Molecular Mechanisms of Programmed Necrotic Death Initiated by Intrinsic Death Signals
|
|
|
- Merry Pierce
- 6 years ago
- Views:
Transcription
1 Washington University in St. Louis Washington University Open Scholarship All Theses and Dissertations (ETDs) Spring Molecular Mechanisms of Programmed Necrotic Death Initiated by Intrinsic Death Signals Gary Xiaoshi Wang Washington University in St. Louis Follow this and additional works at: Recommended Citation Wang, Gary Xiaoshi, "Molecular Mechanisms of Programmed Necrotic Death Initiated by Intrinsic Death Signals" (2014). All Theses and Dissertations (ETDs) This Dissertation is brought to you for free and open access by Washington University Open Scholarship. It has been accepted for inclusion in All Theses and Dissertations (ETDs) by an authorized administrator of Washington University Open Scholarship. For more information, please contact
2 WASHINGTON UNIVERSITY IN ST. LOUIS Division of Biology and Biomedical Sciences Molecular Cell Biology Dissertation Examination Committee: Emily H.-Y. Cheng, Chair Kendall J. Blumer, Co-Chair Ron Bose Shin-ichiro Imai Gerald P. Linette Jason C. Mills Zhongsheng You Molecular Mechanisms of Programmed Necrotic Death Initiated by Intrinsic Death Signals by Gary Xiaoshi Wang A dissertation presented to the Graduate School of Arts and Sciences of Washington University in partial fulfillment of the requirements for the degree of Doctor of Philosophy May 2014 Saint Louis, Missouri 1
3 2014, Gary Xiaoshi Wang
4 TABLE OF CONTENTS List of Figures..iv Acknowledgements vi Abstract...ix Chapter One: Introduction Introduction Apoptosis Autophagy Necrosis Significance Figure Legends Figures References 38 Chapter Two: p63 Inhibits Oxidative Stress-Induced Cell Death by Orchestrating Glutathione Metabolism Abstract Introduction Results Discussion Materials and Methods Figure Legends Figures References 89 Chapter Three: DNA Damage Induces Mitochondrial ROS Production to Execute Programmed Necrotic Death..93 ii
5 3.1 Introduction Results Materials and Methods Discussion Figure Legends Figures References..113 Chapter Four: Conclusions, Discussion, and Future Directions Conclusions Discussion and Future Directions Concluding Remarks References..124 Curriculum Vitae. 127 iii
6 LIST OF FIGURES Chapter One Figure 1.1 Morphological features of apoptosis, autophagy, and necrosis 36 Figure 1.2 Hierarchical regulation of mitochondrion-dependent apoptosis by BCL-2 subfamilies 37 Figure 1.3 Schematic depiction of the autophagy pathway and its core molecular machinery in mammalian cells.. 37 Chapter Two Figure 2.1 Np63 abrogates DNA damage-induced oxidative stress and enhances clonogenic survival of transformed p53 -/- Bax -/- Bak -/- mouse embryonic fibroblasts..77 Figure 2.2 p63 dictates the homeostasis of intracellular redox through transcriptional controls of glutathione metabolism genes..78 Figure 2.3 Deficiency of Np63α generates a more oxidized intracellular redox state and sensitizes cells to oxidative stress 79 Figure 2.4 ΔNp63α mitigates anoikis-induced oxidative stress and cooperates with anti-apoptotic BCL-2 to disrupt the luminal clearance of acini in three-dimensional culture of mammary epithelial cells...80 Supplementary Figures Figure S2.1 DNA damage-induced cell death in Bax -/- Bak -/- DKO MEFs does not require RIP Figure S2.2. p53 -/- Bax -/- Bak -/- TKO MEFs are more resistant to DNA damage-induced cell death than Bax -/- Bak -/- DKO MEFs...81 Figure S2.3 p53 -/- Bax -/- Bak -/- TKO MEFs are protected by antioxidants against DNA damageinduced cell death 82 Figure S2.4 ΔNp63α inhibits DNA damage-induced ROS in p53 -/- Bax -/- Bak -/- TKO MEFs.82 Figure S2.5 Loss of endogenous p63 does not protect Bax -/- Bak -/- DKO or p53 -/- Bax -/- Bak -/- TKO MEFs from DNA damage-induced death.83 iv
7 Figure S2.6. A heatmap representation of glutathione metabolism genes differentially regulated by ΔNp63α 84 Figure S2.7. Immunoblot or qrt-pcr analysis of sirna-mediated knockdown.85 Figure S2.8 Knockdown of p63 by sirna and mir-30-based shrna induces ROS in human cancer cell line.86 Figure S2.9 Deficiency of ΔNp63α sensitizes cells to chemotherapeutic agent-induced cell death..87 Figure S2.10. Upregulation of GCLC by ΔNp63α is Nrf2-independent..88 Chapter Three Figure 3.1 Induction of mitochondrial matrix ROS by DNA damage is an early event that precedes cell death and ATP depletion Figure 3.2 ETC complex III mediates DNA double strand break-induced ROS and cell death 109 Figure 3.3 Figure 3.3 ATM-mediated DNA damage response pathway induces ROS, cell death, and ETC complex III activity following double strand breaks.111 v
8 ACKNOWLEDGEMENTS I would like to sincerely thank my mentor and thesis advisor, Dr. Emily Cheng. Over the five-odd years that I spent in her laboratory, first in St. Louis then in New York City, she has always pointed me towards important and novel scientific questions to address, rescued me when I struggled with my project, helped me remember how much I enjoy scientific discovery, and showed faith in me when I lacked it myself. She taught me that there are always ways to improve my techniques, my writing and communications skills, my way of thinking, my thesis project; in short, she showed me by example what it means to constantly pursue perfection. Without a doubt, completing this Ph.D. thesis is the hardest thing I have ever done, and likely one of the hardest things I will ever do. I would not be here without the lessons she has taught me, and the guidance and encouragement she has given me throughout the past five years. Her lessons have changed me for the better; it has made me more capable, confident, and goal-oriented; and it has helped set the trajectory for the rest of my life. On a similar note, I would also like to thank Dr. James Hsieh, for all the time he took to guide me through personal and professional hurdles and insecurities. Ultimately, through their efforts, Dr. Cheng and Dr. Hsieh showed me a better version of myself than the one I had known before I worked with them, and helped me on my way to becoming that person. For that, I will always be deeply grateful. I am also deeply grateful to Drs. Ho-Chou Tu, Hyungjin Kim, Decheng Ren, David Chen, Satoru Sasagawa, Shugaku Takeda, and Han Liu, as well as Todd Westergard and Hsiu-Fang Chen. They were my introduction to Dr. Cheng and Dr. Hsieh s labs, and it was them as much as my mentor and my rotation project that sealed my decision to carry out my thesis work in Dr. Cheng s lab. Hsiu-Fang I especially thank for taking care of my car in St. Louis while I was in New York City trying to make science work; and for welcoming me to her house and cooking a truly wonderful meal for me when I returned to St. Louis for my final thesis update. I also wish to thank Mr. Hoson Chao and Ms. Smrutiben Mehta, my wonderful laboratory managers in New York. vi
9 I particularly wish to thank Drs. Ho-Chou Tu, Shugaku Takeda, Han Liu, Yiyu Dong, Anders Jacobsen, and Gregory Bean, and Mr. Yogesh Ganesan for their incredible enthusiasm and willingness to help me with many critical experiments during my thesis research. I wish to thank the Medical Scientist Training Program staff at the Washington University in St. Louis School of Medicine for all of their wonderful support to me through the years, and for their patience and guidance. I also would like to acknowledge the financial support I received for my M.D. and Ph.D. training, and for my thesis work, from the MSTP Training Grant, the Washington University in St. Louis School of Medicine, and the National Institutes of Health. My work was also supported by grants to Dr. Cheng from the NIH and the American Cancer Society. Finally, I want to thank my parents, Chenghong and Lu Wang, for all their patience, love, care, and support. They taught me the meaning of hard work and perseverance through their own example; nothing ever seems hard to me when I think about how much they went through to get where they are today. No amount of words will ever do justice to them, so I will not even try; it suffices to say that I could not have asked for more caring, loving parents. vii
10 DEDICATION In loving memory of my maternal grandfather, who passed in the spring of Listening to the music coming from your piano is one of my earliest memories. Our peaceful afternoons of chess and tea, hours happily filled with the clicking of wooden pieces and your frequent warnings of my impending checkmate, are among my best. Lao ye, gan bei! viii
11 ABSTRACT OF THE DISSERTATION Molecular Mechanisms of Programmed Necrotic Death Initiated by Intrinsic Death Signals by Gary Xiaoshi Wang Doctor of Philosophy in Biology & Biomedical Sciences Molecular Cell Biology Washington University in St. Louis, 2014 Professors Emily H.-Y. Cheng, Chair, and Kendall J. Blumer, Co-Chair Proper regulation of cell death is critical for tissue development and homeostasis, with deregulation implicated in many illnesses. Cell death is morphologically categorised as apoptosis, autophagy, and necrosis, with apoptosis being the best studied. BCL-2 family members are critical apoptosis regulators. Death signals induce upstream activator BH3-only molecules to activate the essential mitochondrion apoptosis effectors BAX and BAK to execute death. Yet, combined loss of Bax and Bak causes developmental and homeostatic defects in restricted tissues, indicating the existence of additional death program(s). By using Bax -/- Bak -/- doubleknockout (DKO) mouse embryonic fibroblasts (MEFs) to study non-apoptotic death, we discovered DNA double strand breaks (DSBs) activate a genetic program that upregulates reactive oxygen species (ROS) and the lysosomal protease cathepsin to execute a form of programmed necrotic death (PND). This refutes the long-held view that necrosis is a passive reponse to unmanageable physico-chemical stress. However, the PND execution pathways triggered by death signals and the determinants of death versus long-term survival are unclear. Here, I demonstrated a critical role for the transcription factor ΔNp63α in determining long-term survival in the face of oxidative stress, a central necrotic mediator. ΔNp63α protected DKO MEFs against PND and enhanced long-term survival via a novel ability to control intracellular redox homeostasis by regulating glutathione metabolism. ΔNp63α overexpression protected from oxidative stress induced by oxidants, DNA damage, and anoikis, while deficiency of ΔNp63α increased oxidative stress and sensitized to oxidant-induced death. I further found that ix
12 long-term survival of anchorage-dependent cells following loss of matrix attachment was enhanced by combined inhibition of apoptosis and oxidative stress by BCL-2 and ΔNp63α, respectively. Furthermore, I showed that in DKO MEFs, DSBs triggered an ATM-mediated DNA damage response that increased ROS production by upregulating the activity of complex III of the electron transport chain. My work concluded a key role for oxidative stress in the loss of long-term survival triggered by intrinsic death signals and revealed an important role for ΔNp63α in regulating redox homeostasis. These findings laid the groundwork for future studies on programmed necrotic death initiated by intrinsic death signals. x
13 -CHAPTER ONE- Introduction 1
14 1.1 Introduction For metazoans, the ability of individual cells to respond correctly to pro-survival and prodeath signals is central to embryonic development and tissue homeostasis 1. Accordingly, dysregulated cell death contributes to the pathogenesis of many human diseases 2. Insufficient cell death can lead to cancer and autoimmunity while execessive cell death is implicated in neurodegeneration and ischemic injury 2. Although cell death has been recognized since the 1800s to be a normal part of embryonic development, it was not until the latter half of the 20 th century that fundamental insights on the regulation of cell death began to emerge 3. Not surprisingly, these insights have opened many avenues for therapeutic intervention in a broad range of pathological settings 4. Cell death is categorized into three forms based on morphological features (Fig. 1.1). The first form, apoptosis, has been the most extensively studied and is characterized by prominent nuclear condensation and fragmentation, preservation of plasma and organelle membrane integrity, and formation of apoptotic bodies 5. It is an evolutionarily conserved, highly regulated, genetically encoded process that has long been synonymous with programmed cell death 5. The second form is autophagic cell death, characterized by the formation of double membrane vesicles, i.e. autophagosomes. Autophagy is mainly a prosurvival mechanism to help cells during nutrient deprivation, yet exessive autophagy certainly compromises the survival of cells and leads to cellular demise 5. The third form is necrosis, which features cytoplasmic translucency, irregular chromatin condensation, organellar and cellular swelling, loss of plasma and organelle membrane integrity, and spillage of intracellular contents into the extracellular space 5, 6. Necrosis was traditionally thought to be an unregulated, accidental outcome of exposure to unmanageable physico-chemical stress 7. The first clue to indicate otherwise came in 1988 when it was reported that tumor necrosis factor (TNF) can induce either apoptosis of necrosis, depending on the cell type 8. Accumulating evidence since then, both from our lab and many others, have established the existence of molecular mechanisms that control necrotic death 9, 10. This recent emergence of programmed necrotic death represents a true paradigm shift in a field that has long equated programmed cell death with apoptosis. It is important to note that, in the 2
15 absence of phagocytic clearance, the morphological endpoint of all types of cell death will resemble necrosis due to the eventual loss of plasma membrane integrity 6. This consequence of the gradual degradation of biological materials over time is referred to as secondary necrosis and is distinct from primary necrosis, which describes a cell death mechanism 6. In both apoptosis and programmed necrosis, diverse signals ultimately converge on the molecular mechanisms that integrate and interpret these cues to make critical life or death decisions for the cell 1, 10. In the case of apoptosis, execution of cell death is mediated by caspases, a family of cysteine proteases activated by extrinsic or intrinsic death signals 1. Caspase activation results in the stereotypical biochemical and morphological hallmarks of apoptosis; therefore, caspase-dependent cell death is synonymous to apoptosis 5. Although the pathways that initiate and execute programmed necrosis have not been as extensively elucidated as the ones that control apoptosis, clear themes have emerged. Similar to apoptosis, both death receptor ligation and intrinsic death signals can trigger programmed necrosis 10. However, unlike apoptosis, the execution of programmed necrosis does not rely on caspases 10. Rather, evidence suggest it involves a complex interplay of reactive oxygen species (ROS), organelle dysfunction, and dysregulated intracellular calcium homeostasis 10. The mitochondria and lysosomes have emerged as the target organelles within this network of effectors 7. The aim of this thesis is to further our understanding of the molecular mechanisms that govern programmed necrotic death in response to intrinsic death signals. 1.2 Apoptosis Introduction Though there are reports going as far back as the 1800s describing cells that appeared to die as a normal part of development, cell death was not established to be a normal biological process until Alfred Glucksmann s comprehensive 1951 review 3. In that article, he summarized and categorized instances of cell death observed during vertebrate development and stated that these cells shared a set of morphological characteristics that we now recognize as apoptotic 3. Then in 1965, Richard Lockshin and Carroll Williams, in a report on the death of larval muscles of 3
16 metamorphosing insects, introduced the term programmed cell death to describe this form of death that appeared to be triggered in otherwise healthy cells by a specific series of physiological events versus death elicited by toxic or inhospital environments 11. In that same year, the pathologist John Kerr proposed the term shrinkage necrosis to describe the process that resulted in the appearance of small, condensed, acidophilic, enucleated cytoplasm in the liver under pathological conditions 12. Then, in a landmark 1972 study, Kerr and colleagues changed that term to apoptosis to clearly distinguish it from necrosis and to highlight its presumed importance in the regulation of tissue cell number under both physiological and pathological conditions 13. In that study, apoptosis was described to occur in two discrete, sequential stages. The first stage is marked by condensation of the nucleus and cytoplasm, nuclear fragmentation, and breaking up of the cell into numerous membrane-bound, well-preserved cell remnants i.e. apoptotic bodies. In the second stage, apoptotic cells are ingested by neighboring cells and degraded within phagosomes. Histologically, apoptosis was observed to result in minimal tissue disruption with no evidence of inflammation. Based on previous findings in developmental biology and the histological and ultrastructural features of apoptosis, the authors believed apoptosis to be the regulated process responsible for counterbalancing cell proliferation to ensure normal development and tissue homeostasis. Genetic control of programmed cell death in Caenorhabditis elegans Based on the observation that a wide variety of simuli were able to initiate the stereotyped events of apoptosis, Kerr and colleagues suggested that apoptosis [may depend] on the expression of part of the genome, which is normally repressed in viable cells 13. A series of studies performed using the nematode Caenorhabditis elegans by Sydney Brenner, John Sulston, and H. Robert Horvitz established that programmed cell death in the worm is indeed a genetically regulated process 14. Cell lineage tracings performed by Brenner and Sulston using the C. elegans hermaphrodite demonstrated that over the course its development, the same 131 cells out of the initial 1090 always died. Subsequent mutagenesis studies carried out by Horvitz and 4
17 colleagues identified the genes that constitute the core pathway governing programmed cell death in C. elegans. The first three cell death genes identified in C. elegans were nuc-1, ced-1, and ced-2. Nuc-1 was discovered by Sulston when he observed that in one particular mutant, denselystaining masses of DNA were found at the locations of cells that normally undergo programmed cell death 14. He discovered that the gene defined by this mutant encoded a DNA endonuclease responsible for DNA degradation during cell death. Ced-1 and ced-2 were then identified as two genes responsible for the phagocytosis of dead cells 15. In these mutants, the death process is initiated but the cell remants persist due to defective clearance from the surrounding tissue, allowing the cell corpses to be readily visualized in living animals 15. Horvitz and colleagues carried out subsequent mutagenesis screens of programmed cell death regulators on a ced-1 mutant background to exploit the persistence of dead cells in this mutant and identified a mutant in which no cell corpses could be found 16. The gene defined by this mutation is ced-3 16, later cloned and found to be a cysteine protease homologous to a mammalian protease, the interleukin-1β converting enzyme (ICE) or caspase Subsequent screens uncovered the rest of the core C. elegans cell death regulatory pathways: the pro-death genes egl-1 18 and ced-4 16, and the anti-death gene ced In response to death signals, Egl-1 is transcriptionally upregulated and undergoes mitochondrial translocation, where it displaces CED-4 from its inhibitor CED Once released from CED-9, CED-4 translocates to the perinuclear region, oligomerizes, and interacts with CED-3 to promote its autocatalytic activation 1, 21. For their work, Brenner, Sulston, and Horvitz were honored with the 2002 Nobel Prize in Physiology or Medicine. Mammalian apoptosis: caspases As in C. elegans, the execution of apoptosis in mammalian cells likewise depends on the activity of a family of proteases. The mammalian CED-3 homologue ICE, or caspase-1, is the founding member of this family of proteases that use a cysteine nucleophile to catalyze peptide bond hydrolysis in motifs bearing aspartic acid 22. Thus they are named caspases, a contraction of cysteine-dependent aspartate-specific protease. Synthesized as catalytically inactive zymogens, 5
18 caspases possess an N-terminal prodomain followed by a large and small subunit 22. The human genome encodes 11 caspases, traditionally split into two major subgroups based sequence homology and function 23. The first group are inflammatory caspases, which are primarily activated in cells of the innate immune system to regulate inflammation through the processing of cytokines 23. The second group consist of apoptotic caspases, activated during apoptosis and responsible for the execution of the death program 23. The apoptotic caspases are further subdivided into upstream initiator caspases that are activated by adaptor-induced proximity and downstream effector caspases that are cleaved and activated by initiator caspases 24. Initiator caspases possess a long prodomain that mediates interactions with their adaptor proteins while effector caspases possess a short prodomain 24. There are two varieties of initiator caspase prodomains: the caspase-recruitment domain (CARD) and the death-effector domain (DED) 24. Activation of inactive monomeric initiator caspases requires homodimerization mediated by their recruitment to activation complexes formed following an apoptotic signal 25. According to the induced proximity model, homotypic interactions between the caspase prodomain and adaptor proteins in these complexes activate caspases via an enforced increase in local caspase concentration 25. Three mammalian apoptotic initiator caspase activation complexes have been identified: the apoptosome, the death-inducing signalling complex (DISC), and the PIDDosome 26. The apoptosome describes a complex of cytochrome c, APAF-1 (apoptotic protease activating factor-1), and caspase The APAF-1 CARD domain, which mediates interaction with caspase- 9, is normally bound by two C-terminal WD40 domains and thus unable to bind with the caspase 28. Upstream death signals that culminate in the permeabilization of the mitochondrial outer membrane (MOM) release cytochrome c, which binds to APAF-1 through its WD40 domains, making its CARD domain available to recruit caspase Inactive APAF-1 also binds one molecule of ADP/dADP via its nucleotide binding domain 28. Conformational changes in APAF-1 due to interaction with cytochrome c allows ADP/dADP to be exchanged for ATP/dATP, the hydrolysis of which induces a conformational change that permits its oligomerization into the heptameric apoptosome 29, 30. The prototypical DISC is formed following ligation of the Fas death receptor 31. Fas, which is preassembled in the plasma membrane as a trimer 32, undergoes a 6
19 conformational change mediated by weak protein-protein interactions resulting from ligandinduced receptor clustering, leading to the assembly of the adaptor protein FADD to its cytoplasmic tail, which in turn recruits caspase-8 via the DED 33. This results in high local concentrations of caspase-8, leading to its proximity-induced activation 25. Finally, the PIDDosome is the caspase-2 activating complex composed of caspase-2, the adaptor protein RAIDD, and PIDD, in which proteolytically activated PIDD recruits RAIDD and caspase-2 leading to its activation 34. Effector caspases exist as inactive dimers that are activated following proteolytic cleavage between their large and small subunits by initiator caspases and by the lymphocytespecific serine protease granzyme B 22. Structural studies on caspase-7, a representative effector caspase, demonstrates the mechanistic basis for proteolytic activation 35, 36. Following cleavage, the core structural elements of caspase-7 remain unchanged and the protease remains as a homodimer. However, drastic conformational changes of active site loops shift the active site from a closed state to an open one capable of binding substrate. Once active, effector caspases go on to cleave their intracellular targets to execute apoptosis. A large number of proteins have been reported to be in vivo caspase substrates, with targeted proteomics approaches revealing that over cellular proteins are cleaved in a caspase-dependent manner following induction of apoptosis 37, 38. Of note, the organized degradation of nuclear DNA into 180 bp internucleosomal fragments, one of the key hallmarks of apoptosis, is mediated by the DNase CAD 39. In the absence of apoptotic signals, CAD is sequestered in the cytoplasm by its inhibitor ICAD 39. Once activated either via the DISC or the apoptosome, effector caspase-3 cleaves ICAD, releasing CAD to translocate into the nucleus to degrade chromosomal DNA 40. Besides regulating its activation, cells also possess additional regulatory mechanisms to prevent unwanted, potentially lethal caspase activity 25. First, caspase inhibition is exemplified by the human caspase inhibitor XIAP, the most extensively studied member of the inhibitor of apoptosis (IAP) protein family 41. This multidomain protein achieves selective tight inhibition of both initiator (caspase-9) and effector (caspase-3 and -7) caspases via utilizing a two-site mechanism 41. Caspase inhibition is also mediated by decoy proteins that structurally mimic 7
20 caspase prodomains to compete for the same adaptors in activation platforms. For example, FLIP, a caspase-8 mimic with a nonfunctional catalytic domain, prevents caspase-8 recruitment to the DISC 42, 43. Finally, inhibition of caspase activity can be achieved via proteosomal degradation, with cellular IAPs, which contain domains involved in ubiquitin ligation, proposed to mediate proteosomal removal of caspases 25, 44, 45. Intrinsic and extrinsic mammalian apoptosis pathways In mammals, apoptosis can be initiated via either the intrinsic or the extrinsic pathway 1. The intrinsic pathway is activated by a wide range of stimuli including DNA damage, ER stress, and cytokine/growth factor depletion 1. Upstream pro-survival and pro-death signals are transduced and integrated by the BCL-2 family of proteins, which controls apoptosis by regulating mitochondrial outer membrane (MOM) integrity 1. MOM permeabilization, generally considered to irreversibly commit the cell to death following intrinsic death stimuli, leads to the release of cytochrome c 46 as well as other apoptogenic molecules such as Endo G 47, Smac/DIABLO 48, 49, and HtrA2/OMI 50, and from the intermembrane space (IMS). The extrinsic pathway is triggered by the ligation of cell surface death domain-containing receptors such as Fas, TNFR1, and DR4/5 1. Receptor ligation leads to the formation of the DISC and caspase activation, as described in the previous section. Depending on whether or not MOM permeabilization is required for the DISC to induce effector caspase activation and cell death, cells can be divided into two types 51. Type I cells, typified by T lymphocytes, do not require MOM permeabilization and cytochrome c release for effector caspase activation, and therefore are not protected by inhibition of MOM permeabilization by overexpression of anti-apoptotic BCL-2 family members BCL-2 or BCL-X 51 L. In those cells, a large amount of caspase-8 is recruited to and activated at the DISC, leading to effector caspase activation. In contrast, Type II cells, typified by hepatocytes, have lower levels of caspase-8 and higher levels of XIAP compared to Type I cells, thus abrogating the ability of caspase-8 to directly activate effector caspases 51, 52. In these cells, activation of effector caspases requires amplification of the death signal by the mitochondrial apoptosis pathway 53, 54. In this scenario, proteolytic activation of BID to truncated BID (tbid) by 8
21 caspase-8 results in MOM permeabilization, releasing cytochrome c and Smac/DIABLO, which respectively induces apoptosome formation and neutralizes XIAP, thus leading to effector caspase activation Importantly, the level of XIAP expression has been shown to be a critical determinant of whether a cell is Type I versus Type II 52. In that study, hepatocytes in the liver of Bid -/- mice, which do not engage the mitochondrial pathway downstream of DISC and are thus resistant to apoptosis induced by injection of FasL 56, were rendered sensitive by combined loss of both Bid and XIAP 52. The BCL-2 family The identification of the BCL-2 (B-cell lymphoma 2) proto-oncogene at the chromosomal breakpoint of t(14;18) in human follicular B-cell lymphoma resulted not only in the establishment of the BCL-2 family of proteins, which constitute a critical intracellular checkpoint in intrinsic apoptosis, but also created a new paradigm in cancer biology 1. All oncogenes discovered prior to BCL-2 promoted tumorigenesis by increasing cell growth and proliferation. However, the t(14;18) translocation, in which the immunoglobulin heavy chain promoter and enhancer drive BCL-2 overexpression, did not increase proliferation but rather rendered cells resistant to apoptosis 57. The oncogenic potential of BCL-2 was demonstrated by Stanley J. Korsmeyer and colleagues in two landmark studies published in 1989 and 1991 using the BCL-2-Ig transgenic mice, which carry a minigene that structurally mimics the t(14;18) 58,59. In humans, follicular lymphoma is often an indolent disease comprised of small resting B cells that frequently progresses to high-grade lymphoma. Similarly, in the 1989 study, the investigators reported that BCL-2-Ig mice developed polyclonal follicular hyperplasia of resting B cells. In that study, the investigators also reported the important observation that the BCL-2-Ig transgene endowed mature B cells with a prominent survival advantage when grown in vitro in simple liquid culture. In their subsequent 1991 study, the investigators reported that in the BCL-2-Ig mice, B cell polyclonal follicular hyperplasia progressed to high-grade monoclonal lymphoma following a long latency period, reflecting the clinical couse of human follicular lymphoma. The latency period preceding the appearance of monoclonal disease indicated to the investigators that secondary changes were responsible for 9
22 the progression from hyperplasia to lymphoma. Indeed, half of the high-grade lymphomas harbored rearrangement of c-myc, a proto-oncogene that drives cell growth and proliferation, and which had been shown to synergize with BCL-2 in vitro. In summarizing their findings, the authors proposed BCL-2 to be the founding member of a novel category of proto-oncogenes that drive tumorigenesis not by promoting growth and proliferation, but rather by functioning as an antidote against death. And indeed, the evasion of cell death has been proven to be a hallmark of cancer progression 60. The BCL-2 family contains approximately 20 proteins and is divided into three subfamilies based on their death regulatory activity and homology shared within four conserved BCL-2 homology domains (BH1-4) 61. The anti-apoptotic members include BCL-2, BCL-X L, and MCL-1. Pro-apoptotics are further divided into multidomain members BAX and BAK, and a diverse group of BH3-only members. Studies from our lab have further defined two groups of BH3-only molecules: ones that directly activate BAX/BAK and ones that indirectly activate BAX/BAK through inactivating BCL-2/BCL-X L /MCL Upon apoptosis, the activator BH3-only molecules (BH3s), including tbid, BIM, and PUMA, trigger homo-oligomerization of BAX and BAK to mediate cytochrome c efflux, leading to caspase activation. Conversely, the anti-apoptotic BCL- 2/BCL-X L /MCL-1 sequester activator BH3s into inert complexes, thus preventing the activation of BAX/BAK. The remaining BH3s including BAD, NOXA, BMF, HRK, and BIK/BLK do not activate BAX and BAK directly, but instead prevent the anti-apoptotic BCL-2 members from sequestering the activator BH3s. Thus, the inactivator BH3s can displace activator BH3s from anti-apoptotics to activate BAX and BAK indirectly. The data from this study established a hierarchical regulation of mitochondrion-dependent apoptosis by BCL-2 subfamilies (Fig. 1.2). In the absence of death stimuli, BAX and BAK are both inactive monomers, with BAX in the cytosol and BAK in the mitochondrial outer membrane (MOM) 61. Mitochondrial BAK is kept in an inactive conformation through its association with the MOM protein VDAC2 63. Once activated, BAX inserts into the MOM as homo-oligomers while BAK also homo-oligomerizes, leading to MOM permeabilization, release of cytochrome c from the intermembrane space (IMS), and apoptosome formation 61,
