Congenital Haemoglobinopathies
|
|
|
- Basil Buck Greer
- 5 years ago
- Views:
Transcription


1 Congenital Haemoglobinopathies L. DEDEKEN, MD H O P I T A L U N I V E R S I T A I R E D E S E N F A N T S R E I N E F A B I O L A U N I V E R S I T E L I B R E DE B R U X E L L E S Red Blood Cell Disorders BHS training course 2013

2 Introduction ~ 5% of the world s population carries trait genes for haemoglobin disorders Carriers ~ 25% in some regions > new births with severe haemoglobin disorders each year Lopez et al. Gene 2010
3 Sickle cell disease


4 Epidemiology Rees et al. Lancet 2010 In Brussels 1 st genetic disorder 1/1600 birth Harteveld et al. Orphanet J Rare disease 2010

 Hb SC Hb Sβ-thalassemia Hb SD Punjab Hb")
5 Genetics Autosomal recessive disorder Mutation on β-globin gene sixth aminoacid becomes valine instead of glutamic acid (HbS) Sickle cell trait Hb AS Sickle cell syndrome Hb SS (sickle cell anemia) Hb SC Hb Sβ-thalassemia Hb SD Punjab Hb SO Arab

6 Physiopathology Rees et al. Lancet 2010

7 Subphenotypes of SCD Kato et al. Blood Rev 2007


8 Diagnostic tools Blood cell count Peripheral blood smear Hemoglobin electrophoresis Genetic
9 Survival Quinn. Blood 2010 Platt. NEJM 1994
10 Clinical features

11 Anemia Chronic hemolytic anemia Hemoglobin level AA : g/dl SS : 6-10 g/dl SC : g/dl Acute event Erythroblastopenic anemia < B19 Parvovirus Infection Splenic sequestration G6PD deficiency, folate deficiency, malaria, allo-immunisation,
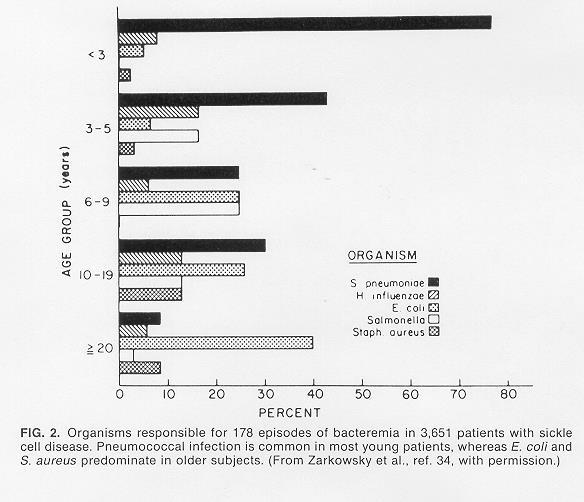
12 Infections


13 Vaso-occlusive crisis Painful event Increased occurence whith Hypoxia, dehydratation, acidosis, cold exposure, Dactylitis Osteo-articular manifestations Pain Swelling Functional impact Differential diagnosis : Osteomyelitis or arthritis

 Differential diagnosis Pulmonary embolism")
14 Acute chest syndrome Fever OR tachypnea OR decreased O 2 saturation with Abnormal pulmonary exam New infiltrate on RX Etiology Infections (30-40%) Fat embolism (7%) No identify cause (40-50%) Differential diagnosis Pulmonary embolism Lancet 2010
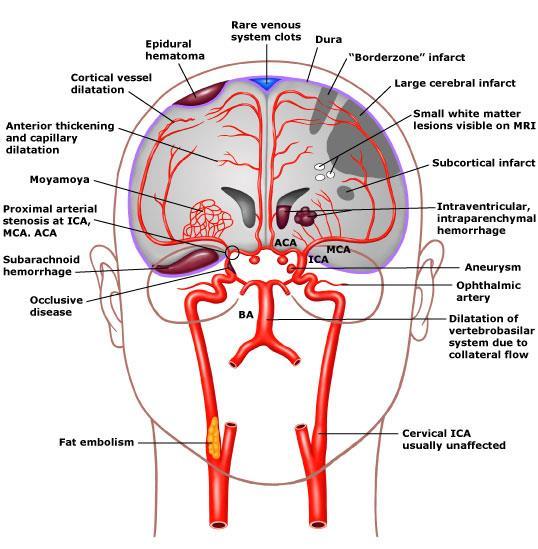
15 Cerebral vasculopathy

16 Lancet Neurol 2006 Blood 1998 Stroke Ischemic or hemorragic Seizures Cognitive impairement Blood 2009


17 How to assess the risk? Blood 2009



18 Chronic complications Retinopathy Leg ulcer Osteonecrosis Pulmonary hypertension
19 Management Education and information Good lifestyle To avoid risk factors (hypoxia, dehydratation, cold exposure, ) To recognize a acute event Folic acid Infection prevention Prophlyactic antibiotics Vaccination Streptococcus pneumonia, Haemophilus influenzae, Flu Emipiric antibiotherapy
20 Symptomatic management Vaso-occlusive crisis Adapted pain management Good hydratation Acute chest syndrome Exchange transfusion Antibiotics Stroke or unexplained neurological symptom Exchange transfusion
 Hydroxyurea The only efficient drug Well tolerated Up to maximal tolerated dosis if needed Ware. Blood 2010")
21 Intensification Chronic (exchange) transfusion program Primary or secondary prevention of stroke Recurrent ACS or VOC despite HU Organ failure (hepatic, renal, ) Hydroxyurea The only efficient drug Well tolerated Up to maximal tolerated dosis if needed Ware. Blood 2010
22 Voskaridou. Blood 2010 Lopes de Castro Lobo. BJH 2013
23 Hematopoietic stem cell transplantation The only curative treatment Severe disease and intra-familial HLA donor Survival Overall survival 92-96% Event free survival 82-86% Death 3-9% In development Matched unrelated donor Halpoidentical transplantation High risk of rejection and GVHD Gene therapy
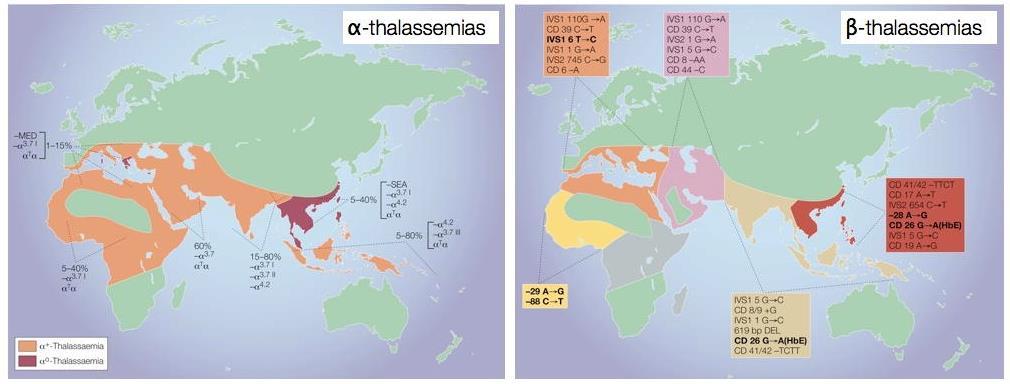
24 Thalassemia

25 Lancet 2012
26 β-thalassemia

27 Physiopathology
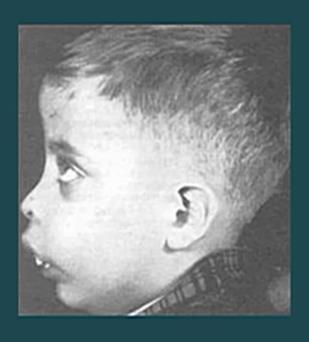
28 Clinical outcome Severe anemia Without appropriate chronic transfusion Asthenia Dilated cardiomyopathy Hepatosplenomegaly Jaundice Bone deformities Specific facial deformation (frontal bossing, maxillary overgrowth,...) Osteoporosis Growth and development delayed Hypermetabolic state Recurrent infections


29 Treatment Chronic transfusion Monthly To keep Hb 9-11 g/dl Hb F inducer (HU) Iron overload Evaluation Ferritine Liver biopsy MRI T2* Clinical outcome

