, Week 4, Week 8) prior to Week 12. For patients who discontinue the study or permanently discontinue the study treatment, the scores collected in the last 7 days prior to discontinuation will be used in the analysis. If a patient undergoes a temporary interruption of study drug at the time of study database lock, this patient will be treated as having permanently discontinued from study drug in the missing data imputation. If there are fewer than 4 nonmissing scores, a similar approach will be used to find 4 nonmissing scores in a 7-day window and the average score will be used in the Week 12 analysis. Patients who do not have at least 4 scores in any postbaseline 7-day window up to Week 12 will have their Day 1 score carried forward to the endpoint for analysis. For patients without a Day 1 score, the last available value up to Day 1 will be used to impute the Day 1 value. If the Day 1 value cannot be obtained through the aforementioned approach, the first available postbaseline score will be used to impute the Day 1 score so that the patient can be included in the analysis. For the analysis at each scheduled visit using the primary analysis method, the same imputation rule for the primary analysis will be used. If there are not at least 4 measurements in that window, we slide the window back 1 day at a time until we obtain 4 measurements. This derivation will be repeated so that 7-day means for Week 11, Week 10,, Week 1 are derived. The imputed value for missing Day 1 measures from postbaseline assessments as described in Section will not be used in the change from Day 1 analysis. For the supportive analysis, if 4 scores during a defined week are missing, the daily scores collected in the week will not be used in the analysis as a protective measure against potentially unreliable or non-representative data Multiple Imputation (MI) For the sensitivity analysis of duration of morning joint stiffness ediary data, a two-step multiple imputation (MI) method will be used to impute any missing weekly durations. The first step will be to create a monotone missing pattern using a Markov chain Monte Carlo method (SAS Proc MI with MCMC option) to handle intermittent missing weekly durations. The second step will be to use a set of Bayesian regressions (using SAS Proc MI with MONOTONE option) for the imputation of monotone dropouts for the weekly durations. The regression models will be fit sequentially starting from the first week with at least one missing response for weekly durations with treatment as a fixed effect and weekly durations in the previous weeks as covariates Analysis of Diary Data The effect of baricitinib compared to placebo up to Week 12 with regard to patient-reported outcomes (PROs) will be evaluated by the following measures: Duration of morning joint stiffness Duration of morning joint stiffness is a patient-administered item that allows for the patients to enter the length of time in minutes that their morning joint stiffness lasted each day (using
397 I4V-MC-JADV Statistical Analysis Plan Version 2 Page 59 an electronic handheld diary) through Visit 7 (Week 12). Duration of morning stiffness will be recorded in minutes. Durations recorded as longer than 12 hours (720 minutes) will be truncated to 720 minutes for analysis. For the analysis used in the graphical multiple testing procedure (Section ), a nonparametric method involving the Hodges-Lehmann estimator will be used to test the treatment difference in the average durations (of morning joint stiffness) collected in the 7 days prior to Week 12. For the comparison between the baricitinib group and the placebo group, the median score and its 100*(1-alpha)% CI from the Hodges-Lehmann estimator will be presented. The p-value for the median difference will be calculated using the Wilcoxon rank-sum test. A 100*(1-alpha)% CI for the median score within each treatment group will be calculated using distribution-free confidence limits. Missing data will be handled as described for the main analysis in Section This analysis of duration of morning joint stiffness will be conducted using the mitt population. The PP population results will be used as supportive evidence to the mitt results. The primary analysis of duration of morning joint stiffness will also be repeated for each scheduled visit using the mitt population. In addition, the average duration (of morning joint stiffness) collected in the 7 days prior to Week 12 change from Day 1 on diary will be compared between the baricitinib group and the placebo group using the same analysis method described. The imputed value for missing Day 1 durations from postbaseline assessments as described in Section will not be used in the change from Day 1 analysis. For the supportive analysis to duration of morning joint stiffness, the weekly ediary data will be analyzed based on using a 3-step process as a supportive analysis. First, missing weekly data will be imputed using a MI approach as described in Section Second, based on the results from the Phase 2 data, it is anticipated that the distribution of data is skewed; therefore, a robust regression model will be used to compare baricitinib to placebo at each week through Week 12 using M-estimation (using SAS Proc ROBUSTREG) with fixed effects and covariates for treatment and Day 1 duration. Finally, the combined mean estimates of the treatment difference with their corresponding SEs, 95% CIs and p-value will be provided through the SAS MIANALYZE procedure using Rubin s Method (Rubin 1987). Severity of morning joint stiffness numeric rating scale (NRS) Severity of morning joint stiffness NRS is a patient-administered, single-item, 11-point horizontal scale anchored at 0 and 10, with 0 representing no joint stiffness and 10 representing joint stiffness as bad as you can imagine. Patients rate their morning joint stiffness each day (using an electronic handheld diary) up to Visit 7 (Week 12) by selecting one number that describes their overall level of joint stiffness at the time they woke up. For the analysis used in the graphical multiple testing procedure (Section ), an ANCOVA model will be used to test for treatment differences in the severity of morning joint stiffness NRS collected in the 7 days prior to Week 12, with treatment, region and baseline joint erosion status (1-2 joint erosions plus seropositivity vs. at least 3 joint erosions)
398 I4V-MC-JADV Statistical Analysis Plan Version 2 Page 60 as fixed effects and Day 1 value as a covariate. Missing data will be handled as described for the primary analysis in Section Type III sums of squares for the LS means will be used for the statistical comparison; the 100*(1-alpha)% CI will also be reported. The alpha level will be defined according to the graphical multiple testing procedure described in Section The LS mean difference, SE, CI, and p-value will be presented for the treatment difference between the baricitinib group and the placebo group. This analysis of severity of morning joint stiffness NRS will be conducted using the mitt population. The PP population results will be used as supportive evidence to the mitt results. The primary analysis of severity of morning joint stiffness NRS will also be repeated for each scheduled visit using the mitt population. A supportive analysis using a MMRM model will be performed to estimate the treatment differences in severity of morning joint stiffness NRS. Missing data will be handled as described for the supportive analysis in Section The severity of morning joint stiffness will be summarized by treatment group using weekly means. Treatment comparisons between baricitinib and placebo will be analyzed and presented through Week 12 using a MMRM model (refer to Section 6.1 for more details) including treatment, Day 1 value as covariate, and study week, and the interactions of baseline-by-week and treatment-by-week as fixed factors. Tiredness severity NRS ( Worst Tiredness ) Worst tiredness NRS is a patient-administered, single-item, 11-point horizontal scale anchored at 0 and 10, with 0 representing no tiredness and 10 representing as bad as you can imagine. Patients rate their tiredness each day (using an electronic handheld diary) through Visit 7 (Week 12) by selecting the 1 number that describes their worst level of tiredness during the past 24 hours. The primary analysis of Worst tiredness will be analyzed in a similar manner as described for severity of morning joint stiffness NRS. This primary analysis of Worst tiredness will be conducted using the mitt population. The PP population results will be used as supportive evidence to the mitt results. The primary analysis of Worst tiredness will also be repeated for each scheduled visit using the mitt population. In addition, supportive analyses will be conducted using the same methods described for severity of morning joint stiffness NRS. Pain severity NRS ( Worst Joint Pain ) Worst joint pain NRS is a patient-administered, single-item, 11-point horizontal scale anchored at 0 and 10, with 0 representing no pain and 10 representing pain as bad as you can imagine. Patients rate their joint pain each day (using an electronic handheld diary) through Visit 7 (Week 12) by selecting the 1 number that describes their worst level of joint pain during the past 24 hours. The primary analysis of Worst joint pain will be analyzed in a similar manner as described for severity of morning joint stiffness NRS. This primary analysis of Worst joint pain will
399 I4V-MC-JADV Statistical Analysis Plan Version 2 Page 61 be conducted using the mitt population. The PP population results will be used as supportive evidence to the mitt results. In addition, supportive analyses will be conducted using the same methods described for severity of morning joint stiffness NRS. Recurrence of joint stiffness during the day Recurrence of joint stiffness throughout the day is a patient-administered single-item question assessed each day (using an electronic handheld diary) through Visit 7 (Week 12) that asks Did you have any joint stiffness throughout today, other than when you woke up? (Yes/No). Recurrence of stiffness during the day will be summarized as a percentage of days with the event at weekly intervals (as defined in Section ) up to Visit 7 (Week 12). Percentage of days will be analyzed by week using ANCOVA with the same model specification described for duration of morning joint stiffness. Missing data imputation will follow what was described for the supportive analysis in Section These analyses may be repeated for the subset of patients who enroll within the study after morning joint stiffness, worst tiredness, and worst pain were added to the measures captured at postbaseline site visits via the epro tablet in Amendment (c) of Study JADV protocol. Formal statistical tests will also be provided for the comparison between the adalimumab group and the placebo group for all the aforementioned diary data analyses. Comparisons between baricitinib and adalimumab will be discussed in the exploratory analysis section (Section 6.15) Analysis of Non-Diary Data The effect of baricitinib compared to placebo from baseline up to Week 24 and to Week 52 in terms of descriptive summaries with regard to patient-reported outcomes (PROs) will be evaluated by the following measures: Duration of Morning Joint Stiffness The duration of morning joint stiffness is a patient-administered item that allows for the patients to enter the length of time in minutes that their morning joint stiffness lasted on the day prior to that visit using an electronic patient-reported outcomes (epro) tablet. Durations recorded as longer than 12 hours (720 minutes) will be truncated to 720 minutes for analysis. Duration of morning joint stiffness will be measured at baseline and at postbaseline visits. Summary statistics of the actual score and change from baseline will be displayed by treatment and protocol-specified visit. The comparison between baricitinib and placebo in change from baseline through Week 24 in duration of morning joint stiffness will be analyzed using a nonparametric method involving the Hodges-Lehmann estimator. For the comparison between baricitinib and placebo, the median difference of change from baseline score and its 95% CI from the Hodges-Lehmann estimator will be presented. The p-value for the median difference of change from baseline will be calculated using the Wilcoxon rank-sum test. A 95% CI for the median change from baseline score within each treatment group will be calculated using
400 I4V-MC-JADV Statistical Analysis Plan Version 2 Page 62 distribution-free confidence limits. The mlocf approach will be used to impute missing data. The observed values will be analyzed as a sensitivity approach to the primary method that utilizes mlocf. Tiredness Severity Numeric Rating Scale (Worst Tiredness NRS) The Worst Tiredness NRS is a patient-administered, single-item, 11-point horizontal scale anchored at 0 and 10, with 0 representing no tiredness and 10 representing as bad as you can imagine. Patients rate their tiredness by selecting the one number that describes their worst level of tiredness during the past 24 hours using an electronic patient-reported outcomes (epro) tablet. Worst Tiredness NRS will be measured at baseline and at postbaseline visits. Summary statistics of the actual score and change from baseline will be displayed by treatment and protocol-specified visit. The change from baseline in Worst Tiredness NRS to Week 24 will be analyzed using ANCOVA. The ANCOVA model will consider treatment, region, and joint erosion status as fixed effects and baseline score as a covariate with change from baseline being the dependent variable. To test the treatment difference between baricitinib and placebo, the LS mean difference, 95% CI, and p-value will be presented. The mlocf approach will be used to impute missing data. The observed values will be analyzed as a sensitivity approach to the primary method that utilizes mlocf. Severity of Joint Pain Numeric Rating Scale (Worst Pain NRS) The Worst Pain NRS is a patient-administered, single-item, 11-point horizontal scale anchored at 0 and 10, with 0 representing no pain and 10 representing pain as bad as you can imagine. Patients rate their pain by selecting the one number that describes their worst level of pain during the past 24 hours using an electronic patient-reported outcomes (epro) tablet. Worst Pain NRS will be measured at baseline and at postbaseline visits. Summary statistics of the actual score and change from baseline will be displayed by treatment and protocolspecified visit. The change from baseline in Worst Pain NRS to Week 24 will be analyzed using ANCOVA. The ANCOVA model will consider treatment, region, and joint erosion status as fixed effects and baseline score as a covariate with change from baseline being the dependent variable. To test the treatment difference between baricitinib and placebo, the LS mean difference, 95% CI, and p-value will be presented. The mlocf approach will be used to impute missing data. The observed values will be analyzed as a sensitivity approach to the primary method that utilizes mlocf. These analyses for change from baseline through Week 24 in duration of morning joint stiffness, severity of morning joint stiffness, worst joint pain, and worst tiredness will be based on the subset of patients who enroll within the study after these measures were added to measures captured at postbaseline site visits via the epro tablet in Amendment (c) of Study JADV
401 I4V-MC-JADV Statistical Analysis Plan Version 2 Page 63 protocol (that is, in patients who have both baseline and postbaseline data). For the complementary subset of patients who enroll within the study before the measures were added to the epro tablet data collection in Amendment (c) of Study JADV protocol (that is, patients who have no baseline data but only postbaseline data), the summary statistics of the observed values of these epro tablets outcomes will be reported. FACIT-F The FACIT-F scale (Cella and Webster 1997) is a 13-item, symptom-specific questionnaire that specifically assesses the self-reported severity of fatigue and its impact upon daily activities and functioning. The FACIT-F uses 0 ( not at all ) to 4 ( very much ) numeric rating scales to assess fatigue and its impact in the past 7 days. Scores range from 0 to 52 with higher scores indicating less fatigue. FACIT-F scores will be measured at baseline and at postbaseline visits. Summary statistics of the actual score and change from baseline will be displayed by treatment and protocol-specified visit. The change from baseline in FACIT-F score to Week 24 will be analyzed using ANCOVA. The ANCOVA model will consider treatment, region, and joint erosion status as fixed effects and baseline score as a covariate with change from baseline being the dependent variable. To test the treatment difference between baricitinib and placebo, the LS mean difference, 95% CI, and p-value will be presented. The mlocf approach will be used to impute missing data. The observed values will be analyzed as a sensitivity approach to the primary method that utilizes mlocf. The Minimum Clinically Important Difference (MCID) for the FACIT-F score is defined as 3.56 improvement (increase) from the baseline value of the Fatigue Subscale Score. The summary and analysis of the FACIT-F score will include the number and percent of patients achieving MCID at each postbaseline visit. The proportion of patients achieving the FACIT- F score MCID will also be analyzed using a logistic regression model with treatment, region, and joint erosion status as fixed effects. The p-value and 95% CI of the odds ratio from the logistic regression model will be reported. The 95% CI of MCID rate difference will be calculated using the Newcombe-Wilson method without continuity correction. The NRI method described in Section will be used to impute missing data. SF-36v2 Acute Summaries of domain and summary scores (scoring details in Appendix 2) will be provided by visit and change from baseline to postbaseline visits. An ANCOVA model will be used to analyze the change from baseline in the physical component score (PCS) and the mental component score (MCS) through Week 24. The ANCOVA model will include treatment, region, and joint erosion status as fixed effects and baseline score as a covariate with change from baseline as the dependent variable. To test the treatment difference between baricitinib and placebo, the LS mean difference, 95% CI, and p-value will be presented. The mlocf approach will be used to impute missing data.
402 I4V-MC-JADV Statistical Analysis Plan Version 2 Page 64 The observed values will be analyzed as a sensitivity approach to the primary method that utilizes mlocf. Analyses will be repeated for the eight health domains (physical functioning, bodily pain, role limitations due to physical problems, role limitations due to emotional problems, general health perceptions, mental health, social function, and vitality). Summary statistics per treatment group will be tabulated for the transformed scale scores (physical functioning, role limitations / physical health, bodily pain, general health perceptions, vitality, social functioning, role limitations / emotional, mental health) and the two summary scores (PCS and MCS). Minimum Clinically Important Differences (MCID): The summary and analyses of the SF-36v2 Acute Score will include the number of patients achieving a MCID, which is defined for each summary score, physical component score (PCS) and the mental component score (MCS), as: MCID at least 5-point improvement (decrease) in PCS MCID at least 5-point improvement (decrease) in MCS. The proportion of patients achieving the SF-36v2 score MCID will also be analyzed using a logistic regression model with treatment, region, and joint erosion status as fixed effects. The p-value and 95% CI of the odds ratio from the logistic regression model will be reported. The 95% CI of the MCID rate difference will be calculated using the Newcombe-Wilson method without continuity correction. The NRI method described in Section will be used to impute missing data. WPAI-RA The WPAI-RA questionnaire was developed to measure the effect of general health and symptom severity on work productivity and regular activities in the last 7 days of the visit. It contains 6 items covering overall work productivity (health), overall work productivity (symptom), impairment of regular activities (health), and impairment of regular activities (symptom). Scores are calculated as impairment percentages (Reilly et al. 1993). The WPAI-RA yields four types of scores: o o o o Absenteeism (work time missed) Presenteeism (impairment at work / reduced on-the-job effectiveness) Work productivity loss (overall work impairment / absenteeism plus presenteeism) Activity Impairment Six questions will be answered: o Q1 = currently employed
403 I4V-MC-JADV Statistical Analysis Plan Version 2 Page 65 o o o o o Q2 = hours missed due to specified problem Q3 = hours missed due to other reasons Q4 = hours actually worked Q5 = degree that RA affected productivity while working Q6 = degree that RA affected regular activities WPAI-RA outcomes are expressed as impairment percentages, with higher numbers indicating greater impairment and less productivity, ie, worse outcomes, as follows: o Percentage work time missed due to RA absenteeism : Q2/(Q2+Q4) o Percentage impairment while working due to RA presenteeism : Q5/10 o Percentage overall work impairment due to RA work productivity loss : Q2/(Q2+Q4)+[(1-Q2/(Q2+Q4))x(Q5/10)] o Percentage activity impairment due to RA activity impairment : Q6/10 o Multiply scores by 100 to express in percents. WPAI-RA scores will be measured at baseline and at postbaseline visits. The proportion of patients who were employed versus not employed at baseline will be summarized, along with by-visit summaries of changes in employment status (i.e. proportion of patients employed at baseline who are still employed at the postbaseline visit and the proportion of patients not employed at baseline that remained unemployed at the postbaseline visit). The analyses of absenteeism, presenteeism, and work productivity loss will be based on patients employed at both baseline and postbaseline visits. The analysis of activity impairment will be based on the mitt population. Summary statistics of the actual and change from baseline scores based on absenteeism, presenteeism, work productivity loss, and activity impairment will be displayed by treatment and protocol-specified visit. The change from baseline in each of the four types of scores of WPAI-RA (absenteeism, presenteeism, work productivity loss, and activity impairment) from baseline to Week 24 will be analyzed using ANCOVA. The ANCOVA model will consider treatment, region, and joint erosion status as independent fixed effects and the baseline score as a covariate with change from baseline being the dependent variable. To test the treatment difference between baricitinib and placebo, the LS mean difference, 95% CI, and p-value will be presented. European Quality of Life 5 Dimensions 5 Level (EQ-5D-5L) The EQ-5D-5L is a standardized measure of health status of the patient at the visit (same day) that provides a simple, generic measure of health for clinical and economic appraisal. The EQ-5D-5L consists of 2 components: a descriptive system of the respondent s health and a rating of his or her current health state using a 0- to 100-mm VAS. The descriptive system comprises the following 5 dimensions: mobility, self-care, usual activities, pain/discomfort, and anxiety/depression. Each dimension has 5 levels: no problems, slight
404 I4V-MC-JADV Statistical Analysis Plan Version 2 Page 66 problems, moderate problems, severe problems, and extreme problems. The respondent is asked to indicate his/her health state by ticking (or placing a cross) in the box associated with the most appropriate statement in each of the 5 dimensions. It should be noted that the numerals 1 to 5 have no arithmetic properties and should not be used as a cardinal score. The VAS records the respondent s self-rated health on a vertical VAS in which the endpoints are labeled best imaginable health state and worst imaginable health state. This information can be used as a quantitative measure of health outcome. The EQ-5D-5L health states, defined by the EQ-5D-5L descriptive system, may be converted into a single summary index by applying a formula that essentially attaches a value (also called weights) to each of the levels in each dimension (Brooks 1996; EuroQol Group 2011 [WWW]; Herdman et al. 2011). The EQ-5D-5L questionnaire is self-completed by the patient in this study at baseline and postbaseline visits through Week 52 (Visit 15). The EQ-5D-5L will be scored according to the developer s instructions (scoring guidelines). Three outcomes are described for each treatment group by visit according to the following: A health profile: summary statistics will be provided, including number of patients and proportions of categorical responses for the 5 dimensions (mobility, self-care, usual activity, pain/discomfort, and anxiety/depression). A health state index score: the UK algorithm (Szende et al. 2006) will be used to produce a patient-level index score between and 1.0 (continuous variable); the same will be done using the US algorithm which produces a patient-level index score between and 1.0. A self-perceived current health score: calculated from patient level EQ VAS responses (continuous variable). The change in health state index scores and EQ VAS score from baseline through Week 24 (Visit 11) will be analyzed using an ANCOVA model with the following terms: treatment, region, joint erosion status, and baseline score. Within each treatment arm, the LS mean, SE of the LS mean, and p-value will be reported. The LS mean difference between baricitinib and placebo, 95% CI for the mean difference, and p-value will be reported. ANCOVA analysis will be computed after imputing the postbaseline missing values using mlocf. The observed values will be analyzed as a sensitivity approach to the primary method that utilizes mlocf. Healthcare Resource Utilization The healthcare resource utilization data will be collected by site staff regarding the number of visits to medical care providers such as general practitioners, specialists, physical or occupational therapists, and other non-physical care providers for services outside of the clinical study; emergency room admissions; hospital admissions; and concomitant medications related to the treatment of RA. These data will be collected to support economic evaluations of treatment. A summary table including the following will be presented for the period of the past two months prior to study treatment as baseline and summarized at
405 I4V-MC-JADV Statistical Analysis Plan Version 2 Page 67 baseline and at postbaseline intervals (Week 0 to Week 12, Week 0 to Week 24, Week 24 to Week 52, and Week 0 to Week 52). Healthcare resource utilization Healthcare providers: A summary of the mean number of consultations of medical care providers for care/treatment of RA will be provided for the period of the past 2 months prior to the study treatment. It will also be summarized at the postbaseline intervals described earlier. The number of consultations will be totaled by days and patients for the categories: o o Healthcare Provider Consultation Emergency Room Consultation The Healthcare Provider Consultation comprises the following: General Practitioner (Such as family doctor or primary care physician) Specialist Rheumatologist Occupational Therapist Physical therapist Non-physician care providers For the Emergency Room Consultation every necessary Emergency Room (ER) or equivalent facility consultation will be counted. Healthcare resource utilization Inpatient Hospitalization: A summary table including the following will be presented from the period of the past two months prior to study treatment and at postbaseline intervals for the following details: o o o Frequency of patient s hospitalization and need for staying overnight because of their rheumatoid arthritis. Frequency of hospitalization type (general ward / intensive care ward). Descriptive statistics for the total number of days in hospital. Healthcare resource utilization Work Status: A frequency table for the non-missing statements on work status for the period of the past 2 months prior to study treatment and at postbaseline intervals will be provided for the following details: o o o Working for pay full-time (> 36 hours per week) Working for pay part-time ( 36 hours per week) Unemployed, unrelated to study disease disability
406 I4V-MC-JADV Statistical Analysis Plan Version 2 Page 68 o o Unemployed due to study disease disability Other [Keeping house (full-time), Student (full-time), Student (part-time), Retired, Volunteer work]. All the aforementioned health outcome analyses will be performed on the mitt population. Descriptive statistics will be provided for all the parameters by visit and treatment group for baseline and all postbaseline visits. Formal statistical tests will also be provided for comparison between the adalimumab group and the placebo group using data through Week 24. The comparison between baricitinib and adalimumab will be discussed in the exploratory analysis section (Section 6.15) Exploratory Analysis Comparisons between baricitinib and adalimumab will be performed as exploratory analyses for all efficacy and health outcome endpoints. In addition, the efficacy of Humira will be summarized as appropriate based on the purchased source of this material (e.g., US, EU, and mixed source) Safety Analyses Safety analyses for this study follow a hierarchy of importance: Of primary importance is the analysis of safety parameters during Part A wherein the comparison between baricitinib and placebo are of primary interest, informing risks to treatment that can be juxtaposed against the potential efficacy advantages shown over the same acute treatment time course. The baricitinib-placebo differences will be formally statistically compared, where applicable. Secondarily, a summary of the safety parameters associated with the patients treated with adalimumab will be provided and compared to placebo to complete the summary of safety for all randomized and treated patients. In addition, statistical comparison between baricitinib and adalimumab will also be provided. Data collected up to the time of rescue (if applicable) will be used in these comparisons. Of secondary importance is the analysis of safety parameters which includes data collected during Part B. If data from all patients participating in Part B are included, the trial is weakly-controlled for the baricitinib-to-adalimumab comparison since the baricitinib group contains rescued (but not randomized) patients and is subject to attrition bias in each treatment relative to Part A. Thus, analysis of Part B will be constrained to the comparison of baricitinib to adalimumab in the subset of patients randomized to those treatments over the entire combined period of Part A and Part B. Formal statistical comparisons are planned. Of tertiary importance will be summarization of safety parameters after inception of treatment with baricitinib, experienced from the moment of randomization, from the time of switching from treatment with either placebo or adalimumab to baricitinib, or from the point of rescue to treatment with baricitinib. As these comparisons do not result from some randomization, no formal statistical comparisons will be performed. These summaries will include all qualifying data from Parts A and B.