23 Targeted gene deletion has demonstrated that BAX and BAK constitute the essential gateway to mitochondrial apoptosis 65. Unlike the stable and ubiquitously-expressed BAX and BAK, a third pro-apoptotic multidomain BCL-2 family member, BOK, is labile with prominent expression restricted to the reproductive organs and is therefore not essential for cytochrome c release 66. The functional redundancy of BAX and BAK is apparent when comparing the mild phenotypes of Bax -/- mice and Bak -/- mice with the severe phenotype of mice deficient for both Bax and Bak 67. Bak -/- mice have no overt phenotype while the Bax -/- mice displays mild lymphoid hyperplasia and male sterility due to defective sperm-cell differentiation. However, most Bax -/- Bak - /- mice die perinatally with fewer than 10% surviving to adulthood. The exact cause of perinatal lethality of the Bax -/- Bak -/- mice is still unknown. The surviving mice demonstrated developmental defects including persistence of interdigital webs, an imperforate vaginal canal, and accumulation of excess cells in the central nervous and hematopoietic systems. Importantly, the elimination of interdigital webs during the formation of digits in higher vertebrates via programmed cell death is a well-described example of its role in tissue remodeling 68 and was completely rescued by just one wild-type Bax or Bak allele. Furthermore, neither intrinsic death signals nor overexpression of BH3-only molecules were able to induce cytochrome c release and apoptotic death in cells deficient for both Bax and Bak 65, 69, 70. The nature of the cytochrome c-releasing pore formed by the BAX and BAK homooligomeric complexes on the MOM is still under investigation. Much more is known about the molecular mechanisms governing BAX and BAK activation, a tightly regulated process mediated by dynamic interactions with activator BH3-only molecules. For both BAX and BAK, activation means that these normally inactive, monomeric proteins undergo conformation changes and homo- oligomerization 61, 71. While BAK always resides in the MOM, BAX is mostly cytosolic with a small fraction loosely associated with the mitochondria 61, 64. Therefore, unlike BAK, BAX activation requires the additional step of mitochondrial translocation. The insertion of BAX and BAK into the MOM is mediated by its C-terminal α9 helix 61, 72. Our lab found that for BAX, in the absence of death signals, the N-terminal α1 helix stabilizes the α9 in the dimerization pocket formed by the BH1-3 domains, thus preventing BAX mitochondrial targeting 64. In the presence of 11
24 upstream death signals, activator BH3-only molecules tbid, BIM, and PUMA transiently bind the α1 helix to drive N- and C-terminal exposure, thereby freeing the α9 helix for MOM insertion. tbid, BIM, and PUMA then drive the homo-oligomerization of mitochondrially-localized BAX. The α1 helix of BAK is always exposed, thus allowing it to be constitutively targeted to the MOM where it is kept inactive through binding with the MOM protein VDAC2 via its dimerization pocket. Death signals induce activator BH3-only molecules to disrupt the inhibitory BAK-VDAC2 interaction to enable BAK homo-oligomerization. The essential role of the activator BH3-only molecules in directly activating BAX and BAK was clearly demonstrated by our studies on knockout mice deficient for Bid, Bim, and PUMA 73. The Bid -/- Bim -/- PUMA -/- mice displayed the same developmental defects associated with Bax -/- Bak -/- mice, including interdigital webbing and imperforate vaginas. Furthermore, despite the presence of other BH3-only molecules, combined deficiency of Bid, Bim, and PUMA prevented BAX and BAK homo-oligomerization, cytochrome c release, and caspase activation in response to intrinsic death signals in neurons and T lymphocytes. Regulation of the BCL-2 family by life and death signals Given its central role in the regulation of apoptosis, the BCL-2 family is understandably under tight control by both pro-death and pro-survival signals at the transcriptional and posttranscriptional level 61. The ratio between pro- and antiapoptotic BCL-2 family members constitutes a rheostat that sets the threshold of susceptibility to apoptosis 1. Pro-death signals shift the ratio to favor BAX/BAK activation while pro-survival signals do the opposite. For example, following death receptor ligation and DISC formation, inactive cytosolic BID is proteolytically converted by caspase-8 to the active, mitochondrially targeted tbid 53, 54. Cleavage of BID by caspase-8 results in its N-myristoylation via a newly-exposed glycine, thus driving its translocation to the MOM. Additionally, genotoxic stress upregulates the pro-apoptotic BH3-only molecules NOXA and PUMA via the transcription factor p Endoplasmic reticulum (ER) stress activates the transcription of BIM via the transcription factor CHOP and induces dephosphorylation of BIM through PP2A, which prevents its ubiquitination and proteasomal 12
25 degradation 77. Furthermore, in lymphocytes, cytokine withdrawal and subsequent shutdown of the pro-survival PI3K/AKT pathway leads to BIM induction via activation of the transcription factor FOXO3A 78. On the other hand, in the presence of growth factors, BIM phosphorylation by ERK1/2 disrupts its ability to bind BAX 79. ERK1/2 phosphorylation of human BIM at Ser69 also promotes its subsequent phosphorylation by Rsk1/2, which mediates its binding to the E3 ligase SCF β TrCP leading to its ubiquitination and proteasomal degradation 80. Antiapoptotic BCL-2 family members are similarly regulated both transcriptionally and post-transcriptionally by pro-death and pro-survival signals. BCL-X L is transcriptionally induced via the JAK-STAT pathway following the activation of cytokine receptors 81. Furthermore, upregulation of BCL-X L by the transcription factor NF-kB mediates the pro-survival effects of CD28 costimulation during T cell receptor (TCR) activation in T lymphocytes 82, 83 and of CD40 activation in B lymphocytes 84. Post-transcriptional regulation of antiapoptotic BCL-2 family members is exemplified by MCL-1. Due to the presence of a PEST region upstream of its BH3 domain, MCL- 1 protein is more labile than BCL-2 and BCL-X L and is rapidly downregulated during apoptosis, suggesting it is important in rapidly shifting the threshold of susceptibility to apoptosis in response to acute environmental changes 85. Upon DNA damage, the HECT-domain-containing E3 ubiquitin ligase MULE ubiquitinates MCL-1, leading to its proteosomal degradation 86. Furthermore, DNA damage and cytokine withdrawal result in phosphorylation of MCL-1 by the kinase GSK Phosphorylated MCL-1 is then ubiquitinated and targeted to the proteosome by the E3 ligase SCF βtrcp. GSK-mediated MCL-1 phosphorylation also targets it for polyubiquitination and degradation by the tumor suppressor E3 ubiquitin ligase SCF FBW7, indicating that the loss of FBW7 observed in diverse human cancers may contribute the tumorigenesis through upregulation of MCL-1 expression 88. Finally, during mitotic arrest induced by antitubulin chemotherapeutic agents, MCL-1 is phosphorylated by GSK-3 and subsequently ubiquitinated and targeted for proteasomal degradation by the tumor suppressor E3 ubiquitin ligase SCF FBW7 89. In contrast, stabilization of MCL-1 and consequent promotion of cell survival is mediated by the deubiquitinase USP9X, which directly interacts with and deubiquitinates MCL Interestingly, 13
26 the same study demonstrated that expression levels of MCL-1, which is known to be abnormally high in a range of human cancers, directly correlates with USP9X expression in human tumors. Caspase-independent cell death Even in the absence of caspase activation, the induction of BAX and BAK homooligomerization by activator BH3-only molecules has lethal consequences for the cell due to mitochondrial dysfunction 69, 91. Indeed, mitochondrial outer membrane permeabilization (MOMP) is generally considered to be the point of no return commiting a cell to death regardless of caspase activation. The occurrence of caspase-independent cell death (CICD) in vivo is apparent upon comparing the phenotype of Apaf-1 -/- 92 and Caspase-9 -/- 93 mice with that of Bax -/- -/- 67 Bak and Bid -/- Bim -/- Puma -/- 73 mice. The elimination of interdigital webs was only delayed by 2 days in Apaf-1 -/- mice 92 and occurred normally in Caspase-9 -/- mice 93. However, interdigital webs persisted into adulthood in both Bax -/- Bak -/- and Bid -/- Bim -/- Puma -/- mice. The accumulation of neuronal cells observed in adult Bax -/- Bak -/- mice was likewise absent in adult mice deficient in Apaf-1 or Caspase-9. Furthermore, deficiency in Apaf-1, Caspase-9, or Caspase-3 provides only transient protection against death induced by BH3-only molecules despite the lack of caspase activation while Bax -/- Bak -/- cells were completely protected 69. In healthy cells, the mitochondrial inner membrane (MIM), which separates the matrix from the intermembrane space (IMS), is highly impermeable to water and ions while the mitochondrial outer membrane (MOM) is generally assumed to be freely permeable to small metabolites and solutes up to ~5 kda 91. The impermeability of the MIM allows the embedded electron transport chain (ETC) to generate a proton gradient across the membrane, which forms the basis of the inner mitochondrial transmembrane potential (ΔΨm) 94. This electrochemical gradient is positively charged and acidic on the side facing the IMS and negatively charged and alkaline on the matrix-facing side. The proton gradient is used by the ATP synthase enzyme complex, located in the MIM, to generate ATP from ADP and is also used by ion-exhange channels to regulate mitochondrial ion homeostasis. The ΔΨm set up by the proton gradient is also required for protein import into the mitochondrial matrix since it both activates the inner 14
27 membrane protein import channel and drives positively charged presequences into the matrix. Accordingly, the loss of ΔΨm that occurs following MOMP leads to loss of ATP production, ion homeostasis, and mitochondrial protein import 91. Mitochondrial dysfunction induced by BAX/BAK activation can be caspase dependent or independent. Caspase-3 can cleave the p75 subunit of the complex I of the electron transport chain, leading to loss of ΔΨm 95. The molecular mechanisms underlying caspase-independent mitochondrial dysfunction are still unclear. Although release of cytochrome c, a component of the ETC, may be expected to inhibit ETC activity thus leading to loss of ΔΨm, there is evidence that the concentration of cytochrome c in the IMS following MOMP is sufficient for ETC function 96. ΔΨm has also been suggested to result from inactivation of ETC enzymes by cytosolic enzymes that are activated and/or gain entry to the IMS following MOMP. In support of this, one study showed the caspase-independent loss of activity of ETC complexes I and IV following MOMP 97. Mitochondrial protein import is also actively shut down following MOMP due to degradation of TIM23, an essential component of the inner membrane protein import machinery, leading to a loss of cell viability 98. The release of IMS proteins AIF, Endo G, and HtrA2/Omi following MOMP has also been implicated in CICD 91. AIF is a phylogenetically conserved flavoprotein with NADH oxidase activity shown to be involved in the assembly and maintenance of Complex I of the ETC 99. Following MOMP, AIF translocates into the nucleus and mediates chromatin condensation and large-scale DNA fragmentation 100. Loss of AIF also impairs oxidative phosphorylation due to a decrease in ETC complex I activity 99. Likewise, Endo G, the predominant mitochondrial endonuclease proposed to mediate mitochondrial biogenesis and DNA synthesis and repair, also translocates to the nucleus following MOMP where it has been shown to degrade chromatin 47. HtrA2/Omi is a serine protease that can both mediate caspase-dependent cell death via inhibitory interactions with IAPs and through caspase-independent mechanisms that depend on it proteolytic activity 50. Evidence indicate that loss of HtrA2/Omi is also deleterious to the mitochondria 101, 102. Under normal circumstances, HtrA2/Omi is proposed to mediate protein quality control in the IMS by degrading denatured proteins. Mice bearing homozygous deletion of HtrA2/Omi or a point 15
28 mutation that disrupts its protease activity die prematurely and exhibit neurodegeneration, possibly due to mitochondrial dysfunction. 1.3 Autophagy Macroautophagy, referred to from here on out as autophagy, is an evolutionarily conserved process in eukaryotes by which proteins, organelles, and cytoplasmic content are sequestered by double-membrane vesicles, termed autophagosomes, and delivered to lysosomes for degradation 103. Basal, constitutive autophagy maintains cellular homeostasis by mediating the elimination and recycling of old or damaged organelles as well as turnover of longlived proteins and protein aggregates 103. Autophagy is also a survival response stimulated by multiple forms of cellular stress such as nutrient depletion, hypoxia, damaged organelles, and pathogen invasion 104. One of the most important roles for autophagy is to provide energy for vital cellular functions via digestion of the cell s own contents 104. Autophagic cell death (ACD) is morphologically defined as cell death accompanied by prominent vacuolization of the cytoplasm due to large-scale autophagosome formation 5. Although the bulk of the evidence indicates that autophagy primarily promotes cell survival, it is conceivable that excessive autophagy can ultimately lead to cellular demise. However, it remains unclear whether there are regulatory mechanisms that specifically activate autophagy to execute cell death. Core mammalian autophagy machinery Although the molecular control of autophagy was initially investigated in yeast, the process of autophagy is largely conserved between yeast and mammals 103. Figure 1.3 depicts a simplified schematic of mammalian autophagosome formation. The first step of mammalian autophagy is autophagosome nucleation, in which a small, flat, crescent-shaped organelle termed the phagophore or isolation membrane is nucleated by core components of the autophagosome machinery. The isolation membrane then elongates and expands around cytoplasmic contents until a double-membraned autophagosome is formed upon complete envelopment of a portion of the cytoplasm or damaged organelles. Studies have shown that membrane components required 16
29 to form the autophagosome membrane can originate from the mitochondria outer membrane 105 and the endoplasmic reticulum 106. The autophagosome then fuses with the lysosome to form the autophagosome, where the inner membrane of the autophagosome and its cargo of cytoplasmderived material are then degraded by lysosomal hydrolases. Monomeric units such as amino acids generated by the degradation of macromolecules are then exported to the cytosol for reuse through permeases that are still poorly characterized. At the peak of autophagy a large percentage of lysosomes may be subsumed into autophagosomes and that, upon cessation of autophagy, proto-lysosomal membrane components are recycled via tubules and vesicles to form new lysosomes 107. The mechanism of autophagosome formation requires a highly orchestrated, evolutionarily-conserved series of events that are still being elucidated. Nutrient depletion is the most typical and potent inducer of autophagy 104. Briefly, the unc-51-like kinase (ULK) complexes are apical inducers of autophagosome formation that, upon nutrient depletion, are activated and localize at the phagophore 108, 109. Nucleation, and also subsequent maturation of the autophagosome, requires the VPS34 class III phosphatidylinositol-3 kinase (PI3K)-Beclin 1 complexes Beclin 1 plays an important role in tumorigenesis that will be discussed in more detail in a later section 110. The PI3K complex generates phosphatidylinositol 3-phosphate (PtdIns(3)P) in the phagophore membrane, an event required for recruitment of other components of the autophagy machinery 103. Phagophore elongation and closure requires the Atg12 and Atg8/LC3 ubiquitin-like protein conjugation systems 103, 114. Atg12 is conjugated to Atg5 in a reaction mediated by the E1-like enzyme Atg7 and the E2-like enzyme Atg10, which then interacts with Atg16 to form the large multimeric Atg16L complex on the cytoplasmic side of the phagophore membrane 103. C-terminal cleavage of Atg8/LC3 by Atg4 generates LC3-I, which is conjugated to phosphatidylethanolamine (PE) to generate LC3-II on the phagophore membrane in an Atg7- and Atg3-dependent manner where it mediates membrane tethering and hemifusion 103,114. The transmembrane protein Atg9 shuttles between the phagophore and peripheral sites to presumably deliver membranes to the developing phagophore, and does so in an ULK- and class III PI3K-dependent manner 115. VMP1, another transmembrane autophagy 17
30 protein, is required for autophagy induced by nutrient depletion, a function that depends on its interaction with Beclin This sugests VMP1 may recruit class III PI3K-Beclin 1 complexes to the phagophore. Upon completion, the proteins that mediate autophagosome formation are released back to the cytoplasm through an unknown mechanism. Autophagy as part of the cellular stress response Autophagy is activated by diverse stresses including nutrient depletion, ER stress, and microbial infection or exposure to microbial products 104. Of these, nutrient depletion is the most typical, best characterized, and most potent trigger of autophagy, which can be induced by lack of any essential nutrient 117. Thus, while autophagy is most potently induced in yeast by nitrogen starvation, the withdrawal of carbon, auxotrophic amino acids and nucleic acids, and sulfate can all initiate autophagy 117. Cultured mammalian cells similarly induce autophagy upon withdrawal of amino acids and glucose 117. For metazoans, though, it is important to note that autophagy is not simply the response of an individual cell to nutrient depletion, but is also a crucial part of an organism s response to starvation 117,118. Therefore, autophagy is also controlled by extracellular growth factors and by the hormones insulin and glucagon 117, 118. Indeed, one of the earliest observations of autophagy, made in 1962 prior even to the coining of the word, was in the hepatocytes of rats that had been exposed to glucagon, a hormone induced during times of fasting 119. Insulin, which is induced during and immediately following a meal, was later shown to inhibit autophagy 119. One of the most striking demonstrations of the importance of autophagy at the organismal level is the phenotype of mice deficient for the genes Atg5 120 or Atg7 121, which are essential for autophagosome formation. Although developmentally normal, they have a mortality rate of >90% within the first 24 period following delivery likely due to an inability to use autophagy to bridge over the starvation period between cessation of trans-placental nutrient supply and onset of suckling. One of the main purposes of autophagy during starvation is to maintain both intracellular and extracellular amino acid levels, both of which decrease in autophagy-deficient mice subjected to starvation 120. The amino acids thus produced may help organisms survive starvation by driving hepatic gluconeogenesis, in which lactate and amino acids from peripheral 18
31 tissues are used by the liver via the glucose-alanine cycle to produce glucose to compensate for depletion of glycogen stores during prolonged starvation 117. Furthermore, amino acids can also be used to produce oxidizable substrates for energy production via the tricarboxylic acid (TCA) cycle 117. The mtor pathway is the best-characterized regulator of starvation-induced autophagy 104. mtor is a serine/threonine kinase and as a component of mtor complex 1 (mtorc1) is central in growth factor signalling and nutrient- and energy-sensing pathways 104. Its activation increases protein synthesis and overall cell growth when metabolic conditions are favorable and/or when stimulated by growth factors 104. In mammalian cells, the first clue that mtor suppresses autophagy was the observation that the mtor inhibitor rapamycin induces autophagy in isolated rat hepatocytes 122. Though the mechanism by which mtor regulates autophagy is still an active area of investigation, mtor-mediated inhibition of the ULK complex is central to this process 103, 108. Sirtuins, NAD-dependent deacetylases that sense environmental stress, have also been implicated to play a crucial role in starvation-induced autophagy 104. Evidence indicates central roles for sirtuins in regulating metabolism and aging-related physiological changes, and that the mammalian sirtuin SIRT1 mediates the lifespan-extending effects of calorie restriction 123. Overexpression of SIRT1 was found capable of inducing autophagy while genetic deletion of Sirt1 impaired autophagy activation in mouse embryonic fibroblasts 124. In that study, SIRT1 was demonstrated to regulate the acetylation status of the core autophagy regulators Atg5, Atg7, and LC3. Furthermore, decreased association between the sirtuin SIRT2 and the transcription factor FoxO1 following serum starvation in human cancer cell lines leads to FoxO1 acetylation 125. Acetylated FoxO1 in the cytosol then binds with Atg7 to promote autophagosome formation. Autophagy as a cellular housekeeper Autophagy is not only induced as part of a cell s integrated response to stress, but also functions at a basal level as a cellular housekeeper that eliminates defective proteins and organelles and prevents the accumulation of abnormal protein aggregates 103. These 19
32 housekeeping functions are viewed as central to the protective role of autophagy observed in neurodegenerative diseases and cancer 126. Mice bearing tissue-specific deletions of Atg5 and Atg7 demonstrate an accumulation of protein aggregates in inclusion bodies associated with cellular degeneration in post-mitotic hepatocytes, cardiomyocytes, and neurons 126. Additionally, Atg7-deficient mouse hepatocytes accumulate peroxisomes, damage mitochondria, and aberrant membranous structures contiguous with the ER 121, 127. Since mitochondrial damage can lead to its overproduction of reactive oxygen species (ROS), some of the pro-survival effects of mitophagy have been attributed to the removal of this potential source of oxidative damage 126. Similarly, peroxisomes generate ROS through their metabolic functions, necessitating turnover of the organelle due to oxidative damage of its constituents 128. A key feature of many neurodegenerative diseases is the formation of intracellular aggregates 129. These include Parkinson s disease, polyglutamine expansion diseases such as Huntington s disease, and forms of dementia. Early observations of autophagosomes in these disease settings suggested involvement of autophagy in disease pathogenesis. However, model organisms have shown that autophagosome formation is either part of a protective response or, in the case of Alzheimer s disease, a consequence of impaired autophagosome maturation 126. For example, mutations of the dynein motor machinery found in human neurodegenerative motor neuron diseases were found to reduce the clearance of aggregate-prone proteins and enhance the toxicity of the causative agent of Huntington s disease, the mutant huntingtin protein, due to impairment of autophagosome-lysosome fusion 130. Furthermore, the mtor inhibitor rapamycin reduces mutant huntingtin toxicity in vitro and in vivo via induction of autophagy 131. Selective autophagic removal of damaged mitochondria, or mitophagy, is of particular interest within the context of cell death since loss of ΔΨm, which occurs following induction of MOMP by diverse death signals, targets mitochondria for mitophagy 132. A pro-survival role for mitophagy in this context has been proposed based on the finding that the glycolytic enzyme GAPDH promotes clonogenic survival following MOMP partly due to a novel ability to enhance autophagy via Atg12 upregulation 133. The recognition and targeting of depolarized mitochondria for autophagic removal is mediated by the kinase PINK1 and the E3 ligase Parkin 132, When 20
33 mitochondria are healthy, PINK1 translocates into the mitochondrial inner membrane via the general mitochondrial import machinery composed of the TOM and TIM23 complexes. There, it undergoes cleavage by the protease PARL, leading to its destabilization and degradation. However, since its import into the inner membrane and subsequent degradation relies on the mitochondrial membrane potential, loss of ΔΨm leads to rapid accumulation of PINK1 on the mitochondrial outer membrane and recruitment of the E3 ubiquitin ligase Parkin in a manner dependent on PINK1 kinase activity. Parkin recruitment to the MOM leads to ubiquitination of MOM substrates including VDAC1, and recruits the autophagy adaptor molecule p62/sqstm1 to initiate mitophagy. Parkin and PINK1 mutations are implicated in the neurodegenerative disorder Parkinson s disease, in which mitochondrial dysfunction is believed to play a casual role 137. Autophagy as a tumor suppressor mechanism Impaired autophagy has also been demonstrated to promote tumorigenesis, possibly through promoting genomic instability and inappropriate cell division 126. The involvement of the autophagy machinery in cancer was initially revealed by the discovery in 1999 that beclin 1, mono-allelically deleted in 40-75% of sporadic human breast and ovarian cancers, is a candidate tumor suppressor 110. In that study, the enforced expression of Beclin 1 in a human breast cancer cell line promoted autophagy, inhibited proliferation and clonogenicity, and impaired tumorigenesis in nude mice. Furthermore, the expression of endogenous Beclin 1 is lower in human breast carcinoma tissue compared to normal mammary epithelia. Subsequent generation of beclin 1 deficient mice revealed that homozygous loss of beclin 1 causes early embryonic lethality while heterozygous loss results in a high incidence of spontaneous tumors 138. Loss of the second beclin 1 allele is not observed in these tumors, indicating that Beclin-1 is a haploinsufficient tumor suppressor. Though beclin 1 was initially discovered during the positional cloning of BRCA1, it was rediscovered independently in a yeast two-hybrid screen for interaction partners of BCL Importantly, ER-localized BCL-2, which is often overexpressed in cancers, can inhibit autophagy by binding to Beclin 1 via the Beclin 1 BH3 domain
34 Autophagy as a cell death mechanism One of the first in vitro demonstrations that autophagy could indeed be a cell death mechanism appeared in 2004 with the report that RNAi-mediated knockdown of the essential autophagy regulators Beclin 1 and Atg7 protects against RIP1-dependent non-apoptotic death induced by caspase-8 inhibition in vitro 141. That same year, another group reported that knockdown of Beclin 1 or Atg5 protects Bax -/- Bak -/- mouse embryonic fibroblasts against intrinsic death signals that induced extensive autophagy 142. Then in 2007, the finding that degradation of larval salivary glands during Drosophila melanogaster metamorphosis depends on autophagy provided the first in vivo evidence that it may be a cell death mechanism 143. In that study, autophagy inhibition via transgenic expression of autophagy inhibitory molecules or removal of autophagy genes results in the persistence of salivary gland cell fragments. Yet, autophagy does not account for all the processes of this developmental death. First, autophagy inhibition only delays tissue degradation by ~24 hours. Also, only the disappearance of cell fragments and not of whole intact cells is delayed. Due to its ability to degrade large portions of the cytoplasm, excessive activation of autophagy can be expected to cause cellular demise. Indeed, expression of Beclin-1 mutants that cannot be regulated by BCL-2 can kill cultured mammalian cells in vitro 140 while transgenic expression in D. melanogaster larvae of a highly active mutant Atg1, an ortholog of mammalian ULKs, results in premature salivary gland degradation in vivo 144. Yet, the in vivo evidence that cell death in a mammalian model organism is executed specifically by autophagy versus by another cell death modality is poor. For example, death induced by the hyperactive Atg1 mutant in D. melanogaster larvae is caspase-dependent and shows the morphological features of apoptosis, leading the authors to suggest that excessive autophagy is an inducer of apoptosis rather than a distinct form of cell death 144. Although neuron-specific genetic deletion of Atg7 has been shown to protect against cell death in a mouse stroke model, the mode of death blocked by Atg7 loss in this scenario likewise displayed characteristic features of apoptosis 145. This suggests that Atg7 is not killing via autophagy; rather, it is activating an apoptotic program either via autophagy or 22
35 another upstream mechanism. Therefore, it is still controversial whether autophagy does indeed constitute a cell death mechanism distinct from apoptosis and necrosis. 1.4 Necrosis Programmed necrotic death: a new cell death paradigm In their seminal report in 1972, Kerr and colleagues explained that they are coining a new term, apoptosis, in order to distinguish an apparently physiologically regulated form of cell death that had until then been called necrobiosis or shrinkage necrosis from necrosis or coagulative necrosis, which was widely regraded as accidental and unregulated 13. Although it was acknowledged that there must exist a programmed form of cell death to counterbalance cell proliferation during development and in the adult, the morphological type of cell death described in almost all standard texts at the time was necrosis 13. At that time, investigators believed that necrosis could not be responsible for physiological cell death as it always appeared to be caused by an irreversible disturbance of cellular homeostasis due to exposure to noxious stimuli 13. And indeed, until the late 1980s, there was scant evidence to suggest otherwise. In 1988, the observation that the death receptor ligand tumor necrosis factor (TNF) can induce either apoptosis or necrosis in vitro depending on the cell type provided the first clue that necrosis may also be subjected to molecular control 8. Subsequent studies demonstrated that the necrotic response to TNF is mediated by increased generation of reactive oxygen species (ROS) by the mitochondria, in part due to increased glutamine metabolism that resulted in increased eletron flux through the mitochondrial electron transport chain A significant step in determining the mechanism of death receptor-induced necrosis was achieved with the finding that caspase inhibition switches the response of death receptor ligation from apoptosis to necrosis 149, 150. Then in 2000, details of the mechanisms regulating death receptor-induced necrosis began to emerge with the finding that necrosis triggered by the Fas death receptor ligand FasL requires its adaptor protein FADD and the enzymatic activity of the serine-threonine kinase RIP The same study found that necrosis induced by the death receptor ligands TNF and TRAIL also require RIP1 kinase activity. The central role of the kinase activity of RIP1 in death receptorinduced necrosis was demonstrated by the 2008 finding that it is the target of necrostatin-1 (nec- 23