30 Iron chelators Neufeld. Blood 2006
31 Survival Adapted from Ladis et al. Ann N Y Acad Sci. 2005

32 HSCT & β-thalassemia Sabloff. Blood 2011

33 α-thalassemia Harteveld & Higgs. Orphanet J Rare Diseases 2010

34 Pathogenesis
35 Clinical features Absence of 1 or 2 α chain(s) -α/αα ; --/αα ; -α/-α Absence of 3 α chains (HbH disease) --/-α Absence of 4 α chains (Hb Bart s Hydrops Fetalis) --/-- Common Asymptomatic or moderate microcytic anemia Does not requires therapy Hemolytic feature (gallbladder lithiasis, HSM) Microcytic anemia (Hb 7-10g/dl) Folic acid, infrequent transfusion Hydrops fetalis Non viable
36 CONCLUSION
37 References Stuart MJ, Nagel RL. Sickle cell disease. Lancet. 2004; 364: Rees DC, Williams TN, Gladwin MT. Sickle cell disease. Lancet. 2010; 376: The management of sickle cell disease. Higgs DR, Engel JD, Stamatoyannopoulos G. Thalassaemia. Lancet. 2012; 379: Harteveld CL, Higgs DR. Alpha-Thalassaemia. Orphanet J Rare Dis May 28;5:13
SICKLE CELL DISEASE. Dr. MUBARAK ABDELRAHMAN MD PEDIATRICS AND CHILD HEALTH. Assistant Professor FACULTY OF MEDICINE -JAZAN
 SICKLE CELL DISEASE Dr. MUBARAK ABDELRAHMAN MD PEDIATRICS AND CHILD HEALTH Assistant Professor FACULTY OF MEDICINE -JAZAN Objective: The student should be able: To identify the presentation, diagnosis,
SICKLE CELL DISEASE Dr. MUBARAK ABDELRAHMAN MD PEDIATRICS AND CHILD HEALTH Assistant Professor FACULTY OF MEDICINE -JAZAN Objective: The student should be able: To identify the presentation, diagnosis,
Anemia s. Troy Lund MSMS PhD MD
 Anemia s Troy Lund MSMS PhD MD lundx072@umn.edu Hemoglobinopathy/Anemia IOM take home points. 1. How do we identify the condtion? Smear, CBC Solubility Test (SCD) 2. How does it present clincally? 3. How
Anemia s Troy Lund MSMS PhD MD lundx072@umn.edu Hemoglobinopathy/Anemia IOM take home points. 1. How do we identify the condtion? Smear, CBC Solubility Test (SCD) 2. How does it present clincally? 3. How
Sickle cell disease. Fareed Omar 10 March 2018
 Sickle cell disease Fareed Omar 10 March 2018 Physiology Haemoglobin structure HbA2: 2α and 2δ chains (2-3%) HbF: 2α and 2γ chains (
Sickle cell disease Fareed Omar 10 March 2018 Physiology Haemoglobin structure HbA2: 2α and 2δ chains (2-3%) HbF: 2α and 2γ chains (
Sickle Cell Disease. Edward Malters, MD
 Sickle Cell Disease Edward Malters, MD Introduction Vaso-occlusive phenomena and hemolysis are the clinical hallmarks of Sickle Cell Disease (SCD) Inherited disorder due to homozygosity for the abnormal
Sickle Cell Disease Edward Malters, MD Introduction Vaso-occlusive phenomena and hemolysis are the clinical hallmarks of Sickle Cell Disease (SCD) Inherited disorder due to homozygosity for the abnormal
How to Write a Life Care Plan for a Child with Hemoglobinopathy
 How to Write a Life Care Plan for a Child with Hemoglobinopathy Tamar Fleischer, BSN, MSN, CNLCP & Mona Yudkoff, RN, MPH, CRRN, CNLCP BalaCare Solutions March 2018 St. Peterburg, Florida What is Hemoglobinopathy?
How to Write a Life Care Plan for a Child with Hemoglobinopathy Tamar Fleischer, BSN, MSN, CNLCP & Mona Yudkoff, RN, MPH, CRRN, CNLCP BalaCare Solutions March 2018 St. Peterburg, Florida What is Hemoglobinopathy?
Rationale for RBC Transfusion in SCD
 Rationale for RBC Transfusion in SCD Dilution of HgbS-containing RBCs via the addition of HgbA-containing cells from the blood of normal donors Suppression of erythropoietin release caused by the rise
Rationale for RBC Transfusion in SCD Dilution of HgbS-containing RBCs via the addition of HgbA-containing cells from the blood of normal donors Suppression of erythropoietin release caused by the rise
Management of Sickle Cell Disease
 Management of Sickle Cell Disease A.Ferster HUDERF-ULB 29 th BHS meeting Friday 31 January 2014 Introduction > 200.000 affected births each year 2000 births in US 15 births/y in Belgium 80.000 Pts in US
Management of Sickle Cell Disease A.Ferster HUDERF-ULB 29 th BHS meeting Friday 31 January 2014 Introduction > 200.000 affected births each year 2000 births in US 15 births/y in Belgium 80.000 Pts in US
In adults, the predominant Hb (HbA) molecule has four chains: two α and two β chains. In thalassemias, the synthesis of either the α or the β chains
 molecule has four chains: two α and two β chains. In thalassemias, the synthesis of either the α or the β chains") Thalassaemias Thalassemia Thalassemia is an inherited autosomal recessive blood disease. Associated with absence or reduction in a or b globin chains. Reduced synthesis of one of the globin chains can
Thalassaemias Thalassemia Thalassemia is an inherited autosomal recessive blood disease. Associated with absence or reduction in a or b globin chains. Reduced synthesis of one of the globin chains can
Haemoglobinophaties EBMT 2011 Data Manager session
 Haemoglobinophaties EBMT 2011 Data Manager session Presentation plan Biological characteristics Clinical characteristics Transplant resuts What is different From transplant in malignancies Between Thalassemia
Haemoglobinophaties EBMT 2011 Data Manager session Presentation plan Biological characteristics Clinical characteristics Transplant resuts What is different From transplant in malignancies Between Thalassemia
Sickle Cell Disease and impact on the society
 Sickle Cell Disease and impact on the society Professor Z.A.Jeremiah Ph.D, FRCPath (London) Professor of Haematology and Blood Transfusion Science Niger Delta University, Wilberforce Island Outline What
Sickle Cell Disease and impact on the society Professor Z.A.Jeremiah Ph.D, FRCPath (London) Professor of Haematology and Blood Transfusion Science Niger Delta University, Wilberforce Island Outline What
Hemolytic anemias (2 of 2)
") Hemolytic anemias (2 of 2) Sickle Cell Anemia The most common familial hemolytic anemia in the world Sickle cell anemia is the prototypical (and most prevalent) hemoglobinopathy Mutation in the β-globin
Hemolytic anemias (2 of 2) Sickle Cell Anemia The most common familial hemolytic anemia in the world Sickle cell anemia is the prototypical (and most prevalent) hemoglobinopathy Mutation in the β-globin
Thalassemia Maria Luz Uy del Rosario, M.D.
 Thalassemia Maria Luz Uy del Rosario, M.D. Philippine Society of Hematology and Blood Transfusion Philippine Society of Pediatric Oncology What is Thalassemia Hereditary Hemoglobin disorder Hemolytic anemia
Thalassemia Maria Luz Uy del Rosario, M.D. Philippine Society of Hematology and Blood Transfusion Philippine Society of Pediatric Oncology What is Thalassemia Hereditary Hemoglobin disorder Hemolytic anemia
Dependance on chronic transfusion
 Dependance on chronic transfusion Pr Saliou Diop Hematology Blood transfusion Dakar- Sénégal diop@cnts-dakar.sn Introduction Chronic transfusion: Regular use of blood transfusion in patients with chronic
Dependance on chronic transfusion Pr Saliou Diop Hematology Blood transfusion Dakar- Sénégal diop@cnts-dakar.sn Introduction Chronic transfusion: Regular use of blood transfusion in patients with chronic
An overview of Thalassaemias and Complications
