SICKLE-CELL CELL DISEASE IN THE FIFTH DECADE
|
|
|
- Frederica Spencer
- 5 years ago
- Views:
Transcription
1 SICKLE-CELL CELL DISEASE IN THE FIFTH DECADE James J. Stark, MD, FACP Medical Director, Cancer Program Maryview Medical Center Professor of Medicine Eastern Virginia Medical School
2 In 1960 Sir John Dacie described SCD as essentially a disease of childhood. * Estimated survival in 1973: 14.3 years 20% of deaths < 2 years old 13% years old 50% % after age 30 By 1994, 85% survival to age 20 *Dacie JV The haemolytic anaemias: congenital and acquired. New York: Grune & Stratton, 1960.
3 SCD in Adulthood Today Described recently as: ongoing complex management issues of a chronic illness of adulthood, characterized by an inexorable accrual of significant health problems that adversely affect the quality of life. Wierenga et al. Survival estimates for patients with homozygous sickle-cell cell disease in Jamaica: a clinic-based population study. Lancet 2001; 357:
4 Today s s Topics What happens to people who survive SCD into the fifth decade? What is new about the treatment of SCD, and what if anything can be done with these new approaches for this subset of patients?
5 Case : SM 48 y.o. A-A A A woman with severe SCD Spent childhood in Carrsville,, VA (Western Hampton Roads, small rural community) Describes sickly childhood with frequent painful episodes of the hands and feet; not diagnosed until moved to New Jersey and became pregnant Had very difficult pregnancy at age 21; hospitalized for three months after delivery; never attempted pregnancy again. Child has sickle trait. Case Case
6 SM, continued Hospitalized at age 24 for unilateral ureteral obstruction?? Renal papillary necrosis Frequent ICU admissions for Acute Chest Syndrome (ACS) gravely ill each time Frequent exacerbations associated with menses; had respite from illness after hysterectomy at age 35. On HRT (premarin( premarin) Has had laser treatments for proliferative retinopathy Tried on Hydroxyurea but could not afford to purchase it Case Case
7 SM, continued Over the last five years became increasingly ill with increasing frequency and duration of hospitalizations Hematocrit unsupported in high teens; gradually slipped into worsening congestive heart failure Several admissions characterized by acute renal failure responsive to hydration and transfusion Case Case
8 SM, continued In conjunction with SCD team from MCV we embarked on a treatment of prophylactic red-cell transfusions Has markedly reduced frequency of admissions Enabled her to slowly gain weight Improved exercise tolerance and general sense of well-being Serum ferritin 1500 pre-transfusion program, now 6500; attempting to initiate chelation program; finances an issue Has frequent severe transfusion reactions despite using leuko-poor product Case Case
9 SM, continued Other problems remained: Moderate hypertension (BP about 160/90) before change in medicine (see below) Creatinine clearance 47ml/min 24-hour urinary protein 1957 mg; started on Fosinopril (Monopril ) about a year ago as replacement for Amlodipine (Norvasc ) to try to reduce proteinuria; ; daily protein excretion now about 450; BP under better control Creatinine has fallen from 3.0 to 1.7 mg/dl Case Case
10 SM: quality of life, continued Married to man with relatively mild SC disease; remarkable synergy and understanding Despite improvement takes Dilaudid 2 mg up to 100 times per month; intolerant of long-acting opiates Pain is now more manageable whereas was in severe pain much of the time despite analgesics Case Case
11 Sickle-Cell Anemia Single base-pair substitution on chromosome 11 resulting in substitution of valine for glutamic acid at the 6 th position on the ß-chain of hemoglobin molecule Mutation results in hemoglobin polymerization and subsequent rigidity of cells Polymerization results in altered morphology of erythrocytes with irreversible sickling at low ph or p0 2 Case Case
12 The Normal ß-chain of Hemoglobin Case Case
13 Case Case
14 SCD: fundamentals, continued Severity of clinical illness variable; depends in part on other secondary inherited mutations in hemoglobin (varies with geography of Africa) Traditional mainstays of therapy during painful (vaso-occlusive) occlusive) crisis: Analgesics Hydration Supplemental oxygen Transfusions traditionally shunned because of the eventual problem of iron overload All of the above are being re-examined examined today Case Case
15 Nitric Oxide Depletion in SCD: concept in evolution Nitric Oxide critical to maintaining vasodilation under a variety of scenarios Pathologic hemolysis is a hallmark of SCD with dramatically shortened RBC survival Hemolysis from SCD or other conditions causing intravascular hemolysis (e.g., PNH) results in release of free hemoglobin into circulation Hgb remains in circulation until RES can eliminate it Bad things result from this. Case Case




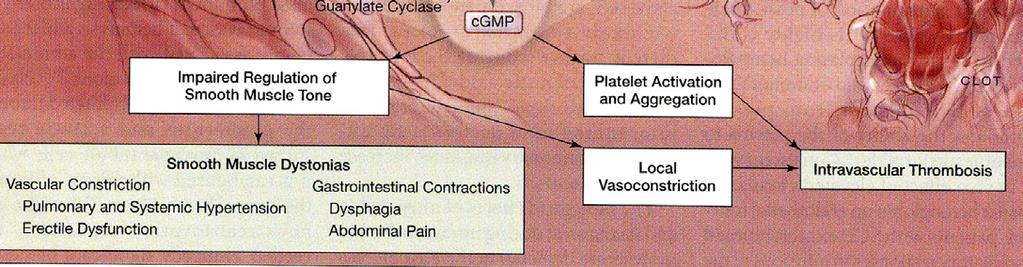
16 Consequences of Hemolysis to Nitric Oxide Metabolism Key event NO converted to Nitrate (NO3) Downstream events
17 Damage from SCD Polymerization of hemoglobin leads to irreversible sickling and occlusion of small blood vessels leading to pain and tissue death Reduction in available Nitric Oxide reduces major mechanism to compensate for vaso- occlusion Leads to irreversible vaso-occlusion occlusion and potentially catastrophic tissue damage, e.g., stroke, myocardial infarction, pulmonary infarction Case Case
18 SCD in Adulthood Defining the problem: excess mortality Case Case
19 Survival with Sickle disease, continued Case Case
20 Sorting Out the Excess Mortality: The Cooperative Study of Sickle Cell Disease Funded by NHLBI Followed 3764 patients with SCD, Sickle C disease and Sickle-cell cell-ß-thalassemia 209 patients died during period of observation (10 years) Causes of death analyzed Case Case
21 Causes of death in 171 patients without known underlying organ failure in CSSCD Case Case
22 Deaths in patients with known organ failure 38 out of 209 deaths in CSSCD Case Case
23 Summary of risk factors for early death in adults with SCD (CSSCD) Case Case
24 Overview of Natural History of SCD in Adults Acute Chest Syndrome Chronic Pulmonary Complications Cardiac Disease Renal Disease Infectious Complications Ophthalmologic Disease Chronic Pain Psychosocial Burden Case Case Natural Natural History/Complications History/Complications
25 Acute Chest Syndrome Acute illness with: fever, respiratory symptoms, and new infiltrate on chest x-rayx Higher incidence in children but higher mortality in adults (4-9% per event). Best data from the National Acute Chest Syndrome Study Group Landmark study published: 671 episodes in 538 patients Case Case Natural Natural History/Complications History/Complications
26 Symptoms at Onset of Acute Chest Syndrome key findings (expressed as %) Case Case Natural Natural History/Complications History/Complications
27 Case Case Natural Natural History/Complications History/Complications
28 Case Case Natural Natural History/Complications History/Complications
29 Case Case Natural Natural History/Complications History/Complications
30 Causes of Acute Chest Syndrome Case Case Natural Natural History/Complications History/Complications
31 Pathogens Isolated in 570 Episodes of Acute Chest Syndrome Case Case Natural Natural History/Complications History/Complications
32 Treatment: ACS, continued Oxygen Transfusion to decrease % sickle cells Empiric antibiotics to include Mycoplasma coverage Incentive spirometry Bronchodilators if evidence of airway hyperreactivity??nitric Oxide Conventional wisdom about hydration during crisis may be counterproductive; may increase likelihood of pulmonary edema especially in older patients Severe complication of SCD, with higher mortality in adults Repeated episodes may lead to chronic respiratory insufficiency and shortened life expectancy Case Case Natural Natural History/Complications History/Complications
33 Chronic Pulmonary Complications May be related to frequency of ACS Pulmonary hypertension Treated with usual measures including nocturnal low-flow oxygen, calcium channel blockers Some enthusiasm for long-term anticoagulation Long-term red-cell transfusion therapy Case Case Natural Natural History/Complications History/Complications
34 Cardiac Complications of SCD Cardiomegaly Elevated cardiac output Hypertension, multifactorial,, including possible role of renal ischemia or focal infarction HBP very deleterious to life-style and outcome Should be treated aggressively especially when associated with proteinuria Autonomic dysfunction contributes to much higher than expected incidence of sudden death Congestive heart failure an ominous development (as with our patient SM) Role of transfusions Case Case Natural Natural History/Complications History/Complications
35 Transfusions in SCD Historically shunned: Iron overload with no convenient way to get rid of burden Alloimmunization with minor blood group antigens making cross- matching increasingly difficult Clearly shorten duration of crisis and can probably lower frequency of crises when given on regular basis Transient enthusiasm for chronic partial exchange transfusions waning in recent years Not usually done prophylactically except in special circumstances (our patient) Oral iron chelating agent in development Case Case Natural Natural History/Complications History/Complications
36 Renal abnormalities in SCD Renal medulla especially susceptible to injury from sickling (hypertonicity,, low ph, hypoxia) Significant loss of vasa recta Hyposthenuria leads to dehydration Renal tubular acidosis Defects in potassium excretion Case Case
