MAST CELL ACTIVATION AND SIGNALING IN THE AUTOIMMUNE DISEASE BULLOUS PEMPHIGOID. Lisa A. Heimbach
|
|
|
- Abigayle Hines
- 6 years ago
- Views:
Transcription
1 MAST CELL ACTIVATION AND SIGNALING IN THE AUTOIMMUNE DISEASE BULLOUS PEMPHIGOID Lisa A. Heimbach A dissertation submitted to the faculty of the University of North Carolina at Chapel Hill in partial fulfillment of the requirements for the degree of Doctor of Philosophy in the Department of Microbiology and Immunology Chapel Hill 2012 Approved by: Zhi Liu, PhD Roland Tisch, PhD Edward Collins, PhD Jenny P-Y Ting, PhD Lishan Su, PhD
2 2012 Lisa A. Heimbach ALL RIGHTS RESERVED ii
3 ASTRACT LISA HEIMBACH: Mast cell activation and signaling in the autoimmune disease bullous pemphigoid (Under the direction of Dr. Zhi Liu) Bullous pemphigoid (BP) is an autoimmune skin blistering disorder primarily observed in the elderly. Autoantibodies directed against the hemidesmosomal protein BP180 trigger a pathological inflammatory response that causes separation of the epidermis from the underlying dermis. Complement, mast cells (MCs), and polymorphonuclear neutrophils (PMNs) are required for disease in experimental BP, an animal model that closely mimics the clinical, immunological and histological features of human BP. In this work, we investigated MC activation and signaling in experimental BP. MC degranulation occurs downstream of complement activation and the generation of complement component 5a (C5a) in experimental BP. We determined that C5a binds to the C5a receptor (C5aR) on MCs in neonatal mice given disease-inducing antibodies. C5a-C5aR interaction significantly increases phosphorylation of the intracellular signaling protein p38 mitogen-activated protein kinase (p38mapk). Pharmacologically blocking p38mapk activation protected mice from MC degranulation and clinical disease. Taken together, we demonstrated that the binding of C5a to C5aR on MCs activates the p38mapk signaling pathway and leads to MC degranulation and skin blistering. Upon degranulation, MCs release bioactive compounds from their secretory granules, including tumor necrosis factor-alpha (TNF-α). Here, we report that MC-derived TNF-α is iii
4 required for disease in experimental BP. Mice lacking TNF-α globally or in MCs alone fail to recruit sufficient numbers of PMNs to the skin and do not develop clinical blisters following injection with pathogenic anti-bp180 antibodies. C5aR-deficient mice are protected from blistering and do not exhibit elevated TNF-α levels or MC degranulation. TNF-α receptor 1 (TNFR1) expression on MCs is required for development of experimental BP, suggesting that TNF-α acts in an autocrine fashion on MCs. The findings described in this dissertation refine our understanding of the mechanisms of MC degranulation in experimental BP. MCs are activated by the binding of C5a to C5aR. C5a-C5aR interaction leads to activation of p38mapk and MC degranulation. MC degranulation releases TNF-α, and TNF-α acts in an autocrine manner on MC TNFR1 to promote disease development. In addition to providing insight into the pathogenesis of BP, our data also suggests that C5a, p38mapk, and TNF-α may be promising therapeutic targets for treatment of human disease. iv
5 This dissertation is dedicated to my husband, Matthew, and my parents, Mike and Kim. Thank you for your love and support. v
6 ACKNOWLEDGEMENTS I would like to thank my advisor, Dr. Zhi Liu, for his support and guidance throughout my graduate school career. From him, I have learned to be an independent thinker, a careful and creative scientist, a critical reviewer, and a thoughtful story teller. I appreciate the opportunities he gave me to explore in science, and I hope to keep his sense of optimism and enthusiasm with me always. To the current and past members of the Liu lab, I would like to thank all of you for your advice, training, and friendship. I would particularly like to thank Flor Evangelista for her enduring friendship and scientific guidance. It is difficult to imagine graduate school without Flor, and I will always be grateful for her support and proud to call her a colleague. I thank Jaime Brozowski and Anne Weaver for their assistance managing our animal colonies. I would also like to thank Lan Lin, Ying Ching Liang, Peng Geng, Elizabeth Lessey, Julia Huang, Ming Lang Zhao, and Megan Meade for their help, ideas, and positive attitudes. I am also grateful to my other graduate school mentors, particularly the members of my thesis committee, Drs. Roland Tisch, Ed Collins, Jenny Ting, and Lishan Su. Their thoughtful suggestions and wise advice were instrumental to my scientific progress. I would like to acknowledge key members of the UNC Department of Dermatology, including Drs. Luis Diaz, David Rubenstein, and Ning Li. Their clinical and scientific expertise was an amazing resource during my training. vi
7 I was very fortunate to make three great friends on my first day of graduate school who have inspired me every step of the way. To Monika Schneider, Mark Johnson, and Rachael Liesman, thank you for sharing this journey with me. Thanks also to Aadra Bhatt and Sean McNally, two friends and colleagues I met a little later but who are just as dear. Finally, I would like to thank my family for their love and encouragement throughout graduate school and my life in general. My parents Mike and Kim Leighty taught me from an early age to be intellectually curious and to pursue my dreams. My sisters Michelle and Laura Leighty have supported and believed in me every step of the way. Most importantly, I would like to thank my husband Matthew for his unwaveringly love and faith in me. vii
8 TABLE OF CONTENTS LIST OF TABLES... x LIST OF FIGURES... xi LIST OF ABBREVIATIONS... xii CHAPTER 1: BACKGROUND AND SIGNIFICANCE BULLOUS PEMPHIGOID Clinical Features of Bullous Pemphigoid in Humans Animal Models of Bullous Pemphigoid Immunopathogenesis of Experimental BP in the Murine Model Current Therapeutic Strategies For Treatment of Human BP MAST CELL BIOLOGY MC Development and Function in the Skin C5aR mediated MC Activation p38mapk Signaling in MCs TNF-α mediated MC Activation MCs in Autoimmunity SUMMARY AND CONCLUDING REMARKS viii
9 1.4 REFERENCES CHAPTER 2: THE C5A RECEPTOR ON MAST CELLS IS CRITICAL FOR THE AUTOIMMUNE DISEASE BULLOUS PEMPHIGOID INTRODUCTION EXPERIMENTAL PROCEDURES RESULTS DISCUSSION REFERENCES CHAPTER 3: FUNCTIONAL INTERACTION BETWEEN COMPLEMENT AND THE PRO-INFLAMMATORY CYTOKINE TNF-α IN EXPERIMENTAL BULLOUS PEMPHIGOID INTRODUCTION EXPERIMENTAL PROCEDURES RESULTS DISCUSSION REFERENES CHAPTER 4: DISCUSSION INTRODUCTION FINDINGS AND IMPLICATIONS REMAINING QUESTIONS AND FUTURE DIRECTIONS CONCLUDING REMARKS REFERENCES ix
10 LIST OF TABLES Table 2.1. Role of C5a receptor and p38 MAPK in experimental BP x
11 LIST OF FIGURES Figure 1.1 Figure 1.2 Figure 2.1 Figure 2.2. Figure 2.3. Figure 2.4 Figure 2.5 Figure 2.6. Figure 3.1. Figure 3.2. Figure 3.3. Figure 3.4. Figure 3.5. Figure 3.6. Figure 3.7. Figure 3.8. Organization of BP180 in human, mouse, and the NC16A+/+ humanized mouse Mechanism of subepidermal blistering in experimental BP.. 7 Level of C5a in the skin is significantly reduced in pathogenic IgG-injected mice lacking the classical pathway of complement Mice lacking C5a receptor are resistant to experimental BP Mice lacking C5aR show impaired p38mapk activation induced by pathogenic antibodies 57 Blocking p38mapk activation abolishes experimental BP C5a activates p38mapk of MCs in vivo and in vitro MC reconstitution restores BP in MC-deficient mice. 60 Pathogenic anti-mbp180 IgG induces BP blisters associated with an elevated level of TNF-α in mouse skin.. 84 TNF-α-deficient mice are resistant to experimental BP. 85 Time-course of TNF-α expression during BP disease development in mice 86 MC reconstitution restores BP in TNF-α-/- and MC-deficient mice C5aR on MCs are required for TNF-α release and skin blistering.. 88 TNFR1 and not TNFR2 is required for experimental BP Local reconstitution of MCs bearing TNFR1 and not TNFR2 restores BP disease in TNFR-deficient mice Experimental BP induced by patient anti-bp180 autoantibodies depends on TNF-α Figure 3.9. `Proposed model for MC regulation of experimental BP xi
12 LIST OF ABBREVIATIONS Ab BMZ BP BPAG BSA C3a C3aR C5a C5aR CD CHAPS CTMC COLXVII DC DEJ ECL ECM F(ab) FADD Fb Fc FcγIIIR Antibody Basement membrane zone Bullous pemphigoid Bullous pemphigoid antigen Bovine serum albumin Complement component 3a Complement component 3a receptor Complement component 5a Complement component 5a receptor Cluster of differentiation 3-(3-cholamidopropyl)dimethylammonio-1-propanesulfonic acid Connective tissue mast cell Collagen seventeen Dendritic cell Dermal-epidermal junction Enhanced chemiluminescence Extracellular matrix Fragment (antigen binding) Fas associated death domain Factor B Fragment, crystallizable Fragment, crystallizable gamma three receptor xii
13 g GPCR GB GM-CSF GST HSC ICD IF Ig IL IVIG H&E KC LC LPS MAPK MAPKK MAPKKK mbp180 MC mg Gravity G protein coupled receptor Gelatinase B Granulocyte macrophage colony stimulating factor Glutathione S-Transferase Hematopoetic stem cell Intracellular domain Immunofluorescence Immunoglobulin Interleukin Intravenous immunoglobulin Hematoxylin and eosin Keratinocyte Langerhans cell Lipopolysaccharide Mitogen-activated protein kinase Mitogen-activated protein kinase kinase Mitogen-activated protein kinase kinase kinase Murine BP180 Mast cell Milligram MIP-2 Macrophage inflammatory protein 2 ml Milliliter xiii
14 μl mm μm Microliter Millimolar Micromolar mmcp4 Mouse mast cell protease 4 MΦ MPO MS MTMC NaCl NC NE NFAT NF-kB Ng OD PMN PVDF rc5a RIP SDS-PAGE SEM SODD Th Macrophage Myeloperoxidase Multiple sclerosis Mucosal tissue mast cell Sodium chloride Non-collagen Neutrophil elastase Nuclear factor of activated T cells Nuclear factor-kappa B Nanogram Optical density Polymorphonuclear neutrophil Polyvinylidene fluoride Recombinant complement component 5a Receptor-interacting protein Sodium dodecyl sulfate polyacrylamide gel electrophoresis Standard error of mean Silencer of death domain T helper xiv
15 TLR TNF TNFR TRADD TRAF Treg WT Toll-like receptor Tumor necrosis factor Tumor necrosis factor receptor TNF-receptor associated death domain TNF-receptor associated factor Regulatory T cell Wild type xv
16 CHAPTER 1 BACKGROUND AND SIGNIFICANCE BULLOUS PEMPHIGOID Bullous pemphigoid (BP) is a human autoimmune and inflammatory disease first described by the dermatologist Walter Lever in 1953 (1). BP is the most prevalent autoimmune skin blistering disease, with 10 new cases per million population reported annually in the United States (2). Unlike many other autoimmune diseases, BP primarily afflicts men and the elderly, with an average age of onset in the 80s (3). BP is associated with significant morbidity and mortality, with between 10% and 40% of patients dying within three years of disease onset (4, 5). The global disease burden of BP is expected to increase over time as the population ages. Although it is a relatively uncommon disease, BP is particularly useful as a paradigm for an organ-specific autoimmune disease based on the well understood mechanism of pathogenesis Clinical Features of Bullous Pemphigoid in Humans BP is characterized by the presence of tense, fluid filled blisters on the skin (1). Blister formation occurs in areas where the basal keratinocytes of the epidermis detach from the underlying dermis (6). This dermal-epidermal junction (DEJ) separation is associated 1 Portions of this chapter have been reproduced/amended from: Heimbach, L., Li, N., Diaz, L.A., and Liu, Z. Experimental animal models of bullous pemphigoid. G Ital Dermatol Venereol (4):
17 with a cellular inflammatory infiltrate in the upper dermis and the bullous cavity. Eosinphils are the predominant cell type present, along with polymorphonuclear neutrophils (PMNs), lymphocytes, mast cells (MCs), and monocytes/macrophages (7-12). Eosinophils, PMNs, and MCs are thought to be locally activated in the dermis by the multiple inflammatory mediators present in the lesional skin and blister fluid. These mediators include granular proteins derived from degranulated leukocytes, such as eosinophil cationic protein, eosinophil major basic protein, and PMN-derived myeloperoxidase (MPO) (13-15), and chemoattractants and cytokines, such as C5a fragments, histamine, leukotriene B4, interleukin (IL)-1, -2, -4, -5, -6, -8, -15, tumor necrosis factor (TNF)-α, interferon-γ, RANTES, and eotaxin (16-22). A hallmark of BP is the deposition of autoantibodies and complement components along the basement membrane zone (BMZ) of the skin (23, 24). The humoral autoimmune reaction in BP is directed against components of the hemidesmosome, a cellular adhesion complex that anchors the basal keratinocytes of the epidermis to the underlying extracellular matrix (ECM) (24, 25). A polyclonal repertoire of both circulating and tissue-bound autoantibodies targets two major hemidesmosomal antigens, the 230 kilodalton (kda) protein termed BP230 or bullous pemphigoid antigen (BPAG) 1, and the 180 kda protein termed BP180, BPAG2, or type XVII collagen (26-29). BP230 localizes to the hemidesmosomal plaque within the intracellular domain of the keratinocyte (30, 31). BP180 is a type II transmembrane protein, and the amino-terminal region also localizes to the intracellular hemidesmosomal plaque (30, 32, 33). The carboxyl-terminal portion of BP180 extends into the extracellular milieu of the basement membrane zone (BMZ) and interacts with laminin- 332 and alpha-6 integrin to promote keratinocyte stability and adhesion (34, 35). The 2
18 ectodomain of human BP180 consists of 15 collagen domains (designated C1 through C15) interspersed by 16 non-collagen sequences (NC1 through NC16A) (Figure 1.1) (32). NC16A is the largest of the non-collagen regions and is adjacent to the transmembrane domain of BP180. Epitope mapping studies demonstrated that the NC16A domain of BP180 harbors multiple epitopes recognized by autoreactive immunoglobulins (Ig) of IgE and IgG isotypes and IgG1 and IgG4 subclasses (36-40). Serum titers of anti-nc16a autoantibodies are correlated with disease severity (37, 41, 42). Together, these data demonstrate that BP180 is the key autoantigen in BP. In addition to the humoral response directed against BP180, most BP patients also exhibit T cell mediated autoimmunity as well. Autoreactive CD4+ T lymphocytes expressing memory cell surface markers recognize epitopes within the extracellular region of BP180, mostly clustering within the NC16A domain (43, 44). The autoreactive T cells exhibit a Th1/Th2 mixed cytokine profile. CD4+CD25+Foxp3+ regulatory T cells (Tregs), a subpopulation of T lymphocytes associated with downregulation of autoimmunity (45), do not appear to play a major role in BP pathogenesis. Tregs are present in normal numbers in the circulation of BP patients, and numerous Tregs are found in the lesional and healthy skin of BP patients (46, 47). Tregs isolated from BP patients are also functionally intact and are able to suppress T cell proliferation (47). A recent study found that elevated levels of Th17 cells, a subset of T lymphocytes associated with inflammation and tissue damage in autoimmunity (48), are found in the lesional skin of BP patients; however, no correlation between Th17 cell levels and disease severity were observed (46). Further research into Th17 cell involvement in the pathogenesis of BP is needed. 3
19 1.1.2 Animal Models of Bullous Pemphigoid Efforts to develop an animal model of BP date back to the early 1970s (49). Based on the strong correlation between BP disease severity and serum anti-bp180 and anti-bp230 antibody titers (24), researchers suspected that autoantibodies triggered blister formation. In the first attempt to demonstrate human patient autoantibody pathogenicity in vivo, researchers transfused Rhesus monkeys with plasma from human BP patients (50). Only limited autoantibody deposition was observed at the BMZ, and all three animals failed to develop blisters. Attempts to elicit disease in mice by passively transferring purified BP patient autoantibodies were also unsuccessful (49). Researchers were puzzled by the failure of the experiments, as two related autoimmune intraepidermal blistering diseases, pemphigus vulgaris and pemphigus foliaceus, were successfully modeled in mice using this same passive transfer strategy (51-53). A breakthrough came in 1993, when Zhi Liu and colleagues determined that the amino acid sequence of NC16A, the human BP180 protein recognized by pathogenic patient autoantibodies, is very poorly conserved in murine BP180 (mbp180) (54). Human patient autoantibodies fail to cross react with the homologous murine antigen, termed NC14A (Figure 1.1), and thus do not trigger disease. Liu et. al. then cloned the NC14A portion of mbp180 and expressed the peptide in DH5α cells as a glutathione S-transferase (GST) fusion protein. New Zealand White rabbits were immunized with the purified mbp180 fusion protein and the polyclonal IgG fraction from the serum was collected. Neonatal Balb/c mice injected intradermally with purified rabbit anti-mbp180 IgG developed the key hallmarks of human disease, including clinical skin lesions, in vivo deposition of rabbit IgG and mouse C3 at the basement membrane by direct 4
20 immunofluorescence (IF), dermal-epidermal separation and an extensive inflammatory cell infiltration by H&E staining, with PMNs being the predominant cells (54). Collagen Domains N C NC16A N C NC14A N C NC16A Figure 1.1 Organization of BP180 in human, mouse, and the NC16a+/+ humanized mouse. The human form of BP180 (top) consists of 15 collagen domains (yellow) interspersed by 16 non-collagen domains. The non-collagen domain closest to the cell membrane, NC16A (red), harbors the epitopes recognized by pathogenic autoantibodies. In mouse (middle), BP180 consists of 13 collagen domains and 14 non-collagen domains. NC14A (black) is the murine homolog of NC16A, but the sequence is sufficiently divergent such that antibodies do not cross-react between the two regions. In the humanized NC16A+/+ mouse, the NC16A region replaces the NC14A sequence in murine BP180. The murine model of experimental bullous pemphigoid developed in 1993 has proven to be an invaluable tool for understanding the etiopathogenesis of disease. The research describing the cellular and inflammatory events that contribute to experimental BP development is reviewed in section 1.3 of this document. However, the rabbit anti-mbp180 IgG passive transfer model does not allow for experimentation with BP autoantibodies isolated from human clinical samples. To assess the pathogenicity of human anti-bp180 5
21 autoantibodies, Liu et. al generated a novel mouse strain in which the murine BP180NC14A was replaced with the homologous human BP180NC16A epitope cluster region (Figure 1.1) (55). The humanized NC16A (NC16+/+) mice injected with anti-bp180nc16a autoantibodies purified from human BP patients exhibit in vivo deposition of human IgG and mouse C3 at the basement membrane, dermal-epidermal separation, clinical blistering, and inflammatory cell infiltration. A second humanized animal model was developed by Nishie et. al. that replaces the entire mbp180 protein with the human BP180 homolog (56). Upon injection of human anti-bp180 autoantibodies, these mice (termed COLXVII-humanized) reproduce the DEJ separation at the lamina lucida, the deposition of human IgG along the DEJ, and the inflammatory cell infiltrate consisting of PMNs and eosinophils seen in human disease Immunopathogenesis of Experimental BP in the Murine Model Development of in vivo systems to study an experimental BP model has allowed for great progress in defining the etiopathogenesis of disease. The rabbit anti-mbp180 IgG passive transfer model is easily adapted from use with Balb/c mice to numerous genetically modified mice to explore the requirement for a wide swath of inflammatory mediators in the development of experimental BP. Specifically, the roles of pathogenic antibodies, the complement system, MCs, PMNs, and proteolytic enzymes have been elucidated in the context of the rabbit anti-mbp180 IgG passive transfer model. Figure 1.2 summarizes the key steps identified in the mechanism of subepidermal blistering in experimental BP. 6



















