Biance Rowe Chris Hani Baragwanath Hospital Paediatric Haematology Oncology University of the Witwatersrand
|
|
|
- Bartholomew Banks
- 5 years ago
- Views:
Transcription



1 Biance Rowe Chris Hani Baragwanath Hospital Paediatric Haematology Oncology University of the Witwatersrand
2

3 SCD affects million people globally million in Africa children with SCD are born annually worldwide of those are born in Sub Saharan Africa (75%) of those are born in Nigeria The incidence in SA is <1%, but migration across borders has increased the SA & North European prevalence of SCD Red cross Children s Hospital in CT showed an annual frequency increase over 10 years by % Patients mainly from the DRC Davidson A, Wonkam A, Ponde C et al. The burden of sickle cell disease in Cape Town. SAMJ 2012;102(9):27-38.
4 Highest incidence in tropical Africa (equatorial) Cameroon, DRC, Gabon, Ghana, Nigeria, Uganda Also in Arabian peninsula, India, Mediterranean, Caribbean, South America Smaller populations in US & UK Mostly from Sub Saharan african descent

5 Malaria hypothesis states that there is a selective advantage for the heterozygous state (sickle cell trait HbAS) as this is protective against Plasmodium falciparum malaria; not true for HbSS Red cells lyse too quickly for the parasite to complete its life cycle Parasites don t survive in sickle polymers


6
7 There is a higher mortality rate in SCD compared to the non SCD population SCD in Africa Greatest burden of disease & highest mortality Under 5 mortality rate is 50-80% Similar to those reported in the early 1960 s in UK, USA Killers: Infection, acute chest syndrome, acute splenic sequestration More developed countries 85.6% survive to age 18yrs in USA 84% in Jamaica, 99% in USA Savers: Early diagnosis, comprehensive care, access to immunisations & prophylactic antibiotics, hydroxyurea, regular transfusion programme, bone marrow transplant



8
9 Ogbanje is an evil spirit that would deliberately plague a family with misfortune Literal translation is children who come and go It was believed that after a certain amount of time from birth the Ogbanje would deliberately die and then come back and be reborn as another child and die again and cause the family grief and so the cycle will continue Strong beliefs in reincarnation theory
10 Reincarnate child (Ogbanje) was well at birth, from early childhood suffered poor growth, body aches and pains, fevers, protuberant abdomen and yellow eyes Seen as a weakling by peers,was thin Died repeatedly (usually before age of 5) and would be reborn to go through the same cycle again Suffered from SCD Initial well period was the time before the switch to HbS occurred. Chronic ill health was the result of chronic haemolytic anaemia, painful crises, fevers from bacterial infection and malaria Reincarnation was the birth of another affected child (25% chance) OGBANJE SICKLE CELL CHILD
11 1846 Autopsy of an executed runaway slave key finding was the absence of a spleen Reports among African slaves in the USA exhibiting a resistance to malaria but are prone to leg ulcers 1910 Dr James Herrick, Chicago Peculiar, elongated and sickle-shaped red blood cells in a dental student from West Africa Presented with anaemia, rheumatism, bilious attacks completed dental studies but died of pneumonia 1933 Distinguish SCD from trait 1950 s Molecular change in HbS described & malaria link described 1954 HB electrophoresis allowed classification of different types of SCD Infectious prophylaxis, antibiotics 1990 Development of Hydroxyurea 2007 Reports of cure through bone marrow transplantation
12

13 HbS results from the substitution of Valine for Glutamic acid at position 6 of the Beta-globin polypeptide chain of Hb Caused by a singlebase mutation in codon 6 within the B-globin gene on chromosome 11

")

14 The abnormal amino acid in the beta chain forms long, insoluble polymers when deoxygenated and the RBC becomes less deformable Abnormal rigid shape occlude small blood vessels PLUS Expression of adhesion molecules (integrins, selectins) interacting with endothelial cells and plasma proteins PLUS Reduced NO levels due to free Hb consuming NO and reducing vasodilator tone










15 Widespread multiorgan damage and dysfunction caused by episodes of tissue ischaemia Leads to a myriad of acute and chronic complications

16


17


18
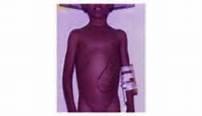
19 Image of man p3 of new opportunities article with clinical consequences

20
, abdomen (mesenteric")

21 The most common acute presentation Caused by microvascular occlusion/sludging and tissue ischaemia and infarction Common in hands & feet of young children Also in arms, legs, axial skeleton (back & shoulders), abdomen (mesenteric vessels), ribs, soft tissues, kidneys, eyes, lungs Can occur in the spleen and lead to an acute splenic sequestration crisis with exsanguination and severe anaemia Episodes of ischaemia & infarction lead to PAIN and MULTI-ORGAN DAMAGE
 F: Folate deficiency G: General surgery H: Hypoxia I:")
22 A: Acidosis, anaesthesia, anxiety, altitude (high) B: Bouts of infection, bad habits (smoking, alcohol) C: Cold exposure D: Dehydration E: Exercise (vigorous) F: Folate deficiency G: General surgery H: Hypoxia I: Infection
 Analgesia should be administered within 30 minutes of arrival at the hospital Pain often excruciating and lead to despair and panic Inadequate pain management can lead to chronic pain")
23 Hydration Oral or IV Pain management Simple oral analgesia NSAIDS, paracetamol, codeine Tilidine Strong opioids Morphine drug of choice parenteral infusion titrated against pain (PCA-devices in older patients) Analgesia should be administered within 30 minutes of arrival at the hospital Pain often excruciating and lead to despair and panic Inadequate pain management can lead to chronic pain syndromes Incentive spirometry NB. especially with chest pain/rib pain to avoid progression to acute chest syndrome Antibiotics Fever often accompany a crisis guided investigations to exclude a source (blood, urine) & empiric antibiotics Individualised plan for managing a painful crisis

24 Avoid / early management of triggers +

25 Cytotoxic agent that inhibits ribonucleotide reductase Used in myeloproliferative disorders Mechanisms of action: INCREASES THE LEVEL OF HbF HbF retards the sickling process & does not participate in the formation of sickle tactoids More HbF = Less HbS DECREASES EXPRESSION OF ADHESION MOLECULES ON TACTOIDS Makes them less sticky UPREGULATES NITRIC OXIDE EXPRESSION Vasodilates CAUSES MILD MYELOSUPPRESSION Reduces adherent leukocytes to the walls of small venules and improves the flow of blood

26 Multicenter Hydroxyurea Trial 1995 Clearly demonstrated the efficacy and safety of hydroxyurea in reducing the frequency of painful crises, acute chest syndrome and transfusions in patients with sickle cell disease of all ages

27 BABY HUG Trial (2011) Multicentre, randomised, double-blind placebo-controlled trial on the use of hydroxyurea in infants (from 9-18 months of age)with SCD Statistically significant lower rates of initial & recurrent episodes of pain, dactylitis, acute chest syndrome, hospitalization and mortality Infants who were asymptomatic at enrollment also had less dactylitis and fewer hospitalisations and transfusions if treated with hydroxyurea It did not show a decrease in splenic and renal damage Mild myelosuppression noted but hydroxyurea was NOT associated with an increased risk of bacteraemia or serious infections nor any longterm complications of carcinogenicity, infertility Provides evidence that hydroxyurea can be safely used even in very young infants regardless of symptoms SA RECOMMENDATION is that hydroxyurea therapy should be instituted in children with SCD >2years of age 10-15mg/kg/d increased to 30mg/kg/d Stop only if neutrophil count below 1000 or when conception planned
28

29 Devastating complication Due to the occlusion of cerebral microvasculature Usually ischaemic in children The incidence is highest between 2-11 years At age 14 years: 10% of children with HbS will have OVERT stroke 20-35% will have silent infarctions with changes on MRI causing cognitive decline, developmental delays, school failure
 High cerebral")

30 Children at high risk for stroke can be identified with transcranial Doppler imaging (TCD) High cerebral vessel bloodflow velocities correlate with an increased risk for stroke Conditional: cm/s Elevated: >200cm/s


