Congenital Hemolytic Anemias
|
|
|
- Tiffany Poole
- 5 years ago
- Views:
Transcription
1 Anemia (4) Congenital Hemolytic Anemias Abdallah Abbadi.MD.FRCP. FRCPath Feras Fararjeh MD
2 Congenital Hemolytic Anemias: Subtypes 1 Membrane defects: HS 2 Enzymopathies: G6PD Deficiency, PK Def 3 Hemoglobinopathies: B Thal, SS



3 Anemia (4): Congenital Hemolytic Anemias Case 4 18 yr old male presented to ER with headches, dizziness, red urine and severe loin pain few hours after he ate fresh fool beans. He looked jaundiced and sweaty. His BP 90/60, Pulse rate 120.He had no splenomegaly. Hb 9 g/dl, WBC 16K, Plt 280K. Retics 9%.LDH 3000, Bilirubin 5 mostly indirect. Urine Bite cells (Bld film) Heinz Bodies
4 Glycolytic pathway Glucose Glucose-6-P Pathways of MetHb reduction: Fructose-6-P Fructose-1,6dP G6PD Oxidative stress GSH GSSG NADP NADPH 1. NADH cyt b5 reductase 2. NADPH MetHbreductase 3. Glutathione reductase Pgluconate Ribose-5-P Pentose Phospate Shunt Pathway Glyceraldehyde 3 phosphate 3phosphoglycerate Pyruvate Lactate Pyruvate kinase NAD NADH ADP ATP ADP ATP 1.
 beans (Favism)) Pentose Phosphate")
5 Diagnosis of case 4: G6PD deficiency: hemolytic anemia induced by fava(broad) beans (Favism)) Pentose Phosphate Pathway

6 Prevalence of G6PD: > 400 mill people/ Malaria belt
7 Clinical Features: Disease from completely asymptomatic to severe intravascular hemolysis upon exposure to oxidant stress. Common precipitating factors: Drugs: Primaquine Methylene Blue Nalidixic acid sulpha drugs pyridium and other. Infections Diabetic ketoacidosis Favism: hemolysis after exposure to Fava beans, occurs in Gd Med variant
8 Clinical Syndromes: G6PD Deficiency 1 Neonatal Jaundice: severe/ Kernicterus /, 1 3 day after birth. 2 Favism: acute intravascular Hemolysis after exposure to broad bean (Vicia fava), the offending agent is divicine, it produces free Oxygen radicals on autoxidation. 3 Infection which promote the formation of H 2 O 2 following oxygen burst in neutrophils and macrophage may result in hemolysis 4 Drug induced hemolysis
9 G6PD variants Genotypes/Isoenzymes G6PD B+ : wild type, whites > blacks G6PD A+ : blacks > whites G6PD A : blacks with mild deficiency G6PD Med : whites Mediterranean, Kurdish, severe def. G6PD Canton : Thailand, Vietnam, Taiwan WHO variants

10 WHO Variants
 Nitrofurantoin Vitamin K analogues Several others Henna can cause a hemolytic crisis in G6PD deficient")
11 Drug Induced Acute Hemolysis Drugs that have been linked to G6PD: Primaquine Sulphonamide antibiotics Sulphones (e.g. dapsone, used against leprosy) Other sulphur containing drugs: glibenclamide (an antidiabetic drug) Nitrofurantoin Vitamin K analogues Several others Henna can cause a hemolytic crisis in G6PD deficient infants
12 Genetics Majority of the variants from a single pointmutation resulting in amino acid substitution in gene encoding for G6PD located at the Xq28 region on the tip of the long arm of the X chromosome G6PD Mediterranean is caused by mutation (563 C >T)
13 Therapy Avoid precipitating factors. Blood transfusion in severe hemolysis. Maintenance of good urine output during hemolytic episodes Folic acid. Exchange transfusion in newborn


14 Case 4 B 36 yr old lady presented with anemia syndrome and splenomegaly. She was mildly jaundiced. Hb 8g/dl, retics 10%, WBC, Plt were normal. LDH 1160, Bilirubin 3mg/dl d 1.DAT ve. Bld film Osmotic fragility test Abd. US
15 Hereditary Spherocytosis Prevalence and inheritance In Northern Europeans prevalence is about 1 in 5,000 Clinical severity is highly variable, but uniform within a given family Typically the autosomal dominant homozygous is very severe or lethal some recessively inherited No consensus for splenectomy indications
16 Hereditary Spherocytosis Molecular pathology Partial deficiency of spectrin Combined deficiency of spectrin and ankyrin Molecular Defects: mutations of ankyrin: most common mutations of band 3 protein mutations of protein 4.2 (common in Japanese) Others: β & α spectrin, protein 4.9 are rare
17 RBC Membrane

18 Splenic Conditioning





19 Case 4 C 18 yr old male complains of acute pain in his back, Dizziness, Fatigue, Shortness of breath and Headaches for the last 6 hours. He has had similar attacks. P/E Xray spine
20 Sickle Cell Disease Inherited as autosomal recessive Point mutation in beta globin gene (β6 Glu Val) Gene occurs in 8% of African Americans
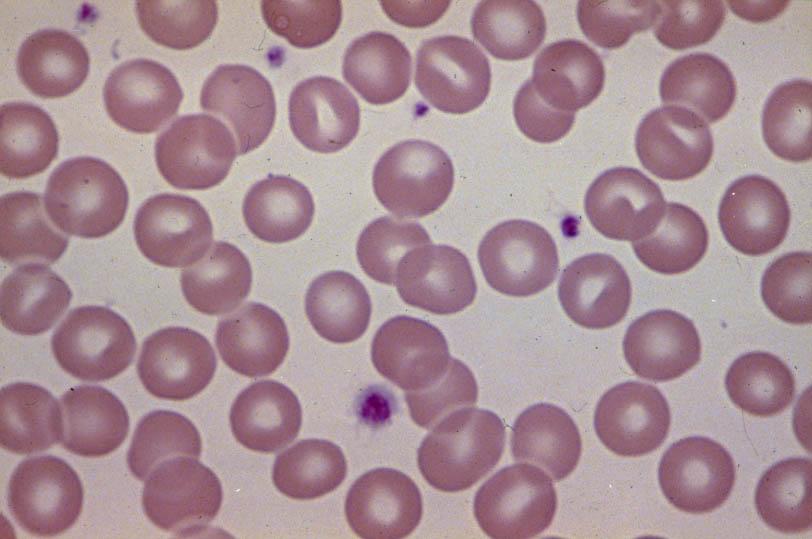
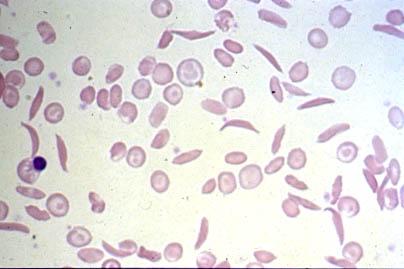
21 Peripheral Blood Smear Normal S S
22 Sickle Cell Anemia Clinical Effects Chronic hemolytic anemia Gallstones (bilirubin) Risk of red cell aplasia (Parvovirus) Decreased vascular tone Susceptible to infection Functional asplenia Infarcted tissue Numerous manipulations Vaso occlusion
23 Vascular beds susceptible to injury Brain Lung Ankle Erectile vasculature of the penis

24 End stage vascular lung disease
25 Infectious complications of Sickle cell anemia Related to absent spleen Pneumococcus infections Hemophilus infections Dramatically improved with the use of prophylactic penicillin in childhood Related to frequent instrumentation Staphyloccocal infections Related to tissue infarction Osteomyelitis
26 Auto-splenectomy occurs in sickle cell disease
27 Sickle Cell Anemia Vaso occlusion: Unique pathophysiologic feature Causes acute and chronic organ damage Acute complications Sickle cell vaso occlusive pain crisis Hepatic crisis Splenic crisis Priapism Chronic organ damage Stroke Chronic lung disease with pulmonary hypertension Renal failure Avascular necrosis of bone

28 Sickle cell: avascular necrosis of the hip