37 Renal abnormalities, continued Hematuria may indicate renal papillary necrosis (as with our patient) Acute renal failure seen often during severe acute painful crisis Proteinuria predicts for development of end-stage renal disease May respond to ACE inhibitors Case Case
38 ACE Inhibitors in SCD with Proteinuria Short-term term clinical trials; no long-term data yet Among SCD patients age 40, 7-10% 7 have elevated serum creatinine 25% have at least mild proteinuria Earliest renal lesion is glomerular enlargement During enalapril administration urinary protein 57%; some benefit remained after drug was discontinued No change in blood pressure noted in this study; no deleterious effect on normotensive patients Case Case Falk et al NEJM 326:910-5, 1992
39 Renal Disease, continued Need long-term data on prevention of ESRD with ACE inhibitors At present best advice is strict BP control with use of ACE inhibitors as part of treatment of hypertension and consideration for any SCD patient with significant proteinuria Case Case
40 Infectious Complications Functional asplenia Opsonizing defect in WBC s Excess mortality in children from Strep. Pneumoniae not seen in adults Salmonella infections more common, including osteomyelitis Excess morbidity and mortality from influenza outbreaks, hepatitis B Parvovirus B19 can cause aplastic crisis Case Case Natural Natural History/Complications History/Complications
41 Ophthalmologic Complications Variety of retinal lesions Natural history depends on type of hemoglobinopathy and perhaps on degree of anemia Patients with SC tend to have more retinal problems Lesions are progressive; natural history can be interrupted Traumatic eye injuries (hyphema( hyphema) ) can cause massive intraocular sickling leading to IOP and central retinal artery occlusion When in doubt in ER setting screen for hemoglobinopathy Case Case Eye Eye Complications Complications
42 Stages of retinal changes in SCD Sickle Cell Stages Stage I Stage II Stage III Stage IV Stage V peripheral arteriolar occlusions peripheral arterio-venular anastomoses neovascularization vitreous hemorrhage retinal detachment Case Case Eye Eye Complications Complications


43 Retinal Lesions: peripheral arteriolar occlusions Case Case Eye Eye Complications Complications

44 Sea-fan neovascularization Case Case Eye Eye Complications Complications

45 Sea-fan neovascularization after laser treatment Case Case Eye Eye Complications Complications
46 Sea-fan neovascularization auto-infarcted Case Case Eye Eye Complications Complications

47 Several lesions together Neovascular frond or Sea Fan Choroidal infarct Pre-retinal hemorrhage Case Case Eye Eye Complications Complications
48 Pain Management Hallmark of acute pain is vaso-occlusive occlusive event Typically required several days of analgesics and often in-hospital care Use of meperidine (Demerol ) a problem; often use has to be proscribed by physician Problem of transient euphoria Problem of toxic metabolites; seizures common Problem of short half life Personal favorite is hydromorphone (Dilaudid ) either oral or parenteral; can be given by continuous infusion or PCA with success in acute painful crisis Case Case Management Management
49 Chronic Pain, continued Much bigger problem in the older sickle cell patient is chronic pain Most patients over 40 take some form of narcotic analgesic daily even if they seem outwardly well Pain thought to be from bone infarcts, compression fractures; high incidence of avascular necrosis Little scholarly work available to guide the clinician in management Long-acting narcotics (e.g., fentanyl patch, slow-release morphine products) very useful if patients will agree to take Use of NSAID s s advocated for narcotic-sparing effect, but prevalence of renal disease in older SCD population makes routine use problematic Case Case Management Management
50 Psychosocial Burden of Disease Inability to keep steady job Living in fear of painful crises Family dynamic as the invalid of the family Perception of pariah status among health-care providers Issues related to narcotic use/abuse Racial overtones between patient and health-care professionals No easy solution to above issues Hint: try to see patients outside of hospital setting; use office as staging area to work on disease management, crisis prevention Case Case Management Management
51 Contemporary Management of Sickle Cell Disease: The Hydroxyurea Story Case Case
52 HU and Fetal Hemoglobin Immediately after birth the body switches from making γ chains to ß chains resulting in loss of fetal hemoglobin Fetal hemoglobin is distributes heterogeneously within erythrocytes: About 1/3 of sickle RBC s can have some HbF Total percentage of HbF is, however, usually only about 5% Case Case
53 The Switch from γ to ß Synthesis of γ chains is repressed by methylation of DNA Hypomethylating agents can stimulate production of γ chains First agent tried was 6-azacytidine6 Hydroxyurea then tried safer, easier to use Initial pilot studies showed: HbF and MCV related to dose of HU MTD had to be determined case by case Magnitude of HbF increase related to initial HbF level Case Case
54 The Multicenter Study of HU (MSH) The Study: 299 adults with >3 crises/year randomized to HU or placebo Dose of HU started at 15 mg/kg; modified by toxicity; duration of trial planned at 2 years End points: Number of painful crises Death rate Development of Chest Syndrome Changes in Hb and HbF levels Differences between groups were so striking that the trial was ended prematurely. Case Case Multicenter Multicenter Hydroxyurea Hydroxyurea Study Study
55 Patients at entry to MSH Charache et al NEJM 332:1317, 1995 Case Case Multicenter Multicenter Hydroxyurea Hydroxyurea Study Study
 Case Case Multicenter Multicenter")
56 Painful Crises: HU vs. placebo (rate/year) Case Case Multicenter Multicenter Hydroxyurea Hydroxyurea Study Study

57 % F Cells over time Case Case Multicenter Multicenter Hydroxyurea Hydroxyurea Study Study

58 Sentinel events during treatment Charache et al Medicine (Balt)75:300, 1996 Case Case Multicenter Multicenter Hydroxyurea Hydroxyurea Study Study
59 Current Recommendations for use of Hydroxyurea in SCD Hydroxyurea in all adults with SCD who experience more than 3 painful crises/yr sufficient to require hospitalization Requires high degree of compliance because of potential of myelosuppression To date no evidence of long-term side effects especially myelodysplasia or myeloid leukemia Most likely to be effective if initial HgF is > 5% Case Case Management Management
60 Cutting-Edge Strategies Gene therapy Stem-cell hematopoietic transplantation from HLA-identical donors Case Case Cutting Cutting Edge Edge
61 Gene Therapy for SCD Goal: introduce gene for normal globin synthesis into erythron of SCD patient HIV-based viruses most efficient transmitters of novel genetic material Mouse models show feasibility of approach Case Case Cutting Cutting Edge Edge
62 Murine experimental SCD Transgenic mice created which express Human ß-globin Custom designed ß-globin variant which would prevent HbS polymerization Introduced into mice via lentiviral vector Anti-sickling protein accumulated in 52% of total hemoglobin and 99% of red cells Case Case Cutting Cutting Edge Edge
63 Gene therapy, continued Rendering HIV (lentiviral( lentiviral) ) sequences harmless yet capable of propagation a huge challenge Amplification of new cell line in vivo remains additional challenge How much regular hemoglobin is required to ameliorate syndrome in humans is unknown Alternative approach is to introduce gene to upregulate fetal hemoglobin synthesis Case Case Cutting Cutting Edge Edge
64 Novel Strategies, continued: Bone-Marrow/Stem Marrow/Stem-Cell Transplantation HLA-identical donor can be found among siblings who are not homozygous SS Histocompatibility genes on different chromosomes from Globin genes Major experience thus far in children (mean age 8) 91% survival; 82% survive without clinical SCD 11% frequency of stable mixed chimerism 25% incidence of graft-versus versus-host disease 25% incidence of major neurologic complications (mostly new onset seizures) Case Case Cutting Cutting Edge Edge
65 Current Status of Transplant Morbidity and mortality from procedure remain quite high, as immunologic barriers are formidable Donor pool (unrelated donors) limited African Americans under-represented represented in International Bone- Marrow Registry Probably should be reserved for sickest patients; key is to proceed before end-organ damage precludes procedure; patients over 40 have probably waited too long Timing is difficult By achieving chimeric state one can prevent future crises without totally ablating marrow of recipient Case Case Cutting Cutting Edge Edge
66 Summary of Management Anticipate potential for end-organ damage: Blood-pressure control ACE inhibitors for proteinuria Regular ophthalmologic examinations Consider hydroxyurea for patients with high frequency of painful crises Consider regular transfusions for sickest patients Keep an open mind about potential cutting-edge technologies such as bone-marrow transplantation Recognize potential for catastrophic complications in hospitalized patients Create a nurturing milieu for patients needing chronic care Case Case Management Management
67 Acknowledgements Mrs. SM and her devoted family Dr. Wally Smith at MCV Dr. Richard Snyder, Dept. of Medicine, EVMS Drs. Peter Mitrev and Mark McCarthy for ophthalmologic advice School of Optometry at IU Medical Center Case Case Acknowledgements Acknowledgements