31 TCD measures the flow velocities in the large intracranial arteries of the circle of Willis Sickle occlusive vasculopathy and resultant large arterial stenosis occurs in the internal carotid artery, middle cerebral and anterior cerebral artery Reduced vessel diameter (stenosis) is inversely proportional to the blood flow through the artery TCD is safe, non-invasive, cost effective and well tolerated by children Recommendation: Screen children with SCD between 2-16years of age ANNUALLY with TCD


32
 Recommendation: Children with abnormal TCD should be on a chronic transfusion programme aiming to keep the")
33 1998 Children with sickle cell disease with a high risk of stroke by transcranial Doppler imaging (>200 cm/s) were randomized to chronic transfusion or usual care Transfusion greatly reduced the risk of a FIRST stroke (90% absolute risk reduction) Recommendation: Children with abnormal TCD should be on a chronic transfusion programme aiming to keep the HbS <30%

34 2005 Randomized controlled trial in which children with sickle cell disease on chronic transfusions as a primary stroke prevention were randomized to continue transfusion or discontinue transfusion. The trial stopped early due to the high rate of reversion to high-risk TCD s % of patients with a first ischaemic stroke had a risk of a second stroke if untreated Chronic transfusions reduce the risk for a second stroke to 10% Recommendation: Patients with abnormal TCD s or following a first stroke should continue indefinitely with long-term transfusions

35 Simple blood transfusions in the acute setting: Splenic sequestration crisis Aplastic crisis Acute chest syndrome Pre-operatively Chronic transfusion therapy refers to regular simple blood transfusions to keep the HbS <30% Prevention of first or subsequent strokes with abnormal TCD Recurrent debilitating VOC Different from exchange transfusions usually done during the acute phase of a stroke, acute chest crisis or priapism to quickly decrease the level of HbS to <30%
36 Red cell and HLA allo-immunisation Red cell allo-antibodies occur in 20-30% of transfused SCD patients Occurs in response to other antigens present on the surface of transfused red cells (E,C,K,Duffy) Cause difficulty in matching blood for transfusion and can cause delayed hemolytic transfusion reactions Cause complications if bone marrow transplantation is considered May be overcome by extended phenotype matching Expensive, availability

 Chelation Indicated when s-ferritin >1000 or >120ml/kg packed cell transfusion Deferioxamine Deferasirox (Exjade) daily oral tablet")
37 Iron overload Each transfused unit of blood has 250mg iron The body has no means to excrete extra iron Iron accumulates in the liver (>>), heart, endocrine organs, lungs etc. Risk of cirrhosis & hepatocellular carcinoma Monitoring for transfusional iron overload s-ferritin MRI (Ferriscan) Measures LIC (liver iron concentration) Chelation Indicated when s-ferritin >1000 or >120ml/kg packed cell transfusion Deferioxamine Deferasirox (Exjade) daily oral tablet dispersed in water or fruit juice Automated erythrocyte apheresis (exchange) Carries less risk of iron overload than top-up simple transfusion
38

39 Children with SCD have an increased susceptibility to infection Functional asplenia Damaged by repeated infarctive VOC episodes Increased susceptibility to infections with encapsulated organisms Streptococcus pneumoniae Neisseria meningitidis Haemophilus influenza (Africa: Additional risk of Staph, Klebsiella, E.coli) Impaired complement activity Zinc deficiency Iron overload Presence of necrotic tissue (leg ulcers) Risk of osteomyelitis Salmonella osteomyelitis Staphylococcus aureus
 Children <2yrs Conjugated vaccine (Prevenar) Conjugated meningococcal C vaccine")
40 Preventive measures: Prophylactic antibiotics Pneumococcal prophylaxis Penicillin VK 125mg bd for children <3years Penicillin VK 250mg bd for children >3years until adolescence Malaria prophylaxis Bed-nets, protective clothing, chemoprophylaxis Pneumococcal vaccine Children >2yrs Unconjugated vaccine (Pneumovax) Children <2yrs Conjugated vaccine (Prevenar) Conjugated meningococcal C vaccine (Menactra) Haemophilus influenza B vaccine Annual influenza vaccine Specific measures: Early recognition and treatment of infections especially if fever 38.5 C Atypical bacterial cover (Mycoplasma) for ACS Incentive spirometry Surgical debridement, IV antibiotics, dressings for leg ulcers

41 Early diagnosis and entry into care programme Newborn screening Genetic councelling/family planning/prenatal diagnosis Identification of heterozygous carriers & pre-marital testing Parental education Folic acid supplementation & good nutrition Early management of complications Appropriate management of pain, anaemia Prophylactic & directed antibiotics Bacterial infection and malaria prophylaxis Immunisations National schedule plus extra Hydroxyurea In all from 9 months Screening TCD, echo in symptomatic patients, opthalmologic checks, microalbuminuria screening Transfusions Exchange Chronic (with ferritin monitoring & chelation)

42

43 The only curative treatment is an allogeneiec bone marrow transplant where the defective host s bone marrow stem cells are replaced with stem cells containing the normal B-globin genotype Major advantage is that SCT controls & stabilises SCD-related organ damage Differs from Hydroxyurea and chronic transfusion therapy which are largely supportive Donor choice historically Best results with matched sibling donor Donor availability a major problem Haploidentical donors.

44 Center for International Blood and Marrow transplant research database 85% of BMT for SCD were from MSD >80% done in children <16 years of age Problem of disease complications and co-morbidities increasing morbidity of BMT in older patients Established nephropathy, pulmonary hypertension, cardiac failure, cerebrovascular disease all worsen the toxicity of the conditioning regimen and increase post-transplant complications Preferred source of stem cells are bone marrow as opposed to peripheral blood stem cells Peripheral blood stem cells have higher risk of GvHD and mortality


45 Clinical variability in the manifestations of sickle cell disease Can range from minimally affected to severely symptomatic BMT generally indicated in individuals with more severe disease but how will one predict which patients will become severely affected before they have too much organ damage? BMT may be considered even in young patients with milder disease with a well-matched sibling donor Want to transplant when the patient is still functionally well with minimal organ damage (young)to minimize the toxicity of the conditioning regimen RISK:BENEFIT RATIO
46

47 Replacing the defective gene (with mutation) with a normal gene Done successfully in the sickle transgenic mouse Progress in humans limited by identification of appropriate vectors & efficacy of gene transfer & low level of expression of globin genes Still in the development phase






