Cystic Fibrosis Diagnosis and Treatment
|
|
|
- Kory Bradley
- 5 years ago
- Views:
Transcription



1 Cystic Fibrosis Diagnosis and Treatment
2 Financial Disclosures Personal financial relationships with commercial interests relevant to medicine, within the past 3 years: NJH site PI for AstraZeneca. As faculty in institutions that are part of the CF Foundation TDN, I have been site PI for Nivalis, Rompex, and PTC Corp. Personal financial support from a non commercial source relevant to medicine, within the past 3 years: I have received grant funding from NHLBI, FDA, Rompex, Nivalis, and Horizon Pharma. Personal relationships with tobacco industry entities within the last 3 years: No relationships to disclose Personal financial support for consulting for Proteostasis, and Abcomm, Inc.
3 Goals and Objectives Review the definitions of classic and non classic Cystic Fibrosis (CF). Understand the genetic and non genetic determinants of CF. Explore the diagnostic tests for CF. Update the understanding of disease pathogenesis and determinants of progression.


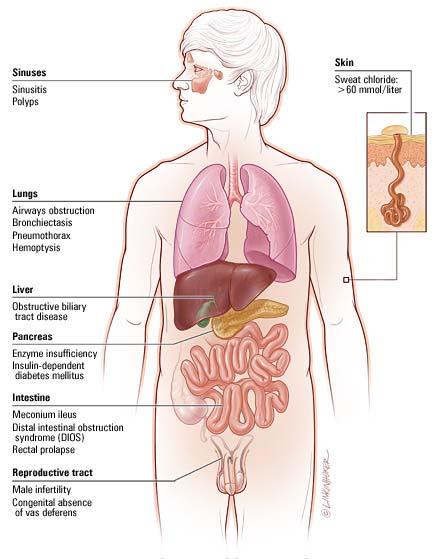
4 Clinical Presentation of CF Pulmonary Symptoms Persistent cough Wheezing Shortness of breath Frequent pulmonary infections Purulent sputum Gastrointestinal Symptoms Pancreatic insufficiency Pancreatitis Malabsorption Bowel obstruction Meconium ileus Rectal prolapse Obstructive cholestasis Other Symptoms Salty sweat or skin Poor growth, weight loss Clubbing Nasal polyps, sinusitis Male infertility
5 Diagnosis by Clinical Triad Elevated Sweat Chloride Pancreatic Insufficiency Chronic Pulmonary Disease Diagnosis by Mutation Analysis F508del Class 1-3 pancreatic insufficiency Class 4-5 pancreatic sufficiency

6 Diagnosis by Sweat Test Sweat Test Pilocarpine iontophoresis > 60 meq/l chloride Inaccurate in first month of life > 30 in newborn and young infant Other causes of elevated sweat chloride Untreated hypothyroidism Glycogen storage disease Addison s disease Ectodermal dysplasia
7 Diagnosis by CFTR Genotyping Greater than 2000 different mutations in CFTR Common mutations in USA: F508del, G542X Mutations in China: I556V, M469V, E527N, F508del Mutations in UAE and Middle Eastern Countries: S4X (7%), S549N, 2043delG, 4016insG, S549R, I1234V* Conventional commercial genotyping Genzyme: 86 mutations Ambry: all coding mutations Many cases are either one or two unknowns at this time
8
9
10 Newborn Screening for CF Mandated nationwide 66% of new diagnoses made in the first year of life Step wise process: Measurement of IRT If elevated, then repeat IRT or CFTR genotyping If still elevated or mutation detected, then sweat chloride test Sweat chloride test = best initial diagnostic test Age 6 mo Age > 6 mo CF is unlikely < 30 mmol/l < 40 mmol/l Intermediate mmol/l mmol/l Consistent with CF 60 mmol/l 60 mmol/l Cystic Fibrosis Foundation, 2015.
11 Disorders affecting the airways with similar characteristics to CF or infections with Pseudomonas Aeruginosa Pan-bronchiolitis Chronic bronchitis Idiopathic bronchiectasis IgA, IgG, IgG subclass deficiencies COPD Patients with tracheostomy tubes
12 Diagnostic Complexity Patients not identified via NBS Normal or equivocal sweat chloride test results 6.8% patients diagnosed 16 years old Clinical presentation Often diagnosed in adolescence or adulthood Genetic mutation less common defect Mild disease respiratory, GI, pancreatic Atypical symptoms infertility, sinusitis Infection with typical CF respiratory pathogens Cystic Fibrosis Foundation, Knowles, et al. N Engl J Med. 2002;347(6):
13 Atypical CF Diagnosis Often occurs in adolescence PMH in retrospect of CF-like symptoms Lung, sinus, liver, male infertility Diagnosis by Mutation Analysis One identifiable mutation plus intron modifier Frequently Class 4-5 pancreatic sufficiency
14 What Has Stayed the Same Since 1996? Sweat testing is still the gold standard test to confirm a CF diagnosis Journal of Pediatrics 1998:132; The CF phenotype remains pretty much the same There are still cases with atypical clinical features and indeterminate laboratory test results that remain hard to classify Nasal PD testing has become standardized but is still used infrequently to confirm or rule out a CF diagnosis


15 F508del is the most common mutation


16 How much is enough? Depends. CFTR Cl channel activity CFTR and Na+ transport CFTR bicarbonate/gsh/other ion transport CF mucociliary transport CF inflammation CF mucus abnormalities CF bacterial susceptibility
17 Lessons from nature in vivo 5T allele reduces CFTR expression 6 11% CBAVD and male infertility 5 10% Partial function CFTR mutations 4.8%
18 Estimates in vitro Chloride transport after gene transfer 6 10% Normalization of sodium transport after gene transfer 100% Correction of inflammation Xx%
19 Nasal Potential Difference Measures the electrical potential across cells and cell membranes Nasal measurements are surrogates for lung airways Baseline PD is negative due to sodium absorption through ENaC PD depolarizes with sodium blockade and hyperpolarizes with chloride secretion
20 Excessive sodium absorption and Pancreatic function CFTR Knowles, et al. Hum. Gene Ther. 1995
21 Chloride secretion and pancreatic function CFTR Knowles, et al. Hum. Gene Ther. 1995
22 100 ** PI (221) Sweat Chloride in Newborns Sweat [Cl ] Mmol/L 50 0 ** PS (24) ** R117H/C (5) F/N (128) Percent CFTR activity * * N/N (184) CFTR * Farrell, 1996 ** Denver, 2005 (F. Accurso; CF-TDN)
23 Sweat Cl vs. Nasal Cl conductance More CFTR Less CFTR From Bishop and Durie Hum Gen. 2005
24 Can we repair the mutant CFTR with drugs alone? Readthrough premature stop codons Correct protein trafficking Potentiate or stimulate chloride conductance Increase synthesis
25 EXAMPLE OF A NON CF NPD Amiloride (to block sodium) Low chloride (to detect open channels) Isoproterenol (to activate CFTR) Basal NPD Ample chloride transport time (min) O ATP
26 EXAMPLE OF A CF NPD mv amiloride Basal NPD chloride free time isoproterenol Absent chloride response
27 Induction of nasal epithelial chloride transport by phenylbutyrate in patient who is homozygous for F508 CF NPD (mv) 0-40 amiloride Low chloride amiloride isoproterenol CF/PBA Non-CF Time (minutes) ATP
28 What Has Changed Since 1996? The number of identified CFTR mutations has increased from 500 in 1996 to in 2017 The number of mutations classified as CF causing has increased from 24 in 1996 to 82 in 2015 Sequencing of the CFTR gene is now widely available and widely used Prenatal and pre conceptual carrier testing recommended by ACOG for routine use in 2001 and now being used with increased frequency.
29 What Has Changed? Newborn screening has gone from 3 states (Colorado, Wisconsin and Wyoming) in 1996 to all 50 states and the District of Columbia in 2011 The number of cases in which the diagnosis is suggested by a NBS test result has gone from 5.7% in 1998 to 42.7% in 2008 to 83% in 2013 Expanded understanding of genotype phenotype correlations Recognition of the CFTR related metabolic syndrome (CRMS) or CFSPID: In 2013, 8.4% of patients entered in the CF registry had a diagnosis of CRMS Resetting of intermediate sweat chloride values in infancy


30 Survival Continues to Improve BUT... Expectancy by Age, In 5 Year Increments CFF Patient Registry, 2015
31 ... We Have More To Do To Advance Quality of Care Intervene Earlier Target CF lung disease before symptoms occur Target genetics to optimize pulmonary and nutritional status Prevent bronchiectasis and loss of lung function Promote good nutrition Develop Comprehensive Treatment Plan (CTP) from Early Age Address comorbidities Add exercise to CTP Address adherence Plan transition to adult care
32 Need to Identify CF and Intervene Early Newborn Screening (NBS) Sweat Cl test Implemented nationwide 2010 Add CFTR genetic analysis If sweat chloride is intermediate Add CFTR physiologic testing If genotype undefined or MVCC Late Diagnosis Normal/equivocal sweat Cl 6.8% diagnosed 16 yo Clinical presentation Mild disease Atypical symptoms Sweat Chloride (mmol/l) CF is unlikely < 30 Intermediate Consistent with CF 60 * mmol/l. MVCC, mutation of varying clinical consequence Cystic Fibrosis Foundation, Farrell PM, White TB, Ren CL, et al. J Ped.181:S4 S15.e11.