68 For more information and for a hard Visit.. copy of this talk.


69
Sickle Cell Disease. Edward Malters, MD
 Sickle Cell Disease Edward Malters, MD Introduction Vaso-occlusive phenomena and hemolysis are the clinical hallmarks of Sickle Cell Disease (SCD) Inherited disorder due to homozygosity for the abnormal
Sickle Cell Disease Edward Malters, MD Introduction Vaso-occlusive phenomena and hemolysis are the clinical hallmarks of Sickle Cell Disease (SCD) Inherited disorder due to homozygosity for the abnormal
SICKLE CELL DISEASE. Dr. MUBARAK ABDELRAHMAN MD PEDIATRICS AND CHILD HEALTH. Assistant Professor FACULTY OF MEDICINE -JAZAN
 SICKLE CELL DISEASE Dr. MUBARAK ABDELRAHMAN MD PEDIATRICS AND CHILD HEALTH Assistant Professor FACULTY OF MEDICINE -JAZAN Objective: The student should be able: To identify the presentation, diagnosis,
SICKLE CELL DISEASE Dr. MUBARAK ABDELRAHMAN MD PEDIATRICS AND CHILD HEALTH Assistant Professor FACULTY OF MEDICINE -JAZAN Objective: The student should be able: To identify the presentation, diagnosis,
Sickle Cell Disease and impact on the society
 Sickle Cell Disease and impact on the society Professor Z.A.Jeremiah Ph.D, FRCPath (London) Professor of Haematology and Blood Transfusion Science Niger Delta University, Wilberforce Island Outline What
Sickle Cell Disease and impact on the society Professor Z.A.Jeremiah Ph.D, FRCPath (London) Professor of Haematology and Blood Transfusion Science Niger Delta University, Wilberforce Island Outline What
Anemia s. Troy Lund MSMS PhD MD
 Anemia s Troy Lund MSMS PhD MD lundx072@umn.edu Hemoglobinopathy/Anemia IOM take home points. 1. How do we identify the condtion? Smear, CBC Solubility Test (SCD) 2. How does it present clincally? 3. How
Anemia s Troy Lund MSMS PhD MD lundx072@umn.edu Hemoglobinopathy/Anemia IOM take home points. 1. How do we identify the condtion? Smear, CBC Solubility Test (SCD) 2. How does it present clincally? 3. How
Rationale for RBC Transfusion in SCD
 Rationale for RBC Transfusion in SCD Dilution of HgbS-containing RBCs via the addition of HgbA-containing cells from the blood of normal donors Suppression of erythropoietin release caused by the rise
Rationale for RBC Transfusion in SCD Dilution of HgbS-containing RBCs via the addition of HgbA-containing cells from the blood of normal donors Suppression of erythropoietin release caused by the rise
Sickle cell disease. Fareed Omar 10 March 2018
 Sickle cell disease Fareed Omar 10 March 2018 Physiology Haemoglobin structure HbA2: 2α and 2δ chains (2-3%) HbF: 2α and 2γ chains (
Sickle cell disease Fareed Omar 10 March 2018 Physiology Haemoglobin structure HbA2: 2α and 2δ chains (2-3%) HbF: 2α and 2γ chains (
Congenital Haemoglobinopathies
 Congenital Haemoglobinopathies L. DEDEKEN, MD H O P I T A L U N I V E R S I T A I R E D E S E N F A N T S R E I N E F A B I O L A U N I V E R S I T E L I B R E DE B R U X E L L E S Red Blood Cell Disorders
Congenital Haemoglobinopathies L. DEDEKEN, MD H O P I T A L U N I V E R S I T A I R E D E S E N F A N T S R E I N E F A B I O L A U N I V E R S I T E L I B R E DE B R U X E L L E S Red Blood Cell Disorders
How to Write a Life Care Plan for a Child with Hemoglobinopathy
 How to Write a Life Care Plan for a Child with Hemoglobinopathy Tamar Fleischer, BSN, MSN, CNLCP & Mona Yudkoff, RN, MPH, CRRN, CNLCP BalaCare Solutions March 2018 St. Peterburg, Florida What is Hemoglobinopathy?
How to Write a Life Care Plan for a Child with Hemoglobinopathy Tamar Fleischer, BSN, MSN, CNLCP & Mona Yudkoff, RN, MPH, CRRN, CNLCP BalaCare Solutions March 2018 St. Peterburg, Florida What is Hemoglobinopathy?
Hemolytic anemias (2 of 2)
") Hemolytic anemias (2 of 2) Sickle Cell Anemia The most common familial hemolytic anemia in the world Sickle cell anemia is the prototypical (and most prevalent) hemoglobinopathy Mutation in the β-globin
Hemolytic anemias (2 of 2) Sickle Cell Anemia The most common familial hemolytic anemia in the world Sickle cell anemia is the prototypical (and most prevalent) hemoglobinopathy Mutation in the β-globin
Health Maintenance and Education for Children and Adults
 Health Maintenance and Education for Children and Adults Richard Ward, MSc, MRCP, FRCPath Director, Red Blood Cell Disorders Program, UHN Assistant Professor, Hematology, University of Toronto Chair, Canadian
Health Maintenance and Education for Children and Adults Richard Ward, MSc, MRCP, FRCPath Director, Red Blood Cell Disorders Program, UHN Assistant Professor, Hematology, University of Toronto Chair, Canadian
Hydroxyurea and Transfusion Therapy for the Treatment of Sickle Cell Disease
 Hydroxyurea and Transfusion Therapy for the Treatment of Sickle Cell Disease A Pocket Guide for the Clinician Susan E. Creary, MD, MSc 1 John J. Strouse, MD, PhD 2 1 The Ohio State University School of
Hydroxyurea and Transfusion Therapy for the Treatment of Sickle Cell Disease A Pocket Guide for the Clinician Susan E. Creary, MD, MSc 1 John J. Strouse, MD, PhD 2 1 The Ohio State University School of
Dr Banu Kaya Consultant Haematologist Barts Health NHS Trust Royal London Hospital, London, UK SICKLE CELL AND THALASSAEMIA OVERVIEW
 Dr Banu Kaya Consultant Haematologist Barts Health NHS Trust Royal London Hospital, London, UK SICKLE CELL AND THALASSAEMIA OVERVIEW Objectives Gain awareness of haemoglobinopathy inheritance, pathophysiology
Dr Banu Kaya Consultant Haematologist Barts Health NHS Trust Royal London Hospital, London, UK SICKLE CELL AND THALASSAEMIA OVERVIEW Objectives Gain awareness of haemoglobinopathy inheritance, pathophysiology
Extra Notes 3. Warm. In the core (center) of the body, where the temperature is 37 C.
 of the body, where the temperature is 37 C.") Extra Notes 3 *The numbers of the slides are according to the last year slides. Slide 33 Autoimmune hemolytic anemia : Abnormal circulating antibodies that target normal antigen on the RBC and cause lysis.
Extra Notes 3 *The numbers of the slides are according to the last year slides. Slide 33 Autoimmune hemolytic anemia : Abnormal circulating antibodies that target normal antigen on the RBC and cause lysis.
Anaemia in Pregnancy
 Anaemia in Pregnancy Definition :anaemia is a pathological condition in which the oxygen-carrying capacity of red blood cells is insufficient to meet the body needs. The WHO : haemoglobin concentration
Anaemia in Pregnancy Definition :anaemia is a pathological condition in which the oxygen-carrying capacity of red blood cells is insufficient to meet the body needs. The WHO : haemoglobin concentration
Putting some hematology into Pediatric Hematology/Oncology: a review of Hemophilia and Sickle Cell Disease in the Pediatric Patient
 Putting some hematology into Pediatric Hematology/Oncology: a review of Hemophilia and Sickle Cell Disease in the Pediatric Patient Kristina Haley, DO March 10, 2012 Jovita Reyes Memorial Pediatric Hematology/Oncology
Putting some hematology into Pediatric Hematology/Oncology: a review of Hemophilia and Sickle Cell Disease in the Pediatric Patient Kristina Haley, DO March 10, 2012 Jovita Reyes Memorial Pediatric Hematology/Oncology
Division of General Internal Medicine and Geriatrics Hospital Medicine 2014
 Division of General Internal Medicine and Geriatrics Hospital Medicine 2014 Objectives Understand workup of acute pain crisis Identify key aspects of management of acute pain crisis in sickle cell patients
Division of General Internal Medicine and Geriatrics Hospital Medicine 2014 Objectives Understand workup of acute pain crisis Identify key aspects of management of acute pain crisis in sickle cell patients
Sickle Cell Disease Why Is A Simple Genetic Disorder So Hard To Treat And How Are We Doing?