22 Figure 1.2 Mechanism of subepidermal blisteringg in experimental BP. Anti-BP180 antibodies bind to the NC14A domain of BP180 on basal keratinocytes. Antibody binding triggers complement activation and the generation off C5a. C5a induces MC degranulation and the release of proinflammatory mediators. MΦ are activated. PMNs are recruited to the site of inflammation, and the FcRs on PMNs interactt with tissue-bound matrix proteins and cause blistering. antibodies. PMNs release proteolytic enzymes, which degrade extracellular -Pathogenic Antibodies- Injection of anti-mbp180 IgG into a normall neonatal mouse is sufficient to initiate subepidermal blister formation, and the levels of circulating anti-mbp180 that IgG is the primary class antibodies tightly control disease onset and severity (54, 57), indicating of antibody that mediates experimental BP. The rabbit immunization protocol used to raise anti-murine BP180 NC14A generates a polyclonal repertoire off antibodies, and not all anti- BP180 NC14A antibodies are pathogenic. The pathogenic epitope of experimental BP was 7
23 mapped using a panel of deletion mutants of the mbp180 ectodomain in concert with immunoadsorption and immunoblotting assays (58). These mapping studies demonstrated that pathogenic anti-bp180 antibodies recognize a 9-12 amino acid stretch within the murine BP180 NC14A region of the antigen (58). This epitope overlaps the region of the human BP180 NC16A containing the immunodominant epitopes recognized by human BP autoantibodies, supporting the relevance of the murine system as a model for human disease. Epitope mapping studies in the humanized mouse are not yet published. Complement- Complement activation occurs via three pathways- the classical, lectin, and alternative pathways (59). The classical pathway initiates when antigen-antibody binding induces a conformational change in the Fc portion of the antibody, enabling the antibody to bind C1. The C1r subunit is converted to a serine protease that cleaves C4 and C2 to C4a + C4b and C2a + C2b. C4b associates with C2a to form the C3 convertase, while C4a and C2b diffuse and promote inflammation. The alternative pathway does not involve antibody, but instead initiates when the labile thioester bond in the C3 molecule spontaneously hydrolyses to produce C3a and C3b. C3b associates with factor B, which is cleaved by factor D to form C3bBb, the C3 convertase. The mannose binding lectin pathway is activated when mannose binding lectin binds sugars on the surface of a bacteria. This binding attracts MASP-1 and MASP-2 to interact with the lectin, and leads to the cleavage of C2 and C4 and generation of the C3 convertase akin to that of the classical pathway. All three pathways of complement activation converge in function at the formation of the C3 convertase capable of cleaving C3 to C3a and C3b. C3b associates with the C3 convertase to form the active C5 convertase; this C5 convertase cleaves C5 into C5a and C5b. The anaphylotoxin C5a is a potent 8
24 chemoattractant and inflammatory mediator, and acts on a wide range of myeloid and lymphoid lineage cells, including skin MCs (60, 61). C5b binds to the surface of a target cell and initiates the assembly of the membrane attack complex. Subsequent binding of additional serum proteins (C6, C7, C8, C9, sequentially and with many C9s binding) generates an amphiphilic helix that inserts into the lipid bilayer and creates a membrane spanning channel. Disruption of the cell membrane integrity leads to lysis of the cell. C3b is also involved in the innate immune response by directly coating particulate antigen and immune complexes, leading to their enhanced opsonization by macrophages. Autoantibodies bind to basement membrane antigens and activate complement in human BP. Lesional skin from BP patients exhibits deposition of complement components along the DEJ, and the murine models of experimental BP recapitulates this feature of human disease (23, 54-56). In the rabbit anti-mbp180 IgG passive transfer model, complement activation is required for disease development. Both BALB/c mice depleted of complement by pretreatment with cobra venom and C5-deficient mice are resistant to experimental BP. In turn, reconstitution of the C5-deficient mice with recombinant C5a restores disease susceptibility (62). F(ab )2 fragments generated from the pathogenic anti-mbp180 IgG cannot induce subepidermal blisters in C5-sufficient mice (62), indicating a key role for the classical pathway of complement activation. Similarly, mice deficient in complement component C4 (specific for the classical and lectin pathways), but not in factor B (specific for the alternative pathway), are resistant to BP (63). Wild-type mice depleted of complement component C1q (also specific for the classical pathway) fail to activate MCs or develop BP skin lesions when injected with pathogenic IgG (63). These data indicate that the classical pathway of the 9
25 complement activation plays a major role in development of BP disease through the generation of C5a (64). -Mast Cells- MCs are key effector cells involved in innate and adaptive immunity, as well as allergic and anaphylactic reactions (65, 66). MCs are found in particularly high concentrations in the connective tissues just below the epithelial surfaces of the body, such as the submucosal tissues of the gastrointestinal and respiratory tracts and the dermis of the skin, positioning them as key sentinels against external challenges. Upon activation, MCs degranulate and release preformed bioactive compounds from their secretory granules, including histamine, cytokines, proteoglycans, and proteases (67). Together, these inflammatory mediators increase the permeability of local blood vessels and recruit effector cells to the site. In addition to their role in protection against environmental stimuli, MCs are also implicated in chronic inflammatory and autoimmune diseases. Mice lacking MCs are partially or completely protected from disease in animal models of multiple sclerosis, collagen-induced arthritis, and BP (64, 68, 69). Intact and degranulated MCs are commonly observed in the dermis of BP patients, and histamine and other MC-derived factors are present in high concentrations in the affected skin (12, 13, 18, 70). Mice injected with pathogenic anti-mbp180 antibodies exhibit extensive MC degranulation in the lesional skin, similar to that observed in human BP (12, 64). MC-deficient mice are resistant to experimental BP, but reconstitution of these mice with MCs restores susceptibility to disease. MC activation precedes PMN infiltration, and either the absence of MCs or the inhibition of MC degranulation prevents PMN infiltration and blister formation. However, MC-deficient mice reconstituted locally with PMNs, or 10
26 injected locally with PMN chemoattractants IL-8 or C5a form blisters in response to antimbp180 IgG. These results suggest that MCs release proinflammatory cytokines critical for PMN recruitment (64). Skin-resident MCs function in both an IgE dependent and IgE independent manner. The IgE independent activation of MCs can be mediated by products of complement activation, proinflammatory cytokines, Toll-like receptors (TLRs), or by Fc receptors (71). Skin MCs express C5a receptor (C5aR, or CD88) (72), and interaction of C5a and C5aR induces MC degranulation and the release of proinflammatory cytokines. Neutrophils- PMNs, produced in vast numbers in the bone marrow, circulate in the blood and act as the front line of defense against microorganisms that breach skin and mucous membrane barriers. PMNs extravasate into tissue under the direction of local endothelial cells, which are activated by inflammatory signals such as TNF-α, IL-1β, or IL-17. (73). The activated local endothelial cells express the adhesion molecules P-selectin, E-selectin, intracellular adhesion molecule (ICAM)-1, ICAM-2, and vascular adhesion molecule (VCAM)-1, which engage their ligands on the PMNs to capture rolling PMNs from the circulation. The endothelial cells also produce the PMN chemoattractant and activators IL-8 and macrophage inflammatory protein 2 (MIP-2) (73). PMNs have a wide range of effector capabilities, including phagocytosis of microbes, production of reactive oxygen species, release of lytic enzymes, synthesis of both proinflammatory and anti-inflammatory mediators, and capture of microbes via fibrillary networks termed neutrophil extracellular traps (NETs) (74-76). PMNs are involved in the pathology of numerous chronic inflammatory and autoimmune disorders, 11
27 including chronic obstructive pulmonary disease, systemic lupus erythematosus, small vessel vasculitis, inflammatory arthritis, and BP (77-82). PMN infiltration is required for the development of experimental BP, and there is a strong correlation between severity of disease and number of infiltrating PMNs (80). Depletion of PMNs with anti-pmn antibodies, or disruption of the events upstream of PMN infiltration (namely complement activation and MC degranulation), renders mice resistant to BP blister formation. However, complement system impairments and MC deficiencies do not block the pathological effects of BP180 autoantibodies unless PMN infiltration is prevented as a consequence of the deficiency. Mice deficient in complement or MCs are still susceptible to experimental BP when PMNs are recruited by injection with IL-8 or rc5a (63, 64). Thus, complement activation and MC degranulation mediate disease progression by promoting PMN recruitment to the skin. These data demonstrate that infiltrating PMNs are the critical cells that directly mediate tissue injury at the DEJ, leading to BP skin blisters. Proteolytic Enzymes- PMNs promote skin injury by release of proteolytic enzymes. The FcγIIIR on the cell surface of infiltrating PMNs interacts with the Fc portion of the tissue-bound pathogenic antimbp180 IgG (83). PMNs activated by the Fc-FcγIIIR molecular interaction secrete proteolytic enzymes known to degrade the ECM, including neutrophil elastase (NE) and gelatinase B (GB, also called matrix metalloproteinase-9) (83). A lack of either of these two enzymes blocks blister formation in mice (84, 85). In vitro, although both GB and NE are capable of degrading the recombinant BP180 protein, only NE produces DEJ separation when incubated with skin sections (84, 86). In vivo, the degradation of BP180 depends on NE activity (87). These findings suggest that GB is the upstream actor that proteolytically 12
28 inactivates the physiological inhibitor of NE (α1-proteinase inhibitor), which allows for unmitigated NE cleavage of ECM proteins (including BP180), resulting in DEJ separation (88). MCs also release proteolytic enzymes upon degranulation (67). Mouse mast cell protease-4, the likely murine homolog of human MC chymase, directly degrades BP180, and also activates GB (89). -Other Inflammatory Cells- Anti-mBP180 autoantibodies trigger experimental BP with equivalent severity in WT mice and mice deficient in T and B lymphocytes (90), suggesting that B and T cells do not play a role in this animal model. The limitations of the passive transfer injection model prevent investigation of the events that precipitate loss of tolerance and the generation of autoreactive B cells. Indeed, little is understood about the etiology of BP or lymphocyte regulation. Macrophages (MΦ) play an accessory role to MCs in PMN recruitment upon pathogenic antibody injection (90). MΦs are activated downstream of MCs, and they serve to amplify the PMN recruitment mediated by MCs Current Therapeutic Strategies For Treatment of Human BP The mortality rate for BP is high, reported to be between 12% and 40% in the first three years after diagnosis (4, 5). The conventional therapy for BP over the past several decades has been high-dose, long-term systemic oral corticosteroids and immunosuppressive agents (91). However, long term use of corticosteroids is associated with deleterious health effects, including severe infection, diabetes mellitus, osteoporosis, hypertension, renal failure, lymphoma, and squamous cell carcinoma (92). The negative side effects of immunosuppression therapy may account for a significant portion of the morbidity and 13
29 mortality associated with BP. Even when disease is successfully treated, up to 45% of patients suffer a relapse within six months of cessation of therapy (93), exacerbating the need for novel therapeutic interventions. In the past ten years, clinical management of BP has shifted away from oral corticosteroids in favor of topical corticosteroids (94). However, topical treatments are not always feasible for cases of extensive blistering, particularly in elderly patients who maybe be bedridden or without the mobility to apply medications regularly. Patients who fail both oral and topical corticosteroid treatment may be treated with biologicals such as intravenous immunoglobulin (IVIG) or rituximab (95). Patients treated with IVIG receive huge amounts of pooled IgG isolated from thousands of healthy human donors (96). The exact mechanism of IVIG is debated, however, there is compelling evidence that IVIG decreases the prevalent of pathogenic antibodies in circulation by increasing the rate of catabolism of IgG (97). Rituximab targets the CD20 molecule on the surface of B cells, depleting the antibody producing cells from circulation (98). No randomized controlled trials have been performed for either of these therapies yet, but anecdotal evidence suggests that these biologic approaches to treatment of BP offer treatment alternatives for patients (99, 100). 1.2 MAST CELL BIOLOGY MCs are innate immune cells primarily distributed in tissues that contact the external environment, such as the skin, airways, and the intestinal mucosa (101). While widely known as the culprits in type I hypersensitivity responses, MCs are now known to participate in host defense, modulation of innate and adaptive immune responses, angiogenesis, wound 14
30 healing, and tissue homeostasis and remodeling (102). Better understanding the mechanisms of MC activation, signaling, and cross-talk with other members of the immune system presents new opportunities for treatment of human disease MC Development and Function in the Skin MCs develop from hematopoietic stem cells (HSC), the common precursor to lymphocytes, granulocytes, and monocytes. The HSC precursors develop into CD34+/CD117 (or ckit)+/cd13+ multipotent hematopoietic progenitors (103, 104). Committed MC progenitors exit the bone marrow, circulate in the peripheral blood, and undergo final maturation under the direction of microenvironmental cues from the tissue where the MCs stake long-term residence ( ). Final MC maturation in the peripheral tissues is regulated by the MC growth factors stem cell factor (SCF, or c-kit ligand) and IL-3 (110, 111), as well as tissue specific factors. MC populations exhibit considerable heterogeneity between tissue sites, based on tissue-specific final maturation. The identities of the factors that promote homeostatic MC progenitor recruitment to the skin and regulate development are not known (112). In mice, nearly all skin MCs are connective tissue MCs (CTMCs) and have granules rich with histamine, tryptase, chymase, carboxypeptidase and cathepsin G-like proteinase, while MCs in the bowel mucosa and airways are predominantly mucosal tissue MC (MTMCs) and have granules that contain either tryptase and histamine or chymase, carboxypeptidase, and histamine but not tryptase ( ). This same pattern of tissue-based protease distinction is observed in human MCs. Skin and peritoneal cavity MCs are categorized as MC CT, indicating that the granules contain both tryptase and chymase (116). 15
31 MCs contribute to host defense by acting as sentinels against pathogens that breach the epidermal barrier. MCs are distributed throughout the dermis, but are greatly enriched in the area immediately below the DEJ, a site dense with small blood vessels (117). This proximity to the microvasculature enables MCs to act rapidly both locally and systemically. Localization to the DEJ also enables MCs to readily communicate with the two main sentinels of the epidermis, the keratinocytes (KCs) and Langerhans cells (LC), a class of specialized dendritic cell (DC) (118). KCs and LCs produce a battery of antimicrobial peptides, chemotactic proteins, and cytokines, including IL-1 (α and β) and TNF-α ( ). IL-1 stimulated MCs secrete IL-3, IL-6, IL-9, and TNF-α, and TNF-α stimulated MCs secrete IL-1 and upregulate FcγR expression (122). Together, these proinflammatory factors can act on endothelial cells to promote local inflammation and recruit leukocytes to the skin, including perhaps additional MC progenitors (112, 123). TLRs enable MCs to directly respond to microbial agents than penetrate beyond the 4 to 5 cell layers of the epidermis and into the dermis (71). Extensive experimental evidence demonstrates that MCs respond to bacterial membrane components such as lipopolysaccharide (LPS) and peptidoglycan (PGN) via TLR-4 and TLR-2. Mouse MCs stimulated with LPS or PGN secrete TNF-α and IL-6 without degranulation in a TLRdependent manner, and mice lacking TLR-4 fail to recruit sufficient PMNs to clear bacterial infections ( ). In vitro evidence suggests roles for TLR-9, TLR-3, TLR-1, and TLR-6 in MC recognition of pathogen associated molecular patterns ( ). MCs are able to indirectly respond to pathogens via their complement receptors (discussed in Section 1.2.2) and their Fc receptors. FcγRs on MCs bind the Fc portion of 16
32 pathogen-specific antibodies, and cross-linking of the FcγRs by the polyvalent antigen on the pathogen results in MC degranulation and release of proinflammatory mediators (135, 136). FcR signaling can also activate MCs in response to bacterial superantigens, such as Streptococcus aureus protein A (137) C5aR mediated MC Activation The anaphylatoxin C5a interacts with two C5a receptors, C5aR (also called CD88) and the C5a receptor-like 2 (C5L2) (61). Both C5aR and C5L2 are broadly expressed by cells of both myeloid and non-myeloid lineages, including MCs (61, ). C5aR is the classically described ligand for C5a, while C5L2 was considered until very recently to be non-signaling decoy receptor that served to dampen C5a-mediated inflammation (141). Recent studies have uncovered a functional signaling role for C5L2 (143, 144), however, the vast majority of the understood C5a-mediated signaling is facilitated by C5aR. C5a is a G protein coupled receptors (GPCRs) (145, 146). Members of the GPCR family are characterized by their seven transmembrane-spanning domains, and they function by coupling to intracellular heterotrimeric G proteins (147). The G protein is comprised of an α, β, and γ subunit. When inactive, the G-α subunit is bound to GDP, and is associated with the β and γ subunits. GPCR ligation causes a conformational change to the intracellular portion of the receptor, enabling it to act as a guanine nucleotide exchange factor. The G-α releases GDP and binds GTP, which causes the β and γ subunits to dissociate from G-α. G-α and G-βγ activate second messengers, including cyclic AMP, phospholipase C, and phosphoinositide 3-kinase. Ultimately, the intracellular signaling cascade leads to production 17
33 of the transcription factors nuclear factor κb (NFκB), nuclear factor of activated T cells (NFAT), and activation protein-1 (AP-1), which modulate gene expression (148). In the case of pathogen assault, complement activation may occur by the classical or mannose binding lectin pathways. While many inflammatory cells (notably PMNs) express complement receptors, MCs are optimally positioned at the DEJ to respond to C5a. MCs express other complement-component receptors, including C3aR (the ligand for C3a) and CR3 (the ligand for C3b), however, degranulation occurs primarily as a result of activation of C5aR (71). Degranulation results in release of TNF-α, histamine, proteases, GM-CSF, IL-6, IL-1, and leukotriene C (71) p38mapk Signaling in MCs p38mapk is a key signaling molecule involved in translating extracellular environmental conditions, particularly inflammatory conditions, into cellular responses (149). There are four isoforms of p38mapk, designated α, -β, -γ, and δ; MCs express the α form exclusively (150). p38mapk is a serine/threonine kinase, thus, it exerts biological effect by phosphorylating proteins. p38mapk can be activated by numerous extracellular stimuli, including endotoxins, reactive oxygen species, ultraviolet radiation, TNF-α, IL-1, and TGF-β (151). Upstream of p38mapk, interaction, mitogen-activated protein kinase kinase kinases (MAPKKKs) are activated in a GTPase-dependent manner. The MAPKKKs phosphorylate the MAPK kinases MKK3, MKK4, and MKK6, which in turn activate p38 via phosphorylation of the Thr180 and Try 182 residues. Activated p38mapk facilitates the stabilization and increased 18
34 transcription of mrnas for proinflammatory cytokines and receptors associated with inflammation (151). The p38mapk pathway plays a key role in numerous immune and inflammatory diseases, including rheumatoid arthritis and the skin autoimmune disease pemphigus vulgaris ( ). In PMNs and monocytes, the interaction of C5a and C5aR leads to activation of p38mapk ( ). MC activation via antigen crosslinking of surface IgE molecules leads to p38mapk activation and cytokine release (158), however, p38mapk activation upon engagement of the C5aR on MCs has not yet been reported TNF-α mediated MC Activation TNF-α has been exhaustively described as a product released by MCs (159). Less is known, however, about the effect of TNF-α on MCs themselves. MCs stimulated by KC and LC-derived TNF-α secrete IL-1 and upregulate FcγR expression (122, 160). In cultured human lung MCs stimulated with SCF and anti-ige, TNF-α was shown to feed back on the MCs to promote enhanced NF-kB activation (161). NF-kB promotes transcription of a wide array of pro-inflammatory cytokines, suggesting that a positive autocrine feedback loop may enable MCs to drive inflammation. TNF-α signals through the TNF-α receptors (TNFRs) TNFR1 and TNFR2, though TNFR1 mediates the majority of the biological activities of TNF-α (162). Ligation of TNFR1 releases the inhibitory protein silencer of death domains (SODD) from TNFR1 s intracellular domain (ICD), which enables ICD to be recognized by the adaptor protein TNF receptor-associated death domain (TRADD) (163). TRADD serves as a dock for additional adaptor proteins that recruit signaling enzymes to the signaling complex. These adaptors 19
35 include TNFR-associated factor 2 (TRAF2), which can activate MAPKKKs, including MEKK1 and ASK1. The adaptor Fas-associated death domain (FADD) recruits caspase-8 to the signaling complex, where it promotes progression of apoptosis. Receptor-interacting protein (RIP) is yet another adaptor recruited by TRADD, and RIP mediates NF-kB activation MCs in Autoimmunity MCs are potent effector cells in the innate immune response to infection in large part due to the many avenues that lead to MC activation. However, inappropriate MC activation outside of the context of pathogen invasion can result in significant pathology and autoimmunity (164). The role of MCs in BP is described in Section In multiple sclerosis, MCs accumulate in the central nervous system plaque lesions and at sites of inflammatory demyelination ( ). MC-deficient mice are protected from experimental autoimmune encephalomyelitis, an animal model of MS, further supporting the case of MC involvement in disease (69). Similarly, MC-deficient mice are not susceptible to autoantibody-induced arthritis (68). In human rheumatoid arthritis patients, elevated numbers of MCs are present in the synovial compartments, along with high levels of MCproducts including TNF-α, IL-1, IL-17, and tryptase.(168, 169). There is also evidence that MCs contribute to pathology in Type 1 diabetes. Cromolyn, a compound that prevents MC degranulation, significantly delays onset of disease in the spontaneously diabetic BioBreeding mouse (170). Furthermore, activated MCs are found in the pancreatic lymph nodes of diseased mice (170). 20
36 In the case of experimental BP, the data are clear: MCs attract PMNs to the skin, and PMNs do the damage (see section 1.1.3). However, MCs interact with many types of immune cells, making the mechanisms by which MCs regulate autoimmunity far more complex than the experimental BP model predicts. MC-derived products are known to influence the migration, maturation, and function of DCs, effector T cells and B cells (171, 172). Intriguingly for the consideration of autoimmunity, MCs also interact with Tregs and Th17 cells (164). One well-defined avenue of Treg-MC interaction occurs via OX40, a molecular constitutively expressed on MCs, and OX40L, a protein constitutively expressed by Tregs. When OX40-OX40L interact, MCs can impair Treg suppression of effector T cells, and Tregs are able to downregulate FcεR1 expression on MCs and prevent FcεR1- mediated degranulation( ). MCs can produce Th17/Treg polarizing cytokines, including IL-6, IL-21, IL-23, and TGFβ. In vitro, MCs can drive Treg or Th17 cell differentiation (175). Conceivably, MCs could perform this same polarizing role at sites of secondary activation and T cell priming. 1.3 SUMMARY AND CONCLUDING REMARKS The murine IgG passive transfer model of BP developed by Liu et al in 1993 has facilitated dissection of the mechanism of disease progression in experimental BP. After nearly 20 years of experimentation, it is clear that BP pathogenesis proceeds through activation of MCs, but the mechanism of MC activation and the consequences of this activation are not clear. MCs express a number of types of receptors capable of responding to proinflammatory ligands, including C5aR. Taken with the evidence that complement 21
37 activation occurs upstream of MC degranulation, the C5aR is a promising candidate for facilitating MC signaling. The primary objective of this dissertation was to elucidate the cellular events that occur immediately upstream and downstream of MC degranulation in experimental BP. Our central hypothesis is that complement components activate MCs, and proinflammatory compounds released from the MCs drive pathology and blistering. In chapters 2 and 3, we described studies that test this hypothesis using a series of knockout mice and MC reconstitution experiments. We found that MCs are activated by the binding of C5a to C5aR. C5a-C5aR interaction leads to activation of p38mapk and MC degranulation. MC degranulation releases TNF-α, and TNF-α acts in an autocrine manner on MC TNFR1 to promote disease development. Our findings highlight the potential for cross-talk between the complement and TNF-α mediated pathways of MC activation, and also support three potential therapeutic targets for treatment of BP. 22
38 1.4 REFERENCES 1. Lever, W. F Pemphigus. Medicine (Baltimore) 32: Korman, N. J Bullous pemphigoid. The latest in diagnosis, prognosis, and therapy. Arch Dermatol 134: Jung, M., W. Kippes, G. Messer, D. Zillikens, and B. Rzany Increased risk of bullous pemphigoid in male and very old patients: A population-based study on incidence. J Am Acad Dermatol 41: Colbert, R. L., D. M. Allen, D. Eastwood, and J. A. Fairley Mortality rate of bullous pemphigoid in a US medical center. J Invest Dermatol 122: Roujeau, J. C., C. Lok, S. Bastuji-Garin, S. Mhalla, V. Enginger, and P. Bernard High risk of death in elderly patients with extensive bullous pemphigoid. Arch Dermatol 134: Stanley, J. R Bullous Pemphigoid. In Fitzpatrick's Dermatology in General Medicine. I. Freedberg, Eisen AZ, Wolff K, Austen KF, Goldsmith LA, Katz SI, ed. McGraw-Hill, New York Emmerson, R. W., and E. Wilson-Jones Eosinophilic spongiosis in pemphigus. A report of an unusual hitological change in pemphigus. Arch Dermatol 97: Iwatsuki, K., H. Tagami, and M. Yamada Induction of leukocyte adherence at the basement membrane zone with subsequent activation of their metabolic pathway by pemphigoid antibodies and complement. Acta Derm Venereol 63: Lever, W. F Pemphigus and Pemphigoid. C. C. Thomas, ed, Springfield, IL Nestor, M. S., A. J. Cochran, and A. R. Ahmed Mononuclear cell infiltrates in bullous disease. J Invest Dermatol 88: Nishioka, K., K. Hashimoto, I. Katayama, C. Sarashi, T. Kubo, and S. Sano Eosinophilic spongiosis in bullous pemphigoid. Arch Dermatol 120:
39 12. Wintroub, B. U., M. C. Mihm, Jr., E. J. Goetzl, N. A. Soter, and K. F. Austen Morphologic and functional evidence for release of mast-cell products in bullous pemphigoid. N Engl J Med 298: Baba, T., H. Sonozaki, K. Seki, M. Uchiyama, Y. Ikesawa, and M. Toriisu An eosinophil chemotactic factor present in blister fluids of bullous pemphigoid patients. J Immunol 116: Borrego, L., B. Maynard, E. A. Peterson, T. George, L. Iglesias, M. S. Peters, W. Newman, G. J. Gleich, and K. M. Leiferman Deposition of eosinophil granule proteins precedes blister formation in bullous pemphigoid. Comparison with neutrophil and mast cell granule proteins. Am J Pathol 148: Czech, W., J. Schaller, E. Schopf, and A. Kapp Granulocyte activation in bullous diseases: release of granular proteins in bullous pemphigoid and pemphigus vulgaris. J Am Acad Dermatol 29: D'Auria, L., P. Cordiali Fei, and F. Ameglio Cytokines and bullous pemphigoid. Eur Cytokine Netw 10: Diaz-Perez, J. L., and R. E. Jordon The complement system in bullous pemphigoid. IV. Chemotactic activity in blister fluid. Clin Immunol Immunopathol 5: Katayama, I., T. Doi, and K. Nishioka High histamine level in the blister fluid of bullous pemphigoid. Arch Dermatol Res 276: Kawana, S., A. Ueno, and S. Nishiyama Increased levels of immunoreactive leukotriene B4 in blister fluids of bullous pemphigoid patients and effects of a selective 5-lipoxygenase inhibitor on experimental skin lesions. Acta Derm Venereol 70: Schmidt, E., A. Ambach, B. Bastian, E. B. Brocker, and D. Zillikens Elevated levels of interleukin-8 in blister fluid of bullous pemphigoid compared with suction blisters of healthy control subjects. J Am Acad Dermatol 34: Schmidt, E., B. Bastian, R. Dummer, H. P. Tony, E. B. Brocker, and D. Zillikens Detection of elevated levels of IL-4, IL-6, and IL-10 in blister fluid of bullous pemphigoid. Arch Dermatol Res 288:
40 22. Wakugawa, M., K. Nakamura, H. Hino, K. Toyama, N. Hattori, H. Okochi, H. Yamada, K. Hirai, K. Tamaki, and M. Furue Elevated levels of eotaxin and interleukin-5 in blister fluid of bullous pemphigoid: correlation with tissue eosinophilia. Br J Dermatol 143: Chorzelski, T., S. Jablonska, and E. H. Beutner Pemphigoid. In Immunopathology of the Skin, 2 ed. E. H. Beutner, T. Chorzelski, and S. F. Bean, eds. John Wiley & Sons, New York. 24. Jordon, R. E., E. H. Beutner, E. Witebsky, G. Blumental, W. L. Hale, and W. F. Lever Basement zone antibodies in bullous pemphigoid. JAMA 200: Borradori, L., and A. Sonnenberg Structure and function of hemidesmosomes: more than simple adhesion complexes. J Invest Dermatol 112: Labib, R. S., G. J. Anhalt, H. P. Patel, D. F. Mutasim, and L. A. Diaz Molecular heterogeneity of the bullous pemphigoid antigens as detected by immunoblotting. J Immunol 136: Mutasim, D. F., Y. Takahashi, R. S. Labib, G. J. Anhalt, H. P. Patel, and L. A. Diaz A pool of bullous pemphigoid antigen(s) is intracellular and associated with the basal cell cytoskeleton-hemidesmosome complex. J Invest Dermatol 84: Stanley, J. R., P. Hawley-Nelson, S. H. Yuspa, E. M. Shevach, and S. I. Katz Characterization of bullous pemphigoid antigen: a unique basement membrane protein of stratified squamous epithelia. Cell 24: Stanley, J. R., D. T. Woodley, and S. I. Katz Identification and partial characterization of pemphigoid antigen extracted from normal human skin. J Invest Dermatol 82: Ishiko, A., H. Shimizu, A. Kikuchi, T. Ebihara, T. Hashimoto, and T. Nishikawa Human autoantibodies against the 230-kD bullous pemphigoid antigen (BPAG1) bind only to the intracellular domain of the hemidesmosome, whereas those against the 180-kD bullous pemphigoid antigen (BPAG2) bind along the plasma membrane of the hemidesmosome in normal human and swine skin. J Clin Invest 91:
41 31. Klatte, D. H., M. A. Kurpakus, K. A. Grelling, and J. C. Jones Immunochemical characterization of three components of the hemidesmosome and their expression in cultured epithelial cells. J Cell Biol 109: Giudice, G. J., D. J. Emery, and L. A. Diaz Cloning and primary structural analysis of the bullous pemphigoid autoantigen BP180. J Invest Dermatol 99: Hopkinson, S. B., K. S. Riddelle, and J. C. Jones Cytoplasmic domain of the 180-kD bullous pemphigoid antigen, a hemidesmosomal component: molecular and cell biologic characterization. J Invest Dermatol 99: Hopkinson, S. B., K. Findlay, G. W. dehart, and J. C. Jones Interaction of BP180 (type XVII collagen) and alpha6 integrin is necessary for stabilization of hemidesmosome structure. J Invest Dermatol 111: Van den Bergh, F., S. L. Eliason, and G. J. Giudice Type XVII collagen (BP180) can function as a cell-matrix adhesion molecule via binding to laminin 332. Matrix Biol 30: Bernard, P., P. Aucouturier, F. Denis, and J. M. Bonnetblanc Immunoblot analysis of IgG subclasses of circulating antibodies in bullous pemphigoid. Clin Immunol Immunopathol 54: Dopp, R., E. Schmidt, I. Chimanovitch, M. Leverkus, E. B. Brocker, and D. Zillikens IgG4 and IgE are the major immunoglobulins targeting the NC16A domain of BP180 in Bullous pemphigoid: serum levels of these immunoglobulins reflect disease activity. J Am Acad Dermatol 42: Giudice, G. J., D. J. Emery, B. D. Zelickson, G. J. Anhalt, Z. Liu, and L. A. Diaz Bullous pemphigoid and herpes gestationis autoantibodies recognize a common non-collagenous site on the BP180 ectodomain. J Immunol 151: Laffitte, E., M. Skaria, F. Jaunin, K. Tamm, J. H. Saurat, B. Favre, and L. Borradori Autoantibodies to the extracellular and intracellular domain of bullous pemphigoid 180, the putative key autoantigen in bullous pemphigoid, belong predominantly to the IgG1 and IgG4 subclasses. Br J Dermatol 144:
42 40. Zillikens, D., P. A. Rose, S. D. Balding, Z. Liu, M. Olague-Marchan, L. A. Diaz, and G. J. Giudice Tight clustering of extracellular BP180 epitopes recognized by bullous pemphigoid autoantibodies. J Invest Dermatol 109: Haase, C., L. Budinger, L. Borradori, C. Yee, H. F. Merk, K. Yancey, and M. Hertl Detection of IgG autoantibodies in the sera of patients with bullous and gestational pemphigoid: ELISA studies utilizing a baculovirus-encoded form of bullous pemphigoid antigen 2. J Invest Dermatol 110: Schmidt, E., K. Obe, E. B. Brocker, and D. Zillikens Serum levels of autoantibodies to BP180 correlate with disease activity in patients with bullous pemphigoid. Arch Dermatol 136: Budinger, L., L. Borradori, C. Yee, R. Eming, S. Ferencik, H. Grosse-Wilde, H. F. Merk, K. Yancey, and M. Hertl Identification and characterization of autoreactive T cell responses to bullous pemphigoid antigen 2 in patients and healthy controls. J Clin Invest 102: Lin, M. S., C. L. Fu, G. J. Giudice, M. Olague-Marchan, A. M. Lazaro, P. Stastny, and L. A. Diaz Epitopes targeted by bullous pemphigoid T lymphocytes and autoantibodies map to the same sites on the bullous pemphigoid 180 ectodomain. J Invest Dermatol 115: Sakaguchi, S Naturally arising Foxp3-expressing CD25+CD4+ regulatory T cells in immunological tolerance to self and non-self. Nat Immunol 6: Arakawa, M., T. Dainichi, N. Ishii, T. Hamada, T. Karashima, T. Nakama, S. Yasumoto, D. Tsuruta, and T. Hashimoto Lesional Th17 cells and regulatory T cells in bullous pemphigoid. Exp Dermatol 20: Rensing-Ehl, A., B. Gaus, L. Bruckner-Tuderman, and S. F. Martin Frequency, function and CLA expression of CD4+CD25+FOXP3+ regulatory T cells in bullous pemphigoid. Exp Dermatol 16: Korn, T., E. Bettelli, M. Oukka, and V. K. Kuchroo IL-17 and Th17 Cells. Annu Rev Immunol 27: Anhalt, G. J., and L. A. Diaz Animal models for bullous pemphigoid. Clin Dermatol 5:
43 50. Sams, W. M., Jr., and G. J. Gleich Failure to transfer bullous pemphigoid with serum from patients. Proc Soc Exp Biol Med 136: Anhalt, G. J., R. S. Labib, J. J. Voorhees, T. F. Beals, and L. A. Diaz Induction of pemphigus in neonatal mice by passive transfer of IgG from patients with the disease. N Engl J Med 306: Roscoe, J. T., L. Diaz, S. A. Sampaio, R. M. Castro, R. S. Labib, Y. Takahashi, H. Patel, and G. J. Anhalt Brazilian pemphigus foliaceus autoantibodies are pathogenic to BALB/c mice by passive transfer. J Invest Dermatol 85: Takahashi, Y., H. P. Patel, R. S. Labib, L. A. Diaz, and G. J. Anhalt Experimentally induced pemphigus vulgaris in neonatal BALB/c mice: a time-course study of clinical, immunologic, ultrastructural, and cytochemical changes. J Invest Dermatol 84: Liu, Z., L. A. Diaz, J. L. Troy, A. F. Taylor, D. J. Emery, J. A. Fairley, and G. J. Giudice A passive transfer model of the organ-specific autoimmune disease, bullous pemphigoid, using antibodies generated against the hemidesmosomal antigen, BP180. J Clin Invest 92: Liu, Z., W. Sui, M. Zhao, Z. Li, N. Li, R. Thresher, G. J. Giudice, J. A. Fairley, C. Sitaru, D. Zillikens, G. Ning, M. P. Marinkovich, and L. A. Diaz Subepidermal blistering induced by human autoantibodies to BP180 requires innate immune players in a humanized bullous pemphigoid mouse model. J Autoimmun 31: Nishie, W., D. Sawamura, M. Goto, K. Ito, A. Shibaki, J. R. McMillan, K. Sakai, H. Nakamura, E. Olasz, K. B. Yancey, M. Akiyama, and H. Shimizu Humanization of autoantigen. Nat Med 13: Liu, Z., D. C. Roopenian, X. Zhou, G. J. Christianson, L. A. Diaz, D. D. Sedmak, and C. L. Anderson Beta2-microglobulin-deficient mice are resistant to bullous pemphigoid. J Exp Med 186: Liu, Z., L. A. Diaz, S. J. Swartz, J. L. Troy, J. A. Fairley, and G. J. Giudice Molecular mapping of a pathogenically relevant BP180 epitope associated with experimentally induced murine bullous pemphigoid. J Immunol 155: Ricklin, D., G. Hajishengallis, K. Yang, and J. D. Lambris Complement: a key system for immune surveillance and homeostasis. Nat Immunol 11:
44 60. Lawrence, I. D., J. A. Warner, V. L. Cohan, W. C. Hubbard, A. Kagey-Sobotka, and L. M. Lichtenstein Purification and characterization of human skin mast cells. Evidence for human mast cell heterogeneity. J Immunol 139: Monk, P. N., A. M. Scola, P. Madala, and D. P. Fairlie Function, structure and therapeutic potential of complement C5a receptors. Br J Pharmacol 152: Liu, Z., G. J. Giudice, S. J. Swartz, J. A. Fairley, G. O. Till, J. L. Troy, and L. A. Diaz The role of complement in experimental bullous pemphigoid. J Clin Invest 95: Nelson, K. C., M. Zhao, P. R. Schroeder, N. Li, R. A. Wetsel, L. A. Diaz, and Z. Liu Role of different pathways of the complement cascade in experimental bullous pemphigoid. J Clin Invest 116: Chen, R., G. Ning, M. L. Zhao, M. G. Fleming, L. A. Diaz, Z. Werb, and Z. Liu Mast cells play a key role in neutrophil recruitment in experimental bullous pemphigoid. J Clin Invest 108: Galli, S. J., and M. Tsai Mast cells in allergy and infection: versatile effector and regulatory cells in innate and adaptive immunity. Eur J Immunol 40: Gurish, M. F., and K. F. Austen The diverse roles of mast cells. J Exp Med 194:F Stevens, R. L., and R. Adachi Protease-proteoglycan complexes of mouse and human mast cells and importance of their beta-tryptase-heparin complexes in inflammation and innate immunity. Immunol Rev 217: Lee, D. M., D. S. Friend, M. F. Gurish, C. Benoist, D. Mathis, and M. B. Brenner Mast cells: a cellular link between autoantibodies and inflammatory arthritis. Science 297: Secor, V. H., W. E. Secor, C. A. Gutekunst, and M. A. Brown Mast cells are essential for early onset and severe disease in a murine model of multiple sclerosis. J Exp Med 191:
45 70. Brockow, K., D. Abeck, K. Hermann, and J. Ring Tryptase concentration in skin blister fluid from patients with bullous skin conditions. Arch Dermatol Res 288: Marshall, J. S Mast-cell responses to pathogens. Nat Rev Immunol 4: Fureder, W., H. Agis, M. Willheim, H. C. Bankl, U. Maier, K. Kishi, M. R. Muller, K. Czerwenka, T. Radaszkiewicz, J. H. Butterfield, G. W. Klappacher, W. R. Sperr, M. Oppermann, K. Lechner, and P. Valent Differential expression of complement receptors on human basophils and mast cells. Evidence for mast cell heterogeneity and CD88/C5aR expression on skin mast cells. J Immunol 155: Borregaard, N Neutrophils, from marrow to microbes. Immunity 33: Brinkmann, V., U. Reichard, C. Goosmann, B. Fauler, Y. Uhlemann, D. S. Weiss, Y. Weinrauch, and A. Zychlinsky Neutrophil extracellular traps kill bacteria. Science 303: Cassatella, M. A Neutrophil-derived proteins: selling cytokines by the pound. Adv Immunol 73: Nathan, C Neutrophils and immunity: challenges and opportunities. Nat Rev Immunol 6: Chen, M., and C. G. Kallenberg ANCA-associated vasculitides--advances in pathogenesis and treatment. Nat Rev Rheumatol 6: Chou, R. C., N. D. Kim, C. D. Sadik, E. Seung, Y. Lan, M. H. Byrne, B. Haribabu, Y. Iwakura, and A. D. Luster Lipid-cytokine-chemokine cascade drives neutrophil recruitment in a murine model of inflammatory arthritis. Immunity 33: Hakkim, A., B. G. Furnrohr, K. Amann, B. Laube, U. A. Abed, V. Brinkmann, M. Herrmann, R. E. Voll, and A. Zychlinsky Impairment of neutrophil extracellular trap degradation is associated with lupus nephritis. Proc Natl Acad Sci U S A 107:
46 80. Liu, Z., G. J. Giudice, X. Zhou, S. J. Swartz, J. L. Troy, J. A. Fairley, G. O. Till, and L. A. Diaz A major role for neutrophils in experimental bullous pemphigoid. J Clin Invest 100: Mantovani, A., M. A. Cassatella, C. Costantini, and S. Jaillon Neutrophils in the activation and regulation of innate and adaptive immunity. Nat Rev Immunol 11: Weathington, N. M., A. H. van Houwelingen, B. D. Noerager, P. L. Jackson, A. D. Kraneveld, F. S. Galin, G. Folkerts, F. P. Nijkamp, and J. E. Blalock A novel peptide CXCR ligand derived from extracellular matrix degradation during airway inflammation. Nat Med 12: Zhao, M., M. E. Trimbeger, N. Li, L. A. Diaz, S. D. Shapiro, and Z. Liu Role of FcRs in animal model of autoimmune bullous pemphigoid. J Immunol 177: Liu, Z., S. D. Shapiro, X. Zhou, S. S. Twining, R. M. Senior, G. J. Giudice, J. A. Fairley, and L. A. Diaz A critical role for neutrophil elastase in experimental bullous pemphigoid. J Clin Invest 105: Liu, Z., J. M. Shipley, T. H. Vu, X. Zhou, L. A. Diaz, Z. Werb, and R. M. Senior Gelatinase B-deficient mice are resistant to experimental bullous pemphigoid. J Exp Med 188: Stahle-Backdahl, M., M. Inoue, G. J. Guidice, and W. C. Parks kD gelatinase is produced by eosinophils at the site of blister formation in bullous pemphigoid and cleaves the extracellular domain of recombinant 180-kD bullous pemphigoid autoantigen. J Clin Invest 93: Lin, L., T. Betsuyaku, L. Heimbach, N. Li, D. Rubenstein, S. D. Shapiro, L. An, G. J. Giudice, L. A. Diaz, R. M. Senior, and Z. Liu Neutrophil elastase cleaves the murine hemidesmosomal protein BP180/type XVII collagen and generates degradation products that modulate experimental bullous pemphigoid. Matrix Biol 31: Liu, Z., X. Zhou, S. D. Shapiro, J. M. Shipley, S. S. Twining, L. A. Diaz, R. M. Senior, and Z. Werb The serpin alpha1-proteinase inhibitor is a critical substrate for gelatinase B/MMP-9 in vivo. Cell 102:
47 89. Lin, L., E. Bankaitis, L. Heimbach, N. Li, M. Abrink, G. Pejler, L. An, L. A. Diaz, Z. Werb, and Z. Liu Dual targets for mouse mast cell protease-4 in mediating tissue damage in experimental bullous pemphigoid. J Biol Chem 286: Chen, R., J. A. Fairley, M. L. Zhao, G. J. Giudice, D. Zillikens, L. A. Diaz, and Z. Liu Macrophages, but not T and B lymphocytes, are critical for subepidermal blister formation in experimental bullous pemphigoid: macrophage-mediated neutrophil infiltration depends on mast cell activation. J Immunol 169: Nousari, H. C., and G. J. Anhalt Pemphigus and bullous pemphigoid. Lancet 354: Williams, L. C., and L. T. Nesbitt, Jr Update on systemic glucocorticosteroids in dermatology. Dermatol Clin 19: Bernard, P., Z. Reguiai, E. Tancrede-Bohin, N. Cordel, P. Plantin, C. Pauwels, L. Vaillant, F. Grange, M. A. Richard-Lallemand, B. Sassolas, J. C. Roujeau, C. Lok, C. Picard-Dahan, O. Chosidow, F. Vitry, and P. Joly Risk factors for relapse in patients with bullous pemphigoid in clinical remission: a multicenter, prospective, cohort study. Arch Dermatol 145: Joly, P., J. C. Roujeau, J. Benichou, C. Picard, B. Dreno, E. Delaporte, L. Vaillant, M. D'Incan, P. Plantin, C. Bedane, P. Young, and P. Bernard A comparison of oral and topical corticosteroids in patients with bullous pemphigoid. N Engl J Med 346: Daniel, B. S., L. Borradori, R. P. Hall, 3rd, and D. F. Murrell Evidence-based management of bullous pemphigoid. Dermatol Clin 29: Kazatchkine, M. D., and S. V. Kaveri Immunomodulation of autoimmune and inflammatory diseases with intravenous immune globulin. N Engl J Med 345: Li, N., M. Zhao, J. Hilario-Vargas, P. Prisayanh, S. Warren, L. A. Diaz, D. C. Roopenian, and Z. Liu Complete FcRn dependence for intravenous Ig therapy in autoimmune skin blistering diseases. J Clin Invest 115: Johnson, P. W., and M. J. Glennie Rituximab: mechanisms and applications. Br J Cancer 85:
48 99. Czernik, A., S. Toosi, J. C. Bystryn, and S. A. Grando Intravenous immunoglobulin in the treatment of autoimmune bullous dermatoses: An update. Autoimmunity 45: Kasperkiewicz, M., I. Shimanovich, R. J. Ludwig, C. Rose, D. Zillikens, and E. Schmidt Rituximab for treatment-refractory pemphigus and pemphigoid: a case series of 17 patients. J Am Acad Dermatol 65: Frossi, B., M. De Carli, and C. Pucillo The mast cell: an antenna of the microenvironment that directs the immune response. J Leukoc Biol 75: Moon, T. C., C. D. St Laurent, K. E. Morris, C. Marcet, T. Yoshimura, Y. Sekar, and A. D. Befus Advances in mast cell biology: new understanding of heterogeneity and function. Mucosal Immunol 3: Chen, C. C., M. A. Grimbaldeston, M. Tsai, I. L. Weissman, and S. J. Galli Identification of mast cell progenitors in adult mice. Proc Natl Acad Sci U S A 102: Rodewald, H. R., M. Dessing, A. M. Dvorak, and S. J. Galli Identification of a committed precursor for the mast cell lineage. Science 271: Abonia, J. P., K. F. Austen, B. J. Rollins, S. K. Joshi, R. A. Flavell, W. A. Kuziel, P. A. Koni, and M. F. Gurish Constitutive homing of mast cell progenitors to the intestine depends on autologous expression of the chemokine receptor CXCR2. Blood 105: Weller, C. L., S. J. Collington, J. K. Brown, H. R. Miller, A. Al-Kashi, P. Clark, P. J. Jose, A. Hartnell, and T. J. Williams Leukotriene B4, an activation product of mast cells, is a chemoattractant for their progenitors. J Exp Med 201: Weller, C. L., S. J. Collington, A. Hartnell, D. M. Conroy, T. Kaise, J. E. Barker, M. S. Wilson, G. W. Taylor, P. J. Jose, and T. J. Williams Chemotactic action of prostaglandin E2 on mouse mast cells acting via the PGE2 receptor 3. Proc Natl Acad Sci U S A 104: Brightling, C. E., P. Bradding, F. A. Symon, S. T. Holgate, A. J. Wardlaw, and I. D. Pavord Mast-cell infiltration of airway smooth muscle in asthma. N Engl J Med 346:
49 109. Mathias, C. B., E. J. Freyschmidt, B. Caplan, T. Jones, D. Poddighe, W. Xing, K. L. Harrison, M. F. Gurish, and H. C. Oettgen IgE influences the number and function of mature mast cells, but not progenitor recruitment in allergic pulmonary inflammation. J Immunol 182: Lantz, C. S., J. Boesiger, C. H. Song, N. Mach, T. Kobayashi, R. C. Mulligan, Y. Nawa, G. Dranoff, and S. J. Galli Role for interleukin-3 in mast-cell and basophil development and in immunity to parasites. Nature 392: Mekori, Y. A., C. K. Oh, and D. D. Metcalfe IL-3-dependent murine mast cells undergo apoptosis on removal of IL-3. Prevention of apoptosis by c-kit ligand. J Immunol 151: Harvima, I. T., and G. Nilsson Mast cells as regulators of skin inflammation and immunity. Acta Derm Venereol 91: Irani, A. M., T. R. Bradford, C. L. Kepley, N. M. Schechter, and L. B. Schwartz Detection of MCT and MCTC types of human mast cells by immunohistochemistry using new monoclonal anti-tryptase and anti-chymase antibodies. J Histochem Cytochem 37: Schechter, N. M., A. M. Irani, J. L. Sprows, J. Abernethy, B. Wintroub, and L. B. Schwartz Identification of a cathepsin G-like proteinase in the MCTC type of human mast cell. J Immunol 145: Weidner, N., and K. F. Austen Heterogeneity of mast cells at multiple body sites. Fluorescent determination of avidin binding and immunofluorescent determination of chymase, tryptase, and carboxypeptidase content. Pathol Res Pract 189: Irani, A. A., N. M. Schechter, S. S. Craig, G. DeBlois, and L. B. Schwartz Two types of human mast cells that have distinct neutral protease compositions. Proc Natl Acad Sci U S A 83: Cowen, T., P. Trigg, and R. A. Eady Distribution of mast cells in human dermis: development of a mapping technique. Br J Dermatol 100: Kupper, T. S., and R. C. Fuhlbrigge Immune surveillance in the skin: mechanisms and clinical consequences. Nat Rev Immunol 4:
50 119. Takashima, A., and P. R. Bergstresser Cytokine-mediated communication by keratinocytes and Langerhans cells with dendritic epidermal T cells. Semin Immunol 8: Wood, L. C., P. M. Elias, C. Calhoun, J. C. Tsai, C. Grunfeld, and K. R. Feingold Barrier disruption stimulates interleukin-1 alpha expression and release from a pre-formed pool in murine epidermis. J Invest Dermatol 106: Yang, D., O. Chertov, and J. J. Oppenheim The role of mammalian antimicrobial peptides and proteins in awakening of innate host defenses and adaptive immunity. Cell Mol Life Sci 58: Hultner, L., S. Kolsch, M. Stassen, U. Kaspers, J. P. Kremer, R. Mailhammer, J. Moeller, H. Broszeit, and E. Schmitt In activated mast cells, IL-1 up-regulates the production of several Th2-related cytokines including IL-9. J Immunol 164: Kneilling, M., R. Mailhammer, L. Hultner, T. Schonberger, K. Fuchs, M. Schaller, D. Bukala, S. Massberg, C. A. Sander, H. Braumuller, M. Eichner, K. L. Maier, R. Hallmann, B. J. Pichler, R. Haubner, M. Gawaz, K. Pfeffer, T. Biedermann, and M. Rocken Direct crosstalk between mast cell-tnf and TNFR1-expressing endothelia mediates local tissue inflammation. Blood 114: Leal-Berumen, I., P. Conlon, and J. S. Marshall IL-6 production by rat peritoneal mast cells is not necessarily preceded by histamine release and can be induced by bacterial lipopolysaccharide. J Immunol 152: McCurdy, J. D., T. J. Lin, and J. S. Marshall Toll-like receptor 4-mediated activation of murine mast cells. J Leukoc Biol 70: McCurdy, J. D., T. J. Olynych, L. H. Maher, and J. S. Marshall Cutting edge: distinct Toll-like receptor 2 activators selectively induce different classes of mediator production from human mast cells. J Immunol 170: Supajatura, V., H. Ushio, A. Nakao, S. Akira, K. Okumura, C. Ra, and H. Ogawa Differential responses of mast cell Toll-like receptors 2 and 4 in allergy and innate immunity. J Clin Invest 109:
51 128. Supajatura, V., H. Ushio, A. Nakao, K. Okumura, C. Ra, and H. Ogawa Protective roles of mast cells against enterobacterial infection are mediated by Tolllike receptor 4. J Immunol 167: Varadaradjalou, S., F. Feger, N. Thieblemont, N. B. Hamouda, J. M. Pleau, M. Dy, and M. Arock Toll-like receptor 2 (TLR2) and TLR4 differentially activate human mast cells. Eur J Immunol 33: Heil, F., H. Hemmi, H. Hochrein, F. Ampenberger, C. Kirschning, S. Akira, G. Lipford, H. Wagner, and S. Bauer Species-specific recognition of singlestranded RNA via toll-like receptor 7 and 8. Science 303: Kulka, M., L. Alexopoulou, R. A. Flavell, and D. D. Metcalfe Activation of mast cells by double-stranded RNA: evidence for activation through Toll-like receptor 3. J Allergy Clin Immunol 114: Matsushima, H., N. Yamada, H. Matsue, and S. Shimada TLR3-, TLR7-, and TLR9-mediated production of proinflammatory cytokines and chemokines from murine connective tissue type skin-derived mast cells but not from bone marrowderived mast cells. J Immunol 173: Okumura, S., J. Kashiwakura, H. Tomita, K. Matsumoto, T. Nakajima, H. Saito, and Y. Okayama Identification of specific gene expression profiles in human mast cells mediated by Toll-like receptor 4 and FcepsilonRI. Blood 102: Zhu, F. G., and J. S. Marshall CpG-containing oligodeoxynucleotides induce TNF-alpha and IL-6 production but not degranulation from murine bone marrowderived mast cells. J Leukoc Biol 69: Kawakami, T., and S. J. Galli Regulation of mast-cell and basophil function and survival by IgE. Nat Rev Immunol 2: Woolhiser, M. R., Y. Okayama, A. M. Gilfillan, and D. D. Metcalfe IgGdependent activation of human mast cells following up-regulation of FcgammaRI by IFN-gamma. Eur J Immunol 31: Genovese, A., J. P. Bouvet, G. Florio, B. Lamparter-Schummert, L. Bjorck, and G. Marone Bacterial immunoglobulin superantigen proteins A and L activate human heart mast cells by interacting with immunoglobulin E. Infect Immun 68:
52 138. Gavrilyuk, V., S. Kalinin, B. S. Hilbush, A. Middlecamp, S. McGuire, D. Pelligrino, G. Weinberg, and D. L. Feinstein Identification of complement 5a-like receptor (C5L2) from astrocytes: characterization of anti-inflammatory properties. J Neurochem 92: Lee, D. K., S. R. George, R. Cheng, T. Nguyen, Y. Liu, M. Brown, K. R. Lynch, and B. F. O'Dowd Identification of four novel human G protein-coupled receptors expressed in the brain. Brain Res Mol Brain Res 86: Ohno, M., T. Hirata, M. Enomoto, T. Araki, H. Ishimaru, and T. A. Takahashi A putative chemoattractant receptor, C5L2, is expressed in granulocyte and immature dendritic cells, but not in mature dendritic cells. Mol Immunol 37: Okinaga, S., D. Slattery, A. Humbles, Z. Zsengeller, O. Morteau, M. B. Kinrade, R. M. Brodbeck, J. E. Krause, H. R. Choe, N. P. Gerard, and C. Gerard C5L2, a nonsignaling C5A binding protein. Biochemistry 42: Otto, M., H. Hawlisch, P. N. Monk, M. Muller, A. Klos, C. L. Karp, and J. Kohl C5a mutants are potent antagonists of the C5a receptor (CD88) and of C5L2: position 69 is the locus that determines agonism or antagonism. J Biol Chem 279: Chen, N. J., C. Mirtsos, D. Suh, Y. C. Lu, W. J. Lin, C. McKerlie, T. Lee, H. Baribault, H. Tian, and W. C. Yeh C5L2 is critical for the biological activities of the anaphylatoxins C5a and C3a. Nature 446: Rittirsch, D., M. A. Flierl, B. A. Nadeau, D. E. Day, M. Huber-Lang, C. R. Mackay, F. S. Zetoune, N. P. Gerard, K. Cianflone, J. Kohl, C. Gerard, J. V. Sarma, and P. A. Ward Functional roles for C5a receptors in sepsis. Nat Med 14: Ames, R. S., Y. Li, H. M. Sarau, P. Nuthulaganti, J. J. Foley, C. Ellis, Z. Zeng, K. Su, A. J. Jurewicz, R. P. Hertzberg, D. J. Bergsma, and C. Kumar Molecular cloning and characterization of the human anaphylatoxin C3a receptor. J Biol Chem 271: Gerard, C., and N. P. Gerard C5A anaphylatoxin and its seven transmembranesegment receptor. Annu Rev Immunol 12: Rajagopal, S., K. Rajagopal, and R. J. Lefkowitz Teaching old receptors new tricks: biasing seven-transmembrane receptors. Nat Rev Drug Discov 9:
53 148. Marinissen, M. J., and J. S. Gutkind G-protein-coupled receptors and signaling networks: emerging paradigms. Trends Pharmacol Sci 22: Han, J., J. D. Lee, L. Bibbs, and R. J. Ulevitch A MAP kinase targeted by endotoxin and hyperosmolarity in mammalian cells. Science 265: Li, Z., Y. Jiang, R. J. Ulevitch, and J. Han The primary structure of p38 gamma: a new member of p38 group of MAP kinases. Biochem Biophys Res Commun 228: Ashwell, J. D The many paths to p38 mitogen-activated protein kinase activation in the immune system. Nat Rev Immunol 6: Berkowitz, P., P. Hu, Z. Liu, L. A. Diaz, J. J. Enghild, M. P. Chua, and D. S. Rubenstein Desmosome signaling. Inhibition of p38mapk prevents pemphigus vulgaris IgG-induced cytoskeleton reorganization. J Biol Chem 280: Berkowitz, P., P. Hu, S. Warren, Z. Liu, L. A. Diaz, and D. S. Rubenstein p38mapk inhibition prevents disease in pemphigus vulgaris mice. Proc Natl Acad Sci U S A 103: Schett, G., J. Zwerina, and G. Firestein The p38 mitogen-activated protein kinase (MAPK) pathway in rheumatoid arthritis. Ann Rheum Dis 67: la Sala, A., M. Gadina, and B. L. Kelsall G(i)-protein-dependent inhibition of IL-12 production is mediated by activation of the phosphatidylinositol 3-kinaseprotein 3 kinase B/Akt pathway and JNK. J Immunol 175: Hawlisch, H., Y. Belkaid, R. Baelder, D. Hildeman, C. Gerard, and J. Kohl C5a negatively regulates toll-like receptor 4-induced immune responses. Immunity 22: Rousseau, S., I. Dolado, V. Beardmore, N. Shpiro, R. Marquez, A. R. Nebreda, J. S. Arthur, L. M. Case, M. Tessier-Lavigne, M. Gaestel, A. Cuenda, and P. Cohen CXCL12 and C5a trigger cell migration via a PAK1/2-p38alpha MAPK-MAPKAP- K2-HSP27 pathway. Cell Signal 18:
54 158. Kalesnikoff, J., N. Baur, M. Leitges, M. R. Hughes, J. E. Damen, M. Huber, and G. Krystal SHIP negatively regulates IgE + antigen-induced IL-6 production in mast cells by inhibiting NF-kappa B activity. J Immunol 168: Gordon, J. R., and S. J. Galli Mast cells as a source of both preformed and immunologically inducible TNF-alpha/cachectin. Nature 346: Nimmerjahn, F., and J. V. Ravetch Fcgamma receptors as regulators of immune responses. Nat Rev Immunol 8: Coward, W. R., Y. Okayama, H. Sagara, S. J. Wilson, S. T. Holgate, and M. K. Church NF-kappa B and TNF-alpha: a positive autocrine loop in human lung mast cells? J Immunol 169: Locksley, R. M., N. Killeen, and M. J. Lenardo The TNF and TNF receptor superfamilies: integrating mammalian biology. Cell 104: Chen, G., and D. V. Goeddel TNF-R1 signaling: a beautiful pathway. Science 296: Walker, M. E., J. K. Hatfield, and M. A. Brown New insights into the role of mast cells in autoimmunity: Evidence for a common mechanism of action? Biochim Biophys Acta 1822: Bebo, B. F., Jr., T. Yong, E. L. Orr, and D. S. Linthicum Hypothesis: a possible role for mast cells and their inflammatory mediators in the pathogenesis of autoimmune encephalomyelitis. J Neurosci Res 45: Ibrahim, M. Z., A. T. Reder, R. Lawand, W. Takash, and S. Sallouh-Khatib The mast cells of the multiple sclerosis brain. J Neuroimmunol 70: Lock, C., G. Hermans, R. Pedotti, A. Brendolan, E. Schadt, H. Garren, A. Langer- Gould, S. Strober, B. Cannella, J. Allard, P. Klonowski, A. Austin, N. Lad, N. Kaminski, S. J. Galli, J. R. Oksenberg, C. S. Raine, R. Heller, and L. Steinman Gene-microarray analysis of multiple sclerosis lesions yields new targets validated in autoimmune encephalomyelitis. Nat Med 8:
55 168. Hueber, A. J., D. L. Asquith, A. M. Miller, J. Reilly, S. Kerr, J. Leipe, A. J. Melendez, and I. B. McInnes Mast cells express IL-17A in rheumatoid arthritis synovium. J Immunol 184: Sandler, C., K. A. Lindstedt, S. Joutsiniemi, J. Lappalainen, T. Juutilainen, J. Kolah, P. T. Kovanen, and K. K. Eklund Selective activation of mast cells in rheumatoid synovial tissue results in production of TNF-alpha, IL-1beta and IL-1Ra. Inflamm Res 56: Geoffrey, R., S. Jia, A. E. Kwitek, J. Woodliff, S. Ghosh, A. Lernmark, X. Wang, and M. J. Hessner Evidence of a functional role for mast cells in the development of type 1 diabetes mellitus in the BioBreeding rat. J Immunol 177: Rao, K. N., and M. A. Brown Mast cells: multifaceted immune cells with diverse roles in health and disease. Ann N Y Acad Sci 1143: Sayed, B. A., A. Christy, M. R. Quirion, and M. A. Brown The master switch: the role of mast cells in autoimmunity and tolerance. Annu Rev Immunol 26: Gri, G., S. Piconese, B. Frossi, V. Manfroi, S. Merluzzi, C. Tripodo, A. Viola, S. Odom, J. Rivera, M. P. Colombo, and C. E. Pucillo CD4+CD25+ regulatory T cells suppress mast cell degranulation and allergic responses through OX40-OX40L interaction. Immunity 29: Kashyap, M., A. M. Thornton, S. K. Norton, B. Barnstein, M. Macey, J. Brenzovich, E. Shevach, W. J. Leonard, and J. J. Ryan Cutting edge: CD4 T cell-mast cell interactions alter IgE receptor expression and signaling. J Immunol 180: Piconese, S., G. Gri, C. Tripodo, S. Musio, A. Gorzanelli, B. Frossi, R. Pedotti, C. E. Pucillo, and M. P. Colombo Mast cells counteract regulatory T-cell suppression through interleukin-6 and OX40/OX40L axis toward Th17-cell differentiation. Blood 114:
56 CHAPTER 2 THE C5A RECEPTOR ON MAST CELLS IS CRITICAL FOR THE AUTOIMMUNE SKIN BLISTERING DISEASE BULLOUS PEMPHIGOID 2 Bullous pemphigoid (BP) is an autoimmune skin blistering disease characterized by the presence of autoantibodies against the hemidesmosomal proteins BP230 and BP180. In the IgG passive transfer mouse model of BP, subepidermal blistering is triggered by anti-bp180 antibodies and depends on the complement system, mast cell (MC) degranulation, and neutrophil infiltration. In this study, we identify the signaling events that connect the activation of the complement system and MC degranulation. We found that mice deficient in MCs or C5a receptor (C5aR) injected with pathogenic anti-bp180 IgG failed to develop subepidermal blisters and exhibited a drastic reduction in p38mapk phosphorylation compared to wild-type (WT) mice. Local reconstitution with MCs from WT but not C5aRdeficient mice restored high levels of p38mapk phosphorylation and subepidermal blistering in MC-deficient mice. Local injection of recombinant C5a (rc5a) induced phosphorylation of p38mapk in WT, but not MC-deficient mice. Cultured mouse MCs treated with rc5a exhibited a significant increase of p38mapk phosphorylation and MC degranulation. Taken together, these data demonstrate that C5a interacts with C5aR on MCs, 2 Reproduced/amended from: Heimbach, L., Li, Z., Berkowitz, P., Zhao, M., Li, N., Rubenstein, D.S., Diaz, L.A., Liu, Z. The C5a receptor on mast cells is critical for the autoimmune skin-blistering disease bullous pemphigoid. J Biol Chem 286(17):
57 and that this C5a-C5aR interaction triggers activation of the p38mapk pathway, subsequent MC degranulation, and ultimately BP blistering. 2.1 INTRODUCTION Bullous pemphigoid (BP) is an acquired skin autoimmune blistering disease characterized by the presence of circulating and tissue bound autoantibodies against two major hemidesmosomal proteins, BP230 (BPAG1) and BP180 (BPAG2 or type XVII collagen) (1-11). These anti-hemidesmosomal autoantibodies, along with complement components, are deposited along the basement membrane at the dermal-epidermal junction (DEJ) of perilesional skin (12-14). Basal keratinocytes detach from the underlying dermis, leading to blister formation (5). Using an animal model of BP in which neonatal BALB/c mice are injected with anti-murine BP180 (mbp180) antibodies, we previously demonstrated that dermal-epidermal separation is triggered by anti-bp180 IgG and depends on complement activation, mast cells (MCs), and neutrophils (PMNs) (15-17). The binding of pathogenic IgG to BP180 activates the classical pathway of the complement and leads to MC degranulation, PMN recruitment, and subsequent skin blistering (18). However, the cellular processes linking complement activation and MC degranulation are not defined in BP. Complement activation via the classical, alternative, and lectin pathways generates the anaphylotoxin C5a (19). A potent chemoattractant and inflammatory mediator, C5a acts on a wide range of myeloid and lymphoid lineage cells, including skin MCs (20, 21). Skin MCs express C5a receptor (C5aR, or CD88) (22), and interaction of C5a and C5aR induces MC degranulation and the release of proinflammatory cytokines such as interleukin 1 (IL-1), IL- 6, tumor necrosis factor alpha (TNF-α), and granulocyte macrophage colony stimulating 42
58 factor (GM-CSF) (23). In PMNs and monocytes, the interaction of C5a and C5aR leads to activation of p38 mitogen activated protein kinase (p38mapk) (24-26). p38mapk is a key signaling molecule involved in translating extracellular environmental conditions, particularly inflammatory conditions, into cellular responses (27, 28). MC activation via antigen crosslinking of surface immunoglobulin E (IgE) molecules leads to p38mapk activation and cytokine release (29), however, p38mapk activation upon engagement of the C5aR on MCs has not yet been reported. In this work, we examined the role of C5a-C5aR interaction in the progression of experimental BP. We found that C5a interacts with C5aR on MCs, and that this C5a-C5aR interaction triggers activation of the p38mapk pathway and ultimately induces MC degranulation and subsequent BP blistering. 2.2 EXPERIMENTAL PROCEDURES Materials. The p38mapk inhibitor SB and the inactive analog SB were purchased from Calbiochem (La Jolla, CA). Rabbit anti-phospho-p38mapk antibody was purchased from Cell Signaling (Danvers, MA), and rabbit anti-p38mapk antibody was purchased from Santa Cruz Biotechnology (Santa Cruz, CA). The recombinant C5a was purchased from R&D System (Minneapolis, MN). Protein concentration was determined with the RC DC protein assay purchased from Bio-Rad Laboratories (Hercules, CA). The ECL Western blotting analysis kit was purchased from GE Healthcare (Piscataway, NJ). Laboratory animals. Breeding pairs of C57BL/6J, C4-deficient (C4-/-), MCdeficient WBB6F1-Kit w /Kit w-v (MC-/-), and C5a receptor-deficient (C5aR-/-) mice were purchased from Jackson Laboratories (Bar Harbor, ME) and maintained at the University of 43
59 North Carolina at Chapel Hill. The MC deficiency in Kit w /Kit w-v mice is caused by distinct mutations in MC c-kit (30). Neonatal mice (24-36 hours old with body weights between 1.4 and 1.6 grams) were used for passive transfer experiments. All animal care and animal experiments were approved by the UNC Animal Care Committee and were in accordance with the National Institutes of Health guidelines. Preparation of pathogenic anti-bp180 IgG. The preparation of recombinant murine BP180 (mbp180) and the immunization of New Zealand white rabbits were performed as previously described (31-33). Briefly, a segment of the ectodomain of the murine BP180 antigen (31) was expressed as a glutathione S-transferase (GST) fusion protein using the pgex prokaryotic expression system (Pharmacia LKB Biotechnology, Piscataway, NJ). The murine BP180 fusion protein, designated GST-mBP180ABC containing NC14A and half of C13 domains (Figure 1.1), was purified to homogeneity by affinity chromatography using a glutathione-agarose column (33). New Zealand White rabbits were immunized with the purified mbp180 fusion protein and the IgG fraction from the serum (designated R621) was purified as previously described (31). The IgG fractions were concentrated, sterilized by ultrafiltration and the protein concentrations determined by OD 280 [E (1%, 1 cm) = 13.6]. The titers of anti-murine BP180 antibodies in both the unfractionated rabbit serum and in the purified IgG fraction were assayed by indirect immunofluorescence (IF) using mouse skin cryosections as substrate. The antibody preparations were also tested by immunoblotting against the GST-mBP180ABC fusion protein. The pathogenicity of these IgG preparations was tested by passive transfer experiment as described below. 44
60 Induction of experimental BP and clinical evaluation of animals. Neonates were given on the back one i.d. injection of a sterile solution of IgG in PBS (50 μl IgG, 2.5 mg IgG/g body weight) as described elsewhere (31). The skin at the IgG-injection site of the mice from the test and control groups was examined at different time points after the IgG injection. The extent of cutaneous disease was scored as follows: "(-)", no detectable skin disease; "1+", mild erythematous reaction with no evidence of the epidermal detachment sign [this sign was elicited by gentle friction of the mouse skin which, when positive, produced fine, persistent wrinkling of the epidermis]; "2+", intense erythema and epidermal detachment sign involving 10-50% of the epidermis in localized areas; and "3+", intense erythema with frank epidermal detachment sign involving more than 50% of the epidermis in the injection site. After clinical examination, the animals were sacrificed, and skin and serum specimens were obtained. Skin sections were used for a) routine histological examination by light microscopy (H/E staining) to localize the lesional site and PMN infiltration, b) toluidine blue staining to quantify MCs and MC degranulation, c) direct IF assays to detect rabbit IgG and mouse C3 deposition at the BMZ, and d) myeloperoxidase (MPO) enzymatic assay to quantify the PMN accumulation at the skin injection site described below. The sera of injected animals were tested by indirect IF techniques to determine the titers of rabbit antimbp180 antibodies using mouse skin cryosections as the substrate. Direct and indirect IF studies were performed as previously described (31) using commercially available FITCconjugated goat anti-rabbit IgG (Kirkeggard & Perry Laboratories Inc.). Monospecific goat anti-mouse C3 IgG was purchased from Cappel Laboratories. 45
61 Quantification of PMN accumulation at the skin site. MPO activity in skin sites of the injected animals was assayed as a measure of PMN infiltration as described (34, 35). A standard reference curve was established using purified MPO (Athens Research and Technology, Inc., Athens, Georgia). The skin samples were extracted by homogenization in an extraction buffer containing 0.1 M Tris-Cl, ph 7.6, 0.15 M NaCl, 0.5% hexadecyl trimethylammonium bromide. MPO activity in the supernatant fraction was measured by the change in optical density at 460 nm resulting from decomposition of H 2 O 2 in the presence of o-dianisidine. MPO content was expressed as units of MPO activity/mg protein. Protein concentrations were determined by the Bio-RAD dye-binding assay using bovine serum albumin (BSA) as a standard. Determination of C5a levels in skin. Skin from the IgG injection site of the diseased and control mice was mechanically homogenized in PBS to extract proteins. The level of mouse C5a in the skin was measured by ELISA (R&D Systems, Minneapolis, MN). Microtiter plates were coated with a rat anti-mouse C5a antibody, incubated with skin protein extracts, followed by goat anti-mouse C5a detection antibody, and then developed and read at OD 492nm. The C5a level was expressed as OD 492 reading/mg protein. Quantification of MCs and MC degranulation. MCs and MC degranulation in skin samples were quantified according to Wershil, et al. (36), with modification. Briefly, lesional and nonlesional skin sections of IgG-injected mice were fixed in 10% formalin. Paraffin sections (5-μm thick) were prepared and stained with toluidine blue and H&E. The total number of MCs was counted and classified as degranulated (>10% of the granules exhibiting fusion or discharge) or normal in five fields under a light microscope as described previously. The results are expressed as percentage of MC degranulation. 46
62 MC reconstitution. Kit w /Kit w-v mice were repaired of their MC deficiency selectively and locally by the injection of growth factor-dependent bone marrow-derived cultured MCs into the skin (15, 37, 38). Briefly, femoral bone marrow cells from WT and C5aR-/- mice were maintained in vitro for 4 weeks in RPMI 1640 complete medium (Life Technologies, Grant Island, NY) supplemented with 20% WEHI-3-conditioned medium until MCs represented >95% of the total cells as determined by toluidine blue staining and flow cytometry analysis using antibodies specific for the MC cell surface markers FcεRI, c-kit, and CD13 (37). Murine IgE and rat anti-mouse IgE were purchased from Southern Biotechnology Associates (Birmingham, AL). FITC-labeled rat anti-mouse c-kit and FITClabeled rat anti-mouse CD13 were obtained from DB Pharmingen (San Diego, CA). MCs (1x10 6 in 20 μl of medium) were injected i.d. into the ears of 8 to 10 week old MC-deficient mice. Medium alone (20 μl) was injected i.d. into the ears of MC-/- and MC+/+ mice to serve as negative controls. This procedure selectively and locally reconstitutes the dermal MC population without systemic effects (38). MC reconstitution was confirmed by staining skin sections from MC-injected sites with toluidine blue. Ten weeks after the adoptive transfer of MCs, when the injected MCs were fully matured into functional cutaneous MCs (15, 37, 38), both ears of the mice were injected i.d. with pathogenic anti-bp180 IgG (2 mg/20 μl/site). 24 hours later, ear skin biopsies were obtained and analyzed by H/E, toluidine blue staining and MPO enzyme assay as described above. In vivo and in vitro inhibition of p38mapk activation. For the in vivo inhibitor studies, mice were pretreated with 50 μl of the p38mapk inhibitor SB or inactive analog SB (i.d., 10 μg/g body weight). Two hours later, the mice were injected i.d. with 50 μl of pathogenic IgG or control IgG (2.5 mg/g body weight) and the mice were 47
63 examined at different time points for BP skin lesions and p38mapk activation (as described below). For in vitro inhibitor studies, cultured bone-marrow-derived MCs from C57BL/6J mice were incubated with C5a (25 ng/ml) in the presence of p38mapk inhibitor SB (20 μm) for 0-60 min and cell extracts were assayed for p38mapk activation. Detection of total and phospho-p38mapk. Protein was extracted from skin samples in lysis buffer (8 M urea, 4% 3-(3-cholamidopropyl)dimethylammonio-1- propanesulfonic acid (CHAPS), 2.5 mm dithiothreitol, 40 mm Tris, 10 µm pepstatin, 100 µm leupeptin, 10 µm E-64, 1 mm phenylmethylsulfonyl fluoride, 0.1 M sodium orthovanadate, 0.5 mm sodium fluoride, and Okadaic acid). For the cell culture studies, bone marrow-derived MCs were incubated with buffer control or C5a (25 ng/ml) in the absence or presence of SB (20 μm) for 0-60 min at 37 C. The cells were harvested and lysed on ice, and 20 µg of each protein extract was loaded on a 10% acrylamide gel and separated by SDS-PAGE. Gels were transferred to PVDF (Millipore) membranes and probed by immunoblot for total p38mapk and phospho-p38mapk. Western blots were developed by enhanced chemiluminescence (ECL) reaction (Amersham Biosciences). The signal intensity was quantified by scanning chemiluminescence on a GeneGnome scanner (Syngene Bio Imaging, Frederick, MD) using GeneSnap software. Statistical analysis. The data are expressed as mean + SEM and were analyzed using the Student's paired t-test. A p value less than 0.05 was considered significant. 48
64 2.3 RESULTS Mice lacking the classical pathway of the complement system are resistant to experimental BP and exhibit a reduced level of skin C5a. We previously reported that the classical pathway of complement activation is critical to the pathogenesis of experimental BP in mice (18). Wild-type (C4+/+) mice and mice deficient in the alternative pathway component factor B (Fb-/-) developed subepidermal blisters following injection with pathogenic anti-mbp180 IgG; however, mice deficient in the classical pathway component C4 (C4-/-) were resistant to blister formation (Fig. 2.1A and Table 2.1). 24 hours following injection, C4-/- mice had significantly lower levels of C5a present in the skin compared to WT and Fb-/- mice (p<0.0001, Fig. 2.1B). Significantly fewer infiltrating PMNs were present in the skin of the C4-/- mice than in the WT or Fb-/- mice, as determined by quantification of the PMN cell marker myeloperoxidase (MPO) enzymatic activity (Fig. 2.1C). These results demonstrate that complement activation via the classical pathway is essential for generation of C5a, recruitment of PMNs to the skin, and blistering in experimental BP. Mice lacking C5a receptor are resistant to experimental BP and exhibit impairment in MC degranulation and p38mapk activation. C5a fragments primarily act through binding to the C5a receptor (C5aR, or CD88) (39). Having determined that high levels of C5a are present in the skin of mice susceptible to experimental BP, we next examined the role of the C5aR in disease development. We injected C5aR-deficient mice (C5aR-/-) and WT mice (C5aR+/+) with pathogenic anti-bp180 antibodies and found that C5aR+/+ mice developed blisters, but C5aR-/- mice did not (Figs. 2.2A and Table 2.1). Although C5aR-/- mice failed to develop skin lesions, the amount of C5a present in their skin was comparable 49
65 to the WT mice at 4 h post IgG injection (Fig. 2.2B), when the early phase of PMN infiltration begins (17). These data demonstrate that C5aR deficiency does not affect C5a generation. However, MC activation in the skin of the pathogenic IgG-injected C5aR-/- mice was severely impaired as evidenced by toluidine blue staining, a MC-specific histochemical staining. At 2 h post-injection when MC degranulation triggered by pathogenic IgG reaches its peak in experimental BP (15), extensive MC degranulation was observed in the dermis of WT mice (Fig. 2.2C, left panel). In contrast, C5aR-/- mice exhibited a minimal level of MC degranulation in the dermis (Fig. 2.2C, right panel). We quantified MC degranulation and found a significant reduction in the number of degranulating MCs in C5aR-/- mice compared to WT mice (p<0.001, Fig. 2.2D). Because MC degranulation releases cytokines that recruit PMNs to the skin during experimental BP pathogenesis (15), we hypothesized that the C5aR- /- mice would be unable to effectively attract PMNs to the skin. Indeed, the pathogenic IgGinjected C5aR-/- mice showed a significantly reduced PMN infiltration in the skin as compared to the diseased WT mice (Fig. 2.2E). In addition to the lack of MC activation, C5aR-/- mice injected with pathogenic antibodies also lacked activation of p38mapk, a critical signaling pathway in inflammation and autoimmunity (28). By immunoblotting, the lesional skin of the pathogenic IgG-injected WT mice exhibited a much more intense phosphorylated p38mapk band as compared to the skin of the pathogenic IgG-injected C5aR-/- mice (Fig. 2.3A, lane 2 vs. lane 4). Quantitative analysis of intensity of each band on immunoblots confirmed that the phospho-p38 levels in the lesional skin of the C5aR+/+ mice were significantly higher than C5aR-/- mice (p<0.0001, Fig. 2.3B, bar 2 vs. bar 4). These results suggest that the molecular interaction 50
66 between C5a and C5aR leads to MC activation and p38mapk signaling, followed subsequently by PMN recruitment and BP blistering. p38mapk activation is required for experimental BP. To determine whether p38mapk activation is involved in the disease development or is simply a secondary stress response, neonatal WT mice were pretreated with the p38mapk inhibitor SB or inactive control SB (i.d., 10 μg/g body weight). Two hours later, the mice were injected i.d. with pathogenic antibodies. The SB treated mice developed clinical and histological subepidermal blisters with IgG deposition at the BMZ (Fig. 2.4A, left panels; also Table 2.1). However, SB treatment completely abolished the BP skin lesions (Fig. 2.4A, right panels). Mice pretreated with SB exhibited significantly higher levels of p38mapk phosphorylation at 2 and 24 hours post IgG injection (Fig. 2.4B, lanes 2 and 5) than mice pretreated with SB (Fig. 2.4B, lanes 3 and 6). Inhibition of p38mapk activation also resulted in significantly reduced MC degranulation (Fig. 2.4C, bar 4 vs. bar 3) and PMN infiltration (Fig. 2.4D, bar 4 vs. bar 3). These results demonstrate that p38mapk is critical for MC activation and subsequent the development of experimental BP. C5aR-mediated p38mapk activation depends on MCs. The complement system and MC activation are indispensable to the pathogenesis of experimental BP (18, 40). Having demonstrated that p38mapk activation is also required for disease development, we next investigated the links between complement, MCs, and p38mapk. MCs express C5aR and can be activated by C5a (41), so we reasoned that the interaction of C5a with the MC C5aR may trigger p38mapk signaling. To test this hypothesis, we injected neonatal MCsufficient (MC+/+) and MC-deficient (MC-/-) mice with PBS or recombinant C5a (rc5a) 51
67 (i.d., 50 ng in PBS/mouse) and analyzed phosphorylated p38mapk levels in the skin at the injection sites. We observed a significantly higher level of p38mapk activation in the rc5atreated MC+/+ mice (Fig. 2.5A, lane 2) as compared to the rc5a-treated MC-/- mice (Fig. 2.5A, lane 4). In vitro, we found that bone marrow-derived MCs, when incubated with rc5a (25 ng/ml) for as little as 5 minutes, exhibited high levels of p38mapk activation (Fig. 2.5B). Addition of SB (20 μm) to MC culture prior to rc5a treatment almost completely blocked p38mapk activation (Fig. 2.5C). These data strongly suggest that p38mapk activation occurs in MCs following exposure to C5a. C5aR is broadly expressed across a number of cell types, and p38mapk is activated in many immune cells in response to extracellular inflammatory signals (42, 43). To strengthen the evidence that MCs are the key cell type that interact with C5a and promote development of experimental BP, we reconstituted MC-/- mice with MCs from C5aR-/- mice, or MCs from C5aR+/+ mice. If p38mapk activation is dependent on the C5aR on MCs, then MC-/- mice reconstituted with MCs expressing C5aR should be susceptible to experimental BP, but mice reconstituted with C5aR-deficient MCs should be protected from experimental BP. To test this hypothesis, we injected the left ears of MC-/- mice with C5aR- /- MCs and the right ears of the same MC-/- mice with C5aR+/+ MCs (1x10 6 cells/site). Ten weeks later, both ears of the mice were injected i.d. with pathogenic anti-bp180 IgG (2 mg/20 μl/site) and examined 24 hour post IgG injection. Similar numbers of MCs were present in the C5aR+/+ MC reconstituted ears and the C5aR-/- MC reconstituted ears. MC-/- mice that were not reconstituted with any MCs and mice reconstituted with C5aR-/- MCs developed no skin lesions, while MC-/- mice reconstituted with C5aR+/+ MCs showed subepidermal blistering (Fig. 2.6A). The number of infiltrating PMNs in the C5aR+/+ MC- 52
68 reconstituted site was significantly higher than the C5aR-/- MC-reconstituted site (Fig. 2.6B, bar 6 vs. bar 5). Pretreatment with p38mapk inhibitor SB abolished BP skin lesions (Fig. 2.6A, 4 th panel) and markedly reduced PMN infiltration in the MC-/- mice reconstituted with C5aR+/+ MCs (Fig. 2.6B, bar 7). Taken together, these results demonstrate that disease progression is dependent on MCs expressing C5aR and p38mapk activation. 2.4 DISCUSSION Activation of the classical pathway of complement generates C5a, a molecule critical for tissue inflammation and the recruitment of PMNs to the cutaneous BMZ. C5a may carry out this role either directly (functioning as a PMN chemoattractant itself) or indirectly (by stimulating local cells to release proinflammatory mediators such as IL-8), or through a combination of these two mechanisms (44). In experimental BP, we found that C5a contributes to disease progression primarily through interaction with C5aR on MCs, as evidenced by the failure of C5aR-/- mice to induce MC degranulation and subsequent PMN accumulation upon passive transfer of pathogenic anti-bp180 antibodies. Without C5aR, activation of p38mapk in the pathogenic anti-mbp180 IgG-injected mouse skin is impaired. p38mapk is an important signaling molecule involved in translating extracellular environmental conditions into cellular responses (27, 28). This signaling pathway plays a key role in numerous immune and inflammatory diseases, including rheumatoid arthritis and skin autoimmune disease pemphigus vulgaris (45-47). Our data show that p38mapk activation is not a secondary stress response and instead is required for 53
69 experimental BP, as mice treated with p38mapk inhibitors are protected from disease progression. It has been reported that in PMNs and monocytes, interaction of C5a and C5aR leads to activation of p38mapk (24-26). In this study, we provide direct evidence that MCs also use the same pathway of C5a-C5aR-mediated p38mapk activation. WT mice injected with rc5a exhibited significantly greater p38mapk phosphorylation than MC-/- mice, indicating that MCs are a key source of the p38mapk signaling required for induction of experimental BP. Compellingly, we found that MC-/- mice reconstituted with MCs expressing C5aR prior to passive transfer of pathogenic antibody develop blisters; however, mice reconstituted with MCs lacking C5aR expression do not exhibit blister formation. Furthermore, rc5a directly activates p38mapk in C5aR-sufficient but not C5aR-deficient cultured MCs. Taken together, these data demonstrate that C5a interaction with C5aR on MCs and the subsequent MC p38mapk activation are indispensable for development of experimental BP. In summary, our present studies identify the C5a-C5aR interaction being a critical molecular linker between basal keratinocyte-based complement activation and MC-based p38mapk activation in experimental BP. Activation of p38mapk is essential for MC activation, PMN infiltration, and blister formation in experimental BP. Inhibitors of p38mapk are currently under investigation for treatment of autoimmune and inflammatory diseases (48-50). We found that BP patients have significantly elevated levels of p38mapk activation in lesional skin, indicating that p38mapk is likely an important mediator of inflammation in the human, as well as the mouse. Thus treatment of BP with these p38mapk inhibitors may prove to be a practical therapy if specific and safe inhibitors can be developed. 54



