World-Wide Distribution of Hemoglobin S. Geographic distribution of hemoglobin S in the world
|
|
|
- Candace Montgomery
- 6 years ago
- Views:
Transcription
1 Sickle Cell Disease Gerald M. Woods, MD Professor of Pediatrics Division Director, Hematology/Oncology/BMT Director of Sickle Cell Program Children s Mercy Hospitals and Clinics Disclaimer Member of DSMB for MAST
2 Objectives To become acquainted with sickle cell center expertise in caring for patients with sickle cell disease To become familiar with the Newborn screening results To understand the role of the primary care provider in the care of patients with Sickle Cell Disease To become familiar with clinical complications in sickle cell children and adolescents Sickle Cell Disease How Common is it? In the US Pediatric population the prevalence of Diabetes Mellitus 1:2,500 Acute Leukemia 1:2,880 Cystic Fibrosis 1:2,940 Muscular Dystrophy 1:5,000 Phenylketonuria (PKU) 1:10,000 Galactosemia 1:17,000
3 SCD Incidence In the US, Sickle Cell Disease affects 1 in every 400 African Americans 1 in 1,000-1,4001,400 Hispanics 1 in 3,000 Native Americans 1 in 60,000 Whites (1993, AFHCPR data) SCD Incidence Disease Impact in our Country Most common genetic defect in the US (autosomal recessive) 100,000 people with SCD in the US 5,000 affected infants born in US each year Hb SS 65% Hb SC 25% Hb Sβ + Thalassemia 8% Hb Sβ 0 Thalassemia 2%




4 World-Wide Distribution of Hemoglobin S Geographic distribution of hemoglobin S in the world




5 Why Do Cells Sickle? Point of mutation on β-globin chain of hemoglobin: Glutamic acid is substituted for valine Allowing the polymerization of sickle hemoglobin when deoxygenated Normal vs. Sickle Red Cells Normal Disc-shapedshaped Deformable Life span of 120 days Sickle Sickle-shaped Rigid Life span of 20 days or less
6 Hemoloysis and Vaso-occlusion Hemoloysis: The anemia in SCD is caused by hemolysis, and the degree of anemia varies widely between patients. The production of red cells by the bone marrow increases dramatically, but is unable to keep pace with the destruction. Vaso-occlusion: occlusion: Occurs when the rigid sickle shaped cells fail to move through the small blood vessels, blocking local blood flow to a microscopic region of tissue. Amplified many times, these episodes produce tissue hypoxia. The result is pain, and often damage to organs. Sickle Cell Manifestations Acute Manifestations Bacterial sepsis or meningitis* Recurrent vaso-occlusive occlusive pain (dactylitis, musculoskeletal or abdominal pain) Splenic sequestration* Aplastic crisis* Acute chest syndrome* Stroke* Priapism Hematuria, including gp papillary p necrosis *Potential cause of mortality Chronic Manifestations Anemia Jaundice Splenomegaly Functional asplenia Cardiomegaly and functional murmurs Hyposthenuria and enuresis Proteinemia Cholelithiasis Delayed grown and sexual maturation Restrictive lung disease* Pulmonary hypertension* Avascular necrosis Proliferative retinopathy Leg ulcers Transfusional hemosiderosis*



7 SCD Incidence Is Newborn Screening Necessary? All 50 states have a newborn screening program to detect sickle cell disease! 1 in 10 African Americans carry one gene (sickle trait)
8 Constituents of Hemoglobin Hemoglobin has 2 Alpha and 2 Beta chains Normal hemoglobin in the red cell consists of Hb A, Hb F, and Hb A2 Chromosome 11 encodes DNA sequences for beta (β), delta (δ) and gamma (λ) chains Chromosome 16 encodes DNA sequences of alpha (α) chains The beta variants such as Hb S, Hb C, and Hb D all occur from a mutation on Chromosome 11 causing a defective beta globin gene Normal Newborn Screening FA F = Fetal hemoglobin 80-90% A = Normal A hemoglobin 10-20%
9 Hemoglobin S Valine substitutes for glutamic acid on position 6 of Beta globin chain Sickle Hb polymerizes with deoxygenation causing distortion of RBC, leading to vasoocclusion and eventual hemolysis Newborn (NBS) Screening Results Hemoglobins are listed in order of percentage Sickle Cell Trait FAS Sickle Cell Disease FS Electrophoresis shows absence of α1, normal α2 2 and F; and presence of Hb S
10 Newborn Screening in Missouri All significant hemoglobinopathies are screened in Missouri Greater responsibility is placed on the physician of record Inform and educate parents Recommend additional appointments to a sickle cell expert All SCA infants should be started on prophylactic penicillin by the age of 2 months All parents are instructed to seek medical attention when there is a fever of > F Hemoglobin C Lysine substitutes for glutamic acid in position 6 of Beta globin chain (Chromosome 11) Higher frequency among Western Africa, Italy, Greece, Turkey, and the Middle East Hb C crystalizes within the red cell The presence of Hb C results in increase viscosity of the blood
11 Ctrait FAC CC hemoglobin FC NBS Results Shortened red cell survival (hemolysis) in HbCC SC disease FSC Sickling complications Beta Thalassemia Absence or decrease of one or more beta globin genes Decreased production of normal Hemoglobin A (Chromosome 11) Microcytosis Unbalance synthesis of alpha and beta globin chains Ineffective erythropoiesis and a hemolytic anemia
12 NBS Results Beta thal major: fatal in early childhood w/o PRBC transfusions Splenomegaly Significant red cell dyscrasia Beta thal intermedia: severe microcytosis; +/- PRBC HEP: Hb F, Hb A 2, Hb A is absent (Beta 0 ) or markedly (Beta + ) Beta Thalassemia Minor/Trait: it heterozygotes t for one abnormal beta globin gene Mild microcytic anemia HEP: + Hb A, slight Hb A2, normal or slightly Hb F and RBC count Alpha Thalassemia Loss of > one alpha globin genes (Chromosome 16) A normal person has four alpha genes (αα/αα) Alpha thal major: lack of all four alpha genes Hydrops fetalis; usually fatal in utero (--/--) Hemoglobin H disease: loss of three alpha genes Moderately severe hemolytic anemia (-α/--) Alpha thal trait: two alpha genes lost Mild anemia with microcytosis; resembles Fe deficiency; normal A2 (α-/α-) or (-- --/αα)
13 Hemoglobin Bart s Silent carrier alpha thal: one alpha gene lost Not clinically significant Hb Bart s on newborn screen (α-/αα) Hb Bart s in cord blood sample may indicate the number of alpha genes lost <5% = most likely one gene deletion; silent carrier alpha thal, (-α/αα) 5-10% = alpha thal minor, loss of two alpha genes (-α/-α) or (-- --/αα) >10% (usually 15-20%) = more severe form of alpha thal; further testing is indicated (--/-α) Newborn Screening Results Confirmation of NBS by HEP must be done Genetic counseling for traits by PCP, Sickle Cell Foundation or hematologist Absence of Hemoglobin A1 confirms it is NOT sickle cell trait : info on NBS results Hb F modifies severity of SCD Alpha thalassemia trait modifies SCA severity
14 Newborn Screening Results FS (Sickle Cell Anemia Hb SS) FS (Sickle beta-0 Thalassemia, no beta globin) FS (Sickle Hereditary Persistence of Fetal Hemoglobin HPFH) FSC (Hemoglobin SC Disease, Hb SC) FSA or FS (Sickle beta -+ Thalassemia, reduced beta- globin) FC (Hemoglobin C Disease, may have hemolysis) FAS (Sickle Cell Trait, must do genetic counseling) FA (Normal newborn screen) FAX or FX (Abnormal hemoglobin present, MUST have confirmatory testing done) F (Homozygous beta Thalassemia) Significant Causes of Morbidity/Mortality in Children with Sickle Cell Disease Aplastic Crisis Splenic Sequestration Stroke Acute Chest Syndrome Painful Episodes Serious Infection
15 Role of the Primary Care Physician ENCOURAGE COMPREHENSIVE CARE AND MEDICAL HOME Check the Newborn Screen Results!!! Start PCN 125mg BID if suspect SCA Refer to hematology Offer genetic counseling for AS, AC, AD Impaired urine concentration AS Gross Hematuria AS Splenic infarct at high altitudes AS Rhabdomyolysis during prolonged strenuous exercise - AS Fever and Infection Fever > 38.5 C (101.5 F) is an EMERGENCY 33% <5y Hb SS will become septic without PCN prophylaxis Basic laboratory evaluation (consider): CBC with differential and reticulocyte count, blood, urine, and throat cultures, urinalysis, chest x-ray Indications for hospitalization & IV antibiotics Child appears ill Any temperature > 40 C Abnormal laboratory values Start t IV antibiotics IMMEDIATELY if child appears ill or temperature > 40 C (DO NOT WAIT FOR LABS)
16 Hyposplenism Functional reduction of splenic activity Intrasplenic sickling altered circulation splenomegaly progressive fibrosis autosplenectomy May see Howell-Jolly bodies on smear Infection risk x more likely to develop overwhelming pneumococcal and H. influenzae sepsis/meningitis Also linked to increased risk of gram-negative enteric organisms and Salmonella Greatest risk is during the first 5 years of life!! Current Recommendations Antibiotic prophylaxis Hgb SS and Hgb Sβ thalassemia patients Pen VK 0-3 yo: 125mg PO BID 3-11 yo: 250mg PO BID > 12 yo: 500mg PO BIX Alternative = Erythromycin Typically stop at 5 CMH (other centers may vary) Longer if surgical splenectomy or pneumococcal sepsis



17 Splenic Sequestration Hemoglobin SS Incidence increased: 6 and 36 months Overall incidence about 16% Hemoglobin SC Incidence increased: 2 and 17 years Mean age 8.9 years Can occur in adolescence and adulthood Incidence about 5%



18 Splenic Sequestration Splenic Sequestration Crisis Rapid splenic enlargement Rapid fall in hemoglobin level Fall in Platelet counts Presence of Nucleated Red Blood Cells Usually febrile and sometimes listless Treatment Close monitor VS, Hb Hydration and transfusion acute and chronic Teach spleen palpation

19 Pneumonias and Acute Chest Syndrome Acute Chest Syndrome May be associated with New pulmonary emerging pulmonary infiltrate on chest hypertension radiograph Treatment +/- chest pain Antibiotics + low oxygen level Bronchodilator/bedsid 2 nd cause for e incentive spirometry hospitalization Respiratory toilet 25% of all deaths in Careful pain control SCD Transfusions Increased association with asthma/atopy Occurs frequently during or after painful episode Acute Chest Syndrome A leading cause of death in sickle cell disease Clinically: Acute onset of fever, respiratory symptoms, new infiltrate on chest x-ray Definition: Acute onset chest symptoms AND new infiltrate on CXR, fever, tachypnea, cough, new onset hypoxia,, increased WOB, chest pain Since you cannot distinguish between acute chest syndrome and pneumonia clinically there is no change in treatment.
