Dr. Ayman Mohsen Mashi, MBBS Consultant Hematology & Blood Transfusion Department Head, Laboratory & Blood Bank King Fahad Central Hospital, Gazan,
|
|
|
- Phillip Holmes
- 5 years ago
- Views:
Transcription

1 Dr. Ayman Mohsen Mashi, MBBS Consultant Hematology & Blood Transfusion Department Head, Laboratory & Blood Bank King Fahad Central Hospital, Gazan, KSA 24/02/2018
2 β-thalassemia syndromes are a group of hereditary disorders characterized by a genetic mutation that leads to a complete absence, severe or mild deficiency of β-globin chains synthesis. The complete absence or severe deficiency results in thalassemia major or thalassemia intermedia.
3 Homozygous and compound heterozygous state (thalassemia major/intermedia) causes severe, transfusion-dependent anemia, whereas heterozygous state (β-thalassemia trait) causes mild-to-moderate microcytic anemia. The molecular bases of β thalassemia are very heterogeneous. The great majorities of β-thalassemia cases are caused by point mutations, affecting the coding region of critical areas of the β-globin gene and are only rarely produced by gross-gene rearrangements.
4 Mutations causing complete inactivation of β genes (such as deletion, initiation codon, nonsense, frameshift, or splicing mutations) will make the gene unable to produce any β-globin chain resulting in β0 thalassemia. Although some other mutations cause partial inactivation of the β genes causing reduction in β-globin chain synthesis resulting in β+ or β++ (silent), thalassemia depends on the degree of reduction of the β chains production.
5 Absence or reduction of β-globin chain will increase the accumulation of free α chain within the erythroblasts and the red blood cells. The major consequences of this pathophysiology are ineffective erythropoiesis, splenomegaly, and tissue hypoxia due to increased hemoglobin F and deformities of the skull and facial bone marrow. The severity of the disease differs according to the ratio between α-globin/non-α-globin chain synthesis and excessing in free α-chain.
6 The β-globin (HBB) genes are located in the short arm of chromosome 11 and controlled by single locus control region. The HBB gene contains three exons, two introns, and both 5 and 3 untranslated regions as well as it contains 146 amino acids with a molecular weight of around 1.6 Kb.
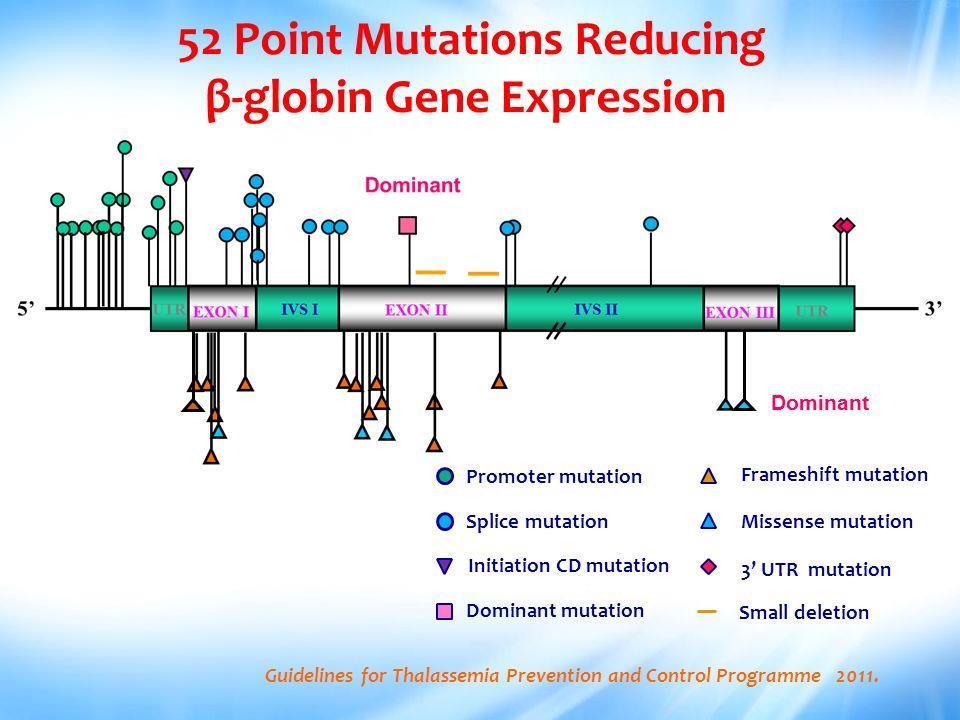
7
8 Worldwide, 3% of the populations (~150 million people) are carriers of the β-thalassemia gene. Over 300 mutations in β-globin gene have been characterized globally with a subset of ~40 mutations responsible for the majority of cases, according to population studies.
9
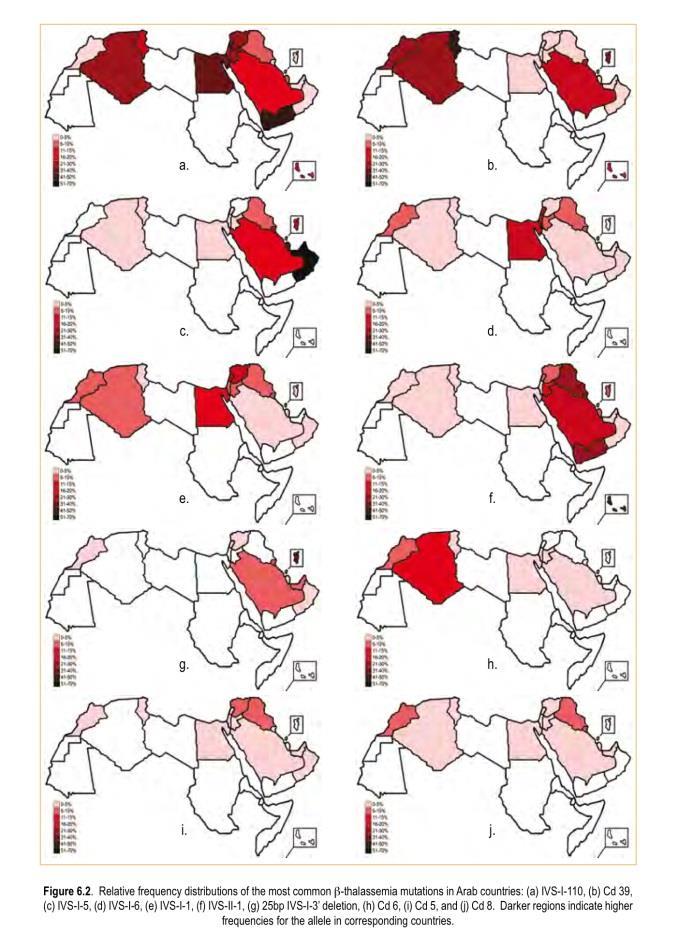
10 The mutations are population specific and each country has its own unique and frequency of β-globin mutations. The prevalence of β thalassemia is high in Mediterranean countries, the Middle East, Central Asia, India, Southern China, and the Far East as well as countries along the north coast of Africa, and in South America.
11
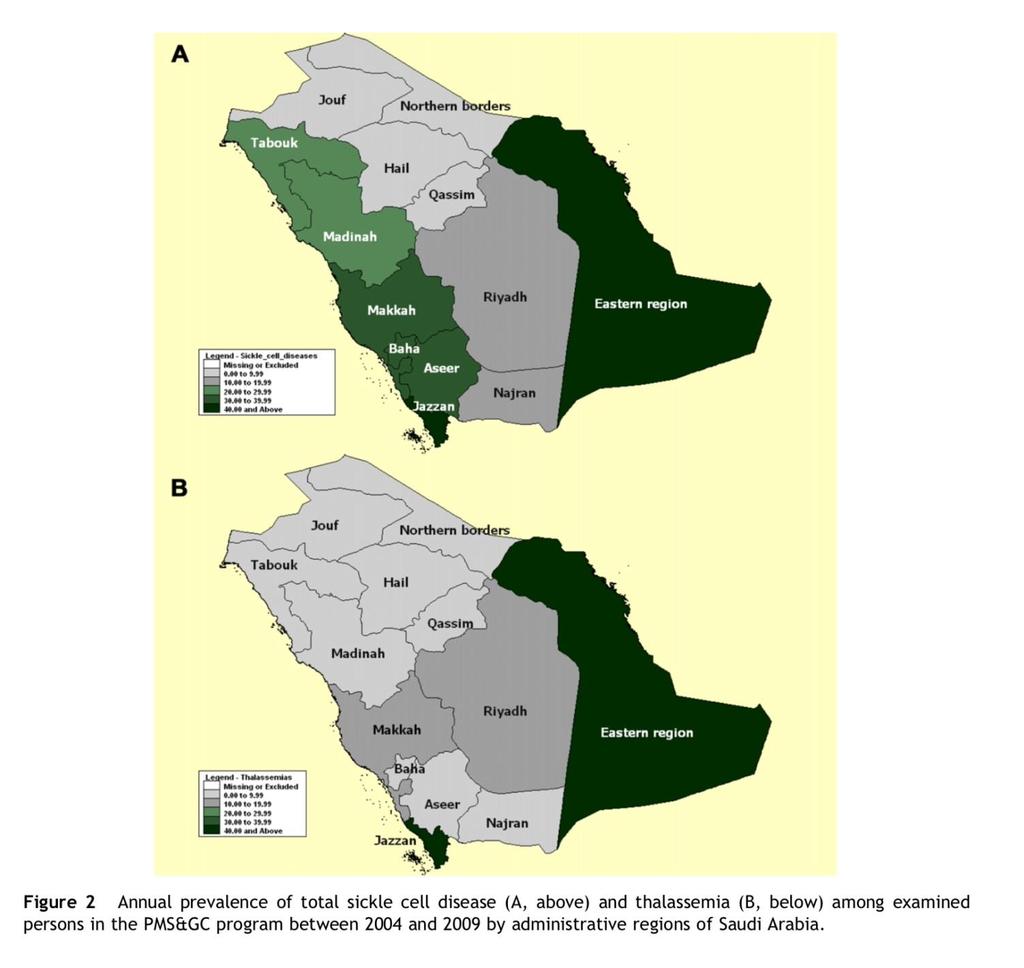
12 In addition, it is encountered in diverse frequencies in all Arab countries with carrier rate of 1 to 11%. The prevalence of β thalassemia in Saudi Arabia varied significantly in different parts of the country, with the highest prevalence being in the Eastern province of the country (around Jubail, Qateef, Dammam, and Hofuf) and along the coastal strip of the Red Sea.