33 Early Diagnosis by Newborn Screening May Improve Survival 27,703 patients reported to CFF Registry 4 diagnostic groups Prenatal or neonatal screening (SCREEN) Meconium ileus (MI) Positive family history (FH) Symptoms other than meconium ileus (SYM) Compared to SCREEN, those in MI and SYM groups had increased risk of Shortened survival Pseudomonas aeruginosa FEV 1 < 70% Lai HJ, et al. Am J Epidemiol
34 Early Growth Predicts Childhood Survival Prospective, observational study using CFF Registry data 3,142 children with CF born Tracked weight for age percentile (WAP) and outcomes Results WAP at age 4 positively associated with height for age throughout childhood If WAP at age 4 > 10%, better lung function at ages 6 18 If WAP at age 4 > 50%, fewer acute pulmonary exacerbations at age 18 Also, fewer total days in hospital Lower rates of impaired glucose tolerance or diabetes Yen EH, et al. J Pediatr. 2013
35 Infants Do Not Catch Up to Length Percentile at 1 Year FIRST study growth was normal at birth Growth (weight, length) declined at 2 months Some catch up growth by 12 months Weight increased Length did not fully recover Weight WHO Percentile Length WHO Percentile At birth At 2 months At 12 months Lai HJ, et al. Abstr # NACFC
36 BONUS Study Also Found Risk for Growth 197 infants ( ) At 3 months: 35% at risk for weight 34% at risk for length At 12 months: 9% < 10th percentile for weight 27% < 10th percentile for length Conclusion: Despite improvements in weight gain in the 1 st year of life, stunting persists in the majority BONUS = Baby Observational and Nutrition Study Leung DH, et al. Abstr #575 NACFC
37 Risk Factors for Mortality < Age 18 in CF 3,880 patients Enrolled in ESCF with at least 1 visit annually at ages 3, 4, & 5 years Also linked to CFFPR data Born (5.7%) died < 18 years of age Median age at death, / 3.1 years Significant Risk Factors for death (regardless of FEV 1 at age 6 8 years) Clubbing Crackles Female sex Unknown CFTR genotype Minority race or ethnicity Medicaid insurance (proxy of low socioeconomic status) Pseudomonas aerguinosa on >2 cultures Weight for age < 50 th percentile ESCF, Epidemiologic Study of Cystic FibrosisMcColley et al. Pediatr Pulmonol Published online ahead of print /earlyview


38 It Is Critical to Develop a Comprehensive Treatment Plan from Early Age: 6 Key Components Treat CF lung disease Add exercise! Target genetics to optimize pulmonary and nutritional status Address adherence Address comorbidities Anxiety and depression Diabetes GI problems Plan transition to adult care
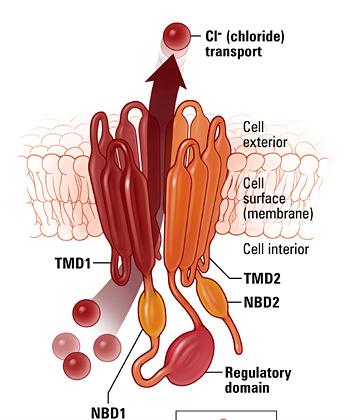
39 CFTR Protein Is Key to Proper Lung Function Cystic fibrosis transmembrane conductance regulator (CFTR) protein: Forms the major transport channel for Cl and HCO 3 Determines mucus viscosity CFTR proteins must be present and functioning well for good lung function Over 1,800 mutations in the CFTR gene Mutated CFTR protein lower or absent for Cl transport Bosch, et al. Eur J Pediatr. 2016;175:1-8.
40

41

42
43 Systems Affected by Mutations in CFTR Protein Lungs Pancreas GI Tract Liver Derichs, et al. Eur Respir Rev. 2013;22:58-65.
44
45


46 Abnormal CFTR Leads to Repeated Infections and Inflammation Abnormal CFTR Reduced airway surface liquid Impaired mucociliary clearance Routine clearance of pathogens from the lungs is compromised Patients at risk of repeat pulmonary infections and prone to exaggerated inflammatory response



47 Downward Spiral of Lung Function from Repeated Infections and Inflammation
48 Changes in Prevalence of Respiratory Pathogens Percentage of Individuals Prevalence of Respiratory Microorganism, Year S. aureus P. aeruginosa MRSA H. influenzae S. maltophilia MDR PA Achromobacter B. cepacia complex CFF Patient Registry, 2015


49 Respiratory Pathogens Difference by Age Prevalence of Respiratory Microorganism, by Age Cohort, 2015 CFF Patient Registry, 2015
50 Airway Microbiome USA CFTDN study 2017 Characterization of microbiota in CF bronchoalveolar lavage fluid (BALF) 146 CF (13 centers) and 45 disease controls If CF < 2yo, Streptococcus predominates; if >6 yo, traditional CF pathogens Sequencing identified a dominant taxon in 24% of culture negative BALF (Streptococcus or Prevatella) Microbial diversity and relative abundance of Streptococcus, Prevotella and Veillonella were inversely associated with airway inflammation Microbial communities were distinct between CF and non CF BALF Zemanick et al, Eur. Respir. J 2017;50:
51 Targeting the Genetic Defect Gene editing Gene therapy RNA repair CFTR modulation mrna modulation Correction of mrna translation to protein Potentiator Inhibitor of proteostasis Corrector Stop codon read through drug Bosch, et al. Eur J Pediatr. 2016;175(1):1 8.
52 CF Mutation Classes Severe disease Mild disease Class Impact on CFTR Mutation Example Potential Therapy I No functional CFTR created G542X W1282X II Processing defect Phe508del Gene therapy RNA correction Read through drug Corrector + Potentiator III Regulation defect G551D Potentiator IV Decreased conductance R117H Potentiator V Reduced synthesis of CFTR kbC T A455E VI Altered channel stability 4326delTC Corrector Potentiator Potentiator Proteostasis inhibitor Bosch, et al. Eur J Pediatr. 2016;175(1):1 8. Derichs, et al. Eur Respir Rev. 2013;22: Boyle, et al. Lancet Resp Med. 2013;1: CFTR2.org
53
54 CFTR Modulators Agent Type Mechanism of Action Phase III Available Ivacaftor Ataluren Lumacaftor VX 661 (Tezacaftor) Potentiator Readthrough Corrector Corrector Increases channel opening Enables the formation of a functioning protein Moves defective CFTR protein to proper place in cell membrane and Improves its function as a chloride channel VX Ivacaftor* *Positive results from two Phase III trials were reported March 28, 2017 NDA to be submitted to FDA in 3rd quarter 2017 Failed Phase III; research program ended Lumacaftor + Ivacaftor Cystic Fibrosis Foundation, Bosch, et al. Eur J Pediatr. 2016;175(1):1 8.
"Management and Treatment of Patients with Cystic fibrosis (CF)
") "Management and Treatment of Patients with Cystic fibrosis (CF) Dr. Malena Cohen-Cymberknoh Pediatric Pulmonology and CF Center Hadassah Hebrew-University Medical Center Jerusalem, Israel Afula, March
"Management and Treatment of Patients with Cystic fibrosis (CF) Dr. Malena Cohen-Cymberknoh Pediatric Pulmonology and CF Center Hadassah Hebrew-University Medical Center Jerusalem, Israel Afula, March
Transformational Treatments. PRESTON W. CAMPBELL, III, M.D. Executive Vice President for Medical Affairs
 Transformational Treatments PRESTON W. CAMPBELL, III, M.D. Executive Vice President for Medical Affairs Symptom-based CF Therapies 45 Median Predicted Survival Age of US Patients with Cystic Fibrosis 41
Transformational Treatments PRESTON W. CAMPBELL, III, M.D. Executive Vice President for Medical Affairs Symptom-based CF Therapies 45 Median Predicted Survival Age of US Patients with Cystic Fibrosis 41
Pediatrics Grand Rounds 13 November University of Texas Health Science Center at San Antonio. Learning Objectives
 Nationwide Newborn Screening for Cystic Fibrosis: Finally Creating an Opportunity for All Patients to Have Better Outcomes Philip M Farrell, MD, PhD* University of Wisconsin-Madison *No disclosures other
Nationwide Newborn Screening for Cystic Fibrosis: Finally Creating an Opportunity for All Patients to Have Better Outcomes Philip M Farrell, MD, PhD* University of Wisconsin-Madison *No disclosures other
Cystic Fibrosis Foundation Patient Registry 2013
 5/9/2015 Targeting CFTR to Treat Cystic Fibrosis: Small Molecule Therapy Mary Ellen Kleinhenz, MD Director, UCSF Adult Cystic Fibrosis Program Professor of Medicine UCSF Division of Pulmonary, Critical
5/9/2015 Targeting CFTR to Treat Cystic Fibrosis: Small Molecule Therapy Mary Ellen Kleinhenz, MD Director, UCSF Adult Cystic Fibrosis Program Professor of Medicine UCSF Division of Pulmonary, Critical
Pediatrics Grand Rounds 18 Sept University of Texas Health Science Center. + Disclosure. + Learning Objectives.