36 1) 152, a potent and specific small molecule inhibitor previously discovered in 1995 by the same investigators to inhibit TNF-induced necrosis in vitro 153. This type of regulated necrotic cell death was named necroptosis by these researchers 153. In the 1995 report, nec-1 was also shown to protect against cerebral necrosis following ischemia-reperfusion in an in vivo stroke model 153. In that same year, which proved to be a particularly fruitful one for the concept of a regulated form of necrosis, three groups independently reported that genetic deletion of Cyclophilin D, a key component of the mitochondrial permeability transition pore that leads to mitochondrial dysfunction and cell death following prolonged calcium overload, protects against necrosis following tissue ischemia-reperfusion in vivo and to specifically protect against necrosis but not apoptosis in vitro Thus, in less than two decades, programmed necrosis went from being a poorly-recognized biological phenomenon to a genetically regulated, physiologically relevant cell death subroutine. Though sometimes used to indicate programmed necrosis in general, necroptosis is defined biochemically as a type of cell death inhibitable by genetic or pharmacological ablation of the kinase activity of RIP1 and its interaction partner RIP3, and is primarily triggered by death receptor activation 10. Reports from our lab and others also indicate the existence of RIP1- and RIP3-independent programmed necrosis elicited by intrinsic death stimuli including genotoxic stress 9, oxidative stress 157, and calcium dysregulation 157. Unlike apoptosis, a linear pathway linking upstream death signals to downstream death effectors has not been established 10. Indeed, physico-chemical stresses can serve as both necrosis initiators and effectors due to amplification by positive feedback loops generated by their deleterious effects on cellular physiology in general and on mitochondrial and lysosomal function in particular 7, 10. Though these downstream death execution events are still an active area of investigation, ATP depletion, oxidative damage, dysregulated intracellular calcium homeostasis, non-caspase proteases, phospholipases, and lipoxygenases have all been implicated. Necroptosis 24
37 RIP1 is a member of the RIP family of serine-threonine kinases involved in innate and adaptive immunity and has both a kinase-dependent function in promoting apoptosis 158 and necrosis 151, 152, and a kinase-independent function in promoting cell survival and inflammation 152, 159, 160. It consists of an N-terminal kinase domain, a RIP homotypic interaction motif (RHIM), and a C-terminal death domain (DD) 160. Ligation of the TNFR1 receptor induces a conformational change in the preformed trimeric receptor complex, leading to recruitment of the adaptor protein TRADD, which in turn recruits RIP1, the E3 ligases ciap1 and ciap2, TRAF2, and TRAF5; this constitutes the pro-survival complex I 161. RIP1 is polyubiquitinated by ciap1 and ciap2, enabling it to act as a scaffold for assembling the TAK1-TAB2-TAB3 complex that initiates the canonical activation pathway of the pro-survival, pro-inflammatory transcription factor NF-κB 162. Internalization of the ligand-bound TNFR1 and deubiquitination of RIP1 by the deubiquitinase CYLD 158 changes the membrane-associated complex I into the cytosolic complex II consisting of RIP1, TRADD, FADD, and caspase Complex II was later discovered independently by three groups to also include RIP Importantly, although RIP1 kinase activity mediates both TNFinduced apoptosis 158 and necrosis 151, RIP3 kinase activity is only required for necrosis 163. Activation of caspase-8 in this complex proteolytically inactivates RIP1 and RIP3 and initiates apoptosis 166, 167. In the absence of caspase-8 activity, mutual RIP1 and RIP3 phosphorylation leads to the formation of the necrosome that constitutes the necroptosis activation complex 160, 164. Although altered mitochondrial metabolism and excessive mitochondrial ROS production has been implicated in TNF-induced necrosis since the mid-1990s , the nature and regulation of these and potentially other lethal events downstream of the necrosome remain poorly characterized. In line with previous findings implicating the mitochondria, RIP3 was reported to mediate TNF-induced necrosis by increasing mitochondrial ROS generation via upregulation of mitochondrial oxidative metabolism. Then in early 2012, a mechanistic breakthrough was revealed by two simultaneous reports from the laboratory of Xiaodong Wang that confirmed a central role for the mitochondria in necroptosis execution 157, 168. In the first report, chemical library screening identified a compound named necrosulfonamide (NSA) that blocked necroptosis downstream of the necrosome 168. NSA functions in this context by disrupting the 25
38 direct interaction between RIP3 and a novel binding partner, mixed lineage kinase domain-like protein (MLKL), via covalent modification of a MLKL cysteine residue. Following initiation of necroptosis, phosphorylated RIP3 recruits and phosphorylates MLKL, an event shown to be crucial for RIP3-mediated necrosis. In the second report, the authors show that MLKL, which does not possess enzymatic activity, acts as an adaptor protein to tether the necrosome to a splice variant of phosphoglycerate mutase 5 (PGAM5S) on the mitochondrial outer membrane via the other PGAM5 splice variant PGAM5L 157. Once formed, the necrosome-pgam5s complex drives mitochondrial fragmentation, an event implicated in cell death, through activation of an essential mediator mitochondrial fission, the GTPase Drp1. Activation of the PGAM5S-Drp1 complex was proposed to constitute necroptosis complex III, which the authors described as a mitochondrial attack complex. Interestingly, complex III but not the necrosome was shown to be required for necrosis induced by dysregulated calcium homeostasis and ROS, raising the possibility that this complex and the induction of mitochondrial fission may be a central convergence point for both extrinsic (death receptor-mediated) and intrinsic necrotic signals. Programmed necrosis initiated by physico-chemical stress In 1997, genetic deletion of the mammalian DNA damage repair enzyme poly(adpribose) polymerase 1 (PARP1) was reported to protect mice against cerebral ischemiareperfusion injury, providing another example of genetic regulation of necrosis in vivo 169. Ischemia-reperfusion injury is an important clinical problem in which necrosis caused by loss of blood supply (ischemia) during a heart attack or stroke, or during solid organ transplantation, is exacerbated by the restoration of blood flow (reperfusion). PARP1 catalyzes the synthesis of poly(adp-ribose) polymers on histones and other chromatin-associated proteins near DNA adducts to promote efficient recognition of damage DNA by repair machinery 170. Overactivation of PARP1 consumes its substrate β-nicotinamide adenine dinucleotide (NAD + ) leading to a depletion of cytosolic NAD + and ATP levels 170. Activation of PARP1 by ischemia-reperfusion has been attributed to oxidative DNA damage induced by the overproduction of ROS observed under this setting 170. In a subsequent study, the same group demonstrated that DNA alkylation and 26
39 excitotoxicity induces PARP1-mediated mitochondrial dysfunction and caspase-independent cell death mediated by the release of AIF from the mitochondria 171. Importantly, a 2004 study using apoptosis-incompetent Bax -/- Bak -/- cells demonstrated that following DNA alkylation-induced PARP1 activation and NAD + depletion, cells that primarily used oxidative phosphorylation for ATP production are protected against ATP depletion and necrosis while those that derive ATP predominantly from glycolysis are sensitive 172. Importantly, since cancer cells prefer to use glycolysis for ATP production even when conditions are permissible for oxidative phosphorylation 173, this findings suggests the clinical efficacy of DNA alkylating chemotherapeutic agents may be due in part to tumor-specific induction of necrosis 172. The opening of the mitochondrial permeability transition pore (MPTP) is also implicated in the initiation of programmed necrosis triggered by oxidative stress, calcium overload, and ischemia-reperfusion injury 91. Mitochondrial permeability transition describes the sudden increase in permeability of the mitochondrial inner membrane (MIM) to solutes with molecular mass up to 1.5 kda 91. This is caused by the opening of the MPTP, a multicomponent complex spanning the mitochondrial outer membrane (MOM) and the MIM at the membrane contact sites 91. Following opening of the MPTP, the osmotic pressure of the matrix leads to an influx of water and solutes, resulting in matrix swelling 91. Since the surface area of the involuted MIM exceeds that of the MOM, matrix swelling can rupture the MOM 174, which releases cytotoxic proteins from the IMS, causes mitochondrial dysfunction, and commits the cell to die 91. The key factor responsible for MPTP opening is a large increase in mitochondrial matrix calcium concentration, with ROS also implicated to play an important role 91. Mitochondrial calcium overload and excessive ROS production are both central to necrosis following ischemiareperfusion and are both a result of ATP depletion secondary to ischemia 175. The MPTP pore has been proposed to consist of the voltage dependent anion channel (VDAC 1, 2, and 3) in the MOM, the adenine nucleotide translocase (ANT1 and 2) in the MIM, and cyclophilin D (CypD) in the mitrochondrial matrix 6. Of these, only CypD has been shown to be an indispensible component of the MPTP. Three groups independently reported in 2005 that necrosis of neuronal and myocardial tissue caused by ischemia-reperfusion injury in the brain and heart can be 27
40 inhibited by genetic deletion of Ppif, the CypD-encoding gene In vitro experiments from those same studies showed that loss of CypD dramatically increased the level of calcium required to trigger MPTP opening. Significantly, though CypD was shown to be critical for necrosis, its deficiency did not impair the induction of apoptosis. In contrast, both VDACs and ANTs are not essential components of the MPTP. Mouse hepatocytes lacking both ANT isoforms displayed only a small increase in the level of calcium needed to trigger permeability transition 176 while mouse fibroblasts deficient for all three VDAC isoforms showed no protection against calciumand oxidative stress-induced MPTP opening and death 177. Indeed, instead of showing resistance against intrinsic death signals, cells deficient for VDAC2 are more sensitive to cell death due to its inhibitory role in BAX activation 178. Finally, our lab reported in 2009 that DNA double-strand breaks induced by topoisomerase inhibitors in apoptosis-defective Bax -/- Bak -/- double knockout mouse embryonic fibroblasts (MEFs) trigger programmed necrosis in a RIP1-, PARP1, and CypD-independent manner 9. We demonstrated that inhibition of transcription and translation protected against death, indicating that the initiation and execution of necrosis in this setting requires de novo gene expression. Furthermore, our study revealed that p53-mediated induction of the lysosomal protease cathepsin Q cooperated with increased production of ROS to execute necrosis. Executioner mechanisms of necrosis It is currently unclear whether diverse death signals induce programmed necrosis via a common, well-defined sequence of executioner mechanisms 10. Indeed, even in the case of the best characterized programmed necrosis pathway, necroptosis, the sequence of events leading from necrosome formation to eventual death is unclear. Nevertheless, most instances of programmed necrosis involve some or all of the following: ROS; mitochondrial and lysosomal dysfunction; dysregulated intracellular calcium homeostasis; and the activation of non-caspase proteases and phospholipases 7, 10. Increasing evidence indicate that mitochondrial damage and dysfunction, induced either directly (e.g. by oxidative damage) or indirectly (e.g. calcium overload 28
41 secondary to energy depletion, as in the case of ischemia-reperfusion), also play central roles in the execution of programmed necrosis under diverse settings 7, 91, 179. Excessive mitochondrial ROS production and consequent oxidative damage is a central execution mechanism of programmed necrosis 7. ROS are derivatives of molecular oxygen that are produced during normal physiological processes such as fatty acid catabolism and aerobic respiration 180. Under healthy states, endogenous ROS production is countered by cellular antioxidant defences 180. Excessive ROS production and/or impaired antioxidant capacity generates a deleterious state of oxidative stress, which damage all cellular constituents 180. In most scenarios, intracellular ROS is primarily produced by the mitochondrial electron transport chain (ETC) as the unavoidable byproduct of aerobic respiration 181. The leakage of electrons passing through the ETC leads to the one-electron reduction of O 2 to O 2 (superoxide), which can then give rise to other ROS such as the hydroxyl radical 181. Oxidative stress may contribute to cellular demise through, for example, inducing autocatalytic lipid peroxidation 180. Among the ROS, superoxide and the hydroxyl radical are common initiators of this process, in which unsaturated membrane lipids are converted into polar lipid hydroperoxides 180. This increases membrane fluidity, damages membrane-associated proteins, and compromises membrane integrity 180. Compromised lysosomal membrane integrity can lead to the harmful release of lysosomal proteases while damage to the ER membrane can result in a deleterious release of ER calcium 179. With regard to proteins, oxidative damage can not only alter or abolish their functions but in some cases can also lead to increased ROS production 180. For proteins containing ironsulfur (Fe-S) clusters, oxidative denaturation of the clusters can release redox-active ferrous iron into the cellular environment 180. There, ferrous iron can catalyze the decomposition of hydrogen peroxide, which is poorly reactive, into the highly reactive hydroxyl radical, which can readily damage all nearby cellular constituents 180. This decomposition of hydrogen peroxide is known as the Fenton reaction. In the case of the ETC, oxidative damage sustained by its components can increase electron leakage, creating a vicious cycle of ROS generation and oxidative damage 181. In pathological settings, elevated cellular calcium levels can contribute to necrosis though triggering mitochondrial membrane permeability transition as previously described, but also by 29
42 inducing ROS production, mitochondrial fragmentation, calcium-dependent calpain protease activation, and cytosolic phospholipase A 2 (cpla 2 ) translocation 7. In a normal, resting state, the concentration of free calcium in the cytosol and the mitochondrial matrix is ~ nm; in contrast the ER calcium concentration is ~ µm while that of the extracellular environment is in the mm range 182, 183. This enables calcium to function as a second messenger in both the cytosol and mitochondria 182. The maintenance of these steep concentration gradients depends on plasma and ER membrane ATPase transporters (respectively, plasma membrane Ca 2+ ATPase or PMCA, and sarco/endoplasmic reticulum Ca 2+ ATPase or SERCA), the plasma membrane sodium-ca 2+ exhanger (NCX), and on the integrity of cellular membranes 182. Therefore, ATP depletion and oxidative damage sustained by cellular membranes, such as that seen during ischemia-reperfusion, can dramatically increase cytosolic and mitochondrial matrix calcium levels. The mitochondrial inner membrane potential drives calcium into the matrix through a highly selective inner membrane Ca 2+ channel 182. Under physiological conditions, upon ER calcium release, matrix [Ca 2+ ] can reach several hundred µm while cytosolic levels can only reach 1-2 µm 183. Thus, mitochondria can buffer cytosolic calcium and tolerate a significant rise in matrix [Ca 2+ ] before underoing permeability transition. Increased mitochondrial matrix calcium concentration upregulates the TCA cycle, leading to increased electron flow into the mitochondrial ETC and thus increased ROS production 182. Pathological rises in calcium can lead to excessive ROS production, leading to oxidative stress. This is the main mechanism proposed for calciuminduced mitochondrial ROS overproduction during programmed necrosis 179. Interestingly, there are hints at another, non-mutually exclusive mechanism. Mitochondria are highly dynamic organelles that can build interconnected networks that continuously move along cytoskeletal tracks and frequently undergo fusion and fission 184. Fusion and fission machinery shape the mitochondrial compartment in response to ever-changing physiological conditions 184. The Drp1 GTPas is a master regulator of mitochondrial fission and can be activated to promote fission by elevated cytosolic [Ca 2+ ] via phosphorylation by Ca 2+ /calmodulin-dependent protein kinase Iα (CaMKIα) at serine and dephosphorylation by calcineurin at serine Increased mitochondrial fission has been shown to mediate increased ROS production following exposure 30
43 to hyperglycemia in vitro, though the mechanism is unclear 187. Importantly, activation of Drp1 by death receptor ligation, elevated calcium levels, and oxidative stress has been implicated in the execution of programmed necrosis 157. This suggests elevated cytosolic [Ca 2+ ] may also induce excessive ROS production as well as other potentially deleterious events during programmed necrosis through promoting mitochondria fission. Calpains are ubiquitously and constitutively expressed cytosolic cysteine proteases activated by an increase in cytosolic calcium concentration 7. Overactivation of calpains can result in proteolysis of the plasma membrane Na + /Ca 2+ exchange pump involved in extrusion of calcium from the cytosol, thus further disrupting cellular calcium homeostasis 7. Calpain overactivation has also been proposed to disrupt lysosomal membranes, leading to lysosomal membrane permeabilization (LMP) and the attendant deleterious release of lysosomal cathepsin proteases into the cytosol 7. Significantly, following ischemic injury, activated µ-calpain was observed to localize at the membranes of damaged lysosomes in monkey hippocampal neurons 188. Furthermore, cathepsin inhibitors were able to protect these neurons from ischemia-induced death 189. Increased cytosolic calcium concentration also promotes translocation of cpla 2 to the nuclear, ER, and Golgi appartus membrane where it hydrolyzes arachidonate-containing phospholipids to release arachidonic acid 7. Both free and esterified arachidonic acid are substrates for lipoxygenases that convert them into lipid hydroperoxides. Lipid hydroperoxidation, which can also occur via oxidative damage as described previously, compromises organelle and plasma membrane integrity. cpla 2 activation is implicated in TNF-induced necrosis while its deficiency has been reported to protect against brain ischemia 7. Finally, lysosomes and cathepsins are also implicated in programmed necrosis. The calpain-cathepsin hypothesis has been previously discussed, as has data from our lab implicating cathepsin Q in executing DNA damage-induced programmed necrosis. Under normal conditions, the lysosome is also the sole intracellular location of redox-active ferrous iron before it is incorporated into the catalytic center of proteins or stored as ferritin 179. Hydrogen peroxide can readily cross cellular membranes to enter the lysosome and generate hydroxyl radicals via the 31
44 Fenton reaction 179. Importantly, the lysosome does not possess enzymes that detoxify hydrogen peroxide 179. Involvement of lysosomal iron in necrosis is indicated by the ability of intralysosomal iron chelators to protect against oxidative stress-induced LMP and cell death. Physiological and pathological roles of programmed necrosis Loss of function animals models have demonstrated the importance of key regulators of programmed necrosis in promoting tissue injury under diverse pathological conditions. Although Rip1 -/- mice are not viable 190 and tissue-specific knockout and kinase-dead knock-in have not yet been reported, its role in brain ischemia 153, myocardial infarction 191, and head trauma 192 are indicated by the protective effects of necrostatin-1 administration in those scenarios. Rip3 -/- mice are viable and protected from pancreatic acinar cell necrosis in a rodent acute pancreatitis model 163. PARP1 loss also protects against acute pancreatitis 193 while loss of CypD reduces tissue infarction in mouse models of stroke and myocardial infarction Though necrosis was traditionally viewed as a pathological form of cell death, the observation that naturally occuring motor neuron death may exhibit necrotic features provided an early indication that necrosis may also constitute a physiological cell death mechanism 194. Then in 2011, three key reports provided evidence suggesting a physiological role for necroptosis during development as well as giving an answer to a conundrum in the cell death field The extrinsic pathway of apoptosis is initiated by the binding of a death ligand to its cognate plasma membrane death receptor, leading to the formation of the death-inducing signalling complex (DISC) previously discussed, with FADD, caspase-8, and the caspase-like regulator molecule FLIP all being components of this complex. Knockout of any of these three molecules results in embryonic lethality, with a failure in yolk sac vascularization and hematopoiesis It is difficult to attribute this phenotype to a defect in apoptosis, which typically leads to defects associated with excessive cells such as those seen with the Bax -/- Bak -/-65 and Bid -/- Bim -/- PUMA -/-73 mice. In 2011, three independent groups of investigators showed that combined genetic deletion of caspase-8 and RIP3 195, 196 or of FADD and RIP1 197 results in normal embryonic development, indicating that the developmental defects and embryonic lethality caused by loss of FADD and 32
45 caspase-8 are due to a failure to suppress necroptosis. Furthermore, one of those groups also used RNAi-mediated knockdown of FLIP in Caspase-8 and Rip3 doubly-deficient mouse embryonic fibroblasts to demonstrate that a caspase-8-flip heterodimer inhibits RIP3-dependent necrosis 196. It is currently unclear how an organism may benefit from being able to readily trigger necroptosis during development or adult life. Rip3 -/- mice develop into healthy and fertile adults 201, indicating that organisms can develop and live successfully without a functional necrosome. Still, the fact that there exists a molecular mechanism to suppress necroptosis during normal embryonic development suggests necroptosis may play roles that are essential for the welfare of the organism under specific, possibly adverse settings. The effect of necrotic death versus apoptosis on the immune system suggests one possible physiological function. With few exceptions, apoptosis in vivo is anti-inflammatory and produces immunological tolerance whereas necrosis triggers inflammation and an immune response 179. Both apoptotic and necrotic cell corpses are efficiently cleared from surrounding tissue by professional and non-professional phagocytes. However, in the case of necrosis, the lack of organized chromatin condensation and loss of cellular membrane integrity results in leakage of pro-inflammatory factors (e.g. histones, the chromatin-associated protein HMGB1, DNA, and RNA) that are detected by and activate immune cells. This suggests that a cell may prefer a necrotic death in settings where an inflammatory response may be beneficial to the organism, such as in cases of pathogenic infection. In support of this, viral infections are known to initiate necroptosis 179. For example, vaccinia virus infection induces the formation of the pro-necrotic RIP1-RIP3 complex 164 and sensitizes cells to TNF-induced death in a RIP1-dependent manner 202. Accordingly, RIP3- deficient mice are resistant to vaccinia virus-induced hepatic inflammation and necrosis, but cannot effectively control viral replication 164. Furthermore, the murine cytomegalovirus protein M45 disrupts RIP1-RIP3 interaction to inhibit necroptosis 203,204. Importantly, viral infection of wildtype mice is severely attenuated by a mutant M45 that cannot interact with RIP1-RIP3. Importantly, attentuation of viral infection is completely rescued in RIP3-deficient mice 204. In aggregate, these studies demonstrate that viruses utilize inhibition of host cell necrosis as a 33
46 strategy to sustain viral replication, whereas host cells undergo necrosis as a way to clear viral infection. 1.5 Significance The past few decades have witnessed a paradigm shift in the field of cell death. The idea that apoptosis is the only form of programmed cell death has been challenged by the emerging evidence that necrosis can be as programmed as apoptosis. Characterization of pathways that initiate programmed necrosis is well under way while animal models are providing increasing insights into the physiological and pathological roles of programmed necrosis. Although the events and processes that ultimately result in cellular demise and the mechanisms of their activation by upstream death initiation events are unclear, these are also beginning to be elucidated. Whether diverse necrotic signals converge on a single sequence and set of execution mechanisms remains to be determined. Given the wide variety of pathologies in which programmed necrosis is implicated, a thorough understanding of the factors that influence the outcome of the life-or-death decision upon exposure to death stimuli can potentially open up broad new avenues for therpeutic intervention for many human injuries and diseases. Importantly, the evasion of apoptosis is a key hallmark of tumorigenesis; thus, delineation of programmed necrosis pathways and the factors that may contribute to resisting necrotic death will provide new strategies for the treatment of human cancers. Oxidative stress is central to both the initiation and execution of programmed necrosis. In Chapter Two, I demonstrated that the transcription factor ΔNp63α protected against programmed necrotic death (PND) and enhanced long-term survival of apoptosis-defective cells in the face of intrinsic death signals via a previously unknown ability to control intracellular redox homeostasis by regulating glutathione metabolism. My findings from Chapter Two also established oxidative stress as a central determinant of death versus long-term survival following exposure to intrinsic death signals. In Chapter Three, I showed that in DKO MEFs, DSBs trigger an ATM/ATR/DNA- PK-mediated DNA damage response that increased ROS production by upregulating the activity of complex III of the electron transport chain. 34
47 1.6 Figure Legends Figure 1.1 Cell death is categorized according to morphological features. Electron microscopy images of a normal (a), autophagic (b), apoptotic (c), and necrotic (d) cell. Scale bar = 1 µm. (Figure adapted from Edinger et al., (2004) Curr Opin Cell Biol. 16: ) Figure 1.2 Hierarchical regulation of mitochondrion-dependent apoptosis by BCL-2 subfamilies. (Figure adapted from Kim et al., (2006) Nat. Cell Biol. 8, ) Figure 1.3 Schematic depiction of the autophagy pathway and its core molecular machinery in mammalian cells. (Figure adapted from Yang et al., (2010) Curr Opin Cell Biol. 22: ) 35

48 1.7 Figures Figure 1.1 Morphological features of apoptosis, autophagy, and necrosis Edinger et al., (2004) Curr Opin Cell Biol. 16:

49 Figure 1.2 Hierarchical regulation of mitochondrion-dependent apoptosis by BCL-2 subfamilies BH3-only Inactivators Bad Bmf Bik/Blk Hrk/Dp5 Noxa Activators Bcl-2 Bid Bax cyt c Bcl-x L Bim Apaf-1 Caspase Bak Mcl-1 Puma Mitochondrial dysfunction Kim et al., (2006) Nat. Cell Biol. 8, Figure 1.3 Schematic depiction of the autophagy pathway and its core molecular machinery in mammalian cells. Yang et al., (2010) Curr Opin Cell Biol. 22:
50 1.8 References 1. Danial, N. N. and Korsmeyer, S. J. Cell death: critical control points. Cell, 116: , Thompson, C. B. Apoptosis in the pathogenesis and treatment of disease. Science, 267: , Lockshin, R. A. and Zakeri, Z. Programmed cell death and apoptosis: origins of the theory. Nat Rev Mol Cell Biol, 2: , Strasser, A., Cory, S., and Adams, J. M. Deciphering the rules of programmed cell death to improve therapy of cancer and other diseases. EMBO J, 30: , Edinger, A. L. and Thompson, C. B. Death by design: apoptosis, necrosis and autophagy. Curr Opin Cell Biol, 16: , Galluzzi, L., Vanden Berghe, T., Vanlangenakker, N., Buettner, S., Eisenberg, T., Vandenabeele, P., Madeo, F., and Kroemer, G. Programmed necrosis from molecules to health and disease. Int Rev Cell Mol Biol, 289: 1-35, Festjens, N., Vanden Berghe, T., and Vandenabeele, P. Necrosis, a well-orchestrated form of cell demise: signalling cascades, important mediators and concomitant immune response. Biochim Biophys Acta, 1757: , Laster, S. M., Wood, J. G., and Gooding, L. R. Tumor necrosis factor can induce both apoptic and necrotic forms of cell lysis. J Immunol, 141: , Tu, H. C., Ren, D., Wang, G. X., Chen, D. Y., Westergard, T. D., Kim, H., Sasagawa, S., Hsieh, J. J., and Cheng, E. H. The p53-cathepsin axis cooperates with ROS to activate programmed necrotic death upon DNA damage. Proc Natl Acad Sci U S A, 106: , Vanlangenakker, N., Vanden Berghe, T., and Vandenabeele, P. Many stimuli pull the necrotic trigger, an overview. Cell Death Differ, 19: 75-86, Lockshin, R. A. and Williams, C. M. Programmed Cell Death--I. Cytology of Degeneration in the Intersegmental Muscles of the Pernyi Silkmoth. J Insect Physiol, 11: ,
51 12. Kerr, J. F. A histochemical study of hypertrophy and ischaemic injury of rat liver with special reference to changes in lysosomes. J Pathol Bacteriol, 90: , Kerr, J. F., Wyllie, A. H., and Currie, A. R. Apoptosis: a basic biological phenomenon with wide-ranging implications in tissue kinetics. Br J Cancer, 26: , Horvitz, H. R. Worms, life, and death (Nobel lecture). Chembiochem, 4: , Hedgecock, E. M., Sulston, J. E., and Thomson, J. N. Mutations affecting programmed cell deaths in the nematode Caenorhabditis elegans. Science, 220: , Ellis, H. M. and Horvitz, H. R. Genetic control of programmed cell death in the nematode C. elegans. Cell, 44: , Yuan, J., Shaham, S., Ledoux, S., Ellis, H. M., and Horvitz, H. R. The C. elegans cell death gene ced-3 encodes a protein similar to mammalian interleukin-1 beta-converting enzyme. Cell, 75: , Trent, C., Tsuing, N., and Horvitz, H. R. Egg-laying defective mutants of the nematode Caenorhabditis elegans. Genetics, 104: , Hengartner, M. O., Ellis, R. E., and Horvitz, H. R. Caenorhabditis elegans gene ced-9 protects cells from programmed cell death. Nature, 356: , Conradt, B. and Horvitz, H. R. The C. elegans protein EGL-1 is required for programmed cell death and interacts with the Bcl-2-like protein CED-9. Cell, 93: , Shaham, S. and Horvitz, H. R. Developing Caenorhabditis elegans neurons may contain both cell-death protective and killer activities. Genes Dev, 10: , Riedl, S. J. and Shi, Y. Molecular mechanisms of caspase regulation during apoptosis. Nat Rev Mol Cell Biol, 5: , Martin, S. J., Henry, C. M., and Cullen, S. P. A perspective on mammalian caspases as positive and negative regulators of inflammation. Mol Cell, 46: , Hyman, B. T. and Yuan, J. Apoptotic and non-apoptotic roles of caspases in neuronal physiology and pathophysiology. Nat Rev Neurosci, 13: , Pop, C. and Salvesen, G. S. Human caspases: activation, specificity, and regulation. J Biol Chem, 284: ,
52 26. Mace, P. D. and Riedl, S. J. Molecular cell death platforms and assemblies. Curr Opin Cell Biol, 22: , Zou, H., Li, Y., Liu, X., and Wang, X. An APAF-1.cytochrome c multimeric complex is a functional apoptosome that activates procaspase-9. J Biol Chem, 274: , Acehan, D., Jiang, X., Morgan, D. G., Heuser, J. E., Wang, X., and Akey, C. W. Threedimensional structure of the apoptosome: implications for assembly, procaspase-9 binding, and activation. Mol Cell, 9: , Kim, H. E., Du, F., Fang, M., and Wang, X. Formation of apoptosome is initiated by cytochrome c-induced datp hydrolysis and subsequent nucleotide exchange on Apaf-1. Proc Natl Acad Sci U S A, 102: , Riedl, S. J., Li, W., Chao, Y., Schwarzenbacher, R., and Shi, Y. Structure of the apoptotic protease-activating factor 1 bound to ADP. Nature, 434: , Kischkel, F. C., Hellbardt, S., Behrmann, I., Germer, M., Pawlita, M., Krammer, P. H., and Peter, M. E. Cytotoxicity-dependent APO-1 (Fas/CD95)-associated proteins form a death-inducing signaling complex (DISC) with the receptor. EMBO J, 14: , Siegel, R. M., Frederiksen, J. K., Zacharias, D. A., Chan, F. K., Johnson, M., Lynch, D., Tsien, R. Y., and Lenardo, M. J. Fas preassociation required for apoptosis signaling and dominant inhibition by pathogenic mutations. Science, 288: , Scott, F. L., Stec, B., Pop, C., Dobaczewska, M. K., Lee, J. J., Monosov, E., Robinson, H., Salvesen, G. S., Schwarzenbacher, R., and Riedl, S. J. The Fas-FADD death domain complex structure unravels signalling by receptor clustering. Nature, 457: , Tinel, A. and Tschopp, J. The PIDDosome, a protein complex implicated in activation of caspase-2 in response to genotoxic stress. Science, 304: ,