 Sickle Cell Disease Why Is A Simple Genetic Disorder So Hard To Treat And How Are We Doing? James R. Eckman, MD Professor of Medicine, Hematology and Oncology Winship Cancer Institute Emory University
Sickle Cell Disease Why Is A Simple Genetic Disorder So Hard To Treat And How Are We Doing? James R. Eckman, MD Professor of Medicine, Hematology and Oncology Winship Cancer Institute Emory University
HU: Myths and Facts. Melanie Kirby Associate Professor of Paediatrics
 HU: Myths and Facts Melanie Kirby Associate Professor of Paediatrics SACGO Hamilton, Ontario March 5, 2016 Declaration of Disclosure I have no actual or potential conflict of interest in relation to this
HU: Myths and Facts Melanie Kirby Associate Professor of Paediatrics SACGO Hamilton, Ontario March 5, 2016 Declaration of Disclosure I have no actual or potential conflict of interest in relation to this
Foamy Urine and Sickled Cells. Margaret Prat Huntwork, MD, MSEd Tulane / Ochsner Residency Program New Orleans, LA
 Foamy Urine and Sickled Cells Margaret Prat Huntwork, MD, MSEd Tulane / Ochsner Residency Program New Orleans, LA Foamy Urine and Sickled Cells Margaret Prat Huntwork, MD, MSEd Tulane University Health
Foamy Urine and Sickled Cells Margaret Prat Huntwork, MD, MSEd Tulane / Ochsner Residency Program New Orleans, LA Foamy Urine and Sickled Cells Margaret Prat Huntwork, MD, MSEd Tulane University Health
1 Kattamis et al. Growth of Children with Thalassemia: Effect of Different Transfusion Regimens. Archives of
 Objectives Sickle Cell Anemia and Thalassemia: Transplantation Provide overview of hemoglobinopathies: Sickle cell disease and Thalassemia Discuss approaches to therapy Review recent registry collaboration
Objectives Sickle Cell Anemia and Thalassemia: Transplantation Provide overview of hemoglobinopathies: Sickle cell disease and Thalassemia Discuss approaches to therapy Review recent registry collaboration
DONE BY : RaSHA RAKAN & Bushra Saleem
 DONE BY : RaSHA RAKAN & Bushra Saleem Hemolytic anemias (2 of 2) Sickle Cell Anemia The most common familial hemolytic anemia in the world Sickle cell anemia is the prototypical (and most prevalent) hemoglobinopathy
DONE BY : RaSHA RAKAN & Bushra Saleem Hemolytic anemias (2 of 2) Sickle Cell Anemia The most common familial hemolytic anemia in the world Sickle cell anemia is the prototypical (and most prevalent) hemoglobinopathy
Introduction reduction in output alter the amino acid sequence combination
 Sickle cell anemia. Introduction Mutations in the globin genes can cause a quantitative reduction in output from that gene or alter the amino acid sequence of the protein produced or a combination of the
Sickle cell anemia. Introduction Mutations in the globin genes can cause a quantitative reduction in output from that gene or alter the amino acid sequence of the protein produced or a combination of the
Hydroxurea: A Novel Approach to Optimizing the Health of Pediatric Patients with Sickle Cell Disease. Maa Ohui Quarmyne September 9 th, 2017
 Hydroxurea: A Novel Approach to Optimizing the Health of Pediatric Patients with Sickle Cell Disease Maa Ohui Quarmyne September 9 th, 2017 Outline Sickle Cell Disease Pathophysiology and Clinical Manifestations
Hydroxurea: A Novel Approach to Optimizing the Health of Pediatric Patients with Sickle Cell Disease Maa Ohui Quarmyne September 9 th, 2017 Outline Sickle Cell Disease Pathophysiology and Clinical Manifestations
CURRENT RESEARCH STUDIES
 CURRENT RESEARCH STUDIES SCAGO SICKLE CELL RESEARCH DAY MAY 12, 2018 REBECCA LEROUX RN, BSCN, CCRP RED BLOOD CELL DISORDERS PROGRAM, UNIVERSITY HEALTH NETWORK MANUELA MERELLES-PULCINI RN, BSCN, MSN, CCRP
CURRENT RESEARCH STUDIES SCAGO SICKLE CELL RESEARCH DAY MAY 12, 2018 REBECCA LEROUX RN, BSCN, CCRP RED BLOOD CELL DISORDERS PROGRAM, UNIVERSITY HEALTH NETWORK MANUELA MERELLES-PULCINI RN, BSCN, MSN, CCRP
DIC. Disseminated intravascular coagulation, is a life threatening pathological process in which clotting factors are abnormally activated.
 Miss. kamlah 1 DIC Disseminated intravascular coagulation, is a life threatening pathological process in which clotting factors are abnormally activated. Resulting in wide spread of clot formation in the
Miss. kamlah 1 DIC Disseminated intravascular coagulation, is a life threatening pathological process in which clotting factors are abnormally activated. Resulting in wide spread of clot formation in the
TRANSFUSION PRACTICES IN THE MANAGEMENT OF SICKLE CELL DISEASE AMONG FLORIDA PHYSICIANS
 TRANSFUSION PRACTICES IN THE MANAGEMENT OF SICKLE CELL DISEASE AMONG FLORIDA PHYSICIANS By LEVETTE NICOLE DUNBAR A THESIS PRESENTED TO THE GRADUATE SCHOOL OF THE UNIVERSITY OF FLORIDA IN PARTIAL FULFILLMENT
TRANSFUSION PRACTICES IN THE MANAGEMENT OF SICKLE CELL DISEASE AMONG FLORIDA PHYSICIANS By LEVETTE NICOLE DUNBAR A THESIS PRESENTED TO THE GRADUATE SCHOOL OF THE UNIVERSITY OF FLORIDA IN PARTIAL FULFILLMENT
COHEM Barcellona 2012 Hemoglobinopathies debate
 COHEM Barcellona 2012 Hemoglobinopathies debate September 8, 2012: h. 10:30-12:00 Hall: A Is it justified to perform BMT in hemoglobinopathies using unrelated and/or partially mismatched donors? HSCT indication
COHEM Barcellona 2012 Hemoglobinopathies debate September 8, 2012: h. 10:30-12:00 Hall: A Is it justified to perform BMT in hemoglobinopathies using unrelated and/or partially mismatched donors? HSCT indication
Acute vaso-occlusive Pain in children
 Acute vaso-occlusive Pain in children Dr François Angoulvant 1 Dr Sebastien Redant 2 Dr Malika Benkerrou 1 Pr Alina Ferster 2 Hôpital Robert Debré - Paris Hôpital des Enfants Reine Fabiola Bruxelles Réseau
Acute vaso-occlusive Pain in children Dr François Angoulvant 1 Dr Sebastien Redant 2 Dr Malika Benkerrou 1 Pr Alina Ferster 2 Hôpital Robert Debré - Paris Hôpital des Enfants Reine Fabiola Bruxelles Réseau
Transfusion Practices and Creation of a Registry for Patients with Sickle Cell Disease within the Atlanta Sickle Cell Consortium
 Transfusion Practices and Creation of a Registry for Patients with Sickle Cell Disease within the Atlanta Sickle Cell Consortium Annie Winkler MD Assistant Professor, Emory University Department of Pathology
Transfusion Practices and Creation of a Registry for Patients with Sickle Cell Disease within the Atlanta Sickle Cell Consortium Annie Winkler MD Assistant Professor, Emory University Department of Pathology
Hemoglobinopathies NORMAL HEMOGLOBINS
 Hemoglobinopathies Millicent Sutton MD October 28, 2005 NORMAL HEMOGLOBINS Consist of 2 alpha chains and 2 non alpha chains Hb A = α2β2 Hb F= α 2γ2 Hb A2 = α2δ2 1 Hemoglobin Variants Altered the conformational
Hemoglobinopathies Millicent Sutton MD October 28, 2005 NORMAL HEMOGLOBINS Consist of 2 alpha chains and 2 non alpha chains Hb A = α2β2 Hb F= α 2γ2 Hb A2 = α2δ2 1 Hemoglobin Variants Altered the conformational
Sickle Cell Disease 101. Objectives. What is SCD? 4/20/2016. Discuss the pathophysiology & genetics of Sickle Cell Disease (SCD).