48
Sickle cell disease. Fareed Omar 10 March 2018
 Sickle cell disease Fareed Omar 10 March 2018 Physiology Haemoglobin structure HbA2: 2α and 2δ chains (2-3%) HbF: 2α and 2γ chains (
Sickle cell disease Fareed Omar 10 March 2018 Physiology Haemoglobin structure HbA2: 2α and 2δ chains (2-3%) HbF: 2α and 2γ chains (
Sickle Cell Disease and impact on the society
 Sickle Cell Disease and impact on the society Professor Z.A.Jeremiah Ph.D, FRCPath (London) Professor of Haematology and Blood Transfusion Science Niger Delta University, Wilberforce Island Outline What
Sickle Cell Disease and impact on the society Professor Z.A.Jeremiah Ph.D, FRCPath (London) Professor of Haematology and Blood Transfusion Science Niger Delta University, Wilberforce Island Outline What
Sickle Cell Disease. Edward Malters, MD
 Sickle Cell Disease Edward Malters, MD Introduction Vaso-occlusive phenomena and hemolysis are the clinical hallmarks of Sickle Cell Disease (SCD) Inherited disorder due to homozygosity for the abnormal
Sickle Cell Disease Edward Malters, MD Introduction Vaso-occlusive phenomena and hemolysis are the clinical hallmarks of Sickle Cell Disease (SCD) Inherited disorder due to homozygosity for the abnormal
Congenital Haemoglobinopathies
 Congenital Haemoglobinopathies L. DEDEKEN, MD H O P I T A L U N I V E R S I T A I R E D E S E N F A N T S R E I N E F A B I O L A U N I V E R S I T E L I B R E DE B R U X E L L E S Red Blood Cell Disorders
Congenital Haemoglobinopathies L. DEDEKEN, MD H O P I T A L U N I V E R S I T A I R E D E S E N F A N T S R E I N E F A B I O L A U N I V E R S I T E L I B R E DE B R U X E L L E S Red Blood Cell Disorders
Hemolytic anemias (2 of 2)
") Hemolytic anemias (2 of 2) Sickle Cell Anemia The most common familial hemolytic anemia in the world Sickle cell anemia is the prototypical (and most prevalent) hemoglobinopathy Mutation in the β-globin
Hemolytic anemias (2 of 2) Sickle Cell Anemia The most common familial hemolytic anemia in the world Sickle cell anemia is the prototypical (and most prevalent) hemoglobinopathy Mutation in the β-globin
SICKLE CELL DISEASE. Dr. MUBARAK ABDELRAHMAN MD PEDIATRICS AND CHILD HEALTH. Assistant Professor FACULTY OF MEDICINE -JAZAN
 SICKLE CELL DISEASE Dr. MUBARAK ABDELRAHMAN MD PEDIATRICS AND CHILD HEALTH Assistant Professor FACULTY OF MEDICINE -JAZAN Objective: The student should be able: To identify the presentation, diagnosis,
SICKLE CELL DISEASE Dr. MUBARAK ABDELRAHMAN MD PEDIATRICS AND CHILD HEALTH Assistant Professor FACULTY OF MEDICINE -JAZAN Objective: The student should be able: To identify the presentation, diagnosis,
Health Maintenance and Education for Children and Adults
 Health Maintenance and Education for Children and Adults Richard Ward, MSc, MRCP, FRCPath Director, Red Blood Cell Disorders Program, UHN Assistant Professor, Hematology, University of Toronto Chair, Canadian
Health Maintenance and Education for Children and Adults Richard Ward, MSc, MRCP, FRCPath Director, Red Blood Cell Disorders Program, UHN Assistant Professor, Hematology, University of Toronto Chair, Canadian
DONE BY : RaSHA RAKAN & Bushra Saleem
 DONE BY : RaSHA RAKAN & Bushra Saleem Hemolytic anemias (2 of 2) Sickle Cell Anemia The most common familial hemolytic anemia in the world Sickle cell anemia is the prototypical (and most prevalent) hemoglobinopathy
DONE BY : RaSHA RAKAN & Bushra Saleem Hemolytic anemias (2 of 2) Sickle Cell Anemia The most common familial hemolytic anemia in the world Sickle cell anemia is the prototypical (and most prevalent) hemoglobinopathy
Anemia s. Troy Lund MSMS PhD MD
 Anemia s Troy Lund MSMS PhD MD lundx072@umn.edu Hemoglobinopathy/Anemia IOM take home points. 1. How do we identify the condtion? Smear, CBC Solubility Test (SCD) 2. How does it present clincally? 3. How
Anemia s Troy Lund MSMS PhD MD lundx072@umn.edu Hemoglobinopathy/Anemia IOM take home points. 1. How do we identify the condtion? Smear, CBC Solubility Test (SCD) 2. How does it present clincally? 3. How
Sickle Cell Anemia. Sickle cell anemia is an inherited disorder of the blood which occurs when just one base pair substitution
 Rose Farrington and Rachel Nash BIOL 362 Lab M. Bulgarella Genetic Diseases 10/14/2008 Sickle Cell Anemia Introduction Sickle cell anemia is an inherited disorder of the blood which occurs when just one
Rose Farrington and Rachel Nash BIOL 362 Lab M. Bulgarella Genetic Diseases 10/14/2008 Sickle Cell Anemia Introduction Sickle cell anemia is an inherited disorder of the blood which occurs when just one
Hydroxyurea and Transfusion Therapy for the Treatment of Sickle Cell Disease
 Hydroxyurea and Transfusion Therapy for the Treatment of Sickle Cell Disease A Pocket Guide for the Clinician Susan E. Creary, MD, MSc 1 John J. Strouse, MD, PhD 2 1 The Ohio State University School of
Hydroxyurea and Transfusion Therapy for the Treatment of Sickle Cell Disease A Pocket Guide for the Clinician Susan E. Creary, MD, MSc 1 John J. Strouse, MD, PhD 2 1 The Ohio State University School of
Sickle Cell Disease 101. Objectives. What is SCD? 4/20/2016. Discuss the pathophysiology & genetics of Sickle Cell Disease (SCD).
.") Sickle Cell Disease 101 Jennifer Young, RN, MS, CPNP-AC Sickle Cell & Thalassemia Nurse Practitioner Nationwide Children s Hospital Objectives Discuss the pathophysiology & genetics of Sickle Cell Disease
Sickle Cell Disease 101 Jennifer Young, RN, MS, CPNP-AC Sickle Cell & Thalassemia Nurse Practitioner Nationwide Children s Hospital Objectives Discuss the pathophysiology & genetics of Sickle Cell Disease
Putting some hematology into Pediatric Hematology/Oncology: a review of Hemophilia and Sickle Cell Disease in the Pediatric Patient
 Putting some hematology into Pediatric Hematology/Oncology: a review of Hemophilia and Sickle Cell Disease in the Pediatric Patient Kristina Haley, DO March 10, 2012 Jovita Reyes Memorial Pediatric Hematology/Oncology
Putting some hematology into Pediatric Hematology/Oncology: a review of Hemophilia and Sickle Cell Disease in the Pediatric Patient Kristina Haley, DO March 10, 2012 Jovita Reyes Memorial Pediatric Hematology/Oncology
Dependance on chronic transfusion
 Dependance on chronic transfusion Pr Saliou Diop Hematology Blood transfusion Dakar- Sénégal diop@cnts-dakar.sn Introduction Chronic transfusion: Regular use of blood transfusion in patients with chronic
Dependance on chronic transfusion Pr Saliou Diop Hematology Blood transfusion Dakar- Sénégal diop@cnts-dakar.sn Introduction Chronic transfusion: Regular use of blood transfusion in patients with chronic
Rationale for RBC Transfusion in SCD
 Rationale for RBC Transfusion in SCD Dilution of HgbS-containing RBCs via the addition of HgbA-containing cells from the blood of normal donors Suppression of erythropoietin release caused by the rise
Rationale for RBC Transfusion in SCD Dilution of HgbS-containing RBCs via the addition of HgbA-containing cells from the blood of normal donors Suppression of erythropoietin release caused by the rise
Introduction reduction in output alter the amino acid sequence combination
 Sickle cell anemia. Introduction Mutations in the globin genes can cause a quantitative reduction in output from that gene or alter the amino acid sequence of the protein produced or a combination of the
Sickle cell anemia. Introduction Mutations in the globin genes can cause a quantitative reduction in output from that gene or alter the amino acid sequence of the protein produced or a combination of the
Dr Banu Kaya Consultant Haematologist Barts Health NHS Trust Royal London Hospital, London, UK SICKLE CELL AND THALASSAEMIA OVERVIEW
 Dr Banu Kaya Consultant Haematologist Barts Health NHS Trust Royal London Hospital, London, UK SICKLE CELL AND THALASSAEMIA OVERVIEW Objectives Gain awareness of haemoglobinopathy inheritance, pathophysiology
Dr Banu Kaya Consultant Haematologist Barts Health NHS Trust Royal London Hospital, London, UK SICKLE CELL AND THALASSAEMIA OVERVIEW Objectives Gain awareness of haemoglobinopathy inheritance, pathophysiology
How to Write a Life Care Plan for a Child with Hemoglobinopathy