13
14
15

16
17
18
19

20

21

22
23

24
25 Samples submitted for molecular screening for β thalassemia during the past 6-year period, 2008 to 2013 from the Medical Genetics and Hematology clinics in King Faisal Specialist Hospital and Research Center (KFSH & RC) (General Organization), Riyadh. Patients who were diagnosed with β thalassemia through clinical suspicion, family history, and have hypochromic microcytic anemia and/or high HbA2 or HbF level in hemoglobin electrophoresis were included for analysis. Family studies were excluded from this cohort. This retrospective review study was approved by the Research Advisory Council and the hospital ethical committee under the RAC# The samples represent cases from all over the kingdom that is referred to KFSH & RC for treatment options. Selection criteria were applied.
26 This study was carried out on a total number of 131 patients who were recruited for clinical investigation from the Hematology clinic at KFSH & RC for a 6-year period. Out of the total population, 28 (21%) were undetectable cases and 103 (79%) were detectable cases for β globin chain mutations.
27 The male gender represented 57/131 patients (43.5%), whereas female patients were 74/131 (56.5%). The pediatric patients (15 years or less) are account for 52% and adult patients (more than 15 years) account for 48%).
28 Detectable cases (103 patients) were categorized into three groups according to allele zygosity: homozygous, heterozygous, and compound heterozygous; 47 (45.6%), 41 (39.8%), and 15 (14.6%), respectively. The majority of homozygous cases are discovered in pediatric pateints, whereas the majority of adult cases are heterozygous.
29 Nineteen mutations were identified in all detectable (103 patients) cases that are illustrated in Table 1.
30
31 Three mutations (c.315+1g>a, c.118c>t, and c.92+5g>c) were detected in the majority of cases (66%). The c.315+1g>a, previously known as IVS-II-1G>A, is a splicing mutation that was most frequently encountered in our study with a frequency of 32%. Followed by truncating nonsense mutation, the c.118c>t (previously known as Q39X or p.gln 40X) 23% and the c.92+5g>c (previously known as IVS-I-5G>C) 11%. The c.315+1g>a and c.118c>t mutations were common in homozygous cases, whereas c.92+5g>c was more common in compound heterozygous cases.
32 Five novel mutations (c.410g>a, c.-31c>t, c.68_74delaagttgg, c.316-3c>a, and c.-151c>t) were identified for the first time in Saudi population. The former three mutations represent 1% of cases for each, whereas the later presented with a frequency of 2%.
33 More recent studies were conducted to provide a precise figures and frequencies of the β-thalassemia mutations in specific region of Saudi Arabia. All previous studies that reported different β-globin gene mutations in Saudi Arabia are summarized in Table 2.
34
35 Abuzenadah et al. have recently identified 23 mutations responsible for β-thalassemia in western region of Saudi Arabia. Of these, there were seven common mutations with the most frequent one being IVS-I-5G>C and the other 16 mutations were less common, including one new mutation that has never been reported in the Saudi Arabia, the FS c20/21 mutation.
36 In other recent studies reported from the Eastern Province of Saudi Arabia by Al-Sultan et al., 14 mutations were identified. Among these, there were five frequent mutations with the more common one being IVS-II-1G>A [Table 2]. The author reported two novel mutations IVS-I-130 (G C) and IVS-I-110 (G A), which have not been previously reported in the population of the Eastern Province.
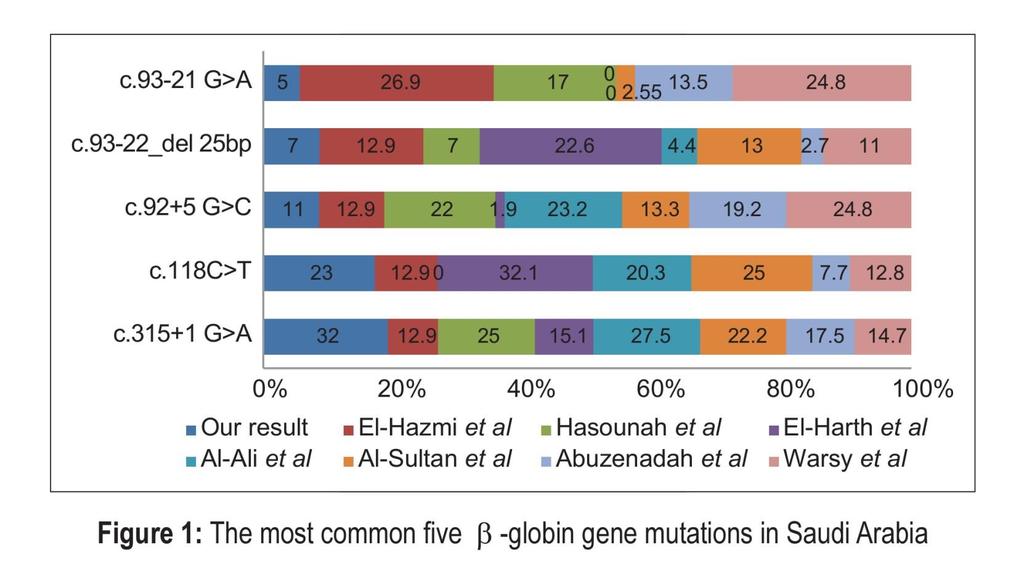
37 A total of 12 different mutations were reported in one more recent study from Riyadh by Warsy et al., in which the IVS-I-5G>C mutation was the most common.

38
39
40 In this study, we present a comprehensive report on the spectrum of β-thalassemia mutations. Given that, KFSH and RC represents a tertiary specialized hospital in Saudi Arabia in which advance molecular technologies are available for detecting wide range of molecular abnormalities.
41 Our study more and less shows similar findings that had been observed by previous studies. A total of 19 mutations were identified in this retrospective report. Out of these, 14 mutations have been reported in the previous studies that have been conducted in Saudi Arabia and five novel mutations were reported for the first time in Saudi population.
42 It has been noticed that the frequencies and prevalence of the 14 mutations are different among the Saudi population who screened for these mutations in the previous studies. This diversity is mainly due to the unique geographical position of Saudi Arabia that lie between the Mediterranean and Southeast Asian region.
43 The most frequent mutations in our study are c.315+1g>a, c.118c>t, and c.92+5g>c. These findings were also reported by most of studies on Saudi population with differences in their frequencies. The clinical significance of c.315+1g>a and c.118c>t mutations is their ability to produce the β0 thalassemia phenotype. These mutations were reported to occur in most of the Mediterranean and Gulf countries (Warsy, Abuzenadah, AlSultan, AlAli), although the c.92+5g>c produces β+ thalassemia phenotype and frequently encountered in Asian Indian.
44 In comparing our data to previous studies, we found that five mutations appear to be fingerprints of Saudi population and were most frequently reported in all studies. These mutations are c.315+1g>a, c.118c>t, c.92+5g>c, c.93-22_del 25bp, and c.93-21g>a.
45
46 We report five novel mutations (c.410g>a, c.-31c>t, c.68_74delaagttgg, c.316-3c>a, and c.-151c>t), which to the best of our knowledge, have not been described in Saudi Arabia.
47 In summary, this comprehensive analysis successfully identified 19 mutations, five of which are novel, and confirmed the previously published mutations among the Saudi population. The potential outcome of this study is to publish a list of more frequent β-globin gene mutations in Saudi β-thalassemia patients.






