Sickle Cell Disease and impact on the society
|
|
|
- Barrie Brown
- 6 years ago
- Views:
Transcription
1 Sickle Cell Disease and impact on the society Professor Z.A.Jeremiah Ph.D, FRCPath (London) Professor of Haematology and Blood Transfusion Science Niger Delta University, Wilberforce Island
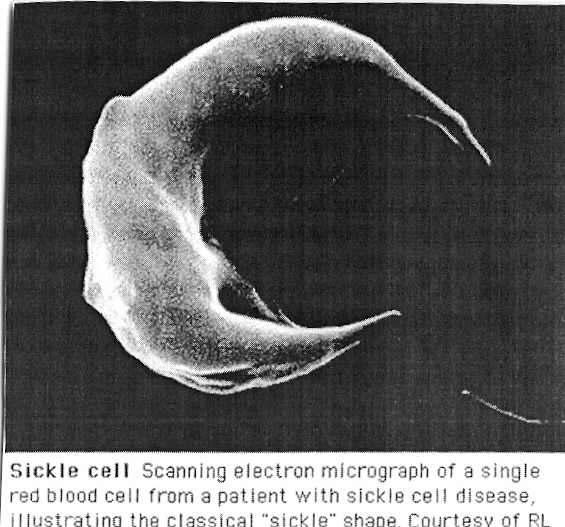
2
3 Outline What is sickle cell disease? How is it formed? Epidemiology Manifestations Laboratory findings Impact on the Society The future
4 What is it? Sickle cell disease is one form of hemoglobinopathy- a structural abnormality in hemoglobin molecule Substitution of glutamic acid by valine at the 6 th position Negatively charged amino acid replaced by neutral amino acid
5 What is it? Hgb S maintains normal function in oxygenated state In de-oxygenated state- induced change in configuration allows valine to interact irregularly Formation of highly ordered polymers Polymers aggregate to rigid rods Spiny brittle RBCs Within vessels, thrombosis/obstruction
6 How it is formed? Sickle cell shape results in decrease cell deformability. Changes also occur in red cell membrane structure and function, disordered cell volume control and increase adherence to vascular endothelium. Vaso-occlusive phenomena and hemolysis are the clinical hallmarks of Sickle Cell Disease (SCD)

7 Red Blood Cells from Sickle Cell Anemia Deoxygenation of SS erythrocytes leads to intracellular hemoglobin polymerization, loss of deformability and changes in cell morphology. OXY-STATE DEOXY-STATE
8 Chromosome 16 5' Sickle Cell Mutation ζ α2 α1 Chromosome 11 ε Gγ Aγ δ β 5' 3' * 3' Normal (HbA) CCT GAG GAG -Pro-Glu-Glu β A α α β β Abnormal (HbS) β S CCT GTG GAG -Pro-Val-Glu O2 +O2 -O2 +O2
9 Epidemiology About 5% of the world s population carries genes responsible for haemoglobinopathies. Each year about infants are born with major haemoglobin disorders including more than cases of sickle-cell anaemia in Africa.
10 Epidemiology Globally, there are more carriers (i.e. healthy people who have inherited only one mutant gene from one parent) of thalassaemia than of sickle-cell anaemia, but the high frequency of the sickle-cell gene in certain areas leads to a high rate of affected newborns.
11 Epidemiology In broad terms, the prevalence of the sickle-cell trait (healthy carriers who have inherited the mutant gene from only one parent) ranges between 10% and 40% across equatorial Africa and decreases to between 1% and 2% on the north African coast and <1% in South Africa.
12 Epidemiology In west African countries such as Ghana and Nigeria, the frequency of the trait is 15% to 30% whereas in Uganda it shows marked tribal variations, reaching 45% among the Baamba tribe in the west of the country.
13 Epidemiology Frequencies of the carrier state determine the prevalence of sickle-cell anaemia at birth. For example, in Nigeria, by far the most populous country in the subregion, 24% of the population are carriers of the mutant gene and the prevalence of sicklecell anaemia is about 20 per 1000 births. This means that in Nigeria alone, about children are born annually with sickle-cell anaemia.
14 Manifestations Generally, no symptoms are seen in the 1 st 6 moths of life due to circulating fetal hemoglobin Dactylitis (aka hand-foot syndrome) Painful, symmetric swelling of hands and feet Due to ischemic necrosis of small bones of hands and feet? Due to rapidly expanding bone marrow, choking of blood supply
15 Manifestations Acute pain episodes Young children- extremities Older patients- head, chest, abdomen, back Recurrence of pain tends to occur in same sites within a particular individual Exacerbated by fever, hypoxia, acidosispromote deoxygenation of Hgb S
16 Manifestations Infarctions Bone/bone marrow Osteomyelitis- concern of salmonella infection Autosplenectomy Increased susceptibility to encapsulated organisms Esp. pneumococcus & H. influenzae Associated with reduction in serum opsonins Pulmonary infarcts Pneumonitis Fat emboli
17 Manifestations Infarcts Stroke Kidney Impaired renal function Hyposthenuria Priapism Avascular necrosis
18 Manifestations Acute Chest Syndrome Fever Tachypnea Chest pain Hypoxia Hypotension X-ray findings
19 Manifestations Splenic seqestration Large amounts of blood pools in spleen Splenic enlargement Criculatory collapse Reason unknown May follow febrile illness Aplastic episodes- may follow infection with parvovirus B 19
20 Manifestations Cardiomegaly Gallstones Body habitus Underweight Delayed puberty
21 Manifestations Laboratory Normocytic anemia- Hgb 5-9 mg/dl Peripheral smear Target cells Poikilocytes Sickled cells Howell Jolly bodies Leukocytosis with neutrophil predominance Thrombocytosis X-ray- expanded marrow spaces, osteoporosis
22 Laboratory findings Moderate anemia Reticulocytosis 3-15% High MCV Unconjugated hyperbilirubinemia Elevetaed LDH Low haptoglobin Folate & iron deficit Peripheral smear shows sickle cells Polychromasia Howell-jolly bodies Elevated WBC Elevated Platelets Low than after 18 yrs high creatinine
23

24
25 Impact of SCD on Society impact on human health may be assessed against the yardsticks of infant and underfive mortality. As not all deaths occur in the first year of life, the most valid measure is under-five deaths. An increasing proportion of affected children now survive past five years of age but remain at risk of premature death.
26 Impact of SCD When health impact is measured by under-five mortality, sickle-cell anaemia contributes the equivalent of 5% of underfive deaths on the African continent, more than 9% of such deaths in west Africa, and up to 16% of under-five deaths in individual west African countries.