29 Sickle cell vaso-occlusive crisis Serious complication of sickle cell anemia Risk of acute event (<48 hours) Acute chest syndrome Splenic sequestration Massive hemolysis Risk of sudden death
30 Sickle Cell Anemia Painful Events: Management Principles Correct fluid/electrolyte abnormalities; use hypotonic fluid and limit volume to avoid overhydration Treat any underlying illness Opioid analgesics (meperidine is not recommended) Blood transfusion is not indicated for an uncomplicated pain episode Incentive spirometry should be used during waking hours
31 Prevention of Painful Episodes Hydroxyurea increases Hgb F Reduces the frequency of painful episodes, acute chest syndrome, RBC transfusions and hospitalizations Non pharmacologic approaches have not been fully evaluated Prophylactic transfusions showed a decreased incidence of painful crisis in pregnancy
32 Sickle Cell Pain Episodes Average duration 5-7 days 30-50% of patients seen in ER are admitted Pain episodes account for ~90% of admissions Account for most of the cost of care
33 Addiction and pseudo addiction Addiction (abuse) Overwhelming involvement with obtaining and using mind altering drug Pseudo addiction Relief seeking behavior misidentified as addictive behavior
34 Acute Chest Syndrome: Clinical Findings Etiology multifactorial Rib infarct causing splinting/atelectasis Pulmonary fat embolism Infection (mycoplasma, chlamydia, viral) Indistinguishable from pneumonia Pleuritic chest pain, fever, cough, tachypnea, hypoxia Laboratory diagnosis Worsening anemia Infiltrate on chest radiograph
35 Acute Chest Syndrome: Outcome Complete recovery 91% Weaned of supplemental O2 3.1±1.9 days Hospital discharge 5.4±2.3 days Chronic respiratory disease 3% Death 6%
36 Acute Chest Syndrome: Prevention and Treatment Incentive spirometry Treat possible underlying infection Bronchodilators and supplemental oxygen RBC transfusion therapy
37 Indication Indications for RBC transfusions in sickle cell disease Outcome Stroke 90% Acute chest syndrome improvement Aplastic crisis Pre operative treatment (Hgb ~10 g/dl) Symptomatic anemia Splenic or hepatic sequestration Initial recovery; decreased recurrence by Rapid May be life saving Decrease post operative complications Clinical improvement Clinical improvement
 OH O N N N * OH * * Fe *3 polar interaction sites in the binding pocket. Nick H, Current Medicinal Chemistry. 2003;10:1065-1076. HO Clinical trial formulation or preparation")
38 Deferasirox :Oral Iron Chelator in chronic blood transfusion Tridentate* iron chelator An oral, dispersible tablet Administered once daily Highly specific for iron Chelated iron excreted mainly in feces (< 10% in urine) OH O N N N * OH * * Fe *3 polar interaction sites in the binding pocket. Nick H, Current Medicinal Chemistry. 2003;10: HO Clinical trial formulation or preparation

39 Sickle Cell Trait Protection against malaria Genitourinary complications Hyposthenuria/ papillary sloughing Painless hematuria UTI during pregnancy Vaso occlusive complications Splenic infarction with hypoxia Sudden death Rhabdomyolysis Sickle cell trait areas shown in orange stripes





40 Case 4 D 13 yr old male complains of skin pigmentation, abdominal swelling and pallor. He has been receiving blood transfusions since the age of 9 months. Stunted growth. Hb 6, MCV 55, retcs16%,s.ferritin P/E Xray


41 Clinical Syndrome Genotype Hb Hb analysis
42 Most commonly reported mutations in the B globin gens in Jordanians Eight mutations constituted about 86% of the Jordanian thalassemic mutations These mutations were IVS1 110 (G>A) (25%), IVS2 1 (G>A) (15%), IVS2 745 (C>G) (14.2%), IVS1 1 (G>A) (10%), IVS1 6 (T>C) (8.3%), codon 37 (G>A) (6.3%), codon 39 (C>T) (4.6%), and codon 5( C) (3.8%)
43 Type of Mutation and severity of defect in B Thal globin gene Mutation Phenotype Ethnic Origin Promoter Region Mutants -101 (C to T) B(+) Turkish -88 (C to A) B(+) Mediterranean -87 (C to G) B(+) Black American Chain Terminator Mutants Codon 1 (-1 bp) B(0) Chinese Codon 6 (-1 bp) B(0) Mediterranean Codon 114 (-2, +1 bp) B(+) French Splice Junction Mutants IVS-1, position 1 (G to A) B(0) Mediterranean IVS-1, position 2 (T to G) B(0) Indian, Chinese New Splice Site IVS (G to A) B(+) Mediterranean RNA Cleavage Defect AATAAA to AACAAA B(+) Black American
44 Beta Thalassemia: Clinical Manifestations/ complication Osteoporosis Extramedullary erythropoiesis/ tumor effect Iron overload: skin, heart, liver, endocrine organs Dilated cardiomyopathy secondary to severe anemia Growth and development delayed Large splenomegaly
45 Treatment/ Prevention of B thal major Blood Transfusion Iron chelation: deferroxamine (parenteral)?splenectomy Allo BMT Supportive Prevention Oral deferasirox
SICKLE CELL DISEASE. Dr. MUBARAK ABDELRAHMAN MD PEDIATRICS AND CHILD HEALTH. Assistant Professor FACULTY OF MEDICINE -JAZAN
 SICKLE CELL DISEASE Dr. MUBARAK ABDELRAHMAN MD PEDIATRICS AND CHILD HEALTH Assistant Professor FACULTY OF MEDICINE -JAZAN Objective: The student should be able: To identify the presentation, diagnosis,
SICKLE CELL DISEASE Dr. MUBARAK ABDELRAHMAN MD PEDIATRICS AND CHILD HEALTH Assistant Professor FACULTY OF MEDICINE -JAZAN Objective: The student should be able: To identify the presentation, diagnosis,
How to Write a Life Care Plan for a Child with Hemoglobinopathy
 How to Write a Life Care Plan for a Child with Hemoglobinopathy Tamar Fleischer, BSN, MSN, CNLCP & Mona Yudkoff, RN, MPH, CRRN, CNLCP BalaCare Solutions March 2018 St. Peterburg, Florida What is Hemoglobinopathy?
How to Write a Life Care Plan for a Child with Hemoglobinopathy Tamar Fleischer, BSN, MSN, CNLCP & Mona Yudkoff, RN, MPH, CRRN, CNLCP BalaCare Solutions March 2018 St. Peterburg, Florida What is Hemoglobinopathy?