, and factor")


.")



 and")


.")



.")

70 Figure 1.1 Level of C5a in the skin is significantly reduced in pathogenic IgG-injected mice lacking the classical pathway of complement. Neonatal wild-type (C4+/+), C4- deficient (C4-/-), and factor b-deficient (Fb-/-) mice were injected i.d. with pathogenic rabbit anti-murine BP180 IgG R621 or control rabbit IgG (2.5 mg/gg body weight). The injected animals were examined 24 h post-injection. (A) Hematoxylin and Eosin-stained skin section from these mice showed a subepidermal vesicle withh neutrophilic infiltrate in the pathogenic IgG-injected C4+ +/+ (left panel) and Fb-/-- (right panel) mice. In contrast, R621-injected C4-/- mice showed no evidence of subepidermal vesiculation at the light microscopic level (middle panel). E, epidermis; D, dermis; V, vesicle. X125 magnifications. (B) A C5a-specific ELISA assay exhibited a significantly reduced levels of C5a in the skin of C4-/- mice as compared to the diseased mice (p<0.0001, bar 4 vs. bars 2, 6). (C) MPO enzymatic assay of skin protein extracts revealed a significantly lower number of infiltrating PMNs in the C4-/- mice as compared to the diseased mice (p<0.001, bar 4 vs. bars 2, 6).. n=8 for each group. Three independent experiments were done for each group of mice. 55


 and")
 mice were")


.")

 Pathogenic R621 IgG")






 Toluidine blue")












71 Figure 2.2. Mice lacking C5a receptorr are resistant to experimental BP. Neonatal wildrabbit type (C5aR+/+) and C5aR-deficient (C5aR-/-) mice were injected i.d. with pathogenic anti-murine BP180 IgG R621 or control rabbit IgG (2.5 mg/gg body weight). The injected animals were examined 2, 4, and 24 h post-injection. (A) Pathogenic R621 IgG induced BP blisters in C5aR+ +/+ mice (left panel), but not in C5aR-/- mice (right panel) at 24 h post injection. (B) A C5a-specific ELISA assay show no differencee in skin C5a levels between C5aR-/- and C5aR+/+ mice at 4 h post IgG injection (p=0.325, bar 2 vs. bar 4). (C) Toluidine blue staining showed an extensive MC degranulation n in the C5aR+/+ mice (left panel) at 2 h post IgG injection, while the C5aR-/- mice (right panel) exhibited a minimal level of MC degranulation. (D) Quantification of degranulated MCs on the toluidine blue-stained skin sections showed a drastically increased MC degranulation in the C5aR+/+ mice as compared to C5aR-/- mice (p<0.001, bar 2 vs. bar 4). (E) MPO enzymatic assay of skin protein extracts revealed a significantly lower number of infiltrating PMNs in the C5aR-/- mice as compared to the diseased mice (p<0.001, bar 2 vs. bar 4). n=8 for each group. Three independent experiments weree done for each group of mice. 56


 mice were injected i.d. with pathogenic (2.")

 were analyzed by")
 and total p38mapk (total p38).")
) Immunoblotting analysis of A much more intense")
 as compared to C5aR-/--")


72 Figure 2.3. Mice lacking C5aR show impaired p38mapk activation inducedd by pathogenic antibodies. Neonatal wild-type rabbit anti-murinee BP180 IgG R621 or control rabbit IgG (C5aR+ +/+) and C5aR-deficient (C5aR-/-) mice were injected i.d. with pathogenic (2.5 mg/g body weight). The skin protein extracts of injected animals were examined 24 h post-injectiop38mapk activation. Skin protein extracts (30 μg/ /lane) were analyzed by immunoblotting with antibodies to phosphorylated p38mapk (phospho-p38) and total p38mapk (total p38). by immunoblotting for p38mapk activation. (A)) Immunoblotting analysis of A much more intense band for phospho-p38 was seen in the skin of the diseased C5aR+/+ mice (lane 2) as compared to C5aR-/-- mice (lane 4). (B) Quantification of p38mapk activation. Intensity of each band on immunoblots was quantified with a GeneGnome scanner and GeneSnap software. The phospho-p38 levels in the skin of the C5aR+/+ mice were significantly higher than C5aR-/- mice (p<0.0001, bar 2 vs. bar 4). n= =8 for each group. Threee independent experiments were done for each group of mice. 57



.")


.")

 Mice pretreated with the")









, while")








73 Figure 2.4 Blocking p38mapk activation abolishes experimental BP. Neonatal C57BL/6J mice were administered i.d. with the p38mapk inhibitor SB or inactive control SB (10 μg/g body weight). Two h later, the mice were injected i.d. with pathogenic rabbit anti-murine BP180 IgG R621 or control rabbit IgG (2.5 mg/g body weight). The injected animals were examined at different time points post IgG injection. (A) Mice pretreated with the inactive control SB prior to injection with pathogenic antibodies developed BP clinically and histologicall ly, with IgG deposition at the BMZ (left panels). In contrast, inhibition of p38mapk with SB completely abolished the skin lesions clinically and histologically without interference with IgG deposition at the BMZ (right panels). (B) Immunoblotting analysis of p38mapk activation. Pathogenic antibodies induced high levels of phospho-p38 in SB treated mice at 2 and 24 h (lanes 2 and 5), while SB treatment induced only background levels of p38mapk phosphorylation (lanes 3 and 6). (C) Quantification of MC degranulation. Pre-treatment with SB (bar 4) significantly reduced MC degranulation at 2 h post pathogenic IgG injection as compared to PBS (bar 2) or SB pre-treatment (bar 3) (p<0.001,, bar 4 vs. bars 2, 3). (D) Quantification of MPO activity revealed significantly lower levels of PMN infiltration in the SB treated mice (bar 4) than SB treated mice (barr 3) at 24 h post IgG injection (p<0.01). n=6 for each group. Three independent experiments were done for each group of mice. Arrowheads indicate basal keratinocytes. 58

 and MC-deficient (MC-/-)")
")


 and C5a-injected MC-/-")

) C5a activates MC p38mapk in vitro.")
 or C5a (25")



 for 0-600 min and cell")
74 Figure 2.5 C5a activates p38mapk of MCs in vivo and in vitro. (A) C5a activates MC p38mapk in vivo. MC-sufficient (MC+/+) and MC-deficient (MC-/-) mice were injected i.d. with PBS or C5a (50 ng in 50 μl PBS/mouse) and the skin extracts were examined 4 h later by immunoblotting with antibodies to phospho-p38 and total p38. Significantly higher levels of phospho-p38 were seen in the C5a-injected d MC+/+ mice (lane 2) than PBS control (lane 1) and C5a-injected MC-/- mice (lane 4). n=6 for each group. Two independent experiments weree done for each group of mice. (B)) C5a activates MC p38mapk in vitro. Bone marrow-derived MCs from C57BL/6 mice were incubated with buffer control (lane 1) or C5a (25 ng/ml, lanes 2-4) for 0-60 min and cell extracts were assayed for p38mapk activation. All C5a-treated MCs showed high levelss of phospho-p38. (C) SB blocks C5a-induced MC p38mapk phosphorylation in vitro. Bone marrow-derived MCs from C57BL/6 mice were incubated with buffer control (lane 1) or C5a (25 ng/ml, lanes 2-4) in the presence of p38mapk inhibitor SB (20 μm) for min and cell extracts were assayed for p38mapk activation. SB treatment almost completely blocked p38mapk activation. A representative of three independent experiments was shown. 59


 Hematoxylin and Eosin staining showed that MC+/+ mice and MC-/-- mice")



