Introduction reduction in output alter the amino acid sequence combination
|
|
|
- Aubrey Ward
- 6 years ago
- Views:
Transcription
1 Sickle cell anemia.
2 Introduction Mutations in the globin genes can cause a quantitative reduction in output from that gene or alter the amino acid sequence of the protein produced or a combination of the two..
3 -Quantitative defects cause thalassaemia syndromes, -Qualitative changes, referred to as haemoglobin variants, cause wide range of problems, including sickle cell disease, unstable haemoglobins, decreased oxygen affinity, increased oxygen affinity, and methaemoglobinaemia.
4 the majority of qualitative mutations cause no significant change in haemoglobin properties or clinical problems.
5 Collectively, the clinical syndromes resulting from disorders of haemoglobin synthesis are referred to as Haemoglobinopathies. They can be grouped into three main categories: 1. Those resulting from a genetically determined structural variant of haemoglobin, such as haemoglobin S. 2. Those owing to failure to synthesise one or more of the globin chains of haemoglobin at a normal rate, as in the thalassaemias..
6 3. Those owing to failure to complete the normal neonatal switch from fetal haemoglobin (haemoglobin F) to adult haemoglobin (haemoglobin A). This category comprises a group of disorders referred to as hereditary persistence of fetal haemoglobin (HPFH)
7 An individual can also have a combination of two or more of these abnormalities (e.g. a variant haemoglobin can be synthesised at a reduced rate).
8 Sickle cell disease The term sickle cell disease is used to denote all entities associated with sickling of haemoglobin within red cells.
9 Sickle cell disease is a group of haemoglobin disorders resulting from the inheritance of the sickle β globin gene.
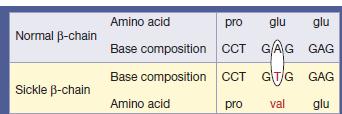
10 The sickle β globin abnormality is caused by substitution of valine for glutamic acid in position 6 in the β chain
11
12 Homozygous sickle cell anaemia (Hb SS) is the most common severe syndrome while the doubly heterozygote conditions of Hb S/C and Hb S/β thal also cause sickling disease.
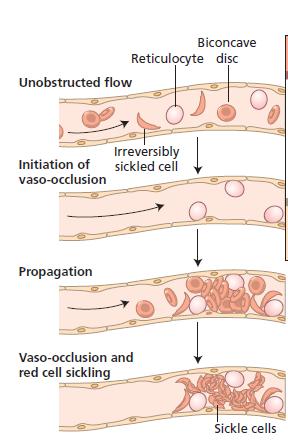
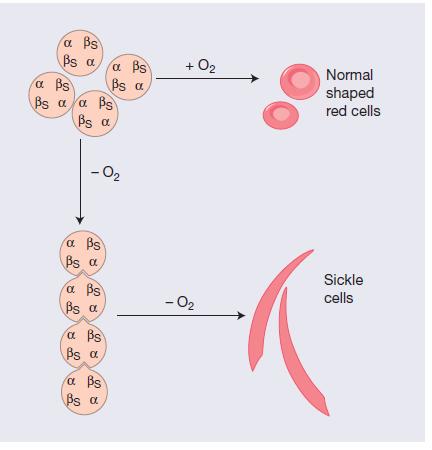
13 Hb S(Hb α2β2 S ) is insoluble and forms crystals when exposed to low oxygen tension. Deoxygenated sickle haemoglobin polymerizes into long fibres, each consisting of seven intertwined double strands with cross linking. The red cells sickle and may block different areas of the microcirculation or large vessels causing infarcts of various organs
14 The polymerization of HbS in the circulating red cells is influenced by the oxygenation status, the intracellular haemoglobin concentration and the presence of non-sickle haemoglobins.
15
16
17 Acidosis and elevated levels of 2,3 diphosphoglycerate (2,3- DPG) promote polymer formation by reducing the oxygen affinity of haemoglobin. The presence of HbA within the red cells, as in sickle trait, inhibits polymerization by diluting HbS. The HbF has an inhibitory effect on polymerization of HbS
18 SCD is characterized by chronic intravascular and extravascular haemolysis. Sickling-induced membrane fragmentation and complement-mediated lysis cause intravascular destruction of red cells. Membrane damage also leads to extravascular haemolysis through entrapment of poorly deformable cells or uptake by macrophages.
19 Individuals with concomitant deletion of one or two α-globin genes, or the Senegal or Arab Indian haplotypes, have higher baseline haemoglobin levels.
20 The carrier state is widespread and is found in up to 30% of West African people,the sickle trait bestows survival benefit in areas endemic for falciparum malaria, and the distribution of SCD historically paralleled this disease. The sickle haemoglobin containing red cells inhibit proliferation of Plasmodium falciparum, and are more likely to become deformed and removed from the circulation.
21 In recent times, the dissemination of the sickle mutation in different areas of the world took place from The movement of populations via trade routes and the slave trade
22 Homozygous disease Clinical features Clinical features are of a severe haemolytic anaemia punctuated by crises. The symptoms of anaemia are often mild in relation to the severity of the anaemia because Hb S gives up oxygen (O2) to tissues relatively easily compared with Hb A (low affinity to oxygen ).
23 The clinical expression of Hb SS is very variable, some patients having an almost normal life, free of crises, but others develop severe crises even as infants and may die in. This is partly due to the effects of inherited modifying factors, such as interaction with α thalassaemia or increased synthesis of haemoglobin F, and partly to socioeconomic conditions and other factors that influence general healthearly childhood or as young adults..
24 Crises may be : 1)vaso occlusive (painful or visceral), 2)aplastic 3) haemolytic. There may be serious damage to many organs
25 Vaso occlusive crises Painful: -These are the most frequent. -They may be sporadic and unpredictable or precipitated by infection, acidosis, dehydration or deoxygenation (e.g. altitude, operations, obstetric delivery, stasis of the circulation, exposure to cold, violent exercise).
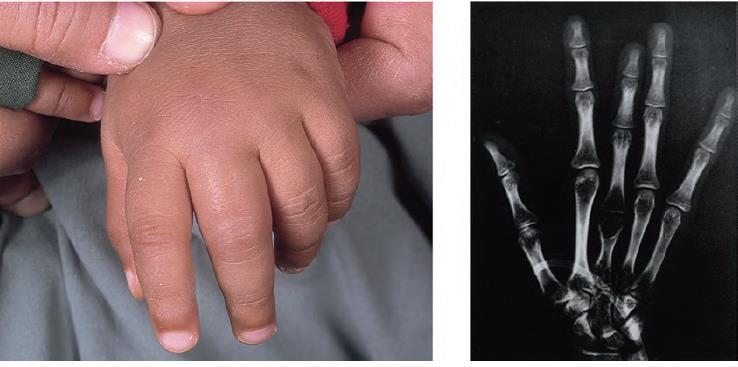
26 -Infarcts causing severe pain occur in the bones (hips, shoulders and vertebrae are commonly affected). -The hand foot syndrome (painful dactylitis caused by infarcts of the small bones) is frequently the first presentation of the disease and may lead to digits of varying lengths
27
28 Visceral These are caused by sickling within organs causing infarction and pooling of blood, often with a severe exacerbation of anaemia.
29 The acute sickle chest syndrome is the most common cause of death both in children and adults. It presents with dyspnoea, falling arterial PO2, chest pain and pulmonary infiltrates on chest X ray..
30 The pathogenesis of acute chest syndrome involves vaso-occlusion, infection or embolization of bone marrow fat...
31 Hypoxia due to acute chest syndrome can result in widespread sickling and vaso-occlusion, with risk of multiorgan failure.
32 --Hepatic and girdle sequestration crises may lead to severe illness requiring exchange transfusions. -Splenic sequestration is typically seen in infants and presents with an enlarging spleen, falling haemoglobin and abdominal pain Attacks tend to be recurrent and splenectomy is often needed..
33 -Priapism and liver and kidney damage due to repeated small infarcts.

34 Aplastic crises These occur as a result of infection with parvovirus or from folate deficiency. They are characterized by a sudden fall in haemoglobin and reticulocytes, usually requiring transfusion.

35 Haemolytic crises These are characterized by an increased rate of haemolysis and fall in haemoglobin but rise in reticulocytes and usually accompany a painful crisis.
