Sickle cell disease. Fareed Omar 10 March 2018
|
|
|
- Lynette Jordan
- 5 years ago
- Views:
Transcription

1 Sickle cell disease Fareed Omar 10 March 2018

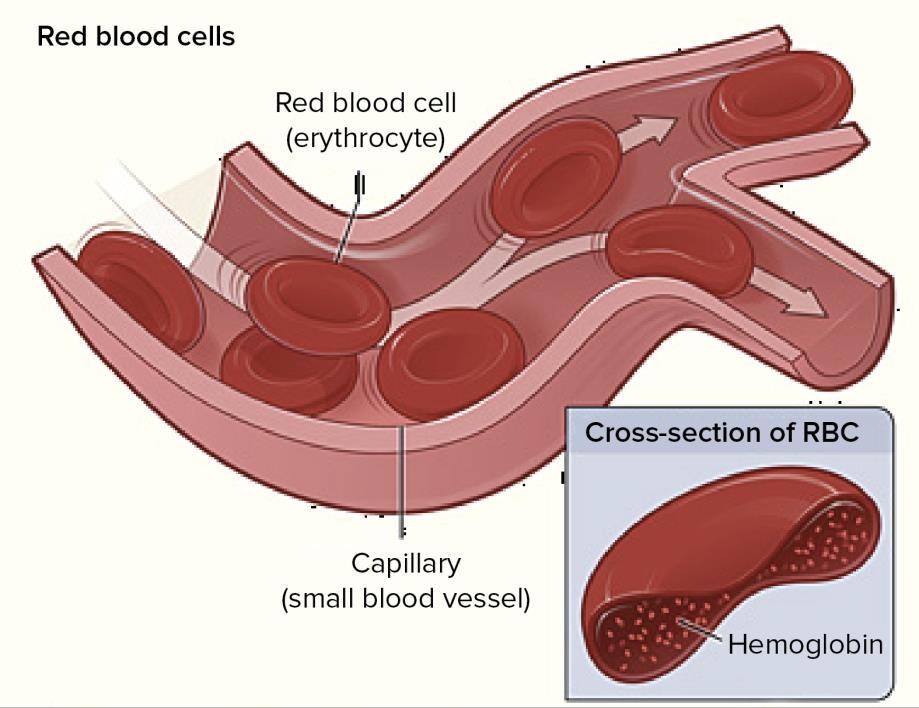
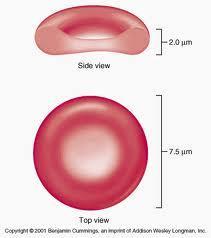
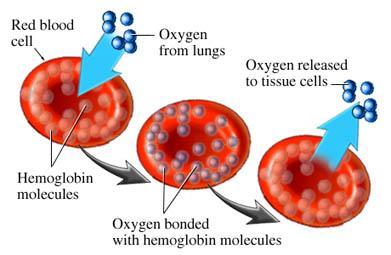
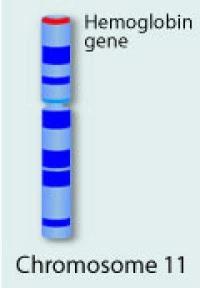
2 Physiology
3
 HbA: 2α and 2β chains")
4 Haemoglobin structure HbA2: 2α and 2δ chains (2-3%) HbF: 2α and 2γ chains (<1%) HbA: 2α and 2β chains (97%)

5
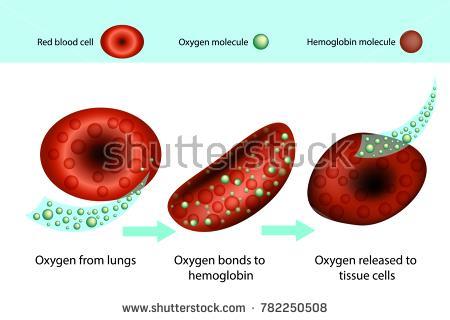
6
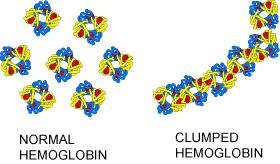
7 Polymerization


8
9 Sickle cell disease Inherited Autosomal recessive genetic disease Two copies of an abnormal gene must be present in order for the disease or trait to develop. If only one copy is present the patient is said to be a carrier or has sickle cell trait If both parents are carriers, the child may inherit both copies of the mutated genes from each parent and is said to be homozygous About 70 million people worldwide affected > infants are born with the disease worldwide

10 Distribution High Risk Medium Risk Low Risk Very Low Risk No Risk
11
12 Diagnosis Hb-Electrophoresis Sickling test: high-performance liquid chromatography (HPLC) This test identifies which type of hemoglobin is present Usually sufficient to confirm diagnosis of both disease & trait Genetic test can be done to confirm Prenatal testing
13 Precipitating factors Oxygen (hypoxia) high altitude Osmolarity (dehydration) Temperature (vasoconstriction) ph Anxiety Exercise Trauma / surgery Folate deficiency Polymerization
14 Systemic Manifestations

15


16 Bone Ischaemia / Infarction of the bone / bone marrow Severe pain Osteopenia Osteomyelitis Avascular necrosis Fat embolism -> Pulmonary embolism Acute aplastic crisis
 Cognitive impairment and learning difficulties")

17 Brain TIA, CVA -> strokes (2-11yrs) (10% before age 20) Cognitive impairment and learning difficulties Undetected microemboli (silent infarcts) Epilepsy: 10 fold increased risk Transcranial Doppler ultrasound
18 Transcranial Doppler (TCD) Annually Non invasive, non ionising, inexpensive, portable and safe technique that uses a pulsed Doppler transducer for assessment of intracerebral blood flow Velocity flow in the distal Internal Carotid Artery (dica) and the proximal Middle Cerebral Artery (MCA) Velocities of > 200 cm/sec increased risk of stroke Once identified, confirmed and treated with transfusion therapy, children with intracranial stenosis showed >90% reduction in stroke risk if haemoglobin S was less than 30%.