53 35. Chai, J., Wu, Q., Shiozaki, E., Srinivasula, S. M., Alnemri, E. S., and Shi, Y. Crystal structure of a procaspase-7 zymogen: mechanisms of activation and substrate binding. Cell, 107: , Riedl, S. J., Fuentes-Prior, P., Renatus, M., Kairies, N., Krapp, S., Huber, R., Salvesen, G. S., and Bode, W. Structural basis for the activation of human procaspase-7. Proc Natl Acad Sci U S A, 98: , Dix, M. M., Simon, G. M., and Cravatt, B. F. Global mapping of the topography and magnitude of proteolytic events in apoptosis. Cell, 134: , Mahrus, S., Trinidad, J. C., Barkan, D. T., Sali, A., Burlingame, A. L., and Wells, J. A. Global sequencing of proteolytic cleavage sites in apoptosis by specific labeling of protein N termini. Cell, 134: , Enari, M., Sakahira, H., Yokoyama, H., Okawa, K., Iwamatsu, A., and Nagata, S. A caspase-activated DNase that degrades DNA during apoptosis, and its inhibitor ICAD. Nature, 391: 43-50, Sakahira, H., Enari, M., and Nagata, S. Cleavage of CAD inhibitor in CAD activation and DNA degradation during apoptosis. Nature, 391: 96-99, Eckelman, B. P., Salvesen, G. S., and Scott, F. L. Human inhibitor of apoptosis proteins: why XIAP is the black sheep of the family. EMBO Rep, 7: , Irmler, M., Thome, M., Hahne, M., Schneider, P., Hofmann, K., Steiner, V., Bodmer, J. L., Schroter, M., Burns, K., Mattmann, C., Rimoldi, D., French, L. E., and Tschopp, J. Inhibition of death receptor signals by cellular FLIP. Nature, 388: , Thome, M., Schneider, P., Hofmann, K., Fickenscher, H., Meinl, E., Neipel, F., Mattmann, C., Burns, K., Bodmer, J. L., Schroter, M., Scaffidi, C., Krammer, P. H., Peter, M. E., and Tschopp, J. Viral FLICE-inhibitory proteins (FLIPs) prevent apoptosis induced by death receptors. Nature, 386: , Schile, A. J., Garcia-Fernandez, M., and Steller, H. Regulation of apoptosis by XIAP ubiquitin-ligase activity. Genes Dev, 22: ,
54 45. Suzuki, Y., Nakabayashi, Y., and Takahashi, R. Ubiquitin-protein ligase activity of X- linked inhibitor of apoptosis protein promotes proteasomal degradation of caspase-3 and enhances its anti-apoptotic effect in Fas-induced cell death. Proc Natl Acad Sci U S A, 98: , Liu, X., Kim, C. N., Yang, J., Jemmerson, R., and Wang, X. Induction of apoptotic program in cell-free extracts: requirement for datp and cytochrome c. Cell, 86: , Li, L. Y., Luo, X., and Wang, X. Endonuclease G is an apoptotic DNase when released from mitochondria. Nature, 412: 95-99, Du, C., Fang, M., Li, Y., Li, L., and Wang, X. Smac, a mitochondrial protein that promotes cytochrome c-dependent caspase activation by eliminating IAP inhibition. Cell, 102: 33-42, Verhagen, A. M., Ekert, P. G., Pakusch, M., Silke, J., Connolly, L. M., Reid, G. E., Moritz, R. L., Simpson, R. J., and Vaux, D. L. Identification of DIABLO, a mammalian protein that promotes apoptosis by binding to and antagonizing IAP proteins. Cell, 102: 43-53, Suzuki, Y., Imai, Y., Nakayama, H., Takahashi, K., Takio, K., and Takahashi, R. A serine protease, HtrA2, is released from the mitochondria and interacts with XIAP, inducing cell death. Mol Cell, 8: , Scaffidi, C., Fulda, S., Srinivasan, A., Friesen, C., Li, F., Tomaselli, K. J., Debatin, K. M., Krammer, P. H., and Peter, M. E. Two CD95 (APO-1/Fas) signaling pathways. EMBO J, 17: , Jost, P. J., Grabow, S., Gray, D., McKenzie, M. D., Nachbur, U., Huang, D. C., Bouillet, P., Thomas, H. E., Borner, C., Silke, J., Strasser, A., and Kaufmann, T. XIAP discriminates between type I and type II FAS-induced apoptosis. Nature, 460: , Li, H., Zhu, H., Xu, C. J., and Yuan, J. Cleavage of BID by caspase 8 mediates the mitochondrial damage in the Fas pathway of apoptosis. Cell, 94: ,
55 54. Luo, X., Budihardjo, I., Zou, H., Slaughter, C., and Wang, X. Bid, a Bcl2 interacting protein, mediates cytochrome c release from mitochondria in response to activation of cell surface death receptors. Cell, 94: , Lavrik, I. N. and Krammer, P. H. Regulation of CD95/Fas signaling at the DISC. Cell Death Differ, 19: 36-41, Yin, X. M., Wang, K., Gross, A., Zhao, Y., Zinkel, S., Klocke, B., Roth, K. A., and Korsmeyer, S. J. Bid-deficient mice are resistant to Fas-induced hepatocellular apoptosis. Nature, 400: , Hockenbery, D., Nunez, G., Milliman, C., Schreiber, R. D., and Korsmeyer, S. J. Bcl-2 is an inner mitochondrial membrane protein that blocks programmed cell death. Nature, 348: , McDonnell, T. J., Deane, N., Platt, F. M., Nunez, G., Jaeger, U., McKearn, J. P., and Korsmeyer, S. J. bcl-2-immunoglobulin transgenic mice demonstrate extended B cell survival and follicular lymphoproliferation. Cell, 57: 79-88, McDonnell, T. J. and Korsmeyer, S. J. Progression from lymphoid hyperplasia to highgrade malignant lymphoma in mice transgenic for the t(14; 18). Nature, 349: , Hanahan, D. and Weinberg, R. A. The hallmarks of cancer. Cell, 100: 57-70, Youle, R. J. and Strasser, A. The BCL-2 protein family: opposing activities that mediate cell death. Nat Rev Mol Cell Biol, 9: 47-59, Kim, H., Rafiuddin-Shah, M., Tu, H. C., Jeffers, J. R., Zambetti, G. P., Hsieh, J. J., and Cheng, E. H. Hierarchical regulation of mitochondrion-dependent apoptosis by BCL-2 subfamilies. Nat Cell Biol, 8: , Cheng, E. H., Sheiko, T. V., Fisher, J. K., Craigen, W. J., and Korsmeyer, S. J. VDAC2 inhibits BAK activation and mitochondrial apoptosis. Science, 301: , Kim, H., Tu, H. C., Ren, D., Takeuchi, O., Jeffers, J. R., Zambetti, G. P., Hsieh, J. J., and Cheng, E. H. Stepwise activation of BAX and BAK by tbid, BIM, and PUMA initiates mitochondrial apoptosis. Mol Cell, 36: ,
56 65. Wei, M. C., Zong, W. X., Cheng, E. H., Lindsten, T., Panoutsakopoulou, V., Ross, A. J., Roth, K. A., MacGregor, G. R., Thompson, C. B., and Korsmeyer, S. J. Proapoptotic BAX and BAK: a requisite gateway to mitochondrial dysfunction and death. Science, 292: , Hsu, S. Y., Kaipia, A., McGee, E., Lomeli, M., and Hsueh, A. J. Bok is a pro-apoptotic Bcl-2 protein with restricted expression in reproductive tissues and heterodimerizes with selective anti-apoptotic Bcl-2 family members. Proc Natl Acad Sci U S A, 94: , Lindsten, T., Ross, A. J., King, A., Zong, W. X., Rathmell, J. C., Shiels, H. A., Ulrich, E., Waymire, K. G., Mahar, P., Frauwirth, K., Chen, Y., Wei, M., Eng, V. M., Adelman, D. M., Simon, M. C., Ma, A., Golden, J. A., Evan, G., Korsmeyer, S. J., MacGregor, G. R., and Thompson, C. B. The combined functions of proapoptotic Bcl-2 family members bak and bax are essential for normal development of multiple tissues. Mol Cell, 6: , Jacobson, M. D., Weil, M., and Raff, M. C. Programmed cell death in animal development. Cell, 88: , Cheng, E. H., Wei, M. C., Weiler, S., Flavell, R. A., Mak, T. W., Lindsten, T., and Korsmeyer, S. J. BCL-2, BCL-X(L) sequester BH3 domain-only molecules preventing BAX- and BAK-mediated mitochondrial apoptosis. Mol Cell, 8: , Zong, W. X., Lindsten, T., Ross, A. J., MacGregor, G. R., and Thompson, C. B. BH3-only proteins that bind pro-survival Bcl-2 family members fail to induce apoptosis in the absence of Bax and Bak. Genes Dev, 15: , Korsmeyer, S. J., Wei, M. C., Saito, M., Weiler, S., Oh, K. J., and Schlesinger, P. H. Proapoptotic cascade activates BID, which oligomerizes BAK or BAX into pores that result in the release of cytochrome c. Cell Death Differ, 7: , Suzuki, M., Youle, R. J., and Tjandra, N. Structure of Bax: coregulation of dimer formation and intracellular localization. Cell, 103: ,
57 73. Ren, D., Tu, H. C., Kim, H., Wang, G. X., Bean, G. R., Takeuchi, O., Jeffers, J. R., Zambetti, G. P., Hsieh, J. J., and Cheng, E. H. BID, BIM, and PUMA are essential for activation of the BAX- and BAK-dependent cell death program. Science, 330: , Han, J., Flemington, C., Houghton, A. B., Gu, Z., Zambetti, G. P., Lutz, R. J., Zhu, L., and Chittenden, T. Expression of bbc3, a pro-apoptotic BH3-only gene, is regulated by diverse cell death and survival signals. Proc Natl Acad Sci U S A, 98: , Nakano, K. and Vousden, K. H. PUMA, a novel proapoptotic gene, is induced by p53. Mol Cell, 7: , Oda, E., Ohki, R., Murasawa, H., Nemoto, J., Shibue, T., Yamashita, T., Tokino, T., Taniguchi, T., and Tanaka, N. Noxa, a BH3-only member of the Bcl-2 family and candidate mediator of p53-induced apoptosis. Science, 288: , Puthalakath, H., O'Reilly, L. A., Gunn, P., Lee, L., Kelly, P. N., Huntington, N. D., Hughes, P. D., Michalak, E. M., McKimm-Breschkin, J., Motoyama, N., Gotoh, T., Akira, S., Bouillet, P., and Strasser, A. ER stress triggers apoptosis by activating BH3-only protein Bim. Cell, 129: , Dijkers, P. F., Medema, R. H., Lammers, J. W., Koenderman, L., and Coffer, P. J. Expression of the pro-apoptotic Bcl-2 family member Bim is regulated by the forkhead transcription factor FKHR-L1. Curr Biol, 10: , Harada, H., Quearry, B., Ruiz-Vela, A., and Korsmeyer, S. J. Survival factor-induced extracellular signal-regulated kinase phosphorylates BIM, inhibiting its association with BAX and proapoptotic activity. Proc Natl Acad Sci U S A, 101: , Dehan, E., Bassermann, F., Guardavaccaro, D., Vasiliver-Shamis, G., Cohen, M., Lowes, K. N., Dustin, M., Huang, D. C., Taunton, J., and Pagano, M. betatrcp- and Rsk1/2- mediated degradation of BimEL inhibits apoptosis. Mol Cell, 33: , Grad, J. M., Zeng, X. R., and Boise, L. H. Regulation of Bcl-xL: a little bit of this and a little bit of STAT. Curr Opin Oncol, 12: ,
58 82. Boise, L. H., Minn, A. J., Noel, P. J., June, C. H., Accavitti, M. A., Lindsten, T., and Thompson, C. B. CD28 costimulation can promote T cell survival by enhancing the expression of Bcl-XL. Immunity, 3: 87-98, Khoshnan, A., Tindell, C., Laux, I., Bae, D., Bennett, B., and Nel, A. E. The NF-kappa B cascade is important in Bcl-xL expression and for the anti-apoptotic effects of the CD28 receptor in primary human CD4+ lymphocytes. J Immunol, 165: , Lee, H. H., Dadgostar, H., Cheng, Q., Shu, J., and Cheng, G. NF-kappaB-mediated upregulation of Bcl-x and Bfl-1/A1 is required for CD40 survival signaling in B lymphocytes. Proc Natl Acad Sci U S A, 96: , Thomas, L. W., Lam, C., and Edwards, S. W. Mcl-1; the molecular regulation of protein function. FEBS Lett, 584: , Zhong, Q., Gao, W., Du, F., and Wang, X. Mule/ARF-BP1, a BH3-only E3 ubiquitin ligase, catalyzes the polyubiquitination of Mcl-1 and regulates apoptosis. Cell, 121: , Ding, Q., He, X., Hsu, J. M., Xia, W., Chen, C. T., Li, L. Y., Lee, D. F., Liu, J. C., Zhong, Q., Wang, X., and Hung, M. C. Degradation of Mcl-1 by beta-trcp mediates glycogen synthase kinase 3-induced tumor suppression and chemosensitization. Mol Cell Biol, 27: , Inuzuka, H., Shaik, S., Onoyama, I., Gao, D., Tseng, A., Maser, R. S., Zhai, B., Wan, L., Gutierrez, A., Lau, A. W., Xiao, Y., Christie, A. L., Aster, J., Settleman, J., Gygi, S. P., Kung, A. L., Look, T., Nakayama, K. I., DePinho, R. A., and Wei, W. SCF(FBW7) regulates cellular apoptosis by targeting MCL1 for ubiquitylation and destruction. Nature, 471: , Wertz, I. E., Kusam, S., Lam, C., Okamoto, T., Sandoval, W., Anderson, D. J., Helgason, E., Ernst, J. A., Eby, M., Liu, J., Belmont, L. D., Kaminker, J. S., O'Rourke, K. M., Pujara, K., Kohli, P. B., Johnson, A. R., Chiu, M. L., Lill, J. R., Jackson, P. K., Fairbrother, W. J., Seshagiri, S., Ludlam, M. J., Leong, K. G., Dueber, E. C., Maecker, H., Huang, D. C., and 46
59 Dixit, V. M. Sensitivity to antitubulin chemotherapeutics is regulated by MCL1 and FBW7. Nature, 471: , Schwickart, M., Huang, X., Lill, J. R., Liu, J., Ferrando, R., French, D. M., Maecker, H., O'Rourke, K., Bazan, F., Eastham-Anderson, J., Yue, P., Dornan, D., Huang, D. C., and Dixit, V. M. Deubiquitinase USP9X stabilizes MCL1 and promotes tumour cell survival. Nature, 463: , Kroemer, G., Galluzzi, L., and Brenner, C. Mitochondrial membrane permeabilization in cell death. Physiol Rev, 87: , Yoshida, H., Kong, Y. Y., Yoshida, R., Elia, A. J., Hakem, A., Hakem, R., Penninger, J. M., and Mak, T. W. Apaf1 is required for mitochondrial pathways of apoptosis and brain development. Cell, 94: , Kuida, K., Haydar, T. F., Kuan, C. Y., Gu, Y., Taya, C., Karasuyama, H., Su, M. S., Rakic, P., and Flavell, R. A. Reduced apoptosis and cytochrome c-mediated caspase activation in mice lacking caspase 9. Cell, 94: , Wallace, D. C. A mitochondrial paradigm of metabolic and degenerative diseases, aging, and cancer: a dawn for evolutionary medicine. Annu Rev Genet, 39: , Ricci, J. E., Munoz-Pinedo, C., Fitzgerald, P., Bailly-Maitre, B., Perkins, G. A., Yadava, N., Scheffler, I. E., Ellisman, M. H., and Green, D. R. Disruption of mitochondrial function during apoptosis is mediated by caspase cleavage of the p75 subunit of complex I of the electron transport chain. Cell, 117: , Waterhouse, N. J., Goldstein, J. C., von Ahsen, O., Schuler, M., Newmeyer, D. D., and Green, D. R. Cytochrome c maintains mitochondrial transmembrane potential and ATP generation after outer mitochondrial membrane permeabilization during the apoptotic process. J Cell Biol, 153: , Lartigue, L., Kushnareva, Y., Seong, Y., Lin, H., Faustin, B., and Newmeyer, D. D. Caspase-independent mitochondrial cell death results from loss of respiration, not cytotoxic protein release. Mol Biol Cell, 20: ,
60 98. Goemans, C. G., Boya, P., Skirrow, C. J., and Tolkovsky, A. M. Intra-mitochondrial degradation of Tim23 curtails the survival of cells rescued from apoptosis by caspase inhibitors. Cell Death Differ, 15: , Vahsen, N., Cande, C., Briere, J. J., Benit, P., Joza, N., Larochette, N., Mastroberardino, P. G., Pequignot, M. O., Casares, N., Lazar, V., Feraud, O., Debili, N., Wissing, S., Engelhardt, S., Madeo, F., Piacentini, M., Penninger, J. M., Schagger, H., Rustin, P., and Kroemer, G. AIF deficiency compromises oxidative phosphorylation. EMBO J, 23: , Susin, S. A., Lorenzo, H. K., Zamzami, N., Marzo, I., Snow, B. E., Brothers, G. M., Mangion, J., Jacotot, E., Costantini, P., Loeffler, M., Larochette, N., Goodlett, D. R., Aebersold, R., Siderovski, D. P., Penninger, J. M., and Kroemer, G. Molecular characterization of mitochondrial apoptosis-inducing factor. Nature, 397: , Vande Walle, L., Lamkanfi, M., and Vandenabeele, P. The mitochondrial serine protease HtrA2/Omi: an overview. Cell Death Differ, 15: , Jones, J. M., Datta, P., Srinivasula, S. M., Ji, W., Gupta, S., Zhang, Z., Davies, E., Hajnoczky, G., Saunders, T. L., Van Keuren, M. L., Fernandes-Alnemri, T., Meisler, M. H., and Alnemri, E. S. Loss of Omi mitochondrial protease activity causes the neuromuscular disorder of mnd2 mutant mice. Nature, 425: , Yang, Z. and Klionsky, D. J. Mammalian autophagy: core molecular machinery and signaling regulation. Curr Opin Cell Biol, 22: , Kroemer, G., Marino, G., and Levine, B. Autophagy and the integrated stress response. Mol Cell, 40: , Hailey, D. W., Rambold, A. S., Satpute-Krishnan, P., Mitra, K., Sougrat, R., Kim, P. K., and Lippincott-Schwartz, J. Mitochondria supply membranes for autophagosome biogenesis during starvation. Cell, 141: , Yla-Anttila, P., Vihinen, H., Jokitalo, E., and Eskelinen, E. L. 3D tomography reveals connections between the phagophore and endoplasmic reticulum. Autophagy, 5: ,
61 107. Yu, L., McPhee, C. K., Zheng, L., Mardones, G. A., Rong, Y., Peng, J., Mi, N., Zhao, Y., Liu, Z., Wan, F., Hailey, D. W., Oorschot, V., Klumperman, J., Baehrecke, E. H., and Lenardo, M. J. Termination of autophagy and reformation of lysosomes regulated by mtor. Nature, 465: , Jung, C. H., Jun, C. B., Ro, S. H., Kim, Y. M., Otto, N. M., Cao, J., Kundu, M., and Kim, D. H. ULK-Atg13-FIP200 complexes mediate mtor signaling to the autophagy machinery. Mol Biol Cell, 20: , Hosokawa, N., Hara, T., Kaizuka, T., Kishi, C., Takamura, A., Miura, Y., Iemura, S., Natsume, T., Takehana, K., Yamada, N., Guan, J. L., Oshiro, N., and Mizushima, N. Nutrient-dependent mtorc1 association with the ULK1-Atg13-FIP200 complex required for autophagy. Mol Biol Cell, 20: , Liang, X. H., Jackson, S., Seaman, M., Brown, K., Kempkes, B., Hibshoosh, H., and Levine, B. Induction of autophagy and inhibition of tumorigenesis by beclin 1. Nature, 402: , Funderburk, S. F., Wang, Q. J., and Yue, Z. The Beclin 1-VPS34 complex--at the crossroads of autophagy and beyond. Trends Cell Biol, 20: , Zhong, Y., Wang, Q. J., Li, X., Yan, Y., Backer, J. M., Chait, B. T., Heintz, N., and Yue, Z. Distinct regulation of autophagic activity by Atg14L and Rubicon associated with Beclin 1- phosphatidylinositol-3-kinase complex. Nat Cell Biol, 11: , Matsunaga, K., Saitoh, T., Tabata, K., Omori, H., Satoh, T., Kurotori, N., Maejima, I., Shirahama-Noda, K., Ichimura, T., Isobe, T., Akira, S., Noda, T., and Yoshimori, T. Two Beclin 1-binding proteins, Atg14L and Rubicon, reciprocally regulate autophagy at different stages. Nat Cell Biol, 11: , Nakatogawa, H., Ichimura, Y., and Ohsumi, Y. Atg8, a ubiquitin-like protein required for autophagosome formation, mediates membrane tethering and hemifusion. Cell, 130: ,
62 115. Young, A. R., Chan, E. Y., Hu, X. W., Kochl, R., Crawshaw, S. G., High, S., Hailey, D. W., Lippincott-Schwartz, J., and Tooze, S. A. Starvation and ULK1-dependent cycling of mammalian Atg9 between the TGN and endosomes. J Cell Sci, 119: , Ropolo, A., Grasso, D., Pardo, R., Sacchetti, M. L., Archange, C., Lo Re, A., Seux, M., Nowak, J., Gonzalez, C. D., Iovanna, J. L., and Vaccaro, M. I. The pancreatitis-induced vacuole membrane protein 1 triggers autophagy in mammalian cells. J Biol Chem, 282: , Mizushima, N. Autophagy: process and function. Genes Dev, 21: , Lum, J. J., DeBerardinis, R. J., and Thompson, C. B. Autophagy in metazoans: cell survival in the land of plenty. Nat Rev Mol Cell Biol, 6: , Yang, Z. and Klionsky, D. J. Eaten alive: a history of macroautophagy. Nat Cell Biol, 12: , Kuma, A., Hatano, M., Matsui, M., Yamamoto, A., Nakaya, H., Yoshimori, T., Ohsumi, Y., Tokuhisa, T., and Mizushima, N. The role of autophagy during the early neonatal starvation period. Nature, 432: , Komatsu, M., Waguri, S., Ueno, T., Iwata, J., Murata, S., Tanida, I., Ezaki, J., Mizushima, N., Ohsumi, Y., Uchiyama, Y., Kominami, E., Tanaka, K., and Chiba, T. Impairment of starvation-induced and constitutive autophagy in Atg7-deficient mice. J Cell Biol, 169: , Blommaart, E. F., Luiken, J. J., Blommaart, P. J., van Woerkom, G. M., and Meijer, A. J. Phosphorylation of ribosomal protein S6 is inhibitory for autophagy in isolated rat hepatocytes. J Biol Chem, 270: , Imai, S. and Guarente, L. Ten years of NAD-dependent SIR2 family deacetylases: implications for metabolic diseases. Trends Pharmacol Sci, 31: , Lee, I. H., Cao, L., Mostoslavsky, R., Lombard, D. B., Liu, J., Bruns, N. E., Tsokos, M., Alt, F. W., and Finkel, T. A role for the NAD-dependent deacetylase Sirt1 in the regulation of autophagy. Proc Natl Acad Sci U S A, 105: ,
63 125. Zhao, Y., Yang, J., Liao, W., Liu, X., Zhang, H., Wang, S., Wang, D., Feng, J., Yu, L., and Zhu, W. G. Cytosolic FoxO1 is essential for the induction of autophagy and tumour suppressor activity. Nat Cell Biol, 12: , Levine, B. and Kroemer, G. Autophagy in the pathogenesis of disease. Cell, 132: 27-42, Iwata, J., Ezaki, J., Komatsu, M., Yokota, S., Ueno, T., Tanida, I., Chiba, T., Tanaka, K., and Kominami, E. Excess peroxisomes are degraded by autophagic machinery in mammals. J Biol Chem, 281: , Smith, J. J. and Aitchison, J. D. Regulation of peroxisome dynamics. Curr Opin Cell Biol, 21: , Ross, C. A. and Poirier, M. A. Protein aggregation and neurodegenerative disease. Nat Med, 10 Suppl: S10-17, Ravikumar, B., Acevedo-Arozena, A., Imarisio, S., Berger, Z., Vacher, C., O'Kane, C. J., Brown, S. D., and Rubinsztein, D. C. Dynein mutations impair autophagic clearance of aggregate-prone proteins. Nat Genet, 37: , Ravikumar, B., Vacher, C., Berger, Z., Davies, J. E., Luo, S., Oroz, L. G., Scaravilli, F., Easton, D. F., Duden, R., O'Kane, C. J., and Rubinsztein, D. C. Inhibition of mtor induces autophagy and reduces toxicity of polyglutamine expansions in fly and mouse models of Huntington disease. Nat Genet, 36: , Narendra, D., Tanaka, A., Suen, D. F., and Youle, R. J. Parkin is recruited selectively to impaired mitochondria and promotes their autophagy. J Cell Biol, 183: , Colell, A., Ricci, J. E., Tait, S., Milasta, S., Maurer, U., Bouchier-Hayes, L., Fitzgerald, P., Guio-Carrion, A., Waterhouse, N. J., Li, C. W., Mari, B., Barbry, P., Newmeyer, D. D., Beere, H. M., and Green, D. R. GAPDH and autophagy preserve survival after apoptotic cytochrome c release in the absence of caspase activation. Cell, 129: , Narendra, D. P., Jin, S. M., Tanaka, A., Suen, D. F., Gautier, C. A., Shen, J., Cookson, M. R., and Youle, R. J. PINK1 is selectively stabilized on impaired mitochondria to activate Parkin. PLoS Biol, 8: e ,
64 135. Matsuda, N., Sato, S., Shiba, K., Okatsu, K., Saisho, K., Gautier, C. A., Sou, Y. S., Saiki, S., Kawajiri, S., Sato, F., Kimura, M., Komatsu, M., Hattori, N., and Tanaka, K. PINK1 stabilized by mitochondrial depolarization recruits Parkin to damaged mitochondria and activates latent Parkin for mitophagy. J Cell Biol, 189: , Jin, S. M., Lazarou, M., Wang, C., Kane, L. A., Narendra, D. P., and Youle, R. J. Mitochondrial membrane potential regulates PINK1 import and proteolytic destabilization by PARL. J Cell Biol, 191: , Lin, M. T. and Beal, M. F. Mitochondrial dysfunction and oxidative stress in neurodegenerative diseases. Nature, 443: , Yue, Z., Jin, S., Yang, C., Levine, A. J., and Heintz, N. Beclin 1, an autophagy gene essential for early embryonic development, is a haploinsufficient tumor suppressor. Proc Natl Acad Sci U S A, 100: , Aita, V. M., Liang, X. H., Murty, V. V., Pincus, D. L., Yu, W., Cayanis, E., Kalachikov, S., Gilliam, T. C., and Levine, B. Cloning and genomic organization of beclin 1, a candidate tumor suppressor gene on chromosome 17q21. Genomics, 59: 59-65, Pattingre, S., Tassa, A., Qu, X., Garuti, R., Liang, X. H., Mizushima, N., Packer, M., Schneider, M. D., and Levine, B. Bcl-2 antiapoptotic proteins inhibit Beclin 1-dependent autophagy. Cell, 122: , Yu, L., Alva, A., Su, H., Dutt, P., Freundt, E., Welsh, S., Baehrecke, E. H., and Lenardo, M. J. Regulation of an ATG7-beclin 1 program of autophagic cell death by caspase-8. Science, 304: , Shimizu, S., Kanaseki, T., Mizushima, N., Mizuta, T., Arakawa-Kobayashi, S., Thompson, C. B., and Tsujimoto, Y. Role of Bcl-2 family proteins in a non-apoptotic programmed cell death dependent on autophagy genes. Nat Cell Biol, 6: , Berry, D. L. and Baehrecke, E. H. Growth arrest and autophagy are required for salivary gland cell degradation in Drosophila. Cell, 131: , Scott, R. C., Juhasz, G., and Neufeld, T. P. Direct induction of autophagy by Atg1 inhibits cell growth and induces apoptotic cell death. Curr Biol, 17: 1-11,
65 145. Koike, M., Shibata, M., Tadakoshi, M., Gotoh, K., Komatsu, M., Waguri, S., Kawahara, N., Kuida, K., Nagata, S., Kominami, E., Tanaka, K., and Uchiyama, Y. Inhibition of autophagy prevents hippocampal pyramidal neuron death after hypoxic-ischemic injury. Am J Pathol, 172: , Goossens, V., Grooten, J., and Fiers, W. The oxidative metabolism of glutamine. A modulator of reactive oxygen intermediate-mediated cytotoxicity of tumor necrosis factor in L929 fibrosarcoma cells. J Biol Chem, 271: , Goossens, V., Grooten, J., De Vos, K., and Fiers, W. Direct evidence for tumor necrosis factor-induced mitochondrial reactive oxygen intermediates and their involvement in cytotoxicity. Proc Natl Acad Sci U S A, 92: , Goossens, V., Stange, G., Moens, K., Pipeleers, D., and Grooten, J. Regulation of tumor necrosis factor-induced, mitochondria- and reactive oxygen species-dependent cell death by the electron flux through the electron transport chain complex I. Antioxid Redox Signal, 1: , Vercammen, D., Beyaert, R., Denecker, G., Goossens, V., Van Loo, G., Declercq, W., Grooten, J., Fiers, W., and Vandenabeele, P. Inhibition of caspases increases the sensitivity of L929 cells to necrosis mediated by tumor necrosis factor. J Exp Med, 187: , Vercammen, D., Brouckaert, G., Denecker, G., Van de Craen, M., Declercq, W., Fiers, W., and Vandenabeele, P. Dual signaling of the Fas receptor: initiation of both apoptotic and necrotic cell death pathways. J Exp Med, 188: , Holler, N., Zaru, R., Micheau, O., Thome, M., Attinger, A., Valitutti, S., Bodmer, J. L., Schneider, P., Seed, B., and Tschopp, J. Fas triggers an alternative, caspase-8- independent cell death pathway using the kinase RIP as effector molecule. Nat Immunol, 1: , Degterev, A., Hitomi, J., Germscheid, M., Ch'en, I. L., Korkina, O., Teng, X., Abbott, D., Cuny, G. D., Yuan, C., Wagner, G., Hedrick, S. M., Gerber, S. A., Lugovskoy, A., and 53
66 Yuan, J. Identification of RIP1 kinase as a specific cellular target of necrostatins. Nat Chem Biol, 4: , Degterev, A., Huang, Z., Boyce, M., Li, Y., Jagtap, P., Mizushima, N., Cuny, G. D., Mitchison, T. J., Moskowitz, M. A., and Yuan, J. Chemical inhibitor of nonapoptotic cell death with therapeutic potential for ischemic brain injury. Nat Chem Biol, 1: , Nakagawa, T., Shimizu, S., Watanabe, T., Yamaguchi, O., Otsu, K., Yamagata, H., Inohara, H., Kubo, T., and Tsujimoto, Y. Cyclophilin D-dependent mitochondrial permeability transition regulates some necrotic but not apoptotic cell death. Nature, 434: , Baines, C. P., Kaiser, R. A., Purcell, N. H., Blair, N. S., Osinska, H., Hambleton, M. A., Brunskill, E. W., Sayen, M. R., Gottlieb, R. A., Dorn, G. W., Robbins, J., and Molkentin, J. D. Loss of cyclophilin D reveals a critical role for mitochondrial permeability transition in cell death. Nature, 434: , Schinzel, A. C., Takeuchi, O., Huang, Z., Fisher, J. K., Zhou, Z., Rubens, J., Hetz, C., Danial, N. N., Moskowitz, M. A., and Korsmeyer, S. J. Cyclophilin D is a component of mitochondrial permeability transition and mediates neuronal cell death after focal cerebral ischemia. Proc Natl Acad Sci U S A, 102: , Wang, Z., Jiang, H., Chen, S., Du, F., and Wang, X. The mitochondrial phosphatase PGAM5 functions at the convergence point of multiple necrotic death pathways. Cell, 148: , Wang, L., Du, F., and Wang, X. TNF-alpha induces two distinct caspase-8 activation pathways. Cell, 133: , Hsu, H., Huang, J., Shu, H. B., Baichwal, V., and Goeddel, D. V. TNF-dependent recruitment of the protein kinase RIP to the TNF receptor-1 signaling complex. Immunity, 4: , Vandenabeele, P., Declercq, W., Van Herreweghe, F., and Vanden Berghe, T. The role of the kinases RIP1 and RIP3 in TNF-induced necrosis. Sci Signal, 3: re4,
67 161. Micheau, O. and Tschopp, J. Induction of TNF receptor I-mediated apoptosis via two sequential signaling complexes. Cell, 114: , Hacker, H. and Karin, M. Regulation and function of IKK and IKK-related kinases. Sci STKE, 2006: re13, He, S., Wang, L., Miao, L., Wang, T., Du, F., Zhao, L., and Wang, X. Receptor interacting protein kinase-3 determines cellular necrotic response to TNF-alpha. Cell, 137: , Cho, Y. S., Challa, S., Moquin, D., Genga, R., Ray, T. D., Guildford, M., and Chan, F. K. Phosphorylation-driven assembly of the RIP1-RIP3 complex regulates programmed necrosis and virus-induced inflammation. Cell, 137: , Zhang, D. W., Shao, J., Lin, J., Zhang, N., Lu, B. J., Lin, S. C., Dong, M. Q., and Han, J. RIP3, an energy metabolism regulator that switches TNF-induced cell death from apoptosis to necrosis. Science, 325: , Feng, S., Yang, Y., Mei, Y., Ma, L., Zhu, D. E., Hoti, N., Castanares, M., and Wu, M. Cleavage of RIP3 inactivates its caspase-independent apoptosis pathway by removal of kinase domain. Cell Signal, 19: , Lin, Y., Devin, A., Rodriguez, Y., and Liu, Z. G. Cleavage of the death domain kinase RIP by caspase-8 prompts TNF-induced apoptosis. Genes Dev, 13: , Sun, L., Wang, H., Wang, Z., He, S., Chen, S., Liao, D., Wang, L., Yan, J., Liu, W., Lei, X., and Wang, X. Mixed lineage kinase domain-like protein mediates necrosis signaling downstream of RIP3 kinase. Cell, 148: , Eliasson, M. J., Sampei, K., Mandir, A. S., Hurn, P. D., Traystman, R. J., Bao, J., Pieper, A., Wang, Z. Q., Dawson, T. M., Snyder, S. H., and Dawson, V. L. Poly(ADP-ribose) polymerase gene disruption renders mice resistant to cerebral ischemia. Nat Med, 3: , van Wijk, S. J. and Hageman, G. J. Poly(ADP-ribose) polymerase-1 mediated caspaseindependent cell death after ischemia/reperfusion. Free Radic Biol Med, 39: 81-90,
68 171. Yu, S. W., Wang, H., Poitras, M. F., Coombs, C., Bowers, W. J., Federoff, H. J., Poirier, G. G., Dawson, T. M., and Dawson, V. L. Mediation of poly(adp-ribose) polymerase-1- dependent cell death by apoptosis-inducing factor. Science, 297: , Zong, W. X., Ditsworth, D., Bauer, D. E., Wang, Z. Q., and Thompson, C. B. Alkylating DNA damage stimulates a regulated form of necrotic cell death. Genes Dev, 18: , DeBerardinis, R. J., Lum, J. J., Hatzivassiliou, G., and Thompson, C. B. The biology of cancer: metabolic reprogramming fuels cell growth and proliferation. Cell Metab, 7: 11-20, Frey, T. G., Renken, C. W., and Perkins, G. A. Insight into mitochondrial structure and function from electron tomography. Biochim Biophys Acta, 1555: , Halestrap, A. P., Clarke, S. J., and Javadov, S. A. Mitochondrial permeability transition pore opening during myocardial reperfusion--a target for cardioprotection. Cardiovasc Res, 61: , Kokoszka, J. E., Waymire, K. G., Levy, S. E., Sligh, J. E., Cai, J., Jones, D. P., MacGregor, G. R., and Wallace, D. C. The ADP/ATP translocator is not essential for the mitochondrial permeability transition pore. Nature, 427: , Baines, C. P., Kaiser, R. A., Sheiko, T., Craigen, W. J., and Molkentin, J. D. Voltagedependent anion channels are dispensable for mitochondrial-dependent cell death. Nat Cell Biol, 9: , Ren, D., Kim, H., Tu, H. C., Westergard, T. D., Fisher, J. K., Rubens, J. A., Korsmeyer, S. J., Hsieh, J. J., and Cheng, E. H. The VDAC2-BAK rheostat controls thymocyte survival. Sci Signal, 2: ra48, Vandenabeele, P., Galluzzi, L., Vanden Berghe, T., and Kroemer, G. Molecular mechanisms of necroptosis: an ordered cellular explosion. Nat Rev Mol Cell Biol, 11: , Avery, S. V. Molecular targets of oxidative stress. Biochem J, 434: ,
69 181. Murphy, M. P. How mitochondria produce reactive oxygen species. Biochem J, 417: 1-13, Clapham, D. E. Calcium signaling. Cell, 131: , Grimm, S. The ER-mitochondria interface: the social network of cell death. Biochim Biophys Acta, 1823: , Westermann, B. Mitochondrial fusion and fission in cell life and death. Nat Rev Mol Cell Biol, 11: , Han, X. J., Lu, Y. F., Li, S. A., Kaitsuka, T., Sato, Y., Tomizawa, K., Nairn, A. C., Takei, K., Matsui, H., and Matsushita, M. CaM kinase I alpha-induced phosphorylation of Drp1 regulates mitochondrial morphology. J Cell Biol, 182: , Cereghetti, G. M., Stangherlin, A., Martins de Brito, O., Chang, C. R., Blackstone, C., Bernardi, P., and Scorrano, L. Dephosphorylation by calcineurin regulates translocation of Drp1 to mitochondria. Proc Natl Acad Sci U S A, 105: , Yu, T., Robotham, J. L., and Yoon, Y. Increased production of reactive oxygen species in hyperglycemic conditions requires dynamic change of mitochondrial morphology. Proc Natl Acad Sci U S A, 103: , Yamashima, T., Tonchev, A. B., Tsukada, T., Saido, T. C., Imajoh-Ohmi, S., Momoi, T., and Kominami, E. Sustained calpain activation associated with lysosomal rupture executes necrosis of the postischemic CA1 neurons in primates. Hippocampus, 13: , Yamashima, T., Kohda, Y., Tsuchiya, K., Ueno, T., Yamashita, J., Yoshioka, T., and Kominami, E. Inhibition of ischaemic hippocampal neuronal death in primates with cathepsin B inhibitor CA-074: a novel strategy for neuroprotection based on 'calpaincathepsin hypothesis'. Eur J Neurosci, 10: , Kelliher, M. A., Grimm, S., Ishida, Y., Kuo, F., Stanger, B. Z., and Leder, P. The death domain kinase RIP mediates the TNF-induced NF-kappaB signal. Immunity, 8: ,
70 191. Lim, S. Y., Davidson, S. M., Mocanu, M. M., Yellon, D. M., and Smith, C. C. The cardioprotective effect of necrostatin requires the cyclophilin-d component of the mitochondrial permeability transition pore. Cardiovasc Drugs Ther, 21: , You, Z., Savitz, S. I., Yang, J., Degterev, A., Yuan, J., Cuny, G. D., Moskowitz, M. A., and Whalen, M. J. Necrostatin-1 reduces histopathology and improves functional outcome after controlled cortical impact in mice. J Cereb Blood Flow Metab, 28: , Burkart, V., Wang, Z. Q., Radons, J., Heller, B., Herceg, Z., Stingl, L., Wagner, E. F., and Kolb, H. Mice lacking the poly(adp-ribose) polymerase gene are resistant to pancreatic beta-cell destruction and diabetes development induced by streptozocin. Nat Med, 5: , Yuan, J., Lipinski, M., and Degterev, A. Diversity in the mechanisms of neuronal cell death. Neuron, 40: , Kaiser, W. J., Upton, J. W., Long, A. B., Livingston-Rosanoff, D., Daley-Bauer, L. P., Hakem, R., Caspary, T., and Mocarski, E. S. RIP3 mediates the embryonic lethality of caspase-8-deficient mice. Nature, 471: , Oberst, A., Dillon, C. P., Weinlich, R., McCormick, L. L., Fitzgerald, P., Pop, C., Hakem, R., Salvesen, G. S., and Green, D. R. Catalytic activity of the caspase-8-flip(l) complex inhibits RIPK3-dependent necrosis. Nature, 471: , Zhang, H., Zhou, X., McQuade, T., Li, J., Chan, F. K., and Zhang, J. Functional complementation between FADD and RIP1 in embryos and lymphocytes. Nature, 471: , Varfolomeev, E. E., Schuchmann, M., Luria, V., Chiannilkulchai, N., Beckmann, J. S., Mett, I. L., Rebrikov, D., Brodianski, V. M., Kemper, O. C., Kollet, O., Lapidot, T., Soffer, D., Sobe, T., Avraham, K. B., Goncharov, T., Holtmann, H., Lonai, P., and Wallach, D. Targeted disruption of the mouse Caspase 8 gene ablates cell death induction by the TNF receptors, Fas/Apo1, and DR3 and is lethal prenatally. Immunity, 9: , Yeh, W. C., Itie, A., Elia, A. J., Ng, M., Shu, H. B., Wakeham, A., Mirtsos, C., Suzuki, N., Bonnard, M., Goeddel, D. V., and Mak, T. W. Requirement for Casper (c-flip) in 58