20 Strokes 10% of children with Hb SS 300 x risk of normal children Mostly between ages 3-12 years 70% risk of reoccurrence without chronic transfusion therapy Treatment is chronic transfusions May be silent manifesting only as learning difficulties or headaches (22%) Stroke Peak incidence b/w 5-10 yo Underlying arterial stenosis or obstruction Internal carotid, MCA or ACA Diagnosis: diffusion-weighted MRI, FLAIR, MRA Treatment: exchange transfusion therapy to maintain sickle hemoglobin at or below 30%




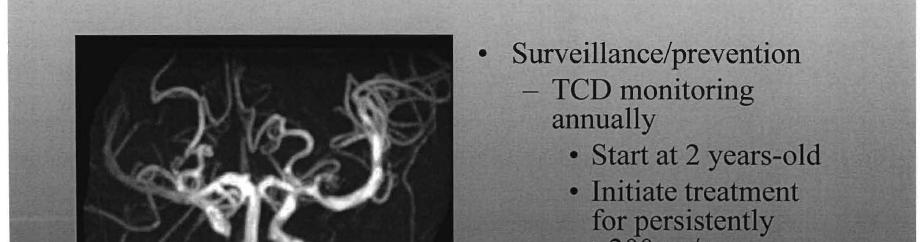

21
 uses ultrasound to")


22 Transcranial Doppler Ultrasonography Transcranial Doppler (TCD) uses ultrasound to examine the arteries, measure blood flow, and look for signs of vasospasm. Courtesy of Mayfield Clinic / University of Cincinnati Department of Neurosurgery, Ohio Transcranial Doppler Ultrasonography
23 Indications for Simple/Acute Transfusion Pre-operatively (Goal to hemoglobin > 10gm/dL Stroke (initial presentation) Acute Chest Syndrome (initial presentation) Aplastic Anemia Splenic sequestration Symptomatic anemia Priapism Indications for Chronic Transfusions Cerebral Vascular Events (stroke) Secondary Stroke Prevention Abnormal Transcranial Doppler (TCD) Primary Stroke Prevention?? Silent Infarct Recurrent Splenic Sequestration Recurrent Acute Chest Syndrome Recurrent Intractable Pain
24 Pain Guidelines Still the hallmark of sickle cell disease All pain is not sickle cell pain episode 20% - frequent pain 30% - rarely have pain 50% - one pain episode per year Painful episode (vasoocclusion episode) Hydration and Anti-inflammatoryinflammatory NSAID Narcotics Oral or parenteral Observe for respiratory compromise Pain Management Pain is an emergency Hospital evaluation: Hydration: 1.5 times maintenance unless acute chest syndrome suspected Assess pain level and treat Do not withhold opioids id Frequently reassess pain control Assess for cause of pain/complications
25 Pain Management Mild moderate pain Acetaminophen Hepatotoxic Non-steroidal anti-inflammatory inflammatory agents (NSAIDs) Contraindicated in patients with gastritis/ulcers and renal failure Monitor renal function if used chronically Pain Management Moderate or less severe pain Opioids are first-line treatment Morphine sulfate or hydromorphone Meperidine NOT recommended (Metabolite causes seizures & renal toxicity) Moderate severe pain Acetaminophen or NSAIDs in combination with opioids Other adjuvant medications (sedatives, anxiolytics) May increase efficacy of analgesics


26 Parenteral Opioids in SCD Other Complications of Sickle Cell Disease Aplastic crisis Priapism Retinopathy Pulmonary hypertension Sleep apnea Cardiomyopathy, heart failure Gall stones, Cholecystitis Enuresis Avascular necrosis, early osteoarthritis Infarcts bone, organs Skin ulcers Increased fetal loss
27 Hydroxyurea Promotes production of Hb F Even a small increase in Hb F can retard sickling inflammation, hemolysis metabolic rate Few side effects (mild neutropenia, thrombocytopenia, hair loss) Treatment with Hydroxyurea has been shown to reduce pain events, hospital admissions and the need for blood transfusions by 50% 15 years experience in adults, not experimental Multicenter Study of Hydroxyurea in Sickle Cell Disease Event Hydroxyurea (n = 152) Placebo (n-147) P value Pain events/year Hospitalizations/y ear for pain Episodes of ACS Patients transfused Transfusion units
:5306.")

28 Hydroxyurea Effect Peripheral Smear Pictures courtesy of Dr. Russell Ware, St. Jude Children s Research Hospital. How I use hydroxyurea to treat young patients with sickle cell anemia. Blood. 2010; 115(26):5306. Sickle Cell Disease and BMT Bone Marrow Transplant Substitutes normal red blood cell progenitors from donor Requires high dose chemotherapy to prevent rejection Donor source either BM or CB Mixed chimera enough to modify course of disease, full ablation may not be needed Success 90% cure, 5% relapse and 5% risk of death
29 Myeloablative Matched Sibling BMT for Sickle Cell Disease Worldwide Walters Atlanta n Survival 93% 95% 96% DFS 85% 85% 96% Rejection 12% 9% 0% agvhd 10% 15% 4% cgvhd 12% 11% 8% Recent and Current Studies BABY HUG (Prospective HU in <18mo) SWITCH (Transfusion to HU for CVA) SITT TWITCH Glutamine Anti-Platelet Drugs Neuroprotective Study Vasodilators for Pulmonary Hypertension
30 Questions Case Scenarios
SICKLE CELL DISEASE. Dr. MUBARAK ABDELRAHMAN MD PEDIATRICS AND CHILD HEALTH. Assistant Professor FACULTY OF MEDICINE -JAZAN
 SICKLE CELL DISEASE Dr. MUBARAK ABDELRAHMAN MD PEDIATRICS AND CHILD HEALTH Assistant Professor FACULTY OF MEDICINE -JAZAN Objective: The student should be able: To identify the presentation, diagnosis,
SICKLE CELL DISEASE Dr. MUBARAK ABDELRAHMAN MD PEDIATRICS AND CHILD HEALTH Assistant Professor FACULTY OF MEDICINE -JAZAN Objective: The student should be able: To identify the presentation, diagnosis,
Anemia s. Troy Lund MSMS PhD MD
 Anemia s Troy Lund MSMS PhD MD lundx072@umn.edu Hemoglobinopathy/Anemia IOM take home points. 1. How do we identify the condtion? Smear, CBC Solubility Test (SCD) 2. How does it present clincally? 3. How
Anemia s Troy Lund MSMS PhD MD lundx072@umn.edu Hemoglobinopathy/Anemia IOM take home points. 1. How do we identify the condtion? Smear, CBC Solubility Test (SCD) 2. How does it present clincally? 3. How
Sickle cell disease. Fareed Omar 10 March 2018
 Sickle cell disease Fareed Omar 10 March 2018 Physiology Haemoglobin structure HbA2: 2α and 2δ chains (2-3%) HbF: 2α and 2γ chains (
Sickle cell disease Fareed Omar 10 March 2018 Physiology Haemoglobin structure HbA2: 2α and 2δ chains (2-3%) HbF: 2α and 2γ chains (
Sickle Cell Disease. Edward Malters, MD
 Sickle Cell Disease Edward Malters, MD Introduction Vaso-occlusive phenomena and hemolysis are the clinical hallmarks of Sickle Cell Disease (SCD) Inherited disorder due to homozygosity for the abnormal
Sickle Cell Disease Edward Malters, MD Introduction Vaso-occlusive phenomena and hemolysis are the clinical hallmarks of Sickle Cell Disease (SCD) Inherited disorder due to homozygosity for the abnormal
Hemolytic anemias (2 of 2)
") Hemolytic anemias (2 of 2) Sickle Cell Anemia The most common familial hemolytic anemia in the world Sickle cell anemia is the prototypical (and most prevalent) hemoglobinopathy Mutation in the β-globin
Hemolytic anemias (2 of 2) Sickle Cell Anemia The most common familial hemolytic anemia in the world Sickle cell anemia is the prototypical (and most prevalent) hemoglobinopathy Mutation in the β-globin
Congenital Haemoglobinopathies
 Congenital Haemoglobinopathies L. DEDEKEN, MD H O P I T A L U N I V E R S I T A I R E D E S E N F A N T S R E I N E F A B I O L A U N I V E R S I T E L I B R E DE B R U X E L L E S Red Blood Cell Disorders
Congenital Haemoglobinopathies L. DEDEKEN, MD H O P I T A L U N I V E R S I T A I R E D E S E N F A N T S R E I N E F A B I O L A U N I V E R S I T E L I B R E DE B R U X E L L E S Red Blood Cell Disorders
How to Write a Life Care Plan for a Child with Hemoglobinopathy
 How to Write a Life Care Plan for a Child with Hemoglobinopathy Tamar Fleischer, BSN, MSN, CNLCP & Mona Yudkoff, RN, MPH, CRRN, CNLCP BalaCare Solutions March 2018 St. Peterburg, Florida What is Hemoglobinopathy?