27 Economic and Social impact In the United States of America median survival was estimated in 1994 to be 42 years for men and 48 years for women, whereas comparable figures for Jamaica published in 2001 suggested 53 years for men and 58.5 years for women. Data for sub saharan Africa not known
28 The future Gene therapy Increase expression of Hb F RNA repair Hematopoietic cell transplantation
Sickle Cell Disease. Edward Malters, MD
 Sickle Cell Disease Edward Malters, MD Introduction Vaso-occlusive phenomena and hemolysis are the clinical hallmarks of Sickle Cell Disease (SCD) Inherited disorder due to homozygosity for the abnormal
Sickle Cell Disease Edward Malters, MD Introduction Vaso-occlusive phenomena and hemolysis are the clinical hallmarks of Sickle Cell Disease (SCD) Inherited disorder due to homozygosity for the abnormal
SICKLE CELL DISEASE. Dr. MUBARAK ABDELRAHMAN MD PEDIATRICS AND CHILD HEALTH. Assistant Professor FACULTY OF MEDICINE -JAZAN
 SICKLE CELL DISEASE Dr. MUBARAK ABDELRAHMAN MD PEDIATRICS AND CHILD HEALTH Assistant Professor FACULTY OF MEDICINE -JAZAN Objective: The student should be able: To identify the presentation, diagnosis,
SICKLE CELL DISEASE Dr. MUBARAK ABDELRAHMAN MD PEDIATRICS AND CHILD HEALTH Assistant Professor FACULTY OF MEDICINE -JAZAN Objective: The student should be able: To identify the presentation, diagnosis,
Hemolytic anemias (2 of 2)
") Hemolytic anemias (2 of 2) Sickle Cell Anemia The most common familial hemolytic anemia in the world Sickle cell anemia is the prototypical (and most prevalent) hemoglobinopathy Mutation in the β-globin
Hemolytic anemias (2 of 2) Sickle Cell Anemia The most common familial hemolytic anemia in the world Sickle cell anemia is the prototypical (and most prevalent) hemoglobinopathy Mutation in the β-globin
Dr Banu Kaya Consultant Haematologist Barts Health NHS Trust Royal London Hospital, London, UK SICKLE CELL AND THALASSAEMIA OVERVIEW
 Dr Banu Kaya Consultant Haematologist Barts Health NHS Trust Royal London Hospital, London, UK SICKLE CELL AND THALASSAEMIA OVERVIEW Objectives Gain awareness of haemoglobinopathy inheritance, pathophysiology
Dr Banu Kaya Consultant Haematologist Barts Health NHS Trust Royal London Hospital, London, UK SICKLE CELL AND THALASSAEMIA OVERVIEW Objectives Gain awareness of haemoglobinopathy inheritance, pathophysiology
Introduction reduction in output alter the amino acid sequence combination
 Sickle cell anemia. Introduction Mutations in the globin genes can cause a quantitative reduction in output from that gene or alter the amino acid sequence of the protein produced or a combination of the
Sickle cell anemia. Introduction Mutations in the globin genes can cause a quantitative reduction in output from that gene or alter the amino acid sequence of the protein produced or a combination of the
DONE BY : RaSHA RAKAN & Bushra Saleem
 DONE BY : RaSHA RAKAN & Bushra Saleem Hemolytic anemias (2 of 2) Sickle Cell Anemia The most common familial hemolytic anemia in the world Sickle cell anemia is the prototypical (and most prevalent) hemoglobinopathy
DONE BY : RaSHA RAKAN & Bushra Saleem Hemolytic anemias (2 of 2) Sickle Cell Anemia The most common familial hemolytic anemia in the world Sickle cell anemia is the prototypical (and most prevalent) hemoglobinopathy
Sickle cell disease. Fareed Omar 10 March 2018
 Sickle cell disease Fareed Omar 10 March 2018 Physiology Haemoglobin structure HbA2: 2α and 2δ chains (2-3%) HbF: 2α and 2γ chains (
Sickle cell disease Fareed Omar 10 March 2018 Physiology Haemoglobin structure HbA2: 2α and 2δ chains (2-3%) HbF: 2α and 2γ chains (
Anemia s. Troy Lund MSMS PhD MD
 Anemia s Troy Lund MSMS PhD MD lundx072@umn.edu Hemoglobinopathy/Anemia IOM take home points. 1. How do we identify the condtion? Smear, CBC Solubility Test (SCD) 2. How does it present clincally? 3. How
Anemia s Troy Lund MSMS PhD MD lundx072@umn.edu Hemoglobinopathy/Anemia IOM take home points. 1. How do we identify the condtion? Smear, CBC Solubility Test (SCD) 2. How does it present clincally? 3. How
Approach to Hemolysis
 Objectives: Approach to Hemolysis To know the function of platelets and the relationship between the platelet count in peripheral blood and the extent of abnormal bleeding. To know about the diseases associated
Objectives: Approach to Hemolysis To know the function of platelets and the relationship between the platelet count in peripheral blood and the extent of abnormal bleeding. To know about the diseases associated
Congenital Haemoglobinopathies
 Congenital Haemoglobinopathies L. DEDEKEN, MD H O P I T A L U N I V E R S I T A I R E D E S E N F A N T S R E I N E F A B I O L A U N I V E R S I T E L I B R E DE B R U X E L L E S Red Blood Cell Disorders
Congenital Haemoglobinopathies L. DEDEKEN, MD H O P I T A L U N I V E R S I T A I R E D E S E N F A N T S R E I N E F A B I O L A U N I V E R S I T E L I B R E DE B R U X E L L E S Red Blood Cell Disorders
RBCs Disorders 1. Dr. Nabila Hamdi MD, PhD
 RBCs Disorders 1 Dr. Nabila Hamdi MD, PhD ILOs Discuss the classification of anemia into hypochromic-microcytic, normochromicnormocytic and macrocytic. Categorize laboratory test procedures used in the
RBCs Disorders 1 Dr. Nabila Hamdi MD, PhD ILOs Discuss the classification of anemia into hypochromic-microcytic, normochromicnormocytic and macrocytic. Categorize laboratory test procedures used in the
HEMOLYTIC ANEMIA DUE TO ABNORMAL HEMOGLOBIN SYNTHESIS
 Hemolytic Anemia Due to Abnormal Hemoglobin Synthesis MODULE 19 HEMOLYTIC ANEMIA DUE TO ABNORMAL HEMOGLOBIN SYNTHESIS 19.1 INTRODUCTION There are two main mechanisms by which anaemia is produced (a) Thalassemia:
Hemolytic Anemia Due to Abnormal Hemoglobin Synthesis MODULE 19 HEMOLYTIC ANEMIA DUE TO ABNORMAL HEMOGLOBIN SYNTHESIS 19.1 INTRODUCTION There are two main mechanisms by which anaemia is produced (a) Thalassemia:
Haemoglobin BY: MUHAMMAD RADWAN WISSAM MUHAMMAD
 Haemoglobin BY: MUHAMMAD RADWAN WISSAM MUHAMMAD Introduction is the iron-containing oxygen transport metalloprotein in the red blood cells Hemoglobin in the blood carries oxygen from the respiratory organs
Haemoglobin BY: MUHAMMAD RADWAN WISSAM MUHAMMAD Introduction is the iron-containing oxygen transport metalloprotein in the red blood cells Hemoglobin in the blood carries oxygen from the respiratory organs
In adults, the predominant Hb (HbA) molecule has four chains: two α and two β chains. In thalassemias, the synthesis of either the α or the β chains
 molecule has four chains: two α and two β chains. In thalassemias, the synthesis of either the α or the β chains") Thalassaemias Thalassemia Thalassemia is an inherited autosomal recessive blood disease. Associated with absence or reduction in a or b globin chains. Reduced synthesis of one of the globin chains can
Thalassaemias Thalassemia Thalassemia is an inherited autosomal recessive blood disease. Associated with absence or reduction in a or b globin chains. Reduced synthesis of one of the globin chains can
Medical and Surgical Complications of Sickle Cell Anemia
 Medical and Surgical Complications of Sickle Cell Anemia Ahmed Al-Salem Medical and Surgical Complications of Sickle Cell Anemia Ahmed Al-Salem Department of Surgery Dar A lalafia Medical Company Qatif