19 Lungs Acute chest syndrome (ACS) Fever, cough, chest pain, dyspnoea, wheeze Pulm infiltrates (CXR) Vaso-occlusion, fat embolism, pulmonary thromboembolism, infection Resp failure, CNS hypoxia, seizures Pulm oedema, multi-organ failure Bed rest, analgesia, hydration, Oxygen, ventilation, IV antibiotics, transfusion, Bronchodilators

20 Spleen Chronic infarcts over many years functional asplenia Higher risk for infection encapsulated organisms Acute crisis: massive splenic infarct splenic sequestration crisis rupture very high mortality Early urgent blood transfusion Splenectomy (after 4-6 years age)
21 Systemic manifestations Heart PHT, LV dysfunction, MI Kidneys Renal papillary necrosis, chronic renal insufficiency, Haematuria, Proteinuria GIT Ischaemia severe pain (Abdominal crisis) Skin ischaemia / infarcts pain and ulceration Priapism Liver fairly resilient relatively spared PAIN!! most painful disorder Don t underestimate pain
22 Management CURATIVE Genetic mutation in erythroid precursor cell erythropeoitic stem cell HSCT: removes the affected erythropeoitic stem cell and replaces it with normal donor stem cells Donor (sibling) who does not have sickle cell disease Risky mortality 7-20% from procedure Infections and GVHD In our setting often not practical and suitable donors not available Worldwide <18% have suitable donors
23 Management CHRONIC MANAGEMENT (AIMS) Preventing Veno-occlusive crisis (VOC) Treat crisis when it occurs Prevent infection (no.1 cause of mortality) Treat anaemia Manage complications (stroke, pulmonary HT)
24 Preventing VOC 1. Hydroxyurea (antisickling agent) Causes a shift in gene expression at the beta globin locus, such that expression from the gamma globin locus is increased relative to that from the beta globin locus Stimulates production of HbF Replaces some HbS with HbF Polymerization and therefore sickling Affects BM cells & can cause BM suppression - risk of infection (dose dependent)
25 N Engl J Med 1990: 322:
26 Am J Hematol. 2017;92:
27 Preventing VOC 2. Chronic transfusions the concentration of HbS For patients that are at increased risk for strokes Abnormal transcranial Doppler Previous strokes 8 10 transfusions / year Iron overload
28 Preventing VOC Avoid and manage conditions that may lead to acidosis Prevent dehydration or treat aggressively Avoid excessive cold Avoid or treat infections quickly and aggressively Vaccine boosters: encapsulated organisms Annual influenza vaccine Prophylactic Pen VK Folic acid (due to continuous haemolysis in these patients)
29 Treat crisis when they occur Goal: aggressive management Vigorous IV fluid management to prevent RBC from dehydration - Polymerization and to prevent acidosis Oxygen therapy to decrease the amount of deoxy Hb If infection suspected treat early Very aggressive pain management (within 30 min) Opioids Transfuse RCC if indicated
30 N Engl J Med 2017;376:848-55
31
32 Summary Hydroxyurea Pen prophylaxis Folic acid Vaccinations Treat crisis aggressively

33
34 Thank You
SICKLE CELL DISEASE. Dr. MUBARAK ABDELRAHMAN MD PEDIATRICS AND CHILD HEALTH. Assistant Professor FACULTY OF MEDICINE -JAZAN
 SICKLE CELL DISEASE Dr. MUBARAK ABDELRAHMAN MD PEDIATRICS AND CHILD HEALTH Assistant Professor FACULTY OF MEDICINE -JAZAN Objective: The student should be able: To identify the presentation, diagnosis,
SICKLE CELL DISEASE Dr. MUBARAK ABDELRAHMAN MD PEDIATRICS AND CHILD HEALTH Assistant Professor FACULTY OF MEDICINE -JAZAN Objective: The student should be able: To identify the presentation, diagnosis,
Anemia s. Troy Lund MSMS PhD MD
 Anemia s Troy Lund MSMS PhD MD lundx072@umn.edu Hemoglobinopathy/Anemia IOM take home points. 1. How do we identify the condtion? Smear, CBC Solubility Test (SCD) 2. How does it present clincally? 3. How
Anemia s Troy Lund MSMS PhD MD lundx072@umn.edu Hemoglobinopathy/Anemia IOM take home points. 1. How do we identify the condtion? Smear, CBC Solubility Test (SCD) 2. How does it present clincally? 3. How
Congenital Haemoglobinopathies
 Congenital Haemoglobinopathies L. DEDEKEN, MD H O P I T A L U N I V E R S I T A I R E D E S E N F A N T S R E I N E F A B I O L A U N I V E R S I T E L I B R E DE B R U X E L L E S Red Blood Cell Disorders
Congenital Haemoglobinopathies L. DEDEKEN, MD H O P I T A L U N I V E R S I T A I R E D E S E N F A N T S R E I N E F A B I O L A U N I V E R S I T E L I B R E DE B R U X E L L E S Red Blood Cell Disorders
Sickle Cell Disease. Edward Malters, MD
 Sickle Cell Disease Edward Malters, MD Introduction Vaso-occlusive phenomena and hemolysis are the clinical hallmarks of Sickle Cell Disease (SCD) Inherited disorder due to homozygosity for the abnormal
Sickle Cell Disease Edward Malters, MD Introduction Vaso-occlusive phenomena and hemolysis are the clinical hallmarks of Sickle Cell Disease (SCD) Inherited disorder due to homozygosity for the abnormal
Hemolytic anemias (2 of 2)
") Hemolytic anemias (2 of 2) Sickle Cell Anemia The most common familial hemolytic anemia in the world Sickle cell anemia is the prototypical (and most prevalent) hemoglobinopathy Mutation in the β-globin
Hemolytic anemias (2 of 2) Sickle Cell Anemia The most common familial hemolytic anemia in the world Sickle cell anemia is the prototypical (and most prevalent) hemoglobinopathy Mutation in the β-globin
How to Write a Life Care Plan for a Child with Hemoglobinopathy
 How to Write a Life Care Plan for a Child with Hemoglobinopathy Tamar Fleischer, BSN, MSN, CNLCP & Mona Yudkoff, RN, MPH, CRRN, CNLCP BalaCare Solutions March 2018 St. Peterburg, Florida What is Hemoglobinopathy?
How to Write a Life Care Plan for a Child with Hemoglobinopathy Tamar Fleischer, BSN, MSN, CNLCP & Mona Yudkoff, RN, MPH, CRRN, CNLCP BalaCare Solutions March 2018 St. Peterburg, Florida What is Hemoglobinopathy?
Introduction reduction in output alter the amino acid sequence combination
 Sickle cell anemia. Introduction Mutations in the globin genes can cause a quantitative reduction in output from that gene or alter the amino acid sequence of the protein produced or a combination of the
Sickle cell anemia. Introduction Mutations in the globin genes can cause a quantitative reduction in output from that gene or alter the amino acid sequence of the protein produced or a combination of the
Health Maintenance and Education for Children and Adults
 Health Maintenance and Education for Children and Adults Richard Ward, MSc, MRCP, FRCPath Director, Red Blood Cell Disorders Program, UHN Assistant Professor, Hematology, University of Toronto Chair, Canadian
Health Maintenance and Education for Children and Adults Richard Ward, MSc, MRCP, FRCPath Director, Red Blood Cell Disorders Program, UHN Assistant Professor, Hematology, University of Toronto Chair, Canadian
Putting some hematology into Pediatric Hematology/Oncology: a review of Hemophilia and Sickle Cell Disease in the Pediatric Patient
 Putting some hematology into Pediatric Hematology/Oncology: a review of Hemophilia and Sickle Cell Disease in the Pediatric Patient Kristina Haley, DO March 10, 2012 Jovita Reyes Memorial Pediatric Hematology/Oncology
Putting some hematology into Pediatric Hematology/Oncology: a review of Hemophilia and Sickle Cell Disease in the Pediatric Patient Kristina Haley, DO March 10, 2012 Jovita Reyes Memorial Pediatric Hematology/Oncology
Sickle Cell Disease and impact on the society
 Sickle Cell Disease and impact on the society Professor Z.A.Jeremiah Ph.D, FRCPath (London) Professor of Haematology and Blood Transfusion Science Niger Delta University, Wilberforce Island Outline What
Sickle Cell Disease and impact on the society Professor Z.A.Jeremiah Ph.D, FRCPath (London) Professor of Haematology and Blood Transfusion Science Niger Delta University, Wilberforce Island Outline What
In adults, the predominant Hb (HbA) molecule has four chains: two α and two β chains. In thalassemias, the synthesis of either the α or the β chains
 molecule has four chains: two α and two β chains. In thalassemias, the synthesis of either the α or the β chains") Thalassaemias Thalassemia Thalassemia is an inherited autosomal recessive blood disease. Associated with absence or reduction in a or b globin chains. Reduced synthesis of one of the globin chains can
Thalassaemias Thalassemia Thalassemia is an inherited autosomal recessive blood disease. Associated with absence or reduction in a or b globin chains. Reduced synthesis of one of the globin chains can
Anaemia in Pregnancy
 Anaemia in Pregnancy Definition :anaemia is a pathological condition in which the oxygen-carrying capacity of red blood cells is insufficient to meet the body needs. The WHO : haemoglobin concentration
Anaemia in Pregnancy Definition :anaemia is a pathological condition in which the oxygen-carrying capacity of red blood cells is insufficient to meet the body needs. The WHO : haemoglobin concentration
DONE BY : RaSHA RAKAN & Bushra Saleem
 DONE BY : RaSHA RAKAN & Bushra Saleem Hemolytic anemias (2 of 2) Sickle Cell Anemia The most common familial hemolytic anemia in the world Sickle cell anemia is the prototypical (and most prevalent) hemoglobinopathy
DONE BY : RaSHA RAKAN & Bushra Saleem Hemolytic anemias (2 of 2) Sickle Cell Anemia The most common familial hemolytic anemia in the world Sickle cell anemia is the prototypical (and most prevalent) hemoglobinopathy
Sickle Cell Disease 101. Objectives. What is SCD? 4/20/2016. Discuss the pathophysiology & genetics of Sickle Cell Disease (SCD).
