Copyright and use of this thesis
|
|
|
- Sharyl Bishop
- 6 years ago
- Views:
Transcription
1 Copyright and use of this thesis This thesis must be used in accordance with the provisions of the Copyright Act Reproduction of material protected by copyright may be an infringement of copyright and copyright owners may be entitled to take legal action against persons who infringe their copyright. Section 51 (2) of the Copyright Act permits an authorized officer of a university library or archives to provide a copy (by communication or otherwise) of an unpublished thesis kept in the library or archives, to a person who satisfies the authorized officer that he or she requires the reproduction for the purposes of research or study. The Copyright Act grants the creator of a work a number of moral rights, specifically the right of attribution, the right against false attribution and the right of integrity. You may infringe the author s moral rights if you: - fail to acknowledge the author of this thesis if you quote sections from the work - attribute this thesis to another author - subject this thesis to derogatory treatment which may prejudice the author s reputation For further information contact the University s Copyright Service. sydney.edu.au/copyright
2 Neuropathy in childhood mitochondrial disease, including riboflavin transporter deficiency: phenotype, neurophysiology and disease-modifying therapy in a recently described treatable disorder Dr Manoj Peter Menezes A thesis submitted for the degree of Doctor of Philosophy in the faculty of Medicine, The University of Sydney, Submitted October
3 DECLARATION I hereby declare that this submission is my own work and that, to the best of my knowledge and belief, it contains no material previously published or written by another person or material which to a substantial extent has been accepted for the award of any other degree or diploma of the university or other institute of higher learning, except where due acknowledgement has been made in the text. Signed Date Manoj Peter Menezes 2
4 ABSTRACT Introduction: Defining the neurophysiological features of the peripheral neuropathy associated with childhood mitochondrial disease, including the neuropathy/neuronopathy associated with Brown-Vialetto-Van Laere syndrome (BVVL), will aid in classifying the mitochondrial syndrome, directing genetic testing and instituting therapy. Methods: Nerve conduction studies and clinical assessments were performed on children with nuclear or mitochondrial genome mutations. The clinical and genetic profile, neurophysiology, histopathology, audiology and response to riboflavin therapy of nine children with BVVL due to RFVT2 deficiency were evaluated. Results: Peripheral neuropathy was more frequent with nuclear gene mutations. SURF1 was associated with a demyelinating neuropathy while PDHc deficiency caused an axonal neuropathy. POLG mutations caused a sensory axonal neuropathy. The neuropathy associated with mitochondrial disease was not length-dependent. Children with RFVT2 deficiency presented with an auditory neuropathy and sensory ataxia, and developed upper limb weakness. Nerve excitability studies showed an increase in myelin permeability. Riboflavin therapy caused partial reversal of these changes and improvement in strength, audiology and respiratory function. Conclusions: Nerve conduction studies should be part of the initial assessment of children with suspected mitochondrial disease. Clinicians need to be familiar with the unique phenotype of RFVT2 deficiency so that affected children are diagnosed early and commenced on appropriate therapy. 3
5 ACKNOWLEDGEMENTS The supervisory team for my PhD is a veritable Who s Who of neuromuscular research in Australia, and I have benefitted greatly from their knowledge, experience and enthusiasm. I would like to thank Prof Kathryn North, for encouraging me to pursue my interest in neuromuscular research, for giving me my first neuromuscular training position, coauthoring my first peer-reviewed paper and suggesting I undertake a postgraduate research degree. She introduced me to Prof Ouvrier and suggested that I pursue peripheral neuropathy as a subspecialty interest, and that interest is now a major part of my clinical and research work. Her support was also invaluable when I took time off clinical work to train in neuromuscular medicine and work on this PhD. I would like to thank my primary supervisor Prof Ouvrier for teaching me both the science and the art of peripheral nerve medicine. This PhD has coincided with probably the most exciting period in peripheral nerve research, with rapid advances in our understanding of the genetics of inherited peripheral nerve disease, due to the availability of next-generation sequencing technologies. I have benefitted from not only the extremely well phenotyped cohort of patients accumulated by Prof Ouvrier but also the national and international collaborations he has established. I would like to thank A/Prof Monique Ryan for not only spending time to carefully edit each chapter of this PhD but also her insightful comments and questions whenever I presented portions of this PhD at conferences. I would also like to thank the many coauthors on the different studies that make up this PhD (some awaiting publication), especially Prof Joshua Burns and Dr Michelle Farrar for teaching me the techniques of CMTPedS evaluation and nerve excitability studies. I would like to thank my wife Riona and son Xavier for accepting without complaint the late evenings and many weekends spent working on this PhD and the missed holidays, swimming lessons and visits to the park. I would also like to thank my parents, Joseph and Muriel, for instilling a hard work ethic and continuing to delight in my every achievement, however minor. Finally, I would like to thank all the patients and their families who participated in the studies on this PhD, especially the families of children with BVVL, and shared the journey from identification of the causative mutations to evaluating the benefits of therapy with riboflavin. 4
6 DEDICATION To Riona, for her unwavering support, and Xavier, for his beautiful smile. 5
7 TABLE OF CONTENTS Declaration... 2 Abstract... 3 Acknowledgements... 4 Dedication... 5 Table of contents... 6 List of abbreviations... 8 Publications, presentations and awards Literature Review Hypothesis Methodology Section I Peripheral neuropathy associated with mitochondrial disease in children Chapter Neurophysiological profile of peripheral neuropathy in childhood mitochondrial disease Section II Brown-Vialetto-Van Laere syndrome due to RFVT2 deficiency Chapter Phenotype of Brown-Vialetto-Van Laere syndrome due to RFVT2 deficiency Chapter Auditory neuropathy in Brown-Vialetto-Van Laere syndrome due to RFVT2 deficiency 6
8 Chapter Pathophysiology of motor nerve dysfunction in Brown-Vialetto-Van Laere syndrome due to RFVT2 deficiency Chapter Outcome after therapy with riboflavin in Brown-Vialetto-Van Laere syndrome due to RFVT2 deficiency Summary and recommendations References Appendix
9 LIST OF COMMON ABBREVIATIONS ABR ANS ANSD APB BVVL CM CMAP CMT CMTPedS FAD FMN GBB HL IH IHC I/V K + KSS LBSL MELAS Automated brainstem responses Ataxia neuropathy spectrum Auditory neuropathy spectrum disorder Abductor pollicis brevis Brown-Vialetto-Van Laere syndrome Cochlear microphonic Compound muscle action potential Charcot-Marie-Tooth disease CMT paediatric scale Flavin adenine dinucleotide Flavin mononucleotide Barrett and Barrett conductance Hearing loss Inward rectifying current Inner hair cells Current threshold relationship Potassium Kearns Sayre syndrome Leukoencephalopathy with brainstem and spinal cord involvement Mitochondrial myopathy, encephalopathy, lactic acidosis and stroke-like episodes MEMSA MERRF MIRAS mtdna Myoclonus, epilepsy, myopathy, sensory ataxia Myoclonic epilepsy and ragged red fibres Mitochondrial recessive ataxia syndrome Mitochondrial DNA 8
10 Na + Na + / K + -ATP NARP OAE OHC PDHc PEO POLG PTA Sodium Sodium/Potassium - adenosine triphosphate pump Neurogenic muscle weakness, ataxia and retinitis pigmentosa Otoacoustic emissions Outer hair cells Pyruvate dehydrogenase complex Progressive external ophthalmoplegia DNA polymerase-γ gene Pure tone audiometry RFVT1/2/3 Riboflavin transporter type 1/2/3 RRP SANDO SCAE SNAP SURF1 Τ SD TE TEd(10-20) TEd(90-100) TEh(10-20) TEh(90-100) Relative refractory period Sensory ataxia, neuropathy, dysarthria, and ophthalmoparesis Spinocerebellar ataxia and epilepsy Sensory nerve action potential Surfeit 1 gene Strength-duration time constant Threshold electrotonus Depolarizing change at 10-20ms in TE Depolarizing change at ms in TE Hyperpolarizing change at 10-20ms in TE Hyperpolarizing change at ms in TE 9
11 PUBLICATIONS, PRESENTATIONS AND AWARDS PUBLICATIONS Literature Review Menezes MP, Ouvrier RA. Peripheral neuropathy associated with mitochondrial disease in children. Dev Med Child Neurol. 2012;54(5): Chapter 2 Foley AR*, Menezes MP*, Pandraud A*, et al. Treatable childhood neuronopathy caused by mutations in riboflavin transporter RFVT2. Brain. 2014;137(Pt 1): *co-first authors Chapter 4 Menezes MP, Farrar MA, Webster R, et al. Pathophysiology of motor nerve dysfunction in a childhood neuronopathy caused by mutations in the riboflavin transporter RFVT2. Clin Neurophysiol Epub ahead of print. PRESENTATIONS The studies in chapters 1-5 have been presented at the following conferences: 1. Peripheral Nerve Society Satellite Meeting, Sydney, Australia, July Peripheral Nerve Society Biennial Meeting, Potomac, USA, July th International Congress of the World Muscle Society, Algarve, Portugal, October nd Annual Scientific Meeting of the Australia and NZ Child Neurology Society, Sydney, Australia, May Peripheral Nerve Society Biennial Meeting, St. Malo, France, June rd Annual Scientific Meeting of the Australia and NZ Child Neurology Society, Adelaide, Australia, July 2014 AWARDS National Health and Medical Research Council, Australia Dora Lush Postgraduate Scholarship ( ) 10
12 Literature Review 11
13 Literature Review This literature review has been modified from a published article (Menezes MP & Ouvrier RA, DMCN 2012). It is a comprehensive review of the characteristics of peripheral neuropathy in childhood mitochondrial disease. A literature review of Brown-Vialetto-Van Laere syndrome, a neurodegenerative syndrome that resembles mitochondrial disease, is provided separately in Chapter 2. INTRODUCTION Mitochondria are subcellular organelles whose most important function is oxidative phosphorylation for the generation of cellular ATP. This is accomplished through four transmembrane respiratory chain complexes that create an electron gradient between them to drive the final complex (ATP synthase) to make ATP. The components of the oxidative phosphorylation pathway are encoded by mitochondrial DNA (mtdna) or by nuclear genes, and mutations in these genes may result in mitochondrial disease. 1,2 Mitochondrial respiratory chain enzyme disorders have a minimum birth prevalence of 1/7634 live births and the onset occurs in childhood in around half of affected individuals. 3 Because of the varied and overlapping clinical features, respiratory chain enzyme analysis on an affected tissue is usually used to confirm a biochemical abnormality and direct genetic testing. However, these tests are invasive and not always abnormal or diagnostic. 2 Mitochondrial diseases are often associated with peripheral nerve dysfunction and neuropathy in adults with mitochondrial disease has been well described. 4,5 While mitochondrial diseases in children are often associated with, and occasionally present as, a peripheral neuropathy, the presence of a neuropathy is frequently under-recognised because of the overwhelming central nervous system involvement. Colomer et al. reported a peripheral neuropathy in 37% of 25 children with mitochondrial disorders. 6 A peripheral neuropathy occurring as part of the 12
14 Literature Review syndrome complex in an individual with a clinical profile, biochemical abnormality or genetic mutation suggestive of mitochondrial disease may be broadly classified as a mitochondrial neuropathy. Such mitochondrial neuropathies are heterogeneous in their clinical, neurophysiological and histopathological characteristics. This chapter provides a comprehensive review of childhood mitochondrial neuropathy. The peripheral neuropathy syndromes caused by different nuclear and mitochondrial DNA mutations have been classified according to the age of onset and clinical phenotype to provide a diagnostically useful guide (Table 1). These childhood mitochondrial neuropathies can also be classified according to whether the neuropathy is a predominant part of the presentation (neuropathy, ataxia, and retinitis pigmentosa [NARP], ataxia neuropathy spectrum [ANS], sensory ataxic, neuropathy, dysarthria, and ophthalmoparesis [SANDO], mitochondrial neurogastrointestinal encephalopathy [MNGIE], Friedreich ataxia, Charcot Marie Tooth disease [CMT] associated with MFN2 and GDAP1 mutations) or occurs as part of a more complex phenotype (mitochondrial depletion syndromes, MTP deficiency, Leigh syndrome, leukoencephalopathy with brainstem and spinal cord involvement and lactate elevation [LBSL], Kearns Sayre syndrome, mitochondrial myopathy, encephalopathy, lactic acidosis, and stroke-like episodes [MELAS], myoclonic epilepsy with ragged red fibres [MERRF], Leber hereditary optic neuropathy). 13
15 Literature Review Table 1. Classification of childhood mitochondrial neuropathy Mitochondrial syndromes with an onset in infancy or early childhood Mitochondrial depletion syndromes (MDS) Hepatocerebral MDS: POLG1, PEO1, DGUOK, MPV17 Encephalomyopathic MDS: SUCLA2 Complete MTP deficiency: HADHA, HADHB Leigh syndrome: Various nuclear and mtdna mutations including SURF1, MTATP6 and PDHA1 NARP: MT-ATP6 LBSL: DARS2 Kearns Sayre syndrome: single large mtdna deletions Mitochondrial syndromes with an onset in late childhood MELAS: various mtdna mutations, most commonly in MT-TL1 MERRF: various mtdna mutations, most commonly in MT-TK Late-onset syndromes due to POLG1 (ANS, MEMSA, and arpeo) MNGIE: TYMP Mitochondrial optic neuropathies Leber hereditary optic neuropathy: various mtdna mutations Dominant optic atrophy: OPA1 Friedreich ataxia: FXN Mitochondrial neuropathies presenting as CMT MFN2, GDAP1 MDS - mitochondrial depletion syndrome, MTP - mitochondrial trifunctional protein, NARP - neuropathy, ataxia, and retinitis pigmentosa, LBSL - leucoencephalopathy with brainstem and spinal cord involvement and lactate elevation, MELAS - mitochondrial myopathy, encephalopathy, lactic acidosis and stroke-like episodes, MERRF - myoclonic epilepsy with ragged red fibres, ANS - ataxia neuropathy spectrum, MEMSA - myoclonus, epilepsy myopathy, sensory ataxia, arpeo - autosomal recessive progressive external ophthalmoplegia, MNGIE - mitochondrial neurogastrointestinal encephalopathy, CMT - Charcot Marie Tooth disease. 14
16 Literature Review Mitochondrial syndromes with an onset in infancy or early childhood Mitochondrial Depletion Syndromes (MDS) Mitochondrial DNA depletion syndromes are disorders of oxidative phosphorylation characterised by a reduction in mtdna copy number in the affected tissues and caused by mutations in genes associated with mitochondrial nucleotide metabolism. The early onset MDS syndromes may be hepatocerebral (POLG1, PEO1, DGUOK, MPV17) or encephalomyopathic (RRM2B, TK2, SUCLA2, SUCLG1). Hepatocerebral MDS Alpers-Huttenlocher syndrome (AHS) is a hepatocerebral mitochondrial DNA depletion syndrome, often with an infantile or early childhood onset, characterised by refractory seizures, psychomotor delay or regression and progressive liver dysfunction. AHS is most commonly caused by recessive POLG1 mutations but mutations in PEO1 have also been implicated. When seizures are not a prominent feature and POLG1 and PEO1 sequencing are negative, mutations in DGUOK and MPV17 should be looked for. 1 POLG1 A peripheral neuropathy is only infrequently described in children with AHS and AHS-like syndrome with homozygous or compound heterozygous POLG1 mutations, possibly due to overwhelming progressive encephalopathy, hepatic failure and early death. 7-9 PEO1 Recessive mutations in PEO1, a gene that codes for mitochondrial Twinkle helicase, result in two similar syndromes: an early-onset encephalopathy resembling AHS with symptom onset 15
17 Literature Review at around 6 months of age and an infantile-onset spinocerebellar ataxia with symptom onset at an average age of 14 months. 10 Reported with both homozygous and compound heterozygous PEO1 mutations, the early onset syndrome is characterised by psychomotor regression, truncal hypotonia, growth failure, ophthalmoparesis, epileptic encephalopathy, lactic acidosis and elevation of liver transaminases with mtdna depletion in the brain and liver. Affected children have an axonal sensory neuropathy with absent SNAPs on nerve conduction studies and loss of large myelinated fibres on sural nerve biopsy. 10,11 Infantile-onset spinocerebellar ataxia is characterised by ataxia, hypotonia, athetosis, hearing loss, ophthalmoplegia and a predominantly sensory axonal neuropathy. The onset of epilepsy is usually beyond the second decade of life. Children with infantile-onset spinocerebellar ataxia have loss of touch, proprioception and vibration sense with preserved pain and temperature responses and may develop distal weakness and pes cavus. 12 SNAPs usually become unrecordable between five and 15 years of age. The motor conduction velocities in the lower limbs are normal initially but show a gradual decline in the second decade of life. The sural nerve biopsy shows an axonal neuropathy with pronounced loss of large myelinated fibres. 13 MtDNA depletion is limited to the CNS in infantile-onset spinocerebellar ataxia. 14 In both syndromes, difficulties with micturition, urinary incontinence, constipation and dry eyes indicate the presence of an autonomic neuropathy. 10,12 DGUOK Children with homozygous or compound heterozygous deoxyguanosine kinase (DGUOK) mutations have progressive liver failure and variable neurological involvement. A sensorimotor neuropathy was identified in two of nine children in a case series of children with DGUOK mutations
18 Literature Review MPV17 Homozygous or compound heterozygous mutations in MPV17 have been described in affected children with an infantile onset of progressive liver failure, ataxia, hypotonia, dystonia and psychomotor regression. Also referred to as Navajo neurohepatopathy, the syndrome is characterised by a severe sensorimotor neuropathy with severe anaesthesia, corneal ulceration, painless fractures, acral mutilation and distal weakness. Nerve conduction studies show reduced motor conduction velocities, with ulnar motor nerve conduction velocities of 28-36m/s reported in two patients aged 8 and 6 years. Sural nerve biopsies show complete loss of myelinated fibres and degeneration and regeneration of unmyelinated fibres. MPV17 encodes for a protein of unknown function located on the inner mitochondrial membrane Encephalomyopathic MDS SUCLA2 Succinyl-coenzyme A ligase (SUCL) is a mitochondrial enzyme associated with the Krebs cycle that catalyses the conversion of succinyl-coa to succinate. Patients with recessive mutations in SUCLA2 and SUCLG1, encoding the β2 and α subunits of SUCL, have an encephalomyopathy and mild methylmalonic aciduria with mitochondrial DNA (mtdna) depletion in muscle and to a lesser extent in the liver. 19 There are multiple reports of children with homozygous or compound heterozygous SUCLA2 mutations, but a peripheral neuropathy has been described in only two children from a consanguineous family from the Faroe Islands, while other affected individuals in the family had normal nerve conduction studies. 20,21 17
19 Literature Review Complete Mitochondrial Trifunctional Protein (MTP) Deficiency MTP is a multienzyme complex bound to the inner mitochondrial membrane that is involved in the β-oxidation of long-chain fatty acids. Homozygous or compound heterozygous mutations in HADHA or HADHB, encoding the α and β-subunits respectively of the Hydroxyacyl-CoA Dehydrogenase/3-Ketoacyl-CoA Thiolase/Enoyl-CoA Hydratase (Trifunctional Protein), have been identified in complete mitochondrial trifunctional protein deficiency. Complete MTP deficiency may present in an early-onset rapidly progressive form with hypoketotic hypoglycaemia and cardiomyopathy, with high mortality due to metabolic decompensation and cardiac failure, or in an insidious neuromyopathic form. In the series described by den Boer et al., 11 of 14 children had a peripheral neuropathy at diagnosis. 22 In the later onset neuromyopathic form, the chronic neuropathic weakness often precedes episodes of recurrent myoglobinuria and can cause distal weakness, foot deformity and loss of ambulation. Nerve conduction studies reveal an axonal sensorimotor neuropathy. 23 Leigh Syndrome/ Neurogenic muscle weakness, Ataxia and Retinitis Pigmentosa (NARP) Leigh syndrome (subacute necrotising encephalopathy) is a clinically and genetically heterogeneous group of neurodegenerative disorders that have similar features on central nervous system imaging and histopathology. The onset of symptoms is usually in infancy or early childhood and children may have progressive psychomotor retardation, hypotonia, weakness, ataxia, optic atrophy, ophthalmoplegia, nystagmus, dystonia, evidence of brainstem dysfunction and lactic acidosis. 24 Neuroimaging demonstrates high T2 signal, most frequently in the caudate nucleus and putamen, but sometimes involving the thalamus, periaqueductal grey matter, tegmentum, red nuclei and dentate nuclei. 25,26 A large number of different mitochondrial and nuclear gene mutations have been reported in Leigh syndrome, of 18
20 Literature Review which SURF1 is the most common nuclear gene involved, and MT-ATP6 the most common mitochondrial gene. 24 NARP syndrome is characterised by neuropathy, ataxia with cerebellar atrophy on MRI, and pigmentary retinopathy and optic atrophy on ophthalmologic examination. MT-ATP6 is the only gene reported to be mutated in this syndrome, with the 8993T>G mutation being the most common, though 8993T>C and 9185T>C mutations have also been described. 27 SURF1 SURFEIT1 or SURF1 is the most commonly mutated nuclear gene in Leigh syndrome, with both homozygous and compound heterozygous mutations being reported. 24 Two case reports of children (16 months and 3 years of age) with Leigh syndrome due to SURF1 mutations described a demyelinating neuropathy with motor conduction velocities in the median and peroneal nerves of 20-27m/s. 28,29 Sural nerve biopsy in one child showed reduced myelinated fibres with a complete absence of fibres of larger diameter and thin myelin sheaths in the remaining fibres. Schwann cell mitochondria were enlarged with rounded cristae. 29 MT-ATP6 In a small case series that included both children and adults with MT-ATP6 mutations, the median age of onset of symptoms was four to five months for those with a 8993T>G mutation compared to four years for the 8993T>C mutation. Around 30% of individuals with 8993T>G and 8993T>C mutations in MT-ATP6 were found to have a peripheral neuropathy. 30 The peripheral neuropathy presents with ataxia, pes cavus and absent ankle jerks. Diminished responses to pinprick and vibration have been described. An axonal sensory neuropathy is described in adults with the 8993T>G mutation, but reports with well 19
21 Literature Review characterised nerve conduction studies in children with this mutation are lacking, probably due to the early onset and prominent central nervous system involvement 31,32. An axonal neuropathy has been reported in children and adults with the 8993T>C and 9185T>C mutations. Sural nerve biopsy shows active Wallerian degeneration of both myelinated and unmyelinated fibres Childs et al. described a family with the 9185T>C mutation with affected individuals having either an axonal or demyelinating neuropathy, but the nerve conduction velocities were not reported. 37 PDHA1 The pyruvate dehydrogenase complex (PDHc) is a multi-enzymatic complex with three catalytic subunits. PDHc deficiency is most frequently caused by a deficit in the α subunit of E1, due to mutations in the E1 α subunit gene, PDHA1. 38 Affected children may present acutely with weakness and motor regression. Symptoms and nerve conduction velocities may improve after treatment with thiamine in deficiency of the PDHc E1subunit, a thiaminepyrophosphate dependent decarboxylase. 39,40 Seven percent of reported cases with PDHc deficiency had an associated peripheral neuropathy. 38 PDHc deficiency is associated with an axonal sensorimotor neuropathy, with normal or mildly reduced motor conduction velocity (>= 30m/s), low amplitude or absent SNAPs and evidence of denervation distally on electromyography The sural nerve biopsy usually shows only a mild reduction in myelinated fibre density. 42,45 Di Rocco et al. reported a five-year-old male with more significant reduction in motor conduction velocity (19.8m/s in the median nerve) which improved to 37.9m/s after treatment with thiamine. 20
22 Literature Review Leukoencephalopathy with Brainstem and Spinal cord involvement and Lactate elevation (LBSL) LBSL is characterised by a childhood onset of slowly progressive cerebellar ataxia, spasticity and dorsal column dysfunction. Mild cognitive decline is seen in some affected individuals. The MRI shows signal changes in the cortical white matter and in specific tracts through the brainstem and spinal cord. LBSL occurs due to compound heterozygous, and rarely homozygous, mutations in DARS2 that encodes mitochondrial aspartyl-trna synthetase. 46,47 A neuropathy, often subclinical, is associated with LBSL though it may be symptomatic with pes cavus and distal weakness. Nerve conduction studies show an axonal neuropathy with preserved or mildly reduced SNAPs, reduced CMAPs and preserved or mildly reduced conduction velocities with denervation in distal muscles on EMG. 48,49 Interestingly, mutations in genes encoding cytoplasmic aminoacyl-trna synthetases have been reported to cause CMT, of which only KARS, a lysyl-trna synthetase, appears to have an additional role in mitochondrial protein translation. 50 Kearns Sayre Syndrome (KSS) KSS is a multi-system disorder caused by single, large deletions in mtdna and is associated with retinitis pigmentosa and progressive external ophthalmoplegia developing before the age of twenty years. In addition, affected individuals have at least one of the following: cardiac conduction block, cerebrospinal fluid protein concentration greater than 100 mg/dl or cerebellar ataxia. Cognitive impairment, sensorineural deafness, cardiomyopathy, short stature, endocrinopathies and dysphagia are associated features. 2 Reports in adults with KSS document either normal nerve conduction studies or an axonal peripheral neuropathy. 51,52 An axonal sensorimotor peripheral neuropathy has been described 21
23 Literature Review in childhood with gait ataxia, glove and stocking paresthesia, distal weakness and areflexia. The muscle biopsy may show evidence of denervation with type 2 atrophy, and sural nerve biopsy in severe neuropathy shows prominent endoneurial collagen with few remaining axons. In sural nerve biopsies in adults, enlarged mitochondria with abnormal cristae may be seen on electron microscopy in the Schwann cells as well as in endoneurial capillaries and small arterioles of the sural nerves MITOCHONDRIAL SYNDROMES WITH AN ONSET IN LATE CHILDHOOD Mitochondrial Myopathy, Encephalopathy, Lactic Acidosis, and Stroke-like episodes (MELAS) While 80% of patients with MELAS have the 3243A>G mutation in the mtdna MT-TL1 gene, encoding mitochondrial trna for leucine, mutations in several other mitochondrial genes have also been described as causing this syndrome characterised by stroke-like episodes with seizures, intermittent episodes of encephalopathy, vomiting, migraine like headaches, ataxia and cognitive impairment. 2 Between 22 and 77% of adults with the 3243A>G mutation have neurophysiological evidence of a peripheral neuropathy which is predominantly axonal. 56,57 Stickler et al. reported that children with MELAS commonly had a mixed neuropathy. 58 Myoclonic Epilepsy and Ragged Red Fibres (MERRF) MERRF is caused most commonly by an 8344A>G point mutation in the mtdna MT-TK gene, encoding mitochondrial trna for lysine, though mutations in several other mitochondrial genes can be associated with this progressive neurodegenerative disorder characterised by myoclonic seizures among other seizure types, myopathy with ragged red fibres on muscle biopsy, ataxia, optic atrophy, pyramidal signs and hearing loss. 2 An axonal 22
24 Literature Review or mixed neuropathy is most commonly reported in adults. 59 Erol et al. reported a child with the 8344A>G mutation and combined central and peripheral demyelination, with reduced motor conduction velocities (31m/s in the peroneal and 41m/s in the median nerves) and absent SNAPs. 60 Late-onset syndromes associated with POLG1 mutations Homozygous or compound heterozygous mutations in DNA polymerase-γ (POLG1) have been identified as causing varied late-onset disease phenotypes that include: (1) Ataxia neuropathy spectrum (ANS) disorders that include the mitochondrial recessive ataxia syndrome (MIRAS) and spinocerebellar ataxia and epilepsy (SCAE), (2) Myoclonus, epilepsy, myopathy, sensory ataxia (MEMSA) and (3) autosomal recessive progressive external ophthalmoplegia (arpeo) that includes the sensory ataxia, neuropathy, dysarthria, and ophthalmoparesis (SANDO) syndrome. 61 POLG1 is required for the genetic stability of mtdna, and its exonuclease domain increases the fidelity of mtdna replication by conferring a proofreading activity on the enzyme. 62 A peripheral neuropathy is commonly described as a component of the clinical phenotype in a number of overlapping syndromes including ANS, MEMSA and arpeo. 61,63-65 These syndromes with predominant ataxia and peripheral neuropathy have an onset in late adolescence or adulthood, while those with a prominent encephalopathy or hepatopathy usually have a much earlier onset in infancy or early childhood. 61,66,67 Older children and young adults with recessive POLG1 mutations present with deterioration of their gait and progressive unsteadiness and demonstrate loss of kinesthetic and vibration sensation, positive Romberg sign, ataxia and areflexia on examination, consistent with a sensory ataxic neuropathy. Nerve conduction studies show a predominantly sensory axonal neuropathy. 23
25 Literature Review Sural nerve biopsy shows loss of large and small myelinated and unmyelinated fibres. Most patients have progressive ataxia due to a combination of cerebellar and sensory deficits and this results in significant disability and wheelchair dependence. 63,64 Mitochondrial Neurogastrointestinal Encephalomyopathy (MNGIE) MNGIE is caused by homozygous or compound heterozygous mutations in TYMP encoding thymidine phosphorylase and is associated with multiple deletions and depletion of mtdna in skeletal muscle MNGIE is characterised clinically by severe gastrointestinal dysmotility, cachexia, ptosis, ophthalmoparesis, peripheral neuropathy and leukoencephalopathy. The thymidine phosphorylase enzyme catabolizes thymidine to thymine and 2-deoxy-D-ribose 1-phosphate. In patients with MNGIE, reduced thymidine phosphorylase activity alters deoxynucleoside and nucleotide pools and consequently impairs mtdna replication and repair. 71 Recently, mutations in RRM2B, encoding cytosolic p53- inducible ribonucleoside reductase small subunit (RIR2B), have been described as causing the MNGIE-like phenotype. 72 The age of onset of symptoms is usually in the 2 nd decade of life (median 18.7 years) and commonly with gastrointestinal symptoms, though a small number may have a peripheral neuropathy as the first symptom. All patients will ultimately develop a peripheral neuropathy. 71 Patients often present with numbness, paresthesiae and a burning pain in their feet and toes. Deterioration in gait and hand weakness may also be presenting symptoms or develop soon afterwards. Examination reveals distal weakness and atrophy, areflexia and sensory loss, including pain and vibration, in a glove-stocking distribution. 71,
26 Literature Review On nerve conduction studies, a demyelinating sensorimotor neuropathy is seen Prolongation of F-waves and presence of conduction block may lead to a misdiagnosis of chronic inflammatory demyelinating neuropathy. 73,75 Biopsies from the lumbar and brachial plexus show lost or markedly attenuated myelin sheaths with comparatively little axonal loss. Sural nerve biopsy shows thin myelin sheaths around axons and axonal loss. While ultrastructural mitochondrial abnormalities have not been reported in the peripheral nerve biopsy, the cytoplasm of ganglion cells on rectal biopsy may show numerous megamitochondria with an expanded matrix and reduced cristae. 74,75 Sustained biochemical and clinical improvement has been described after allogeneic bone marrow transplantation for MNGIE. 76 It is recommended that transplantation be carried out early in the disease course where the potential for optimum recovery may be higher Mitochondrial Optic Neuropathies 3.1. Leber Hereditary Optic Neuropathy (LHON) LHON usually has an onset between 15 and 30 years of age and is characterised by bilateral acute or subacute visual loss. Ninety-five percent of cases of LHON are caused by the 3460G>A, 11778G>A and 14484T>C mutations in mtdna. Cardiac arrhythmias, postural tremor, myopathy and movement disorder may be associated. 78 A peripheral neuropathy with mild to moderate reduction of motor conduction velocities may be seen in up to 20% of adults with LHON but is often mild or subclinical. 79, Dominant Optic Atrophy (OPA1 gene) Dominant mutations in OPA1, encoding a dynamin-related ATPase, are a common cause of autosomal dominant optic atrophy. Affected patients present with visual loss in the first two 25