 An overview of Thalassaemias and Complications Haemoglobin Haemoglobin is the most abundant protein in blood, and exists as three main types in normal adults: HbA ( ) - 97% HbA 2 ( ) - 2.5% HbF ( ) - 0.5%
An overview of Thalassaemias and Complications Haemoglobin Haemoglobin is the most abundant protein in blood, and exists as three main types in normal adults: HbA ( ) - 97% HbA 2 ( ) - 2.5% HbF ( ) - 0.5%
Dr Banu Kaya Consultant Haematologist Barts Health NHS Trust Royal London Hospital, London, UK SICKLE CELL AND THALASSAEMIA OVERVIEW
 Dr Banu Kaya Consultant Haematologist Barts Health NHS Trust Royal London Hospital, London, UK SICKLE CELL AND THALASSAEMIA OVERVIEW Objectives Gain awareness of haemoglobinopathy inheritance, pathophysiology
Dr Banu Kaya Consultant Haematologist Barts Health NHS Trust Royal London Hospital, London, UK SICKLE CELL AND THALASSAEMIA OVERVIEW Objectives Gain awareness of haemoglobinopathy inheritance, pathophysiology
Medical and Surgical Complications of Sickle Cell Anemia
 Medical and Surgical Complications of Sickle Cell Anemia Ahmed Al-Salem Medical and Surgical Complications of Sickle Cell Anemia Ahmed Al-Salem Department of Surgery Dar A lalafia Medical Company Qatif
Medical and Surgical Complications of Sickle Cell Anemia Ahmed Al-Salem Medical and Surgical Complications of Sickle Cell Anemia Ahmed Al-Salem Department of Surgery Dar A lalafia Medical Company Qatif
SICKLE CELL DISEASE TO TREAT OR
 SICKLE CELL DISEASE TO TREAT OR NOT TO TREAT COHEM Barcelona September 8, 2012 Sujit Sheth, M.D. Pediatric Hematology Oncology Disclosures None Outline Morbidity and mortality Definitive therapies Risk
SICKLE CELL DISEASE TO TREAT OR NOT TO TREAT COHEM Barcelona September 8, 2012 Sujit Sheth, M.D. Pediatric Hematology Oncology Disclosures None Outline Morbidity and mortality Definitive therapies Risk
4 Jumana Jihad Dr. Ahmad Mansour Dr. Ahmad Mansour
 4 Jumana Jihad Dr. Ahmad Mansour Dr. Ahmad Mansour Anemia Decreased blood production Increased blood loss Hemolytic Hemorrhage Extravascular Intravascular Hemolytic (Further classification( Extrinsic Intrinsic
4 Jumana Jihad Dr. Ahmad Mansour Dr. Ahmad Mansour Anemia Decreased blood production Increased blood loss Hemolytic Hemorrhage Extravascular Intravascular Hemolytic (Further classification( Extrinsic Intrinsic
Introduction reduction in output alter the amino acid sequence combination
 Sickle cell anemia. Introduction Mutations in the globin genes can cause a quantitative reduction in output from that gene or alter the amino acid sequence of the protein produced or a combination of the
Sickle cell anemia. Introduction Mutations in the globin genes can cause a quantitative reduction in output from that gene or alter the amino acid sequence of the protein produced or a combination of the
Health Maintenance and Education for Children and Adults
 Health Maintenance and Education for Children and Adults Richard Ward, MSc, MRCP, FRCPath Director, Red Blood Cell Disorders Program, UHN Assistant Professor, Hematology, University of Toronto Chair, Canadian
Health Maintenance and Education for Children and Adults Richard Ward, MSc, MRCP, FRCPath Director, Red Blood Cell Disorders Program, UHN Assistant Professor, Hematology, University of Toronto Chair, Canadian
DONE BY : RaSHA RAKAN & Bushra Saleem
 DONE BY : RaSHA RAKAN & Bushra Saleem Hemolytic anemias (2 of 2) Sickle Cell Anemia The most common familial hemolytic anemia in the world Sickle cell anemia is the prototypical (and most prevalent) hemoglobinopathy
DONE BY : RaSHA RAKAN & Bushra Saleem Hemolytic anemias (2 of 2) Sickle Cell Anemia The most common familial hemolytic anemia in the world Sickle cell anemia is the prototypical (and most prevalent) hemoglobinopathy
Hydroxyurea and Transfusion Therapy for the Treatment of Sickle Cell Disease
 Hydroxyurea and Transfusion Therapy for the Treatment of Sickle Cell Disease A Pocket Guide for the Clinician Susan E. Creary, MD, MSc 1 John J. Strouse, MD, PhD 2 1 The Ohio State University School of
Hydroxyurea and Transfusion Therapy for the Treatment of Sickle Cell Disease A Pocket Guide for the Clinician Susan E. Creary, MD, MSc 1 John J. Strouse, MD, PhD 2 1 The Ohio State University School of
Heme Questions and Derivatives for the USMLE Step One Exam. Winter Storm Skylar Edition
 Heme Questions and Derivatives for the USMLE Step One Exam Winter Storm Skylar Edition Howard J. Sachs, MD Howard@12DaysinMarch.com www.12daysinmarch.com Patient presents for routine preoperative evaluation
Heme Questions and Derivatives for the USMLE Step One Exam Winter Storm Skylar Edition Howard J. Sachs, MD Howard@12DaysinMarch.com www.12daysinmarch.com Patient presents for routine preoperative evaluation
General Characterisctics
 Anemia General Characterisctics Definition: anemia is a decrease in red blood cells. Happens due to underproduction, increased destruction or loss of red cells. Diagnosis of anemia: Hgb < 135 (men) Hgb
Anemia General Characterisctics Definition: anemia is a decrease in red blood cells. Happens due to underproduction, increased destruction or loss of red cells. Diagnosis of anemia: Hgb < 135 (men) Hgb
Fetal Anemia 02/13/13. Anjulika Chawla, M.D. Assistant Professor Division of Pediatric Hematology/Oncology
 Fetal Anemia 02/13/13 Anjulika Chawla, M.D. Assistant Professor Division of Pediatric Hematology/Oncology Objectives Definition of anemia Diagnosis of fetal anemia Normal developmental hematopoiesis Etiology
Fetal Anemia 02/13/13 Anjulika Chawla, M.D. Assistant Professor Division of Pediatric Hematology/Oncology Objectives Definition of anemia Diagnosis of fetal anemia Normal developmental hematopoiesis Etiology
Anaemia in Pregnancy
 Anaemia in Pregnancy Definition :anaemia is a pathological condition in which the oxygen-carrying capacity of red blood cells is insufficient to meet the body needs. The WHO : haemoglobin concentration
Anaemia in Pregnancy Definition :anaemia is a pathological condition in which the oxygen-carrying capacity of red blood cells is insufficient to meet the body needs. The WHO : haemoglobin concentration
CURRENT RESEARCH STUDIES
 CURRENT RESEARCH STUDIES SCAGO SICKLE CELL RESEARCH DAY MAY 12, 2018 REBECCA LEROUX RN, BSCN, CCRP RED BLOOD CELL DISORDERS PROGRAM, UNIVERSITY HEALTH NETWORK MANUELA MERELLES-PULCINI RN, BSCN, MSN, CCRP
CURRENT RESEARCH STUDIES SCAGO SICKLE CELL RESEARCH DAY MAY 12, 2018 REBECCA LEROUX RN, BSCN, CCRP RED BLOOD CELL DISORDERS PROGRAM, UNIVERSITY HEALTH NETWORK MANUELA MERELLES-PULCINI RN, BSCN, MSN, CCRP
Screening for haemoglobinopathies in pregnancy
 Policy Statement All Southern Health patients will receive clinical care that reflects best practice and is based on the best available evidence. Index of chapters within background 1. Prevalence of haemoglobinopathies
Policy Statement All Southern Health patients will receive clinical care that reflects best practice and is based on the best available evidence. Index of chapters within background 1. Prevalence of haemoglobinopathies
Third Visit Posttest
 Test code 03C Patient s name: Third Visit Posttest Patient s birth date: Your name and relationship to patient: Today s date: Please mark only one answer for each of the following questions: 1. Which one