.") Sickle Cell Disease 101 Jennifer Young, RN, MS, CPNP-AC Sickle Cell & Thalassemia Nurse Practitioner Nationwide Children s Hospital Objectives Discuss the pathophysiology & genetics of Sickle Cell Disease
Sickle Cell Disease 101 Jennifer Young, RN, MS, CPNP-AC Sickle Cell & Thalassemia Nurse Practitioner Nationwide Children s Hospital Objectives Discuss the pathophysiology & genetics of Sickle Cell Disease
Extra Notes 3. Warm. In the core (center) of the body, where the temperature is 37 C.
 of the body, where the temperature is 37 C.") Extra Notes 3 *The numbers of the slides are according to the last year slides. Slide 33 Autoimmune hemolytic anemia : Abnormal circulating antibodies that target normal antigen on the RBC and cause lysis.
Extra Notes 3 *The numbers of the slides are according to the last year slides. Slide 33 Autoimmune hemolytic anemia : Abnormal circulating antibodies that target normal antigen on the RBC and cause lysis.
Rationale for RBC Transfusion in SCD
 Rationale for RBC Transfusion in SCD Dilution of HgbS-containing RBCs via the addition of HgbA-containing cells from the blood of normal donors Suppression of erythropoietin release caused by the rise
Rationale for RBC Transfusion in SCD Dilution of HgbS-containing RBCs via the addition of HgbA-containing cells from the blood of normal donors Suppression of erythropoietin release caused by the rise
Medical and Surgical Complications of Sickle Cell Anemia
 Medical and Surgical Complications of Sickle Cell Anemia Ahmed Al-Salem Medical and Surgical Complications of Sickle Cell Anemia Ahmed Al-Salem Department of Surgery Dar A lalafia Medical Company Qatif
Medical and Surgical Complications of Sickle Cell Anemia Ahmed Al-Salem Medical and Surgical Complications of Sickle Cell Anemia Ahmed Al-Salem Department of Surgery Dar A lalafia Medical Company Qatif
Hydroxyurea and Transfusion Therapy for the Treatment of Sickle Cell Disease
 Hydroxyurea and Transfusion Therapy for the Treatment of Sickle Cell Disease A Pocket Guide for the Clinician Susan E. Creary, MD, MSc 1 John J. Strouse, MD, PhD 2 1 The Ohio State University School of
Hydroxyurea and Transfusion Therapy for the Treatment of Sickle Cell Disease A Pocket Guide for the Clinician Susan E. Creary, MD, MSc 1 John J. Strouse, MD, PhD 2 1 The Ohio State University School of
Biance Rowe Chris Hani Baragwanath Hospital Paediatric Haematology Oncology University of the Witwatersrand
 Biance Rowe Chris Hani Baragwanath Hospital Paediatric Haematology Oncology University of the Witwatersrand SCD affects 20-25 million people globally 12-15 million in Africa 300 000 children with SCD
Biance Rowe Chris Hani Baragwanath Hospital Paediatric Haematology Oncology University of the Witwatersrand SCD affects 20-25 million people globally 12-15 million in Africa 300 000 children with SCD
Aneurin Bevan University Health Board Sickle Cell Anaemia and Haemoglobinopathy Screening and Management in Pregnancy Guidelines
 Sickle Cell Anaemia and Haemoglobinopathy Screening and Management in Pregnancy Guidelines N.B. Staff should be discouraged from printing this document. This is to avoid the risk of out of date printed
Sickle Cell Anaemia and Haemoglobinopathy Screening and Management in Pregnancy Guidelines N.B. Staff should be discouraged from printing this document. This is to avoid the risk of out of date printed
Atlantic Provinces Pediatric Hematology Oncology Network Réseau d Oncologie et Hématologie Pédiatrique des Provinces Atlantiques
 Atlantic Provinces Pediatric Hematology Oncology Network Réseau d Oncologie et Hématologie Pédiatrique des Provinces Atlantiques 5850/5980 University Avenue, PO Box 9700 Halifax, NS, B3K 6R8 1.902.470.7429
Atlantic Provinces Pediatric Hematology Oncology Network Réseau d Oncologie et Hématologie Pédiatrique des Provinces Atlantiques 5850/5980 University Avenue, PO Box 9700 Halifax, NS, B3K 6R8 1.902.470.7429
Dr Banu Kaya Consultant Haematologist Barts Health NHS Trust Royal London Hospital, London, UK SICKLE CELL AND THALASSAEMIA OVERVIEW
 Dr Banu Kaya Consultant Haematologist Barts Health NHS Trust Royal London Hospital, London, UK SICKLE CELL AND THALASSAEMIA OVERVIEW Objectives Gain awareness of haemoglobinopathy inheritance, pathophysiology
Dr Banu Kaya Consultant Haematologist Barts Health NHS Trust Royal London Hospital, London, UK SICKLE CELL AND THALASSAEMIA OVERVIEW Objectives Gain awareness of haemoglobinopathy inheritance, pathophysiology
Screening for haemoglobinopathies in pregnancy
 Policy Statement All Southern Health patients will receive clinical care that reflects best practice and is based on the best available evidence. Index of chapters within background 1. Prevalence of haemoglobinopathies
Policy Statement All Southern Health patients will receive clinical care that reflects best practice and is based on the best available evidence. Index of chapters within background 1. Prevalence of haemoglobinopathies
RBCs Disorders 1. Dr. Nabila Hamdi MD, PhD
 RBCs Disorders 1 Dr. Nabila Hamdi MD, PhD ILOs Discuss the classification of anemia into hypochromic-microcytic, normochromicnormocytic and macrocytic. Categorize laboratory test procedures used in the
RBCs Disorders 1 Dr. Nabila Hamdi MD, PhD ILOs Discuss the classification of anemia into hypochromic-microcytic, normochromicnormocytic and macrocytic. Categorize laboratory test procedures used in the
Hemoglobinopathies NORMAL HEMOGLOBINS
 Hemoglobinopathies Millicent Sutton MD October 28, 2005 NORMAL HEMOGLOBINS Consist of 2 alpha chains and 2 non alpha chains Hb A = α2β2 Hb F= α 2γ2 Hb A2 = α2δ2 1 Hemoglobin Variants Altered the conformational
Hemoglobinopathies Millicent Sutton MD October 28, 2005 NORMAL HEMOGLOBINS Consist of 2 alpha chains and 2 non alpha chains Hb A = α2β2 Hb F= α 2γ2 Hb A2 = α2δ2 1 Hemoglobin Variants Altered the conformational