36 Other organ damage -The most serious is of the brain or spinal cord. - Stroke, cognitive impairment in children
37 Ulcers of the lower legs are common. Ulcers arise near the medial or lateral malleolus and may be single or multiple. Occlusion of skin microvasculature from sickle red cells predisposes to ulcers, which are made worse by trauma, infection or warm climate.

38 Leg Ulcer
39 The spleen is enlarged in infancy and early childhood but later is often reduced in size as a result of infarcts (autosplenectomy).
40 -Infections are frequent partly due to hyposplenism. -Pneumonia, urinary tract infections and Gram negative septicaemia are common. -Osteomyelitis may also occur, usually from Salmonella spp
41 -Vaso-occlusion of retinal and other vascular beds in the eye can lead to grave complications.
42 Growth and development Children with SCD are born with normal weight, but fall behind other children by the end of the first year. The pubertal growth spurt is delayed by 1 2 years, but the final adult height is normal. Delays also occur in skeletal maturation and onset of puberty, and female patients achieve menarche 1 2 years later than their peers.
43 Pregnancy -The steady-state haemoglobin level falls in SCD during pregnancy, similar to the decline in haemoglobin observed in normal pregnant women. -Folate deficiency can exacerbate the anaemia and supplements should be -provided throughout pregnancy.
44 -Painful episodes become more common in the last trimester. -The incidence of pre-eclampsia is higher than normal in SCD patients and there is a slight increase in maternal mortality. -
45 Risk to the fetus from abortion, stillbirth, low birth weight and neonatal death is also increased.
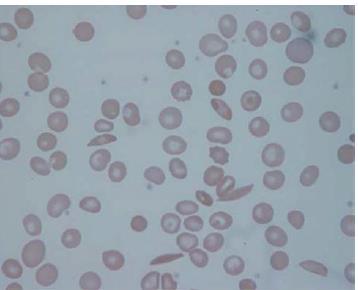
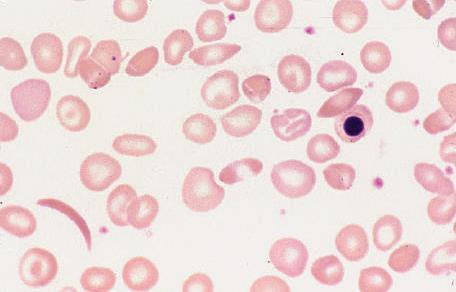
46 Laboratory findings 1 -The haemoglobin is usually g/l low in comparison to mild or no symptoms of anaemia. 2 -Sickle cells and target cells occur in the blood film. Features of splenic atrophy (e.g. Howell Jolly bodies) may also be present. 3- Screening tests for sickling are positive when the blood is deoxygenated..
47 -Sickling of red cells can be induced by sealing a drop of blood under a coverslip to exclude oxygen or by adding 2% sodium metabisulfite -
48 -The solubility test for HbS utilizes a reducing agent such as sodium dithionite, which is added to the haemolysate. Deoxy-HbS is insoluble and renders the solution turbid. Both these tests are unable to distinguish sickle cell trait from sickle cell anaemia and cannot be used for primary diagnosis. They are useful aids in the identification of an abnormal electrophoretic band as HbS and for identifying sickle cell trait in units of red cells prior to transfusion.
49
50

51 Sickle cell solubility test. In this test, whole blood is added to a high phosphate buffer with saponin and sodium dithionite, which causes the hemoglobin to become deoxyhemoglobin. Deoxyhemoglobin S is insoluble. The turbidity of the sample on the left indicates the presence of HbS. The clear sample on the right contains no HbS.
52 4.HbS can be identified by cellulose acetate electrophoresis at ph 8.4.HbD and HbG have the same electrophoretic mobility with this method, but can be distinguished using citrate agar electrophoresis at ph 6.2 or thinlayer isoelectric focusing.
53 HPLC or haemoglobin electrophoresis in Hb SS, no Hb A is detected. The amount of Hb F is variable and is usually 5 15%, larger amounts are usually associated with a milder disorder
54
55
Sickle Cell Disease and impact on the society
 Sickle Cell Disease and impact on the society Professor Z.A.Jeremiah Ph.D, FRCPath (London) Professor of Haematology and Blood Transfusion Science Niger Delta University, Wilberforce Island Outline What
Sickle Cell Disease and impact on the society Professor Z.A.Jeremiah Ph.D, FRCPath (London) Professor of Haematology and Blood Transfusion Science Niger Delta University, Wilberforce Island Outline What
Sickle Cell Disease. Edward Malters, MD
 Sickle Cell Disease Edward Malters, MD Introduction Vaso-occlusive phenomena and hemolysis are the clinical hallmarks of Sickle Cell Disease (SCD) Inherited disorder due to homozygosity for the abnormal
Sickle Cell Disease Edward Malters, MD Introduction Vaso-occlusive phenomena and hemolysis are the clinical hallmarks of Sickle Cell Disease (SCD) Inherited disorder due to homozygosity for the abnormal
Sickle cell disease. Fareed Omar 10 March 2018
 Sickle cell disease Fareed Omar 10 March 2018 Physiology Haemoglobin structure HbA2: 2α and 2δ chains (2-3%) HbF: 2α and 2γ chains (
Sickle cell disease Fareed Omar 10 March 2018 Physiology Haemoglobin structure HbA2: 2α and 2δ chains (2-3%) HbF: 2α and 2γ chains (
Hemolytic anemias (2 of 2)
") Hemolytic anemias (2 of 2) Sickle Cell Anemia The most common familial hemolytic anemia in the world Sickle cell anemia is the prototypical (and most prevalent) hemoglobinopathy Mutation in the β-globin
Hemolytic anemias (2 of 2) Sickle Cell Anemia The most common familial hemolytic anemia in the world Sickle cell anemia is the prototypical (and most prevalent) hemoglobinopathy Mutation in the β-globin
Anemia s. Troy Lund MSMS PhD MD
 Anemia s Troy Lund MSMS PhD MD lundx072@umn.edu Hemoglobinopathy/Anemia IOM take home points. 1. How do we identify the condtion? Smear, CBC Solubility Test (SCD) 2. How does it present clincally? 3. How
Anemia s Troy Lund MSMS PhD MD lundx072@umn.edu Hemoglobinopathy/Anemia IOM take home points. 1. How do we identify the condtion? Smear, CBC Solubility Test (SCD) 2. How does it present clincally? 3. How
HEMOLYTIC ANEMIA DUE TO ABNORMAL HEMOGLOBIN SYNTHESIS
 Hemolytic Anemia Due to Abnormal Hemoglobin Synthesis MODULE 19 HEMOLYTIC ANEMIA DUE TO ABNORMAL HEMOGLOBIN SYNTHESIS 19.1 INTRODUCTION There are two main mechanisms by which anaemia is produced (a) Thalassemia:
Hemolytic Anemia Due to Abnormal Hemoglobin Synthesis MODULE 19 HEMOLYTIC ANEMIA DUE TO ABNORMAL HEMOGLOBIN SYNTHESIS 19.1 INTRODUCTION There are two main mechanisms by which anaemia is produced (a) Thalassemia:
DONE BY : RaSHA RAKAN & Bushra Saleem
 DONE BY : RaSHA RAKAN & Bushra Saleem Hemolytic anemias (2 of 2) Sickle Cell Anemia The most common familial hemolytic anemia in the world Sickle cell anemia is the prototypical (and most prevalent) hemoglobinopathy
DONE BY : RaSHA RAKAN & Bushra Saleem Hemolytic anemias (2 of 2) Sickle Cell Anemia The most common familial hemolytic anemia in the world Sickle cell anemia is the prototypical (and most prevalent) hemoglobinopathy
SICKLE CELL DISEASE. Dr. MUBARAK ABDELRAHMAN MD PEDIATRICS AND CHILD HEALTH. Assistant Professor FACULTY OF MEDICINE -JAZAN
 SICKLE CELL DISEASE Dr. MUBARAK ABDELRAHMAN MD PEDIATRICS AND CHILD HEALTH Assistant Professor FACULTY OF MEDICINE -JAZAN Objective: The student should be able: To identify the presentation, diagnosis,
SICKLE CELL DISEASE Dr. MUBARAK ABDELRAHMAN MD PEDIATRICS AND CHILD HEALTH Assistant Professor FACULTY OF MEDICINE -JAZAN Objective: The student should be able: To identify the presentation, diagnosis,