 Disclosure Dr Donna Willey Courand receives research support from Cystic Fibrosis Therapeutics The Cystic Fibrosis Foundation Children with Special Health Care Needs Cystic Fibrosis 05: Improving Survival
Disclosure Dr Donna Willey Courand receives research support from Cystic Fibrosis Therapeutics The Cystic Fibrosis Foundation Children with Special Health Care Needs Cystic Fibrosis 05: Improving Survival
The Future of CF Therapy
 The Future of CF Therapy Peter J. Mogayzel, Jr., M.D., Ph.D. Eudowood Division of Pediatric Respiratory Sciences The Johns Hopkins School of Medicine Overview The Future of CF Therapy Personalized therapy
The Future of CF Therapy Peter J. Mogayzel, Jr., M.D., Ph.D. Eudowood Division of Pediatric Respiratory Sciences The Johns Hopkins School of Medicine Overview The Future of CF Therapy Personalized therapy
THE ROLE OF CFTR MUTATIONS IN CAUSING CYSTIC FIBROSIS (CF)
") THE ROLE OF CFTR MUTATIONS IN CAUSING CYSTIC FIBROSIS (CF) Vertex Pharmaceuticals Incorporated, 50 Northern Avenue, Boston, MA 02210. Vertex and the Vertex triangle logo are registered trademarks for Vertex
THE ROLE OF CFTR MUTATIONS IN CAUSING CYSTIC FIBROSIS (CF) Vertex Pharmaceuticals Incorporated, 50 Northern Avenue, Boston, MA 02210. Vertex and the Vertex triangle logo are registered trademarks for Vertex
CYSTIC FIBROSIS OBJECTIVES NO CONFLICT OF INTEREST TO DISCLOSE
 CYSTIC FIBROSIS Madhu Pendurthi MD MPH Staff Physician, Mercy Hospital Springfield, MO NO CONFLICT OF INTEREST TO DISCLOSE OBJECTIVES Epidemiology of Cystic Fibrosis (CF) Genetic basis and pathophysiology
CYSTIC FIBROSIS Madhu Pendurthi MD MPH Staff Physician, Mercy Hospital Springfield, MO NO CONFLICT OF INTEREST TO DISCLOSE OBJECTIVES Epidemiology of Cystic Fibrosis (CF) Genetic basis and pathophysiology
Enabling CF Therapeutic Development
 Enabling CF Therapeutic Development PRESTON W. CAMPBELL, III, M.D. Executive Vice President for Medical Affairs No Disclosures Cystic Fibrosis In 1955 In 1955 most children with CF did not live long enough
Enabling CF Therapeutic Development PRESTON W. CAMPBELL, III, M.D. Executive Vice President for Medical Affairs No Disclosures Cystic Fibrosis In 1955 In 1955 most children with CF did not live long enough
You Can Observe a Lot By Just Watching. Wayne J. Morgan, MD, CM
 You Can Observe a Lot By Just Watching Wayne J. Morgan, MD, CM Disclosures Genentech Epidemiological Study of Cystic Fibrosis, Scientific Advisory Group CF Foundation Data Safety Monitoring Board Registry/Comparative
You Can Observe a Lot By Just Watching Wayne J. Morgan, MD, CM Disclosures Genentech Epidemiological Study of Cystic Fibrosis, Scientific Advisory Group CF Foundation Data Safety Monitoring Board Registry/Comparative
Evaluation of Patients with Diffuse Bronchiectasis
 Evaluation of Patients with Diffuse Bronchiectasis Dr. Patricia Eshaghian, MD Assistant Clinical Professor of Medicine Director, UCLA Adult Cystic Fibrosis Affiliate Program UCLA Division of Pulmonary
Evaluation of Patients with Diffuse Bronchiectasis Dr. Patricia Eshaghian, MD Assistant Clinical Professor of Medicine Director, UCLA Adult Cystic Fibrosis Affiliate Program UCLA Division of Pulmonary
Medical Policy An independent licensee of the Blue Cross Blue Shield Association
 Cystic Fibrosis Transmembrane Page 1 of 11 Medical Policy An independent licensee of the Blue Cross Blue Shield Association Title: Cystic Fibrosis Transmembrane Conductance Regulator (CFTR) Prime Therapeutics
Cystic Fibrosis Transmembrane Page 1 of 11 Medical Policy An independent licensee of the Blue Cross Blue Shield Association Title: Cystic Fibrosis Transmembrane Conductance Regulator (CFTR) Prime Therapeutics
What is Cystic Fibrosis? CYSTIC FIBROSIS. Genetics of CF
 What is Cystic Fibrosis? CYSTIC FIBROSIS Lynne M. Quittell, M.D. Director, CF Center Columbia University Chronic, progressive and life limiting autosomal recessive genetic disease characterized by chronic
What is Cystic Fibrosis? CYSTIC FIBROSIS Lynne M. Quittell, M.D. Director, CF Center Columbia University Chronic, progressive and life limiting autosomal recessive genetic disease characterized by chronic
Medical Policy An independent licensee of the Blue Cross Blue Shield Association
 Cystic Fibrosis Transmembrane Page 1 of 13 Medical Policy An independent licensee of the Blue Cross Blue Shield Association Title: Cystic Fibrosis Transmembrane Conductance Regulator (CFTR) Prime Therapeutics
Cystic Fibrosis Transmembrane Page 1 of 13 Medical Policy An independent licensee of the Blue Cross Blue Shield Association Title: Cystic Fibrosis Transmembrane Conductance Regulator (CFTR) Prime Therapeutics
A Genetic Approach to the Treatment of Cystic Fibrosis
 A Genetic Approach to the Treatment of Cystic Fibrosis Peter Mueller, PhD Chief Scientific Officer and Executive Vice President Global Research and Development Vertex Pharmaceuticals, Incorporated March
A Genetic Approach to the Treatment of Cystic Fibrosis Peter Mueller, PhD Chief Scientific Officer and Executive Vice President Global Research and Development Vertex Pharmaceuticals, Incorporated March
TIMELINESS IN NEWBORN SCREENING: CONSIDERATIONS FOR CYSTIC FIBROSIS
 TIMELINESS IN NEWBORN SCREENING: CONSIDERATIONS FOR CYSTIC FIBROSIS Susanna A. McColley, MD Northwestern University Feinberg School of Medicine Ann & Robert H. Lurie Children s Hospital of Chicago Stanley
TIMELINESS IN NEWBORN SCREENING: CONSIDERATIONS FOR CYSTIC FIBROSIS Susanna A. McColley, MD Northwestern University Feinberg School of Medicine Ann & Robert H. Lurie Children s Hospital of Chicago Stanley
Cystic Fibrosis. Jennifer McDaniel, BS, RRT-NPS
 Cystic Fibrosis Jennifer McDaniel, BS, RRT-NPS Overview Cystic fibrosis is the most common fatal, inherited disease in the U. S. CF results from a defective autosomal recessive gene One copy of gene =
Cystic Fibrosis Jennifer McDaniel, BS, RRT-NPS Overview Cystic fibrosis is the most common fatal, inherited disease in the U. S. CF results from a defective autosomal recessive gene One copy of gene =
A review of Cystic Fibrosis
 A review of Cystic Fibrosis Jennifer Landry md F.R.C.P.(C) Pulmonary & Critical Care Medicine McGill University Health Center Cystic Fibrosis One of the most common lethal inherited AR disorders in the
A review of Cystic Fibrosis Jennifer Landry md F.R.C.P.(C) Pulmonary & Critical Care Medicine McGill University Health Center Cystic Fibrosis One of the most common lethal inherited AR disorders in the
Cystic fibrosis: hitting the target
 Cystic fibrosis: hitting the target Heartland Collaborative Annual Meeting Friday, October 5, 2012 Thomas Ferkol MD 1938 1953 Cystic fibrosis: a historical timeline Cystic fibrosis (CF) of the pancreas
Cystic fibrosis: hitting the target Heartland Collaborative Annual Meeting Friday, October 5, 2012 Thomas Ferkol MD 1938 1953 Cystic fibrosis: a historical timeline Cystic fibrosis (CF) of the pancreas
Cystic Fibrosis the future
 Cystic Fibrosis the future Pathophysiologic cascade Abnormal Gene Abnormal CFTR Therapy Gene replacement Protein replacement Gene read through therapy Abnormal sodium chloride & water movement through
Cystic Fibrosis the future Pathophysiologic cascade Abnormal Gene Abnormal CFTR Therapy Gene replacement Protein replacement Gene read through therapy Abnormal sodium chloride & water movement through
Disclosures. Advances in the Management of Cystic Fibrosis: A Closer Look at the Roles of CFTR Modulation Therapy 10/28/2016
 Advances in the Management of Cystic Fibrosis: A Closer Look at the Roles of CFTR Modulation Therapy Susanna A McColley, MD Associate Chief Research Officer Stanley Manne Children s Research Institute
Advances in the Management of Cystic Fibrosis: A Closer Look at the Roles of CFTR Modulation Therapy Susanna A McColley, MD Associate Chief Research Officer Stanley Manne Children s Research Institute
Cystic Fibrosis. Cystic Fibrosis. Cystic Fibrosis 5/01/2011 CYSTIC FIBROSIS OF THE PANCREAS AND ITS RELATION TO CELIAC DISEASE. D ANDERSEN.