Haemoglobin BY: MUHAMMAD RADWAN WISSAM MUHAMMAD
 Haemoglobin BY: MUHAMMAD RADWAN WISSAM MUHAMMAD Introduction is the iron-containing oxygen transport metalloprotein in the red blood cells Hemoglobin in the blood carries oxygen from the respiratory organs
Haemoglobin BY: MUHAMMAD RADWAN WISSAM MUHAMMAD Introduction is the iron-containing oxygen transport metalloprotein in the red blood cells Hemoglobin in the blood carries oxygen from the respiratory organs
4 Jumana Jihad Dr. Ahmad Mansour Dr. Ahmad Mansour
 4 Jumana Jihad Dr. Ahmad Mansour Dr. Ahmad Mansour Anemia Decreased blood production Increased blood loss Hemolytic Hemorrhage Extravascular Intravascular Hemolytic (Further classification( Extrinsic Intrinsic
4 Jumana Jihad Dr. Ahmad Mansour Dr. Ahmad Mansour Anemia Decreased blood production Increased blood loss Hemolytic Hemorrhage Extravascular Intravascular Hemolytic (Further classification( Extrinsic Intrinsic
Managing Emergencies
 Managing Emergencies Richard Ward, MSc, MRCP, FRCPath Director, Red Blood Cell Disorders Program, UHN Assistant Professor, Hematology, University of Toronto Chair, Canadian Hemoglobinopathy Association
Managing Emergencies Richard Ward, MSc, MRCP, FRCPath Director, Red Blood Cell Disorders Program, UHN Assistant Professor, Hematology, University of Toronto Chair, Canadian Hemoglobinopathy Association
Emergency Presentations of Sickle cell disease. Dr S Pancham Sickle Cell & Thalassaemia Centre City Hospital
 Emergency Presentations of Sickle cell disease Dr S Pancham Sickle Cell & Thalassaemia Centre City Hospital Aims Key features to elicit at presentation What complications to look for When to phone for
Emergency Presentations of Sickle cell disease Dr S Pancham Sickle Cell & Thalassaemia Centre City Hospital Aims Key features to elicit at presentation What complications to look for When to phone for
SICKLE CELL DISEASE TO TREAT OR
 SICKLE CELL DISEASE TO TREAT OR NOT TO TREAT COHEM Barcelona September 8, 2012 Sujit Sheth, M.D. Pediatric Hematology Oncology Disclosures None Outline Morbidity and mortality Definitive therapies Risk
SICKLE CELL DISEASE TO TREAT OR NOT TO TREAT COHEM Barcelona September 8, 2012 Sujit Sheth, M.D. Pediatric Hematology Oncology Disclosures None Outline Morbidity and mortality Definitive therapies Risk
4 Fahed Al Karmi Sufian Alhafez Dr nayef karadsheh
 4 Fahed Al Karmi Sufian Alhafez Dr nayef karadsheh Genetic variants of hemoglobin Hemoglobinopathies (abnormal variants of hemoglobin) are divided into: 1. Structural abnormalities: Any change in the genes
4 Fahed Al Karmi Sufian Alhafez Dr nayef karadsheh Genetic variants of hemoglobin Hemoglobinopathies (abnormal variants of hemoglobin) are divided into: 1. Structural abnormalities: Any change in the genes
Comprehensive Care for Children and Adolescents with Sickle Cell Diseases
 Comprehensive Care for Children and Adolescents with Sickle Cell Diseases Objectives To review the system for newborn screening of infants for sickling diseases To provide the framework for a comprehensive
Comprehensive Care for Children and Adolescents with Sickle Cell Diseases Objectives To review the system for newborn screening of infants for sickling diseases To provide the framework for a comprehensive
The anaesthetic management of children with sickle cell disease
 The anaesthetic management of children with sickle cell disease Based in part upon Locke, C. Anaesthetic management of sickle cell disease in children. Anaesthesia Tutorial of the Week 153 (2009). Tanya
The anaesthetic management of children with sickle cell disease Based in part upon Locke, C. Anaesthetic management of sickle cell disease in children. Anaesthesia Tutorial of the Week 153 (2009). Tanya
HbSC disease is it different and how should we manage it? David Rees Department of Paediatric Haematology, King s College Hospital, London
 HbSC disease is it different and how should we manage it? David Rees Department of Paediatric Haematology, King s College Hospital, London Different types of sickle cell disesease Severe sickle cell disease
HbSC disease is it different and how should we manage it? David Rees Department of Paediatric Haematology, King s College Hospital, London Different types of sickle cell disesease Severe sickle cell disease
- Ensherah Mokheemer. - Rama Nada. - Tareq Aladily. 1 P a g e
 -3 - Ensherah Mokheemer - Rama Nada - Tareq Aladily 1 P a g e In this lecture we will continue talking about autoimmune hemolytic anemia. Autoimmune hemolytic anemia - There are several types that shares
-3 - Ensherah Mokheemer - Rama Nada - Tareq Aladily 1 P a g e In this lecture we will continue talking about autoimmune hemolytic anemia. Autoimmune hemolytic anemia - There are several types that shares
TRANSFUSION PRACTICES IN THE MANAGEMENT OF SICKLE CELL DISEASE AMONG FLORIDA PHYSICIANS
 TRANSFUSION PRACTICES IN THE MANAGEMENT OF SICKLE CELL DISEASE AMONG FLORIDA PHYSICIANS By LEVETTE NICOLE DUNBAR A THESIS PRESENTED TO THE GRADUATE SCHOOL OF THE UNIVERSITY OF FLORIDA IN PARTIAL FULFILLMENT
TRANSFUSION PRACTICES IN THE MANAGEMENT OF SICKLE CELL DISEASE AMONG FLORIDA PHYSICIANS By LEVETTE NICOLE DUNBAR A THESIS PRESENTED TO THE GRADUATE SCHOOL OF THE UNIVERSITY OF FLORIDA IN PARTIAL FULFILLMENT
Sickle Cell Disease- Case studies
 Sickle Cell Disease- Case studies Subarna Chakravorty King s College Hospital 2017 Conditions requiring immediate admission Agonising pain (i.e. requiring opiate analgesia) Increased pallor, breathlessness,
Sickle Cell Disease- Case studies Subarna Chakravorty King s College Hospital 2017 Conditions requiring immediate admission Agonising pain (i.e. requiring opiate analgesia) Increased pallor, breathlessness,
Full Case: Questions: What is sickle cell crisis?