Anaemia in Pregnancy
 Anaemia in Pregnancy Definition :anaemia is a pathological condition in which the oxygen-carrying capacity of red blood cells is insufficient to meet the body needs. The WHO : haemoglobin concentration
Anaemia in Pregnancy Definition :anaemia is a pathological condition in which the oxygen-carrying capacity of red blood cells is insufficient to meet the body needs. The WHO : haemoglobin concentration
Approach to Hemolysis
 Objectives: Approach to Hemolysis To know the function of platelets and the relationship between the platelet count in peripheral blood and the extent of abnormal bleeding. To know about the diseases associated
Objectives: Approach to Hemolysis To know the function of platelets and the relationship between the platelet count in peripheral blood and the extent of abnormal bleeding. To know about the diseases associated
RBCs Disorders 1. Dr. Nabila Hamdi MD, PhD
 RBCs Disorders 1 Dr. Nabila Hamdi MD, PhD ILOs Discuss the classification of anemia into hypochromic-microcytic, normochromicnormocytic and macrocytic. Categorize laboratory test procedures used in the
RBCs Disorders 1 Dr. Nabila Hamdi MD, PhD ILOs Discuss the classification of anemia into hypochromic-microcytic, normochromicnormocytic and macrocytic. Categorize laboratory test procedures used in the
Congenital Haemoglobinopathies
 Congenital Haemoglobinopathies L. DEDEKEN, MD H O P I T A L U N I V E R S I T A I R E D E S E N F A N T S R E I N E F A B I O L A U N I V E R S I T E L I B R E DE B R U X E L L E S Red Blood Cell Disorders
Congenital Haemoglobinopathies L. DEDEKEN, MD H O P I T A L U N I V E R S I T A I R E D E S E N F A N T S R E I N E F A B I O L A U N I V E R S I T E L I B R E DE B R U X E L L E S Red Blood Cell Disorders
Screening for haemoglobinopathies in pregnancy
 Policy Statement All Southern Health patients will receive clinical care that reflects best practice and is based on the best available evidence. Index of chapters within background 1. Prevalence of haemoglobinopathies
Policy Statement All Southern Health patients will receive clinical care that reflects best practice and is based on the best available evidence. Index of chapters within background 1. Prevalence of haemoglobinopathies
HAEMOLYTIC ANAEMIA. Dr. Hasan Fahmawi, MRCP(London), FRCP(Edin) Consultant Physician
, FRCP(Edin) Consultant Physician") HAEMOLYTIC ANAEMIA Dr. Hasan Fahmawi, MRCP(London), FRCP(Edin) Consultant Physician Haemolysis Definition shortening of the normal red blood lifespan of 120 days Increase in unconjugated bilirubin, increased
HAEMOLYTIC ANAEMIA Dr. Hasan Fahmawi, MRCP(London), FRCP(Edin) Consultant Physician Haemolysis Definition shortening of the normal red blood lifespan of 120 days Increase in unconjugated bilirubin, increased
In adults, the predominant Hb (HbA) molecule has four chains: two α and two β chains. In thalassemias, the synthesis of either the α or the β chains
 molecule has four chains: two α and two β chains. In thalassemias, the synthesis of either the α or the β chains") Thalassaemias Thalassemia Thalassemia is an inherited autosomal recessive blood disease. Associated with absence or reduction in a or b globin chains. Reduced synthesis of one of the globin chains can
Thalassaemias Thalassemia Thalassemia is an inherited autosomal recessive blood disease. Associated with absence or reduction in a or b globin chains. Reduced synthesis of one of the globin chains can
Dr Banu Kaya Consultant Haematologist Barts Health NHS Trust Royal London Hospital, London, UK SICKLE CELL AND THALASSAEMIA OVERVIEW
 Dr Banu Kaya Consultant Haematologist Barts Health NHS Trust Royal London Hospital, London, UK SICKLE CELL AND THALASSAEMIA OVERVIEW Objectives Gain awareness of haemoglobinopathy inheritance, pathophysiology
Dr Banu Kaya Consultant Haematologist Barts Health NHS Trust Royal London Hospital, London, UK SICKLE CELL AND THALASSAEMIA OVERVIEW Objectives Gain awareness of haemoglobinopathy inheritance, pathophysiology
Haemoglobinopathies case studies 11 th Annual Sickle Cell and Thalassaemia Conference October 2017
 Haemoglobinopathies case studies 11 th Annual Sickle Cell and Thalassaemia Conference 11 13 October 2017 Chris Lambert Haematology Service Delivery Manager Viapath Laboratories Kings College Hospital HUMAN
Haemoglobinopathies case studies 11 th Annual Sickle Cell and Thalassaemia Conference 11 13 October 2017 Chris Lambert Haematology Service Delivery Manager Viapath Laboratories Kings College Hospital HUMAN
Extra Notes 3. Warm. In the core (center) of the body, where the temperature is 37 C.
 of the body, where the temperature is 37 C.") Extra Notes 3 *The numbers of the slides are according to the last year slides. Slide 33 Autoimmune hemolytic anemia : Abnormal circulating antibodies that target normal antigen on the RBC and cause lysis.
Extra Notes 3 *The numbers of the slides are according to the last year slides. Slide 33 Autoimmune hemolytic anemia : Abnormal circulating antibodies that target normal antigen on the RBC and cause lysis.
- Ensherah Mokheemer. - Rama Nada. - Tareq Aladily. 1 P a g e
 -3 - Ensherah Mokheemer - Rama Nada - Tareq Aladily 1 P a g e In this lecture we will continue talking about autoimmune hemolytic anemia. Autoimmune hemolytic anemia - There are several types that shares
-3 - Ensherah Mokheemer - Rama Nada - Tareq Aladily 1 P a g e In this lecture we will continue talking about autoimmune hemolytic anemia. Autoimmune hemolytic anemia - There are several types that shares
HEMOGLOBIN ELECTROPHORESIS DR ARASH ALGHASI SHAFA HOSPITAL-AHWAZ
 HEMOGLOBIN ELECTROPHORESIS DR ARASH ALGHASI SHAFA HOSPITAL-AHWAZ Hemoglobin Hemoglobin (Hb), protein constituting 1/3 of the red blood cells Each red cell has 640 million molecules of Hb sites in the cells:
HEMOGLOBIN ELECTROPHORESIS DR ARASH ALGHASI SHAFA HOSPITAL-AHWAZ Hemoglobin Hemoglobin (Hb), protein constituting 1/3 of the red blood cells Each red cell has 640 million molecules of Hb sites in the cells:
Done by :Aseel Twaijer & Laith Sorour Hemolytic Anemias
 Hemolytic Anemias Hemolytic anemias share the following features: - A shortened red cell life < 120 days - Elevated erythropoietin levels (compensatory increase in erythropoiesis) - Accumulation of hemoglobin
Hemolytic Anemias Hemolytic anemias share the following features: - A shortened red cell life < 120 days - Elevated erythropoietin levels (compensatory increase in erythropoiesis) - Accumulation of hemoglobin
4 Fahed Al Karmi Sufian Alhafez Dr nayef karadsheh
 4 Fahed Al Karmi Sufian Alhafez Dr nayef karadsheh Genetic variants of hemoglobin Hemoglobinopathies (abnormal variants of hemoglobin) are divided into: 1. Structural abnormalities: Any change in the genes
4 Fahed Al Karmi Sufian Alhafez Dr nayef karadsheh Genetic variants of hemoglobin Hemoglobinopathies (abnormal variants of hemoglobin) are divided into: 1. Structural abnormalities: Any change in the genes