General Characterisctics
 Anemia General Characterisctics Definition: anemia is a decrease in red blood cells. Happens due to underproduction, increased destruction or loss of red cells. Diagnosis of anemia: Hgb < 135 (men) Hgb
Anemia General Characterisctics Definition: anemia is a decrease in red blood cells. Happens due to underproduction, increased destruction or loss of red cells. Diagnosis of anemia: Hgb < 135 (men) Hgb
Sickle Cell Disease and impact on the society
 Sickle Cell Disease and impact on the society Professor Z.A.Jeremiah Ph.D, FRCPath (London) Professor of Haematology and Blood Transfusion Science Niger Delta University, Wilberforce Island Outline What
Sickle Cell Disease and impact on the society Professor Z.A.Jeremiah Ph.D, FRCPath (London) Professor of Haematology and Blood Transfusion Science Niger Delta University, Wilberforce Island Outline What
4 Jumana Jihad Dr. Ahmad Mansour Dr. Ahmad Mansour
 4 Jumana Jihad Dr. Ahmad Mansour Dr. Ahmad Mansour Anemia Decreased blood production Increased blood loss Hemolytic Hemorrhage Extravascular Intravascular Hemolytic (Further classification( Extrinsic Intrinsic
4 Jumana Jihad Dr. Ahmad Mansour Dr. Ahmad Mansour Anemia Decreased blood production Increased blood loss Hemolytic Hemorrhage Extravascular Intravascular Hemolytic (Further classification( Extrinsic Intrinsic
Management. (By the World Health Organization according to the magnitude of the enzyme deficiency and the severity of hemolysis)
") Glucose-6-Phosphate Dehydrogenase (G6PD) Deficiency Management Definition: Glucose-6-phosphate dehydrogenase (G6PD) deficiency is an inherited disorder caused by a genetic defect in the red blood cell
Glucose-6-Phosphate Dehydrogenase (G6PD) Deficiency Management Definition: Glucose-6-phosphate dehydrogenase (G6PD) deficiency is an inherited disorder caused by a genetic defect in the red blood cell
Anemia s. Troy Lund MSMS PhD MD
 Anemia s Troy Lund MSMS PhD MD lundx072@umn.edu Hemoglobinopathy/Anemia IOM take home points. 1. How do we identify the condtion? Smear, CBC Solubility Test (SCD) 2. How does it present clincally? 3. How
Anemia s Troy Lund MSMS PhD MD lundx072@umn.edu Hemoglobinopathy/Anemia IOM take home points. 1. How do we identify the condtion? Smear, CBC Solubility Test (SCD) 2. How does it present clincally? 3. How
Sickle cell disease. Fareed Omar 10 March 2018
 Sickle cell disease Fareed Omar 10 March 2018 Physiology Haemoglobin structure HbA2: 2α and 2δ chains (2-3%) HbF: 2α and 2γ chains (
Sickle cell disease Fareed Omar 10 March 2018 Physiology Haemoglobin structure HbA2: 2α and 2δ chains (2-3%) HbF: 2α and 2γ chains (
Sickle Cell Disease. Edward Malters, MD
 Sickle Cell Disease Edward Malters, MD Introduction Vaso-occlusive phenomena and hemolysis are the clinical hallmarks of Sickle Cell Disease (SCD) Inherited disorder due to homozygosity for the abnormal
Sickle Cell Disease Edward Malters, MD Introduction Vaso-occlusive phenomena and hemolysis are the clinical hallmarks of Sickle Cell Disease (SCD) Inherited disorder due to homozygosity for the abnormal
Congenital Haemoglobinopathies
 Congenital Haemoglobinopathies L. DEDEKEN, MD H O P I T A L U N I V E R S I T A I R E D E S E N F A N T S R E I N E F A B I O L A U N I V E R S I T E L I B R E DE B R U X E L L E S Red Blood Cell Disorders
Congenital Haemoglobinopathies L. DEDEKEN, MD H O P I T A L U N I V E R S I T A I R E D E S E N F A N T S R E I N E F A B I O L A U N I V E R S I T E L I B R E DE B R U X E L L E S Red Blood Cell Disorders
Anemia (3).ms Hemolytic Anemia. Abdallah Abbadi Feras Fararjeh
.ms Hemolytic Anemia. Abdallah Abbadi Feras Fararjeh") Anemia (3).ms4.26.2.18 Hemolytic Anemia Abdallah Abbadi Feras Fararjeh Case 3 24 yr old female presented with anemia syndrome and jaundice. She was found to have splenomegaly. Hb 8, wbc 12k, Plt 212k,
Anemia (3).ms4.26.2.18 Hemolytic Anemia Abdallah Abbadi Feras Fararjeh Case 3 24 yr old female presented with anemia syndrome and jaundice. She was found to have splenomegaly. Hb 8, wbc 12k, Plt 212k,
Pediatrics. Pyruvate Kinase Deficiency (PKD) Symptoms and Treatment. Definition. Epidemiology of Pyruvate Kinase Deficiency.
 Symptoms and Treatment. Definition. Epidemiology of Pyruvate Kinase Deficiency.") Pediatrics Pyruvate Kinase Deficiency (PKD) Symptoms and Treatment See online here Pyruvate kinase deficiency is an inherited metabolic disorder characterized by a deficiency in the enzyme "pyruvate kinase"
Pediatrics Pyruvate Kinase Deficiency (PKD) Symptoms and Treatment See online here Pyruvate kinase deficiency is an inherited metabolic disorder characterized by a deficiency in the enzyme "pyruvate kinase"
Anemia (3).ms4.25.Oct.15 Hemolytic Anemia. Abdallah Abbadi
.ms4.25.Oct.15 Hemolytic Anemia. Abdallah Abbadi") Anemia (3).ms4.25.Oct.15 Hemolytic Anemia Abdallah Abbadi Case 3 24 yr old female presented with anemia syndrome and jaundice. She was found to have splenomegaly. Hb 8, wbc 12k, Plt 212k, retics 12%, LDH
Anemia (3).ms4.25.Oct.15 Hemolytic Anemia Abdallah Abbadi Case 3 24 yr old female presented with anemia syndrome and jaundice. She was found to have splenomegaly. Hb 8, wbc 12k, Plt 212k, retics 12%, LDH
Glucose-6-Phosphate Dehydrogenase
 Glucose-6-Phosphate Dehydrogenase Is the major enzyme in the pentose phosphate pathway (also called the phosphogluconate pathway or the hexose monophosphate shunt) which is a metabolic pathway parallel
Glucose-6-Phosphate Dehydrogenase Is the major enzyme in the pentose phosphate pathway (also called the phosphogluconate pathway or the hexose monophosphate shunt) which is a metabolic pathway parallel
Symptoms and Signs in Hematology (2)/ 2013
/ 2013") Symptoms and Signs in Hematology (2)/ 2013 Abdallah Abbadi.MD.FRCP Professor of Medicine,Hematology & Oncology University of Jordan & JUH Email: abdalla.awidi@gmail.com Case one: A 24 yr old female complains
Symptoms and Signs in Hematology (2)/ 2013 Abdallah Abbadi.MD.FRCP Professor of Medicine,Hematology & Oncology University of Jordan & JUH Email: abdalla.awidi@gmail.com Case one: A 24 yr old female complains
RBCs Disorders 1. Dr. Nabila Hamdi MD, PhD
 RBCs Disorders 1 Dr. Nabila Hamdi MD, PhD ILOs Discuss the classification of anemia into hypochromic-microcytic, normochromicnormocytic and macrocytic. Categorize laboratory test procedures used in the
RBCs Disorders 1 Dr. Nabila Hamdi MD, PhD ILOs Discuss the classification of anemia into hypochromic-microcytic, normochromicnormocytic and macrocytic. Categorize laboratory test procedures used in the
Zeina Al-Assaf. Mustafa Khader. Nayef Karadsheh
 6 Zeina Al-Assaf Mustafa Khader Nayef Karadsheh 1 P a g e Metabolism in mature erythrocytes: During the maturation of RBCs most of its intracellular organelles are lost such as the nucleus and the mitochondria,
6 Zeina Al-Assaf Mustafa Khader Nayef Karadsheh 1 P a g e Metabolism in mature erythrocytes: During the maturation of RBCs most of its intracellular organelles are lost such as the nucleus and the mitochondria,
HAEMOLYTIC ANAEMIA. Dr. Hasan Fahmawi, MRCP(London), FRCP(Edin) Consultant Physician
, FRCP(Edin) Consultant Physician") HAEMOLYTIC ANAEMIA Dr. Hasan Fahmawi, MRCP(London), FRCP(Edin) Consultant Physician Haemolysis Definition shortening of the normal red blood lifespan of 120 days Increase in unconjugated bilirubin, increased
HAEMOLYTIC ANAEMIA Dr. Hasan Fahmawi, MRCP(London), FRCP(Edin) Consultant Physician Haemolysis Definition shortening of the normal red blood lifespan of 120 days Increase in unconjugated bilirubin, increased
Dr Banu Kaya Consultant Haematologist Barts Health NHS Trust Royal London Hospital, London, UK SICKLE CELL AND THALASSAEMIA OVERVIEW
 Dr Banu Kaya Consultant Haematologist Barts Health NHS Trust Royal London Hospital, London, UK SICKLE CELL AND THALASSAEMIA OVERVIEW Objectives Gain awareness of haemoglobinopathy inheritance, pathophysiology
Dr Banu Kaya Consultant Haematologist Barts Health NHS Trust Royal London Hospital, London, UK SICKLE CELL AND THALASSAEMIA OVERVIEW Objectives Gain awareness of haemoglobinopathy inheritance, pathophysiology
C. treatment with Desferal (deferoxamine mesylate USP, iron-chelating agent)
") HEMOLYTIC ANEMIAS Single choice tests 1. Select the clinical manifestation that is not characteristic for the hemolytic crisis: A. decrease of the red blood cell count B. reticulocytosis C. jaundice D.
HEMOLYTIC ANEMIAS Single choice tests 1. Select the clinical manifestation that is not characteristic for the hemolytic crisis: A. decrease of the red blood cell count B. reticulocytosis C. jaundice D.