Medical and Surgical Complications of Sickle Cell Anemia Ahmed Al-Salem Medical and Surgical Complications of Sickle Cell Anemia Ahmed Al-Salem Department of Surgery Dar A lalafia Medical Company Qatif
HbSC disease is it different and how should we manage it? David Rees Department of Paediatric Haematology, King s College Hospital, London
 HbSC disease is it different and how should we manage it? David Rees Department of Paediatric Haematology, King s College Hospital, London Different types of sickle cell disesease Severe sickle cell disease
HbSC disease is it different and how should we manage it? David Rees Department of Paediatric Haematology, King s College Hospital, London Different types of sickle cell disesease Severe sickle cell disease
Rationale for RBC Transfusion in SCD
 Rationale for RBC Transfusion in SCD Dilution of HgbS-containing RBCs via the addition of HgbA-containing cells from the blood of normal donors Suppression of erythropoietin release caused by the rise
Rationale for RBC Transfusion in SCD Dilution of HgbS-containing RBCs via the addition of HgbA-containing cells from the blood of normal donors Suppression of erythropoietin release caused by the rise
4 Jumana Jihad Dr. Ahmad Mansour Dr. Ahmad Mansour
 4 Jumana Jihad Dr. Ahmad Mansour Dr. Ahmad Mansour Anemia Decreased blood production Increased blood loss Hemolytic Hemorrhage Extravascular Intravascular Hemolytic (Further classification( Extrinsic Intrinsic
4 Jumana Jihad Dr. Ahmad Mansour Dr. Ahmad Mansour Anemia Decreased blood production Increased blood loss Hemolytic Hemorrhage Extravascular Intravascular Hemolytic (Further classification( Extrinsic Intrinsic
Non-malignant hematologic disorders associated arthropathies: hemoglobinopathy-associated musculoskeletal manifestations, hemophilia
 Non-malignant hematologic disorders associated arthropathies: hemoglobinopathy-associated musculoskeletal manifestations, hemophilia HAEMOGLOBINOPATHIES = inherited disorders of globin divided into: Thalassaemia
Non-malignant hematologic disorders associated arthropathies: hemoglobinopathy-associated musculoskeletal manifestations, hemophilia HAEMOGLOBINOPATHIES = inherited disorders of globin divided into: Thalassaemia
Chem*3560 Lecture 4: Inherited modifications in hemoglobin
 Chem*3560 Lecture 4: Inherited modifications in hemoglobin Genetic modifications fall into two classes: Thalassemias, which are the result of failure to express globin genes. Thalassa is Greek for the
Chem*3560 Lecture 4: Inherited modifications in hemoglobin Genetic modifications fall into two classes: Thalassemias, which are the result of failure to express globin genes. Thalassa is Greek for the
Done by :Aseel Twaijer & Laith Sorour Hemolytic Anemias
 Hemolytic Anemias Hemolytic anemias share the following features: - A shortened red cell life < 120 days - Elevated erythropoietin levels (compensatory increase in erythropoiesis) - Accumulation of hemoglobin
Hemolytic Anemias Hemolytic anemias share the following features: - A shortened red cell life < 120 days - Elevated erythropoietin levels (compensatory increase in erythropoiesis) - Accumulation of hemoglobin
Screening for haemoglobinopathies in pregnancy
 Policy Statement All Southern Health patients will receive clinical care that reflects best practice and is based on the best available evidence. Index of chapters within background 1. Prevalence of haemoglobinopathies
Policy Statement All Southern Health patients will receive clinical care that reflects best practice and is based on the best available evidence. Index of chapters within background 1. Prevalence of haemoglobinopathies
Hemoglobinopathies NORMAL HEMOGLOBINS
 Hemoglobinopathies Millicent Sutton MD October 28, 2005 NORMAL HEMOGLOBINS Consist of 2 alpha chains and 2 non alpha chains Hb A = α2β2 Hb F= α 2γ2 Hb A2 = α2δ2 1 Hemoglobin Variants Altered the conformational
Hemoglobinopathies Millicent Sutton MD October 28, 2005 NORMAL HEMOGLOBINS Consist of 2 alpha chains and 2 non alpha chains Hb A = α2β2 Hb F= α 2γ2 Hb A2 = α2δ2 1 Hemoglobin Variants Altered the conformational
Anaemia in Pregnancy
 Anaemia in Pregnancy Definition :anaemia is a pathological condition in which the oxygen-carrying capacity of red blood cells is insufficient to meet the body needs. The WHO : haemoglobin concentration
Anaemia in Pregnancy Definition :anaemia is a pathological condition in which the oxygen-carrying capacity of red blood cells is insufficient to meet the body needs. The WHO : haemoglobin concentration
How to Write a Life Care Plan for a Child with Hemoglobinopathy
 How to Write a Life Care Plan for a Child with Hemoglobinopathy Tamar Fleischer, BSN, MSN, CNLCP & Mona Yudkoff, RN, MPH, CRRN, CNLCP BalaCare Solutions March 2018 St. Peterburg, Florida What is Hemoglobinopathy?
How to Write a Life Care Plan for a Child with Hemoglobinopathy Tamar Fleischer, BSN, MSN, CNLCP & Mona Yudkoff, RN, MPH, CRRN, CNLCP BalaCare Solutions March 2018 St. Peterburg, Florida What is Hemoglobinopathy?
Newborn Screening for Sickle Cell Disease in Africa: Public health meets reality
 13 th September 2016 Newborn Screening for Sickle Cell Disease in Africa: Public health meets reality Kwaku Ohene-Frempong. MD Children s Hospital of Philadelphia Sickle Cell Foundation of Ghana Disclosure
13 th September 2016 Newborn Screening for Sickle Cell Disease in Africa: Public health meets reality Kwaku Ohene-Frempong. MD Children s Hospital of Philadelphia Sickle Cell Foundation of Ghana Disclosure
Putting some hematology into Pediatric Hematology/Oncology: a review of Hemophilia and Sickle Cell Disease in the Pediatric Patient
 Putting some hematology into Pediatric Hematology/Oncology: a review of Hemophilia and Sickle Cell Disease in the Pediatric Patient Kristina Haley, DO March 10, 2012 Jovita Reyes Memorial Pediatric Hematology/Oncology
Putting some hematology into Pediatric Hematology/Oncology: a review of Hemophilia and Sickle Cell Disease in the Pediatric Patient Kristina Haley, DO March 10, 2012 Jovita Reyes Memorial Pediatric Hematology/Oncology
are associated with sickle cell disease and carriers: a study of patients from the southeastregion of Iran
 CXC chemokines CXCL1, CXCL9, CXCL10 and CXCL12 are associated with sickle cell disease and carriers: a study of patients from the southeastregion of Iran Mojgan Noroozi Karimabad Molecular Medicine Research
CXC chemokines CXCL1, CXCL9, CXCL10 and CXCL12 are associated with sickle cell disease and carriers: a study of patients from the southeastregion of Iran Mojgan Noroozi Karimabad Molecular Medicine Research
World-Wide Distribution of Hemoglobin S. Geographic distribution of hemoglobin S in the world
 Sickle Cell Disease Gerald M. Woods, MD Professor of Pediatrics Division Director, Hematology/Oncology/BMT Director of Sickle Cell Program Children s Mercy Hospitals and Clinics Disclaimer Member of DSMB
Sickle Cell Disease Gerald M. Woods, MD Professor of Pediatrics Division Director, Hematology/Oncology/BMT Director of Sickle Cell Program Children s Mercy Hospitals and Clinics Disclaimer Member of DSMB
Sickle Cell Disease 101. Objectives. What is SCD? 4/20/2016. Discuss the pathophysiology & genetics of Sickle Cell Disease (SCD).