How to Write a Life Care Plan for a Child with Hemoglobinopathy Tamar Fleischer, BSN, MSN, CNLCP & Mona Yudkoff, RN, MPH, CRRN, CNLCP BalaCare Solutions March 2018 St. Peterburg, Florida What is Hemoglobinopathy?
Sickle Cell Disease and impact on the society
 Sickle Cell Disease and impact on the society Professor Z.A.Jeremiah Ph.D, FRCPath (London) Professor of Haematology and Blood Transfusion Science Niger Delta University, Wilberforce Island Outline What
Sickle Cell Disease and impact on the society Professor Z.A.Jeremiah Ph.D, FRCPath (London) Professor of Haematology and Blood Transfusion Science Niger Delta University, Wilberforce Island Outline What
DONE BY : RaSHA RAKAN & Bushra Saleem
 DONE BY : RaSHA RAKAN & Bushra Saleem Hemolytic anemias (2 of 2) Sickle Cell Anemia The most common familial hemolytic anemia in the world Sickle cell anemia is the prototypical (and most prevalent) hemoglobinopathy
DONE BY : RaSHA RAKAN & Bushra Saleem Hemolytic anemias (2 of 2) Sickle Cell Anemia The most common familial hemolytic anemia in the world Sickle cell anemia is the prototypical (and most prevalent) hemoglobinopathy
Sickle Cell Disease 101. Objectives. What is SCD? 4/20/2016. Discuss the pathophysiology & genetics of Sickle Cell Disease (SCD).
.") Sickle Cell Disease 101 Jennifer Young, RN, MS, CPNP-AC Sickle Cell & Thalassemia Nurse Practitioner Nationwide Children s Hospital Objectives Discuss the pathophysiology & genetics of Sickle Cell Disease
Sickle Cell Disease 101 Jennifer Young, RN, MS, CPNP-AC Sickle Cell & Thalassemia Nurse Practitioner Nationwide Children s Hospital Objectives Discuss the pathophysiology & genetics of Sickle Cell Disease
Hydroxyurea and Transfusion Therapy for the Treatment of Sickle Cell Disease
 Hydroxyurea and Transfusion Therapy for the Treatment of Sickle Cell Disease A Pocket Guide for the Clinician Susan E. Creary, MD, MSc 1 John J. Strouse, MD, PhD 2 1 The Ohio State University School of
Hydroxyurea and Transfusion Therapy for the Treatment of Sickle Cell Disease A Pocket Guide for the Clinician Susan E. Creary, MD, MSc 1 John J. Strouse, MD, PhD 2 1 The Ohio State University School of
Introduction reduction in output alter the amino acid sequence combination
 Sickle cell anemia. Introduction Mutations in the globin genes can cause a quantitative reduction in output from that gene or alter the amino acid sequence of the protein produced or a combination of the
Sickle cell anemia. Introduction Mutations in the globin genes can cause a quantitative reduction in output from that gene or alter the amino acid sequence of the protein produced or a combination of the
In adults, the predominant Hb (HbA) molecule has four chains: two α and two β chains. In thalassemias, the synthesis of either the α or the β chains
 molecule has four chains: two α and two β chains. In thalassemias, the synthesis of either the α or the β chains") Thalassaemias Thalassemia Thalassemia is an inherited autosomal recessive blood disease. Associated with absence or reduction in a or b globin chains. Reduced synthesis of one of the globin chains can
Thalassaemias Thalassemia Thalassemia is an inherited autosomal recessive blood disease. Associated with absence or reduction in a or b globin chains. Reduced synthesis of one of the globin chains can
Hemoglobinopathies NORMAL HEMOGLOBINS
 Hemoglobinopathies Millicent Sutton MD October 28, 2005 NORMAL HEMOGLOBINS Consist of 2 alpha chains and 2 non alpha chains Hb A = α2β2 Hb F= α 2γ2 Hb A2 = α2δ2 1 Hemoglobin Variants Altered the conformational
Hemoglobinopathies Millicent Sutton MD October 28, 2005 NORMAL HEMOGLOBINS Consist of 2 alpha chains and 2 non alpha chains Hb A = α2β2 Hb F= α 2γ2 Hb A2 = α2δ2 1 Hemoglobin Variants Altered the conformational
Disclosure. Hemoglobinopathies. Screening for Hemoglobinopthies. Learning Objectives. Screening for Hemoglobinopthies. Interpreting Reports
 Disclosure Hemoglobinopathies (everything you wanted to know but were afraid to ask) Melissa Frei-Jones, MD MSCI Pediatric Grand Rounds February 21, 2014 I have no relationships with commercial companies
Disclosure Hemoglobinopathies (everything you wanted to know but were afraid to ask) Melissa Frei-Jones, MD MSCI Pediatric Grand Rounds February 21, 2014 I have no relationships with commercial companies
Dr Banu Kaya Consultant Haematologist Barts Health NHS Trust Royal London Hospital, London, UK SICKLE CELL AND THALASSAEMIA OVERVIEW
 Dr Banu Kaya Consultant Haematologist Barts Health NHS Trust Royal London Hospital, London, UK SICKLE CELL AND THALASSAEMIA OVERVIEW Objectives Gain awareness of haemoglobinopathy inheritance, pathophysiology
Dr Banu Kaya Consultant Haematologist Barts Health NHS Trust Royal London Hospital, London, UK SICKLE CELL AND THALASSAEMIA OVERVIEW Objectives Gain awareness of haemoglobinopathy inheritance, pathophysiology
4 Jumana Jihad Dr. Ahmad Mansour Dr. Ahmad Mansour
 4 Jumana Jihad Dr. Ahmad Mansour Dr. Ahmad Mansour Anemia Decreased blood production Increased blood loss Hemolytic Hemorrhage Extravascular Intravascular Hemolytic (Further classification( Extrinsic Intrinsic
4 Jumana Jihad Dr. Ahmad Mansour Dr. Ahmad Mansour Anemia Decreased blood production Increased blood loss Hemolytic Hemorrhage Extravascular Intravascular Hemolytic (Further classification( Extrinsic Intrinsic
Atlantic Provinces Pediatric Hematology Oncology Network Réseau d Oncologie et Hématologie Pédiatrique des Provinces Atlantiques
 Atlantic Provinces Pediatric Hematology Oncology Network Réseau d Oncologie et Hématologie Pédiatrique des Provinces Atlantiques 5850/5980 University Avenue, PO Box 9700 Halifax, NS, B3K 6R8 1.902.470.7429
Atlantic Provinces Pediatric Hematology Oncology Network Réseau d Oncologie et Hématologie Pédiatrique des Provinces Atlantiques 5850/5980 University Avenue, PO Box 9700 Halifax, NS, B3K 6R8 1.902.470.7429
Comprehensive Care for Children and Adolescents with Sickle Cell Diseases
 Comprehensive Care for Children and Adolescents with Sickle Cell Diseases Objectives To review the system for newborn screening of infants for sickling diseases To provide the framework for a comprehensive
Comprehensive Care for Children and Adolescents with Sickle Cell Diseases Objectives To review the system for newborn screening of infants for sickling diseases To provide the framework for a comprehensive
Genetics of Thalassemia
 Genetics of Thalassemia Submitted by : Raya Samir Al- Hayaly Sura Zuhair Salih Saad Ghassan Al- Dulaimy Saad Farouq Kassir Sama Naal Salouha Zahraa Jasim Al- Aarajy Supervised by : Dr. Kawkab Adris Mahmod
Genetics of Thalassemia Submitted by : Raya Samir Al- Hayaly Sura Zuhair Salih Saad Ghassan Al- Dulaimy Saad Farouq Kassir Sama Naal Salouha Zahraa Jasim Al- Aarajy Supervised by : Dr. Kawkab Adris Mahmod
Hemoglobinopathies Diagnosis and management
 Hemoglobinopathies Diagnosis and management Morgan L. McLemore, M.D. Hematology/Leukemia Department of Hematology and Oncology Winship Cancer Institute at Emory University mlmclem@emory.edu Disclosures
Hemoglobinopathies Diagnosis and management Morgan L. McLemore, M.D. Hematology/Leukemia Department of Hematology and Oncology Winship Cancer Institute at Emory University mlmclem@emory.edu Disclosures
Health Maintenance and Education for Children and Adults
 Health Maintenance and Education for Children and Adults Richard Ward, MSc, MRCP, FRCPath Director, Red Blood Cell Disorders Program, UHN Assistant Professor, Hematology, University of Toronto Chair, Canadian