Dana Alsulaibi. - Ahmad Almuhtaseb. - Tariq Al - Adaily
 - 2 - Dana Alsulaibi - Ahmad Almuhtaseb - Tariq Al - Adaily This sheet will talk about 4 diseases that cause hemolytic anemia, best of luck! 1) Hereditary Spherocytosis Transferred through inheritance
- 2 - Dana Alsulaibi - Ahmad Almuhtaseb - Tariq Al - Adaily This sheet will talk about 4 diseases that cause hemolytic anemia, best of luck! 1) Hereditary Spherocytosis Transferred through inheritance
Hemolytic anemias (2 of 2)
") Hemolytic anemias (2 of 2) Sickle Cell Anemia The most common familial hemolytic anemia in the world Sickle cell anemia is the prototypical (and most prevalent) hemoglobinopathy Mutation in the β-globin
Hemolytic anemias (2 of 2) Sickle Cell Anemia The most common familial hemolytic anemia in the world Sickle cell anemia is the prototypical (and most prevalent) hemoglobinopathy Mutation in the β-globin
Red Blood Cell s Metabolism: HMP Pathway
 Click to edit Master title style Edit Master text styles Second level Third level Fourth level Fifth level Red Blood Cell s Metabolism: HMP Pathway Prof. Samar Kassim Prof. Reem Sallam 2017-2018 1. Recognize
Click to edit Master title style Edit Master text styles Second level Third level Fourth level Fifth level Red Blood Cell s Metabolism: HMP Pathway Prof. Samar Kassim Prof. Reem Sallam 2017-2018 1. Recognize
THE UNIVERSITY OF JORDAN FACULTY OF MEDICINE DEPARTMENT OF PATHOLOGY HEMOLYTIC ANEMIAS. Dr. Tariq Aladily
 THE UNIVERSITY OF JORDAN FACULTY OF MEDICINE DEPARTMENT OF PATHOLOGY HEMOLYTIC ANEMIAS Third year medical students First semester Faculty 2018/2019 of Medicine Hereditary Spherocytosis Intrinsic defects
THE UNIVERSITY OF JORDAN FACULTY OF MEDICINE DEPARTMENT OF PATHOLOGY HEMOLYTIC ANEMIAS Third year medical students First semester Faculty 2018/2019 of Medicine Hereditary Spherocytosis Intrinsic defects
Glucose 6 phosphate dehydrogenase deficiency. Prof. Renzo Galanello Pediatric Clinic 2 University of Cagliari Ospedale Regionale Microcitemie-ASL8
 Glucose 6 phosphate dehydrogenase deficiency Prof. Renzo Galanello Pediatric Clinic 2 University of Cagliari Ospedale Regionale Microcitemie-ASL8 Role of G6PD in glucose RBC metabolism Glucose 1 % use
Glucose 6 phosphate dehydrogenase deficiency Prof. Renzo Galanello Pediatric Clinic 2 University of Cagliari Ospedale Regionale Microcitemie-ASL8 Role of G6PD in glucose RBC metabolism Glucose 1 % use
Dr. Shiva Nazari Assistant Professor of Pediatric Oncologist & Hematologist Shahid Beheshti Medical Science University Mofid Children s Hospital
 Dr. Shiva Nazari Assistant Professor of Pediatric Oncologist & Hematologist Shahid Beheshti Medical Science University Mofid Children s Hospital Reduction in the normal red cell survival (120 days) RBC
Dr. Shiva Nazari Assistant Professor of Pediatric Oncologist & Hematologist Shahid Beheshti Medical Science University Mofid Children s Hospital Reduction in the normal red cell survival (120 days) RBC
Approach to Hemolysis
 Objectives: Approach to Hemolysis To know the function of platelets and the relationship between the platelet count in peripheral blood and the extent of abnormal bleeding. To know about the diseases associated
Objectives: Approach to Hemolysis To know the function of platelets and the relationship between the platelet count in peripheral blood and the extent of abnormal bleeding. To know about the diseases associated
Atlantic Provinces Pediatric Hematology Oncology Network Réseau d Oncologie et Hématologie Pédiatrique des Provinces Atlantiques
 Atlantic Provinces Pediatric Hematology Oncology Network Réseau d Oncologie et Hématologie Pédiatrique des Provinces Atlantiques 5850/5980 University Avenue, PO Box 9700 Halifax, NS, B3K 6R8 1.902.470.7429
Atlantic Provinces Pediatric Hematology Oncology Network Réseau d Oncologie et Hématologie Pédiatrique des Provinces Atlantiques 5850/5980 University Avenue, PO Box 9700 Halifax, NS, B3K 6R8 1.902.470.7429
In adults, the predominant Hb (HbA) molecule has four chains: two α and two β chains. In thalassemias, the synthesis of either the α or the β chains
 molecule has four chains: two α and two β chains. In thalassemias, the synthesis of either the α or the β chains") Thalassaemias Thalassemia Thalassemia is an inherited autosomal recessive blood disease. Associated with absence or reduction in a or b globin chains. Reduced synthesis of one of the globin chains can
Thalassaemias Thalassemia Thalassemia is an inherited autosomal recessive blood disease. Associated with absence or reduction in a or b globin chains. Reduced synthesis of one of the globin chains can
The Child with a Hematologic Alteration
 47 The Child with a Hematologic Alteration HELPFUL HINT Review the anatomy and physiology of the hematologic system in an anatomy and physiology textbook. MATCHING KEY TERMS Match the term with the correct
47 The Child with a Hematologic Alteration HELPFUL HINT Review the anatomy and physiology of the hematologic system in an anatomy and physiology textbook. MATCHING KEY TERMS Match the term with the correct
Anaemia due to a red blood cell membrane defect
 Anaemia due to a red blood cell membrane defect BHS training course 2013 1 Red blood cell membrane defect Pathologies Clinical signs Diagnostic criteria Treatment (HS) 2 The pathologies Structural organisation
Anaemia due to a red blood cell membrane defect BHS training course 2013 1 Red blood cell membrane defect Pathologies Clinical signs Diagnostic criteria Treatment (HS) 2 The pathologies Structural organisation
High Hemoglobin F in a Saudi Child Presenting with Pancytopenia
 Case Report imedpub Journals http://www.imedpub.com Journal of Pediatric Care ISSN 2471-805X DOI: 10.21767/2471-805X.100002 High Hemoglobin F in a Saudi Child Presenting with Pancytopenia Abstract Saudi
Case Report imedpub Journals http://www.imedpub.com Journal of Pediatric Care ISSN 2471-805X DOI: 10.21767/2471-805X.100002 High Hemoglobin F in a Saudi Child Presenting with Pancytopenia Abstract Saudi
DONE BY : RaSHA RAKAN & Bushra Saleem
 DONE BY : RaSHA RAKAN & Bushra Saleem Hemolytic anemias (2 of 2) Sickle Cell Anemia The most common familial hemolytic anemia in the world Sickle cell anemia is the prototypical (and most prevalent) hemoglobinopathy
DONE BY : RaSHA RAKAN & Bushra Saleem Hemolytic anemias (2 of 2) Sickle Cell Anemia The most common familial hemolytic anemia in the world Sickle cell anemia is the prototypical (and most prevalent) hemoglobinopathy
Sickle Cell Disease 101. Objectives. What is SCD? 4/20/2016. Discuss the pathophysiology & genetics of Sickle Cell Disease (SCD).