407 I4V-MC-JADV Statistical Analysis Plan Version 2 Page 69 Thus, safety results will generally be prepared according to 4 formats: Part A, Week 0 (Visit 2) to Week 12 (Visit 9) covering exposures to randomized treatments prior to the primary timepoint at Week 12, Part A, Week 0 (Visit 2) to Week 24 (Visit 11) covering all of Part A but with censoring for patients after the point of rescue, Parts A and B combined from Week 0 (Visit 2) to Week 52 (Visit 15) for patients randomized to adalimumab or baricitinib but with censoring after the point of rescue, Parts A and B combined from Week 0 (Visit 2) to Week 52 (Visit 15) for patients randomized to baricitinib and for patients who change treatment to baricitinib from either placebo or adalimumab covering all exposure to baricitinib. Safety analyses will take place using the Safety Population defined in Section Safety topics that will be addressed include the following: adverse events (AEs), clinical laboratory evaluations, vital signs and physical characteristics, safety in special groups and circumstances, including Adverse Events of Special Interest (AESI) (see Section ), and investigational product interruptions Adverse Events (AEs) Adverse events are recorded in the ecrfs. Each AE will be coded to system organ class (SOC) and preferred term (PT) using the Medical Dictionary for Regulatory Activities (MedDRA) using the MedDRA version that is current at the time of each database lock. Severity of AEs is recorded as mild, moderate, or severe. A treatment-emergent adverse event (TEAE) is defined as an event that either first occurred or worsened in severity after the first dose of study treatment and on or prior to the date of the last visit within the treatment period being analyzed. Adverse events are classified based upon the MedDRA PT. The MedDRA Lowest Level Term (LLT) will be used in defining which events are treatment-emergent. The maximum severity for each LLT during the baseline period until the first dose of the study medication will be used as baseline. The treatment period (Parts A and B) will be included as postbaseline for the analysis. If an event is pre-existing during the baseline period but it has missing severity, and the event persists or reoccurs during the treatment period, then the baseline severity will be considered mild for determining any postbaseline treatment-emergence (ie, the event is treatment-emergent unless the severity is coded as mild postbaseline). If an event occurring postbaseline has a missing severity rating, then the event is considered treatment-emergent. Should there be insufficient data for adverse event start date to make this comparison (for example, the adverse event start year is the same as the treatment start year, but the adverse event start month and day are missing), the adverse event will be considered treatment-emergent. For events occurring on the day of the first dose of study treatment, a case report form (CRF) will be used to document whether the onset of the event was pre-treatment versus post-treatment in order to derive treatment-emergence.
408 I4V-MC-JADV Statistical Analysis Plan Version 2 Page 70 For patients who were randomized to placebo and required rescue therapy or who were switched to baricitinib at the end of Part A, the AE severity just prior to rescue or switch to baricitinib will be used as baseline for TEAE during baricitinib treatment. Similarly, for patients who were randomized to adalimumab and required rescue therapy, the AE severity just prior to rescue to baricitinib will be used as baseline for TEAE during baricitinib treatment. Further, for some AEs of interest, duration of the TEAE may be summarized or listed within patient AE listings. The duration of each event will be defined as the end date of AE minus the start date of treatment-emergence of the AE plus 1. If an AE has not ended by the date of study drug discontinuation, early discontinuation of the study/period or completion of the study, it will be censored as of that appropriate final date. Duration of AEs may also be expressed conversely as the time to resolution of the AE. In general, summaries of TEAEs, SAEs, AEs that lead to discontinuation from the study and AEs that lead to temporary interruption or permanent discontinuation of study drug will include the number of patients in the safety population (N), frequency of patients experiencing the event (n), percent of patients experiencing the event (n/n*100), and the exposure-adjusted incidence rate (EAIR). The EAIR will be expressed as the number of patients experiencing an AE (n) per 100 patient years (PY) of exposure to treatment and are derived as 100 times the number of patients with the event divided by the sum of all patient exposure times (in years) to the treatment for the study/period. For any events that are gender-specific based on the displayed PT, the denominator used to compute the percentage and the total patient-years used for EAIR will only include patients from the given gender. Treatment comparisons between treatment groups will be made using the Fisher exact test. In an overview table, the number and percentage of patients in the safety population who experienced a death, an SAE (ICH-defined, protocol-defined, and ICH- or protocol-defined), discontinuation from the study due to an AE or death, permanent discontinuation from study drug due to an AE or death, temporary interruption from study drug due to an AE, temporary interruption of study drug due to a lab abnormality, a TEAE, a TEAE rated as severe, or an AE possibly related to study drug based on the opinion of the investigator will be summarized by treatment. The number and percent of patients with TEAEs will be summarized by treatment group using MedDRA PT nested within SOC. The SOCs will be ordered alphabetically, and events will be ordered within each SOC by decreasing frequency in the baricitinib group (or combined baricitinib group, as appropriate). As additional tables, the number and percent of patients with TEAEs will be summarized by treatment using MedDRA PT (without regard to SOC) and the number and percent of patients with TEAEs will be summarized by treatment using only the MedDRA SOC. Events will be ordered by decreasing frequency in the baricitinib group. Exposure-adjusted incidence rates will be displayed for these outcomes. Adverse events considered possibly related to study drug by the investigator will be summarized by treatment group using MedDRA PT nested within SOC. A possibly-related event is defined as an event recorded on the ecrf as Yes to the question Is the event possibly related to study
409 I4V-MC-JADV Statistical Analysis Plan Version 2 Page 71 drug? Events will be ordered by decreasing frequency in the baricitinib group (or combined baricitinib group, where appropriate), within alphabetical SOC. If the investigator s determination of possible relationship to study drug is missing, then the event will be considered as related. Follow-up emergent adverse events (FEAEs) will be established for the post-treatment follow-up period. An FEAE is an event that either first occurred or worsened in severity after the last visit during the treatment period. The baseline severity for FEAE is the severity level at the last visit from the treatment period; otherwise, FEAE processing is similar to TEAE processing. Followup AEs will be summarized in a similar manner as TEAEs (using MedDRA PT within SOC) according to groups defined by the treatment the patients were receiving prior to entering the follow-up period. An individual listing of all AEs including preexisting conditions will be provided. A separate listing will include all AEs that led to discontinuation from the study Common Adverse Events (AEs) The number and percent of patients with TEAEs, including the EAIR, will be summarized by treatment group using MedDRA PT nested within SOC for common TEAEs (i.e., TEAEs that occurred in 2% (before rounding) of any treatment group. Events will be ordered by decreasing frequency in the baricitinib group (or combined baricitinib group, where appropriate) within alphabetical SOC. The number and percent of patients with TEAEs will be summarized by maximum severity by treatment using MedDRA PT nested within SOC for the common TEAEs. For each patient and TEAE, the maximum severity for the MedDRA level being displayed (PT and SOC) is the maximum postbaseline severity observed from all associated LLTs mapping to that MedDRA level. For the "Severe" category, treatment comparisons between treatment groups will be made using the Fisher exact test for each PT and SOC Serious Adverse Events (SAEs) Consistent with the ICH E2A guideline, an SAE is any AE that results in one of the following outcomes: Death Initial or prolonged inpatient hospitalization A life-threating experience (ie, immediate risk of dying) Persistent or significant disability/incapacity Congenital anomaly/birth defect Considered significant by the investigator for any other reason. In addition, if any patient experiences an AE that requires permanent discontinuation from study drug, for an AE or abnormal laboratory result, that AE or abnormal laboratory value will be reported as an SAE (reported as Other reason serious ). These protocol-defined specific SAEs
410 I4V-MC-JADV Statistical Analysis Plan Version 2 Page 72 were defined as an AE or laboratory value that results in permanent study drug discontinuation or discontinuation from the study. These will be distinguished from ICH-defined SAEs using a scripted Clinical Data Question and Answer (CDQA) form to be completed by the investigator. The CDQA form will be sent as a query to the investigative site whenever an SAE is designated as serious based only on other reason serious. The site will provide a response that will explain if the SAE was serious by conventional ICH criteria or if it was designated as serious only by the protocol requirement. In the absence of a CDQA response, the SAE cannot be considered as serious only by the protocol requirement and will be included in SAEs that are serious according to conventional ICH criteria. The number and percent of patients who experienced any SAE (also ICH-defined and protocoldefined SAE) will be summarized by treatment group using MedDRA PT nested within SOC. Events will be ordered by decreasing frequency in the baricitinib group (or combined baricitinib group, where appropriate) within alphabetical SOC. Serious AEs overall and of each type will be summarized by treatment period, and during the post-treatment follow-up period. An individual listing of all SAEs will be provided Other Significant Adverse Events The number and percent of patients who discontinued from the study because of an AE or death, the number and percent of patients who permanently discontinued study drug because of an AE or death, and the number and percent of patients who temporarily interrupted study drug because of an AE will be summarized by treatment group using MedDRA PT nested within SOC. Events will be ordered by decreasing frequency in the baricitinib group (or combined baricitinib group, where appropriate) within alphabetical SOC. A summary of temporary interruptions of study drug (tablets and injections) will also be provided, showing the number of patients who experienced at least one temporary interruption and the number of temporary interruptions per patient with an interruption. Further, the duration of each temporary interruption (in days) and the cumulative duration of dose interruption (in days) using basic descriptive statistics (n, mean, SD, minimum, Q1, median, Q3 and maximum) will be displayed. Individual listings of all AEs leading to study discontinuation and to permanent discontinuation from the study drug will be provided. A listing of all temporary study drug interruptions, including interruptions for reasons other than AEs, will be provided Vital Signs and Physical Characteristics Vital signs and physical characteristics include systolic blood pressure (SBP), diastolic blood pressure (DBP), pulse, weight, BMI, and waist circumference. Triplicate assessments of SBP and DBP are collected at each visit. When replicate assessments are recorded on the ecrf, the average will be derived; otherwise, the average which is recorded on the ecrf will be used for the summaries. Only the average across the replicates or the average recorded on the ecrf will be analyzed. When these parameters are analyzed as continuous variables at each visit, unscheduled measurements will be excluded. When these parameters are analyzed as categorical outcomes, as treatment-emergent abnormalities, and/or as continuous variables ignoring the visit
411 I4V-MC-JADV Statistical Analysis Plan Version 2 Page 73 (ie, change from minimum or maximum baseline value to minimum or maximum value during the treatment period), scheduled and unscheduled measurements will be included. In general, vital signs and physical characteristics will be summarized with descriptive statistics (n, mean, SD, minimum, Q1, median, Q3, and maximum). Summaries will be provided for baseline and for each postbaseline visit by treatment group, including change from baseline, percent change from baseline, mlocf change from baseline and mlocf percent change from baseline for postbaseline visits. These summaries will be based on patients who have both baseline and at least one postbaseline assessment. The observed change from baseline and the mlocf change from baseline will be analyzed using an ANCOVA model with explanatory terms for treatment and the baseline value as a covariate. Treatment comparisons will be provided showing the LS means for each treatment, the treatment differences in LS means, the 95% CI for the treatment differences, within-treatment p-values, and p-values for the treatment differences obtained from the model. In addition, three approaches to the evaluation of change from baseline will be taken: Change from baseline to last observation during the treatment period considering baseline as the last non-missing observation from the baseline period; missing data will be imputed using mlocf; Change from baseline to the minimum value during the treatment period considering baseline as the minimum value of the non-missing observations in the baseline period; Change from baseline to the maximum value during the treatment considering baseline as the maximum value of the non-missing observations in the baseline period. Change from baseline in vital signs and physical characteristics parameters, using each approach above, will be summarized using standard descriptive statistics by visit and analyzed by visit through Week 52 using an ANCOVA model with explanatory terms for treatment and the baseline value as a covariate. Treatment comparisons will be provided showing the LS mean for each treatment, the pairwise differences in LS means, 95% CI for pairwise treatment differences, within-treatment p-values, and p-values for the treatment differences obtained from the model. Missing data will be imputed using mlocf imputation. Vital signs and physical characteristics parameters will be summarized across time points using box plots which include accompanying summary statistics. The number and percent of patients with treatment-emergent high or low vital signs and weight at any time will be summarized by treatment group. Comparisons between treatments through Week 52 will be made using the Fisher exact test. The treatment-emergent high or low vital signs and weight outcomes are:
412 I4V-MC-JADV Statistical Analysis Plan Version 2 Page 74 A treatment-emergent high result is defined as a change from a value less than or equal to the high limit at all baseline visits to a value greater than the high limit at any time that meets the specified change criteria during the treatment period. A treatment-emergent low result is defined as a change from a value greater than or equal to the low limit at all baseline visits to a value less than the low limit at any time that meets the specified change criteria during the treatment period. Treatment-emergent weight gain/loss is based on percent increase/decrease from baseline. The number and percent of patients with treatment-emergent high or low vital signs will be further summarized in terms of shift tables by time point and last visit and by treatment group. Only scheduled measurements will be included in these analyses. Table JADV.6.2 defines the low and high baseline values as well as the criteria used to define treatment-emergence based on postbaseline values. Table JADV.6.2. Categorical Criteria for Abnormal Treatment-Emergent Blood Pressure and Heart Rate Measurement, and Categorical Criteria for Weight Changes for Adults Parameter Low High SBP (mm Hg) (sitting) 90 (low limit) and decrease from lowest value during baseline 20 if > 90 at each baseline visit 140 (high limit) and increase from highest value during baseline 20 if < 140 at each baseline visit DBP (mm Hg) (sitting) Heart rate (bpm) (sitting) Weight (kg) 50 (low limit) and decrease from lowest value during baseline 10 if > 50 at each baseline visit < 50 (low limit) and decrease from lowest value during baseline 15 if 50 at each baseline visit (Loss) decrease 7% from lowest value during baseline 90 (high limit) and increase from highest value during baseline 10 if < 90 at each baseline visit > 100 (high limit) and increase from highest value during baseline 15 if 100 at each baseline visit (Gain) increase 7% from highest value during baseline Abbreviations: bpm = beats per minute; DBP = diastolic blood pressure; SBP = systolic blood pressure Laboratory Measurements Two sets of tables for laboratory tests will be presented for standard clinical laboratory evaluations. All laboratory tests will be presented using units conventional to the United States (CN) and also using the Système International of units (SI). For topics of safety in special groups and circumstances, laboratory test units will be specified for each analysis. Lilly large clinical trial population based reference limits (Lilly-defined; LCTPB) will be used to define the low and high limits since it is generally desirable for limits used for analyses to have greater specificity (identify fewer false positive cases) than reference limits used for individual patient management. For the hepatic laboratory assessments (alanine aminotransferase [ALT], aspartate aminotransferase [AST], total bilirubin, and alkaline phosphatase [ALP]), central
413 I4V-MC-JADV Statistical Analysis Plan Version 2 Page 75 laboratory reference ranges (Quintiles) will be used and all results pertaining to these assessments will be included as a separate analysis to address the risk of liver injury as a special safety topic (see Section ). Central laboratory reference ranges (Quintiles) will also be used to evaluate immunoglobulins and lymphocyte cell subsets (see Section ). The ratio of the low-density lipoprotein (LDL) cholesterol value to the high-density lipoprotein (HDL) cholesterol, referred to as the LDL/HDL ratio, will be derived for each patient at each visit where both values are simultaneously available. This lab parameter was not included in the standard lab processing for the Phase 3 RA studies and, as such, will not have valid reference ranges for comparison of the ratio to a standard for characterization as treatment-emergent abnormal. The ratio of total iron to total iron binding capacity (TIBC) expressed as a percent (multiplied by 100) and referred to as the transferrin saturation will also be derived for each patient at each visit where both values are simultaneously available. Central lab reference ranges will be applied to transferrin saturation values for determining treatment-emergent high/low transferrin saturation as a safety outcome. The gender-specific reference ranges are 20% to 50% for males and 15% to 50% for females. Change from baseline to last observation for laboratory tests will be summarized for patients who have both baseline and at least one postbaseline result. Baseline will be the last nonmissing observation in the baseline period prior to receiving the first dose of the study drug. The last nonmissing observation in the treatment period will be used for this analysis. Data will be analyzed in original scale units. Unscheduled measurements will be excluded. Change from the minimum value during the baseline period to the minimum value during the treatment period for selected laboratory tests will be summarized for patients who have both baseline and at least one postbaseline result. Baseline will be the minimum of non-missing observations in the baseline period. The minimum value in the treatment period will be used for this analysis. Similarly, change from the maximum value during the baseline period to the maximum value during the treatment period for selected laboratory tests will be summarized for patients who have both baseline and at least one postbaseline result. Baseline will be the maximum of non-missing observations in the baseline period. The maximum value in the treatment period will be used for this analysis. Original-scale data will be analyzed. Scheduled and unscheduled measurements will be included. The change from baseline in laboratory assessments, using each of the three approaches above, will be summarized using standard descriptive statistics and analyzed using an ANCOVA model with explanatory terms for treatment and the baseline value as a covariate. Treatment comparisons will be provided showing the LS means for each treatment, the treatment differences in LS means, the 95% CI for the treatment differences, within-treatment p-values, and p-values for the treatment differences obtained from the model. In addition, laboratory assessments will be summarized with descriptive statistics (n, mean, SD, minimum, Q1, median, Q3, and maximum) by time point. Summaries will be provided for baseline and for each postbaseline visit by treatment group, including change from baseline,
414 I4V-MC-JADV Statistical Analysis Plan Version 2 Page 76 percent change from baseline, mlocf change from baseline, and mlocf percent change from baseline for postbaseline visits. These summaries will be based on patients who have both baseline and at least one postbaseline assessment. The change from baseline and the mlocf change from baseline will be analyzed using an ANCOVA model with explanatory terms for treatment and the baseline value as a covariate. Treatment comparisons will be provided showing the LS means for each treatment, the treatment differences in LS means, the 95% CI for the treatment differences, within-treatment p-values, and p-values for the treatment differences obtained from the model. The number and percent of patients with treatment-emergent abnormal, high, or low laboratory results at any time will be summarized by treatment group. A treatment-emergent abnormal result is defined as a change from normal at all baseline visits to abnormal at any time during the treatment period. A treatment-emergent high result is defined as a change from a value less than or equal to the high limit at all baseline visits to a value greater than the high limit at any time during the treatment period. A treatment-emergent low result is defined as a change from a value greater than or equal to the low limit at all baseline visits to a value less than the low limit at any time during the treatment period. Scheduled and unscheduled measurements will be included in analysis of treatment-emergent abnormal, high, or low laboratory results. Differences between treatments (when appropriate) in incidence of treatment-emergent abnormal, high or low laboratory results will be assessed using the Fisher exact test. The number and percent of patients with treatment-emergent high, normal or low laboratory results will be further summarized in terms of shift tables by time point and last visit and by treatment group. Only scheduled measurements will be included in these analyses. Additionally, values at each visit (starting at randomization) for laboratory tests will be displayed in box plots (with outliers displayed). Baseline will be the last non-missing observation in the baseline period. Original-scale data will be used for the display but box plots for some analytes that are typically characterized by skewed distributions (e.g. immunoglobulins) will be displayed on logarithmic axes to aid in viewing the measures of central tendency and dispersion (i.e., the box part of the box-and-whisker plot). Unscheduled measurements will be excluded. In each box-plot, lines indicating the appropriate reference limits will be added; in cases where limits vary across age, race and/or gender, the lowest of the high limits and the highest of the low limits will be used. Displays using both SI and conventional units will be provided. The following summary statistics will be included in a table after the box plot presentation: N, mean, SD, minimum, Q1, median, Q3, and maximum. P-values and confidence limits will not be included in the summary statistics after the box plot. Box plots will be used to evaluate trends over time and to assess a potential impact of outliers on central tendency summaries.
415 I4V-MC-JADV Statistical Analysis Plan Version 2 Page 77 Change from study baseline to final treatment visit, from final treatment visit to follow-up visit, and from study baseline to follow-up visit in laboratory assessments will be summarized using standard descriptive statistics. The number and percent of patients with treatment-emergent high, normal or low laboratory results will be further summarized comparing study baseline to last follow-up visit and follow-up baseline to last follow-up visit by treatment group. Individual listings of laboratory results which include hematology, chemistry, urinalysis, immunoglobulin, and lymphocyte cell subsets will be provided. Additional listings that restrict to abnormal results will also be provided for both the treatment period and the post-treatment follow-up period. All lab listings will be performed separately using SI units and CN units Safety in Special Groups and Circumstances, including Adverse Events of Special Interest (AESI) In addition to general safety parameters, safety information on specific topics of special interest will also be presented. Topics will be identified by laboratory results, standardized MedDRA queries (SMQs), Lilly-defined MedDRA PT listings, or a combination of these methods. The primary focus of analyses will be on laboratory test values, where applicable, with a secondary focus on reported adverse events related to laboratory values. Additional special safety topics may be added as warranted. The topics outlined in this section include the protocol-specified AESI: severe or opportunistic infections myelosuppressive events of anemia, leukopenia, neutropenia, lymphopenia, and thrombocytopenia thrombocytosis elevations in ALT or AST (>3 times ULN) with total bilirubin (>2 times ULN). In general, for topics regarding safety in special groups and circumstances, patient profiles and/or patient listings, where applicable, will be provided when needed to allow medical review of the time course of cases/events, related parameters, patient demographics, study drug treatment and meaningful concomitant medication use. In addition to the safety topics for which provision or review of patient data is specified, these will be provided when summary data are insufficient to permit adequate understanding of the safety topic Abnormal Hepatic Tests Analyses for abnormal hepatic tests involve 5 lab analytes: ALT, AST, total bilirubin, ALP, and albumin. Analyses for change from baseline to last observation, change from the minimum value during the baseline period to the minimum value during the treatment period, change from the maximum value during the baseline period to the maximum value during the treatment period, and treatment-emergent high or low laboratory results at any time are described in Section This section describes additional analyses for the topic. Recall also that central laboratory reference ranges (Quintiles) will be used for the some hepatic laboratory assessments
416 I4V-MC-JADV Statistical Analysis Plan Version 2 Page 78 (alanine transaminase [ALT], aspartate aminotransferase [AST], total bilirubin, and alkaline phosphatase [ALP]). The number and percent of patients with the following elevations in hepatic laboratory tests at any time will be summarized between treatment groups: The percentages of patients with an ALT measurement greater than or equal to 3 times (3 ), 5 times (5 ), and 10 times (10 ) the central laboratory upper limit of normal (ULN) during the treatment period will be summarized for all patients with a postbaseline value and for subsets based on various levels of baseline. o o o The analysis of 3 ULN will contain four subsets: patients whose non-missing maximum baseline value is less than or equal to 1 ULN, patients whose maximum baseline is greater than 1 ULN but less than 3 ULN, patients whose maximum baseline value is greater than or equal 3 ULN, and patients whose baseline values are missing. The analysis of 5 ULN will contain five subsets: patients whose non-missing maximum baseline value is less than or equal to 1 ULN, patients whose maximum baseline is greater than 1 ULN but less than 3 ULN, patients whose maximum baseline is greater than or equal to 3 ULN but less than 5 ULN, patients whose maximum baseline value is greater than or equal to 5 ULN, and patients whose baseline values are missing. The analysis of 10 ULN will contain six subsets: patients whose non-missing maximum baseline value is less than or equal to 1 ULN, patients whose maximum baseline is greater than 1 ULN but less than 3 ULN, patients whose maximum baseline is greater than or equal to 3 ULN but less than 5 ULN, patients whose maximum baseline is greater than or equal to 5 ULN but less than 10 ULN, patients whose maximum baseline value is greater than or equal to 10 ULN, and patients whose baseline values are missing. The percentages of patients with an AST measurement greater than or equal to 3, 5, and 10 the central laboratory ULN during the treatment period will be summarized for all patients with a postbaseline value and for subsets based on various levels of baseline. Analyses will be constructed as described above for ALT. The percentages of patients with a total bilirubin measurement greater than or equal to 2 times (2 ) the central laboratory ULN during the treatment period will be summarized for all patients with a postbaseline value, and subset into four subsets: patients whose nonmissing maximum baseline value is less than or equal to 1 ULN, patients whose maximum baseline is greater than 1 ULN but less than 2 ULN, patients whose maximum baseline value is greater than or equal to 2 ULN, and patients whose baseline values are missing. The percentages of patients with an ALP measurement greater than or equal to 1.5 times (1.5 ) the central laboratory ULN during the treatment period will be summarized for all
417 I4V-MC-JADV Statistical Analysis Plan Version 2 Page 79 patients with a postbaseline value, and subset into four subsets: patients whose nonmissing maximum baseline value is less than or equal to 1 ULN, patients whose maximum baseline is greater than 1 ULN but less than 1.5 ULN, patients whose maximum baseline value is greater than or equal to 1.5 ULN, and patients whose baseline values are missing. Narratives for patients with an ALT or AST measurement greater than or equal to 3 ULN or with an ALP measurement greater than or equal to 2 ULN will be prepared. The review for these subjects includes an assessment of the proximity of any ALT or AST elevation to any total bilirubin elevation, alkaline phosphatase levels, other potential causes, and the temporal association with events such as nausea, vomiting, anorexia, abdominal pain, or fatigue. To facilitate this review, a patient profile will be created for each notable case. Components of the patient profile will include relevant demographic, study drug, concomitant therapy, AEs, and laboratory data. To further evaluate potential hepatotoxicity, an edish (Evaluation of Drug-Induced Serious Hepatotoxicity) plot will be created. Each patient with at least one postbaseline ALT and total bilirubin contributes one point to the plot. The points correspond to maximum total bilirubin and maximum ALT, even if not obtained from the same blood draw. Criteria for AE grading from the CTCAE will be applied for the hepatic laboratory tests AST, ALT, ALP, total bilirubin and albumin (Table JADV.6.3). These CTCAE grading schemes are consistent with both Version 3.0 and Version 4.03 of the CTCAE guidelines. When deriving CTCAE grades for lab analytes with absolute criteria for grading, such as albumin, an adjustment to the CTCAE-defined approach was made. The use of a lower limit of normal/upper limit of normal (LLN/ULN) from appropriate reference ranges was replaced with a simpler approach that uses a single value. This adjustment to the CTCAE grading criteria overcomes the irresolute derivation of the grade that can occur when some demographically-specific LLN/ULN values (from either the Lilly LCTPB or the Quintiles reference ranges) overlap with the CTCAEdefined value that separates normal (Grade 0) and Grade 1. Treatment-emergent lab abnormalities related to hepatic abnormalities occurring at any time during the treatment period and shift tables of baseline to maximum during the treatment period will be tabulated. Treatment-emergence will be characterized using two criteria (as appropriate to the grading scheme): any increase in postbaseline CTCAE grade from baseline grade an increase from baseline less than Grade 3 to Grade 3 or 4 at worst postbaseline. Pairwise comparisons between treatments will be made using the Fisher exact test. Scheduled and unscheduled measurements will be included. Shift tables will show the number and percent of patients with measurement in each category based on baseline to maximum CTCAE grade during the treatment period, with baseline depicted by the most extreme grade during the baseline period. With each shift table, a shift table
418 I4V-MC-JADV Statistical Analysis Plan Version 2 Page 80 summary displaying the number and percent of patients with maximum postbaseline results will be presented overall and by treatment group within the following categories: Decreased; postbaseline category < baseline category, Increased; postbaseline category > baseline category, Same; postbaseline category = baseline category. Treatment differences for the shift table summary categorizations will be assessed with the Likelihood Ratio Chi-Square test, using the exact version of this test whenever at least one cell has an expected cell count <5. Scheduled and unscheduled measurements will be included. Furthermore, a summary of grade increases by each maximum grade will be provided. The number and percent of patients with measurement in each category based on baseline to Common Terminology Criteria for Adverse Event (CTCAE) grade at each scheduled visit during the treatment period, with baseline depicted by the last grade during the baseline period, will be further summarized in terms of shift tables by time point and last visit and by treatment group. Only scheduled measurements will be included in these analyses. Following the shift table for the last visit, a shift table summary of the last visit will be included. Furthermore, a summary of grade increases by each grade at last visit will be provided.