, but not MC-/- mice reconstituted with C5aR-/- MCs (bar 5) and")
75 Figure 2.6. MC reconstitution restores BP in MC-deficient mice. Pathogenic antibodies were injected into ears of MC+/+, MC-/-, MC-/- reconstituted with 1x10 6 MCs from C5aR-/- mice, and MC-/- reconstituted with MCs from C5aR+/+ mice with or without pretreatment with SB The ears were examined 24 h post IgG injection. (A) Hematoxylin and Eosin staining showed that MC+/+ mice and MC-/-- mice reconstituted with wild-type MCs developed DEJ separation following antibody injection. MC-/- mice reconstituted with C5aR-/- MCs and MC-/- mice reconstituted with C5aR+/+ MCs that weree pretreated with SB prior to antibody injection failed to develop DEJ separation. (B) Quantification of PMN infiltration. Pathogenic antibodies induced BP with significantly increased PMN infiltration in MC+/+ mice (bar 2), and MC-/- mice reconstituted wild-type MCs (bar 6), but not MC-/- mice reconstituted with C5aR-/- MCs (bar 5) and MC-/- mice reconstituted with wild-typexperiments weree done for each group of MCs plus SB (bar 7). p<0.01, 6 mice perr group. Three independent mice. 60
76 Strain IgG injected Treatment Number of mice Mean disease activity Wild-type R R R621 + SB R621 + SB C4-/- R R Fb-/- R R C5aR-/- R R Table 2.1. Role of C5a receptor and p38 MAPK in experimental BP. Neonatal wildtype, C4-/-, factor B (Fb)-/-, and C5aR-/- mice were injected intradermally with either control IgG R50 or pathogenic rabbit anti-mbp180 IgG R621 alone (2.5 mg/g body weight) or pretreated with the p38mapk inhibitor SB or inactive control SB (10 μg/g body weight). All mice were examined 24 h after IgG injection. The extent of cutaneous disease was scored as follows: "(-)", no detectable skin disease; "1+", mild erythematous reaction with no evidence of the "epidermal detachment sign [this sign was elicited by gentle friction of the mouse skin which, when positive, produced fine, persistent wrinkling of the epidermis]; "2+", intense erythema and "epidermal detachment" sign involving 10-50% of the epidermis in localized areas; and "3+", intense erythema with frank "epidermal detachment" sign involving more than 50% of the epidermis in the injection site. The clinical disease severity is expressed as Mean + SE and analyzed by paired Student t-test. 61
77 2.5 REFERENCES 1. Diaz, L. A., H. Ratrie, 3rd, W. S. Saunders, S. Futamura, H. L. Squiquera, G. J. Anhalt, and G. J. Giudice Isolation of a human epidermal cdna corresponding to the 180-kD autoantigen recognized by bullous pemphigoid and herpes gestationis sera. Immunolocalization of this protein to the hemidesmosome. J Clin Invest 86: Giudice, G. J., D. J. Emery, and L. A. Diaz Cloning and primary structural analysis of the bullous pemphigoid autoantigen BP180. J Invest Dermatol 99: Hopkinson, S. B., K. S. Riddelle, and J. C. Jones Cytoplasmic domain of the 180-kD bullous pemphigoid antigen, a hemidesmosomal component: molecular and cell biologic characterization. J Invest Dermatol 99: Jordon, R. E., E. H. Beutner, E. Witebsky, G. Blumental, W. L. Hale, and W. F. Lever Basement zone antibodies in bullous pemphigoid. JAMA 200: Lever, W. F Pemphigus. Medicine (Baltimore) 32: Li, K., G. J. Guidice, K. Tamai, H. C. Do, D. Sawamura, L. A. Diaz, and J. Uitto Cloning of partial cdna for mouse 180-kDa bullous pemphigoid antigen (BPAG2), a highly conserved collagenous protein of the cutaneous basement membrane zone. J Invest Dermatol 99: Nishizawa, Y., J. Uematsu, and K. Owaribe HD4, a 180 kda bullous pemphigoid antigen, is a major transmembrane glycoprotein of the hemidesmosome. J Biochem 113: Stanley, J. R Bullous Pemphigoid. In Fitzpatrick's Dermatology in General Medicine. I. Freedberg, Eisen AZ, Wolff K, Austen KF, Goldsmith LA, Katz SI, ed. McGraw-Hill, New York Stanley, J. R., P. Hawley-Nelson, S. H. Yuspa, E. M. Shevach, and S. I. Katz Characterization of bullous pemphigoid antigen: a unique basement membrane protein of stratified squamous epithelia. Cell 24:
78 10. Stanley, J. R., D. T. Woodley, and S. I. Katz Identification and partial characterization of pemphigoid antigen extracted from normal human skin. J Invest Dermatol 82: Tanaka, T., N. J. Korman, H. Shimizu, R. A. Eady, V. Klaus-Kovtun, K. Cehrs, and J. R. Stanley Production of rabbit antibodies against carboxy-terminal epitopes encoded by bullous pemphigoid cdna. J Invest Dermatol 94: Jordon, R. E Complement activation in bullous skin diseases. J Invest Dermatol 65: Jordon, R. E., A. L. Schroeter, R. A. Good, and N. K. Day The complement system in bullous pemphigoid. II. Immunofluorescent evidence for both classical and alternate-pathway activation. Clin Immunol Immunopathol 3: Provost, T. T., and T. B. Tomasi, Jr Evidence for complement activation via the alternate pathway in skin diseases, I. Herpes gestationis, systemic lupus erythematosus, and bullous pemphigoid. J Clin Invest 52: Chen, R., G. Ning, M. L. Zhao, M. G. Fleming, L. A. Diaz, Z. Werb, and Z. Liu Mast cells play a key role in neutrophil recruitment in experimental bullous pemphigoid. J Clin Invest 108: Liu, Z., G. J. Giudice, S. J. Swartz, J. A. Fairley, G. O. Till, J. L. Troy, and L. A. Diaz The role of complement in experimental bullous pemphigoid. J Clin Invest 95: Liu, Z., G. J. Giudice, X. Zhou, S. J. Swartz, J. L. Troy, J. A. Fairley, G. O. Till, and L. A. Diaz A major role for neutrophils in experimental bullous pemphigoid. J Clin Invest 100: Nelson, K. C., M. Zhao, P. R. Schroeder, N. Li, R. A. Wetsel, L. A. Diaz, and Z. Liu Role of different pathways of the complement cascade in experimental bullous pemphigoid. J Clin Invest 116: Zipfel, P. F., and C. Skerka Complement regulators and inhibitory proteins. Nat Rev Immunol 9:
79 20. Monk, P. N., A. M. Scola, P. Madala, and D. P. Fairlie Function, structure and therapeutic potential of complement C5a receptors. Br J Pharmacol 152: Lawrence, I. D., J. A. Warner, V. L. Cohan, W. C. Hubbard, A. Kagey-Sobotka, and L. M. Lichtenstein Purification and characterization of human skin mast cells. Evidence for human mast cell heterogeneity. J Immunol 139: Fureder, W., H. Agis, M. Willheim, H. C. Bankl, U. Maier, K. Kishi, M. R. Muller, K. Czerwenka, T. Radaszkiewicz, J. H. Butterfield, G. W. Klappacher, W. R. Sperr, M. Oppermann, K. Lechner, and P. Valent Differential expression of complement receptors on human basophils and mast cells. Evidence for mast cell heterogeneity and CD88/C5aR expression on skin mast cells. J Immunol 155: Marshall, J. S Mast-cell responses to pathogens. Nat Rev Immunol 4: la Sala, A., M. Gadina, and B. L. Kelsall G(i)-protein-dependent inhibition of IL-12 production is mediated by activation of the phosphatidylinositol 3-kinaseprotein 3 kinase B/Akt pathway and JNK. J Immunol 175: Hawlisch, H., Y. Belkaid, R. Baelder, D. Hildeman, C. Gerard, and J. Kohl C5a negatively regulates toll-like receptor 4-induced immune responses. Immunity 22: Rousseau, S., I. Dolado, V. Beardmore, N. Shpiro, R. Marquez, A. R. Nebreda, J. S. Arthur, L. M. Case, M. Tessier-Lavigne, M. Gaestel, A. Cuenda, and P. Cohen CXCL12 and C5a trigger cell migration via a PAK1/2-p38alpha MAPK-MAPKAP- K2-HSP27 pathway. Cell Signal 18: Ashwell, J. D The many paths to p38 mitogen-activated protein kinase activation in the immune system. Nat Rev Immunol 6: Johnson, G. L., and R. Lapadat Mitogen-activated protein kinase pathways mediated by ERK, JNK, and p38 protein kinases. Science 298: Kalesnikoff, J., N. Baur, M. Leitges, M. R. Hughes, J. E. Damen, M. Huber, and G. Krystal SHIP negatively regulates IgE + antigen-induced IL-6 production in mast cells by inhibiting NF-kappa B activity. J Immunol 168:
80 30. Galli, S. J., and Y. Kitamura Genetically mast-cell-deficient W/Wv and Sl/Sld mice. Their value for the analysis of the roles of mast cells in biologic responses in vivo. Am J Pathol 127: Liu, Z., L. A. Diaz, J. L. Troy, A. F. Taylor, D. J. Emery, J. A. Fairley, and G. J. Giudice A passive transfer model of the organ-specific autoimmune disease, bullous pemphigoid, using antibodies generated against the hemidesmosomal antigen, BP180. J Clin Invest 92: Li, K., K. Tamai, E. M. Tan, and J. Uitto Cloning of type XVII collagen. Complementary and genomic DNA sequences of mouse 180-kilodalton bullous pemphigoid antigen (BPAG2) predict an interrupted collagenous domain, a transmembrane segment, and unusual features in the 5'-end of the gene and the 3'- untranslated region of the mrna. J Biol Chem 268: Liu, Z., L. A. Diaz, A. L. Haas, and G. J. Giudice cdna cloning of a novel human ubiquitin carrier protein. An antigenic domain specifically recognized by endemic pemphigus foliaceus autoantibodies is encoded in a secondary reading frame of this human epidermal transcript. J Biol Chem 267: Bradley, P. P., D. A. Priebat, R. D. Christensen, and G. Rothstein Measurement of cutaneous inflammation: estimation of neutrophil content with an enzyme marker. J Invest Dermatol 78: Mulligan, M. S., M. L. Jones, M. A. Bolanowski, M. P. Baganoff, C. L. Deppeler, D. M. Meyers, U. S. Ryan, and P. A. Ward Inhibition of lung inflammatory reactions in rats by an anti-human IL-8 antibody. J Immunol 150: Wershil, B. K., Z. S. Wang, J. R. Gordon, and S. J. Galli Recruitment of neutrophils during IgE-dependent cutaneous late phase reactions in the mouse is mast cell-dependent. Partial inhibition of the reaction with antiserum against tumor necrosis factor-alpha. J Clin Invest 87: Ryan, J. J., S. DeSimone, G. Klisch, C. Shelburne, L. J. McReynolds, K. Han, R. Kovacs, P. Mirmonsef, and T. F. Huff IL-4 inhibits mouse mast cell Fc epsilonri expression through a STAT6-dependent mechanism. J Immunol 161: Wershil, B. K., Y. A. Mekori, T. Murakami, and S. J. Galli I-fibrin deposition in IgE-dependent immediate hypersensitivity reactions in mouse skin. 65
81 Demonstration of the role of mast cells using genetically mast cell-deficient mice locally reconstituted with cultured mast cells. J Immunol 139: Gerard, N. P., and C. Gerard The chemotactic receptor for human C5a anaphylatoxin. Nature 349: Chen, R., J. A. Fairley, M. L. Zhao, G. J. Giudice, D. Zillikens, L. A. Diaz, and Z. Liu Macrophages, but not T and B lymphocytes, are critical for subepidermal blister formation in experimental bullous pemphigoid: macrophage-mediated neutrophil infiltration depends on mast cell activation. J Immunol 169: Church, M. K., and G. F. Clough Human skin mast cells: in vitro and in vivo studies. Ann Allergy Asthma Immunol 83: Klos, A., A. J. Tenner, K. O. Johswich, R. R. Ager, E. S. Reis, and J. Kohl The role of the anaphylatoxins in health and disease. Mol Immunol 46: Ono, K., and J. Han The p38 signal transduction pathway: activation and function. Cell Signal 12: Muller-Eberhard, H. J Molecular organization and function of the complement system. Annu Rev Biochem 57: Berkowitz, P., P. Hu, Z. Liu, L. A. Diaz, J. J. Enghild, M. P. Chua, and D. S. Rubenstein Desmosome signaling. Inhibition of p38mapk prevents pemphigus vulgaris IgG-induced cytoskeleton reorganization. J Biol Chem 280: Berkowitz, P., P. Hu, S. Warren, Z. Liu, L. A. Diaz, and D. S. Rubenstein p38mapk inhibition prevents disease in pemphigus vulgaris mice. Proc Natl Acad Sci U S A 103: Schett, G., J. Zwerina, and G. Firestein The p38 mitogen-activated protein kinase (MAPK) pathway in rheumatoid arthritis. Ann Rheum Dis 67: Adcock, I. M., K. F. Chung, G. Caramori, and K. Ito Kinase inhibitors and airway inflammation. Eur J Pharmacol 533:
82 49. Perez, O. A., and T. Patton Novel therapies for pemphigus vulgaris: an overview. Drugs Aging 26: Cohen, P Targeting protein kinases for the development of anti-inflammatory drugs. Curr Opin Cell Biol 21:
83 CHAPTER 3 FUNCTIONAL INTERACTION BETWEEN COMPLEMENT AND THE PRO- INFLAMMATORY CYTOKINE TNF-α IN EXPERIMENTAL BULLOUS PEMPHGOID Bullous pemphigoid (BP) is a chronic inflammatory skin blistering disease characterized by autoantibodies against two major hemidesmosomal proteins, BP230 and BP180. Existing therapeutic strategies rely heavily on corticosteroids; hence there is urgent need for new treatments. High levels of TNF-α are reported in the sera of BP patients, providing a possible intervention target. However, the role of TNF- α in BP is unknown. In this study, we examined TNF-α s activity using two IgG passive-transfer mouse models of BP. One of the models is a humanized model, where human anti-bp180 is injected into a knock-in mouse which expresses the human pathogenic portion of BP180. Upon injection with pathogenic anti-bp180 antibodies, mice exhibit sharp mast cell (MC)-dependent increases in TNF-α in the skin and blister fluid. Mice lacking TNF-α or MCs fail to recruit sufficient numbers of neutrophils to the skin and do not develop blisters. TNFR1 but not TNFR2 expression on MCs is required for experimental BP. Additionally, anti-tnf-α therapies abrogated disease pathogenesis. These data indicate that pathogenic antibodies trigger TNFα release from MCs, and that TNF-α mediates neutrophil infiltration and disease by acting in an autocrine fashion on MCs. Additionally, this study provides rationale for considering anti-tnf therapy for the treatment of BP.
84 3.1 INTRODUCTION Bullous pemphigoid (BP) is a chronic, autoimmune skin blistering disease that predominantly occurs in the elderly, but also can affect younger people (1-3). BP is associated with high mortality rates, ranging from 12% and 40% in the first three years after diagnosis (4, 5). The conventional therapy for BP over the past several decades has been high-dose, long-term systemic oral corticosteroids and immunosuppressive agents (6). However, long term use of corticosteroids is associated with deleterious health effects, including severe infection, diabetes mellitus, osteoporosis, hypertension, renal failure, lymphoma, and squamous cell carcinoma (7). The negative side effects of immunosuppression therapy may account for a significant portion of the morbidity and mortality associated with BP. Even when disease is successfully treated, up to 45% of patients suffer a relapse within six months of cessation of therapy (8), underscoring the urgent need for novel therapeutic interventions. BP patients develop circulating and tissue bound autoantibodies against two major hemidesmosomal proteins, BP230 (BPAG1) and BP180 (BPAG2, type XVII collagen) (9-14). These anti-hemidesmosomal autoantibodies, along with complement components, are deposited along the basement membrane at the dermal-epidermal junction (DEJ) of perilesional skin (15). Basal keratinocytes detach from the underlying dermis, leading to the formation of tense, fluid filled blisters (1). Blister fluid contains cellular dermal inflammatory infiltrates, including eosinophils, neutrophils (PMNs), lymphocytes, degranulated mast cells (MCs), and monocyte/macrophages (16-21). In addition, various inflammatory mediators that can recruit/activate MCs or leukocytes have been identified in lesional skin and/or blister 69
85 fluids of BP patients, including C5a, eosinophilic/neutrophilic chemotactic factors, histamine, leukotrienes, and various cytokines (3, 22-28). The cytokine TNF-α is a potent and pleiotropic mediator of biological responses and signals through the TNF-α receptors (TNFRs) TNFR1 and TNFR2 (29). TNF-α is implicated in the pathogenesis of a number of inflammatory diseases, including rheumatoid arthritis, inflammatory bowel disease, psoriasis, and pemphigus vulgaris (30). Elevated levels of TNF-α have been reported in the serum and blister fluid of BP patients, and disease severity correlates with TNF-α serum levels (31, 32). In one case study, BP in a patient with severe psoriatic background was successfully treated with a combination therapy of rituximab and etanercept (29), while etanercept alone was used to control psoriasis and BP in another patient (30). These studies are limited in scope but provide the attractive possibility of utilizing established anti-tnf-α therapies to treat BP patients However, prior to considering the broad application of TNF- α therapies to BP, it is vital to understand the role of this cytokine in the pathogenesis of BP. To address this issue, we used two IgG passive transfer mouse models of BP that mimic the key features of human BP to investigate the role of TNF-α in the pathogenesis of experimental BP. In the first of these models, neonatal C57BL/6J mice are injected with polyclonal rabbit antibodies that recognize the immunodominant domain of murine BP180, called NC14A (33). NC14A is the murine homolog of NC16A, the domain of human BP180 that harbors multiple epitopes recognized by human BP autoantibodies (34, 35). Animals injected with rabbit anti-bp180nc14a IgG develop skin blistering that depends on complement activation, MC degranulation, and PMN infiltration to the skin (36-38). In the second model, mice humanized to express NC16A instead of NC14A are injected with 70
86 BP180-specific IgG isolated from human BP patients. The injected patient antibodies trigger blistering that also requires complement, MC degranulation, and PMN recruitment (39). Using these two mouse models, we describe a key role for TNF-α via TNFRI in the pathogenesis of BP. 3.2 EXPERIMENTAL PROCEDURES Laboratory animals. Breeding pairs of C57BL/6J, MC-deficient WBB6F1- Kit w /Kit w-v (MC -/- ), C5a receptor-deficient (C5aR -/- ), TNFR1-deficient (TNFR1 -/- ), TNFR2- deficient (TNFR2 -/- ), and TNFR1 and TNFR2 deficient (TNFR1,2 -/- ) mice were purchased from Jackson Laboratories (Bar Harbor, ME) and maintained at the University of North Carolina at Chapel Hill. The MC deficiency in Kit w /Kit w-v mice are caused by distinct mutations in MC c-kit (40). Humanized NC16A mice were generated as described (39). In these mice, the mouse BP180NC14A domain was replaced with human BP180NC16A domain. Neonatal mice (24-36 hours old with body weights between 1.4 and 1.6 g) were used for passive transfer experiments. All animal care and animal experiments were approved by the UNC Animal Care Committee and were in accordance with the National Institutes of Health guidelines. Preparation of pathogenic anti-bp180 IgG. The preparation of recombinant murine BP180 (mbp180) and the immunization of New Zealand White rabbits were performed as previously described (33, 41, 42). Briefly, New Zealand White rabbits were immunized with the GST-hBP180NC16A or GST-mNC14A fusion protein. IgG fractions from sera of the immunized rabbits were purified using a protein G Sepharose column (Sigma). The pathogenicity of these IgG preparations was tested by passive transfer 71
87 experiment as described below. A pathogenic anti-mbp180 IgG preparation (R530) and a control IgG preparation were used in this study (38). For experiments with human patient IgG, BP180NC16A-specific autoantibodies from BP patients were purified using a BP180NC16A-glutathione Sepharose column (39). Induction of experimental BP and clinical evaluation of animals. Neonates were given on the back one i.d. injection of a sterile solution of IgG in PBS (50 μl IgG, 2.5 mg IgG/g body weight) as described elsewhere (33). The skin at the IgG-injection site of the mice from the test and control groups was examined at different time points after the IgG injection. The extent of cutaneous disease was scored as follows: "(-)", no detectable skin disease; "1+", mild erythematous reaction with no evidence of the epidermal detachment sign [this sign was elicited by gentle friction of the mouse skin which, when positive, produced fine, persistent wrinkling of the epidermis]; "2+", intense erythema and epidermal detachment sign involving 10-50% of the epidermis in localized areas; and "3+", intense erythema with frank epidermal detachment sign involving more than 50% of the epidermis in the injection site. After clinical examination, the animals were sacrificed, and skin and serum specimens were obtained. Skin sections were used for a) routine histological examination by light microscopy (H&E staining) to localize the lesional site and PMN infiltration, b) toluidine blue staining to quantify MCs and MC degranulation, c) direct IF assays to detect rabbit IgG and mouse C3 deposition at the BMZ, and d) myeloperoxidase (MPO) enzymatic assay to quantify the PMN accumulation at the skin injection site described below. The sera of injected animals were tested by indirect IF techniques to determine the titers of rabbit anti- 72
88 mbp180 antibodies using mouse skin cryosections as the substrate. Direct and indirect IF studies were performed as previously described (33) using commercially available FITCconjugated goat anti-rabbit IgG (Kirkeggard& Perry Laboratories Inc.). Monospecific goat anti-mouse C3 IgG was purchased from Cappel Laboratories. Pretreatment with TNF-α neutralizing antibodies and rtnf-α. Neonatal WT and TNF-α-deficient (TNF-α -/- ) mice were injected i.d. with 50 μl of BSA control (1 μg/50 μl PBS ), TNF-α neutralizing antibodies (1 μg/50 μl PBS, R&D System), or rtnf-α (50 ng/50 μl PBS, R&D System). Two hours later, the mice were given i.d. with pathogenic antimbp180 antibodies and examined 24 h post IgG injection. Identification of TNF-α in blister fluid. One hundred microliters of PBS was injected into the skin blisters (formed 12 hours after pathogenic IgG injection) and nonlesional sites, and withdrawn 1 minute later. The washout PBS was centrifuged at low speed (1,000 g) for 5 minutes to remove cells and then at high speed (12,000 g) for 5 minutes to remove cell debris. TNF-α in the supernatant was measured by a mouse TNF-α-specific ELISA kit (R&D System). TNF-α level were expressed as pg/ml. RT-PCR. mrna was isolated from skin samples using an RNeasy kit (Qiagen). TNFα, TNFR1, TNFR2 messages were amplified from the cdna using the following primers: Forward:5 -TAGTGGTGCCAGCCGATGGGT-3 and Reverse: 5 - GACCTGCCCGGACTCCGCAA-3 for mouse TNF-α, Forward: 5 - GTGCGTCCCTTGCAGCCACT -3 and reverse 5 -AGGGTCAGCTCCCTGGGTTGG-3 for mouse TNFR1, Forward: 5 - GTTTCAGGAGGCCCGTGCCAG -3 and Reverse:5-73
89 CCATATCCGGCACACCAAGGGGCA -3 for mouse TNFR2. Sizes of PCR products were 305, 439, and 293 base pairs for TNF-α, TNFR1, and TNFR2, respectively. Quantification of PMN accumulation at the skin site. Myeloperoxidase (MPO) activity in skin sites of the injected animals was assayed as a measure of PMN infiltration as described (43, 44). A standard reference curve was established using purified MPO (Athens Research and Technology, Inc., Athens, Georgia). The skin samples were extracted by homogenization in an extraction buffer containing 0.1 M Tris-Cl, ph 7.6, 0.15 M NaCl, 0.5% hexadecyltrimethylammonium bromide. MPO activity in the supernatant fraction was measured by the change in optical density at 460 nm resulting from decomposition of H 2 O 2 in the presence of o-dianisidine. MPO content was expressed as units of MPO activity/mg protein. Protein concentrations were determined by the Bio-RAD dye-binding assay using BSA as a standard. Quantification of MCs and MC degranulation. MCs and MC degranulation in skin samples were quantified according to Wershil, et al. (45) with modification. Briefly, lesional and nonlesional skin sections of IgG-injected mice were fixed in 10% formalin. Paraffin sections (5-μm thick) were prepared and stained with toluidine blue and H&E. Toluidine blue stains histamine in the MC granules (46). Total number of MC was counted and classified as degranulated (>10% of the granules exhibiting fusion or discharge) or normal in five fields under a light microscope as described previously. The results were expressed as percentage of MC degranulating. Determination of C5a levels in skin. Skin from the IgG injection site of the diseased and control mice was mechanically homogenized in PBS to extract proteins. The level of mouse C5a (mc5a) in the skin was measured by ELISA (R&D Systems, Minneapolis, MN). 74
90 Microtiter plates were coated with a rat anti-mouse C5a antibody, incubated with skin protein extracts, followed by goat anti-mc5a detection antibody, and then developed and read at OD 492nm. The C5a level was expressed as OD 492 reading/mg protein. MC reconstitution. Kit w /Kit w-v mice were repaired of their MC deficiency selectively and locally by the injection of growth factor-dependent bone marrow-derived cultured MC into the skin (47, 48). Briefly, femoral bone marrow cells from WT and C5aR -/- mice were maintained in vitro for 4 wk in RPMI 1640 complete medium (Gibco/Life Technologies, Grant Island, NY) supplemented with 20% WEHI-3-conditioned medium until MCs represented >95% of the total cells as determined by toluidine blue staining and flow cytometry analysis using antibodies specific for MC cell surface markers FcεRI, c-kit, and CD13 (47). Murine IgE and rat anti-mouse IgE were purchased from Southern Biotechnology Associates (Birmingham, AL). FITC-labeled rat anti-mouse c-kit and FITClabeled rat anti-mouse CD13 were obtained from BD Biosciences (San Diego, CA). MCs (1x10 6 in 20 μl of medium) were injected i.d. into right ears of MC-deficient mice. Medium alone (20 μl) were injected i.d. into left ears of the same mice as negative control. This procedure selectively and locally reconstitute dermal MC population without systemic effects (48). To confirm MC reconstitution, skin sections from MC-injected sites were stained by toluidine blue. Ten weeks after adoptive transfer of MCs, both ears of the mice were injected i.d. with pathogenic anti-bp180 IgG (2 mg/20 μl/site). Twenty four hours later, ear skin biopsies were obtained and analyzed by H&E, toluidine blue staining and MPO enzyme assay as described above. Statistical analysis. The data are expressed as mean + SEM and were analyzed using the Student's paired t-test. A p value less than 0.05 was considered significant. 75
91 3.3 RESULTS Pathogenic anti-bp180 antibodies induce local expression of TNF-α. As we previously reported (33), wild-type (WT) C57BL/6J mice injected with pathogenic rabbit anti-mbp180 IgG R530 (2.5 mg/g body weight) develop intense clinical blisters after 24 hours (Figure 3.1A, top row, arrow denotes blistered area). A linear pattern of IgG binding along the DEJ is detected by direct immunofluorescence (Figure 1A, middle, arrow marks DEJ), and separation of the dermis from the epidermis is apparent in H&E stained skin sections (Figure 3.1A, bottom). Mice injected with control IgG lack blisters and antibody deposition. Levels of myeloperoxidase (MPO) in the skin are significantly higher in mice given R530 than in mice given control IgG, indicating that PMNs are recruited to the skin in the pathogenic antibody-injected animals (Figure 3.1B). To investigate the role of TNF-α in experimental BP, we collected skin biopsies from the affected area of R530-injected mice and from the equivalent, unblistered areas of control IgG-injected mice and measured TNF-α protein, which was significantly higher (over 25 fold) in the blister fluid from the R530 injected mice than in the wash-out of control IgG injected mice (Figure 3.1C). These results demonstrate that expression of TNF-α is greatly augmented in experimental BP. TNF-α is required for the development of experimental BP. Having established that TNF-α levels rise dramatically in mice given pathogenic anti-bp180 antibodies, we next investigated whether TNF-α is required for experimental BP disease pathogenesis. TNF-α deficient mice (TNF-α -/- ) and WT mice were pretreated locally with PBS, recombinant TNFα (rtnf-α), or anti-tnf-α neutralizing antibody. Two hours later, the mice were injected i.d. with control IgG or R hours post IgG injection, TNF-α -/- mice showed no skin lesions clinically or histologically (Figure 3.2A, left column). However, pretreatment with 76
92 rtnf-α restored clinical and histological lesions in TNF-α -/- mice (Figure 3.2A, middle column). WT mice pretreated with TNF-α neutralizing antibody were also resistant to experimental BP (Figure 3.2A, right column). Taken together, these data demonstrate an essential role for TNF-α in experimental BP. Experimental BP pathogenesis proceeds through the activation of complement, followed by MC degranulation, and then PMN infiltration to the skin (49). We next examined the effect of TNF-α deficiency on each of these processes. Four hours after IgG injection, similar levels of C5a were detected by ELISA in the skin of WT and TNF-α -/- animals injected with control IgG or R530 IgG (Figure 3.2B). Toluidine blue stained skin sections from mice sacrificed two hours after R530 antibody injection, when MC degranulation reaches its peak level (37), revealed equivalent levels of MC degranulation in the dermis of WT and TNF-α -/- animals (Figure 3.2C). While complement activation and MC degranulation were unaffected by the loss of TNF-α, PMN recruitment was significantly impacted (Figure 3.2D). Twenty-four hours after R530 injection, WT mice exhibit a significant increase in skin PMN levels. Pretreatment with the TNF-α neutralizing antibody ablated the PMN recruitment in WT mice. TNF-α -/- animals recruit significantly fewer PMNs to the skin than the WT mice, but PMN recruitment was completely restored in TNFα -/- animal pretreated with rtnf-α. These experiments demonstrate that TNF-α is required for PMN recruitment in experimental BP. MCs are the source of TNF-α needed for experimental BP pathogenesis. Upon degranulation, MCs release large amounts of pre-formed TNF-α (50). However, a number of other skin-resident cell populations can produce TNF-α, including keratinocytes and Langerhans cells (8). To determine if MCs are the critical cellular source of TNF-α in the 77
93 development of experimental BP, we first examined the kinetics of TNF-α production in the skin of WT and WBB6F1-Kit w /Kit w-v mice which lacked MC (referred to as MC -/- ) postinjection with R530 or control IgG. Mice (n=6 per group) were sacrificed 0, 1, 2, 4, 8, and 24 hours post IgG injection, and blisters or the area of skin where blisters would form were washed with 100 μl of PBS to collect protein. TNF-α levels were measured in the washout fluids by ELISA. Injection with R530 IgG induced an initial peak in TNF-α levels 2 hours post injection, and a second peak 24 hours post-injection in WT mice (Figure 3.3). In contrast, TNF-α levels did not significantly increase in mice injected with control IgG at all time-points. The TNF-α protein level peaked 2 h post injection which coincides with the peak in MC degranulation, suggesting that MCs release of preformed TNF-α may be the key source of the cytokine in the early stages of experimental BP. To further investigate this hypothesis, we tested whether TNF-α was produced in MC -/- mice following R530 injection. TNF-α levels in the washouts from MC -/- mice injected with R530 were not significantly different from the levels observed in mice injected with control IgG (Figure 3.3, closed triangles) indicating that MC is the source of TNF-α in response to R530 injection. MC reconstitution experiments further confirmed that MCs are the critical source of TNF-α in experimental BP pathogenesis. TNF-α -/- mice did not develop blisters following injection with pathogenic IgG. Furthermore, reconstitution with 1x10 6 MC from TNF-α -/- mice prior to R530 injection does not restore histological blistering. However, TNF-α -/- mice reconstituted with WT MCs prior to R530 injection develop histological blistering (Figure 3.4A). Similarly, MC -/- mice and MC -/- mice reconstituted with TNF-α -/- MCs fail to develop blisters following R530 injection, but MC -/- mice reconstituted with WT MCs develop blisters (Figure 3.4B). Only mice with WT MCs, but not TNF-α -/- MCs are able to recruit 78
94 PMNs to the skin and develop blisters following injection with pathogenic antibodies (Figure 3.4C). Taken together, these data demonstrate that TNF-α released by degranulating MCs is required for experimental BP. C5a-C5a receptor interaction is required for TNF-α release. We recently reported that C5a interaction with the C5a receptor (C5aR, or CD88) on MCs triggers activation of p38mapk in MCs and MC degranulation (51). To assess if TNF-α released by degranulating MCs is also affected by the C5a pathway, we injected WT and C5aR -/- neonatal mice with R530 and examined various stages of disease progression. C5aR -/- mice fail to develop clinical blisters after 24 hours (Figure 3.5A), despite generating levels of C5a equivalent to the WT mice (Figure 3.5B). MC degranulation is significantly impaired in the C5aR -/- mice injected with R530 compared to WT mice injected with R530, and washout fluid from C5aR -/- mice contains significantly lower levels of TNF-α than washout from diseased WT mice (Figures 3.5C, 3.5D). C5aR -/- mice fail to recruit PMNs to the skin following R530 injection, and reconstitution with C5aR -/- MCs or TNF-α -/- MCs does not restore PMNs. Only reconstitution with WT MCs restores PMN infiltration to the skin (Figure 3.5E). These experiments place C5a upstream of TNF-α production and PMN recruitment to the skin. TNF-α receptor 1 mediates the downstream effects of TNF-α. TNF-α acts via TNFR1 and TNFR2. To determine which TNF receptors are required for experimental BP pathogenesis, we first looked at receptor expression following pathogenic antibody injection. Over 8 hours post injection, TNFR1 is expressed constitutively, whereas TNFR2 mrna expression increased over time (Figure 3.6A). We next injected TNFR1 -/-, TNFR2 -/-, and TNFR1,2 -/- mice with R530 and checked for clinical blistering after 24 hours. TNFR2 -/- mice 79
95 developed extensive blisters equivalent to those seen on WT mice, but the TNFR1 -/- and TNFR1,2 -/- were protected from disease (Figure 3.6B). We found that PMN infiltration is impaired in both TNFR1 -/- and TNFR1,2 -/- mice, indicating that TNFR1 is required for the transition from MC degranulation to PMN recruitment (Figure 3.6C). TNFR1 is broadly expressed across cell types, including MCs, keratinocytes, fibroblasts, endothelial cells, and PMNs (52). To investigate the possibility that TNF-α acts on MCs in an autocrine fashion, we performed a series of MC reconstitution experiments. TNFR1,2 -/- and MC -/- mice were reconstituted with MCs from TNFR1,2 -/-, TNFR1 -/-, TNFR2 - /-, or WT and then injected with R530 IgG. The TNFR1,2 -/- and the MC -/- mice recruited PMNs to the skin and developed BP blisters only when they were reconstituted with TNFR2 - /- or WT MCs (Figure 3.7). These data indicate that TNF-α acts on TNFR1 expressed by MCs in an autocrine fashion to promote experimental BP. Human BP patient IgG induces TNF-α production in humanized mouse model. To extend these findings to the human BP disease, we used a humanized animal model of experimental BP (39). The humanized mice, called NC16A+/+, express the human sequence of the pathogenic portion of BP180 in place of the murine homolog. In these mice, passive transfer of anti-bp180nc16a autoantibodies purified from human patients triggers experimental BP. Similar to the rabbit anti-mouse IgG model, disease development depends on complement activation, MC degranulation and PMN infiltration (39). To test whether human BP autoantibody-induced skin disease also depends on TNF-α, we injected human control IgG or NC16A-specific IgG into NC16A+/+ mice. The anti-nc16a antibodies induced clinical blisters in NC16A mice, and these blisters were prevented by pretreatment with anti-tnf-α neutralizing antibodies (Figure 3.8A). Significantly higher levels of TNF-α 80
96 were present in blister washout of the humanized mice given pathogenic antibodies as compared to the humanized mice injected with human control IgG (Figure 3.8B). Pretreatment with anti-tnf-α reduced the total amount of TNF-α present in the washout fluid of the anti-nc16a-injected animals. As expected, increased PMN recruitment was associated with higher levels of TNF-α in the diseased NC16+/+ mice, and blocked by anti- TNF-α neutralizing antibodies (Figure 3.8C). These results suggest that TNF-α also acts to increase skin blistering and granulocyte infiltration in a humanized model of BP. 3.4 DISCUSSION TNF-α is a pleiotropic cytokine with a broad range of biological effects in regulating cell growth and death, development, oncogenesis, immune function and the inflammatory response (53). Various TNF-α inhibitors are used in the clinic to treat Crohns disease, colitis and rheumatoid arthritis. Here, we show evidence for TNF-α s autocrine effect on MCs in experimental BP. We found that TNF-α is required for development of experimental BP. Mice injected with anti-bp180 antibodies develop a significant increase in serum levels of TNF-α, and TNF-α is detected in the blister fluid from diseased animals. Upon injection with pathogenic antibodies, animals lacking TNF-α fail to recruit PMNs to the skin and are protected from blister formation, but exhibit normal levels of complement activation and MC degranulation. Mice lacking MCs do not exhibit increased serum TNF-α levels upon pathogenic antibody injection and are resistant to experimental BP. Reconstitution with TNF-α sufficient MCs restores disease susceptibility in TNF-α -/- or MC -/- mice, whereas TNF-α -/- MCs do not. Activation of the C5aR on MCs by C5a is required for TNF-α release. We also found that TNF-α acts on MCs in an autocrine fashion through TNFR1. MC -/- mice 81
97 develop experimental BP only when they are reconstituted with MCs that express TNFR1. Thus, we conclude that MCs are both the critical source of TNF-α and the key target of TNFα in experimental BP. The binding of C5a to C5aR on MCs leads to activation of the p38mapk signaling pathway and MC degranulation (51), which releases preformed TNF-α from intracellular stores (Figure 3.9). This initial wave of TNF-α binds to MC TNFR1 and initiates an intracellular signaling cascade that leads to activation of the transcription factors NF-kB and c-jun (54). NF-kB and c-jun mediate production of pro-inflammatory factors, and our laboratory is currently investigating roles for these compounds in PMN recruitment and BP pathogenesis. While TNF-α-TNF-R1 interaction on MCs is critical for disease pathogenesis, it is possible that TNF-α has other secondary effects during disease. TNF-α expression in the skin upregulates expression of VCAM-1, ICAM-1, and E-selectin, which promotes extravasation of inflammatory cells to the skin (55-57). PMNs and macrophages also express high levels of TNFR1; the effects of TNF-α on these cell types remains to be investigated. TNF-α also upregulates MC production of matrix metalloproteinase-9 (MMP-9) (58), an enzyme critical for the secondary wave of PMN recruitment in experimental BP (59). While PMNs are the key source of MMP-9 in experimental BP, TNF-α stimulated MCs may augment these levels. Anti-TNF-α therapies, including anti-tnf-α antibodies and TNF-α blocking peptidesetanercept (Enbrel), infliximab (Remicade), and adalimumab (Humira), are effective in treating a number of inflammatory diseases (30). Limited case reports indicate that 82
98 treatment with anti-tnf-α therapies is therapeutically beneficial to BP patients (60, 61). Our work makes a strong case for expansion of anti-tnf-α therapies for the treatment of BP. 83





 or pathogenic")


 Top row:")







 MPO assay")





99 Figure 3.1. Pathogenic anti-mbp18 80 IgG induces BP blisters associated with an elevated level of TNF-α in mouse skin. Neonatall C57BL/6J mice were injected i.d. with normal rabbit IgG (control IgG) or pathogenic rabbitt anti-mbp180 IgGR530 (2.5 mg/g body weight) and examined 24 h post injection. (A) Top row: Clinicall evaluation of mice 24 hours after antibody injection. Arrow, blister. Middle row: Directt IF for rabbit IgG on skin sections. Bottom row: H&E staining on skin sections. E, epidermis. D, dermis. V, vesicle. Arrow, basal keratinocytes. 100x magnification. (B) MPO assay exhibited a significantly increased number of PMNs in the lesional skin relative to the non-lesional skin of control mice. N=8 for each group. *p<0.01 (bar 1 vs. bar 2). (C) TNF-α ELISA showed a drastic increase in TNF-α protein level in the blister fluid of the diseased mice as compared to that in the wash-out of control mice. 84










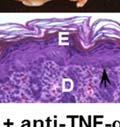
















100 Figure 3.2. TNF-α-deficient mice are resistant to experimental BP. Neonatal WT and TNF-α-deficienanti-TNF-α neutralizing antibody. Two hours later, the mice were injected i.d. with control IgG or pathogenic rabbit anti-mbp180 IgG R530 (2.5 mg/g body weight) (A) Clinicall (top (TNF-α -/- ) mice were pretreated locally with PBS, recombinant TNF-α, or row, arrow) and histological (bottom row) blisterss 24 h post IgG injection; (B) Relative levels of C5a in skin extracts from WT and TNF-α -/- mice at 4 h post IgG injection, measured by ELISA. (C) Toluidine blue staining for MC degranulation in the dermis between WT and TNF-α -/- mice at 2 h post IgG injection, when MC degranulationn reaches the peak level. (D) Relative MPO activity assay demonstrated that anti-tnf-α while local reconstitution of TNF-α restored BP antibody treatment resulted in a significant reduction of PMN infiltration, disease with an increased level of PMN infiltration similar to the diseased mice at 24 h post IgG injection. n =8 for each group. *p<0.01 (bar 2 vs. bar 3; bar 5 vs. bar 6). 85

 and")

.")

101 Figure 3.3. Time-course of TNF-α expression during BP disease development in mice. Neonatal wild-type (WT) and MC-deficient (MC-/-) mice were injected i.d. with control IgG or pathogenic rabbit anti-mbp180 IgG (2.5 mg/g body weight). At 0, 1, 2, 4, 8, and 24 h post injection, mice were given 50 μl of PBS and the levels of TNF-α in the PBS wash-out were quantified by ELISA. N=6. *p<


 or 1x10 6 MCs from")




 H&E staining showed that pathogenic")
 Quantification of PMN infiltration.")



102 Figure 3.4. MC reconstitution restores BP in TNF-α-/- and MC-deficient mice. TNF-α-/- and MC-/- mice were reconstituted at the ear with buffer control (none) or 1x10 6 MCs from TNF-α-/reconstituted mice and the ears were examined after 24 h. (A)) H&E staining showed that or WT mice. Ten weeks later, pathogenicc abs were injected into ears of the MC- pathogenic IgG induced blistering in TNF-α-/- mice reconstituted with WT MCs (right panel), but not with buffer control or TNF-α-/- MCss (left, middle panels). (B) H&E staining showed that pathogenic IgG induced blistering in MC-/- mice reconstituted with WT MCs (right panel), but not with buffer control or TNF-α-/- MCss (left, middle panels). (C) Quantification of PMN infiltration. Pathogenic IgG induced blistering and PMN infiltration in WT mice and TNF-α-/- or MC-/- mice reconstituted with WT MCs but not TNF-α-/- MCs. *p<0.01, 6 mice per group. Three independentt experiments were done for each group. 87






 ELISA assay showed")





, 6 mice per group.")