Test code 03C Patient s name: Third Visit Posttest Patient s birth date: Your name and relationship to patient: Today s date: Please mark only one answer for each of the following questions: 1. Which one
Epidemiology, Care and Prevention of Hemoglobinopathies
 Epidemiology, Care and Prevention of Hemoglobinopathies Nasir Al-Allawi MBChB, PhD. Professor of Hematology College of Medicine University of Dohuk, IRAQ From Research to Practice Training Course in Sexual
Epidemiology, Care and Prevention of Hemoglobinopathies Nasir Al-Allawi MBChB, PhD. Professor of Hematology College of Medicine University of Dohuk, IRAQ From Research to Practice Training Course in Sexual
HEMOLYTIC ANEMIA DUE TO ABNORMAL HEMOGLOBIN SYNTHESIS
 Hemolytic Anemia Due to Abnormal Hemoglobin Synthesis MODULE 19 HEMOLYTIC ANEMIA DUE TO ABNORMAL HEMOGLOBIN SYNTHESIS 19.1 INTRODUCTION There are two main mechanisms by which anaemia is produced (a) Thalassemia:
Hemolytic Anemia Due to Abnormal Hemoglobin Synthesis MODULE 19 HEMOLYTIC ANEMIA DUE TO ABNORMAL HEMOGLOBIN SYNTHESIS 19.1 INTRODUCTION There are two main mechanisms by which anaemia is produced (a) Thalassemia:
Red cell disorder. Dr. Ahmed Hasan
 Red cell disorder Dr. Ahmed Hasan Things to be learned in this lecture Definition and clinical feature of anemia. Classification of anemia. Know some details of microcytic anemia Question of the lecture:
Red cell disorder Dr. Ahmed Hasan Things to be learned in this lecture Definition and clinical feature of anemia. Classification of anemia. Know some details of microcytic anemia Question of the lecture:
Putting some hematology into Pediatric Hematology/Oncology: a review of Hemophilia and Sickle Cell Disease in the Pediatric Patient
 Putting some hematology into Pediatric Hematology/Oncology: a review of Hemophilia and Sickle Cell Disease in the Pediatric Patient Kristina Haley, DO March 10, 2012 Jovita Reyes Memorial Pediatric Hematology/Oncology
Putting some hematology into Pediatric Hematology/Oncology: a review of Hemophilia and Sickle Cell Disease in the Pediatric Patient Kristina Haley, DO March 10, 2012 Jovita Reyes Memorial Pediatric Hematology/Oncology
Quiz. What percentage of the world s population is a carrier of a hemoglobinopathy? Hemoglobinopathies in Pregnancy 1-2% 5-7% 8-12% 10-15%
 Hemoglobinopathies in Pregnancy Emily Parkhurst, MS, LCGC Kaiser West Los Angeles November 2017 Genetics Department Quiz What percentage of the world s population is a carrier of a hemoglobinopathy? 1-2%
Hemoglobinopathies in Pregnancy Emily Parkhurst, MS, LCGC Kaiser West Los Angeles November 2017 Genetics Department Quiz What percentage of the world s population is a carrier of a hemoglobinopathy? 1-2%
Transfusion support in Thalassaemia. Dr.A.keerti 1 st year PG DEPT. OF TRANSFUSION MEDICINE
 Transfusion support in Thalassaemia Dr.A.keerti 1 st year PG DEPT. OF TRANSFUSION MEDICINE Structure of hemoglobin Types of hemoglobins Hemoglobin-Development Switching Thalassaemia- introduction Classification
Transfusion support in Thalassaemia Dr.A.keerti 1 st year PG DEPT. OF TRANSFUSION MEDICINE Structure of hemoglobin Types of hemoglobins Hemoglobin-Development Switching Thalassaemia- introduction Classification
Compassionate-use Voxelotor (GBT440) for up to 2 Years in Patients With Severe Sickle Cell Disease and Life-Threatening Comorbidities
 for up to 2 Years in Patients With Severe Sickle Cell Disease and Life-Threatening Comorbidities") Compassionate-use Voxelotor (GBT440) for up to 2 Years in Patients With Severe Sickle Cell Disease and Life-Threatening Comorbidities Gershwin Blyden, MD 1, Kenneth Bridges, MD 2, Lanetta Bronté, MD 1
Compassionate-use Voxelotor (GBT440) for up to 2 Years in Patients With Severe Sickle Cell Disease and Life-Threatening Comorbidities Gershwin Blyden, MD 1, Kenneth Bridges, MD 2, Lanetta Bronté, MD 1
1 Kattamis et al. Growth of Children with Thalassemia: Effect of Different Transfusion Regimens. Archives of
 Objectives Sickle Cell Anemia and Thalassemia: Transplantation Provide overview of hemoglobinopathies: Sickle cell disease and Thalassemia Discuss approaches to therapy Review recent registry collaboration
Objectives Sickle Cell Anemia and Thalassemia: Transplantation Provide overview of hemoglobinopathies: Sickle cell disease and Thalassemia Discuss approaches to therapy Review recent registry collaboration
Genetics of Thalassemia
 Genetics of Thalassemia Submitted by : Raya Samir Al- Hayaly Sura Zuhair Salih Saad Ghassan Al- Dulaimy Saad Farouq Kassir Sama Naal Salouha Zahraa Jasim Al- Aarajy Supervised by : Dr. Kawkab Adris Mahmod
Genetics of Thalassemia Submitted by : Raya Samir Al- Hayaly Sura Zuhair Salih Saad Ghassan Al- Dulaimy Saad Farouq Kassir Sama Naal Salouha Zahraa Jasim Al- Aarajy Supervised by : Dr. Kawkab Adris Mahmod
COHEM Barcellona 2012 Hemoglobinopathies debate
 COHEM Barcellona 2012 Hemoglobinopathies debate September 8, 2012: h. 10:30-12:00 Hall: A Is it justified to perform BMT in hemoglobinopathies using unrelated and/or partially mismatched donors? HSCT indication
COHEM Barcellona 2012 Hemoglobinopathies debate September 8, 2012: h. 10:30-12:00 Hall: A Is it justified to perform BMT in hemoglobinopathies using unrelated and/or partially mismatched donors? HSCT indication
World-Wide Distribution of Hemoglobin S. Geographic distribution of hemoglobin S in the world
 Sickle Cell Disease Gerald M. Woods, MD Professor of Pediatrics Division Director, Hematology/Oncology/BMT Director of Sickle Cell Program Children s Mercy Hospitals and Clinics Disclaimer Member of DSMB
Sickle Cell Disease Gerald M. Woods, MD Professor of Pediatrics Division Director, Hematology/Oncology/BMT Director of Sickle Cell Program Children s Mercy Hospitals and Clinics Disclaimer Member of DSMB
DIC. Disseminated intravascular coagulation, is a life threatening pathological process in which clotting factors are abnormally activated.
 Miss. kamlah 1 DIC Disseminated intravascular coagulation, is a life threatening pathological process in which clotting factors are abnormally activated. Resulting in wide spread of clot formation in the
Miss. kamlah 1 DIC Disseminated intravascular coagulation, is a life threatening pathological process in which clotting factors are abnormally activated. Resulting in wide spread of clot formation in the
The Thalassemias in Clinical Practice. Ashutosh Lal, MD Director Comprehensive Thalassemia Program UCSF Benioff Children s Hospital Oakland
 The Thalassemias in Clinical Practice Ashutosh Lal, MD Director Comprehensive Thalassemia Program UCSF Benioff Children s Hospital Oakland Outline Thalassemia: definitions and pathophysiology Epidemiology
The Thalassemias in Clinical Practice Ashutosh Lal, MD Director Comprehensive Thalassemia Program UCSF Benioff Children s Hospital Oakland Outline Thalassemia: definitions and pathophysiology Epidemiology
Aneurin Bevan University Health Board Sickle Cell Anaemia and Haemoglobinopathy Screening and Management in Pregnancy Guidelines