Health Maintenance and Education for Children and Adults Richard Ward, MSc, MRCP, FRCPath Director, Red Blood Cell Disorders Program, UHN Assistant Professor, Hematology, University of Toronto Chair, Canadian
Hematology/Oncology/BMT
 The University of Arizona Pediatric Residency Program Primary Goals for Rotation Hematology/Oncology/BMT 1. GOAL: Understand the role of the pediatrician in preventing hematologic or oncologic conditions,
The University of Arizona Pediatric Residency Program Primary Goals for Rotation Hematology/Oncology/BMT 1. GOAL: Understand the role of the pediatrician in preventing hematologic or oncologic conditions,
1 Kattamis et al. Growth of Children with Thalassemia: Effect of Different Transfusion Regimens. Archives of
 Objectives Sickle Cell Anemia and Thalassemia: Transplantation Provide overview of hemoglobinopathies: Sickle cell disease and Thalassemia Discuss approaches to therapy Review recent registry collaboration
Objectives Sickle Cell Anemia and Thalassemia: Transplantation Provide overview of hemoglobinopathies: Sickle cell disease and Thalassemia Discuss approaches to therapy Review recent registry collaboration
Newborn Screening and Followup for Hemoglobinopathies
 Newborn Screening and Followup for Hemoglobinopathies October 4, 2012 Monica Hulbert, MD Director, Sickle Cell Disease Program Washington University School of Medicine St. Louis Children s Hospital Disclosures
Newborn Screening and Followup for Hemoglobinopathies October 4, 2012 Monica Hulbert, MD Director, Sickle Cell Disease Program Washington University School of Medicine St. Louis Children s Hospital Disclosures
Putting some hematology into Pediatric Hematology/Oncology: a review of Hemophilia and Sickle Cell Disease in the Pediatric Patient
 Putting some hematology into Pediatric Hematology/Oncology: a review of Hemophilia and Sickle Cell Disease in the Pediatric Patient Kristina Haley, DO March 10, 2012 Jovita Reyes Memorial Pediatric Hematology/Oncology
Putting some hematology into Pediatric Hematology/Oncology: a review of Hemophilia and Sickle Cell Disease in the Pediatric Patient Kristina Haley, DO March 10, 2012 Jovita Reyes Memorial Pediatric Hematology/Oncology
Family Centered Pediatric Emergency Department Sickle Cell Assessment of Needs and Strengths (FC-Peds-ED-SCANS) Overall Algorithm
 Overall Algorithm") Family Centered Pediatric Emergency Department Sickle Cell Assessment of Needs and Strengths (FC-Peds-ED-SCANS) Overall Algorithm Decision 1: Triage Decision 2: Analgesic Management Decision 3: Diagnostic
Family Centered Pediatric Emergency Department Sickle Cell Assessment of Needs and Strengths (FC-Peds-ED-SCANS) Overall Algorithm Decision 1: Triage Decision 2: Analgesic Management Decision 3: Diagnostic
Medical and Surgical Complications of Sickle Cell Anemia
 Medical and Surgical Complications of Sickle Cell Anemia Ahmed Al-Salem Medical and Surgical Complications of Sickle Cell Anemia Ahmed Al-Salem Department of Surgery Dar A lalafia Medical Company Qatif
Medical and Surgical Complications of Sickle Cell Anemia Ahmed Al-Salem Medical and Surgical Complications of Sickle Cell Anemia Ahmed Al-Salem Department of Surgery Dar A lalafia Medical Company Qatif
Thalassemia Maria Luz Uy del Rosario, M.D.
 Thalassemia Maria Luz Uy del Rosario, M.D. Philippine Society of Hematology and Blood Transfusion Philippine Society of Pediatric Oncology What is Thalassemia Hereditary Hemoglobin disorder Hemolytic anemia
Thalassemia Maria Luz Uy del Rosario, M.D. Philippine Society of Hematology and Blood Transfusion Philippine Society of Pediatric Oncology What is Thalassemia Hereditary Hemoglobin disorder Hemolytic anemia
The Overlap Between Sickle Cell and Effective Chronic Disease Treatment and Management
 The Overlap Between Sickle Cell and Effective Chronic Disease Treatment and Management Nirmish Shah, MD Assistant Professor Director of the Sickle Cell Transition Program Division of Pediatric Hematology/Oncology
The Overlap Between Sickle Cell and Effective Chronic Disease Treatment and Management Nirmish Shah, MD Assistant Professor Director of the Sickle Cell Transition Program Division of Pediatric Hematology/Oncology
Haemoglobin BY: MUHAMMAD RADWAN WISSAM MUHAMMAD
 Haemoglobin BY: MUHAMMAD RADWAN WISSAM MUHAMMAD Introduction is the iron-containing oxygen transport metalloprotein in the red blood cells Hemoglobin in the blood carries oxygen from the respiratory organs
Haemoglobin BY: MUHAMMAD RADWAN WISSAM MUHAMMAD Introduction is the iron-containing oxygen transport metalloprotein in the red blood cells Hemoglobin in the blood carries oxygen from the respiratory organs
High Hemoglobin F in a Saudi Child Presenting with Pancytopenia
 Case Report imedpub Journals http://www.imedpub.com Journal of Pediatric Care ISSN 2471-805X DOI: 10.21767/2471-805X.100002 High Hemoglobin F in a Saudi Child Presenting with Pancytopenia Abstract Saudi
Case Report imedpub Journals http://www.imedpub.com Journal of Pediatric Care ISSN 2471-805X DOI: 10.21767/2471-805X.100002 High Hemoglobin F in a Saudi Child Presenting with Pancytopenia Abstract Saudi
CURRENT RESEARCH STUDIES
 CURRENT RESEARCH STUDIES SCAGO SICKLE CELL RESEARCH DAY MAY 12, 2018 REBECCA LEROUX RN, BSCN, CCRP RED BLOOD CELL DISORDERS PROGRAM, UNIVERSITY HEALTH NETWORK MANUELA MERELLES-PULCINI RN, BSCN, MSN, CCRP
CURRENT RESEARCH STUDIES SCAGO SICKLE CELL RESEARCH DAY MAY 12, 2018 REBECCA LEROUX RN, BSCN, CCRP RED BLOOD CELL DISORDERS PROGRAM, UNIVERSITY HEALTH NETWORK MANUELA MERELLES-PULCINI RN, BSCN, MSN, CCRP
1. Adequate diet and iron intake to prevent iron deficiency 2. Signs and symptoms of malignant disease
 Hematology/Oncology Description: The pediatric hematology-oncology division sees a wide spectrum of pediatric disease including but not limited to leukemia, hemophilia, solid tumors, ITP, and other blood
Hematology/Oncology Description: The pediatric hematology-oncology division sees a wide spectrum of pediatric disease including but not limited to leukemia, hemophilia, solid tumors, ITP, and other blood
RBCs Disorders 1. Dr. Nabila Hamdi MD, PhD
 RBCs Disorders 1 Dr. Nabila Hamdi MD, PhD ILOs Discuss the classification of anemia into hypochromic-microcytic, normochromicnormocytic and macrocytic. Categorize laboratory test procedures used in the
RBCs Disorders 1 Dr. Nabila Hamdi MD, PhD ILOs Discuss the classification of anemia into hypochromic-microcytic, normochromicnormocytic and macrocytic. Categorize laboratory test procedures used in the
HbSC disease is it different and how should we manage it? David Rees Department of Paediatric Haematology, King s College Hospital, London
 HbSC disease is it different and how should we manage it? David Rees Department of Paediatric Haematology, King s College Hospital, London Different types of sickle cell disesease Severe sickle cell disease
HbSC disease is it different and how should we manage it? David Rees Department of Paediatric Haematology, King s College Hospital, London Different types of sickle cell disesease Severe sickle cell disease
Dr.Abdolreza Afrasiabi
 Dr.Abdolreza Afrasiabi Thalassemia & Heamophilia Genetic Reaserch Center Shiraz Medical University Hemoglobin tetramer Hemoglobin Structure % A 1 α 2 β 2 94-97% A 2 α 2 δ 2 2.5% A 1C α 2 (β-n-glucose)
Dr.Abdolreza Afrasiabi Thalassemia & Heamophilia Genetic Reaserch Center Shiraz Medical University Hemoglobin tetramer Hemoglobin Structure % A 1 α 2 β 2 94-97% A 2 α 2 δ 2 2.5% A 1C α 2 (β-n-glucose)