 1938 OF THE PANCREAS AND ITS RELATION TO CELIAC DISEASE. D ANDERSEN. American Journal Diseases Children. : The beginning May 1938: 49 cases 25 20 15 Nos of cases 10 5 0 0.5 1 1.5 2 2.5 3 3.5 4 4.5 5 Age
1938 OF THE PANCREAS AND ITS RELATION TO CELIAC DISEASE. D ANDERSEN. American Journal Diseases Children. : The beginning May 1938: 49 cases 25 20 15 Nos of cases 10 5 0 0.5 1 1.5 2 2.5 3 3.5 4 4.5 5 Age
A Quick Guide to the. I507del. Mutation CFTR SCIENCE
 A Quick Guide to the I507del Mutation CFTR SCIENCE 2016 Vertex Pharmaceuticals Incorporated VXR-HQ-02-00045a(1) 03/2016 Loss of CFTR activity is the underlying cause of cystic fibrosis (CF) 1 Spectrum
A Quick Guide to the I507del Mutation CFTR SCIENCE 2016 Vertex Pharmaceuticals Incorporated VXR-HQ-02-00045a(1) 03/2016 Loss of CFTR activity is the underlying cause of cystic fibrosis (CF) 1 Spectrum
Cystic Fibrosis: Progress in Treatment Management. Patrick A. Flume, M.D. Medical University of South Carolina
 Cystic Fibrosis: Progress in Treatment Management Patrick A. Flume, M.D. Medical University of South Carolina Disclosures Grant support Mpex Pharmaceuticals, Inc Gilead Sciences, Inc Bayer Healthcare AG
Cystic Fibrosis: Progress in Treatment Management Patrick A. Flume, M.D. Medical University of South Carolina Disclosures Grant support Mpex Pharmaceuticals, Inc Gilead Sciences, Inc Bayer Healthcare AG
Cystic Fibrosis a) Diagnostic Dilemmas b) The New Problems. Dr Mark Rosenthal Royal Brompton Hospital London
 Diagnostic Dilemmas b) The New Problems. Dr Mark Rosenthal Royal Brompton Hospital London") Cystic Fibrosis a) Diagnostic Dilemmas b) The New Problems Dr Mark Rosenthal Royal Brompton Hospital London So what exactly is CF? A genetic diagnosis An electrolyte diagnosis An electrical diagnosis A
Cystic Fibrosis a) Diagnostic Dilemmas b) The New Problems Dr Mark Rosenthal Royal Brompton Hospital London So what exactly is CF? A genetic diagnosis An electrolyte diagnosis An electrical diagnosis A
Respiratory Pharmacology: Treatment of Cystic Fibrosis
 Respiratory Pharmacology: Treatment of Cystic Fibrosis Dr. Tillie-Louise Hackett Department of Anesthesiology, Pharmacology and Therapeutics University of British Columbia Associate Head, Centre of Heart
Respiratory Pharmacology: Treatment of Cystic Fibrosis Dr. Tillie-Louise Hackett Department of Anesthesiology, Pharmacology and Therapeutics University of British Columbia Associate Head, Centre of Heart
Pharmacogenomics in Rare Diseases: Development Strategy for Ivacaftor as a Therapy for Cystic Fibrosis
 Pharmacogenomics in Rare Diseases: Development Strategy for Ivacaftor as a Therapy for Cystic Fibrosis Federico Goodsaid Vice President Strategic Regulatory Intelligence Vertex Pharmaceuticals Is there
Pharmacogenomics in Rare Diseases: Development Strategy for Ivacaftor as a Therapy for Cystic Fibrosis Federico Goodsaid Vice President Strategic Regulatory Intelligence Vertex Pharmaceuticals Is there
North American Cystic Fibrosis Conference 27 October Noreen R Henig, MD Chief Development Officer ProQR Therapeutics
 Proof of Concept Study to Demonstrate the Effects of QR-010 on Nasal Potential Difference in Subjects With Cystic Fibrosis with the F508del CFTR Mutation Noreen R Henig, MD Chief Development Officer ProQR
Proof of Concept Study to Demonstrate the Effects of QR-010 on Nasal Potential Difference in Subjects With Cystic Fibrosis with the F508del CFTR Mutation Noreen R Henig, MD Chief Development Officer ProQR
Goals Basic defect Pathophysiology Clinical i l signs and symptoms Therapy
 CYSTIC FIBROSIS Lynne M. Quittell, M.D. Director, CF Center Columbia University Goals Basic defect Pathophysiology Clinical i l signs and symptoms Therapy What is Cystic Fibrosis? Chronic, progressive
CYSTIC FIBROSIS Lynne M. Quittell, M.D. Director, CF Center Columbia University Goals Basic defect Pathophysiology Clinical i l signs and symptoms Therapy What is Cystic Fibrosis? Chronic, progressive
Case Study What is the Relationship Between the Cell Membrane and Cystic Fibrosis?
 Names: Date: Case Study What is the Relationship Between the Cell Membrane and Cystic Fibrosis? Dr. Weyland examined a six month old infant that had been admitted to University Hospital earlier in the
Names: Date: Case Study What is the Relationship Between the Cell Membrane and Cystic Fibrosis? Dr. Weyland examined a six month old infant that had been admitted to University Hospital earlier in the
Cystic Fibrosis as it relates to the neonate MARIANNE MUHLEBACH, MD PROFESSOR, DEPT. PEDIATRICS UNC CHAPEL HILL
 Cystic Fibrosis as it relates to the neonate MARIANNE MUHLEBACH, MD PROFESSOR, DEPT. PEDIATRICS UNC CHAPEL HILL Objectives: At the end of the presentation the listeners will Be able to describe neonatal
Cystic Fibrosis as it relates to the neonate MARIANNE MUHLEBACH, MD PROFESSOR, DEPT. PEDIATRICS UNC CHAPEL HILL Objectives: At the end of the presentation the listeners will Be able to describe neonatal
Characterizing aggressiveness and predicting future progression of CF lung disease
 Journal of Cystic Fibrosis Volume 8 Suppl 1 (2009) S15 S19 www.elsevier.com/locate/jcf Characterizing aggressiveness and predicting future progression of CF lung disease Michael W. Konstan a, *, Jeffrey
Journal of Cystic Fibrosis Volume 8 Suppl 1 (2009) S15 S19 www.elsevier.com/locate/jcf Characterizing aggressiveness and predicting future progression of CF lung disease Michael W. Konstan a, *, Jeffrey
THE ROLE OF CFTR MUTATIONS IN CAUSING CYSTIC FIBROSIS (CF)
") THE ROLE OF CFTR MUTATIONS IN CAUSING CYSTIC FIBROSIS (CF) Vertex Pharmaceuticals Incorporated, 50 Northern Avenue, Boston, MA 02210. Vertex and the Vertex triangle logo are registered trademarks of Vertex
THE ROLE OF CFTR MUTATIONS IN CAUSING CYSTIC FIBROSIS (CF) Vertex Pharmaceuticals Incorporated, 50 Northern Avenue, Boston, MA 02210. Vertex and the Vertex triangle logo are registered trademarks of Vertex
A Case of Cystic Fibrosis
 Name(s) Date A Case of Cystic Fibrosis Dr. Weyland examined a six month old infant that had been admitted to University Hospital earlier in the day. The baby's parents had brought young Zoey to the emergency
Name(s) Date A Case of Cystic Fibrosis Dr. Weyland examined a six month old infant that had been admitted to University Hospital earlier in the day. The baby's parents had brought young Zoey to the emergency
Cystic Fibrosis Care at the University of Florida
 Cystic Fibrosis Care at the University of Florida Objectives To introduce you to the University of Florida CF Center To review center specific data for the UF pediatric CF Center To review current status
Cystic Fibrosis Care at the University of Florida Objectives To introduce you to the University of Florida CF Center To review center specific data for the UF pediatric CF Center To review current status
North American Cystic Fibrosis Conference 27 October Noreen R Henig, MD Chief Development Officer ProQR Therapeutics
 Proof of Concept Study to Demonstrate the Effects of QR-010 on Nasal Potential Difference in Subjects With Cystic Fibrosis with the F508del CFTR Mutation Noreen R Henig, MD Chief Development Officer ProQR
Proof of Concept Study to Demonstrate the Effects of QR-010 on Nasal Potential Difference in Subjects With Cystic Fibrosis with the F508del CFTR Mutation Noreen R Henig, MD Chief Development Officer ProQR
Supplementary appendix
 Supplementary appendix This appendix formed part of the original submission and has been peer reviewed. We post it as supplied by the authors. Supplement to: Moss RB, Flume PA, Elborn JS, et al, on behalf
Supplementary appendix This appendix formed part of the original submission and has been peer reviewed. We post it as supplied by the authors. Supplement to: Moss RB, Flume PA, Elborn JS, et al, on behalf
Atypical cystic fibrosis: from the genetic causes to current and future treatments
 Boston University OpenBU Theses & Dissertations http://open.bu.edu Boston University Theses & Dissertations 2016 Atypical cystic fibrosis: from the genetic causes to current and future treatments Quinn,
Boston University OpenBU Theses & Dissertations http://open.bu.edu Boston University Theses & Dissertations 2016 Atypical cystic fibrosis: from the genetic causes to current and future treatments Quinn,
FACTS ABOUT. Cystic Fibrosis. What Is Cystic Fibrosis. What Are the Signs and Symptoms of CF?