27 Literature Review decades of life, and have abnormalities in colour vision, central or paracentral scotomas and optic disc pallor. 78,81 Yu-Wai-Man et al. reported an axonal sensory and/or motor neuropathy in 31 out of 104 individuals with OPA1. In this cohort, one-third of children below 18 years of age had evidence of a neuropathy Friedreich Ataxia Friedreich ataxia is caused by hyperexpansion of GAA repeats in the first intron of the FXN gene that codes for frataxin, a protein that is a component of iron-sulphur clusters and involved in mitochondrial respiratory chain activity. 83 Friedreich ataxia is characterised by ataxia, sensory neuropathy, muscle weakness, scoliosis, hypertrophic cardiomyopathy, optic atrophy, sensorineural hearing loss and diabetes. 84 Though onset is commonly in the second decade, onset before the age of 10 years is well described. 85 Nerve conduction studies reveal an axonal sensory neuropathy with severely reduced or absent SNAPs, and sural nerve biopsy shows loss of myelinated fibres, particularly those of large diameter. Santoro et al. have shown that, on multiple regression analysis, the number and percentage of sural myelinated fibres as well as the sural and tibial SAP are significantly related to the size of the GAA repeats but not to disease duration Mitochondrial neuropathies presenting as Charcot-Marie-Tooth disease (CMT) The characteristics of the peripheral nerve disorders associated with abnormalities of genes that are known to be important for mitochondrial aggregation provide a framework to understand peripheral nerve dysfunction in mitochondrial disease. The coordinated action of the mitofusins, mfn1 and mfn2, located on the outer mitochondrial membrane and OPA1, located on the inner mitochondrial membrane, is required for mitochondrial fusion. 87,88 Dominant mutations in the gene encoding mitofusin 2 (MFN2), a large transmembrane 26
28 Literature Review GTPase, are the most common cause of Charcot-Marie-Tooth disease type 2, an axonal sensorimotor peripheral neuropathy. 89 Some individuals with MFN2 mutations have associated optic atrophy. 90 Abnormal small, round and aggregated mitochondria have been demonstrated on electron microscopy of sural nerve biopsies of children with MFN2 mutations. 91 Mutations in the ganglioside-induced differentiation-associated protein 1 gene (GDAP1) are associated with both axonal and demyelinating types of autosomal recessive CMT (CMT4A) and are also a rare cause of autosomal dominant axonal CMT (CMT2H/K). 92,93 GDAP1 is located on the outer mitochondrial membrane and expressed by both neurons and myelinating Schwann cells. It promotes mitochondrial fission and over-expression results in mitochondrial fragmentation. Fibroblasts from patients with GDAP1 mutations show reduced complex 1 activity, a fragmented mitochondrial network and abnormally large mitochondria. 94 MFN2 and GDAP1 thus have opposing effects on mitochondria and their equilibrium is necessary for normal mitochondrial dynamics. 95 The clinical features and neurophysiological abnormalities associated with these two mutations have been well described and will not be repeated here. CONCLUSION Peripheral nervous system involvement has been described in a number of mitochondrial syndromes and may be a significant part of the presenting phenotype. However, the clinical, neurophysiological and histopathological characteristics of these mitochondrial neuropathies are poorly described, often only in the form of individual case reports and occasional children in mainly adult cohorts. The quality of the studies in this cohort of children is poor, with level 3 studies (non-random sample) available for children with IOSCA, MTP deficiency, MT- 27
29 Literature Review ATP6 mutations, PDHc deficiency, LHON and Friedreich Ataxia, and only case series or case reports (level 4 studies) being available for other mutations. Early recognition of the associated neuropathy (in NARP, ANS, MNGIE and the optic neuropathies) may help identification of the mitochondrial syndrome. The characteristics of the neuropathy may help direct genetic testing without the requirement for invasive skin, muscle and liver biopsies in mitochondrial syndromes where a common phenotype is caused by different mutations. This could be useful in Leigh syndrome, where the common nuclear SURF1 mutation causes a demyelinating neuropathy while mtdna MT-ATP6 mutations cause an axonal neuropathy. POLG1 mutations, especially when associated with late-onset phenotypes, cause a predominantly sensory neuropathy with prominent ataxia. The identification of the peripheral neuropathy also helps target genetic testing in the mitochondrial optic neuropathies. Though often subclinical, the peripheral neuropathy associated with mitochondrial disease may occasionally be symptomatic and cause significant disability. Recognition of the neuropathy, where symptomatic, will help early institution of rehabilitative therapy. I also suggest that nerve conduction studies should be a part of the early evaluation of children with suspected mitochondrial disease. This literature review indicates that there is a need for a systematic study investigating the characteristics of the peripheral neuropathy in children with different genetic changes causing mitochondrial disease, to determine more accurately the different characteristics of the peripheral neuropathy associated with each of these genetic changes. This thesis presents the results of such a study with data from patients at three tertiary level paediatric hospitals. 28
30 Hypotheses Hypotheses (and structure of thesis) 29
31 Hypotheses Hypotheses 1. The peripheral neuropathies associated with the common genetic causes of childhood mitochondrial disease have unique neurophysiological characteristics that help direct genetic testing. 2. Brown-Vialetto-Van Laere syndrome (BVVL) due to RFVT2 deficiency has a unique clinical, audiological, neurophysiological and biochemical profile that resembles mitochondrial disease and is different from that of RFVT3 deficiency. 3. Treatment with high-dose riboflavin is effective in stabilising or reversing the progress of auditory neuropathy and peripheral neuropathy in Brown-Vialetto-Van Laere syndrome due to RFVT2 deficiency. Structure of this thesis Hypothesis 1 I performed prospective nerve conduction studies and reviewed previously performed nerve conduction studies in a total of 26 children with mitochondrial disease with identified nuclear or mitochondrial mutations. The results were classified according to the underlying genetic cause before assessment. The results of this study are detailed in Chapter 1. Hypothesis 2 BVVL is a neurodegenerative syndrome with a motor and sensory neuropathy. The introduction to Chapter 2 provides a literature review on BVVL. I studied the clinical phenotype, audiology, neurophysiology, biochemical defect and histopathology of 9 children from 4 families with BVVL with mutations in SLC52A2. This phenotypic characterisation is detailed in Chapter 2, and the audiological profile in Chapter 3. 30
32 Hypotheses Hypothesis 3 As SLC52A2 encodes a neuronal riboflavin transporter RFVT2, it has been suggested that high-dose riboflavin may be beneficial in BVVL due to RFVT2 deficiency, and there have been reports of benefit with riboflavin on short-term follow-up. I treated seven of the affected children with high-dose riboflavin over a 2-year period. Clinical, audiological, neurophysiological and biochemical assessments were performed at baseline and after 12 and 24 months of therapy. The neurophysiological assessments included the use of novel nerve excitability techniques. The nerve excitability tests at baseline helped understand the pathophysiological basis of motor nerve dysfunction in BVVL, and this study, including the change after riboflavin therapy, is detailed in Chapter 4. Chapter 5 details the change in clinical, audiological and biochemical parameters after therapy, including assessments on the validated CMT Pediatric Scale (CMTPedS). 31
33 Methodology 32
34 Methodology Subjects in the mitochondrial neuropathies study were confirmed to have mitochondrial disease by the identification of pathogenic nuclear or mitochondrial genome mutations on genetic testing or, in the case of pyruvate dehydrogenase complex (PDHc) deficiency, by the identification of a biochemical defect on PDHc enzyme assay that was consistent with PDHc deficiency. The diagnosis of Brown-Vialetto-Van Laere syndrome due to RFVT2 deficiency in the study subjects was confirmed by the identification of homozygous or compound heterozygous mutations in the SLC52A2 gene. All subjects and/or their parental guardian gave informed consent to the study and procedures, which were approved by the relevant human research ethics committee. Details of specific ethics approvals are noted in the individual chapters. The information sheet and consent forms for the neuropathy in children with mitochondrial disease study are available in the Appendix. Other consents were taken as part of larger genetic, natural history and nerve excitability studies. NEUROPHYSIOLOGICAL ASSESSMENTS Nerve conduction tests Nerve conduction tests were performed using a Viking On Nicolet EDX Electrodiagnostic System from CareFusion Nicolet with surface electrodes. Sedation with nitrous oxide was used in some children above the age of 12 months. For the motor studies, a supramaximal rectangular pulse direct current stimulus was delivered using a two-prong stimulator, to elicit a compound muscle action potential (CMAP). The median nerve was stimulated at the wrist and elbow, and the CMAP was recorded over the bulk of the abductor pollicis brevis (APB) muscle. The ulnar nerve was stimulated over the wrist and behind the elbow, and the CMAP was recorded over the bulk of the abductor digiti minimi (ADM) 33
35 Methodology muscle. The common peroneal nerve was stimulated over the ankle and knee, and the CMAP was recorded over the bulk of the extensor digitorum brevis (EDB) muscle. The tibial nerve was stimulated over the ankle and behind the knee, and the CMAP was recorded over the bulk of the abductor hallucis (AH) muscle. The sweep speed and sensitivities were 5 ms/division and 5 mv/division respectively. The duration of the stimulus was 0.1ms. The sensory nerve action potential (SNAP) was recorded by an orthodromic technique for the upper limb (median and ulnar) nerves and by an antidromic technique for the lower limb (sural) nerve. For the median nerve, the stimulus was delivered by ring electrodes over the index finger (digital branch of the median nerve) and SNAPs were recorded over the wrist. For the ulnar nerve, the stimulus was delivered by ring electrodes over the little finger (digital branch of the ulnar nerve) and SNAPs were recorded over the wrist. For the sural nerve, the stimulus was delivered above the lateral malleolus, approximately 14 cm from the proximal recording electrode which was placed on the lateral border of the foot. All sensory potentials were recorded using a recurrent stimulus and with averaging using a supramaximal stimulus. The stimulus duration was 0.1ms and the sweep speed and sensitivities were 1 ms/division and 10 µv/division respectively. The distal motor latencies, CMAP amplitudes and conduction velocities were recorded using conventional methods. CMAP and SNAP amplitudes were measured from baseline to the negative peak of the action potential. The distal motor latency was calculated from the stimulus artefact to the initial negative deflection from the baseline. Audiological tests The methodology of the audiological tests is detailed in Chapter 3. 34
36 Methodology Nerve excitability studies The methodology of the nerve excitability studies is detailed in Chapter 4. CMT PAEDIATRIC SCALE (CMTPedS) The Charcot-Marie-Tooth Paediatric Scale (CMTPedS) is a reliable, valid and sensitive global measure of function and disability for children with CMT. The CMTPedS was developed by Prof Joshua Burns at The Children s Hospital at Westmead and The University of Sydney, Australia, through the Inherited Neuropathies Consortium and has undergone a rigorous process of development and validation. 96 Most forms of CMT have an onset in childhood and this scale was devised because of the need for a clinical tool to measure impairment in affected children. The scale includes test items relevant to neuropathy and disability that are sensitive to change. The test items in the CMTPedS include 1. Functional dexterity test 2. Nine hole peg test 3. Grip strength 4. Plantarflexion strength 5. Dorsiflexion strength 6. Pinprick 7. Vibration 8. Balance: Bruininks-Oseretsky Test of Motor Proficiency, 2nd Ed (BOT-2) 9. Gait 10. Long jump 11. Six-minute walk test 35
37 Methodology The CMTPedS scale has test items that measure function (upper limb - functional dexterity test and nine hole peg test; lower limb - balance, gait, long jump and six-minute walk test), strength (upper limb grip strength; lower limb plantarflexion and dorsiflexion strength) and sensation (pinprick and vibration). All items are measured in the dominant limb only, as item analysis has shown that left and right paired items are highly correlated. In order to rate performance across age and gender, item scores are converted to dimensionless z-scores based on age and gender-specific normative reference values. This offsets the improvement in strength and function seen with increasing growth and development. Based on z-scores, each item is thus scored between 0 and 4. The 11-item CMTPedS total score has a possible range of 0 (unaffected) to 44 (severely affected). CMTPedS equipment and training resource kit is available from a link in the article detailing the validation of the CMTPedS scale ( An online calculator that calculates the individual item z-score, individual item category score and total CMTPedS score is available at Prior to use in study subjects, I was trained in the application of the individual test items by Prof Joshua Burns. Reliability for the test items was compared with other trained users and found to be excellent. The CMTPedS scale has been shown to have excellent inter-rater reliability. Rasch analysis has shown that the CMTPedS is a viable global measure of disability in children with CMT. 96 Since its validation, the CMTPedS has been used by the Inherited Neuropathies Consortium as the primary measure for the measurement of disability in children aged 3-20 years with CMT, in natural history studies as well as intervention trials. Analysis of longitudinal data 36
38 Methodology has shown that the CMTPedS is sensitive to change in children with CMT aged 3-20 years over a 2-year period. 97 The CMTPedS scale can be used to (1) measure baseline disability (2) perform longitudinal measurements as part of a natural history study or to determine response to an intervention, in children with CMT. I have used the CMTPedS to measure baseline disability and response to treatment with riboflavin in children with BVVL due to RFVT2 deficiency. The phenotype of BVVL due to RFVT2 includes generalised abnormality in sensation, differential motor involvement in upper and lower limb strength, and significant functional abnormalities. The CMTPedS has items that are able to measure each of these deficits. As discussed in Chapter 3, the CMTPedS is not currently validated as an outcome measure in BVVL. 37
39 SECTION I Peripheral neuropathy associated with mitochondrial disease in children 38
40 Chapter 1 Neurophysiological profile of peripheral neuropathy in childhood mitochondrial disease 39
41 Mitochondrial Neuropathy INTRODUCTION Childhood mitochondrial diseases have a heterogeneous phenotype with many different systems being affected including the peripheral nervous system. Around 30% of children with a mitochondrial disease have an associated peripheral neuropathy, 6 but the neuropathy is often unrecognised due to the overwhelming central nervous system manifestations. Mutations in nuclear genes responsible for mitochondrial fission, fusion and axonal transport, including MFN2 and GDAP1, are recognised causes of Charcot-Marie-Tooth disease (CMT). 89,95 Recently, mutations in MT-ATP6 and SURF1, genes known to cause Leigh syndrome and other multisystemic mitochondrial diseases, have been shown to cause phenotypes characterised predominantly by a peripheral neuropathy. 98,99 As evident from the literature review, the neurophysiological characteristics of the associated peripheral neuropathy often differ among the various mitochondrial syndromes. Identifying the presence of a peripheral neuropathy and defining its characteristics may help with classifying the mitochondrial syndrome and targeting genetic testing. The associated peripheral neuropathy may be symptomatic and disabling, and specific treatments and rehabilitative interventions may be useful. METHODS (a) Children with mitochondrial disease and an identified genetic mutation who had previously undergone nerve conduction studies were identified from the mitochondrial diseases database at the Murdoch Childrens Research Institute, Melbourne, Australia, the records of the Metabolic Diseases Clinic at the Children s Hospital at Westmead and the Neurophysiology Department at the Great Ormond Street Children s Hospital. The Metabolic Diseases Clinic is the primary clinic for children with mitochondrial disease at The 40
42 Mitochondrial Neuropathy Children s Hospital at Westmead. The Murdoch Childrens Research Institute is the premier diagnostic and research institute for mitochondrial disease in Australia, and almost all diagnostic testing for mitochondrial disease (muscle enzymology and genetic testing) on samples from patients in Australia takes place at this institute. The institute maintains a clinical database on samples referred from all over Australia. I was working as an Honorary Fellow at the National Hospital for Neurology and Neurosurgery at Queens Square and Great Ormond Street Children s Hospital in as part of the Churchill Fellowship. The Great Ormond Street Children s Hospital has a clinic and research program for childhood mitochondrial disease. The Neurophysiology Department has a database of previously performed nerve conduction studies and I compared this database to the patient list from the GOSH mitochondrial disease clinic to identify those affected children who had nerve conduction studies. (b) Prospective nerve conduction studies were performed on children from the Metabolic Diseases Clinic at the Children s Hospital at Westmead who had an identified mitochondrial mutation and consented to inclusion in the study. Both studies were approved by the Sydney Children s Hospital ethics committee (10/56). The methodology of the prospective nerve conduction studies is detailed in the Methodology section on page 13. Normative data was sourced from Cai et al. 100 The neuropathy was designated as demyelinating when the nerve conduction velocity was reduced to < 70% of the lower limit for that age range (lower limit = mean-2sd). The neuropathy was designated as axonal when there was a reduction in the CMAP amplitude and there was no reduction in the conduction velocity, or the reduction did not satisfy criteria for demyelinating neuropathy. Care was taken when the CMAP amplitude was <1mV as it is known that measures of 41
43 Mitochondrial Neuropathy conduction velocity in this setting can be erroneous and appear pseudo-demyelinating due to preferential loss of faster conduction fibres. All prospective nerve conduction studies (designated with P in column 5 of the tables and column 6 for table 1.4) were performed by me according to a previously defined protocol (see Methodology section). I also reviewed, tabulated and compared all nerve conduction study results with age matched norms. RESULTS Nerve conduction data was available from 27 studies on 26 children with a genetically identified mitochondrial disease. This included retrospective nerve conduction study data on 20 children and prospectively performed nerve conduction studies on seven children. One child in the prospective study also had data included from another nerve conduction study performed four years prior. The results were classified according to the underlying genetic abnormality. 42
44 Mitochondrial Neuropathy PDHC Deficiency The pyruvate dehydrogenase complex (PDHc) is a mitochondrial complex that catalyzes the rate-limiting step in aerobic glucose oxidation and is made up of three enzymatic subunits, all of which are nuclear encoded. Most affected individuals have a mutation in the PDHA1 gene on the X-chromosome, encoding the E1 α subunit. Diagnosis is established by showing reduced overall PDHc activity or reduced activity of one of its component parts in muscle, skin fibroblasts, lymphoblasts or other tissues. 38 Review of five nerve conduction studies (3 prospective, 2 retrospective) from three children with PDHc deficiency showed the presence of a patchy axonal, non-length dependent sensorimotor neuropathy. CMAP amplitudes were low in the upper and lower limbs with mild or no slowing of the nerve conduction velocity, consistent with axonal loss. Sensory responses were universally absent, even in studies done at a young age. There was no clear correlation of severity of motor neuropathy with increasing age or disease duration, and nerve conduction studies performed 4 years apart in patient 1 did not show a significant deterioration. The study in patient 4 was performed 8 weeks into an episode of acute weakness. Previously published reports of children with PDHC deficiency and a neuropathy show a predominantly axonal neuropathy with significantly reduced CMAP amplitudes and mildly reduced nerve conduction velocities (32-38m/s) ,101 In these reports, the axonal neuropathy was recognised in both those investigated prior to institution of treatment with thiamine as well as those on treatment. Only a single individual had a more significant reduction of median motor conduction velocity, which improved from 19.8m/s to 37.9m/s following treatment with thiamine
45 Mitochondrial Neuropathy Table 1.1: Neurophysiological profile of children with PDHc deficiency Pt./Sex Mutation Age at presentation Possible neuropathic features Age at NCS, Retrospective /Prospective Median CMAP (mv) Median CV (m/s) Ulnar CMAP (mv) Ulnar CV (m/s) Peroneal CMAP (mv) Peroneal CV (m/s) Tibial CMAP (mv) Tibial CV (m/s) Median SNAP (µv) Ulnar SNAP (µv) Sural SNAP (µv) 1 yr/r NR NR NR 1/M c.787c>g 11 m mild generalised weakness, areflexia, acute episodic weakness 5 yrs/p NR NR 6 yrs/p NR - NR 2/M NT 5 m - 7 yrs/p NR NR NR 3/M NT 3 yrs generalised distal predominant weakness, areflexic during acute episode 11 yrs/r (8 weeks into acute episode) NR NR NR - NR Abnormal results (< 2SD) in bold. Reference values from Cai et al. 100 R retrospective, P prospective, CMAP compound muscle action potential, CV conduction velocity, SNAP sensory nerve action potential, m months, NR not recordable, NT not tested 44
46 Mitochondrial Neuropathy SURF1 Leigh syndrome is a neurodegenerative condition characterised by progressive psychomotor deterioration, characteristic basal ganglia and brainstem changes on neuroimaging or neuropathology, and raised CSF or serum lactate. Leigh syndrome can be caused by mutations in the nuclear or mitochondrial genomes. The SURF1 (surfeit-1) gene encodes one of the assembly factors of COX, the terminal component of the mitochondrial respiratory chain, and mutations in SURF1 can cause Leigh syndrome. I reviewed 11 nerve conduction studies from 10 children with homozygous or compound heterozygous SURF1 mutations. All SURF1 mutations identified in my study have been previously reported except for the c.792_793delag mutation seen in patient 13. Seven children had a demyelinating sensorimotor neuropathy, while three showed predominantly axonal changes with a mild reduction in motor conduction velocity. The cohort had a median CV of 28m/s (range 14-43m/s). Unlike typical forms of CMT, the nerve conduction abnormalities were not length-dependent. Three children had only motor involvement on nerve conduction studies, while both sensory and motor nerves were involved in the rest. Ataxia is a prominent feature of children with Leigh syndrome due to SURF1 deficiency, and these nerve conduction studies indicate a neuropathic component to the ataxia. Six of the children described here (patients 5,6,7,8,9,11) were also included in a description of 44 individuals with SURF1 mutations by Wedatilake et al. 102 In that report, 13 of 16 individuals undergoing nerve conduction studies had a neuropathy, which was demyelinating in seven cases. 45
47 Mitochondrial Neuropathy Table 1.2: Neurophysiological profile of children SURF1 mutations Pt./ Sex Mutation Age at presentation Possible neuropathic features Age at NCS, Retrospective/ Prospective Median CMAP (mv) Median CV (m/s) Ulnar CMAP (mv) Ulnar CV (m/s) Peroneal CMAP (mv) Peroneal CV (m/s) Tibial CMAP (mv) Tibial CV (m/s) Median SNAP (µv) Ulnar SNAP (µv) Sural SNAP (µv) 4/M c.312_320del10insat/ c.532_535delaata 10 m areflexia 12 m/r NR 5/M Hz c.516-2a>g 10 m ataxia, tremor 14 m/r /M Hz c t>g 9 m ataxia, hypotonia 18 m/r NR 7/M Hz c.312_320del10insat 10 m ataxia, hypotonia, tremor 18 m/r /M Hz c.516-2a>g 4 m ataxia, hypotonia 21 m/r /F Hz c.751c>t 9 m ataxia, hypotonia, tremor 2 yrs/r NR yrs/r NR - NR 10/M Hz c.312_320del10insat 18 m ataxia, tremor, areflexia 2 yrs/r NR 11/F c.240+1g >T, c.575g>a 18 m ataxia, hypotonia, tremor 2 yrs/r (radial) - 21 (medial plantar) 12/M Hz c.516-2a>g 3 d 4 yrs/r (radial) - NR 13/F Hz c.792_793delag 5 yrs ataxia, tremor, areflexia 5 yrs/r Abnormal results (< 2SD) in bold. Reference values from Cai et al. 100 R retrospective, P prospective, CMAP compound muscle action potential, CV conduction velocity, SNAP sensory nerve action potential, Hz homozygous, m months, NR not recordable 46
48 Mitochondrial Neuropathy Echaniz-Laguna and colleagues have reported three individuals from two families presenting with a childhood-onset demyelinating sensorimotor neuropathy, initially diagnosed as CMT, in whom recessive mutations in SURF1 were identified. 99 The individuals also had nystagmus, hearing loss, lactic acidosis and MRI brain lesions suggestive of Leigh syndrome, and developed cerebellar ataxia with disease progression. POLG1 DNA polymerase-γ (POLG1) mutations in children have a wide spectrum of onset and phenotype, ranging from infantile hepato-encephalopathy [including the Alpers-Huttenlocher syndrome and childhood myocerebrohepatopathy (MCHS)], to syndromes with an onset in young adulthood including MEMSA (myoclonic epilepsy, myopathy, sensory ataxia), ANS (ataxia neuropathy spectrum) and recessive and dominantly inherited PEO (progressive external ophthalmoplegia). 61 In my study, previously performed nerve conduction studies in six children with compound heterozygous and homozygous mutations in POLG1 showed a severe axonal sensory neuropathy, irrespective of the age at which nerve conduction studies were performed. Additional axonal motor involvement was seen in older children. While hypotonia and areflexia are listed as features of POLG1 mutations in the first year of life, there are very few reports of the nerve conduction findings of affected children in this age-group. My study included a 2-week old term infant in whom the nerve conduction velocities were significantly reduced (8-10m/s). In a cohort of eight children with Alpers syndrome, Ferrari et al. reported two infants with neurophysiological or biopsy evidence of a demyelinating neuropathy. 9 One five-month-old had a demyelinating neuropathy with nerve conduction velocities of 3.4 6m/s, while another had a marked reduction of myelinated fibres. 47
49 Mitochondrial Neuropathy Table 1.3: Neurophysiological profile of children with POLG1 mutations Pt./ Sex Mutation Age at presentation Possible neuropathic features Age at NCS, Retrospective/ Prospective Median CMAP (mv) Median CV (m/s) Ulnar CMAP (mv) Ulnar CV (m/s) Peroneal CMAP (mv) Peroneal CV (m/s) Tibial CMAP (mv) Tibial CV (m/s) Median SNAP (µv) Ulnar SNAP (µv) Sural SNAP (µv) 21/M c.1399g>a/c.695g>a 2 weeks - 2 weeks/r /F Hz c.911t>g 4 yrs ataxia, areflexia 10 yrs/r NR NR NR 23/F Hz c.1399g>a 7 yrs ataxia, tremor, areflexia 13 yrs/r NR NR (sup peroneal) 24/M c.1943c>g/c.926g>a 12 yrs - 16 yrs/r NR - NR /M c.2551a>g/c.3140g>a 17 yrs - 17 yrs/r NR NR NR 26/F Hz c.1399g>a 15 yrs - 19 yrs/r NR NR NR NR (sup peroneal) Abnormal results (< 2SD) in bold. Reference values from Cai et al. 100 R retrospective, P prospective, CMAP compound muscle action potential, CV conduction velocity, SNAP sensory nerve action potential, Hz homozygous, m months, NR not recordable, sup - superficial 48
50 Mitochondrial Neuropathy While a sensory ataxic neuropathy almost universally develops in individuals with lateonset POLG syndromes, its onset is usually in the late second decade or beyond, though an earlier onset has rarely been described. 61,63 Isohanni and colleagues sequenced POLG1 in 136 children with varying central and peripheral nervous system features and found mutations in seven children, all of whom had an encephalopathy. 7 A peripheral neuropathy was not listed as a feature in any of these seven children, though nerve conduction study results were not specifically reported as part of this study. The neuropathy associated with the late-onset POLG1 syndromes is often a severe axonal sensory neuropathy, with motor studies varying from normal to showing a motor axonal neuropathy predominantly affecting the lower limbs, 104,106 similar to the findings from the children with POLG1 mutations in this study. As suggested by the single neonate in my cohort and the previously published by Ferrari et al., very young children with POLG1 mutations may have a demyelinating neuropathy. 49
51 Mitochondrial Neuropathy Mitochondrial genome mutations Neurophysiologic findings in seven children with mutations in the mitochondrial genome were evaluated. Three had Leigh/NARP syndrome due to 8993T>C mutation in MT-ATP6, two had MELAS due to the 3243A>G mutation in MT-TL1, and two individuals had single large mitochondrial deletions. When compared to the prominent nerve conduction study abnormalities in children with nuclear mutations affecting mitochondrial function, this group of children had normal studies or mild abnormalities, usually in the form of an axonal sensorimotor neuropathy. The 3243A>G mutation in MT-TL1 is the most common cause of MELAS syndrome. Between 22 and 77% of individuals with MELAS have a peripheral neuropathy 56,107 and the incidence of a peripheral neuropathy is likely to be lower in children. Out of the nine children in the cohort of adults and children with MELAS described by Kaufmann et al., five had normal nerve conduction studies while another three had only borderline reductions in peroneal CMAP amplitudes. 107 This study included two siblings with MELAS, and nerve conduction tests in one sibling were done when he was admitted with an acute stroke-like episode with acute sensorineural hearing loss, ataxia, bilateral intention tremor and reduced reflexes in the lower limbs. Nerve conduction studies during that acute episode showed an axonal predominantly motor neuropathy. The studies were repeated a year later, when he was well, and showed almost complete resolution of neuropathy with only borderline reduction of lower limb motor amplitudes. 50
52 Mitochondrial Neuropathy Table 1.4: Neurophysiological profile of children with mitochondrial genome mutations Pt./ Sex Syndrome Mutation Age at presentation Possible neuropathic features Age at NCS, Retrospective/ Prospective Median CMAP (mv) Median CV (m/s) Ulnar CMAP (mv) Ulnar CV (m/s) Peroneal CMAP (mv) Peroneal CV (m/s) Tibial CMAP (mv) Tibial CV (m/s) Median SNAP (µv) Ulnar SNAP (µv) Sural SNAP (µv) 14/M Leigh MTATP6 8993T>C 2 yrs - 5 yrs/r /F Leigh MTATP6 8993T>C 12 yrs - 12 yrs/p NR 16/F NARP MTATP6 8993T>C 16 yrs - 16 yrs/r /M MELAS MTTL1 3243A>G 9 yrs - 11 yrs/p /M MELAS MTTL1 3243A>G 10 yrs mild ataxia during acute episode 12 yrs/p NR NR NR NR NR NR NR 13 yrs/p NR 19/M Pearson single mtdna deletion 7 m - 4 yrs/p /M Kearns- Sayre single mtdna deletion 11 yrs - 15 yrs/p Abnormal results (< 2SD) in bold. Reference values from Cai et al. 100 R retrospective, P prospective, CMAP compound muscle action potential, CV conduction velocity, SNAP sensory nerve action potential, m months, NR not recordable 51