 Full Case: 30 y/o with avascular necrosis of her right hip was admitted for a total hip arthroplasty. Her hematocrit was 22%, blood pressure was 130/90 mm Hg, and pulse was 107 beats per minute. She had
Full Case: 30 y/o with avascular necrosis of her right hip was admitted for a total hip arthroplasty. Her hematocrit was 22%, blood pressure was 130/90 mm Hg, and pulse was 107 beats per minute. She had
Division of General Internal Medicine and Geriatrics Hospital Medicine 2014
 Division of General Internal Medicine and Geriatrics Hospital Medicine 2014 Objectives Understand workup of acute pain crisis Identify key aspects of management of acute pain crisis in sickle cell patients
Division of General Internal Medicine and Geriatrics Hospital Medicine 2014 Objectives Understand workup of acute pain crisis Identify key aspects of management of acute pain crisis in sickle cell patients
World-Wide Distribution of Hemoglobin S. Geographic distribution of hemoglobin S in the world
 Sickle Cell Disease Gerald M. Woods, MD Professor of Pediatrics Division Director, Hematology/Oncology/BMT Director of Sickle Cell Program Children s Mercy Hospitals and Clinics Disclaimer Member of DSMB
Sickle Cell Disease Gerald M. Woods, MD Professor of Pediatrics Division Director, Hematology/Oncology/BMT Director of Sickle Cell Program Children s Mercy Hospitals and Clinics Disclaimer Member of DSMB
Newborn Screening and Followup for Hemoglobinopathies
 Newborn Screening and Followup for Hemoglobinopathies October 4, 2012 Monica Hulbert, MD Director, Sickle Cell Disease Program Washington University School of Medicine St. Louis Children s Hospital Disclosures
Newborn Screening and Followup for Hemoglobinopathies October 4, 2012 Monica Hulbert, MD Director, Sickle Cell Disease Program Washington University School of Medicine St. Louis Children s Hospital Disclosures
Sickle cell disease (SCD) and other hemoglobinopathies
 and other hemoglobinopathies") Sickle cell disease (SCD) and other hemoglobinopathies You have received this leaflet, because your child has been diagnosed with sickle cell disease. We can imagine how overwhelming such a diagnosis must
Sickle cell disease (SCD) and other hemoglobinopathies You have received this leaflet, because your child has been diagnosed with sickle cell disease. We can imagine how overwhelming such a diagnosis must
The Management of Acute Chest Syndrome in Children with Sickle Cell Disease
 The Management of Acute Chest Syndrome in Children with Sickle Cell Disease Document Information Version: 4 Date: Dec 2013 Authors (incl. job title): Professor David Rees and Dr Sue Height, consultant
The Management of Acute Chest Syndrome in Children with Sickle Cell Disease Document Information Version: 4 Date: Dec 2013 Authors (incl. job title): Professor David Rees and Dr Sue Height, consultant
SICKLE CELL BROCHURE
 SICKLE CELL BROCHURE SICKLE CELL DIESEASE According to CDC, Sickle cell disease (SCD) is a group of inherited red blood cell disorders. Healthy red blood cells are round and SCD C -shaped farm tool called
SICKLE CELL BROCHURE SICKLE CELL DIESEASE According to CDC, Sickle cell disease (SCD) is a group of inherited red blood cell disorders. Healthy red blood cells are round and SCD C -shaped farm tool called
Dependance on chronic transfusion
 Dependance on chronic transfusion Pr Saliou Diop Hematology Blood transfusion Dakar- Sénégal diop@cnts-dakar.sn Introduction Chronic transfusion: Regular use of blood transfusion in patients with chronic
Dependance on chronic transfusion Pr Saliou Diop Hematology Blood transfusion Dakar- Sénégal diop@cnts-dakar.sn Introduction Chronic transfusion: Regular use of blood transfusion in patients with chronic
Haemoglobinophaties EBMT 2011 Data Manager session
 Haemoglobinophaties EBMT 2011 Data Manager session Presentation plan Biological characteristics Clinical characteristics Transplant resuts What is different From transplant in malignancies Between Thalassemia
Haemoglobinophaties EBMT 2011 Data Manager session Presentation plan Biological characteristics Clinical characteristics Transplant resuts What is different From transplant in malignancies Between Thalassemia
HEMOGLOBINOPATHIES LECTURE OUTLINE. An overview of the structure of hemoglobin. Different types of hemoglobin. Definition of hemoglobinopathies
 Slide 1 HEOGLOBINOPATHIES Slide 2 LETURE OUTLINE An overview of the structure of hemoglobin. Different types of hemoglobin. Definition of hemoglobinopathies Sickle ell Disease and Hemoglobin Slide 3 HEOGLOBIN
Slide 1 HEOGLOBINOPATHIES Slide 2 LETURE OUTLINE An overview of the structure of hemoglobin. Different types of hemoglobin. Definition of hemoglobinopathies Sickle ell Disease and Hemoglobin Slide 3 HEOGLOBIN
Guideline for the Management of Acute Chest Syndrome in Children with Sickle Cell Disease
 Guideline for the Management of Acute Chest Syndrome in Children with Sickle Cell Disease Definition Acute chest syndrome (ACS) is defined as an acute illness characterized by fever and/or respiratory
Guideline for the Management of Acute Chest Syndrome in Children with Sickle Cell Disease Definition Acute chest syndrome (ACS) is defined as an acute illness characterized by fever and/or respiratory
Blood Transfusion Guidelines in Clinical Practice
 Blood Transfusion Guidelines in Clinical Practice Salwa Hindawi Director of Blood Transfusion Services Associate Professor in Haematology and Transfusion Medicine King Abdalaziz University, Jeddah Saudi
Blood Transfusion Guidelines in Clinical Practice Salwa Hindawi Director of Blood Transfusion Services Associate Professor in Haematology and Transfusion Medicine King Abdalaziz University, Jeddah Saudi
DIC. Disseminated intravascular coagulation, is a life threatening pathological process in which clotting factors are abnormally activated.
 Miss. kamlah 1 DIC Disseminated intravascular coagulation, is a life threatening pathological process in which clotting factors are abnormally activated. Resulting in wide spread of clot formation in the
Miss. kamlah 1 DIC Disseminated intravascular coagulation, is a life threatening pathological process in which clotting factors are abnormally activated. Resulting in wide spread of clot formation in the
Sickle cell disease: Complications in adult patients
 Sickle cell disease: Complications in adult patients Dr Sara Stuart-Smith Haematology Consultant Sickle cell and thalassaemia Nurses, AHP and Junior Doctor Training Days September 2017 SCD is caused by
Sickle cell disease: Complications in adult patients Dr Sara Stuart-Smith Haematology Consultant Sickle cell and thalassaemia Nurses, AHP and Junior Doctor Training Days September 2017 SCD is caused by
Pediatric Red Cell Exchange Indications, Benefits, Barriers. View from California Saturday May 9 th ASFA 2015
 Pediatric Red Cell Exchange Indications, Benefits, Barriers View from California Saturday May 9 th ASFA 2015 Red Cell Exchange: Not SCD Recommendations for Red Cell Exchange Indication Procedure Recommendation
Pediatric Red Cell Exchange Indications, Benefits, Barriers View from California Saturday May 9 th ASFA 2015 Red Cell Exchange: Not SCD Recommendations for Red Cell Exchange Indication Procedure Recommendation
SICKLE CELL DISEASE 1
 SICKLE CELL DISEASE 1 Your topic: My topic is about sickle cell disease, I need someone to write an introduction about the disease and the effects it has on the individual and family, also have to include
SICKLE CELL DISEASE 1 Your topic: My topic is about sickle cell disease, I need someone to write an introduction about the disease and the effects it has on the individual and family, also have to include
CURRENT RESEARCH STUDIES
 CURRENT RESEARCH STUDIES SCAGO SICKLE CELL RESEARCH DAY MAY 12, 2018 REBECCA LEROUX RN, BSCN, CCRP RED BLOOD CELL DISORDERS PROGRAM, UNIVERSITY HEALTH NETWORK MANUELA MERELLES-PULCINI RN, BSCN, MSN, CCRP
CURRENT RESEARCH STUDIES SCAGO SICKLE CELL RESEARCH DAY MAY 12, 2018 REBECCA LEROUX RN, BSCN, CCRP RED BLOOD CELL DISORDERS PROGRAM, UNIVERSITY HEALTH NETWORK MANUELA MERELLES-PULCINI RN, BSCN, MSN, CCRP
General Characterisctics
 Anemia General Characterisctics Definition: anemia is a decrease in red blood cells. Happens due to underproduction, increased destruction or loss of red cells. Diagnosis of anemia: Hgb < 135 (men) Hgb
Anemia General Characterisctics Definition: anemia is a decrease in red blood cells. Happens due to underproduction, increased destruction or loss of red cells. Diagnosis of anemia: Hgb < 135 (men) Hgb
Interleukin-1ß and Interleukin-6 Genetic Polymorphisms and Sickle Cell Disease: An Egyptian Study
 Interleukin-1ß and Interleukin-6 Genetic Polymorphisms and Sickle Cell Disease: An Egyptian Study MONA EL-GHAMRAWY, MD, PROFESSOR OF PEDIATRICS & PEDIATRIC HEMATOLOGY, CAIRO UNIVERSITY melghamrawy95@gmail.com
Interleukin-1ß and Interleukin-6 Genetic Polymorphisms and Sickle Cell Disease: An Egyptian Study MONA EL-GHAMRAWY, MD, PROFESSOR OF PEDIATRICS & PEDIATRIC HEMATOLOGY, CAIRO UNIVERSITY melghamrawy95@gmail.com
1 Kattamis et al. Growth of Children with Thalassemia: Effect of Different Transfusion Regimens. Archives of
 Objectives Sickle Cell Anemia and Thalassemia: Transplantation Provide overview of hemoglobinopathies: Sickle cell disease and Thalassemia Discuss approaches to therapy Review recent registry collaboration