Putting some hematology into Pediatric Hematology/Oncology: a review of Hemophilia and Sickle Cell Disease in the Pediatric Patient
 Putting some hematology into Pediatric Hematology/Oncology: a review of Hemophilia and Sickle Cell Disease in the Pediatric Patient Kristina Haley, DO March 10, 2012 Jovita Reyes Memorial Pediatric Hematology/Oncology
Putting some hematology into Pediatric Hematology/Oncology: a review of Hemophilia and Sickle Cell Disease in the Pediatric Patient Kristina Haley, DO March 10, 2012 Jovita Reyes Memorial Pediatric Hematology/Oncology
4 Jumana Jihad Dr. Ahmad Mansour Dr. Ahmad Mansour
 4 Jumana Jihad Dr. Ahmad Mansour Dr. Ahmad Mansour Anemia Decreased blood production Increased blood loss Hemolytic Hemorrhage Extravascular Intravascular Hemolytic (Further classification( Extrinsic Intrinsic
4 Jumana Jihad Dr. Ahmad Mansour Dr. Ahmad Mansour Anemia Decreased blood production Increased blood loss Hemolytic Hemorrhage Extravascular Intravascular Hemolytic (Further classification( Extrinsic Intrinsic
Year 2004 Paper two: Questions supplied by Megan 1
 Year 2004 Paper two: Questions supplied by Megan 1 QUESTION 93 A 16yo adolescent male presents with lethargy and lower respiratory tract infection. Physical examination shows him to be febrile, icteric
Year 2004 Paper two: Questions supplied by Megan 1 QUESTION 93 A 16yo adolescent male presents with lethargy and lower respiratory tract infection. Physical examination shows him to be febrile, icteric
Haemoglobin BY: MUHAMMAD RADWAN WISSAM MUHAMMAD
 Haemoglobin BY: MUHAMMAD RADWAN WISSAM MUHAMMAD Introduction is the iron-containing oxygen transport metalloprotein in the red blood cells Hemoglobin in the blood carries oxygen from the respiratory organs
Haemoglobin BY: MUHAMMAD RADWAN WISSAM MUHAMMAD Introduction is the iron-containing oxygen transport metalloprotein in the red blood cells Hemoglobin in the blood carries oxygen from the respiratory organs
Dr.Abdolreza Afrasiabi
 Dr.Abdolreza Afrasiabi Thalassemia & Heamophilia Genetic Reaserch Center Shiraz Medical University Hemoglobin tetramer Hemoglobin Structure % A 1 α 2 β 2 94-97% A 2 α 2 δ 2 2.5% A 1C α 2 (β-n-glucose)
Dr.Abdolreza Afrasiabi Thalassemia & Heamophilia Genetic Reaserch Center Shiraz Medical University Hemoglobin tetramer Hemoglobin Structure % A 1 α 2 β 2 94-97% A 2 α 2 δ 2 2.5% A 1C α 2 (β-n-glucose)
HbSC disease is it different and how should we manage it? David Rees Department of Paediatric Haematology, King s College Hospital, London
 HbSC disease is it different and how should we manage it? David Rees Department of Paediatric Haematology, King s College Hospital, London Different types of sickle cell disesease Severe sickle cell disease
HbSC disease is it different and how should we manage it? David Rees Department of Paediatric Haematology, King s College Hospital, London Different types of sickle cell disesease Severe sickle cell disease
Genetic Modifiers of Sickle Cell Disease Severity. Kunle Adekile, MD, PhD Professor Department of Pediatrics Kuwait University
 Genetic Modifiers of Sickle Cell Disease Severity Kunle Adekile, MD, PhD Professor Department of Pediatrics Kuwait University Outline Hb Molecule and Genetic control of globin synthesis Pathophysiology
Genetic Modifiers of Sickle Cell Disease Severity Kunle Adekile, MD, PhD Professor Department of Pediatrics Kuwait University Outline Hb Molecule and Genetic control of globin synthesis Pathophysiology
Hemoglobin and anemia BCH 471
 Hemoglobin and anemia BCH 471 OBJECTIVES Quantitative determination of hemoglobin in a blood sample. Hemoglobin structure Hemoglobin (Hb) is a porphyrin iron (II) protein in RBCs that transport oxygen
Hemoglobin and anemia BCH 471 OBJECTIVES Quantitative determination of hemoglobin in a blood sample. Hemoglobin structure Hemoglobin (Hb) is a porphyrin iron (II) protein in RBCs that transport oxygen
Investigation of sickle cell disease
 Investigation of sickle cell disease Sickle cell disease Sickle cell disease is an inherited disorder which affects people from Africa, India, Middle East and the Mediterranean region The main forms of
Investigation of sickle cell disease Sickle cell disease Sickle cell disease is an inherited disorder which affects people from Africa, India, Middle East and the Mediterranean region The main forms of
Medical and Surgical Complications of Sickle Cell Anemia
 Medical and Surgical Complications of Sickle Cell Anemia Ahmed Al-Salem Medical and Surgical Complications of Sickle Cell Anemia Ahmed Al-Salem Department of Surgery Dar A lalafia Medical Company Qatif
Medical and Surgical Complications of Sickle Cell Anemia Ahmed Al-Salem Medical and Surgical Complications of Sickle Cell Anemia Ahmed Al-Salem Department of Surgery Dar A lalafia Medical Company Qatif
Interleukin-1ß and Interleukin-6 Genetic Polymorphisms and Sickle Cell Disease: An Egyptian Study
 Interleukin-1ß and Interleukin-6 Genetic Polymorphisms and Sickle Cell Disease: An Egyptian Study MONA EL-GHAMRAWY, MD, PROFESSOR OF PEDIATRICS & PEDIATRIC HEMATOLOGY, CAIRO UNIVERSITY melghamrawy95@gmail.com
Interleukin-1ß and Interleukin-6 Genetic Polymorphisms and Sickle Cell Disease: An Egyptian Study MONA EL-GHAMRAWY, MD, PROFESSOR OF PEDIATRICS & PEDIATRIC HEMATOLOGY, CAIRO UNIVERSITY melghamrawy95@gmail.com
Chem*3560 Lecture 4: Inherited modifications in hemoglobin
 Chem*3560 Lecture 4: Inherited modifications in hemoglobin Genetic modifications fall into two classes: Thalassemias, which are the result of failure to express globin genes. Thalassa is Greek for the
Chem*3560 Lecture 4: Inherited modifications in hemoglobin Genetic modifications fall into two classes: Thalassemias, which are the result of failure to express globin genes. Thalassa is Greek for the
Hemoglobinopathies Diagnosis and management
 Hemoglobinopathies Diagnosis and management Morgan L. McLemore, M.D. Hematology/Leukemia Department of Hematology and Oncology Winship Cancer Institute at Emory University mlmclem@emory.edu Disclosures
Hemoglobinopathies Diagnosis and management Morgan L. McLemore, M.D. Hematology/Leukemia Department of Hematology and Oncology Winship Cancer Institute at Emory University mlmclem@emory.edu Disclosures
The Distribution of Human Differences. If all this genetic variation is so recent and continuous, why do we think of it in categorical terms?
 Expansion Routes of Homo sapiens ~40-25,000 b.p. The Distribution of Human Differences ~120-100,000 b.p. ~50-40,000 b.p. ~20-15,000 b.p. - - - Coastal Route Circa 10-3,500 b.p. If all this genetic variation
Expansion Routes of Homo sapiens ~40-25,000 b.p. The Distribution of Human Differences ~120-100,000 b.p. ~50-40,000 b.p. ~20-15,000 b.p. - - - Coastal Route Circa 10-3,500 b.p. If all this genetic variation
The Distribution of Human Differences. If all this genetic variation is so recent and continuous, why do we think of it in categorical terms?