53 Mitochondrial Neuropathy Of the three children in this study with MT-ATP6 mutations, one child had an absent sural SNAP and otherwise normal study, while another had an axonal motor neuropathy affecting the lower limbs. 30% of cases of Leigh syndrome are associated with a mutation in the mitochondrial genome (mitochondrial (mt)-dna associated Leigh syndrome or maternally inherited Leigh syndrome [MILS]). A third of those with mt-dna associated Leigh syndrome have an 8993T>C or 8993T>G mutation in MT-ATP6. 108,109 Mt-DNA associated Leigh syndrome usually has an onset in the first year of life. Of the 67 individuals with Leigh and Leigh-like syndrome described by Rahman et al, only four had a peripheral neuropathy (one each with PDHC deficiency, Complex-I deficiency, Complex-IV deficiency and no defect identified). 109 None of 12 individuals with a mutation involving the mitochondrial genome had a peripheral neuropathy. In contrast to Leigh syndrome, NARP (neurogenic muscle weakness, ataxia, and retinitis pigmentosa) usually presents in young adulthood, though childhood onset is known. Presentation in childhood is usually with ataxia and learning difficulties. The neurogenic muscle weakness is often seen only in adulthood and with disease progression, 31 though those with the 9185T>C mutation may have a neuropathy at presentation or in the first two decades of life. 37 MT-ATP6 is the only gene associated with NARP, with more than half of those with NARP having the 8993T>C or 8993T>G mutations. Recently, the 9185T>C mutation in MT-ATP6, usually associated with NARP and Leigh syndrome, has been reported to cause CMT, with 1.1% of a cohort of undiagnosed CMT shown to have the mutation. 98 Affected individuals were all homoplasmic for the mutation; 52
54 Mitochondrial Neuropathy most had a pure motor or motor-predominant neuropathy with onset often in the first or second decade of life and variable sensory involvement with disease progression. In addition to a peripheral neuropathy, learning difficulties, sensorineural hearing loss, retinal degeneration and Leigh syndrome-like acute deteriorations with intercurrent illness were seen in some affected individuals. None of the 442 probands with CMT screened in this published report by Pitceathly et al. had the 8993T>C mutation. Similarly, in another study of 96 individuals with CMT, none was found to have the 8993T>C mutation. 34 Overall, it appears that presentations predominantly with a peripheral neuropathy or the presence of a peripheral neuropathy in the first two decades of life in individuals with the NARP phenotype are specific to the 9185T>C mutation, and the occurrence of a neuropathy in childhood is rare with the 8993T>C mutation. Single large mitochondrial deletions usually cause three overlapping syndromes, progressive external ophthalmoplegia (PEO), Kearns-Sayre syndrome (onset before age 20, pigmentary retinopathy and PEO, often with cardiac arrhythmias and cerebellar involvement) and Pearson syndrome (sideroblastic anemia and exocrine pancreatic insufficiency). A peripheral neuropathy in childhood has been described with the Kearns-Sayre syndrome in only a single case report. 53 This study includes prospective nerve conduction studies in two individuals with single large mitochondrial deletions, neither of whom had neurophysiological evidence of a peripheral neuropathy. 53
55 DISCUSSION The characteristics of the mitochondrial disease- associated peripheral neuropathies differ depending on the underlying genetic defect Previous investigations and prospectively performed nerve conduction studies in a total of 27 children with mitochondrial disease and an identified molecular genetic defect were reviewed as part of this study. As the retrospective group included only those known to have a peripheral neuropathy, this study was not designed to characterise the frequency of peripheral neuropathy in different mitochondrial diseases. Studies data collected in the retrospective group were performed at different centres and hence there was variation in the protocol used. It is, however, the largest study to date of peripheral neuropathy in childhood mitochondrial disease and provides valuable data on the characteristics of the peripheral neuropathy associated with different mitochondrial diseases. SURF1 mutations are associated with a predominantly demyelinating sensorimotor neuropathy. MNGIE (mitochondrial neurogastrointestinal encephalomyopathy) due to mutations in the TYMP gene is also reported to be associated with a childhood-onset demyelinating neuropathy. 71,110 PDHc deficiency is characterised by axonal sensorimotor neuropathy. POLG1 mutations are associated with a axonal sensory neuropathy with variable motor involvement. Recessive mutations in PEO1, the gene encoding mitochondrial Twinkle helicase, present with a similar phenotypic spectrum to POLG1 and are also associated with an early-onset sensory neuropathy. 14 In this study, children with a mitochondrial genome mutation had either normal studies or a mild neuropathy when not acutely ill. Similar to the findings in our cohort, Horga et al. found that peripheral neuropathy had the highest specificity (91%), negative predictive value (83%) and positive likelihood ratio (5.87) for 54
56 Mitochondrial Neuropathy the diagnosis of a nuclear DNA defect as opposed to a mitochondrial gene defect, in individuals with mitochondrial ophthalmoplegia. 111 The neuropathy associated with CMT is usually length-dependent, with weakness starting and being more pronounced distally, and the lower limbs being earlier and more severely affected than the upper limbs In contrast, the nerve conduction abnormalities in this cohort of children with mitochondrial disease did not show a length-dependent pattern. Acute neuropathy in mitochondrial disease An acute neuropathy, either as a presenting feature or during the disease course, has only rarely been described with mitochondrial disease. 45,115,116 Acute weakness associated with mitochondrial disease appears to be predominantly myopathic in origin. Aure et al. described acute weakness in a cohort of individuals with MT-ATP6/8 mutations, who also had evidence of a late-onset distal motor neuropathy. 117 The acute weakness was triggered by prolonged sitting or rest after exercise, lasted less than 24 hours and resolved with acetazolamide, resembling the periodic paralysis seen with a channelopathy. In this study, one of the children with MELAS due to 3243 A>G mutation in MT-TL1 developed an acute axonal neuropathy during an acute stroke-like episode. An acute axonal neuropathy has previously been reported in a 30 year-old who presented with acute weakness, leading to the diagnosis of MELAS. 116 As seen in the child in this study, it is possible that an acute neuropathy accompanies stroke-like episodes with MELAS but is under-recognised due to the prominent central nervous system features. 55
57 Mitochondrial Neuropathy Debray et al. provided a summary of individuals with PDHc deficiency and acute weakness, describing 13 individuals (11 of whom had previously been reported). Of the seven who had undergone nerve conduction studies, five had neurophysiological evidence of a peripheral neuropathy. 118 However, the lack of studies in these individuals before the onset of weakness or after recovery makes it difficult to determine if the neuropathy was chronic and unrelated to the episode of muscle weakness or if an acute metabolic neuropathy or worsening of a pre-existing neuropathy was responsible for the acute weakness. Two children in this study (patients1 and 2) with PDHc deficiency had evidence of an axonal sensorimotor neuropathy on studies performed when they were not acutely ill or weak. Patient 4 had very low CMAP amplitudes while recovering from an episode of acute weakness, raising the possibility that a worsening of a preexisting neuropathy may have contributed to the acute symptomatology. Mechanism of peripheral nerve dysfunction in mitochondrial disease Normal mitochondrial function is essential for neuronal growth, survival and function. 119 The genes that affect mitochondrial function may cause a peripheral neuropathy by alteration in the mitochondrial dynamics of fusion, fission and axonal transport, or due to abnormalities in energy production. Our understanding of the pathophysiology of peripheral neuropathy caused by genes associated with CMT and known to be associated with maintaining mitochondrial function (MFN2, OPA1, GDAP1), provides a template for explaining this dysfunction. Dominant and recessive mutations in mitofusin 2 (MFN2), encoding the outer mitochondrial membrane GTPase mfn2, cause CMT2A, the most common form of inherited axonal neuropathy. 90 Some affected individuals with MFN2 mutations also have acute or chronic optic atrophy. Dominant mutations in OPA1 cause inherited optic atrophy, and a third of affected individuals have an axonal 56
58 Mitochondrial Neuropathy peripheral neuropathy. 82 The coordinated function of the mitofusins (mfn1 and mfn2) and OPA1(located on the inner mitochondrial membrane) is required for mitochondrial fusion. 87,88 Small, rounded and abnormally aggregated axonal mitochondria are seen in sural nerve biopsies of individuals with MFN2 mutations. 91 Mutations in the ganglioside-induced differentiation associated protein 1 gene (GDAP1) cause recessive (CMT4A) and dominant (CMT2H/K) forms of axonal and demyelinating neuropathy. GDAP1 is an outer mitochondrial membrane protein expressed in both myelinating Schwann cells and axons and known to play a role in mitochondrial fission. Fibroblasts from individuals with GDAP1 mutations show abnormally large mitochondria, a fragmented mitochondrial network and reduced complex I activity. 94 In addition to abnormalities with mitochondrial fission, defective mitochondrial axonal transport has been demonstrated in cultured neurons from MFN2 knock-out mice or neurons with MFN2 disease mutants. 120 Mitochondria supply the vast axonal energy requirement for maintaining the axonal gradient required for impulse transmission and are known to be positioned at areas of high energy demand along the axon. 121 Mitochondria move both anterogradely and retrogradely, predominantly along the axonal microtubules using kinesin and cytoplasmic dynein motors and for short distances along actin filaments using myosin motors. 121 Mfn2 interacts with the molecular complex (Milton/Miro) that links mitochondria to the kinesin motors. Disruption of this interaction impairs transport of mitochondria along the axon and this may be the cause of the length-dependent nature of neuropathy due to MFN2 mutations. Mitochondrial ATP production supports synapse assembly, action potential generation and synaptic transmission in peripheral nerves. 122 Another hypothesis for the nerve dysfunction 57
59 Mitochondrial Neuropathy associated with mitochondrial diseases is energy failure due to the failure of the Na + /K + -ATPase pump. Because of their long axons, peripheral nerves are especially susceptible to energy failure. Nerve excitability studies during an acute stroke-like episode in MELAS are consistent with membrane depolarisation, possibly due to dysfunction of the Na + /K + -ATPase pump 123. However, nerve excitability testing in the non-acute setting, in a cohort of adults with either MELAS 3243A>G mutation or muscle biopsy evidence of a mitochondrial myopathy, did not show an abnormality, indicating that this is unlikely to be the cause of the chronic neuropathy in mitochondrial disease. 124 In a mouse model where mitochondrial metabolism was disrupted solely in Schwann cells, there was activation of an abnormal integrated stress response through actions of heme-regulated inhibitor kinase (HRI). This resulted in a shift of lipid metabolism away from fatty acid synthesis towards oxidation, causing depletion of myelin lipid components and accumulation of acylcarnitines which can cause axonal degeneration. 125 CONCLUSION Peripheral neuropathy is more common in childhood mitochondrial disease due to a nuclear gene mutation when compared to mitochondrial genome mutations. The identification of a peripheral neuropathy and definition of its characteristics may help classify the mitochondrial syndrome and direct genetic testing. SURF1 mutations are associated with a demyelinating neuropathy while PDHc deficiency is associated with an axonal neuropathy. POLG mutations are associated with an axonal sensory neuropathy. In contrast to that seen with CMT, the neuropathy of mitochondrial disease is non-length dependent. Nerve conduction studies should be an integral component of the diagnostic evaluation of suspected childhood mitochondrial disease. 58
60 SECTION II Brown-Vialetto-Van Laere syndrome due to RFVT2 deficiency 59
61 Chapter 2 Phenotype of Brown-Vialetto-Van Laere syndrome due to RFVT2 deficiency 60
62 RFVT2 Phenotype INTRODUCTION This chapter describes a cohort of previously genetically unclassified families with a common phenotype suggestive of a mitochondrial disorder. Subsequent genetic testing revealed mutations in a gene (SLC52A2) encoding the neuronal transporter (Riboflavin transporter type 2) for a cofactor essential for mitochondrial metabolism and energy transport. I have characterised the phenotype of this disorder, the features that identify it as a mitochondrial disorder and the features that differentiate it from another riboflavin transporter disorder (RFVT3 deficiency due to mutations in SLC52A3). As hearing loss is an initial and characteristic feature of BVVL and has unique characteristics that aid early diagnosis, the audiological profile of my cohort is detailed separately in Chapter 3. Brown-Vialetto-Van Laere syndrome (BVVL) is a progressive inherited neurodegenerative disorder characterised by bilateral sensorineural deafness and pontobulbar palsy. The syndrome is named after Brown, 126 Vialetto 127 and Van Laere, 128 who described individuals affected with the disorder. Onset is usually in childhood but may vary from infancy to the third decade of life. 129 The course is often progressive, though episodic deterioration or long periods of stability have been described. 130 Hearing loss is often the initial symptom, is progressive and ultimately severe. Affected individuals go on to develop lower (VII-XII) and less commonly upper (II-VI) cranial nerve abnormalities, neck, shoulder and limb muscle weakness and respiratory failure. 131 Associated features may include epilepsy, autonomic disturbances, cerebellar ataxia, retinitis pigmentosa and upper motor neuron signs. Sensory abnormalities have not been described as a feature of this disorder. 131 Cognition is usually preserved. Electromyography shows chronic or active denervation in affected muscles. The onset of symptoms, or a clinical deterioration, may 61
63 RFVT2 Phenotype occur in association with intercurrent illness. 132 Spontaneous significant improvement in the clinical state has not been described. Mortality from the disease is usually from respiratory failure, occasionally precipitated by an intercurrent infection. 132 As elucidated in this chapter, the neuropathy associated with RFVT2 deficiency can be classified as a mitochondrial neuropathy. The biochemical defect in BVVL is an abnormality of mitochondrial fatty acid beta-oxidation and branched-chain amino acid catabolism, resulting in a deficiency in energy production. Affected individuals with BVVL have multisystemic involvement including optic atrophy and sensorineural hearing, similar to other mitochondrial diseases. The peripheral neuropathy is non-length dependent, as it is with other mitochondrial neuropathies, and the nerve biopsy shows a number of mitochondrial abnormalities. The Fazio-Londe syndrome has a very similar clinical profile to BVVL, but affected individuals do not have hearing loss. 133 Madras motor neuron disease (childhood onset, sensorineural hearing loss, cranial nerve palsies, limb weakness), Nathalie syndrome (deafness, cataract, muscle weakness, hypogonadism, electrocardiographic abnormalities) and Boltshauser syndrome (hearing loss, vocal cord paralysis, distal muscular atrophy) share phenotypic similarities with BVVL Both familial and sporadic cases of BVVL have been described with the inheritance suggested to be autosomal recessive with reports of affected siblings in families with unaffected parents and occurrence in consanguineous families, though other modes of inheritance have occasionally been reported. 130,137,138 62
64 RFVT2 Phenotype Post-mortem examination of affected individuals shows neurogenic atrophy in the limb, neck and laryngeal muscles, which are clinically weak. The lower (VII-XII) cranial nerves, and sometimes the III, V and VI cranial nerves, show severe axonal loss with depletion of neurons and gliosis in the brainstem nuclei (including the tractus solitarius and nucleus ambiguus). The cochlear nucleus shows loss of neurons, and the lateral lemniscus and inferior colliculus show gliosis. 130,139 The medial lemniscus and the gracile and cuneate nuclei also show degeneration. 139 The cerebral cortex, basal ganglia, thalami, white matter and corticospinal tracts are usually spared. The spinal cord shows loss of neurons in both the anterior and posterior horns with axonal loss and degeneration of the spinothalamic and spinocerebellar tracts, though pyramidal fibres are relatively spared. 139 Recently, autosomal recessive mutations in SLC52A3 (formerly C20orf54) and SLC52A2, encoding the riboflavin transporters RFVT3 (formerly RFT2) and RFVT2 (formerly RFT3) respectively have been identified in individuals with BVVL. 140,141 The clinical and genetic profile, neurophysiology, biochemical abnormalities and histopathology of nine individuals from four families with homozygous and compound heterozygous mutations in SLC52A2 are described in this chapter. METHODS Ethics Patients were enrolled with informed consent from the patient and/or parental guardian. This study had Ethics Committee/IRB approval from the Sydney Children s Hospitals Network (10/CHW/45) and the University Of Miami Miller School Of Medicine. 63
65 RFVT2 Phenotype Exome and Sanger Sequencing Families 3 and 4 were independently selected for exome sequencing as they had an undiagnosed familial neuropathy. There exists a research collaboration between the Peripheral Neuropathy Service at The Children s Hospital at Westmead and the Center for Human Molecular Genomics, John P. Hussman Institute for Human Genomics University of Miami Miller School of Medicine (led by Prof. Stephan Zuchner). Exome sequencing was performed in family 3 (including patients 3.1 and 3.2) and family 4 (including patients 4.1, 4.2 and 4.3) at the Center for Human Molecular Genomics, Miami and Western Sydney Genetics Program, The Children s Hospital at Westmead respectively. Sanger sequencing of SLC52A2 was performed in the affected members of family 2 (including patients 2.1 and 2.2) at the Center for Human Molecular Genomics, Miami as their phenotype was similar to that recently described with SLC52A2 mutations. Sanger sequencing for only the founder Lebanese mutation was performed in family 1 (including patients 1.1 and 1.2) at the Department of Molecular Genetics, The Children s Hospital at Westmead, also based on the phenotypic resemblance to individuals with SLC52A2 mutations. The medical records, results of previous nerve conduction testing, medical imaging, hearing and ophthalmology assessments and histopathology of all individuals identified to have SLC52A2 mutations were reviewed. The acylcarnitine profiles, including the level of medium-chain acylcarnitines, were also reviewed. Assessments were repeated at the time of diagnosis. These assessments included a clinical examination, nerve conduction testing and formal hearing assessment. The acylcarnitine profile and FAD levels were also measured at baseline. Newborn screening cards, where available, were tested for the level of medium-chain acylcarnitines. 64
66 RFVT2 Phenotype Portions of this chapter have been modified from the previously published article (Foley AR, Menezes M et al. Brain 2014) on which I was a co-first author. I was the treating neurologist for Families 1, 2 and 3, and shared care for family 4. I reviewed previous clinical notes and investigations (including nerve conduction studies) on all the study patients. I performed baseline clinical assessments and nerve conduction tests on all the study patients, tabulated the nerve conduction results and acylcarnitine profile results and compared with age-matched and CHW laboratory norms respectively. I reviewed the biopsies of patients 3.1 and 4.2. I assisted with segregating the variants found in the exome sequencing of Family 3. The molecular genetic tests including exome sequencing and sanger sequencing and functional assays were performed by collaborators and I had no role in performing these tests. RESULTS Nine individuals from four families with either compound heterozygous or homozygous mutations in SLC52A2 were identified. The pedigrees and genotype of the affected individuals is illustrated in Figure 2.1. Families 2, 3 and 4 have previously been reported as part of a published cohort of 18 children with SLC52A2 mutations
67 RFVT2 Phenotype Figure 2.1: Pedigrees and corresponding SLC52A2 mutations for affected patients Squares denote males, circles females and completely shaded shapes affected individuals. Half-shaded shapes indicate unaffected carriers. 66
68 RFVT2 Phenotype SLC52A2 mutations The location of each published SLC52A2 mutation, including those in our cohort, is shown in Fig. 2.2A. Both the SLC52A2 mutations identified in my cohort, p.g306r (c.916g>a) and p.l339p (c.1016t>c) have been reported previously 140,143 and have been predicted as not tolerated by the SIFT prediction program and as probably damaging by Polyphen-2. Both mutations alter amino acids evolutionarily conserved from humans to D. rerio and are also conserved in RFVT1 and RFVT3 (Fig. 2.2B). Array based genotyping Patients from families 1, 2 and 4, like the affected members of the family described by Megarbane et al. in 2000, 137 are of Lebanese origin and all carry homozygous p.g306r SLC52A2 mutations. Haplotype analysis was carried out by researchers at the Western Sydney Genetics Program using SNP based arrays or genotyping of SNPs around the SLC52A2 gene in patients from families 2 and 4 and patient L1 in the larger published cohort to determine whether this was a shared ancestral allele. All of them, as well as the two affected members of the family described by Megarbane et al., were found to be homozygous for the p.g306r mutation and all 11 SNPs studied indicating that the mutation arose on a common haplotype within the Lebanese population as a founder mutation. 67
69 RFVT2 Phenotype Figure 2.2: Mutations in SLC52A2 in Brown-Vialetto-Van Laere syndrome A B (A) Predicted transmembrane domains in RFVT2, gene structure and location of mutations identified in SLC52A2, including those in this patient cohort. (B) Structural conservation of relevant amino acid residues in RFVT2 across species and in RFVT1 and RFVT3. Dark blue, medium blue and light blue colors correspond to amino acids conserved in 6, 5 or 3 out of the 7 sequences, respectively. 68
70 RFVT2 Phenotype Clinical Phenotype Nine individuals from four families were found to harbour either compound heterozygous or homozygous mutations in SLC52A2. The clinical and genetic features of these patients are listed in Table 2.1. Three of the four families in my cohort were of Lebanese origin while the other was of English and Scottish descent. Three of the four families had a history of consanguinity. Affected children presented between 2 and 8 years of age with an ataxic gait due to a severe sensory ganglionopathy, sometimes associated with a sensorineural hearing loss. Hearing was normal on newborn screening (except in patient 1.1). The hearing loss had the characteristics of an auditory neuropathy and progressed rapidly after diagnosis. Hearing aids did not provide any benefit. A detailed description of the hearing loss associated with riboflavin transporter deficiency is available in Chapter 3. 69
71 RFVT2 Phenotype Table 2.1: Clinical profile of affected individuals with SLC52A2 mutations Family 1 Family 2 Patient SLC52A2 mutations homozygous: c.916g>a homozygous: c.916g>a homozygous: c.916g>a homozygous: c.916g>a p. G306R p. G306R p. G306R p. G306R Sex F M F F Ethnicity Lebanese Lebanese Lebanese Lebanese Consanguinity yes yes yes yes First symptom ataxic gait ataxic gait ataxic gait ataxic gait Age at first symptom hearing loss (1 month) ataxic gait (4 yrs) 8 yrs 8 yrs 3 yrs Optic atrophy no NT no yes Sensorineural hearing loss Sensorimotor neuropathy Distribution of weakness yes NT yes yes sensory only NT sensory only yes none detected none detected none detected UL Overall maximal motor function independent ambulation independent ambulation independent ambulation independent ambulation Maximal motor function at the time of diagnosis independent ambulation independent ambulation independent ambulation independent ambulation Respiratory function normal normal normal normal Feeding by mouth by mouth by mouth by mouth Age at genetic diagnosis 5 yrs 8 yrs 10 yrs 9 yrs UL upper limb, NT not tested, NA not applicable, NIV non-invasive ventilation 70
72 RFVT2 Phenotype Table 2.1: Clinical profile of affected individuals with SLC52A2 mutations (continued) Family 3 Family 4 Patient compound compound homozygous: homozygous: homozygous: heterozygous: heterozygous: c.916g>a c.916g>a c.916g>a c.916 G>A; 1016T>C c.916 G>A; 1016T>C SLC52A2 mutations p.g306r; L339P p.g306r; L339P p. G306R p. G306R p. G306R Sex F F M M M Ethnicity English and Scottish English and Scottish Lebanese Lebanese Lebanese Consanguinity no no yes yes yes First symptom ataxic gait and UL weakness ataxic gait and hearing loss ataxic gait and hearing loss ataxic gait and hearing loss ataxic gait Age at first symptom 2 yrs 5 yrs 3 yrs 3 yrs 3 yrs Optic atrophy no yes yes yes yes Sensorineural hearing loss Sensorimotor neuropathy Distribution of weakness NT yes yes yes yes yes yes yes yes yes UL UL; neck extension UL; neck extension UL; neck extension UL Overall maximal motor function independent ambulation independent ambulation independent ambulation independent ambulation independent ambulation Maximal motor function at the time of diagnosis Respiratory function NA decreased; on ventilator at the time of death independent ambulation decreased; nocturnal NIV recommended non-ambulant ventilator dependent non-ambulant nocturnal NIV independent ambulation normal Feeding by mouth by mouth by gastrostomy only by gastrostomy only by mouth Age at genetic diagnosis deceased (3.5 yrs) 15 yrs 16 yrs 16yrs 21 yrs UL upper limb, NT not tested, NA not applicable, NIV non-invasive ventilation 71
73 RFVT2 Phenotype Sensory ataxia was noted in all patients at presentation. The gait ataxia, severe generalised large-fibre sensory loss, lack of regeneration on nerve biopsy (see below) and absence of a clear length-dependent abnormality on nerve conduction studies indicate that the sensory abnormalities were most likely due to a sensory ganglionopathy. Upper limb strength was normal at first presentation but weakness was usually noticed in the second decade of life and then progressed rapidly. Patient 3.2 had an earlier onset of upper limb and respiratory muscle weakness at 2 years of age, which progressed rapidly resulting in death from acute on chronic respiratory failure. Older affected children had a combination of proximal and distal upper limb weakness with no movement in the small muscles of the hand, only a flicker of movement on shoulder abduction, and neck extension weakness. Strength in the lower limbs was normal or only mildly reduced with even those late in the disease course being able to walk on their heels and toes when supported, though they were confined to a wheelchair because of the sensory ataxia. Mild optic atrophy was identified in five of the eight children who had a formal ophthalmological evaluation. Respiratory failure developed in four children. Patient 3.2 died within 6 months of presentation due to rapidly progressive respiratory failure. In the others, respiratory muscle weakness was identified on polysomnography between years of age and was associated with longer disease duration and paralleled the proximal upper limb weakness. Cognition appeared preserved but was difficult to test in the most severely affected children (patients 4.1 and 4.2) because of their speech, writing and visual deficits. Two children had evidence of severe bulbar weakness necessitating gastrostomy insertion at the age of 16 years. 72
74 RFVT2 Phenotype Biochemical Studies Plasma acylcarnitine profiles were performed in eight children after diagnosis and prior to highdose oral riboflavin therapy and were abnormal in five (Table 2.2). Acylcarnitine profiles previously performed in patients 3.1 and 4.2 were also reviewed and showed the mild increase in medium-chain acylcarnitines characteristic of this disorder. However, as these two profiles were performed six and nine years prior to recognition that these mild abnormalities were suggestive of riboflavin transporter deficiency, they had been reported normal at that time. Urine metabolic screens done just prior to high-dose riboflavin therapy, as well as previously, were all normal. FAD levels done just prior to high-dose riboflavin therapy were within the normal range for our laboratory. Medium-chain acylcarnitines were normal on newborn screening in all five individuals for whom this testing had been undertaken. 73
75 RFVT2 Phenotype Table 2.2: Biochemical profile of affected individuals with SLC52A2 mutations Family 1 Family 2 Family 3 Family 4 Acylcarnitine species Reference range Free Carnitine C C C5 < C6 < C8 < C10:1 < C10 < C12 < C14:1 < C14 < C16 < Riboflavin level ND ND UMS ND ND Normal Normal Normal ND Normal Normal Normal Newborn screening Normal Normal Normal Normal Normal NA NA NA NA Abnormal values indicated in bold. ND not done, NA not available 74
76 RFVT2 Phenotype Neurophysiology Nerve conduction studies performed early in the disease course showed absent or very low sensory amplitudes in the upper and lower limbs and normal motor studies (Table 2.3). This pattern of generalised sensory abnormalities on nerve conduction studies, severe sensory ataxia and absent reflexes is consistent with a sensory ganglionopathy. Sequential studies showed a steady decline of upper limb motor amplitudes consistent with a motor axonal neuropathy. Lower limb CMAP amplitudes were normal or mildy reduced and did not show a clear progression with disease duration. This overall pattern is in contrast to inherited sensorimotor polyneuropathies which are typically length-dependent, with sensory symptoms and weakness in the lower limbs preceding and progressing to a greater degree than in the upper limbs
77 RFVT2 Phenotype Table 2.3: Nerve conduction studies in affected individuals with SLC52A2 mutations Patient Age at NCS 4 yrs 8 yrs 10 yrs 3 yrs 5yrs 9 yrs 8 yrs 16 yrs 15 yrs 16 yrs 16 yrs Motor Median Nerve Ulnar Nerve Peroneal Nerve Tibial Nerve Sensory CMAP (mv) NR NR NR 1.3 CV (m/s) CMAP (mv) NR CV (m/s) CMAP (mv) NR NR CV (m/s) CMAP (mv) CV (m/s) Sural (µv) NR NR NR NR NR NR NR NR NR NR NR Median (µv) NR NR NR 2 NR NR 3 5 NR NR NR Ulnar (µv) NR NR (radial) NR NR NR NR 3 NR NR NR Abnormal values (< 2SD) indicated in bold. Reference values from Cai et al. 100 CMAP compound muscle action potential, CV conduction velocity, NR not recordable 76
78 RFVT2 Phenotype Histopathology The histopathological characteristics, including electron microscopy, of sural nerve biopsies from 6 patients with BVVL have been reported in the published article, and showed a severe chronic axonal neuropathy with accompanying increase in connective tissue but no evidence of ongoing degeneration (Fig. 2.3). Large myelinated fibres were more severely affected and regeneration was strikingly absent. Unmyelinated fibres were generally better preserved. There was no inflammation, pathological hypomyelination or demyelination. This included slides of sural nerve biopsies from patients 3.1 and 4.2 which I have reviewed. An adequately preserved sample was only available from the biopsy from patient 3.1, and electron microscopy was performed by Prof J-M Vallat at the Reference Centre for Peripheral Neuropathies, CHU Limoges, France and reviewed by me at the University of Sydney Nerve Laboratory (Fig. 2.4). This revealed a focal proliferation of microfilaments in myelinated axons, abnormal glycogen accumulation in axons, Schwann cells and fibroblasts, and abnormalities in the number and shape of mitochondria. These unique features have not been reported previously. Neuroimaging Brain magnetic resonance imaging (MRI) was performed in four patients and revealed no abnormality in the cerebral cortex, brainstem, internal auditory structures or cochlear nerve. This finding is in contrast to reports of hyperintensity of brainstem nuclei 145,146, atrophy of the brainstem 139,146,147 and atrophy of the cerebellum 139,147 in genetically undifferentiated cohorts of patients with BVVL and those with SLC52A3 mutations. This is consistent with the pure lower motor neuron phenotype of SLC52A2 mutations which contrasts with the upper motor neuron signs seen with SLC52A3 mutations and in genetically undifferentiated cohorts. 77
.")