The Thalassemias in Clinical Practice. Ashutosh Lal, MD Director Comprehensive Thalassemia Program UCSF Benioff Children s Hospital Oakland
 The Thalassemias in Clinical Practice Ashutosh Lal, MD Director Comprehensive Thalassemia Program UCSF Benioff Children s Hospital Oakland Outline Thalassemia: definitions and pathophysiology Epidemiology
The Thalassemias in Clinical Practice Ashutosh Lal, MD Director Comprehensive Thalassemia Program UCSF Benioff Children s Hospital Oakland Outline Thalassemia: definitions and pathophysiology Epidemiology
The Child with a Hematologic Alteration
 47 The Child with a Hematologic Alteration HELPFUL HINT Review the anatomy and physiology of the hematologic system in an anatomy and physiology textbook. MATCHING KEY TERMS Match the term with the correct
47 The Child with a Hematologic Alteration HELPFUL HINT Review the anatomy and physiology of the hematologic system in an anatomy and physiology textbook. MATCHING KEY TERMS Match the term with the correct
Extra Notes 3. Warm. In the core (center) of the body, where the temperature is 37 C.
 of the body, where the temperature is 37 C.") Extra Notes 3 *The numbers of the slides are according to the last year slides. Slide 33 Autoimmune hemolytic anemia : Abnormal circulating antibodies that target normal antigen on the RBC and cause lysis.
Extra Notes 3 *The numbers of the slides are according to the last year slides. Slide 33 Autoimmune hemolytic anemia : Abnormal circulating antibodies that target normal antigen on the RBC and cause lysis.
Acute Complications of Sickle Cell Disease
 Management of Acute Complications of Sickle Cell Disease A Pocket Guide for the Clinician Timothy McCavit, MD, MSCS 1 Payal Desai, MD 1 University of Texas Southwestern Medical Center The Ohio State University,
Management of Acute Complications of Sickle Cell Disease A Pocket Guide for the Clinician Timothy McCavit, MD, MSCS 1 Payal Desai, MD 1 University of Texas Southwestern Medical Center The Ohio State University,
Division of General Internal Medicine and Geriatrics Hospital Medicine 2014
 Division of General Internal Medicine and Geriatrics Hospital Medicine 2014 Objectives Understand workup of acute pain crisis Identify key aspects of management of acute pain crisis in sickle cell patients
Division of General Internal Medicine and Geriatrics Hospital Medicine 2014 Objectives Understand workup of acute pain crisis Identify key aspects of management of acute pain crisis in sickle cell patients
- Ensherah Mokheemer. - Rama Nada. - Tareq Aladily. 1 P a g e
 -3 - Ensherah Mokheemer - Rama Nada - Tareq Aladily 1 P a g e In this lecture we will continue talking about autoimmune hemolytic anemia. Autoimmune hemolytic anemia - There are several types that shares
-3 - Ensherah Mokheemer - Rama Nada - Tareq Aladily 1 P a g e In this lecture we will continue talking about autoimmune hemolytic anemia. Autoimmune hemolytic anemia - There are several types that shares
Assessing the follow-up of BART hemoglobin reported by the Wisconsin Newborn Screening Laboratory
 Assessing the follow-up of BART hemoglobin reported by the Wisconsin Newborn Screening Laboratory Bao Yang MPH Candidate Background The Wisconsin Newborn Screening (NBS) Laboratory screens 70,000 babies
Assessing the follow-up of BART hemoglobin reported by the Wisconsin Newborn Screening Laboratory Bao Yang MPH Candidate Background The Wisconsin Newborn Screening (NBS) Laboratory screens 70,000 babies
HEMOLYTIC ANEMIA DUE TO ABNORMAL HEMOGLOBIN SYNTHESIS
 Hemolytic Anemia Due to Abnormal Hemoglobin Synthesis MODULE 19 HEMOLYTIC ANEMIA DUE TO ABNORMAL HEMOGLOBIN SYNTHESIS 19.1 INTRODUCTION There are two main mechanisms by which anaemia is produced (a) Thalassemia:
Hemolytic Anemia Due to Abnormal Hemoglobin Synthesis MODULE 19 HEMOLYTIC ANEMIA DUE TO ABNORMAL HEMOGLOBIN SYNTHESIS 19.1 INTRODUCTION There are two main mechanisms by which anaemia is produced (a) Thalassemia:
Screening for haemoglobinopathies in pregnancy
 Policy Statement All Southern Health patients will receive clinical care that reflects best practice and is based on the best available evidence. Index of chapters within background 1. Prevalence of haemoglobinopathies
Policy Statement All Southern Health patients will receive clinical care that reflects best practice and is based on the best available evidence. Index of chapters within background 1. Prevalence of haemoglobinopathies
THALASSEMIA AND COMPREHENSIVE CARE
 1 THALASSEMIA AND COMPREHENSIVE CARE Melanie Kirby MBBS, FRCP (C), Hospital for Sick Children, Toronto Associate Professor of Paediatrics, University of Toronto. Objectives 2 By the end of this presentation,
1 THALASSEMIA AND COMPREHENSIVE CARE Melanie Kirby MBBS, FRCP (C), Hospital for Sick Children, Toronto Associate Professor of Paediatrics, University of Toronto. Objectives 2 By the end of this presentation,
Rationale for RBC Transfusion in SCD
 Rationale for RBC Transfusion in SCD Dilution of HgbS-containing RBCs via the addition of HgbA-containing cells from the blood of normal donors Suppression of erythropoietin release caused by the rise
Rationale for RBC Transfusion in SCD Dilution of HgbS-containing RBCs via the addition of HgbA-containing cells from the blood of normal donors Suppression of erythropoietin release caused by the rise
MI Newborn Screening Program: An Overview of Follow-up & Case Management for Sickle Cell Conditions
 Michigan Department of Community Health MI Newborn Screening Program: An Overview of Follow-up & Case Management for Sickle Cell Conditions Dominic Smith, MSA - Linda Carter, BSW - Ben Frazier, BSW - Ruth
Michigan Department of Community Health MI Newborn Screening Program: An Overview of Follow-up & Case Management for Sickle Cell Conditions Dominic Smith, MSA - Linda Carter, BSW - Ben Frazier, BSW - Ruth
DAYTON CHILDREN S HOSPITAL CLINICAL PRACTICE GUIDELINES
 DAYTON CHILDREN S HOSPITAL CLINICAL PRACTICE GUIDELINES DISCLAIMER: This Clinical Practice Guideline (CPG) generally describes a recommended course of treatment for patients with the identified health
DAYTON CHILDREN S HOSPITAL CLINICAL PRACTICE GUIDELINES DISCLAIMER: This Clinical Practice Guideline (CPG) generally describes a recommended course of treatment for patients with the identified health
TRANSFUSION PRACTICES IN THE MANAGEMENT OF SICKLE CELL DISEASE AMONG FLORIDA PHYSICIANS
 TRANSFUSION PRACTICES IN THE MANAGEMENT OF SICKLE CELL DISEASE AMONG FLORIDA PHYSICIANS By LEVETTE NICOLE DUNBAR A THESIS PRESENTED TO THE GRADUATE SCHOOL OF THE UNIVERSITY OF FLORIDA IN PARTIAL FULFILLMENT
TRANSFUSION PRACTICES IN THE MANAGEMENT OF SICKLE CELL DISEASE AMONG FLORIDA PHYSICIANS By LEVETTE NICOLE DUNBAR A THESIS PRESENTED TO THE GRADUATE SCHOOL OF THE UNIVERSITY OF FLORIDA IN PARTIAL FULFILLMENT
Pitfalls in the premarital testing for thalassaemia
 Pitfalls in the premarital testing for thalassaemia Dr. Riad Amer MB ChB, MSc, FRCP, FRCPath, JBH Assistant Professor of Medicine Al Najah University Consultant Haematologist Case 1 Husband and Wife are
Pitfalls in the premarital testing for thalassaemia Dr. Riad Amer MB ChB, MSc, FRCP, FRCPath, JBH Assistant Professor of Medicine Al Najah University Consultant Haematologist Case 1 Husband and Wife are
Hydroxyurea. for Sickle Cell Disease. A Guide for Starting Treatment. Hydroxyurea is a medicine proven to prevent pain from sickle cell disease.