 FACTS ABOUT Cystic Fibrosis What Is Cystic Fibrosis Cystic fibrosis (CF) is a chronic, progressive, and frequently fatal genetic (inherited) disease of the body s mucus glands. CF primarily affects the
FACTS ABOUT Cystic Fibrosis What Is Cystic Fibrosis Cystic fibrosis (CF) is a chronic, progressive, and frequently fatal genetic (inherited) disease of the body s mucus glands. CF primarily affects the
REGISTRY ANNUAL DATA REPORT
 216 PAT I E N T REGISTRY ANNUAL DATA REPORT MISSION OF THE CYSTIC FIBROSIS FOUNDATION The mission of the Cystic Fibrosis Foundation is to cure cystic fibrosis and to provide all people with the disease
216 PAT I E N T REGISTRY ANNUAL DATA REPORT MISSION OF THE CYSTIC FIBROSIS FOUNDATION The mission of the Cystic Fibrosis Foundation is to cure cystic fibrosis and to provide all people with the disease
CF: Understanding the Biology Curing the Disease
 CF: Understanding the Biology Curing the Disease Scott H. Donaldson, MD Associate Professor of Medicine Director, Adult CF Care Center University of North Carolina at Chapel Hill Defining the path Drilling
CF: Understanding the Biology Curing the Disease Scott H. Donaldson, MD Associate Professor of Medicine Director, Adult CF Care Center University of North Carolina at Chapel Hill Defining the path Drilling
Key Points: References: Canadian data from the Canadian Cystic Fibrosis Registry 2015 Annual Report normal
 1 2 3 Cystic fibrosis is a rare life-long genetic disease that affects approximately 4,000 people in Canada and about 70,000 worldwide regardless of race or ethnicity but is more common in Caucasians 1,2
1 2 3 Cystic fibrosis is a rare life-long genetic disease that affects approximately 4,000 people in Canada and about 70,000 worldwide regardless of race or ethnicity but is more common in Caucasians 1,2
Cystic Fibrosis 8/23/2014 GROWTH DEFICIENCY IN CYSTIC FIBROSIS IS
 8/23/214 GROWTH DEFICIENCY IN CYSTIC FIBROSIS IS OBSERVABLE AT BIRTH AND PREDICTIVE OF EARLY PULMONARY FUNCTION by Rebecca Joan Nelson Case Western Reserve University Cleveland, Ohio Thesis Advisor: Rebecca
8/23/214 GROWTH DEFICIENCY IN CYSTIC FIBROSIS IS OBSERVABLE AT BIRTH AND PREDICTIVE OF EARLY PULMONARY FUNCTION by Rebecca Joan Nelson Case Western Reserve University Cleveland, Ohio Thesis Advisor: Rebecca
Briefing Document. FDA Pulmonary - Allergy Drugs Advisory Committee
 FDA Advisory Committee Briefing Materials Page 1 of 157 Briefing Document FDA Pulmonary - Allergy Drugs Advisory Committee Ivacaftor for the Treatment of Cystic Fibrosis in Patients Age 6 Years and Older
FDA Advisory Committee Briefing Materials Page 1 of 157 Briefing Document FDA Pulmonary - Allergy Drugs Advisory Committee Ivacaftor for the Treatment of Cystic Fibrosis in Patients Age 6 Years and Older
Targeted therapies to improve CFTR function in cystic fibrosis
 Brodlie et al. Genome Medicine (2015) 7:101 DOI 10.1186/s13073-015-0223-6 REVIEW Targeted therapies to improve CFTR function in cystic fibrosis Malcolm Brodlie 1*, Iram J. Haq 2, Katie Roberts 2 and J.
Brodlie et al. Genome Medicine (2015) 7:101 DOI 10.1186/s13073-015-0223-6 REVIEW Targeted therapies to improve CFTR function in cystic fibrosis Malcolm Brodlie 1*, Iram J. Haq 2, Katie Roberts 2 and J.
HOW DISEASE ALTERING THERAPY IS CHANGING THE GOALS OF TREATMENT IN CF
 HOW DISEASE ALTERING THERAPY IS CHANGING THE GOALS OF TREATMENT IN CF Peter D. Sly MBBS, MD, FRACP, DSc OUTLINE Goals of CF treatment Drivers of early disease neutrophilic inflammation oxidative stress
HOW DISEASE ALTERING THERAPY IS CHANGING THE GOALS OF TREATMENT IN CF Peter D. Sly MBBS, MD, FRACP, DSc OUTLINE Goals of CF treatment Drivers of early disease neutrophilic inflammation oxidative stress
Disclosures. Learning Objectives. What is Cystic Fibrosis? Background
 39 th National Conference on Pediatric Health Care March 19-22, 2018 CHICAGO Disclosures The Vision and the Journey of Cystic Fibrosis: Newborn Screening to Breakthrough Therapy March 20, 2018 Cynthia
39 th National Conference on Pediatric Health Care March 19-22, 2018 CHICAGO Disclosures The Vision and the Journey of Cystic Fibrosis: Newborn Screening to Breakthrough Therapy March 20, 2018 Cynthia
What is the inheritance pattern (e.g., autosomal, sex-linked, dominant, recessive, etc.)?
?") Module I: Introduction to the disease Give a brief introduction to the disease, considering the following: the symptoms that define the syndrome, the range of phenotypes exhibited by individuals with the
Module I: Introduction to the disease Give a brief introduction to the disease, considering the following: the symptoms that define the syndrome, the range of phenotypes exhibited by individuals with the
Focus on Cystic Fibrosis. Cystic Fibrosis. Cystic Fibrosis
 Focus on (Relates to Chapter 29, Nursing Management: Obstructive Pulmonary Diseases, in the textbook) Copyright 2011, 2007 by Mosby, Inc., an affiliate of Elsevier Inc. Autosomal recessive, multisystem
Focus on (Relates to Chapter 29, Nursing Management: Obstructive Pulmonary Diseases, in the textbook) Copyright 2011, 2007 by Mosby, Inc., an affiliate of Elsevier Inc. Autosomal recessive, multisystem
CYSTIC FIBROSIS Risk Factors Epidemiology Pathogenesis Defective protein synthesis (10%) Abnormal protein folding, processing & trafficking
 Abnormal protein folding, processing & trafficking") CYSTIC FIBROSIS Risk Factors Caucasian Family history of CF Infection Exposure to allergens and tobacco Epidemiology Carrier frequency of 1 in 25 for Caucasians The most common lethal genetic disease affecting
CYSTIC FIBROSIS Risk Factors Caucasian Family history of CF Infection Exposure to allergens and tobacco Epidemiology Carrier frequency of 1 in 25 for Caucasians The most common lethal genetic disease affecting
A Cure for All: Leaving No One Behind. Assuring Effective Therapies for All Patients with Cystic Fibrosis
 A Cure for All: Leaving No One Behind Assuring Effective Therapies for All Patients with Cystic Fibrosis Topics for Today s Presentation Demographics of the CF patient population in the modulator era
A Cure for All: Leaving No One Behind Assuring Effective Therapies for All Patients with Cystic Fibrosis Topics for Today s Presentation Demographics of the CF patient population in the modulator era
Changes in the management of children with Cystic Fibrosis. Caroline Murphy & Deirdre O Donovan CF Nurses
 Changes in the management of children with Cystic Fibrosis Caroline Murphy & Deirdre O Donovan CF Nurses What Is Cystic Fibrosis? Cystic fibrosis (CF) is an inherited chronic disease that primarily affects
Changes in the management of children with Cystic Fibrosis Caroline Murphy & Deirdre O Donovan CF Nurses What Is Cystic Fibrosis? Cystic fibrosis (CF) is an inherited chronic disease that primarily affects
Cost-effectiveness of Ivacaftor (Kalydeco ) for the treatment of cystic fibrosis in patients age 6 years and older who have the G551D mutation
 for the treatment of cystic fibrosis in patients age 6 years and older who have the G551D mutation") Cost-effectiveness of Ivacaftor (Kalydeco ) for the treatment of cystic fibrosis in patients age 6 years and older who have the G551D mutation January 2013 1. A rapid review submission on the drug ivacaftor
Cost-effectiveness of Ivacaftor (Kalydeco ) for the treatment of cystic fibrosis in patients age 6 years and older who have the G551D mutation January 2013 1. A rapid review submission on the drug ivacaftor
PA Update: Oral Cystic Fibrosis Modulators
 Copyright 2012 Oregon State University. All Rights Reserved Drug Use Research & Management Program Oregon State University, 500 Summer Street NE, E35 Salem, Oregon 97301-1079 Phone 503-947-5220 Fax 503-947-1119
Copyright 2012 Oregon State University. All Rights Reserved Drug Use Research & Management Program Oregon State University, 500 Summer Street NE, E35 Salem, Oregon 97301-1079 Phone 503-947-5220 Fax 503-947-1119