419 I4V-MC-JADV Statistical Analysis Plan Version 2 Page 81 Table JADV.6.3. Common Terminology Criteria for Adverse Events (CTCAEs) to Hepatic Tests Lab Test Lab Units Grade Criteria Albumin SI 0 (normal) g/l < 35 g/land 30 g/l < 30 g/l and 20 g/l < 20 g/l ALT CN 0 (normal) AST CN 0 (normal) ALP CN 0 (normal) Total bilirubin CN 0 (normal) ULN > ULN and 2.5 ULN > 2.5 ULN and 5 ULN > 5 ULN and 20 ULN > 20 ULN ULN > ULN and 2.5 ULN > 2.5 ULN and 5 ULN > 5 ULN and 20 ULN > 20 ULN ULN > ULN and 2.5 ULN > 2.5 ULN and 5 ULN > 5 ULN and 20 ULN > 20 ULN ULN > ULN and 1.5 ULN > 1.5 ULN and 3 ULN > 3 ULN and 10 ULN > 10 ULN Abbreviations: ALP = alkaline phosphatase; ALT = alanine transaminase; AST = aspartate aminotransferase; LLN = lower limit of normal; ULN = upper limit of normal. The number and percent of patients with treatment-emergent hepatic laboratory abnormalities based on CTCAE grades will be summarized in terms of shift tables descriptively comparing baseline to maximum and last post-treatment follow-up grade and follow-up baseline to maximum and last post-treatment follow-up grade by treatment group. A shift table summary for each analysis will also be included. Furthermore, a summary of grade increases by each maximum and last grade will be provided. First shift table summary: Decreased; maximum post-treatment follow-up category < baseline category Increased; maximum post-treatment follow-up category > baseline category Same; maximum post-treatment follow-up category = baseline category. Second shift table summary: Decreased; maximum post-treatment follow-up category < follow-up baseline category
420 I4V-MC-JADV Statistical Analysis Plan Version 2 Page 82 Increased; maximum post-treatment follow-up category > follow-up baseline category Same; maximum post-treatment follow-up category = follow-up baseline category. Third shift table summary: Decreased; last post-treatment follow-up category < follow-up baseline category Increased; last post-treatment follow-up category > follow-up baseline category Same; last post-treatment follow-up category = follow-up baseline category. Fourth shift table summary: Decreased; last post-treatment follow-up category < follow-up baseline category Increased; last post-treatment follow-up category > follow-up baseline category Same; last post-treatment follow-up category = follow-up baseline category Myelosuppressive Events Using clinical laboratory assessments, the myelosuppressive events of anemia, leukopenia, neutropenia, lymphopenia, and thrombocytopenia will be assessed through analysis of decreased hemoglobin, decreased white cell count, decreased neutrophils absolute count, decreased lymphocytes absolute count, and decreased platelet count, respectively. Laboratory tests for these measures will be summarized across time points using box plots which include accompanying summary statistics. Criteria for AE grading from the CTCAEs will be applied for laboratory tests related to myelosuppressive events (Table JADV.6.4). These CTCAE grading schemes are consistent with both Version 3.0 and Version 4.03 of the CTCAE guidelines. When deriving CTCAE grades for lab analytes with absolute criteria for grading, an adjustment to the CTCAE-defined approach was made. The use of an LLN/ULN from appropriate reference ranges was replaced with a simpler approach that uses a single value (or a sex-specific pair of values for hemoglobin). This adjustment to the CTCAE grading criteria overcomes the irresolute derivation of the grade that can occur when some demographically-specific LLN/ULN values overlap with the CTCAEdefined value that separates normal (Grade 0) and Grade 1. Treatment-emergent lab abnormalities related to myelosuppression occurring at any time during the treatment period and shift tables of baseline to maximum during the treatment period will be tabulated. Treatment-emergence will be characterized using two criteria:
421 I4V-MC-JADV Statistical Analysis Plan Version 2 Page 83 any increase in postbaseline CTCAE grade from worst baseline grade an increase from worst baseline less than Grade 3 to Grade 3 or Grade 4 at worst postbaseline. Pairwise comparisons between treatment groups will be made using the Fisher exact test.
422 I4V-MC-JADV Statistical Analysis Plan Version 2 Page 84 Table JADV.6.4. Common Terminology Criteria for Adverse Events (CTCAEs) Related to Myelosuppressive Events Event Lab Test Grade Criteria in SI Units Criteria in Conventional Units Anemia Hemoglobin 0 (normal) mmol (Fe)/L for females and 8.18 mmol (Fe)/L for males < 7.27 mmol (Fe)/L for females and < 8.18 mmol (Fe)/L for males and 6.2 mmol (Fe)/L for both females and males < 6.2 mmol (Fe)/L and 4.9 mmol (Fe)/L < 4.9 mmol (Fe)/L and 4.0 mmol (Fe)/L < 4.0 mmol (Fe)/L 12 g/dl for females and 13.5 g/dl for males < 12 g/dl for females and< 13.5 g/dl for males and 10 g/dl for both females and males < 10 g/dl and 8.0 g/dl < 8.0 g/dl and 6.5 g/dl < 6.5 g/dl Leukopenia White blood cell (WBC) count 0 (normal) GI/L < 4.0 GI/L and 3.0 GI/L < 3.0 GI/L and 2.0 GI/L < 2.0 GI/L and 1.0 GI/L < 1.0 GI/L cells/l < cells/l and cells/l < cells/l and cells/l < cells/l and cells/l < cells/l Neutropenia Lymphopenia Absolute neutrophil count (ANC) Lymphocyte count 0 (normal) (normal) GI/L < 2 GI/L and 1.5 GI/L < 1.5 GI/L and 1.0 GI/L < 1.0 GI/L and 0.5 GI/L < 0.5 GI/L 1.1 GI/L < 1.1 GI/L and 0.8 GI/L < 0.8 GI/L and 0.5 GI/L < 0.5 GI/L and 0.2 GI/L < 0.2 GI/L cells/l < cells/l and cells/l < cells/l and cells/l < cells/l and cells/l < cells/l cells/l < cells/l and cells/l < cells/l and cells/l < cells/l and cells/l < cells/l
423 I4V-MC-JADV Statistical Analysis Plan Version 2 Page 85 Common Terminology Criteria for Adverse Events (CTCAEs) Related to Myelosuppressive Events Event Lab Test Grade Criteria in SI Units Criteria in Conventional Units Thrombocytopenia Platelet count 0 (normal) GI/L < 150 GI/L and 75 GI/L < 75 GI/L and 50 GI/L < 50 GI/L and 25 GI/L < 25 GI/L /L < /L and /L < /L and /L < /L and /L < /L Abbreviations: ANC = absolute neutrophil count; dl = deciliters; Fe = iron; g = grams; GI/L = Giga/Liter; mmol = millimoles; SI = Système International; WBC = white blood cell.
424 I4V-MC-JADV Statistical Analysis Plan Version 2 Page 86 Shift tables will show the number and percent of patients with measurement in each category based on baseline to maximum grade during the treatment period, with baseline depicted by the most extreme grade during the baseline period. With each shift table, a shift table summary displaying the number and percent of patients with maximum postbaseline results overall and by treatment group within the following categories: Decreased; postbaseline category < baseline category, Increased; postbaseline category > baseline category, Same; postbaseline category = baseline category. Scheduled and unscheduled measurements will be included. Treatment differences for the shift table summary categorizations will be assessed with the Likelihood Ratio Chi-Square test, using the exact version of this test whenever at least one cell has an expected cell count <5. Furthermore, a summary of grade increases by each maximum grade will be provided. The number and percent of patients with measurement in each category based on baseline to CTCAE grade at each scheduled visit during the treatment period, with baseline depicted by the last grade during the baseline period, will be further summarized in terms of shift tables by time point and last visit and by treatment group. Only scheduled measurements will be included in these analyses. Following the shift table for the last visit, a shift table summary of the last visit will be included. Furthermore, a summary of grade increases by each grade at last visit will be provided. A laboratory-based treatment emergent outcome related to increased platelet count will be summarized in similar fashion. Treatment-emergent thrombocytosis as a laboratory-based abnormality will be defined as an increase in platelet count from a maximum baseline value 600,000/μL to any postbaseline value >600,000/μL. A frequency table showing the number and percent of patients meeting criteria for thrombocytosis by treatment group will be provided. Scheduled and unscheduled measurements will be included. The number and percent of patients with treatment-emergent lab abnormalities related to myelosuppression based on CTCAE grades will be summarized in terms of shift tables descriptively comparing baseline to maximum and last post-treatment follow-up grade and follow-up baseline to maximum and last post-treatment follow-up grade by treatment group. A shift table summary for each analysis will also be included. Furthermore, a summary of grade increases by each maximum and last grade will be provided. First shift table summary: Decreased; maximum post-treatment follow-up category < baseline category Increased; maximum post-treatment follow-up category > baseline category Same; maximum post-treatment follow-up category = baseline category. Second shift table summary:
425 I4V-MC-JADV Statistical Analysis Plan Version 2 Page 87 Decreased; maximum post-treatment follow-up category < follow-up baseline category Increased; maximum post-treatment follow-up category > follow-up baseline category Same; maximum post-treatment follow-up category = follow-up baseline category. Third shift table summary: Decreased; last post-treatment follow-up category < baseline category Increased; last post-treatment follow-up category > baseline category Same; last post-treatment follow-up category = baseline category. Fourth shift table summary: Decreased; last post-treatment follow-up category < baseline category Increased; last post-treatment follow-up category > baseline category Same; last post-treatment follow-up category = baseline category Lymphocyte Subset Cell Counts The following lymphocyte subsets will be analyzed; those subsets marked with a ( ) will be evaluated only in patients from the United Kingdom: CD3+ T cells CD3+CD4+ T cells (CD4) CD3+CD8+ T cells (CD8) CD4+CD45RA-CCR7- effector memory T cells ( ) CD4+CD45RA+CCR7- effector memory T cells ( ) CD4+CD45RA+CCR7+ naïve T cells ( ) CD4+CD45RA-CCR7+ central memory T cells ( ) CD4+CXCR3+CCR6- Th1 cells CD4+CXCR3-CCR6+ Th17 cells CD3+CD4+CD127-/loCD25+FoxP3+ (CD4+) T regulatory cells CD3+CD4+CD127-/loCD25+ (CD4+) IL2-producing naïve and central memory Helper T cells CD8+CD45RA+CCR7+ naïve T cells ( ) CD8+CD45RA-CCR7+ central memory T cells ( ) CD8+CD45RA-CCR7- effector memory T cells( )
426 I4V-MC-JADV Statistical Analysis Plan Version 2 Page 88 CD8+CD45RA+CCR7- effector memory T cells ( ) CD56+/CD16+ NK cells CD19+ B cells CD20+ B cells CD19+CD27-IgD+ mature naïve B cells CD19+CD27+IgD- switched memory B cells CD19+CD27+IgD+ non-switched memory B cells CD19+CD27-IgD- Immature/transitional B cells. For each type of cells, both the absolute count and the relative count (i.e. as a percentage of the total lymphocyte population) will be analyzed. In addition, the ratio of CD4 cell counts to CD8 cell counts will be analyzed. For these parameters, the analyses of: change from baseline to last assessment, change from minimum at baseline to minimum postbaseline, change from maximum at baseline to maximum postbaseline, and the number and percentage of patients with treatment-emergent abnormal, high, or low cell counts at any time, will be performed using the same approaches as described for analysis of clinical laboratory measurements in Section For determining treatment-emergent abnormal, high, or low lymphocyte subset cell counts, central laboratory reference ranges (Quintiles) will be used when available; note that reference ranges are available only for some of the lymphocyte subset cell count analytes Lipids Effects Lipids effects will be assessed through analysis of elevated total cholesterol, elevated lowdensity lipoprotein cholesterol (LDL cholesterol), decreased high-density lipoprotein cholesterol (HDL cholesterol), and elevated triglycerides. National Cholesterol Education Program (NCEP) Adult Treatment Panel III (ATP III) guidelines will be applied for laboratory tests related to lipids effects (see Table JADV.6.5). The grade-like categories shown in this table are ordered from lowest to highest for the purposes of these analyses. Shift tables will show the number and percent of patients with measurement in each category based on baseline to maximum category during the treatment period, with baseline depicted by the most extreme category during the baseline period. With each shift table, a shift table summary displaying the number and percentage of patients with maximum postbaseline results overall and by treatment group within the following categories: Decreased; postbaseline category < baseline category,
427 I4V-MC-JADV Statistical Analysis Plan Version 2 Page 89 Increased; postbaseline category > baseline category, Same; postbaseline category = baseline category. Pairwise treatment differences for the shift table summary categorizations will be assessed with the Likelihood Ratio Chi-Square test, using the exact version of this test whenever at least one cell has an expected cell count <5. Furthermore, a summary of category increases by each maximum category will be provided.
428 I4V-MC-JADV Statistical Analysis Plan Version 2 Page 90 Table JADV.6.5. National Cholesterol Education Adult Treatment Panel III (NCEP ATP III) Criteria Lab Test Category Criteria in SI Units Criteria in Conventional Units Total Cholesterol Desirable Borderline high High < 5.17 mmol/l 5.17 mmol/l and < 6.21 mmol/l 6.21 mmol/l < 200 mg/dl 200 mg/dl and < 240 mg/dl 240 mg/dl LDL C Optimal Near optimal Borderline high High Very high < 2.59 mmol/l 2.59 mmol/l and < 3.36 mmol/l 3.36 mmol/l and < 4.14 mmol/l 4.14 mmol/l and < 4.91 mmol/l 4.91 mmol/l < 100 mg/dl 100 mg/dl and < 130 mg/dl 130 mg/dl and < 160 mg/dl 160 mg/dl and < 190 mg/dl 190 mg/dl HDL-C High Normal Low 1.55 mmol/l 1.03 mmol/l and < 1.55 mmol/l < 1.03 mmol/l 60 mg/dl 40 mg/dl and < 60 mg/dl < 40 mg/dl Triglycerides Normal Borderline high High Very high < 1.69 mmol/l 1.69 mmol/l and < 2.26 mmol/l 2.26 mmol/l and < 5.65 mmol/l 5.65 mmol/l < 150 mg/dl 150 mg/dl and < 200 mg/dl 200 mg/dl and < 500 mg/dl 500 mg/dl Abbreviations: HDL-C = high-density lipoprotein cholesterol; LDL-C = low-density lipoprotein cholesterol.
429 I4V-MC-JADV Statistical Analysis Plan Version 2 Page 91 In addition, the number and percent of patients with measurement in each category based on baseline to NCEP category at each scheduled time point, with baseline depicted by the last category during the baseline period, will be further summarized in terms of shift tables by time point and last visit and by treatment group. Only scheduled measurements will be included in these analyses. Following the shift table for the last visit, a shift table summary of the last visit will be included. Furthermore, a summary of category increases by each category at last visit will be provided. Treatment-emergent lab abnormalities related to elevated total cholesterol, elevated LDL-C, decreased HDL-C and elevated triglycerides occurring at any time during the treatment period will be tabulated using the NCEP categories shown in Table JADV.6.5. Treatment-emergent elevated total cholesterol will be characterized as movement from categories Desirable or Borderline High to category High considering patients who are at risk for the abnormality, that is, not High based on the maximum baseline total cholesterol category. Treatmentemergent elevated LDL-C will be characterized as movement from categories Optimal, Near optimal, or Borderline high to categories High or Very high considering patients who are at risk for the abnormality, that is, neither High nor Very high based on the maximum baseline LDL-C category. Treatment-emergent decreased HDL-C will be characterized as movement from categories High or Normal to category Low considering patients who are at risk for the abnormality, that is, not Low based on the maximum baseline HDL-C category. Treatment-emergent elevated triglycerides will be characterized as movement from categories Normal or Borderline high to categories High or Very high considering patients who are at risk for the abnormality, that is, neither High nor Very high based on the maximum baseline triglycerides category. Pairwise comparisons between treatment groups will be made using the Fisher exact test. Scheduled and unscheduled measurements will be included Renal Function Effects Effects on renal function will be assessed through analysis of elevated creatinine. Laboratory tests for creatinine will be summarized across time points using box plots which include accompanying summary statistics. Unscheduled measurements will be excluded from these analyses. Common Terminology Criteria for Adverse Events will be applied for laboratory tests related to renal effects (Table JADV.6.6). The creatinine grades come from CTCAE Version 3.0 guidelines. Shift tables will show the number and percent of patients with measurement in each category based on baseline to maximum CTCAE grade during the treatment period, with baseline depicted by the most extreme grade during the baseline period. With each shift table, a shift table summary displaying the number and percent of patients with maximum postbaseline results by treatment group within the following categories: Decreased; postbaseline category < baseline category, Increased; postbaseline category > baseline category,
430 I4V-MC-JADV Statistical Analysis Plan Version 2 Page 92 Same; postbaseline category = baseline category. Pairwise treatment differences for the shift table summary categorizations will be assessed with the Likelihood Ratio Chi-Square test, using the exact version of this test whenever at least one cell has an expected cell count <5. Scheduled and unscheduled measurements will be included. Furthermore, a summary of grade increases by each maximum grade will be provided. Treatment-emergent lab abnormalities related to elevated creatinine occurring at any time during the treatment period will be tabulated using the CTCAE grades shown in Table JADV.6.6. Scheduled and unscheduled measurements will be included. Treatment-emergence will be characterized using two characterizations: any increase in postbaseline CTCAE grade from baseline grade an increase from baseline less than Grade 3 to Grade 3 or Grade 4 at worst postbaseline. Pairwise comparisons between treatment groups will be made using the Fisher exact test. In addition, the number and percent of patients with measurement in each category based on baseline to CTCAE grade at each scheduled time point, with baseline depicted by the last grade during the baseline period, will be further summarized in terms of shift tables by time point and last visit and by treatment group. Only scheduled measurements will be included in these analyses. Following the shift table for the last visit, a shift table summary of the last visit will be included. Furthermore, a summary of grade increases by each grade at last visit will be provided. Table JADV.6.6. Common Terminology Criteria for Adverse Events (CTCAEs) Related to Renal Effects Lab Test CTCAE Version Grade Criteria Elevated creatinine (nomal) ULN > ULN and 1.5 ULN > 1.5 ULN and 3 ULN > 3 ULN and 6 ULN > 6 ULN Abbreviations: ULN = upper limit of normal. The number and percent of patients with treatment-emergent lab abnormalities related to elevated creatinine based on CTCAE grades will be summarized in terms of shift tables descriptively comparing baseline to maximum and last post-treatment follow-up and follow-up baseline to maximum and last post-treatment follow-up by treatment group. A shift table summary for each analysis will also be included. Furthermore, a summary of grade increases by each maximum and last grade will be provided. First shift table summary:
431 I4V-MC-JADV Statistical Analysis Plan Version 2 Page 93 Decreased; maximum post-treatment follow-up category < baseline category Increased; maximum post-treatment follow-up category > baseline category Same; maximum post-treatment follow-up category = baseline category. Second shift table summary: Decreased; maximum post-treatment follow-up category < follow-up baseline category Increased; maximum post-treatment follow-up category > follow-up baseline category Same; maximum post-treatment follow-up category = follow-up baseline category. Third shift table summary: Decreased; last post-treatment follow-up category < baseline category Increased; last post-treatment follow-up category > baseline category Same; last post-treatment follow-up category = baseline category. Fourth shift table summary: Decreased; last post-treatment follow-up category < baseline category Increased; last post-treatment follow-up category > baseline category Same; last post-treatment follow-up category = baseline category Elevations in Creatine Phosphokinase (CPK) Laboratory tests for CPK will be summarized across time points using box plots which include accompanying summary statistics. Unscheduled measurements will be excluded from these analyses. Elevations in CPK will be addressed using CTCAE criteria (Table JADV.6.7) from CTCAE Version 3.0 guidelines. Shift tables will show the number and percent of patients with measurement in each category based on baseline to maximum CTCAE grade during the treatment period, with baseline depicted by the most extreme grade during the baseline period. With each shift table, a shift table summary displaying the number and percent of patients with maximum postbaseline results, by treatment group within the following categories: Decreased; postbaseline category < baseline category Increased; postbaseline category > baseline category Same; postbaseline category = baseline category. Treatment differences for the shift table summary categorizations will be assessed with the Likelihood Ratio Chi-Square test, using the exact version of this test whenever at least one cell
432 I4V-MC-JADV Statistical Analysis Plan Version 2 Page 94 has an expected cell count <5. Scheduled and unscheduled measurements will be included. Furthermore, a summary of grade increases by each maximum grade will be provided. Treatment-emergent lab abnormalities related to elevated CPK occurring at any time during the treatment period will be tabulated using the CTCAE grades shown in Table JADV.6.7. Treatment-emergence will be characterized using two characterizations: any increase in postbaseline CTCAE grade from worst baseline grade an increase from worst baseline less than Grade 3 to Grade 3 or Grade 4 at worst postbaseline. Pairwise comparisons between treatment groups will be made using the Fisher exact test. Scheduled and unscheduled measurements will be included. In addition, the number and percent of patients with measurement in each category based on baseline to CTCAE grade at each scheduled visit, with baseline depicted by the last grade during the baseline period, will be further summarized in terms of shift tables by time point and last visit and by treatment group. Only scheduled measurements will be included in these analyses. Following the shift table for the last visit, a shift table summary of the last visit will be included. Furthermore, a summary of grade increases by each grade at last visit will be provided. Table JADV.6.7. Common Terminology Criteria for Adverse Events (CTCAE) Related to Elevations in CPK Lab Test CTCAE Version Grade Criteria Elevated CPK (normal) ULN > ULN and 2.5 ULN > 2.5 ULN and 5 ULN > 5 ULN and 10 ULN > 10 ULN Abbreviations: CPK = creatine phosphokinase; LCTPB = large clinical trial population based; ULN = upper limit of normal based on Lilly LCTPB reference limits. The number and percent of patients with treatment-emergent lab abnormalities related to elevated CPK based on CTCAE grades will be summarized in terms of shift tables descriptively comparing baseline to maximum and last post-treatment follow-up and follow-up baseline to maximum and last post-treatment follow-up by treatment group. A shift table summary for each analysis will also be included. Furthermore, a summary of grade increases by each maximum and last grade will be provided. First shift table summary: Decreased; maximum post-treatment follow-up category < baseline category Increased; maximum post-treatment follow-up category > baseline category Same; maximum post-treatment follow-up category = baseline category.