 Sickle Cell Anaemia and Haemoglobinopathy Screening and Management in Pregnancy Guidelines N.B. Staff should be discouraged from printing this document. This is to avoid the risk of out of date printed
Sickle Cell Anaemia and Haemoglobinopathy Screening and Management in Pregnancy Guidelines N.B. Staff should be discouraged from printing this document. This is to avoid the risk of out of date printed
RBCs Disorders 1. Dr. Nabila Hamdi MD, PhD
 RBCs Disorders 1 Dr. Nabila Hamdi MD, PhD ILOs Discuss the classification of anemia into hypochromic-microcytic, normochromicnormocytic and macrocytic. Categorize laboratory test procedures used in the
RBCs Disorders 1 Dr. Nabila Hamdi MD, PhD ILOs Discuss the classification of anemia into hypochromic-microcytic, normochromicnormocytic and macrocytic. Categorize laboratory test procedures used in the
Approach to Hemolysis
 Objectives: Approach to Hemolysis To know the function of platelets and the relationship between the platelet count in peripheral blood and the extent of abnormal bleeding. To know about the diseases associated
Objectives: Approach to Hemolysis To know the function of platelets and the relationship between the platelet count in peripheral blood and the extent of abnormal bleeding. To know about the diseases associated
Sickle Cell Disease 101. Objectives. What is SCD? 4/20/2016. Discuss the pathophysiology & genetics of Sickle Cell Disease (SCD).
.") Sickle Cell Disease 101 Jennifer Young, RN, MS, CPNP-AC Sickle Cell & Thalassemia Nurse Practitioner Nationwide Children s Hospital Objectives Discuss the pathophysiology & genetics of Sickle Cell Disease
Sickle Cell Disease 101 Jennifer Young, RN, MS, CPNP-AC Sickle Cell & Thalassemia Nurse Practitioner Nationwide Children s Hospital Objectives Discuss the pathophysiology & genetics of Sickle Cell Disease
SICKLE CELL BROCHURE
 SICKLE CELL BROCHURE SICKLE CELL DIESEASE According to CDC, Sickle cell disease (SCD) is a group of inherited red blood cell disorders. Healthy red blood cells are round and SCD C -shaped farm tool called
SICKLE CELL BROCHURE SICKLE CELL DIESEASE According to CDC, Sickle cell disease (SCD) is a group of inherited red blood cell disorders. Healthy red blood cells are round and SCD C -shaped farm tool called
Haemoglobin BY: MUHAMMAD RADWAN WISSAM MUHAMMAD
 Haemoglobin BY: MUHAMMAD RADWAN WISSAM MUHAMMAD Introduction is the iron-containing oxygen transport metalloprotein in the red blood cells Hemoglobin in the blood carries oxygen from the respiratory organs
Haemoglobin BY: MUHAMMAD RADWAN WISSAM MUHAMMAD Introduction is the iron-containing oxygen transport metalloprotein in the red blood cells Hemoglobin in the blood carries oxygen from the respiratory organs
Genetic Modifiers of Sickle Cell Disease Severity. Kunle Adekile, MD, PhD Professor Department of Pediatrics Kuwait University
 Genetic Modifiers of Sickle Cell Disease Severity Kunle Adekile, MD, PhD Professor Department of Pediatrics Kuwait University Outline Hb Molecule and Genetic control of globin synthesis Pathophysiology
Genetic Modifiers of Sickle Cell Disease Severity Kunle Adekile, MD, PhD Professor Department of Pediatrics Kuwait University Outline Hb Molecule and Genetic control of globin synthesis Pathophysiology
Interleukin-1ß and Interleukin-6 Genetic Polymorphisms and Sickle Cell Disease: An Egyptian Study
 Interleukin-1ß and Interleukin-6 Genetic Polymorphisms and Sickle Cell Disease: An Egyptian Study MONA EL-GHAMRAWY, MD, PROFESSOR OF PEDIATRICS & PEDIATRIC HEMATOLOGY, CAIRO UNIVERSITY melghamrawy95@gmail.com
Interleukin-1ß and Interleukin-6 Genetic Polymorphisms and Sickle Cell Disease: An Egyptian Study MONA EL-GHAMRAWY, MD, PROFESSOR OF PEDIATRICS & PEDIATRIC HEMATOLOGY, CAIRO UNIVERSITY melghamrawy95@gmail.com
Extra Notes 3. Warm. In the core (center) of the body, where the temperature is 37 C.
 of the body, where the temperature is 37 C.") Extra Notes 3 *The numbers of the slides are according to the last year slides. Slide 33 Autoimmune hemolytic anemia : Abnormal circulating antibodies that target normal antigen on the RBC and cause lysis.
Extra Notes 3 *The numbers of the slides are according to the last year slides. Slide 33 Autoimmune hemolytic anemia : Abnormal circulating antibodies that target normal antigen on the RBC and cause lysis.
HAEMOGLOBINOPATHIES. Editing file. References: 436 girls & boys slides 435 teamwork slides. Color code: Important. Extra.
 HAEMOGLOBINOPATHIES Objectives: normal structure and function of haemoglobin. how the globin components of haemoglobin change during development, and postnatally. the mechanisms by which the thalassaemias
HAEMOGLOBINOPATHIES Objectives: normal structure and function of haemoglobin. how the globin components of haemoglobin change during development, and postnatally. the mechanisms by which the thalassaemias
Blood Transfusions in Children with Haemoglobinopathies
 Blood Transfusions in Children with Haemoglobinopathies Version: 2 Date: 22 nd April 2010 Authors: Responsible committee or Director: Review date: Target audience: Stakeholders/ committees involved in
Blood Transfusions in Children with Haemoglobinopathies Version: 2 Date: 22 nd April 2010 Authors: Responsible committee or Director: Review date: Target audience: Stakeholders/ committees involved in
THALASSEMIA AND COMPREHENSIVE CARE
 1 THALASSEMIA AND COMPREHENSIVE CARE Melanie Kirby MBBS, FRCP (C), Hospital for Sick Children, Toronto Associate Professor of Paediatrics, University of Toronto. Objectives 2 By the end of this presentation,
1 THALASSEMIA AND COMPREHENSIVE CARE Melanie Kirby MBBS, FRCP (C), Hospital for Sick Children, Toronto Associate Professor of Paediatrics, University of Toronto. Objectives 2 By the end of this presentation,
Hypochromic Anaemias
 Hypochromic Anaemias Dr Mere Kende MBBS, MMED (Path), MAACB, MACTM, MACRRM LECTURER-SMHS Anaemia LOW HEMOGLOBIN Anaemia Definition: Hb
Hypochromic Anaemias Dr Mere Kende MBBS, MMED (Path), MAACB, MACTM, MACRRM LECTURER-SMHS Anaemia LOW HEMOGLOBIN Anaemia Definition: Hb
Biance Rowe Chris Hani Baragwanath Hospital Paediatric Haematology Oncology University of the Witwatersrand
 Biance Rowe Chris Hani Baragwanath Hospital Paediatric Haematology Oncology University of the Witwatersrand SCD affects 20-25 million people globally 12-15 million in Africa 300 000 children with SCD
Biance Rowe Chris Hani Baragwanath Hospital Paediatric Haematology Oncology University of the Witwatersrand SCD affects 20-25 million people globally 12-15 million in Africa 300 000 children with SCD
Non-transfusion-dependent thalassemia (NTDT) Bor-Sheng Ko, M.D. Ph.D. Meng-Yao Lu, M.D. National Taiwan University Hospital
 Bor-Sheng Ko, M.D. Ph.D. Meng-Yao Lu, M.D. National Taiwan University Hospital") Non-transfusion-dependent thalassemia (NTDT) Bor-Sheng Ko, M.D. Ph.D. Meng-Yao Lu, M.D. National Taiwan University Hospital Introduction Transfusion dependency in thalassemia Transfusions seldom required
Non-transfusion-dependent thalassemia (NTDT) Bor-Sheng Ko, M.D. Ph.D. Meng-Yao Lu, M.D. National Taiwan University Hospital Introduction Transfusion dependency in thalassemia Transfusions seldom required