Advances in CF therapies and their effect on GI manifestations. Presenter Disclosure Daniel Gelfond, MD Relationship related to this presentation
 Advances in CF therapies and their effect on GI manifestations Daniel Gelfond, MD University of Rochester WNY Pediatric Gastroenterology Presenter Disclosure Daniel Gelfond, MD Relationship related to
Advances in CF therapies and their effect on GI manifestations Daniel Gelfond, MD University of Rochester WNY Pediatric Gastroenterology Presenter Disclosure Daniel Gelfond, MD Relationship related to
IVACAFTOR THE ISRAELI EXPERIENCE ADI DAGAN MD THE ISRAELI CF CENTER SHEBA MEDICAL CENTER, TEL-HASHOMER
 IVACAFTOR THE ISRAELI EXPERIENCE ADI DAGAN MD THE ISRAELI CF CENTER SHEBA MEDICAL CENTER, TEL-HASHOMER February 21, 2014 U.S. Food and Drug Administration Approves KALYDECO (ivacaftor) for Use in Eight
IVACAFTOR THE ISRAELI EXPERIENCE ADI DAGAN MD THE ISRAELI CF CENTER SHEBA MEDICAL CENTER, TEL-HASHOMER February 21, 2014 U.S. Food and Drug Administration Approves KALYDECO (ivacaftor) for Use in Eight
Cystic Fibrosis. Presented by: Chris Belanger & Dylan Medd
 Cystic Fibrosis Presented by: Chris Belanger & Dylan Medd Outline What is Cystic Fibrosis? Signs, Symptoms & Diagnosis Who does it effect? General effects on daily life Managing Cystic Fibrosis Exercise
Cystic Fibrosis Presented by: Chris Belanger & Dylan Medd Outline What is Cystic Fibrosis? Signs, Symptoms & Diagnosis Who does it effect? General effects on daily life Managing Cystic Fibrosis Exercise
Efficacy of NaCl nebulized hypertonic solutions in cystic fibrosis
 Acta Biomed 2014; Vol. 85, Supplement 4: 10-18 Mattioli 1885 Original article Efficacy of NaCl nebulized hypertonic solutions in cystic fibrosis Azienda Ospedaliero Universitaria Policlinico, DAI Scienze
Acta Biomed 2014; Vol. 85, Supplement 4: 10-18 Mattioli 1885 Original article Efficacy of NaCl nebulized hypertonic solutions in cystic fibrosis Azienda Ospedaliero Universitaria Policlinico, DAI Scienze
Diagnostic challenges after NBS for CF
 Diagnostic challenges after NBS for CF A greater degree of complexity than was anticipated Kevin Southern University of Liverpool 2000 2006 2016 Processing a positive result Multi-agency working is key
Diagnostic challenges after NBS for CF A greater degree of complexity than was anticipated Kevin Southern University of Liverpool 2000 2006 2016 Processing a positive result Multi-agency working is key
CF: Information for Case Managers. Cindy Capen MSN, RN Pediatric Pulmonary Division University of Florida
 CF: Information for Case Managers Cindy Capen MSN, RN capencl@peds.ufl.edu Pediatric Pulmonary Division University of Florida About your speaker 25+ years in pulmonary Coordinator for CF Center and CF
CF: Information for Case Managers Cindy Capen MSN, RN capencl@peds.ufl.edu Pediatric Pulmonary Division University of Florida About your speaker 25+ years in pulmonary Coordinator for CF Center and CF
Pharmacy Policy Bulletin
 Pharmacy Policy Bulletin Title: Policy #: Cystic Fibrosis Agents (Kalydeco, Orkambi ) Rx.01.117 Application of pharmacy policy is determined by benefits and contracts. Benefits may vary based on product
Pharmacy Policy Bulletin Title: Policy #: Cystic Fibrosis Agents (Kalydeco, Orkambi ) Rx.01.117 Application of pharmacy policy is determined by benefits and contracts. Benefits may vary based on product
Oral Cystic Fibrosis Modulators
 Oral Cystic Fibrosis Modulators Goals: To ensure appropriate drug use and limit to patient populations in which they have demonstrated to be effective and safe. To monitor for clinical response for appropriate
Oral Cystic Fibrosis Modulators Goals: To ensure appropriate drug use and limit to patient populations in which they have demonstrated to be effective and safe. To monitor for clinical response for appropriate
Kalydeco. Kalydeco (ivacaftor) Description
 Description") Federal Employee Program 1310 G Street, N.W. Washington, D.C. 20005 202.942.1000 Fax 202.942.1125 5.45.03 Subject: Kalydeco Page: 1 of 6 Last Review Date: November 30, 2018 Kalydeco Description Kalydeco
Federal Employee Program 1310 G Street, N.W. Washington, D.C. 20005 202.942.1000 Fax 202.942.1125 5.45.03 Subject: Kalydeco Page: 1 of 6 Last Review Date: November 30, 2018 Kalydeco Description Kalydeco
A Quick Guide to the G A. Mutation CFTR SCIENCE
 A Quick Guide to the 1717-1G A Mutation FR SIENE 2016 Vertex Pharmaceuticals Incorporated VXR-HQ-02-00045a(1) 03/2016 Loss of FR activity is the underlying cause of cystic fibrosis (F) 1 Spectrum of Phenotypes
A Quick Guide to the 1717-1G A Mutation FR SIENE 2016 Vertex Pharmaceuticals Incorporated VXR-HQ-02-00045a(1) 03/2016 Loss of FR activity is the underlying cause of cystic fibrosis (F) 1 Spectrum of Phenotypes
Microbiome and Asthma
 제 12 차천식연구회 COPD 연구회공동심포지엄 Microbiome and Asthma 한양대학교병원호흡기알레르기내과 김상헌 Disclosure 내용 1 Lung Microbiome 2 Lung Microbiome and Asthma 3 Gut Microbiome and Asthma Microbiome and Microbiota human microbiome
제 12 차천식연구회 COPD 연구회공동심포지엄 Microbiome and Asthma 한양대학교병원호흡기알레르기내과 김상헌 Disclosure 내용 1 Lung Microbiome 2 Lung Microbiome and Asthma 3 Gut Microbiome and Asthma Microbiome and Microbiota human microbiome
A Quick Guide to the. CFTRdele2,3. Mutation CFTR SCIENCE
 Quick uide to the FRdele2,3 Mutation FR SIENE 2016 Vertex Pharmaceuticals Incorporated VXR-HQ-02-00045a(1) 03/2016 Loss of FR activity is the underlying cause of cystic fibrosis (F) 1 Spectrum of Phenotypes
Quick uide to the FRdele2,3 Mutation FR SIENE 2016 Vertex Pharmaceuticals Incorporated VXR-HQ-02-00045a(1) 03/2016 Loss of FR activity is the underlying cause of cystic fibrosis (F) 1 Spectrum of Phenotypes
Rhianna Cenci, Sodexo Dietetic Intern
 Rhianna Cenci, Sodexo Dietetic Intern Objectives Overview of CF and Treatments CF Medical Nutrition Therapy CF Case Study Cystic Fibrosis (CF) Overview Inherited chronic disease Produces unusually thick
Rhianna Cenci, Sodexo Dietetic Intern Objectives Overview of CF and Treatments CF Medical Nutrition Therapy CF Case Study Cystic Fibrosis (CF) Overview Inherited chronic disease Produces unusually thick
Cystic Fibrosis. Parkland College. Monica Rahman Parkland College. Recommended Citation
 Parkland College A with Honors Projects Honors Program 2013 Cystic Fibrosis Monica Rahman Parkland College Recommended Citation Rahman, Monica, "Cystic Fibrosis" (2013). A with Honors Projects. 98. http://spark.parkland.edu/ah/98
Parkland College A with Honors Projects Honors Program 2013 Cystic Fibrosis Monica Rahman Parkland College Recommended Citation Rahman, Monica, "Cystic Fibrosis" (2013). A with Honors Projects. 98. http://spark.parkland.edu/ah/98
Diseases of the gastrointestinal system. H Awad Lecture 2: small intestine/ part 2 and appendix
 Diseases of the gastrointestinal system H Awad Lecture 2: small intestine/ part 2 and appendix Malabsorption most important causes of malabsorption: Celiac disease tropical sprue Lactase deficiency Whipple
Diseases of the gastrointestinal system H Awad Lecture 2: small intestine/ part 2 and appendix Malabsorption most important causes of malabsorption: Celiac disease tropical sprue Lactase deficiency Whipple
PULMONARY SURFACTANT, ALPHA 1 ANTITRYPSIN INHIBITOR DEFICIENCY, AND CYSTIC FIBROSIS DR. NABIL BASHIR BIOCHEMISTRY/RESPIRATORY SYSTEM
 PULMONARY SURFACTANT, ALPHA 1 ANTITRYPSIN INHIBITOR DEFICIENCY, AND CYSTIC FIBROSIS DR. NABIL BASHIR BIOCHEMISTRY/RESPIRATORY SYSTEM Pulmonary surfactant Pulmonary surfactant is (phospholipoprotein) complex
PULMONARY SURFACTANT, ALPHA 1 ANTITRYPSIN INHIBITOR DEFICIENCY, AND CYSTIC FIBROSIS DR. NABIL BASHIR BIOCHEMISTRY/RESPIRATORY SYSTEM Pulmonary surfactant Pulmonary surfactant is (phospholipoprotein) complex
THE JOURNAL OF PEDIATRICS
 SUPPLEMENT www.jpeds.com THE JOURNAL OF PEDIATRICS Diagnosis of Cystic Fibrosis: Consensus Guidelines from the Cystic Fibrosis Foundation Philip M. Farrell, MD, PhD 1, Terry B. White, PhD 2, Clement L.