.") Sickle Cell Disease 101 Jennifer Young, RN, MS, CPNP-AC Sickle Cell & Thalassemia Nurse Practitioner Nationwide Children s Hospital Objectives Discuss the pathophysiology & genetics of Sickle Cell Disease
Sickle Cell Disease 101 Jennifer Young, RN, MS, CPNP-AC Sickle Cell & Thalassemia Nurse Practitioner Nationwide Children s Hospital Objectives Discuss the pathophysiology & genetics of Sickle Cell Disease
Hydroxyurea. for Sickle Cell Disease. A Guide for Starting Treatment. Hydroxyurea is a medicine proven to prevent pain from sickle cell disease.
 Hydroxyurea for Sickle Cell Disease A Guide for Starting Treatment Hydroxyurea is a medicine proven to prevent pain from sickle cell disease. This handbook was created to help answer common questions about
Hydroxyurea for Sickle Cell Disease A Guide for Starting Treatment Hydroxyurea is a medicine proven to prevent pain from sickle cell disease. This handbook was created to help answer common questions about
4 Jumana Jihad Dr. Ahmad Mansour Dr. Ahmad Mansour
 4 Jumana Jihad Dr. Ahmad Mansour Dr. Ahmad Mansour Anemia Decreased blood production Increased blood loss Hemolytic Hemorrhage Extravascular Intravascular Hemolytic (Further classification( Extrinsic Intrinsic
4 Jumana Jihad Dr. Ahmad Mansour Dr. Ahmad Mansour Anemia Decreased blood production Increased blood loss Hemolytic Hemorrhage Extravascular Intravascular Hemolytic (Further classification( Extrinsic Intrinsic
Sickle Cell Disease: How Should YOU Reassess Management & Treatment?
 Transcript Details This is a transcript of an educational program accessible on the ReachMD network. Details about the program and additional media formats for the program are accessible by visiting: https://reachmd.com/programs/changing-conversation-sickle-cell-disease/sickle-cell-disease-howshould-you-reassess-management-treatment/10184/
Transcript Details This is a transcript of an educational program accessible on the ReachMD network. Details about the program and additional media formats for the program are accessible by visiting: https://reachmd.com/programs/changing-conversation-sickle-cell-disease/sickle-cell-disease-howshould-you-reassess-management-treatment/10184/
Renal Unit. Renal complications of sickle cell disease. Claire Sharpe Reader in Renal medicine King s College London and King s College Hospital
 Renal complications of sickle cell disease Academy for Sickle Cell and Thalassaemia 10th Anniversary Conference Renal Unit Claire Sharpe Reader in Renal medicine King s College London and King s College
Renal complications of sickle cell disease Academy for Sickle Cell and Thalassaemia 10th Anniversary Conference Renal Unit Claire Sharpe Reader in Renal medicine King s College London and King s College
Myeloproliferative Disorders: Diagnostic Enigmas, Therapeutic Dilemmas. James J. Stark, MD, FACP
 Myeloproliferative Disorders: Diagnostic Enigmas, Therapeutic Dilemmas James J. Stark, MD, FACP Medical Director, Cancer Program and Palliative Care Maryview Medical Center Professor of Medicine, EVMS
Myeloproliferative Disorders: Diagnostic Enigmas, Therapeutic Dilemmas James J. Stark, MD, FACP Medical Director, Cancer Program and Palliative Care Maryview Medical Center Professor of Medicine, EVMS
Medical and Surgical Complications of Sickle Cell Anemia
 Medical and Surgical Complications of Sickle Cell Anemia Ahmed Al-Salem Medical and Surgical Complications of Sickle Cell Anemia Ahmed Al-Salem Department of Surgery Dar A lalafia Medical Company Qatif
Medical and Surgical Complications of Sickle Cell Anemia Ahmed Al-Salem Medical and Surgical Complications of Sickle Cell Anemia Ahmed Al-Salem Department of Surgery Dar A lalafia Medical Company Qatif
Full Case: Questions: What is sickle cell crisis?
 Full Case: 30 y/o with avascular necrosis of her right hip was admitted for a total hip arthroplasty. Her hematocrit was 22%, blood pressure was 130/90 mm Hg, and pulse was 107 beats per minute. She had
Full Case: 30 y/o with avascular necrosis of her right hip was admitted for a total hip arthroplasty. Her hematocrit was 22%, blood pressure was 130/90 mm Hg, and pulse was 107 beats per minute. She had
Are We There Yet? Gene Therapy and BMT as Curative Therapies in Sickle Cell. Ann Haight, MD 9 Sept 2017
 Are We There Yet? Gene Therapy and BMT as Curative Therapies in Sickle Cell Ann Haight, MD 9 Sept 2017 Spoiler alert Yes (we have a cure) And No Work to do! 2 Sickle Cell Treatment Options Supportive Care
Are We There Yet? Gene Therapy and BMT as Curative Therapies in Sickle Cell Ann Haight, MD 9 Sept 2017 Spoiler alert Yes (we have a cure) And No Work to do! 2 Sickle Cell Treatment Options Supportive Care
Haemoglobin BY: MUHAMMAD RADWAN WISSAM MUHAMMAD
 Haemoglobin BY: MUHAMMAD RADWAN WISSAM MUHAMMAD Introduction is the iron-containing oxygen transport metalloprotein in the red blood cells Hemoglobin in the blood carries oxygen from the respiratory organs
Haemoglobin BY: MUHAMMAD RADWAN WISSAM MUHAMMAD Introduction is the iron-containing oxygen transport metalloprotein in the red blood cells Hemoglobin in the blood carries oxygen from the respiratory organs
Fetal Anemia 02/13/13. Anjulika Chawla, M.D. Assistant Professor Division of Pediatric Hematology/Oncology
 Fetal Anemia 02/13/13 Anjulika Chawla, M.D. Assistant Professor Division of Pediatric Hematology/Oncology Objectives Definition of anemia Diagnosis of fetal anemia Normal developmental hematopoiesis Etiology
Fetal Anemia 02/13/13 Anjulika Chawla, M.D. Assistant Professor Division of Pediatric Hematology/Oncology Objectives Definition of anemia Diagnosis of fetal anemia Normal developmental hematopoiesis Etiology
CRISPR-mediated Editing of Hematopoietic Stem Cells for the Treatment of β-hemoglobinopathies
 CRISPR-mediated Editing of Hematopoietic Stem Cells for the Treatment of β-hemoglobinopathies Jennifer Gori American Society of Gene & Cell Therapy May 11, 2017 editasmedicine.com 1 Highlights Developed
CRISPR-mediated Editing of Hematopoietic Stem Cells for the Treatment of β-hemoglobinopathies Jennifer Gori American Society of Gene & Cell Therapy May 11, 2017 editasmedicine.com 1 Highlights Developed
SICKLE CELL DISEASE TO TREAT OR
 SICKLE CELL DISEASE TO TREAT OR NOT TO TREAT COHEM Barcelona September 8, 2012 Sujit Sheth, M.D. Pediatric Hematology Oncology Disclosures None Outline Morbidity and mortality Definitive therapies Risk
SICKLE CELL DISEASE TO TREAT OR NOT TO TREAT COHEM Barcelona September 8, 2012 Sujit Sheth, M.D. Pediatric Hematology Oncology Disclosures None Outline Morbidity and mortality Definitive therapies Risk
Making Hope A Reality December 10, Nasdaq : BLUE
 Making Hope A Reality December 10, 2014 Nasdaq : BLUE Forward Looking Statement These slides and the accompanying oral presentation contain forward-looking statements and information. The use of words
Making Hope A Reality December 10, 2014 Nasdaq : BLUE Forward Looking Statement These slides and the accompanying oral presentation contain forward-looking statements and information. The use of words
ANEMIA & HEMODIALYSIS
 ANEMIA & HEMODIALYSIS The anemia of CKD is, in most patients, normocytic and normochromic, and is due primarily to reduced production of erythropoietin by the kidney and to shortened red cell survival.
ANEMIA & HEMODIALYSIS The anemia of CKD is, in most patients, normocytic and normochromic, and is due primarily to reduced production of erythropoietin by the kidney and to shortened red cell survival.