 Hydroxyurea for Sickle Cell Disease A Guide for Starting Treatment Hydroxyurea is a medicine proven to prevent pain from sickle cell disease. This handbook was created to help answer common questions about
Hydroxyurea for Sickle Cell Disease A Guide for Starting Treatment Hydroxyurea is a medicine proven to prevent pain from sickle cell disease. This handbook was created to help answer common questions about
DIC. Disseminated intravascular coagulation, is a life threatening pathological process in which clotting factors are abnormally activated.
 Miss. kamlah 1 DIC Disseminated intravascular coagulation, is a life threatening pathological process in which clotting factors are abnormally activated. Resulting in wide spread of clot formation in the
Miss. kamlah 1 DIC Disseminated intravascular coagulation, is a life threatening pathological process in which clotting factors are abnormally activated. Resulting in wide spread of clot formation in the
THE UNIVERSITY OF JORDAN FACULTY OF MEDICINE DEPARTMENT OF PATHOLOGY HEMOLYTIC ANEMIAS. Dr. Tariq Aladily
 THE UNIVERSITY OF JORDAN FACULTY OF MEDICINE DEPARTMENT OF PATHOLOGY HEMOLYTIC ANEMIAS Third year medical students First semester Faculty 2018/2019 of Medicine Hereditary Spherocytosis Intrinsic defects
THE UNIVERSITY OF JORDAN FACULTY OF MEDICINE DEPARTMENT OF PATHOLOGY HEMOLYTIC ANEMIAS Third year medical students First semester Faculty 2018/2019 of Medicine Hereditary Spherocytosis Intrinsic defects
Quiz. What percentage of the world s population is a carrier of a hemoglobinopathy? Hemoglobinopathies in Pregnancy 1-2% 5-7% 8-12% 10-15%
 Hemoglobinopathies in Pregnancy Emily Parkhurst, MS, LCGC Kaiser West Los Angeles November 2017 Genetics Department Quiz What percentage of the world s population is a carrier of a hemoglobinopathy? 1-2%
Hemoglobinopathies in Pregnancy Emily Parkhurst, MS, LCGC Kaiser West Los Angeles November 2017 Genetics Department Quiz What percentage of the world s population is a carrier of a hemoglobinopathy? 1-2%
Corporate Medical Policy
 Corporate Medical Policy Genetic Testing for Alpha Thalassemia File Name: Origination: Last CAP Review: Next CAP Review: Last Review: genetic_testing_for_alpha_thalassemia 9/2013 7/2017 7/2018 7/2017 Description
Corporate Medical Policy Genetic Testing for Alpha Thalassemia File Name: Origination: Last CAP Review: Next CAP Review: Last Review: genetic_testing_for_alpha_thalassemia 9/2013 7/2017 7/2018 7/2017 Description
Hydroxurea: A Novel Approach to Optimizing the Health of Pediatric Patients with Sickle Cell Disease. Maa Ohui Quarmyne September 9 th, 2017
 Hydroxurea: A Novel Approach to Optimizing the Health of Pediatric Patients with Sickle Cell Disease Maa Ohui Quarmyne September 9 th, 2017 Outline Sickle Cell Disease Pathophysiology and Clinical Manifestations
Hydroxurea: A Novel Approach to Optimizing the Health of Pediatric Patients with Sickle Cell Disease Maa Ohui Quarmyne September 9 th, 2017 Outline Sickle Cell Disease Pathophysiology and Clinical Manifestations
Fetal Anemia 02/13/13. Anjulika Chawla, M.D. Assistant Professor Division of Pediatric Hematology/Oncology
 Fetal Anemia 02/13/13 Anjulika Chawla, M.D. Assistant Professor Division of Pediatric Hematology/Oncology Objectives Definition of anemia Diagnosis of fetal anemia Normal developmental hematopoiesis Etiology
Fetal Anemia 02/13/13 Anjulika Chawla, M.D. Assistant Professor Division of Pediatric Hematology/Oncology Objectives Definition of anemia Diagnosis of fetal anemia Normal developmental hematopoiesis Etiology
Hydroxyurea Treatment for Sickle Cell Disease
 Hydroxyurea Treatment for Sickle Cell Disease Before Hydroxyurea After Hydroxyurea Hydroxyurea Treatment for Sickle Cell Disease This document is not intended to take the place of the care and attention
Hydroxyurea Treatment for Sickle Cell Disease Before Hydroxyurea After Hydroxyurea Hydroxyurea Treatment for Sickle Cell Disease This document is not intended to take the place of the care and attention
Good afternoon and thank you for joining us today as we discuss hydroxyurea for the treatment of sickle cell disease. Dr. Emily Meier is a pediatric
 Good afternoon and thank you for joining us today as we discuss hydroxyurea for the treatment of sickle cell disease. Dr. Emily Meier is a pediatric hematologist at the Indiana Hemophilia & Thrombosis
Good afternoon and thank you for joining us today as we discuss hydroxyurea for the treatment of sickle cell disease. Dr. Emily Meier is a pediatric hematologist at the Indiana Hemophilia & Thrombosis
Managing Emergencies
 Managing Emergencies Richard Ward, MSc, MRCP, FRCPath Director, Red Blood Cell Disorders Program, UHN Assistant Professor, Hematology, University of Toronto Chair, Canadian Hemoglobinopathy Association
Managing Emergencies Richard Ward, MSc, MRCP, FRCPath Director, Red Blood Cell Disorders Program, UHN Assistant Professor, Hematology, University of Toronto Chair, Canadian Hemoglobinopathy Association
Acute Complications of Sickle Cell Disease Case Study 5 year old girl with Hemoglobin SS, weakness and slurred speech
 Acute Complications of Sickle Cell Disease Case Study 5 year old girl with Hemoglobin SS, weakness and slurred speech Beatrice E. Gee, MD Medical Director, Sickle Cell and Hematology Program Children s
Acute Complications of Sickle Cell Disease Case Study 5 year old girl with Hemoglobin SS, weakness and slurred speech Beatrice E. Gee, MD Medical Director, Sickle Cell and Hematology Program Children s
A Counseling Guide for Sickle Cell and Other Hemoglobin Variants
 A Counseling Guide for Sickle Cell and Other Hemoglobin Variants The Virginia Sickle Cell Awareness Program Virginia Department of Health Division of Women s and Infant s Health 109 Governor Street Richmond,
A Counseling Guide for Sickle Cell and Other Hemoglobin Variants The Virginia Sickle Cell Awareness Program Virginia Department of Health Division of Women s and Infant s Health 109 Governor Street Richmond,
Approach to Hemolysis
 Objectives: Approach to Hemolysis To know the function of platelets and the relationship between the platelet count in peripheral blood and the extent of abnormal bleeding. To know about the diseases associated
Objectives: Approach to Hemolysis To know the function of platelets and the relationship between the platelet count in peripheral blood and the extent of abnormal bleeding. To know about the diseases associated
What is Thalassaemia?
 What is Thalassaemia? Introduction The thalassaemias are a diverse group of genetic blood diseases characterized by absent or decreased production of normal hemoglobin, resulting in a microcytic anemia
What is Thalassaemia? Introduction The thalassaemias are a diverse group of genetic blood diseases characterized by absent or decreased production of normal hemoglobin, resulting in a microcytic anemia
Hydroxyurea in Pediatric Patients With Sickle Cell Disease: What Nurses Need to Know
 614962JPOXXX10.1177/1043454215614962Journal of Pediatric Oncology NursingRees research-article2015 Article Hydroxyurea in Pediatric Patients With Sickle Cell Disease: What Nurses Need to Know Journal of
614962JPOXXX10.1177/1043454215614962Journal of Pediatric Oncology NursingRees research-article2015 Article Hydroxyurea in Pediatric Patients With Sickle Cell Disease: What Nurses Need to Know Journal of
Pediatrics. Pyruvate Kinase Deficiency (PKD) Symptoms and Treatment. Definition. Epidemiology of Pyruvate Kinase Deficiency.