Hemoglobinopathies NORMAL HEMOGLOBINS
 Hemoglobinopathies Millicent Sutton MD October 28, 2005 NORMAL HEMOGLOBINS Consist of 2 alpha chains and 2 non alpha chains Hb A = α2β2 Hb F= α 2γ2 Hb A2 = α2δ2 1 Hemoglobin Variants Altered the conformational
Hemoglobinopathies Millicent Sutton MD October 28, 2005 NORMAL HEMOGLOBINS Consist of 2 alpha chains and 2 non alpha chains Hb A = α2β2 Hb F= α 2γ2 Hb A2 = α2δ2 1 Hemoglobin Variants Altered the conformational
Non-transfusion-dependent thalassemia (NTDT) Bor-Sheng Ko, M.D. Ph.D. Meng-Yao Lu, M.D. National Taiwan University Hospital
 Bor-Sheng Ko, M.D. Ph.D. Meng-Yao Lu, M.D. National Taiwan University Hospital") Non-transfusion-dependent thalassemia (NTDT) Bor-Sheng Ko, M.D. Ph.D. Meng-Yao Lu, M.D. National Taiwan University Hospital Introduction Spectrum of thalassemia: Resulting from unbalanced α/β chains α-thalassemias
Non-transfusion-dependent thalassemia (NTDT) Bor-Sheng Ko, M.D. Ph.D. Meng-Yao Lu, M.D. National Taiwan University Hospital Introduction Spectrum of thalassemia: Resulting from unbalanced α/β chains α-thalassemias
Hydroxycarbamide. Sickle and Thalassaemia Training days. September Dr Sara Stuart-Smith. Why do sickle cells cause pain and organ damage?
 Sickle and Thalassaemia Training days September 2017 Hydroxycarbamide Dr Sara Stuart-Smith Why do sickle cells cause pain and organ damage? Under certain conditions, haemoglobin S forms long rigid strands
Sickle and Thalassaemia Training days September 2017 Hydroxycarbamide Dr Sara Stuart-Smith Why do sickle cells cause pain and organ damage? Under certain conditions, haemoglobin S forms long rigid strands
TRANSFUSION PRACTICES IN THE MANAGEMENT OF SICKLE CELL DISEASE AMONG FLORIDA PHYSICIANS
 TRANSFUSION PRACTICES IN THE MANAGEMENT OF SICKLE CELL DISEASE AMONG FLORIDA PHYSICIANS By LEVETTE NICOLE DUNBAR A THESIS PRESENTED TO THE GRADUATE SCHOOL OF THE UNIVERSITY OF FLORIDA IN PARTIAL FULFILLMENT
TRANSFUSION PRACTICES IN THE MANAGEMENT OF SICKLE CELL DISEASE AMONG FLORIDA PHYSICIANS By LEVETTE NICOLE DUNBAR A THESIS PRESENTED TO THE GRADUATE SCHOOL OF THE UNIVERSITY OF FLORIDA IN PARTIAL FULFILLMENT
COEXISTENCE OF β-thalassemia AND POLYCYTHEMIA VERA: A CHICKEN-AND-EGG DEBATE?
 COEXISTENCE OF β-thalassemia AND POLYCYTHEMIA VERA: A CHICKEN-AND-EGG DEBATE? M. DE SLOOVERE (1), L. HARLET (2), S. VAN STEENWEGHEN (3), E. MOREAU (1), D. DE SMET (1) (1) DEPARTMENT OF LABORATORY MEDICINE,
COEXISTENCE OF β-thalassemia AND POLYCYTHEMIA VERA: A CHICKEN-AND-EGG DEBATE? M. DE SLOOVERE (1), L. HARLET (2), S. VAN STEENWEGHEN (3), E. MOREAU (1), D. DE SMET (1) (1) DEPARTMENT OF LABORATORY MEDICINE,
RBCs Disorders 2. Dr. Nabila Hamdi MD, PhD
 RBCs Disorders 2 Dr. Nabila Hamdi MD, PhD ILOs Discuss the classification of anemia into hypochromic-microcytic, normochromicnormocytic and macrocytic. Categorize laboratory test procedures used in the
RBCs Disorders 2 Dr. Nabila Hamdi MD, PhD ILOs Discuss the classification of anemia into hypochromic-microcytic, normochromicnormocytic and macrocytic. Categorize laboratory test procedures used in the
Index. Note: Page numbers of article titles are in boldface type.
 Note: Page numbers of article titles are in boldface type. A Abdominal tumors, in children, 530 531 Alkalinization, in tumor lysis syndrome, 516 Allopurinol, in tumor lysis syndrome, 515 Anaphylaxis, drug
Note: Page numbers of article titles are in boldface type. A Abdominal tumors, in children, 530 531 Alkalinization, in tumor lysis syndrome, 516 Allopurinol, in tumor lysis syndrome, 515 Anaphylaxis, drug
Susan Stegman, MD Medical Director AXA Equitable Life May 3, 2016
 Susan Stegman, MD Medical Director AXA Equitable Life May 3, 2016 Underwriting impact Anemia overview Classification of anemia Specific anemia topics Iron deficiency anemia Thalassemia Megaloblastic anemia
Susan Stegman, MD Medical Director AXA Equitable Life May 3, 2016 Underwriting impact Anemia overview Classification of anemia Specific anemia topics Iron deficiency anemia Thalassemia Megaloblastic anemia
Laura M. De Castro, MD Duke Comprehensive Sickle Cell Center November 2012
 Laura M. De Castro, MD Duke Comprehensive Sickle Cell Center November 2012 32 yo AA F; Hgb SS Hx: CVA w transient hemiparesis -16 yo Recurrent ACS/Pneumonia Pulm HTN O 2 dependent- Osteomyelitis Chronic
Laura M. De Castro, MD Duke Comprehensive Sickle Cell Center November 2012 32 yo AA F; Hgb SS Hx: CVA w transient hemiparesis -16 yo Recurrent ACS/Pneumonia Pulm HTN O 2 dependent- Osteomyelitis Chronic
INFLUENCE OF DELETIONAL ALPHA THALASSEMIA ON CLINICAL AND LABORATORY PARAMETERS OF YOUNG NIGERIANS WITH SICKLE CELL ANAEMIA
 INFLUENCE OF DELETIONAL ALPHA THALASSEMIA ON CLINICAL AND LABORATORY PARAMETERS OF YOUNG NIGERIANS WITH SICKLE CELL ANAEMIA Authors:Oladele Simeon Olatunya, 1,2,3, Dulcineia Martins de Albuquerque, 2,
INFLUENCE OF DELETIONAL ALPHA THALASSEMIA ON CLINICAL AND LABORATORY PARAMETERS OF YOUNG NIGERIANS WITH SICKLE CELL ANAEMIA Authors:Oladele Simeon Olatunya, 1,2,3, Dulcineia Martins de Albuquerque, 2,
Pediatric Red Cell Exchange Indications, Benefits, Barriers. View from California Saturday May 9 th ASFA 2015
 Pediatric Red Cell Exchange Indications, Benefits, Barriers View from California Saturday May 9 th ASFA 2015 Red Cell Exchange: Not SCD Recommendations for Red Cell Exchange Indication Procedure Recommendation
Pediatric Red Cell Exchange Indications, Benefits, Barriers View from California Saturday May 9 th ASFA 2015 Red Cell Exchange: Not SCD Recommendations for Red Cell Exchange Indication Procedure Recommendation
Corporate Medical Policy
 Corporate Medical Policy Genetic Testing for Alpha Thalassemia File Name: Origination: Last CAP Review: Next CAP Review: Last Review: genetic_testing_for_alpha_thalassemia 9/2013 7/2017 7/2018 7/2017 Description
Corporate Medical Policy Genetic Testing for Alpha Thalassemia File Name: Origination: Last CAP Review: Next CAP Review: Last Review: genetic_testing_for_alpha_thalassemia 9/2013 7/2017 7/2018 7/2017 Description
Hydroxurea: A Novel Approach to Optimizing the Health of Pediatric Patients with Sickle Cell Disease. Maa Ohui Quarmyne September 9 th, 2017
 Hydroxurea: A Novel Approach to Optimizing the Health of Pediatric Patients with Sickle Cell Disease Maa Ohui Quarmyne September 9 th, 2017 Outline Sickle Cell Disease Pathophysiology and Clinical Manifestations
Hydroxurea: A Novel Approach to Optimizing the Health of Pediatric Patients with Sickle Cell Disease Maa Ohui Quarmyne September 9 th, 2017 Outline Sickle Cell Disease Pathophysiology and Clinical Manifestations
Anemia (3).ms4.25.Oct.15 Hemolytic Anemia. Abdallah Abbadi
.ms4.25.Oct.15 Hemolytic Anemia. Abdallah Abbadi") Anemia (3).ms4.25.Oct.15 Hemolytic Anemia Abdallah Abbadi Case 3 24 yr old female presented with anemia syndrome and jaundice. She was found to have splenomegaly. Hb 8, wbc 12k, Plt 212k, retics 12%, LDH
Anemia (3).ms4.25.Oct.15 Hemolytic Anemia Abdallah Abbadi Case 3 24 yr old female presented with anemia syndrome and jaundice. She was found to have splenomegaly. Hb 8, wbc 12k, Plt 212k, retics 12%, LDH