SUPPLEMENT www.jpeds.com THE JOURNAL OF PEDIATRICS Diagnosis of Cystic Fibrosis: Consensus Guidelines from the Cystic Fibrosis Foundation Philip M. Farrell, MD, PhD 1, Terry B. White, PhD 2, Clement L.
We strive to develop innovative solutions that improve and extend the lives of people with cystic fibrosis
 Nivalis Therapeutics, Inc., October 2015 Analyst Breakfast at NACFC We strive to develop innovative solutions that improve and extend the lives of people with cystic fibrosis Disclaimer Regarding Forward
Nivalis Therapeutics, Inc., October 2015 Analyst Breakfast at NACFC We strive to develop innovative solutions that improve and extend the lives of people with cystic fibrosis Disclaimer Regarding Forward
Caregiver burden and quality of life of parents of young children with cystic fibrosis
 Caregiver burden and quality of life of parents of young children with cystic fibrosis Professor Patricia Fitzpatrick 1 S George 1, R Somerville 1, B Linnane 2, C Fitzgerald 1 1 UCD School of Public Health,
Caregiver burden and quality of life of parents of young children with cystic fibrosis Professor Patricia Fitzpatrick 1 S George 1, R Somerville 1, B Linnane 2, C Fitzgerald 1 1 UCD School of Public Health,
Newborn Screening for Cystic Fibrosis in Switzerland - Evaluation after two years
 Newborn Screening for Cystic Fibrosis in Switzerland - Evaluation after two years R. Fingerhut, T. Torresani, S. Gallati, M.H. Schoeni, C. Kuehni, C. Rueeg, M. Baumgartner, J. Barben and the Swiss CF Screening
Newborn Screening for Cystic Fibrosis in Switzerland - Evaluation after two years R. Fingerhut, T. Torresani, S. Gallati, M.H. Schoeni, C. Kuehni, C. Rueeg, M. Baumgartner, J. Barben and the Swiss CF Screening
Recently, the cystic fibrosis (CF) community celebrated the 25th
 community celebrated the 25th") focused review A new era of personalized medicine for cystic fibrosis at last! Bradley S Quon MD MSc FRCPC 1,2, Pearce G Wilcox MD FRCPC 1,2 BS Quon, PG Wilcox. A new era of personalized medicine for cystic
focused review A new era of personalized medicine for cystic fibrosis at last! Bradley S Quon MD MSc FRCPC 1,2, Pearce G Wilcox MD FRCPC 1,2 BS Quon, PG Wilcox. A new era of personalized medicine for cystic
Bronchiectasis Domiciliary treatment. Prof. Adam Hill Royal Infirmary and University of Edinburgh
 Bronchiectasis Domiciliary treatment Prof. Adam Hill Royal Infirmary and University of Edinburgh Plan of talk Background of bronchiectasis Who requires IV antibiotics Domiciliary treatment Results to date.
Bronchiectasis Domiciliary treatment Prof. Adam Hill Royal Infirmary and University of Edinburgh Plan of talk Background of bronchiectasis Who requires IV antibiotics Domiciliary treatment Results to date.
Symdeko. Symdeko (tezacaftor and ivacaftor) Description
 Description") Federal Employee Program 1310 G Street, N.W. Washington, D.C. 20005 202.942.1000 Fax 202.942.1125 5.45.10 Subject: Symdeko Page: 1 of 5 Last Review Date: June 22, 2018 Symdeko Description Symdeko (tezacaftor
Federal Employee Program 1310 G Street, N.W. Washington, D.C. 20005 202.942.1000 Fax 202.942.1125 5.45.10 Subject: Symdeko Page: 1 of 5 Last Review Date: June 22, 2018 Symdeko Description Symdeko (tezacaftor
T he cystic fibrosis transmembrane conductance regulator
 971 RESPIRATORY PHYSIOLOGY Nasal airway ion transport is linked to the cystic fibrosis phenotype in adult patients I Fajac, D Hubert, D Guillemot, I Honoré, T Bienvenu, F Volter, J Dall Ava-Santucci, D
971 RESPIRATORY PHYSIOLOGY Nasal airway ion transport is linked to the cystic fibrosis phenotype in adult patients I Fajac, D Hubert, D Guillemot, I Honoré, T Bienvenu, F Volter, J Dall Ava-Santucci, D
UK Cystic Fibrosis Registry
 UK Cystic Fibrosis Registry Annual Data Report 2017 Scotland UK Cystic Fibrosis Registry 2017 Annual Data Report - Scotland Report prepared by Andrew Lee Statistician Cystic Fibrosis Trust Susan Charman
UK Cystic Fibrosis Registry Annual Data Report 2017 Scotland UK Cystic Fibrosis Registry 2017 Annual Data Report - Scotland Report prepared by Andrew Lee Statistician Cystic Fibrosis Trust Susan Charman
Cystic Fibrosis. Information for Caregivers
 Cystic Fibrosis Information for Caregivers Arkansas Children s Hospital is an accredited Cystic Fibrosis Care Center by the National Cystic Fibrosis Foundation Cystic Fibrosis: Information for Caregivers
Cystic Fibrosis Information for Caregivers Arkansas Children s Hospital is an accredited Cystic Fibrosis Care Center by the National Cystic Fibrosis Foundation Cystic Fibrosis: Information for Caregivers
A Quick Guide to the 621+1G T. Mutation CFTR SCIENCE
 Quick uide to the 621+1 Mutation FR SIENE 2016 Vertex Pharmaceuticals Incorporated VXR-HQ-02-00045a(1) 03/2016 Loss of FR activity is the underlying cause of cystic fibrosis (F) 1 Spectrum of Phenotypes
Quick uide to the 621+1 Mutation FR SIENE 2016 Vertex Pharmaceuticals Incorporated VXR-HQ-02-00045a(1) 03/2016 Loss of FR activity is the underlying cause of cystic fibrosis (F) 1 Spectrum of Phenotypes
Cystic fibrosis (CF) is the most frequent. Ivacaftor treatment in patients with cystic REVIEW. Isabelle Sermet-Gaudelus
 is the most frequent. Ivacaftor treatment in patients with cystic REVIEW. Isabelle Sermet-Gaudelus") Eur Respir Rev 2013; 22: 127, 66 71 DOI: 10.1183/09059180.00008512 CopyrightßERS 2013 REVIEW Ivacaftor treatment in patients with cystic fibrosis and the G551D-CFTR mutation Isabelle Sermet-Gaudelus ABSTRACT:
Eur Respir Rev 2013; 22: 127, 66 71 DOI: 10.1183/09059180.00008512 CopyrightßERS 2013 REVIEW Ivacaftor treatment in patients with cystic fibrosis and the G551D-CFTR mutation Isabelle Sermet-Gaudelus ABSTRACT:
Cystic fibrosis and survival to 40 years: a study of cystic fibrosis transmembrane conductance regulator function
 Eur Respir J 2011; 37: 1076 1082 DOI: 10.1183/09031936.00079010 CopyrightßERS 2011 Cystic fibrosis and survival to 40 years: a study of cystic fibrosis transmembrane conductance regulator function N.J.
Eur Respir J 2011; 37: 1076 1082 DOI: 10.1183/09031936.00079010 CopyrightßERS 2011 Cystic fibrosis and survival to 40 years: a study of cystic fibrosis transmembrane conductance regulator function N.J.