.") Sickle Cell Disease 101 Jennifer Young, RN, MS, CPNP-AC Sickle Cell & Thalassemia Nurse Practitioner Nationwide Children s Hospital Objectives Discuss the pathophysiology & genetics of Sickle Cell Disease
Sickle Cell Disease 101 Jennifer Young, RN, MS, CPNP-AC Sickle Cell & Thalassemia Nurse Practitioner Nationwide Children s Hospital Objectives Discuss the pathophysiology & genetics of Sickle Cell Disease
Sickle Cell Anemia A Fictional Reconstruction Answer Key
 We have made it easy for you to find a PDF Ebooks without any digging. And by having access to our ebooks online or by storing it on your computer, you have convenient answers with sickle cell anemia a
We have made it easy for you to find a PDF Ebooks without any digging. And by having access to our ebooks online or by storing it on your computer, you have convenient answers with sickle cell anemia a
Biology 2C03: Genetics What is a Gene?
 Biology 2C03: Genetics What is a Gene? September 9 th, 2013 Model Organisms - E. coli - Yeast - Worms - Arabodopsis - Fruitflie - Mouse What is a Gene? - Define, recognize, describe and apply Mendel s
Biology 2C03: Genetics What is a Gene? September 9 th, 2013 Model Organisms - E. coli - Yeast - Worms - Arabodopsis - Fruitflie - Mouse What is a Gene? - Define, recognize, describe and apply Mendel s
O 2 O 2 O 2. Haemoglobin
 O 2 O 2 O 2 Haemoglobin O 2 O 2 O 2 98% travels in oxyhaemoglobin (in red blood cells) 2% is dissolved in plasma (compared to carbon dioxide, oxygen is relatively insoluble in plasma) O 2 is not very soluble
O 2 O 2 O 2 Haemoglobin O 2 O 2 O 2 98% travels in oxyhaemoglobin (in red blood cells) 2% is dissolved in plasma (compared to carbon dioxide, oxygen is relatively insoluble in plasma) O 2 is not very soluble
Interleukin-1ß and Interleukin-6 Genetic Polymorphisms and Sickle Cell Disease: An Egyptian Study
 Interleukin-1ß and Interleukin-6 Genetic Polymorphisms and Sickle Cell Disease: An Egyptian Study MONA EL-GHAMRAWY, MD, PROFESSOR OF PEDIATRICS & PEDIATRIC HEMATOLOGY, CAIRO UNIVERSITY melghamrawy95@gmail.com
Interleukin-1ß and Interleukin-6 Genetic Polymorphisms and Sickle Cell Disease: An Egyptian Study MONA EL-GHAMRAWY, MD, PROFESSOR OF PEDIATRICS & PEDIATRIC HEMATOLOGY, CAIRO UNIVERSITY melghamrawy95@gmail.com
Genetics of Thalassemia
 Genetics of Thalassemia Submitted by : Raya Samir Al- Hayaly Sura Zuhair Salih Saad Ghassan Al- Dulaimy Saad Farouq Kassir Sama Naal Salouha Zahraa Jasim Al- Aarajy Supervised by : Dr. Kawkab Adris Mahmod
Genetics of Thalassemia Submitted by : Raya Samir Al- Hayaly Sura Zuhair Salih Saad Ghassan Al- Dulaimy Saad Farouq Kassir Sama Naal Salouha Zahraa Jasim Al- Aarajy Supervised by : Dr. Kawkab Adris Mahmod
COEXISTENCE OF β-thalassemia AND POLYCYTHEMIA VERA: A CHICKEN-AND-EGG DEBATE?
 COEXISTENCE OF β-thalassemia AND POLYCYTHEMIA VERA: A CHICKEN-AND-EGG DEBATE? M. DE SLOOVERE (1), L. HARLET (2), S. VAN STEENWEGHEN (3), E. MOREAU (1), D. DE SMET (1) (1) DEPARTMENT OF LABORATORY MEDICINE,
COEXISTENCE OF β-thalassemia AND POLYCYTHEMIA VERA: A CHICKEN-AND-EGG DEBATE? M. DE SLOOVERE (1), L. HARLET (2), S. VAN STEENWEGHEN (3), E. MOREAU (1), D. DE SMET (1) (1) DEPARTMENT OF LABORATORY MEDICINE,
Genetic Modifiers of Sickle Cell Disease Severity. Kunle Adekile, MD, PhD Professor Department of Pediatrics Kuwait University
 Genetic Modifiers of Sickle Cell Disease Severity Kunle Adekile, MD, PhD Professor Department of Pediatrics Kuwait University Outline Hb Molecule and Genetic control of globin synthesis Pathophysiology
Genetic Modifiers of Sickle Cell Disease Severity Kunle Adekile, MD, PhD Professor Department of Pediatrics Kuwait University Outline Hb Molecule and Genetic control of globin synthesis Pathophysiology
Hydroxyurea Treatment for Sickle Cell Disease
 Hydroxyurea Treatment for Sickle Cell Disease Before Hydroxyurea After Hydroxyurea Hydroxyurea Treatment for Sickle Cell Disease This document is not intended to take the place of the care and attention
Hydroxyurea Treatment for Sickle Cell Disease Before Hydroxyurea After Hydroxyurea Hydroxyurea Treatment for Sickle Cell Disease This document is not intended to take the place of the care and attention
Batool Emad. Marah Karablieh. - Nayef Karadsheh
 4 4 1 P a g e Batool Emad Marah Karablieh - Nayef Karadsheh ***Topics that will be discussed in this Lecture: 1) Globin gene organization 2) Hemoglobinopathies 3) HbS (sickle cell disease) 4) HbC and HbSC
4 4 1 P a g e Batool Emad Marah Karablieh - Nayef Karadsheh ***Topics that will be discussed in this Lecture: 1) Globin gene organization 2) Hemoglobinopathies 3) HbS (sickle cell disease) 4) HbC and HbSC
2018 Biochemistry 110 California Institute of Technology Lecture 7: Molecular Disease: Sickle-Cell Anemia
 2018 Biochemistry 110 California Institute of Technology Lecture 7: Molecular Disease: Sickle-Cell Anemia James Herrick (1861-1954) Phase-Contrast microscopy image of Sickle Cells intermingled with erythrocytes.
2018 Biochemistry 110 California Institute of Technology Lecture 7: Molecular Disease: Sickle-Cell Anemia James Herrick (1861-1954) Phase-Contrast microscopy image of Sickle Cells intermingled with erythrocytes.
Sickle Cell Anemia. Sickle cell anemia is an inherited disorder of the blood which occurs when just one base pair substitution
 Rose Farrington and Rachel Nash BIOL 362 Lab M. Bulgarella Genetic Diseases 10/14/2008 Sickle Cell Anemia Introduction Sickle cell anemia is an inherited disorder of the blood which occurs when just one
Rose Farrington and Rachel Nash BIOL 362 Lab M. Bulgarella Genetic Diseases 10/14/2008 Sickle Cell Anemia Introduction Sickle cell anemia is an inherited disorder of the blood which occurs when just one
The Child with a Hematologic Alteration
 47 The Child with a Hematologic Alteration HELPFUL HINT Review the anatomy and physiology of the hematologic system in an anatomy and physiology textbook. MATCHING KEY TERMS Match the term with the correct
47 The Child with a Hematologic Alteration HELPFUL HINT Review the anatomy and physiology of the hematologic system in an anatomy and physiology textbook. MATCHING KEY TERMS Match the term with the correct
Atlantic Provinces Pediatric Hematology Oncology Network Réseau d Oncologie et Hématologie Pédiatrique des Provinces Atlantiques
 Atlantic Provinces Pediatric Hematology Oncology Network Réseau d Oncologie et Hématologie Pédiatrique des Provinces Atlantiques 5850/5980 University Avenue, PO Box 9700 Halifax, NS, B3K 6R8 1.902.470.7429
Atlantic Provinces Pediatric Hematology Oncology Network Réseau d Oncologie et Hématologie Pédiatrique des Provinces Atlantiques 5850/5980 University Avenue, PO Box 9700 Halifax, NS, B3K 6R8 1.902.470.7429
4 Fahed Al Karmi Sufian Alhafez Dr nayef karadsheh
 4 Fahed Al Karmi Sufian Alhafez Dr nayef karadsheh Genetic variants of hemoglobin Hemoglobinopathies (abnormal variants of hemoglobin) are divided into: 1. Structural abnormalities: Any change in the genes
4 Fahed Al Karmi Sufian Alhafez Dr nayef karadsheh Genetic variants of hemoglobin Hemoglobinopathies (abnormal variants of hemoglobin) are divided into: 1. Structural abnormalities: Any change in the genes
Below are the sections of the DNA sequences of a normal hemoglobin gene and the mutated gene that causes sickle cell disease.