71 regulation of death receptor-induced apoptosis and embryonic development. Immunity, 12: , Yeh, W. C., Pompa, J. L., McCurrach, M. E., Shu, H. B., Elia, A. J., Shahinian, A., Ng, M., Wakeham, A., Khoo, W., Mitchell, K., El-Deiry, W. S., Lowe, S. W., Goeddel, D. V., and Mak, T. W. FADD: essential for embryo development and signaling from some, but not all, inducers of apoptosis. Science, 279: , Newton, K., Sun, X., and Dixit, V. M. Kinase RIP3 is dispensable for normal NF-kappa Bs, signaling by the B-cell and T-cell receptors, tumor necrosis factor receptor 1, and Toll-like receptors 2 and 4. Mol Cell Biol, 24: , Chan, F. K., Shisler, J., Bixby, J. G., Felices, M., Zheng, L., Appel, M., Orenstein, J., Moss, B., and Lenardo, M. J. A role for tumor necrosis factor receptor-2 and receptorinteracting protein in programmed necrosis and antiviral responses. J Biol Chem, 278: , Upton, J. W., Kaiser, W. J., and Mocarski, E. S. Cytomegalovirus M45 cell death suppression requires receptor-interacting protein (RIP) homotypic interaction motif (RHIM)-dependent interaction with RIP1. J Biol Chem, 283: , Upton, J. W., Kaiser, W. J., and Mocarski, E. S. Virus inhibition of RIP3-dependent necrosis. Cell Host Microbe, 7: ,
72 -CHAPTER TWO- p63 Inhibits Oxidative Stress-Induced Cell Death by Orchestrating Glutathione Metabolism 60
73 2.1 Abstract Despite the importance of ROS in executing cell death, its regulation remains elusive. Here, we report that p63 dictates the homeostasis of intracellular redox through transcriptional controls of glutathione metabolism genes. Overexpression of ΔNp63α, the major isoform of p63, protected cells from oxidative stress induced by thiol or lipid oxidants, DNA damage, and anoikis. Conversely, deficiency of ΔNp63α increased oxidative stress. Strikingly, overexpression of ΔNp63α rendered clonogenic survival of p53 -/- Bax -/- Bak -/- triple-knockout cells against DNA damage. Along the same line, co-expression of BCL-2 and ΔNp63α, which abrogated apoptosis and oxidative stress, respectively, produced clonogenic survival against matrix detachment and disrupted the luminal clearance of mammary acini. In conclusion, inhibiting apoptosis only offers short-term survival, whereas a simultaneous blockade of apoptosis and oxidative stress is central for long-term survival. 2.2 Introduction Proper execution of cell death ensures normal biological processes, and its deregulation causes human illness, ranging from cancer to neurodegenerative disorders 1. Three forms of cell death have been recognized based on morphological features: apoptosis, autophagic cell death, and necrosis 2. Among the three distinct forms of cell death, apoptosis is best studied. The BCL-2 family proteins are central regulators of apoptosis 3. Genetic loss-of-function studies reveal an essential axis of upstream activator BH3-only molecules and downstream BAX and BAK in activating mitochondrion-dependent apoptosis 4-7. In response to apoptotic signals, the activator BH3-only molecules, including BID, BIM, and PUMA, trigger the homo-oligomerization of multidomain proapoptotic BAX and BAK to permeabilize mitochondria, leading to the efflux of cytochrome c which in turn initiates the assembly of apoptosome complex for caspase activation Although apoptosis has long been considered as the major cell death mechanism required for the successful development and maintenance of tissue homeostasis in metazoans 2, double deficiency of Bax and Bak only disrupts the development and homeostasis in restricted sets of tissues 4, suggesting the existence of BAX and BAK-independent cell death mechanism(s) in 61
74 maintaining the tissue homeostatic state. To explore this cell death conundrum, we utilized transformed Bax -/- Bak -/- double-knockout (DKO) mouse embryonic fibroblasts (MEFs) and discovered that these apoptosis-deficient cells underwent a regulated form of necrotic cell death in response to DNA damage induced by topoisomerase inhibitors 11. Surprisingly, this type of necrotic cell death requires active transcription/translation 11, which is in stark contrast with a general perception that necrosis is not programmed. As this DNA damage-induced programmed necrotic death (PND) does not require RIP1, it is distinct from TNF-induced necroptosis 12 (fig. S2.1). Furthermore, we have reported that this death is independent of caspases, mitochondrial permeability transition pore (PTP), autophagy, or PARP 11. Notably, DNA alkylation induces PARP-dependent necrotic death whereas double-strand DNA breaks induces PARP-independent necrotic death in DKO cells 11,13. Mechanistically, we delineated a p53-cathepsin axis that cooperates with ROS (reactive oxygen species) to activate PND in DKO cells undergoing doublestrand DNA breaks 11. By analogy to DNA damage-induced cell death, it was reported that inhibition of apoptosis by BCL-2 is insufficient to provide long-term clonogenic survival against anoikis 14,15, a form of cell death that is induced by detachment from extracellular matrix in anchorage-dependent cells. Remarkably, antioxidant cooperates with BCL-2 to enhance clonogenic survival and prevent the luminal clearance of acini in three-dimensional culture of mammary epithelial cells 15. Thus, although apoptosis is the fastest mechanism for eliminating cells in response to death stimuli, inhibition of apoptosis is insufficient to render long-term clonogenic survival, which is indeed required for the prevention of pathological loss of cells during disease processes. ROS is one of the most important second messengers in executing necrosis 16 and appears to play a critical role in abrogating long-term clonogenic survival. Despite its essence, how ROS is regulated remains largely uncharacterized. 2.3 Results Because our data indicated that genotoxic stress-induced necrotic death requires active transcription 11, we hypothesized that ROS could be regulated by transcription factor(s). A series of gain-of-function and loss-of-function screens identified p63, the primordial member of the p53 62
75 family 17, as a master transcriptional regulator of ROS. Overexpression of the major isoform of p63, ΔNp63α, prevented etoposide-induced ROS accumulation in transformed DKO MEFs (Fig. 2.1A). Three ROS-sensitive dyes were employed to interrogate intracellular ROS levels H 2 DCFDA, a broad-spectrum ROS indicator; dihydroethidium (DHE), a specific indicator for superoxide; and MitoSOX Red, a specific indicator for mitochondrial matrix superoxide as mitochondria are the major source of ROS within the cells 16,18. Consistent with our previous identification of p53 and ROS as two independent effectors in executing necrotic death 11, overexpression of a dominant negative mutant of p53 (p53 DN) failed to block ROS induction triggered by genotoxic stress (Fig. 2.1 A). Overexpression of either ΔNp63α or p53 DN provided DKO cells a short-term survival advantage against etoposide (Fig. 2.1 B). Importantly, coexpression of both ΔNp63α and p53 DN enhanced clonogenic survival of DKO cells against etoposide (Fig. 2.1 C). To further demonstrate p53-independent regulation of ROS by ΔNp63α, p53 -/- Bax -/- Bak -/- triple-knockout (TKO) MEFs were generated. Transformed p53 -/- Bax -/- Bak -/- TKO MEFs were more resistant to DNA damage than Bax -/- Bak -/- DKO MEFs (fig. S2.2). However, TKO cells were eventually killed by DNA damage due to ROS-induced oxidative stress that was abrogated by antioxidant N-acetyl-L-cysteine (NAC) or diphenyleneiodonium (DPI) (fig. S2.3). Strikingly, overexpression of ΔNp63α protected TKO from etoposide-induced ROS accumulation and cell death (Fig. 2.1, D to F, and fig. S2.4). It was reported that another p63 isoform, TAp63, is induced by DNA damage in MEFs 19 and deficiency of TAp63 but not p53 protects oocytes from ionizing irradiation-induced cell death 20. Of note, we could only detect TAp63 but not ΔNp63 in MEFs by PCR (fig. S2.5). In fact, the level of TAp63 in MEFs was too low to be detected by immunoblots. Knockdown of TAp63 in DKO or TKO MEFs had no effect on etoposide-induced cell death (fig. S2.5), indicating that endogenous TAp63 is not involved in DNA damage-induced necrotic death and ΔNp63α does not inhibit oxidative stress through antagonizing endogenous TAp63. In summary, our gain-of-function approach discovered a previously unrecognized role of ΔNp63α in regulating oxidative stress. Moreover, the inhibition of oxidative stress by ΔNp63α rendered clonogenic survival of p53 -/- Bax -/- Bak -/- TKO cells against DNA damage. 63
76 Oxidative stress is caused by an imbalance between the production of ROS and the ability of detoxifying reactive intermediates 16,21. If ΔNp63α prevents DNA damage-induced oxidative stress by increasing antioxidant capacity, ΔNp63α should also protect cells against exogenous oxidants. Indeed, overexpression of ΔNp63α protected wild-type cells from thiol oxidant diamide and lipid oxidant tert-butyl hydroperoxide (TBH) (Fig. 2.2A). These findings are consistent with a notion that ΔNp63α alters the intracellular redox state by increasing the antioxidant capacity of cells and thereby protects cells against diverse sources of oxidative stress. Given that glutathione is the major antioxidant produced by the cell and plays a central role in regulating intracellular redox state 16,21,22, one testable thesis is that ΔNp63α mitigates oxidative stress through transcriptional regulation of glutathione metabolism genes. Glutathione is synthesized in two sequential steps, catalyzed by glutamate-cysteine ligase (GCL) and glutathione synthetase (GSS), respectively 22 (Fig. 2.2B). It exists in both reduced (GSH) and oxidized (GSSG) states 22. GSH can act as a free radical scavenger and an inhibitor of lipid peroxidation. GSH also participates in the detoxification of hydrogen peroxide through various glutathione peroxidases (GPX) (Fig. 2.2B). During this process, GSH is oxidized to GSSG, which can then be recycled back to GSH through the action of glutathione reductase (GSR) at the expense of reduced nicotinamide adenine dinucleotide phosphate (NADPH). NADPH can be generated by several enzymes, including isocitrate dehydrogenase 2 (IDH2). Interestingly, gene expression profiling revealed that genes involved in glutathione redox pathway were upregulated in ΔNp63α-overexpressing cells both before and after DNA damage (fig. S2.6). Quantitative RT- PCR analysis confirmed that GCLC (the catalytic subunit of GCL), GSS, IDH2, and GPX2 were upregulated by ΔNp63α (Fig. 2.2C). Hence, it appears that DNp63a orchestrates a genetic program to integrate the biosynthesis of glutathione, the utilization of GSH as an antioxidant, and the regeneration of GSH from GSSG (Fig. 2.2B), the outcome of which would increase the ratio of GSH to GSSG, an indicator of a more reduced intracellular redox state. Indeed, overexpression of ΔNp63α significantly increased the GSH/GSSG ratio in cells both before and after etoposide treatment (Fig. 2.2D). The functional significance of these ΔNp63α-induced glutathione metabolism genes in regulating necrotic cell death was interrogated by sirna-mediated 64
77 knockdown (fig. S2.7). Deficiency of GCLC, GSS, GPX2, or IDH2 compromised, to varying extents, the ability of ΔNp63α to protect DKO cells against DNA damage (Fig. 2.2, E to H). Of note, deficiency of these genes also enhanced etoposide-induced cell death in control DKO cells, but to a lesser degree (Fig. 2.2, E to H), supporting the involvement of oxidative stress in this form of cell death. Collectively, our data demonstrate that p63 orchestrates a tightly coupled glutathione metabolic circuit to alleviate oxidative stress and thereby protects apoptosis-defective cells from ROS-mediated necrotic cell death (Fig. 2.2B). To investigate the role of endogenous ΔNp63α in regulating redox homeostasis, knockdown of p63 using a published shrna 23 was performed in cell lines where ΔNp63α is the predominant isoform (Fig. 2.3A). In agreement with our gain-of-function study, knockdown of endogenous ΔNp63α led to increased intracellular ROS levels in both human mammary epithelial cell line MCF-10A and human cervical carcinoma cell line ME-180 (Fig. 2.3B). Furthermore, knockdown of p63 using sirna or mir30-based shrna also increased ROS in these cells (Fig. S2.8). Importantly, quantitative RT-PCR demonstrated that deficiency of ΔNp63α downregulated GCLC, GSS, IDH2, and GPX2 (Fig. 2.3C) the same set of genes that was upregulated by ΔNp63α (Fig. 2.2C). Of note, GSR was suppressed by the loss of endogenous ΔNp63α but not induced by the overexpression of ΔNp63α (Fig. 2.3C and data not shown), which might reflect cell type-specific regulation. Consistent with the downregulation of glutathione metabolism genes caused by the knockdown of ΔNp63α, the GSH/GSSG ratio was reduced in cells deficient for ΔNp63α (Fig. 2.3D), indicating a more oxidized intracellular redox state. Consequently, deficiency of ΔNp63α enhanced diamide-induced cell death, which was protected by the antioxidant NAC (Fig. 2.3E). Furthermore, deficiency of ΔNp63α sensitized cells to chemotherapeutic agentinduced cell death (fig. S2.9). Altogether, our loss-of-function studies demonstrate an important role of endogenous ΔNp63α in gauging redox homeostasis. The observation that antioxidant and anti-apoptotic BCL-2 work in concert to promote long-term clonogenic survival against matrix detachment 15 prompted us to investigate whether p63 can mitigate oxidative stress associated with anoikis and whether p63 can cooperate with BCL-2 to prevent the luminal clearance of acini in three-dimensional culture of MCF-10A cells. 65
78 Overexpression of ΔNp63α protected MCF-10A cells from anoikis, but to a lesser degree than BCL-2 (Fig. 2.4A). Importantly, overexpression of ΔNp63α but not BCL-2 prevented ROS induction in cells undergoing anoikis (Fig. 2.4B). Because BCL-2 profoundly blocked anoikis up to 5 days, the combined effect of BCL-2 and ΔNp63α was not evident in short-term death assays (Fig. 2.4A). To investigate long-term survival in response to the loss of matrix attachment, MCF- 10A cells were seeded onto reconstituted basement membrane under conditions that enable the formation of three-dimensional acini where the inner, matrix-deprived cells are eliminated to facilitate lumen formation 14,15. Overexpression of either BCL-2 or ΔNp63α only weakly whereas co-expression of BCL-2 and ΔNp63α markedly inhibited luminal clearance (Fig. 2.4, C and D). Luminal clearance of MCF-10A acini was evident within two weeks after seeding, whereas coexpression of BCL-2 and ΔNp63α near completely blocked luminal clearance for up to a month (Fig. 2.4, C and D). Importantly, overexpression of ΔNp63α but not BCL-2 suppressed ROS accumulation in the matrix-deprived luminal cells (Fig. 2.4E). We presented evidence that ΔNp63α enhances endogenous antioxidant capacity through a coordinated regulation of glutathione metabolism genes. Among these identified ΔNp63α targets, GPX2 is the only reported direct transcriptional target of ΔNp63α 24. It is conceivable that ΔNp63α may regulate the remaining targets directly or indirectly. Since Nrf2 is the most recognized transcription factor that controls the expression of many antioxidant genes, including GCLC 25,26, and Nrf2 mrna was slightly induced by ΔNp63α (fig. S2.10), we investigated whether ΔNp63α regulates GCLC through Nrf2 and whether Nrf2 is required for the antioxidant effect of ΔNp63α. Although knockdown of Nrf2 partially reduced GCLC (fig. S2.10), ΔNp63α-mediated upregulation of GCLC was not affected by the loss of Nrf2 (fig. S2.10). Accordingly, the protective effect of ΔNp63α against DNA damage-induced necrotic death was not affected by the deficiency of Nrf2 (fig. S2.10). These data not only illustrate an Nrf2-independent regulation of GCLC by ΔNp63α but also indicate that ΔNp63α is a bona fide central regulator of glutathione biogenesis. It remains to be determined how ΔNp63α integrates the glutathione metabolic program. 66
79 2.4 Discussion Here, we report that ΔNp63α prevents oxidative stress and cooperates with apoptotic inhibition to promote long-term cellular survival against DNA damage and extracellular matrix detachment, highlighting the quintessence of suppressing oxidative stress in promoting long-term survival. Foreseeably, inhibition of oxidative stress may be especially critical to the wellbeing of long-lived cells such as stem cells. Notably, ΔNp63α is important in maintaining the stem cell populations of stratified epithelia 17,27. In addition to sustaining the proliferative capacity of epithelial stem cells 27, ΔNp63α may also promote stem cell survival through its antioxidant function. p63 +/- mice have a shortened life span and display features of accelerated aging, and p63-deficient cells exhibit heightened cellular senescence 28, all of which could be caused by oxidative damage 18 and may be linked to oxidative stress resulted from the loss of ΔNp63α. In contrast to the tumor suppressor function of p53, ΔNp63α has been implicated as an oncogene and is frequently amplified and/or overexpressed in human squamous cell carcinoma of the lung and head and neck 17,29. Because cancers often have an increased demand for antioxidant capacity compared to normal tissues likely due to inherent metabolic derangements that increase ROS production 21, the Nrf2 antioxidant program has thus been shown to contribute to tumorigenesis 30. By analogy, ΔNp63α might promote tumorigenesis by reducing oxidative stress. Although a detailed blueprint of the core apoptotic pathway has been formulated through biochemical and genetic studies over the past two decades, we are still unable to fully rescue cells from most death stimuli. The identification of oxidative stress as a critical mechanism compromising long-term survival of apoptosis-defective cells likely open new avenues for the therapeutic intervention aimed at preventing cellular loss during pathological processes. Conversely, targeting the antioxidant pathway may hold promise for the future development of anticancer therapeutics to eliminate cancer cells that evade apoptotic checkpoints. 67
80 2.5 Materials and Methods Plasmid construction, retrovirus production, and sirna Murine ΔNp63α was dually tagged with FLAG and HA at the N-terminus and cloned into MSCV- IRES-Puro or tagged with HA at the N-terminus and cloned into MSCV-IRES-GFP. A dominant negative mutant of p53 (aa 1-14 and aa ) was dually tagged with FLAG and HA at the N- terminus and cloned into MSCV-Puro (Clontech). Human BCL-2 was cloned into MSCV-IRES- GFP or MSCV-Puro. Retrovirus-mediated knockdown constructs were generated using psuperior-retro-puro according to the manufacture s instructions (Oligoengine). The target sequence of shrna against mouse p63 is 5 -AGCACACGATCGAAACGTA-3. The target sequence of scramble shrna is 5 - GCGCGCTTTGTAGGATTCG-3. The production of retroviruses was described previously 6. Lentivirus-mediated knockdown constructs for human p63 and GFP were previously described 23, 31. All constructs were confirmed by DNA sequencing. Lentiviral tetracycline-inducible mir30-based shrna against human p63 was obtained from Open Biosystems. The target sequence is 5 -TCCGAGCTATGTCAGTACTATT-3. To induce the expression of shrnamir, cells were treated with 2 µg/ml doxycycline (Sigma) for 3 days. Lentivirus was produced by cotransfection with pcmvdr8.2 and phcmv.vsvg using Lipofectamine 2000 (Invitrogen). sirna oligos were reverse-transfected using Lipofectamine TM RNAiMAX (Invitrogen) to a final concentration of 10 or 20 nm. The sirna oligos were purchased from Ambion Silencer Select oligos (Applied Biosystems) or Dharmacon: mouse IDH2 #1, 5 - AGACUGACUUCGACAGGAAtt (Ambion s114463); mouse IDH2 #2, 5 - GGAAUAAGAUCUGGUAUGAtt (Ambion 95489); mouse GCLC #1, 5 - CGGUAUGACUCAAUAGAUAtt (Ambion s66718); mouse GCLC #2, 5 - GGGUGAUCCUCUCAUACAAtt (Ambion s201400); mouse GSS #1, 5 - GCCCAGUCAGUAUAAUUCAtt (Ambion s67110); mouse GSS #2, 5 - CCAUCAAAAAGGACGACUAtt (Ambion s67112); mouse GPX2 #1, 5 - UCAAUGAGCUGCAAUGUCG (Dharmacon sigenome-02); mouse GPX2 #2 5 - CAACUACCCGGGACUACAA (Dharmacon sigenome-03); mouse Nrf2, 5 - CAUUUUUACUCAUCGAUCUtt (Ambion s70523); mouse p63, 5 -CCACCGAACUGAAGAAGCU 68
81 (Dharmacon sigenome-04); human p63, 5 -CGACAGUCUUGUACAAUUU (Dharmacon sigenome-08); mouse RIP1 #1, 5 -GGUGGUACCCUUUACUACAtt (Ambion s72975); mouse RIP1 #2, 5 -GGAUUUGGAACUACAGGUAtt (Ambion s72977); Ambion Silencer Select Negative Control #1; Dharmacon sigenome Negative Control #2. Cell culture and viability assays SV40-transformed Bax -/- Bak -/- double-knockout (DKO) mouse embryonic fibroblasts (MEFs) were previously described 6. E1A/Ras and c-myc/ras transformation was performed by infecting primary MEFs within the first 2 passages with retrovirus expressing E1A-IRES-Ras or c-myc- IRES-Ras as described previously 32. For E1A/Ras transformation, primary MEFs were generated from E12.5 Bax -/- Bak -/- DKO embryos and E12.5 p53 -/- Bax -/- Bak -/- triple-knockout (TKO) embryos. E12.5 p53 -/- Bax -/- Bak -/- TKO MEFs were transformed with SV40 as described previously 6. ME- 180 and MCF-10A cell lines were obtained from ATCC and cultured according the instructions. To induce anoikis, cells were grown on poly-hema (Sigma)-coated 6 well plates in the presence of 1% methylcellulose (Sigma). Cell death was quantified by annexin-v (BioVision) or propidium iodide (Sigma) staining, followed by flow cytometric analysis using either a FACSCalibur (BD Biosciences) or a LSRFortessa (BD Biosciences). Data were analyzed using CellQuest Pro (BD Biosciences) or FACSDiva (BD Biosciences). P values for statistical analysis were obtained using Student's t test. Measurement of ROS Production of ROS was monitored by flow cytometric analysis using the redox-sensitive dyes. Cells were incubated with 1 µm 2,7 -dichlorofluorescein diacetate (H 2 DCFDA, Invitrogen), 1 µm 5-(and-6)-chloromethyl-2',7'-dichlorodihydrofluorescein diacetate (CM-H 2 DCFDA, Invitrogen), or 1 µm dihydroethidium (DHE, Invitrogen) at 37 C for 30 minutes, or with 2.5 µm MitoSOX Red (Invitrogen) at 37 C for 10 minutes. Oxidation of the ROS-sensitive dyes was quantified by flow cytometric analysis using either a FACSCalibur (BD Biosciences) or a LSRFortessa (BD Biosciences). Median fluorescence in the appropriate detection channels was assessed by 69
82 FlowJo (Tree Star). Fold induction of ROS was determined by dividing the median fluorescence of experimental samples with that of control samples. P values for statistical analysis were obtained using Student s t-test. Clonogenic assays SV40-transformed MEFs were treated with etoposide (10 µg/ml) for 14 hours. E1A/Ras- and c- Myc/Ras-transformed p53 -/- Bax -/- Bak -/- TKO MEFs, transduced with retrovirus expressing GFP or GFP-IRES-ΔNp63α, were subjected to MoFlo (Beckman Coulter) sorting for GFP-positive cells before treatment with etoposide for 9 hours. 5 x 10 4 or 10 5 cells were washed with PBS and plated in 24-well tissue culture plates. Colonies were fixed with methanol and stained with 0.5% crystal violet in 25% methanol after days. Reverse transcription and quantitative PCR Total RNA was extracted from cells using TRIZOL (Invitrogen) according to the manufacturer s instructions. Reverse transcription was performed with oligo-dt plus random decamer primers (Ambion) using Superscript II (Invitrogen). Quantitative PCR was performed with SYBR green master mix (Applied Biosystems) in duplicates using the indicated gene specific primers. For TaqMan probes, quantitative PCR was performed with TaqMan PCR Master Mix (Applied Biosystems). Quantitative PCR was performed on an ABI Prism 7300 sequence detection system (Applied Biosystems) or a ViiA 7 Real-Time PCR System (Applied Biosystems). Data were analyzed as described previously by normalization against GAPDH or 18S rrna that was detected using TaqMan probes (Applied Biosystems) 11. Primers for qrt-pcr are listed as follows: mouse p63 pan-isoform, 5 - CCTTTCCGTCAGAATACACACGGA-3 and 5 - GTTTCTGAAGTAGGTGCTGGTGCT-3 ; mouse p63 TA isoform, 5 - CCTATATGCTCAGTACAGCCCATCG-3 and 5 - CTATTCTGTGCGTGGTCTGTGTTGT-3 ; mouse p63 N isoform, 5 - TCTTAGAAGATTCGCAGCGCAAGG-3 and 5 - CTATTCTGTGCGTGGTCTGTGTTGT-3 ; mouse GCLC, 5 -TACACCTGGATGATGCCAACGA-3 and 3 -GTCAACCTTGGACAGCGGAATG; mouse GSS, 5 -CCTGATGCTAGAGAGATCTCGTGC 70
83 and 3 -CTCTCTCCTCACTGTCCTTCAGC; mouse IDH2, 5 -CCTGATGACATCTGTGCTGGTCT and 3 -GAGCTCTGTCCAGGTTGCTCTT; human GSS, 5'-GACCAGCGTGCCATAGAGAATGA and 3 -CATGTGACCTCTCCAGCAGTAGAC; human IDH2, 5 - GATGGGAAGACGATTGAGGCTGA and 3 -TCAGGAAGTGCTCGTTCAGCTT; human GPX2, 5'- TCTATGACCTCAGTGCCATCAGC and 3'-CCAGGACGGACATACTTGAGACTG; human GSR, 5'-CCAACGTCAAAGGCATCTATGCAG and 3'-ATCTTCCGTGAGTCCCACTGTC; mouse Nrf2, 5 -ATCCAGACAGACACCAGTGGATC-3 and 5 -CAAACTTGCTCCATGTCCTGCTC-3. The TaqMan probes for mouse GPX2 (TaqMan Assay ID Mm _g1) and human GCLC (TaqMan Assay ID Hs _m1) were obtained from Invitrogen. Three-dimensional culture of MCF-10A and indirect immunofluorescence microscopy MCF-10A cells were seeded onto reconstituted basement membrane (Matrigel) under conditions that enable formation of three-dimensional acini and were then assessed by indirect immunofluorescence microscopy as previously described 33, 34. Briefly, 10 4 cells were seeded onto one chamber of a 4-well chamber slide pre-coated with Matrigel (BD Biosciences). After 31 days, cells were fixed using 4% paraformaldehyde (Fisher) and permeabilized using 0.5% Triton X-100, and sequentially incubated with anti-laminin-5 (D4B5, EMD Millipore), Alexa Fluor 568 conjugated goat anti-mouse IgG (Invitrogen), anti-gfp (ab13970, Abcam), Alexa Fluor 488 conjugated goat anti-chicken IgG (Invitrogen), and nuclear stain 4,6-diamidino-2-phenylindole (DAPI, Sigma). Images were acquired using the Leica TCS SP5-II Confocal Upright and the PerkinElmer UltraVIEW ERS confocal microscopes at the Molecular Cytology Core Facility at the Memorial Sloan-Kettering Cancer Center and analyzed by MetaMorph software (Molecular Devices). Cellularity of the equitorial cross-sections was scored as clear (~0-25% of the luminal space filled with cell nuclei), mostly clear (~25-50% cellularity), mostly filled (~50-75% cellularity), and filled (~75-100% cellularity). To measure ROS levels, three-dimensional culture of MCF-10A cells after 6-8 days were incubated with Hank s Balanced Salt Solution (HBSS, Invitrogen) containing 5 µm dihydroethidium (DHE, Invitrogen) and 3 µg/ml Hoechst (Invitrogen) at 71
84 37 C for 30 minutes in a humidified 5% CO 2 incubator, washed once with HBSS, then immediately imaged using the Leica TCS SP5-II confocal microscope. Reagents, antibodies, and immunoblot analysis Chemicals used are listed as follows: etoposide (Sigma), diamide (Sigma), tert-butyl hydroperoxide (Sigma), diphenyleneiodonium (Sigma), doxorubicin (Sigma), cisplatin (Sigma), and N-acetyl-L-cysteine (Sigma). Antibodies used for immunoblot analysis are listed as follows: anti-flag (M2, Sigma), anti-p53 (FL393, Santa Cruz Biotechnology), anti-p63 (4A4, Santa Cruz Biotechnology), anti-gclc (HPA036359, Sigma), anti-idh2 (ab55271, Abcam), anti-rip (610458, BD Biosciences), and anti-β-actin (A1978, Sigma). Cell lysates were resolved by 10% or 4-12% NuPAGE gels (Invitrogen), transferred onto PVDF membrane (Immobilon-P, Millipore). Antibody detection was accomplished using enhanced chemiluminescence method (Western Lightning, PerkinElmer) and LAS-3000 Imaging system (FUJIFILM). Glutathione assays Measurement of intracellular reduced (GSH) and oxidized (GSSG) glutathione concentration was performed using the 5,5 -dithio-bis(2-nitrobenzoic acid)-glutathione reductase recycling method as previously described 35. Briefly, the rate of 5-thio-2-nitrobenzoic acid (TNB) formation was determined by measuring the rate of change of absorbance at 412 nm. Linear regressions based on values obtained from standard curves were used to calculate concentrations of total and oxidized glutathione, which were then used to calculate concentration of reduced glutathione. Glutathione concentrations were normalized based on protein concentration determined using the BCA Kit (Pierce). Reagents for glutathione assays were obtained from Sigma and include 5- sulfosalicylic acid dihydrate, DTNB, β-nadph, glutathione reductase, L-glutathione reduced, L- glutathione oxidized, triethanolamine, and 2-vinylpyridine. P values for statistical analysis were obtained using Student s t-test. Gene expression profiling and heatmap plot Total RNA was extracted from cells using TRIZOL (Invitrogen) and was subjected to cleanup using the RNeasy Mini Kit (Qiagen) according to the manufacturers instructions. RNA samples were submitted to the Laboratory for Clinical Genomics at the Washington University in St. Louis 72
85 for gene expression microarray analysis using the GeneChip Mouse Gene 1.0 ST array (Affymetrix). Heatmap plot was generated using Partek Genomics Suite (Partek). 2.6 Figure Legends Fig Np63 abrogates DNA damage-induced oxidative stress and enhances clonogenic survival of transformed p53 -/- Bax -/- Bak -/- triple-knockout (TKO) mouse embryonic fibroblasts (MEFs). (A) Np63 but not a dominant negative mutant of p53 prevents DNA damage-induced ROS accumulation. SV40-transformed Bax -/- Bak -/- double-knockout (DKO) MEFs transduced with control retrovirus or retrovirus expressing ΔNp63α or dominant negative p53 (p53 DN) were mock treated or treated with etoposide (10 µg/ml). Oxidation of the ROS-sensitive dyes H 2 DCFDA, DHE, or MitoSOX Red was quantified by flow cytometric analysis. Data shown are fold increase of ROS after etoposide treatment. (B) Cell death was quantified by flow cytometric analysis following propidium iodide staining of DKO cells described in (A) at 3 days after etoposide treatment. Data are the mean percentage of PI-positive cells ± SD from three independent experiments. The expression of N-terminal Flag-HA-tagged Np63 and dominant negative p53 was assessed by anti-flag immunoblot (right panel). Asterisk denotes a cross-reactive band serving as a loading control. (C) SV40-transformed Bax -/- Bak -/- DKO MEFs transduced with the indicated retrovirus were treated with etoposide (10 µg/ml) for 14 hours. Colonies were stained with crystal violet after 12 days. The data shown are representative of three independent experiments. (D) SV40-transformed p53 -/- Bax -/- Bak -/- TKO MEFs transduced with control or ΔNp63α-expressing retrovirus were mock treated or treated with etoposide (10 µg/ml). Oxidation of the ROS-sensitive dye DHE was quantified by flow cytometric analysis. Data shown are fold increase of ROS after etoposide treatment. (E) Cell death was quantified by flow cytometric analysis following propidium iodide staining of TKO cells described in (D) at the indicated times after etoposide treatment. Data are the mean percentage of PI-positive cells ± SD from three independent experiments. (F) c-myc/ras or E1A/Ras transformed p53 -/- Bax -/- Bak -/- TKO MEFs transduced with control retrovirus or retrovirus expressing ΔNp63α were treated with etoposide for 9 hours. Colonies were stained with crystal violet after 10 days. *, P<