433 I4V-MC-JADV Statistical Analysis Plan Version 2 Page 95 Second shift table summary: Decreased; maximum post-treatment follow-up category < follow-up baseline category Increased; maximum post-treatment follow-up category > follow-up baseline category Same; maximum post-treatment follow-up category = follow-up baseline category. Third shift table summary: Decreased; last post-treatment follow-up category < baseline category Increased; last post-treatment follow-up category > baseline category Same; last post-treatment follow-up category = baseline category. Fourth shift table summary: Decreased; last post-treatment follow-up category < baseline category Increased; last post-treatment follow-up category > baseline category Same; last post-treatment follow-up category = baseline category Infections, Including Potential Opportunistic Infections (POIs) Infections will be defined using the PTs from the MedDRA Infections and Infestations SOC, with additional terms from the Investigations SOC being used in selected instances, as described below. Treatment-emergent infections will be analyzed according to various groups of infectious events including: all infections o all PTs in the Infections and Infestations SOC, serious infections o all PTs in the Infections and Infestations SOC that are SAEs, infections that require therapeutic intervention (antibiotics, antivirals, antifungals, etc.) o all PTs in the Infections and Infestations System Organ Class for which there is an antimicrobial concomitant medication associated with that event for that patient, herpes zoster o specific Lilly-defined PTs from the Herpes Viral Infections HLT in the Infections and Infestations SOC, shown in the appendices of the Baricitinib Program Safety Analysis Plan (PSAP), Version 3
434 I4V-MC-JADV Statistical Analysis Plan Version 2 Page 96 tuberculosis o specific Lilly-defined PTs from the Tuberculous Infections high-level term (HLT) and the Atypical Mycobacterial infections HLT of the Infections and Infestations SOC and the Investigations SOC, shown in the appendices of the current version of the Baricitinib PSAP, Version 3 viral hepatitis o all PTs from the Hepatitis Viral Infections HLT (HLT code ) in the Infections and Infestations SOC, potential opportunistic infections, as described in detail below. For each of all infections, serious infections (overall and on each approach to identifying SAEs), infections that require therapeutic intervention, herpes zoster infections, tuberculosis, viral hepatitis, and potential opportunistic infections, the frequency and relative frequency for each PT will be provided, ordered by decreasing frequency in the baricitinib group (or combined baricitinib group, where appropriate). In addition to the incidence of infectious AEs by MedDRA PT as described above, the number and percent of patients with treatment-emergent infectious AEs by infecting organism by treatment group will be summarized. Infecting organism is defined as any of the primary, secondary, or tertiary infecting organisms as entered on the ecrf. In addition, the frequency counts and percents of patients experiencing treatment-emergent infectious AEs will also be presented by treatment group and primary site of infection. The site of infection used in summary displays is based on a grouping of sites from the ecrf that is more meaningful than the many specific sites allowed on the ecrf. The Program SAP (PSAP) shows how the ecrfavailable entries are bundled into site categories for some reporting purposes. Summaries that display the infecting site within PT will use the actual site entered into the ecrf. Potential Opportunistic Infections: Potential Opportunistic Infections (POIs) will be identified according to two different approaches. First, POIs are identified from treatment-emergent adverse events based on a Lilly-defined list of MedDRA PTs shown in the PSAP, Version 3. These PTs are a subset of terms from the Infections and Infestations SOC. Note that the current list is based on MedDRA version 17.0, but it will be updated for use as each MedDRA version is released, including versions released after SAP finalization but before database lock. Second, POIs are identified using a Lilly-defined list of combinations of the infectious organism and site of infection, as recorded on the specialized ecrf designed to provide additional details about infections; this list is contained in the PSAP, Version 3. Up to three infecting organisms are collected (primary, second and third), and any of them can contribute to a POI. Only a single (primary) site is collected.
435 I4V-MC-JADV Statistical Analysis Plan Version 2 Page 97 For the POIs identified from MedDRA PTs, the number and percent of patients overall and for each specific PT will be summarized by treatment group, with specific event terms ordered by decreasing frequency in the baricitinib group (or combined baricitinib group, where appropriate). For the POI identified from organism/site combinations, the number and percent of patients overall and for each specific combination will be summarized by treatment group, with specific organism/site pairs ordered by decreasing frequency of organism based on frequency in the baricitinib group (or combined baricitinib group, where appropriate) and then alphabetically by site within organism. Treatment-emergent infectious events will be reviewed in context of other clinical and laboratory parameters. A listing of patients experiencing treatment-emergent infectious AEs will be provided, which will include patient demographics, treatment group, treatment start and stop dates, infectious event, event start and stop dates, event severity and possible relationship to the study drug, total leukocytes, total lymphocytes, absolute neutrophils, event seriousness, event outcome, whether the patient was immunized for tuberculosis and/or herpes zoster, and whether anti-microbial treatment was recorded Major Adverse Cardiovascular Events (MACE) Potential MACE and other cardiovascular events requiring adjudication will be summarized. Categories and subcategories analyzed will include the following: MACE events o o o Cardiovascular death Myocardial infarction (MI) Stroke, Other cardiovascular events o o o o o o Hospitalization for unstable angina Hospitalization for heart failure Serious arrhythmia Resuscitated sudden death Cardiogenic shock Coronary interventions (such as coronary artery bypass graft or percutaneous coronary intervention), Non-cardiovascular death. In general, events requiring adjudication are documented by investigative sites using an endpoint reporting CRF. This CRF is then sent to the adjudication center which uses a cardiovascular adjudication reporting CRF to document the final assessment of the event as a MACE, as some other cardiovascular event, or as no event (according to the Clinical Endpoint Committee
436 I4V-MC-JADV Statistical Analysis Plan Version 2 Page 98 Charter). In some cases, however, the investigator may not have deemed that an event had met the endpoint criteria but the event was still sent for adjudication as a potential MACE, other cardiovascular event, or no event. In these instances, the adjudication reporting CRF will not have a matching endpoint reporting CRF from the investigator. Events generated from these circumstances will be considered as events sent for adjudication in the absence of an investigator s endpoint reporting form. First, the number and percent of patients with potential MACE and other potential cardiovascular events as sent for adjudication will be summarized by treatment group based on the categories and subcategories above. If an event is adjudicated without a matching endpoint reporting CRF from the investigative site, then these events will be separately summarized using the MedDRA PT for the reported event as documented on the adjudication reporting CRF. This term will be used in lieu of the term that would have been included on an endpoint data collection form. Pairwise treatment comparisons will be made using the Fisher exact test. Second, the number and percent of patients with MACE, other cardiovascular events, noncardiovascular death, and all-cause death, as adjudicated, will be summarized by treatment group based on the categories and subcategories above. Pairwise treatment group comparisons will be made using the Fisher exact test. Third, an overall summary of the number of events and deaths sent for adjudication by adjudicated outcome will be produced. The count of events (including death) sent for adjudication will be summarized along with how many of these were adjudicated as MACE, other cardiovascular event, or no event. A separate summary will be displayed for the numbers of deaths sent for adjudication and whether they were adjudicated as cardiovascular or noncardiovascular. A listing of all MACE or other cardiovascular events sent for adjudication will be provided and will include data concerning the reason the event was sent for adjudication, the adjudicated outcome and the related MedDRA PT Malignancies Malignancies will be identified using terms from the malignant tumours SMQ (SMQ ). The number and percent of patients with TEAEs associated with malignant tumors will be summarized by treatment group using MedDRA PT. Events will be ordered by decreasing frequency in the baricitinib group (or combined baricitinib group, where appropriate). Additional summaries will give an overview of malignancies. First, the total number of patients with malignant tumors which are SAEs by ICH GCP or by protocol, by ICH GCP, and by protocol only, will be provided. Second, the number of patients with TEAEs associated with malignant tumors that were rated as severe, and that were considered related to study drug in the opinion of the investigator, will be provided. Third, the number of patients with TEAEs associated with malignant tumors that led to discontinuation from the study, that led to permanent study drug discontinuation, and that led to temporary study drug interruption will be provided. EAIR will also be calculated for each PT. Tumors that are gender-specific, based on
437 I4V-MC-JADV Statistical Analysis Plan Version 2 Page 99 PT, will have the denominators and patient-years of exposure limited to patients of that gender. Pairwise comparisons between treatment groups will be made using the Fisher exact test. A by-patient listing for all patients having a malignancy event at any time will be provided Allergic Reactions/Hypersensitivities Allergic reactions/hypersensitivities will be examined within the context of anaphylactic reactions. Patients with a treatment-emergent anaphylactic reaction will be identified using the MedDRA Anaphylactic Reaction SMQ (SMQ ). This SMQ is unique compared to most SMQs in that an algorithmic approach is taken to identify events. Anaphylaxis has been broadly defined as a serious allergic reaction that is rapid in onset and may cause death (Sampson et al. 2006). Sampson described a two-tiered approach to identify cases of anaphylactic reaction, an approach which is mimicked by the algorithmic approach taken for the Anaphylactic Reaction SMQ. The algorithm is defined in the following manner within the Introductory Guide for Standardized MedDRA Queries (SMQs) Version 17.0: All narrow search terms as well as: if one term from category B and one term from category C is present, or if one term from (category B or category C) plus one term from category D. This algorithm is also expressed as A or (B and C) or (D and (B or C)). The narrow search terms represent core anaphylactic reaction terms whereas Categories B, C and D represent signs and symptoms that are possibly indicative of anaphylactic reaction, with Category B terms representing Upper Airway/Respiratory signs and symptoms, Category C representing Angioedema/Urticaria/Prutitis/Flush signs and symptoms and Category D representing Cardiovascular/Hypertension signs and symptoms. Note that all terms within Categories B, C and D are broad terms. The use of the narrow search terms match roughly with Sampson s first tier approach, whereas the broad terms of Categories B, C and D match roughly with Sampson s second tier approach. Within this second approach using broad terms, it is important to recognize that occurrence of these events should be nearly coincident and develop rapidly after exposure to an antigen; based on recording of events on CRFs, a window wherein the events occur within 2 days of one another (based on the onset dates of the events) is allowed. The number and percent of patients with treatment-emergent anaphylactic reactions will be summarized by treatment group. Results for each PT will be provided along with subtotals that first pool narrow and broad terms together, and then, second, for narrow terms only, ordered by decreasing frequency in the baricitinib group (or combined baricitinib group, where appropriate). Additional summaries will give an overview of anaphylactic reactions. First, the total number of patients with anaphylactic reactions which are SAEs by ICH GCP or by protocol, by ICH GCP, and by protocol only, will be provided. Second, the number of patients with TEAEs associated with anaphylactic reactions that were rated as severe, and that were considered related to study drug in the opinion of the investigator, will be provided. Third, the number of patients with TEAEs associated with anaphylactic reactions that led to discontinuation from the study, that led to permanent study drug discontinuation, and that led to temporary study drug interruption will be provided. Exposure-adjusted incidence rates will also be provided for each PT. Pairwise comparisons between treatment groups will be made using the Fisher exact test.
438 I4V-MC-JADV Statistical Analysis Plan Version 2 Page Gastrointestinal (GI) Perforations Treatment-emergent adverse events potentially related to GI perforations will be analyzed using reported AEs. Identification of these events will be based on the PTs of the MedDRA Gastrointestinal Perforations SMQ (SMQ ); note that this SMQ holds only narrow terms and has no broad terms. Frequency and relative frequency for each PT will be provided, ordered by decreasing frequency in the baricitinib group (or combined baricitinib group, where appropriate). Additional summaries within this analysis will give an overview of events related to GI perforations. First, the total number of patients with GI perforations which are SAEs by ICH GCP or by protocol, by ICH GCP, and by protocol only, will be provided. Second, the number of patients with TEAEs associated with GI perforations that were rated as severe, and that were considered related to study drug in the opinion of the investigator, will be provided. Third, the number of patients with TEAEs associated with GI perforations that led to discontinuation from the study, that led to permanent study drug discontinuation, and that led to temporary study drug interruption will be provided. Exposure-adjusted incidence rates will also be provided for each PT. Pairwise comparisons between treatment groups will be made using the Fisher exact test Symptoms of Depression (QIDS-SR16) The Quick Inventory of Depressive Symptomatology Self-Rated (QIDS-SR 16 ) is a 16-item, selfreport instrument intended to assess the existence and severity of symptoms of depression as listed in the American Psychiatric Association s Diagnostic and Statistical Manual of Mental Disorders, 4th Edition (DSM-IV; APA 1994). Patients are asked to consider each statement as it relates to the way they have felt for the past 7 days. There is a unique 4-point ordinal scale for each item with scores ranging from 0 to 3 reflecting increasing depressive symptoms as the item score increases. Additional information and the QIDS-SR 16 questions may be found on the University of Pittsburgh Epidemiology Data Center web site ( A key reference for this instrument covers its psychometric properties for use in patients with chronic major depression. (Rush et al. 2003). The QIDS-SR 16 total score is derived as the sum of the scores across the 9 scale domains. For scale domains that contain more than one item, the domain score is the highest item rating given across the items within that domain): Sleep: the highest score on any one of the four sleep items (Items 1 to 4) Depressed mood (Item 5) Weight/appetite change: the highest score on any one of the four weight items (Items 6 to 9) Psychomotor changes: the highest score on either of the two psychomotor items (Items 15 and 16) Concentration (Item 10) Worthlessness/guilt (Item 11) Suicidal ideation (Item 12)
439 I4V-MC-JADV Statistical Analysis Plan Version 2 Page 101 Decreased interest (Item 13) Decreased energy (Item 14). In the presence of missing data, the following rules will be employed to derive the total score. First, considering the three multi-item domains (sleep, weight/appetite change, psychomotor change), the domain score should be based on the maximum value across the appropriate items, and it should be missing only if each item is missing. Further, considering the nine domain scores, the total score should be derived as missing if there are 3 or more domains that are missing; if 1 or 2 domain scores are missing, then the total score should be derived using a total score that is pro-rated to the full scale range (0 to 27) based on the available domain scores, retaining 1 decimal place in the total score derived in the presence of missing data. The QIDS-SR 16 total scores will also be categorized in the following severity classes: Table JADV.6.8. Quick Inventory of Depressive Symptomatology Self-Rated 16 (QIDS-SR 16 ) Severity of Depressive Symptoms Categories based on the QIDS-SR 16 Total Score QIDS-SR 16 Severity of Depressive Symptoms Category QIDS-SR 16 Total Score 0=None 0-5 1=Mild =Moderate =Severe =Very Severe Treatment differences in mean change in the QIDS-SR 16 total score will be analyzed by time point using an ANCOVA model with explanatory terms for treatment and the baseline value as a covariate. Treatment comparisons will be provided showing the LS means for each treatment, the treatment differences in LS means, the 95% CI for treatment differences, within-treatment p- values, and p-values for the treatment differences obtained from the model. Summary statistics for percent change from baseline will also be presented. Missing data will be imputed using mlocf imputation. Frequency counts, and percentages will be used to summarize the QIDS-SR 16 total score severity categories and the item responses for the QIDS-SR 16 suicidal ideation item at each visit by treatment group. Scheduled measurements will be included in these analyses. Using the QIDS-SR 16 Severity of Depressive Symptoms Categories shown in Table JADV.6.8, shift tables will show the number and percent of patients with total score in each category based on baseline to maximum category during the treatment period, with baseline depicted by the most extreme category during the baseline period, with further summarization of change from baseline in severity using categories of any improvement, no change, and any worsening, by treatment. Similarly, shift tables will be created for the QIDS-SR 16 suicidal ideation item (Item 12) responses. Pairwise treatment differences for the shift table summary categorizations will be assessed with the Likelihood Ratio Chi-Square test, using the exact version of this test whenever at least one cell has an expected cell count <5. Scheduled and unscheduled
440 I4V-MC-JADV Statistical Analysis Plan Version 2 Page 102 measurements will be included in these analyses. Furthermore, a summary of increases by each maximum severity category will be provided. In addition, the number and percent of patients with total score in each category based on baseline to each scheduled visit, with baseline depicted by the last category during the baseline period, will be further summarized in terms of shift tables by time point and last visit and by treatment group. Following the shift table for the last visit, a shift table summary of the last visit will be included. Furthermore, a summary of increases by each category at last visit will be provided. Similarly, shift tables will be created for the QIDS-SR 16 suicidal ideation item (Item 12) responses by time point, including last visit. Shift tables for the QIDS-SR 16 total score severity categories and for the suicidal ideation item descriptively comparing baseline to maximum and last post-treatment follow-up and follow-up baseline to maximum and last post-treatment follow-up by treatment group will be performed. A shift table summary for each analysis will also be included. Furthermore, a summary of increases by each maximum and last severity category will be provided. First shift table summary: Decreased; maximum post-treatment follow-up category < baseline category Increased; maximum post-treatment follow-up category > baseline category Same; maximum post-treatment follow-up category = baseline category. Second shift table summary: Decreased; maximum post-treatment follow-up category < follow-up baseline category Increased; maximum post-treatment follow-up category > follow-up baseline category Same; maximum post-treatment follow-up category = follow-up baseline category. Third shift table summary: Decreased; last post-treatment follow-up category < baseline category Increased; last post-treatment follow-up category > baseline category Same; last post-treatment follow-up category = baseline category. Fourth shift table summary: Decreased; last post-treatment follow-up category < baseline category Increased; last post-treatment follow-up category > baseline category Same; last post-treatment follow-up category = baseline category.
441 I4V-MC-JADV Statistical Analysis Plan Version 2 Page Other Safety Analyses The Framingham risk score represents the likelihood of experiencing a CVD event in the next 10 years for an individual patient. It is calculated for men and women aged 30 and 74 years with no prior history of CVD. The outcome CVD comprises of coronary death, myocardial infarction, coronary insufficiency, angina, ischemic stroke, hemorrhagic stroke, transient ischemic attack, peripheral artery disease, and heart failure. It will be analyzed as continuous and categorical variables (ordinal endpoint of FRS). The difference between treatment groups in the changes from baseline through Week 24 will be analyzed by time point using ANCOVA which includes treatment as a fixed effect, the baseline measurement as a covariate, and change from baseline as the dependent variable. The ordinal endpoint of FRS is defined as low risk (risk <10%), intermediate risk (risk between 10% and 20%), and high risk (risk >20%). Shift tables will be used to summarize the number and percent of patients with calculated risk in each category based on baseline to maximum category during the treatment period, with baseline depicted by the most extreme category during the baseline period, with further summarization of change from baseline in severity using categories of decreased, same, and increased, by treatment. Pairwise treatment differences for the shift table summary categorizations will be assessed with the Likelihood Ratio Chi-Square test, using the exact version of this test whenever at least one cell has an expected cell count <5. Scheduled and unscheduled measurements will be included in these analyses. Furthermore, a summary of increases by each maximum severity category will be provided. In addition, shift tables by time point and last visit will be used to summarize the FRS categories. Scheduled measurements will be used in these analyses. Following the shift table for the last visit, a shift table summary of the last visit will be included. Furthermore, a summary of increases by each category at last visit will be provided. The RRS is an alternative characterization to the FRS for non-diabetic men and women aged 45 and 80 years with no prior history of CVD and/ or cerebrovascular events. The RRS will be analyzed similarly to what was described for the FRS. Separate analyses will be performed for all patients who meet the criteria at baseline and for patients with a baseline hscrp 20 mg/l. The ordinal endpoint of the RRS is defined as low risk (risk <5%), low to moderate risk (risk 5% to <10%), moderate to high risk (risk 10% to <20%), and high risk (risk 20%) Subgroup Analysis Subgroup analyses comparing baricitinib and adalimumab to placebo will be performed on the mitt population using ACR20, ACR50, DAS28-hsCRP <2.6, DAS28-hsCRP 3.2, SDAI 3.3, HAQ-DI Improvement 0.22, and change from baseline in HAQ-DI and DAS28-hsCRP at Week 12 (Visit 7) and Week 24 (Visit 11) and using mtss at Week 24 (Visit 11). The following subgroups (but not limited to only these) will be categorized into disease-related characteristics and demographic characteristics and will be evaluated: Gender: (Male, Female) Age: (<65 years, 65 years)
442 I4V-MC-JADV Statistical Analysis Plan Version 2 Page 104 Age: (<75 years, 75 years) Age: (<85 years, 85 years) Age: (<65 years, 65 to <75 years, 75 to <85 years, 85 years) Baseline weight: ( median, > median) Baseline weight group: (<60 kg, 60 to <100 kg, 100 kg) Baseline body mass index (BMI) category: (<18.5 kg/m 2, 18.5 to <25 kg/m 2, 25 to <30 kg/m 2, 30 kg/m 2 ) Baseline BMI category: ( median, >median) Race: (Asian, Black/African American, White, Other [American Indian/Alaska Native, Native Hawaiian or other Pacific Islander], Multiple) Region (as defined in Section 5.3) Specific region: (Europe, other) Specific country: (USA, other) Specific country: (Japan, other) Screening renal function status: impaired (egfr <60 ml/min/1.73 m2) or not impaired (egfr 60 ml/min/1.73 m2) Duration of RA from diagnosis: (<1 year, 1 to <5 years, 5 to < 10 years, 10 years) Baseline disease severity: (DAS28-hsCRP 5.1, DAS28-hsCRP >5.1) Joint erosion status: (1-2 joint erosions plus seropositivity, at least 3 joint erosions) Baseline Seropositivity and Joint Erosion Status (Seropositive (RF positive/ ACPA positive) and 3 joint erosion; others) Seropositivity: (either rheumatoid factor [RF] positive or anti-citrullinated protein antibody [ACPA] positive, both RF negative and ACPA negative) Seropositivity: (RF positive, RF negative) Seropositivity: (ACPA positive, ACPA negative) Background therapy: (MTX only, MTX + Other cdmards) Baseline mtss: ( median, > median) Corticosteroid use at baseline: (yes, no). The subgroup analyses for categorical outcomes will be performed using logistic regression. The model will include the categorical outcome based on NRI as the dependent variable and treatment, subgroup, and treatment-by-subgroup interaction as explanatory variables. The treatment-by-subgroup interaction comparing treatment groups will be tested at the 0.1
443 I4V-MC-JADV Statistical Analysis Plan Version 2 Page 105 significance level. The p-value from the logistic regression will be reported for the interaction test. When logistic regression sample size requirements (<5 responders in any category for any subgroup) are not met, the p-value will not be produced. Counts, percentages, the odds ratio, and the 95% CI of the odds ratio within a subgroup obtained from the same model with the subgroup and interaction terms removed will be presented for comparing treatment group differences. When logistic regression sample size requirements are not met, the p-value from the Fisher exact test will be produced instead of the odds ratio and associated 95% CI. An ANCOVA will be used to analyze the change from baseline based on mbocf in HAQ-DI and DAS28-hsCRP at Week 12, based on mlocf in HAQ-DI and DAS28-hsCRP at Week 24, and based on LE in mtss at Week 24 with baseline, treatment, subgroup, and treatment-bysubgroup as explanatory variables. The treatment-by-subgroup interaction will be tested at the 0.1 significance level. The LS mean, LS mean difference, SE, and 95% CI within subgroups obtained from the same ANCOVA model with the interaction and subgroup terms removed will be presented. Descriptive statistics will be provided for each treatment and stratum of a subgroup as outlined, regardless of sample size. In addition, the aforementioned formal statistical analysis will be performed when there are a minimum of 30 patients per treatment group per stratum of the subgroup. In some cases, select strata within the subgroups may need to be pooled in order to achieve 30 patients per treatment group per stratum for analysis. The descriptive and analysis statistics for the pooled strata will be displayed in addition to the descriptive statistics for the initial strata. The detailed pooling strategy is described below: For subgroups with two strata: gender, specific age group (<65 vs. 65; <75 vs. 75; <85 vs. 85), baseline weight group, baseline body mass index category, screening renal function status, specific region (Europe vs. complement), specific country (US vs. complement; Japan vs. complement), disease severity, seropositivity (RF+ vs. RF-; ACPA+ vs. ACPA-; either RF+ or ACPA+ vs. both RF+ and ACPA+), baseline seropositivity and joint erosion status: (Seropositive [RF or ACPA positive] and 3 joint erosions, other), corticosteroid use, background therapy, baseline mtss If one of the strata fails to meet the required minimum number, then no formal statistical analysis will be provided. Age group (four strata) If one of the strata fails to meet the required minimum number, the stratum will be pooled with the next contiguous older age stratum except for the oldest age stratum which will be pooled with the next oldest age stratum. The pooling will continue until all strata including pooled strata meet the required minimum number. In the event that the pooling strategy results in levels that are the same as the two-level age groups, the pooling method will not be used.