 How to Write a Life Care Plan for a Child with Hemoglobinopathy Tamar Fleischer, BSN, MSN, CNLCP & Mona Yudkoff, RN, MPH, CRRN, CNLCP BalaCare Solutions March 2018 St. Peterburg, Florida What is Hemoglobinopathy?
How to Write a Life Care Plan for a Child with Hemoglobinopathy Tamar Fleischer, BSN, MSN, CNLCP & Mona Yudkoff, RN, MPH, CRRN, CNLCP BalaCare Solutions March 2018 St. Peterburg, Florida What is Hemoglobinopathy?
SICKLE CELL DISEASE TO TREAT OR
 SICKLE CELL DISEASE TO TREAT OR NOT TO TREAT COHEM Barcelona September 8, 2012 Sujit Sheth, M.D. Pediatric Hematology Oncology Disclosures None Outline Morbidity and mortality Definitive therapies Risk
SICKLE CELL DISEASE TO TREAT OR NOT TO TREAT COHEM Barcelona September 8, 2012 Sujit Sheth, M.D. Pediatric Hematology Oncology Disclosures None Outline Morbidity and mortality Definitive therapies Risk
Hydroxycarbamide. Sickle and Thalassaemia Training days. September Dr Sara Stuart-Smith. Why do sickle cells cause pain and organ damage?
 Sickle and Thalassaemia Training days September 2017 Hydroxycarbamide Dr Sara Stuart-Smith Why do sickle cells cause pain and organ damage? Under certain conditions, haemoglobin S forms long rigid strands
Sickle and Thalassaemia Training days September 2017 Hydroxycarbamide Dr Sara Stuart-Smith Why do sickle cells cause pain and organ damage? Under certain conditions, haemoglobin S forms long rigid strands
Thalassaemia. What is thalassaemia? What causes thalassaemia? What are the different types of thalassaemia?
 Thalassaemia Thalassaemia is an inherited condition affecting the blood. There are different types, which vary from a mild condition with no symptoms, to a serious or lifethreatening condition. For the
Thalassaemia Thalassaemia is an inherited condition affecting the blood. There are different types, which vary from a mild condition with no symptoms, to a serious or lifethreatening condition. For the
Sickle cell disease: Complications in adult patients
 Sickle cell disease: Complications in adult patients Dr Sara Stuart-Smith Haematology Consultant Sickle cell and thalassaemia Nurses, AHP and Junior Doctor Training Days September 2017 SCD is caused by
Sickle cell disease: Complications in adult patients Dr Sara Stuart-Smith Haematology Consultant Sickle cell and thalassaemia Nurses, AHP and Junior Doctor Training Days September 2017 SCD is caused by
1 Kattamis et al. Growth of Children with Thalassemia: Effect of Different Transfusion Regimens. Archives of
 Objectives Sickle Cell Anemia and Thalassemia: Transplantation Provide overview of hemoglobinopathies: Sickle cell disease and Thalassemia Discuss approaches to therapy Review recent registry collaboration
Objectives Sickle Cell Anemia and Thalassemia: Transplantation Provide overview of hemoglobinopathies: Sickle cell disease and Thalassemia Discuss approaches to therapy Review recent registry collaboration
Anaemia in Pregnancy
 Anaemia in Pregnancy Definition :anaemia is a pathological condition in which the oxygen-carrying capacity of red blood cells is insufficient to meet the body needs. The WHO : haemoglobin concentration
Anaemia in Pregnancy Definition :anaemia is a pathological condition in which the oxygen-carrying capacity of red blood cells is insufficient to meet the body needs. The WHO : haemoglobin concentration
Lessons in Management of Sickle Cell Disease. CME Conference, Lloyd Erskine Sandiford Centre, November 15, 2015
 Lessons in Management of Sickle Cell Disease CME Conference, Lloyd Erskine Sandiford Centre, November 15, 2015 Age at Death in African SS Disease The greatest chance of death is in the first is in the
Lessons in Management of Sickle Cell Disease CME Conference, Lloyd Erskine Sandiford Centre, November 15, 2015 Age at Death in African SS Disease The greatest chance of death is in the first is in the
THALASSEMIA AND COMPREHENSIVE CARE
 1 THALASSEMIA AND COMPREHENSIVE CARE Melanie Kirby MBBS, FRCP (C), Hospital for Sick Children, Toronto Associate Professor of Paediatrics, University of Toronto. Objectives 2 By the end of this presentation,
1 THALASSEMIA AND COMPREHENSIVE CARE Melanie Kirby MBBS, FRCP (C), Hospital for Sick Children, Toronto Associate Professor of Paediatrics, University of Toronto. Objectives 2 By the end of this presentation,
Comprehensive Care for Children and Adolescents with Sickle Cell Diseases
 Comprehensive Care for Children and Adolescents with Sickle Cell Diseases Objectives To review the system for newborn screening of infants for sickling diseases To provide the framework for a comprehensive
Comprehensive Care for Children and Adolescents with Sickle Cell Diseases Objectives To review the system for newborn screening of infants for sickling diseases To provide the framework for a comprehensive
Transfusion in Sickle Cell Disease What the guidelines [are likely to] say. Dr Bernard Davis Whittington Hospital, London
![Transfusion in Sickle Cell Disease What the guidelines [are likely to] say. Dr Bernard Davis Whittington Hospital, London](/thumbs/85/91702350.jpg "Transfusion in Sickle Cell Disease What the guidelines [are likely to] say. Dr Bernard Davis Whittington Hospital, London") Transfusion in Sickle Cell Disease What the guidelines [are likely to] say Dr Bernard Davis Whittington Hospital, London Background to BCSH Guideline Rationale Current guidance in disparate publications
Transfusion in Sickle Cell Disease What the guidelines [are likely to] say Dr Bernard Davis Whittington Hospital, London Background to BCSH Guideline Rationale Current guidance in disparate publications
Hydroxyurea Treatment for Sickle Cell Disease
 Hydroxyurea Treatment for Sickle Cell Disease Before Hydroxyurea After Hydroxyurea Hydroxyurea Treatment for Sickle Cell Disease This document is not intended to take the place of the care and attention
Hydroxyurea Treatment for Sickle Cell Disease Before Hydroxyurea After Hydroxyurea Hydroxyurea Treatment for Sickle Cell Disease This document is not intended to take the place of the care and attention
HbSC disease is it different and how should we manage it? David Rees Department of Paediatric Haematology, King s College Hospital, London
 HbSC disease is it different and how should we manage it? David Rees Department of Paediatric Haematology, King s College Hospital, London Different types of sickle cell disesease Severe sickle cell disease
HbSC disease is it different and how should we manage it? David Rees Department of Paediatric Haematology, King s College Hospital, London Different types of sickle cell disesease Severe sickle cell disease
Managing Emergencies
 Managing Emergencies Richard Ward, MSc, MRCP, FRCPath Director, Red Blood Cell Disorders Program, UHN Assistant Professor, Hematology, University of Toronto Chair, Canadian Hemoglobinopathy Association
Managing Emergencies Richard Ward, MSc, MRCP, FRCPath Director, Red Blood Cell Disorders Program, UHN Assistant Professor, Hematology, University of Toronto Chair, Canadian Hemoglobinopathy Association
CURRENT RESEARCH STUDIES
 CURRENT RESEARCH STUDIES SCAGO SICKLE CELL RESEARCH DAY MAY 12, 2018 REBECCA LEROUX RN, BSCN, CCRP RED BLOOD CELL DISORDERS PROGRAM, UNIVERSITY HEALTH NETWORK MANUELA MERELLES-PULCINI RN, BSCN, MSN, CCRP
CURRENT RESEARCH STUDIES SCAGO SICKLE CELL RESEARCH DAY MAY 12, 2018 REBECCA LEROUX RN, BSCN, CCRP RED BLOOD CELL DISORDERS PROGRAM, UNIVERSITY HEALTH NETWORK MANUELA MERELLES-PULCINI RN, BSCN, MSN, CCRP
The anaesthetic management of children with sickle cell disease
 The anaesthetic management of children with sickle cell disease Based in part upon Locke, C. Anaesthetic management of sickle cell disease in children. Anaesthesia Tutorial of the Week 153 (2009). Tanya
The anaesthetic management of children with sickle cell disease Based in part upon Locke, C. Anaesthetic management of sickle cell disease in children. Anaesthesia Tutorial of the Week 153 (2009). Tanya
Aneurin Bevan University Health Board Sickle Cell Anaemia and Haemoglobinopathy Screening and Management in Pregnancy Guidelines