4 Fahed Al Karmi Sufian Alhafez Dr nayef karadsheh
 4 Fahed Al Karmi Sufian Alhafez Dr nayef karadsheh Genetic variants of hemoglobin Hemoglobinopathies (abnormal variants of hemoglobin) are divided into: 1. Structural abnormalities: Any change in the genes
4 Fahed Al Karmi Sufian Alhafez Dr nayef karadsheh Genetic variants of hemoglobin Hemoglobinopathies (abnormal variants of hemoglobin) are divided into: 1. Structural abnormalities: Any change in the genes
Hemoglobinopathies Diagnosis and management
 Hemoglobinopathies Diagnosis and management Morgan L. McLemore, M.D. Hematology/Leukemia Department of Hematology and Oncology Winship Cancer Institute at Emory University mlmclem@emory.edu Disclosures
Hemoglobinopathies Diagnosis and management Morgan L. McLemore, M.D. Hematology/Leukemia Department of Hematology and Oncology Winship Cancer Institute at Emory University mlmclem@emory.edu Disclosures
Anemia (3).ms Hemolytic Anemia. Abdallah Abbadi Feras Fararjeh
.ms Hemolytic Anemia. Abdallah Abbadi Feras Fararjeh") Anemia (3).ms4.26.2.18 Hemolytic Anemia Abdallah Abbadi Feras Fararjeh Case 3 24 yr old female presented with anemia syndrome and jaundice. She was found to have splenomegaly. Hb 8, wbc 12k, Plt 212k,
Anemia (3).ms4.26.2.18 Hemolytic Anemia Abdallah Abbadi Feras Fararjeh Case 3 24 yr old female presented with anemia syndrome and jaundice. She was found to have splenomegaly. Hb 8, wbc 12k, Plt 212k,
Pediatrics. Pyruvate Kinase Deficiency (PKD) Symptoms and Treatment. Definition. Epidemiology of Pyruvate Kinase Deficiency.
 Symptoms and Treatment. Definition. Epidemiology of Pyruvate Kinase Deficiency.") Pediatrics Pyruvate Kinase Deficiency (PKD) Symptoms and Treatment See online here Pyruvate kinase deficiency is an inherited metabolic disorder characterized by a deficiency in the enzyme "pyruvate kinase"
Pediatrics Pyruvate Kinase Deficiency (PKD) Symptoms and Treatment See online here Pyruvate kinase deficiency is an inherited metabolic disorder characterized by a deficiency in the enzyme "pyruvate kinase"
Compassionate-use Experience With Voxelotor (GBT440) for Patients With Severe Sickle Cell Disease (SCD) and Life-Threatening Comorbidities
 for Patients With Severe Sickle Cell Disease (SCD) and Life-Threatening Comorbidities") Compassionate-use Experience With Voxelotor (GBT440) for Patients With Severe Sickle Cell Disease (SCD) and Life-Threatening Comorbidities Gershwin Blyden, MD 1, Kenneth R. Bridges, MD 2, Lanetta Bronté,
Compassionate-use Experience With Voxelotor (GBT440) for Patients With Severe Sickle Cell Disease (SCD) and Life-Threatening Comorbidities Gershwin Blyden, MD 1, Kenneth R. Bridges, MD 2, Lanetta Bronté,
UNRELATED DONOR TRANSPLANTATION FOR SICKLE CELL DISEASE AN UPDATE
 UNRELATED DONOR TRANSPLANTATION FOR SICKLE CELL DISEASE AN UPDATE Naynesh Kamani, M.D. Children s National Medical Center GW University School of Medicine Washington, DC SCD scope of problem in USA Commonest
UNRELATED DONOR TRANSPLANTATION FOR SICKLE CELL DISEASE AN UPDATE Naynesh Kamani, M.D. Children s National Medical Center GW University School of Medicine Washington, DC SCD scope of problem in USA Commonest
Non-malignant hematologic disorders associated arthropathies: hemoglobinopathy-associated musculoskeletal manifestations, hemophilia
 Non-malignant hematologic disorders associated arthropathies: hemoglobinopathy-associated musculoskeletal manifestations, hemophilia HAEMOGLOBINOPATHIES = inherited disorders of globin divided into: Thalassaemia
Non-malignant hematologic disorders associated arthropathies: hemoglobinopathy-associated musculoskeletal manifestations, hemophilia HAEMOGLOBINOPATHIES = inherited disorders of globin divided into: Thalassaemia
Acute vaso-occlusive Pain in children
 Acute vaso-occlusive Pain in children Dr François Angoulvant 1 Dr Sebastien Redant 2 Dr Malika Benkerrou 1 Pr Alina Ferster 2 Hôpital Robert Debré - Paris Hôpital des Enfants Reine Fabiola Bruxelles Réseau
Acute vaso-occlusive Pain in children Dr François Angoulvant 1 Dr Sebastien Redant 2 Dr Malika Benkerrou 1 Pr Alina Ferster 2 Hôpital Robert Debré - Paris Hôpital des Enfants Reine Fabiola Bruxelles Réseau
Historic and Current Complications in Children with Sickle Cell Disease
 Historic and Current Complications in Children with Sickle Cell Disease Trish McMahon Peterson RN, MSN, CPNP Thomas C. Hofstra MD Children's Hospital Los Angeles Comprehensive Sickle Cell Program Children's
Historic and Current Complications in Children with Sickle Cell Disease Trish McMahon Peterson RN, MSN, CPNP Thomas C. Hofstra MD Children's Hospital Los Angeles Comprehensive Sickle Cell Program Children's
HPLC profile of sickle cell disease in central India
 Original Research Article HPLC profile of sickle cell disease in central India Shweta P. Bijwe * Department of Pathology, IGGMC, Nagpur, Maharashtra, India * Corresponding author email: dr.shwetabijwe@gmail.com
Original Research Article HPLC profile of sickle cell disease in central India Shweta P. Bijwe * Department of Pathology, IGGMC, Nagpur, Maharashtra, India * Corresponding author email: dr.shwetabijwe@gmail.com
C. treatment with Desferal (deferoxamine mesylate USP, iron-chelating agent)
") HEMOLYTIC ANEMIAS Single choice tests 1. Select the clinical manifestation that is not characteristic for the hemolytic crisis: A. decrease of the red blood cell count B. reticulocytosis C. jaundice D.
HEMOLYTIC ANEMIAS Single choice tests 1. Select the clinical manifestation that is not characteristic for the hemolytic crisis: A. decrease of the red blood cell count B. reticulocytosis C. jaundice D.
Acute Complications of Sickle Cell Disease
 Management of Acute Complications of Sickle Cell Disease A Pocket Guide for the Clinician Timothy McCavit, MD, MSCS 1 Payal Desai, MD 1 University of Texas Southwestern Medical Center The Ohio State University,
Management of Acute Complications of Sickle Cell Disease A Pocket Guide for the Clinician Timothy McCavit, MD, MSCS 1 Payal Desai, MD 1 University of Texas Southwestern Medical Center The Ohio State University,
Medical Complications of Pregnancy
 Medical Complications of Pregnancy Systems Cardiovascular Pulmonary Endocrine Gastrointestinal Urologic Neurologic Cardiovascular System Physiologic anemia 3:1 increase of plasma volume:rbc mass Treat
Medical Complications of Pregnancy Systems Cardiovascular Pulmonary Endocrine Gastrointestinal Urologic Neurologic Cardiovascular System Physiologic anemia 3:1 increase of plasma volume:rbc mass Treat
Sickle Cell Diseasechronic. curable disease? Objectives. Why would a family ask about cure for SCD?
 Sickle Cell Diseasechronic illness or curable disease? Gregory M.T. Guilcher MD, FRCPC, FAAP Objectives To review the general principles of hematopoietic stem cell transplantation (HSCT), including risks
Sickle Cell Diseasechronic illness or curable disease? Gregory M.T. Guilcher MD, FRCPC, FAAP Objectives To review the general principles of hematopoietic stem cell transplantation (HSCT), including risks