 Expansion Routes of Homo sapiens ~40-25,000 b.p. The Distribution of Human Differences ~120-100,000 b.p. ~50-40,000 b.p. ~20-15,000 b.p. - - - Coastal Route Circa 10-3,500 b.p. If all this genetic variation
Expansion Routes of Homo sapiens ~40-25,000 b.p. The Distribution of Human Differences ~120-100,000 b.p. ~50-40,000 b.p. ~20-15,000 b.p. - - - Coastal Route Circa 10-3,500 b.p. If all this genetic variation
High Hemoglobin F in a Saudi Child Presenting with Pancytopenia
 Case Report imedpub Journals http://www.imedpub.com Journal of Pediatric Care ISSN 2471-805X DOI: 10.21767/2471-805X.100002 High Hemoglobin F in a Saudi Child Presenting with Pancytopenia Abstract Saudi
Case Report imedpub Journals http://www.imedpub.com Journal of Pediatric Care ISSN 2471-805X DOI: 10.21767/2471-805X.100002 High Hemoglobin F in a Saudi Child Presenting with Pancytopenia Abstract Saudi
HEMOGLOBINOPATHIES LECTURE OUTLINE. An overview of the structure of hemoglobin. Different types of hemoglobin. Definition of hemoglobinopathies
 Slide 1 HEOGLOBINOPATHIES Slide 2 LETURE OUTLINE An overview of the structure of hemoglobin. Different types of hemoglobin. Definition of hemoglobinopathies Sickle ell Disease and Hemoglobin Slide 3 HEOGLOBIN
Slide 1 HEOGLOBINOPATHIES Slide 2 LETURE OUTLINE An overview of the structure of hemoglobin. Different types of hemoglobin. Definition of hemoglobinopathies Sickle ell Disease and Hemoglobin Slide 3 HEOGLOBIN
Batool Emad. Marah Karablieh. - Nayef Karadsheh
 4 4 1 P a g e Batool Emad Marah Karablieh - Nayef Karadsheh ***Topics that will be discussed in this Lecture: 1) Globin gene organization 2) Hemoglobinopathies 3) HbS (sickle cell disease) 4) HbC and HbSC
4 4 1 P a g e Batool Emad Marah Karablieh - Nayef Karadsheh ***Topics that will be discussed in this Lecture: 1) Globin gene organization 2) Hemoglobinopathies 3) HbS (sickle cell disease) 4) HbC and HbSC
RBCs Disorders 2. Dr. Nabila Hamdi MD, PhD
 RBCs Disorders 2 Dr. Nabila Hamdi MD, PhD ILOs Discuss the classification of anemia into hypochromic-microcytic, normochromicnormocytic and macrocytic. Categorize laboratory test procedures used in the
RBCs Disorders 2 Dr. Nabila Hamdi MD, PhD ILOs Discuss the classification of anemia into hypochromic-microcytic, normochromicnormocytic and macrocytic. Categorize laboratory test procedures used in the
Sickle Cell Anemia. Sickle cell anemia is an inherited disorder of the blood which occurs when just one base pair substitution
 Rose Farrington and Rachel Nash BIOL 362 Lab M. Bulgarella Genetic Diseases 10/14/2008 Sickle Cell Anemia Introduction Sickle cell anemia is an inherited disorder of the blood which occurs when just one
Rose Farrington and Rachel Nash BIOL 362 Lab M. Bulgarella Genetic Diseases 10/14/2008 Sickle Cell Anemia Introduction Sickle cell anemia is an inherited disorder of the blood which occurs when just one
Haemoglobinopathy and sickle cell disease
 Key points Sickle cell disease (SCD) is a congenital haemoglobinopathy characterized by a mutation on chromosome 11, resulting in the production of the unstable and relatively insoluble haemoglobin S.
Key points Sickle cell disease (SCD) is a congenital haemoglobinopathy characterized by a mutation on chromosome 11, resulting in the production of the unstable and relatively insoluble haemoglobin S.
Hemoglobinopathies NORMAL HEMOGLOBINS
 Hemoglobinopathies Millicent Sutton MD October 28, 2005 NORMAL HEMOGLOBINS Consist of 2 alpha chains and 2 non alpha chains Hb A = α2β2 Hb F= α 2γ2 Hb A2 = α2δ2 1 Hemoglobin Variants Altered the conformational
Hemoglobinopathies Millicent Sutton MD October 28, 2005 NORMAL HEMOGLOBINS Consist of 2 alpha chains and 2 non alpha chains Hb A = α2β2 Hb F= α 2γ2 Hb A2 = α2δ2 1 Hemoglobin Variants Altered the conformational
The unstable haemoglobins and haemolysis. Name Robin Carrell Haematology Department, University of Cambridge.
 The unstable haemoglobins and haemolysis Name Robin Carrell Haematology Department, University of Cambridge. The unstable haemoglobins and haemolysis 1966-2016 : and the lessons they have taught us Köln
The unstable haemoglobins and haemolysis Name Robin Carrell Haematology Department, University of Cambridge. The unstable haemoglobins and haemolysis 1966-2016 : and the lessons they have taught us Köln
RBCs Disorders 2. Dr. Nabila Hamdi MD, PhD
 RBCs Disorders 2 Dr. Nabila Hamdi MD, PhD ILOs Discuss the classification of anemia into hypochromic-microcytic, normochromicnormocytic and macrocytic. Categorize laboratory test procedures used in the
RBCs Disorders 2 Dr. Nabila Hamdi MD, PhD ILOs Discuss the classification of anemia into hypochromic-microcytic, normochromicnormocytic and macrocytic. Categorize laboratory test procedures used in the
How to Write a Life Care Plan for a Child with Hemoglobinopathy
 How to Write a Life Care Plan for a Child with Hemoglobinopathy Tamar Fleischer, BSN, MSN, CNLCP & Mona Yudkoff, RN, MPH, CRRN, CNLCP BalaCare Solutions March 2018 St. Peterburg, Florida What is Hemoglobinopathy?
How to Write a Life Care Plan for a Child with Hemoglobinopathy Tamar Fleischer, BSN, MSN, CNLCP & Mona Yudkoff, RN, MPH, CRRN, CNLCP BalaCare Solutions March 2018 St. Peterburg, Florida What is Hemoglobinopathy?
HPLC profile of sickle cell disease in central India
 Original Research Article HPLC profile of sickle cell disease in central India Shweta P. Bijwe * Department of Pathology, IGGMC, Nagpur, Maharashtra, India * Corresponding author email: dr.shwetabijwe@gmail.com
Original Research Article HPLC profile of sickle cell disease in central India Shweta P. Bijwe * Department of Pathology, IGGMC, Nagpur, Maharashtra, India * Corresponding author email: dr.shwetabijwe@gmail.com
Genetics of Thalassemia
 Genetics of Thalassemia Submitted by : Raya Samir Al- Hayaly Sura Zuhair Salih Saad Ghassan Al- Dulaimy Saad Farouq Kassir Sama Naal Salouha Zahraa Jasim Al- Aarajy Supervised by : Dr. Kawkab Adris Mahmod
Genetics of Thalassemia Submitted by : Raya Samir Al- Hayaly Sura Zuhair Salih Saad Ghassan Al- Dulaimy Saad Farouq Kassir Sama Naal Salouha Zahraa Jasim Al- Aarajy Supervised by : Dr. Kawkab Adris Mahmod
George R. Honig Junius G. Adams III. Human Hemoglobin. Genetics. Springer-Verlag Wien New York
 George R. Honig Junius G. Adams III Human Hemoglobin Genetics Springer-Verlag Wien New York George R. Honig, M.D., Ph.D. Professor and Head Department of Pediatrics, College of Medicine University of Illinois
George R. Honig Junius G. Adams III Human Hemoglobin Genetics Springer-Verlag Wien New York George R. Honig, M.D., Ph.D. Professor and Head Department of Pediatrics, College of Medicine University of Illinois
HAEMATOLOGY CASE STUDIES SICKLE CELL SS
 OPEN EDUCATIONAL RESOURCES @ DE MONTFORT UNIVERSITY, Leicester UK HAEMATOLOGY CASE STUDIES SICKLE CELL SS Here are a series of diagnostic case studies comparing normal neonate and adult haematology results
OPEN EDUCATIONAL RESOURCES @ DE MONTFORT UNIVERSITY, Leicester UK HAEMATOLOGY CASE STUDIES SICKLE CELL SS Here are a series of diagnostic case studies comparing normal neonate and adult haematology results
General Characterisctics
 Anemia General Characterisctics Definition: anemia is a decrease in red blood cells. Happens due to underproduction, increased destruction or loss of red cells. Diagnosis of anemia: Hgb < 135 (men) Hgb
Anemia General Characterisctics Definition: anemia is a decrease in red blood cells. Happens due to underproduction, increased destruction or loss of red cells. Diagnosis of anemia: Hgb < 135 (men) Hgb