86 Fig p63 dictates the homeostasis of intracellular redox through transcriptional controls of glutathione metabolism genes. (A) Np63 protects against cell death induced by exogenous oxidants. Wild-type MEFs transduced with control or Np63-expressing retrovirus were mock treated or treated with diamide (100 µm) for 4 hours or tert-butyl hydroperoxide (TBH, 50 µm) for 7 hours. Cell death was quantified by propidium iodide (PI) staining. Data are the mean percentage of PI-positive cells ± SD from three independent experiments. (B) Schematic representation of the effect of ΔNp63α on glutathione metabolism. Overexpression of ΔNp63α in cells generates a more reduced intracellular redox state and increases resistance to oxidative damage through a coordinated regulation of glutathione metabolism genes. Specifically, ΔNp63α upregulates enzymes responsible for (1) glutathione biosynthesis, (2) ROS detoxification by glutathione, and (3) regeneration of GSH from GSSG. (C) Np63 upregulates genes involved in glutathione metabolism. Bax -/- Bak -/- DKO cells transduced with control or Np63-expressing retrovirus were mock treated or treated with etoposide (10 µg/ml) for 6 hours. mrna levels of the indicated genes were analyzed by qrt-pcr and presented as mean ± SD from three independent experiments. (D) Np63 generates a more reduced intracellular redox state. Bax -/- Bak -/- DKO MEFs transduced with control or Np63-expressing retrovirus were mock treated or treated with etoposide for 24 hours. Intracellular reduced (GSH) and oxidized (GSSG) glutathione concentrations were determined using an enzyme-cycling assay and normalized against protein concentration. Normalized [GSH] and [GSSG] were used to calculate the GSH/GSSG ratio. Data presented are mean ± SD from at least three independent experiments. (E to H) The ability of Np63 to prevent DNA damage-induced cell death is compromised by the deficiency of its transcriptional targets engaged in glutathione metabolism. Bax -/- Bak -/- DKO cells stably expressing GFP or ΔNp63α were transfected with scrambled sirna (siscr) or sirna against GCLC (E), GSS (F), GPX2 (G), or IDH2 (H). Cells were subsequently mock treated or treated with etoposide for 3 days. Cell death was quantified by propidium iodide (PI) staining. Data are the mean percentage of PI-positive cells ± SD from three independent experiments. *, P < Fig Deficiency of Np63α generates a more oxidized intracellular redox state and sensitizes cells to oxidative stress. (A) The indicated human cell lines transduced with lentivirus expressing 74
87 shrna against GFP or p63 were analyzed by Western blots using antibodies specific for p63 and actin. (B) Deficiency of Np63α leads to increased ROS levels. MCF-10A and ME-180 cell lines, transduced with lentivirus expressing shrna against GFP or p63, were stained with CM- H 2 DCFDA or H 2 DCFDA. Oxidation of the respective dyes was quantified by flow cytometric analysis. Data shown are fold increase of ROS induced by knockdown of p63 from three independent experiments. (C) Deficiency of ΔNp63α downregulates genes involved in glutathione metabolism. The indicated human cells lines were transduced with lentivirus expressing shrna against GFP or p63. mrna levels of the indicated genes were analyzed by qrt-pcr and presented as mean ± SD from three independent experiments. (D) Deficiency of Np63α generates a more oxidized intracellular redox state. The indicated human cell lines transduced with lentivirus expressing shrna against GFP or p63 were analyzed for intracellular reduced (GSH) and oxidized (GSSG) glutathione concentrations using an enzyme-cycling assay. Normalized [GSH] and [GSSG] were used to calculate the GSH/GSSG ratio. Data presented are mean ± SD from three independent experiments. (E) Deficiency of Np63α sensitizes cells to oxidant-induced cell death. The indicated human cell lines transduced with lentivirus expressing shrna against GFP or p63 were mock treated or treated with oxidant diamide in combination with antioxidant N-acetyl-L-cysteine (NAC). Cell death was quantified by propidium iodide (PI) staining. Data are the mean percentage of PI-positive cells ± SD from three independent experiments. *, P < Fig ΔNp63α mitigates anoikis-induced oxidative stress and cooperates with anti-apoptotic BCL-2 to disrupt the luminal clearance of acini in three-dimensional culture of mammary epithelial cells. (A) ΔNp63α protects MCF-10A cells from anoikis. MCF-10A cells transduced with retrovirus expressing GFP, BCL-2, ΔNp63α, or both BCL-2 and ΔNp63α were grown on poly-hema-coated plates in the presence of 1% methylcellulose to induce anoikis. Cell death was quantified at the indicated times by annexin-v staining. Data are the mean percentage of annexin-v-positive cells ± SD from three independent experiments. *, P < (B) ΔNp63α but not BCL-2 prevents anoikis-induced ROS accumulation. MCF-10A cells transduced with control retrovirus or retrovirus expressing BCL-2, ΔNp63α, or both BCL-2 and ΔNp63α were grown on poly-hema- 75
88 coated plates in the presence of 1% methylcellulose to induce anoikis. Paired samples grown in either regular or detachment condition were stained with DHE at 1.5 days. Oxidation of DHE was quantified by flow cytometric analysis. Data shown are fold increase of ROS induced by anoikis from three independent experiments. *, P < (C and D) Coexpression of BCL-2 and ΔNp63α disrupts the luminal clearance of acini in three-dimensional culture. MCF-10A cells transduced with retrovirus expressing GFP, BCL-2, ΔNp63α, or both BCL-2 and ΔNp63α were cultured in reconstituted basement membrane (Matrigel) for 31 days. Acini were fixed and stained for laminin 5 (red) and nuclear stain 4,6-diamidino-2-phenylindole (DAPI, blue). Luminal cellularity of acini was scored using confocal microscopy. Clear, 0-25% of the luminal space filled with cell nuclei; mostly clear, 25-50%; mostly filled, 50-75%; filled, %. Representative confocal microscopy images from three independent experiments are shown (C). Scale bar indicates 25 µm. Data shown in (D) are average of three independent experiments. (E) ΔNp63α prevents intraluminal ROS accumulation in three-dimensional acini. MCF-10A cells transduced with retrovirus expressing GFP, BCL-2, p63, or both BCL-2 and p63 were cultured in Matrigel for 6-8 days. Acini were stained with DHE (red) and Hoechst (blue). Representative confocal microscopy images are shown. Arrowheads indicate DHE-high cells. Scale bar indicates 25 µm. 76
89 2.7 Figures Figure 2.1 Np63 abrogates DNA damage-induced oxidative stress and enhances -/- -/- -/- clonogenic survival of transformed p53 Bax Bak mouse embryonic fibroblasts. 77
90 Figure 2.2 p63 dictates the homeostasis of intracellular redox through transcriptional controls of glutathione metabolism genes. 78
91 Figure 2.3 Deficiency of Np63α generates a more oxidized intracellular redox state and sensitizes cells to oxidative stress. 79

92 Fig. 2.4 ΔNp63α mitigates anoikis-induced oxidative stress and cooperates with antiapoptotic BCL-2 to disrupt the luminal clearance of acini in three-dimensional culture of mammary epithelial cells. 80
93 Fig. S2.1. (A) SV40-transformed Bax -/- Bak -/- DKO MEFs were transfected with scrambled sirna (siscr) or sirna against RIP1. Cell lysates were analyzed by Western blots using the antibodies specific for RIP1 or actin. (B) SV40-transformed Bax -/- Bak -/- DKO MEFs, transfected with scrambled sirna (siscr) or sirna against RIP1 for 3 days, were treated with etoposide (10 µg/ml). Cell death was quantified by propidium iodide staining at 3 days after etoposide treatment. Data are the mean percentage of PI-positive cells ± SD from three independent experiments. Data courtesy of Dr. Ho-Chou Tu Fig. S2.2. p53 -/- Bax -/- Bak -/- TKO MEFs are more resistant to DNA damage-induced cell death than Bax -/- Bak -/- DKO MEFs. (A) Primary MEFs isolated from two Bax -/- Bak -/- DKO embryos and two p53 -/- Bax -/- Bak -/- TKO embryos were transformed by E1a and Ras. E1a/Ras-transformed Bax -/- Bak -/- DKO and p53 -/- Bax -/- Bak -/- TKO MEFs were treated with etoposide (10 µg/ml). Cell death was quantified by annexin-v staining at the indicated times. Data are the mean percentage of annexin-v-positive cells ± SD from three independent experiments. (B) SV40-transformed Bax -/- Bak -/- DKO or p53 -/- Bax -/- Bak -/- TKO MEFs were treated with etoposide (10 µg/ml). Cell death was 81
94 quantified by propidium iodide staining at the indicated times. Data are the mean percentage of PI-positive cells ± SD from three independent experiments. *, P<0.05. Fig. S2.3. p53 -/- Bax -/- Bak -/- TKO cells were treated with etoposide or etoposide plus N-acetyl-Lcysteine (NAC, 20 mm) or etoposide plus diphenyleneiodonium (DPI, 100 nm) for the indicated times. Cell death was quantified by propidium iodide staining at the indicated times. Data are the mean percentage of PI-positive cells ± SD from three independent experiments. Fig. S2.4. SV40-transformed p53 -/- Bax -/- Bak -/- TKO MEFs transduced with control or ΔNp63αexpressing retrovirus were mock treated or treated with etoposide (10 µg/ml). Oxidation of the ROS-sensitive dye CM-H 2 DCFDA or MitoSOX Red was quantified by flow cytometric analysis. Data shown are fold increase of ROS after etoposide treatment. *, P<
95 Fig. S2.5. (A) cdna prepared from Bax -/- Bak -/- DKO MEFs transduced with control or ΔNp63αexpressing retrovirus was subjected to PCR using primers specific for all isoforms of p63 (Pan), TA isoforms of p63 (TA), or N isoforms of p63 ( N). (B) SV40-transformed Bax -/- Bak -/- DKO MEFs transduced with retrovirus expressing shrna against luciferase or p63, or transfected with scrambled sirna (siscr) or sirna against p63 were subjected to quantitative RP-PCR analysis for p63 expression. Data presented as mean ± SD from three independent experiments. (C) SV40-transformed Bax -/- Bak -/- DKO MEFs, tranfected with scrambled sirna (siscr) or sirna against p63 for 3 days, were mock treated or treated with etoposide (10 µg/ml) for 3 days (left panel). DKO MEFs transduced with retrovirus expressing shrna against luciferase or p63 were mock treated or treated with etoposide (10 µg/ml) for 3 days (right panel). Cell death was 83
 SV40-transformed p53 -/- Bax -/- Bak -/- TKO MEFs, tranfected with scrambled sirna (siscr) or sirna against p63 for 3 days, were mock treated or treated with etoposide (10 µg/ml) for 5 days (left")
96 quantified by propidium iodide staining. Data are the mean percentage of PI-positive cells ± SD from three independent experiments. (D) SV40-transformed p53 -/- Bax -/- Bak -/- TKO MEFs, tranfected with scrambled sirna (siscr) or sirna against p63 for 3 days, were mock treated or treated with etoposide (10 µg/ml) for 5 days (left panel). TKO MEFs transduced with retrovirus expressing shrna against luciferase or p63 were mock treated or treated with etoposide (10 µg/ml) for 5 days (right panel). Cell death was quantified by propidium iodide staining. Data are the mean percentage of PI-positive cells ± SD from three independent experiments. Fig. S2.6. A heatmap representation of glutathione metabolism genes differentially regulated by ΔNp63α. DKO cells transduced with retrovirus expressing GFP or GFP-IRES-ΔNp63α were mock treated or treated with etoposide (10 µg/ml) for 6 hours. The gene expression profiles were assessed using the Affymetrix GeneChip Mouse Gene 1.0 ST array and analyzed for genes involved in glutathione metabolism. 84
97 Fig. S2.7. Immunoblot or qrt-pcr analysis of sirna-mediated knockdown. (A) Bax -/- Bak -/- DKO MEFs transfected with scrambled sirna (siscr) or sirna against IDH2 were analyzed by immunoblots using antibodies specific for IDH2 or actin. (B to D) Bax -/- Bak -/- DKO MEFs transfected with scrambled sirna (siscr) or sirna against GCLC, GSS, or GPX2 were analyzed by qrt-pcr using the indicated gene-specific primers. Data presented as mean ± SD from three independent experiments. *, P<
98 Fig. S2.8. (A) ME-180 cells transfected with scrambled sirna (siscr) or sirna against p63 for 3 days were subjected to analysis for ROS production or immunoblots using antibodies specific for p63 or actin. Oxidation of the ROS-sensitive dye H 2 DCFDA was quantified by flow cytometric analysis. (B) ME-180 cells were transduced with lentivirus expressing doxycycline-inducible mir30-based shrna against p63. Cells were mock-treated or treated with doxycycline (2 µg/ml) for three days and analyzed for ROS production and immunoblots using antibodies specific for p63 or actin. Oxidation of the ROS-sensitive dye H 2 DCFDA was quantified by flow cytometric analysis. *, P<
99 Fig. S2.9. (A) MCF-10A cells transduced with lentivirus expressing shrna against GFP or p63 for 48 hours were mock treated or treated with doxorubicin (1 µg/ml) for 27 hours. Cell death was quantified by propidium iodide (PI) staining. Data are the mean percentage of PI-positive cells ± SD from three independent experiments. (B) ME-180 cells transduced with lentivirus expressing shrna against GFP or p63 for 48 hours were mock treated or treated with cisplatin (10 µm) for 32 hours. Cell death was quantified by propidium iodide (PI) staining. Data are the mean percentage of PI-positive cells ± SD from three independent experiments. *, P<
100 Fig. S2.10. Upregulation of GCLC by ΔNp63α is Nrf2-independent. (A and B) Bax -/- Bak -/- DKO MEFs transduced with retrovirus expressing GFP or GFP-IRES-ΔNp63α were transfected with scrambled sirna (siscr) or sirna against Nrf2. mrna levels of Nrf2 and GCLC were analyzed by qrt-pcr and presented as mean ± SD from three independent experiments (A). Cell lysates were analyzed by Western blots using antibodies specific for GCLC or actin (B). (C) Cells described above were mock treated or treated with etoposide (10 µg/ml) for 3 days. Cell death was quantified by propidium iodide staining. Data are the mean percentage of PI-positive cells ± SD from three independent experiments. *, P <
101 2.8 References 1. Thompson, C. B. Apoptosis in the pathogenesis and treatment of disease. Science, 267: , Yuan, J., Lipinski, M., and Degterev, A. Diversity in the mechanisms of neuronal cell death. Neuron, 40: , Youle, R. J. and Strasser, A. The BCL-2 protein family: opposing activities that mediate cell death. Nat Rev Mol Cell Biol, 9: 47-59, Lindsten, T., Ross, A. J., King, A., Zong, W.-X., Rathmell, J. C., Shiels, H. A., Ulrich, E., Waymire, K. G., Mahar, P., Frauwirth, K., Chen, Y., Wei, M., Eng, V. M., Adelman, D. M., Simon, M. C., Ma, A., Golden, J. A., Evan, G., Korsmeyer, S. J., MacGregor, G. R., and Thompson, C. B. The combined functions of proapoptotic Bcl-2 family members Bak and Bax are essential for normal development of multiple tissues. Mol. Cell, 6: , Wei, M. C., Zong, W. X., Cheng, E. H., Lindsten, T., Panoutsakopoulou, V., Ross, A. J., Roth, K. A., MacGregor, G. R., Thompson, C. B., and Korsmeyer, S. J. Proapoptotic BAX and BAK: a requisite gateway to mitochondrial dysfunction and death. Science, 292: , Cheng, E. H., Wei, M. C., Weiler, S., Flavell, R. A., Mak, T. W., Lindsten, T., and Korsmeyer, S. J. BCL-2, BCL-X(L) sequester BH3 domain-only molecules preventing BAX- and BAK-mediated mitochondrial apoptosis. Mol Cell, 8: , Ren, D., Tu, H. C., Kim, H., Wang, G. X., Bean, G. R., Takeuchi, O., Jeffers, J. R., Zambetti, G. P., Hsieh, J. J., and Cheng, E. H. BID, BIM, and PUMA are essential for activation of the BAX- and BAK-dependent cell death program. Science, 330: , Kim, H., Rafiuddin-Shah, M., Tu, H. C., Jeffers, J. R., Zambetti, G. P., Hsieh, J. J., and Cheng, E. H. Hierarchical regulation of mitochondrion-dependent apoptosis by BCL-2 subfamilies. Nat Cell Biol, 8: ,
102 9. Kim, H., Tu, H. C., Ren, D., Takeuchi, O., Jeffers, J. R., Zambetti, G. P., Hsieh, J. J., and Cheng, E. H. Stepwise activation of BAX and BAK by tbid, BIM, and PUMA initiates mitochondrial apoptosis. Mol Cell, 36: , Wang, X. The expanding role of mitochondria in apoptosis. Genes Dev, 15: , Tu, H. C., Ren, D., Wang, G. X., Chen, D. Y., Westergard, T. D., Kim, H., Sasagawa, S., Hsieh, J. J., and Cheng, E. H. The p53-cathepsin axis cooperates with ROS to activate programmed necrotic death upon DNA damage. Proc Natl Acad Sci U S A, 106: , Vandenabeele, P., Galluzzi, L., Vanden Berghe, T., and Kroemer, G. Molecular mechanisms of necroptosis: an ordered cellular explosion. Nat Rev Mol Cell Biol, 11: , Zong, W. X., Ditsworth, D., Bauer, D. E., Wang, Z. Q., and Thompson, C. B. Alkylating DNA damage stimulates a regulated form of necrotic cell death. Genes Dev, 18: , Debnath, J., Mills, K. R., Collins, N. L., Reginato, M. J., Muthuswamy, S. K., and Brugge, J. S. The role of apoptosis in creating and maintaining luminal space within normal and oncogene-expressing mammary acini. Cell, 111: 29-40, Schafer, Z. T., Grassian, A. R., Song, L., Jiang, Z., Gerhart-Hines, Z., Irie, H. Y., Gao, S., Puigserver, P., and Brugge, J. S. Antioxidant and oncogene rescue of metabolic defects caused by loss of matrix attachment. Nature, 461: , Festjens, N., Vanden Berghe, T., and Vandenabeele, P. Necrosis, a well-orchestrated form of cell demise: signalling cascades, important mediators and concomitant immune response. Biochim Biophys Acta, 1757: , Dotsch, V., Bernassola, F., Coutandin, D., Candi, E., and Melino, G. p63 and p73, the ancestors of p53. Cold Spring Harb Perspect Biol, 2: a004887, Balaban, R. S., Nemoto, S., and Finkel, T. Mitochondria, oxidants, and aging. Cell, 120: ,
103 19. Flores, E. R., Tsai, K. Y., Crowley, D., Sengupta, S., Yang, A., McKeon, F., and Jacks, T. p63 and p73 are required for p53-dependent apoptosis in response to DNA damage. Nature, 416: , Suh, E. K., Yang, A., Kettenbach, A., Bamberger, C., Michaelis, A. H., Zhu, Z., Elvin, J. A., Bronson, R. T., Crum, C. P., and McKeon, F. p63 protects the female germ line during meiotic arrest. Nature, 444: , Trachootham, D., Alexandre, J., and Huang, P. Targeting cancer cells by ROS-mediated mechanisms: a radical therapeutic approach? Nat Rev Drug Discov, 8: , Meister, A. Glutathione metabolism and its selective modification. J Biol Chem, 263: , Godar, S., Ince, T. A., Bell, G. W., Feldser, D., Donaher, J. L., Bergh, J., Liu, A., Miu, K., Watnick, R. S., Reinhardt, F., McAllister, S. S., Jacks, T., and Weinberg, R. A. Growthinhibitory and tumor- suppressive functions of p53 depend on its repression of CD44 expression. Cell, 134: 62-73, Yan, W. and Chen, X. GPX2, a direct target of p63, inhibits oxidative stress-induced apoptosis in a p53-dependent manner. J Biol Chem, 281: , Motohashi, H. and Yamamoto, M. Nrf2-Keap1 defines a physiologically important stress response mechanism. Trends Mol Med, 10: , Hayes, J. D. and McMahon, M. NRF2 and KEAP1 mutations: permanent activation of an adaptive response in cancer. Trends Biochem Sci, 34: , Senoo, M., Pinto, F., Crum, C. P., and McKeon, F. p63 Is essential for the proliferative potential of stem cells in stratified epithelia. Cell, 129: , Keyes, W. M., Wu, Y., Vogel, H., Guo, X., Lowe, S. W., and Mills, A. A. p63 deficiency activates a program of cellular senescence and leads to accelerated aging. Genes Dev, 19: , Keyes, W. M., Pecoraro, M., Aranda, V., Vernersson-Lindahl, E., Li, W., Vogel, H., Guo, X., Garcia, E. L., Michurina, T. V., Enikolopov, G., Muthuswamy, S. K., and Mills, A. A. 91
104 DeltaNp63alpha is an oncogene that targets chromatin remodeler Lsh to drive skin stem cell proliferation and tumorigenesis. Cell Stem Cell, 8: , DeNicola, G. M., Karreth, F. A., Humpton, T. J., Gopinathan, A., Wei, C., Frese, K., Mangal, D., Yu, K. H., Yeo, C. J., Calhoun, E. S., Scrimieri, F., Winter, J. M., Hruban, R. H., Iacobuzio-Donahue, C., Kern, S. E., Blair, I. A., and Tuveson, D. A. Oncogeneinduced Nrf2 transcription promotes ROS detoxification and tumorigenesis. Nature, 475: , Sancak, Y., Peterson, T. R., Shaul, Y. D., Lindquist, R. A., Thoreen, C. C., Bar-Peled, L., and Sabatini, D. M. The Rag GTPases bind raptor and mediate amino acid signaling to mtorc1. Science, 320: , Takeda, S., Chen, D. Y., Westergard, T. D., Fisher, J. K., Rubens, J. A., Sasagawa, S., Kan, J. T., Korsmeyer, S. J., Cheng, E. H., and Hsieh, J. J. Proteolysis of MLL family proteins is essential for taspase1-orchestrated cell cycle progression. Genes Dev, 20: , Debnath, J., Muthuswamy, S. K., and Brugge, J. S. Morphogenesis and oncogenesis of MCF-10A mammary epithelial acini grown in three-dimensional basement membrane cultures. Methods, 30: , Nelson, C. M., Inman, J. L., and Bissell, M. J. Three-dimensional lithographically defined organotypic tissue arrays for quantitative analysis of morphogenesis and neoplastic progression. Nat Protoc, 3: , Rahman, I., Kode, A., and Biswas, S. K. Assay for quantitative determination of glutathione and glutathione disulfide levels using enzymatic recycling method. Nat Protoc, 1: ,
105 -CHAPTER THREE- DNA Damage Induces Mitochondrial ROS Production to Execute Programmed Necrotic Death 93
106 3.1 Introduction Reactive oxygn species (ROS) are potentially harmful reactive derivatives of molecular oxygen that can damage all intracellular constituents 1. Although they are proposed to be central initiators and effectors of programmed necrosis under diverse settings 2, much about their regulation in this setting remain unknown. Mitochondria are the primary sources of ROS in most cell types and are proposed to be major sources of ROS production during programmed necrosis 2. ROS production occurs during normal physiological processes such as fatty acid catabolism and aerobic respiration, and are countered by cellular antioxidant defences 3. Excessive production and/or impaired antioxidant capacity generates a deleterious state of oxidative stress, which can lead to cellular dysfunction and demise 1. Intracellular ROS is primarily produced by the mitochondrial electron transport chain (ETC) as the unavoidable byproduct of aerobic respiration 3. The leakage of electrons passing through the ETC leads to the one-electron reduction of O 2 to O 2 (superoxide), which can then give rise to other ROS such as the hydroxyl radical 3. Depending on the nature of the initial death signal, broadly similar models have emerged to explain the mechanism underlying increased mitochondrial ROS production during programmed necrosis, with the commonality being that increased electron flow into the ETC is proposed to cause increased rate of electron leakage, and hence excessive ROS production 2. With death receptor ligation-induced RIP1 kinase-dependent necroptosis, this can be caused by increased entry of energy substrates into the tricarboxylic acid (TCA) cycle due to upregulation of glycolysis and glutamine metabolism, itself due to phosphorylation-mediated stimulation of metabolic enzymes by the RIP1 interacting partner RIP3 kinase 4. TNF receptor ligation can also induce superoxide production by the NAPDH oxidase Nox1 to execute necrosis 5. In the case of ischemia-reperfusion injury, ATP depletion during ischemia impairs the ability of ATP-dependent calcium pumps to extrude calcium from the cytosol into the endoplasmic reticulum (ER) or the extracellular space 6. Increased cytosolic [Ca 2+ ] leads to elevated mitochondrial matrix [Ca 2+ ], which stimulates TCA cycle enzymes and thus electron entry into the ETC 6. This acute increase in mitochondrial oxidative metabolism leads to a burst of ROS generation following reperfusion 6. 94
107 We have reported that DNA double strand break (DSB)-induced programmed necrotic death (PND) requires de novo gene expression and is executed by the p53-mediated induction of the lysosomal protease cathepsin Q in cooperation with p53-independent induction of reactive oxygen species (ROS) 7. DNA DSB-induced PND in Bax -/- Bak -/- double knockout (DKO) mouse embryonic fibroblasts (MEFs) does not rely on RIP1 7, thus making it unlikley for RIP3-mediated metabolic upregulation to be responsible for ROS production since RIP3 is phosphorylated and activated by RIP1 in the necrosome. Genotoxic stress induced by DNA alkylation has previously been shown to deplete cellular ATP due to overactivation of the DNA repair enzyme PARP1 8. However, we found that PND induced by DSBs is not dependent on PARP1 7. Furthermore, in our setting, DNA damage-induced ROS production and death requires de novo gene expression 7, an ATP-dependent process. Additionally, this form of programmed necrosis is also independent of cyclophilin D, a primary mediator of calcium-induced necrosis Thus, it is unlikely for increased ROS production to be secondary to ATP depletion and calcium dysregulation, as in the case of ischemia-reperfusion injury. Although it is clear that there is a regulated production of ROS in this setting, the underlying mechanism(s) are unknown and will be the focus of this chapter. 3.2 Results Though it is unlikely for ATP depletion and subsequent calcium dysregulation to be responsible for double strand break-induced ROS production, we nevertheless sought to rule out this possible by comparing the kinetics of mitochondrial ROS production and cell death with changes in cellular ATP levels. Following treatment of transformed Bax and Bak DKO MEFs with the topoisomerase II inhibitor etoposide, which generates DNA double strand breaks, an early and sustained rise in mitochondrial matrix superoxide levels was detected by the matrix-localized superoxide-specific indicator dye MitoSOX Red (Fig. 3.1A) prior to any detectable cell death (Fig. 3.1B). Surprisingly, there was a burst of ATP production at 12 hours following etoposide treatment, coinciding with the time frame during which superoxide started to increase (Fig. 3.1C). Subsequently, ATP levels continued to decline following ROS induction (Fig. 3.1C). These data indicate that mitochondrial ROS production is not caused by bioenergetic collapse, as in the case 95
108 of ischemia-reperfusion injury. Rather, the early increase in ATP levels suggest that double strand breaks may induce an early upregulation of ETC activity, leading to ATP and superoxide generation. The latter then cooperates with cathepsin Q, with deleterious and eventually lethal consequences for the cell. The ETC is located in the mitochondrial inner membrane and generates the electrochemical gradient required for ATP generation 12. Electrons enter the ETC via complex I or II and are carried from both by ubiquinone to complex III, then transferred via cytochrome c to complex IV, where they leave via the reduction of molecular oxygen to water 12 (Fig. 3.2A). Of these, complex I and III are accepted as the major sources of electron leakage and hence mitochondrial superoxide production, with a minor contribution by complex II implicated in specific settings 3. To determine whether or not the ETC plays a role in DNA damage-induced ROS production, we assayed for the intrinsic enzymatic activity of the ETC complexes I, II, III, and IV in isolated mitochondria. Indeed, treatment with etoposide increased the enzymatic activity of complexes III and IV, but not of the others. Notably, etoposide did not affect the activity of the mitochondrial matrix enzyme citrate synthase (Fig. 3.2, B to F). Since complex IV is not a site of superoxide formation, this suggests DNA damage increases mitochondrial ROS production via induction of ETC complex III. Complex III of the ETC is composed of 11 subunits 12. The mechanism of superoxide production at complex III is as follows 13 : 1) complexes I and II transfer two electrons to the mobile electron carrier ubiquinone, forming the reduced ubiquinol; 2) the Rieske iron-sulfur protein (RISP) subunit of complex III, encoded by the gene Uqcrfs1, sequentially transfers one electron at a time from ubiquinol to cytochrome c; 3) sequential single electron oxidation of ubiquinol transiently creates the free radical ubisemiquinone, which can then either donate its remaining electron to other complex III electron carriers, or donate directly to oxygen to generate superoxide. RNAi-mediated knockdown of the RISP subunit prevents ubisemiquinone formation, thus abolishing complex III ROS production Therefore, to test the role of complex III in DNA damage-induced ROS and cell death, we used RNAi to knockdown the RISP. We found that knockdown of RISP was able to prevent both ROS induction (Fig. 3.2G) and cell death (Fig. 3.2H) 96
109 following treatment with etoposide. In contrast, knockdown of the essential ETC complex I chaperon protein Ndufaf1, loss of which decreases complex I abundance and activity 16 and inhibits antigen receptor-induced ROS production in T lymphocytes 17, did not protect against etoposide-induced ROS (Fig. 3.2H) and cell death (Fig. 3.2H), in line with our observation that etoposide did not alter complex I activity. We next sought to determine whether the canonical ATM-mediated DNA damage response to double strand breaks is involved in the induction of ETC complex III activity and ROS by etoposide. ATM is a phosphatidylinositol 3-kinase (PI3K)-related kinase (PIKK) whose activity can be inhibited using the compound LY This compound also inhibits PI3Ks as well as ATR and DNA-PK, two other PIKKs involved in the DNA damage checkpoint. Though both ATR and DNA-PK have been implicated in the response to double strand breaks, ATM is widely considered to be the primary mediator Importantly, we demonstrated that LY blocked etoposide-induced ROS as measured by the broad-spectrum ROS indicator H 2 DCFDA, the nonmitochondria-specific superoxide indicator dihydroethidium (DHE), and MitoSOX Red (Fig. 3.3A); and protected against cell death (Fig. 3.3B). Furthermore, RNAi-mediated knockdown of ATM using two sirna oligos targeting distinct regions of ATM mrna provided modest but significant inhibition of etoposide-induced ROS using all three ROS indicator dyes (Fig. 3.3C), and also protected against death (Fig. 3.3D). Knockdown of ATM was confirmed by qrt-pcr (Fig. 3.3E). The difference in protection afforded by knockdown of ATM versus treatment with LY suggests that ATR and DNA-PK are also involved. Notably, ATM provided much greater protection against cell death (Fig. 3.3D) than against ROS induction (Fig. 3.3C). This is most likely due to abrogation of ATM-mediated activation of p53, whose transcriptional target cathepsin Q cooperates in parallel with ROS to execute programmed necrotic death. These results provided clear indication that the canonical double strand break-induced DNA damage signaling pathway may be involved in upregulating ETC complex III activity in response to etoposide. Indeed, LY was able to completely prevent the induction of ETC complex III activity triggered by treatment with etoposide (Fig. 3.3F). Immunoblot analysis of the mitochondrial outer membrane protein VDAC1 showed it was present in equal amounts between 97
110 all samples of isolated mitochondria used for the complex III activity assay (Fig. 3.3G), indicating differences in its activity was not due to differences in purity of the mitochondria preparations. 3.3 Materials and Methods sirna sirna oligos were reverse-transfected using LipofectamineTM RNAiMAX (Invitrogen) to a final concentration of 10. The mouse sirna oligos were purchased from Ambion Silencer Select oligos (Applied Biosystems): Ndufaf1 #1, 5 -GACACGGAGUUUAUCCAGAtt (Ambion s88121); Ndufaf1 #2, 5 -GGCAGUUUCGUGAAAAAGAtt (Ambion s206245); Uqcrfs1 #1, 5 - CGACUUCUCUGACUAUCGUtt (Ambion s83717); Uqcrfs1 #2, 5 - GCAUGAUUUAGAUCGAGUAtt (Ambion s83718); ATM #1, 5 -CAUCUAAUGGUCUAACGUAtt (Ambion s62693); ATM #2, 5 -GGCUUUUUCCUACGAUUAUtt (Ambion s62694; Ambion Silencer Select Negative Control #1. Reverse transcription and quantitative PCR Total RNA was extracted from cells using TRIZOL (Invitrogen) according to the manufacturer s instruction. Reverse transcription was performed with oligo-dt plus random decamer primers (Ambion) using Superscript II (Invitrogen). Quantitative PCR was performed with SYBR green master mix (Applied Biosystems) in duplicates using indicated gene specific primers. For TaqMan probes, quantitative PCR was performed with TaqMan PCR Master Mix (Applied Biosystems). Quantitative PCR was performed on an ABI Prism 7300 sequence detection system (Applied Biosystems). Data were analyzed as described previously by normalization against GAPDH that was detected using TaqMan probes (Applied Biosystems) 7. Cell culture and viability assays SV40-transformed Bax -/- Bak -/- double-knockout (DKO) mouse embryonic fibroblasts (MEFs) were previously described 7. Cell death was quantified by propidium iodide (Sigma) staining, followed by flow cytometric analysis using either a FACSCalibur (BD Biosciences) or a LSRFortessa (BD 98