103 Figure 3.5. C5aR on MCs are required for TNF-α rabbit anti-mbp180 IgG and examined at release and skin blistering. WT and C5aR-/- mice were injected i.d. with pathogenic post injection. (A) Pathogenic IgG induced clinical blisters in WT and not C5aR-/- mice. (B) ELISA assay showed compatible skin C5a levels in WT and C5aR-/- mice 4 h post IgG injection. (C) Toluidine blue staining showed a significant reduction of MC degranulation in pathogenic IgG-injected C5aR-/- mice compared to WT mice 2 h post injection. (D) ELISA showed a significantly reduced TNF-α in the wash-out 2 vs. barr 4), 6 mice per group. (E) C5aR-/- mice of C5aR-/- mice as compared to the blister fluid of the WT mice. *p<0.01 (bar were reconstituted at the ear with buffer control, 1x10 6 MCs from C5aR-/-- mice, TNF-α-/- mice or WT mice. Ten weeks later, pathogenic abs were injected into ears of the MCreconstituted mice and the ears were examined 24 h later by H& &E and MPO assay. C5aR-/- mice reconstituted with WT MCs and not with C5aR-/- or TNF-α-/- showed restored PMN infiltration and dermal-epidermal blistering. n=6 per group. *p<0.01 (bar 7 vs. bars 5, 6). 88






")



")













104 Figure 3.6. TNFR1 and not TNFR2 is required for experimental BP. Neonatal WT and mice lacking TNFR1 (TNFR1-/-), TNFR2 (TNFR2-/-), and both TNFR1 and TNFR2 (TNFR1,2-/-) were injected i.d. with pathogenic rabbit anti-mbp180 IgG (2.5 mg/g body weight) and examined post injection. (A) RT-PCR demonstratedd that TNFR-1 was expressed constitutively, while TNFR2 expression was induced in the pathogenic IgG-injected skin of WT mice. (B) Pathogenic IgG induced BP blisters in WT, TNFR2-/-, but not TNFR1-/ /- and TNFR1,2-/- mice. (C) Pathogenic IgG induced BP with significantly increased PMN infiltration in WT mice and TNFR2-/ - mice, butt not TNFR1-/- and TNFR1,2-/- mice. 89






 or WT MCs (bar 8) but not with")
 showed restored PMN infiltration and dermal-epide")
105 *p<0.01 (bar 2 vs. bars 6, 8) ), 6 mice per group. Three independent experiments were for each group of mice. done Figure 3.7. Local reconstitution of MCs bearingg TNFR1 and not TNFR2 restores BP disease in TNFR-deficient mice. WT, TNFR1,2 -/-, and MC -/- mice were reconstituted at the ear with buffer control (none), 1x10 6 MCs from WT, TNFR1 -/-, TNFR2 -/-, or TNFR1,2 -/- mice. Ten weeks later, pathogenic antibodies were injected into ears of the MC-reconstituted mice and the ears were examined 24 h post IgG injection by H& &E staining and MPO assay. TNFR1,2 -/- and MC -/- mice reconstituted with TNFR22 -/- MCs (bars 7, 11 ) or WT MCs (bar 8) but not with TNFR1,2 -/- MCs (bars 5, 9) or TNFR1 -/-- MCs (bars 6, 10) showed restored PMN infiltration and dermal-epide rmal blistering. n= 6 forr each group. *p<0.01 ( bars 7, 8 vs. bars 5, 6; bar 11 vs. bars 9, 10). 90

")




 ELISA showed a significant fluid of")