48 Thank you,
Thalassemias. Emanuela Veras, M.D. 01/08/2006
 Thalassemias Emanuela Veras, M.D. 01/08/2006 Structure and Function of normal Hemoglobin molecules: 2/3 1/3 β: increases from 6 th week of fetal life to 12 months of age At birth: HbF: 75-90% HbA: 10-25%
Thalassemias Emanuela Veras, M.D. 01/08/2006 Structure and Function of normal Hemoglobin molecules: 2/3 1/3 β: increases from 6 th week of fetal life to 12 months of age At birth: HbF: 75-90% HbA: 10-25%
Comprehensive Hemoglobin Analysis HBA1/2 (
 Comprehensive Hemoglobin Analysis HBA1/2 ( α-globin) and HBB (β-globin) mutation and deletion/duplication analysis and HBD (δ-globin) and HBG1/2 (γ-globin) mutation analysis Description: Hemoglobin (Hb)
Comprehensive Hemoglobin Analysis HBA1/2 ( α-globin) and HBB (β-globin) mutation and deletion/duplication analysis and HBD (δ-globin) and HBG1/2 (γ-globin) mutation analysis Description: Hemoglobin (Hb)
In adults, the predominant Hb (HbA) molecule has four chains: two α and two β chains. In thalassemias, the synthesis of either the α or the β chains
 molecule has four chains: two α and two β chains. In thalassemias, the synthesis of either the α or the β chains") Thalassaemias Thalassemia Thalassemia is an inherited autosomal recessive blood disease. Associated with absence or reduction in a or b globin chains. Reduced synthesis of one of the globin chains can
Thalassaemias Thalassemia Thalassemia is an inherited autosomal recessive blood disease. Associated with absence or reduction in a or b globin chains. Reduced synthesis of one of the globin chains can
SICKLE CELL DISEASE. Dr. MUBARAK ABDELRAHMAN MD PEDIATRICS AND CHILD HEALTH. Assistant Professor FACULTY OF MEDICINE -JAZAN
 SICKLE CELL DISEASE Dr. MUBARAK ABDELRAHMAN MD PEDIATRICS AND CHILD HEALTH Assistant Professor FACULTY OF MEDICINE -JAZAN Objective: The student should be able: To identify the presentation, diagnosis,
SICKLE CELL DISEASE Dr. MUBARAK ABDELRAHMAN MD PEDIATRICS AND CHILD HEALTH Assistant Professor FACULTY OF MEDICINE -JAZAN Objective: The student should be able: To identify the presentation, diagnosis,
Educational Items Section
 Atlas of Genetics and Cytogenetics in Oncology and Haematology OPEN ACCESS JOURNAL AT INIST-CNRS Educational Items Section Hemoglobin genes; Sickle-cell anemia - Thalassemias Jean-Loup Huret, Xavier Troussard
Atlas of Genetics and Cytogenetics in Oncology and Haematology OPEN ACCESS JOURNAL AT INIST-CNRS Educational Items Section Hemoglobin genes; Sickle-cell anemia - Thalassemias Jean-Loup Huret, Xavier Troussard
Thalassemias:general aspects and molecular pathology
 Thalassemias:general aspects and molecular pathology Prof. Renzo Galanello Pediatric Clinic 2 University of Cagliari Ospedale Regionale Microcitemie-ASL8 HEMOGLOBINOPATHIES CLASSIFICATION Structurally
Thalassemias:general aspects and molecular pathology Prof. Renzo Galanello Pediatric Clinic 2 University of Cagliari Ospedale Regionale Microcitemie-ASL8 HEMOGLOBINOPATHIES CLASSIFICATION Structurally
Anemia s. Troy Lund MSMS PhD MD
 Anemia s Troy Lund MSMS PhD MD lundx072@umn.edu Hemoglobinopathy/Anemia IOM take home points. 1. How do we identify the condtion? Smear, CBC Solubility Test (SCD) 2. How does it present clincally? 3. How
Anemia s Troy Lund MSMS PhD MD lundx072@umn.edu Hemoglobinopathy/Anemia IOM take home points. 1. How do we identify the condtion? Smear, CBC Solubility Test (SCD) 2. How does it present clincally? 3. How
Thalassemia Maria Luz Uy del Rosario, M.D.
 Thalassemia Maria Luz Uy del Rosario, M.D. Philippine Society of Hematology and Blood Transfusion Philippine Society of Pediatric Oncology What is Thalassemia Hereditary Hemoglobin disorder Hemolytic anemia
Thalassemia Maria Luz Uy del Rosario, M.D. Philippine Society of Hematology and Blood Transfusion Philippine Society of Pediatric Oncology What is Thalassemia Hereditary Hemoglobin disorder Hemolytic anemia
Beta Thalassemia Case Study Introduction to Bioinformatics
 Beta Thalassemia Case Study Sami Khuri Department of Computer Science San José State University San José, California, USA sami.khuri@sjsu.edu www.cs.sjsu.edu/faculty/khuri Outline v Hemoglobin v Alpha
Beta Thalassemia Case Study Sami Khuri Department of Computer Science San José State University San José, California, USA sami.khuri@sjsu.edu www.cs.sjsu.edu/faculty/khuri Outline v Hemoglobin v Alpha
Corporate Medical Policy
 Corporate Medical Policy Genetic Testing for Alpha Thalassemia File Name: Origination: Last CAP Review: Next CAP Review: Last Review: genetic_testing_for_alpha_thalassemia 9/2013 7/2017 7/2018 7/2017 Description
Corporate Medical Policy Genetic Testing for Alpha Thalassemia File Name: Origination: Last CAP Review: Next CAP Review: Last Review: genetic_testing_for_alpha_thalassemia 9/2013 7/2017 7/2018 7/2017 Description
Report of Beta Thalassemia in Newar Ethnicity
 Report of Beta Thalassemia in Newar Ethnicity Rajendra Dev Bhatt 1*, Surendra Koju 2, Prabodh Risal 1 Affiliations: 1 Department of Clinical Biochemistry, Dhulikhel Hospital, Kathmandu University Hospital
Report of Beta Thalassemia in Newar Ethnicity Rajendra Dev Bhatt 1*, Surendra Koju 2, Prabodh Risal 1 Affiliations: 1 Department of Clinical Biochemistry, Dhulikhel Hospital, Kathmandu University Hospital
Cover Page. The handle holds various files of this Leiden University dissertation.
 Cover Page The handle http://hdl.handle.net/1887/35456 holds various files of this Leiden University dissertation. Author: Hassan, Suha Mustafa Title: Toward prevention of Hemoglobinopathies in Oman Issue
Cover Page The handle http://hdl.handle.net/1887/35456 holds various files of this Leiden University dissertation. Author: Hassan, Suha Mustafa Title: Toward prevention of Hemoglobinopathies in Oman Issue
Diagnostic difficulties in prevention and control program for thalassemia in Thailand: atypical thalassemia carriers
 Diagnostic difficulties in prevention and control program for thalassemia in Thailand: atypical thalassemia carriers Pranee Winichagoon Fucharoen Thalassemia Research Center Institute of Molecular Biosciences
Diagnostic difficulties in prevention and control program for thalassemia in Thailand: atypical thalassemia carriers Pranee Winichagoon Fucharoen Thalassemia Research Center Institute of Molecular Biosciences
MOLECULAR BASIS OF THALASSEMIA IN SLOVENIA
 MOLECULAR BASIS OF THALASSEMIA IN SLOVENIA Dijana Plaseska-Karanfilska, MD, PhD Research Centre for Genetic Engineering and Biotechnology Georgi D. Efremov, Macedonian Academy of Sciences and Arts, Skopje,
MOLECULAR BASIS OF THALASSEMIA IN SLOVENIA Dijana Plaseska-Karanfilska, MD, PhD Research Centre for Genetic Engineering and Biotechnology Georgi D. Efremov, Macedonian Academy of Sciences and Arts, Skopje,
THALASSEMIA AND COMPREHENSIVE CARE
 1 THALASSEMIA AND COMPREHENSIVE CARE Melanie Kirby MBBS, FRCP (C), Hospital for Sick Children, Toronto Associate Professor of Paediatrics, University of Toronto. Objectives 2 By the end of this presentation,
1 THALASSEMIA AND COMPREHENSIVE CARE Melanie Kirby MBBS, FRCP (C), Hospital for Sick Children, Toronto Associate Professor of Paediatrics, University of Toronto. Objectives 2 By the end of this presentation,
Genetics of Thalassemia
 Genetics of Thalassemia Submitted by : Raya Samir Al- Hayaly Sura Zuhair Salih Saad Ghassan Al- Dulaimy Saad Farouq Kassir Sama Naal Salouha Zahraa Jasim Al- Aarajy Supervised by : Dr. Kawkab Adris Mahmod
Genetics of Thalassemia Submitted by : Raya Samir Al- Hayaly Sura Zuhair Salih Saad Ghassan Al- Dulaimy Saad Farouq Kassir Sama Naal Salouha Zahraa Jasim Al- Aarajy Supervised by : Dr. Kawkab Adris Mahmod
Extra Notes 3. Warm. In the core (center) of the body, where the temperature is 37 C.
 of the body, where the temperature is 37 C.") Extra Notes 3 *The numbers of the slides are according to the last year slides. Slide 33 Autoimmune hemolytic anemia : Abnormal circulating antibodies that target normal antigen on the RBC and cause lysis.
Extra Notes 3 *The numbers of the slides are according to the last year slides. Slide 33 Autoimmune hemolytic anemia : Abnormal circulating antibodies that target normal antigen on the RBC and cause lysis.
Beta Thalassemia Frequency in Bahrain: A Ten Year Study. Shaikha Salim Al-Arrayed, MB,ChB, DHCG, PhD*
 Bahrain Medical Bulletin, Vol. 2, No. 2, June 200 Beta Thalassemia Frequency in Bahrain: A Ten Year Study Shaikha Salim Al-Arrayed, MB,ChB, DHCG, PhD* Background: Sickle-cell disease and Thalassanemia
Bahrain Medical Bulletin, Vol. 2, No. 2, June 200 Beta Thalassemia Frequency in Bahrain: A Ten Year Study Shaikha Salim Al-Arrayed, MB,ChB, DHCG, PhD* Background: Sickle-cell disease and Thalassanemia
Hemolytic anemias (2 of 2)
") Hemolytic anemias (2 of 2) Sickle Cell Anemia The most common familial hemolytic anemia in the world Sickle cell anemia is the prototypical (and most prevalent) hemoglobinopathy Mutation in the β-globin
Hemolytic anemias (2 of 2) Sickle Cell Anemia The most common familial hemolytic anemia in the world Sickle cell anemia is the prototypical (and most prevalent) hemoglobinopathy Mutation in the β-globin
HAEMOGLOBINOPATHIES. Editing file. References: 436 girls & boys slides 435 teamwork slides. Color code: Important. Extra.