111 Biosciences). Data were analyzed using CellQuest Pro (BD Biosciences) or FACSDiva (BD Biosciences). P values for statistical analyses were obtained using Student's t test. Measurement of ROS Production of ROS was monitored by flow cytometric analyses using ROS-sensitive dyes. Cells were incubated with 1 µm 2,7 -dichlorofluorescein diacetate (H2DCFDA, Invitrogen) or 1 µm dihydroethidium (DHE, Invitrogen) at 37 C for 30 minutes, or with 2.5 µm MitoSOX Red (Invitrogen) at 37 C for 10 minutes. Oxidation of the ROS-sensitive dye was quantified by flow cytometric analysis using either a FACSCalibur (BD Biosciences) or a LSRFortessa (BD Biosciences). Median fluorescence in the appropriate detection channels was assessed by FlowJo (Tree Star). Fold induction of ROS was determined by dividing the median fluorescence of experimental samples with that of control samples. P values for statistical analyses were obtained using Student s t-test. Reagents Chemicals used are listed as follows: etoposide, acetyl-coa, malonate, cytochrome C, DCPIP, sodium succinate, DTNB, oxaloacetic acid, rotenone, decylubiquinone, antimycin A, sodium borohydride, propidium iodide, LY (all Sigma); NADH (Roche). Mitochondrial isolation, antibodies, and immunoblot analysis Isolation of mitochondria was performed using trituration and differential centrifugation as previously described 21, using SHE buffer (250 mm D-sucrose, 10 mm HEPES ph 7.4, 1 mm EGTA, 0.5 mm PMSF). For electron transport chain activity assays, disruption of mitochondrial membranes was achieved with 3 freeze-thaw cycles followed by sonication; mitochondria were aliquoted and stored at -80C until use in assays. For immunoblot analysis, lysate was prepared from an aliquot of intact mitochondria taken prior to free-thaw and sonication and was resolved by 10% NuPAGE gel (Invitrogen) and transferred onto PVDF membrane (Immobilon-P, Millipore). Antibody detection was accomplished using enhanced chemiluminescence method (Western 99
112 Lightning, PerkinElmer) and LAS-3000 Imaging system (FUJIFILM). Anti-VDAC1 antibody (Mouse anti-porin 31HL, Calbiochem) was used for immunblot analysis. Electron transport chain activity assays Spectrophotometric assays for electron transport chain complex I-IV and citrate synthase activities using isolated mitochondria have been previously described 22 and are summarized below. Complex I: NADH:ubiquinone oxidoreductase activity was determined by incubating mitochondria in 25 mm K-phosphate buffer ph 7.5 containing 2.5 mg/ml BSA, 5 mm MgCl2, 0.1 mm NADH, 0.6 mm KCN, and 130 mm decylubiquinone, and either with or without 10 mm rotenone in order to determine Complex I-specific, rotenone-sensitive activity. Oxidoreductase activity was calculated from the rate of decrease in absorbance at 340 nm due to NADH oxidation. Molar extinction coefficient for NADH = 6290 M -1 cm -1. Complex II: Succinate dehydrogenase activity was determined by incubating mitochondria in 30 mm K-phosphate buffer ph 7.5 containing 20 mm sodium succinate, 3 mm KCN, 50 mm DCPIP, and 130 mm decylubiquinone, and either with or without 5 mm malonate in order to determine Complex IIspecific, malonate-sensitive activity. Dehydrogenase activity was calculated from the rate of decrease in absorbance at 600 nm due to DCPIP oxidation. Molar extinction coefficient for DCPIP = M -1 cm -1. Complex III: Ubiquinol:cytochrome c oxidoreductase activity was determined by incubating mitochondria in buffer containing 26.7 mm KH 2 PO 4, 2.58 mm EDTA, mm KCN, 8.6% DMSO, 92.8 mm decylubiquinol, 0.1% oxidized cytochrome c, ph 7.5, either with or without 50 nm antimycin A to determine Complex III-specific, antimycin A-sensitive activity. Oxidoreductase activity was calculated from the rate of increase in absorbance at 550 nm due to cytochrome c reduction. Molar extinction coefficient for cytochrome c = M -1 cm -1. Complex IV: Cytochrome c oxidase activity was determined by incubating mitochondria in 10 mm K- phosphate buffer ph 7 containing 0.1% reduced cytochrome c, with non-specific activity determined by performing the reaction without mitochondria. Oxidase activity was calculated from the rate of decrease in absorbance at 550 mm due to cytochrome c oxidation. Molar extinction coefficient for cytochrome c = M -1 cm -1. Citrate synthase: Activity was determined by 100
113 incubating mitochondria in 40.4 mm Tris-Cl buffer ph 8.1 containing 0.06 mm Acetyl-CoA, 0.1 mm DTNB, 0.048% Triton-X 100, and 0.5 mm oxaloacetic acid. Citrate synthase activity was calculated from the rate of increase in absorbance at 412 nm due to formation of 5-thio-2- nitrobenzoate anion from DTNB, with non-specific formation determined by performing the reaction without oxaloacetic acid. Molar extinction coefficient for 5-thio-2-nitrobenzoate anion = M -1 cm -1. Enzyme activity for all assays were calculated as follows: Activity (nmol / min / mg protein) = ΔAbs/(Molar extinction coefficient x protein concentration x volume sample). Mitochondria protein concentration was determined using BCA Kit (Pierce). ATP assay Cellular ATP levels were measured by the luciferin/luciferase method using ATP Bioluminescence Assay Kit HS II (Roche) per manufacturer s instructions. Lysates used for ATP determination were prepared from 10 4 cells for each condition, as determined by staining with Trypan blue and counting on a hemocytometer. Upon automatic injection of 50 µl of luciferase reagents to 50 µl of diluted samples, the luminescence signal was integrated for 10 sec after a delay of 1 sec using a Dynex MLX96-well Plate Luminometer (MTX Lab Systems, Inc). Control samples were harvested from subconfluent cell populations. 3.4 Discussion We previously reported that DNA double strand breaks activate programmed necrotic death via induction of the lysosomal protease cathepsin Q and ROS in a manner dependent on de novo gene expression 7. Although that study revealed p53 as the transcription factor responsible for cathepsin Q upregulation, the mechanism underlying ROS induction was unclear. Here, we conclude that the DNA damage signalling pathway mediated by ATM/ATR/DNA-PK is responsible for etoposide-induced ROS via increasing the activity of the ETC complex III. The mechanism of ETC complex III activity induction will be the focus of future investigation. Given the rapid rise in matrix superoxide (almost two-fold 18 hours following etoposide treatment) following etoposide treatment and the dependence of ROS induction on de novo gene 101
114 expression, the most likely mechanism for ROS induction is the transcriptional upregulation of a regulatory protein that alters the activity of existing complex III subunits via protein modification. This is more likely than direct transcriptional upregulation of complex III subunits due to the kinetics of mitochondrial superoxide induction and the complexity of this enzyme complex. ETC complex III consists of 10 subunits encoded by nuclear DNA and 1 subunit encoded by mitochondrial DNA 12. The mature complex functions as a homodimer and is assembled and embedded into the mitochondrial inner membrane in a multi-step process 12. Assembly involves the formation of three subcomplexes that assemble into a membrane-embedded precomplex, which homodimerize with another precomplex before full assembly is completed with the addition of the final subunits 12. Two likely candidates that may mediate ATM/ATR/DNA-PK-induced upregulation of complex III activity are p73 and PGC-1α. p73 is a member of the p53/p63/p73 family of transcription factors and is known to mediate cell cycle arrest and apoptosis in response to genotoxic stress 23. Furthermore, loss of function studies revealed that deficiency of p73 protects against DNA damage-induced cell death 24. Genotoxic stress can activate p73 via the ATM and ATR effector kinases Chk2 and Chk1 25 ; and also via the p38 MAP kinase (MAPK) pathway 26, which can be activated in an ATM/ATR-dependent manner in response to genotoxic stress 18. p38 MAPK can also activate the transcriptional co-activator PGC-1α 27, a master regulator of oxidative metabolism with an important role in regulating mitochondrial gene expression and biogenesis 28. Phosphorylation of PGC-1α by the p38 MAPK leads to its stabilization and activation, resulting in increased mitochondrial respiration 27. PGC-1α can also be activated via phosphorylation by AMPK 29. Intriguingly, etoposide was recently shown to activate PGC-1α and mitochondrial biogenesis via AMPK in an ATM-dependent manner 30. Therefore, double strand breaks may induce increased ETC complex III activity via activation of PGC-1α. The identity of the downstream transcriptional targets that are ultimately responsible for regulating ETC complex III activity in response to double strand breaks is unclear due to a lack of characterization of complex III post-translational regulation. However, existing data on the bettercharacterized ETC complexes I and IV provides some insight into possible regulatory 102
115 mechanisms. Both complex I and IV have been demonstrated to be regulated via phosphorylation 31. Phosphorylation of human complex I NDUFS4 subunit at serine 173 promotes its mitochondrial matrix import and maturation 32 while phosphorylation of bovine complex IV catalytic subunit I at tyrosine 304 leads to its inactivation 33. Other phosphorylation sites have also been identified for complex I and complex IV, though the functional consequences of those are unclear. Furthermore, complex I is positively regulated through deacetylation 34 and inhibited by S- nitrosylation of its 75 kda subunit 35, 36. Although proteomics studies suggest complex III also undergoes phosphorylation, the residues, effects, and mediators of this are unclear 31. However, the emerging prominence of post-translational as a mechanism for regulating complex I and IV activity suggests complex III is likely to be regulated in a similar manner. Especially provocative is our finding that complex IV activity is also upregulated following DNA double strand breaks to a similar extent as complex III (Fig. 3.2E), suggesting possible post-translational co-regulation of these complexes. Impaired apoptosis is frequently observed in cancers, with evasion of cell death being a hallmark of cancer progression 37. Recently, cancerous cells have also been proposed to exhibit an increased need for antioxidant defences compared to noncancerous cells, likey due to metabolic derangements and microenvironmental factors that increase ROS production 38. Therefore, pharmacological induction of oxidative is currently being evaluated for use as chemotherapy 38. Our study demonstrates a novel mechanism by which DNA double strand breaks can induce increased mitochondrial ROS production in apoptosis incompetent cells. This suggests that chemotherapeutic strategies combining genotoxic agents with therapeutics that increase mitochondrial oxidative stress may prove particularly beneficial in the clinical setting. 3.5 Figure Legends Figure 3.1 Induction of mitochondrial matrix ROS by DNA damage is an early event that precedes cell death and ATP depletion (A) DNA damage induces early increase of mitochondrial matrix ROS in Bax and Bak DKO MEFs. SV40-transformed DKO MEFs were mock treated or treated with etoposide (10 103
116 µg/ml). At the indicated time points following treatment, oxidation of the mitochondrial matrix-targeted ROS-sensitive dye MitoSOX Red was quantified by flow cytometric analysis. Data shown are fold increase of ROS after etoposide treatment. Data presented are mean ± SD from three independent experiments. (B) DNA damage-induced death of DKO MEFs is observed following rise in mitochondrial matrix ROS. DKO MEFs were mock treated or treated with etoposide (10 µg/ml) as in (A). At the indicated time points following treatment, cell viability was assessed by propidium iodide (PI) staining followed by flow cytometric analysis. Data presented are mean percentage of PI positive cells ± SD from three independent experiments. (C) DNA damage-induced ATP depletion in DKO MEFs occurs later than ROS induction. DKO MEFs were mock-treated or treated with etoposide (10 µg/ml) as in (A). At indicated time points following treatment, equal numbers of cells were taken from mock-treated or etoposide-treated samples for the determination of [ATP]. Data presented are mean ± SD from three independent experiments. Figure 3.2 ETC complex III mediates DNA double strand break-induced ROS and necrosis (A) Flow of electrons through the mitochondrial electron transport chain. Electrons enter the chain via complex I or compex II; they are transferred to ubiquinol (Q) which carries them to complex III; cytochrome c carries electrons from complex III to complex IV, where they reduce molecular oxygen to water. (B) DKO MEFs were mock-treated or treated with etoposide (10 µg/ml). 40 hours after etoposide treatment, mitochondria were isolated and used to determine the activity of electron transport chain complex I. Data presented are mean ± SD from three independent experiments. (C) Mitochondria isolated from MEFs following 40 hours of mock or etoposide (10 µg/ml) treatment, as in (B), were used to determine activity of electron transport chain complex II. Data presented are mean ± SD from three independent experiments. 104
117 (D) Mitochondria isolated from MEFs following 40 hours of mock or etoposide (10 µg/ml) treatment, as in (B), were used to determine activity of electron transport chain complex III. Data presented are mean ± SD from three independent experiments. *, P < (E) Mitochondria isolated from MEFs following 40 hours of mock or etoposide (10 µg/ml) treatment, as in (B), were used to determine activity of electron transport chain complex IV. Data presented are mean ± SD from three independent experiments. *, P < (F) Mitochondria isolated from MEFs following 40 hours of mock or etoposide (10 µg/ml) treatment, as in (B), were used to determine activity of citrate synthase. Data presented are mean ± SD from three independent experiments. (G) Deficiency of complex III but not complex I activity protects DKO MEFs against DNA damage-induced ROS. DKO MEFs were transfected with scrambled sirna (siscr), or sirna against the Ndufaf1 subunit of electron transport chain complex I or against the RISP subunit of complex III (encoded by the gene Uqcrfs1). Cells were subsequently mock-treated or treated with etoposide (10 µg/ml) for two days before oxidation of the ROS-sensitive dye H 2 DCFDA was quantified by flow cytometric analysis. Data shown are fold increase of ROS after etoposide treatment. Data presented are mean ± SD from three independent experiments. (H) Deficiency of complex III but not complex I activity protects DKO MEFs against DNA damage-induced death. DKO MEFs were transfected with scrambled sirna (siscr), or sirna against the Ndufaf1 subunit of electron transport chain complex I or against the RISP subunit of complex III (encoded by the gene Uqcrfs1). Cells were subsequently mock-treated or treated with etoposide (10 µg/ml) for three days before cell viability was assessed by staining with PI followed by flow cytometric analysis. Data are presented as mean percentage of PI-positive cells ± SD from three independent experiments. Figure 3.3 DNA damage response pathway induces ROS, necrotic death, and ETC complex III activity 105
118 (A) Inhibition of ATM/ATR/DNA-PK-mediated DNA damage signaling protects against DNA damage-induced ROS. DKO MEFs were treated with LY (50 mm), etoposide (10 mg/ml), or both. Conditions using LY were pre-treated with the drug for 30 minutes before mock or etoposide treatment. Two days after drug treatment, oxidation of the redox-sensitive dyes H 2 DCFDA, dihydroethidium (DHE), and MitoSOX Red were quantified by flow cytometric analysis. Data shown are fold increase of ROS after etoposide treatment. Data presented are mean ± SD from three independent experiments. (B) Inhibition of ATM/ATR/DNA-PK-mediated DNA damage signaling protects against DNA damage-induced death. DKO MEFs were treated as in (A). Three days after drug treatment, cell viability was assessed using PI staining followed by flow cytometric analysis. Data presented are mean percentage of PI positive cells ± SD from three independent experiments. (C) Loss of ATM protects DKO MEFs against DNA damage-induced ROS. DKO MEFs were transfected with scrambled sirna (siscr) or sirna against ATM. Cells were subsequently mock-treated or treated with etoposide (10 µg/ml) for two days before oxidation of the ROS-sensitive dyes H 2 DCFDA, DHE, and MitoSOX Red were quantified by flow cytometric analysis. Data shown are fold increase of ROS after etoposide treatment. Data presented are mean ± SD from three independent experiments. *, P < (D) Loss of ATM protects DKO MEFs against DNA damage-induced death. DKO MEFs were transfected as in (C) and mock-treated or treated with etoposide (10 µg/ml). Three days following etoposide treatment, cell viability was assessed using PI staining followed by flow cytometric analysis. Data presented are mean percentage of PI positive cells ± SD from three independent experiments (E) (F) DKO MEFs transfected as in (C) were analyzed for ATM expression by qrt-pcr. Inhibition of ATM/ATR/DNA-PK-mediated DNA damage signaling protects DKO MEFs against DNA damage-induced upregulation of ETC complex III activity. DKO MEFs were 106
119 treated with LY (50 mm), etoposide (10 mg/ml), or both, as in (A). 24 hours after etoposide treatment, mitochondria were isolated and used to determine the activity of the electron transport chain complex III. Data presented are mean ± SD from three independent experiments. *, P < (G) Protein lysates prepared from mitochondrial samples described in (F) were assessed for VDAC1 expression by anti-vdac1 immunoblot. LY = LY Etop = etoposide. 107
120 3.6 Figures 108
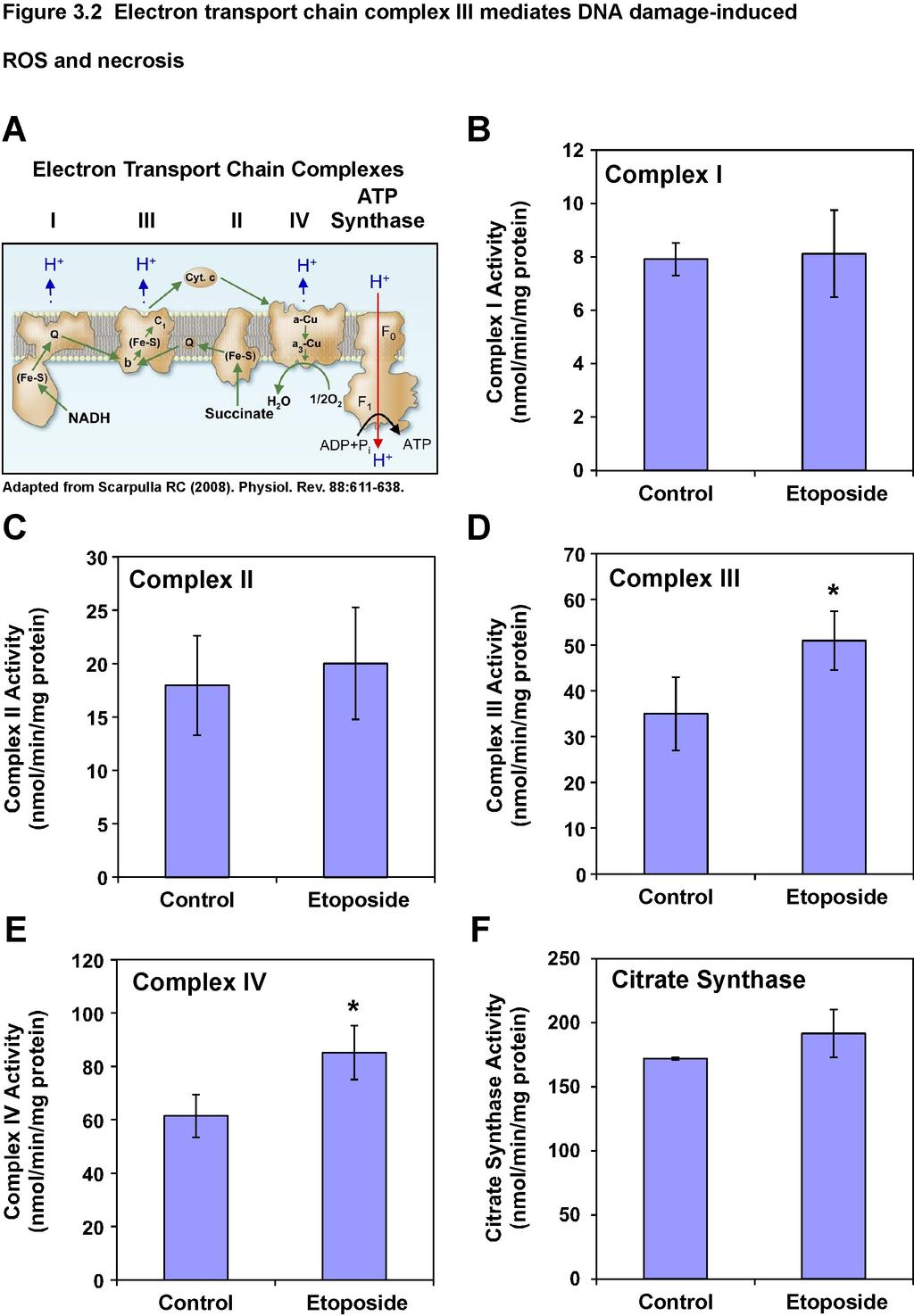
121 109
Programmed Cell Death (apoptosis)
") Programmed Cell Death (apoptosis) Stereotypic death process includes: membrane blebbing nuclear fragmentation chromatin condensation and DNA framentation loss of mitochondrial integrity and release of
Programmed Cell Death (apoptosis) Stereotypic death process includes: membrane blebbing nuclear fragmentation chromatin condensation and DNA framentation loss of mitochondrial integrity and release of
Apoptosis Chapter 9. Neelu Yadav PhD
 Apoptosis Chapter 9 Neelu Yadav PhD Neelu.Yadav@Roswellpark.org 1 Apoptosis: Lecture outline Apoptosis a programmed cell death pathway in normal homeostasis Core Apoptosis cascade is conserved Compare
Apoptosis Chapter 9 Neelu Yadav PhD Neelu.Yadav@Roswellpark.org 1 Apoptosis: Lecture outline Apoptosis a programmed cell death pathway in normal homeostasis Core Apoptosis cascade is conserved Compare
Apoptotic Pathways in Mammals Dr. Douglas R. Green
 Apoptotic Pathways in Mammals Douglas R. Green 1 Apoptosis A form of cell death that is defined morphologically, and features a number of biochemical events Programmed cell death Cell death that occurs
Apoptotic Pathways in Mammals Douglas R. Green 1 Apoptosis A form of cell death that is defined morphologically, and features a number of biochemical events Programmed cell death Cell death that occurs
#19 Apoptosis Chapter 9. Neelu Yadav PhD
 #19 Apoptosis Chapter 9 Neelu Yadav PhD Neelu.Yadav@Roswellpark.org Why cells decide to die? - Stress, harmful, not needed - Completed its life span Death stimulation or Stress Cell Survival Death Functions
#19 Apoptosis Chapter 9 Neelu Yadav PhD Neelu.Yadav@Roswellpark.org Why cells decide to die? - Stress, harmful, not needed - Completed its life span Death stimulation or Stress Cell Survival Death Functions
Introduction to pathology lecture 5/ Cell injury apoptosis. Dr H Awad 2017/18
 Introduction to pathology lecture 5/ Cell injury apoptosis Dr H Awad 2017/18 Apoptosis = programmed cell death = cell suicide= individual cell death Apoptosis cell death induced by a tightly regulated
Introduction to pathology lecture 5/ Cell injury apoptosis Dr H Awad 2017/18 Apoptosis = programmed cell death = cell suicide= individual cell death Apoptosis cell death induced by a tightly regulated
Molecular biology :- Cancer genetics lecture 11
 Molecular biology :- Cancer genetics lecture 11 -We have talked about 2 group of genes that is involved in cellular transformation : proto-oncogenes and tumour suppressor genes, and it isn t enough to
Molecular biology :- Cancer genetics lecture 11 -We have talked about 2 group of genes that is involved in cellular transformation : proto-oncogenes and tumour suppressor genes, and it isn t enough to
Mechanisms of Cell Death
 Mechanisms of Cell Death CELL DEATH AND FORMATION OF THE SEMICIRCULAR CANALS Carol M. Troy August 25, 2008 FROM: Fekete et al., Development 124: 2451 (1997) PHENOMENOLOGY OF CELL DEATH I. DEVELOPMENT A.
Mechanisms of Cell Death CELL DEATH AND FORMATION OF THE SEMICIRCULAR CANALS Carol M. Troy August 25, 2008 FROM: Fekete et al., Development 124: 2451 (1997) PHENOMENOLOGY OF CELL DEATH I. DEVELOPMENT A.
34 Apoptosis Programmed cell death is vital to the health and development of multicellular organisms.
 Principles of Biology contents 34 Apoptosis Programmed cell death is vital to the health and development of multicellular organisms. Apoptosis is the reason we have separate fingers and toes. During embryonic
Principles of Biology contents 34 Apoptosis Programmed cell death is vital to the health and development of multicellular organisms. Apoptosis is the reason we have separate fingers and toes. During embryonic
Cell Quality Control. Peter Takizawa Department of Cell Biology
 Cell Quality Control Peter Takizawa Department of Cell Biology Cellular quality control reduces production of defective proteins. Cells have many quality control systems to ensure that cell does not build
Cell Quality Control Peter Takizawa Department of Cell Biology Cellular quality control reduces production of defective proteins. Cells have many quality control systems to ensure that cell does not build
GMS 6644: Apoptosis. Introduction
 GMS 6644: Apoptosis Introduction (Feb. 15, 2006) Lei Xiao, Ph.D. Department of Anatomy & Cell Biology UF Shands Cancer Center ARB Rm R4-250, 846-1199, lxiao@ufl.edu Outline of the Lecture Different types
GMS 6644: Apoptosis Introduction (Feb. 15, 2006) Lei Xiao, Ph.D. Department of Anatomy & Cell Biology UF Shands Cancer Center ARB Rm R4-250, 846-1199, lxiao@ufl.edu Outline of the Lecture Different types
Cell cycle and apoptosis
 Cell cycle and apoptosis Cell cycle Definition Stages and steps Cell cycle Interphase (G1/G0, S, and G2) Mitosis (prophase, metaphase, anaphase, telophase, karyokinesis, cytokinesis) Control checkpoints
Cell cycle and apoptosis Cell cycle Definition Stages and steps Cell cycle Interphase (G1/G0, S, and G2) Mitosis (prophase, metaphase, anaphase, telophase, karyokinesis, cytokinesis) Control checkpoints
Apoptotic cell signaling in cancer progression and therapyw
 Integrative Biology Dynamic Article Links Cite this: Integr. Biol., 2011, 3, 279 296 www.rsc.org/ibiology REVIEW ARTICLE Apoptotic cell signaling in cancer progression and therapyw Jessica Plati, a Octavian
Integrative Biology Dynamic Article Links Cite this: Integr. Biol., 2011, 3, 279 296 www.rsc.org/ibiology REVIEW ARTICLE Apoptotic cell signaling in cancer progression and therapyw Jessica Plati, a Octavian
#19 Apoptosis Chapter 9. Neelu Yadav PhD
 #19 Apoptosis Chapter 9 Neelu Yadav PhD Neelu.Yadav@Roswellpark.org Why cells decide to die? - Stress, harmful, not needed - Completed its life span Death stimulation or Stress Cell Survival Death Functions
#19 Apoptosis Chapter 9 Neelu Yadav PhD Neelu.Yadav@Roswellpark.org Why cells decide to die? - Stress, harmful, not needed - Completed its life span Death stimulation or Stress Cell Survival Death Functions
Cancer. Throughout the life of an individual, but particularly during development, every cell constantly faces decisions.
 Cancer Throughout the life of an individual, but particularly during development, every cell constantly faces decisions. Should it divide? Yes No--> Should it differentiate? Yes No-->Should it die? Yes-->Apoptosis
Cancer Throughout the life of an individual, but particularly during development, every cell constantly faces decisions. Should it divide? Yes No--> Should it differentiate? Yes No-->Should it die? Yes-->Apoptosis
p53 and Apoptosis: Master Guardian and Executioner Part 2
 p53 and Apoptosis: Master Guardian and Executioner Part 2 p14arf in human cells is a antagonist of Mdm2. The expression of ARF causes a rapid increase in p53 levels, so what would you suggest?.. The enemy
p53 and Apoptosis: Master Guardian and Executioner Part 2 p14arf in human cells is a antagonist of Mdm2. The expression of ARF causes a rapid increase in p53 levels, so what would you suggest?.. The enemy
Cell Injury MECHANISMS OF CELL INJURY
 Cell Injury MECHANISMS OF CELL INJURY The cellular response to injurious stimuli depends on the following factors: Type of injury, Its duration, and Its severity. Thus, low doses of toxins or a brief duration
Cell Injury MECHANISMS OF CELL INJURY The cellular response to injurious stimuli depends on the following factors: Type of injury, Its duration, and Its severity. Thus, low doses of toxins or a brief duration
Signaling Apoptosis. Scott André Oakes, M.D. Dept. of Pathology Univ. of Calif-San Francisco. Cyt c Release BAX/BAK. Apoptosome Formation
 Signaling Apoptosis Cyt c Release BAX/BAK Apoptosome Formation Caspase Activation Scott André Oakes, M.D. Dept. of Pathology Univ. of Calif-San Francisco Why do we need cell death? Sculpt Organs Control
Signaling Apoptosis Cyt c Release BAX/BAK Apoptosome Formation Caspase Activation Scott André Oakes, M.D. Dept. of Pathology Univ. of Calif-San Francisco Why do we need cell death? Sculpt Organs Control
The death receptors: signaling and modulation
 The death receptors: signaling and modulation 1 1 The extrinsic cell death pathway 2 Nat Rev Drug Discov. 2008 Dec;7(12):1001-12. 2 Death receptors Belong to the tumor necrosis factor (TNF) receptor gene
The death receptors: signaling and modulation 1 1 The extrinsic cell death pathway 2 Nat Rev Drug Discov. 2008 Dec;7(12):1001-12. 2 Death receptors Belong to the tumor necrosis factor (TNF) receptor gene
A particular set of insults induces apoptosis (part 1), which, if inhibited, can switch to autophagy. At least in some cellular settings, autophagy se
, which, if inhibited, can switch to autophagy. At least in some cellular settings, autophagy se") A particular set of insults induces apoptosis (part 1), which, if inhibited, can switch to autophagy. At least in some cellular settings, autophagy serves as a defence mechanism that prevents or retards
A particular set of insults induces apoptosis (part 1), which, if inhibited, can switch to autophagy. At least in some cellular settings, autophagy serves as a defence mechanism that prevents or retards
Lecture 14 - The cell cycle and cell death
 02.17.10 Lecture 14 - The cell cycle and cell death The cell cycle: cells duplicate their contents and divide The cell cycle may be divided into 4 phases The cell cycle triggers essential processes (DNA
02.17.10 Lecture 14 - The cell cycle and cell death The cell cycle: cells duplicate their contents and divide The cell cycle may be divided into 4 phases The cell cycle triggers essential processes (DNA
Part I Molecular Cell Biology
 1 Part I Molecular Cell Biology RNA Regulation: Advances in Molecular Biology and Medicine, First Edition. Edited by Robert A. Meyers. 2014 Wiley-VCH Verlag GmbH & Co. KGaA. Published 2014 by Wiley-VCH
1 Part I Molecular Cell Biology RNA Regulation: Advances in Molecular Biology and Medicine, First Edition. Edited by Robert A. Meyers. 2014 Wiley-VCH Verlag GmbH & Co. KGaA. Published 2014 by Wiley-VCH
Enzyme-coupled Receptors. Cell-surface receptors 1. Ion-channel-coupled receptors 2. G-protein-coupled receptors 3. Enzyme-coupled receptors
 Enzyme-coupled Receptors Cell-surface receptors 1. Ion-channel-coupled receptors 2. G-protein-coupled receptors 3. Enzyme-coupled receptors Cell-surface receptors allow a flow of ions across the plasma
Enzyme-coupled Receptors Cell-surface receptors 1. Ion-channel-coupled receptors 2. G-protein-coupled receptors 3. Enzyme-coupled receptors Cell-surface receptors allow a flow of ions across the plasma
The discovery of Bcl-2
 The discovery of Bcl-2 Bcl-2: B-cell lymphoma 2 The pro-survival subfamily of Bcl-2 protein family Cloning of Bcl-2 as the oncogene which is deregulated at t(14;18) lymphomas Pioneer works by Tsujimoto
The discovery of Bcl-2 Bcl-2: B-cell lymphoma 2 The pro-survival subfamily of Bcl-2 protein family Cloning of Bcl-2 as the oncogene which is deregulated at t(14;18) lymphomas Pioneer works by Tsujimoto
Problem Set 8 Key 1 of 8
 7.06 2003 Problem Set 8 Key 1 of 8 7.06 2003 Problem Set 8 Key 1. As a bright MD/PhD, you are interested in questions about the control of cell number in the body. Recently, you've seen three patients
7.06 2003 Problem Set 8 Key 1 of 8 7.06 2003 Problem Set 8 Key 1. As a bright MD/PhD, you are interested in questions about the control of cell number in the body. Recently, you've seen three patients
Follicular Lymphoma. ced3 APOPTOSIS. *In the nematode Caenorhabditis elegans 131 of the organism's 1031 cells die during development.
 Harvard-MIT Division of Health Sciences and Technology HST.176: Cellular and Molecular Immunology Course Director: Dr. Shiv Pillai Follicular Lymphoma 1. Characterized by t(14:18) translocation 2. Ig heavy
Harvard-MIT Division of Health Sciences and Technology HST.176: Cellular and Molecular Immunology Course Director: Dr. Shiv Pillai Follicular Lymphoma 1. Characterized by t(14:18) translocation 2. Ig heavy
MOLECULAR CELL BIOLOGY
 1 Lodish Berk Kaiser Krieger scott Bretscher Ploegh Matsudaira MOLECULAR CELL BIOLOGY SEVENTH EDITION CHAPTER 13 Moving Proteins into Membranes and Organelles Copyright 2013 by W. H. Freeman and Company
1 Lodish Berk Kaiser Krieger scott Bretscher Ploegh Matsudaira MOLECULAR CELL BIOLOGY SEVENTH EDITION CHAPTER 13 Moving Proteins into Membranes and Organelles Copyright 2013 by W. H. Freeman and Company
Apoptosome dysfunction in human cancer
 Apoptosis 2004; 9: 691 704 C 2004 Kluwer Academic Publishers Apoptosome dysfunction in human cancer K. M. Hajra and J. R. Liu Department of Obstetrics and Gynecology, University of Michigan Medical School,
Apoptosis 2004; 9: 691 704 C 2004 Kluwer Academic Publishers Apoptosome dysfunction in human cancer K. M. Hajra and J. R. Liu Department of Obstetrics and Gynecology, University of Michigan Medical School,
shehab Moh Tarek ... ManarHajeer
 3 shehab Moh Tarek... ManarHajeer In the previous lecture we discussed the accumulation of oxygen- derived free radicals as a mechanism of cell injury, we covered their production and their pathologic
3 shehab Moh Tarek... ManarHajeer In the previous lecture we discussed the accumulation of oxygen- derived free radicals as a mechanism of cell injury, we covered their production and their pathologic
Objectives. Abbas Chapter 11: Immunological Tolerance. Question 1. Question 2. Question 3. Definitions
 Objectives Abbas Chapter 11: Immunological Tolerance Christina Ciaccio, MD Children s Mercy Hospitals and Clinics February 1, 2010 To introduce the concept of immunologic tolerance To understand what factors
Objectives Abbas Chapter 11: Immunological Tolerance Christina Ciaccio, MD Children s Mercy Hospitals and Clinics February 1, 2010 To introduce the concept of immunologic tolerance To understand what factors
Apoptosis Oncogenes. Srbová Martina
 Apoptosis Oncogenes Srbová Martina Cell Cycle Control point Cyclin B Cdk1 Cyclin D Cdk4 Cdk6 Cyclin A Cdk2 Cyclin E Cdk2 Cyclin-dependent kinase (Cdk) have to bind a cyclin to become active Regulation
Apoptosis Oncogenes Srbová Martina Cell Cycle Control point Cyclin B Cdk1 Cyclin D Cdk4 Cdk6 Cyclin A Cdk2 Cyclin E Cdk2 Cyclin-dependent kinase (Cdk) have to bind a cyclin to become active Regulation
BIOL 4374/BCHS 4313 Cell Biology Exam #1 February 13, 2001
 BIOL 4374/BCHS 4313 Cell Biology Exam #1 February 13, 2001 SS# Name This exam is worth a total of 100 points. The number of points each question is worth is shown in parentheses. Good luck! 1. (2) The
BIOL 4374/BCHS 4313 Cell Biology Exam #1 February 13, 2001 SS# Name This exam is worth a total of 100 points. The number of points each question is worth is shown in parentheses. Good luck! 1. (2) The
T cell maturation. T-cell Maturation. What allows T cell maturation?
 T-cell Maturation What allows T cell maturation? Direct contact with thymic epithelial cells Influence of thymic hormones Growth factors (cytokines, CSF) T cell maturation T cell progenitor DN DP SP 2ry
T-cell Maturation What allows T cell maturation? Direct contact with thymic epithelial cells Influence of thymic hormones Growth factors (cytokines, CSF) T cell maturation T cell progenitor DN DP SP 2ry
AperTO - Archivio Istituzionale Open Access dell'università di Torino
 AperTO - Archivio Istituzionale Open Access dell'università di Torino From the nucleus to the mitochondria and backthe odyssey of a multitask STAT3 This is the author's manuscript Original Citation: From
AperTO - Archivio Istituzionale Open Access dell'università di Torino From the nucleus to the mitochondria and backthe odyssey of a multitask STAT3 This is the author's manuscript Original Citation: From
RAS Genes. The ras superfamily of genes encodes small GTP binding proteins that are responsible for the regulation of many cellular processes.