Objectives Sickle Cell Anemia and Thalassemia: Transplantation Provide overview of hemoglobinopathies: Sickle cell disease and Thalassemia Discuss approaches to therapy Review recent registry collaboration
HAEMOLYTIC ANAEMIA. Dr. Hasan Fahmawi, MRCP(London), FRCP(Edin) Consultant Physician
, FRCP(Edin) Consultant Physician") HAEMOLYTIC ANAEMIA Dr. Hasan Fahmawi, MRCP(London), FRCP(Edin) Consultant Physician Haemolysis Definition shortening of the normal red blood lifespan of 120 days Increase in unconjugated bilirubin, increased
HAEMOLYTIC ANAEMIA Dr. Hasan Fahmawi, MRCP(London), FRCP(Edin) Consultant Physician Haemolysis Definition shortening of the normal red blood lifespan of 120 days Increase in unconjugated bilirubin, increased
Acute Complications of Sickle Cell Disease Case Study 5 year old girl with Hemoglobin SS, weakness and slurred speech
 Acute Complications of Sickle Cell Disease Case Study 5 year old girl with Hemoglobin SS, weakness and slurred speech Beatrice E. Gee, MD Medical Director, Sickle Cell and Hematology Program Children s
Acute Complications of Sickle Cell Disease Case Study 5 year old girl with Hemoglobin SS, weakness and slurred speech Beatrice E. Gee, MD Medical Director, Sickle Cell and Hematology Program Children s
Acute Complications of Sickle Cell Disease
 Management of Acute Complications of Sickle Cell Disease A Pocket Guide for the Clinician Timothy McCavit, MD, MSCS 1 Payal Desai, MD 1 University of Texas Southwestern Medical Center The Ohio State University,
Management of Acute Complications of Sickle Cell Disease A Pocket Guide for the Clinician Timothy McCavit, MD, MSCS 1 Payal Desai, MD 1 University of Texas Southwestern Medical Center The Ohio State University,
Transfusion in Sickle Cell Disease What the guidelines [are likely to] say. Dr Bernard Davis Whittington Hospital, London
![Transfusion in Sickle Cell Disease What the guidelines [are likely to] say. Dr Bernard Davis Whittington Hospital, London](/thumbs/85/91702350.jpg "Transfusion in Sickle Cell Disease What the guidelines [are likely to] say. Dr Bernard Davis Whittington Hospital, London") Transfusion in Sickle Cell Disease What the guidelines [are likely to] say Dr Bernard Davis Whittington Hospital, London Background to BCSH Guideline Rationale Current guidance in disparate publications
Transfusion in Sickle Cell Disease What the guidelines [are likely to] say Dr Bernard Davis Whittington Hospital, London Background to BCSH Guideline Rationale Current guidance in disparate publications
Guidelines for Shared Care Centres and Community Staff
 Reference: CG1411 Written by: Dr Jenny Welch Peer reviewer Dr Jeanette Payne Approved: September 2015 Approved by D&TC: 10 th July 2015 Review Due: September 2018 Intended Audience This document contains
Reference: CG1411 Written by: Dr Jenny Welch Peer reviewer Dr Jeanette Payne Approved: September 2015 Approved by D&TC: 10 th July 2015 Review Due: September 2018 Intended Audience This document contains
Thalassemia Maria Luz Uy del Rosario, M.D.
 Thalassemia Maria Luz Uy del Rosario, M.D. Philippine Society of Hematology and Blood Transfusion Philippine Society of Pediatric Oncology What is Thalassemia Hereditary Hemoglobin disorder Hemolytic anemia
Thalassemia Maria Luz Uy del Rosario, M.D. Philippine Society of Hematology and Blood Transfusion Philippine Society of Pediatric Oncology What is Thalassemia Hereditary Hemoglobin disorder Hemolytic anemia
Multidisciplinary care. Michael Angastiniotis
 Multidisciplinary care Michael Angastiniotis Pathopysiology of β-thalassaemia Thalassaemia syndromes are inherited haemoglobin disorders caused by defective and imbalanced globin production Excess free
Multidisciplinary care Michael Angastiniotis Pathopysiology of β-thalassaemia Thalassaemia syndromes are inherited haemoglobin disorders caused by defective and imbalanced globin production Excess free
Approach to Hemolysis
 Objectives: Approach to Hemolysis To know the function of platelets and the relationship between the platelet count in peripheral blood and the extent of abnormal bleeding. To know about the diseases associated
Objectives: Approach to Hemolysis To know the function of platelets and the relationship between the platelet count in peripheral blood and the extent of abnormal bleeding. To know about the diseases associated
COHEM Barcellona 2012 Hemoglobinopathies debate
 COHEM Barcellona 2012 Hemoglobinopathies debate September 8, 2012: h. 10:30-12:00 Hall: A Is it justified to perform BMT in hemoglobinopathies using unrelated and/or partially mismatched donors? HSCT indication
COHEM Barcellona 2012 Hemoglobinopathies debate September 8, 2012: h. 10:30-12:00 Hall: A Is it justified to perform BMT in hemoglobinopathies using unrelated and/or partially mismatched donors? HSCT indication
Done by :Aseel Twaijer & Laith Sorour Hemolytic Anemias
 Hemolytic Anemias Hemolytic anemias share the following features: - A shortened red cell life < 120 days - Elevated erythropoietin levels (compensatory increase in erythropoiesis) - Accumulation of hemoglobin
Hemolytic Anemias Hemolytic anemias share the following features: - A shortened red cell life < 120 days - Elevated erythropoietin levels (compensatory increase in erythropoiesis) - Accumulation of hemoglobin
HEMOLYTIC ANEMIA DUE TO ABNORMAL HEMOGLOBIN SYNTHESIS
 Hemolytic Anemia Due to Abnormal Hemoglobin Synthesis MODULE 19 HEMOLYTIC ANEMIA DUE TO ABNORMAL HEMOGLOBIN SYNTHESIS 19.1 INTRODUCTION There are two main mechanisms by which anaemia is produced (a) Thalassemia:
Hemolytic Anemia Due to Abnormal Hemoglobin Synthesis MODULE 19 HEMOLYTIC ANEMIA DUE TO ABNORMAL HEMOGLOBIN SYNTHESIS 19.1 INTRODUCTION There are two main mechanisms by which anaemia is produced (a) Thalassemia:
The Thalassemias in Clinical Practice. Ashutosh Lal, MD Director Comprehensive Thalassemia Program UCSF Benioff Children s Hospital Oakland
 The Thalassemias in Clinical Practice Ashutosh Lal, MD Director Comprehensive Thalassemia Program UCSF Benioff Children s Hospital Oakland Outline Thalassemia: definitions and pathophysiology Epidemiology
The Thalassemias in Clinical Practice Ashutosh Lal, MD Director Comprehensive Thalassemia Program UCSF Benioff Children s Hospital Oakland Outline Thalassemia: definitions and pathophysiology Epidemiology
Historic and Current Complications in Children with Sickle Cell Disease
 Historic and Current Complications in Children with Sickle Cell Disease Trish McMahon Peterson RN, MSN, CPNP Thomas C. Hofstra MD Children's Hospital Los Angeles Comprehensive Sickle Cell Program Children's
Historic and Current Complications in Children with Sickle Cell Disease Trish McMahon Peterson RN, MSN, CPNP Thomas C. Hofstra MD Children's Hospital Los Angeles Comprehensive Sickle Cell Program Children's
Routine Management of Sickle Cell Anaema
 Routine Management of Sickle Cell Anaema By Date: 1 St March 2014 Prof. E. O. Temiye, Department of Paediatrics, College of Medicine University of Lagos & Consultant Paediatrician, Lagos University Teaching
Routine Management of Sickle Cell Anaema By Date: 1 St March 2014 Prof. E. O. Temiye, Department of Paediatrics, College of Medicine University of Lagos & Consultant Paediatrician, Lagos University Teaching
Disclosure. Hemoglobinopathies. Screening for Hemoglobinopthies. Learning Objectives. Screening for Hemoglobinopthies. Interpreting Reports
 Disclosure Hemoglobinopathies (everything you wanted to know but were afraid to ask) Melissa Frei-Jones, MD MSCI Pediatric Grand Rounds February 21, 2014 I have no relationships with commercial companies
Disclosure Hemoglobinopathies (everything you wanted to know but were afraid to ask) Melissa Frei-Jones, MD MSCI Pediatric Grand Rounds February 21, 2014 I have no relationships with commercial companies
Chem*3560 Lecture 4: Inherited modifications in hemoglobin
 Chem*3560 Lecture 4: Inherited modifications in hemoglobin Genetic modifications fall into two classes: Thalassemias, which are the result of failure to express globin genes. Thalassa is Greek for the
Chem*3560 Lecture 4: Inherited modifications in hemoglobin Genetic modifications fall into two classes: Thalassemias, which are the result of failure to express globin genes. Thalassa is Greek for the
The Child with a Hematologic Alteration
 47 The Child with a Hematologic Alteration HELPFUL HINT Review the anatomy and physiology of the hematologic system in an anatomy and physiology textbook. MATCHING KEY TERMS Match the term with the correct
47 The Child with a Hematologic Alteration HELPFUL HINT Review the anatomy and physiology of the hematologic system in an anatomy and physiology textbook. MATCHING KEY TERMS Match the term with the correct
Quiz. What percentage of the world s population is a carrier of a hemoglobinopathy? Hemoglobinopathies in Pregnancy 1-2% 5-7% 8-12% 10-15%
 Hemoglobinopathies in Pregnancy Emily Parkhurst, MS, LCGC Kaiser West Los Angeles November 2017 Genetics Department Quiz What percentage of the world s population is a carrier of a hemoglobinopathy? 1-2%
Hemoglobinopathies in Pregnancy Emily Parkhurst, MS, LCGC Kaiser West Los Angeles November 2017 Genetics Department Quiz What percentage of the world s population is a carrier of a hemoglobinopathy? 1-2%
Haemoglobinopathy and sickle cell disease
 Key points Sickle cell disease (SCD) is a congenital haemoglobinopathy characterized by a mutation on chromosome 11, resulting in the production of the unstable and relatively insoluble haemoglobin S.