Are We There Yet? Gene Therapy and BMT as Curative Therapies in Sickle Cell. Ann Haight, MD 9 Sept 2017
 Are We There Yet? Gene Therapy and BMT as Curative Therapies in Sickle Cell Ann Haight, MD 9 Sept 2017 Spoiler alert Yes (we have a cure) And No Work to do! 2 Sickle Cell Treatment Options Supportive Care
Are We There Yet? Gene Therapy and BMT as Curative Therapies in Sickle Cell Ann Haight, MD 9 Sept 2017 Spoiler alert Yes (we have a cure) And No Work to do! 2 Sickle Cell Treatment Options Supportive Care
Thalassaemia. What is thalassaemia? What causes thalassaemia? What are the different types of thalassaemia?
 Thalassaemia Thalassaemia is an inherited condition affecting the blood. There are different types, which vary from a mild condition with no symptoms, to a serious or lifethreatening condition. For the
Thalassaemia Thalassaemia is an inherited condition affecting the blood. There are different types, which vary from a mild condition with no symptoms, to a serious or lifethreatening condition. For the
Done by :Aseel Twaijer & Laith Sorour Hemolytic Anemias
 Hemolytic Anemias Hemolytic anemias share the following features: - A shortened red cell life < 120 days - Elevated erythropoietin levels (compensatory increase in erythropoiesis) - Accumulation of hemoglobin
Hemolytic Anemias Hemolytic anemias share the following features: - A shortened red cell life < 120 days - Elevated erythropoietin levels (compensatory increase in erythropoiesis) - Accumulation of hemoglobin
Hydroxyurea Treatment for Sickle Cell Disease
 Hydroxyurea Treatment for Sickle Cell Disease Before Hydroxyurea After Hydroxyurea Hydroxyurea Treatment for Sickle Cell Disease This document is not intended to take the place of the care and attention
Hydroxyurea Treatment for Sickle Cell Disease Before Hydroxyurea After Hydroxyurea Hydroxyurea Treatment for Sickle Cell Disease This document is not intended to take the place of the care and attention
Biology 2C03: Genetics What is a Gene?
 Biology 2C03: Genetics What is a Gene? September 9 th, 2013 Model Organisms - E. coli - Yeast - Worms - Arabodopsis - Fruitflie - Mouse What is a Gene? - Define, recognize, describe and apply Mendel s
Biology 2C03: Genetics What is a Gene? September 9 th, 2013 Model Organisms - E. coli - Yeast - Worms - Arabodopsis - Fruitflie - Mouse What is a Gene? - Define, recognize, describe and apply Mendel s
Blood Transfusion Guidelines in Clinical Practice
 Blood Transfusion Guidelines in Clinical Practice Salwa Hindawi Director of Blood Transfusion Services Associate Professor in Haematology and Transfusion Medicine King Abdalaziz University, Jeddah Saudi
Blood Transfusion Guidelines in Clinical Practice Salwa Hindawi Director of Blood Transfusion Services Associate Professor in Haematology and Transfusion Medicine King Abdalaziz University, Jeddah Saudi
HAEMOLYTIC ANAEMIA. Dr. Hasan Fahmawi, MRCP(London), FRCP(Edin) Consultant Physician
, FRCP(Edin) Consultant Physician") HAEMOLYTIC ANAEMIA Dr. Hasan Fahmawi, MRCP(London), FRCP(Edin) Consultant Physician Haemolysis Definition shortening of the normal red blood lifespan of 120 days Increase in unconjugated bilirubin, increased
HAEMOLYTIC ANAEMIA Dr. Hasan Fahmawi, MRCP(London), FRCP(Edin) Consultant Physician Haemolysis Definition shortening of the normal red blood lifespan of 120 days Increase in unconjugated bilirubin, increased
High Hemoglobin F in a Saudi Child Presenting with Pancytopenia
 Case Report imedpub Journals http://www.imedpub.com Journal of Pediatric Care ISSN 2471-805X DOI: 10.21767/2471-805X.100002 High Hemoglobin F in a Saudi Child Presenting with Pancytopenia Abstract Saudi
Case Report imedpub Journals http://www.imedpub.com Journal of Pediatric Care ISSN 2471-805X DOI: 10.21767/2471-805X.100002 High Hemoglobin F in a Saudi Child Presenting with Pancytopenia Abstract Saudi
are associated with sickle cell disease and carriers: a study of patients from the southeastregion of Iran
 CXC chemokines CXCL1, CXCL9, CXCL10 and CXCL12 are associated with sickle cell disease and carriers: a study of patients from the southeastregion of Iran Mojgan Noroozi Karimabad Molecular Medicine Research
CXC chemokines CXCL1, CXCL9, CXCL10 and CXCL12 are associated with sickle cell disease and carriers: a study of patients from the southeastregion of Iran Mojgan Noroozi Karimabad Molecular Medicine Research
Beta Thalassemia Frequency in Bahrain: A Ten Year Study. Shaikha Salim Al-Arrayed, MB,ChB, DHCG, PhD*
 Bahrain Medical Bulletin, Vol. 2, No. 2, June 200 Beta Thalassemia Frequency in Bahrain: A Ten Year Study Shaikha Salim Al-Arrayed, MB,ChB, DHCG, PhD* Background: Sickle-cell disease and Thalassanemia
Bahrain Medical Bulletin, Vol. 2, No. 2, June 200 Beta Thalassemia Frequency in Bahrain: A Ten Year Study Shaikha Salim Al-Arrayed, MB,ChB, DHCG, PhD* Background: Sickle-cell disease and Thalassanemia
Making Hope A Reality December 10, Nasdaq : BLUE
 Making Hope A Reality December 10, 2014 Nasdaq : BLUE Forward Looking Statement These slides and the accompanying oral presentation contain forward-looking statements and information. The use of words
Making Hope A Reality December 10, 2014 Nasdaq : BLUE Forward Looking Statement These slides and the accompanying oral presentation contain forward-looking statements and information. The use of words
7 Medical Genetics. Hemoglobinopathies. Hemoglobinopathies. Protein and Gene Structure. and Biochemical Genetics
 SESSION 7 Medical Genetics Hemoglobinopathies and Biochemical Genetics J a v a d F a s a J a m s h i d i U n i v e r s i t y o f M e d i c a l S c i e n c e s, N o v e m b e r 2 0 1 7 Hemoglobinopathies
SESSION 7 Medical Genetics Hemoglobinopathies and Biochemical Genetics J a v a d F a s a J a m s h i d i U n i v e r s i t y o f M e d i c a l S c i e n c e s, N o v e m b e r 2 0 1 7 Hemoglobinopathies
Emergency Presentations of Sickle cell disease. Dr S Pancham Sickle Cell & Thalassaemia Centre City Hospital
 Emergency Presentations of Sickle cell disease Dr S Pancham Sickle Cell & Thalassaemia Centre City Hospital Aims Key features to elicit at presentation What complications to look for When to phone for
Emergency Presentations of Sickle cell disease Dr S Pancham Sickle Cell & Thalassaemia Centre City Hospital Aims Key features to elicit at presentation What complications to look for When to phone for
Transfusion in Sickle Cell Disease What the guidelines [are likely to] say. Dr Bernard Davis Whittington Hospital, London
![Transfusion in Sickle Cell Disease What the guidelines [are likely to] say. Dr Bernard Davis Whittington Hospital, London](/thumbs/85/91702350.jpg "Transfusion in Sickle Cell Disease What the guidelines [are likely to] say. Dr Bernard Davis Whittington Hospital, London") Transfusion in Sickle Cell Disease What the guidelines [are likely to] say Dr Bernard Davis Whittington Hospital, London Background to BCSH Guideline Rationale Current guidance in disparate publications
Transfusion in Sickle Cell Disease What the guidelines [are likely to] say Dr Bernard Davis Whittington Hospital, London Background to BCSH Guideline Rationale Current guidance in disparate publications