106 Figure 3.8. Experimental BP induced by patient anti-bp180 autoantibodies depends on TNF-αpathogenic anti-bp180nc16a autoantibodies (anti-nc16a IgG) and examined 2 and 24 h Neonatal NC16A mice were injected i.d. with normal human IgG (control IgG) or post injection. (A) Pathogenic anti-nc16a and not control IgG induced BP blisters in NC16A mice 24 h post injection. Pretreatment with anti-tnf-increase in TNF-α protein in the blister antibody abolished experimental BP. (B) ELISA showed a significant fluid of diseased mice compared to that in the wash-out of control mice. Blocking TNF-α significantly reduced TNF-αα in the skin. (C) MPO assay demonstratedd a significantly increased number of PMNs in the lesional skin relative to the non-lesional skin of control mice and anti-tnf-α-treated mice. *p<0.01 (bar 1 vs. bar 2), 8 mice for each group. 91
 MCs degranulate, releasing pre-formed TNF-to transcription of unidentified pro-inflammatory factors. (6) The MC-derivedd pro- extracellularly. (4) TNF-α binds to TNFR1 on MCs.")
107 Figure 3.9. Proposed model for MC regulation of experimental BP. (1) C5a binds to C5aR on MCs. (2) p38mapk is phosphorylated andd signals. (3) MCs degranulate, releasing pre-formed TNF-to transcription of unidentified pro-inflammatory factors. (6) The MC-derivedd pro- extracellularly. (4) TNF-α binds to TNFR1 on MCs. (5) TNFR1 signaling leads inflammatory mediators lead to recruitment of PMNs to the skin and ultimately blister formation. 92
Cutaneous Immunology: Innate Immune Responses. Skin Biology Lecture Series
 Cutaneous Immunology: Innate Immune Responses Skin Biology Lecture Series The Immune Response: Innate and Adaptive Components Source: Wolff, Goldsmith, Katz, Gilchrest, Paller, Leffell. Fitzpatrick s Dermatology
Cutaneous Immunology: Innate Immune Responses Skin Biology Lecture Series The Immune Response: Innate and Adaptive Components Source: Wolff, Goldsmith, Katz, Gilchrest, Paller, Leffell. Fitzpatrick s Dermatology
Cytokines modulate the functional activities of individual cells and tissues both under normal and pathologic conditions Interleukins,
 Cytokines http://highered.mcgraw-hill.com/sites/0072507470/student_view0/chapter22/animation the_immune_response.html Cytokines modulate the functional activities of individual cells and tissues both under
Cytokines http://highered.mcgraw-hill.com/sites/0072507470/student_view0/chapter22/animation the_immune_response.html Cytokines modulate the functional activities of individual cells and tissues both under
Question 1. Kupffer cells, microglial cells and osteoclasts are all examples of what type of immune system cell?
 Abbas Chapter 2: Sarah Spriet February 8, 2015 Question 1. Kupffer cells, microglial cells and osteoclasts are all examples of what type of immune system cell? a. Dendritic cells b. Macrophages c. Monocytes
Abbas Chapter 2: Sarah Spriet February 8, 2015 Question 1. Kupffer cells, microglial cells and osteoclasts are all examples of what type of immune system cell? a. Dendritic cells b. Macrophages c. Monocytes
Complement. History. Chapter 7. Complement Components. Complement Pathways. Pathways of complement activation
 History Chapter 7 Complement Jules Border in 1890 s discovered complement Paul Ehrlich coined the term complement The activity of blood serum that completes the action of antibody Now: Set of serum proteins
History Chapter 7 Complement Jules Border in 1890 s discovered complement Paul Ehrlich coined the term complement The activity of blood serum that completes the action of antibody Now: Set of serum proteins
Topic (6): The Complement System
: The Complement System") Topic (6): The Complement System Introduction The complement system is a complex system of many different glycoproteins. It comprises several plasma proteins that sequentially activate each other by proteolytic
Topic (6): The Complement System Introduction The complement system is a complex system of many different glycoproteins. It comprises several plasma proteins that sequentially activate each other by proteolytic
ACTIVATION OF T LYMPHOCYTES AND CELL MEDIATED IMMUNITY
 ACTIVATION OF T LYMPHOCYTES AND CELL MEDIATED IMMUNITY The recognition of specific antigen by naïve T cell induces its own activation and effector phases. T helper cells recognize peptide antigens through
ACTIVATION OF T LYMPHOCYTES AND CELL MEDIATED IMMUNITY The recognition of specific antigen by naïve T cell induces its own activation and effector phases. T helper cells recognize peptide antigens through
Subject Index. Bcl-2, apoptosis regulation Bone marrow, polymorphonuclear neutrophil release 24, 26
 Subject Index A1, apoptosis regulation 217, 218 Adaptive immunity, polymorphonuclear neutrophil role 31 33 Angiogenesis cancer 178 endometrium remodeling 172 HIV Tat induction mechanism 176 inflammatory
Subject Index A1, apoptosis regulation 217, 218 Adaptive immunity, polymorphonuclear neutrophil role 31 33 Angiogenesis cancer 178 endometrium remodeling 172 HIV Tat induction mechanism 176 inflammatory
History. Chapter 13. Complement Components. Complement Pathways
 History Chapter 13 Complement Jules Border in 1890 s discovered complement Paul Ehrlich coined the term complement The activity of blood serum that completes the action of antibody Now: Set of serum proteins
History Chapter 13 Complement Jules Border in 1890 s discovered complement Paul Ehrlich coined the term complement The activity of blood serum that completes the action of antibody Now: Set of serum proteins
1. The scavenger receptor, CD36, functions as a coreceptor for which TLR? a. TLR ½ b. TLR 3 c. TLR 4 d. TLR 2/6
 Allergy and Immunology Review Corner: Cellular and Molecular Immunology, 8th Edition By Abul K. Abbas, MBBS, Andrew H. H. Lichtman, MD, PhD and Shiv Pillai, MBBS, PhD. Chapter 4 (pages 62-74): Innate Immunity
Allergy and Immunology Review Corner: Cellular and Molecular Immunology, 8th Edition By Abul K. Abbas, MBBS, Andrew H. H. Lichtman, MD, PhD and Shiv Pillai, MBBS, PhD. Chapter 4 (pages 62-74): Innate Immunity
HYPERSENSITIVITY REACTIONS D R S H O AI B R AZ A
 HYPERSENSITIVITY REACTIONS D R S H O AI B R AZ A HYPERSENSITIVITY REACTIONS Are exaggerated immune response upon antigenic stimulation Individuals who have been previously exposed to an antigen are said
HYPERSENSITIVITY REACTIONS D R S H O AI B R AZ A HYPERSENSITIVITY REACTIONS Are exaggerated immune response upon antigenic stimulation Individuals who have been previously exposed to an antigen are said
The Adaptive Immune Response. B-cells
 The Adaptive Immune Response B-cells The innate immune system provides immediate protection. The adaptive response takes time to develop and is antigen specific. Activation of B and T lymphocytes Naive
The Adaptive Immune Response B-cells The innate immune system provides immediate protection. The adaptive response takes time to develop and is antigen specific. Activation of B and T lymphocytes Naive
The Skinny of the Immune System
 The Skinny of the Immune System Robert Hostoffer, DO, FACOP, FAAP Associate Professor of Pediatrics Case Western Reserve University, Cleveland, Ohio Overview 1. Immune system of the skin 2. Immune Players
The Skinny of the Immune System Robert Hostoffer, DO, FACOP, FAAP Associate Professor of Pediatrics Case Western Reserve University, Cleveland, Ohio Overview 1. Immune system of the skin 2. Immune Players
Medical Virology Immunology. Dr. Sameer Naji, MB, BCh, PhD (UK) Head of Basic Medical Sciences Dept. Faculty of Medicine The Hashemite University
 Head of Basic Medical Sciences Dept. Faculty of Medicine The Hashemite University") Medical Virology Immunology Dr. Sameer Naji, MB, BCh, PhD (UK) Head of Basic Medical Sciences Dept. Faculty of Medicine The Hashemite University Human blood cells Phases of immune responses Microbe Naïve
Medical Virology Immunology Dr. Sameer Naji, MB, BCh, PhD (UK) Head of Basic Medical Sciences Dept. Faculty of Medicine The Hashemite University Human blood cells Phases of immune responses Microbe Naïve
CYTOKINE RECEPTORS AND SIGNAL TRANSDUCTION
 CYTOKINE RECEPTORS AND SIGNAL TRANSDUCTION What is Cytokine? Secreted popypeptide (protein) involved in cell-to-cell signaling. Acts in paracrine or autocrine fashion through specific cellular receptors.
CYTOKINE RECEPTORS AND SIGNAL TRANSDUCTION What is Cytokine? Secreted popypeptide (protein) involved in cell-to-cell signaling. Acts in paracrine or autocrine fashion through specific cellular receptors.
T-cell activation T cells migrate to secondary lymphoid tissues where they interact with antigen, antigen-presenting cells, and other lymphocytes:
 Interactions between innate immunity & adaptive immunity What happens to T cells after they leave the thymus? Naïve T cells exit the thymus and enter the bloodstream. If they remain in the bloodstream,
Interactions between innate immunity & adaptive immunity What happens to T cells after they leave the thymus? Naïve T cells exit the thymus and enter the bloodstream. If they remain in the bloodstream,
T-cell activation T cells migrate to secondary lymphoid tissues where they interact with antigen, antigen-presenting cells, and other lymphocytes:
 Interactions between innate immunity & adaptive immunity What happens to T cells after they leave the thymus? Naïve T cells exit the thymus and enter the bloodstream. If they remain in the bloodstream,
Interactions between innate immunity & adaptive immunity What happens to T cells after they leave the thymus? Naïve T cells exit the thymus and enter the bloodstream. If they remain in the bloodstream,
Third line of Defense
 Chapter 15 Specific Immunity and Immunization Topics -3 rd of Defense - B cells - T cells - Specific Immunities Third line of Defense Specific immunity is a complex interaction of immune cells (leukocytes)
Chapter 15 Specific Immunity and Immunization Topics -3 rd of Defense - B cells - T cells - Specific Immunities Third line of Defense Specific immunity is a complex interaction of immune cells (leukocytes)
Immunology Part II. Innate Immunity. 18. April 2018, Ruhr-Universität Bochum Marcus Peters,
 Immunology Part II Innate Immunity 18. April 2018, Ruhr-Universität Bochum Marcus Peters, marcus.peters@rub.de Conserved structures of pathogens PAMPs are detected by Pattern Recognition Receptors PRRs
Immunology Part II Innate Immunity 18. April 2018, Ruhr-Universität Bochum Marcus Peters, marcus.peters@rub.de Conserved structures of pathogens PAMPs are detected by Pattern Recognition Receptors PRRs
MACROPHAGE "MONOCYTES" SURFACE RECEPTORS
 LECTURE: 13 Title: MACROPHAGE "MONOCYTES" SURFACE RECEPTORS LEARNING OBJECTIVES: The student should be able to: Describe the blood monocytes (size, and shape of nucleus). Enumerate some of the monocytes
LECTURE: 13 Title: MACROPHAGE "MONOCYTES" SURFACE RECEPTORS LEARNING OBJECTIVES: The student should be able to: Describe the blood monocytes (size, and shape of nucleus). Enumerate some of the monocytes
T Cell Effector Mechanisms I: B cell Help & DTH
 T Cell Effector Mechanisms I: B cell Help & DTH Ned Braunstein, MD The Major T Cell Subsets p56 lck + T cells γ δ ε ζ ζ p56 lck CD8+ T cells γ δ ε ζ ζ Cα Cβ Vα Vβ CD3 CD8 Cα Cβ Vα Vβ CD3 MHC II peptide
T Cell Effector Mechanisms I: B cell Help & DTH Ned Braunstein, MD The Major T Cell Subsets p56 lck + T cells γ δ ε ζ ζ p56 lck CD8+ T cells γ δ ε ζ ζ Cα Cβ Vα Vβ CD3 CD8 Cα Cβ Vα Vβ CD3 MHC II peptide
Structure and Function of Antigen Recognition Molecules
 MICR2209 Structure and Function of Antigen Recognition Molecules Dr Allison Imrie allison.imrie@uwa.edu.au 1 Synopsis: In this lecture we will examine the major receptors used by cells of the innate and
MICR2209 Structure and Function of Antigen Recognition Molecules Dr Allison Imrie allison.imrie@uwa.edu.au 1 Synopsis: In this lecture we will examine the major receptors used by cells of the innate and
11/25/2017. THE IMMUNE SYSTEM Chapter 43 IMMUNITY INNATE IMMUNITY EXAMPLE IN INSECTS BARRIER DEFENSES INNATE IMMUNITY OF VERTEBRATES
 THE IMMUNE SYSTEM Chapter 43 IMMUNITY INNATE IMMUNITY EXAMPLE IN INSECTS Exoskeleton made of chitin forms the first barrier to pathogens Digestive system is protected by a chitin-based barrier and lysozyme,
THE IMMUNE SYSTEM Chapter 43 IMMUNITY INNATE IMMUNITY EXAMPLE IN INSECTS Exoskeleton made of chitin forms the first barrier to pathogens Digestive system is protected by a chitin-based barrier and lysozyme,
History. Chapter 13. Complement Components. Complement Pathways
 History Chapter 13 Complement Jules Border in 1890 s discovered complement Paul Ehrlich coined the term complement The activity of blood serum that completes the action of antibody Now: Set of serum proteins
History Chapter 13 Complement Jules Border in 1890 s discovered complement Paul Ehrlich coined the term complement The activity of blood serum that completes the action of antibody Now: Set of serum proteins
Adaptive Immunity. Jeffrey K. Actor, Ph.D. MSB 2.214,
 Adaptive Immunity Jeffrey K. Actor, Ph.D. MSB 2.214, 500-5344 Lecture Objectives: Understand role of various molecules including cytokines, chemokines, costimulatory and adhesion molecules in the development
Adaptive Immunity Jeffrey K. Actor, Ph.D. MSB 2.214, 500-5344 Lecture Objectives: Understand role of various molecules including cytokines, chemokines, costimulatory and adhesion molecules in the development
Macrophage Activation & Cytokine Release. Dendritic Cells & Antigen Presentation. Neutrophils & Innate Defense
 Macrophage Activation & Cytokine Release Dendritic Cells & Antigen Presentation Neutrophils & Innate Defense Neutrophils Polymorphonuclear cells (PMNs) are recruited to the site of infection where they
Macrophage Activation & Cytokine Release Dendritic Cells & Antigen Presentation Neutrophils & Innate Defense Neutrophils Polymorphonuclear cells (PMNs) are recruited to the site of infection where they
The recruitment of leukocytes and plasma proteins from the blood to sites of infection and tissue injury is called inflammation
 The migration of a particular type of leukocyte into a restricted type of tissue, or a tissue with an ongoing infection or injury, is often called leukocyte homing, and the general process of leukocyte
The migration of a particular type of leukocyte into a restricted type of tissue, or a tissue with an ongoing infection or injury, is often called leukocyte homing, and the general process of leukocyte
Adaptive Immunity: Humoral Immune Responses
 MICR2209 Adaptive Immunity: Humoral Immune Responses Dr Allison Imrie 1 Synopsis: In this lecture we will review the different mechanisms which constitute the humoral immune response, and examine the antibody
MICR2209 Adaptive Immunity: Humoral Immune Responses Dr Allison Imrie 1 Synopsis: In this lecture we will review the different mechanisms which constitute the humoral immune response, and examine the antibody
Allergic rhinitis (Hay fever) Asthma Anaphylaxis Urticaria Atopic dermatitis
 Asthma Anaphylaxis Urticaria Atopic dermatitis") Hypersensitivity Disorders Hypersensitivity Disorders Immune Response IgE Disease Example Ragweed hay fever IgG Cytotoxic Immune complex T Cell Hemolytic anemia Serum sickness Poison ivy IgE-mediated Diseases
Hypersensitivity Disorders Hypersensitivity Disorders Immune Response IgE Disease Example Ragweed hay fever IgG Cytotoxic Immune complex T Cell Hemolytic anemia Serum sickness Poison ivy IgE-mediated Diseases
All animals have innate immunity, a defense active immediately upon infection Vertebrates also have adaptive immunity
 1 2 3 4 5 6 7 8 9 The Immune System All animals have innate immunity, a defense active immediately upon infection Vertebrates also have adaptive immunity Figure 43.2 In innate immunity, recognition and
1 2 3 4 5 6 7 8 9 The Immune System All animals have innate immunity, a defense active immediately upon infection Vertebrates also have adaptive immunity Figure 43.2 In innate immunity, recognition and
Effector T Cells and
 1 Effector T Cells and Cytokines Andrew Lichtman, MD PhD Brigham and Women's Hospital Harvard Medical School 2 Lecture outline Cytokines Subsets of CD4+ T cells: definitions, functions, development New
1 Effector T Cells and Cytokines Andrew Lichtman, MD PhD Brigham and Women's Hospital Harvard Medical School 2 Lecture outline Cytokines Subsets of CD4+ T cells: definitions, functions, development New
االستاذ المساعد الدكتور خالد ياسين الزاملي \مناعة \المرحلة الثانية \ التحليالت المرضية \ المعهد التقني كوت
 Complement System The term complement refers to the ability of a system of some nonspecific proteins in normal human serum to complement, i.e., augment the effects of other components of immune system,
Complement System The term complement refers to the ability of a system of some nonspecific proteins in normal human serum to complement, i.e., augment the effects of other components of immune system,
Newly Recognized Components of the Innate Immune System
 Newly Recognized Components of the Innate Immune System NOD Proteins: Intracellular Peptidoglycan Sensors NOD-1 NOD-2 Nod Protein LRR; Ligand Recognition CARD RICK I-κB p50 p65 NF-κB Polymorphisms in Nod-2
Newly Recognized Components of the Innate Immune System NOD Proteins: Intracellular Peptidoglycan Sensors NOD-1 NOD-2 Nod Protein LRR; Ligand Recognition CARD RICK I-κB p50 p65 NF-κB Polymorphisms in Nod-2
Immunology of Asthma. Kenneth J. Goodrum,Ph. Ph.D. Ohio University College of Osteopathic Medicine
 Immunology of Asthma Kenneth J. Goodrum,Ph Ph.D. Ohio University College of Osteopathic Medicine Outline Consensus characteristics/incidence data Immune/inflammatory basis Etiology/Genetic basis Hygiene
Immunology of Asthma Kenneth J. Goodrum,Ph Ph.D. Ohio University College of Osteopathic Medicine Outline Consensus characteristics/incidence data Immune/inflammatory basis Etiology/Genetic basis Hygiene
Chapter 3, Part A (Pages 37-45): Leukocyte Migration into Tissues
: Leukocyte Migration into Tissues") Allergy and Immunology Review Corner: Chapter 3, Part A (pages 37-45) of Cellular and Molecular Immunology (Seventh Edition), by Abul K. Abbas, Andrew H. Lichtman and Shiv Pillai. Chapter 3, Part A (Pages
Allergy and Immunology Review Corner: Chapter 3, Part A (pages 37-45) of Cellular and Molecular Immunology (Seventh Edition), by Abul K. Abbas, Andrew H. Lichtman and Shiv Pillai. Chapter 3, Part A (Pages
Immunology for the Rheumatologist
 Immunology for the Rheumatologist Rheumatologists frequently deal with the immune system gone awry, rarely studying normal immunology. This program is an overview and discussion of the function of the
Immunology for the Rheumatologist Rheumatologists frequently deal with the immune system gone awry, rarely studying normal immunology. This program is an overview and discussion of the function of the
Innate Immunity. Chapter 3. Connection Between Innate and Adaptive Immunity. Know Differences and Provide Examples. Antimicrobial peptide psoriasin
 Chapter Know Differences and Provide Examples Innate Immunity kin and Epithelial Barriers Antimicrobial peptide psoriasin -Activity against Gram (-) E. coli Connection Between Innate and Adaptive Immunity
Chapter Know Differences and Provide Examples Innate Immunity kin and Epithelial Barriers Antimicrobial peptide psoriasin -Activity against Gram (-) E. coli Connection Between Innate and Adaptive Immunity
Innate Immunity. Natural or native immunity
 Innate Immunity 1 Innate Immunity Natural or native immunity 2 When microbes enter in the body 3 Secondly, it also stimulates the adaptive immune system 4 Immunologic memory 5 Components of Innate Immunity
Innate Immunity 1 Innate Immunity Natural or native immunity 2 When microbes enter in the body 3 Secondly, it also stimulates the adaptive immune system 4 Immunologic memory 5 Components of Innate Immunity
Immune System. Presented by Kazzandra Anton, Rhea Chung, Lea Sado, and Raymond Tanaka
 Immune System Presented by Kazzandra Anton, Rhea Chung, Lea Sado, and Raymond Tanaka Content Standards 35.1 In innate immunity, recognition and response rely on traits common to groups of pathogens 35.2
Immune System Presented by Kazzandra Anton, Rhea Chung, Lea Sado, and Raymond Tanaka Content Standards 35.1 In innate immunity, recognition and response rely on traits common to groups of pathogens 35.2
Adaptive immune responses: T cell-mediated immunity
 MICR2209 Adaptive immune responses: T cell-mediated immunity Dr Allison Imrie allison.imrie@uwa.edu.au 1 Synopsis: In this lecture we will discuss the T-cell mediated immune response, how it is activated,
MICR2209 Adaptive immune responses: T cell-mediated immunity Dr Allison Imrie allison.imrie@uwa.edu.au 1 Synopsis: In this lecture we will discuss the T-cell mediated immune response, how it is activated,
Innate Immunity. Natural or native immunity
 Innate Immunity 1 Innate Immunity Natural or native immunity 2 When microbes enter in the body 3 Secondly, it also stimulates the adaptive immune system 4 Immunologic memory 5 Components of Innate Immunity
Innate Immunity 1 Innate Immunity Natural or native immunity 2 When microbes enter in the body 3 Secondly, it also stimulates the adaptive immune system 4 Immunologic memory 5 Components of Innate Immunity
Third line of Defense. Topic 8 Specific Immunity (adaptive) (18) 3 rd Line = Prophylaxis via Immunization!
 (18) 3 rd Line = Prophylaxis via Immunization!") Topic 8 Specific Immunity (adaptive) (18) Topics - 3 rd Line of Defense - B cells - T cells - Specific Immunities 1 3 rd Line = Prophylaxis via Immunization! (a) A painting of Edward Jenner depicts a cow
Topic 8 Specific Immunity (adaptive) (18) Topics - 3 rd Line of Defense - B cells - T cells - Specific Immunities 1 3 rd Line = Prophylaxis via Immunization! (a) A painting of Edward Jenner depicts a cow
The Innate Immune Response
 The Innate Immune Response FUNCTIONS OF THE IMMUNE SYSTEM: Recognize, destroy and clear a diversity of pathogens. Initiate tissue and wound healing processes. Recognize and clear damaged self components.
The Innate Immune Response FUNCTIONS OF THE IMMUNE SYSTEM: Recognize, destroy and clear a diversity of pathogens. Initiate tissue and wound healing processes. Recognize and clear damaged self components.
Cell-Derived Inflammatory Mediators
 Cell-Derived Inflammatory Mediators Introduction about chemical mediators in inflammation Mediators may be Cellular mediators cell-produced or cell-secreted derived from circulating inactive precursors,
Cell-Derived Inflammatory Mediators Introduction about chemical mediators in inflammation Mediators may be Cellular mediators cell-produced or cell-secreted derived from circulating inactive precursors,
Overview of the immune system
 Overview of the immune system Immune system Innate (nonspecific) 1 st line of defense Adaptive (specific) 2 nd line of defense Cellular components Humoral components Cellular components Humoral components
Overview of the immune system Immune system Innate (nonspecific) 1 st line of defense Adaptive (specific) 2 nd line of defense Cellular components Humoral components Cellular components Humoral components
THE COMPLEMENT SYSTEM OBJECTIVES:
 Dr Mohammed Al- ani THE COMPLEMENT SYSTEM OBJECTIVES: When you finish this section, you should be able to: 1. Describe the effects of complement activation. 2. Outline the Classical, Mannan-Binding (MB)
Dr Mohammed Al- ani THE COMPLEMENT SYSTEM OBJECTIVES: When you finish this section, you should be able to: 1. Describe the effects of complement activation. 2. Outline the Classical, Mannan-Binding (MB)
Current concepts of autoimmune bullous diseases Advances in pathogenesis. Luca Borradori
 Current concepts of autoimmune bullous diseases Advances in pathogenesis Luca Borradori Dept. of Dermatology Inselspital, University Hospital of Berne Switzerland Luca.Borradori@insel.ch Autoimmune bullous
Current concepts of autoimmune bullous diseases Advances in pathogenesis Luca Borradori Dept. of Dermatology Inselspital, University Hospital of Berne Switzerland Luca.Borradori@insel.ch Autoimmune bullous
Hematopoiesis. Hematopoiesis. Hematopoiesis
 Chapter. Cells and Organs of the Immune System Hematopoiesis Hematopoiesis- formation and development of WBC and RBC bone marrow. Hematopoietic stem cell- give rise to any blood cells (constant number,
Chapter. Cells and Organs of the Immune System Hematopoiesis Hematopoiesis- formation and development of WBC and RBC bone marrow. Hematopoietic stem cell- give rise to any blood cells (constant number,
The Adaptive Immune Response. T-cells
 The Adaptive Immune Response T-cells T Lymphocytes T lymphocytes develop from precursors in the thymus. Mature T cells are found in the blood, where they constitute 60% to 70% of lymphocytes, and in T-cell
The Adaptive Immune Response T-cells T Lymphocytes T lymphocytes develop from precursors in the thymus. Mature T cells are found in the blood, where they constitute 60% to 70% of lymphocytes, and in T-cell
Immune System AP SBI4UP
 Immune System AP SBI4UP TYPES OF IMMUNITY INNATE IMMUNITY ACQUIRED IMMUNITY EXTERNAL DEFENCES INTERNAL DEFENCES HUMORAL RESPONSE Skin Phagocytic Cells CELL- MEDIATED RESPONSE Mucus layer Antimicrobial
Immune System AP SBI4UP TYPES OF IMMUNITY INNATE IMMUNITY ACQUIRED IMMUNITY EXTERNAL DEFENCES INTERNAL DEFENCES HUMORAL RESPONSE Skin Phagocytic Cells CELL- MEDIATED RESPONSE Mucus layer Antimicrobial
Scott Abrams, Ph.D. Professor of Oncology, x4375 Kuby Immunology SEVENTH EDITION
 Scott Abrams, Ph.D. Professor of Oncology, x4375 scott.abrams@roswellpark.org Kuby Immunology SEVENTH EDITION CHAPTER 13 Effector Responses: Cell- and Antibody-Mediated Immunity Copyright 2013 by W. H.
Scott Abrams, Ph.D. Professor of Oncology, x4375 scott.abrams@roswellpark.org Kuby Immunology SEVENTH EDITION CHAPTER 13 Effector Responses: Cell- and Antibody-Mediated Immunity Copyright 2013 by W. H.
Basis of Immunology and
 Basis of Immunology and Immunophysiopathology of Infectious Diseases Jointly organized by Institut Pasteur in Ho Chi Minh City and Institut Pasteur with kind support from ANRS & Université Pierre et Marie
Basis of Immunology and Immunophysiopathology of Infectious Diseases Jointly organized by Institut Pasteur in Ho Chi Minh City and Institut Pasteur with kind support from ANRS & Université Pierre et Marie
Fc receptors, phagocytosis role 128
 Subject Index Adaptive immunity dependence on innate immunity 9, 10 evolution 10 Aging anti-inflammatory agents in counteraction 202 beneficial polymorphisms 199 201 definition 18, 189 innate immunity
Subject Index Adaptive immunity dependence on innate immunity 9, 10 evolution 10 Aging anti-inflammatory agents in counteraction 202 beneficial polymorphisms 199 201 definition 18, 189 innate immunity
The term complement refers to the ability of a system of some nonspecific proteins in normal human serum to complement, i.e., augment the effects of
 COMPLEMENT SYSTEM The term complement refers to the ability of a system of some nonspecific proteins in normal human serum to complement, i.e., augment the effects of other components of immune system,
COMPLEMENT SYSTEM The term complement refers to the ability of a system of some nonspecific proteins in normal human serum to complement, i.e., augment the effects of other components of immune system,
T cell maturation. T-cell Maturation. What allows T cell maturation?
 T-cell Maturation What allows T cell maturation? Direct contact with thymic epithelial cells Influence of thymic hormones Growth factors (cytokines, CSF) T cell maturation T cell progenitor DN DP SP 2ry
T-cell Maturation What allows T cell maturation? Direct contact with thymic epithelial cells Influence of thymic hormones Growth factors (cytokines, CSF) T cell maturation T cell progenitor DN DP SP 2ry
Secretory antibodies in the upper respiratory tract
 Secretory antibodies in the upper respiratory tract B lymphocytes IgM (pneumococcus) Dimeric IgA J chain Polymeric immunoglobulin receptor (PigR) Polysaccharide capsule Epithelial cell Basolateral Secretory
Secretory antibodies in the upper respiratory tract B lymphocytes IgM (pneumococcus) Dimeric IgA J chain Polymeric immunoglobulin receptor (PigR) Polysaccharide capsule Epithelial cell Basolateral Secretory
Target cell lysis Opsonization Activation of the inflammatory response (e.g. degranulation, extravasation) Clearance of immune complexes
 Clearance of immune complexes") Immunology Dr. John J. Haddad Chapter 13 Complement Major roles of complement (Figure 13-1): Target cell lysis Opsonization Activation of the inflammatory response (e.g. degranulation, extravasation) Clearance
Immunology Dr. John J. Haddad Chapter 13 Complement Major roles of complement (Figure 13-1): Target cell lysis Opsonization Activation of the inflammatory response (e.g. degranulation, extravasation) Clearance
10. Which of the following immune cell is unable to phagocytose (a) neutrophils (b) eosinophils (c) macrophages (d) T-cells (e) monocytes
 neutrophils (b) eosinophils (c) macrophages (d) T-cells (e) monocytes") Chapter 2. Acute and chronic inflammation(6): 1. In acute inflammation, which events occur in the correct chronological order? (Remembered from 2000, 2004 exam.) p50 (a) transient vasoconstriction, stasis
Chapter 2. Acute and chronic inflammation(6): 1. In acute inflammation, which events occur in the correct chronological order? (Remembered from 2000, 2004 exam.) p50 (a) transient vasoconstriction, stasis
General Overview of Immunology. Kimberly S. Schluns, Ph.D. Associate Professor Department of Immunology UT MD Anderson Cancer Center
 General Overview of Immunology Kimberly S. Schluns, Ph.D. Associate Professor Department of Immunology UT MD Anderson Cancer Center Objectives Describe differences between innate and adaptive immune responses
General Overview of Immunology Kimberly S. Schluns, Ph.D. Associate Professor Department of Immunology UT MD Anderson Cancer Center Objectives Describe differences between innate and adaptive immune responses
NEUTROPHIL, BASOPHIL, EOSINOPHIL, AND PLATELETS SURFACE RECEPTORS
 LECTURE: 15 Title NEUTROPHIL, BASOPHIL, EOSINOPHIL, AND PLATELETS SURFACE RECEPTORS LEARNING OBJECTIVES: The student should be able to: Determine the relative percentages in blood for the various types
LECTURE: 15 Title NEUTROPHIL, BASOPHIL, EOSINOPHIL, AND PLATELETS SURFACE RECEPTORS LEARNING OBJECTIVES: The student should be able to: Determine the relative percentages in blood for the various types
The Adaptive Immune Responses
 The Adaptive Immune Responses The two arms of the immune responses are; 1) the cell mediated, and 2) the humoral responses. In this chapter we will discuss the two responses in detail and we will start
The Adaptive Immune Responses The two arms of the immune responses are; 1) the cell mediated, and 2) the humoral responses. In this chapter we will discuss the two responses in detail and we will start
How the Innate Immune System Profiles Pathogens
 How the Innate Immune System Profiles Pathogens Receptors on macrophages, neutrophils, dendritic cells for bacteria and viruses Broad specificity - Two main groups of bacteria: gram positive, gram-negative
How the Innate Immune System Profiles Pathogens Receptors on macrophages, neutrophils, dendritic cells for bacteria and viruses Broad specificity - Two main groups of bacteria: gram positive, gram-negative
Blood and Immune system Acquired Immunity
 Blood and Immune system Acquired Immunity Immunity Acquired (Adaptive) Immunity Defensive mechanisms include : 1) Innate immunity (Natural or Non specific) 2) Acquired immunity (Adaptive or Specific) Cell-mediated
Blood and Immune system Acquired Immunity Immunity Acquired (Adaptive) Immunity Defensive mechanisms include : 1) Innate immunity (Natural or Non specific) 2) Acquired immunity (Adaptive or Specific) Cell-mediated
M.Sc. III Semester Biotechnology End Semester Examination, 2013 Model Answer LBTM: 302 Advanced Immunology
 Code : AS-2246 M.Sc. III Semester Biotechnology End Semester Examination, 2013 Model Answer LBTM: 302 Advanced Immunology A. Select one correct option for each of the following questions:- 2X10=10 1. (b)
Code : AS-2246 M.Sc. III Semester Biotechnology End Semester Examination, 2013 Model Answer LBTM: 302 Advanced Immunology A. Select one correct option for each of the following questions:- 2X10=10 1. (b)
DNA vaccine, peripheral T-cell tolerance modulation 185
 Subject Index Airway hyperresponsiveness (AHR) animal models 41 43 asthma inhibition 45 overview 41 mast cell modulation of T-cells 62 64 respiratory tolerance 40, 41 Tregs inhibition role 44 respiratory
Subject Index Airway hyperresponsiveness (AHR) animal models 41 43 asthma inhibition 45 overview 41 mast cell modulation of T-cells 62 64 respiratory tolerance 40, 41 Tregs inhibition role 44 respiratory
IMMUNE CELL SURFACE RECEPTORS AND THEIR FUNCTIONS
 LECTURE: 07 Title: IMMUNE CELL SURFACE RECEPTORS AND THEIR FUNCTIONS LEARNING OBJECTIVES: The student should be able to: The chemical nature of the cellular surface receptors. Define the location of the
LECTURE: 07 Title: IMMUNE CELL SURFACE RECEPTORS AND THEIR FUNCTIONS LEARNING OBJECTIVES: The student should be able to: The chemical nature of the cellular surface receptors. Define the location of the
Cellular Pathology of immunological disorders
 Cellular Pathology of immunological disorders SCBM344 Cellular and Molecular Pathology Witchuda Payuhakrit, Ph.D (Pathobiology) witchuda.pay@mahidol.ac.th Objectives Describe the etiology of immunological
Cellular Pathology of immunological disorders SCBM344 Cellular and Molecular Pathology Witchuda Payuhakrit, Ph.D (Pathobiology) witchuda.pay@mahidol.ac.th Objectives Describe the etiology of immunological
Immunology - Lecture 2 Adaptive Immune System 1
 Immunology - Lecture 2 Adaptive Immune System 1 Book chapters: Molecules of the Adaptive Immunity 6 Adaptive Cells and Organs 7 Generation of Immune Diversity Lymphocyte Antigen Receptors - 8 CD markers
Immunology - Lecture 2 Adaptive Immune System 1 Book chapters: Molecules of the Adaptive Immunity 6 Adaptive Cells and Organs 7 Generation of Immune Diversity Lymphocyte Antigen Receptors - 8 CD markers
The Immune System. These are classified as the Innate and Adaptive Immune Responses. Innate Immunity
 The Immune System Biological mechanisms that defend an organism must be 1. triggered by a stimulus upon injury or pathogen attack 2. able to counteract the injury or invasion 3. able to recognise foreign
The Immune System Biological mechanisms that defend an organism must be 1. triggered by a stimulus upon injury or pathogen attack 2. able to counteract the injury or invasion 3. able to recognise foreign
Innate Immunity. Connection Between Innate and Adaptive Immunity. Know Differences and Provide Examples Chapter 3. Antimicrobial peptide psoriasin
 Know Differences and Provide Examples Chapter * Innate Immunity * kin and Epithelial Barriers * Antimicrobial peptide psoriasin -Activity against Gram (-) E. coli Connection Between Innate and Adaptive
Know Differences and Provide Examples Chapter * Innate Immunity * kin and Epithelial Barriers * Antimicrobial peptide psoriasin -Activity against Gram (-) E. coli Connection Between Innate and Adaptive
Principles of Adaptive Immunity
 Principles of Adaptive Immunity Chapter 3 Parham Hans de Haard 17 th of May 2010 Agenda Recognition molecules of adaptive immune system Features adaptive immune system Immunoglobulins and T-cell receptors
Principles of Adaptive Immunity Chapter 3 Parham Hans de Haard 17 th of May 2010 Agenda Recognition molecules of adaptive immune system Features adaptive immune system Immunoglobulins and T-cell receptors
ACTIVATION AND EFFECTOR FUNCTIONS OF CELL-MEDIATED IMMUNITY AND NK CELLS. Choompone Sakonwasun, MD (Hons), FRCPT
, FRCPT") ACTIVATION AND EFFECTOR FUNCTIONS OF CELL-MEDIATED IMMUNITY AND NK CELLS Choompone Sakonwasun, MD (Hons), FRCPT Types of Adaptive Immunity Types of T Cell-mediated Immune Reactions CTLs = cytotoxic T lymphocytes
ACTIVATION AND EFFECTOR FUNCTIONS OF CELL-MEDIATED IMMUNITY AND NK CELLS Choompone Sakonwasun, MD (Hons), FRCPT Types of Adaptive Immunity Types of T Cell-mediated Immune Reactions CTLs = cytotoxic T lymphocytes
IL-17 in health and disease. March 2014 PSO13-C051n
 IL-17 in health and disease March 2014 PSO13-C051n Originally Researchers Suggested That IL-12 and IL-4 drove Th Cell Differentiation Naïve CD4 + T cell Question: Which of these cell types is responsible
IL-17 in health and disease March 2014 PSO13-C051n Originally Researchers Suggested That IL-12 and IL-4 drove Th Cell Differentiation Naïve CD4 + T cell Question: Which of these cell types is responsible
Chapter 1. Chapter 1 Concepts. MCMP422 Immunology and Biologics Immunology is important personally and professionally!
 MCMP422 Immunology and Biologics Immunology is important personally and professionally! Learn the language - use the glossary and index RNR - Reading, Note taking, Reviewing All materials in Chapters 1-3
MCMP422 Immunology and Biologics Immunology is important personally and professionally! Learn the language - use the glossary and index RNR - Reading, Note taking, Reviewing All materials in Chapters 1-3
C. Incorrect! MHC class I molecules are not involved in the process of bridging in ADCC.
 Immunology - Problem Drill 13: T- Cell Mediated Immunity Question No. 1 of 10 1. During Antibody-dependent cell mediated cytotoxicity (ADCC), the antibody acts like a bridge between the specific antigen
Immunology - Problem Drill 13: T- Cell Mediated Immunity Question No. 1 of 10 1. During Antibody-dependent cell mediated cytotoxicity (ADCC), the antibody acts like a bridge between the specific antigen
PBS Class #2 Introduction to the Immune System part II Suggested reading: Abbas, pgs , 27-30
 PBS 803 - Class #2 Introduction to the Immune System part II Suggested reading: Abbas, pgs. 15-25, 27-30 Learning Objectives Compare and contrast the maturation of B and T lymphocytes Compare and contrast
PBS 803 - Class #2 Introduction to the Immune System part II Suggested reading: Abbas, pgs. 15-25, 27-30 Learning Objectives Compare and contrast the maturation of B and T lymphocytes Compare and contrast
Clinical Basis of the Immune Response and the Complement Cascade
 Clinical Basis of the Immune Response and the Complement Cascade Bryan L. Martin, DO, MMAS, FACAAI, FAAAAI, FACOI, FACP Emeritus Professor of Medicine and Pediatrics President, American College of Allergy,
Clinical Basis of the Immune Response and the Complement Cascade Bryan L. Martin, DO, MMAS, FACAAI, FAAAAI, FACOI, FACP Emeritus Professor of Medicine and Pediatrics President, American College of Allergy,
The Immune System. by Dr. Carmen Rexach Physiology Mt San Antonio College
 The Immune System by Dr. Carmen Rexach Physiology Mt San Antonio College What is the immune system? defense system found in vertebrates Two categories Nonspecific specific provides protection from pathogens
The Immune System by Dr. Carmen Rexach Physiology Mt San Antonio College What is the immune system? defense system found in vertebrates Two categories Nonspecific specific provides protection from pathogens
ANATOMY OF THE IMMUNE SYSTEM
 Immunity Learning objectives Explain what triggers an immune response and where in the body the immune response occurs. Understand how the immune system handles exogenous and endogenous antigen differently.
Immunity Learning objectives Explain what triggers an immune response and where in the body the immune response occurs. Understand how the immune system handles exogenous and endogenous antigen differently.
INFLAMMATION & REPAIR
 INFLAMMATION & REPAIR Lecture 7 Chemical Mediators of Inflammation Winter 2013 Chelsea Martin Special thanks to Drs. Hanna and Forzan Course Outline i. Inflammation: Introduction and generalities (lecture
INFLAMMATION & REPAIR Lecture 7 Chemical Mediators of Inflammation Winter 2013 Chelsea Martin Special thanks to Drs. Hanna and Forzan Course Outline i. Inflammation: Introduction and generalities (lecture
Natural Defense Mechanisms
 Color code: Important in red Extra in blue For team error adjustments, click here Natural Defense Mechanisms Objectives To know First (non-specific immunity) and second (adaptive immunity) lines of defense
Color code: Important in red Extra in blue For team error adjustments, click here Natural Defense Mechanisms Objectives To know First (non-specific immunity) and second (adaptive immunity) lines of defense
Physiology Unit 3. ADAPTIVE IMMUNITY The Specific Immune Response
 Physiology Unit 3 ADAPTIVE IMMUNITY The Specific Immune Response In Physiology Today The Adaptive Arm of the Immune System Specific Immune Response Internal defense against a specific pathogen Acquired
Physiology Unit 3 ADAPTIVE IMMUNITY The Specific Immune Response In Physiology Today The Adaptive Arm of the Immune System Specific Immune Response Internal defense against a specific pathogen Acquired
Time course of immune response
 Time course of immune response Route of entry Route of entry (cont.) Steps in infection Barriers to infection Mf receptors Facilitate engulfment Glucan, mannose Scavenger CD11b/CD18 Allows immediate response
Time course of immune response Route of entry Route of entry (cont.) Steps in infection Barriers to infection Mf receptors Facilitate engulfment Glucan, mannose Scavenger CD11b/CD18 Allows immediate response
Autoimmunity. Autoimmunity arises because of defects in central or peripheral tolerance of lymphocytes to selfantigens
 Autoimmunity Autoimmunity arises because of defects in central or peripheral tolerance of lymphocytes to selfantigens Autoimmune disease can be caused to primary defects in B cells, T cells and possibly
Autoimmunity Autoimmunity arises because of defects in central or peripheral tolerance of lymphocytes to selfantigens Autoimmune disease can be caused to primary defects in B cells, T cells and possibly
Molecular and Cellular Basis of Immune Protection of Mucosal Surfaces
 Molecular and Cellular Basis of Immune Protection of Mucosal Surfaces Department of Biologic & Materials Sciences School of Dentistry University of Michigan Ann Arbor, Michigan 48109-1078 1 Image quality
Molecular and Cellular Basis of Immune Protection of Mucosal Surfaces Department of Biologic & Materials Sciences School of Dentistry University of Michigan Ann Arbor, Michigan 48109-1078 1 Image quality
Cell Mediated Immunity CELL MEDIATED IMMUNITY. Basic Elements of Cell Mediated Immunity (CMI) Antibody-dependent cell-mediated cytotoxicity (ADCC)
 Antibody-dependent cell-mediated cytotoxicity (ADCC)") Chapter 16 CELL MEDIATED IMMUNITY Cell Mediated Immunity Also known as Cellular Immunity or CMI The effector phase T cells Specificity for immune recognition reactions TH provide cytokines CTLs do the
Chapter 16 CELL MEDIATED IMMUNITY Cell Mediated Immunity Also known as Cellular Immunity or CMI The effector phase T cells Specificity for immune recognition reactions TH provide cytokines CTLs do the
The Immune System: Innate and Adaptive Body Defenses Outline PART 1: INNATE DEFENSES 21.1 Surface barriers act as the first line of defense to keep
 The Immune System: Innate and Adaptive Body Defenses Outline PART 1: INNATE DEFENSES 21.1 Surface barriers act as the first line of defense to keep invaders out of the body (pp. 772 773; Fig. 21.1; Table
The Immune System: Innate and Adaptive Body Defenses Outline PART 1: INNATE DEFENSES 21.1 Surface barriers act as the first line of defense to keep invaders out of the body (pp. 772 773; Fig. 21.1; Table
Chapter 13: Cytokines
 Chapter 13: Cytokines Definition: secreted, low-molecular-weight proteins that regulate the nature, intensity and duration of the immune response by exerting a variety of effects on lymphocytes and/or
Chapter 13: Cytokines Definition: secreted, low-molecular-weight proteins that regulate the nature, intensity and duration of the immune response by exerting a variety of effects on lymphocytes and/or
immunity defenses invertebrates vertebrates chapter 48 Animal defenses --
 defenses Animal defenses -- immunity chapter 48 invertebrates coelomocytes, amoebocytes, hemocytes sponges, cnidarians, etc. annelids basophilic amoebocytes, acidophilic granulocytes arthropod immune systems
defenses Animal defenses -- immunity chapter 48 invertebrates coelomocytes, amoebocytes, hemocytes sponges, cnidarians, etc. annelids basophilic amoebocytes, acidophilic granulocytes arthropod immune systems
Effector mechanisms of cell-mediated immunity: Properties of effector, memory and regulatory T cells
 ICI Basic Immunology course Effector mechanisms of cell-mediated immunity: Properties of effector, memory and regulatory T cells Abul K. Abbas, MD UCSF Stages in the development of T cell responses: induction
ICI Basic Immunology course Effector mechanisms of cell-mediated immunity: Properties of effector, memory and regulatory T cells Abul K. Abbas, MD UCSF Stages in the development of T cell responses: induction
Topics. Humoral Immune Response Part II Accessory cells Fc Receptors Opsonization and killing mechanisms of phagocytes NK, mast, eosynophils
 Topics Humoral Immune Response Part II Accessory cells Fc Receptors Opsonization and killing mechanisms of phagocytes NK, mast, eosynophils Immune regulation Idiotypic network 2/15/2005 MICR 415 / 515
Topics Humoral Immune Response Part II Accessory cells Fc Receptors Opsonization and killing mechanisms of phagocytes NK, mast, eosynophils Immune regulation Idiotypic network 2/15/2005 MICR 415 / 515
Innate Immunity: Nonspecific Defenses of the Host
 PowerPoint Lecture Presentations prepared by Bradley W. Christian, McLennan Community College C H A P T E R 16 Innate Immunity: Nonspecific Defenses of the Host Host Response to Disease Resistance- ability
PowerPoint Lecture Presentations prepared by Bradley W. Christian, McLennan Community College C H A P T E R 16 Innate Immunity: Nonspecific Defenses of the Host Host Response to Disease Resistance- ability
WHY IS THIS IMPORTANT?
 CHAPTER 16 THE ADAPTIVE IMMUNE RESPONSE WHY IS THIS IMPORTANT? The adaptive immune system protects us from many infections The adaptive immune system has memory so we are not infected by the same pathogen
CHAPTER 16 THE ADAPTIVE IMMUNE RESPONSE WHY IS THIS IMPORTANT? The adaptive immune system protects us from many infections The adaptive immune system has memory so we are not infected by the same pathogen
Immune response to infection
 Immune response to infection Dr. Sandra Nitsche (Sandra.Nitsche@rub.de ) 20.06.2018 1 Course of acute infection Typical acute infection that is cleared by an adaptive immune reaction 1. invasion of pathogen
Immune response to infection Dr. Sandra Nitsche (Sandra.Nitsche@rub.de ) 20.06.2018 1 Course of acute infection Typical acute infection that is cleared by an adaptive immune reaction 1. invasion of pathogen
Components of the innate immune system
 Components of the innate immune system Before our discussion about innate immunity Differences between innate and adaptive systems: Innate immune system = natural = native -Germline: prepared before exposure
Components of the innate immune system Before our discussion about innate immunity Differences between innate and adaptive systems: Innate immune system = natural = native -Germline: prepared before exposure
The Immune System All animals have innate immunity, a defense active immediately
 The Immune System All animals have innate immunity, a defense active immediately upon infection Vertebrates also have adaptive immunity Figure 43.2 INNATE IMMUNITY (all animals) Recognition of traits shared
The Immune System All animals have innate immunity, a defense active immediately upon infection Vertebrates also have adaptive immunity Figure 43.2 INNATE IMMUNITY (all animals) Recognition of traits shared
Cytokines, adhesion molecules and apoptosis markers. A comprehensive product line for human and veterinary ELISAs
 Cytokines, adhesion molecules and apoptosis markers A comprehensive product line for human and veterinary ELISAs IBL International s cytokine product line... is extremely comprehensive. The assays are
Cytokines, adhesion molecules and apoptosis markers A comprehensive product line for human and veterinary ELISAs IBL International s cytokine product line... is extremely comprehensive. The assays are
محاضرة مناعت مدرس المادة :ا.م. هدى عبدالهادي علي النصراوي Immunity to Infectious Diseases
 محاضرة مناعت مدرس المادة :ا.م. هدى عبدالهادي علي النصراوي Immunity to Infectious Diseases Immunity to infection depends on a combination of innate mechanisms (phagocytosis, complement, etc.) and antigen
محاضرة مناعت مدرس المادة :ا.م. هدى عبدالهادي علي النصراوي Immunity to Infectious Diseases Immunity to infection depends on a combination of innate mechanisms (phagocytosis, complement, etc.) and antigen
The T cell receptor for MHC-associated peptide antigens
 1 The T cell receptor for MHC-associated peptide antigens T lymphocytes have a dual specificity: they recognize polymporphic residues of self MHC molecules, and they also recognize residues of peptide
1 The T cell receptor for MHC-associated peptide antigens T lymphocytes have a dual specificity: they recognize polymporphic residues of self MHC molecules, and they also recognize residues of peptide
Immunology. T-Lymphocytes. 16. Oktober 2014, Ruhr-Universität Bochum Karin Peters,
 Immunology T-Lymphocytes 16. Oktober 2014, Ruhr-Universität Bochum Karin Peters, karin.peters@rub.de The role of T-effector cells in the immune response against microbes cellular immunity humoral immunity
Immunology T-Lymphocytes 16. Oktober 2014, Ruhr-Universität Bochum Karin Peters, karin.peters@rub.de The role of T-effector cells in the immune response against microbes cellular immunity humoral immunity
生命科学基础 (21)- 动物的免疫器官. The Immune System. KE, Yuehai 柯越海. Zhejiang University, School of Basic Medical Sciences (BMS-ZJU) 浙江大学基础医学院
- 动物的免疫器官. The Immune System. KE, Yuehai 柯越海. Zhejiang University, School of Basic Medical Sciences (BMS-ZJU) 浙江大学基础医学院") 生命科学基础 (21)- 动物的免疫器官 The Immune System KE, Yuehai 柯越海 Zhejiang University, School of Basic Medical Sciences (BMS-ZJU) 浙江大学基础医学院 Outlines The Immune System 1. Innate immunity 2. Adaptive immunity 3. Immune
生命科学基础 (21)- 动物的免疫器官 The Immune System KE, Yuehai 柯越海 Zhejiang University, School of Basic Medical Sciences (BMS-ZJU) 浙江大学基础医学院 Outlines The Immune System 1. Innate immunity 2. Adaptive immunity 3. Immune