.") Sickle Cell Disease 101 Jennifer Young, RN, MS, CPNP-AC Sickle Cell & Thalassemia Nurse Practitioner Nationwide Children s Hospital Objectives Discuss the pathophysiology & genetics of Sickle Cell Disease
Sickle Cell Disease 101 Jennifer Young, RN, MS, CPNP-AC Sickle Cell & Thalassemia Nurse Practitioner Nationwide Children s Hospital Objectives Discuss the pathophysiology & genetics of Sickle Cell Disease
Hemoglobinopathies Diagnosis and management
 Hemoglobinopathies Diagnosis and management Morgan L. McLemore, M.D. Hematology/Leukemia Department of Hematology and Oncology Winship Cancer Institute at Emory University mlmclem@emory.edu Disclosures
Hemoglobinopathies Diagnosis and management Morgan L. McLemore, M.D. Hematology/Leukemia Department of Hematology and Oncology Winship Cancer Institute at Emory University mlmclem@emory.edu Disclosures
HEMOLYTIC ANEMIA Anemia of increased destruction Normochromic, normochromic anemia Shortened RBC survival Reticulocytosis - Response to increased RBC
 HEMOLYTIC ANEMIAS Edited by: GR. Bahoush, MD. HEMOLYTIC ANEMIA Anemia of increased destruction Normochromic, normochromic anemia Shortened RBC survival Reticulocytosis - Response to increased RBC destruction
HEMOLYTIC ANEMIAS Edited by: GR. Bahoush, MD. HEMOLYTIC ANEMIA Anemia of increased destruction Normochromic, normochromic anemia Shortened RBC survival Reticulocytosis - Response to increased RBC destruction
Non-malignant hematologic disorders associated arthropathies: hemoglobinopathy-associated musculoskeletal manifestations, hemophilia
 Non-malignant hematologic disorders associated arthropathies: hemoglobinopathy-associated musculoskeletal manifestations, hemophilia HAEMOGLOBINOPATHIES = inherited disorders of globin divided into: Thalassaemia
Non-malignant hematologic disorders associated arthropathies: hemoglobinopathy-associated musculoskeletal manifestations, hemophilia HAEMOGLOBINOPATHIES = inherited disorders of globin divided into: Thalassaemia
Hemoglobin and anemia BCH 471
 Hemoglobin and anemia BCH 471 OBJECTIVES Quantitative determination of hemoglobin in a blood sample. Hemoglobin structure Hemoglobin (Hb) is a porphyrin iron (II) protein in RBCs that transport oxygen
Hemoglobin and anemia BCH 471 OBJECTIVES Quantitative determination of hemoglobin in a blood sample. Hemoglobin structure Hemoglobin (Hb) is a porphyrin iron (II) protein in RBCs that transport oxygen
RBCs Disorders 2. Dr. Nabila Hamdi MD, PhD
 RBCs Disorders 2 Dr. Nabila Hamdi MD, PhD ILOs Discuss the classification of anemia into hypochromic-microcytic, normochromicnormocytic and macrocytic. Categorize laboratory test procedures used in the
RBCs Disorders 2 Dr. Nabila Hamdi MD, PhD ILOs Discuss the classification of anemia into hypochromic-microcytic, normochromicnormocytic and macrocytic. Categorize laboratory test procedures used in the
Done by :Aseel Twaijer & Laith Sorour Hemolytic Anemias
 Hemolytic Anemias Hemolytic anemias share the following features: - A shortened red cell life < 120 days - Elevated erythropoietin levels (compensatory increase in erythropoiesis) - Accumulation of hemoglobin
Hemolytic Anemias Hemolytic anemias share the following features: - A shortened red cell life < 120 days - Elevated erythropoietin levels (compensatory increase in erythropoiesis) - Accumulation of hemoglobin
Deficiencies of Glycolytic Pathway
 Deficiencies of Glycolytic Pathway -Mature RBCs have the capacity for a limited number of enzymatic reactions -The mature RBC is completely dependent on glucose as a source of energy. Glucose usually (90%)
Deficiencies of Glycolytic Pathway -Mature RBCs have the capacity for a limited number of enzymatic reactions -The mature RBC is completely dependent on glucose as a source of energy. Glucose usually (90%)
Putting some hematology into Pediatric Hematology/Oncology: a review of Hemophilia and Sickle Cell Disease in the Pediatric Patient
 Putting some hematology into Pediatric Hematology/Oncology: a review of Hemophilia and Sickle Cell Disease in the Pediatric Patient Kristina Haley, DO March 10, 2012 Jovita Reyes Memorial Pediatric Hematology/Oncology
Putting some hematology into Pediatric Hematology/Oncology: a review of Hemophilia and Sickle Cell Disease in the Pediatric Patient Kristina Haley, DO March 10, 2012 Jovita Reyes Memorial Pediatric Hematology/Oncology
HEMOLYTIC ANEMIA DUE TO ABNORMAL HEMOGLOBIN SYNTHESIS
 Hemolytic Anemia Due to Abnormal Hemoglobin Synthesis MODULE 19 HEMOLYTIC ANEMIA DUE TO ABNORMAL HEMOGLOBIN SYNTHESIS 19.1 INTRODUCTION There are two main mechanisms by which anaemia is produced (a) Thalassemia:
Hemolytic Anemia Due to Abnormal Hemoglobin Synthesis MODULE 19 HEMOLYTIC ANEMIA DUE TO ABNORMAL HEMOGLOBIN SYNTHESIS 19.1 INTRODUCTION There are two main mechanisms by which anaemia is produced (a) Thalassemia:
Genetics of Thalassemia
 Genetics of Thalassemia Submitted by : Raya Samir Al- Hayaly Sura Zuhair Salih Saad Ghassan Al- Dulaimy Saad Farouq Kassir Sama Naal Salouha Zahraa Jasim Al- Aarajy Supervised by : Dr. Kawkab Adris Mahmod
Genetics of Thalassemia Submitted by : Raya Samir Al- Hayaly Sura Zuhair Salih Saad Ghassan Al- Dulaimy Saad Farouq Kassir Sama Naal Salouha Zahraa Jasim Al- Aarajy Supervised by : Dr. Kawkab Adris Mahmod
Thalassemia Maria Luz Uy del Rosario, M.D.
 Thalassemia Maria Luz Uy del Rosario, M.D. Philippine Society of Hematology and Blood Transfusion Philippine Society of Pediatric Oncology What is Thalassemia Hereditary Hemoglobin disorder Hemolytic anemia
Thalassemia Maria Luz Uy del Rosario, M.D. Philippine Society of Hematology and Blood Transfusion Philippine Society of Pediatric Oncology What is Thalassemia Hereditary Hemoglobin disorder Hemolytic anemia
BCM 317 LECTURE OJEMEKELE O.
 BCM 317 LECTURE BY OJEMEKELE O. JAUNDICE Jaundice is yellowish discoloration of the skin, sclera and mucous membrane, resulting from an increased bilirubin concentration in the body fluid. It is usually
BCM 317 LECTURE BY OJEMEKELE O. JAUNDICE Jaundice is yellowish discoloration of the skin, sclera and mucous membrane, resulting from an increased bilirubin concentration in the body fluid. It is usually
Heme Questions and Derivatives for the USMLE Step One Exam. Winter Storm Skylar Edition
 Heme Questions and Derivatives for the USMLE Step One Exam Winter Storm Skylar Edition Howard J. Sachs, MD Howard@12DaysinMarch.com www.12daysinmarch.com Patient presents with RUQ pain. HIDA scan fails
Heme Questions and Derivatives for the USMLE Step One Exam Winter Storm Skylar Edition Howard J. Sachs, MD Howard@12DaysinMarch.com www.12daysinmarch.com Patient presents with RUQ pain. HIDA scan fails
Rationale for RBC Transfusion in SCD
 Rationale for RBC Transfusion in SCD Dilution of HgbS-containing RBCs via the addition of HgbA-containing cells from the blood of normal donors Suppression of erythropoietin release caused by the rise
Rationale for RBC Transfusion in SCD Dilution of HgbS-containing RBCs via the addition of HgbA-containing cells from the blood of normal donors Suppression of erythropoietin release caused by the rise
Approach to a pale child
 Approach to a pale child Dr. Dafalla Ahmed Babiker Jazan university objectives Definition of anemia Classification and causes Important points in history and physical examination Investigations. Definition
Approach to a pale child Dr. Dafalla Ahmed Babiker Jazan university objectives Definition of anemia Classification and causes Important points in history and physical examination Investigations. Definition
RBCs Disorders 2. Dr. Nabila Hamdi MD, PhD
 RBCs Disorders 2 Dr. Nabila Hamdi MD, PhD ILOs Discuss the classification of anemia into hypochromic-microcytic, normochromicnormocytic and macrocytic. Categorize laboratory test procedures used in the
RBCs Disorders 2 Dr. Nabila Hamdi MD, PhD ILOs Discuss the classification of anemia into hypochromic-microcytic, normochromicnormocytic and macrocytic. Categorize laboratory test procedures used in the
Moh Tarek + Faisal Massad. Tala Saleh ... Naif
 19 Moh Tarek + Faisal Massad Tala Saleh... Naif Last lecture we ve talked about the main antioxidant system which are the enzymes found in our body, mainly: 1. Glutathione peroxidase 2. Super oxide dismutase(sod)
19 Moh Tarek + Faisal Massad Tala Saleh... Naif Last lecture we ve talked about the main antioxidant system which are the enzymes found in our body, mainly: 1. Glutathione peroxidase 2. Super oxide dismutase(sod)
Extra Notes 3. Warm. In the core (center) of the body, where the temperature is 37 C.
 of the body, where the temperature is 37 C.") Extra Notes 3 *The numbers of the slides are according to the last year slides. Slide 33 Autoimmune hemolytic anemia : Abnormal circulating antibodies that target normal antigen on the RBC and cause lysis.