Key points Sickle cell disease (SCD) is a congenital haemoglobinopathy characterized by a mutation on chromosome 11, resulting in the production of the unstable and relatively insoluble haemoglobin S.
Dr. MUNEER ALBAGSHI Consultant Pediatric Hematologist Oncologist- HBDC, Al-Ahsa. Saudi Arabia
 Dr. MUNEER ALBAGSHI Consultant Pediatric Hematologist Oncologist- HBDC, Al-Ahsa. Saudi Arabia Sickle cell is global disease of old world and immigrants to the new world. Sickle cell anemia to predict that
Dr. MUNEER ALBAGSHI Consultant Pediatric Hematologist Oncologist- HBDC, Al-Ahsa. Saudi Arabia Sickle cell is global disease of old world and immigrants to the new world. Sickle cell anemia to predict that
Pitfalls in the premarital testing for thalassaemia
 Pitfalls in the premarital testing for thalassaemia Dr. Riad Amer MB ChB, MSc, FRCP, FRCPath, JBH Assistant Professor of Medicine Al Najah University Consultant Haematologist Case 1 Husband and Wife are
Pitfalls in the premarital testing for thalassaemia Dr. Riad Amer MB ChB, MSc, FRCP, FRCPath, JBH Assistant Professor of Medicine Al Najah University Consultant Haematologist Case 1 Husband and Wife are
Report of Beta Thalassemia in Newar Ethnicity
 Report of Beta Thalassemia in Newar Ethnicity Rajendra Dev Bhatt 1*, Surendra Koju 2, Prabodh Risal 1 Affiliations: 1 Department of Clinical Biochemistry, Dhulikhel Hospital, Kathmandu University Hospital
Report of Beta Thalassemia in Newar Ethnicity Rajendra Dev Bhatt 1*, Surendra Koju 2, Prabodh Risal 1 Affiliations: 1 Department of Clinical Biochemistry, Dhulikhel Hospital, Kathmandu University Hospital
An overview of Thalassaemias and Complications
 An overview of Thalassaemias and Complications Haemoglobin Haemoglobin is the most abundant protein in blood, and exists as three main types in normal adults: HbA ( ) - 97% HbA 2 ( ) - 2.5% HbF ( ) - 0.5%
An overview of Thalassaemias and Complications Haemoglobin Haemoglobin is the most abundant protein in blood, and exists as three main types in normal adults: HbA ( ) - 97% HbA 2 ( ) - 2.5% HbF ( ) - 0.5%
COEXISTENCE OF β-thalassemia AND POLYCYTHEMIA VERA: A CHICKEN-AND-EGG DEBATE?
 COEXISTENCE OF β-thalassemia AND POLYCYTHEMIA VERA: A CHICKEN-AND-EGG DEBATE? M. DE SLOOVERE (1), L. HARLET (2), S. VAN STEENWEGHEN (3), E. MOREAU (1), D. DE SMET (1) (1) DEPARTMENT OF LABORATORY MEDICINE,
COEXISTENCE OF β-thalassemia AND POLYCYTHEMIA VERA: A CHICKEN-AND-EGG DEBATE? M. DE SLOOVERE (1), L. HARLET (2), S. VAN STEENWEGHEN (3), E. MOREAU (1), D. DE SMET (1) (1) DEPARTMENT OF LABORATORY MEDICINE,
Sickle Cell Anemia. Sickle cell anemia is an inherited disorder of the blood which occurs when just one base pair substitution
 Rose Farrington and Rachel Nash BIOL 362 Lab M. Bulgarella Genetic Diseases 10/14/2008 Sickle Cell Anemia Introduction Sickle cell anemia is an inherited disorder of the blood which occurs when just one
Rose Farrington and Rachel Nash BIOL 362 Lab M. Bulgarella Genetic Diseases 10/14/2008 Sickle Cell Anemia Introduction Sickle cell anemia is an inherited disorder of the blood which occurs when just one
Genetics of Thalassemia
 Genetics of Thalassemia Submitted by : Raya Samir Al- Hayaly Sura Zuhair Salih Saad Ghassan Al- Dulaimy Saad Farouq Kassir Sama Naal Salouha Zahraa Jasim Al- Aarajy Supervised by : Dr. Kawkab Adris Mahmod
Genetics of Thalassemia Submitted by : Raya Samir Al- Hayaly Sura Zuhair Salih Saad Ghassan Al- Dulaimy Saad Farouq Kassir Sama Naal Salouha Zahraa Jasim Al- Aarajy Supervised by : Dr. Kawkab Adris Mahmod
Marie-Christine Hendriks PGY 5 Pediatric Respirology CHU Sainte-Justine
 Marie-Christine Hendriks PGY 5 Pediatric Respirology CHU Sainte-Justine Hemoglobinopathy (AR) Amino acid substitution in the beta globin chain of hemoglobin = HbS Hb S is poorly soluble when deoxygenated
Marie-Christine Hendriks PGY 5 Pediatric Respirology CHU Sainte-Justine Hemoglobinopathy (AR) Amino acid substitution in the beta globin chain of hemoglobin = HbS Hb S is poorly soluble when deoxygenated
Hydroxycarbamide. Sickle and Thalassaemia Training days. September Dr Sara Stuart-Smith. Why do sickle cells cause pain and organ damage?