 HAEMOGLOBINOPATHIES Objectives: normal structure and function of haemoglobin. how the globin components of haemoglobin change during development, and postnatally. the mechanisms by which the thalassaemias
HAEMOGLOBINOPATHIES Objectives: normal structure and function of haemoglobin. how the globin components of haemoglobin change during development, and postnatally. the mechanisms by which the thalassaemias
Hemoglobinopathies Diagnosis and management
 Hemoglobinopathies Diagnosis and management Morgan L. McLemore, M.D. Hematology/Leukemia Department of Hematology and Oncology Winship Cancer Institute at Emory University mlmclem@emory.edu Disclosures
Hemoglobinopathies Diagnosis and management Morgan L. McLemore, M.D. Hematology/Leukemia Department of Hematology and Oncology Winship Cancer Institute at Emory University mlmclem@emory.edu Disclosures
High Hemoglobin F in a Saudi Child Presenting with Pancytopenia
 Case Report imedpub Journals http://www.imedpub.com Journal of Pediatric Care ISSN 2471-805X DOI: 10.21767/2471-805X.100002 High Hemoglobin F in a Saudi Child Presenting with Pancytopenia Abstract Saudi
Case Report imedpub Journals http://www.imedpub.com Journal of Pediatric Care ISSN 2471-805X DOI: 10.21767/2471-805X.100002 High Hemoglobin F in a Saudi Child Presenting with Pancytopenia Abstract Saudi
Genetic Modulation on the Phenotypic Diversity of Sickle Cell Disease
 Genetic Modulation on the Phenotypic Diversity of Sickle Cell Disease Malay B. Mukherjee Abstract Sickle cell hemoglobin is a β chain structural variant where valine is substituted for glutamic acid in
Genetic Modulation on the Phenotypic Diversity of Sickle Cell Disease Malay B. Mukherjee Abstract Sickle cell hemoglobin is a β chain structural variant where valine is substituted for glutamic acid in
DONE BY : RaSHA RAKAN & Bushra Saleem
 DONE BY : RaSHA RAKAN & Bushra Saleem Hemolytic anemias (2 of 2) Sickle Cell Anemia The most common familial hemolytic anemia in the world Sickle cell anemia is the prototypical (and most prevalent) hemoglobinopathy
DONE BY : RaSHA RAKAN & Bushra Saleem Hemolytic anemias (2 of 2) Sickle Cell Anemia The most common familial hemolytic anemia in the world Sickle cell anemia is the prototypical (and most prevalent) hemoglobinopathy
Screening for haemoglobinopathies in pregnancy
 Policy Statement All Southern Health patients will receive clinical care that reflects best practice and is based on the best available evidence. Index of chapters within background 1. Prevalence of haemoglobinopathies
Policy Statement All Southern Health patients will receive clinical care that reflects best practice and is based on the best available evidence. Index of chapters within background 1. Prevalence of haemoglobinopathies
Prevalence of Thalassemia in Patients With Microcytosis Referred for Hemoglobinopathy Investigation in Ontario A Prospective Cohort Study
 Hematopathology / PREVALENCE OF THALASSEMIA IN ONTARIO Prevalence of Thalassemia in Patients With Microcytosis Referred for Hemoglobinopathy Investigation in Ontario A Prospective Cohort Study John D.
Hematopathology / PREVALENCE OF THALASSEMIA IN ONTARIO Prevalence of Thalassemia in Patients With Microcytosis Referred for Hemoglobinopathy Investigation in Ontario A Prospective Cohort Study John D.
Next Generation Sequencing as a tool for breakpoint analysis in rearrangements of the globin-gene clusters
 Next Generation Sequencing as a tool for breakpoint analysis in rearrangements of the globin-gene clusters XXXth International Symposium on Technical Innovations in Laboratory Hematology Honolulu, Hawaii
Next Generation Sequencing as a tool for breakpoint analysis in rearrangements of the globin-gene clusters XXXth International Symposium on Technical Innovations in Laboratory Hematology Honolulu, Hawaii
HEMOLYTIC ANEMIA DUE TO ABNORMAL HEMOGLOBIN SYNTHESIS
 Hemolytic Anemia Due to Abnormal Hemoglobin Synthesis MODULE 19 HEMOLYTIC ANEMIA DUE TO ABNORMAL HEMOGLOBIN SYNTHESIS 19.1 INTRODUCTION There are two main mechanisms by which anaemia is produced (a) Thalassemia:
Hemolytic Anemia Due to Abnormal Hemoglobin Synthesis MODULE 19 HEMOLYTIC ANEMIA DUE TO ABNORMAL HEMOGLOBIN SYNTHESIS 19.1 INTRODUCTION There are two main mechanisms by which anaemia is produced (a) Thalassemia:
Beta Thalassemia Sami Khuri Department of Computer Science San José State University Spring 2015
 Bioinformatics in Medical Product Development SMPD 287 Three Beta Thalassemia Sami Khuri Department of Computer Science San José State University Hemoglobin Outline Anatomy of a gene Hemoglobinopathies
Bioinformatics in Medical Product Development SMPD 287 Three Beta Thalassemia Sami Khuri Department of Computer Science San José State University Hemoglobin Outline Anatomy of a gene Hemoglobinopathies
Haemoglobin BY: MUHAMMAD RADWAN WISSAM MUHAMMAD
 Haemoglobin BY: MUHAMMAD RADWAN WISSAM MUHAMMAD Introduction is the iron-containing oxygen transport metalloprotein in the red blood cells Hemoglobin in the blood carries oxygen from the respiratory organs
Haemoglobin BY: MUHAMMAD RADWAN WISSAM MUHAMMAD Introduction is the iron-containing oxygen transport metalloprotein in the red blood cells Hemoglobin in the blood carries oxygen from the respiratory organs
POLICY PRODUCT VARIATIONS DESCRIPTION/BACKGROUND RATIONALE DEFINITIONS BENEFIT VARIATIONS DISCLAIMER CODING INFORMATION REFERENCES POLICY HISTORY
 Original Issue Date (Created): November 26, 2013 Most Recent Review Date (Revised): November 26, 2013 Effective Date: April 1, 2014 POLICY PRODUCT VARIATIONS DESCRIPTION/BACKGROUND RATIONALE DEFINITIONS
Original Issue Date (Created): November 26, 2013 Most Recent Review Date (Revised): November 26, 2013 Effective Date: April 1, 2014 POLICY PRODUCT VARIATIONS DESCRIPTION/BACKGROUND RATIONALE DEFINITIONS
got anemia? IT COULD BE THALASSEMIA TRAIT
 got anemia? IT COULD BE THALASSEMIA TRAIT 1 in 20 people worldwide carry some type of thalassemia trait You are especially at risk of carrying the thalassemia trait if you have ancestry from: Southeast
got anemia? IT COULD BE THALASSEMIA TRAIT 1 in 20 people worldwide carry some type of thalassemia trait You are especially at risk of carrying the thalassemia trait if you have ancestry from: Southeast
4 Jumana Jihad Dr. Ahmad Mansour Dr. Ahmad Mansour
 4 Jumana Jihad Dr. Ahmad Mansour Dr. Ahmad Mansour Anemia Decreased blood production Increased blood loss Hemolytic Hemorrhage Extravascular Intravascular Hemolytic (Further classification( Extrinsic Intrinsic
4 Jumana Jihad Dr. Ahmad Mansour Dr. Ahmad Mansour Anemia Decreased blood production Increased blood loss Hemolytic Hemorrhage Extravascular Intravascular Hemolytic (Further classification( Extrinsic Intrinsic
Dr.Abdolreza Afrasiabi
 Dr.Abdolreza Afrasiabi Thalassemia & Heamophilia Genetic Reaserch Center Shiraz Medical University Hemoglobin tetramer Hemoglobin Structure % A 1 α 2 β 2 94-97% A 2 α 2 δ 2 2.5% A 1C α 2 (β-n-glucose)
Dr.Abdolreza Afrasiabi Thalassemia & Heamophilia Genetic Reaserch Center Shiraz Medical University Hemoglobin tetramer Hemoglobin Structure % A 1 α 2 β 2 94-97% A 2 α 2 δ 2 2.5% A 1C α 2 (β-n-glucose)
HEMOGLOBIN ELECTROPHORESIS DR ARASH ALGHASI SHAFA HOSPITAL-AHWAZ
 HEMOGLOBIN ELECTROPHORESIS DR ARASH ALGHASI SHAFA HOSPITAL-AHWAZ Hemoglobin Hemoglobin (Hb), protein constituting 1/3 of the red blood cells Each red cell has 640 million molecules of Hb sites in the cells:
HEMOGLOBIN ELECTROPHORESIS DR ARASH ALGHASI SHAFA HOSPITAL-AHWAZ Hemoglobin Hemoglobin (Hb), protein constituting 1/3 of the red blood cells Each red cell has 640 million molecules of Hb sites in the cells:
Detecting and Reporting Alpha Thalassemia In Newborns