79 RFVT2 Phenotype Figure 2.3: Sural nerve pathology with mutations in SLC52A2 Resin-semithin section, stained with toluidine blue; x 40 magnification (A) in patient 3.1 at 3 years of age demonstrates a moderate loss of myelinated axons, preferentially of large diameters (8-12µm). There is an increase of collagen in the endoneurium, and there are no inflammatory infiltrates. (B) Unaffected control. Scale bar represents 25nm. 78





, and abnormal accumulation of")
80 RFVT2 Phenotype Figure 2.4: Sural nerve pathology with mutations in SLC52A2: electron microscopy A B C D E F Electron microscopy examination of transverse (A, C, E and F) and longitudinal sections (B, D) of the sural nerve biopsy from patient 3.2 at 3 years of age demonstrates focal proliferation of microfilaments in myelinated axons (A, B), mitochondrial abnormalities with an increased number of intra-axonal mitochondria (C), abnormally shaped mitochondria (black arrowheads, D), and abnormal accumulation of black glycogen granules in axons (E), Schwann cell cytoplasm and fibroblasts (F). 79
81 RFVT2 Phenotype Functional Analyses of SLC52A2 mutations Seven SLC52A2 mutations [p.w31s (c.92g>c), p.q234x (c.700c>t), p.a284d (c.851c>a), p.y305c (c.914a>g), p.g306r (c.916g>a), p.l312p (c.935t>c) and p.l339p (c.1016t>c)] were analysed in an in vitro transient expression system at the Department of Clinical Pharmacology and Therapeutics, Kyoto University Hospital (Kumiko Sugano, Kazuo Matsubara and Atsushi Yonezawa). This department has previously performed functional studies on other riboflavin transporter mutations and has a research collaboration with the Western Sydney Genetics Program, The Children s Hospital at Westmead. 148 [ 3 H]riboflavin transport activity and protein expression of SLC52A2 92G>C, SLC52A2 700C>T, SLC52A2 851C>A, SLC52A2 914A>G, SLC52A2 916G>A, SLC52A2 935T>C and SLC52A2 1016T>C were assessed (Fig. 2.5A). [ 3 H]Riboflavin uptake by the SLC52A2 mutations p.w31s, p.q234x, p.a284d, p.y305c and p.l339p was completely abolished, and SLC52A2 mutations p.g306r and p.l312p showed a moderate but significant decrease in [ 3 H]riboflavin transport activity compared with wild-type SLC52A2. To determine whether the transport activity reduction was due to the decreased expression of transporter proteins in the plasma membranes, Western blot analysis was carried out using the crude membrane of HEK293 cells transiently transfected with these variants (Fig. 2.5B). The expression levels of SLC52A2 mutants except for p.w31s were decreased compared with wild-type SLC52A2, which are well correlated with the reduction ratios of the transport activity for these variants. The p.w31s mutant was expressed in the plasma membrane but was dysfunctional as evidenced by completely abolished riboflavin uptake in vitro. 80
 Uptake of [ 3 H]riboflavin by HEK293 cells transfected with empty vector (Vector), wild-type SLC52A2 (WT), SLC52A2 92G>C (W31S), SLC52A2 700C>T (Q234X),")
82 RFVT2 Phenotype Figure 2.5: Functional studies of SLC52A2 mutations (A) Uptake of [ 3 H]riboflavin by HEK293 cells transfected with empty vector (Vector), wild-type SLC52A2 (WT), SLC52A2 92G>C (W31S), SLC52A2 700C>T (Q234X), SLC52A2 851C>A (A284D), SLC52A2 914A>G (Y305C), SLC52A2 916G>A (G306R), SLC52A2 935T>C (L312P) and SLC52A2 1016T>C (L339P). The cells were incubated with 5 nm [ 3 H]riboflavin (ph 7.4) for 1 min at 37 C. Each bar represents the mean ± SEM, n=3. Data were analyzed by Dunnett s twotailed test after one-way analysis of variance (ANOVA). *P<0.05, ***P<0.001, significantly different from vector-transfected cells. #P<0.05, ###P<0.001, significantly different from SLC52A2 WT -transfected cells. 81
 (B) Western blot analysis performed using the crude membrane of HEK293 cells expressing empty vector, SLC52A2 WT and SLC52A2 variants.")
83 RFVT2 Phenotype Figure 2.5: Functional studies of SLC52A2 mutations (continued) (B) Western blot analysis performed using the crude membrane of HEK293 cells expressing empty vector, SLC52A2 WT and SLC52A2 variants. The crude membrane fractions were subjected to Western blotting using antibodies against FLAG and Na + /K + -ATPase. Na + /K + - ATPase was used as an internal standard. fmol = femtomole 82
84 RFVT2 Phenotype DISCUSSION The unique clinical, neurophysiological, histopathological, biochemical and molecular genetic profile of nine cases of Brown-Vialetto-Van Laere syndrome due to mutations in the riboflavin transporter gene SLC52A2 has been characterised in this chapter. While the age of onset and severity of disease progression may vary among those affected, the phenotype is unique and striking. Initial presentation is with sensory ataxia and hearing loss. Progressive upper limb weakness, optic atrophy, respiratory failure and bulbar weakness emerge with disease progression. The phenotype is caused by an early-onset generalised sensory ganglionopathy, a progressive upper limb motor axonal neuropathy and cranial neuropathy affecting II, VIII and bulbar cranial nerves. This unique phenotype may be described as a child-in-the-barrel, reminiscent of the man-in-the-barrel phenotype, with severe weakness in the upper limbs and preserved strength in the lower limbs. A similar phenotype has been previously reported with the flail-arm variant of motor neuron disease and in the pharyngo-cervico-brachial variant of Guillain-Barre syndrome in children. 149,150 The phenotype of SLC52A2 mutations has characteristic features distinguishing this condition from that previously described with genetically unclassified BVVL or with SLC52A3 mutations, another cause of riboflavin transporter deficiency (Table 2.4). My cohort of affected individuals with SLC52A2 mutations presented with sensory ataxia and had a generalised sensory ganglionopathy. Sensory loss and abnormalities in sensory nerve conduction studies are not a feature of SLC52A3 mutations and have only rarely been described with genetically unclassified BVVL. 131,141,151 83
85 RFVT2 Phenotype Table 2.4: Phenotypical and biochemical differences between RFVT2 and RFVT3 deficiency Feature Sensory ataxia Motor weakness Upper motor neuron features Urine organic acid profile RFVT2 deficiency (SLC52A2 mutations) Presenting feature and universally present. Due to a generalised sensory ganglionopathy Predominantly involves the upper limb with relative sparing of the lower limbs, leading to the child-in-the-barrel phenotype Not reported Normal RFVT3 deficiency (SLC52A3 mutations) Not reported Generalised weakness Brisk reflexes, ankle clonus and extensor plantar responses have been reported Abnormal. Resembles mild multiple acyl-coa dehydrogenation defect (MADD) Plasma flavin levels Tissue expression of encoded riboflavin transporter Normal Mainly in brain and spinal cord. Responsible for the neuronal transport of riboflavin Reduced when compared to controls Mainly in small intestine. Responsible for intestinal absorption of riboflavin 84
86 RFVT2 Phenotype The muscle weakness associated with SLC52A2 mutations predominantly affects the upper limbs and neck while that described with SLC52A3 mutations and genetically unclassified BVVL is generalised. 141,152 Upper motor neurone signs such as brisk reflexes, clonus and extensor plantar responses and abnormalities on brain imaging have been described with both genetically unclassified BVVL and affected individuals with SLC52A3 mutations. 131,141 In contrast, SLC52A2 mutations appear to cause a pure lower motor neuron syndrome. One of the possible explanations for the different phenotypes of SLC52A2 and SLC52A3 mutations could be the differential tissue expression. It is known that RFVT3 and RFVT1, the riboflavin transporters encoded by SLC52A3 and SLC52A1, are predominantly expressed in the small intestine and responsible for intestinal transport of riboflavin. Preliminary human tissue studies of SLC52A2 encoded RFVT2 demonstrate a relatively higher expression in the brain and spinal cord than in the small intestine. 153,154 It is possible that there are further differences in the neuronal expression of RFVT2 in the anterior horn cells along the spinal cord. The initial description by C.H. Brown in 1894 captures the salient features of this disorder. 126 He described a fifteen-year-old boy in whom symptoms were first seen at 3 years of age and in whom the onset was rather rapid and grew more pronounced every day so that in a week he could not move his tongue, whistle or swallow without an effort. He also noted that He grew hard of hearing and on least exertion he had difficulty in breathing. This description of an early childhood onset, rapidly progressive neurodegeneration characterised by hearing loss, pontobulbar weakness and respiratory failure is characteristic of the disorder subsequently known as Brown-Vialetto-Van Laere syndrome. However, it can be argued that the case described was likely to be specifically of RFVT2 deficiency, as Brown reports that the boy found it difficult to button or unbutton his clothes, write or carry weights and was 85
87 RFVT2 Phenotype an exceedingly emaciated boy, more markedly so, however, in the upper parts of the body and had apparent paresis and atrophy of most of the arm muscles. This description evokes the proprioceptive loss (contributing to difficulty in fine motor tasks), upper limbpredominant weakness and child-in-the-barrel phenotype that has described in association with RFVT2 deficiency. Interestingly, he notes previous description of similar cases by Fazio (1892) and Londe (1894), among others. 155,156 He also recognised this was a neuronopathy, concluding his report by remarking on similarities with amyotrophic lateral sclerosis in adults. BVVL associated with SLC52A2 mutations is inherited as an autosomal recessive condition, which helps to differentiate it from other optico-acoustic neuropathies including autosomal dominantly inherited OPA1 or MFN2 mutations, X-linked PRPS1 mutations and mitochondrially inherited neuropathy due to mitochondrial DNA mutations (such as Leber hereditary optic neuropathy or the syndrome of neuropathy, ataxia and retinitis pigmentosa known as NARP ). While it is an inherited neuropathy, the prominent cranial nerve involvement, upper-limb predominance of motor weakness, sensory ganglionopathy and nonlength dependent nature of peripheral nerve involvement differentiate it from most types of Charcot-Marie-Tooth disease. 157 In the past, hearing loss has been used to distinguish BVVL as a diagnostic entity from other forms of childhood onset motor neuron diseases including Fazio-Londe disease. 158 It now appears that BVVL and Fazio-Londe disease are allelic conditions. 133,152 Sequencing of all three riboflavin transporter genes in a cohort of patients with Madras motor neuron disease did not reveal a causative mutation, though the close similarity in the clinical phenotype suggests that the disease may share a common pathophysiological pathway with BVVL
88 RFVT2 Phenotype While the phenotype associated with SLC52A2 mutations is relatively homogeneous, the rate of progression may vary, even within families. One affected individual in my cohort (patient 3.1) had a rapid deterioration, dying from respiratory failure within nine months of onset of symptoms. A trial of intravenous immunoglobulin therapy was ineffective. Her elder sibling (patient 3.2), who shares the same SLC52A2 mutations, remains able to ambulate independently at 15 years of age and respiratory muscle weakness requiring non-invasive ventilation was first identified only at 15 years of age. Patient 4.3 shares the same SLC52A2 genotype and similar age of onset as his more severely affected siblings (patients 4.1 and 4.2) but remains ambulant at 21 years of age with only mild weakness in the small muscles of the hand and no evidence of respiratory failure. His younger siblings have severe proximal and distal upper limb weakness, are wheelchair-bound and require assisted ventilation. This suggests that there may be other genetic or environmental factors that modulate disease severity. Nomenclature of riboflavin transporter disorders Because of the different names being used for the genes, transporters and diseases caused by mutations in the genes encoding riboflavin transporters, the group involved in reporting the large international cohort has made suggestions for a new nomenclature. 142 Using the new protein nomenclature and aiming to achieve improved clarity, the group recommended that the term riboflavin transporter deficiency, type 2 be used to correspond to the SLC52A2 encoded RFVT2 (formerly RFT3) and riboflavin transporter deficiency, type 3 to correspond to the SLC52A3 encoded RFVT3 (formerly RFT2). 87
89 RFVT2 Phenotype Diagnostic pathway for riboflavin transporter deficiency Riboflavin is a precursor of flavin mononucleotide (FMN) and flavin adenine dinucleotide (FAD), and FAD acts as an electron acceptor for a number of acyl-coa dehydrogenation reactions involved in mitochondrial fatty acid beta-oxidation and branched-chain amino acid catabolism. 160 Both FMN and FAD are required for normal mitochondrial respiratory chain function. Acylcarnitine profiles need to be carefully reviewed for the mild elevations of medium-chain acylcarnitines that are diagnostic of this disorder. While elevation of mediumchain acylcarnitines on an acylcarnitine profile is a useful clue to the diagnosis, abnormalities were noted on this test in only 60% of patients in this study and in the larger cohort, showing that this test is not an adequately sensitive screening test. 142 Unlike patients with SLC52A3 mutations, patients with SLC52A2 mutations have normal urine organic acids on a urine metabolic screen and normal plasma FAD levels. 152 As the condition is potentially treatable, it is important that treating clinicians maintain a continued suspicion and consider genetic testing despite a normal acylcarnitine profile if the clinical profile is suggestive of SLC52A2 mutations. The neuropathy associated with RFVT2 deficiency can be classified as a mitochondrial neuropathy. While the pathophysiological basis of the motor nerve dysfunction in this disorder remains to be explored, the biochemical defect is due to abnormalities of mitochondrial fatty acid beta-oxidation and branched-chain amino acid catabolism, resulting in an abnormality in energy production. BVVL also shares a number of phenotypic similarities with other mitochondrial diseases, especially with multisystemic involvement including optic atrophy and sensorineural hearing loss. As is characteristic of mitochondrial neuropathies (Chapter 1), the peripheral neuropathy is non-length dependent and 88
90 RFVT2 Phenotype neuropathological examination of nerve biopsies in this disorder has revealed a number of mitochondrial abnormalities. Treatment of RFVT2 deficiency Functional studies have demonstrated that SLC52A2 mutations cause decreased riboflavin transporter expression and reduced riboflavin uptake. This raised the possibility that therapy with riboflavin may be a useful therapeutic intervention in this group of patients for whom no disease-modifying therapy has previously been available. 131 Doses of up to 25 mg/kg/day of riboflavin have been used in patients with SLC52A3 mutations, and treated individuals have shown improvement in muscle strength, motor function, vision and hearing, and a decrease in the amount of respiratory support required. 151 However, the optimum dose and frequency of riboflavin administration has not yet been defined. The intestinal riboflavin transporters, RFVT1 and RFVT3, are expected to remain functional in patients with RFVT2 deficiency and further studies are required to define how to optimise riboflavin transport via these transporters as well as uptake via diffusion once these transporters are saturated. Similarly, the extent of potential improvement, especially in affected individuals with a significant sensory and motor neuronopathy, also needs to be established. Given the likely benefit of high-dose riboflavin in this otherwise progressive and often fatal disorder, clinicians need to be made aware of the phenotype of this rare disorder and institute riboflavin therapy in all individuals with a phenotype suggestive of riboflavin transporter deficiency, even while mutational analysis results are pending. Siblings of affected individuals should be promptly screened for the disorder, as benefit is likely to be the highest when treatment is started in the pre-symptomatic phase. 89
91 Chapter 3 Auditory neuropathy in Brown-Vialetto- Van Laere syndrome due to RFVT2 deficiency 90
92 RFVT2 Audiology INTRODUCTION While the clinical phenotype of children with BVVL due to RFVT2 deficiency has been reported in detail, the characteristics of the hearing loss have not been explored, despite it being a presenting or early feature in affected individuals. Reports of individuals with RFVT2 deficiency and of genetically undifferentiated cohorts of individuals with BVVL classify the hearing impairment as a sensorineural hearing loss without a clear description of its characteristics. 131,142 As this progressive neurodegenerative disorder is treatable, it is important that treating clinicians are able to diagnose and commence treatment early in children with riboflavin transporter deficiency. It is therefore important for clinicians to identify those children warranting further investigation, for possible riboflavin transporter deficiency, among the many children who present with new-onset hearing loss. Otoacoustic emissions The cochlea can generate several different kinds of sounds that reflect back into the middle ear, known as otoacoustic emissions (OAEs). Self-oscillation of the outer hair cells (OHCs), without a provoking stimulus, produces a pure tone sound called the spontaneous otoacoustic emission (SPOE). The transient evoked otoacoustic emission (TEOAE) is the cochlear echo that occurs after a transient sound is presented to the ear. Distortion product otoacoustic emissions (DPOAEs) are used for diagnostic purposes and are generated by presenting two tones simultaneously to the ear. These reflect function of the OHCs. DPOAEs are of low amplitude or absent when the OHCs are injured or destroyed. Auditory brainstem responses and cochlear microphonics The auditory brainstem responses (ABR) are evoked potentials from the auditory nervous system generated after the presentation of a transient sound, usually a click sound or a tone 91
93 RFVT2 Audiology burst. When recorded as the potential difference between a vertex electrode and a mastoid or earlobe electrode ipsilateral to the stimulus sound, the ABR typically consists of five to seven vertex positive waves. The ABR is commonly elicited by click stimulus presented to one ear at a time. Condensation clicks move the tympanic membrane inward initially, while rarefaction clicks cause the opposite movement. In simple terms, the ABR components are generated by the activity in sequentially activated structures along the auditory pathway. The cochlear microphonic (CM), recorded from an electrode at the round window, is the sum of potentials generated by the cochlear hair cells, mostly by the OHCs at the basal portion of the cochlea. Patients with auditory neuropathy spectrum disorder (ANSD) typically have abnormal or absent ABRs with preserved otoacoustic emissions. With an ANSD, a cochlear microphonic may be present at the beginning of the click recording without the presence of synchronous neural activity in the form of the normal ABR waves. While evaluating for ANSD, recordings from a click stimulus are used and responses are collected from one rarefaction and one condensation run. The cochlear microphonic should be in complete phase cancellation ( ringing cochlear microphonic). To rule out a stimulus artefact, the earphone tubing is pinched; this should cause disappearance of all recorded responses. Auditory neuropathy spectrum disorder The hearing loss reported in most genetic conditions associated with a peripheral neuropathy, including Charcot-Marie-Tooth disease and Friedreich ataxia, is due to an auditory neuropathy spectrum disorder. 161 Auditory neuropathy spectrum disorder (ANSD) is a term used to describe the type of hearing impairment in which OHC function (the cochlear amplifier) is normal but there is a disruption in afferent conduction along the auditory neural 92
94 RFVT2 Audiology pathway. 162,163 The diagnosis of ANSD is based on a characteristic pattern of results on hearing assessments: absent or abnormal ABR with the presence of a CM. OAEs are often, but not always, present and acoustic reflexes may be elevated or absent. 164 Depending on the underlying cause, the disruption could be at the level of the inner hair cells, the spiral ganglion fibres, the myelinated cochlear nerve fibres or the brainstem. Hearing thresholds on pure tone audiometry (PTA) in individuals with ANSD may vary from normal to levels indicating a profound hearing loss, and speech perception may be much worse than predicted from the pure tone audiogram. 165,166 In addition, understanding speech in the presence of background noise has been reported to be especially difficult for individuals with ANSD. 167 Some of this difficulty in speech perception is due to temporal (rate and sequence of processing auditory information) disruption of the auditory signal. 168,169 This study explores the characteristics of the hearing impairment in seven children with BVVL due to RFVT2 deficiency, and the benefits of cochlear implantation in one affected child. METHODS Seven children (from four families) with RFVT2 deficiency due to homozygous or compound heterozygous mutations in SLC52A2 were enrolled in this study. I reviewed the results of previous hearing tests including OAE, PTA, ABR, impedance audiometry and speech perception tests, which had been performed at my hospital and in other centres. These tests were repeated at the time of genetic diagnosis and prior to the commencement of riboflavin therapy. Prospective audiological tests were performed by Katherine O Brien, Department of Audiology, The Children s Hospital at Westmead. Pre- and post-surgical audiological assessments on patient 2.2 were performed by Mandy Hill, Sydney Cochlear Implant Centre 93
95 RFVT2 Audiology and cochlear implantation was performed by A/Prof Catherine Birman, The Children s Hospital at Westmead and The University of Sydney. I reviewed the pure-tone audiogram and ABR recordings and re-interpreted the results so that there was uniformity in the interpretation of the retrospectively reviewed and prospectively performed tests. When there was a discrepancy between the reported result and my interpretation, the test was discussed with audiologist Katherine O Brien to develop a consensus. I did not perform any of the audiological assessments. Otoacoustic emissions (OAE) Distortion Product Otoacoustic Emissions (DPOAEs) were acquired using the Interacoustics Titan, Interacoustics OtoRead or the GSI Audera systems. Protocols were consistent between the three systems and as described. Two primary tones at frequency F1 and F2 with F2/F1 = 1.22 and intensity levels of L1 and L2 = 60/50 db SPL (SPL sound pressure level) were delivered through an emission probe. DPOAE levels were recorded at F2 frequencies between Hz to Hz. DPOAE was considered present if DPOAE level was 8 db greater than the level of the noise floor at each tested F2 frequency. Audiometry Pure Tone Audiometry was conducted in a sound-attenuated test room with an ambient noise level of less than 30 dba (decibels with an A scale filter). Pure tones were presented via THD39 head phones, EAR Tone 3A insert phones and B71 bone conductor using an Interacoustics AC33 Audiometer. Thresholds were obtained using the modified Hughson- Westlake technique as described by Carhart and Jerger. 170 Where masking was required, Hood s plateau procedure was used
96 RFVT2 Audiology Impedance Audiometry Tympanometry and acoustic reflexes were obtained using the Interacoustics Titan or the GSI TympStar systems. Acoustic Reflexes were reported as absent if no repeatable threshold was obtained to a maximum stimulus of 100 db. Electrophysiology ABRs were recorded using the Vivosonic Integrity evoked potential system. Acoustic click stimuli (100 µs) were presented to each ear individually at 80 dbnhl. Stimulus presentation rate was 37.7 Hz and EEG samples following at least 2000 clicks were averaged using Kalman Weighting to produce each test run. Initial assessment in each ear was carried out using rarefaction polarity clicks. In cases where an ABR could not be observed, the testing was repeated using condensation polarity stimuli to investigate the presence of the cochlear microphonic. Speech perception assessments At the baseline assessment prior to riboflavin therapy, speech audiometry was conducted using the NAL-AB Word Lists, which are based on the Consonant Vowel Consonant Words (CVC words) lists by Arthur Boothroyd and recorded by a native Australian speaker. Stimulus was presented via THD39 headphones or EAR Tone 3A insert phones. Tests of expressive and receptive language ability were performed on one individual (patient 2.2) prior to cochlear implantation. The tests included the Peabody Picture Vocabulary test (PPVT) and Clinical Evaluation of Language Fundamentals Fourth Edition (CELF-4). Speech production was assessed using the Diagnostic Evaluation of Articulation and Phonology (DEAP). Tests of speech perception performed on patient 2.2 prior to and following cochlear implantation included the Bench-Kowal-Bamford Sentence test (BKB 95
97 RFVT2 Audiology sentences), DeVault Common Phrases test, Manchester Junior Words test, CVC words and Glendonald Auditory Screening Procedure (GASP) Phoneme Detection and Imitation test. RESULTS The clinical profile, pedigree, and molecular genetic profile of all the individuals in this study is presented in Chapter 1, and that of six individuals (patients 2.1, 2,2, 3.2, 4.1,4.2 and 4.3) has been previously published. 142 All individuals had homozygous p.g306r mutations in SLC52A2, except for patient 3.2 who had a compound heterozygous p.g306r/p.l339p genotype. All patients had presented between the ages of three and eight years with either an ataxic gait or hearing loss. Upper limb weakness was identified in the early second decade of life. Patients 2.1, 2.2 and 4.3 had mild distal upper limb weakness while patients 3.2, 4.1 and 4.2 had severe proximal and distal weakness with no movement at the shoulder or in the small muscles of the hand. Respiratory weakness requiring non-invasive ventilation (patients 3.2 and 4.2) or tracheostomy (patient 4.1) and bulbar weakness requiring gastrostomy (patients 4.1 and 4.2) were seen with those more severely affected. Optic atrophy, usually mild, was seen in five of the seven individuals. Hearing loss Onset The median age of onset of hearing loss was 3 years (range 1 st month of life 10 years) (Table 3.1). One child (patient 1.1), whose parents were consanguineous, had hearing loss identified in the first month of life. She had an older brother who also had congenital hearing loss but did not carry the homozygous SLC52A2 mutations. This raised the possibility that her hearing loss and its congenital onset were also influenced by another unidentified recessive genetic cause for hearing loss. Onset of hearing loss in infancy was also noted in 96
98 RFVT2 Audiology one individual in the cohort described by Vialetto. 127 Hearing loss was the presenting feature in five of the seven affected children, and occurred two and six years after a presentation with gait ataxia in the other two. MRI imaging was normal without evidence of VIII nerve hypoplasia. Characteristics of hearing loss Hearing loss in BVVL due to RFVT2 deficiency had the characteristics of an ANSD. Otoacoustic emissions were present at onset in most individuals, consistent with normal outer hair cell (OHC) function. The ABR was abnormal or absent at onset with a large ringing cochlear microphonic that inverted with change in polarity of the click stimulus. Progression of hearing loss Hearing loss progressed rapidly in all affected individuals, with most progressing to severe or profound hearing loss, and lip reading, within 2 years of identification of the hearing loss. Interventions Hearing aids and FM amplification were used in all 6 individuals with poor speech discrimination but were of limited or no benefit. As described in Chapter 4, six of the individuals in this study (patients 2.1, 2,2, 3.2, 4.1,4.2 and 4.3) were treated with high-dose riboflavin (up to 1600 mg daily) and assessed after 24 months of therapy. Riboflavin therapy resulted in an improvement in auditory function in moderately affected patients (Table 5.1 and Fig. 5.1 from Chapter 5). 97