 Sickle Cell Anaemia and Haemoglobinopathy Screening and Management in Pregnancy Guidelines N.B. Staff should be discouraged from printing this document. This is to avoid the risk of out of date printed
Sickle Cell Anaemia and Haemoglobinopathy Screening and Management in Pregnancy Guidelines N.B. Staff should be discouraged from printing this document. This is to avoid the risk of out of date printed
Historic and Current Complications in Children with Sickle Cell Disease
 Historic and Current Complications in Children with Sickle Cell Disease Trish McMahon Peterson RN, MSN, CPNP Thomas C. Hofstra MD Children's Hospital Los Angeles Comprehensive Sickle Cell Program Children's
Historic and Current Complications in Children with Sickle Cell Disease Trish McMahon Peterson RN, MSN, CPNP Thomas C. Hofstra MD Children's Hospital Los Angeles Comprehensive Sickle Cell Program Children's
TRANSFUSION PRACTICES IN THE MANAGEMENT OF SICKLE CELL DISEASE AMONG FLORIDA PHYSICIANS
 TRANSFUSION PRACTICES IN THE MANAGEMENT OF SICKLE CELL DISEASE AMONG FLORIDA PHYSICIANS By LEVETTE NICOLE DUNBAR A THESIS PRESENTED TO THE GRADUATE SCHOOL OF THE UNIVERSITY OF FLORIDA IN PARTIAL FULFILLMENT
TRANSFUSION PRACTICES IN THE MANAGEMENT OF SICKLE CELL DISEASE AMONG FLORIDA PHYSICIANS By LEVETTE NICOLE DUNBAR A THESIS PRESENTED TO THE GRADUATE SCHOOL OF THE UNIVERSITY OF FLORIDA IN PARTIAL FULFILLMENT
4 Fahed Al Karmi Sufian Alhafez Dr nayef karadsheh
 4 Fahed Al Karmi Sufian Alhafez Dr nayef karadsheh Genetic variants of hemoglobin Hemoglobinopathies (abnormal variants of hemoglobin) are divided into: 1. Structural abnormalities: Any change in the genes
4 Fahed Al Karmi Sufian Alhafez Dr nayef karadsheh Genetic variants of hemoglobin Hemoglobinopathies (abnormal variants of hemoglobin) are divided into: 1. Structural abnormalities: Any change in the genes
HU: Myths and Facts. Melanie Kirby Associate Professor of Paediatrics
 HU: Myths and Facts Melanie Kirby Associate Professor of Paediatrics SACGO Hamilton, Ontario March 5, 2016 Declaration of Disclosure I have no actual or potential conflict of interest in relation to this
HU: Myths and Facts Melanie Kirby Associate Professor of Paediatrics SACGO Hamilton, Ontario March 5, 2016 Declaration of Disclosure I have no actual or potential conflict of interest in relation to this
Hydroxyurea. for Sickle Cell Disease. A Guide for Starting Treatment. Hydroxyurea is a medicine proven to prevent pain from sickle cell disease.
 Hydroxyurea for Sickle Cell Disease A Guide for Starting Treatment Hydroxyurea is a medicine proven to prevent pain from sickle cell disease. This handbook was created to help answer common questions about
Hydroxyurea for Sickle Cell Disease A Guide for Starting Treatment Hydroxyurea is a medicine proven to prevent pain from sickle cell disease. This handbook was created to help answer common questions about
RBCs Disorders 1. Dr. Nabila Hamdi MD, PhD
 RBCs Disorders 1 Dr. Nabila Hamdi MD, PhD ILOs Discuss the classification of anemia into hypochromic-microcytic, normochromicnormocytic and macrocytic. Categorize laboratory test procedures used in the
RBCs Disorders 1 Dr. Nabila Hamdi MD, PhD ILOs Discuss the classification of anemia into hypochromic-microcytic, normochromicnormocytic and macrocytic. Categorize laboratory test procedures used in the
Good afternoon and thank you for joining us today as we discuss hydroxyurea for the treatment of sickle cell disease. Dr. Emily Meier is a pediatric
 Good afternoon and thank you for joining us today as we discuss hydroxyurea for the treatment of sickle cell disease. Dr. Emily Meier is a pediatric hematologist at the Indiana Hemophilia & Thrombosis
Good afternoon and thank you for joining us today as we discuss hydroxyurea for the treatment of sickle cell disease. Dr. Emily Meier is a pediatric hematologist at the Indiana Hemophilia & Thrombosis
Sickle Cell Diseasechronic. curable disease? Objectives. Why would a family ask about cure for SCD?
 Sickle Cell Diseasechronic illness or curable disease? Gregory M.T. Guilcher MD, FRCPC, FAAP Objectives To review the general principles of hematopoietic stem cell transplantation (HSCT), including risks
Sickle Cell Diseasechronic illness or curable disease? Gregory M.T. Guilcher MD, FRCPC, FAAP Objectives To review the general principles of hematopoietic stem cell transplantation (HSCT), including risks
Pediatric Red Cell Exchange Indications, Benefits, Barriers. View from California Saturday May 9 th ASFA 2015
 Pediatric Red Cell Exchange Indications, Benefits, Barriers View from California Saturday May 9 th ASFA 2015 Red Cell Exchange: Not SCD Recommendations for Red Cell Exchange Indication Procedure Recommendation
Pediatric Red Cell Exchange Indications, Benefits, Barriers View from California Saturday May 9 th ASFA 2015 Red Cell Exchange: Not SCD Recommendations for Red Cell Exchange Indication Procedure Recommendation
Vaccination and prophylaxis for asplenia: Guideline for clinicians
 Vaccination and prophylaxis for asplenia: Guideline for clinicians Adults better health * better care * better value Acknowledgements The Western Australian Committee for Antimicrobials (WACA) would like
Vaccination and prophylaxis for asplenia: Guideline for clinicians Adults better health * better care * better value Acknowledgements The Western Australian Committee for Antimicrobials (WACA) would like
Newborn Screening for Sickle Cell Disease in Africa: Public health meets reality
 13 th September 2016 Newborn Screening for Sickle Cell Disease in Africa: Public health meets reality Kwaku Ohene-Frempong. MD Children s Hospital of Philadelphia Sickle Cell Foundation of Ghana Disclosure
13 th September 2016 Newborn Screening for Sickle Cell Disease in Africa: Public health meets reality Kwaku Ohene-Frempong. MD Children s Hospital of Philadelphia Sickle Cell Foundation of Ghana Disclosure
UNRELATED DONOR TRANSPLANTATION FOR SICKLE CELL DISEASE AN UPDATE
 UNRELATED DONOR TRANSPLANTATION FOR SICKLE CELL DISEASE AN UPDATE Naynesh Kamani, M.D. Children s National Medical Center GW University School of Medicine Washington, DC SCD scope of problem in USA Commonest
UNRELATED DONOR TRANSPLANTATION FOR SICKLE CELL DISEASE AN UPDATE Naynesh Kamani, M.D. Children s National Medical Center GW University School of Medicine Washington, DC SCD scope of problem in USA Commonest
Hydroxurea: A Novel Approach to Optimizing the Health of Pediatric Patients with Sickle Cell Disease. Maa Ohui Quarmyne September 9 th, 2017
 Hydroxurea: A Novel Approach to Optimizing the Health of Pediatric Patients with Sickle Cell Disease Maa Ohui Quarmyne September 9 th, 2017 Outline Sickle Cell Disease Pathophysiology and Clinical Manifestations
Hydroxurea: A Novel Approach to Optimizing the Health of Pediatric Patients with Sickle Cell Disease Maa Ohui Quarmyne September 9 th, 2017 Outline Sickle Cell Disease Pathophysiology and Clinical Manifestations
Blood Transfusion Guidelines in Clinical Practice
 Blood Transfusion Guidelines in Clinical Practice Salwa Hindawi Director of Blood Transfusion Services Associate Professor in Haematology and Transfusion Medicine King Abdalaziz University, Jeddah Saudi
Blood Transfusion Guidelines in Clinical Practice Salwa Hindawi Director of Blood Transfusion Services Associate Professor in Haematology and Transfusion Medicine King Abdalaziz University, Jeddah Saudi
Hydroxyurea Treatment for Sickle Cell Disease
 Hydroxyurea Treatment for Sickle Cell Disease Hydroxyurea Treatment for Sickle Cell Disease 1 This document is not intended to take the place of the care and attention of your personal physician. Our aim
Hydroxyurea Treatment for Sickle Cell Disease Hydroxyurea Treatment for Sickle Cell Disease 1 This document is not intended to take the place of the care and attention of your personal physician. Our aim
An overview of Thalassaemias and Complications
 An overview of Thalassaemias and Complications Haemoglobin Haemoglobin is the most abundant protein in blood, and exists as three main types in normal adults: HbA ( ) - 97% HbA 2 ( ) - 2.5% HbF ( ) - 0.5%