 Detecting and Reporting Alpha Thalassemia In Newborns T. Davis, C. Moore, L. Nayak, M.C. Dorley, M. del Pilar Aguinaga, M. Chan, J. Ubaike, C. Yusuf Alpha Thalassemia Screening Status in the US Clinical
Detecting and Reporting Alpha Thalassemia In Newborns T. Davis, C. Moore, L. Nayak, M.C. Dorley, M. del Pilar Aguinaga, M. Chan, J. Ubaike, C. Yusuf Alpha Thalassemia Screening Status in the US Clinical
RBCs Disorders 2. Dr. Nabila Hamdi MD, PhD
 RBCs Disorders 2 Dr. Nabila Hamdi MD, PhD ILOs Discuss the classification of anemia into hypochromic-microcytic, normochromicnormocytic and macrocytic. Categorize laboratory test procedures used in the
RBCs Disorders 2 Dr. Nabila Hamdi MD, PhD ILOs Discuss the classification of anemia into hypochromic-microcytic, normochromicnormocytic and macrocytic. Categorize laboratory test procedures used in the
Natural Selection In Humans (Sickle Cell Anemia)
") Natural Selection In Humans (Sickle Cell Anemia) Background Information Hemoglobin is a protein found in red blood cells Transports oxygen to body tissues Individuals homozygous for the sickle cell allele
Natural Selection In Humans (Sickle Cell Anemia) Background Information Hemoglobin is a protein found in red blood cells Transports oxygen to body tissues Individuals homozygous for the sickle cell allele
Genetic Modifiers of Sickle Cell Disease Severity. Kunle Adekile, MD, PhD Professor Department of Pediatrics Kuwait University
 Genetic Modifiers of Sickle Cell Disease Severity Kunle Adekile, MD, PhD Professor Department of Pediatrics Kuwait University Outline Hb Molecule and Genetic control of globin synthesis Pathophysiology
Genetic Modifiers of Sickle Cell Disease Severity Kunle Adekile, MD, PhD Professor Department of Pediatrics Kuwait University Outline Hb Molecule and Genetic control of globin synthesis Pathophysiology
Hematologic Features of Alpha Thalassemia Carriers
 IJMCM Summer 2012, Vol 1, No 3 Original Article Hematologic Features of Alpha Thalassemia Carriers Haleh Akhavan-Niaki 1,2, Reza Youssefi Kamangari 2, Ali Banihashemi 2, Vahid Kholghi Oskooei 1, Mandana
IJMCM Summer 2012, Vol 1, No 3 Original Article Hematologic Features of Alpha Thalassemia Carriers Haleh Akhavan-Niaki 1,2, Reza Youssefi Kamangari 2, Ali Banihashemi 2, Vahid Kholghi Oskooei 1, Mandana
Sickle Cell Anemia. Sickle cell anemia is an inherited disorder of the blood which occurs when just one base pair substitution
 Rose Farrington and Rachel Nash BIOL 362 Lab M. Bulgarella Genetic Diseases 10/14/2008 Sickle Cell Anemia Introduction Sickle cell anemia is an inherited disorder of the blood which occurs when just one
Rose Farrington and Rachel Nash BIOL 362 Lab M. Bulgarella Genetic Diseases 10/14/2008 Sickle Cell Anemia Introduction Sickle cell anemia is an inherited disorder of the blood which occurs when just one
An overview of Thalassaemias and Complications
 An overview of Thalassaemias and Complications Haemoglobin Haemoglobin is the most abundant protein in blood, and exists as three main types in normal adults: HbA ( ) - 97% HbA 2 ( ) - 2.5% HbF ( ) - 0.5%
An overview of Thalassaemias and Complications Haemoglobin Haemoglobin is the most abundant protein in blood, and exists as three main types in normal adults: HbA ( ) - 97% HbA 2 ( ) - 2.5% HbF ( ) - 0.5%
Neonatal Screening for Genetic Blood Diseases. Shaikha Al-Arayyed, PhD* A Aziz Hamza, MD** Bema Sultan*** D. K. Shome, MRCPath**** J. P.
 Bahrain Medical Bulletin, Vol. 29, No. 3, September, 2007 Neonatal Screening for Genetic Blood Diseases Shaikha Al-Arayyed, PhD* A Aziz Hamza, MD** Bema Sultan*** D. K. Shome, MRCPath**** J. P. Bapat,PhD****
Bahrain Medical Bulletin, Vol. 29, No. 3, September, 2007 Neonatal Screening for Genetic Blood Diseases Shaikha Al-Arayyed, PhD* A Aziz Hamza, MD** Bema Sultan*** D. K. Shome, MRCPath**** J. P. Bapat,PhD****
SHORT COMMUNICATION. of Medical Sciences, Isfahan, Iran 2 MRC Molecular Haematology Unit, Weatherall Institute of Molecular Medicine, John
 Hemoglobin, 34(1):115 120, (2010) Copyright Informa UK Ltd. ISSN: 0363-0269 print/1532-432x online DOI: 10.3109/03630260903554894 LHEM 0363-0269 1532-432X Hemoglobin, Vol. 34, No. 1, Dec 2009: pp. 0 0
Hemoglobin, 34(1):115 120, (2010) Copyright Informa UK Ltd. ISSN: 0363-0269 print/1532-432x online DOI: 10.3109/03630260903554894 LHEM 0363-0269 1532-432X Hemoglobin, Vol. 34, No. 1, Dec 2009: pp. 0 0
George R. Honig Junius G. Adams III. Human Hemoglobin. Genetics. Springer-Verlag Wien New York
 George R. Honig Junius G. Adams III Human Hemoglobin Genetics Springer-Verlag Wien New York George R. Honig, M.D., Ph.D. Professor and Head Department of Pediatrics, College of Medicine University of Illinois
George R. Honig Junius G. Adams III Human Hemoglobin Genetics Springer-Verlag Wien New York George R. Honig, M.D., Ph.D. Professor and Head Department of Pediatrics, College of Medicine University of Illinois
Red cell disorder. Dr. Ahmed Hasan
 Red cell disorder Dr. Ahmed Hasan Things to be learned in this lecture Definition and clinical feature of anemia. Classification of anemia. Know some details of microcytic anemia Question of the lecture:
Red cell disorder Dr. Ahmed Hasan Things to be learned in this lecture Definition and clinical feature of anemia. Classification of anemia. Know some details of microcytic anemia Question of the lecture:
Evaluation of hemoglobinopathy screening results of a six year period in Turkey
 Evaluation of hemoglobinopathy screening results of a six year period in Turkey Seçil Gunher Arıca, Ebru Turhan, Cahit Özer, Vefik Arıca, Dilek Benk Şilfeler, Đbrahim Şilfeler, Ayşe Betül Altun Vol. 4
Evaluation of hemoglobinopathy screening results of a six year period in Turkey Seçil Gunher Arıca, Ebru Turhan, Cahit Özer, Vefik Arıca, Dilek Benk Şilfeler, Đbrahim Şilfeler, Ayşe Betül Altun Vol. 4
Chem*3560 Lecture 4: Inherited modifications in hemoglobin
 Chem*3560 Lecture 4: Inherited modifications in hemoglobin Genetic modifications fall into two classes: Thalassemias, which are the result of failure to express globin genes. Thalassa is Greek for the
Chem*3560 Lecture 4: Inherited modifications in hemoglobin Genetic modifications fall into two classes: Thalassemias, which are the result of failure to express globin genes. Thalassa is Greek for the
Dr B Lal Clinical Laboratory Pvt Ltd. Jaipur, Rajasthan, India
 Volume 1, Issue 1, pp: 1-5 Research Article Introduction Open Access Role of Hematological Indices in the Screening of Β-Thalassemia Minor (Trait) and Iron Deficiency Shaily Garg, Anshika Srivastava, Sanjeev
Volume 1, Issue 1, pp: 1-5 Research Article Introduction Open Access Role of Hematological Indices in the Screening of Β-Thalassemia Minor (Trait) and Iron Deficiency Shaily Garg, Anshika Srivastava, Sanjeev
Clinical Characteristics of Pediatric Thalassemia in Korea: A Single Institute Experience
 ORIGINAL ARTICLE Pediatrics http://dx.doi.org/10.3346/jkms.2013.28.11.1645 J Korean Med Sci 2013; 28: 1645-1649 Clinical Characteristics of Pediatric Thalassemia in Korea: A Single Institute Experience
ORIGINAL ARTICLE Pediatrics http://dx.doi.org/10.3346/jkms.2013.28.11.1645 J Korean Med Sci 2013; 28: 1645-1649 Clinical Characteristics of Pediatric Thalassemia in Korea: A Single Institute Experience
6.1 Extended family screening
 CHAPTER 6 CONCLUSION Cost benefit analysis of thalassemia screening programs have shown that the single years treatment for a β-thalassemia major patient was much higher than a total cost per case prevented.
CHAPTER 6 CONCLUSION Cost benefit analysis of thalassemia screening programs have shown that the single years treatment for a β-thalassemia major patient was much higher than a total cost per case prevented.