99 RFVT2 Audiology Table 3.1: Characteristics of individuals with hearing loss in RFVT2 deficiency Patient Presenting feature hearing loss (1 month) ataxic gait (4 yrs) ataxic gait (8 years) ataxic gait (3 yrs) hearing loss Hearing loss Age of onset of hearing loss 1 st month of life 10 yrs 9 yrs 5 yrs Audiological characteristics at initial hearing loss diagnosis OAE absent B/L present B/L present B/L present B/L ABR abnormal ABR Audiometry - abnormal ABR CM present B/L mild lowfrequency HL abnormal ABR CM present R - moderate to severe HL L - severe to profound HL abnormal ABR CM present B/L mild to moderate HL Auditory discrimination tests (NAL AB word lists) - excellent speech perception poor discrimination without visual cues poor discrimination without visual cues MRI Brain and IAMS - - normal normal CT Petrous Temporal bones normal Audiological characteristics prior to riboflavin therapy 5 yrs 10 yrs 9 yrs 15 yrs OAE absent B/L present B/L present B/L present B/L ABR Audiometry Auditory discrimination tests (NAL AB word lists) - unaided abnormal ABR CM present in R ear R - moderate HL L - moderate to severe HL - abnormal ABR CM present B/L mild lowfrequency HL excellent speech perception abnormal ABR CM present R - moderate to severe HL L - severe to profound HL poor discrimination without visual cues abnormal ABR CM present B/L moderate severe HL extremely poor speech discrimination even in aided condition Sensory ataxia present present present present Optic atrophy absent absent present present Previous treatment Hearing aids limited benefit - no benefit limited benefit FM device some benefit OAE otoacoustic emissions, ABR automated brainstem responses, B/L bilateral, R right, L left, HL hearing loss, TROG - Test of Reception and Grammar, CM cochlear microphonic, FM frequency modulation, CT computerised tomography, MRI magnetic resonance imaging, IAMS internal auditory meati. All audiology tests are performed unaided unless specified. 98
100 RFVT2 Audiology Table 3.1: Characteristics of individuals with hearing loss in RFVT2 deficiency (continued) Patient Presenting feature ataxic gait and hearing loss ataxic gait and hearing loss ataxic gait and hearing loss Hearing loss Age of onset of hearing loss 3 yrs 3 yrs 3 yrs Audiological characteristics at initial hearing loss diagnosis OAE R - present, L - absent absent B/L present B/L Audiometry Auditory discrimination tests (NAL AB word lists) ABR abnormal ABR abnormal ABR abnormal ABR B/L severe to profound HL B/L severe to profound HL - - R - moderate lowfrequency HL L- moderate to mild HL (6 yrs) TROG - poor discrimination without visual cues MRI Brain and IAMS normal - normal CT Petrous Temporal bones normal - normal Audiological characteristics prior to riboflavin therapy 15 yrs 15 yrs 20 yrs OAE reduced or absent B/L present B/L - ABR Audiometry abnormal ABR CM present B/L profound to severe HL abnormal ABR CM present B/L profound to severe HL - B/l severe to mild HL Auditory discrimination tests (NAL AB word lists) unaided Sensory ataxia present present present Optic atrophy present present present Previous treatment Hearing aids no benefit no benefit no benefit FM device - - no benefit OAE otoacoustic emissions, ABR automated brainstem responses, B/L bilateral, R right, L left, HL hearing loss, TROG - Test of Reception and Grammar, CM cochlear microphonic, FM frequency modulation. CT computerised tomography, MRI magnetic resonance imaging, IAMS internal auditory meati. All audiology tests are performed unaided unless specified. 99
101 RFVT2 Audiology Cochlear implantation Patient 2.2 received a cochlear implant on the left side after 18 months of riboflavin therapy. Auditory assessment, six months prior to cochlear implant surgery, showed a mild moderate hearing loss in the right ear and a severe to profound hearing loss in the left ear. OAEs were present bilaterally. The ABR was abnormal with a large ringing cochlear microphonic, absent ABR waveforms on maximum testing at 110dBHL and bilateral absent acoustic reflexes. Tests of receptive and expressive language ability were performed prior to cochlear implantation and showed poor speech perception, which correlated with the patient s description that she could not understand what was said to her (Table 3.2). She received a Cochlear Nucleus CI422 implant on the left side. The left side was chosen as it was the side with poor hearing and poorer speech perception. Intra-operative recordings of neural auditory function on the left side showed electrically-evoked auditory brainstem responses which were poorly formed but present. The cochlear implant was switched on two weeks after surgery. The results of audiometry and speech perception testing pre-, 6 months and 12 months post- implantation are in Table 3.3. These tests showed a significant and progressive improvement in her speech perception after cochlear implantation, enabling her to return to a normal class room and continue with verbal language. 100
102 RFVT2 Audiology Table 3.2: Speech and language, and speech production ability prior to cochlear implantation for patient 2.2 Measure Peabody Picture Vocabulary Test (PPVT) Percentile rank score 14 th percentile Clinical Evaluation of Language Fundamentals 4 th edition (CELF-4) Concepts & following directions Recalling sentences Formulated sentences 5 th percentile 25 th percentile 63 rd percentile Word classes: Receptive Expressive Total language score: Diagnostic Evaluation of Articulation & 9 th percentile 63 rd percentile 23 rd percentile 100% Phonology (DEAP) 101
103 RFVT2 Audiology Table 3.3: Audiometry and speech perception testing prior to and after cochlear implantation in patient 2.2 Speech Perception Pre-CI Pre-CI 6 months post-ci 12 months post-ci 12 months post-ci 12 months post-ci Condition Live Voice Live Voice Live Voice Live Voice Live Voice Recorded Recorded Condition Quiet Quiet Quiet Quiet Quiet Quiet Quiet Device/s Left HA Right HA Left CI Left CI Left CI CI & HA DeVault Common Phrases - words correct BKB sentences - words correct 35% 0% NT 46% 85% 78% NT 94% NT 70% NT 82% Manchester Junior - words correct - phonemes correct CVC words - words correct - phonemes correct 10% 45% NT NT NT NT 40% 69% 65% 81% 40% 65% NT NT 52% 75% NT NT NT NT NT NT 72% 89% GASP Phoneme Detection and Imitation - vowel detection - consonant detection - vowel identification - consonant identification 100% 66% 18% 16% 100% 100% 66% 50% 100% 100% 100% 58% 100% 100% 100% 83% NT NT NT NT NT NT NT NT CI cochlear implant, HA hearing aid, NT not tested, BKB sentences - Bench-Kowal-Bamford Sentence test, CVC words - Consonant Vowel Consonant Words, GASP Glendonald Auditory Screening Procedure 102
104 RFVT2 Audiology DISCUSSION This study has evaluated the characteristics of the hearing loss in seven children with BVVL due to RFVT2 deficiency and shown that this hearing loss is due to an ANSD. To date, 29 individuals from 17 families have been reported with BVVL due to RFVT2 deficiency (including families 2, 3 and 4 in this study). 137,140,142,143,172,173 In this group, hearing loss was present in all affected individuals, with an earliest reported age of onset of two years, and was the presenting feature in five of the 17 probands. The hearing loss was rapidly progressive with many children progressing to severe deafness and communicating via lip reading within two years of identification of the hearing loss. ANSD is not uncommon and makes up approximately 8% of newly diagnosed cases of hearing loss in children per year. 174 ANSD was identified in 15% of children with severe hearing loss identified through a universal newborn screening program and approximately 13% of children aged 6-32 months with severe to profound hearing loss. 162,163 Auditory neuropathy may be caused by acquired or genetic factors. Acquired causes and risk factors include hyperbilirubinemia, sepsis, prematurity, low birth weight, ototoxic drug exposure and hypoxic-ischaemic encephalopathy. 175,176 Hereditary causes include primary non-syndromic genetic causes of auditory neuropathy and genetic disorders in which auditory neuropathy is an associated feature. The non-syndromic genetic causes of auditory neuropathy include genes with an autosomal dominant (AUNA1, PCDH), autosomal recessive (OTOF/DFNB9, Pejvakin/DFNB59) and X-linked (GJB2, AUNX1) inheritance. 161 ANSD may be a feature of Charcot-Marie-Tooth disease, 177 Friedreich ataxia, 178 autosomal dominant optic atrophy due to OPA1 mutations, 179 Refsum disease, 180 Mohr-Tranebjaerg syndrome, 181 ARTS syndrome, 182 Leber hereditary optic neuropathy 183 and other mitochondrial disorders. 184 Interestingly, like BVVL, many of these disorders have an associated peripheral neuropathy 103
105 RFVT2 Audiology and optic neuropathy and are often referred to as optico-acoustic neuropathies. Clinicians must now consider BVVL due to riboflavin transporter mutations in the differential diagnoses of any child who presents with new-onset auditory neuropathy, as this progressive neurodegenerative disorder is treatable with high-dose riboflavin and response to therapy is best with early treatment. Otoacoustic emissions may reduce or disappear over time in a small number of individuals with auditory neuropathy, as was the case in patient 4.1. Their retention should not be considered the only marker of auditory neuropathy, and individuals with acquired hearing loss should also be assessed with ABR. 185 All children with new-onset ANSD should undergo testing of their serum acylcarnitine profile as part of their baseline assessment. In addition, as medium-chain acylcarnitines are raised on acylcarnitine profiles in only 60% of those with RFVT2 deficiency, 142 genetic testing for SLC52A2 mutations may need to be considered despite a negative acylcarnitine profile when an alternative explanation for the hearing loss has not been identified. It has been shown previously that the p.g306r mutation in SLC52A2 is a founder mutation in those of Lebanese heritage, and genetic testing for riboflavin transporter mutations should be considered in children of Lebanese origin who present with ANSD. 142 Details of laboratories in Australia and overseas offering SLC52A2 sequencing is provided in the recommendations section in the Summary and Recommendations chapter. The emergence of other signs of BVVL, such as gait ataxia, should encourage the commencement of riboflavin therapy while waiting for the results of genetic testing. It is not yet clear if BVVL due to riboflavin transporter deficiency can present with congenital (present at birth) hearing loss. Patient 1.1 in my cohort had hearing loss identified 104
106 RFVT2 Audiology soon after birth, though its early occurrence may have been caused by a different recessive genetic cause of hearing loss prevalent in the family. Of the children in this cohort, there is definite evidence of a normal ABR at birth only in siblings 2.1 and 2.2. As it is a low-cost test, and the benefits of early treatment are significant, an acylcarnitine profile should be considered even in those with congenital-onset ANSD. As with other forms of sensorineural hearing loss, hearing aids, FM systems and cochlear implants are some of the technologies available for the management of children with ANSD. While most children with ANSD, especially those younger than a year of age, will be initially managed with hearing aids, these are often of limited benefit as they do not address the underlying temporal processing deficits. 164,186 There is increasing evidence that children with ANSD may achieve greater benefit with cochlear implantation, especially when the cochlear nerves are normal on imaging In addition, children who do not respond to amplification may benefit from cochlear implantation when this is done at a young age Hearing aids were tried in six of the children in this cohort with limited or no benefit. The benefits of cochlear implantation have also been demonstrated in patients with ANSD associated with other inherited peripheral neuropathies. 179 These good results in ANSD are possibly due to the ability of cochlear implants to improve auditory processing by aiding synchronous firing of the auditory nerve. 193 Studies on children with pre-lingual deafness indicate that those implanted prior to 24 months of age and before the development of a substantial delay in language development show a greater benefit when compared to those implanted after 24 months of age. 194,195 A similar benefit with implantation before 12 months of age has been seen with children with ANSD. 196,197 There has been only a single previous report regarding the use of cochlear implantation in a family with genetically unclassified 105
107 RFVT2 Audiology BVVL, in which no benefit had been derived from hearing aids. 198 In that kindred, cochlear implantation at the age of 38 and 45 years, 29 years after onset of hearing loss in both siblings, did not result in an improvement in speech perception. My study describes the first patient with BVVL reported in the literature to benefit from a cochlear implant. She was treated early in the disease course, within two years of identification of the hearing loss, and has had excellent speech perception outcomes. There is little experience of cochlear implant surgery for children with BVVL due to RFVT2 deficiency and impaired speech perception. The success of patient 2.2 may be aided by the continued use of riboflavin therapy, possibly slowing or halting auditory nerve damage. Hearing aids are often tried initially for ANSD, and for some there is limited benefit. Mild low-frequency loss may recover completely with high-dose riboflavin therapy. Cochlear implant surgery should be performed early in children with BVVL due to RFVT2 deficiency, based on their impaired speech perception results, when PTA still shows moderate to severe hearing loss. In addition, I recommend continued use of riboflavin therapy. 106
108 Chapter 4 Pathophysiology of motor nerve dysfunction in Brown-Vialetto-Van Laere syndrome due to RFVT2 deficiency 107
109 RFVT2 Nerve Excitability INTRODUCTION Brown-Vialetto-Van Laere (BVVL) syndrome is a progressive neurodegenerative disorder characterised by pontobulbar palsy and sensorineural hearing loss. 131 Recently, a significant number of patients with BVVL have been shown to harbour homozygous or compound heterozygous mutations in SLC52A2 and SLC52A3. 140,141 The SLC52A1, -A2 and -A3 genes, encoding the riboflavin transporters RFVT1, RFVT2 and RFVT3, are members of the solute carrier family 52 and are localised within the cytoplasm and endosomal vesicles. While the riboflavin transporters RFVT1 and RFVT3 are highly expressed in the small intestine, RFVT2 expression is most pronounced in foetal brain and spinal cord. 153 Patients with RFVT2 deficiency develop a motor and sensory neuronopathy characterised by childhoodonset pontobulbar palsy, sensory ataxia, upper limb weakness, respiratory insufficiency, sensorineural deafness and optic atrophy. 140 This rare neurodegenerative disorder has a poor prognosis, resulting in loss of ambulation, respiratory failure requiring ventilation and early death. 142 Functional studies have demonstrated that SLC52A2 mutations reduce both riboflavin uptake and riboflavin transporter expression. 142,143 Riboflavin is critical for the biosynthesis of flavin mononucleotide (FMN) and flavin adenine dinucleotide (FAD), important cofactors for carbohydrate, amino acid and lipid metabolism. FAD acts as an electron acceptor in acyl- CoA dehydrogenation reactions in mitochondrial fatty acid oxidation and branched chain amino acid catabolism. Untreated patients with BVVL exhibit an increase in blood mediumchain acylcarnitine levels, which normalizes rapidly after riboflavin supplementation. In addition, supplementation with riboflavin may exert a neuroprotective effect by slowing the disease course and improving motor function. 140,142,152 While identification of mutations in 108

110 RFVT2 Nerve Excitability the riboflavin transporters have opened the way to therapy for BVVL, the pathophysiological basis of motor neuron dysfunction in this condition remains unclear. Figure 4.1: Severe distal upper limb weakness in BVVL due to RFVT2 deficiency. There is clawing of the hands and prominent wasting of the lumbricals and interossei, including the first dorsal interosseous muscle. The development of sensory and motor neuronopathy is a common clinical feature of BVVL. Nerve conduction studies in BVVL due to RFVT2 deficiency reveal a severe axonal sensory neuronopathy at presentation, followed by the development of a progressive motor neuronopathy. 142 Pathological studies in BVVL have disclosed degeneration of sensory and motor neurons, in keeping with the neurophysiological findings. 139,142 Novel 109
111 RFVT2 Nerve Excitability neurophysiologic tests of axonal excitability have been recently applied to various neurodegenerative disorders, including motor neuron disease, and have identified significant abnormalities in axonal ion channel function in these conditions In contrast to conventional nerve conduction studies that explore the conduction of an impulse along a segment of a motor or sensory nerve, axonal excitability studies explore the properties at one point of the motor nerve. While conventional studies seek to classify peripheral nerve involvement as axonal or demyelinating, these classifications are not associated with characteristic findings on nerve excitability studies. Rather, nerve excitability studies investigate the changes in membrane potential and ion channel function that result in the motor nerve dysfunction. The aim of the present study was to use these novel means of testing axonal excitability to better characterise the pathophysiological basis of motor axon dysfunction in BVVL. The studies were repeated after 12 months to determine the extent to which any identifiable abnormalities of axonal excitability might respond to treatment with riboflavin and hence be included as one of the outcomes in the study evaluating the benefit of high-dose riboflavin therapy in BVVL due to RFVT2 deficiency. METHODS Patients with BVVL due to mutations in the SLC52A2 gene were prospectively recruited from a specialised neuropathy clinic. Henceforth, the term BVVL will be used for this group. The nerve excitability tests were performed by me along with Dr Michelle Farrar, and both of us are co-first authors on the resulting publication (Menezes MP et al. Clin Neurophysiol 2015). All patients or their parent guardian gave informed consent for the procedures which were approved by the South Eastern Sydney and Illawarra Area Health Service (SCH/08/215) and The Children s Hospital at Westmead Human Research Ethics Committees (11/CHW/7). Assessments were undertaken at baseline, at the time of initiation of riboflavin treatment, and 110
112 RFVT2 Nerve Excitability after 12 months of treatment. All patients were treated with 1000mg/day of oral riboflavin (Herbs of Gold, Riboflavin 200mg tablets, Kirrawee, Australia), equating to a dose of 20-26mg/kg/day. Nerve excitability studies The nerve excitability studies were performed by me along with Dr Michelle Farrar, Sydney Children s Hospital, Randwick, Australia and data analysis was performed under the supervision of Dr Michelle Farrar, Prof Steve Vucic, Westmead Hospital and The University of Sydney and Prof Matthew Kiernan, Brain and Mind Research Institute, University of Sydney. Nerve excitability studies were undertaken according to a previously described protocol. 200 Stimulus current was applied at the wrist to the median nerve, recording over the abductor pollicis brevis muscle (APB). Skin temperature was measured at the site of stimulation and was stable at or above 32 C. The studies were undertaken using TRONDNF protocol of the multiple nerve excitability QTRACs software (Institute of Neurology, London, England). Multiple nerve excitability variables were recorded and measured, including: 1. Stimulus-response relationship To commence the protocol, a stimulus-response curve was generated by increasing the stimulus intensity in a stepwise fashion from zero, until a maximal CMAP amplitude was achieved. Threshold (current) refers to the stimulus current required to evoke a compound potential that was 40% of the maximum. 111
113 RFVT2 Nerve Excitability 2. Strength-Duration relationship Strength-duration time constant (SDTC), a surrogate biomarker of nodal persistent sodium (Na + ) conductances, 204,205 was calculated according to Weiss s formula. 206 The rheobase, defined as the threshold for stimuli of infinitely long duration, 207 was also calculated. 3. Threshold Electrotonus (TE) and current/threshold (I/V) relationship Threshold electrotonus (TE) provides information about internodal membrane properties and conductances in addition to an estimate of resting membrane potential. 207,208 In this protocol, threshold tracking was used to record changes in excitability in response to prolonged (100ms) subthreshold polarising currents, set to ±40% of the control threshold current. 200,207 The changes in threshold at different time intervals before, during and after the subthreshold polarising (±40% of threshold) conditioning stimuli were measured. For example, depolarising TE was assessed at 10 and 20ms (TEd 10-20ms) and 90 and 100ms (TEd ms). The parameter accommodation half-time was defined as the time from the start of the 40% depolarising current until the threshold reduction returned to half-way between the peak and plateau levels. The threshold change to hyperpolarising subthreshold conditioning current was assessed at 10 and 20ms (TEh 10-20ms) and 90 and 100ms (TEh ms). With the onset or offset of the polarising current, there is a fast change in threshold due to rapid depolarisation or hyperpolarisation at the node of Ranvier, followed by a slower change in threshold in the same direction, due to slower change in the potential across the internodal axonal membrane affecting the nodal membrane potential. Activation of the slow K + channels at the node then brings the threshold slowly back to baseline. A third slow component, due to the activation of the inward rectifier channel by hyperpolarization, results in excitation. The membrane potential is the most important variable affecting the threshold 112
114 RFVT2 Nerve Excitability electrotonus, and the threshold electrotonus can hence be used as a marker of the membrane potential. 207 For example, ischemia inhibits the Na + pump, resulting in membrane depolarization and a fall or fanning in of the threshold electrotonus. Release of the ischemia results in hyperpolarization and an increase or fanning out of the threshold electrotonus. The current-threshold relationship, a biomarker of inward and outward rectifying axonal currents, was also assessed. Long duration (200ms) polarising conditioning current was altered in 10% steps, from +50% (depolarising) to -100% (hyperpolarising) of the threshold stimulus, and the change in threshold was measured 200ms after the onset of the conditioning current. The hyperpolarising I/V gradient was calculated from polarising current between -80 and -100% while the resting I/V slope was calculated from polarising currents +10% to -10%. 4. Recovery cycle The recovery cycle was determined utilising a paired pulse paradigm with a supramaximal conditioning stimulus preceding a test stimulus at decreasing interstimulus intervals from 200 to 2ms. 200,209 The following parameters were measured from the recovery cycle: (i) relative refractory period (RRP, ms), defined as the first intercept at which the recovery curve crosses the x-axis. Refractoriness is primarily due to Na + channel inactivation and the RRP is a biomarker of transient Na + channel function; (ii) superexcitability (%), defined as the minimum mean threshold change of three adjacent points. Superexcitability correlates well with the depolarising afterpotential, caused due to long-lasting passive depolarisation of the internodal axon by the action potential, and is a biomarker of paranodal fast K + channel conduction; (iii) late subexcitability (%), defined as the largest increase in threshold in three adjacent points following the superexcitability period. The late subexcitability correlates with 113
115 RFVT2 Nerve Excitability the late hyperpolarising afterpotential due to the slow turn-off of slow K + channel and represents a biomarker of nodal K + channel conduction. 210 Mathematical model of nerve excitability To model the excitability changes in motor axons in patients with BVVL, and the functional effects of altered axonal conductances and passive membrane properties in these subjects, mathematical simulations were undertaken using a model of the human axon. 199, Transient Na + channels were modelled using the voltage-clamp data, 215 and persistent Na + currents were added. 205 The equations for a single node and internode, representing a spatially uniform axon, were assessed by integration over successive small time steps (Euler s method). 153,216 At times corresponding to those in human nerve excitability recordings, the excitability of the model nerve was tested repeatedly to determine threshold with an accuracy of 0.5%. The discrepancy between the thresholds determined for the model and those determined from a sample of real nerves was scored as the weighted sum of the error terms: [(x m -x n )/s n ] 2, where x m is the threshold of the model, x n the mean and s n the standard deviation of the thresholds for the real nerves. The weights were the same for all threshold measurements of the same type (e.g. recovery cycle), chosen to give an equal total weight to the different types of threshold measurement: current/threshold relationship, threshold electrotonus and the recovery cycle. The standard model was obtained by minimising the discrepancy between the model and the normal control data with an iterative least squares procedure, so that alteration of any of the above parameters would increase the discrepancy. 114
116 RFVT2 Nerve Excitability Statistical analysis Nerve excitability and grip strength dynamometry on the CMTPedS were used as the primary outcomes to evaluate the clinical and pathophysiological impact of 12 months of riboflavin therapy on motor nerve function in subjects with BVVL. The grip strength item was chosen as it is the subscale on the CMT Pediatric Scale (CMTPedS) corresponding best with distal upper limb motor nerve function. Measurements from BVVL patients at baseline were compared to the 95% confidence intervals (CI) of healthy controls (and considered significant if outside this range). Control data was obtained from 17 age-matched participants (nine males, eight females; age range 9-18 years, mean 12.4 years). This control data was sourced by including all those in the age range of 9-18 years from previously collected control data from a larger healthy cohort. Paired Student t-tests were used to determine differences between BVVL patients before and after treatment. A p-value of <0.05 was considered statistically significant. Results in BVVL patients are expressed as mean ± standard error of the mean (SEM). The results in healthy controls are expressed as mean and 95% confidence interval (CI), as the patient sample size was small. RESULTS Clinical features A total of six patients, aged between 10 and 21 years, from 3 different families (patients 2.1 and 2.2 from family 2, 3.2 from family 3, and 4.1, 4.2 and 4.3 from family 4) were recruited for this study. All patients were homozygous for the p.g306r mutation in SLC52A2, except for patient 3.2 who had a compound heterozygous p.g306r/p.l339p genotype. The clinical phenotype in this patient cohort was typical of BVVL and characterised by sensory ataxia, progressive upper limb, axial and respiratory weakness along with cranial neuropathy affecting cranial nerves II (optic atrophy) and VIII (sensorineural hearing loss). 142 Hearing 115
117 RFVT2 Nerve Excitability deteriorated rapidly, with patients progressing to lip-reading within 2 years of onset of hearing loss. Upper limb weakness and wasting was not present at onset but was identified in the early second decade of life and progressed rapidly. The weakness involved both the proximal and distal upper limb and was ultimately severe, resulting in a flail-arm phenotype. Bulbar symptoms included dysarthria, tongue weakness and dysphagia. Respiratory weakness requiring invasive or non-invasive respiratory support was present in 2 patients. Cognition was preserved in all patients. Baseline nerve excitability Conventional neurophysiological testing disclosed the presence of an axonal sensorimotor polyneuropathy in subjects with BVVL (Table 2.3 from Chapter 2). Nerve excitability was assessed in three patients with recordable median CMAP responses (patients 2.1, 2.2 and 4.3) 2, 6 and 16 years after symptom onset respectively. The other three subjects exhibited absent or significantly reduced median motor responses, such that axonal excitability studies could not be undertaken on these patients. In the tested group, the CMAP amplitude was similar to healthy controls (BVVL, 5.4 ± 1.2mV; controls, 6.3 mv [95% CI ]). The threshold currents required to elicit a response were similar in BVVL patients and controls (BVVL, 2.6 ± 1.2mA; controls, 3.2mA [95% CI ]). The strength duration time constant (BVVL, 0.42 ± 0.03ms; controls, 0.40ms [95% CI ]) and rheobase (BVVL, 1.9 ± 1.1mA; controls, 2.2 ma [95% CI ]) were also similar. A fanned out appearance of threshold electrotonus was evident in the BVVL patients, signifying greater threshold change to both depolarisation and hyperpolarisation (Table 4.1, Fig. 4.2A). Specifically, TEd ms (BVVL, 56.1 ± 2.3%; controls 42.6% [95% CI ]) and TEh ms (BVVL, ± 10.2%; controls % [95% CI ]) were significantly increased when compared to controls. The changes in TE were accompanied by abnormalities 116