444 I4V-MC-JADV Statistical Analysis Plan Version 2 Page 106 Baseline weight group (three strata) If one of the strata fails to meet the required minimum number, the stratum will be pooled with the next contiguous heavier weight stratum, except for the heaviest weight stratum which will be pooled with the next heaviest weight stratum. The pooling will continue until all strata including pooled strata meet the required minimum number. Baseline body mass index (four strata) If one of the strata fails to meet the required minimum number, the stratum will be pooled with the next contiguous heavier BMI stratum, except for the heaviest BMI stratum which will be pooled with the next heaviest BMI stratum. The pooling will continue until all strata including pooled strata meet the required minimum number. Race (five strata) If one of the race strata fails to meet the required minimum number, the 2 race strata with the least number of patients will be pooled, and pooling of the smallest strata will continue until all strata including pooled strata meet the required minimum number. Geographic region (seven strata) Pool eastern Europe and western Europe first, if needed. Pool US and Canada with the combined Europe if necessary. Pool Asia ex-japan with Japan next, if needed. If Central and South America and Mexico fails to meet the required minimum number, then Central and South America and Mexico will be pooled with the Rest of World stratum. If after the above pooling there are still strata not meeting the requirement, then the 2 strata including the pooled strata with the least number of patients will be pooled. The pooling will continue until all strata including pooled strata meet the required minimum number. Duration of RA from diagnosis (four strata): <1 year, 1 to <5 years, 5 to <10 years, or 10 years If one of the strata fails to meet the required minimum number, the stratum will be pooled with the next contiguous longer duration stratum except that the longest duration stratum will be pooled with the next longest duration stratum. The pooling will continue until all strata including pooled strata meet the required minimum number. If any of the pooling methods result in all subgroup levels being pooled into one level, the pooling method will not be used and no formal comparisons will be generated Interim Analysis and Data Monitoring A DMC will oversee the conduct of all the Phase 3 clinical trials evaluating baricitinib in patients with RA. The DMC will consist of members external to Lilly. This DMC will follow the rules defined in the DMC charter, focusing on potential and identified risks for this molecule and for this class of compounds. The DMC membership will include, at a minimum, specialists with expertise in rheumatology, statistics, and other appropriate specialties. The DMC will review and evaluate up to 3 planned interim analyses on an approximate semiannual basis and the coordination of these analyses will be documented in the DMC Charter. Access to the unblinded interim data will be limited to the statisticians who conduct the interim analyses and the DMC.
445 I4V-MC-JADV Statistical Analysis Plan Version 2 Page 107 The statisticians conducting the interim analyses will be independent from the study team. The study team will not have access to the unblinded data. Study sites will receive information about interim results ONLY if they need to know for the safety of their patients. The DMC will review study discontinuation data, AEs including SAEs, clinical laboratory data, vital sign data, etc. The DMC may recommend continuation of the study, as designed, temporary suspension of enrollment, or the discontinuation of a particular treatment regimen or the entire study. The DMC may request to review efficacy data to investigate the benefit/risk relationship in the context of safety observations for ongoing patients in the study. However, the study will not be stopped for positive efficacy results, and there is no planned futility assessment. Hence, there will be no alpha adjustment for these interim analyses. Details of the DMC and interim safety analyses will be documented in a DMC charter and DMC analysis plan. Information that may unblind the study during the analyses will not be reported to study sites or the blinded study team until the database lock for the primary and major secondary efficacy analyses.
446 I4V-MC-JADV Statistical Analysis Plan Version 2 Page Unblinding Plan There will be two planned database locks for this study: a primary database lock after all patients complete Part A and a final database lock at end of the study. A database lock is planned after all patients complete Part A in which by-visit data through Week 24 (Visit 11) will be locked but ongoing log-type entry forms for ongoing patients such as adverse events and concomitant medications will remain unlocked. Analyses will include the primary and key secondary endpoints (included in the graphical multiple testing procedure except for x-ray data), efficacy, health outcomes measures, and safety endpoints that are collected through Week 24 according to the treatment groups defined in Section 6.1. This analysis will not be considered an interim analysis. Information that may unblind the study during the analyses will not be shared with study team and study site personnel who still remain blinded at the time. Members of the study team not in direct contact with the study sites may have access to the unblinded data from this study after all patients complete Week 24 (Visit 11) and the database has been locked. Members of the study team who become unblinded will have no further contact with study sites until after the final data lock. A final database lock is planned at the end of the study when all patients complete the study. The final analysis will include all data that are collected during the study. All investigators, study site personnel, and patients will remain blinded to treatment assignments until the final database lock.
447 I4V-MC-JADV Statistical Analysis Plan Version 2 Page References Alosh M, Bretz F, Huque M. Advanced multiplicity adjustment methods in clinical trials. Stat Med. 2014;33: [APA] American Psychiatric Association. Diagnostic and Statistical manual of Mental Disorders. 4 th ed. DSM-IV. Washington, DC: American Psychiatric Association; Bretz F, Maurer W, Brannath W, Posch M. A graphical approach to sequentially rejective multiple test procedures. Stat Med. 2009;28(4): Brooks R. EuroQol: The current state of play. Health Policy. 1996;37: Bruynesteyn K, Boers M, Kostense P, van der Linden S, van der Heijde D. Decidingon progression of joint damage in paired films of individual patients: smallest detectable difference or change. Ann Rheum Dis. 2005;64(2): Cancer Therapy Evaluation Program, Common Terminology Criteria for Adverse Events, Version 3.0, DCTD, NCI, NIH, DHHS, March 31, Cella D, Webster K. Linking outcomes management to quality-of-life measurement. Oncology (Willeston Park). 1997;11(11A): Cohen SB, Emery P, Greenwald MW, Dougados M, Furie RA, Genovese MC, Keystone EC, Loveless JE, Burmester GR, Cravets MW, Hessey EW, Shaw T, Totoritis MC. Rituximab for rheumatoid arthritis refractory to anti-tumor necrosis factor therapy. Results of a multicenter, randomized, double-blind, placebo-controlled phase III trial evaluating primary efficacy and safety at twenty-four weeks. Arthritis Rheum. 2006;54(9): Common Terminology Criteria for Adverse Events, Version 4.03, DHHS NIH NCI, NIH Publication No , June 14, D'Agostino, Vasan, Pencina, Wolf, Cobain, Massaro, Kannel, A General Cardiovascular Risk Profile for Use in Primary Care: The Framingham Heart Study. Circulation The EuroQol Group. EQ-5D-5L User Guide. Version 1.0. April Available at: EQ-5D-5L.pdf. Accessed 01 Jul Felson DT, Smolen JS, Wells G, Zhang B, van Tuyl LH, Funovits J, Aletaha D, Allaart CF, Bathon J, Bombardieri S, Brooks P, Brown A, Matucci-Cerinic M, Choi H, Combe B, de Wit M, Dougados M, Emery P, Furst D, Gomez-Teino J, Hawker G, Keystone E, Khanna D, Kirwan J, Kvien TK, Landewé R, Listing J, Michaud K, Martin-Mola E, Montie P, Pincus T, Richards P, Siegel JN, Simon LS, Sokka T, Strand V, Tugwell P, Tyndall A, van der Heijde D, Verstappen S, White B, Wolfe F, Zink A, Boers M. American College of Rheumatology/European League against Rheumatism provisional definition of remission in rheumatoid arthritis for clinical trials. Ann Rheum Dis. 2011;70: Fries JF, Spitz P, Kraines RG, Holman HR. Measurement of patient outcome in arthritis. Arthritis Rheum. 1980;23(2): Fries JF, Spitz PW, Young DY. The dimensions of health outcomes: the health assessment questionnaire, disability and pain scales. J Rheumatol. 1982;9(5):
448 I4V-MC-JADV Statistical Analysis Plan Version 2 Page 110 Herdman M, Gudex C, Lloyd A, Janssen MF, Kind P, Parkin D, Bonsel G, Badia X. Development and preliminary testing of the new five-level version of EQ-5D (EQ-5D-5L). Qual Life Res. 2011;20(10): Keystone EC, Kavanaugh AF, Sharp JT, Tannenbaum H, Hua Y, Teoh LS, Fischkoff SA, Chartash EK. Radiographic, clinical, and functional outcomes of treatment with adalimumab (a human anti tumor necrosis factor monoclonal antibody) in patients with active rheumatoid arthritis receiving concomitant methotrexate therapy: a randomized, placebo-controlled, 52-week trial. Arthritis Rheum. 2004;50(5): Keystone E, Heijde D, Mason D Jr, Landewé R, Vollenhoven RV, Combe B, Emery P, Strand V, Mease P, Desai C, Pavelka K. Certolizumab pegol plus methotrexate is significantly more effective than placebo plus methotrexate in active rheumatoid arthritis: findings of a fifty-twoweek, phase III, multicenter, randomized, double-blind, placebo-controlled, parallel-group study. Arthritis Rheum. 2008;58(11): Keystone E, Emery P, Peterfy CG, Tak PP, Cohen S, Genovese MC, Dougados M, Burmester GR, Greenwald M, Kvien TK, Williams S, Hagerty D, Cravets MW, Shaw T. Rituximab inhibits structural joint damage in patients with rheumatoid arthritis with an inadequate response to tumour necrosis factor inhibitor therapies. Ann Rheum Dis. 2009;68(2): National Kidney Foundation. K/DOQI Clinical Practice Guidelines for Chronic KidneyDisease: Evaluation, Classification and Stratification. Am J Kidney Dis ; 39(2)(suppl 1):S1-S266. Newcombe RG. Interval estimation for the difference between independent proportions: comparison of eleven methods. Stat Med. 1998;17: Erratum in Stat Med. 1999;18:1293. Protocol I4V-MC-JADV. A Randomized, Double-Blind, Placebo- and Active-Controlled, Phase 3 Study Evaluating the Efficacy and Safety of Baricitinib in Patients with Moderately to Severely Active Rheumatoid Arthritis Who Have Had an Inadequate Response to Methotrexate Therapy. Ramey DR, Fries JF, Singh G. The Health Assessment Questionnaire 1995: status and review. In: Spiker B, editor. Quality of life and pharmacoeconomics in clinical trials. 2nd ed. Philadelphia: Lippincott-Raven, 1996: p Reilly MC, Zbrozek AS, Dukes EM. The validity and reproducibility of a work productivity and activity impairment instrument. Pharmacoeconomics. 1993;4(5): Ridker PM, Buring JE, Rifari N, Cook NF. Development and Validation of Improved Algorithms for the Assessment of Global Cardiovascular Risk in Women: The Reynolds Risk Score. JAMA. 2007;(297):6. Ridker PM, Paynter NP, Rifai N, Gaziano JM, Cook NR. C-Reactive Protein and Parental History Improve Global Cardiovascular Risk Prediction: The Reynolds Risk Score for Men. Circulation. 2008;118: Rubin DB. Multiple imputation for nonresponse in surveys. New York: John Wiley & Sons;1987. Rush AJ, Trivedi MH, Ibrahim HM, Carmody TJ, Arnow B, Klein DN, Markowitz JC,Ninan PT, Kornstein S, Manber R, Thase ME, Kocsis JH, Keller MB. The 16-item Quick Inventory of
449 I4V-MC-JADV Statistical Analysis Plan Version 2 Page 111 Depressive Symptomatology (QIDS) Clinician Rating (QIDS-C) and Self-Report (QIDS-SR): A psychometric evaluation in patients with chronic major depression. Biological Psychiatry. 2003;54: Sampson, HA, Muñoz-Furlong, A, Campbell RL, Adkinson NF Jr, Bock SA, Branum A, Brown SG, Camargo CA Jr, Cydulka R, Galli SJ, Gidudu J, Gruchalla RS, Harlor AD Jr, Hepner DL, Lewis LM, Liberman PL, Metcalfe DD, O Connor R, Muraro A, Rudman A, Schmitt C, Scherrer D, Simons FE, Thomas S, Wood JP, Decker WW. Second symposium on the definition and management of anaphylaxis: summary report - second National Institute of Allergy and Infectious Disease/Food Allergy and Anaphylaxis Network symposium. J Allergy Clin Immunol. 2006;117(2): Schiff M, Fleischmann R, Weinblatt M, Valente R, van der Heijde D, Citera G, Zhao C, Maldonado M. Abatacept sc versus adalimumab on background methotrexate in RA: one year results from the AMPLE study. Ann Rheum Dis. 2012;71(suppl 3):60. Smolen J, Landewé RB, Mease P, Brzezicki J, Mason D, Luijtens K, van Vollenhoven RF, Kavanaugh A, Schiff M, Burmester GR, Strand V, Vencovskỳ J, van der Heijde D. Efficacy and safety of certolizumab pegol plus methotrexate in active rheumatoid arthritis: the RAPID 2 study. A randomised controlled trial. Ann Rheum Dis. 2009;68(6): Smolen JS, Beaulieu A, Rubbert-Roth A, Ramos-Remus C, Rovensky J, Alecock E, Woodworth T, Alten R, OPTION investigators. Effect of interleukin-6 receptor inhibition with tocilizumab in patients with rheumatoid arthritis (OPTION study): a double-blind, placebocontrolled, randomised trial. Lancet. 2008;371(9617): Szende A, Oppe M, Devlin N. EQ-5D value sets: Inventory, comparative review and user guide. EuroQol Group Monographs. Volume 2. Springer, Third Report of the National Cholesterol Education Program (NCEP) Expert Panel on Detection, Evaluation, and Treatment of High Blood Cholesterol in Adults (Adult Treatment Panel III) Final Report, National Cholesterol Education Program, National Heart, Lung, and Blood Institute, National Institutes of Health, NIH Publication No , September van der Heijde D, Tanaka Y, Fleischmann R, Keystone E, Kremer J, Zerbini C, Cardiel MH, Cohen S, Nash P, Song YW, Tegzová D, Wyman BT, Gruben D, Benda B, Wallenstein G, Krishnaswami S, Zwillich SH, Bradley JD, Connell CA. Tofacitinib (CP-690,550) in patients with rheumatoid arthritis receiving methotrexate: twelve-month data from a twenty-fourmonth phase III randomized radiographic study. Arthritis Rheum. 2013;65(3): van der Heijde D. How to read radiographs according to the Sharp/van der Heijde method. J Rheumatol. 2000;27(1): University of Pittsburgh IDS/QIDS page. IDS/QIDS web site. Available at: Accessed June 13, 2012.
450 I4V-MC-JADV Statistical Analysis Plan Version 2 Page Appendices
451 I4V-MC-JADV Statistical Analysis Plan Version 2 Page 113 Appendix 1. Algorithm for Determining ACR Response Details presented in this appendix will use x as a generic symbol, and the appropriate number (either 20, 50, or 70) is to be filled in when implementing in dataset programming code. ACRx response is defined as x% improvement from baseline in tender joint count (68 joint counts), and x% improvement in swollen joint count (66 joint counts), and x% improvement in at least three of the following five items: Patient s global assessment of arthritis pain; Patient s global assessment of disease activity; Physician s global assessment of disease activity; HAQ-DI; hscrp. The following abbreviations will be used throughout this appendix to refer to the items needed in the algorithm definitions: Parameter Abbreviation for the Parameter % improvement in tender joint count TJC68 % improvement in swollen joint count SJC66 % improvement in patient s assessment pain PATPAIN % improvement in patient s global assessment of disease PATGA activity % improvement in physician s global assessment of disease PHYGA activity % improvement in HAQ-DI HAQ % improvement in hscrp hscrp For all seven parameters mentioned above, % improvement at a visit is calculated as: (baseline value value at visit) * 100 / baseline value. To calculate the observed ACRx response at a visit: Step 1: If the patient discontinued from the study prior to reaching the visit, then STOP assign ACRx response as blank (ie, missing). Otherwise, calculate the % improvement at the visit for all seven parameters as described above. Step 2: o o If TJC68 AND SJC66 are BOTH x%, then proceed to step3. If both are non-missing but one or both is <x%, then STOP assign the patient as a non-responder for ACRx.
452 I4V-MC-JADV Statistical Analysis Plan Version 2 Page 114 o If either or both are missing, proceed as follows: a. If both are missing, then STOP assign ACRx response as blank (ie, missing). b. If one of TJC68 or SJC66 is missing and the non-missing value is <x%, then STOP assign the patient as a non-responder for ACRx. c. If one of TJC68 or SJC66 is missing and the non-missing value is x%, then STOP assign ACRx response as blank (ie, missing). Step 3: Consider the following five variables: PATPAIN, PTGA, PHYGA, HAQ, and hscrp. o o If three or more items are missing, then STOP assign ACRx response as blank (ie, missing). If three or more items are non-missing, then proceed with the following order: a. If at least three items are x%, then STOP assign the patient as a responder for ACRx. b. If at least three items are <x%, then STOP assign the patient as a nonresponder for ACRx. c. If less than three items are x%, then STOP assign ACRx response as blank. To calculate the ACRx response at post baseline visits up to Week 52 using NRI: Step 1: Calculate the % improvement at the visit for all seven parameters as described above. Step 2: If all seven parameters are missing at the visit, or if the patient discontinued from the study prior to reaching the visit, then STOP assign the patient as a nonresponder for ACRx. Step 3: If at least one of the seven parameters is non-missing at the visit and the patient is still enrolled in the study, then use LOCF to fill in any missing values. Step 4: If TJC68 AND SJC66 are BOTH x%, then proceed to step 5. If both are nonmissing but one or both is <x%, then STOP assign the patient as a nonresponder for ACRx. If either or both are missing, then STOP assign the patient as a non-responder for ACRx. Step 5: Consider the following five variables: PATPAIN, PATGA, PHYGA, HAQ, hscrp. o If three or more of the five items are non-missing, then: If three or more of the five items are x%, then STOP assign the patient as a responder for ACRx. If less than three of those five items are x%, then STOP assign the patient as a non-responder for ACRx. o If three or more of the five items are missing, then STOP assign the patient as a non-responder for ACRx.
453 I4V-MC-JADV Statistical Analysis Plan Version 2 Page 115 Appendix 2. Details of Efficacy and Health Outcomes Measures van der Heijde Modified Total Sharp Score X-rays of the hands/wrists and feet will be scored (according to the Synarc predefined imaging charter) for structural progression as measured using the mtss (van der Heijde, 2000). This methodology quantifies the extent of bone erosions for 44 joints and joint space narrowing for 42 joints with higher scores representing greater damage. Structural progression will be measured using the mtss. During screening, all patients meeting entry criteria will have a single posteroanterior radiographic assessment of the left and right hand/wrist and a single dorsoplantar radiographic image taken of the left and right foot. Alternatively, images taken within 4 weeks prior to baseline may be used. Images taken at screening (or within 4 weeks of screening) will be reviewed centrally by qualified readers for evidence of erosive bony change(s). These initial radiographs will serve as the baseline radiographs for comparison throughout the study. Additional radiographs (ie, a single posteroanterior view of each hand and a single dorsoplantar view of each foot) will be obtained at Week 16, Week 24 (the primary evaluation), and Week 52 (for the purposes of demonstrating durability of structural preservation as requested by regulatory authorities), as shown in the study schedule (See Protocol Appendix). For patients who discontinue prematurely from the study, x-rays will be performed at the early termination visit only if the previous x-rays were taken more than 12 weeks earlier. Bone Joint Erosion Score The joint erosion score is a summary of erosion severity in 32 joints of the hands and 12 joints of the feet. The maximum erosion score for a hand joint is 5 and for a foot joint is 10. Thus, the maximal erosion score is 280 for a timepoint (160 for both hands/ wrists and 120 for both feet). Each joint is scored according to the surface area involved from 0 to 5 for hand joints and 0 to 10 for the foot joints. The highest score (5 for the hand and 10 for the foot) indicates extensive loss of bone from more than one half of the articulating bone. A score of 0 in either the hand or foot joints indicates no erosion. Erosion for each hand joint (16 joints per hand) is graded on the following scale. 0 Normal (no erosion) 1 Discrete small erosion 2 Two discrete erosions or one large erosion not passing the joint middle line 3 One large erosion passing the joint middle line of the bone or combination of the above 4 Combinations of discrete and/or large erosions adding up to 4
454 I4V-MC-JADV Statistical Analysis Plan Version 2 Page Combinations of discrete and/or large erosions adding up to 5 or more NA - Not assessable due to poor radiographic depiction S - Surgically modified (e.g. joint replacement, amputation or other surgery) Discrete erosions will be graded 1 if small and 2 or 3 if larger, with a score of 3 if the erosion is large and extends across the imaginary middle of the bone. Discrete erosions of each grade will then be summed over both sides of the joint to a maximum of 5 to give a total score for each location in the hands. When erosions in the carpal bones of the wrist are confluent rather than discrete and focal, the percent surface eroded will be scored from 0 to 5 in approximately 20% intervals. A score of NA will be assigned in the case that the location is not evaluable due to advanced subluxation or poor radiographic depiction. A score of S will be assigned in the case of joint replacement, amputation or other surgery. Erosion for each side of a foot joint (6 joints per foot) is graded on the following scale. 0 Normal (no erosion) 1 Discrete small erosion 2 Two discrete erosions or one large erosion not passing the joint middle line 3 One large erosion passing the joint middle line 4-10 Combination of the above conditions. NA - Not assessable due to poor radiographic depiction S - Surgically modified (e.g. joint replacement, amputation or other surgery) Since both sides of each foot joint are graded, the maximum score for a foot joint is 10 (a score of 5 on each side). Joint Space Narrowing (JSN) Score The JSN score summarizes the severity of JSN in 30 joints of both hands and 12 joints of both feet. Assessment of JSN for each hand (15 joints per hand) and foot (6 joints per foot), including subluxation, is scored from 0 to 4, with 0 indicating no (normal) JSN and 4 indicating complete loss of joint space, bony ankylosis or luxation. Thus, the maximum JSN score is 168 (120 for both hands/ wrists and 48 for both feet) at a time point. JSN for each hand and foot joint is graded on the following scale: 0 Normal or no narrowing 1 Asymmetrical and/ or minimal narrowing- up to 25% 2 Definite narrowing with loss of up to 50% of the normal space 3 Definite narrowing with loss of 50-99% of the normal space or subluxation 4 Absence of a joint space, presumptive evidence of ankylosis or complete luxation NA - Not assessable due to poor radiographic depiction
455 I4V-MC-JADV Statistical Analysis Plan Version 2 Page 117 S - Surgically modified (e.g. joint replacement, amputation or other surgery) Total Modified Sharp Score (mtss) The total mtss score at a time point is the sum of the erosion (maximum of 280) and JSN (maximum of 168) scores, for a maximum score of 448. Handling of Missing Joint Data Refer to a separate document for a detailed description of imputation of missing joint data. Adjudication Process The read of x-ray images will be performed by 2 independent readers. An adjudication of results will be in place to show any discrepancy between the two independent readers. The adjudication process will involve re-reading by a third independent reader. Cases that require adjudication will be identified once the two primary independent reviews are completed. To ensure a consistent read by the two independent readers, an inter-/intra-reader variability assessment will be evaluated. Refer to the Bioclinica imaging charter and a separate document describing missing data imputation for more details. ACR Core Set The individual components that make up the ACR Core Set of measures for RA are: Tender Joint Count (TJC) For ACR measures, the number of tender and painful joints will be determined by examination of 68 joints (34 joints on each side of the patient s body). These 68 joints will be assessed and classified as tender, not tender, or not evaluable. The number of tender joints ranges from 0 to 68. The algorithm to calculate TJC is in the Joint Assessment section of this Appendix. Swollen Joint Count (SJC) For ACR measures, the number of swollen joints will be determined by examination of 66 joints (33 joints on each side of the patient s body). These 66 joints will be assessed and classified as swollen, not swollen, or not evaluable. The number of swollen joints ranges from 0 to 66. Refer to the Joint Assessment section of this Appendix for the algorithm to calculate SJC. Patient s Assessment of Pain (VAS) The patient will be asked to assess his or her current level of pain by marking a vertical tick on a 100-mm horizontal VAS with the left end marked as no pain and the right end marked as worst possible pain. Results will be expressed in millimeters measured between the left end of the scale and the crossing point of the vertical line of the tick. This procedure is applicable for all VAS scales used in the trial. Patient s Global Assessment of Disease Activity (VAS) The patients will be asked to give an overall assessment of how their rheumatoid arthritis is affecting them at present by marking a vertical tick on a VAS from very well to very poor.