Counselling and prenatal diagnosis. Antonis Kattamis, Greece
 Counselling and prenatal diagnosis Antonis Kattamis, Greece Epidemiology of Hemoglobinopathies 7% of world population carriers of hemoglobinopathies 500.000 newborns annually affected 300.000 : Thalassemias
Counselling and prenatal diagnosis Antonis Kattamis, Greece Epidemiology of Hemoglobinopathies 7% of world population carriers of hemoglobinopathies 500.000 newborns annually affected 300.000 : Thalassemias
7 Medical Genetics. Hemoglobinopathies. Hemoglobinopathies. Protein and Gene Structure. and Biochemical Genetics
 SESSION 7 Medical Genetics Hemoglobinopathies and Biochemical Genetics J a v a d F a s a J a m s h i d i U n i v e r s i t y o f M e d i c a l S c i e n c e s, N o v e m b e r 2 0 1 7 Hemoglobinopathies
SESSION 7 Medical Genetics Hemoglobinopathies and Biochemical Genetics J a v a d F a s a J a m s h i d i U n i v e r s i t y o f M e d i c a l S c i e n c e s, N o v e m b e r 2 0 1 7 Hemoglobinopathies
RBCs Disorders 2. Dr. Nabila Hamdi MD, PhD
 RBCs Disorders 2 Dr. Nabila Hamdi MD, PhD ILOs Discuss the classification of anemia into hypochromic-microcytic, normochromicnormocytic and macrocytic. Categorize laboratory test procedures used in the
RBCs Disorders 2 Dr. Nabila Hamdi MD, PhD ILOs Discuss the classification of anemia into hypochromic-microcytic, normochromicnormocytic and macrocytic. Categorize laboratory test procedures used in the
Thalassemia intermedia in HbH-CS disease with compound heterozygosity for β-thalassemia: Challenges in hemoglobin analysis and clinical diagnosis
 Genes Genet. Syst. (2009) 84, p. 67 71 Thalassemia intermedia in HbH-CS disease with compound heterozygosity for β-thalassemia: Challenges in hemoglobin analysis and clinical diagnosis Jin Ai Mary Anne
Genes Genet. Syst. (2009) 84, p. 67 71 Thalassemia intermedia in HbH-CS disease with compound heterozygosity for β-thalassemia: Challenges in hemoglobin analysis and clinical diagnosis Jin Ai Mary Anne
International Journal of Drug Research and Technology
 Int. J. Drug Res. Tech. 2012, Vol. 2 (7), 472-478 ISSN 2277-1506 International Journal of Drug Research and Technology Available online at http://www.ijdrt.com Original Research Paper SCREENING, ANALYSIS
Int. J. Drug Res. Tech. 2012, Vol. 2 (7), 472-478 ISSN 2277-1506 International Journal of Drug Research and Technology Available online at http://www.ijdrt.com Original Research Paper SCREENING, ANALYSIS
Heme Questions and Derivatives for the USMLE Step One Exam. Winter Storm Skylar Edition
 Heme Questions and Derivatives for the USMLE Step One Exam Winter Storm Skylar Edition Howard J. Sachs, MD Howard@12DaysinMarch.com www.12daysinmarch.com Patient presents for routine preoperative evaluation
Heme Questions and Derivatives for the USMLE Step One Exam Winter Storm Skylar Edition Howard J. Sachs, MD Howard@12DaysinMarch.com www.12daysinmarch.com Patient presents for routine preoperative evaluation
JMSCR Vol 06 Issue 01 Page January 2018
 www.jmscr.igmpublication.org Impact Factor 5.84 Index Copernicus Value: 71.58 ISSN (e)-2347-176x ISSN (p) 2455-0450 DOI: https://dx.doi.org/10.18535/jmscr/v6i1.102 HPLC based evaluation of Haemoglobinopathies
www.jmscr.igmpublication.org Impact Factor 5.84 Index Copernicus Value: 71.58 ISSN (e)-2347-176x ISSN (p) 2455-0450 DOI: https://dx.doi.org/10.18535/jmscr/v6i1.102 HPLC based evaluation of Haemoglobinopathies
Line Probe Assay for Detection of Alpha Thalassemia: A Pilot Study
 Line Probe Assay for Detection of Alpha Thalassemia: A Pilot Study Menon PK *, Nimmakayalu M, Bylappa SK, Kumar M, Abdalhaleem HM Center for Advanced Biomedical Research and Innovation, Gulf Medical University,
Line Probe Assay for Detection of Alpha Thalassemia: A Pilot Study Menon PK *, Nimmakayalu M, Bylappa SK, Kumar M, Abdalhaleem HM Center for Advanced Biomedical Research and Innovation, Gulf Medical University,
The Beats of Natural Sciences Issue 3-4 (September-December) Vol. 3 (2016)
 Vol. 3 (2016)") Frequency of β (Beta Thalassaemia) Trait and Haemaglobin E (HbE) Trait: Case Study in a Thalassaemia Carrier Detection Camp in Gurudas College, West Bengal, India Mitu De Department of Botany, Gurudas
Frequency of β (Beta Thalassaemia) Trait and Haemaglobin E (HbE) Trait: Case Study in a Thalassaemia Carrier Detection Camp in Gurudas College, West Bengal, India Mitu De Department of Botany, Gurudas
Genetic Testing for α-thalassemia
 Medical Policy Manual Genetic Testing, Policy No. 52 Genetic Testing for α-thalassemia Next Review: January 2019 Last Review: January 2018 Effective: February 1, 2018 IMPORTANT REMINDER Medical Policies
Medical Policy Manual Genetic Testing, Policy No. 52 Genetic Testing for α-thalassemia Next Review: January 2019 Last Review: January 2018 Effective: February 1, 2018 IMPORTANT REMINDER Medical Policies
Spectrum of Haemoglobinopathies in a Suburb of Indore (India): A Two Year Study
: A Two Year Study") Original 378 Article Indian Journal of Pathology: Research and Practice Volume 6 Number 2, April - June 2017 (Part 2) DOI: http://dx.doi.org/10.21088/ijprp.2278.148x.6217.6 Spectrum of Haemoglobinopathies
Original 378 Article Indian Journal of Pathology: Research and Practice Volume 6 Number 2, April - June 2017 (Part 2) DOI: http://dx.doi.org/10.21088/ijprp.2278.148x.6217.6 Spectrum of Haemoglobinopathies
- Ensherah Mokheemer. - Rama Nada. - Tareq Aladily. 1 P a g e
 -3 - Ensherah Mokheemer - Rama Nada - Tareq Aladily 1 P a g e In this lecture we will continue talking about autoimmune hemolytic anemia. Autoimmune hemolytic anemia - There are several types that shares
-3 - Ensherah Mokheemer - Rama Nada - Tareq Aladily 1 P a g e In this lecture we will continue talking about autoimmune hemolytic anemia. Autoimmune hemolytic anemia - There are several types that shares
The Meaning of Genetic Variation
 Activity 2 The Meaning of Genetic Variation Focus: Students investigate variation in the beta globin gene by identifying base changes that do and do not alter function, and by using several CD-ROM-based
Activity 2 The Meaning of Genetic Variation Focus: Students investigate variation in the beta globin gene by identifying base changes that do and do not alter function, and by using several CD-ROM-based
The pros and cons of the fourth revision of thalassaemia screening programme in Iran
 Original Article The pros and cons of the fourth revision of thalassaemia screening programme in Iran J Med Screen 2017, Vol. 24(1) 1 5! The Author(s) 2016 Reprints and permissions: sagepub.co.uk/journalspermissions.nav
Original Article The pros and cons of the fourth revision of thalassaemia screening programme in Iran J Med Screen 2017, Vol. 24(1) 1 5! The Author(s) 2016 Reprints and permissions: sagepub.co.uk/journalspermissions.nav
THE UNIVERSITY OF JORDAN FACULTY OF MEDICINE DEPARTMENT OF PATHOLOGY HEMOLYTIC ANEMIAS. Dr. Tariq Aladily
 THE UNIVERSITY OF JORDAN FACULTY OF MEDICINE DEPARTMENT OF PATHOLOGY HEMOLYTIC ANEMIAS Third year medical students First semester Faculty 2018/2019 of Medicine Hereditary Spherocytosis Intrinsic defects
THE UNIVERSITY OF JORDAN FACULTY OF MEDICINE DEPARTMENT OF PATHOLOGY HEMOLYTIC ANEMIAS Third year medical students First semester Faculty 2018/2019 of Medicine Hereditary Spherocytosis Intrinsic defects
The Prevalence and Heterogeneity of Beta Thalassemia Mutations in The Western Maharashtra Population: A Hospital Based Study
 Kamla-Raj 2001 IJHG 1(3): 219-223 (2001) The Prevalence and Heterogeneity of Beta Thalassemia Mutations in The Western Maharashtra Population: A Hospital Based Study S.S. Ambekar, M.A. Phadke, D.N. Balpande,
Kamla-Raj 2001 IJHG 1(3): 219-223 (2001) The Prevalence and Heterogeneity of Beta Thalassemia Mutations in The Western Maharashtra Population: A Hospital Based Study S.S. Ambekar, M.A. Phadke, D.N. Balpande,
Haemoglobinopathy Case Studies. Dr Jill Finlayson Department of Haematology Pathwest Laboratory Medicine
 Haemoglobinopathy Case Studies Dr Jill Finlayson Department of Haematology Pathwest Laboratory Medicine Case 1 KB, 36y M Refugee Afghanistan Screening bloods Hb 101 g/l RCC 3.75 x10 12 /L MCV 90 fl MCH
Haemoglobinopathy Case Studies Dr Jill Finlayson Department of Haematology Pathwest Laboratory Medicine Case 1 KB, 36y M Refugee Afghanistan Screening bloods Hb 101 g/l RCC 3.75 x10 12 /L MCV 90 fl MCH
When do you have to perform the molecular biology in the hemoglobinopathies diagnosis
 When do you have to perform the molecular biology in the hemoglobinopathies diagnosis Maria Domenica Cappellini MD, FRCP;FACP Fondazione Ca Granda Policlinico IRCCS University of Milan Disclosure Member
When do you have to perform the molecular biology in the hemoglobinopathies diagnosis Maria Domenica Cappellini MD, FRCP;FACP Fondazione Ca Granda Policlinico IRCCS University of Milan Disclosure Member
Epidemiological Study among Thalassemia Intermedia Pediatric Patients