SICKLE CELL AWARENESS. The Sickle Cell Society has produced the following information leaflets available at sicklecellsociety.org
 sickle cell disease in the UK Sickle cell disease (SCD) affects around 15,000 people in the UK People with Sickle Cell Disease have Sickle haemoglobin (HbS) which can make red blood cells rigid and sickle-shaped
sickle cell disease in the UK Sickle cell disease (SCD) affects around 15,000 people in the UK People with Sickle Cell Disease have Sickle haemoglobin (HbS) which can make red blood cells rigid and sickle-shaped
Quiz. What percentage of the world s population is a carrier of a hemoglobinopathy? Hemoglobinopathies in Pregnancy 1-2% 5-7% 8-12% 10-15%
 Hemoglobinopathies in Pregnancy Emily Parkhurst, MS, LCGC Kaiser West Los Angeles November 2017 Genetics Department Quiz What percentage of the world s population is a carrier of a hemoglobinopathy? 1-2%
Hemoglobinopathies in Pregnancy Emily Parkhurst, MS, LCGC Kaiser West Los Angeles November 2017 Genetics Department Quiz What percentage of the world s population is a carrier of a hemoglobinopathy? 1-2%
THE UNIVERSITY OF JORDAN FACULTY OF MEDICINE DEPARTMENT OF PATHOLOGY HEMOLYTIC ANEMIAS. Dr. Tariq Aladily
 THE UNIVERSITY OF JORDAN FACULTY OF MEDICINE DEPARTMENT OF PATHOLOGY HEMOLYTIC ANEMIAS Third year medical students First semester Faculty 2018/2019 of Medicine Hereditary Spherocytosis Intrinsic defects
THE UNIVERSITY OF JORDAN FACULTY OF MEDICINE DEPARTMENT OF PATHOLOGY HEMOLYTIC ANEMIAS Third year medical students First semester Faculty 2018/2019 of Medicine Hereditary Spherocytosis Intrinsic defects
The anaesthetic management of children with sickle cell disease
 The anaesthetic management of children with sickle cell disease Based in part upon Locke, C. Anaesthetic management of sickle cell disease in children. Anaesthesia Tutorial of the Week 153 (2009). Tanya
The anaesthetic management of children with sickle cell disease Based in part upon Locke, C. Anaesthetic management of sickle cell disease in children. Anaesthesia Tutorial of the Week 153 (2009). Tanya
A Counseling Guide for Sickle Cell and Other Hemoglobin Variants
 A Counseling Guide for Sickle Cell and Other Hemoglobin Variants The Virginia Sickle Cell Awareness Program Virginia Department of Health Division of Women s and Infant s Health 109 Governor Street Richmond,
A Counseling Guide for Sickle Cell and Other Hemoglobin Variants The Virginia Sickle Cell Awareness Program Virginia Department of Health Division of Women s and Infant s Health 109 Governor Street Richmond,
High Prevalence of Sickle Haemoglobin in Mehra Caste of District Betul, Madhya Pradesh
 High Prevalence of Sickle Haemoglobin in Mehra Caste of District Betul, Madhya Pradesh R.B. Gupta, Subhash Godbole, Rajiv Yadav, M.P.S.S. Singh, Ujwala Das, V.S. Gadge, Ashok Gupta, Anil Gwal, C.P. Vishwakarma
High Prevalence of Sickle Haemoglobin in Mehra Caste of District Betul, Madhya Pradesh R.B. Gupta, Subhash Godbole, Rajiv Yadav, M.P.S.S. Singh, Ujwala Das, V.S. Gadge, Ashok Gupta, Anil Gwal, C.P. Vishwakarma
Epidemiology, Care and Prevention of Hemoglobinopathies
 Epidemiology, Care and Prevention of Hemoglobinopathies Nasir Al-Allawi MBChB, PhD. Professor of Hematology College of Medicine University of Dohuk, IRAQ From Research to Practice Training Course in Sexual
Epidemiology, Care and Prevention of Hemoglobinopathies Nasir Al-Allawi MBChB, PhD. Professor of Hematology College of Medicine University of Dohuk, IRAQ From Research to Practice Training Course in Sexual
Rationale for RBC Transfusion in SCD
 Rationale for RBC Transfusion in SCD Dilution of HgbS-containing RBCs via the addition of HgbA-containing cells from the blood of normal donors Suppression of erythropoietin release caused by the rise
Rationale for RBC Transfusion in SCD Dilution of HgbS-containing RBCs via the addition of HgbA-containing cells from the blood of normal donors Suppression of erythropoietin release caused by the rise
Genetic Modulation on the Phenotypic Diversity of Sickle Cell Disease
 Genetic Modulation on the Phenotypic Diversity of Sickle Cell Disease Malay B. Mukherjee Abstract Sickle cell hemoglobin is a β chain structural variant where valine is substituted for glutamic acid in
Genetic Modulation on the Phenotypic Diversity of Sickle Cell Disease Malay B. Mukherjee Abstract Sickle cell hemoglobin is a β chain structural variant where valine is substituted for glutamic acid in
 HbF
HbF Biology 2C03: Genetics What is a Gene?
 Biology 2C03: Genetics What is a Gene? September 9 th, 2013 Model Organisms - E. coli - Yeast - Worms - Arabodopsis - Fruitflie - Mouse What is a Gene? - Define, recognize, describe and apply Mendel s
Biology 2C03: Genetics What is a Gene? September 9 th, 2013 Model Organisms - E. coli - Yeast - Worms - Arabodopsis - Fruitflie - Mouse What is a Gene? - Define, recognize, describe and apply Mendel s
Voxelotor for sickle cell disease
 NIHR Innovation Observatory Evidence Briefing: November 2017 Voxelotor for sickle cell disease NIHRIO (HSRIC) ID: 10748 NICE ID: 9700 LAY SUMMARY Sickle cell disease (SCD) describes a group of inherited
NIHR Innovation Observatory Evidence Briefing: November 2017 Voxelotor for sickle cell disease NIHRIO (HSRIC) ID: 10748 NICE ID: 9700 LAY SUMMARY Sickle cell disease (SCD) describes a group of inherited
Chapter 3 Diseases of the Blood and Bloodforming Organs and Certain Disorders Involving the Immune Mechanism D50-D89
 Chapter 3 Diseases of the Blood and Bloodforming Organs and Certain Disorders Involving the Immune Mechanism D50-D89 Presented by Jennifer Kurkulonis 1 FOUR MAJOR TYPES OF BLOOD CELLS White blood cells
Chapter 3 Diseases of the Blood and Bloodforming Organs and Certain Disorders Involving the Immune Mechanism D50-D89 Presented by Jennifer Kurkulonis 1 FOUR MAJOR TYPES OF BLOOD CELLS White blood cells
Instructor s Manual Chapter 26 Hematological Alterations. 1. A man and woman both test positive for the sickle cell trait. The couple asks the
 1 Instructor s Manual Chapter 26 Hematological Alterations Answers to Study Questions 1. A man and woman both test positive for the sickle cell trait. The couple asks the nurse how many of their children
1 Instructor s Manual Chapter 26 Hematological Alterations Answers to Study Questions 1. A man and woman both test positive for the sickle cell trait. The couple asks the nurse how many of their children
Tenth Visit posttest
 Test Code 10C Patient s name: Tenth Visit posttest Patient s birth date: Your name and relationship to patient: Today s date: 1. Which one of the medications listed below should every child with a sickle
Test Code 10C Patient s name: Tenth Visit posttest Patient s birth date: Your name and relationship to patient: Today s date: 1. Which one of the medications listed below should every child with a sickle
270,000,000 hemoglobin units are. hemoglobin has 4 heme units; 2 α and 2 β units. Active site of a heme unit has an Iron ion
 RBC strange shape a biconcave disc that is round and flat RBC has no nucleus. The nucleus is extruded from the cell as it matures. An RBC can change shape to an amazing extent, without breaking, as it
RBC strange shape a biconcave disc that is round and flat RBC has no nucleus. The nucleus is extruded from the cell as it matures. An RBC can change shape to an amazing extent, without breaking, as it
Death due to sickle cell anemia: autopsy diagnosis