Proceedings of the 34th World Small Animal Veterinary Congress WSAVA 2009
 www.ivis.org Proceedings of the 34th World Small Animal Veterinary Congress WSAVA 2009 São Paulo, Brazil - 2009 Next WSAVA Congress : Reprinted in IVIS with the permission of the Congress Organizers PROTEINURIA
www.ivis.org Proceedings of the 34th World Small Animal Veterinary Congress WSAVA 2009 São Paulo, Brazil - 2009 Next WSAVA Congress : Reprinted in IVIS with the permission of the Congress Organizers PROTEINURIA
The Management of Acute Chest Syndrome in Children with Sickle Cell Disease
 The Management of Acute Chest Syndrome in Children with Sickle Cell Disease Document Information Version: 4 Date: Dec 2013 Authors (incl. job title): Professor David Rees and Dr Sue Height, consultant
The Management of Acute Chest Syndrome in Children with Sickle Cell Disease Document Information Version: 4 Date: Dec 2013 Authors (incl. job title): Professor David Rees and Dr Sue Height, consultant
Chem*3560 Lecture 4: Inherited modifications in hemoglobin
 Chem*3560 Lecture 4: Inherited modifications in hemoglobin Genetic modifications fall into two classes: Thalassemias, which are the result of failure to express globin genes. Thalassa is Greek for the
Chem*3560 Lecture 4: Inherited modifications in hemoglobin Genetic modifications fall into two classes: Thalassemias, which are the result of failure to express globin genes. Thalassa is Greek for the
Instructor s Manual Chapter 26 Hematological Alterations. 1. A man and woman both test positive for the sickle cell trait. The couple asks the
 1 Instructor s Manual Chapter 26 Hematological Alterations Answers to Study Questions 1. A man and woman both test positive for the sickle cell trait. The couple asks the nurse how many of their children
1 Instructor s Manual Chapter 26 Hematological Alterations Answers to Study Questions 1. A man and woman both test positive for the sickle cell trait. The couple asks the nurse how many of their children
Compassionate-use Experience With Voxelotor (GBT440) for Patients With Severe Sickle Cell Disease (SCD) and Life-Threatening Comorbidities
 for Patients With Severe Sickle Cell Disease (SCD) and Life-Threatening Comorbidities") Compassionate-use Experience With Voxelotor (GBT440) for Patients With Severe Sickle Cell Disease (SCD) and Life-Threatening Comorbidities Gershwin Blyden, MD 1, Kenneth R. Bridges, MD 2, Lanetta Bronté,
Compassionate-use Experience With Voxelotor (GBT440) for Patients With Severe Sickle Cell Disease (SCD) and Life-Threatening Comorbidities Gershwin Blyden, MD 1, Kenneth R. Bridges, MD 2, Lanetta Bronté,
Hydroxycarbamide. Sickle and Thalassaemia Training days. September Dr Sara Stuart-Smith. Why do sickle cells cause pain and organ damage?
 Sickle and Thalassaemia Training days September 2017 Hydroxycarbamide Dr Sara Stuart-Smith Why do sickle cells cause pain and organ damage? Under certain conditions, haemoglobin S forms long rigid strands
Sickle and Thalassaemia Training days September 2017 Hydroxycarbamide Dr Sara Stuart-Smith Why do sickle cells cause pain and organ damage? Under certain conditions, haemoglobin S forms long rigid strands
General Characterisctics
 Anemia General Characterisctics Definition: anemia is a decrease in red blood cells. Happens due to underproduction, increased destruction or loss of red cells. Diagnosis of anemia: Hgb < 135 (men) Hgb
Anemia General Characterisctics Definition: anemia is a decrease in red blood cells. Happens due to underproduction, increased destruction or loss of red cells. Diagnosis of anemia: Hgb < 135 (men) Hgb
In adults, the predominant Hb (HbA) molecule has four chains: two α and two β chains. In thalassemias, the synthesis of either the α or the β chains
 molecule has four chains: two α and two β chains. In thalassemias, the synthesis of either the α or the β chains") Thalassaemias Thalassemia Thalassemia is an inherited autosomal recessive blood disease. Associated with absence or reduction in a or b globin chains. Reduced synthesis of one of the globin chains can
Thalassaemias Thalassemia Thalassemia is an inherited autosomal recessive blood disease. Associated with absence or reduction in a or b globin chains. Reduced synthesis of one of the globin chains can
Original Research Article Ssafety and efficacy of prolonged hydroxycarbamide administration in adults with
 1 1 2 3 Original Research Article Ssafety and efficacy of prolonged hydroxycarbamide administration in adults with sickle cell disease in Northwestern Greece 4 5 6 7 8 9 10 11 12 13 14 15 16 17 18 19 20
1 1 2 3 Original Research Article Ssafety and efficacy of prolonged hydroxycarbamide administration in adults with sickle cell disease in Northwestern Greece 4 5 6 7 8 9 10 11 12 13 14 15 16 17 18 19 20
Haemoglobinophaties EBMT 2011 Data Manager session
 Haemoglobinophaties EBMT 2011 Data Manager session Presentation plan Biological characteristics Clinical characteristics Transplant resuts What is different From transplant in malignancies Between Thalassemia
Haemoglobinophaties EBMT 2011 Data Manager session Presentation plan Biological characteristics Clinical characteristics Transplant resuts What is different From transplant in malignancies Between Thalassemia
Screening for haemoglobinopathies in pregnancy
 Policy Statement All Southern Health patients will receive clinical care that reflects best practice and is based on the best available evidence. Index of chapters within background 1. Prevalence of haemoglobinopathies
Policy Statement All Southern Health patients will receive clinical care that reflects best practice and is based on the best available evidence. Index of chapters within background 1. Prevalence of haemoglobinopathies
SCD Advocacy Talking Points!
 "#$!%&'()*)+!,!-././-1!! SCD Advocacy Talking Points! * 23!4*'3!53*673&!84*8!#*59:(679*!#495&637;!9
"#$!%&'()*)+!,!-././-1!! SCD Advocacy Talking Points! * 23!4*'3!53*673&!84*8!#*59:(679*!#495&637;!9
Sickle Cell Anemia A Fictional Reconstruction Answer Key
 We have made it easy for you to find a PDF Ebooks without any digging. And by having access to our ebooks online or by storing it on your computer, you have convenient answers with sickle cell anemia a
We have made it easy for you to find a PDF Ebooks without any digging. And by having access to our ebooks online or by storing it on your computer, you have convenient answers with sickle cell anemia a
Transfusion Pitfalls. Objectives. Packed Red Blood Cells. TRICC trial (subgroups): Is transfusion always good? Components
: Is transfusion always good? Components") Objectives Transfusion Pitfalls Gregory W. Hendey, MD, FACEP Professor and Chief UCSF Fresno, Emergency Medicine To list risks and benefits of various blood products To discuss controversy over liberal
Objectives Transfusion Pitfalls Gregory W. Hendey, MD, FACEP Professor and Chief UCSF Fresno, Emergency Medicine To list risks and benefits of various blood products To discuss controversy over liberal
Good afternoon and thank you for joining us today as we discuss hydroxyurea for the treatment of sickle cell disease. Dr. Emily Meier is a pediatric
 Good afternoon and thank you for joining us today as we discuss hydroxyurea for the treatment of sickle cell disease. Dr. Emily Meier is a pediatric hematologist at the Indiana Hemophilia & Thrombosis
Good afternoon and thank you for joining us today as we discuss hydroxyurea for the treatment of sickle cell disease. Dr. Emily Meier is a pediatric hematologist at the Indiana Hemophilia & Thrombosis
RBCs Disorders 1. Dr. Nabila Hamdi MD, PhD
 RBCs Disorders 1 Dr. Nabila Hamdi MD, PhD ILOs Discuss the classification of anemia into hypochromic-microcytic, normochromicnormocytic and macrocytic. Categorize laboratory test procedures used in the
RBCs Disorders 1 Dr. Nabila Hamdi MD, PhD ILOs Discuss the classification of anemia into hypochromic-microcytic, normochromicnormocytic and macrocytic. Categorize laboratory test procedures used in the
Hemoglobinopathies Diagnosis and management
 Hemoglobinopathies Diagnosis and management Morgan L. McLemore, M.D. Hematology/Leukemia Department of Hematology and Oncology Winship Cancer Institute at Emory University mlmclem@emory.edu Disclosures
Hemoglobinopathies Diagnosis and management Morgan L. McLemore, M.D. Hematology/Leukemia Department of Hematology and Oncology Winship Cancer Institute at Emory University mlmclem@emory.edu Disclosures
Genetics of Thalassemia
 Genetics of Thalassemia Submitted by : Raya Samir Al- Hayaly Sura Zuhair Salih Saad Ghassan Al- Dulaimy Saad Farouq Kassir Sama Naal Salouha Zahraa Jasim Al- Aarajy Supervised by : Dr. Kawkab Adris Mahmod
Genetics of Thalassemia Submitted by : Raya Samir Al- Hayaly Sura Zuhair Salih Saad Ghassan Al- Dulaimy Saad Farouq Kassir Sama Naal Salouha Zahraa Jasim Al- Aarajy Supervised by : Dr. Kawkab Adris Mahmod
Sickle Cell Anemia. Sickle cell anemia is an inherited disorder of the blood which occurs when just one base pair substitution
 Rose Farrington and Rachel Nash BIOL 362 Lab M. Bulgarella Genetic Diseases 10/14/2008 Sickle Cell Anemia Introduction Sickle cell anemia is an inherited disorder of the blood which occurs when just one
Rose Farrington and Rachel Nash BIOL 362 Lab M. Bulgarella Genetic Diseases 10/14/2008 Sickle Cell Anemia Introduction Sickle cell anemia is an inherited disorder of the blood which occurs when just one