 Med. J. Cairo Univ., Vol. 78, No. 2, December 651-655, 2010 www.medicaljournalofcairouniversity.com Epidemiological Study among Thalassemia Intermedia Pediatric Patients NERMEEN KADDAH, M.D.; KHALED SALAMA,
Med. J. Cairo Univ., Vol. 78, No. 2, December 651-655, 2010 www.medicaljournalofcairouniversity.com Epidemiological Study among Thalassemia Intermedia Pediatric Patients NERMEEN KADDAH, M.D.; KHALED SALAMA,
Genetic Diversity of 3-thalassemia Mutations in Pakistani Population
 Genetic Diversity of 3-thalassemia Mutations in Pakistani Population Bushra Khateeb,Tariq Moatter,Asim M. Shaghil,Sarwat Haroon,Ghulam N. Kakepoto ( Department of Pathology, The Aga Khan University Hospital,
Genetic Diversity of 3-thalassemia Mutations in Pakistani Population Bushra Khateeb,Tariq Moatter,Asim M. Shaghil,Sarwat Haroon,Ghulam N. Kakepoto ( Department of Pathology, The Aga Khan University Hospital,
db-thalassemia Patients With Homozygous Xmn-1 Polymorphism That Are Characterized By A Milder Phenotype
 ISPUB.COM The Internet Journal of Hematology Volume 7 Number 2 db-thalassemia Patients With Homozygous Xmn-1 Polymorphism That Are Characterized By A Milder S Ashraf Citation S Ashraf. db-thalassemia Patients
ISPUB.COM The Internet Journal of Hematology Volume 7 Number 2 db-thalassemia Patients With Homozygous Xmn-1 Polymorphism That Are Characterized By A Milder S Ashraf Citation S Ashraf. db-thalassemia Patients
1) Anemias Dr. Anwar Sheikha
 Anemias Dr. Anwar Sheikha") 1) Anemias Dr. Anwar Sheikha DEFINITION: Anemia can be defined as a reduction in the concentration of hemoglobin below what is normal for age and sex of the patient. This reduction of hemoglobin is usually
1) Anemias Dr. Anwar Sheikha DEFINITION: Anemia can be defined as a reduction in the concentration of hemoglobin below what is normal for age and sex of the patient. This reduction of hemoglobin is usually
Molecular epidemiological survey of haemoglobinopathies in the Guangxi Zhuang Autonomous Region of southern China
 Clin Genet 2010: 78: 139 148 Printed in Singapore. All rights reserved Original Article 2010 John Wiley & Sons A/S CLINICAL GENETICS doi: 10.1111/j.1399-0004.2010.01430.x Molecular epidemiological survey
Clin Genet 2010: 78: 139 148 Printed in Singapore. All rights reserved Original Article 2010 John Wiley & Sons A/S CLINICAL GENETICS doi: 10.1111/j.1399-0004.2010.01430.x Molecular epidemiological survey
Dr Banu Kaya Consultant Haematologist Barts Health NHS Trust Royal London Hospital, London, UK SICKLE CELL AND THALASSAEMIA OVERVIEW
 Dr Banu Kaya Consultant Haematologist Barts Health NHS Trust Royal London Hospital, London, UK SICKLE CELL AND THALASSAEMIA OVERVIEW Objectives Gain awareness of haemoglobinopathy inheritance, pathophysiology
Dr Banu Kaya Consultant Haematologist Barts Health NHS Trust Royal London Hospital, London, UK SICKLE CELL AND THALASSAEMIA OVERVIEW Objectives Gain awareness of haemoglobinopathy inheritance, pathophysiology
Changes in hematological parameters in α-thalassemia individuals co-inherited with erythroid Krüppel-like factor mutations
 Clin Genet 2015: 88: 56 61 Printed in Singapore. All rights reserved Short Report 2014 John Wiley & Sons A/S. Published by John Wiley & Sons Ltd CLINICAL GENETICS doi: 10.1111/cge.12443 Changes in hematological
Clin Genet 2015: 88: 56 61 Printed in Singapore. All rights reserved Short Report 2014 John Wiley & Sons A/S. Published by John Wiley & Sons Ltd CLINICAL GENETICS doi: 10.1111/cge.12443 Changes in hematological
Research Article Clinical Features and Molecular Analysis of Hb H Disease in Taiwan
 BioMed Research International, Article ID 271070, 5 pages http://dx.doi.org/10.1155/2014/271070 Research Article Clinical Features and Molecular Analysis of Hb H Disease in Taiwan Yu-Hua Chao, 1,2,3 Kang-Hsi
BioMed Research International, Article ID 271070, 5 pages http://dx.doi.org/10.1155/2014/271070 Research Article Clinical Features and Molecular Analysis of Hb H Disease in Taiwan Yu-Hua Chao, 1,2,3 Kang-Hsi
Ch 4: Mendel and Modern evolutionary theory
 Ch 4: Mendel and Modern evolutionary theory 1 Mendelian principles of inheritance Mendel's principles explain how traits are transmitted from generation to generation Background: eight years breeding pea
Ch 4: Mendel and Modern evolutionary theory 1 Mendelian principles of inheritance Mendel's principles explain how traits are transmitted from generation to generation Background: eight years breeding pea
Original Paper. Inherited Haemoglobin Disorders Among Apparently Healthy Individuals- An Analysis of 105 Cases. Abstract
 Original Paper Inherited Haemoglobin Disorders Among Apparently Healthy Individuals- An Analysis of 105 Cases Salsabil MA 1, Islam M 2, Jahan D 3, Khan MA 4 Abstract Introduction: Inherited hemoglobin
Original Paper Inherited Haemoglobin Disorders Among Apparently Healthy Individuals- An Analysis of 105 Cases Salsabil MA 1, Islam M 2, Jahan D 3, Khan MA 4 Abstract Introduction: Inherited hemoglobin
Haemoglobinopathies case studies 11 th Annual Sickle Cell and Thalassaemia Conference October 2017
 Haemoglobinopathies case studies 11 th Annual Sickle Cell and Thalassaemia Conference 11 13 October 2017 Chris Lambert Haematology Service Delivery Manager Viapath Laboratories Kings College Hospital HUMAN
Haemoglobinopathies case studies 11 th Annual Sickle Cell and Thalassaemia Conference 11 13 October 2017 Chris Lambert Haematology Service Delivery Manager Viapath Laboratories Kings College Hospital HUMAN
The Thalassemias in Clinical Practice. Ashutosh Lal, MD Director Comprehensive Thalassemia Program UCSF Benioff Children s Hospital Oakland
 The Thalassemias in Clinical Practice Ashutosh Lal, MD Director Comprehensive Thalassemia Program UCSF Benioff Children s Hospital Oakland Outline Thalassemia: definitions and pathophysiology Epidemiology
The Thalassemias in Clinical Practice Ashutosh Lal, MD Director Comprehensive Thalassemia Program UCSF Benioff Children s Hospital Oakland Outline Thalassemia: definitions and pathophysiology Epidemiology
Article Preimplantation diagnosis and HLA typing for haemoglobin disorders
 RBMOnline - Vol 11. No 3. 2005 362-370 Reproductive BioMedicine Online; www.rbmonline.com/article/1853 on web 20 July 2005 Article Preimplantation diagnosis and HLA typing for haemoglobin disorders Dr
RBMOnline - Vol 11. No 3. 2005 362-370 Reproductive BioMedicine Online; www.rbmonline.com/article/1853 on web 20 July 2005 Article Preimplantation diagnosis and HLA typing for haemoglobin disorders Dr
REVIEWS PHENOTYPE GENOTYPE RELATIONSHIPS IN MONOGENIC DISEASE: LESSONS FROM THE THALASSAEMIAS. D. J. Weatherall
 PHENOTYPE GENOTYPE RELATIONSHIPS IN MONOGENIC DISEASE: LESSONS FROM THE THALASSAEMIAS D. J. Weatherall The remarkable phenotypic diversity of the β-thalassaemias reflects the heterogeneity of mutations
PHENOTYPE GENOTYPE RELATIONSHIPS IN MONOGENIC DISEASE: LESSONS FROM THE THALASSAEMIAS D. J. Weatherall The remarkable phenotypic diversity of the β-thalassaemias reflects the heterogeneity of mutations
Type and frequency of hemoglobinopathies, diagnosed in the area of Karachi, in Pakistan
 HEMATOLOGY RESEARCH ARTICLE Type and frequency of hemoglobinopathies, diagnosed in the area of Karachi, in Pakistan Received: 09 March 2016 Accepted: 09 May 2016 First Published: 13 May 2016 *Corresponding
HEMATOLOGY RESEARCH ARTICLE Type and frequency of hemoglobinopathies, diagnosed in the area of Karachi, in Pakistan Received: 09 March 2016 Accepted: 09 May 2016 First Published: 13 May 2016 *Corresponding
Nuovi strumenti diagnostici : NGS era
 Nuovi strumenti diagnostici : NGS era Achille Iolascon, MD, PhD Dept. Molecular Medicine and Medical Biotechnology, University Federico II, Naples, Italy achille.iolascon@unina.it madre padre figlio neonato
Nuovi strumenti diagnostici : NGS era Achille Iolascon, MD, PhD Dept. Molecular Medicine and Medical Biotechnology, University Federico II, Naples, Italy achille.iolascon@unina.it madre padre figlio neonato
ß-Thalassemia Major: Experience at King Fahad Hofuf Hospital, Al-Hassa, Saudi Arabia
 Case Reports Adewale Ayodele Laditan, FRCP; Mohamed Amin El-Agib, DCH; Saad Al-Naeem, ABDP; Michael Georgeos, CES; Sameera Khabour, DCH From the Departments of Pediatrics (Drs. Laditan, El-Agib, Al-Naeem,
Case Reports Adewale Ayodele Laditan, FRCP; Mohamed Amin El-Agib, DCH; Saad Al-Naeem, ABDP; Michael Georgeos, CES; Sameera Khabour, DCH From the Departments of Pediatrics (Drs. Laditan, El-Agib, Al-Naeem,
S-Beta Thalassemia leading to avascular necrosis of left hip joint in a young male - A rare case report
 Case Report S-Beta Thalassemia leading to avascular necrosis of left hip joint in a young male - A rare case report Shubhi Saxena 1*, Nishant Saxena 1, R.M Jaiswal 2 1 PG Student, 2 Associate Professor,
Case Report S-Beta Thalassemia leading to avascular necrosis of left hip joint in a young male - A rare case report Shubhi Saxena 1*, Nishant Saxena 1, R.M Jaiswal 2 1 PG Student, 2 Associate Professor,
Cover Page. The handle holds various files of this Leiden University dissertation.