Extra Notes 3 *The numbers of the slides are according to the last year slides. Slide 33 Autoimmune hemolytic anemia : Abnormal circulating antibodies that target normal antigen on the RBC and cause lysis.
Structure of G6PD. Glucose-6-Phosphate Dehydrogenase (G6PD) and Malaria. Function of G6PD. Familial Genetics of G6PD. Mendelian Transmission
 and Malaria. Function of G6PD. Familial Genetics of G6PD. Mendelian Transmission") (G6PD) and Malaria Structure of G6PD The enzyme,, is comprised of a dimer or tetramer of identical polypeptide chains Each unit consists of 515 amino acids The single G6PD locus in humans is located on
(G6PD) and Malaria Structure of G6PD The enzyme,, is comprised of a dimer or tetramer of identical polypeptide chains Each unit consists of 515 amino acids The single G6PD locus in humans is located on
An overview of Thalassaemias and Complications
 An overview of Thalassaemias and Complications Haemoglobin Haemoglobin is the most abundant protein in blood, and exists as three main types in normal adults: HbA ( ) - 97% HbA 2 ( ) - 2.5% HbF ( ) - 0.5%
An overview of Thalassaemias and Complications Haemoglobin Haemoglobin is the most abundant protein in blood, and exists as three main types in normal adults: HbA ( ) - 97% HbA 2 ( ) - 2.5% HbF ( ) - 0.5%
Medical and Surgical Complications of Sickle Cell Anemia
 Medical and Surgical Complications of Sickle Cell Anemia Ahmed Al-Salem Medical and Surgical Complications of Sickle Cell Anemia Ahmed Al-Salem Department of Surgery Dar A lalafia Medical Company Qatif
Medical and Surgical Complications of Sickle Cell Anemia Ahmed Al-Salem Medical and Surgical Complications of Sickle Cell Anemia Ahmed Al-Salem Department of Surgery Dar A lalafia Medical Company Qatif
Hemoglobinopathies NORMAL HEMOGLOBINS
 Hemoglobinopathies Millicent Sutton MD October 28, 2005 NORMAL HEMOGLOBINS Consist of 2 alpha chains and 2 non alpha chains Hb A = α2β2 Hb F= α 2γ2 Hb A2 = α2δ2 1 Hemoglobin Variants Altered the conformational
Hemoglobinopathies Millicent Sutton MD October 28, 2005 NORMAL HEMOGLOBINS Consist of 2 alpha chains and 2 non alpha chains Hb A = α2β2 Hb F= α 2γ2 Hb A2 = α2δ2 1 Hemoglobin Variants Altered the conformational
Year 2004 Paper two: Questions supplied by Megan 1
 Year 2004 Paper two: Questions supplied by Megan 1 QUESTION 93 A 16yo adolescent male presents with lethargy and lower respiratory tract infection. Physical examination shows him to be febrile, icteric
Year 2004 Paper two: Questions supplied by Megan 1 QUESTION 93 A 16yo adolescent male presents with lethargy and lower respiratory tract infection. Physical examination shows him to be febrile, icteric
Hydroxyurea and Transfusion Therapy for the Treatment of Sickle Cell Disease
 Hydroxyurea and Transfusion Therapy for the Treatment of Sickle Cell Disease A Pocket Guide for the Clinician Susan E. Creary, MD, MSc 1 John J. Strouse, MD, PhD 2 1 The Ohio State University School of
Hydroxyurea and Transfusion Therapy for the Treatment of Sickle Cell Disease A Pocket Guide for the Clinician Susan E. Creary, MD, MSc 1 John J. Strouse, MD, PhD 2 1 The Ohio State University School of
Managing Emergencies
 Managing Emergencies Richard Ward, MSc, MRCP, FRCPath Director, Red Blood Cell Disorders Program, UHN Assistant Professor, Hematology, University of Toronto Chair, Canadian Hemoglobinopathy Association
Managing Emergencies Richard Ward, MSc, MRCP, FRCPath Director, Red Blood Cell Disorders Program, UHN Assistant Professor, Hematology, University of Toronto Chair, Canadian Hemoglobinopathy Association
Haemoglobin BY: MUHAMMAD RADWAN WISSAM MUHAMMAD
 Haemoglobin BY: MUHAMMAD RADWAN WISSAM MUHAMMAD Introduction is the iron-containing oxygen transport metalloprotein in the red blood cells Hemoglobin in the blood carries oxygen from the respiratory organs
Haemoglobin BY: MUHAMMAD RADWAN WISSAM MUHAMMAD Introduction is the iron-containing oxygen transport metalloprotein in the red blood cells Hemoglobin in the blood carries oxygen from the respiratory organs
Definitions: Hematocrit <41% in men or <36 in women Hemoglobin <13.5 gm\dl in men or <12 gm\dl in women
 1 Nadanotes.com Definitions: Hematocrit
1 Nadanotes.com Definitions: Hematocrit
THE UNIVERSITY OF JORDAN FACULTY OF MEDICINE DEPARTMENT OF PATHOLOGY
 THE UNIVERSITY OF JORDAN FACULTY OF MEDICINE DEPARTMENT OF PATHOLOGY INTRODUCTION TO ANEMIA Third year medical students First semester 2018/2019 Dr. RBC DISORDERS Lecturer: Dr. Tariq Al-Adaily Email: TNALADILY@ju.edu.jo
THE UNIVERSITY OF JORDAN FACULTY OF MEDICINE DEPARTMENT OF PATHOLOGY INTRODUCTION TO ANEMIA Third year medical students First semester 2018/2019 Dr. RBC DISORDERS Lecturer: Dr. Tariq Al-Adaily Email: TNALADILY@ju.edu.jo
Acute Complications of Sickle Cell Disease
 Management of Acute Complications of Sickle Cell Disease A Pocket Guide for the Clinician Timothy McCavit, MD, MSCS 1 Payal Desai, MD 1 University of Texas Southwestern Medical Center The Ohio State University,
Management of Acute Complications of Sickle Cell Disease A Pocket Guide for the Clinician Timothy McCavit, MD, MSCS 1 Payal Desai, MD 1 University of Texas Southwestern Medical Center The Ohio State University,
4 Fahed Al Karmi Sufian Alhafez Dr nayef karadsheh
 4 Fahed Al Karmi Sufian Alhafez Dr nayef karadsheh Genetic variants of hemoglobin Hemoglobinopathies (abnormal variants of hemoglobin) are divided into: 1. Structural abnormalities: Any change in the genes
4 Fahed Al Karmi Sufian Alhafez Dr nayef karadsheh Genetic variants of hemoglobin Hemoglobinopathies (abnormal variants of hemoglobin) are divided into: 1. Structural abnormalities: Any change in the genes
Pediatric د.رياض العبيدي
 Fifth stage Lec-1 Pediatric د.رياض العبيدي 9/11/2015 Hematology Hemoglobin; normal HB consists of 4globin chains and 4 heme groups. These globins chains are inhereted from parents.. At birth newborn has
Fifth stage Lec-1 Pediatric د.رياض العبيدي 9/11/2015 Hematology Hemoglobin; normal HB consists of 4globin chains and 4 heme groups. These globins chains are inhereted from parents.. At birth newborn has
Heme Questions and Derivatives for the USMLE Step One Exam. Winter Storm Skylar Edition
 Heme Questions and Derivatives for the USMLE Step One Exam Winter Storm Skylar Edition Howard J. Sachs, MD Howard@12DaysinMarch.com www.12daysinmarch.com Patient presents for routine preoperative evaluation
Heme Questions and Derivatives for the USMLE Step One Exam Winter Storm Skylar Edition Howard J. Sachs, MD Howard@12DaysinMarch.com www.12daysinmarch.com Patient presents for routine preoperative evaluation
HEMOLYSIS AND JAUNDICE:
 1 University of Papua New Guinea School of Medicine and Health Sciences Division of Basic Medical Sciences Discipline of Biochemistry and Molecular Biology PBL SEMINAR HEMOLYSIS AND JAUNDICE: An overview
1 University of Papua New Guinea School of Medicine and Health Sciences Division of Basic Medical Sciences Discipline of Biochemistry and Molecular Biology PBL SEMINAR HEMOLYSIS AND JAUNDICE: An overview
Thalassemias. Emanuela Veras, M.D. 01/08/2006
 Thalassemias Emanuela Veras, M.D. 01/08/2006 Structure and Function of normal Hemoglobin molecules: 2/3 1/3 β: increases from 6 th week of fetal life to 12 months of age At birth: HbF: 75-90% HbA: 10-25%
Thalassemias Emanuela Veras, M.D. 01/08/2006 Structure and Function of normal Hemoglobin molecules: 2/3 1/3 β: increases from 6 th week of fetal life to 12 months of age At birth: HbF: 75-90% HbA: 10-25%
Around million aged erythrocytes/hour are broken down.