 Symptoms and Treatment. Definition. Epidemiology of Pyruvate Kinase Deficiency.") Pediatrics Pyruvate Kinase Deficiency (PKD) Symptoms and Treatment See online here Pyruvate kinase deficiency is an inherited metabolic disorder characterized by a deficiency in the enzyme "pyruvate kinase"
Pediatrics Pyruvate Kinase Deficiency (PKD) Symptoms and Treatment See online here Pyruvate kinase deficiency is an inherited metabolic disorder characterized by a deficiency in the enzyme "pyruvate kinase"
AMERICAN ACADEMY OF PEDIATRICS. Health Supervision for Children With Sickle Cell Disease
 AMERICAN ACADEMY OF PEDIATRICS Section on Hematology/Oncology Committee on Genetics Health Supervision for Children With Sickle Cell Disease ABSTRACT. Sickle cell disease (SCD) is a group of complex genetic
AMERICAN ACADEMY OF PEDIATRICS Section on Hematology/Oncology Committee on Genetics Health Supervision for Children With Sickle Cell Disease ABSTRACT. Sickle cell disease (SCD) is a group of complex genetic
4 Fahed Al Karmi Sufian Alhafez Dr nayef karadsheh
 4 Fahed Al Karmi Sufian Alhafez Dr nayef karadsheh Genetic variants of hemoglobin Hemoglobinopathies (abnormal variants of hemoglobin) are divided into: 1. Structural abnormalities: Any change in the genes
4 Fahed Al Karmi Sufian Alhafez Dr nayef karadsheh Genetic variants of hemoglobin Hemoglobinopathies (abnormal variants of hemoglobin) are divided into: 1. Structural abnormalities: Any change in the genes
Haemoglobinophaties EBMT 2011 Data Manager session
 Haemoglobinophaties EBMT 2011 Data Manager session Presentation plan Biological characteristics Clinical characteristics Transplant resuts What is different From transplant in malignancies Between Thalassemia
Haemoglobinophaties EBMT 2011 Data Manager session Presentation plan Biological characteristics Clinical characteristics Transplant resuts What is different From transplant in malignancies Between Thalassemia
Comprehensive Hemoglobin Analysis HBA1/2 (
 Comprehensive Hemoglobin Analysis HBA1/2 ( α-globin) and HBB (β-globin) mutation and deletion/duplication analysis and HBD (δ-globin) and HBG1/2 (γ-globin) mutation analysis Description: Hemoglobin (Hb)
Comprehensive Hemoglobin Analysis HBA1/2 ( α-globin) and HBB (β-globin) mutation and deletion/duplication analysis and HBD (δ-globin) and HBG1/2 (γ-globin) mutation analysis Description: Hemoglobin (Hb)
7 Medical Genetics. Hemoglobinopathies. Hemoglobinopathies. Protein and Gene Structure. and Biochemical Genetics
 SESSION 7 Medical Genetics Hemoglobinopathies and Biochemical Genetics J a v a d F a s a J a m s h i d i U n i v e r s i t y o f M e d i c a l S c i e n c e s, N o v e m b e r 2 0 1 7 Hemoglobinopathies
SESSION 7 Medical Genetics Hemoglobinopathies and Biochemical Genetics J a v a d F a s a J a m s h i d i U n i v e r s i t y o f M e d i c a l S c i e n c e s, N o v e m b e r 2 0 1 7 Hemoglobinopathies
General Characterisctics
 Anemia General Characterisctics Definition: anemia is a decrease in red blood cells. Happens due to underproduction, increased destruction or loss of red cells. Diagnosis of anemia: Hgb < 135 (men) Hgb
Anemia General Characterisctics Definition: anemia is a decrease in red blood cells. Happens due to underproduction, increased destruction or loss of red cells. Diagnosis of anemia: Hgb < 135 (men) Hgb
HEMOGLOBINOPATHIES LECTURE OUTLINE. An overview of the structure of hemoglobin. Different types of hemoglobin. Definition of hemoglobinopathies
 Slide 1 HEOGLOBINOPATHIES Slide 2 LETURE OUTLINE An overview of the structure of hemoglobin. Different types of hemoglobin. Definition of hemoglobinopathies Sickle ell Disease and Hemoglobin Slide 3 HEOGLOBIN
Slide 1 HEOGLOBINOPATHIES Slide 2 LETURE OUTLINE An overview of the structure of hemoglobin. Different types of hemoglobin. Definition of hemoglobinopathies Sickle ell Disease and Hemoglobin Slide 3 HEOGLOBIN
Susan Stegman, MD Medical Director AXA Equitable Life May 3, 2016
 Susan Stegman, MD Medical Director AXA Equitable Life May 3, 2016 Underwriting impact Anemia overview Classification of anemia Specific anemia topics Iron deficiency anemia Thalassemia Megaloblastic anemia
Susan Stegman, MD Medical Director AXA Equitable Life May 3, 2016 Underwriting impact Anemia overview Classification of anemia Specific anemia topics Iron deficiency anemia Thalassemia Megaloblastic anemia
Sickle cell disease (SCD) and other hemoglobinopathies
 and other hemoglobinopathies") Sickle cell disease (SCD) and other hemoglobinopathies You have received this leaflet, because your child has been diagnosed with sickle cell disease. We can imagine how overwhelming such a diagnosis must
Sickle cell disease (SCD) and other hemoglobinopathies You have received this leaflet, because your child has been diagnosed with sickle cell disease. We can imagine how overwhelming such a diagnosis must
Heme Questions and Derivatives for the USMLE Step One Exam. Winter Storm Skylar Edition
 Heme Questions and Derivatives for the USMLE Step One Exam Winter Storm Skylar Edition Howard J. Sachs, MD Howard@12DaysinMarch.com www.12daysinmarch.com Patient presents for routine preoperative evaluation
Heme Questions and Derivatives for the USMLE Step One Exam Winter Storm Skylar Edition Howard J. Sachs, MD Howard@12DaysinMarch.com www.12daysinmarch.com Patient presents for routine preoperative evaluation
Dr. MUNEER ALBAGSHI Consultant Pediatric Hematologist Oncologist- HBDC, Al-Ahsa. Saudi Arabia
 Dr. MUNEER ALBAGSHI Consultant Pediatric Hematologist Oncologist- HBDC, Al-Ahsa. Saudi Arabia Sickle cell is global disease of old world and immigrants to the new world. Sickle cell anemia to predict that
Dr. MUNEER ALBAGSHI Consultant Pediatric Hematologist Oncologist- HBDC, Al-Ahsa. Saudi Arabia Sickle cell is global disease of old world and immigrants to the new world. Sickle cell anemia to predict that
Thalassemias. Emanuela Veras, M.D. 01/08/2006
 Thalassemias Emanuela Veras, M.D. 01/08/2006 Structure and Function of normal Hemoglobin molecules: 2/3 1/3 β: increases from 6 th week of fetal life to 12 months of age At birth: HbF: 75-90% HbA: 10-25%
Thalassemias Emanuela Veras, M.D. 01/08/2006 Structure and Function of normal Hemoglobin molecules: 2/3 1/3 β: increases from 6 th week of fetal life to 12 months of age At birth: HbF: 75-90% HbA: 10-25%
Historic and Current Complications in Children with Sickle Cell Disease
 Historic and Current Complications in Children with Sickle Cell Disease Trish McMahon Peterson RN, MSN, CPNP Thomas C. Hofstra MD Children's Hospital Los Angeles Comprehensive Sickle Cell Program Children's
Historic and Current Complications in Children with Sickle Cell Disease Trish McMahon Peterson RN, MSN, CPNP Thomas C. Hofstra MD Children's Hospital Los Angeles Comprehensive Sickle Cell Program Children's
Newborn Screening for Sickle Cell Disease in Africa: Public health meets reality
 13 th September 2016 Newborn Screening for Sickle Cell Disease in Africa: Public health meets reality Kwaku Ohene-Frempong. MD Children s Hospital of Philadelphia Sickle Cell Foundation of Ghana Disclosure
13 th September 2016 Newborn Screening for Sickle Cell Disease in Africa: Public health meets reality Kwaku Ohene-Frempong. MD Children s Hospital of Philadelphia Sickle Cell Foundation of Ghana Disclosure
Done by :Aseel Twaijer & Laith Sorour Hemolytic Anemias
 Hemolytic Anemias Hemolytic anemias share the following features: - A shortened red cell life < 120 days - Elevated erythropoietin levels (compensatory increase in erythropoiesis) - Accumulation of hemoglobin
Hemolytic Anemias Hemolytic anemias share the following features: - A shortened red cell life < 120 days - Elevated erythropoietin levels (compensatory increase in erythropoiesis) - Accumulation of hemoglobin