 ۱ RAS Genes The ras superfamily of genes encodes small GTP binding proteins that are responsible for the regulation of many cellular processes. Oncogenic ras genes in human cells include H ras, N ras,
۱ RAS Genes The ras superfamily of genes encodes small GTP binding proteins that are responsible for the regulation of many cellular processes. Oncogenic ras genes in human cells include H ras, N ras,
7.06 Cell Biology EXAM #3 April 24, 2003
 7.06 Spring 2003 Exam 3 Name 1 of 8 7.06 Cell Biology EXAM #3 April 24, 2003 This is an open book exam, and you are allowed access to books and notes. Please write your answers to the questions in the
7.06 Spring 2003 Exam 3 Name 1 of 8 7.06 Cell Biology EXAM #3 April 24, 2003 This is an open book exam, and you are allowed access to books and notes. Please write your answers to the questions in the
Zool 3200: Cell Biology Exam 4 Part I 2/3/15
 Name: Key Trask Zool 3200: Cell Biology Exam 4 Part I 2/3/15 Answer each of the following questions in the space provided, explaining your answers when asked to do so; circle the correct answer or answers
Name: Key Trask Zool 3200: Cell Biology Exam 4 Part I 2/3/15 Answer each of the following questions in the space provided, explaining your answers when asked to do so; circle the correct answer or answers
1. to understand how proteins find their destination in prokaryotic and eukaryotic cells 2. to know how proteins are bio-recycled
 Protein Targeting Objectives 1. to understand how proteins find their destination in prokaryotic and eukaryotic cells 2. to know how proteins are bio-recycled As a protein is being synthesized, decisions
Protein Targeting Objectives 1. to understand how proteins find their destination in prokaryotic and eukaryotic cells 2. to know how proteins are bio-recycled As a protein is being synthesized, decisions
The Cell Organelles. Eukaryotic cell. The plasma membrane separates the cell from the environment. Plasma membrane: a cell s boundary
 Eukaryotic cell The Cell Organelles Enclosed by plasma membrane Subdivided into membrane bound compartments - organelles One of the organelles is membrane bound nucleus Cytoplasm contains supporting matrix
Eukaryotic cell The Cell Organelles Enclosed by plasma membrane Subdivided into membrane bound compartments - organelles One of the organelles is membrane bound nucleus Cytoplasm contains supporting matrix
PCB 3023 Exam 4 - Form A First and Last Name
 PCB 3023 Exam 4 - Form A First and Last Name Student ID # (U Number) A Before beginning this exam, please complete the following instructions: 1) Write your name and U number on the first page of this
PCB 3023 Exam 4 - Form A First and Last Name Student ID # (U Number) A Before beginning this exam, please complete the following instructions: 1) Write your name and U number on the first page of this
Signal Transduction Cascades
 Signal Transduction Cascades Contents of this page: Kinases & phosphatases Protein Kinase A (camp-dependent protein kinase) G-protein signal cascade Structure of G-proteins Small GTP-binding proteins,
Signal Transduction Cascades Contents of this page: Kinases & phosphatases Protein Kinase A (camp-dependent protein kinase) G-protein signal cascade Structure of G-proteins Small GTP-binding proteins,
REGULATION OF ENZYME ACTIVITY. Medical Biochemistry, Lecture 25
 REGULATION OF ENZYME ACTIVITY Medical Biochemistry, Lecture 25 Lecture 25, Outline General properties of enzyme regulation Regulation of enzyme concentrations Allosteric enzymes and feedback inhibition
REGULATION OF ENZYME ACTIVITY Medical Biochemistry, Lecture 25 Lecture 25, Outline General properties of enzyme regulation Regulation of enzyme concentrations Allosteric enzymes and feedback inhibition
Receptor mediated Signal Transduction
 Receptor mediated Signal Transduction G-protein-linked receptors adenylyl cyclase camp PKA Organization of receptor protein-tyrosine kinases From G.M. Cooper, The Cell. A molecular approach, 2004, third
Receptor mediated Signal Transduction G-protein-linked receptors adenylyl cyclase camp PKA Organization of receptor protein-tyrosine kinases From G.M. Cooper, The Cell. A molecular approach, 2004, third
4. Lysosomes, Smooth Endoplasmic Reticulum, Mitochondria, and Inclusions
 4. Lysosomes, Smooth Endoplasmic Reticulum, Mitochondria, and Inclusions Undergraduate Graduate Histology Lecture Series Larry Johnson, Professor Veterinary Integrative Biosciences Texas A&M University
4. Lysosomes, Smooth Endoplasmic Reticulum, Mitochondria, and Inclusions Undergraduate Graduate Histology Lecture Series Larry Johnson, Professor Veterinary Integrative Biosciences Texas A&M University
Signaling Through Immune System Receptors (Ch. 7)
") Signaling Through Immune System Receptors (Ch. 7) 1. General principles of signal transduction and propagation. 2. Antigen receptor signaling and lymphocyte activation. 3. Other receptors and signaling
Signaling Through Immune System Receptors (Ch. 7) 1. General principles of signal transduction and propagation. 2. Antigen receptor signaling and lymphocyte activation. 3. Other receptors and signaling
Kinase Inhibitor p21 WAF1/CIP1 in Apoptosis and Autophagy
 Pivotal Role of the Cyclin-dependent Kinase Inhibitor p21 WAF1/CIP1 in Apoptosis and Autophagy Keishi Fujiwara, Shigeru Daido, Akitsugu Yamamoto, Ryuji Kobayash, Tomohisa Yokoyama, Hiroshi Aok, Eiji Iwado,
Pivotal Role of the Cyclin-dependent Kinase Inhibitor p21 WAF1/CIP1 in Apoptosis and Autophagy Keishi Fujiwara, Shigeru Daido, Akitsugu Yamamoto, Ryuji Kobayash, Tomohisa Yokoyama, Hiroshi Aok, Eiji Iwado,
19 Oxidative Phosphorylation and Photophosphorylation W. H. Freeman and Company
 19 Oxidative Phosphorylation and Photophosphorylation 2013 W. H. Freeman and Company CHAPTER 19 Oxidative Phosphorylation and Photophosphorylation Key topics: Electron transport chain in mitochondria Capture
19 Oxidative Phosphorylation and Photophosphorylation 2013 W. H. Freeman and Company CHAPTER 19 Oxidative Phosphorylation and Photophosphorylation Key topics: Electron transport chain in mitochondria Capture
BIO360 Fall 2013 Quiz 1
 BIO360 Fall 2013 Quiz 1 1. Examine the diagram below. There are two homologous copies of chromosome one and the allele of YFG carried on the light gray chromosome has undergone a loss-of-function mutation.
BIO360 Fall 2013 Quiz 1 1. Examine the diagram below. There are two homologous copies of chromosome one and the allele of YFG carried on the light gray chromosome has undergone a loss-of-function mutation.
mirna Dr. S Hosseini-Asl
 mirna Dr. S Hosseini-Asl 1 2 MicroRNAs (mirnas) are small noncoding RNAs which enhance the cleavage or translational repression of specific mrna with recognition site(s) in the 3 - untranslated region
mirna Dr. S Hosseini-Asl 1 2 MicroRNAs (mirnas) are small noncoding RNAs which enhance the cleavage or translational repression of specific mrna with recognition site(s) in the 3 - untranslated region
TRANSPORT PROCESSES. 1b. moving proteins into membranes and organelles
 1b. moving proteins into membranes and organelles SLIDE 1 A typical mammalian cell contains up to 10,000 different kinds of proteins. The vast majority of these proteins are synthesized by cytosolic ribosomes,
1b. moving proteins into membranes and organelles SLIDE 1 A typical mammalian cell contains up to 10,000 different kinds of proteins. The vast majority of these proteins are synthesized by cytosolic ribosomes,
Regulation of Gene Expression in Eukaryotes
 Ch. 19 Regulation of Gene Expression in Eukaryotes BIOL 222 Differential Gene Expression in Eukaryotes Signal Cells in a multicellular eukaryotic organism genetically identical differential gene expression
Ch. 19 Regulation of Gene Expression in Eukaryotes BIOL 222 Differential Gene Expression in Eukaryotes Signal Cells in a multicellular eukaryotic organism genetically identical differential gene expression
Apoptosis-based Therapies: Mechanisms and Applications
 Apoptosis-based Therapies: Mechanisms and Applications Perspective Bcl-2 family members, potential usage of BH3 domains as drug targets Bcl-2/xL inhibitors - Antisense, inhibitors of protein-protein interactions,
Apoptosis-based Therapies: Mechanisms and Applications Perspective Bcl-2 family members, potential usage of BH3 domains as drug targets Bcl-2/xL inhibitors - Antisense, inhibitors of protein-protein interactions,
Cell Communication. Chapter 11. Biology Eighth Edition Neil Campbell and Jane Reece. PowerPoint Lecture Presentations for
 Chapter 11 Cell Communication PowerPoint Lecture Presentations for Biology Eighth Edition Neil Campbell and Jane Reece Lectures by Chris Romero, updated by Erin Barley with contributions from Joan Sharp
Chapter 11 Cell Communication PowerPoint Lecture Presentations for Biology Eighth Edition Neil Campbell and Jane Reece Lectures by Chris Romero, updated by Erin Barley with contributions from Joan Sharp
Cell Communication. Chapter 11. Biology Eighth Edition Neil Campbell and Jane Reece. PowerPoint Lecture Presentations for
 Chapter 11 Cell Communication PowerPoint Lecture Presentations for Biology Eighth Edition Neil Campbell and Jane Reece Lectures by Chris Romero, updated by Erin Barley with contributions from Joan Sharp
Chapter 11 Cell Communication PowerPoint Lecture Presentations for Biology Eighth Edition Neil Campbell and Jane Reece Lectures by Chris Romero, updated by Erin Barley with contributions from Joan Sharp
Chapter 14 - Electron Transport and Oxidative Phosphorylation
 Chapter 14 - Electron Transport and Oxidative Phosphorylation The cheetah, whose capacity for aerobic metabolism makes it one of the fastest animals Prentice Hall c2002 Chapter 14 1 14.4 Oxidative Phosphorylation
Chapter 14 - Electron Transport and Oxidative Phosphorylation The cheetah, whose capacity for aerobic metabolism makes it one of the fastest animals Prentice Hall c2002 Chapter 14 1 14.4 Oxidative Phosphorylation
Oxidative phosphorylation & Photophosphorylation
 Oxidative phosphorylation & Photophosphorylation Oxidative phosphorylation is the last step in the formation of energy-yielding metabolism in aerobic organisms. All oxidative steps in the degradation of
Oxidative phosphorylation & Photophosphorylation Oxidative phosphorylation is the last step in the formation of energy-yielding metabolism in aerobic organisms. All oxidative steps in the degradation of
Molecular Cell Biology Problem Drill 16: Intracellular Compartment and Protein Sorting
 Molecular Cell Biology Problem Drill 16: Intracellular Compartment and Protein Sorting Question No. 1 of 10 Question 1. Which of the following statements about the nucleus is correct? Question #01 A. The
Molecular Cell Biology Problem Drill 16: Intracellular Compartment and Protein Sorting Question No. 1 of 10 Question 1. Which of the following statements about the nucleus is correct? Question #01 A. The
Regulators of Cell Cycle Progression
 Regulators of Cell Cycle Progression Studies of Cdk s and cyclins in genetically modified mice reveal a high level of plasticity, allowing different cyclins and Cdk s to compensate for the loss of one
Regulators of Cell Cycle Progression Studies of Cdk s and cyclins in genetically modified mice reveal a high level of plasticity, allowing different cyclins and Cdk s to compensate for the loss of one
Mechanisms of Cell Injury
 Causes of Cell Injury 1- oxygen deprivation (anoxia) 2- physical agents 3- chemical agents 4- infections agents 5- immunologic reactions 6- genetic defects 7- nutritional imbalances Mechanisms of Cell
Causes of Cell Injury 1- oxygen deprivation (anoxia) 2- physical agents 3- chemical agents 4- infections agents 5- immunologic reactions 6- genetic defects 7- nutritional imbalances Mechanisms of Cell
Chapter 9. Cellular Signaling
 Chapter 9 Cellular Signaling Cellular Messaging Page 215 Cells can signal to each other and interpret the signals they receive from other cells and the environment Signals are most often chemicals The
Chapter 9 Cellular Signaling Cellular Messaging Page 215 Cells can signal to each other and interpret the signals they receive from other cells and the environment Signals are most often chemicals The
Section B: The Process of Cellular Respiration
 CHAPTER 9 CELLULAR RESPIRATION: HARVESTING CHEMICAL ENERGY Section B: The Process of Cellular Respiration 1. Respiration involves glycolysis, the Krebs cycle, and electron transport: an overview 2. Glycolysis
CHAPTER 9 CELLULAR RESPIRATION: HARVESTING CHEMICAL ENERGY Section B: The Process of Cellular Respiration 1. Respiration involves glycolysis, the Krebs cycle, and electron transport: an overview 2. Glycolysis
The Biochemistry of apoptosis
 The Biochemistry of apoptosis 1 1 The apoptosis is composed of multiple biochemical events 2 2 Biochemical, cellular, and molecular events in Apoptosis 1. Membrane blebbing; phosphatidyl serine exposure
The Biochemistry of apoptosis 1 1 The apoptosis is composed of multiple biochemical events 2 2 Biochemical, cellular, and molecular events in Apoptosis 1. Membrane blebbing; phosphatidyl serine exposure
Principles of Anatomy and Physiology
 Principles of Anatomy and Physiology 14 th Edition CHAPTER 3 The Cellular Level of Organization Introduction The purpose of the chapter is to: 1. Introduce the parts of a cell 2. Discuss the importance
Principles of Anatomy and Physiology 14 th Edition CHAPTER 3 The Cellular Level of Organization Introduction The purpose of the chapter is to: 1. Introduce the parts of a cell 2. Discuss the importance
NFκB What is it and What s the deal with radicals?
 The Virtual Free Radical School NFκB What is it and What s the deal with radicals? Emily Ho, Ph.D Linus Pauling Institute Scientist Department of Nutrition and Food Management Oregon State University 117
The Virtual Free Radical School NFκB What is it and What s the deal with radicals? Emily Ho, Ph.D Linus Pauling Institute Scientist Department of Nutrition and Food Management Oregon State University 117
Cell Communication. Chapter 11. Overview: The Cellular Internet
 Chapter 11 Cell Communication Overview: The Cellular Internet Cell-to-cell communication is essential for multicellular organisms Biologists have discovered some universal mechanisms of cellular regulation
Chapter 11 Cell Communication Overview: The Cellular Internet Cell-to-cell communication is essential for multicellular organisms Biologists have discovered some universal mechanisms of cellular regulation
C-Phycocyanin (C-PC) is a n«sjfc&c- waefc-jduble phycobiliprotein. pigment isolated from Spirulina platensis. This water- soluble protein pigment is
 is a n«sjfc&c- waefc-jduble phycobiliprotein. pigment isolated from Spirulina platensis. This water- soluble protein pigment is") ' ^Summary C-Phycocyanin (C-PC) is a n«sjfc&c- waefc-jduble phycobiliprotein pigment isolated from Spirulina platensis. This water- soluble protein pigment is of greater importance because of its various
' ^Summary C-Phycocyanin (C-PC) is a n«sjfc&c- waefc-jduble phycobiliprotein pigment isolated from Spirulina platensis. This water- soluble protein pigment is of greater importance because of its various
Cell Communication. Biology Eighth Edition Neil Campbell and Jane Reece. PowerPoint Lecture Presentations for
 Chapter 11 Cell Communication PowerPoint Lecture Presentations for Biology Eighth Edition Neil Campbell and Jane Reece Lectures by Chris Romero, updated by Erin Barley with contributions from Joan Sharp
Chapter 11 Cell Communication PowerPoint Lecture Presentations for Biology Eighth Edition Neil Campbell and Jane Reece Lectures by Chris Romero, updated by Erin Barley with contributions from Joan Sharp
BIOLOGY 111. CHAPTER 3: The Cell: The Fundamental Unit of Life
 BIOLOGY 111 CHAPTER 3: The Cell: The Fundamental Unit of Life The Cell: The Fundamental Unit of Life Learning Outcomes 3.1 Explain the similarities and differences between prokaryotic and eukaryotic cells
BIOLOGY 111 CHAPTER 3: The Cell: The Fundamental Unit of Life The Cell: The Fundamental Unit of Life Learning Outcomes 3.1 Explain the similarities and differences between prokaryotic and eukaryotic cells
Lecture 10. G1/S Regulation and Cell Cycle Checkpoints. G1/S regulation and growth control G2 repair checkpoint Spindle assembly or mitotic checkpoint
 Lecture 10 G1/S Regulation and Cell Cycle Checkpoints Outline: G1/S regulation and growth control G2 repair checkpoint Spindle assembly or mitotic checkpoint Paper: The roles of Fzy/Cdc20 and Fzr/Cdh1
Lecture 10 G1/S Regulation and Cell Cycle Checkpoints Outline: G1/S regulation and growth control G2 repair checkpoint Spindle assembly or mitotic checkpoint Paper: The roles of Fzy/Cdc20 and Fzr/Cdh1
Citric Acid Cycle and Oxidative Phosphorylation
 Citric Acid Cycle and Oxidative Phosphorylation Page by: OpenStax Summary The Citric Acid Cycle In eukaryotic cells, the pyruvate molecules produced at the end of glycolysis are transported into mitochondria,
Citric Acid Cycle and Oxidative Phosphorylation Page by: OpenStax Summary The Citric Acid Cycle In eukaryotic cells, the pyruvate molecules produced at the end of glycolysis are transported into mitochondria,
number Done by Corrected by Doctor Maha Shomaf
 number 19 Done by Waseem Abo-Obeida Corrected by Abdullah Zreiqat Doctor Maha Shomaf Carcinogenesis: the molecular basis of cancer. Non-lethal genetic damage lies at the heart of carcinogenesis and leads
number 19 Done by Waseem Abo-Obeida Corrected by Abdullah Zreiqat Doctor Maha Shomaf Carcinogenesis: the molecular basis of cancer. Non-lethal genetic damage lies at the heart of carcinogenesis and leads
Immunology - Lecture 2 Adaptive Immune System 1
 Immunology - Lecture 2 Adaptive Immune System 1 Book chapters: Molecules of the Adaptive Immunity 6 Adaptive Cells and Organs 7 Generation of Immune Diversity Lymphocyte Antigen Receptors - 8 CD markers
Immunology - Lecture 2 Adaptive Immune System 1 Book chapters: Molecules of the Adaptive Immunity 6 Adaptive Cells and Organs 7 Generation of Immune Diversity Lymphocyte Antigen Receptors - 8 CD markers
ACTIVATION OF T LYMPHOCYTES AND CELL MEDIATED IMMUNITY
 ACTIVATION OF T LYMPHOCYTES AND CELL MEDIATED IMMUNITY The recognition of specific antigen by naïve T cell induces its own activation and effector phases. T helper cells recognize peptide antigens through
ACTIVATION OF T LYMPHOCYTES AND CELL MEDIATED IMMUNITY The recognition of specific antigen by naïve T cell induces its own activation and effector phases. T helper cells recognize peptide antigens through
Early cell death (FGF) B No RunX transcription factor produced Yes No differentiation
 B No RunX transcription factor produced Yes No differentiation") Solution Key - Practice Questions Question 1 a) A recent publication has shown that the fat stem cells (FSC) can act as bone stem cells to repair cavities in the skull, when transplanted into immuno-compromised
Solution Key - Practice Questions Question 1 a) A recent publication has shown that the fat stem cells (FSC) can act as bone stem cells to repair cavities in the skull, when transplanted into immuno-compromised
A. Major parts 1. Nucleus 2. Cytoplasm a. Contain organelles (see below) 3. Plasma membrane (To be discussed in Cellular Transport Lecture)
 3. Plasma membrane (To be discussed in Cellular Transport Lecture)") Lecture 5: Cellular Biology I. Cell Theory Concepts: 1. Cells are the functional and structural units of living organisms 2. The activity of an organism is dependent on both the individual and collective
Lecture 5: Cellular Biology I. Cell Theory Concepts: 1. Cells are the functional and structural units of living organisms 2. The activity of an organism is dependent on both the individual and collective
Cell morphology. Cell organelles structure and function. Chapter 1: UNIT 1. Dr. Charushila Rukadikar
 UNIT 1 Cell morphology Cell organelles structure and function Chapter 1: Dr. Charushila Rukadikar Assistant Professor Department Of Physiology ZMCH, Dahod Physiology The science that is concerned with
UNIT 1 Cell morphology Cell organelles structure and function Chapter 1: Dr. Charushila Rukadikar Assistant Professor Department Of Physiology ZMCH, Dahod Physiology The science that is concerned with
Oxidative Phosphorylation
 Oxidative Phosphorylation Energy from Reduced Fuels Is Used to Synthesize ATP in Animals Carbohydrates, lipids, and amino acids are the main reduced fuels for the cell. Electrons from reduced fuels are
Oxidative Phosphorylation Energy from Reduced Fuels Is Used to Synthesize ATP in Animals Carbohydrates, lipids, and amino acids are the main reduced fuels for the cell. Electrons from reduced fuels are
Homework Hanson section MCB Course, Fall 2014
 Homework Hanson section MCB Course, Fall 2014 (1) Antitrypsin, which inhibits certain proteases, is normally secreted into the bloodstream by liver cells. Antitrypsin is absent from the bloodstream of
Homework Hanson section MCB Course, Fall 2014 (1) Antitrypsin, which inhibits certain proteases, is normally secreted into the bloodstream by liver cells. Antitrypsin is absent from the bloodstream of
Supplementary Figures
 Supplementary Figures Figure S1. Validation of kinase regulators of ONC201 sensitivity. Validation and screen results for changes in cell viability associated with the combination of ONC201 treatment (1
Supplementary Figures Figure S1. Validation of kinase regulators of ONC201 sensitivity. Validation and screen results for changes in cell viability associated with the combination of ONC201 treatment (1
Transformation of Normal HMECs (Human Mammary Epithelial Cells) into Metastatic Breast Cancer Cells: Introduction - The Broad Picture:
 into Metastatic Breast Cancer Cells: Introduction - The Broad Picture:") Transformation of Normal HMECs (Human Mammary Epithelial Cells) into Metastatic Breast Cancer Cells: Introduction - The Broad Picture: Spandana Baruah December, 2016 Cancer is defined as: «A disease caused
Transformation of Normal HMECs (Human Mammary Epithelial Cells) into Metastatic Breast Cancer Cells: Introduction - The Broad Picture: Spandana Baruah December, 2016 Cancer is defined as: «A disease caused
Proteasomes. When Death Comes a Knock n. Warren Gallagher Chem412, Spring 2001
 Proteasomes When Death Comes a Knock n Warren Gallagher Chem412, Spring 2001 I. Introduction Introduction The central dogma Genetic information is used to make proteins. DNA RNA Proteins Proteins are the
Proteasomes When Death Comes a Knock n Warren Gallagher Chem412, Spring 2001 I. Introduction Introduction The central dogma Genetic information is used to make proteins. DNA RNA Proteins Proteins are the
KEY CONCEPT QUESTIONS IN SIGNAL TRANSDUCTION
 Signal Transduction - Part 2 Key Concepts - Receptor tyrosine kinases control cell metabolism and proliferation Growth factor signaling through Ras Mutated cell signaling genes in cancer cells are called
Signal Transduction - Part 2 Key Concepts - Receptor tyrosine kinases control cell metabolism and proliferation Growth factor signaling through Ras Mutated cell signaling genes in cancer cells are called
T-cell activation T cells migrate to secondary lymphoid tissues where they interact with antigen, antigen-presenting cells, and other lymphocytes:
 Interactions between innate immunity & adaptive immunity What happens to T cells after they leave the thymus? Naïve T cells exit the thymus and enter the bloodstream. If they remain in the bloodstream,
Interactions between innate immunity & adaptive immunity What happens to T cells after they leave the thymus? Naïve T cells exit the thymus and enter the bloodstream. If they remain in the bloodstream,
T-cell activation T cells migrate to secondary lymphoid tissues where they interact with antigen, antigen-presenting cells, and other lymphocytes:
 Interactions between innate immunity & adaptive immunity What happens to T cells after they leave the thymus? Naïve T cells exit the thymus and enter the bloodstream. If they remain in the bloodstream,
Interactions between innate immunity & adaptive immunity What happens to T cells after they leave the thymus? Naïve T cells exit the thymus and enter the bloodstream. If they remain in the bloodstream,
Lysosomes, Peroxisomes and Centrioles. Hüseyin Çağsın
 Lysosomes, Peroxisomes and Centrioles Hüseyin Çağsın Lysosomes Outline Endosomes Molecule transport to the lysosomes Endocytosis Exocytosis Autophagy Vacuoles Peroxisomes Centrioles Lysosomes Lysosomes
Lysosomes, Peroxisomes and Centrioles Hüseyin Çağsın Lysosomes Outline Endosomes Molecule transport to the lysosomes Endocytosis Exocytosis Autophagy Vacuoles Peroxisomes Centrioles Lysosomes Lysosomes
Electron Transport Chain and Oxidative phosphorylation
 Electron Transport Chain and Oxidative phosphorylation So far we have discussed the catabolism involving oxidation of 6 carbons of glucose to CO 2 via glycolysis and CAC without any oxygen molecule directly
Electron Transport Chain and Oxidative phosphorylation So far we have discussed the catabolism involving oxidation of 6 carbons of glucose to CO 2 via glycolysis and CAC without any oxygen molecule directly
4/12/17. Cells. Cell Structure. Ch. 2 Cell Structure and Func.on. Range of Cell Sizes BIOL 100
 Ch. 2 Cell Structure and Func.on BIOL 100 Cells Fundamental units of life Cell theory All living things are composed of one or more cells. The cell is the most basic unit of life. All cells come from pre-existing
Ch. 2 Cell Structure and Func.on BIOL 100 Cells Fundamental units of life Cell theory All living things are composed of one or more cells. The cell is the most basic unit of life. All cells come from pre-existing
Cell Adaptation, Cell Injury and Cell Death
 Cell Adaptation, Cell Injury and Cell Death Pathology:- is the study of structural and functional abnormalities that are expressed as diseases of organs and systems. Modern pathology, proposed that injury
Cell Adaptation, Cell Injury and Cell Death Pathology:- is the study of structural and functional abnormalities that are expressed as diseases of organs and systems. Modern pathology, proposed that injury
1- Which of the following statements is TRUE in regards to eukaryotic and prokaryotic cells?
 Name: NetID: Exam 3 - Version 1 October 23, 2017 Dr. A. Pimentel Each question has a value of 4 points and there are a total of 160 points in the exam. However, the maximum score of this exam will be capped
Name: NetID: Exam 3 - Version 1 October 23, 2017 Dr. A. Pimentel Each question has a value of 4 points and there are a total of 160 points in the exam. However, the maximum score of this exam will be capped
Introduction: 年 Fas signal-mediated apoptosis. PI3K/Akt
 Fas-ligand (CD95-L; Fas-L) Fas (CD95) Fas (apoptosis) 年 了 不 度 Fas Fas-L 力 不 Fas/Fas-L T IL-10Fas/Fas-L 不 年 Fas signal-mediated apoptosis 度降 不 不 力 U-118, HeLa, A549, Huh-7 MCF-7, HepG2. PI3K/Akt FasPI3K/Akt
Fas-ligand (CD95-L; Fas-L) Fas (CD95) Fas (apoptosis) 年 了 不 度 Fas Fas-L 力 不 Fas/Fas-L T IL-10Fas/Fas-L 不 年 Fas signal-mediated apoptosis 度降 不 不 力 U-118, HeLa, A549, Huh-7 MCF-7, HepG2. PI3K/Akt FasPI3K/Akt
Cell Signaling part 2
 15 Cell Signaling part 2 Functions of Cell Surface Receptors Other cell surface receptors are directly linked to intracellular enzymes. The largest family of these is the receptor protein tyrosine kinases,
15 Cell Signaling part 2 Functions of Cell Surface Receptors Other cell surface receptors are directly linked to intracellular enzymes. The largest family of these is the receptor protein tyrosine kinases,
COURSE: Medical Microbiology, MBIM 650/720 - Fall TOPIC: Antigen Processing, MHC Restriction, & Role of Thymus Lecture 12
 COURSE: Medical Microbiology, MBIM 650/720 - Fall 2008 TOPIC: Antigen Processing, MHC Restriction, & Role of Thymus Lecture 12 FACULTY: Dr. Mayer Office: Bldg. #1, Rm B32 Phone: 733-3281 Email: MAYER@MED.SC.EDU
COURSE: Medical Microbiology, MBIM 650/720 - Fall 2008 TOPIC: Antigen Processing, MHC Restriction, & Role of Thymus Lecture 12 FACULTY: Dr. Mayer Office: Bldg. #1, Rm B32 Phone: 733-3281 Email: MAYER@MED.SC.EDU
Zool 3200: Cell Biology Exam 4 Part II 2/3/15
 Name:Key Trask Zool 3200: Cell Biology Exam 4 Part II 2/3/15 Answer each of the following questions in the space provided, explaining your answers when asked to do so; circle the correct answer or answers
Name:Key Trask Zool 3200: Cell Biology Exam 4 Part II 2/3/15 Answer each of the following questions in the space provided, explaining your answers when asked to do so; circle the correct answer or answers
Prepared by Cyrus H. Nozad, MD, University of Tennessee and John Seyerle, MD, Ohio State University
 Allergy and Immunology Review Corner: Chapter 21 of Middleton s Allergy Principles and Practice, Seventh Edition, edited by N. Franklin Adkinson, et al. Chapter 21: Antigen-Presenting Dendritic Cells (Pages
Allergy and Immunology Review Corner: Chapter 21 of Middleton s Allergy Principles and Practice, Seventh Edition, edited by N. Franklin Adkinson, et al. Chapter 21: Antigen-Presenting Dendritic Cells (Pages
The functional investigation of the interaction between TATA-associated factor 3 (TAF3) and p53 protein
 and p53 protein") THESIS BOOK The functional investigation of the interaction between TATA-associated factor 3 (TAF3) and p53 protein Orsolya Buzás-Bereczki Supervisors: Dr. Éva Bálint Dr. Imre Miklós Boros University of
THESIS BOOK The functional investigation of the interaction between TATA-associated factor 3 (TAF3) and p53 protein Orsolya Buzás-Bereczki Supervisors: Dr. Éva Bálint Dr. Imre Miklós Boros University of
CELL BIOLOGY - CLUTCH CH AEROBIC RESPIRATION.
 !! www.clutchprep.com CONCEPT: OVERVIEW OF AEROBIC RESPIRATION Cellular respiration is a series of reactions involving electron transfers to breakdown molecules for (ATP) 1. Glycolytic pathway: Glycolysis
!! www.clutchprep.com CONCEPT: OVERVIEW OF AEROBIC RESPIRATION Cellular respiration is a series of reactions involving electron transfers to breakdown molecules for (ATP) 1. Glycolytic pathway: Glycolysis
Campbell Biology in Focus (Urry) Chapter 9 The Cell Cycle. 9.1 Multiple-Choice Questions
 Chapter 9 The Cell Cycle. 9.1 Multiple-Choice Questions") Campbell Biology in Focus (Urry) Chapter 9 The Cell Cycle 9.1 Multiple-Choice Questions 1) Starting with a fertilized egg (zygote), a series of five cell divisions would produce an early embryo with how
Campbell Biology in Focus (Urry) Chapter 9 The Cell Cycle 9.1 Multiple-Choice Questions 1) Starting with a fertilized egg (zygote), a series of five cell divisions would produce an early embryo with how
Metabolism is regulated by the rate of ATP production
 BCHM2972 Human Biochemistry Introduction to Metabolism Metabolism is regulated by the rate of ATP production Anabolism/Catabolism Anabolism Reactions that build macromolecules Use energy from catabolism
BCHM2972 Human Biochemistry Introduction to Metabolism Metabolism is regulated by the rate of ATP production Anabolism/Catabolism Anabolism Reactions that build macromolecules Use energy from catabolism
Introduction. Biochemistry: It is the chemistry of living things (matters).
.") Introduction Biochemistry: It is the chemistry of living things (matters). Biochemistry provides fundamental understanding of the molecular basis for the function and malfunction of living things. Biochemistry
Introduction Biochemistry: It is the chemistry of living things (matters). Biochemistry provides fundamental understanding of the molecular basis for the function and malfunction of living things. Biochemistry
Summary of Endomembrane-system
 Summary of Endomembrane-system 1. Endomembrane System: The structural and functional relationship organelles including ER,Golgi complex, lysosome, endosomes, secretory vesicles. 2. Membrane-bound structures
Summary of Endomembrane-system 1. Endomembrane System: The structural and functional relationship organelles including ER,Golgi complex, lysosome, endosomes, secretory vesicles. 2. Membrane-bound structures
Chapter 2 The Bcl-2 Family Proteins
 Chapter 2 The Bcl-2 Family Proteins Wen-Xing Ding and Xiao-Ming Yin Abstract The Bcl-2 family proteins are a group of evolutionarily conserved molecules that regulate apoptosis mainly at the site of mitochondria.
Chapter 2 The Bcl-2 Family Proteins Wen-Xing Ding and Xiao-Ming Yin Abstract The Bcl-2 family proteins are a group of evolutionarily conserved molecules that regulate apoptosis mainly at the site of mitochondria.