 Sickle Cell Analysis Directions: Read the information below to complete the two tables. A person with sickle-cell disease has the genotype: Hb s Hb s. People who have this condition have two abnormal genes,
Sickle Cell Analysis Directions: Read the information below to complete the two tables. A person with sickle-cell disease has the genotype: Hb s Hb s. People who have this condition have two abnormal genes,
Tenth Visit posttest
 Test Code 10C Patient s name: Tenth Visit posttest Patient s birth date: Your name and relationship to patient: Today s date: 1. Which one of the medications listed below should every child with a sickle
Test Code 10C Patient s name: Tenth Visit posttest Patient s birth date: Your name and relationship to patient: Today s date: 1. Which one of the medications listed below should every child with a sickle
Year 2003 Paper two: Questions supplied by Tricia
 QUESTION 65 A 36-year-old man presents in a post-ictal state after an observed generalised seizure. Full blood investigation shows: haemoglobin 0 g/l [128-175] mean corpuscular volume (MCV) 106 fl [80-7]
QUESTION 65 A 36-year-old man presents in a post-ictal state after an observed generalised seizure. Full blood investigation shows: haemoglobin 0 g/l [128-175] mean corpuscular volume (MCV) 106 fl [80-7]
Extra Notes 3. Warm. In the core (center) of the body, where the temperature is 37 C.
 of the body, where the temperature is 37 C.") Extra Notes 3 *The numbers of the slides are according to the last year slides. Slide 33 Autoimmune hemolytic anemia : Abnormal circulating antibodies that target normal antigen on the RBC and cause lysis.
Extra Notes 3 *The numbers of the slides are according to the last year slides. Slide 33 Autoimmune hemolytic anemia : Abnormal circulating antibodies that target normal antigen on the RBC and cause lysis.
CURRENT RESEARCH STUDIES
 CURRENT RESEARCH STUDIES SCAGO SICKLE CELL RESEARCH DAY MAY 12, 2018 REBECCA LEROUX RN, BSCN, CCRP RED BLOOD CELL DISORDERS PROGRAM, UNIVERSITY HEALTH NETWORK MANUELA MERELLES-PULCINI RN, BSCN, MSN, CCRP
CURRENT RESEARCH STUDIES SCAGO SICKLE CELL RESEARCH DAY MAY 12, 2018 REBECCA LEROUX RN, BSCN, CCRP RED BLOOD CELL DISORDERS PROGRAM, UNIVERSITY HEALTH NETWORK MANUELA MERELLES-PULCINI RN, BSCN, MSN, CCRP
Hydroxyurea and Transfusion Therapy for the Treatment of Sickle Cell Disease
 Hydroxyurea and Transfusion Therapy for the Treatment of Sickle Cell Disease A Pocket Guide for the Clinician Susan E. Creary, MD, MSc 1 John J. Strouse, MD, PhD 2 1 The Ohio State University School of
Hydroxyurea and Transfusion Therapy for the Treatment of Sickle Cell Disease A Pocket Guide for the Clinician Susan E. Creary, MD, MSc 1 John J. Strouse, MD, PhD 2 1 The Ohio State University School of
DIC. Disseminated intravascular coagulation, is a life threatening pathological process in which clotting factors are abnormally activated.
 Miss. kamlah 1 DIC Disseminated intravascular coagulation, is a life threatening pathological process in which clotting factors are abnormally activated. Resulting in wide spread of clot formation in the
Miss. kamlah 1 DIC Disseminated intravascular coagulation, is a life threatening pathological process in which clotting factors are abnormally activated. Resulting in wide spread of clot formation in the
Hematological profile among Sudanese patients with sickle cell anemia
 EUROPEAN ACADEMIC RESEARCH Vol. III, Issue 4/ July 2015 ISSN 2286-4822 www.euacademic.org Impact Factor: 3.4546 (UIF) DRJI Value: 5.9 (B+) Hematological profile among Sudanese patients with sickle cell
EUROPEAN ACADEMIC RESEARCH Vol. III, Issue 4/ July 2015 ISSN 2286-4822 www.euacademic.org Impact Factor: 3.4546 (UIF) DRJI Value: 5.9 (B+) Hematological profile among Sudanese patients with sickle cell
Blood Cell Identification Graded
 Blood Cell Identification Graded Case History The patient is a 20-year-old female with sickle cell disease who presents with bilateral leg pain for 3 days. She is scheduled to have bilateral hip and leg
Blood Cell Identification Graded Case History The patient is a 20-year-old female with sickle cell disease who presents with bilateral leg pain for 3 days. She is scheduled to have bilateral hip and leg
Haemoglobinophaties EBMT 2011 Data Manager session
 Haemoglobinophaties EBMT 2011 Data Manager session Presentation plan Biological characteristics Clinical characteristics Transplant resuts What is different From transplant in malignancies Between Thalassemia
Haemoglobinophaties EBMT 2011 Data Manager session Presentation plan Biological characteristics Clinical characteristics Transplant resuts What is different From transplant in malignancies Between Thalassemia
Year 2004 Paper two: Questions supplied by Megan 1
 Year 2004 Paper two: Questions supplied by Megan 1 QUESTION 93 A 16yo adolescent male presents with lethargy and lower respiratory tract infection. Physical examination shows him to be febrile, icteric
Year 2004 Paper two: Questions supplied by Megan 1 QUESTION 93 A 16yo adolescent male presents with lethargy and lower respiratory tract infection. Physical examination shows him to be febrile, icteric
General Characterisctics
 Anemia General Characterisctics Definition: anemia is a decrease in red blood cells. Happens due to underproduction, increased destruction or loss of red cells. Diagnosis of anemia: Hgb < 135 (men) Hgb
Anemia General Characterisctics Definition: anemia is a decrease in red blood cells. Happens due to underproduction, increased destruction or loss of red cells. Diagnosis of anemia: Hgb < 135 (men) Hgb
270,000,000 hemoglobin units are. hemoglobin has 4 heme units; 2 α and 2 β units. Active site of a heme unit has an Iron ion
 RBC strange shape a biconcave disc that is round and flat RBC has no nucleus. The nucleus is extruded from the cell as it matures. An RBC can change shape to an amazing extent, without breaking, as it
RBC strange shape a biconcave disc that is round and flat RBC has no nucleus. The nucleus is extruded from the cell as it matures. An RBC can change shape to an amazing extent, without breaking, as it
Good afternoon and thank you for joining us today as we discuss hydroxyurea for the treatment of sickle cell disease. Dr. Emily Meier is a pediatric
 Good afternoon and thank you for joining us today as we discuss hydroxyurea for the treatment of sickle cell disease. Dr. Emily Meier is a pediatric hematologist at the Indiana Hemophilia & Thrombosis
Good afternoon and thank you for joining us today as we discuss hydroxyurea for the treatment of sickle cell disease. Dr. Emily Meier is a pediatric hematologist at the Indiana Hemophilia & Thrombosis
Other labs 4/24/2012. N 24: Pediatric Hematological Alterations & Cancer Intro. Cabrillo College ADN Program C. Madsen RN, MSN 1.