 Anemia Degradation ofheme Around 100 200 million aged erythrocytes/hour are broken down. The degradation process starts in reticuloendothelial cells in the spleen, liver, and bone marrow. [1] The tetrapyrrole
Anemia Degradation ofheme Around 100 200 million aged erythrocytes/hour are broken down. The degradation process starts in reticuloendothelial cells in the spleen, liver, and bone marrow. [1] The tetrapyrrole
Introduction reduction in output alter the amino acid sequence combination
 Sickle cell anemia. Introduction Mutations in the globin genes can cause a quantitative reduction in output from that gene or alter the amino acid sequence of the protein produced or a combination of the
Sickle cell anemia. Introduction Mutations in the globin genes can cause a quantitative reduction in output from that gene or alter the amino acid sequence of the protein produced or a combination of the
Thalassemias:general aspects and molecular pathology
 Thalassemias:general aspects and molecular pathology Prof. Renzo Galanello Pediatric Clinic 2 University of Cagliari Ospedale Regionale Microcitemie-ASL8 HEMOGLOBINOPATHIES CLASSIFICATION Structurally
Thalassemias:general aspects and molecular pathology Prof. Renzo Galanello Pediatric Clinic 2 University of Cagliari Ospedale Regionale Microcitemie-ASL8 HEMOGLOBINOPATHIES CLASSIFICATION Structurally
DIC. Disseminated intravascular coagulation, is a life threatening pathological process in which clotting factors are abnormally activated.
 Miss. kamlah 1 DIC Disseminated intravascular coagulation, is a life threatening pathological process in which clotting factors are abnormally activated. Resulting in wide spread of clot formation in the
Miss. kamlah 1 DIC Disseminated intravascular coagulation, is a life threatening pathological process in which clotting factors are abnormally activated. Resulting in wide spread of clot formation in the
رناد زكريا Dr. ahmad Dr. ahmad. P a g e 1
 5 رناد زكريا Dr. ahmad Dr. ahmad P a g e 1 Before we start. -This sheet was written according to section 2 s record and reviewed according to section 1 s record by Ruba Hussien with all thanks and I referred
5 رناد زكريا Dr. ahmad Dr. ahmad P a g e 1 Before we start. -This sheet was written according to section 2 s record and reviewed according to section 1 s record by Ruba Hussien with all thanks and I referred
SCD Advocacy Talking Points!
 "#$!%&'()*)+!,!-././-1!! SCD Advocacy Talking Points! * 23!4*'3!53*673&!84*8!#*59:(679*!#495&637;!9
"#$!%&'()*)+!,!-././-1!! SCD Advocacy Talking Points! * 23!4*'3!53*673&!84*8!#*59:(679*!#495&637;!9
Sickle Cell Anemia. Sickle cell anemia is an inherited disorder of the blood which occurs when just one base pair substitution
 Rose Farrington and Rachel Nash BIOL 362 Lab M. Bulgarella Genetic Diseases 10/14/2008 Sickle Cell Anemia Introduction Sickle cell anemia is an inherited disorder of the blood which occurs when just one
Rose Farrington and Rachel Nash BIOL 362 Lab M. Bulgarella Genetic Diseases 10/14/2008 Sickle Cell Anemia Introduction Sickle cell anemia is an inherited disorder of the blood which occurs when just one
Course: Nutrition and Metabolism
 Course: Nutrition and Metabolism Part (1): Metabolism of Carbohydrates Lecture (8): Pentose Phosphate Pathway Dr. Rihab Siddig Mobile: +249918191982 Glucose Uses Energy Stores Glycogen Glucose Pentose
Course: Nutrition and Metabolism Part (1): Metabolism of Carbohydrates Lecture (8): Pentose Phosphate Pathway Dr. Rihab Siddig Mobile: +249918191982 Glucose Uses Energy Stores Glycogen Glucose Pentose
QUESTIONS OF HEMATOLOGY AND THEIR ANSWERS
 QUESTIONS OF HEMATOLOGY AND THEIR ANSWERS WHAT IS TRUE AND WHAT IS FALSE? Questions 1 Iron deficiency anemia a) Is usually associated with a raised MCV. b) The MCH is usually low. c) Is most commonly due
QUESTIONS OF HEMATOLOGY AND THEIR ANSWERS WHAT IS TRUE AND WHAT IS FALSE? Questions 1 Iron deficiency anemia a) Is usually associated with a raised MCV. b) The MCH is usually low. c) Is most commonly due
World-Wide Distribution of Hemoglobin S. Geographic distribution of hemoglobin S in the world
 Sickle Cell Disease Gerald M. Woods, MD Professor of Pediatrics Division Director, Hematology/Oncology/BMT Director of Sickle Cell Program Children s Mercy Hospitals and Clinics Disclaimer Member of DSMB
Sickle Cell Disease Gerald M. Woods, MD Professor of Pediatrics Division Director, Hematology/Oncology/BMT Director of Sickle Cell Program Children s Mercy Hospitals and Clinics Disclaimer Member of DSMB
Division of General Internal Medicine and Geriatrics Hospital Medicine 2014
 Division of General Internal Medicine and Geriatrics Hospital Medicine 2014 Objectives Understand workup of acute pain crisis Identify key aspects of management of acute pain crisis in sickle cell patients
Division of General Internal Medicine and Geriatrics Hospital Medicine 2014 Objectives Understand workup of acute pain crisis Identify key aspects of management of acute pain crisis in sickle cell patients
Screening for haemoglobinopathies in pregnancy
 Policy Statement All Southern Health patients will receive clinical care that reflects best practice and is based on the best available evidence. Index of chapters within background 1. Prevalence of haemoglobinopathies
Policy Statement All Southern Health patients will receive clinical care that reflects best practice and is based on the best available evidence. Index of chapters within background 1. Prevalence of haemoglobinopathies
(anemia) ก hemoglobin concentration, hematocrit deviation 1 1 ก hemoglobin, hematocrit mean corpuscular volume (MCV) 2
 ก hemoglobin concentration, hematocrit deviation 1 1 ก hemoglobin, hematocrit mean corpuscular volume (MCV) 2") ก ก. ก ก.. ก (anemia) ก hemoglobin concentration, hematocrit ก ก ก 2 Standard deviation 1 1 ก hemoglobin, hematocrit mean corpuscular volume (MCV) 2 Hemoglobin hematocrit MCV (g/dl) (%) (fl) ( ) 0.5-1.9
ก ก. ก ก.. ก (anemia) ก hemoglobin concentration, hematocrit ก ก ก 2 Standard deviation 1 1 ก hemoglobin, hematocrit mean corpuscular volume (MCV) 2 Hemoglobin hematocrit MCV (g/dl) (%) (fl) ( ) 0.5-1.9
Full Case: Questions: What is sickle cell crisis?