456 I4V-MC-JADV Statistical Analysis Plan Version 2 Page 118 Physician s Global Assessment of Disease Activity (VAS) The physician s assessment of a patient s disease activity assessed at the visit will be recorded using the VAS from none to extremely active arthritis. Patient s Assessment of Physical Function (HAQ-DI) The patient will be asked to complete the HAQ-DI to measure the impact of arthritis on their ability to function in daily life. The HAQ-DI functional disability index score ranges from 0 to 3, with lower scores indicating better physical functioning. See Section HAQ-DI of this Appendix for the algorithm to calculate the HAQ-DI functional disability index score. High sensitivity C-Reactive Protein (hscrp) hscrp will be the ACR Core Set measure of acute phase reactant. It will be measured at the central laboratory. Joint Assessment Each of 28 or 68 joints will be evaluated for tenderness and 28 or 66 joints will be evaluated for swelling (hips are excluded for swelling) at the specified visits as shown in the schedule of events of the protocol. The 68/66 joint count will be performed at screening, Week 0 (baseline), Week 1 to Week 24 in Part A, Week 24 to Week 52 in Part B, end of treatment and at the followup visit. For scores using 28 joints, the following subset will be assessed (both right and left side): Shoulder, Elbow, Wrist, Metacarpophalangeal (MCP) I, II, III, IV, and V, Thumb interphalangeal, Proximal interphalangeal (PIP) II, III, IV, and V, Knee. Joints will be assessed for tenderness by pressure and joint manipulation on physical examination. Any positive response on pressure, movement, or both will then be translated into a single tender-versus non-tender dichotomy. Joints will be classified as either swollen or not swollen. Swelling is defined as palpable fluctuating synovitis of the joint. Swelling secondary to osteoarthrosis will be assessed as not swollen, unless there is unmistakable fluctuation. The following joint count imputation rules will be applied. (1) Joints that had any of the following procedures or disease conditions that occur at the screening (Visit 1) up to and including baseline (Visit 2) will be imputed as follows:
457 I4V-MC-JADV Statistical Analysis Plan Version 2 Page 119 Arthroplasty, fusion, synovectomy, ankylosis, amputation, injury, fracture and infection: these will be considered non-evaluable from baseline up to end of the study. Joints that include non-evaluable joints will be prorated from baseline up to end of the study. For example, the swollen joint count score for 6 swollen plus 2 non-evaluable joints for DAS28 is calculated as [6/(28-2)]*28 = Injection: joints that have received intra-articular and bursal injections will be collected in the data but set to painful/ tender or swollen after the injection for 6 months (up to Week 24 [Visit 11]). (2) Joints that had any of the following procedures or disease conditions that occur postbaseline (anytime during treatment with the study drugs) will be imputed as follows: Amputation: amputation that occurs postbaseline will be considered non-evaluable from the visit after amputation up to end of the study. Joints that include non-evaluable joints will be prorated from the visit after amputation up to end of the study. For example, the swollen joint count score for 6 swollen plus 2 non-evaluable joints for DAS28 is calculated as [6/(28-2)]*28 = Arthroplasty, fusion, synovectomy and ankylosis: if any of these procedures occur postbaseline, joints will be set to painful/tender or swollen from the visit after the procedure up to end of the study. Infection, injury and fracture: if any of these conditions occur postbaseline, joints will be set to painful/tender or swollen from the visit after the condition until the condition is resolved. Injection: joints that have received intra-articular and bursal injections postbaseline will be collected in the data but set to painful/tender or swollen after the injection for 6 months or end of study (whichever comes first). The number of tender and swollen joints will be calculated by summing all joints. For patients who have an incomplete set of joints evaluated, the joint count will be adjusted to a 28- or 68- joint count for tenderness and a 28- or 66-joint count for swelling by dividing the number of affected joints by the number of evaluated joints and multiplying by 28 or 68 for tenderness and 28 or 66 for swelling. Disease Activity Score-Erythrocyte Sedimentation Rate (DAS28- ESR) and Disease Activity Score-High Sensitivity C-Reactive Protein (DAS28-hsCRP) The DAS28 is a measure of disease activity in 28 joints that consists of a composite numeric score of the following variables: TJC, SJC, hscrp or ESR, and Patient s Global Assessment of Disease Activity (Vander Cruyssen et al. 2005). The 28 joints to be examined and assessed as tender or not tender for TJC and as swollen or not swollen for SJC include 14 joints on each side of the patient s body: the 2 shoulders, the 2 elbows, the 2 wrists, the 10 metacarpophalangeal joints, the 2 interphalangeal joints of the thumb, the 8 proximal interphalangeal joints, and the 2 knees (Smolen et al. 1995). The following equation will be used to calculate the DAS28-hsCRP (Vander Cruyssen et al. 2005):
458 I4V-MC-JADV Statistical Analysis Plan Version 2 Page 120 DAS28-hsCRP = 0.56(TJC28) 1/ ( SJC28) 1/ (ln(hsCRP +1)) (VAS) The hscrp value in mg/l must be used. In order to calculate DAS28-hsCRP data must be present for all 4 components: TJC28, SJC28, patient s global assessment (VAS) and hscrp. Component-level imputation will be performed for the calculation of DAS28-hsCRP for the imputed visits summaries. If at least one of the 4 components is nonmissing at the visit and the patient is still enrolled in the study, then use LOCF to fill in any missing values. Based on the DAS- hscrp, the following will be calculated: DAS28-hsCRP 3.2 DAS28-hsCRP <2.6 Similarly, DAS28-ESR=0.56(TJC28) 1/ (SJC28) 1/ ln(esr) (VAS) [If ESR = 0 in analysis data, set ln(esr)=0] The ESR (Erythrocyte sedimentation rate) in mm/first hour must be used. In order to calculate DAS28-ESR data must be present for all 4 components: TJC28, SJC28, patient s global assessment (VAS) and ESR. Component-level imputation will be performed for the calculation of DAS28-ESR for the imputed visits summaries. If at least one of the 4 components is nonmissing at the visit and the patient is still enrolled in the study, then use LOCF to fill in any missing values. Based on the DAS-ESR, the following will be calculated: DAS28-ESR 3.2 DAS28-ESR <2.6 A NRI rule for the DAS28 binary endpoints will be applied for each visit as follows: if the DAS28 is missing at the visit or if the patient discontinued treatment or the study prior to the visit, then assign the patient as DAS28 non-responder. Hybrid American College of Rheumatology Response Measure The hybrid ACR (bounded) response measure will be obtained as described by the ACR Committee to Reevaluate Improvement Criteria (2007);
459 I4V-MC-JADV Statistical Analysis Plan Version 2 Page 121 Table APP.1. Scoring Method for the Hybrid American College of Rheumatology Response Measure Mean % Change in Core Set Measuresa ACR Status <20 20, <50 50, <70 70 Not ACR20 Mean % change ACR20 but not ACR50 20 Mean % change ACR50 but not ACR Mean % change ACR Mean % change Abbreviations: ACR = American College of Rheumatology; ACR20 = 20% improvement in ACR criteria; ACR50 = 50% improvement in ACR criteria; ACR70 = 70% improvement in ACR criteria. a 1) Calculate the average percentage change in core set measures. For each core set measure, subtract score after treatment from baseline score and determine percentage improvement in each measure. Next, if a core set measure worsened by >100%, limit that percentage change to 100% (set equal to -100% bound). Then, average the percentage changes for all core set measures. 2) Determine whether the patient has achieved ACR20, ACR50, or ACR70. 3) Using the table above, obtain the hybrid ACR response measure. To use the table, take the ACR20, ACR50, or ACR70 status of the patient (left column) and the mean percentage improvement in core set items; the hybrid ACR score is where they intersect in the table. Simplified Disease Activity Index (SDAI) The SDAI is a tool for measurement of disease activity in RA that integrates measures of physical examination, acute phase response, patient self-assessment, and evaluator assessment. The SDAI is calculated by adding together scores from the following assessments: number of swollen joints (0 to 28), number of tender joints (0 to 28), hscrp in mg/dl (0.1 to 10.0), patient global assessment of DAS on VAS (0 to 10.0, measured in cm), and evaluator or physician global assessment of DAS on VAS (0 to 10.0, measured in cm) (Aletaha and Smolen 2005) Disease remission according to ACR/EULAR index-based definition of remission is defined as an SDAI score of 3.3 (Felson et al. 2011). Low disease activity according to SDAI is defined as a SDAI score 11 (Aletaha and Smolen 2005). Clinical Disease Activity Index (CDAI) The CDAI is similar to the SDAI, but it allows for immediate scoring because it does not use a laboratory result. The CDAI is calculated by adding together scores from the following assessments: number of swollen joints (0 to 28), number of tender joints (0 to 28), patient global assessment of DAS on VAS (0 to 10.0, measured in cm), and
460 I4V-MC-JADV Statistical Analysis Plan Version 2 Page 122 evaluator or physician global assessment of DAS on VAS (0 to 10.0, measured in cm) (Aletaha and Smolen 2005) Remission according to CDAI is defined as a CDAI score of 2.8 (Felson et al. 2011). Low disease activity according to CDAI is defined as a CDAI score 10 (Aletaha and Smolen 2005). European League Against Rheumatism (EULAR) Responder Index Assessments of patients with RA by the EULAR Responder Index based on the 28-joint count will be used to categorize patients as nonresponders, moderate responders, good responders, or responders (moderate + good responders) according to van Gestel et al Table APP.2. Postbaseline Level of DAS28-hsCRP DAS28-hsCRP 3.2 Categorization of Patients as Nonresponders, Moderate Responders, or Good Responders Improvement Since Baseline in DAS28-hsCRP (Decline in DAS28-hsCRP) > and > Good response 3.2 < DAS28-hsCRP 5.1 Moderate response DAS28-hsCRP >5.1 No response Abbreviation: DAS28-hsCRP = Disease Activity Score modified to include the 28 diarthroidal joint count used for hscrp. There are 3 categories, no improvement (or no response), moderate improvement (or moderate response), and good improvement (highest kind of improvement or good response). The category of no improvement also includes a worsening of RA. Hence, relative to baseline, a patient can only have a 0 change in categories, a change of 1 category or a change of 2 categories. EULAR response based on DAS28-ESR may also be evaluated. ACR/EULAR Rheumatoid Arthritis Remission Two new ACR/EULAR definitions of RA remission will be evaluated, a boolean-based definition and an index-based definition (Felson et al. 2011). Boolean-based Definition of Remission: All 4 criteria below must be met (at the same visit): o o o o TJC28 1 SJC28 1 hscrp 1 mg/dl (10 mg/l) Patient Global Assessment of Disease Activity (on a mm scale) / 10 1
461
462 I4V-MC-JADV Statistical Analysis Plan Version 2 Page 124 The FACIT-F uses a 13-item symptom-specific questionnaire to assess the burden of selfreported fatigue caused by a chronic disease and its impact upon daily activities and function. A 5-point Likert-type scale (0 = not at all; 1 = a little bit; 2 = somewhat; 3 = quite a bit; 4 = very much) is used. As each of the 13 items of the FACIT-Fatigue scale ranges from 0 4, the range of possible scores is 0 52, with 0 being the worst possible score and 52 the best. To obtain the 0 52 score, each negatively worded item response is transformed so that 0 is a bad response and 4 is good response. All responses are added with equal weight to obtain the total score. Lower values of the FACIT-Fatigue score denote higher fatigue. Specifically, all questions with the exception of I have energy and I am able to perform usual activities will have their responses recoded so that 0 = Very much, 1 = Quite a Bit, 2 = Somewhat, 3 = A little bit, 4 = Not at all) prior to calculation of the FACIT-Fatigue scale score. To score FACIT-Fatigue Subscale (FACIT_F), do the following: 1. Perform reversals for 11 out of the 13 individual items, namely HI7, HI12, An1, An2, An3, An4, An8, An12, An14, An15, An16: item score = 4 minus item response 2. For the individual items An5 and An7: items score = item response 3. Sum the individual items to obtain a score 4. Multiply the sum of the items scores by 13 (the number of items in the subscale), then divide by the number of items answered which produces the Fatigue Subscale score. Missing items are acceptable as long as more than 50% of the items are answered (i.e. a minimum of 7 out of 13 items). If less than 7 items are answered, the Fatigue Subscale score will be set to missing. SF-36v2 Acute: The SF-36v2 Acute measure is a subjective, generic, health-related quality of life instrument that is patient-reported and consists of 36 questions covering 8 health domains: physical functioning, bodily pain, role limitations due to physical problems, role limitations due to emotional problems, general health perceptions, mental health, social function, and vitality. Each domain is scored by summing the individual items and transforming the scores into a 0 to 100 scale with higher scores indicating better health status or functioning. In addition, 2 summary scores, the PCS (physical component score) and the MCS (mental component score), will be evaluated based on the 8 SF-36v2 Acute domains (Brazier et al. 1992; Ware and Sherbourne 1992). How to Calculate the SF-36v2 Scores This protocol uses the SF-36 version 2 acute (1-week recall) form. Scoring for the form is as follows, based on the manual How to Score version 2 of the SF-36 Health Survey (Ware JE, et al. 2000):
463

464

465
466 I4V-MC-JADV Statistical Analysis Plan Version 2 Page 128 European Quality of Life-5 Dimensions-5 Level (EQ-5D-5L) Scores: The European Quality of Life - 5 dimensions (EQ-5D) questionnaire is a widely used, generic questionnaire that assesses health status (The EuroQol Group 1990/Herdman et al. 2011). The questionnaire consists of 2 parts: The first part assesses 5 dimensions: mobility, self-care, usual activities, pain/discomfort and anxiety/depression. The levels of response for mobility, self-care and usual activities are no problems, slight problems, moderate problems, severe problems or unable to. For pain/discomfort, the levels of response are no pain/discomfort, slight pain/discomfort, moderate pain/discomfort, severe pain/discomfort and extreme pain/discomfort. Lastly, for anxiety/depression, the levels of response are not anxious or depressed, slightly anxious or depressed, moderately anxious or depressed, severely anxious or depressed, and extremely anxious or depressed. This part of the EQ-5D can be used to generate a health state index score, which is often used to compute quality-adjusted life years (QALY) for utilization in health economic analyses. The health state index score is calculated based on the responses to the 5 dimensions, providing a single value on a scale from less than 0 (where zero is a health state equivalent to death; negative values are valued as worse than dead) to 1 (perfect health), with higher scores indicating better health utility. The second part of the questionnaire consists of a visual analog scale (VAS) on which the patient rates their perceived health state from 0 (worst imaginable health state/the worst health you can imagine) to 100 (best imaginable health state/the best health you can imagine).
467 I4V-MC-JADV Statistical Analysis Plan Version 2 Page 129 Health Assessment Questionnaire Disability Index (HAQ-DI) The HAQ-DI is a patient-reported questionnaire that is commonly used in RA to measure disease-associated disability (assessment of physical function). It consists of 24 questions referring to 8 domains: dressing/grooming, arising, eating, walking, hygiene, reach, grip, and activities (Fries et al. 1980, 1982; Ramey et al. 1996). 1. Dressing and grooming (C1. Dress yourself, including tying shoelaces and doing buttons, C2. Shampooing your Hair) Includes 2 component questions, 1 device checkbox (devices used for dressing), 1 help checkbox. 2. Arising (C1. Stand up from straight chair, C2. Get in and out of bed) Includes 2 component questions, 1 device checkbox (built-up or special chair), 1 help checkbox. 3. Eating (C1. Cut your meat, C2. Lift a cup or glass to your mouth, C3. Open a new carton of milk) Includes 3 component questions, 1 device checkbox (built-up or special utensils), 1 help checkbox. 4. Walking (C1. Walk outdoors on flat ground, C2. Climb up five steps) Includes 2 component questions, 4 device checkboxes (cane, walker, crutches, wheelchair), 1 help checkbox. 5. Hygiene (C1. Wash and dry your body, C2. Take a tub bath, C3. Get on and off the toilet) Includes 3 component questions, 4 device checkboxes (raised toilet seat, bathtub seat, bathtub bar, long-handled appliances in bathroom), 1 help checkbox. 6. Reach (C1. Reach and get down a 5 pound object (such as a bag of sugar) from just above your head, C2. Bend down to pick up clothing from the floor) Includes 2 component questions, 1 device checkbox (long-handled appliances for reach), 1 help checkbox. 7. Grip (C1. Open car doors, C2. Open jars which have been previously opened, C3. Turn faucets on and off) Includes 3 component questions, 1 device checkbox (jar opener), 1 help checkbox. 8. Activities (C1. Run errands and shop, C2. Get in and out of a car, C3. Do chores such as vacuuming or yard work) Includes 3 component questions, 1 help checkbox. In order to compute the HAQ-DI (Standard Disability Index) score, the following scores are assigned to the responses: Without any difficulty = 0 With some difficulty = 1 With much difficulty = 2
468 I4V-MC-JADV Statistical Analysis Plan Version 2 Page 130 Unable to do = 3. The disability section of the questionnaire scores the patient s self-perception on the degree of difficulty (0 = without any difficulty, 1 = with some difficulty, 2 = with much difficulty, and 3 = unable to do) when dressing and grooming, arising, eating, walking, hygiene, reach, grip, and performing other daily activities. The reported use of special aids or devices and/or the need for assistance of another person to perform these activities is also assessed. The scores for each of the functional domains will be averaged to calculate the functional disability index. Calculating the HAQ-DI: The patient must have a score for at least 6 of the 8 categories. If there are less than six categories completed, a HAQ-DI cannot be computed. A category score is determined from the highest score of the sub-categories, or components, in that category. (For example, in the category EATING there are three sub-category items. If a patient responds with a 1, 2, and 0, respectively; the category score is 2.) Adjust for use of aids/devices and/or help from another person when indicated: o When there are no aids or devices or help indicated for a category, the category s score is not modified. o When aids or devices or help ARE indicated by the patient, adjust the score for a category by increasing a zero or a one to a two. If a patient's highest score for that sub-category is a two it remains a two, and if a three, it remains a three. o Sum the eight category scores o Divide the sum by the number of categories answered (range 6-8) The scale is not truly continuous but has 25 possible values (i.e., 0, 0.125, 0.250, ). The mapping of the aids or devices to the categories is the following: HAQ-DI Category Companion aids or devices item Dressing and Grooming Devices used for dressing (button hook, zipper pull, long handled shoe horn, etc.) Arising Built up or special chair Eating Built up or special utensils Walking Cane, walker, crutches Hygiene Long handled appliances in bathroom Reach Long handled appliances for reach Grip Jar opener (for jars previously opened) Minimum Clinically Important Differences: The summary and analyses of the HAQ-DI Score will include the number of patients achieving a MCID, which is defined as: MCID 0.22 reduction in HAQ-DI Score. Also, the number of patients having 0.3 reduction in HAQ-DI Score will be assessed.
469 I4V-MC-JADV Statistical Analysis Plan Version 2 Page 131 Appendix 3. Framingham Risk Assessment and Reynolds Risk Assessment: 10-Year Risk of Cardiovascular Disease Events Framingham Risk Assessment The Framingham risk score is a 10-year prediction of cardiovascular disease (CVD) events (D Agostino et al. 2008). The outcome CVD comprises: coronary death, myocardial infarction, coronary insufficiency, angina, ischemic stroke, hemorrhagic stroke, transient ischemic attack, peripheral artery disease, and heart failure. Following predictors will be assessed in the primary model: Age [years] Diabetes [Yes / No] Smoking [Yes / No] Treated and untreated Systolic Blood Pressure [mmhg] Total cholesterol [mg/dl] HDL cholesterol [mg/dl] The following data will be used to calculate the Framingham score: Age - year of birth recorded in the CRF. Diabetes - coded pre-existing condition will be evaluated. Smoking - current tobacco usage recorded in the CRF. Treated and untreated hypertension - the coded concomitant therapy will be evaluated to distinguish between treated and untreated hypertension. Systolic Blood Pressure - vital signs data. Total cholesterol - LAB data. HDL cholesterol - LAB data. The measurements must be entered into the calculation using the units mentioned above. The 10-year risk for women can be calculated as exp(σßx ) and the risk for men is given as exp(σßx ) where ß is the regression coefficient and X is the level (value) for each risk factor. The estimated regression coefficients (Beta) are shown in the following table.

470
471 I4V-MC-JADV Statistical Analysis Plan Version 2 Page 133 Reynolds Risk Assessment The RRS is derived for non-diabetic men and for non-diabetic women aged between 45 and 80 years old with no prior history of CVD and/ or cerebrovascular events. The following predictors will be used to calculate the RRS: Age (years) Smoking status (Yes / No) SBP (mmhg) Total cholesterol (mg/dl) HDL cholesterol (mg/dl) hscrp (mg/l) Patient s parent history of myocardial infarction diagnosis before the age of 60 years (Yes/ No) The 10-year RRS (%) for men is calculated as: [ exp(σßx ) ] * 100 The 10-year RRS (%) for women is calculated as: [ exp(σßx ) ] * 100 (where ß is the regression coefficient and X is the value for each risk factor) Additional information and the estimated regression coefficients (ß) used for the calculation of RRS can be found in Ridker et al for women and to Ridker et al for men.
472 I4V-MC-JADV Statistical Analysis Plan Version 2 Page 134 Appendix 4. Corticosteroids Corticosteroids The following ATC code will be used to select all possible corticosteroids: H02 Corticosteroids for systemic use with route of Oral. All unique preferred terms in the database falling under the above ATC code will be reviewed by the medical group in order to determine which ones should be included. All corticosteroid doses need to be converted to prednisone equivalent doses. If additional conversion factors are required, these will be added to the table below in a SAP prior to database lock. The following table should be used for converting non-prednisone medications to prednisone equivalent: Multiply the dose of the corticosteroid taken by the patient (in milligrams) in Column 1 by the conversion factor in Column 2 to get the equivalent dose of prednisone (in milligrams). Example: Patient is taking 16 mg of methylprednisolone orally daily. To convert to prednisone: 16 mg methylprednisolone X 1.25 = 20 mg prednisone. 16 mg of methylprednisolone taken orally daily is equivalent to 20 mg of prednisone taken orally daily.

473
New Evidence reports on presentations given at EULAR Safety and Efficacy of Tocilizumab as Monotherapy and in Combination with Methotrexate
 New Evidence reports on presentations given at EULAR 2009 Safety and Efficacy of Tocilizumab as Monotherapy and in Combination with Methotrexate Report on EULAR 2009 presentations Tocilizumab inhibits
New Evidence reports on presentations given at EULAR 2009 Safety and Efficacy of Tocilizumab as Monotherapy and in Combination with Methotrexate Report on EULAR 2009 presentations Tocilizumab inhibits
Efficacy and Safety of Tocilizumab in the Treatment of Rheumatoid Arthritis and Juvenile Idiopathic Arthritis
 New Evidence reports on presentations given at EULAR 2010 Efficacy and Safety of Tocilizumab in the Treatment of Rheumatoid Arthritis and Juvenile Idiopathic Arthritis Report on EULAR 2010 presentations
New Evidence reports on presentations given at EULAR 2010 Efficacy and Safety of Tocilizumab in the Treatment of Rheumatoid Arthritis and Juvenile Idiopathic Arthritis Report on EULAR 2010 presentations
Individual Study Table Referring to Part of Dossier: Use Only) Name of Study Drug:
 Name of Study Drug:") 2.0 Synopsis AbbVie Inc. Individual Study Table Referring to Part of Dossier: (For National Authority Use Only) Name of Study Drug: Volume: Adalimumab (Humira ) Page: Name of Active Ingredient: Adalimumab
2.0 Synopsis AbbVie Inc. Individual Study Table Referring to Part of Dossier: (For National Authority Use Only) Name of Study Drug: Volume: Adalimumab (Humira ) Page: Name of Active Ingredient: Adalimumab
2.0 Synopsis. Adalimumab (HUMIRA ) W Clinical Study Report R&D/15/0629. Individual Study Table Referring to Part of Dossier: Volume:
 W Clinical Study Report R&D/15/0629. Individual Study Table Referring to Part of Dossier: Volume:") 2.0 Synopsis AbbVie Inc. Name of Study Drug: Adalimumab / HUMIRA Name of Active Ingredient: Adalimumab Individual Study Table Referring to Part of Dossier: Volume: Page: (For National Authority Use Only)
2.0 Synopsis AbbVie Inc. Name of Study Drug: Adalimumab / HUMIRA Name of Active Ingredient: Adalimumab Individual Study Table Referring to Part of Dossier: Volume: Page: (For National Authority Use Only)
PFIZER INC. These results are supplied for informational purposes only. Prescribing decisions should be made based on the approved package insert.
 PFIZER INC. These results are supplied for informational purposes only. Prescribing decisions should be made based on the approved package insert. GENERIC DRUG NAME / COMPOUND NUMBER: Tofacitinib / CP-690,550
PFIZER INC. These results are supplied for informational purposes only. Prescribing decisions should be made based on the approved package insert. GENERIC DRUG NAME / COMPOUND NUMBER: Tofacitinib / CP-690,550
Annual Rheumatology & Therapeutics Review for Organizations & Societies
 Annual Rheumatology & Therapeutics Review for Organizations & Societies Comparative Effectiveness Studies of Biologics Learning Objectives Understand the motivation for comparative effectiveness research
Annual Rheumatology & Therapeutics Review for Organizations & Societies Comparative Effectiveness Studies of Biologics Learning Objectives Understand the motivation for comparative effectiveness research
PFIZER INC. These results are supplied for informational purpose only. Prescribing decisions should be made based on the approved package insert.
 Public Disclosure Synopsis Protocol A3924 4 November 24 Final PFIZER INC. These results are supplied for informational purpose only. Prescribing decisions should be made based on the approved package insert.
Public Disclosure Synopsis Protocol A3924 4 November 24 Final PFIZER INC. These results are supplied for informational purpose only. Prescribing decisions should be made based on the approved package insert.