MI Newborn Screening Program: An Overview of Follow-up & Case Management for Sickle Cell Conditions
 Michigan Department of Community Health MI Newborn Screening Program: An Overview of Follow-up & Case Management for Sickle Cell Conditions Dominic Smith, MSA - Linda Carter, BSW - Ben Frazier, BSW - Ruth
Michigan Department of Community Health MI Newborn Screening Program: An Overview of Follow-up & Case Management for Sickle Cell Conditions Dominic Smith, MSA - Linda Carter, BSW - Ben Frazier, BSW - Ruth
Aneurin Bevan University Health Board Sickle Cell Anaemia and Haemoglobinopathy Screening and Management in Pregnancy Guidelines
 Sickle Cell Anaemia and Haemoglobinopathy Screening and Management in Pregnancy Guidelines N.B. Staff should be discouraged from printing this document. This is to avoid the risk of out of date printed
Sickle Cell Anaemia and Haemoglobinopathy Screening and Management in Pregnancy Guidelines N.B. Staff should be discouraged from printing this document. This is to avoid the risk of out of date printed
An overview of Thalassaemias and Complications
 An overview of Thalassaemias and Complications Haemoglobin Haemoglobin is the most abundant protein in blood, and exists as three main types in normal adults: HbA ( ) - 97% HbA 2 ( ) - 2.5% HbF ( ) - 0.5%
An overview of Thalassaemias and Complications Haemoglobin Haemoglobin is the most abundant protein in blood, and exists as three main types in normal adults: HbA ( ) - 97% HbA 2 ( ) - 2.5% HbF ( ) - 0.5%
4 Fahed Al Karmi Sufian Alhafez Dr nayef karadsheh
 4 Fahed Al Karmi Sufian Alhafez Dr nayef karadsheh Genetic variants of hemoglobin Hemoglobinopathies (abnormal variants of hemoglobin) are divided into: 1. Structural abnormalities: Any change in the genes
4 Fahed Al Karmi Sufian Alhafez Dr nayef karadsheh Genetic variants of hemoglobin Hemoglobinopathies (abnormal variants of hemoglobin) are divided into: 1. Structural abnormalities: Any change in the genes
SICKLE CELL BROCHURE
 SICKLE CELL BROCHURE SICKLE CELL DIESEASE According to CDC, Sickle cell disease (SCD) is a group of inherited red blood cell disorders. Healthy red blood cells are round and SCD C -shaped farm tool called
SICKLE CELL BROCHURE SICKLE CELL DIESEASE According to CDC, Sickle cell disease (SCD) is a group of inherited red blood cell disorders. Healthy red blood cells are round and SCD C -shaped farm tool called
ASH Draft Recommendations for SCD Related Transfusion Support
 ASH Draft Recommendations for SCD Related Transfusion Support INTRODUCTION American Society of Hematology (ASH) guidelines are based on a systematic review of available evidence. Through a structured process,
ASH Draft Recommendations for SCD Related Transfusion Support INTRODUCTION American Society of Hematology (ASH) guidelines are based on a systematic review of available evidence. Through a structured process,
Overview of Aplastic Anemia. Overview of Aplastic Anemia. Epidemiology of aplastic anemia. Normal hematopoiesis 10/6/2017
 Overview of Aplastic Anemia Overview of Aplastic Anemia Peter Westervelt, MD, PhD Professor of Medicine Chief, BMT/Leukemia Section Washington University School of Medicine Epidemiology Normal hematopoiesis
Overview of Aplastic Anemia Overview of Aplastic Anemia Peter Westervelt, MD, PhD Professor of Medicine Chief, BMT/Leukemia Section Washington University School of Medicine Epidemiology Normal hematopoiesis
Sickle Cell Diseasechronic. curable disease? Objectives. Why would a family ask about cure for SCD?
 Sickle Cell Diseasechronic illness or curable disease? Gregory M.T. Guilcher MD, FRCPC, FAAP Objectives To review the general principles of hematopoietic stem cell transplantation (HSCT), including risks
Sickle Cell Diseasechronic illness or curable disease? Gregory M.T. Guilcher MD, FRCPC, FAAP Objectives To review the general principles of hematopoietic stem cell transplantation (HSCT), including risks
THALASSEMIA AND COMPREHENSIVE CARE
 1 THALASSEMIA AND COMPREHENSIVE CARE Melanie Kirby MBBS, FRCP (C), Hospital for Sick Children, Toronto Associate Professor of Paediatrics, University of Toronto. Objectives 2 By the end of this presentation,
1 THALASSEMIA AND COMPREHENSIVE CARE Melanie Kirby MBBS, FRCP (C), Hospital for Sick Children, Toronto Associate Professor of Paediatrics, University of Toronto. Objectives 2 By the end of this presentation,
Clinical Policy: Allogenic Hematopoietic Cell Transplants for Sickle Cell Anemia and β-thalassemia
 Clinical Policy: Allogenic Hematopoietic Cell Transplants for Sickle Cell Anemia and β-thalassemia Reference Number: CP.MP.108 Effective Date: 03/16 Last Review Date: 03/17 See Important Reminder at the
Clinical Policy: Allogenic Hematopoietic Cell Transplants for Sickle Cell Anemia and β-thalassemia Reference Number: CP.MP.108 Effective Date: 03/16 Last Review Date: 03/17 See Important Reminder at the
High Hemoglobin F in a Saudi Child Presenting with Pancytopenia
 Case Report imedpub Journals http://www.imedpub.com Journal of Pediatric Care ISSN 2471-805X DOI: 10.21767/2471-805X.100002 High Hemoglobin F in a Saudi Child Presenting with Pancytopenia Abstract Saudi
Case Report imedpub Journals http://www.imedpub.com Journal of Pediatric Care ISSN 2471-805X DOI: 10.21767/2471-805X.100002 High Hemoglobin F in a Saudi Child Presenting with Pancytopenia Abstract Saudi
World-Wide Distribution of Hemoglobin S. Geographic distribution of hemoglobin S in the world
 Sickle Cell Disease Gerald M. Woods, MD Professor of Pediatrics Division Director, Hematology/Oncology/BMT Director of Sickle Cell Program Children s Mercy Hospitals and Clinics Disclaimer Member of DSMB
Sickle Cell Disease Gerald M. Woods, MD Professor of Pediatrics Division Director, Hematology/Oncology/BMT Director of Sickle Cell Program Children s Mercy Hospitals and Clinics Disclaimer Member of DSMB
Guideline for the Management of Acute Chest Syndrome in Children with Sickle Cell Disease
 Guideline for the Management of Acute Chest Syndrome in Children with Sickle Cell Disease Definition Acute chest syndrome (ACS) is defined as an acute illness characterized by fever and/or respiratory
Guideline for the Management of Acute Chest Syndrome in Children with Sickle Cell Disease Definition Acute chest syndrome (ACS) is defined as an acute illness characterized by fever and/or respiratory
Transfusions in Sickle Cell Disease: How, When and Why
 Transfusions in Sickle Cell Disease: How, When and Why James R. Eckman, MD Professor Emeritus of Hematology and Medical Oncology Emory University School of Medicine This work is supported by the Centers
Transfusions in Sickle Cell Disease: How, When and Why James R. Eckman, MD Professor Emeritus of Hematology and Medical Oncology Emory University School of Medicine This work is supported by the Centers
Managing Emergencies
 Managing Emergencies Richard Ward, MSc, MRCP, FRCPath Director, Red Blood Cell Disorders Program, UHN Assistant Professor, Hematology, University of Toronto Chair, Canadian Hemoglobinopathy Association
Managing Emergencies Richard Ward, MSc, MRCP, FRCPath Director, Red Blood Cell Disorders Program, UHN Assistant Professor, Hematology, University of Toronto Chair, Canadian Hemoglobinopathy Association
Chronic kidney disease in cats
 Chronic kidney disease in cats What is chronic kidney disease (CKD)? Chronic kidney disease (CKD) is the name now used to refer to cats with kidney failure (or chronic kidney failure). CKD is one of the
Chronic kidney disease in cats What is chronic kidney disease (CKD)? Chronic kidney disease (CKD) is the name now used to refer to cats with kidney failure (or chronic kidney failure). CKD is one of the
Heart disease remains the leading cause of morbidity and mortality in industrialized nations. It accounts for nearly 40% of all deaths in the United
 Heart disease remains the leading cause of morbidity and mortality in industrialized nations. It accounts for nearly 40% of all deaths in the United States, totaling about 750,000 individuals annually
Heart disease remains the leading cause of morbidity and mortality in industrialized nations. It accounts for nearly 40% of all deaths in the United States, totaling about 750,000 individuals annually
Hydroxyurea Treatment for Sickle Cell Disease
 Hydroxyurea Treatment for Sickle Cell Disease Before Hydroxyurea After Hydroxyurea Hydroxyurea Treatment for Sickle Cell Disease This document is not intended to take the place of the care and attention
Hydroxyurea Treatment for Sickle Cell Disease Before Hydroxyurea After Hydroxyurea Hydroxyurea Treatment for Sickle Cell Disease This document is not intended to take the place of the care and attention
Natural History Of Tricuspid Regurgitant Jet Velocity And A New Association With Proteinuria In Children With Sickle Cell Disease
 Yale University EliScholar A Digital Platform for Scholarly Publishing at Yale Yale Medicine Thesis Digital Library School of Medicine January 2013 Natural History Of Tricuspid Regurgitant Jet Velocity
Yale University EliScholar A Digital Platform for Scholarly Publishing at Yale Yale Medicine Thesis Digital Library School of Medicine January 2013 Natural History Of Tricuspid Regurgitant Jet Velocity
Your sickle cell disease story
 YOUR STORY Not actual patients. Your sickle cell disease story From the very beginning of sickle cell disease (SCD) to your role in the next chapter Visit GenSickleCell.com to get involved with the movement.