 Full Case: 30 y/o with avascular necrosis of her right hip was admitted for a total hip arthroplasty. Her hematocrit was 22%, blood pressure was 130/90 mm Hg, and pulse was 107 beats per minute. She had
Full Case: 30 y/o with avascular necrosis of her right hip was admitted for a total hip arthroplasty. Her hematocrit was 22%, blood pressure was 130/90 mm Hg, and pulse was 107 beats per minute. She had
Dr.Abdolreza Afrasiabi
 Dr.Abdolreza Afrasiabi Thalassemia & Heamophilia Genetic Reaserch Center Shiraz Medical University Hemoglobin tetramer Hemoglobin Structure % A 1 α 2 β 2 94-97% A 2 α 2 δ 2 2.5% A 1C α 2 (β-n-glucose)
Dr.Abdolreza Afrasiabi Thalassemia & Heamophilia Genetic Reaserch Center Shiraz Medical University Hemoglobin tetramer Hemoglobin Structure % A 1 α 2 β 2 94-97% A 2 α 2 δ 2 2.5% A 1C α 2 (β-n-glucose)
Quiz. What percentage of the world s population is a carrier of a hemoglobinopathy? Hemoglobinopathies in Pregnancy 1-2% 5-7% 8-12% 10-15%
 Hemoglobinopathies in Pregnancy Emily Parkhurst, MS, LCGC Kaiser West Los Angeles November 2017 Genetics Department Quiz What percentage of the world s population is a carrier of a hemoglobinopathy? 1-2%
Hemoglobinopathies in Pregnancy Emily Parkhurst, MS, LCGC Kaiser West Los Angeles November 2017 Genetics Department Quiz What percentage of the world s population is a carrier of a hemoglobinopathy? 1-2%
Anaemia in Pregnancy
 Anaemia in Pregnancy Definition :anaemia is a pathological condition in which the oxygen-carrying capacity of red blood cells is insufficient to meet the body needs. The WHO : haemoglobin concentration
Anaemia in Pregnancy Definition :anaemia is a pathological condition in which the oxygen-carrying capacity of red blood cells is insufficient to meet the body needs. The WHO : haemoglobin concentration
Genetic Modifiers of Sickle Cell Disease Severity. Kunle Adekile, MD, PhD Professor Department of Pediatrics Kuwait University
 Genetic Modifiers of Sickle Cell Disease Severity Kunle Adekile, MD, PhD Professor Department of Pediatrics Kuwait University Outline Hb Molecule and Genetic control of globin synthesis Pathophysiology
Genetic Modifiers of Sickle Cell Disease Severity Kunle Adekile, MD, PhD Professor Department of Pediatrics Kuwait University Outline Hb Molecule and Genetic control of globin synthesis Pathophysiology
O 2 O 2 O 2. Haemoglobin
 O 2 O 2 O 2 Haemoglobin O 2 O 2 O 2 98% travels in oxyhaemoglobin (in red blood cells) 2% is dissolved in plasma (compared to carbon dioxide, oxygen is relatively insoluble in plasma) O 2 is not very soluble
O 2 O 2 O 2 Haemoglobin O 2 O 2 O 2 98% travels in oxyhaemoglobin (in red blood cells) 2% is dissolved in plasma (compared to carbon dioxide, oxygen is relatively insoluble in plasma) O 2 is not very soluble
Tenth Visit posttest
 Test Code 10C Patient s name: Tenth Visit posttest Patient s birth date: Your name and relationship to patient: Today s date: 1. Which one of the medications listed below should every child with a sickle
Test Code 10C Patient s name: Tenth Visit posttest Patient s birth date: Your name and relationship to patient: Today s date: 1. Which one of the medications listed below should every child with a sickle
Symptoms and Signs in Hematology/ 2013
 Symptoms and Signs in Hematology/ 2013 Abdallah Abbadi.MD.FRCP Professor of Hematology & Oncology University of Jordan & JUH Email: abdalla.awidi@gmail.com Diseases of Blood & Blood forming organs A- Benign
Symptoms and Signs in Hematology/ 2013 Abdallah Abbadi.MD.FRCP Professor of Hematology & Oncology University of Jordan & JUH Email: abdalla.awidi@gmail.com Diseases of Blood & Blood forming organs A- Benign
Red cell disorder. Dr. Ahmed Hasan
 Red cell disorder Dr. Ahmed Hasan Things to be learned in this lecture Definition and clinical feature of anemia. Classification of anemia. Know some details of microcytic anemia Question of the lecture:
Red cell disorder Dr. Ahmed Hasan Things to be learned in this lecture Definition and clinical feature of anemia. Classification of anemia. Know some details of microcytic anemia Question of the lecture:
CHERYL KOVALSKI, DO FACOI NO DISCLOSURES ACOI BOARD REVIEW 2018
 CHERYL KOVALSKI, DO FACOI NO DISCLOSURES ACOI BOARD REVIEW 2018 ANEMIA Hemoglobin
CHERYL KOVALSKI, DO FACOI NO DISCLOSURES ACOI BOARD REVIEW 2018 ANEMIA Hemoglobin
THALASSEMIA AND COMPREHENSIVE CARE
 1 THALASSEMIA AND COMPREHENSIVE CARE Melanie Kirby MBBS, FRCP (C), Hospital for Sick Children, Toronto Associate Professor of Paediatrics, University of Toronto. Objectives 2 By the end of this presentation,
1 THALASSEMIA AND COMPREHENSIVE CARE Melanie Kirby MBBS, FRCP (C), Hospital for Sick Children, Toronto Associate Professor of Paediatrics, University of Toronto. Objectives 2 By the end of this presentation,
HEMOLYSIS & JAUNDICE: An Overview
 HEMOLYSIS & JAUNDICE: An Overview University of Papua New Guinea School of Medicine and Health Sciences Division of Basic Medical Sciences Discipline of Biochemistry and Molecular Biology PBL MBBS III
HEMOLYSIS & JAUNDICE: An Overview University of Papua New Guinea School of Medicine and Health Sciences Division of Basic Medical Sciences Discipline of Biochemistry and Molecular Biology PBL MBBS III
Educational Items Section
 Atlas of Genetics and Cytogenetics in Oncology and Haematology OPEN ACCESS JOURNAL AT INIST-CNRS Educational Items Section Hemoglobin genes; Sickle-cell anemia - Thalassemias Jean-Loup Huret, Xavier Troussard
Atlas of Genetics and Cytogenetics in Oncology and Haematology OPEN ACCESS JOURNAL AT INIST-CNRS Educational Items Section Hemoglobin genes; Sickle-cell anemia - Thalassemias Jean-Loup Huret, Xavier Troussard
Miss. kamlah ahmed 1
 Miss. kamlah ahmed 1 Anatomy & Physiology Blood has two compartments: 1- a fluid portion called plasma. 2- a cellular portion known as the formed elements of the blood. Which are RBC (erythrocytes), WBC
Miss. kamlah ahmed 1 Anatomy & Physiology Blood has two compartments: 1- a fluid portion called plasma. 2- a cellular portion known as the formed elements of the blood. Which are RBC (erythrocytes), WBC
Dependance on chronic transfusion
 Dependance on chronic transfusion Pr Saliou Diop Hematology Blood transfusion Dakar- Sénégal diop@cnts-dakar.sn Introduction Chronic transfusion: Regular use of blood transfusion in patients with chronic
Dependance on chronic transfusion Pr Saliou Diop Hematology Blood transfusion Dakar- Sénégal diop@cnts-dakar.sn Introduction Chronic transfusion: Regular use of blood transfusion in patients with chronic
Health Maintenance and Education for Children and Adults
 Health Maintenance and Education for Children and Adults Richard Ward, MSc, MRCP, FRCPath Director, Red Blood Cell Disorders Program, UHN Assistant Professor, Hematology, University of Toronto Chair, Canadian
Health Maintenance and Education for Children and Adults Richard Ward, MSc, MRCP, FRCPath Director, Red Blood Cell Disorders Program, UHN Assistant Professor, Hematology, University of Toronto Chair, Canadian
It should be noted that in the initial stage, nearly all anemias are normocytic. The major primary causes of normocytic anemia are given in Table 1.
 Normocytic Anemia JOHN R. BRILL, M.D., and DENNIS J. BAUMGARDNER, M.D., University of Wisconsin Medical School, Milwaukee Clinical Campus, Milwaukee, Wisconsin Am Fam Physician. 2000 Nov 15;62(10):2255
Normocytic Anemia JOHN R. BRILL, M.D., and DENNIS J. BAUMGARDNER, M.D., University of Wisconsin Medical School, Milwaukee Clinical Campus, Milwaukee, Wisconsin Am Fam Physician. 2000 Nov 15;62(10):2255
Instructor s Manual Chapter 26 Hematological Alterations. 1. A man and woman both test positive for the sickle cell trait. The couple asks the
 1 Instructor s Manual Chapter 26 Hematological Alterations Answers to Study Questions 1. A man and woman both test positive for the sickle cell trait. The couple asks the nurse how many of their children
1 Instructor s Manual Chapter 26 Hematological Alterations Answers to Study Questions 1. A man and woman both test positive for the sickle cell trait. The couple asks the nurse how many of their children