Synopsis Style Clinical Study Report SAR ACT sarilumab Version number : 1 (electronic 1.0)
") SYNOPSIS Title of the study: A randomized, double-blind, parallel-group, placebo- and active calibrator-controlled study assessing the clinical benefit of SAR153191 subcutaneous (SC) on top of methotrexate
SYNOPSIS Title of the study: A randomized, double-blind, parallel-group, placebo- and active calibrator-controlled study assessing the clinical benefit of SAR153191 subcutaneous (SC) on top of methotrexate
New Evidence reports on presentations given at EULAR Tocilizumab for the Treatment of Rheumatoid Arthritis
 New Evidence reports on presentations given at EULAR 2012 Tocilizumab for the Treatment of Rheumatoid Arthritis Report on EULAR 2012 presentations Tocilizumab monotherapy is superior to adalimumab monotherapy
New Evidence reports on presentations given at EULAR 2012 Tocilizumab for the Treatment of Rheumatoid Arthritis Report on EULAR 2012 presentations Tocilizumab monotherapy is superior to adalimumab monotherapy
New Evidence reports on presentations given at ACR Improving Radiographic, Clinical, and Patient-Reported Outcomes with Rituximab
 New Evidence reports on presentations given at ACR 2009 Improving Radiographic, Clinical, and Patient-Reported Outcomes with Rituximab From ACR 2009: Rituximab Rituximab in combination with methotrexate
New Evidence reports on presentations given at ACR 2009 Improving Radiographic, Clinical, and Patient-Reported Outcomes with Rituximab From ACR 2009: Rituximab Rituximab in combination with methotrexate
2.0 Synopsis. Adalimumab DE019 OLE (5-year) Clinical Study Report Amendment 1 R&D/06/095. (For National Authority Use Only)
 Clinical Study Report Amendment 1 R&D/06/095. (For National Authority Use Only)") 2.0 Synopsis Abbott Laboratories Name of Study Drug: Humira Name of Active Ingredient: Adalimumab Individual Study Table Referring to Part of Dossier: Volume: Page: (For National Authority Use Only) Title
2.0 Synopsis Abbott Laboratories Name of Study Drug: Humira Name of Active Ingredient: Adalimumab Individual Study Table Referring to Part of Dossier: Volume: Page: (For National Authority Use Only) Title
Supplemental Table 1. Key Inclusion Criteria Inclusion Criterion OPTIMA PREMIER 18 years old with RA (per 1987 revised American College of General
 Supplemental Table 1. Key Inclusion Criteria Inclusion Criterion OPTIMA PREMIER 18 years old with RA (per 1987 revised American College of General Rheumatology classification criteria) 34 ; erythrocyte
Supplemental Table 1. Key Inclusion Criteria Inclusion Criterion OPTIMA PREMIER 18 years old with RA (per 1987 revised American College of General Rheumatology classification criteria) 34 ; erythrocyte
Page: 17 December 2012 (Study M13-692) 22 October 2013 (Study M13-692)
 22 October 2013 (Study M13-692)") 2.0 Synopsis AbbVie Inc. Name of Study Drug: Adalimumab Name of Active Ingredient: D2E7 Individual Study Table Referring to Part of Dossier: Volume: Page: (For National Authority Use Only) Title of Studies:
2.0 Synopsis AbbVie Inc. Name of Study Drug: Adalimumab Name of Active Ingredient: D2E7 Individual Study Table Referring to Part of Dossier: Volume: Page: (For National Authority Use Only) Title of Studies:
This clinical study synopsis is provided in line with Boehringer Ingelheim s Policy on Transparency and Publication of Clinical Study Data.
 abcd Clinical Study Synopsis for Public Disclosure This clinical study synopsis is provided in line with Boehringer Ingelheim s Policy on Transparency and Publication of Clinical Study Data. The synopsis
abcd Clinical Study Synopsis for Public Disclosure This clinical study synopsis is provided in line with Boehringer Ingelheim s Policy on Transparency and Publication of Clinical Study Data. The synopsis
Olumiant (baricitinib) NEW PRODUCT SLIDESHOW
 NEW PRODUCT SLIDESHOW") Olumiant (baricitinib) NEW PRODUCT SLIDESHOW Introduction Brand name: Olumiant Generic name: Baricitinib Pharmacological class: Janus kinase (JAK) inhibitor Strength and Formulation: 2mg; tabs Manufacturer:
Olumiant (baricitinib) NEW PRODUCT SLIDESHOW Introduction Brand name: Olumiant Generic name: Baricitinib Pharmacological class: Janus kinase (JAK) inhibitor Strength and Formulation: 2mg; tabs Manufacturer:
New Evidence reports on presentations given at EULAR Tocilizumab for the Treatment of Rheumatoid Arthritis and Juvenile Idiopathic Arthritis
 New Evidence reports on presentations given at EULAR 2011 Tocilizumab for the Treatment of Rheumatoid Arthritis and Juvenile Idiopathic Arthritis Report on EULAR 2011 presentations Benefit of continuing
New Evidence reports on presentations given at EULAR 2011 Tocilizumab for the Treatment of Rheumatoid Arthritis and Juvenile Idiopathic Arthritis Report on EULAR 2011 presentations Benefit of continuing
2.0 Synopsis. Adalimumab M Clinical Study Report Final R&D/15/1093. (For National Authority Use Only)
") 2.0 Synopsis AbbVie Inc. Name of Study Drug: Adalimumab Name of Active Ingredient: Adalimumab Individual Study Table Referring to Part of Dossier: Volume: Page: (For National Authority Use Only) Title
2.0 Synopsis AbbVie Inc. Name of Study Drug: Adalimumab Name of Active Ingredient: Adalimumab Individual Study Table Referring to Part of Dossier: Volume: Page: (For National Authority Use Only) Title
Tocilizumab F. Hoffmann-La Roche Ltd. Protocol MA 27950, Version 3.0 1
 Protocol MA 27950, Version 3.0 1 Protocol MA 27950, Version 3.0 2 TABLE OF CONTENTS PROTOCOL ACCEPTANCE FORM... 8 PROTOCOL SYNOPSIS... 9 1. BACKGROUND... 15 1.1 Background on Rheumatoid Arthritis... 15
Protocol MA 27950, Version 3.0 1 Protocol MA 27950, Version 3.0 2 TABLE OF CONTENTS PROTOCOL ACCEPTANCE FORM... 8 PROTOCOL SYNOPSIS... 9 1. BACKGROUND... 15 1.1 Background on Rheumatoid Arthritis... 15
1.0 Abstract. Title. Keywords. Adalimumab, Rheumatoid Arthritis, Effectiveness, Safety. Rationale and Background
 1.0 Abstract Title Assessment of the safety of adalimumab in rheumatoid arthritis (RA) patients showing rapid progression of structural damage of the joints, who have no prior history of treatment with
1.0 Abstract Title Assessment of the safety of adalimumab in rheumatoid arthritis (RA) patients showing rapid progression of structural damage of the joints, who have no prior history of treatment with
Criteria Inclusion criteria Exclusion criteria. despite treatment with csdmards, NSAIDs, and/or previous anti-tnf therapy and/or
 Supplementary Material Table S1 Eligibility criteria (PICOS) for the SLR Criteria Inclusion criteria Exclusion criteria Population Adults (aged 18 years) with active PsA despite treatment with csdmards,
Supplementary Material Table S1 Eligibility criteria (PICOS) for the SLR Criteria Inclusion criteria Exclusion criteria Population Adults (aged 18 years) with active PsA despite treatment with csdmards,
Synopsis (C0524T12 GO LIVE)
") Protocol: EudraCT No.: 2005-003232-21 Title of the study: A Multicenter, Randomized, Double-blind, Placebo-controlled Trial of Golimumab, a Fully Human Anti-TNFα Monoclonal Antibody, Administered Intravenously,
Protocol: EudraCT No.: 2005-003232-21 Title of the study: A Multicenter, Randomized, Double-blind, Placebo-controlled Trial of Golimumab, a Fully Human Anti-TNFα Monoclonal Antibody, Administered Intravenously,
(For National Authority Use Only) Page:
 Page:") 2.0 Synopsis AbbVie Individual Study Table Referring to Part of Dossier: Name of Study Drug: Volume: HUMIRA 40 mg/0.8 ml for subcutaneous injection Page: (For National Authority Use Only) Name of Active
2.0 Synopsis AbbVie Individual Study Table Referring to Part of Dossier: Name of Study Drug: Volume: HUMIRA 40 mg/0.8 ml for subcutaneous injection Page: (For National Authority Use Only) Name of Active
This clinical study synopsis is provided in line with Boehringer Ingelheim s Policy on Transparency and Publication of Clinical Study Data.
 abcd Clinical Study Synopsis for Public Disclosure This clinical study synopsis is provided in line with Boehringer Ingelheim s Policy on Transparency and Publication of Clinical Study Data. The synopsis
abcd Clinical Study Synopsis for Public Disclosure This clinical study synopsis is provided in line with Boehringer Ingelheim s Policy on Transparency and Publication of Clinical Study Data. The synopsis
WARNING: RISK OF SERIOUS INFECTIONS
 RA PROGRESSION INTERRUPTED 1 DOSAGE AND ADMINISTRATION GUIDE No structural damage progression was observed at week 52 in 55.6% and in 47.8% of patients receiving KEVZARA 200 mg + MTX or 150 mg + MTX, compared
RA PROGRESSION INTERRUPTED 1 DOSAGE AND ADMINISTRATION GUIDE No structural damage progression was observed at week 52 in 55.6% and in 47.8% of patients receiving KEVZARA 200 mg + MTX or 150 mg + MTX, compared
Kevzara (sarilumab) NEW PRODUCT SLIDESHOW
 NEW PRODUCT SLIDESHOW") Kevzara (sarilumab) NEW PRODUCT SLIDESHOW Introduction Brand name: Kevzara Generic name: Sarilumab Pharmacological class: Interleukin-6 antagonist Strength and Formulation: 150mg/1.14mL, 200mg/1.14mL;
Kevzara (sarilumab) NEW PRODUCT SLIDESHOW Introduction Brand name: Kevzara Generic name: Sarilumab Pharmacological class: Interleukin-6 antagonist Strength and Formulation: 150mg/1.14mL, 200mg/1.14mL;
golimumab Principal Investigator(s): Principal Investigator: Michael E. Weinblatt, MD Brigham and Women s
: Principal Investigator: Michael E. Weinblatt, MD Brigham and Women s") Module 5.3.5.1 Rheumatoid Arthritis IV (24-Week submission) 24-Week CNTO148ART3001Clinical Study Report SYNOPSIS Issue Date: 07 Nov 2011 Document No.: EDMS-ERI-22836553 Name of Sponsor/Company Name of
Module 5.3.5.1 Rheumatoid Arthritis IV (24-Week submission) 24-Week CNTO148ART3001Clinical Study Report SYNOPSIS Issue Date: 07 Nov 2011 Document No.: EDMS-ERI-22836553 Name of Sponsor/Company Name of
SYNOPSIS. Issue Date: 25 Oct 2011
 SYNOPSIS Issue Date: 25 Oct 2011 Name of Sponsor/Company Name of Finished Product Name of Active Ingredient(s) Janssen Research & Development STELARA Ustekinumab Protocol No.: Title of Study: Study Name:
SYNOPSIS Issue Date: 25 Oct 2011 Name of Sponsor/Company Name of Finished Product Name of Active Ingredient(s) Janssen Research & Development STELARA Ustekinumab Protocol No.: Title of Study: Study Name:
1.0 Abstract. Title. Keywords. Rationale and Background
 1.0 Abstract Title A Prospective, Multi-Center Study in Rheumatoid Arthritis Patients on Adalimumab to Evaluate its Effect on Synovitis Using Ultrasonography in an Egyptian Population Keywords Synovitis
1.0 Abstract Title A Prospective, Multi-Center Study in Rheumatoid Arthritis Patients on Adalimumab to Evaluate its Effect on Synovitis Using Ultrasonography in an Egyptian Population Keywords Synovitis
SYNOPSIS. Issue Date: 17 Jan 2013
 STELARA (ustekinumab) Clinical Study Report CNTO1275PSA3002 24-Week CSR SYNOPSIS Issue Date: 17 Jan 2013 Name of Sponsor/Company Name of Finished Product Name of Active Ingredient(s) Janssen Research &
STELARA (ustekinumab) Clinical Study Report CNTO1275PSA3002 24-Week CSR SYNOPSIS Issue Date: 17 Jan 2013 Name of Sponsor/Company Name of Finished Product Name of Active Ingredient(s) Janssen Research &
London, 1 June 2006 Product name: REMICADE Procedure number: Remicade-H-240-II-73-AR SCIENTIFIC DISCUSSION 1/8
 London, 1 June 2006 Product name: REMICADE Procedure number: Remicade-H-240-II-73-AR SCIENTIFIC DISCUSSION 1/8 1. Introduction Infliximab is a chimeric human-murine IgG1κ monoclonal antibody, which binds
London, 1 June 2006 Product name: REMICADE Procedure number: Remicade-H-240-II-73-AR SCIENTIFIC DISCUSSION 1/8 1. Introduction Infliximab is a chimeric human-murine IgG1κ monoclonal antibody, which binds
Adalimumab M Clinical Study Report Final R&D/14/1263. Page:
 Synopsis AbbVie Inc. Name of Study Drug: Adalimumab Name of Active Ingredient: Adalimumab Individual Study Table Referring to Part of Dossier: Volume: Page: (For National Authority Use Only) Title of Study:
Synopsis AbbVie Inc. Name of Study Drug: Adalimumab Name of Active Ingredient: Adalimumab Individual Study Table Referring to Part of Dossier: Volume: Page: (For National Authority Use Only) Title of Study:
Abatacept (Orencia) for active rheumatoid arthritis. August 2009
 for active rheumatoid arthritis. August 2009") Abatacept (Orencia) for active rheumatoid arthritis August 2009 This technology summary is based on information available at the time of research and a limited literature search. It is not intended to
Abatacept (Orencia) for active rheumatoid arthritis August 2009 This technology summary is based on information available at the time of research and a limited literature search. It is not intended to
2.0 Synopsis. Adalimumab DE018 OLE (5 year) Clinical Study Report R&D/06/369. (For National Authority Use Only) to Part of Dossier: Volume:
 Clinical Study Report R&D/06/369. (For National Authority Use Only) to Part of Dossier: Volume:") 2.0 Synopsis Abbott Laboratories Name of Study Drug: Name of Active Ingredient: Individual Study Table Referring to Part of Dossier: Volume: Page: (For National Authority Use Only) Title of Study: A Multi-center
2.0 Synopsis Abbott Laboratories Name of Study Drug: Name of Active Ingredient: Individual Study Table Referring to Part of Dossier: Volume: Page: (For National Authority Use Only) Title of Study: A Multi-center
Mipomersen (ISIS ) Page 2 of 1979 Clinical Study Report ISIS CS3
 Page 2 of 1979 Clinical Study Report ISIS CS3") (ISIS 301012) Page 2 of 1979 2 SYNOPSIS ISIS 301012-CS3 synopsis Page 1 Title of Study: A Phase 2, Randomized, Double-Blind, Placebo-Controlled Study to Assess the Safety, Tolerability, Pharmacokinetics,
(ISIS 301012) Page 2 of 1979 2 SYNOPSIS ISIS 301012-CS3 synopsis Page 1 Title of Study: A Phase 2, Randomized, Double-Blind, Placebo-Controlled Study to Assess the Safety, Tolerability, Pharmacokinetics,
GSK 165: anti-gm-csf antibody
 GSK 165: anti-gm-csf antibody A novel mechanism with potentially differentiated impact on pain in the treatment of Rheumatoid Arthritis 23 October 2018 Cautionary statement regarding forward-looking statements
GSK 165: anti-gm-csf antibody A novel mechanism with potentially differentiated impact on pain in the treatment of Rheumatoid Arthritis 23 October 2018 Cautionary statement regarding forward-looking statements
Synopsis (C0743T10) CNTO 1275 Module 5.3 C0743T10. Associated with Module 5.3 of the Dossier
 CNTO 1275 Module 5.3 C0743T10. Associated with Module 5.3 of the Dossier") Module 5.3 Protocol: EudraCT No.: 2005-003525-92 Title of the study: A Phase 2, Multicenter, Randomized, Double-blind, Placebo-controlled Trial of, a Fully Human Anti-IL-12 Monoclonal Antibody, Administered
Module 5.3 Protocol: EudraCT No.: 2005-003525-92 Title of the study: A Phase 2, Multicenter, Randomized, Double-blind, Placebo-controlled Trial of, a Fully Human Anti-IL-12 Monoclonal Antibody, Administered
Summary ID# Clinical Study Summary: Study B4Z-MC-LYBX
 CT Registry ID#7068 Page 1 Summary ID# 7068 Clinical Study Summary: Study B4Z-MC-LYBX A Randomized, Double-Blind Comparison of Hydrochloride and Placebo in Child and Adolescent Outpatients with Attention-
CT Registry ID#7068 Page 1 Summary ID# 7068 Clinical Study Summary: Study B4Z-MC-LYBX A Randomized, Double-Blind Comparison of Hydrochloride and Placebo in Child and Adolescent Outpatients with Attention-
Adalimumab M Clinical Study Report Final R&D/16/0603
 Methodology (Continued): The 70-day safety follow-up period started from the last dose of study drug, but was not required for any subject who initiated commercial Humira after study completion. Additional
Methodology (Continued): The 70-day safety follow-up period started from the last dose of study drug, but was not required for any subject who initiated commercial Humira after study completion. Additional
SYNOPSIS. Clinical Study Report IM Double-blind Period
 Name of Sponsor/Company: Bristol-Myers Squibb Name of Finished Product: Abatacept () Name of Active Ingredient: Abatacept () Individual Study Table Referring to the Dossier SYNOPSIS (For National Authority
Name of Sponsor/Company: Bristol-Myers Squibb Name of Finished Product: Abatacept () Name of Active Ingredient: Abatacept () Individual Study Table Referring to the Dossier SYNOPSIS (For National Authority
2.0 Synopsis. Choline fenofibrate capsules (ABT-335) M Clinical Study Report R&D/06/772. (For National Authority Use Only) Name of Study Drug:
 M Clinical Study Report R&D/06/772. (For National Authority Use Only) Name of Study Drug:") 2.0 Synopsis Abbott Laboratories Individual Study Table Referring to Part of Dossier: (For National Authority Use Only) Name of Study Drug: Volume: Choline Fenofibrate (335) Name of Active Ingredient:
2.0 Synopsis Abbott Laboratories Individual Study Table Referring to Part of Dossier: (For National Authority Use Only) Name of Study Drug: Volume: Choline Fenofibrate (335) Name of Active Ingredient:
(For National Authority Use Only) Name of Study Drug: to Part of Dossier:
 Name of Study Drug: to Part of Dossier:") 2.0 Synopsis Abbott Laboratories Individual Study Table Referring to Part of Dossier: (For National Authority Use Only) Name of Study Drug: Volume: ABT-335 Name of Active Ingredient: Page: ABT-335, A-7770335.115
2.0 Synopsis Abbott Laboratories Individual Study Table Referring to Part of Dossier: (For National Authority Use Only) Name of Study Drug: Volume: ABT-335 Name of Active Ingredient: Page: ABT-335, A-7770335.115
PDF of Trial CTRI Website URL -
 Clinical Trial Details (PDF Generation Date :- Sun, 20 Jan 2019 21:39:27 GMT) CTRI Number CTRI/2009/091/000777 [Registered on: 11/01/2010] - Last Modified On Post Graduate Thesis Type of Trial Type of
Clinical Trial Details (PDF Generation Date :- Sun, 20 Jan 2019 21:39:27 GMT) CTRI Number CTRI/2009/091/000777 [Registered on: 11/01/2010] - Last Modified On Post Graduate Thesis Type of Trial Type of
2.0 Synopsis. ABT-333 M Clinical Study Report R&D/09/956
 2.0 Synopsis Abbott Laboratories Individual Study Table Referring to Part of Dossier: (For National Authority Use Only) Name of Study Drug: ABT-333 Volume: Name of Active Ingredient: Page: Sodium N-{6-[3-tert-butyl-5-(2,4-
2.0 Synopsis Abbott Laboratories Individual Study Table Referring to Part of Dossier: (For National Authority Use Only) Name of Study Drug: ABT-333 Volume: Name of Active Ingredient: Page: Sodium N-{6-[3-tert-butyl-5-(2,4-
SYNOPSIS. Approved Date: 9 November 2015 Prepared by: Status: Janssen Research & Development, LLC
 SYNOPSIS Name of Sponsor/Company Janssen Research & Development* Name of Investigational Product STELARA (ustekinumab) * Janssen Research & Development is a global organization that operates through different
SYNOPSIS Name of Sponsor/Company Janssen Research & Development* Name of Investigational Product STELARA (ustekinumab) * Janssen Research & Development is a global organization that operates through different
PRODUCT INFORMATION HUMIRA
 NAME OF THE MEDICINE Adalimumab (rch) DESCRIPTION PRODUCT INFORMATION HUMIRA (adalimumab) is a recombinant human immunoglobulin (IgG1) monoclonal antibody containing only human peptide sequences. was created
NAME OF THE MEDICINE Adalimumab (rch) DESCRIPTION PRODUCT INFORMATION HUMIRA (adalimumab) is a recombinant human immunoglobulin (IgG1) monoclonal antibody containing only human peptide sequences. was created
Clinical Study Synopsis
 Clinical Study Synopsis This Clinical Study Synopsis is provided for patients and healthcare professionals to increase the transparency of Bayer's clinical research. This document is not intended to replace
Clinical Study Synopsis This Clinical Study Synopsis is provided for patients and healthcare professionals to increase the transparency of Bayer's clinical research. This document is not intended to replace
ORENCIA (abatacept) Demonstrates Comparable Efficacy to Humira ( adalimumab
 Demonstrates Comparable Efficacy to Humira ( adalimumab") ORENCIA (abatacept) Demonstrates Comparable Efficacy to Humira (adalimumab) in Patients with Moderate to Severe Rheumatoid Arthritis in First Head-to-Head Study of These Agents ORENCIA demonstrated comparable
ORENCIA (abatacept) Demonstrates Comparable Efficacy to Humira (adalimumab) in Patients with Moderate to Severe Rheumatoid Arthritis in First Head-to-Head Study of These Agents ORENCIA demonstrated comparable
Horizon Scanning Research & Intelligence Centre. Baricitinib for moderate to severe rheumatoid arthritis. May 2015 SUMMARY NIHR HSRIC ID: 5270
 May 2015 Horizon Scanning Research & Intelligence Centre Baricitinib for moderate to severe rheumatoid arthritis SUMMARY NIHR HSRIC ID: 5270 This briefing is based on information available at the time
May 2015 Horizon Scanning Research & Intelligence Centre Baricitinib for moderate to severe rheumatoid arthritis SUMMARY NIHR HSRIC ID: 5270 This briefing is based on information available at the time
PFIZER INC. These results are supplied for informational purposes only.
 PFIZER INC. These results are supplied for informational purposes only. COMPOUND NUMBER: PF-05212374 PROTOCOL NO.: 3206K1-2203 (B2051001) PROTOCOL TITLE: A Randomized, Parallel, Double-Blind, Placebo-Controlled
PFIZER INC. These results are supplied for informational purposes only. COMPOUND NUMBER: PF-05212374 PROTOCOL NO.: 3206K1-2203 (B2051001) PROTOCOL TITLE: A Randomized, Parallel, Double-Blind, Placebo-Controlled
QUALITATIVE AND QUANTITATIVE COMPOSITION
 This medicinal product is subject to additional monitoring in Australia. This will allow quick identification of new safety information. Healthcare professionals are asked to report any suspected adverse
This medicinal product is subject to additional monitoring in Australia. This will allow quick identification of new safety information. Healthcare professionals are asked to report any suspected adverse
SCIENTIFIC DISCUSSION. London, 27 April 2006 Product name: HUMIRA/TRUDEXA Procedure number: EMEA/H/C/ /II/26
 SCIENTIFIC DISCUSSION London, 27 April 2006 Product name: HUMIRA/TRUDEXA Procedure number: EMEA/H/C/481-482/II/26 3.1. Introduction Adalimumab is a recombinant human immunoglobulin (IgG 1 ) monoclonal
SCIENTIFIC DISCUSSION London, 27 April 2006 Product name: HUMIRA/TRUDEXA Procedure number: EMEA/H/C/481-482/II/26 3.1. Introduction Adalimumab is a recombinant human immunoglobulin (IgG 1 ) monoclonal
American College of Rheumatology Analyst and Investor Meeting November 6, 2011
 American College of Rheumatology 2011 Analyst and Investor Meeting November 6, 2011 Chuck Triano Senior Vice President, Investor Relations Forward-Looking Statements Our discussions during this meeting
American College of Rheumatology 2011 Analyst and Investor Meeting November 6, 2011 Chuck Triano Senior Vice President, Investor Relations Forward-Looking Statements Our discussions during this meeting
Guideline on clinical investigation of medicinal products for the treatment of rheumatoid arthritis
 14 December 2017 CPMP/EWP/556/95 Rev. 2 Committee for Medicinal Products for Human Use (CHMP) Guideline on clinical investigation of medicinal products for the treatment of Draft Agreed by Rheumatology-Immunology
14 December 2017 CPMP/EWP/556/95 Rev. 2 Committee for Medicinal Products for Human Use (CHMP) Guideline on clinical investigation of medicinal products for the treatment of Draft Agreed by Rheumatology-Immunology
24-Week CNTO1275PSA3001 Clinical Study Report
 24-Week CNTO1275PSA3001 Clinical Study Report SYNOPSIS Issue Date: 17 Jan 2013 Name of Sponsor/Company Name of Finished Product Name of Active Ingredient(s) Janssen Research & Development, Inc Ustekinumab
24-Week CNTO1275PSA3001 Clinical Study Report SYNOPSIS Issue Date: 17 Jan 2013 Name of Sponsor/Company Name of Finished Product Name of Active Ingredient(s) Janssen Research & Development, Inc Ustekinumab
Study synopsis of the global non-interventional study SWITCH-RA
 Study synopsis of the global non-interventional study SWITCH-RA Protocol number: MA22401 Title of Study: A global multi-centre observational study in RA patients who are non-responders or intolerant to
Study synopsis of the global non-interventional study SWITCH-RA Protocol number: MA22401 Title of Study: A global multi-centre observational study in RA patients who are non-responders or intolerant to
Referring to Part of Dossier: Volume: Page:
 Synopsis Abbott Laboratories Name of Study Drug: Adalimumab Name of Active Ingredient: Adalimumab Title of Study: Individual Study Table Referring to Part of Dossier: Volume: Page: (For National Authority
Synopsis Abbott Laboratories Name of Study Drug: Adalimumab Name of Active Ingredient: Adalimumab Title of Study: Individual Study Table Referring to Part of Dossier: Volume: Page: (For National Authority
The clinical trial information provided in this public disclosure synopsis is supplied for informational purposes only.