 International Journal of Research in Medical Sciences Dhawle MS et al. Int J Res Med Sci. 2017 Jul;5(7):3185-3189 www.msjonline.org pissn 2320-6071 eissn 2320-6012 Original Research Article DOI: http://dx.doi.org/10.18203/2320-6012.ijrms20173010
International Journal of Research in Medical Sciences Dhawle MS et al. Int J Res Med Sci. 2017 Jul;5(7):3185-3189 www.msjonline.org pissn 2320-6071 eissn 2320-6012 Original Research Article DOI: http://dx.doi.org/10.18203/2320-6012.ijrms20173010
SICKLE CELL BROCHURE
 SICKLE CELL BROCHURE SICKLE CELL DIESEASE According to CDC, Sickle cell disease (SCD) is a group of inherited red blood cell disorders. Healthy red blood cells are round and SCD C -shaped farm tool called
SICKLE CELL BROCHURE SICKLE CELL DIESEASE According to CDC, Sickle cell disease (SCD) is a group of inherited red blood cell disorders. Healthy red blood cells are round and SCD C -shaped farm tool called
Transfusion in Sickle Cell Disease What the guidelines [are likely to] say. Dr Bernard Davis Whittington Hospital, London
![Transfusion in Sickle Cell Disease What the guidelines [are likely to] say. Dr Bernard Davis Whittington Hospital, London](/thumbs/85/91702350.jpg "Transfusion in Sickle Cell Disease What the guidelines [are likely to] say. Dr Bernard Davis Whittington Hospital, London") Transfusion in Sickle Cell Disease What the guidelines [are likely to] say Dr Bernard Davis Whittington Hospital, London Background to BCSH Guideline Rationale Current guidance in disparate publications
Transfusion in Sickle Cell Disease What the guidelines [are likely to] say Dr Bernard Davis Whittington Hospital, London Background to BCSH Guideline Rationale Current guidance in disparate publications
Beta Thalassemia Frequency in Bahrain: A Ten Year Study. Shaikha Salim Al-Arrayed, MB,ChB, DHCG, PhD*
 Bahrain Medical Bulletin, Vol. 2, No. 2, June 200 Beta Thalassemia Frequency in Bahrain: A Ten Year Study Shaikha Salim Al-Arrayed, MB,ChB, DHCG, PhD* Background: Sickle-cell disease and Thalassanemia
Bahrain Medical Bulletin, Vol. 2, No. 2, June 200 Beta Thalassemia Frequency in Bahrain: A Ten Year Study Shaikha Salim Al-Arrayed, MB,ChB, DHCG, PhD* Background: Sickle-cell disease and Thalassanemia
Carrying Beta Thalassaemia A carrier can use this booklet to
 Carrying Beta Thalassaemia A carrier can use this booklet to help explain carrying beta to their partner, blood relatives and others. show to any health professional (doctor, nurse or midwife) they see
Carrying Beta Thalassaemia A carrier can use this booklet to help explain carrying beta to their partner, blood relatives and others. show to any health professional (doctor, nurse or midwife) they see
Non-malignant hematologic disorders associated arthropathies: hemoglobinopathy-associated musculoskeletal manifestations, hemophilia
 Non-malignant hematologic disorders associated arthropathies: hemoglobinopathy-associated musculoskeletal manifestations, hemophilia HAEMOGLOBINOPATHIES = inherited disorders of globin divided into: Thalassaemia
Non-malignant hematologic disorders associated arthropathies: hemoglobinopathy-associated musculoskeletal manifestations, hemophilia HAEMOGLOBINOPATHIES = inherited disorders of globin divided into: Thalassaemia
7 Medical Genetics. Hemoglobinopathies. Hemoglobinopathies. Protein and Gene Structure. and Biochemical Genetics
 SESSION 7 Medical Genetics Hemoglobinopathies and Biochemical Genetics J a v a d F a s a J a m s h i d i U n i v e r s i t y o f M e d i c a l S c i e n c e s, N o v e m b e r 2 0 1 7 Hemoglobinopathies
SESSION 7 Medical Genetics Hemoglobinopathies and Biochemical Genetics J a v a d F a s a J a m s h i d i U n i v e r s i t y o f M e d i c a l S c i e n c e s, N o v e m b e r 2 0 1 7 Hemoglobinopathies
Sickle Cell Anemia A Fictional Reconstruction Answer Key
 We have made it easy for you to find a PDF Ebooks without any digging. And by having access to our ebooks online or by storing it on your computer, you have convenient answers with sickle cell anemia a
We have made it easy for you to find a PDF Ebooks without any digging. And by having access to our ebooks online or by storing it on your computer, you have convenient answers with sickle cell anemia a
Full Case: Questions: What is sickle cell crisis?
 Full Case: 30 y/o with avascular necrosis of her right hip was admitted for a total hip arthroplasty. Her hematocrit was 22%, blood pressure was 130/90 mm Hg, and pulse was 107 beats per minute. She had
Full Case: 30 y/o with avascular necrosis of her right hip was admitted for a total hip arthroplasty. Her hematocrit was 22%, blood pressure was 130/90 mm Hg, and pulse was 107 beats per minute. She had
An overview of Thalassaemias and Complications
 An overview of Thalassaemias and Complications Haemoglobin Haemoglobin is the most abundant protein in blood, and exists as three main types in normal adults: HbA ( ) - 97% HbA 2 ( ) - 2.5% HbF ( ) - 0.5%
An overview of Thalassaemias and Complications Haemoglobin Haemoglobin is the most abundant protein in blood, and exists as three main types in normal adults: HbA ( ) - 97% HbA 2 ( ) - 2.5% HbF ( ) - 0.5%
Thalassemias:general aspects and molecular pathology
 Thalassemias:general aspects and molecular pathology Prof. Renzo Galanello Pediatric Clinic 2 University of Cagliari Ospedale Regionale Microcitemie-ASL8 HEMOGLOBINOPATHIES CLASSIFICATION Structurally
Thalassemias:general aspects and molecular pathology Prof. Renzo Galanello Pediatric Clinic 2 University of Cagliari Ospedale Regionale Microcitemie-ASL8 HEMOGLOBINOPATHIES CLASSIFICATION Structurally
Neonatal Screening for Genetic Blood Diseases. Shaikha Al-Arayyed, PhD* A Aziz Hamza, MD** Bema Sultan*** D. K. Shome, MRCPath**** J. P.
 Bahrain Medical Bulletin, Vol. 29, No. 3, September, 2007 Neonatal Screening for Genetic Blood Diseases Shaikha Al-Arayyed, PhD* A Aziz Hamza, MD** Bema Sultan*** D. K. Shome, MRCPath**** J. P. Bapat,PhD****
Bahrain Medical Bulletin, Vol. 29, No. 3, September, 2007 Neonatal Screening for Genetic Blood Diseases Shaikha Al-Arayyed, PhD* A Aziz Hamza, MD** Bema Sultan*** D. K. Shome, MRCPath**** J. P. Bapat,PhD****
Comprehensive Hemoglobin Analysis HBA1/2 (
 Comprehensive Hemoglobin Analysis HBA1/2 ( α-globin) and HBB (β-globin) mutation and deletion/duplication analysis and HBD (δ-globin) and HBG1/2 (γ-globin) mutation analysis Description: Hemoglobin (Hb)
Comprehensive Hemoglobin Analysis HBA1/2 ( α-globin) and HBB (β-globin) mutation and deletion/duplication analysis and HBD (δ-globin) and HBG1/2 (γ-globin) mutation analysis Description: Hemoglobin (Hb)
Guidelines for Shared Care Centres and Community Staff
 Reference: CG1411 Written by: Dr Jenny Welch Peer reviewer Dr Jeanette Payne Approved: September 2015 Approved by D&TC: 10 th July 2015 Review Due: September 2018 Intended Audience This document contains
Reference: CG1411 Written by: Dr Jenny Welch Peer reviewer Dr Jeanette Payne Approved: September 2015 Approved by D&TC: 10 th July 2015 Review Due: September 2018 Intended Audience This document contains
Some Observations on Haemoglobin A 2
 Some Observations on Haemoglobin A 2 Barbara J Bain St Mary s Hospital and Imperial College London Image from www.dsc.discovery.com Haemoglobin A 2 5 HBE1 HBG2 HBG1 HBD HBB LCRB ε G γ A γ ψβ δ β 3 5 LCRA
Some Observations on Haemoglobin A 2 Barbara J Bain St Mary s Hospital and Imperial College London Image from www.dsc.discovery.com Haemoglobin A 2 5 HBE1 HBG2 HBG1 HBD HBB LCRB ε G γ A γ ψβ δ β 3 5 LCRA
The hemoglobin (Hb) can bind a maximum of 220 ml O2 per liter.