 Sickle and Thalassaemia Training days September 2017 Hydroxycarbamide Dr Sara Stuart-Smith Why do sickle cells cause pain and organ damage? Under certain conditions, haemoglobin S forms long rigid strands
Sickle and Thalassaemia Training days September 2017 Hydroxycarbamide Dr Sara Stuart-Smith Why do sickle cells cause pain and organ damage? Under certain conditions, haemoglobin S forms long rigid strands
Sickle cell anaemia. GP Education Update 19 th July 2018 Croydon University Hospital. Arne de Kreuk Consultant Haematologist
 Sickle cell anaemia GP Education Update 19 th July 201 Croydon University Hospital Arne de Kreuk Consultant Haematologist Outline: Prevention of sickle cell related complications; annual review Identification
Sickle cell anaemia GP Education Update 19 th July 201 Croydon University Hospital Arne de Kreuk Consultant Haematologist Outline: Prevention of sickle cell related complications; annual review Identification
Dr.Abdolreza Afrasiabi
 Dr.Abdolreza Afrasiabi Thalassemia & Heamophilia Genetic Reaserch Center Shiraz Medical University Hemoglobin tetramer Hemoglobin Structure % A 1 α 2 β 2 94-97% A 2 α 2 δ 2 2.5% A 1C α 2 (β-n-glucose)
Dr.Abdolreza Afrasiabi Thalassemia & Heamophilia Genetic Reaserch Center Shiraz Medical University Hemoglobin tetramer Hemoglobin Structure % A 1 α 2 β 2 94-97% A 2 α 2 δ 2 2.5% A 1C α 2 (β-n-glucose)
Apheresis red cell exchange and transfusion therapy for management of sickle cell disease. Dr Paul Telfer
 Apheresis red cell exchange and transfusion therapy for management of sickle cell disease Dr Paul Telfer OVERVIEW Pathophysiology and epidemiology Physiological and clinical considerations prior to transfusion
Apheresis red cell exchange and transfusion therapy for management of sickle cell disease Dr Paul Telfer OVERVIEW Pathophysiology and epidemiology Physiological and clinical considerations prior to transfusion
THALASSEMIA AND COMPREHENSIVE CARE
 1 THALASSEMIA AND COMPREHENSIVE CARE Melanie Kirby MBBS, FRCP (C), Hospital for Sick Children, Toronto Associate Professor of Paediatrics, University of Toronto. Objectives 2 By the end of this presentation,
1 THALASSEMIA AND COMPREHENSIVE CARE Melanie Kirby MBBS, FRCP (C), Hospital for Sick Children, Toronto Associate Professor of Paediatrics, University of Toronto. Objectives 2 By the end of this presentation,
LOOK BEYOND THE OBVIOUS DR. PADMASANI L. N DR. ARUNA RAJENDRAN PRESENTOR : DR. VIGNATHA SAJJA
 LOOK BEYOND THE OBVIOUS DR. PADMASANI L. N DR. ARUNA RAJENDRAN PRESENTOR : DR. VIGNATHA SAJJA HISTORY Baby X, 5 year old, Girl from Madhya Pradesh CHIEF COMPLAINTS : Fever and body pain x 5 days Abdomen
LOOK BEYOND THE OBVIOUS DR. PADMASANI L. N DR. ARUNA RAJENDRAN PRESENTOR : DR. VIGNATHA SAJJA HISTORY Baby X, 5 year old, Girl from Madhya Pradesh CHIEF COMPLAINTS : Fever and body pain x 5 days Abdomen
Transfusion support in Thalassaemia. Dr.A.keerti 1 st year PG DEPT. OF TRANSFUSION MEDICINE
 Transfusion support in Thalassaemia Dr.A.keerti 1 st year PG DEPT. OF TRANSFUSION MEDICINE Structure of hemoglobin Types of hemoglobins Hemoglobin-Development Switching Thalassaemia- introduction Classification
Transfusion support in Thalassaemia Dr.A.keerti 1 st year PG DEPT. OF TRANSFUSION MEDICINE Structure of hemoglobin Types of hemoglobins Hemoglobin-Development Switching Thalassaemia- introduction Classification
6.1 Extended family screening
 CHAPTER 6 CONCLUSION Cost benefit analysis of thalassemia screening programs have shown that the single years treatment for a β-thalassemia major patient was much higher than a total cost per case prevented.
CHAPTER 6 CONCLUSION Cost benefit analysis of thalassemia screening programs have shown that the single years treatment for a β-thalassemia major patient was much higher than a total cost per case prevented.
Gaining perspective on the spectrum of sickle cell anemia: Question: What do all these real life presentations have in common?
 Sickle-Cell Management Objectives: After reading this lecture the participant should be able to: 1. Explain the basic disease process of Sickle Cell Anemia 2. Have an understanding of the precipitants
Sickle-Cell Management Objectives: After reading this lecture the participant should be able to: 1. Explain the basic disease process of Sickle Cell Anemia 2. Have an understanding of the precipitants
UNRELATED DONOR TRANSPLANTATION FOR SICKLE CELL DISEASE AN UPDATE
 UNRELATED DONOR TRANSPLANTATION FOR SICKLE CELL DISEASE AN UPDATE Naynesh Kamani, M.D. Children s National Medical Center GW University School of Medicine Washington, DC SCD scope of problem in USA Commonest
UNRELATED DONOR TRANSPLANTATION FOR SICKLE CELL DISEASE AN UPDATE Naynesh Kamani, M.D. Children s National Medical Center GW University School of Medicine Washington, DC SCD scope of problem in USA Commonest
HU: Myths and Facts. Melanie Kirby Associate Professor of Paediatrics
 HU: Myths and Facts Melanie Kirby Associate Professor of Paediatrics SACGO Hamilton, Ontario March 5, 2016 Declaration of Disclosure I have no actual or potential conflict of interest in relation to this
HU: Myths and Facts Melanie Kirby Associate Professor of Paediatrics SACGO Hamilton, Ontario March 5, 2016 Declaration of Disclosure I have no actual or potential conflict of interest in relation to this
Perioperative Management of Patients with Sickle Cell Disease
 Perioperative Management of Patients with Sickle Cell Disease November 29 th, 2012 David Vivas, MD Case - History CC: RLQ pain HPI: 14 y/o female with h/o Hg SC presented to ED with 4 days h/o abdominal
Perioperative Management of Patients with Sickle Cell Disease November 29 th, 2012 David Vivas, MD Case - History CC: RLQ pain HPI: 14 y/o female with h/o Hg SC presented to ED with 4 days h/o abdominal
Blood transfusions in sepsis, the elderly and patients with TBI
 Blood transfusions in sepsis, the elderly and patients with TBI Shabbir Alekar MICU, CH Baragwanath Academic Hospital & The University of the Witwatersrand CCSSA Congress 11 June 2015 Packed RBC - complications
Blood transfusions in sepsis, the elderly and patients with TBI Shabbir Alekar MICU, CH Baragwanath Academic Hospital & The University of the Witwatersrand CCSSA Congress 11 June 2015 Packed RBC - complications
Compassionate-use Experience With Voxelotor (GBT440) for Patients With Severe Sickle Cell Disease (SCD) and Life-Threatening Comorbidities
 for Patients With Severe Sickle Cell Disease (SCD) and Life-Threatening Comorbidities") Compassionate-use Experience With Voxelotor (GBT440) for Patients With Severe Sickle Cell Disease (SCD) and Life-Threatening Comorbidities Gershwin Blyden, MD 1, Kenneth R. Bridges, MD 2, Lanetta Bronté,
Compassionate-use Experience With Voxelotor (GBT440) for Patients With Severe Sickle Cell Disease (SCD) and Life-Threatening Comorbidities Gershwin Blyden, MD 1, Kenneth R. Bridges, MD 2, Lanetta Bronté,
Thalassaemia. What is thalassaemia? What causes thalassaemia? What are the different types of thalassaemia?
 Thalassaemia Thalassaemia is an inherited condition affecting the blood. There are different types, which vary from a mild condition with no symptoms, to a serious or lifethreatening condition. For the
Thalassaemia Thalassaemia is an inherited condition affecting the blood. There are different types, which vary from a mild condition with no symptoms, to a serious or lifethreatening condition. For the