118 RFVT2 Nerve Excitability of the I/V gradient, whereby the resting I/V gradient was significantly reduced in BVVL patients (BVVL, 0.44 ± 0.03; controls 0.68 [95% CI ], Fig 4.2B). Table 4.1: Results of motor nerve excitability testing Normal BVVL baseline BVVL on riboflavin n = 17 n = 3 n = 3 CMAP amplitude 6.3 [ ] 5.4 ± ± 1.1 Strength duration time constant (ms) 0.40[ ] 0.42 ± ± 0.02 Threshold electrotonus Depolarizing TE peak (%) 65.1 [ ] 76.2 ± ± 2.6 Depolarising TE 40-60ms (%) 50.6[ ] 61.5 ± ± 1.7 Depolarising TE ms (%) 42.6[ ] 56.1 ± ± 1.3 Hyperpolarising TE 10-20ms (%) -70.8[ ] ± ± 1.9 Hyperpolarising TE ms (%) [ ] ± ± 5.5 Current-voltage relationship Resting I/V slope 0.68 [ ] 0.44 ± ± 0.03 Recovery cycle Refractoriness at 2.5ms (%) 23.8[ ] -4.8 ± ± 12.3 Superexcitabilty (%) -26.2[ ] ± ± 2.1 Late subexcitability (%) 13.9[ ] 11.8 ± ± 0.9 Results in BVVL patients are expressed as mean ± standard error of the mean (SEM) and in healthy controls as mean and 95% confidence intervals (CI). p-values comparing BVVL baseline to BVVL on riboflavin. Significant results (p < 0.05) in bold. 117
119 RFVT2 Nerve Excitability Figure 4.2: Nerve excitability measures in BVVL patients and normal controls Comparison of multiple measures of nerve excitability in median motor nerves in BVVL patients at baseline (filled circles) and normal controls (dashed lines) plotted as mean and standard errors of mean or 95% confidence intervals respectively. Riboflavin therapy resulted in significant modulation of excitability with partial reversibility of baseline changes (open circles = BVVL patients ON riboflavin therapy; filled circles = BVVL patients baseline). (A) Threshold electrotonus, depicting TEh ms and TEd ms. 118
120 RFVT2 Nerve Excitability Figure 4.2: Nerve excitability measures in BVVL patients and normal controls (continued) Comparison of multiple measures of nerve excitability in median motor nerves in BVVL patients at baseline (filled circles) and normal controls (dashed lines) plotted as mean and standard errors of mean or 95% confidence intervals respectively. Riboflavin therapy resulted in significant modulation of excitability with partial reversibility of baseline changes (open circles = BVVL patients ON riboflavin therapy; filled circles = BVVL patients baseline). (B) Current threshold relationship. 119
121 RFVT2 Nerve Excitability Figure 4.2: Nerve excitability measures in BVVL patients and normal controls (continued) Comparison of multiple measures of nerve excitability in median motor nerves in BVVL patients at baseline (filled circles) and normal controls (dashed lines) plotted as mean and standard errors of mean or 95% confidence intervals respectively. Riboflavin therapy resulted in significant modulation of excitability with partial reversibility of baseline changes (open circles = BVVL patients ON riboflavin therapy; filled circles = BVVL patients baseline). (C) Recovery cycle of excitability, demonstrating reduced refractoriness and increased superexcitability at baseline. 120
122 RFVT2 Nerve Excitability The recovery cycle of axonal excitability curves was markedly shifted downwards in BVVL patients when compared to controls (Fig. 4.2C). Specifically, there was a significant reduction in refractoriness at 2.5 ms (BVVL, -4.8 ± 4.8%; controls, 23.8 [95% CI ]) and an increase in superexcitability (BVVL, ± 1.7; controls [95% CI ], Table 4.1, Fig 4.2C) in subjects with BVVL. Late subexcitability was similar between the groups (BVVL, 11.8 ± 3.1; controls 13.9 [95% CI ]). Mathematical modeling of abnormal excitability properties To assist in interpreting the complex changes observed in clinical nerve excitability measures, a mathematical model of the human motor axon was adjusted to provide a close match to the age-matched control group. The model was then used to explore whether changes in any membrane parameter could reproduce the changes seen in BVVL patient recordings (3 patients). Alterations in membrane conductances or potential, in isolation, could not account satisfactorily for the changes seen in subjects with BVVL. Membrane hyperpolarisation reduced the discrepancy by 67%, yet did not support similarities in late subexcitability, Τ SD and threshold between BVVL patients and controls. Rather, increasing the Barrett and Barrett conductance (GBB) from 35.2 to 50.5 units reduced the discrepancy by 76.7% (Fig. 4.3). The GBB refers to passive membrane property related to applied currents accessing and crossing the internodal compartment of the axon. 207 An increase in GBB may be caused by abnormal myelin permeability, secondary to thin or leaky myelin, or by a loosening of paranodal seal. Modeling of changes in two parameters improved the fit to 77.1% by means of increasing GBB from 35.2 to 47.5 units and reducing leak conductances from 1 to 0.82 units. Consequently, the mathematical modeling suggested that an increase in myelin permeability may account for the abnormalities of axonal excitability in BVVL
123 RFVT2 Nerve Excitability Figure 4.3: Simulation of the excitability changes in clinical nerve excitability in BVVL patients using the mathematical model Open circles represent the model generated by the normal control group. Black lines were generated by the model by increasing GBB from 35.2 units to 50.5 units, which reduced the discrepancy by 76.7%. (A) Threshold electrotonus for 100-millisecond polarising currents ±40% of the resting threshold. (B) Current-threshold relationship. (C) Recovery cycles. 122
124 RFVT2 Nerve Excitability ASSESSMENTS AFTER 12-MONTHS OF RIBOFLAVIN THERAPY Improvement in motor nerve function Treatment with riboflavin was accompanied by partial normalisation of motor nerve function (Table 4.1, Fig. 4.2). Specifically, there was a significant improvement in threshold electrotonus, with reduced TEd (90-100ms) and TEh (90-100ms) (TEd90-100ms: BVVL baseline, 56.1 ± 2.3, BVVL on riboflavin, 49.3 ± 1.3, p<0.05, TEh90-100ms: BVVL baseline, ± 10.2; BVVL on riboflavin, ± 5.5, p<0.05). There was also an increase in resting I/V slope (BVVL baseline, 0.44 ± 0.03; BVVL on riboflavin, 0.54 ± 0.03, p<0.05). Prominent changes were also noted in the recovery cycle of nerve excitability, with reduction in superexcitability (BVVL baseline, ± 1.7; BVVL on riboflavin, ± 2.1, p<0.05) and an increase in refractoriness at 2.5ms (BVVL baseline, -4.8 ± 4.8; BVVL on riboflavin, 15.8 ± 12.3, p=0.06). The excitability changes suggested that riboflavin therapy exerted a stabilising effect on myelin permeability in BVVL patients. Similarly, there was an improvement in absolute scores for grip strength on dynamometry (Table 5.2 from Chapter 5), contrasting with the natural history outcomes in untreated BVVL patients, who typically experience progressive upperlimb weakness. This was paralleled by improvement on audiometry in the younger affected individuals, and stable or improved respiratory function in all six individuals. DISCUSSION Utilising the axonal excitability technique, the present study established that abnormalities of passive membrane properties were a feature of BVVL. Specifically, an increase in depolarising and hyperpolarising threshold electrotonus, termed the fanned-out appearance, was evident in BVVL. The changes in TE were accompanied by a reduction in the current threshold slope and refractoriness, along with an increase in superexcitability. Mathematical modeling suggested that abnormalities of axonal excitability were best explained by 123
125 RFVT2 Nerve Excitability alteration in passive membrane properties, namely an increase in the Barrett and Barrett conductance (GBB), while axonal ion channel function and resting membrane potential were maintained. The increase in GBB could be attributed to an increase in myelin permeability, either due to thin or leaky myelin or an increased permeability of the paranodal region, a pathophysiological process that may be secondary to riboflavin deficiency. Pathophysiological mechanisms underlying BVVL neuropathy Although riboflavin deficiency secondary to SLC52A2 gene mutations is postulated to underlie the BVVL phenotype, the pathophysiological processes mediating this condition have not yet been fully elucidated. Importantly, BVVL is characterised by a sensorimotor axonal neuropathy. 142 Given that riboflavin is an important co-factor in the synthesis of myelin, 217 abnormalities of myelin permeability may account for the development of the BVVL neuropathy. Schwann cells are critical in maintaining axonal integrity, and axonal degeneration due to primary Schwann cell and myelin dysfunction has been demonstrated in other inherited neuropathies Myelin adheres to axons at the paranodal junction, an axonal region that is permeable to small molecules, thereby forming a pathway to short-circuit the nodal currents. 222 The paranodal currents may traverse three possible pathways including (i) along the paranodal periaxonal space, (ii) through the obliquely-oriented transverse bands and (iii) between adjacent lamellae of myelin sheaths. 223 While structural changes in peripheral nerve myelination have not been previously described in BVVL, the findings in the present study would suggest that dysfunction at the paranodal region, particularly loosening of the paranodal seal, may lead to an increase in GBB and superexcitability in this condition. The changes demonstrated here are due to functional alteration in the myelin (increased 124
126 RFVT2 Nerve Excitability permeability) rather than demyelination. Consequently, significant changes in motor conduction velocity would not be expected, a notion underscored by the nerve conduction study findings in the BVVL cohort (Table 5.1 from Chapter 5). It should be highlighted that the axonal excitability abnormalities evident in BVVL are different from those previously described in dysmyelination of maturing nerves, 214,224,225 mitigating against the possibility that the abnormalities evident in the present study are relate simply to delayed nerve maturation. Underscoring this notion are pathological studies in riboflavin-deficient animal models which develop a similar neuropathy to BVVL patients, disclosing abnormalities of myelin with dissociation of both the inner and outer spirals of the myelin lamellae. 226,227 In a mouse model of mitochondrial dysfunction, cerebroside depletion was shown to interfere with the maintenance of Schwann cell-axon contacts on electron microscopic analysis, with enlarged nodal gaps and axons that had pulled away from their myelin sheaths. 125 It could be argued that age was a confounding factor in the present study, particularly given that GBB changes with nerve maturation. This seems unlikely given that an age-matched reference model was used and that maturational changes in axonal excitability reach a plateau between 8-15 years, the very age range of two of the patients modeled (1.1 and 1.2). Separately, it may be expected that an increase in GBB should also lead to reduction in Τ SD, although this was not observed in the current study. Τ SD depends on many membrane parameters and a possible explanation may relate to subtle differences in these (for example leak conductance, nodal capacitance, nodal K + and Na + conductances or nodal width). 206 This notion is supported by the mathematical simulations of excitability changes in the present study, as a reduction in leak conductance should lead to an increase in Τ SD, opposing the effect of increasing GBB. 125
127 RFVT2 Nerve Excitability Progress with riboflavin therapy The natural history of BVVL is of progressive neurodegeneration together with decline in motor amplitudes. 142 Previous excitability studies in motor neuron disease have demonstrated progressive axonal dysfunction paralleling clinical impairment and also established the reproducibility of demonstrated abnormalities of axonal excitability. 199,228 The longitudinal improvement in nerve excitability profiles with riboflavin treatment in all BVVL patients in this series, which was accompanied by improvement in grip strength and stable motor amplitudes (Chapter 5), appears incongruous with the natural history, suggesting that riboflavin exerts positive effects on peripheral nerve function with modulation of myelin dysfunction. Stabilisation of the myelin sheath would in turn be expected to prevent further axonal degeneration and promote recovery, which is supported by the motor amplitudes remaining stable. In my cohort, the established and severe sensory neuronopathy did not improve with therapy. In contrast, neuropathy of more recent onset (motor neuropathy of onset in early 2 nd decade and mild in the younger siblings, VIII nerve involvement hearing loss in the younger children) improved significantly with therapy. This emphasises the need for clinicians to diagnose and commence therapy early in children with the disorder. Taken together, findings from the present study suggest that abnormalities of myelin function are a feature of BVVL and these may be partially normalised with riboflavin therapy. These findings also suggest that nerve excitability studies may be further developed in larger cohorts as a potential biomarker, to identify and monitor treatment response and to guide more specific and tailored treatment strategies for BVVL patients. 126
128 Chapter 5 Outcome after therapy with riboflavin in Brown-Vialetto-Van Laere syndrome due to RFVT2 deficiency 127
129 RFVT2 Riboflavin Therapy INTRODUCTION Brown-Vialetto-Van Laere (BVVL) syndrome due to riboflavin transporter type 2 (RFVT2) deficiency is a neurodegenerative disorder with a poor prognosis in which disease progression results in loss of ambulation, respiratory failure and early death. 142 Nerve excitability studies (Chapter 4) suggest that motor dysfunction may be more amenable to therapy commenced early in the course of BVVL. The sensory deficit is due to an early-onset ganglionopathy which is already established at diagnosis and unlikely to improve with disease-modifying therapy. In contrast, strength and motor amplitudes on nerve conduction studies may be normal at presentation and subsequently deteriorate. Early evidence from a small number of patients on short-term treatment demonstrates that supplementation with riboflavin may be beneficial in slowing disease progression and may improve motor function. 140,142,152 The possible pathophysiology of motor nerve dysfunction and partial recovery with riboflavin therapy has been discussed in Chapter 4. The variable progression and lack of a sensitive and relevant assessment scale remains a challenge in objectively evaluating efficacy of treatment in BVVL. While the phenotype of BVVL due to RFVT2 mutations is unique, there is significant variability in age of onset and rate of progression. 140,142,143,172,173 The age of onset ranges from infancy to late childhood. While some patients become non-ambulant in early childhood, others continue to ambulate independently into early adulthood. Developing a robust clinical and functional scale that can characterise the heterogeneity in disease severity and course, including sensory and motor deficits, is critical to mapping the natural history of neurodegeneration in this disorder. A sensitive scale is also necessary to evaluate the efficacy of therapeutic interventions. 128
130 RFVT2 Riboflavin Therapy The aim of the present study was to define the baseline motor and sensory deficits in a diverse cohort of patients with BVVL due to RFVT2 deficiency using the Charcot-Marie- Tooth Pediatric scale (CMTPedS). 96 The study also aimed to monitor progression of disease on riboflavin therapy using the CMTPedS and other clinical assessments. METHODS Patients with BVVL due to mutations in the SLC52A2 gene were prospectively recruited from a specialised neuropathy clinic. Clinical (CMTPedS, respiratory function testing and audiometry) and biochemical (acylcarnitine profile) assessments were combined with conventional and specialised neurophysiological tests. Clinical and functional retrospective data were utilised, to enable comparison with untreated patients. Research assessments on the CMTPedS scale were part of a study approved by The Children s Hospital at Westmead Human Research Ethics Committee (11/CHW/7). I performed and interpreted all the clinical assessments, nerve excitability tests and CMTPedS assessments that are detailed in Chapters 4 and 5, both at baseline as well as after 12 and 24 months of therapy. The nerve excitability tests were performed by me along with Dr Michelle Farrar, and both of us are co-first authors on the resulting publication. I performed the CMTPedS assessments under the supervision of Prof. Joshua Burns, Pediatric Gait Analysis Service, The Sydney Children s Hospital Network and The University of Sydney. Assessments were undertaken at baseline, at the time of initiation of riboflavin treatment, and compared with those after 12 and 24 months of treatment. All patients were treated with 1000mg/day of oral riboflavin (Herbs of Gold, Riboflavin 200mg tablets, Kirrawee, Australia), equating to a dose of 20-26mg/kg/day, for the first 12 months, and with 1600mg of riboflavin (32-42mg/kg/day) for the next 12 months. The results of axonal nerve 129
131 RFVT2 Riboflavin Therapy excitability testing at baseline and after 12 months of therapy with riboflavin are reported in Chapter 4. Clinical and functional grading All patients were clinically assessed using the Charcot-Marie-Tooth Pediatric Scale (CMTPedS), an 11-item linearly weighted scale that has been validated as a reliable and sensitive global measure of disability in children with a variety of CMT types from the age of 3 years, including those genetic types that are more severe than CMT1A. 96 The CMTPedS was chosen as it includes subscales that evaluate upper limb and lower limb strength (grip, ankle dorsiflexion and plantarflexion), sensation (vibration and pinprick) and function (upper limb - functional dexterity and 9-hole peg tests, lower limb balance, gait manoeuvres, long jump and 6-minute walk test). According to the original publication, raw scores for each item (seconds, Newtons, metres) are converted to dimensionless z-scores based on age- and sexmatched normative reference values. The z-scores for each item are then categorised into individual point scores of 0 (normal) to 4 (severe). Summation of individual scores generates a total score from 0 (normal) to 44 (severe disability). While more severely affected subjects with BVVL are non-ambulant, and hence unable to attempt some items, the range of items covered by the CMTPedS and their contribution to the total score make both the total score and comparison separately on various items a useful mode of monitoring those affected. The CMTPedS has also been shown to be sensitive to change over a 1-year period in children with CMT. 97 RESULTS As described in Chapter 4, a total of six patients, aged between 10 and 21 years, from three different families (patients 2.1 and 2.2 from family 2, 3.2 from family 3, and 4.1, 4.2 and
132 RFVT2 Riboflavin Therapy from family 4) were recruited for this study. Patients from family 4 were only assessed at 12 months as their care was transitioned to an adult facility after this time. All patients were homozygous for the p.g306r mutation in SLC52A2, except for patient 3.2 who had compound heterozygous p.g306r/p.l339p genotype. Overall, there was a broad spectrum of clinical severity, ranging from mild to profound weakness and functional impairment, as reflected by the variation in baseline CMTPedS scores, respiratory function testing and audiometry. At baseline before riboflavin therapy, two patients were wheelchair-bound while four patients continued to walk despite being unsteady due to severe sensory ataxia. Neurophysiological features All patients demonstrated a sensorimotor axonal neuropathy with relative preservation of conduction velocities (Table 5.1). Historical data for patient 2.2 demonstrated a progressive decline in distal median CMAPs over a period of seven years (11.1mV at 3 years of age to 3.1mV at 9 years of age). 131
133 RFVT2 Riboflavin Therapy Table 5.1: Comparison of assessments in patients with BVVL at baseline and after 12 and 24 months of riboflavin therapy Patient Gender F F F Disease duration (yrs) Ambulation independently ambulant independently ambulant independently ambulant Previous Baseline 12m 24m Previous Baseline 12m 24m Previous Baseline 12m 24m Age at assessment (yrs) CMTPedS total score Nerve conduction studies 5 yrs 8 yrs Median motor (APB) CMAP (mv) NR NR CV (m/s) Tibial motor (AH) CMAP (mv) CV (m/s) Median SAP (µv) NR NR NR NR NR NR Sural SAP (µv) NR NR NR NR NR NR NR NR NR Audiology 8 yrs 8 yrs Pure tone audiometry B/L mild lowfrequency normal - normalised at 6 months B/L mild lowfrequency normal R - moderatesevere L - severeprofound R - mildmoderate L - severe to moderate R - mildmoderate L - severe to moderate B/L moderate B/L moderatesevere B/L moderatesevere B/L moderatesevere Respiratory function 6 months 6 months 8 yrs 13 yrs testing (% predicted) of therapy of therapy FEV ND FVC ND BiPAP/Ventilation pressure (cm of H2O) Acylcarnitine profile abnormal a normal normal abnormal a normal abnormal a abnormal a normal ND FAD level (nmol/l) (407) (383) (324) ND M - male, F - female, CMTPedS - CMT pediatric scale, CMAP - compound muscle action potential, SAP - sensory action potential, NR - not recordable, ND - not done, L - left, R - right, FAD - flavin adenine dinucleotide, B/L bilateral, a elevated medium chain acylcarnitines, b baseline data not available. Abnormal nerve conduction values (< 2SD) indicated in bold. 100 Riboflavin levels at 6 weeks in parentheses. 132
134 RFVT2 Riboflavin Therapy Table 5.1: Comparison of assessments in patients with BVVL at baseline and after 12 and 24 months of riboflavin therapy (continued) Patient b Gender M M M Disease duration (yrs) Ambulation non-ambulant non-ambulant independently ambulant Previous Baseline 12m Previous Baseline 12m Baseline 12m Age at assessment (yrs) CMTPedS total score Nerve conduction studies 15 yrs Median motor (APB) CMAP (mv) NR NR NR CV (m/s) Tibial motor (AH) CMAP (mv) CV (m/s) Median SAP (µv) NR NR NR NR NR Sural SAP (µv) NR NR NR NR NR Audiology 11 yrs 11 yrs Pure tone audiometry B/L severe to profound B/L profound to severe B/L profound R - severeprofound L - profound B/L profound to severe R - profound to severe L- profound B/L severe to mild B/L profound to mild Respiratory function testing (% predicted) ventilated nocturnal BiPAP FEV1 71 FVC 15 yrs 15 yrs 77 BiPAP/Ventilation pressure (cm of H2O) 16/5 16/5 12/5 10/5 20/8 18/8 Acylcarnitine profile normal normal normal normal normal FAD level (nmol/l) (396) (380) 324 M - male, F - female, CMTPedS - CMT pediatric scale, CMAP - compound muscle action potential, SAP - sensory action potential, NR - not recordable, ND - not done, L - left, R - right, FAD - flavin adenine dinucleotide, B/L bilateral, a elevated medium chain acylcarnitines, b baseline data not available. Abnormal nerve conduction values (< 2SD) indicated in bold. 100 Riboflavin levels at 6 weeks in parentheses. 133
135 RFVT2 Riboflavin Therapy Table 5.2: CMT Pediatric Scale (CMTPedS) in patients with BVVL at baseline and after 12 and 24 months of riboflavin therapy Patient Baseline 12m 24m Prev Baseline 12m 24m Age at assessment (years) Functional dexterity test (sec) Point score Nine-hole peg test (sec) Point score Grip (Newtons) Point score Ankle plantarflexion (Newtons) Point score Ankle dorsiflexion (Newtons) Point score Pinprick (point score) Vibration (point score) BOT-2 score (raw) Point score Gait (point score) Long Jump (cm) Point score minute walk test (metres) Point score Total score Balance BOT-2- Bruininks Oseretsky test of Motor Proficiency 2 nd Edition, b baseline data not available 134
136 RFVT2 Riboflavin Therapy Table 5.2: CMT Pediatric Scale (CMTPedS) in patients with BVVL at baseline and after 12 and 24 months of riboflavin therapy (continued) Patient b Baseline 12m 24m Baseline 12m Baseline 12m 12m Age at assessment (years) Functional dexterity test (sec) Point score Nine-hole peg test (sec) Point score Grip (Newtons) Point score Ankle plantarflexion (Newtons) Point score Ankle dorsiflexion (Newtons) Point score Pinprick (point score) Vibration (point score) BOT-2 score (raw) Point score Gait (point score) Long Jump (cm) Point score minute walk test (metres) Point score Total score Balance BOT-2- Bruininks Oseretsky test of Motor Proficiency 2 nd Edition, b baseline data not available 135
137 RFVT2 Riboflavin Therapy CMTPedS Total CMTPedS scores at baseline ranged from 18-36, with two individuals (2.1, 2.2) being moderately affected (CMTPedS score between 15-29) and three (3.2, 4.1, 4.2) severely affected (CMTPedS score between 30-44). Review of the CMTPedS subscales (Table 5.2) at baseline confirms many of the unique features of the RFVT2 phenotype. Affected individuals had worse scores on the subscales that measure sensation (vibration, BOT-2) and functional scales that rely predominantly on normal proprioception (functional dexterity, nine hole peg test), irrespective of disease duration, consistent with their clinical presentation with earlyonset severe ataxia and absent sensory responses in the upper and lower limbs on nerve conduction studies (Table 5.1). The sensory loss associated with RFVT2 deficiency is consistent with a large-diameter fibre sensory neuronopathy (ganglionopathy). The early-onset sensory ataxia and positive Romberg sign, absent reflexes in the upper and lower limbs early in disease course and nonlength dependent pure sensory abnormalities on nerve conduction studies with none or limited motor involvement at disease onset are all consistent with a sensory ganglionopathy. 229 The finding is also consistent with the severe proprioceptive abnormality causing difficulty with functional tasks like the functional dexterity test and nine hole peg test in the younger affected individuals despite preserved grip strength. The preserved sensation to pinprick, irrespective of disease duration, shows that small-fibre sensory nerves remain unaffected even late in disease course. In contrast, motor involvement showed differential involvement of the upper and lower limbs and progression of upper limb weakness with disease duration. Those early in the disease course had normal grip strength or only mild weakness while those with advanced disease 136
138 RFVT2 Riboflavin Therapy had only minimal movement of the upper limbs. In contrast, ankle dorsiflexion and plantarflexion strength were preserved even late in disease course. This parallels the change in motor amplitudes on nerve conduction studies (Table 5.1) with normal upper and lower limb CMAPs at presentation, and a progressive decline in upper limb CMAPs with disease progression. Change with riboflavin therapy The mean change in the CMTPedS total score after treatment with riboflavin was a deterioration of 0.66 points (range +3 to -2, n=5) after 1 year and an improvement of 1.66 points (range -1 to -2, n=3) after 2 years. Interpreting the magnitude of this change is difficult due to the lack of natural history data in large cohorts of subjects with this condition. The rate of progression in CMT1A, a more common but less severe neuropathy, is +0.6 points/year. 97 In addition, BVVL is a progressive neurodegenerative condition in which improvement without therapy has not previously been described. 131 Historical CMTPedS data was available for patient 2.2 and showed that her total CMTPedS score had deteriorated from 19 to 27 over the preceding 12 months prior to riboflavin therapy, with worsening in functional dexterity, ankle dorsiflexion strength, long jump distance and six-minute walk test (6MWT) distance. Scores on the subscales measuring sensory function showed no change over the two year period of treatment. In contrast, absolute scores for grip strength improved, especially in the less severely affected, consistent with the improvement in motor nerve function on axonal excitability studies (Chapter 4). Absolute scores for ankle dorsiflexion and plantarflexion varied, but Z-scores were stable. Similarly, long jump distance improved or remained stable, both on absolute as well as Z-scores, in those who were able to attempt the test. However, the 137
139 RFVT2 Riboflavin Therapy 6MWT distance, a functional performance measure that requires both balance and strength, deteriorated in the second year of therapy. Audiology Riboflavin therapy resulted in significant improvement in auditory function in moderately affected patients (Table 5.1 and Fig. 5.1). Patient 2.1 had low frequency hearing loss identified at baseline which resolved after a year of riboflavin therapy. The hearing loss was identified again when evaluated after 24 months of therapy; this apparent relapse may have related to poor compliance with riboflavin. Patient 2.2 had moderate to severe hearing loss in the right ear and severe profound hearing loss in the left ear, which improved after a year of riboflavin therapy and then remained stable. Respiratory function testing Respiratory function testing was possible in 3 patients at baseline (Table 5.1). Forced vital capacity (FVC) remained normal in the 2 patients in whom baseline studies were normal, and remained stable in patient 3.2 during the period of observation, after deteriorating in the 2 years prior to therapy initiation. In those patients who required ventilation, pressures decreased significantly in patient 3.1 and stabilised, after a year of significant increase, in patient
 and 1.1 (D,E) showing deterioration during disease course, and improvement following riboflavin therapy. Patient 1.")