Chapter 3 The Role of Nutrition in CF Care
 Chapter 3 The Role of Nutrition in CF Care S. King, N. Saxby & N. Sander Cystic fibrosis is the most common lethal autosomal recessive genetic condition affecting Caucasians 186,187. Over 3500 Australians
Chapter 3 The Role of Nutrition in CF Care S. King, N. Saxby & N. Sander Cystic fibrosis is the most common lethal autosomal recessive genetic condition affecting Caucasians 186,187. Over 3500 Australians
Cystic Fibrosis Update
 Cystic Fibrosis Update More than Decades of Discovery Dr. Raj Padman Professor of Pediatric TJU Board certified in Pediatrics, Pulmonology, and Sleep Medicine Dupont, Kosciusko Community, Lutheran, Rehabilitation,
Cystic Fibrosis Update More than Decades of Discovery Dr. Raj Padman Professor of Pediatric TJU Board certified in Pediatrics, Pulmonology, and Sleep Medicine Dupont, Kosciusko Community, Lutheran, Rehabilitation,
CLINICAL MEDICAL POLICY
 Policy Name: Policy Number: Approved By: CLINICAL MEDICAL POLICY Genetic Testing for Cystic Fibrosis MP-006-MD-DE Provider Notice Date: 11/1/2016 Original Effective Date: 12/1/2016 Annual Approval Date:
Policy Name: Policy Number: Approved By: CLINICAL MEDICAL POLICY Genetic Testing for Cystic Fibrosis MP-006-MD-DE Provider Notice Date: 11/1/2016 Original Effective Date: 12/1/2016 Annual Approval Date:
**Cystic Fibrosis** Notes :
 **Cystic Fibrosis** Notes : - the slides are included in the lecture. - everything in Italic is just important things from the old lecture. - It s easy lecture, even it seems long.. GOOD LUCK Cystic fibrosis
**Cystic Fibrosis** Notes : - the slides are included in the lecture. - everything in Italic is just important things from the old lecture. - It s easy lecture, even it seems long.. GOOD LUCK Cystic fibrosis
Liver Disease in Cystic Fibrosis
 Liver Disease in Cystic Fibrosis Basic Overview Clinical Aspects Management What Is Cystic Fibrosis? Autosomal recessive disease W-1:3000, H-1:10,000, AA-1:15,000 Mutations of CFTR defective Cl - transport
Liver Disease in Cystic Fibrosis Basic Overview Clinical Aspects Management What Is Cystic Fibrosis? Autosomal recessive disease W-1:3000, H-1:10,000, AA-1:15,000 Mutations of CFTR defective Cl - transport
CYSTIC FIBROSIS (CF): THE CHALLENGE OF EARLY, SYSTEMIC PROGRESSION
: THE CHALLENGE OF EARLY, SYSTEMIC PROGRESSION") CYSTIC FIBROSIS (CF): THE CHALLENGE OF EARLY, SYSTEMIC PROGRESSION A Program for the CF Center Care Team Vertex Pharmaceuticals Incorporated, 50 Northern Avenue, Boston, MA 02210. Vertex and the Vertex
CYSTIC FIBROSIS (CF): THE CHALLENGE OF EARLY, SYSTEMIC PROGRESSION A Program for the CF Center Care Team Vertex Pharmaceuticals Incorporated, 50 Northern Avenue, Boston, MA 02210. Vertex and the Vertex
Application to be an additional provider for existing test on the NHS Directory of Molecular Genetic Testing Additional Provider form
 Application to be an additional provider for existing test on the NHS Directory of Molecular Genetic Testing Additional Provider form Disease: Gene: Cystic Fibrosis (CF) (carrier testing in reproductive
Application to be an additional provider for existing test on the NHS Directory of Molecular Genetic Testing Additional Provider form Disease: Gene: Cystic Fibrosis (CF) (carrier testing in reproductive
THE CANADIAN CYSTIC FIBROSIS REGISTRY
 THE CANADIAN CYSTIC FIBROSIS REGISTRY 15 ANNUAL REPORTCanadian Cystic Fibrosis Registry 15 Annual Report 1 CYSTIC FIBROSIS Cystic fibrosis (CF) is a rare disease affecting almost 4, Canadians or roughly
THE CANADIAN CYSTIC FIBROSIS REGISTRY 15 ANNUAL REPORTCanadian Cystic Fibrosis Registry 15 Annual Report 1 CYSTIC FIBROSIS Cystic fibrosis (CF) is a rare disease affecting almost 4, Canadians or roughly
Evening Case studies, Tuesday April 30, Vijay L. Grey McMaster University
 Evening Case studies, Tuesday April 30, 2013 Vijay L. Grey McMaster University Case 1 Gus Diaz was born to a 28-year-old gravida I mom who had evidence of gestational diabetes. It was managed with attention
Evening Case studies, Tuesday April 30, 2013 Vijay L. Grey McMaster University Case 1 Gus Diaz was born to a 28-year-old gravida I mom who had evidence of gestational diabetes. It was managed with attention
Cystic Fibrosis. Na+ 2Cl - K+ Na+ Na+
 1 Cystic Fibrosis I. Overview of cystic fibrosis Among Caucasians, about one out of twenty people carry the gene for cystic fibrosis (CF), and one of 2,000 to 4,000 people is afflicted with the recessive
1 Cystic Fibrosis I. Overview of cystic fibrosis Among Caucasians, about one out of twenty people carry the gene for cystic fibrosis (CF), and one of 2,000 to 4,000 people is afflicted with the recessive
CYSTIC FIBROSIS IN AUSTRALIA 2008
 CYSTIC FIBROSIS IN AUSTRALIA 2008 11th Annual Report from the Australian Cystic Fibrosis Data Registry Cystic Fibrosis Australia 2010 This work is copyright. Apart from any use as permitted under the Copyright
CYSTIC FIBROSIS IN AUSTRALIA 2008 11th Annual Report from the Australian Cystic Fibrosis Data Registry Cystic Fibrosis Australia 2010 This work is copyright. Apart from any use as permitted under the Copyright
Title: Lack of correlation between pulmonary disease and CFTR dysfunction in cystic fibrosis -- the case of 3791delC
 Author's response to reviews Title: Lack of correlation between pulmonary disease and CFTR dysfunction in cystic fibrosis -- the case of 3791delC Authors: Hara Levy (hlevy@mcw.edu) Carolyn L. Cannon (cannon_c@kids.wustl.edu)
Author's response to reviews Title: Lack of correlation between pulmonary disease and CFTR dysfunction in cystic fibrosis -- the case of 3791delC Authors: Hara Levy (hlevy@mcw.edu) Carolyn L. Cannon (cannon_c@kids.wustl.edu)
Exploring New Advances and Best Practices to Personalize Therapy and Improve Lung Function in Cystic Fibrosis
 CME/CNE/CPE Exploring New Advances and Best Practices to Personalize Therapy and Improve Lung Function in Cystic Fibrosis Course Director Jerry Nick, MD National Jewish Health University of Colorado Denver,
CME/CNE/CPE Exploring New Advances and Best Practices to Personalize Therapy and Improve Lung Function in Cystic Fibrosis Course Director Jerry Nick, MD National Jewish Health University of Colorado Denver,
Caring for a person with cystic fibrosis
 Caring for a person with cystic fibrosis Item Type Article Authors McDonagh, Yvonne;Meagher, Catherine Publisher Green Cross Publishing Journal Nursing in General Practice Download date 01/09/2018 03:18:31
Caring for a person with cystic fibrosis Item Type Article Authors McDonagh, Yvonne;Meagher, Catherine Publisher Green Cross Publishing Journal Nursing in General Practice Download date 01/09/2018 03:18:31
CYSTIC FIBROSIS REGISTRY
 THE CANADIAN CYSTIC FIBROSIS REGISTRY 16 ANNUAL DATA REPORT Canadian Cystic Fibrosis Registry 16 Annual Data Report 1 CYSTIC FIBROSIS Cystic fibrosis (CF) is a rare disease affecting over 4, Canadians
THE CANADIAN CYSTIC FIBROSIS REGISTRY 16 ANNUAL DATA REPORT Canadian Cystic Fibrosis Registry 16 Annual Data Report 1 CYSTIC FIBROSIS Cystic fibrosis (CF) is a rare disease affecting over 4, Canadians
Supplementary appendix
 Supplementary appendix This appendix formed part of the original submission and has been peer reviewed. We post it as supplied by the authors. Supplement to: Wells JM, Farris RF, Gosdin TA, et al. Pulmonary
Supplementary appendix This appendix formed part of the original submission and has been peer reviewed. We post it as supplied by the authors. Supplement to: Wells JM, Farris RF, Gosdin TA, et al. Pulmonary
1. Introduction. Obstructive lung disease remains the leading cause of morbidity and mortality in cystic fibrosis
 1. Introduction Obstructive lung disease remains the leading cause of morbidity and mortality in cystic fibrosis (CF) [1]. With time it has become increasingly clear that CF lung disease is present very
1. Introduction Obstructive lung disease remains the leading cause of morbidity and mortality in cystic fibrosis (CF) [1]. With time it has become increasingly clear that CF lung disease is present very