 Evaluation of CBC Evaluate type of WBCs Reticulocyte count RBC size, shape, color MCV: size RBC color (hypo or normo chromic) Mean corpuscular hemoglobin concentration (MCHC) Mean corpuscular hemoglobin
Evaluation of CBC Evaluate type of WBCs Reticulocyte count RBC size, shape, color MCV: size RBC color (hypo or normo chromic) Mean corpuscular hemoglobin concentration (MCHC) Mean corpuscular hemoglobin
SICKLE CELL DISEASE- AN AYURVEDIC PERSPECTIVE
 INTERNATIONAL AYURVEDIC MEDICAL JOURNAL International Ayurvedic Medical Journal, (ISSN: 2320 5091) (March, 2017) 5 (3) SICKLE CELL DISEASE- AN AYURVEDIC PERSPECTIVE Sickle cell disease or Sickle cell anemia
INTERNATIONAL AYURVEDIC MEDICAL JOURNAL International Ayurvedic Medical Journal, (ISSN: 2320 5091) (March, 2017) 5 (3) SICKLE CELL DISEASE- AN AYURVEDIC PERSPECTIVE Sickle cell disease or Sickle cell anemia
HEMOGLOBINOPATHIES LECTURE OUTLINE. An overview of the structure of hemoglobin. Different types of hemoglobin. Definition of hemoglobinopathies
 Slide 1 HEOGLOBINOPATHIES Slide 2 LETURE OUTLINE An overview of the structure of hemoglobin. Different types of hemoglobin. Definition of hemoglobinopathies Sickle ell Disease and Hemoglobin Slide 3 HEOGLOBIN
Slide 1 HEOGLOBINOPATHIES Slide 2 LETURE OUTLINE An overview of the structure of hemoglobin. Different types of hemoglobin. Definition of hemoglobinopathies Sickle ell Disease and Hemoglobin Slide 3 HEOGLOBIN
Hemoglobinopathies Diagnosis and management
 Hemoglobinopathies Diagnosis and management Morgan L. McLemore, M.D. Hematology/Leukemia Department of Hematology and Oncology Winship Cancer Institute at Emory University mlmclem@emory.edu Disclosures
Hemoglobinopathies Diagnosis and management Morgan L. McLemore, M.D. Hematology/Leukemia Department of Hematology and Oncology Winship Cancer Institute at Emory University mlmclem@emory.edu Disclosures
Human Cell Diagram, Parts, Pictures, Structure and Functions
 Human Cell Diagram, Parts, Pictures, Structure and Functions The cell is the basic functional in a human meaning that it is a self-contained and fully operational living entity. Humans are multicellular
Human Cell Diagram, Parts, Pictures, Structure and Functions The cell is the basic functional in a human meaning that it is a self-contained and fully operational living entity. Humans are multicellular
SCD Advocacy Talking Points!
 "#$!%&'()*)+!,!-././-1!! SCD Advocacy Talking Points! * 23!4*'3!53*673&!84*8!#*59:(679*!#495&637;!9
"#$!%&'()*)+!,!-././-1!! SCD Advocacy Talking Points! * 23!4*'3!53*673&!84*8!#*59:(679*!#495&637;!9
Dr.Abdolreza Afrasiabi
 Dr.Abdolreza Afrasiabi Thalassemia & Heamophilia Genetic Reaserch Center Shiraz Medical University Hemoglobin tetramer Hemoglobin Structure % A 1 α 2 β 2 94-97% A 2 α 2 δ 2 2.5% A 1C α 2 (β-n-glucose)
Dr.Abdolreza Afrasiabi Thalassemia & Heamophilia Genetic Reaserch Center Shiraz Medical University Hemoglobin tetramer Hemoglobin Structure % A 1 α 2 β 2 94-97% A 2 α 2 δ 2 2.5% A 1C α 2 (β-n-glucose)
Index. Note: Page numbers of article titles are in boldface type.
 Note: Page numbers of article titles are in boldface type. A Acute lymphoblastic leukemia, in India, 439 440 pediatric, global approach to, 420 424 core resources in low- and middle-income countries, 423
Note: Page numbers of article titles are in boldface type. A Acute lymphoblastic leukemia, in India, 439 440 pediatric, global approach to, 420 424 core resources in low- and middle-income countries, 423
Around million aged erythrocytes/hour are broken down.
 Anemia Degradation ofheme Around 100 200 million aged erythrocytes/hour are broken down. The degradation process starts in reticuloendothelial cells in the spleen, liver, and bone marrow. [1] The tetrapyrrole
Anemia Degradation ofheme Around 100 200 million aged erythrocytes/hour are broken down. The degradation process starts in reticuloendothelial cells in the spleen, liver, and bone marrow. [1] The tetrapyrrole
namib la UnIVERSITY OF SCIEnCE AnD TECHnOLOGY FACULTY OF HEALTH AND APPLIED SCIENCES DEPARTMENT OF HEALTH SCIENCES
 namib la UnIVERSITY OF SCIEnCE AnD TECHnOLOGY FACULTY OF HEALTH AND APPLIED SCIENCES DEPARTMENT OF HEALTH SCIENCES QUALIFICATION: BACHELOR OF BIOMEDICAL SCIENCES QUALIFICATION CODE: SOBBMS LEVEL: 6 COURSE
namib la UnIVERSITY OF SCIEnCE AnD TECHnOLOGY FACULTY OF HEALTH AND APPLIED SCIENCES DEPARTMENT OF HEALTH SCIENCES QUALIFICATION: BACHELOR OF BIOMEDICAL SCIENCES QUALIFICATION CODE: SOBBMS LEVEL: 6 COURSE
SICKLE CELL BROCHURE
 SICKLE CELL BROCHURE SICKLE CELL DIESEASE According to CDC, Sickle cell disease (SCD) is a group of inherited red blood cell disorders. Healthy red blood cells are round and SCD C -shaped farm tool called
SICKLE CELL BROCHURE SICKLE CELL DIESEASE According to CDC, Sickle cell disease (SCD) is a group of inherited red blood cell disorders. Healthy red blood cells are round and SCD C -shaped farm tool called
Thalassemias:general aspects and molecular pathology
 Thalassemias:general aspects and molecular pathology Prof. Renzo Galanello Pediatric Clinic 2 University of Cagliari Ospedale Regionale Microcitemie-ASL8 HEMOGLOBINOPATHIES CLASSIFICATION Structurally
Thalassemias:general aspects and molecular pathology Prof. Renzo Galanello Pediatric Clinic 2 University of Cagliari Ospedale Regionale Microcitemie-ASL8 HEMOGLOBINOPATHIES CLASSIFICATION Structurally
Congenital Hemolytic Anemias
 Anemia (4) Congenital Hemolytic Anemias 20.02.2019 Abdallah Abbadi.MD.FRCP. FRCPath Feras Fararjeh MD Congenital Hemolytic Anemias: Subtypes 1 Membrane defects: HS 2 Enzymopathies: G6PD Deficiency, PK
Anemia (4) Congenital Hemolytic Anemias 20.02.2019 Abdallah Abbadi.MD.FRCP. FRCPath Feras Fararjeh MD Congenital Hemolytic Anemias: Subtypes 1 Membrane defects: HS 2 Enzymopathies: G6PD Deficiency, PK
Approach to a pale child
 Approach to a pale child Dr. Dafalla Ahmed Babiker Jazan university objectives Definition of anemia Classification and causes Important points in history and physical examination Investigations. Definition
Approach to a pale child Dr. Dafalla Ahmed Babiker Jazan university objectives Definition of anemia Classification and causes Important points in history and physical examination Investigations. Definition
C. treatment with Desferal (deferoxamine mesylate USP, iron-chelating agent)
") HEMOLYTIC ANEMIAS Single choice tests 1. Select the clinical manifestation that is not characteristic for the hemolytic crisis: A. decrease of the red blood cell count B. reticulocytosis C. jaundice D.