140 RFVT2 Riboflavin Therapy Figure 5.1: Audiometry in BVVL patients before and after riboflavin therapy Audiometry in patient 1.2 (A,B,C) and 1.1 (D,E) showing deterioration during disease course, and improvement following riboflavin therapy. Patient 1.2 progressed from normal (A) to severe hearing loss (B) over 12 months with improvement after 12 months of riboflavin therapy (C). Patient 1.1 was diagnosed with low frequency hearing loss at baseline (D), returning to normal after 12 months of riboflavin therapy (E). 139
141 RFVT2 Riboflavin Therapy Biochemical profile Untreated patients with BVVL have an elevation of medium-chain acylcarnitines which normalises rapidly after riboflavin supplementation. Elevation of medium chain acylcarnitines was evident in 60% of BVVL patients at baseline. Riboflavin therapy led to normalisation of medium-chain acylcarnitine levels within six weeks of commencement of treatment (Table 5.1). No significant adverse effects were reported during the two years of riboflavin therapy. Patients 2.1 and 2.2 reported poor compliance with the riboflavin during the second year of therapy due to the need to take large doses (three capsules three times a day). This was reflected in recurrence of mild abnormalities in the acylcarnitine profile of patient 2.2. DISCUSSION In this study, the CMTPedS has been used as an effective tool in characterising disease burden in affected individuals with RFVT2 deficiency. As CMTPedS has subscales that evaluate strength, sensation and function, it is useful in defining baseline deficits and monitoring progression in BVVL, particularly as each of these domains may be differentially involved at different stages of the disease. The individuals studied in this series showed wide variation in disease severity as measured by the CMTPedS, partly related to disease duration, though there were outliers to this rule; patient 4.3 had less severe disease when compared to his twin brothers (patients 4.1 and 4.2) who carried the same mutation, indicating that there are other genetic, epigenetic or environmental factors moderating disease severity in BVVL. Evaluation of the CMTPedS at baseline characterised the unique features of BVVL due to RFVT2 deficiency: differential motor and sensory involvement, severe large fibre sensory 140
142 RFVT2 Riboflavin Therapy ganglionopathy early in disease course and progressive upper limb muscle weakness with relative sparing of the lower limbs. At baseline, the CMTPedS was sensitive to increased upper limb weakness and development of restrictive lung disease and hence could be used to identify when surveillance for restrictive lung disease should begin. The CMTPedS appears sensitive to baseline disability and progression of disease in BVVL and further studies formally validating it for clinical trials in BVVL are warranted. This study does provide face/content validation and shows that the CMTPedS does hold promise as a useful way to monitor disease progression in this group and in related motor and sensory neur(on)opathies. Long-term effects of riboflavin To date, 29 patients from 17 families have been reported with BVVL due to RFVT2 deficiency. 140,142,143,172,173 A poor prognosis may be expected, with 20% (6 patients) patients dying between the ages of three and 22 years, most commonly due to respiratory failure. Approximately half of the remaining patients required respiratory support and half had lost independent ambulation by the age of 21 years. In the present study, riboflavin therapy stabilised or improved clinical, biochemical and neurophysiological parameters in subjects with BVVL, in contrast to the progressive neurodegeneration seen in untreated patients. Following a two year therapeutic course with high-dose riboflavin, there were improvements in total CMTPedS score, and the items of grip strength and long jump distance. The youngest patients demonstrated significant improvement in their hearing and older patients showed stabilisation or improvement in their respiratory function, both aspects in which clinical deterioration was relatively recent and hence reversible. This is consistent with earlier reports of short-term (up to seven months) riboflavin therapy, where greater gains in gross motor functions were seen in younger patients. 142 In contrast, clinical deficits related to underlying 141
143 RFVT2 Riboflavin Therapy neuronal death (sensory ataxia linked to dorsal root ganglionopathy) were irreversible. As expected, all patients received maximal scores for those components of the CMTPedS relying on large fibre sensory function (vibration, balance) and these did not change during the treatment trial. This places responsibility on the treating physician to recognise the early clinical features of BVVL (sensory ataxia and sensorineural hearing loss) and initiate riboflavin therapy as early as possible. There was sustained improvement in the biochemical parameters and no significant side effects were reported. Taken together, this suggests that outcome measures focusing on motor dysfunction and audiology tests monitoring change in hearing may be the most sensitive and relevant to include in future treatment trials. While this was a prospective study, its findings are to some extent limited by the small patient numbers and by the lack of historical data and of an untreated patient group. Most patients with a suggestive clinical phenotype are commenced on riboflavin prior to genetic confirmation, due to the lack of a sensitive biochemical test (the acylcarnitine profile is abnormal in only two-thirds of those affected) and difficulty in accessing genetic testing. It would also be unethical to have a trial with a placebo arm in this ultimately fatal rapidly progressive disorder while a low-cost treatment with no side-effects is available. While this study has shown that therapy with high-dose riboflavin is beneficial in BVVL due to RFVT2 deficiency, a multicentre dose-escalating cohort study including recruiting children recently diagnosed and early in their disease course will be required to better delineate the benefits of riboflavin therapy. 142
144 SUMMARY AND RECOMMENDATIONS 143
145 Summary SUMMARY The studies contained in this thesis have explored the neurophysiological characteristics of a broad group of childhood mitochondrial diseases, and specifically studied the clinical, neurophysiological, biochemical and histopathological profile, pathophysiology and response to disease-modifying therapy in affected children with Brown-Vialetto-Van Laere syndrome due to riboflavin transporter -type 2 (RFVT2) deficiency. These studies demonstrate the enormous progress made over the last three years in understanding this disorder, commencing with the identification of the causative gene mutations and extending onto the evaluation of a disease-modifying therapy. BVVL remains one of the few inherited neuropathies to have an effective treatment. Peripheral neuropathy in childhood mitochondrial disease Peripheral nerve involvement is seen in a number of childhood mitochondrial diseases but is often under-recognised due to the clinical prominence of the signs of central nervous system involvement. The study detailed in Chapter 1 investigated the neurophysiological features of the peripheral neuropathy associated with the common genetic childhood mitochondrial diseases and showed that the results of nerve conduction studies may help direct genetic testing. In those who were investigated as part of the study, a peripheral neuropathy was uncommon with mitochondrial genome mutations causing mitochondrial disease. In contrast, large-fibre peripheral nerve involvement was frequently identified in those with nuclear gene mutations affecting mitochondrial function. Of the nuclear genes associated with Leigh syndrome, SURF1 mutations were associated with a demyelinating neuropathy while PDHc deficiency was associated with an axonal sensorimotor neuropathy. Individuals with POLG mutations 144
146 Summary had a generalised sensory axonal neuropathy with variable motor involvement. Overall, the neuropathy associated with mitochondrial disease was not length-dependent, in contrast to Charcot-Marie-Tooth disease, and clear progression with age or disease duration was not identified in this study. BVVL due to RFVT2 mutations In addition to studying this group of children with varying genetic causes of mitochondrial disease, studies also specifically evaluated a cohort of children with a clinical profile that closely resembled that seen with mitochondrial disease, and who had homozygous and compound heterozygous mutations in SLC52A2, a gene that encodes the transporter responsible for riboflavin transport in the nervous system. This very variable phenotype has been previously described as Brown-Vialetto-Van Laere syndrome (BVVL), and recently mutations in SLC52A2 and SLC52A3, encoding the riboflavin transporters RFVT2 and RFVT3 respectively, have been described in a subset of individuals with BVVL. Riboflavin is a precursor of FMN and FAD, important cofactors for oxidative phosphorylation and energy transport. FAD is also a cofactor for mitochondrial fatty acid β-oxidation and branched-chain amino acid catabolism. Examination of this cohort of children with BVVL due to RFVT2 deficiency revealed a unique and striking phenotype. These children were normal at birth and had normal newborn hearing tests. The most common presenting features were sensory ataxia and hearing loss, which often developed in early childhood. Weakness involving the proximal and distal muscles of the upper limbs was usually seen during the late first decade or early second decade of life and was rapidly progressive, while strength in the lower limbs was relatively preserved, resulting in the child-in-the-barrel phenotype. Bulbar weakness and optic atrophy 145
147 Summary were part of the phenotype and progressive respiratory weakness required assisted ventilation. There was wide variability in the rate of disease progression even within families. Evaluation of the audiological tests of this cohort of children revealed that the hearing loss was due to an auditory neuropathy spectrum disorder (ANSD) with the defect expected to be along the auditory pathway (involving the inner hair cells, cochlear nerve and brainstem cochlear nucleus). Automated brainstem responses were abnormal or absent with a present cochlear microphonic. Unlike the common forms of sensorineural hearing loss, otoacoustic emissions (also derived from outer hair cells like the cochlear microphonic) were usually present. Pure tone audiometry did not always correspond well with the severe impairment in speech perception. Hearing aids did not provide a benefit. Therapy with high-dose riboflavin resulted in improvement in hearing thresholds in those early in the disease course and with recent-onset hearing loss. One child underwent cochlear implantation with a significant improvement in her speech perception. Nerve conduction studies showed that BVVL due to RFVT2 causes both a sensory ganglionopathy of early onset and a progressive motor axonal neuropathy affecting the upper limbs. There was a progressive decline in upper limb motor potential amplitudes in the early second decade of life, paralleling the rapidly progressive upper limb weakness. Examination of sural nerve biopsies shows a chronic axonal neuropathy without evidence of regeneration. The acylcarnitine profile was a useful but not sensitive marker of RFVT2 deficiency with only 60% of our cohort showing a characteristic elevation of medium-chain acylcarnitines. The urine metabolic screen, plasma flavin levels and acylcarnitine profiles on newborn screening cards were all normal. Both FMN and FAD are required for normal mitochondrial 146
148 Summary respiratory chain function. While the other known riboflavin transporters, RFVT3 and RFVT1, are primarily involved in the intestinal absorption of riboflavin, RFVT2 is predominantly expressed in the brain. The SLC52A2 mutations identified in affected individuals either abolish or significantly diminish riboflavin uptake in an in vitro model. Overall, BVVL due to RFVT2 mutations had clinical (multisystemic disease with non-length dependent neuropathy, optic atrophy and auditory neuropathy), biochemical (defect in energy transport) and neuropathological defects that would classify it as a mitochondrial disease. While the metabolic abnormality caused by RFVT2 deficiency has been identified, the pathophysiology of neuronal dysfunction is not yet clearly understood and was hence explored using novel nerve excitability techniques. Abnormalities at baseline, prior to riboflavin therapy, included greater threshold change to depolarisation and hyperpolarisation with a fanning out of the threshold electrotonus, reduction in refractoriness and an increase in superexcitability. Modeling on a mathematical model of a human motor axon suggested that the abnormality was due to an increase in the Barrett and Barrett conductance (GBB), due to an increase in myelin permeability at the paranode. Assessment on the CMT Pediatric scale (CMTPedS) at baseline showed differential motor and sensory involvement. There were poor scores on the subscales that measured sensation, irrespective of disease duration, consistent with the severe generalised sensory ganglionopathy. Scores on subscales that measured strength and motor performance revealed predominantly upper limb weakness consistent with the child-in-the-barrel phenotype. Treatment with high-dose riboflavin for 12 months resulted in a partial normalisation of the axonal excitability findings. Following a 24 month therapeutic course with riboflavin, there were improvements in total CMTPedS score, specifically in the items of grip strength and long jump distance. The youngest patients also demonstrated significant improvement in 147
149 Summary their hearing and older patients showed stabilisation or improvement in their respiratory function, both being aspects in which clinical deterioration was relatively recent. In contrast, clinical deficits related to underlying neuronal death (sensory ataxia linked to dorsal root ganglionopathy) were irreversible. RECOMMENDATIONS ARISING FROM THIS THESIS Nerve conduction tests should be an integral part of the initial evaluation of children with suspected mitochondrial disease. It is essential that physicians and audiologists are made aware of the clinical phenotype of BVVL due to RFVT2 mutations so that appropriate therapy with highdose riboflavin can be commenced early in affected individuals. While elevation of medium-chain acylcarnitines on the acylcarnitine profile is a useful marker for the disorder, it is not a sensitive test, and genetic testing should be considered in those with a suggestive phenotype even if the acylcarnitine profile is normal. The disorder should be considered during the initial screening of those with a new-onset auditory neuropathy or those presenting with sensory ataxia. It is planned that the results detailed in thesis will be disseminated widely by publication in high impact peerreviewed journals and presentation at national and international paediatric neurology conferences. In addition to the Paediatric Neurology and Neurogenetics departments, I have also discussed these findings with our hospital audiology staff and Hearing Loss Clinic staff so that affected children may be identified and treated early. 148
150 Summary There should be a low threshold for genetic testing for riboflavin transporter deficiency and commencing riboflavin. The rapid progress in next-generation sequencing technology has made genetic testing cheaper and more accessible. Previously available only at research laboratories, sequencing of the riboflavin transporter genes SLC52A1, -A2, -A3 is now available on a number of diagnostic multi-gene metabolic and neuromuscular panels, including the latest version of the Neuromuscular (NM) Gene Panel at PathWest in Western Australia. Sequencing for the SLC52A2 p.g306r mutation, a founder mutation in those of Lebanese heritage, is also available through the Molecular Genetics Department at The Children s Hospital at Westmead. It is my practice to sequence affected children of Lebanese ethnicity for this mutation before referring for a multi-gene panel or whole exome sequencing. A list of international laboratories offering sequencing of SLC52A2 is available at (accessed 1 st October 2015). In those with suspected BVVL, consideration should also be given to commencing riboflavin prior to the availability of genetic testing results, especially if sequencing is expected to take time. In addition to therapy with riboflavin, cochlear implantation should be considered early in affected children with ANSD and impaired speech perception. Prior to commencing riboflavin, affected individuals should have a detailed clinical examination including accurate strength and sensory assessments, hearing assessment including an ABR, ophthalmology review, respiratory function testing, nerve conduction tests and evaluation on the CMTPedS scale. The CMTPedS scale is sensitive to the baseline deficits in motor and sensory modalities and is a useful tool in measuring the response to riboflavin. Nerve excitability studies hold promise as an 149
151 Summary effective outcome measure in research studies involving affected children with riboflavin transporter mutations, as well as other anterior horn cell disorders. FUTURE DIRECTIONS The initial presentation of children with riboflavin transporter mutations is with hearing loss or sensory ataxia and hence likely to be to a hearing clinic or paediatric neurologist. The initial assessment of children with a new-onset ANSD does not currently include an acylcarnitine profile. Though the disorder is rare, it is one of the few treatable causes of an ANSD. In addition, the audiological management of children diagnosed with RFVT2 deficiency is likely to change, with those whose speech perception does not respond adequately to riboflavin therapy being considered early for cochlear implantation. It is therefore necessary to determine if testing of all children with a new-onset ANSD using an acylcarnitine profile is a useful way of identifying those very early in the disease course. This will also determine if RFVT2 deficiency can present with infantile or even congenital hearing loss. A prospective evaluation of acylcarnitine profiles in all children diagnosed with ANSD, including those with congenital hearing loss, will provide an answer to these questions. While the benefits of riboflavin therapy are evident, the optimal dose of riboflavin therapy has not yet been determined. Initial doses were extrapolated from trials of riboflavin in migraine (up to 400mg/day), but as shown in my study, doses of 1600mg/day are safe and effective. Because of the progressive nature of BVVL due to RFVT2 deficiency and its poor ultimate outcome, it will not be possible to perform a randomised trial with a placebo arm to accurately determine the benefits of riboflavin therapy or optimum dose. Patient numbers at individual centres are likely to be small and an international randomised dose-escalation study remains the best way to identify the optimum dose of riboflavin in this disorder. 150
152 Summary While progress has been made in understanding the biochemical and pathophysiological basis of BVVL due to riboflavin transporter mutations, including through the use of nerve excitability studies as reported in Chapter 4, the process through which riboflavin affects myelin permeability remains unexplored. Efforts to develop an animal model of this disease (in mice) are ongoing in a number of international centres and offer the best hope of further insights into the pathophysiology of BVVL. Understanding the pathophysiology of motor nerve dysfunction in BVVL would also enhance our understanding of the process of neurodegeneration in other inherited and metabolic neuronopathies. 151
153 REFERENCES 152
154 References 1. McFarland R, Taylor RW, Turnbull DM. A neurological perspective on mitochondrial disease. Lancet Neurol. 2010;9(8): Tuppen HA, Blakely EL, Turnbull DM, Taylor RW. Mitochondrial DNA mutations and human disease. Biochim. Biophys. Acta. 2010;1797(2): Skladal D, Halliday J, Thorburn DR. Minimum birth prevalence of mitochondrial respiratory chain disorders in children. Brain. 2003;126(8): Funalot B. Peripheral neuropathies due to mitochondrial disorders. Rev. Neurol. (Paris). 2009;165(12): Finsterer J. Inherited mitochondrial neuropathies. J. Neurol. Sci. 2011;304(1-2): Colomer J, Iturriaga C, Bestue M, et al. Aspects of neuropathy in mitochondrial diseases. Rev. Neurol. 2000;30(12): Isohanni P, Hakonen AH, Euro L, et al. POLG1 manifestations in childhood. Neurology. 2011;76(9): Darin N, Oldfors A, Moslemi A-R, Holme E, Tulinius M. The incidence of mitochondrial encephalomyopathies in childhood: Clinical features and morphological, biochemical, and DNA abnormalities. Ann. Neurol. 2001;49(3): Ferrari G, Lamantea E, Donati A, et al. Infantile hepatocerebral syndromes associated with mutations in the mitochondrial DNA polymerase-γa. Brain. 2005;128(4): Hakonen AH, Isohanni P, Paetau A, Herva R, Suomalainen A, Lönnqvist T. Recessive Twinkle mutations in early onset encephalopathy with mtdna depletion. Brain. 2007;130(11): Sarzi E, Goffart S, Serre V, et al. Twinkle helicase (PEO1) gene mutation causes mitochondrial DNA depletion. Ann. Neurol. 2007;62(6):
155 References 12. Lönnqvist T, Paetau A, Nikali K, von Boguslawski K, Pihko H. Infantile onset spinocerebellar ataxia with sensory neuropathy (IOSCA): neuropathological features. J. Neurol. Sci. 1998;161(1): Koskinen T, Sainio K, Rapola J, Pihko H, Paetau A. Sensory neuropathy in infantile onset spinocerebellar ataxia (IOSCA). Muscle Nerve. 1994;17(5): Lönnqvist T, Paetau A, Valanne L, Pihko H. Recessive twinkle mutations cause severe epileptic encephalopathy. Brain. 2009;132(6): Dimmock DP, Zhang Q, Dionisi-Vici C, et al. Clinical and molecular features of mitochondrial DNA depletion due to mutations in deoxyguanosine kinase. Hum. Mutat. 2008;29(2): Appenzeller O, Kornfeld M, Snyder R. Acromutilating, Paralyzing Neuropathy With Corneal Ulceration in Navajo Children. Arch. Neurol. 1976;33(11): Karadimas CL, Vu TH, Holve SA, et al. Navajo Neurohepatopathy Is Caused by a Mutation in the MPV17 Gene. Am. J. Hum. Genet. 2006;79(3): Spinazzola A, Santer R, Akman OH, et al. Hepatocerebral Form of Mitochondrial DNA Depletion Syndrome: Novel MPV17 Mutations. Arch. Neurol. 2008;65(8): Ostergaard E. Disorders caused by deficiency of succinate-coa ligase. J. Inherit. Metab. Dis. 2008;31(2): Carrozzo R, Dionisi-Vici C, Steuerwald U, et al. SUCLA2 mutations are associated with mild methylmalonic aciduria, Leigh-like encephalomyopathy, dystonia and deafness. Brain. 2007;130(3): Morava E, Steuerwald U, Carrozzo R, et al. Dystonia and deafness due to SUCLA2 defect; Clinical course and biochemical markers in 16 children. Mitochondrion. 2009;9(6):
156 References 22. den Boer MEJ, Dionisi-Vici C, Chakrapani A, van Thuijl AOJ, Wanders RJA, Wijburg FA. Mitochondrial trifunctional protein deficiency: A severe fatty acid oxidation disorder with cardiac and neurologic involvement. J. Pediatr. 2003;142(6): Spiekerkoetter U, Bennett MJ, Ben-Zeev B, Strauss AW, Tein I. Peripheral neuropathy, episodic myoglobinuria, and respiratory failure in deficiency of the mitochondrial trifunctional protein. Muscle Nerve. 2004;29(1): Finsterer J. Leigh and Leigh-Like Syndrome in Children and Adults. Pediatr. Neurol. 2008;39(4): Barkovich AJ. An approach to MRI of metabolic disorders in children. J. Neuroradiol. 2007;34(2): Saneto RP, Friedman SD, Shaw DW. Neuroimaging of mitochondrial disease. Mitochondrion. 2008;8(5-6): Thorburn DR, Rahman S. Mitochondrial DNA-Associated Leigh Syndrome and NARP In: Pagon RA, Bird TD, Dolan CR, Stephens K, eds. GeneReviews. Seattle (WA): University of Washington, Seattle; 2003 (updated 2011). 28. Bruno C, Biancheri R, Garavaglia B, et al. A Novel Mutation in the SURF1 Gene in a Child With Leigh Disease, Peripheral Neuropathy, and Cytochrome-c Oxidase Deficiency. J. Child Neurol. 2002;17(3): Santoro L, Carrozzo R, Malandrini A, et al. A novel SURF1 mutation results in Leigh syndrome with peripheral neuropathy caused by cytochrome c oxidase deficiency. Neuromuscul. Disord. 2000;10(6): Fujii T, Hattori H, Higuchi Y, Tsuji M, Mitsuyoshi I. Phenotypic Differences Between T C and T G Mutations at nt 8993 of Mitochondrial DNA in Leigh Syndrome. Pediatr. Neurol. 1998;18(3):
157 References 31. Holt IJ, Harding AE, Petty RK, Morgan-Hughes JA. A new mitochondrial disease associated with mitochondrial DNA heteroplasmy. Am. J. Hum. Genet. 1990;46(3): Rojo A, Campos Y, Sánchez J, et al. NARP-MILS syndrome caused by 8993 T > G mitochondrial DNA mutation: a clinical, genetic and neuropathological study. Acta Neuropathol. (Berl.). 2006;111(6): Castagna AE, Addis J, McInnes RR, et al. Late onset Leigh syndrome and ataxia due to a T to C mutation at bp 9,185 of mitochondrial DNA. Am. J. Med. Genet. A. 2007;143A(8): Craig K, Elliott HR, Keers SM, et al. Episodic ataxia and hemiplegia caused by the 8993T->C mitochondrial DNA mutation. J. Med. Genet. 2007;44(12): de Vries DD, van Engelen BG, Gabreels FJ, Ruitenbeek W, van Oost BA. A second missense mutation in the mitochondrial ATPase 6 gene in Leigh's syndrome. Ann. Neurol. 1993;34(3): van Erven PM, Gabreels FJ, Ruitenbeek W, Renier WO, Lamers KJ, Sloof JL. Familial Leigh's syndrome: association with a defect in oxidative metabolism probably restricted to brain. J. Neurol. 1987;234(4): Childs AM, Hutchin T, Pysden K, et al. Variable phenotype including Leigh syndrome with a 9185T>C mutation in the MTATP6 gene. Neuropediatrics. 2007;38(6): Patel KP, O'Brien TW, Subramony SH, Shuster J, Stacpoole PW. The spectrum of pyruvate dehydrogenase complex deficiency: Clinical, biochemical and genetic features in 371 patients. Mol. Genet. Metab. 2012;105(1):
158 References 39. Di Rocco M, Doria Lamba L, Minniti G, Caruso U, Naito E. Outcome of thiamine treatment in a child with Leigh disease due to thiamine-responsive pyruvate dehydrogenase deficiency. Eur. J. Paediatr. Neurol. 2000;4(3): Bonne G, Benelli C, De Meirleir L, et al. E1 pyruvate dehydrogenase deficiency in a child with motor neuropathy. Pediatr. Res. 1993;33(3): Chabrol B, Mancini J, Benelli C, Gire C, Munnich A. Leigh syndrome: pyruvate dehydrogenase defect. A case with peripheral neuropathy. J. Child Neurol. 1994;9(1): Federico A, Dotti MT, Fabrizi GM, et al. Congenital lactic acidosis due to a defect of pyruvate dehydrogenase complex (E1). Clinical, biochemical, nerve biopsy study and effect of therapy. Eur. Neurol. 1990;30(3): Marsac C, Benelli C, Desguerre I, et al. Biochemical and genetic studies of four patients with pyruvate dehydrogenase E1 alpha deficiency. Hum. Genet. 1997;99(6): Sedel F, Challe G, Mayer JM, et al. Thiamine responsive pyruvate dehydrogenase deficiency in an adult with peripheral neuropathy and optic neuropathy. J. Neurol. Neurosurg. Psychiatry. 2008;79(7): Stickler DE, Carney PR, Valenstein ER. Juvenile-onset Leigh syndrome with an acute polyneuropathy at presentation. J. Child Neurol. 2003;18(8): Miyake N, Yamashita S, Kurosawa K, et al. A novel homozygous mutation of DARS2 may cause a severe LBSL variant. Clin. Genet. 2011;80(3): Scheper GC, van der Klok T, van Andel RJ, et al. Mitochondrial aspartyl-trna synthetase deficiency causes leukoencephalopathy with brain stem and spinal cord involvement and lactate elevation. Nat. Genet. 2007;39(4):
159 References 48. Isohanni P, Linnankivi T, Buzkova J, et al. DARS2 mutations in mitochondrial leucoencephalopathy and multiple sclerosis. J. Med. Genet. 2010;47(1): Tavora DGF, Nakayama M, Gama RL, Alvim TCdL, Portugal D, Comerlato EA. Leukoencephalopathy with brainstem and spinal cord involvement and high brain lactate: report of three Brazilian patients. Arq. Neuropsiquiatr. 2007;65(2B): McLaughlin HM, Sakaguchi R, Liu C, et al. Compound heterozygosity for loss-offunction lysyl-trna synthetase mutations in a patient with peripheral neuropathy. Am. J. Hum. Genet. 2010;87(4): Eymard B, Penicaud A, Leger JM, et al. Clinical and electrophysiologic study of the peripheral nerve in 28 cases of mitochondrial disease. Rev. Neurol. (Paris). 1991;147(6-7): Zanssen S, Molnar M, Buse G, Schroder JM. Mitochondrial cytochrome b gene deletion in Kearns-Sayre syndrome associated with a subclinical type of peripheral neuropathy. Clin. Neuropathol. 1998;17(6): McDonald DG, McMenamin JB, Farrell MA, Droogan O, Green AJ. Familial childhood onset neuropathy and cirrhosis with the 4977bp mitochondrial DNA deletion. Am. J. Med. Genet. 2002;111(2): Molnar M, Neudecker S, Schroder JM. Increase of mitochondria in vasa nervorum of cases with mitochondrial myopathy, Kearns-Sayre syndrome, progressive external ophthalmoplegia and MELAS. Neuropathol. Appl. Neurobiol. 1995;21(5): Schroder JM. Neuropathy associated with mitochondrial disorders. Brain Pathol. 1993;3(2): Kärppä M, Syrjälä P, Tolonen U, Majamaa K. Peripheral neuropathy in patients with the 3243A>G mutation in mitochondrial DNA. J. Neurol. 2003;250(2):
160 References 57. Kaufmann P, Engelstad K, Wei Y, et al. Dichloroacetate causes toxic neuropathy in MELAS: a randomized, controlled clinical trial. Neurology. 2006;66(3): Stickler DE, Valenstein E, Neiberger RE, et al. Peripheral Neuropathy in Genetic Mitochondrial Diseases. Pediatr. Neurol. 2006;34(2): Bouillot S, Martin-Negrier ML, Vital A, et al. Peripheral neuropathy associated with mitochondrial disorders: 8 cases and review of the literature. J. Peripher. Nerv. Syst. 2002;7(4): Erol I, Alehan F, Horvath R, Schneiderat P, Talim B. Demyelinating disease of central and peripheral nervous systems associated with a A8344G mutation in trnalys. Neuromuscul. Disord. 2009;19(4): Wong LJ, Naviaux RK, Brunetti-Pierri N, et al. Molecular and clinical genetics of mitochondrial diseases due to POLG mutations. Hum. Mutat. 2008;29(9):E Longley MJ, Nguyen D, Kunkel TA, Copeland WC. The Fidelity of Human DNA Polymerase γ with and without Exonucleolytic Proofreading and the p55 Accessory Subunit. J. Biol. Chem. 2001;276(42): Tzoulis C, Engelsen BA, Telstad W, et al. The spectrum of clinical disease caused by the A467T and W748S POLG mutations: a study of 26 cases. Brain. 2006;129(7): Van Goethem G, Martin JJ, Dermaut B, et al. Recessive POLG mutations presenting with sensory and ataxic neuropathy in compound heterozygote patients with progressive external ophthalmoplegia. Neuromuscul. Disord. 2003;13(2): Van Goethem G, Mercelis R, Lofgren A, et al. Patient homozygous for a recessive POLG mutation presents with features of MERRF. Neurology. 2003;61(12):
161 References 66. Horvath R, Hudson G, Ferrari G, et al. Phenotypic spectrum associated with mutations of the mitochondrial polymerase gene. Brain. 2006;129(7): Stewart JD, Tennant S, Powell H, et al. Novel POLG1 mutations associated with neuromuscular and liver phenotypes in adults and children. J. Med. Genet. 2009;46(3): Hirano M, Garcia-de-Yebenes J, Jones AC, et al. Mitochondrial Neurogastrointestinal Encephalomyopathy Syndrome Maps to Chromosome 22q13.32-qter. Am. J. Hum. Genet. 1998;63(2): Nishino I, Spinazzola A, Hirano M. Thymidine Phosphorylase Gene Mutations in MNGIE, a Human Mitochondrial Disorder. Science. 1999;283(5402): Papadimitriou A, Comi G, Hadjigeorgiou G, et al. Partial depletion and multiple deletions of muscle mtdna in familial MNGIE syndrome. Neurology. 1998;51(4): Hirano M, Nishigaki Y, Marti R. Mitochondrial neurogastrointestinal encephalomyopathy (MNGIE): a disease of two genomes. Neurologist. 2004;10(1): Shaibani A, Shchelochkov OA, Zhang S, et al. Mitochondrial Neurogastrointestinal Encephalopathy Due to Mutations in RRM2B. Arch. Neurol. 2009;66(8): Bedlack RS, Vu T, Hammans S, et al. MNGIE neuropathy: Five cases mimicking chronic inflammatory demyelinating polyneuropathy. Muscle Nerve. 2004;29(3): Simon LT, Horoupian DS, Dorfman LJ, et al. Polyneuropathy, ophthalmoplegia, leukoencephalopathy, and intestinal pseudo-obstruction: POLIP syndrome. Ann. Neurol. 1990;28(3):