An overview of Thalassaemias and Complications Haemoglobin Haemoglobin is the most abundant protein in blood, and exists as three main types in normal adults: HbA ( ) - 97% HbA 2 ( ) - 2.5% HbF ( ) - 0.5%
Acute Complications of Sickle Cell Disease Case Study 5 year old girl with Hemoglobin SS, weakness and slurred speech
 Acute Complications of Sickle Cell Disease Case Study 5 year old girl with Hemoglobin SS, weakness and slurred speech Beatrice E. Gee, MD Medical Director, Sickle Cell and Hematology Program Children s
Acute Complications of Sickle Cell Disease Case Study 5 year old girl with Hemoglobin SS, weakness and slurred speech Beatrice E. Gee, MD Medical Director, Sickle Cell and Hematology Program Children s
Making Hope A Reality December 10, Nasdaq : BLUE
 Making Hope A Reality December 10, 2014 Nasdaq : BLUE Forward Looking Statement These slides and the accompanying oral presentation contain forward-looking statements and information. The use of words
Making Hope A Reality December 10, 2014 Nasdaq : BLUE Forward Looking Statement These slides and the accompanying oral presentation contain forward-looking statements and information. The use of words
Screening for haemoglobinopathies in pregnancy
 Policy Statement All Southern Health patients will receive clinical care that reflects best practice and is based on the best available evidence. Index of chapters within background 1. Prevalence of haemoglobinopathies
Policy Statement All Southern Health patients will receive clinical care that reflects best practice and is based on the best available evidence. Index of chapters within background 1. Prevalence of haemoglobinopathies
In adults, the predominant Hb (HbA) molecule has four chains: two α and two β chains. In thalassemias, the synthesis of either the α or the β chains
 molecule has four chains: two α and two β chains. In thalassemias, the synthesis of either the α or the β chains") Thalassaemias Thalassemia Thalassemia is an inherited autosomal recessive blood disease. Associated with absence or reduction in a or b globin chains. Reduced synthesis of one of the globin chains can
Thalassaemias Thalassemia Thalassemia is an inherited autosomal recessive blood disease. Associated with absence or reduction in a or b globin chains. Reduced synthesis of one of the globin chains can
COHEM Barcellona 2012 Hemoglobinopathies debate
 COHEM Barcellona 2012 Hemoglobinopathies debate September 8, 2012: h. 10:30-12:00 Hall: A Is it justified to perform BMT in hemoglobinopathies using unrelated and/or partially mismatched donors? HSCT indication
COHEM Barcellona 2012 Hemoglobinopathies debate September 8, 2012: h. 10:30-12:00 Hall: A Is it justified to perform BMT in hemoglobinopathies using unrelated and/or partially mismatched donors? HSCT indication
Hemoglobinopathies NORMAL HEMOGLOBINS
 Hemoglobinopathies Millicent Sutton MD October 28, 2005 NORMAL HEMOGLOBINS Consist of 2 alpha chains and 2 non alpha chains Hb A = α2β2 Hb F= α 2γ2 Hb A2 = α2δ2 1 Hemoglobin Variants Altered the conformational
Hemoglobinopathies Millicent Sutton MD October 28, 2005 NORMAL HEMOGLOBINS Consist of 2 alpha chains and 2 non alpha chains Hb A = α2β2 Hb F= α 2γ2 Hb A2 = α2δ2 1 Hemoglobin Variants Altered the conformational
SICKLE CELL BROCHURE
 SICKLE CELL BROCHURE SICKLE CELL DIESEASE According to CDC, Sickle cell disease (SCD) is a group of inherited red blood cell disorders. Healthy red blood cells are round and SCD C -shaped farm tool called
SICKLE CELL BROCHURE SICKLE CELL DIESEASE According to CDC, Sickle cell disease (SCD) is a group of inherited red blood cell disorders. Healthy red blood cells are round and SCD C -shaped farm tool called
Acute vaso-occlusive Pain in children
 Acute vaso-occlusive Pain in children Dr François Angoulvant 1 Dr Sebastien Redant 2 Dr Malika Benkerrou 1 Pr Alina Ferster 2 Hôpital Robert Debré - Paris Hôpital des Enfants Reine Fabiola Bruxelles Réseau
Acute vaso-occlusive Pain in children Dr François Angoulvant 1 Dr Sebastien Redant 2 Dr Malika Benkerrou 1 Pr Alina Ferster 2 Hôpital Robert Debré - Paris Hôpital des Enfants Reine Fabiola Bruxelles Réseau
Are We There Yet? Gene Therapy and BMT as Curative Therapies in Sickle Cell. Ann Haight, MD 9 Sept 2017
 Are We There Yet? Gene Therapy and BMT as Curative Therapies in Sickle Cell Ann Haight, MD 9 Sept 2017 Spoiler alert Yes (we have a cure) And No Work to do! 2 Sickle Cell Treatment Options Supportive Care
Are We There Yet? Gene Therapy and BMT as Curative Therapies in Sickle Cell Ann Haight, MD 9 Sept 2017 Spoiler alert Yes (we have a cure) And No Work to do! 2 Sickle Cell Treatment Options Supportive Care
Disclosure. Hemoglobinopathies. Screening for Hemoglobinopthies. Learning Objectives. Screening for Hemoglobinopthies. Interpreting Reports
 Disclosure Hemoglobinopathies (everything you wanted to know but were afraid to ask) Melissa Frei-Jones, MD MSCI Pediatric Grand Rounds February 21, 2014 I have no relationships with commercial companies
Disclosure Hemoglobinopathies (everything you wanted to know but were afraid to ask) Melissa Frei-Jones, MD MSCI Pediatric Grand Rounds February 21, 2014 I have no relationships with commercial companies
Is there a rationale for treatment of sickle cell anemia, except for acute complications?
 Is there a rationale for treatment of sickle cell anemia, NO, but JL Vives Corrons Red Cell Pathology Unit Hospital Clnic. University of Barcelona Head of ENERCA Project EUROPEAN NETWORK FOR RARE AND CONGENITAL
Is there a rationale for treatment of sickle cell anemia, NO, but JL Vives Corrons Red Cell Pathology Unit Hospital Clnic. University of Barcelona Head of ENERCA Project EUROPEAN NETWORK FOR RARE AND CONGENITAL
SICKLE CELL DISEASE 1
 SICKLE CELL DISEASE 1 Your topic: My topic is about sickle cell disease, I need someone to write an introduction about the disease and the effects it has on the individual and family, also have to include
SICKLE CELL DISEASE 1 Your topic: My topic is about sickle cell disease, I need someone to write an introduction about the disease and the effects it has on the individual and family, also have to include
Sickle Cell Disease- Case studies
 Sickle Cell Disease- Case studies Subarna Chakravorty King s College Hospital 2017 Conditions requiring immediate admission Agonising pain (i.e. requiring opiate analgesia) Increased pallor, breathlessness,
Sickle Cell Disease- Case studies Subarna Chakravorty King s College Hospital 2017 Conditions requiring immediate admission Agonising pain (i.e. requiring opiate analgesia) Increased pallor, breathlessness,
4 Jumana Jihad Dr. Ahmad Mansour Dr. Ahmad Mansour
 4 Jumana Jihad Dr. Ahmad Mansour Dr. Ahmad Mansour Anemia Decreased blood production Increased blood loss Hemolytic Hemorrhage Extravascular Intravascular Hemolytic (Further classification( Extrinsic Intrinsic
4 Jumana Jihad Dr. Ahmad Mansour Dr. Ahmad Mansour Anemia Decreased blood production Increased blood loss Hemolytic Hemorrhage Extravascular Intravascular Hemolytic (Further classification( Extrinsic Intrinsic
MI Newborn Screening Program: An Overview of Follow-up & Case Management for Sickle Cell Conditions
 Michigan Department of Community Health MI Newborn Screening Program: An Overview of Follow-up & Case Management for Sickle Cell Conditions Dominic Smith, MSA - Linda Carter, BSW - Ben Frazier, BSW - Ruth
Michigan Department of Community Health MI Newborn Screening Program: An Overview of Follow-up & Case Management for Sickle Cell Conditions Dominic Smith, MSA - Linda Carter, BSW - Ben Frazier, BSW - Ruth
Clinical Guidelines on the Use of Iron Chelation in Children Receiving Regular Blood Transfusions
 Clinical Guidelines on the Use of Iron Chelation in Children Receiving Regular Blood Transfusions Version: 1 Date: 4 th May 2010 Authors: Responsible committee or Director: Review date: Target audience:
Clinical Guidelines on the Use of Iron Chelation in Children Receiving Regular Blood Transfusions Version: 1 Date: 4 th May 2010 Authors: Responsible committee or Director: Review date: Target audience:
Original Article:http://www.mayoclinic.com/health/sickle-cell-anemia/DS00324
 1 Original Article:http://www.mayoclinic.com/health/sickle-cell-anemia/DS00324 Sickle Cell Anemia [Mayo Clinic Staff. Sickle cell anemia. MayoClinic.com; Mar 28, 2007: http://www.mayoclinic.com/health/sickle-cell-anemia/ds00324]
1 Original Article:http://www.mayoclinic.com/health/sickle-cell-anemia/DS00324 Sickle Cell Anemia [Mayo Clinic Staff. Sickle cell anemia. MayoClinic.com; Mar 28, 2007: http://www.mayoclinic.com/health/sickle-cell-anemia/ds00324]
Extra Notes 3. Warm. In the core (center) of the body, where the temperature is 37 C.
 of the body, where the temperature is 37 C.") Extra Notes 3 *The numbers of the slides are according to the last year slides. Slide 33 Autoimmune hemolytic anemia : Abnormal circulating antibodies that target normal antigen on the RBC and cause lysis.
Extra Notes 3 *The numbers of the slides are according to the last year slides. Slide 33 Autoimmune hemolytic anemia : Abnormal circulating antibodies that target normal antigen on the RBC and cause lysis.
The Distribution of Human Differences. If all this genetic variation is so recent and continuous, why do we think of it in categorical terms?
 Expansion Routes of Homo sapiens ~40-25,000 b.p. The Distribution of Human Differences ~120-100,000 b.p. ~50-40,000 b.p. ~20-15,000 b.p. - - - Coastal Route Circa 10-3,500 b.p. If all this genetic variation
Expansion Routes of Homo sapiens ~40-25,000 b.p. The Distribution of Human Differences ~120-100,000 b.p. ~50-40,000 b.p. ~20-15,000 b.p. - - - Coastal Route Circa 10-3,500 b.p. If all this genetic variation
Tenth Visit posttest
 Test Code 10C Patient s name: Tenth Visit posttest Patient s birth date: Your name and relationship to patient: Today s date: 1. Which one of the medications listed below should every child with a sickle
Test Code 10C Patient s name: Tenth Visit posttest Patient s birth date: Your name and relationship to patient: Today s date: 1. Which one of the medications listed below should every child with a sickle
If these vaccines haven t been given, please follow guidelines below for emergency procedures.
 MANAGEMENT OF PATIIENTS POST SPLENECTOMY & HYPOSPLENIIC PATIIENTS Splenectomised and hyposplenic patients are at increased risk of life-threatening infections due to encapsulated micro-organisms such as
MANAGEMENT OF PATIIENTS POST SPLENECTOMY & HYPOSPLENIIC PATIIENTS Splenectomised and hyposplenic patients are at increased risk of life-threatening infections due to encapsulated micro-organisms such as
The Distribution of Human Differences. If all this genetic variation is so recent and continuous, why do we think of it in categorical terms?
 Expansion Routes of Homo sapiens ~40-25,000 b.p. The Distribution of Human Differences ~120-100,000 b.p. ~50-40,000 b.p. ~20-15,000 b.p. - - - Coastal Route Circa 10-3,500 b.p. If all this genetic variation
Expansion Routes of Homo sapiens ~40-25,000 b.p. The Distribution of Human Differences ~120-100,000 b.p. ~50-40,000 b.p. ~20-15,000 b.p. - - - Coastal Route Circa 10-3,500 b.p. If all this genetic variation
Sickle Cell Anemia A Fictional Reconstruction Answer Key
 We have made it easy for you to find a PDF Ebooks without any digging. And by having access to our ebooks online or by storing it on your computer, you have convenient answers with sickle cell anemia a
We have made it easy for you to find a PDF Ebooks without any digging. And by having access to our ebooks online or by storing it on your computer, you have convenient answers with sickle cell anemia a
Carrying Beta Thalassaemia A carrier can use this booklet to
 Carrying Beta Thalassaemia A carrier can use this booklet to help explain carrying beta to their partner, blood relatives and others. show to any health professional (doctor, nurse or midwife) they see
Carrying Beta Thalassaemia A carrier can use this booklet to help explain carrying beta to their partner, blood relatives and others. show to any health professional (doctor, nurse or midwife) they see
Chem*3560 Lecture 4: Inherited modifications in hemoglobin
 Chem*3560 Lecture 4: Inherited modifications in hemoglobin Genetic modifications fall into two classes: Thalassemias, which are the result of failure to express globin genes. Thalassa is Greek for the
Chem*3560 Lecture 4: Inherited modifications in hemoglobin Genetic modifications fall into two classes: Thalassemias, which are the result of failure to express globin genes. Thalassa is Greek for the
The Child with a Hematologic Alteration
 47 The Child with a Hematologic Alteration HELPFUL HINT Review the anatomy and physiology of the hematologic system in an anatomy and physiology textbook. MATCHING KEY TERMS Match the term with the correct
47 The Child with a Hematologic Alteration HELPFUL HINT Review the anatomy and physiology of the hematologic system in an anatomy and physiology textbook. MATCHING KEY TERMS Match the term with the correct
SICKLE CELL AWARENESS. The Sickle Cell Society has produced the following information leaflets available at sicklecellsociety.org
 sickle cell disease in the UK Sickle cell disease (SCD) affects around 15,000 people in the UK People with Sickle Cell Disease have Sickle haemoglobin (HbS) which can make red blood cells rigid and sickle-shaped
sickle cell disease in the UK Sickle cell disease (SCD) affects around 15,000 people in the UK People with Sickle Cell Disease have Sickle haemoglobin (HbS) which can make red blood cells rigid and sickle-shaped
Division of General Internal Medicine and Geriatrics Hospital Medicine 2014
 Division of General Internal Medicine and Geriatrics Hospital Medicine 2014 Objectives Understand workup of acute pain crisis Identify key aspects of management of acute pain crisis in sickle cell patients
Division of General Internal Medicine and Geriatrics Hospital Medicine 2014 Objectives Understand workup of acute pain crisis Identify key aspects of management of acute pain crisis in sickle cell patients
Guidelines for Shared Care Centres and Community Staff
 Reference: CG1411 Written by: Dr Jenny Welch Peer reviewer Dr Jeanette Payne Approved: September 2015 Approved by D&TC: 10 th July 2015 Review Due: September 2018 Intended Audience This document contains
Reference: CG1411 Written by: Dr Jenny Welch Peer reviewer Dr Jeanette Payne Approved: September 2015 Approved by D&TC: 10 th July 2015 Review Due: September 2018 Intended Audience This document contains
Original Research Article Ssafety and efficacy of prolonged hydroxycarbamide administration in adults with
 1 1 2 3 Original Research Article Ssafety and efficacy of prolonged hydroxycarbamide administration in adults with sickle cell disease in Northwestern Greece 4 5 6 7 8 9 10 11 12 13 14 15 16 17 18 19 20
1 1 2 3 Original Research Article Ssafety and efficacy of prolonged hydroxycarbamide administration in adults with sickle cell disease in Northwestern Greece 4 5 6 7 8 9 10 11 12 13 14 15 16 17 18 19 20
Haemoglobinophaties EBMT 2011 Data Manager session
 Haemoglobinophaties EBMT 2011 Data Manager session Presentation plan Biological characteristics Clinical characteristics Transplant resuts What is different From transplant in malignancies Between Thalassemia
Haemoglobinophaties EBMT 2011 Data Manager session Presentation plan Biological characteristics Clinical characteristics Transplant resuts What is different From transplant in malignancies Between Thalassemia
Atlantic Provinces Pediatric Hematology Oncology Network Réseau d Oncologie et Hématologie Pédiatrique des Provinces Atlantiques
 Atlantic Provinces Pediatric Hematology Oncology Network Réseau d Oncologie et Hématologie Pédiatrique des Provinces Atlantiques 5850/5980 University Avenue, PO Box 9700 Halifax, NS, B3K 6R8 1.902.470.7429
Atlantic Provinces Pediatric Hematology Oncology Network Réseau d Oncologie et Hématologie Pédiatrique des Provinces Atlantiques 5850/5980 University Avenue, PO Box 9700 Halifax, NS, B3K 6R8 1.902.470.7429
Medical and Surgical Complications of Sickle Cell Anemia