Anaemia in Pregnancy
 Anaemia in Pregnancy Definition :anaemia is a pathological condition in which the oxygen-carrying capacity of red blood cells is insufficient to meet the body needs. The WHO : haemoglobin concentration
Anaemia in Pregnancy Definition :anaemia is a pathological condition in which the oxygen-carrying capacity of red blood cells is insufficient to meet the body needs. The WHO : haemoglobin concentration
Epidemiology, Care and Prevention of Hemoglobinopathies
 Epidemiology, Care and Prevention of Hemoglobinopathies Nasir Al-Allawi MBChB, PhD. Professor of Hematology College of Medicine University of Dohuk, IRAQ From Research to Practice Training Course in Sexual
Epidemiology, Care and Prevention of Hemoglobinopathies Nasir Al-Allawi MBChB, PhD. Professor of Hematology College of Medicine University of Dohuk, IRAQ From Research to Practice Training Course in Sexual
Peripheral Blood Smear Examination. Momtazmanesh MD. Ped. Hematologist & Oncologist Loghman General Hospital
 1395 Peripheral Blood Smear Examination Momtazmanesh MD. Ped. Hematologist & Oncologist Loghman General Hospital Peripheral Blood Smear A peripheral blood smear is a snapshot of the cells that are present
1395 Peripheral Blood Smear Examination Momtazmanesh MD. Ped. Hematologist & Oncologist Loghman General Hospital Peripheral Blood Smear A peripheral blood smear is a snapshot of the cells that are present
Chapter 3 Diseases of the Blood and Bloodforming Organs and Certain Disorders Involving the Immune Mechanism D50-D89
 Chapter 3 Diseases of the Blood and Bloodforming Organs and Certain Disorders Involving the Immune Mechanism D50-D89 Presented by Jennifer Kurkulonis 1 FOUR MAJOR TYPES OF BLOOD CELLS White blood cells
Chapter 3 Diseases of the Blood and Bloodforming Organs and Certain Disorders Involving the Immune Mechanism D50-D89 Presented by Jennifer Kurkulonis 1 FOUR MAJOR TYPES OF BLOOD CELLS White blood cells
SICKLE CELL DISEASE TO TREAT OR
 SICKLE CELL DISEASE TO TREAT OR NOT TO TREAT COHEM Barcelona September 8, 2012 Sujit Sheth, M.D. Pediatric Hematology Oncology Disclosures None Outline Morbidity and mortality Definitive therapies Risk
SICKLE CELL DISEASE TO TREAT OR NOT TO TREAT COHEM Barcelona September 8, 2012 Sujit Sheth, M.D. Pediatric Hematology Oncology Disclosures None Outline Morbidity and mortality Definitive therapies Risk
Sickle Cell Disease Overview/Transfusion Support Wednesday, August 29, :00 p.m. 3:30 p.m. (ET) / 6:00p.m.-7:30 p.m. (GMT)
 / 6:00p.m.-7:30 p.m. (GMT)") Sickle Cell Disease Overview/Transfusion Support Wednesday, August 29, 2012 2:00 p.m. 3:30 p.m. (ET) / 6:00p.m.-7:30 p.m. (GMT) When this file is opened, Acrobat Reader will, by default, display the slides
Sickle Cell Disease Overview/Transfusion Support Wednesday, August 29, 2012 2:00 p.m. 3:30 p.m. (ET) / 6:00p.m.-7:30 p.m. (GMT) When this file is opened, Acrobat Reader will, by default, display the slides
Sickle Cell Anemia. Sickle cell anemia is an inherited disorder of the blood which occurs when just one base pair substitution
 Rose Farrington and Rachel Nash BIOL 362 Lab M. Bulgarella Genetic Diseases 10/14/2008 Sickle Cell Anemia Introduction Sickle cell anemia is an inherited disorder of the blood which occurs when just one
Rose Farrington and Rachel Nash BIOL 362 Lab M. Bulgarella Genetic Diseases 10/14/2008 Sickle Cell Anemia Introduction Sickle cell anemia is an inherited disorder of the blood which occurs when just one
COHEM Barcellona 2012 Hemoglobinopathies debate
 COHEM Barcellona 2012 Hemoglobinopathies debate September 8, 2012: h. 10:30-12:00 Hall: A Is it justified to perform BMT in hemoglobinopathies using unrelated and/or partially mismatched donors? HSCT indication
COHEM Barcellona 2012 Hemoglobinopathies debate September 8, 2012: h. 10:30-12:00 Hall: A Is it justified to perform BMT in hemoglobinopathies using unrelated and/or partially mismatched donors? HSCT indication
THE UNIVERSITY OF JORDAN FACULTY OF MEDICINE DEPARTMENT OF PATHOLOGY
 THE UNIVERSITY OF JORDAN FACULTY OF MEDICINE DEPARTMENT OF PATHOLOGY INTRODUCTION TO ANEMIA Third year medical students First semester 2018/2019 Dr. RBC DISORDERS Lecturer: Dr. Tariq Al-Adaily Email: TNALADILY@ju.edu.jo
THE UNIVERSITY OF JORDAN FACULTY OF MEDICINE DEPARTMENT OF PATHOLOGY INTRODUCTION TO ANEMIA Third year medical students First semester 2018/2019 Dr. RBC DISORDERS Lecturer: Dr. Tariq Al-Adaily Email: TNALADILY@ju.edu.jo
Report of Beta Thalassemia in Newar Ethnicity
 Report of Beta Thalassemia in Newar Ethnicity Rajendra Dev Bhatt 1*, Surendra Koju 2, Prabodh Risal 1 Affiliations: 1 Department of Clinical Biochemistry, Dhulikhel Hospital, Kathmandu University Hospital
Report of Beta Thalassemia in Newar Ethnicity Rajendra Dev Bhatt 1*, Surendra Koju 2, Prabodh Risal 1 Affiliations: 1 Department of Clinical Biochemistry, Dhulikhel Hospital, Kathmandu University Hospital
Lessons in Management of Sickle Cell Disease. CME Conference, Lloyd Erskine Sandiford Centre, November 15, 2015
 Lessons in Management of Sickle Cell Disease CME Conference, Lloyd Erskine Sandiford Centre, November 15, 2015 Age at Death in African SS Disease The greatest chance of death is in the first is in the
Lessons in Management of Sickle Cell Disease CME Conference, Lloyd Erskine Sandiford Centre, November 15, 2015 Age at Death in African SS Disease The greatest chance of death is in the first is in the
Thalassemias:general aspects and molecular pathology
 Thalassemias:general aspects and molecular pathology Prof. Renzo Galanello Pediatric Clinic 2 University of Cagliari Ospedale Regionale Microcitemie-ASL8 HEMOGLOBINOPATHIES CLASSIFICATION Structurally
Thalassemias:general aspects and molecular pathology Prof. Renzo Galanello Pediatric Clinic 2 University of Cagliari Ospedale Regionale Microcitemie-ASL8 HEMOGLOBINOPATHIES CLASSIFICATION Structurally
Genetic Modifiers of Sickle Cell Disease Severity. Kunle Adekile, MD, PhD Professor Department of Pediatrics Kuwait University
 Genetic Modifiers of Sickle Cell Disease Severity Kunle Adekile, MD, PhD Professor Department of Pediatrics Kuwait University Outline Hb Molecule and Genetic control of globin synthesis Pathophysiology
Genetic Modifiers of Sickle Cell Disease Severity Kunle Adekile, MD, PhD Professor Department of Pediatrics Kuwait University Outline Hb Molecule and Genetic control of globin synthesis Pathophysiology
HPLC profile of sickle cell disease in central India
 Original Research Article HPLC profile of sickle cell disease in central India Shweta P. Bijwe * Department of Pathology, IGGMC, Nagpur, Maharashtra, India * Corresponding author email: dr.shwetabijwe@gmail.com
Original Research Article HPLC profile of sickle cell disease in central India Shweta P. Bijwe * Department of Pathology, IGGMC, Nagpur, Maharashtra, India * Corresponding author email: dr.shwetabijwe@gmail.com
SCD Advocacy Talking Points!
 "#$!%&'()*)+!,!-././-1!! SCD Advocacy Talking Points! * 23!4*'3!53*673&!84*8!#*59:(679*!#495&637;!9
"#$!%&'()*)+!,!-././-1!! SCD Advocacy Talking Points! * 23!4*'3!53*673&!84*8!#*59:(679*!#495&637;!9
A group of inherited disorders characterized by reduced or absent amounts of hemoglobin, the oxygencarrying protein inside the red blood cells.
 Thalassemia A group of inherited disorders characterized by reduced or absent amounts of hemoglobin, the oxygencarrying protein inside the red blood cells. Types of Thallasemia 1) Thalassemia trait 2)
Thalassemia A group of inherited disorders characterized by reduced or absent amounts of hemoglobin, the oxygencarrying protein inside the red blood cells. Types of Thallasemia 1) Thalassemia trait 2)