 The clinical trial information provided in this public disclosure synopsis is supplied for informational purposes only. Please note that the results reported in any single trial may not reflect the overall
The clinical trial information provided in this public disclosure synopsis is supplied for informational purposes only. Please note that the results reported in any single trial may not reflect the overall
Bringing the clinical experience with anakinra to the patient
 Rheumatology 2003;42(Suppl. 2):ii36 ii40 doi:10.1093/rheumatology/keg331, available online at www.rheumatology.oupjournals.org Bringing the clinical experience with anakinra to the patient S. B. Cohen
Rheumatology 2003;42(Suppl. 2):ii36 ii40 doi:10.1093/rheumatology/keg331, available online at www.rheumatology.oupjournals.org Bringing the clinical experience with anakinra to the patient S. B. Cohen
Efficacy and Safety of Rituximab in the Treatment of Rheumatoid Arthritis and ANCA-associated Vasculitis
 New Evidence reports on presentations given at ACR/ARHP 2010 Efficacy and Safety of Rituximab in the Treatment of Rheumatoid Arthritis and ANCA-associated Vasculitis Report on ACR/ARHP 2010 presentations
New Evidence reports on presentations given at ACR/ARHP 2010 Efficacy and Safety of Rituximab in the Treatment of Rheumatoid Arthritis and ANCA-associated Vasculitis Report on ACR/ARHP 2010 presentations
ABT-493/ABT-530 M Clinical Study Report Post-Treatment Week 12 Primary Data R&D/16/0145. Referring to Part of Dossier: Volume: Page:
 2.0 Synopsis AbbVie Inc. Name of Study Drug: ABT-493/ABT-530 Name of Active Ingredient: ABT-493: (3aR,7S,10S,12R,21E,24aR)- 7-tert-butyl-N-{(1R,2R)-2- (difluoromethyl)-1-[(1- methylcyclopropane-1- sulfonyl)carbamoyl]cyclopropyl}-20,20-
2.0 Synopsis AbbVie Inc. Name of Study Drug: ABT-493/ABT-530 Name of Active Ingredient: ABT-493: (3aR,7S,10S,12R,21E,24aR)- 7-tert-butyl-N-{(1R,2R)-2- (difluoromethyl)-1-[(1- methylcyclopropane-1- sulfonyl)carbamoyl]cyclopropyl}-20,20-
Individual Study Table Referring to Part of Dossier: Volume: Page:
 Synopsis Abbott Laboratories Name of Study Drug: Paricalcitol Capsules (ABT-358) (Zemplar ) Name of Active Ingredient: Paricalcitol Individual Study Table Referring to Part of Dossier: Volume: Page: (For
Synopsis Abbott Laboratories Name of Study Drug: Paricalcitol Capsules (ABT-358) (Zemplar ) Name of Active Ingredient: Paricalcitol Individual Study Table Referring to Part of Dossier: Volume: Page: (For
CDEC FINAL RECOMMENDATION
 CDEC FINAL RECOMMENDATION TOFACITINIB (Xeljanz Pfizer Canada Inc.) Indication: Rheumatoid Arthritis Recommendation: The Canadian Drug Expert Committee (CDEC) recommends that tofacitinib be listed, in combination
CDEC FINAL RECOMMENDATION TOFACITINIB (Xeljanz Pfizer Canada Inc.) Indication: Rheumatoid Arthritis Recommendation: The Canadian Drug Expert Committee (CDEC) recommends that tofacitinib be listed, in combination
ClinialTrials.gov Identifier: Sponsor/company: sanofi-aventis
 These results are supplied for informational purposes only. Prescribing decisions should be made based on the approved package insert in the country of prescription Sponsor/company: sanofi-aventis ClinialTrials.gov
These results are supplied for informational purposes only. Prescribing decisions should be made based on the approved package insert in the country of prescription Sponsor/company: sanofi-aventis ClinialTrials.gov
Summary ID#7029. Clinical Study Summary: Study F1D-MC-HGKQ
 CT Registry ID# 7029 Page 1 Summary ID#7029 Clinical Study Summary: Study F1D-MC-HGKQ Clinical Study Report: Versus Divalproex and Placebo in the Treatment of Mild to Moderate Mania Associated with Bipolar
CT Registry ID# 7029 Page 1 Summary ID#7029 Clinical Study Summary: Study F1D-MC-HGKQ Clinical Study Report: Versus Divalproex and Placebo in the Treatment of Mild to Moderate Mania Associated with Bipolar
ClinialTrials.gov Identifier: HOE901_4020 Insulin Glargine Date: Study Code: This was a multicenter study that was conducted at 59 US sites
 These results are supplied for informational purposes only. Prescribing decisions should be made based on the approved package insert in the country of prescription Sponsor/company: Generic drug name:
These results are supplied for informational purposes only. Prescribing decisions should be made based on the approved package insert in the country of prescription Sponsor/company: Generic drug name:
STELARA (ustekinumab) Clinical Study Report CNTO1275CRD3001
 Clinical Study Report CNTO1275CRD3001") SYNOPSIS Name of Sponsor/Company Janssen Research & Development* Name of Investigational Product STELARA (ustekinumab) * Janssen Research & Development is a global organization that operates through different
SYNOPSIS Name of Sponsor/Company Janssen Research & Development* Name of Investigational Product STELARA (ustekinumab) * Janssen Research & Development is a global organization that operates through different
Adalimumab M Clinical Study Report Final R&D/13/224
 Diagnosis and Main Criteria for Inclusion: A parent or guardian had voluntarily signed and dated an informed consent form, approved by an institutional review board/independent ethics committee, after
Diagnosis and Main Criteria for Inclusion: A parent or guardian had voluntarily signed and dated an informed consent form, approved by an institutional review board/independent ethics committee, after
1 Executive summary. Background
 1 Executive summary Background Rheumatoid Arthritis (RA) is the most common inflammatory polyarthropathy in the UK affecting between.5% and 1% of the population. The mainstay of RA treatment interventions
1 Executive summary Background Rheumatoid Arthritis (RA) is the most common inflammatory polyarthropathy in the UK affecting between.5% and 1% of the population. The mainstay of RA treatment interventions
ABT-493/ABT-530 M Clinical Study Report Post-Treatment Week 12 Primary Data R&D/16/0162. Referring to Part of Dossier: Volume: Page:
 2.0 Synopsis AbbVie Inc. Name of Study Drug: ABT-493/ABT-530 Name of Active Ingredient: ABT-493: (3aR,7S,10S,12R,21E,24aR)- 7-tert-butyl-N-{(1R,2R)-2- (difluoromethyl)-1-[(1- methylcyclopropane-1- sulfonyl)carbamoyl]cyclopropyl}-20,20-
2.0 Synopsis AbbVie Inc. Name of Study Drug: ABT-493/ABT-530 Name of Active Ingredient: ABT-493: (3aR,7S,10S,12R,21E,24aR)- 7-tert-butyl-N-{(1R,2R)-2- (difluoromethyl)-1-[(1- methylcyclopropane-1- sulfonyl)carbamoyl]cyclopropyl}-20,20-
10/28/2013. Disclosures. Objectives. Background. Study Design. Key Inclusion Criteria
 Randomization (1:1:1:1) /28/13 Tocilizumab in Combination Therapy and Monotherapy Versus Methotrexate in Methotrexate-Naive Patients With Early Rheumatoid Arthritis: Clinical and Radiographic Outcomes
Randomization (1:1:1:1) /28/13 Tocilizumab in Combination Therapy and Monotherapy Versus Methotrexate in Methotrexate-Naive Patients With Early Rheumatoid Arthritis: Clinical and Radiographic Outcomes
Canadian Society of Internal Medicine Annual Meeting 2016 Montreal, QC
 Canadian Society of Internal Medicine Annual Meeting 2016 Montreal, QC Update on the Treatment of Rheumatoid Arthritis Sabrina Fallavollita MDCM McGill University Canadian Society of Internal Medicine
Canadian Society of Internal Medicine Annual Meeting 2016 Montreal, QC Update on the Treatment of Rheumatoid Arthritis Sabrina Fallavollita MDCM McGill University Canadian Society of Internal Medicine
Charité - University Hospital, Free University and Humboldt University of Berlin, Berlin, Germany; 2 Sanofi Genzyme, Bridgewater, NJ, USA; 3
 Efficacy and Safety of Sarilumab Versus Adalimumab in a Phase 3, Randomized, Double-blind, Monotherapy Study in Patients With Active Rheumatoid Arthritis With Intolerance or Inadequate Response to Methotrexate
Efficacy and Safety of Sarilumab Versus Adalimumab in a Phase 3, Randomized, Double-blind, Monotherapy Study in Patients With Active Rheumatoid Arthritis With Intolerance or Inadequate Response to Methotrexate
2.0 Synopsis. Adalimumab M Clinical Study Report Final R&D/15/1054. (For National Authority Use Only)
") 2.0 Synopsis AbbVie Inc. Name of Study Drug: Adalimumab Name of Active Ingredient: Adalimumab Individual Study Table Referring to Part of Dossier: Volume: Page: (For National Authority Use Only) Title
2.0 Synopsis AbbVie Inc. Name of Study Drug: Adalimumab Name of Active Ingredient: Adalimumab Individual Study Table Referring to Part of Dossier: Volume: Page: (For National Authority Use Only) Title
adalimumab, 40mg/0.8mL, solution for injection (Humira ) SMC No. (858/13) AbbVie Ltd (previously part of Abbott)
 SMC No. (858/13) AbbVie Ltd (previously part of Abbott)") adalimumab, 40mg/0.8mL, solution for injection (Humira ) SMC No. (858/13) AbbVie Ltd (previously part of Abbott) 08 March 2013 The Scottish Medicines Consortium (SMC) has completed its assessment of the
adalimumab, 40mg/0.8mL, solution for injection (Humira ) SMC No. (858/13) AbbVie Ltd (previously part of Abbott) 08 March 2013 The Scottish Medicines Consortium (SMC) has completed its assessment of the
certolizumab pegol (Cimzia )
") Applies to all products administered or underwritten by Blue Cross and Blue Shield of Louisiana and its subsidiary, HMO Louisiana, Inc.(collectively referred to as the Company ), unless otherwise provided
Applies to all products administered or underwritten by Blue Cross and Blue Shield of Louisiana and its subsidiary, HMO Louisiana, Inc.(collectively referred to as the Company ), unless otherwise provided
Synopsis. Clinical Report Synopsis for Protocol Name of Company: Otsuka Pharmaceutical Development & Commercialization, Inc.
 Synopsis Clinical Report Synopsis for Protocol 197-02-220 Name of Company: Otsuka Pharmaceutical Development & Commercialization, Inc. Name of Product: Tetomilast (OPC-6535) Study Title: A Phase 3, Multicenter,
Synopsis Clinical Report Synopsis for Protocol 197-02-220 Name of Company: Otsuka Pharmaceutical Development & Commercialization, Inc. Name of Product: Tetomilast (OPC-6535) Study Title: A Phase 3, Multicenter,
Baricitinib versus Placebo or Adalimumab in Rheumatoid Arthritis
 The new england journal of medicine Original Article Baricitinib versus Placebo or Adalimumab in Rheumatoid Arthritis Peter C. Taylor, M.D., Ph.D., Edward C. Keystone, M.D., Désirée van der Heijde, M.D.,
The new england journal of medicine Original Article Baricitinib versus Placebo or Adalimumab in Rheumatoid Arthritis Peter C. Taylor, M.D., Ph.D., Edward C. Keystone, M.D., Désirée van der Heijde, M.D.,
2.0 Synopsis. Adalimumab M Clinical Study Report R&D/13/1011. (For National Authority Use Only)
") 2.0 Synopsis AbbVie Inc. Name of Study Drug: Name of Active Ingredient: Individual Study Table Referring to Part of Dossier: Volume: Page: (For National Authority Use Only) Title of Study: A Phase 3 Multicenter
2.0 Synopsis AbbVie Inc. Name of Study Drug: Name of Active Ingredient: Individual Study Table Referring to Part of Dossier: Volume: Page: (For National Authority Use Only) Title of Study: A Phase 3 Multicenter
Individual Study Table Referring to Part of Dossier: Volume: Page:
 2.0 Synopsis Abbott Laboratories Eisai Co., Ltd Name of Study Drug: Humira 40 mg/ 0.8 ml (To be determined for 20 mg product) Humira 20 mg/ 0.4 ml (To be determined for 20 mg product) Name of Active Ingredient:
2.0 Synopsis Abbott Laboratories Eisai Co., Ltd Name of Study Drug: Humira 40 mg/ 0.8 ml (To be determined for 20 mg product) Humira 20 mg/ 0.4 ml (To be determined for 20 mg product) Name of Active Ingredient:
Sponsor / Company: Sanofi Drug substance(s): HOE901-U300 (insulin glargine) According to template: QSD VERSION N 4.0 (07-JUN-2012) Page 1
: HOE901-U300 (insulin glargine) According to template: QSD VERSION N 4.0 (07-JUN-2012) Page 1") These results are supplied for informational purposes only. Prescribing decisions should be made based on the approved package insert in the country of prescription. Sponsor / Company: Sanofi Drug substance(s):
These results are supplied for informational purposes only. Prescribing decisions should be made based on the approved package insert in the country of prescription. Sponsor / Company: Sanofi Drug substance(s):
ABSTRACT. Keywords: Africa; Efficacy; Etanercept; Maintenance therapy; Middle East; Rheumatoid arthritis ORIGINAL RESEARCH
 Rheumatol Ther (2018) 5:149 158 https://doi.org/10.1007/s40744-018-0094-6 ORIGINAL RESEARCH Maintenance of Remission with Etanercept DMARD Combination Therapy Compared with DMARDs Alone in African and
Rheumatol Ther (2018) 5:149 158 https://doi.org/10.1007/s40744-018-0094-6 ORIGINAL RESEARCH Maintenance of Remission with Etanercept DMARD Combination Therapy Compared with DMARDs Alone in African and
23-Aug-2011 Lixisenatide (AVE0010) - EFC6014 Version number: 1 (electronic 1.0)
 - EFC6014 Version number: 1 (electronic 1.0)") SYNOPSIS Title of the study: A randomized, double-blind, placebo-controlled, parallel-group, multicenter 24-week study followed by an extension assessing the efficacy and safety of AVE0010 on top of metformin
SYNOPSIS Title of the study: A randomized, double-blind, placebo-controlled, parallel-group, multicenter 24-week study followed by an extension assessing the efficacy and safety of AVE0010 on top of metformin
BRIEFING DOCUMENT. human, recombinant fusion protein: extracellular domain of CTLA-4 and Fc domain of human IgG1
 BRIEFING DOCUMENT Application Type BLA Submission Number 125118/0 Reviewer Name Team Leader Division Director Established Name (Proposed) Trade Name Applicant Formulation Dosing Regimen Indication Intended
BRIEFING DOCUMENT Application Type BLA Submission Number 125118/0 Reviewer Name Team Leader Division Director Established Name (Proposed) Trade Name Applicant Formulation Dosing Regimen Indication Intended
PFIZER INC. These results are supplied for informational purposes only. Prescribing decisions should be made based on the approved package insert.
 PFIZER INC. These results are supplied for informational purposes only. Prescribing decisions should be made based on the approved package insert. PROPRIETARY DRUG NAME / GENERIC DRUG NAME: Lyrica / Pregabalin
PFIZER INC. These results are supplied for informational purposes only. Prescribing decisions should be made based on the approved package insert. PROPRIETARY DRUG NAME / GENERIC DRUG NAME: Lyrica / Pregabalin
Safety and effectiveness of biologic Disease-Modifying Antirheumatic Drugs in elderly patients with rheumatoid arthritis
 1. Title: Safety and effectiveness of biologic Disease-Modifying Antirheumatic Drugs in elderly patients with rheumatoid arthritis 2. Background: The population of older individuals with rheumatoid arthritis
1. Title: Safety and effectiveness of biologic Disease-Modifying Antirheumatic Drugs in elderly patients with rheumatoid arthritis 2. Background: The population of older individuals with rheumatoid arthritis
ACTEMRA Risk Mitigation Strategy Presenter Name, Degree
 ACTEMRA Risk Mitigation Strategy Presenter Name, Degree Medical Science Liaison Genentech, Inc. 1 Indications and Dosage Rheumatoid Arthritis (RA) (1 of 2) Indication in RA ACTEMRA (tocilizumab) is indicated
ACTEMRA Risk Mitigation Strategy Presenter Name, Degree Medical Science Liaison Genentech, Inc. 1 Indications and Dosage Rheumatoid Arthritis (RA) (1 of 2) Indication in RA ACTEMRA (tocilizumab) is indicated
NIHR Innovation Observatory Evidence Briefing: November 2017
 NIHR Innovation Observatory Evidence Briefing: November 2017 Upadacitinib for adults with moderate to severe active rheumatoid arthritis after conventional synthetic disease-modifying anti-rheumatic drugs
NIHR Innovation Observatory Evidence Briefing: November 2017 Upadacitinib for adults with moderate to severe active rheumatoid arthritis after conventional synthetic disease-modifying anti-rheumatic drugs
Adalimumab M Clinical Study Report Final R&D/17/0360
 Methodology (Continued): Beginning at Week 8, subjects who developed a flare while receiving ew therapy or had a PCDAI 15 points higher than Baseline at any time during this study may have been discontinued
Methodology (Continued): Beginning at Week 8, subjects who developed a flare while receiving ew therapy or had a PCDAI 15 points higher than Baseline at any time during this study may have been discontinued
SYNOPSIS. The study results and synopsis are supplied for informational purposes only.
 SYNOPSIS INN : LEFLUNOMIDE Study number : HMR486/1037 et HMR486/3503 Study title : Population pharmacokinetics of A77 1726 (M1) after oral administration of leflunomide in pediatric subjects with polyarticular
SYNOPSIS INN : LEFLUNOMIDE Study number : HMR486/1037 et HMR486/3503 Study title : Population pharmacokinetics of A77 1726 (M1) after oral administration of leflunomide in pediatric subjects with polyarticular
Efficacy and safety of GLPG0634, a selective JAK1 inhibitor, after short-term treatment of rheumatoid arthritis; results of a phase IIA trial
 Efficacy and safety of GLPG0634, a selective JAK1 inhibitor, after short-term treatment of rheumatoid arthritis; results of a phase IIA trial Frédéric Vanhoutte, MD Minodora Mazur, MD, PhD EULAR 09 June
Efficacy and safety of GLPG0634, a selective JAK1 inhibitor, after short-term treatment of rheumatoid arthritis; results of a phase IIA trial Frédéric Vanhoutte, MD Minodora Mazur, MD, PhD EULAR 09 June
Golimumab: a novel anti-tumor necrosis factor
 Golimumab: a novel anti-tumor necrosis factor Rossini M, De Vita S, Ferri C, et al. Biol Ther. 2013. This slide deck represents the opinions of the authors, and not necessarily the opinions of the publisher
Golimumab: a novel anti-tumor necrosis factor Rossini M, De Vita S, Ferri C, et al. Biol Ther. 2013. This slide deck represents the opinions of the authors, and not necessarily the opinions of the publisher
Sponsor Novartis. Generic Drug Name Vildagliptin/Metformin. Therapeutic Area of Trial Type 2 diabetes. Approved Indication Type 2 diabetes
 Clinical Trial Results Database Page 1 Sponsor Novartis Generic Drug Name Vildagliptin/Metformin Therapeutic Area of Trial Type 2 diabetes Approved Indication Type 2 diabetes Study Number CLMF237A2309
Clinical Trial Results Database Page 1 Sponsor Novartis Generic Drug Name Vildagliptin/Metformin Therapeutic Area of Trial Type 2 diabetes Approved Indication Type 2 diabetes Study Number CLMF237A2309
Synopsis. Adalimumab M Clinical Study Report R&D/09/060. (For National Authority Use Only) to Part of Dossier: Name of Study Drug:
 to Part of Dossier: Name of Study Drug:") Synopsis Abbott Laboratories Name of Study Drug: Individual Study Table Referring to Part of Dossier: Volume: (For National Authority Use Only) Name of Active Ingredient: Page: Title of Study: A Multi-Center,
Synopsis Abbott Laboratories Name of Study Drug: Individual Study Table Referring to Part of Dossier: Volume: (For National Authority Use Only) Name of Active Ingredient: Page: Title of Study: A Multi-Center,
Figure 1. Study flow diagram. Reasons for withdrawal during the extension phase are included in the flow diagram. 508 patients enrolled and randomized
 508 patients enrolled and randomized 126 assigned to sequential monotherapy (group 1) 121 assigned to step-up combination therapy (group 2) 133 assigned to initial combination therapy with prednisone (group
508 patients enrolled and randomized 126 assigned to sequential monotherapy (group 1) 121 assigned to step-up combination therapy (group 2) 133 assigned to initial combination therapy with prednisone (group
The clinical trial information provided in this public disclosure synopsis is supplied for informational purposes only.
 The clinical trial information provided in this public disclosure synopsis is supplied for informational purposes only. Please note that the results reported in any single trial may not reflect the overall
The clinical trial information provided in this public disclosure synopsis is supplied for informational purposes only. Please note that the results reported in any single trial may not reflect the overall
Orencia (abatacept) for Rheumatoid Arthritis. Media backgrounder
 for Rheumatoid Arthritis. Media backgrounder") Orencia (abatacept) for Rheumatoid Arthritis Media backgrounder What is Orencia (abatacept)? Orencia (abatacept) is the first biologic agent to be available in both an intravenous (IV) and a self-injectable,
Orencia (abatacept) for Rheumatoid Arthritis Media backgrounder What is Orencia (abatacept)? Orencia (abatacept) is the first biologic agent to be available in both an intravenous (IV) and a self-injectable,
Sponsor. Novartis Pharmaceuticals Corporation Generic Drug Name. Agomelatine Therapeutic Area of Trial. Major depressive disorder Approved Indication
 Clinical Trial Results Database Page 1 Sponsor Novartis Pharmaceuticals Corporation Generic Drug Name Therapeutic Area of Trial Major depressive disorder Approved Indication Investigational drug Study
Clinical Trial Results Database Page 1 Sponsor Novartis Pharmaceuticals Corporation Generic Drug Name Therapeutic Area of Trial Major depressive disorder Approved Indication Investigational drug Study
This clinical study synopsis is provided in line with Boehringer Ingelheim s Policy on Transparency and Publication of Clinical Study Data.
 abcd Clinical Study Synopsis for Public Disclosure This clinical study synopsis is provided in line with s Policy on Transparency and Publication of Clinical Study Data. The synopsis which is part of the
abcd Clinical Study Synopsis for Public Disclosure This clinical study synopsis is provided in line with s Policy on Transparency and Publication of Clinical Study Data. The synopsis which is part of the
ACTEMRA (tocilizumab)
") RATIONALE FOR INCLUSION IN PA PROGRAM Background Actemra is an agent in the class of drugs known as biologic disease modifiers. It is used to treat adult onset rheumatoid (RA) arthritis, polyarticular
RATIONALE FOR INCLUSION IN PA PROGRAM Background Actemra is an agent in the class of drugs known as biologic disease modifiers. It is used to treat adult onset rheumatoid (RA) arthritis, polyarticular