 Medical and Surgical Complications of Sickle Cell Anemia Ahmed Al-Salem Medical and Surgical Complications of Sickle Cell Anemia Ahmed Al-Salem Department of Surgery Dar A lalafia Medical Company Qatif
Medical and Surgical Complications of Sickle Cell Anemia Ahmed Al-Salem Medical and Surgical Complications of Sickle Cell Anemia Ahmed Al-Salem Department of Surgery Dar A lalafia Medical Company Qatif
Dr. MUNEER ALBAGSHI Consultant Pediatric Hematologist Oncologist- HBDC, Al-Ahsa. Saudi Arabia
 Dr. MUNEER ALBAGSHI Consultant Pediatric Hematologist Oncologist- HBDC, Al-Ahsa. Saudi Arabia Sickle cell is global disease of old world and immigrants to the new world. Sickle cell anemia to predict that
Dr. MUNEER ALBAGSHI Consultant Pediatric Hematologist Oncologist- HBDC, Al-Ahsa. Saudi Arabia Sickle cell is global disease of old world and immigrants to the new world. Sickle cell anemia to predict that
Moving from child to adult sickle cell disease services
 What will happen when I move from child to adult? When you move over from child to adult sickle cell services you will be under the care of the Sickle Cell and Thalassaemia (SCaT) team at City Hospital.
What will happen when I move from child to adult? When you move over from child to adult sickle cell services you will be under the care of the Sickle Cell and Thalassaemia (SCaT) team at City Hospital.
Sickle cell anaemia. GP Education Update 19 th July 2018 Croydon University Hospital. Arne de Kreuk Consultant Haematologist
 Sickle cell anaemia GP Education Update 19 th July 201 Croydon University Hospital Arne de Kreuk Consultant Haematologist Outline: Prevention of sickle cell related complications; annual review Identification
Sickle cell anaemia GP Education Update 19 th July 201 Croydon University Hospital Arne de Kreuk Consultant Haematologist Outline: Prevention of sickle cell related complications; annual review Identification
Sickle cell disease (SCD) and other hemoglobinopathies
 and other hemoglobinopathies") Sickle cell disease (SCD) and other hemoglobinopathies You have received this leaflet, because your child has been diagnosed with sickle cell disease. We can imagine how overwhelming such a diagnosis must
Sickle cell disease (SCD) and other hemoglobinopathies You have received this leaflet, because your child has been diagnosed with sickle cell disease. We can imagine how overwhelming such a diagnosis must
Bridging the Gap: Improving Sickle Cell Disease Transition from Pediatric- to Adult-Focused Care ASFA 2017 Annual Meeting
 Bridging the Gap: Improving Sickle Cell Disease Transition from Pediatric- to Adult-Focused Care ASFA 2017 Annual Meeting Kim Smith-Whitley, MD Director Comprehensive Sickle Cell Center The Children s
Bridging the Gap: Improving Sickle Cell Disease Transition from Pediatric- to Adult-Focused Care ASFA 2017 Annual Meeting Kim Smith-Whitley, MD Director Comprehensive Sickle Cell Center The Children s
The use of red cell genotyping in the management of sickle cell disease
 The use of red cell genotyping in the management of sickle cell disease Dr Sara Trompeter Consultant Haematologist UCLH and NHSBT Dr Keir Pickard Core Medical Trainee UCLH British Blood Transfusion Society
The use of red cell genotyping in the management of sickle cell disease Dr Sara Trompeter Consultant Haematologist UCLH and NHSBT Dr Keir Pickard Core Medical Trainee UCLH British Blood Transfusion Society
World-Wide Distribution of Hemoglobin S. Geographic distribution of hemoglobin S in the world
 Sickle Cell Disease Gerald M. Woods, MD Professor of Pediatrics Division Director, Hematology/Oncology/BMT Director of Sickle Cell Program Children s Mercy Hospitals and Clinics Disclaimer Member of DSMB
Sickle Cell Disease Gerald M. Woods, MD Professor of Pediatrics Division Director, Hematology/Oncology/BMT Director of Sickle Cell Program Children s Mercy Hospitals and Clinics Disclaimer Member of DSMB
Sickle Cell Disease Patient Handbook
 Sickle Cell Disease Patient Handbook , Important Contact Information Office/Unit Phone Number: Outpatient Clinic: 708.684.3898 Make an appointment: 708.684.3113 Please leave a message and a receptionist
Sickle Cell Disease Patient Handbook , Important Contact Information Office/Unit Phone Number: Outpatient Clinic: 708.684.3898 Make an appointment: 708.684.3113 Please leave a message and a receptionist
Blood Transfusions in Children with Haemoglobinopathies
 Blood Transfusions in Children with Haemoglobinopathies Version: 2 Date: 22 nd April 2010 Authors: Responsible committee or Director: Review date: Target audience: Stakeholders/ committees involved in
Blood Transfusions in Children with Haemoglobinopathies Version: 2 Date: 22 nd April 2010 Authors: Responsible committee or Director: Review date: Target audience: Stakeholders/ committees involved in
Thalassemia Maria Luz Uy del Rosario, M.D.
 Thalassemia Maria Luz Uy del Rosario, M.D. Philippine Society of Hematology and Blood Transfusion Philippine Society of Pediatric Oncology What is Thalassemia Hereditary Hemoglobin disorder Hemolytic anemia
Thalassemia Maria Luz Uy del Rosario, M.D. Philippine Society of Hematology and Blood Transfusion Philippine Society of Pediatric Oncology What is Thalassemia Hereditary Hemoglobin disorder Hemolytic anemia
Approach to Hemolysis
 Objectives: Approach to Hemolysis To know the function of platelets and the relationship between the platelet count in peripheral blood and the extent of abnormal bleeding. To know about the diseases associated
Objectives: Approach to Hemolysis To know the function of platelets and the relationship between the platelet count in peripheral blood and the extent of abnormal bleeding. To know about the diseases associated
Bill S-211: Recognizing June 19 as the Canadian Sickle Cell Awareness Day
 Bill S-211: Recognizing June 19 as the Canadian Sickle Cell Awareness Day Sickle Cell Disease (SCD) is the most common genetic disease in the world. According to the World Health Organization (WHO) estimates,
Bill S-211: Recognizing June 19 as the Canadian Sickle Cell Awareness Day Sickle Cell Disease (SCD) is the most common genetic disease in the world. According to the World Health Organization (WHO) estimates,
Every day matters. To help you stay out of the hospital, you can: Live healthy with sickle cell disease
 Living with sickle cell disease (SCD) can be difficult. There can be severe pain and discomfort. AmeriHealth Caritas Louisiana wants to help you manage this life-threatening disease. To help you stay out
Living with sickle cell disease (SCD) can be difficult. There can be severe pain and discomfort. AmeriHealth Caritas Louisiana wants to help you manage this life-threatening disease. To help you stay out
Human Cell Diagram, Parts, Pictures, Structure and Functions
 Human Cell Diagram, Parts, Pictures, Structure and Functions The cell is the basic functional in a human meaning that it is a self-contained and fully operational living entity. Humans are multicellular
Human Cell Diagram, Parts, Pictures, Structure and Functions The cell is the basic functional in a human meaning that it is a self-contained and fully operational living entity. Humans are multicellular
Siklos (hydroxyurea) NEW PRODUCT SLIDESHOW
 NEW PRODUCT SLIDESHOW") Siklos (hydroxyurea) NEW PRODUCT SLIDESHOW Introduction Brand name: Siklos Generic name: Hydroxyurea Pharmacological class: Antimetabolite Strength and Formulation: 100mg, 1000mg+; tabs; +triple-scored
Siklos (hydroxyurea) NEW PRODUCT SLIDESHOW Introduction Brand name: Siklos Generic name: Hydroxyurea Pharmacological class: Antimetabolite Strength and Formulation: 100mg, 1000mg+; tabs; +triple-scored
Post Transplant Management for Sickle Cell. Title
 Post Transplant Management for Sickle Cell Title Kimberly Kasow, DO October 14, 2016 Thank you for this opportunity to present this information I have no financial interests to disclose. Goal of Transplant
Post Transplant Management for Sickle Cell Title Kimberly Kasow, DO October 14, 2016 Thank you for this opportunity to present this information I have no financial interests to disclose. Goal of Transplant