YOUR STORY Not actual patients. Your sickle cell disease story From the very beginning of sickle cell disease (SCD) to your role in the next chapter Visit GenSickleCell.com to get involved with the movement.
Mantle-Cell Leukemia: Lessons in Life and Death
 Mantle-Cell Leukemia: Lessons in Life and Death James J. Stark, MD, FACP Medical Director, Cancer Program and Palliative Care Maryview Medical Center Professor of Medicine, EVMS Case Presentation 60 y.o.
Mantle-Cell Leukemia: Lessons in Life and Death James J. Stark, MD, FACP Medical Director, Cancer Program and Palliative Care Maryview Medical Center Professor of Medicine, EVMS Case Presentation 60 y.o.
Index. Note: Page numbers of article titles are in boldface type.
 Note: Page numbers of article titles are in boldface type. A Abdominal tumors, in children, 530 531 Alkalinization, in tumor lysis syndrome, 516 Allopurinol, in tumor lysis syndrome, 515 Anaphylaxis, drug
Note: Page numbers of article titles are in boldface type. A Abdominal tumors, in children, 530 531 Alkalinization, in tumor lysis syndrome, 516 Allopurinol, in tumor lysis syndrome, 515 Anaphylaxis, drug
Post Transplant Management for Sickle Cell. Title
 Post Transplant Management for Sickle Cell Title Kimberly Kasow, DO October 14, 2016 Thank you for this opportunity to present this information I have no financial interests to disclose. Goal of Transplant
Post Transplant Management for Sickle Cell Title Kimberly Kasow, DO October 14, 2016 Thank you for this opportunity to present this information I have no financial interests to disclose. Goal of Transplant
Thalassemia Maria Luz Uy del Rosario, M.D.
 Thalassemia Maria Luz Uy del Rosario, M.D. Philippine Society of Hematology and Blood Transfusion Philippine Society of Pediatric Oncology What is Thalassemia Hereditary Hemoglobin disorder Hemolytic anemia
Thalassemia Maria Luz Uy del Rosario, M.D. Philippine Society of Hematology and Blood Transfusion Philippine Society of Pediatric Oncology What is Thalassemia Hereditary Hemoglobin disorder Hemolytic anemia
Comprehensive Care for Children and Adolescents with Sickle Cell Diseases
 Comprehensive Care for Children and Adolescents with Sickle Cell Diseases Objectives To review the system for newborn screening of infants for sickling diseases To provide the framework for a comprehensive
Comprehensive Care for Children and Adolescents with Sickle Cell Diseases Objectives To review the system for newborn screening of infants for sickling diseases To provide the framework for a comprehensive
Dependance on chronic transfusion
 Dependance on chronic transfusion Pr Saliou Diop Hematology Blood transfusion Dakar- Sénégal diop@cnts-dakar.sn Introduction Chronic transfusion: Regular use of blood transfusion in patients with chronic
Dependance on chronic transfusion Pr Saliou Diop Hematology Blood transfusion Dakar- Sénégal diop@cnts-dakar.sn Introduction Chronic transfusion: Regular use of blood transfusion in patients with chronic
Aplastic Anemia: Current Thinking
 Aplastic Anemia: Current Thinking ANDREW C. DIETZ, MD, MSCR PEDIATRIC BLOOD AND MARROW TRANSPLANTATION CHILDREN S HOSPITAL LOS ANGELES, UNIVERSITY OF SOUTHERN CALIFORNIA Outline Ø What is Aplastic Anemia?
Aplastic Anemia: Current Thinking ANDREW C. DIETZ, MD, MSCR PEDIATRIC BLOOD AND MARROW TRANSPLANTATION CHILDREN S HOSPITAL LOS ANGELES, UNIVERSITY OF SOUTHERN CALIFORNIA Outline Ø What is Aplastic Anemia?
RENAL & HEMATOLOGY EMERGENCIES JEFF SIMONS B.S. F-PC
 RENAL & HEMATOLOGY EMERGENCIES JEFF SIMONS B.S. F-PC GOALS Overview of renal system anatomy / physiology Discuss common medical / trauma renal issues Identify associated assessment keys GOALS Introduction
RENAL & HEMATOLOGY EMERGENCIES JEFF SIMONS B.S. F-PC GOALS Overview of renal system anatomy / physiology Discuss common medical / trauma renal issues Identify associated assessment keys GOALS Introduction
The Distribution of Human Differences. If all this genetic variation is so recent and continuous, why do we think of it in categorical terms?
 Expansion Routes of Homo sapiens ~40-25,000 b.p. The Distribution of Human Differences ~120-100,000 b.p. ~50-40,000 b.p. ~20-15,000 b.p. - - - Coastal Route Circa 10-3,500 b.p. If all this genetic variation
Expansion Routes of Homo sapiens ~40-25,000 b.p. The Distribution of Human Differences ~120-100,000 b.p. ~50-40,000 b.p. ~20-15,000 b.p. - - - Coastal Route Circa 10-3,500 b.p. If all this genetic variation
Transfusion in Sickle Cell Disease What the guidelines [are likely to] say. Dr Bernard Davis Whittington Hospital, London
![Transfusion in Sickle Cell Disease What the guidelines [are likely to] say. Dr Bernard Davis Whittington Hospital, London](/thumbs/85/91702350.jpg "Transfusion in Sickle Cell Disease What the guidelines [are likely to] say. Dr Bernard Davis Whittington Hospital, London") Transfusion in Sickle Cell Disease What the guidelines [are likely to] say Dr Bernard Davis Whittington Hospital, London Background to BCSH Guideline Rationale Current guidance in disparate publications
Transfusion in Sickle Cell Disease What the guidelines [are likely to] say Dr Bernard Davis Whittington Hospital, London Background to BCSH Guideline Rationale Current guidance in disparate publications
Historic and Current Complications in Children with Sickle Cell Disease
 Historic and Current Complications in Children with Sickle Cell Disease Trish McMahon Peterson RN, MSN, CPNP Thomas C. Hofstra MD Children's Hospital Los Angeles Comprehensive Sickle Cell Program Children's
Historic and Current Complications in Children with Sickle Cell Disease Trish McMahon Peterson RN, MSN, CPNP Thomas C. Hofstra MD Children's Hospital Los Angeles Comprehensive Sickle Cell Program Children's
HbSC disease is it different and how should we manage it? David Rees Department of Paediatric Haematology, King s College Hospital, London
 HbSC disease is it different and how should we manage it? David Rees Department of Paediatric Haematology, King s College Hospital, London Different types of sickle cell disesease Severe sickle cell disease
HbSC disease is it different and how should we manage it? David Rees Department of Paediatric Haematology, King s College Hospital, London Different types of sickle cell disesease Severe sickle cell disease
Carrying Beta Thalassaemia A carrier can use this booklet to
 Carrying Beta Thalassaemia A carrier can use this booklet to help explain carrying beta to their partner, blood relatives and others. show to any health professional (doctor, nurse or midwife) they see
Carrying Beta Thalassaemia A carrier can use this booklet to help explain carrying beta to their partner, blood relatives and others. show to any health professional (doctor, nurse or midwife) they see
Biology 2C03: Genetics What is a Gene?
 Biology 2C03: Genetics What is a Gene? September 9 th, 2013 Model Organisms - E. coli - Yeast - Worms - Arabodopsis - Fruitflie - Mouse What is a Gene? - Define, recognize, describe and apply Mendel s
Biology 2C03: Genetics What is a Gene? September 9 th, 2013 Model Organisms - E. coli - Yeast - Worms - Arabodopsis - Fruitflie - Mouse What is a Gene? - Define, recognize, describe and apply Mendel s
Dr. MUNEER ALBAGSHI Consultant Pediatric Hematologist Oncologist- HBDC, Al-Ahsa. Saudi Arabia
 Dr. MUNEER ALBAGSHI Consultant Pediatric Hematologist Oncologist- HBDC, Al-Ahsa. Saudi Arabia Sickle cell is global disease of old world and immigrants to the new world. Sickle cell anemia to predict that
Dr. MUNEER ALBAGSHI Consultant Pediatric Hematologist Oncologist- HBDC, Al-Ahsa. Saudi Arabia Sickle cell is global disease of old world and immigrants to the new world. Sickle cell anemia to predict that