HEMOLYTIC ANEMIAS Single choice tests 1. Select the clinical manifestation that is not characteristic for the hemolytic crisis: A. decrease of the red blood cell count B. reticulocytosis C. jaundice D.
Hydroxyurea Treatment for Sickle Cell Disease
 Hydroxyurea Treatment for Sickle Cell Disease Hydroxyurea Treatment for Sickle Cell Disease 1 This document is not intended to take the place of the care and attention of your personal physician. Our aim
Hydroxyurea Treatment for Sickle Cell Disease Hydroxyurea Treatment for Sickle Cell Disease 1 This document is not intended to take the place of the care and attention of your personal physician. Our aim
HPLC profile of sickle cell disease in central India
 Original Research Article HPLC profile of sickle cell disease in central India Shweta P. Bijwe * Department of Pathology, IGGMC, Nagpur, Maharashtra, India * Corresponding author email: dr.shwetabijwe@gmail.com
Original Research Article HPLC profile of sickle cell disease in central India Shweta P. Bijwe * Department of Pathology, IGGMC, Nagpur, Maharashtra, India * Corresponding author email: dr.shwetabijwe@gmail.com
Compassionate-use Experience With Voxelotor (GBT440) for Patients With Severe Sickle Cell Disease (SCD) and Life-Threatening Comorbidities
 for Patients With Severe Sickle Cell Disease (SCD) and Life-Threatening Comorbidities") Compassionate-use Experience With Voxelotor (GBT440) for Patients With Severe Sickle Cell Disease (SCD) and Life-Threatening Comorbidities Gershwin Blyden, MD 1, Kenneth R. Bridges, MD 2, Lanetta Bronté,
Compassionate-use Experience With Voxelotor (GBT440) for Patients With Severe Sickle Cell Disease (SCD) and Life-Threatening Comorbidities Gershwin Blyden, MD 1, Kenneth R. Bridges, MD 2, Lanetta Bronté,
Biance Rowe Chris Hani Baragwanath Hospital Paediatric Haematology Oncology University of the Witwatersrand
 Biance Rowe Chris Hani Baragwanath Hospital Paediatric Haematology Oncology University of the Witwatersrand SCD affects 20-25 million people globally 12-15 million in Africa 300 000 children with SCD
Biance Rowe Chris Hani Baragwanath Hospital Paediatric Haematology Oncology University of the Witwatersrand SCD affects 20-25 million people globally 12-15 million in Africa 300 000 children with SCD
Education Visit #1 *** All Sickle Cell Patients*** from A Parent s Handbook for Sickle Cell Disease Booklet.
 Education Visit #1 *** All Sickle Cell Patients*** Step 1: Administer Pretest A. Step 2: Education Watch DVD: Education Visit #1 For All Patients Handout So You Have Sickle Cell Disorder Handout Infection
Education Visit #1 *** All Sickle Cell Patients*** Step 1: Administer Pretest A. Step 2: Education Watch DVD: Education Visit #1 For All Patients Handout So You Have Sickle Cell Disorder Handout Infection
Guidelines for Shared Care Centres and Community Staff
 Reference: CG1411 Written by: Dr Jenny Welch Peer reviewer Dr Jeanette Payne Approved: September 2015 Approved by D&TC: 10 th July 2015 Review Due: September 2018 Intended Audience This document contains
Reference: CG1411 Written by: Dr Jenny Welch Peer reviewer Dr Jeanette Payne Approved: September 2015 Approved by D&TC: 10 th July 2015 Review Due: September 2018 Intended Audience This document contains
Educational Items Section
 Atlas of Genetics and Cytogenetics in Oncology and Haematology OPEN ACCESS JOURNAL AT INIST-CNRS Educational Items Section Hemoglobin genes; Sickle-cell anemia - Thalassemias Jean-Loup Huret, Xavier Troussard
Atlas of Genetics and Cytogenetics in Oncology and Haematology OPEN ACCESS JOURNAL AT INIST-CNRS Educational Items Section Hemoglobin genes; Sickle-cell anemia - Thalassemias Jean-Loup Huret, Xavier Troussard
HU: Myths and Facts. Melanie Kirby Associate Professor of Paediatrics
 HU: Myths and Facts Melanie Kirby Associate Professor of Paediatrics SACGO Hamilton, Ontario March 5, 2016 Declaration of Disclosure I have no actual or potential conflict of interest in relation to this
HU: Myths and Facts Melanie Kirby Associate Professor of Paediatrics SACGO Hamilton, Ontario March 5, 2016 Declaration of Disclosure I have no actual or potential conflict of interest in relation to this
THE KENYA POLYTECHNIC UNIVERSITY COLLEGE
 THE KENYA POLYTECHNIC UNIVERSITY COLLEGE SCHOOL OF HEALTH SCIENCES AND TECHNOLOGY DEPARTMENT OF BIOMEDICAL LABORATORY SCIENCES AND TECHNOLOGY DIPLOMA IN MEDICAL LABORATORY SCIENCE END OF YEAR 1 EXAMINATION
THE KENYA POLYTECHNIC UNIVERSITY COLLEGE SCHOOL OF HEALTH SCIENCES AND TECHNOLOGY DEPARTMENT OF BIOMEDICAL LABORATORY SCIENCES AND TECHNOLOGY DIPLOMA IN MEDICAL LABORATORY SCIENCE END OF YEAR 1 EXAMINATION
Bill S-211: Recognizing June 19 as the Canadian Sickle Cell Awareness Day
 Bill S-211: Recognizing June 19 as the Canadian Sickle Cell Awareness Day Sickle Cell Disease (SCD) is the most common genetic disease in the world. According to the World Health Organization (WHO) estimates,
Bill S-211: Recognizing June 19 as the Canadian Sickle Cell Awareness Day Sickle Cell Disease (SCD) is the most common genetic disease in the world. According to the World Health Organization (WHO) estimates,
What is Thalassaemia?