 Cover Page The handle http://hdl.handle.net/1887/346 holds various files of this Leiden University dissertation. Author: Hassan, Suha Mustafa Title: Toward prevention of Hemoglobinopathies in Oman Issue
Cover Page The handle http://hdl.handle.net/1887/346 holds various files of this Leiden University dissertation. Author: Hassan, Suha Mustafa Title: Toward prevention of Hemoglobinopathies in Oman Issue
Hypochromic Anaemias
 Hypochromic Anaemias Dr Mere Kende MBBS, MMED (Path), MAACB, MACTM, MACRRM LECTURER-SMHS Anaemia LOW HEMOGLOBIN Anaemia Definition: Hb
Hypochromic Anaemias Dr Mere Kende MBBS, MMED (Path), MAACB, MACTM, MACRRM LECTURER-SMHS Anaemia LOW HEMOGLOBIN Anaemia Definition: Hb
Approach to Hemolysis
 Objectives: Approach to Hemolysis To know the function of platelets and the relationship between the platelet count in peripheral blood and the extent of abnormal bleeding. To know about the diseases associated
Objectives: Approach to Hemolysis To know the function of platelets and the relationship between the platelet count in peripheral blood and the extent of abnormal bleeding. To know about the diseases associated
Research Article Pattern of β-thalassemia and Other Haemoglobinopathies: A Cross-Sectional Study in Bangladesh
 International Scholarly Research Network ISRN Hematology Volume 2012, Article ID 659191, 6 pages doi:10.5402/2012/659191 Research Article Pattern of β-thalassemia and Other Haemoglobinopathies: A Cross-Sectional
International Scholarly Research Network ISRN Hematology Volume 2012, Article ID 659191, 6 pages doi:10.5402/2012/659191 Research Article Pattern of β-thalassemia and Other Haemoglobinopathies: A Cross-Sectional
Evaluation of the Molecular basis of KLF1 Gene in Iranian Thalassemia individuals with borderline hemoglobin A2
 Advances in Bioresearch Adv. Biores., Vol 7 (5) September 2016: 11-15 2016 Society of Education, India Print ISSN 0976-4585; Online ISSN 2277-1573 Journal s URL:http://www.soeagra.com/abr.html CODEN: ABRDC3
Advances in Bioresearch Adv. Biores., Vol 7 (5) September 2016: 11-15 2016 Society of Education, India Print ISSN 0976-4585; Online ISSN 2277-1573 Journal s URL:http://www.soeagra.com/abr.html CODEN: ABRDC3
Haemoglobinophaties EBMT 2011 Data Manager session
 Haemoglobinophaties EBMT 2011 Data Manager session Presentation plan Biological characteristics Clinical characteristics Transplant resuts What is different From transplant in malignancies Between Thalassemia
Haemoglobinophaties EBMT 2011 Data Manager session Presentation plan Biological characteristics Clinical characteristics Transplant resuts What is different From transplant in malignancies Between Thalassemia
G.Madhu Latha et al., Asian Journal of Pharmaceutical Technology & Innovation, 02 (08); 2014; Review Article
; 2014; Review Article") Asian Journal of Pharmaceutical Technology & Innovation ISSN: 2347-8810 Review Article Received on: 29-07-2014 Accepted on: 19-08-2014 Published on: 15-10-2014 A New Era in Thalassemia Disorder: An Overview
Asian Journal of Pharmaceutical Technology & Innovation ISSN: 2347-8810 Review Article Received on: 29-07-2014 Accepted on: 19-08-2014 Published on: 15-10-2014 A New Era in Thalassemia Disorder: An Overview
ijifm case report ABSTRACT
 ijifm case report 1p36 Deletions 10.5005/jp-journals-10016-1085 in Two Cases with Thalassemia 1p36 Deletions in Two Cases with Thalassemia 1 Puspal De, 2 Sudipa Chakravarty, 3 Amit Chakravarty ABSTRACT
ijifm case report 1p36 Deletions 10.5005/jp-journals-10016-1085 in Two Cases with Thalassemia 1p36 Deletions in Two Cases with Thalassemia 1 Puspal De, 2 Sudipa Chakravarty, 3 Amit Chakravarty ABSTRACT
Guideline developed by Shelley Crary, MD, MS,* in collaboration with the ANGELS team. Last reviewed by Shelley Crary, MD, MS, January 19, 2017.
 Microcytic Anemia Guideline developed by Shelley Crary, MD, MS,* in collaboration with the ANGELS team. Last reviewed by Shelley Crary, MD, MS, January 19, 2017. Dr. Crary is a member of the hemophilia
Microcytic Anemia Guideline developed by Shelley Crary, MD, MS,* in collaboration with the ANGELS team. Last reviewed by Shelley Crary, MD, MS, January 19, 2017. Dr. Crary is a member of the hemophilia
Level of Hemoglobin F and G g Gene Expression in Sickle Cell Disease and Their Association with Haplotype and XmnI Polymorphic Site in South of Iran
 IJMS Vol 32, No 4, December 2007 Original Article Level of Hemoglobin F and G g Gene Expression in Sickle Cell Disease and Their Association with Haplotype and XmnI Polymorphic Site in South of Iran Z.
IJMS Vol 32, No 4, December 2007 Original Article Level of Hemoglobin F and G g Gene Expression in Sickle Cell Disease and Their Association with Haplotype and XmnI Polymorphic Site in South of Iran Z.
Chapter 2 Genetic Basis and Genetic Modifiers of β-thalassemia and Sickle Cell Disease
 Chapter 2 Genetic Basis and Genetic Modifiers of β-thalassemia and Sickle Cell Disease Swee Lay Thein Abstract β-thalassemia and sickle cell disease (SCD) are prototypical Mendelian single gene disorders,
Chapter 2 Genetic Basis and Genetic Modifiers of β-thalassemia and Sickle Cell Disease Swee Lay Thein Abstract β-thalassemia and sickle cell disease (SCD) are prototypical Mendelian single gene disorders,
Cancer Incidence and Mortality in the Kingdom of Bahrain Statistics and Trends
 Cancer Incidence and Mortality in the Kingdom of Bahrain Statistics and Trends Mohammed Amin Al Awadhi, MDCM, FRCSC, FRCSI* Najat Mohammed Abulfateh, MD, Arab Board Family Medicine, MSc** Fatema Abu-Hassan,
Cancer Incidence and Mortality in the Kingdom of Bahrain Statistics and Trends Mohammed Amin Al Awadhi, MDCM, FRCSC, FRCSI* Najat Mohammed Abulfateh, MD, Arab Board Family Medicine, MSc** Fatema Abu-Hassan,
Study of distribution of ABO blood groups in ß-thalassemia patients
 International Journal of Research in Medical Sciences Sinha PA et al. Int J Res Med Sci. 2017 Aug;5(8):3479-3483 www.msjonline.org pissn 2320-6071 eissn 2320-6012 Original Research Article DOI: http://dx.doi.org/10.18203/2320-6012.ijrms20173545
International Journal of Research in Medical Sciences Sinha PA et al. Int J Res Med Sci. 2017 Aug;5(8):3479-3483 www.msjonline.org pissn 2320-6071 eissn 2320-6012 Original Research Article DOI: http://dx.doi.org/10.18203/2320-6012.ijrms20173545