 can bind a maximum of 220 ml O2 per liter.") Hemoglobin Hemoglobin The most important function of the red blood cells is totransport (O2) from the lungs into the tissues, and carbon dioxide (CO2) from the tissues back into the lungs. O2 is poorly
Hemoglobin Hemoglobin The most important function of the red blood cells is totransport (O2) from the lungs into the tissues, and carbon dioxide (CO2) from the tissues back into the lungs. O2 is poorly
The Child with a Hematologic Alteration
 47 The Child with a Hematologic Alteration HELPFUL HINT Review the anatomy and physiology of the hematologic system in an anatomy and physiology textbook. MATCHING KEY TERMS Match the term with the correct
47 The Child with a Hematologic Alteration HELPFUL HINT Review the anatomy and physiology of the hematologic system in an anatomy and physiology textbook. MATCHING KEY TERMS Match the term with the correct
Educational Items Section
 Atlas of Genetics and Cytogenetics in Oncology and Haematology OPEN ACCESS JOURNAL AT INIST-CNRS Educational Items Section Hemoglobin genes; Sickle-cell anemia - Thalassemias Jean-Loup Huret, Xavier Troussard
Atlas of Genetics and Cytogenetics in Oncology and Haematology OPEN ACCESS JOURNAL AT INIST-CNRS Educational Items Section Hemoglobin genes; Sickle-cell anemia - Thalassemias Jean-Loup Huret, Xavier Troussard
Lec.2 Medical Physiology Blood Physiology Z.H.Kamil
 Destruction of Red Blood Cells When red blood cells are delivered from the bone marrow into the circulatory system, they normally circulate an average of 120 days before being destroyed. Even though mature
Destruction of Red Blood Cells When red blood cells are delivered from the bone marrow into the circulatory system, they normally circulate an average of 120 days before being destroyed. Even though mature
World-Wide Distribution of Hemoglobin S. Geographic distribution of hemoglobin S in the world
 Sickle Cell Disease Gerald M. Woods, MD Professor of Pediatrics Division Director, Hematology/Oncology/BMT Director of Sickle Cell Program Children s Mercy Hospitals and Clinics Disclaimer Member of DSMB
Sickle Cell Disease Gerald M. Woods, MD Professor of Pediatrics Division Director, Hematology/Oncology/BMT Director of Sickle Cell Program Children s Mercy Hospitals and Clinics Disclaimer Member of DSMB
Sickle cell disease: Complications in adult patients
 Sickle cell disease: Complications in adult patients Dr Sara Stuart-Smith Haematology Consultant Sickle cell and thalassaemia Nurses, AHP and Junior Doctor Training Days September 2017 SCD is caused by
Sickle cell disease: Complications in adult patients Dr Sara Stuart-Smith Haematology Consultant Sickle cell and thalassaemia Nurses, AHP and Junior Doctor Training Days September 2017 SCD is caused by
Joanne Mallon, Nora O Neill, Dr D Hull Antenatal midwife coordinator, Consultant Haematologist
 Title: CLINICAL GUIDELINES ID TAG Guideline for Antenatal Screening for Haemoglobinopathies Author: Designation: Speciality / Division: Directorate: Joanne Mallon, Nora O Neill, Dr D Hull Antenatal midwife
Title: CLINICAL GUIDELINES ID TAG Guideline for Antenatal Screening for Haemoglobinopathies Author: Designation: Speciality / Division: Directorate: Joanne Mallon, Nora O Neill, Dr D Hull Antenatal midwife
Beta thalassaemia traits in Nigerian patients with sickle cell anaemia
 JMBR: A Peer-review Journal of Biomedical Sciences June 2005 Vol. 4 No.1 pp-37-43 Beta thalassaemia traits in Nigerian patients with sickle cell anaemia CE Omoti ABSTRACT Haematological values were determined
JMBR: A Peer-review Journal of Biomedical Sciences June 2005 Vol. 4 No.1 pp-37-43 Beta thalassaemia traits in Nigerian patients with sickle cell anaemia CE Omoti ABSTRACT Haematological values were determined
We are IntechOpen, the world s leading publisher of Open Access books Built by scientists, for scientists. International authors and editors
 We are IntechOpen, the world s leading publisher of Open Access books Built by scientists, for scientists 3,350 108,000 1.7 M Open access books available International authors and editors Downloads Our
We are IntechOpen, the world s leading publisher of Open Access books Built by scientists, for scientists 3,350 108,000 1.7 M Open access books available International authors and editors Downloads Our
CURRENT RESEARCH STUDIES
 CURRENT RESEARCH STUDIES SCAGO SICKLE CELL RESEARCH DAY MAY 12, 2018 REBECCA LEROUX RN, BSCN, CCRP RED BLOOD CELL DISORDERS PROGRAM, UNIVERSITY HEALTH NETWORK MANUELA MERELLES-PULCINI RN, BSCN, MSN, CCRP
CURRENT RESEARCH STUDIES SCAGO SICKLE CELL RESEARCH DAY MAY 12, 2018 REBECCA LEROUX RN, BSCN, CCRP RED BLOOD CELL DISORDERS PROGRAM, UNIVERSITY HEALTH NETWORK MANUELA MERELLES-PULCINI RN, BSCN, MSN, CCRP
Hydroxyurea and Transfusion Therapy for the Treatment of Sickle Cell Disease
 Hydroxyurea and Transfusion Therapy for the Treatment of Sickle Cell Disease A Pocket Guide for the Clinician Susan E. Creary, MD, MSc 1 John J. Strouse, MD, PhD 2 1 The Ohio State University School of
Hydroxyurea and Transfusion Therapy for the Treatment of Sickle Cell Disease A Pocket Guide for the Clinician Susan E. Creary, MD, MSc 1 John J. Strouse, MD, PhD 2 1 The Ohio State University School of
HAEMOGLOBINOPATHIES. Editing file. References: 436 girls & boys slides 435 teamwork slides. Color code: Important. Extra.
 HAEMOGLOBINOPATHIES Objectives: normal structure and function of haemoglobin. how the globin components of haemoglobin change during development, and postnatally. the mechanisms by which the thalassaemias
HAEMOGLOBINOPATHIES Objectives: normal structure and function of haemoglobin. how the globin components of haemoglobin change during development, and postnatally. the mechanisms by which the thalassaemias
FUNCTIONS OF HEMOGLOBIN:
 HEMOGLOBIN: Conjugated protein Simple protein combined with a non-protein substance Hemoglobin HEME +GLOBIN nonprotein substance HEME( prosthetic group) Red colour of blood is due to Hb in RBCs Normal
HEMOGLOBIN: Conjugated protein Simple protein combined with a non-protein substance Hemoglobin HEME +GLOBIN nonprotein substance HEME( prosthetic group) Red colour of blood is due to Hb in RBCs Normal
Welcome to Sickle Cell Disease by Connie Martin, MS, RDN; Lolita McLean, MPH, RDN; and Claire Stephens, MS, RDN of the Alabama Department of
 Welcome to Sickle Cell Disease by Connie Martin, MS, RDN; Lolita McLean, MPH, RDN; and Claire Stephens, MS, RDN of the Alabama Department of Rehabilitation Services, Children s Rehabilitation Service.
Welcome to Sickle Cell Disease by Connie Martin, MS, RDN; Lolita McLean, MPH, RDN; and Claire Stephens, MS, RDN of the Alabama Department of Rehabilitation Services, Children s Rehabilitation Service.
Haemoglobinopathy Case Studies. Dr Jill Finlayson Department of Haematology Pathwest Laboratory Medicine
 Haemoglobinopathy Case Studies Dr Jill Finlayson Department of Haematology Pathwest Laboratory Medicine Case 1 KB, 36y M Refugee Afghanistan Screening bloods Hb 101 g/l RCC 3.75 x10 12 /L MCV 90 fl MCH
Haemoglobinopathy Case Studies Dr Jill Finlayson Department of Haematology Pathwest Laboratory Medicine Case 1 KB, 36y M Refugee Afghanistan Screening bloods Hb 101 g/l RCC 3.75 x10 12 /L MCV 90 fl MCH
Good afternoon and thank you for joining us today as we discuss hydroxyurea for the treatment of sickle cell disease. Dr. Emily Meier is a pediatric
 Good afternoon and thank you for joining us today as we discuss hydroxyurea for the treatment of sickle cell disease. Dr. Emily Meier is a pediatric hematologist at the Indiana Hemophilia & Thrombosis
Good afternoon and thank you for joining us today as we discuss hydroxyurea for the treatment of sickle cell disease. Dr. Emily Meier is a pediatric hematologist at the Indiana Hemophilia & Thrombosis