 What is Thalassaemia? Introduction The thalassaemias are a diverse group of genetic blood diseases characterized by absent or decreased production of normal hemoglobin, resulting in a microcytic anemia
What is Thalassaemia? Introduction The thalassaemias are a diverse group of genetic blood diseases characterized by absent or decreased production of normal hemoglobin, resulting in a microcytic anemia
JIHS. The Journal of Integrated Health Sciences. Role of Hydroxyurea In Management of Sickle Cell Disease. Original Article
 JIHS Available online at www.jihs.in The Journal of Integrated Health Sciences Role of Hydroxyurea In Management of Sickle Cell Disease Rakesh Amroliwala 1, Vishruti Gandhi* 2, Niyati Parikh 3, Arti Gupta
JIHS Available online at www.jihs.in The Journal of Integrated Health Sciences Role of Hydroxyurea In Management of Sickle Cell Disease Rakesh Amroliwala 1, Vishruti Gandhi* 2, Niyati Parikh 3, Arti Gupta
Hydroxyurea in Pediatric Patients With Sickle Cell Disease: What Nurses Need to Know
 614962JPOXXX10.1177/1043454215614962Journal of Pediatric Oncology NursingRees research-article2015 Article Hydroxyurea in Pediatric Patients With Sickle Cell Disease: What Nurses Need to Know Journal of
614962JPOXXX10.1177/1043454215614962Journal of Pediatric Oncology NursingRees research-article2015 Article Hydroxyurea in Pediatric Patients With Sickle Cell Disease: What Nurses Need to Know Journal of
Genetic Modulation on the Phenotypic Diversity of Sickle Cell Disease
 Genetic Modulation on the Phenotypic Diversity of Sickle Cell Disease Malay B. Mukherjee Abstract Sickle cell hemoglobin is a β chain structural variant where valine is substituted for glutamic acid in
Genetic Modulation on the Phenotypic Diversity of Sickle Cell Disease Malay B. Mukherjee Abstract Sickle cell hemoglobin is a β chain structural variant where valine is substituted for glutamic acid in
Arginine as an Example of a Conditionally Essential Nutrient: Sickle Cell Disease & Trauma Claudia R. Morris MD, FAAP
 Arginine as an Example of a Conditionally Essential Nutrient: Sickle Cell Disease & Trauma Claudia R. Morris MD, FAAP Examining Special Nutritional Requirements in Disease States, A Workshop April 1, 2018
Arginine as an Example of a Conditionally Essential Nutrient: Sickle Cell Disease & Trauma Claudia R. Morris MD, FAAP Examining Special Nutritional Requirements in Disease States, A Workshop April 1, 2018
Perioperative Management of Patients with Sickle Cell Disease
 Perioperative Management of Patients with Sickle Cell Disease November 29 th, 2012 David Vivas, MD Case - History CC: RLQ pain HPI: 14 y/o female with h/o Hg SC presented to ED with 4 days h/o abdominal
Perioperative Management of Patients with Sickle Cell Disease November 29 th, 2012 David Vivas, MD Case - History CC: RLQ pain HPI: 14 y/o female with h/o Hg SC presented to ED with 4 days h/o abdominal
1 Kattamis et al. Growth of Children with Thalassemia: Effect of Different Transfusion Regimens. Archives of
 Objectives Sickle Cell Anemia and Thalassemia: Transplantation Provide overview of hemoglobinopathies: Sickle cell disease and Thalassemia Discuss approaches to therapy Review recent registry collaboration
Objectives Sickle Cell Anemia and Thalassemia: Transplantation Provide overview of hemoglobinopathies: Sickle cell disease and Thalassemia Discuss approaches to therapy Review recent registry collaboration
Anemia (3).ms4.25.Oct.15 Hemolytic Anemia. Abdallah Abbadi
.ms4.25.Oct.15 Hemolytic Anemia. Abdallah Abbadi") Anemia (3).ms4.25.Oct.15 Hemolytic Anemia Abdallah Abbadi Case 3 24 yr old female presented with anemia syndrome and jaundice. She was found to have splenomegaly. Hb 8, wbc 12k, Plt 212k, retics 12%, LDH
Anemia (3).ms4.25.Oct.15 Hemolytic Anemia Abdallah Abbadi Case 3 24 yr old female presented with anemia syndrome and jaundice. She was found to have splenomegaly. Hb 8, wbc 12k, Plt 212k, retics 12%, LDH
Health Maintenance and Education for Children and Adults
 Health Maintenance and Education for Children and Adults Richard Ward, MSc, MRCP, FRCPath Director, Red Blood Cell Disorders Program, UHN Assistant Professor, Hematology, University of Toronto Chair, Canadian
Health Maintenance and Education for Children and Adults Richard Ward, MSc, MRCP, FRCPath Director, Red Blood Cell Disorders Program, UHN Assistant Professor, Hematology, University of Toronto Chair, Canadian
Hemoglobin and anemia BCH 471
 Hemoglobin and anemia BCH 471 OBJECTIVES Quantitative determination of hemoglobin in a blood sample. Hemoglobin structure Hemoglobin (Hb) is a porphyrin iron (II) protein in RBCs that transport oxygen
Hemoglobin and anemia BCH 471 OBJECTIVES Quantitative determination of hemoglobin in a blood sample. Hemoglobin structure Hemoglobin (Hb) is a porphyrin iron (II) protein in RBCs that transport oxygen
THE UNIVERSITY OF JORDAN FACULTY OF MEDICINE DEPARTMENT OF PATHOLOGY HEMOLYTIC ANEMIAS. Dr. Tariq Aladily
 THE UNIVERSITY OF JORDAN FACULTY OF MEDICINE DEPARTMENT OF PATHOLOGY HEMOLYTIC ANEMIAS Third year medical students First semester Faculty 2018/2019 of Medicine Hereditary Spherocytosis Intrinsic defects
THE UNIVERSITY OF JORDAN FACULTY OF MEDICINE DEPARTMENT OF PATHOLOGY HEMOLYTIC ANEMIAS Third year medical students First semester Faculty 2018/2019 of Medicine Hereditary Spherocytosis Intrinsic defects
Thalassaemia. What is thalassaemia? What causes thalassaemia? What are the different types of thalassaemia?
 Thalassaemia Thalassaemia is an inherited condition affecting the blood. There are different types, which vary from a mild condition with no symptoms, to a serious or lifethreatening condition. For the
Thalassaemia Thalassaemia is an inherited condition affecting the blood. There are different types, which vary from a mild condition with no symptoms, to a serious or lifethreatening condition. For the
Diseases Of The Blood
 Diseases Of The Blood DR. Associate Professor Of Pathology Faculty Of Medicine Ain Shams University Red Blood Cells and Anemia RBC=4-6 million/mm 2 Hb=12-18 g/dl Oxygen Carrying Molecule Hemoglobin Tetramer:
Diseases Of The Blood DR. Associate Professor Of Pathology Faculty Of Medicine Ain Shams University Red Blood Cells and Anemia RBC=4-6 million/mm 2 Hb=12-18 g/dl Oxygen Carrying Molecule Hemoglobin Tetramer:
Utility of hemoglobin electrophoresis to detect hemoglobinopathies in adults not presenting with hematological problems
 Original Research Article Utility of hemoglobin electrophoresis to detect hemoglobinopathies in adults not presenting with hematological problems G. J. Vani Padmaja 1*, S. S. S. Quadri 1, O. Shravan Kumar
Original Research Article Utility of hemoglobin electrophoresis to detect hemoglobinopathies in adults not presenting with hematological problems G. J. Vani Padmaja 1*, S. S. S. Quadri 1, O. Shravan Kumar
HAEMOGLOBINOPATHIES. Editing file. References: 436 girls & boys slides 435 teamwork slides. Color code: Important. Extra.
 HAEMOGLOBINOPATHIES Objectives: normal structure and function of haemoglobin. how the globin components of haemoglobin change during development, and postnatally. the mechanisms by which the thalassaemias
HAEMOGLOBINOPATHIES Objectives: normal structure and function of haemoglobin. how the globin components of haemoglobin change during development, and postnatally. the mechanisms by which the thalassaemias
FUNCTIONS OF HEMOGLOBIN:
 HEMOGLOBIN: Conjugated protein Simple protein combined with a non-protein substance Hemoglobin HEME +GLOBIN nonprotein substance HEME( prosthetic group) Red colour of blood is due to Hb in RBCs Normal
HEMOGLOBIN: Conjugated protein Simple protein combined with a non-protein substance Hemoglobin HEME +GLOBIN nonprotein substance HEME( prosthetic group) Red colour of blood is due to Hb in RBCs Normal