162 References 75. Perez-Atayde AR, Fox V, Teitelbaum JE, et al. Mitochondrial neurogastrointestinal encephalomyopathy: diagnosis by rectal biopsy. Am. J. Surg. Pathol. 1998;22(9): Hirano M, Marti R, Casali C, et al. Allogeneic stem cell transplantation corrects biochemical derangements in MNGIE. Neurology. 2006;67(8): Halter J, Schupbach WMM, Casali C, et al. Allogeneic hematopoietic SCT as treatment option for patients with mitochondrial neurogastrointestinal encephalomyopathy (MNGIE): a consensus conference proposal for a standardized approach. Bone Marrow Transplant. 2011;46(3): Yu-Wai-Man P, Griffiths PG, Hudson G, Chinnery PF. Inherited mitochondrial optic neuropathies. J. Med. Genet. 2009;46(3): Gilhuis HJ, Schelhaas HJ, Cruysberg JRM, Zwarts MJ. Demyelinating polyneuropathy in Leber hereditary optic neuropathy. Neuromuscul. Disord. 2006;16(6): Nikoskelainen EK, Marttila RJ, Huoponen K, et al. Leber's "plus": neurological abnormalities in patients with Leber's hereditary optic neuropathy. J. Neurol. Neurosurg. Psychiatry. 1995;59(2): Alexander C, Votruba M, Pesch UEA, et al. OPA1, encoding a dynamin-related GTPase, is mutated in autosomal dominant optic atrophy linked to chromosome 3q28. Nat. Genet. 2000;26(2): Yu-Wai-Man P, Griffiths PG, Gorman GS, et al. Multi-system neurological disease is common in patients with OPA1 mutations. Brain. 2010;133(3): Pandolfo M, Pastore A. The pathogenesis of Friedreich ataxia and the structure and function of frataxin. J. Neurol. 2009;256(1): Pandolfo M. Friedreich ataxia: the clinical picture. J. Neurol. 2009;256 Suppl 1:
163 References 85. Ouvrier RA, McLeod JG, Conchin TE. Friedreich's ataxia: Early detection and progression of peripheral nerve abnormalities. J. Neurol. Sci. 1982;55(2): Santoro L, De Michele G, Perretti A, et al. Relation between trinucleotide GAA repeat length and sensory neuropathy in Friedreich s ataxia. J. Neurol. Neurosurg. Psychiatry. 1999;66(1): Song Z, Ghochani M, McCaffery JM, Frey TG, Chan DC. Mitofusins and OPA1 mediate sequential steps in mitochondrial membrane fusion. Mol. Biol. Cell. 2009;20(15): Chen H, Detmer SA, Ewald AJ, Griffin EE, Fraser SE, Chan DC. Mitofusins Mfn1 and Mfn2 coordinately regulate mitochondrial fusion and are essential for embryonic development. J. Cell Biol. 2003;160(2): Züchner S, Mersiyanova IV, Muglia M, et al. Mutations in the mitochondrial GTPase mitofusin 2 cause Charcot-Marie-Tooth neuropathy type 2A. Nat. Genet. 2004;36(5): Züchner S, Jonghe PD, Jordanova A, et al. Axonal neuropathy with optic atrophy is caused by mutations in mitofusin 2. Ann. Neurol. 2006;59(2): Vallat J-M, Ouvrier RA, Pollard JD, et al. Histopathological findings in hereditary motor and sensory neuropathy of axonal type with onset in early childhood associated with mitofusin 2 mutations. J. Neuropathol. Exp. Neurol. 2008;67(11): Nelis E, Erdem S, Van Den Bergh PY, et al. Mutations in GDAP1: autosomal recessive CMT with demyelination and axonopathy. Neurology. 2002;59(12): Claramunt R, Pedrola L, Sevilla T, et al. Genetics of Charcot-Marie-Tooth disease type 4A: mutations, inheritance, phenotypic variability, and founder effect. J. Med. Genet. 2005;42(4):
164 References 94. Cassereau J, Chevrollier A, Gueguen N, et al. Mitochondrial complex I deficiency in GDAP1-related autosomal dominant Charcot-Marie-Tooth disease (CMT2K). Neurogenetics. 2009;10(2): Niemann A, Ruegg M, La Padula V, Schenone A, Suter U. Ganglioside-induced differentiation associated protein 1 is a regulator of the mitochondrial network: new implications for Charcot-Marie-Tooth disease. J. Cell Biol. 2005;170(7): Burns J, Ouvrier R, Estilow T, et al. Validation of the Charcot-Marie-Tooth disease pediatric scale as an outcome measure of disability. Ann. Neurol. 2012;71(5): Burns J, Menezes MP, Shy R, et al. Two year natural history of disease progression in childhood Charcot-Marie-Tooth disease. J. Peripher. Nerv. Syst. 2013;18: Pitceathly RD, Murphy SM, Cottenie E, et al. Genetic dysfunction of MT-ATP6 causes axonal Charcot-Marie-Tooth disease. Neurology. 2012;79(11): Echaniz-Laguna A, Ghezzi D, Chassagne M, et al. SURF1 deficiency causes demyelinating Charcot-Marie-Tooth disease. Neurology. 2013;81(17): Cai F, Zhang J. Study of nerve conduction and late responses in normal Chinese infants, children, and adults. J. Child Neurol. 1997;12(1): Koga Y, Povalko N, Katayama K, et al. Beneficial effect of pyruvate therapy on Leigh syndrome due to a novel mutation in PDH E1alpha gene. Brain Dev. 2012;34(2): Wedatilake Y, Brown RM, McFarland R, et al. SURF1 deficiency: a multi-centre natural history study. Orphanet J. Rare Dis. 2013;8: Hakonen AH, Heiskanen S, Juvonen V, et al. Mitochondrial DNA polymerase W748S mutation: a common cause of autosomal recessive ataxia with ancient European origin. Am. J. Hum. Genet. 2005;77(3):
165 References 104. Schulte C, Synofzik M, Gasser T, Schols L. Ataxia with ophthalmoplegia or sensory neuropathy is frequently caused by POLG mutations. Neurology. 2009;73(11): Neeve VC, Samuels DC, Bindoff LA, et al. What is influencing the phenotype of the common homozygous polymerase-gamma mutation p.ala467thr? Brain. 2012;135(Pt 12): Synofzik M, Srulijes K, Godau J, Berg D, Schols L. Characterizing POLG ataxia: clinics, electrophysiology and imaging. Cerebellum. 2012;11(4): Kaufmann P, Pascual JM, Anziska Y, et al. Nerve Conduction Abnormalities in Patients With MELAS and the A3243G Mutation. Arch. Neurol. 2006;63(5): Makino M, Horai S, Goto Y, Nonaka I. Confirmation that a T-to-C mutation at 9176 in mitochondrial DNA is an additional candidate mutation for Leigh's syndrome. Neuromuscul. Disord. 1998;8(3-4): Rahman S, Blok RB, Dahl HH, et al. Leigh syndrome: clinical features and biochemical and DNA abnormalities. Ann. Neurol. 1996;39(3): Garone C, Tadesse S, Hirano M. Clinical and genetic spectrum of mitochondrial neurogastrointestinal encephalomyopathy. Brain. 2011;134(Pt 11): Horga A, Pitceathly RD, Blake JC, et al. Peripheral neuropathy predicts nuclear gene defect in patients with mitochondrial ophthalmoplegia. Brain. 2014;137(Pt 12): Kamholz J, Menichella D, Jani A, et al. Charcot-Marie-Tooth disease type 1: molecular pathogenesis to gene therapy. Brain. 2000;123 ( Pt 2): Krajewski KM, Lewis RA, Fuerst DR, et al. Neurological dysfunction and axonal degeneration in Charcot-Marie-Tooth disease type 1A. Brain. 2000;123 ( Pt 7):
166 References 114. Scherer S. Axonal pathology in demyelinating diseases. Ann. Neurol. 1999;45(1): Coker SB. Leigh disease presenting as Guillain-Barre syndrome. Pediatr. Neurol. 1993;9(1): Hara H, Wakayama Y, Kouno Y, Yamada H, Tanaka M, Ozawa T. Acute peripheral neuropathy, rhabdomyolysis, and severe lactic acidosis associated with 3243 A to G mitochondrial DNA mutation. J. Neurol. Neurosurg. Psychiatry. 1994;57(12): Aure K, Dubourg O, Jardel C, et al. Episodic weakness due to mitochondrial DNA MT-ATP6/8 mutations. Neurology. 2013;81(21): Debray FG, Lambert M, Vanasse M, et al. Intermittent peripheral weakness as the presenting feature of pyruvate dehydrogenase deficiency. Eur. J. Pediatr. 2006;165(7): Sheng ZH. Mitochondrial trafficking and anchoring in neurons: New insight and implications. J. Cell Biol. 2014;204(7): Misko A, Jiang S, Wegorzewska I, Milbrandt J, Baloh RH. Mitofusin 2 is necessary for transport of axonal mitochondria and interacts with the Miro/Milton complex. J. Neurosci. 2010;30(12): Hollenbeck PJ, Saxton WM. The axonal transport of mitochondria. J. Cell Sci. 2005;118(Pt 23): Sheng ZH, Cai Q. Mitochondrial transport in neurons: impact on synaptic homeostasis and neurodegeneration. Nat. Rev. Neurosci. 2012;13(2): Farrar MA, Lin CS, Krishnan AV, Park SB, Andrews PI, Kiernan MC. Acute, reversible axonal energy failure during stroke-like episodes in MELAS. Pediatrics. 2010;126(3):e
167 References 124. Ng K, Winter S, Sue C, Burke D. Preserved motor axonal membrane potential in mitochondrial disease. J. Neurol. Neurosurg. Psychiatry. 2010;81(8): Viader A, Sasaki Y, Kim S, et al. Aberrant Schwann cell lipid metabolism linked to mitochondrial deficits leads to axon degeneration and neuropathy. Neuron. 2013;77(5): Brown CH. Infantile amyotrophic lateral sclerosis of the family. Journal of the Nervous and Mental Disease. 1894;19(11): Vialetto E. Contributo alia forma ereditaria della paralisi bulbare progressiva. Riv. sper. Freniat. 1936;40: Van Laere JE. A new case of chronic progressive bulbo-pontine paralysis and deafness. Rev. Neurol. (Paris). 1977;133(2): Piccolo G, Marchioni E, Maurelli M, Simonetti F, Bizzetti F, Savoldi F. Recovery from respiratory muscle failure in a sporadic case of Brown-Vialetto-Van Laere syndrome with unusually late onset. J. Neurol. 1992;239(6): Gallai V, Hockaday JM, Hughes JT, Lane DJ, Oppenheimer DR, Rushworth G. Ponto-bulbar palsy with deafness (Brown-Vialetto-Van Laere syndrome). J. Neurol. Sci. 1981;50(2): Sathasivam S. Brown-Vialetto-Van Laere syndrome. Orphanet J. Rare Dis. 2008;3: Sathasivam S, O'Sullivan S, Nicolson A, Tilley PJ, Shaw PJ. Brown-Vialetto-Van Laere syndrome: case report and literature review. Amyotroph. Lateral Scler. Other Motor Neuron Disord. 2000;1(4): Dipti S, Childs AM, Livingston JH, et al. Brown-Vialetto-Van Laere syndrome; variability in age at onset and disease progression highlighting the phenotypic overlap with Fazio-Londe disease. Brain Dev. 2005;27(6):
168 References 134. Cremers CW, Ter Haar BG, Van Rens TJ. The Nathalie syndrome. A new hereditary syndrome. Clin. Genet. 1975;8(5): Boltshauser E, Lang W, Spillmann T, Hof E. Hereditary distal muscular atrophy with vocal cord paralysis and sensorineural hearing loss: a dominant form of spinal muscular atrophy? J. Med. Genet. 1989;26(2): Nalini A, Thennarasu K, Yamini BK, Shivashankar D, Krishna N. Madras motor neuron disease (MMND): clinical description and survival pattern of 116 patients from Southern India seen over 36 years ( ). J. Neurol. Sci. 2008;269(1-2): Megarbane A, Desguerres I, Rizkallah E, et al. Brown-Vialetto-Van Laere syndrome in a large inbred Lebanese family: confirmation of autosomal recessive inheritance? Am. J. Med. Genet. 2000;92(2): Hawkins SA, Nevin NC, Harding AE. Pontobulbar palsy and neurosensory deafness (Brown-Vialetto-Van Laere syndrome) with possible autosomal dominant inheritance. J. Med. Genet. 1990;27(3): Francis DA, Ponsford JR, Wiles CM, Thomas PK, Duchen LW. Brown-Vialetto-Van Laere Syndrome. Neuropathol. Appl. Neurobiol. 1993;19(1): Johnson JO, Gibbs JR, Megarbane A, et al. Exome sequencing reveals riboflavin transporter mutations as a cause of motor neuron disease. Brain. 2012;135: Green P, Wiseman M, Crow YJ, et al. Brown-Vialetto-Van Laere syndrome, a pontobulbar palsy with deafness, is caused by mutations in c20orf54. Am. J. Hum. Genet. 2010;86(3): Foley AR, Menezes MP, Pandraud A, et al. Treatable childhood neuronopathy caused by mutations in riboflavin transporter RFVT2. Brain. 2014;137:
169 References 143. Haack TB, Makowski C, Yao Y, et al. Impaired riboflavin transport due to missense mutations in SLC52A2 causes Brown-Vialetto-Van Laere syndrome. J. Inherit. Metab. Dis. 2012;35(6): Lindh J, Tondel M, Osterberg A, Vrethem M. Cryptogenic polyneuropathy: clinical and neurophysiological findings. J. Peripher. Nerv. Syst. 2005;10(1): Koul R, Jain R, Chacko A, Alfutaisi A, Hashim J, Chacko J. Pontobulbar palsy and neurosensory deafness (Brown-Vialetto-van Laere syndrome) with hyperintense brainstem nuclei on magnetic resonance imaging: new finding in three siblings. J. Child Neurol. 2006;21(6): Malheiros JA, Camargos ST, Oliveira JT, Cardoso FE. A Brazilian family with Brown-Vialetto-van Laere syndrome with autosomal recessive inheritance. Arq. Neuropsiquiatr. 2007;65(1): Koc AF, Bozdemir H, Sarica Y. Mental retardation associated with Brown-Vialetto- Van Laere syndrome. Amyotroph. Lateral Scler. Other Motor Neuron Disord. 2003;4(1): Ho G, Yonezawa A, Masuda S, et al. Maternal riboflavin deficiency, resulting in transient neonatal-onset glutaric aciduria Type 2, is caused by a microdeletion in the riboflavin transporter gene GPR172B. Hum. Mutat. 2011;32(1):E Orsini M, Catharino AM, Catharino FM, et al. Man-in-the-barrel syndrome, a symmetrical proximal brachial amyotrophic diplegia related to motor neuron diseases: a survey of nine cases. Rev. Assoc. Med. Bras. 2009;55(6): Rousseff RT, Khuraibet AJ, Neubauer D. The "Child in the Barrel syndrome"--severe pharyngeal-cervical-brachial variant of Guillain-Barre Syndrome in a toddler. Neuropediatrics. 2008;39(6):
170 References 151. Bosch AM, Stroek K, Abeling NG, Waterham HR, Ijlst L, Wanders RJ. The Brown- Vialetto-Van Laere and Fazio Londe syndrome revisited: natural history, genetics, treatment and future perspectives. Orphanet J. Rare Dis. 2012;7: Bosch AM, Abeling NG, Ijlst L, et al. Brown-Vialetto-Van Laere and Fazio Londe syndrome is associated with a riboflavin transporter defect mimicking mild MADD: a new inborn error of metabolism with potential treatment. J. Inherit. Metab. Dis. 2011;34(1): Yao Y, Yonezawa A, Yoshimatsu H, Masuda S, Katsura T, Inui K-i. Identification and Comparative Functional Characterization of a New Human Riboflavin Transporter hrft3 Expressed in the Brain1-3. J. Nutr. 2010;140(7): Yonezawa A, Inui K. Novel riboflavin transporter family RFVT/SLC52: Identification, nomenclature, functional characterization and genetic diseases of RFVT/SLC52. Mol. Aspects Med. 2013;34(2 3): Londe P. Paralysie bulbaire progressive, infantile et familiale. Rev. Med. 1894;14: Fazio M. Ereditarietá della paralisi bulbare progressiva. Riforma Med. 1892;8: Pareyson D, Marchesi C. Diagnosis, natural history, and management of Charcot- Marie-Tooth disease. Lancet Neurol. 2009;8(7): McShane MA, Boyd S, Harding B, Brett EM, Wilson J. Progressive bulbar paralysis of childhood. A reappraisal of Fazio-Londe disease. Brain. 1992;115 ( Pt 6): Nalini A, Pandraud A, Mok K, Houlden H. Madras motor neuron disease (MMND) is distinct from the riboflavin transporter genetic defects that cause Brown-Vialetto-Van Laere syndrome. J. Neurol. Sci. 2013;334(1-2):
171 References 160. Gregersen N, Andresen BS, Pedersen CB, Olsen RK, Corydon TJ, Bross P. Mitochondrial fatty acid oxidation defects--remaining challenges. J. Inherit. Metab. Dis. 2008;31(5): Manchaiah VK, Zhao F, Danesh AA, Duprey R. The genetic basis of auditory neuropathy spectrum disorder (ANSD). Int. J. Pediatr. Otorhinolaryngol. 2011;75(2): Kirkim G, Serbetcioglu B, Erdag TK, Ceryan K. The frequency of auditory neuropathy detected by universal newborn hearing screening program. Int. J. Pediatr. Otorhinolaryngol. 2008;72(10): Sanyelbhaa Talaat H, Kabel AH, Samy H, Elbadry M. Prevalence of auditory neuropathy (AN) among infants and young children with severe to profound hearing loss. Int. J. Pediatr. Otorhinolaryngol. 2009;73(7): Starr A, Picton TW, Sininger Y, Hood LJ, Berlin CI. Auditory neuropathy. Brain. 1996; Rance G, McKay C, Grayden D. Perceptual characterization of children with auditory neuropathy. Ear Hear. 2004;25(1): Starr A, Sininger YS, Pratt H. The varieties of auditory neuropathy. J. Basic Clin. Physiol. Pharmacol. 2000;11(3): Rance G, Barker E, Mok M, Dowell R, Rincon A, Garratt R. Speech perception in noise for children with auditory neuropathy/dys-synchrony type hearing loss. Ear Hear. 2007;28(3): Zeng FG, Oba S, Garde S, Sininger Y, Starr A. Temporal and speech processing deficits in auditory neuropathy. Neuroreport. 1999;10(16): Zeng FG, Kong YY, Michalewski HJ, Starr A. Perceptual consequences of disrupted auditory nerve activity. J. Neurophysiol. 2005;93(6):
172 References 170. Carhart R, Jerger JF. Preferred Method For Clinical Determination Of Pure-Tone Thresholds. J. Speech Hear. Disord. 1959;24(4): Hood JD. The principles and practice of bone conduction audiometry: A review of the present position. Laryngoscope. 1960;70: Ciccolella M, Corti S, Catteruccia M, et al. Riboflavin transporter 3 involvement in infantile Brown-Vialetto-Van Laere disease: two novel mutations. J. Med. Genet. 2013;50(2): Srour M, Putorti ML, Schwartzentruber J, et al. Mutations in riboflavin transporter present with severe sensory loss and deafness in childhood. Muscle Nerve. 2014;50(5): Vlastarakos PV, Nikolopoulos TP, Tavoulari E, Papacharalambous G, Korres S. Auditory neuropathy: endocochlear lesion or temporal processing impairment? Implications for diagnosis and management. Int. J. Pediatr. Otorhinolaryngol. 2008;72(8): Uhler K, Heringer A, Thompson N, Yoshinaga-Itano C. A tutorial on auditory neuropathy/dyssynchrony for the speech-language pathologist and audiologist. Semin. Speech Lang. 2012;33(4): Dowley AC, Whitehouse WP, Mason SM, Cope Y, Grant J, Gibbin KP. Auditory neuropathy: unexpectedly common in a screened newborn population. Dev. Med. Child Neurol. 2009;51(8): Rance G, Ryan MM, Bayliss K, Gill K, O'Sullivan C, Whitechurch M. Auditory function in children with Charcot-Marie-Tooth disease. Brain. 2012;135(Pt 5): Rance G, Corben L, Delatycki M. Auditory processing deficits in children with Friedreich ataxia. J. Child Neurol. 2012;27(9):
173 References 179. Santarelli R, Rossi R, Scimemi P, et al. OPA1-related auditory neuropathy: site of lesion and outcome of cochlear implantation. Brain. 2015;138: Oysu C, Aslan I, Basaran B, Baserer N. The site of the hearing loss in Refsum's disease. Int. J. Pediatr. Otorhinolaryngol. 2001;61(2): Bamiou DE, Spraggs PR, Gibberd FB, Sidey MC, Luxon LM. Hearing loss in adult Refsum's disease. Clin. Otolaryngol. Allied. Sci. 2003;28(3): de Brouwer AP, Williams KL, Duley JA, et al. Arts syndrome is caused by loss-offunction mutations in PRPS1. Am. J. Hum. Genet. 2007;81(3): Ćeranić B, Luxon LM. Progressive auditory neuropathy in patients with Leber s hereditary optic neuropathy. J. Neurol. Neurosurg. Psychiatry. 2004;75(4): Forli F, Mancuso M, Santoro A, Dotti MT, Siciliano G, Berrettini S. Auditory neuropathy in a patient with mitochondrial myopathy and multiple mtdna deletions. J. Laryngol. Otol. 2006;120(10): Sanyelbhaa Talaat H, Khalil LH, Khafagy AH, Alkandari MM, Zein AM. Persistence of otoacoustic emissions in children with auditory neuropathy spectrum disorders. Int. J. Pediatr. Otorhinolaryngol. 2013;77(5): Berlin CI, Morlet T, Hood LJ. Auditory neuropathy/dyssynchrony: its diagnosis and management. Pediatr. Clin. North Am. 2003;50(2): Berlin CI, Hood LJ, Morlet T, et al. Multi-site diagnosis and management of 260 patients with Auditory Neuropathy/Dys-synchrony (Auditory Neuropathy Spectrum Disorder*). Int. J. Audiol. 2010;49(1): Breneman AI, Gifford RH, Dejong MD. Cochlear implantation in children with auditory neuropathy spectrum disorder: long-term outcomes. J. Am. Acad. Audiol. 2012;23(1):
174 References 189. Humphriss R, Hall A, Maddocks J, Macleod J, Sawaya K, Midgley E. Does cochlear implantation improve speech recognition in children with auditory neuropathy spectrum disorder? A systematic review. Int. J. Audiol. 2013;52(7): Dean C, Felder G, Kim AH. Analysis of speech perception outcomes among patients receiving cochlear implants with auditory neuropathy spectrum disorder. Otol. Neurotol. 2013;34(9): Pelosi S, Wanna G, Hayes C, et al. Cochlear implantation versus hearing amplification in patients with auditory neuropathy spectrum disorder. Otolaryngol. Head Neck Surg. 2013;148(5): Kontorinis G, Lloyd SK, Henderson L, et al. Cochlear implantation in children with auditory neuropathy spectrum disorders. Cochlear Implants Int. 2014;15 Suppl 1:S Carvalho AC, Bevilacqua MC, Sameshima K, Costa Filho OA. Auditory neuropathy/auditory dyssynchrony in children with Cochlear Implants. Braz. J. Otorhinolaryngol. 2011;77(4): May-Mederake B. Early intervention and assessment of speech and language development in young children with cochlear implants. Int. J. Pediatr. Otorhinolaryngol. 2012;76(7): McConkey Robbins A, Koch DB, Osberger MJ, Zimmerman-Phillips S, Kishon- Rabin L. Effect of age at cochlear implantation on auditory skill development in infants and toddlers. Arch. Otolaryngol. Head Neck Surg. 2004;130(5): Liu Y, Dong R, Li Y, Xu T, Chen X, Gong S. Effect of age at cochlear implantation on auditory and speech development of children with auditory neuropathy spectrum disorder. Auris. Nasus. Larynx. 2014;41(6):
175 References 197. Ching TY, Dillon H. Major findings of the LOCHI study on children at 3 years of age and implications for audiological management. Int. J. Audiol. 2013;52 Suppl 2:S Sinnathuray AR, Watson DR, Fruhstorfer B, Olarte JR, Toner JG. Cochlear Implantation in Brown-Vialetto-Van-Laere syndrome. J. Laryngol. Otol. 2011;125(3): Farrar MA, Vucic S, Lin CS, et al. Dysfunction of axonal membrane conductances in adolescents and young adults with spinal muscular atrophy. Brain. 2011;134(Pt 11): Kiernan MC, Burke D, Andersen KV, Bostock H. Multiple measures of axonal excitability: a new approach in clinical testing. Muscle Nerve. 2000;23(3): Vucic S, Kiernan MC. Axonal excitability properties in amyotrophic lateral sclerosis. Clin. Neurophysiol. 2006;117(7): Vucic S, Kiernan MC. Abnormalities in cortical and peripheral excitability in flail arm variant amyotrophic lateral sclerosis. J. Neurol. Neurosurg. Psychiatry. 2007;78(8): Vucic S, Kiernan MC. Pathophysiologic insights into motor axonal function in Kennedy disease. Neurology. 2007;69(19): Mogyoros I, Kiernan MC, Burke D. Strength-duration properties of human peripheral nerve. Brain. 1996;119: Bostock H, Rothwell JC. Latent addition in motor and sensory fibres of human peripheral nerve. J. Physiol. 1997;498: Bostock H. The strength-duration relationship for excitation of myelinated nerve: computed dependence on membrane parameters. J. Physiol. 1983;341:
176 References 207. Bostock H, Cikurel K, Burke D. Threshold tracking techniques in the study of human peripheral nerve. Muscle Nerve. 1998;21(2): Bostock H, Baker M. Evidence for two types of potassium channel in human motor axons in vivo. Brain Res. 1988;462(2): Kiernan MC, Lin CS, Andersen KV, Murray NM, Bostock H. Clinical evaluation of excitability measures in sensory nerve. Muscle Nerve. 2001;24(7): Burke D, Kiernan MC, Bostock H. Excitability of human axons. Clin. Neurophysiol. 2001;112(9): Kiernan MC, Isbister GK, Lin CS, Burke D, Bostock H. Acute tetrodotoxin-induced neurotoxicity after ingestion of puffer fish. Ann. Neurol. 2005;57(3): Bostock H, Baker M, Reid G. Changes in excitability of human motor axons underlying post-ischaemic fasciculations: evidence for two stable states. J. Physiol. 1991;441: Bostock H, Sharief MK, Reid G, Murray NM. Axonal ion channel dysfunction in amyotrophic lateral sclerosis. Brain. 1995; Farrar MA, Park SB, Lin CS, Kiernan MC. Evolution of peripheral nerve function in humans: novel insights from motor nerve excitability. J. Physiol. 2013;591(Pt 1): Schwarz JR, Reid G, Bostock H. Action potentials and membrane currents in the human node of Ranvier. Pflugers Arch. 1995;430(2): Boland RA, Bostock H, Kiernan MC. Plasticity of lower limb motor axons after cervical cord injury. Clin. Neurophysiol. 2009;120(1): Champe PC, Harvey RA, Ferrier DR. Complex Lipid Metabolism. In: Champe PC, Harvey RA, eds. Lippincott's Illustrated Reviews: Biochmistry. 4th ed. Philadelphia: Lippincott Williams & Wilkins; 2008:
177 References 218. Sahenk Z. Abnormal Schwann cell-axon interactions in CMT neuropathies. The effects of mutant Schwann cells on the axonal cytoskeleton and regenerationassociated myelination. Ann. N. Y. Acad. Sci. 1999;883: Beirowski B. Concepts for regulation of axon integrity by enwrapping glia. Front Cell Neurosci. 2013;7: Nave KA, Sereda MW, Ehrenreich H. Mechanisms of disease: inherited demyelinating neuropathies--from basic to clinical research. Nat. Clin. Pract. Neurol. 2007;3(8): Scherer SS, Wrabetz L. Molecular mechanisms of inherited demyelinating neuropathies. Glia. 2008;56(14): Rosenbluth J, Mierzwa A, Shroff S. Molecular architecture of myelinated nerve fibers: leaky paranodal junctions and paranodal dysmyelination. Neuroscientist. 2013;19(6): Mierzwa A, Shroff S, Rosenbluth J. Permeability of the paranodal junction of myelinated nerve fibers. J. Neurosci. 2010;30(47): Nodera H, Bostock H, Kuwabara S, et al. Nerve excitability properties in Charcot- Marie-Tooth disease type 1A. Brain. 2004;127(Pt 1): Farrar MA, Park SB, Krishnan AV, Kiernan MC, Lin CS. Axonal dysfunction, dysmyelination and conduction failure in HNPP. Muscle Nerve. 2014;49: Norton WN, Daskal I, Savage HE, Seibert RA, Lane M. Effects of riboflavin deficiency on the ultrastructure of rat sciatic nerve fibers. Am. J. Pathol. 1976;85(3): Jortner BS, Cherry J, Lidsky TI, Manetto C, Shell L. Peripheral neuropathy of dietary riboflavin deficiency in chickens. J. Neuropathol. Exp. Neurol. 1987;46(5):
178 References 228. Cheah BC, Lin CS, Park SB, Vucic S, Krishnan AV, Kiernan MC. Progressive axonal dysfunction and clinical impairment in amyotrophic lateral sclerosis. Clin. Neurophysiol. 2012;123(12): Sghirlanzoni A, Pareyson D, Lauria G. Sensory neuron diseases. Lancet Neurol. 2005;4(6):
179 APPENDIX 178
180 PARENT INFORMATION SHEET The peripheral neuropathy in children with mitochondrial disease study Corner Hawkesbury Road and Hainsworth Street Locked Bag 4001 Westmead NSW 2145 Sydney Australia DX 8213 Parramatta Tel Fax ABN Investigators: Dr Manoj Menezes, Paediatric Neurologist, The Institute for Neuroscience and Muscle Research Tel: (02) , Prof. John Christodoulou, Director, Western Sydney Genetics Program Tel: (02) We would like you to consider your child participating in a research study that will be conducted in The Institute for Neuroscience and Muscle Research at The Children s Hospital at Westmead. What is the study about? The study investigates if the nerves that carry sensation and impulses to muscles are affected in different mitochondrial diseases. The goal of this research is to improve diagnosis of children with mitochondrial disorders. The study is being conducted by investigators at The Institute for Neuroscience and Muscle Research (based at The Children s Hospital at Westmead) in collaboration with the Western Sydney Genetics Program and The Children s Hospital at Westmead Metabolic Clinic. You are invited to contribute your child s personal details and medical history to the project, and enrol your child for a one-off medical examination and nerve conduction study. Who can participate in the study? Any child less than 18 years of age, who has been diagnosed with a mitochondrial disease, either on the basis of a positive genetic test, or after respiratory chain enzyme testing on a skin, muscle or liver biopsy, is eligible to participate in the study. Why should we look at peripheral nerve involvement in mitochondrial disease? While peripheral nerve involvement (neuropathy) is common in mitochondrial disease, the frequency with which it occurs in children in different mitochondrial diseases and the clinical features and nerve conduction findings of these mitochondrial diseases has not been well understood. Mitochondrial diseases are often difficult to diagnose. Similar clinical characteristics may be shared by different mitochondrial diseases, and reaching a diagnosis usually requires skin, muscle or liver biopsies. This study attempts to find out if the presence of a neuropathy and the characteristics of the neuropathy on nerve conduction studies differ among the different mitochondrial diseases, and if this information helps with identifying the mitochondrial disease and the underlying genetic abnormality without the need for invasive biopsies. Occasionally, children with mitochondrial disease may have difficulties because of the associated neuropathy, and this may manifest as weakness, abnormalities with sensation or difficulty with walking. In these cases, the identification of a neuropathy will help early rehabilitation. What does participating in the study involve? Your child was chosen to participate in this study as the records of the Metabolic Clinic identified him/her as having a mitochondrial disease. The study includes a medical assessment (looking for evidence of a neuropathy) and nerve conduction study that will usually be scheduled on a day you are visiting The Children s Hospital at Westmead either for a clinic visit or to have another test done. One of the investigators will meet with you to talk to you about the study and answer any questions you may have.
181 The investigator will ask you some personal details about your child, such as name, date of birth, address and diagnosis. The investigators require these details to access information from your child s medical record and so that the report from the study is stored under the appropriate name. During the nerve conduction tests, small electrode dots are placed on the skin of an arm or a leg and a small electrical impulse is given by a probe over the skin. The doctor may also occasionally need to test a muscle by gently inserting a small needle (like an acupuncture needle) into the muscle. As some children find the nerve conduction study uncomfortable, they can be performed under sedation with nitrous oxide in children above one year of age. This pack contains separate leaflets that explain the nerve conduction study and nitrous oxide sedation. Both the medical assessment and the nerve conduction study will together take around 45 minutes. The study requires only a single visit and no further follow-up or repeat nerve conductions are required. The results of your child s nerve conduction study will be sent to your child s doctor at the Metabolic Clinic. The investigators thank you and your child for participating in this study. We expect 30 children to take part in this study. There is no external funding for the study and no payments or reimbursements will be made to participants. Are there any benefits for my child from participating in the study? We hope that information gained from the study will improve our knowledge about peripheral nerve involvement in mitochondrial disease, and help to improve our ability to diagnose mitochondrial disease and characterise the associated neuropathy. For some children, the early recognition of the neuropathy could help with early rehabilitation. In the short term however, there may be no direct benefit to you or your child from participating in the study. Are there any side-effects associated with the study? The nerve conduction study may be uncomfortable, and children over one year of age can be given nitrous oxide gas (laughing gas) through a mask to relax during the test. The needle, used occasionally during the nerve conduction test, may cause some bruising. There are no other risks from being involved in this study. Is the information collected during the study secure and confidential? To ensure that all information about your child is kept strictly confidential, access to the information collected as part of the study will only be available to the investigators of this project. Investigators will have a security password that will allow them to enter and access data. Data from the study will be stored for 15 years after the completion of the study and will then be destroyed by shredding of paper documents and permanently erasing all computer files. The information collected during this study could be used in reports or papers about the research. Your child will not be able to be identified in these reports or papers. The information collected during this study may be used in further ethics-committee approved studies. Other information Participation in this project is entirely voluntary and if you decide not to take part or decide to withdraw at any time this will not otherwise affect your child s care at the Hospital. This copy of the Information Sheet is for you to keep. We will also give you a copy of the signed consent form. For questions about the study please call Dr Manoj Menezes at This project has been approved by The Sydney Children s Hospital Network Human Research Ethics Committee. If you have any concerns about the conduct of this study, please do not hesitate to contact the Secretary of the Ethics Committee ( ) and quote the approval code 12/SCHN/195.
182 PARENT/GUARDIAN CONSENT FORM The peripheral neuropathy in children with mitochondrial disease study Corner Hawkesbury Road and Hainsworth Street Locked Bag 4001 Westmead NSW 2145 Sydney Australia DX 8213 Parramatta Tel Fax ABN Investigators: Dr Manoj Menezes, The Institute for Neuroscience and Muscle Research, Tel: (02) , Prof. John Christodoulou, Western Sydney Genetics Program, Tel: (02) I have read and understood the Parent Information Sheet, the Nerve Conduction Study Fact Sheet and the Nitrous Oxide Fact Sheet, and give my consent for my child to participate in this research study, which has been explained to me by I consent for the information about my child, collected during this study, to be used in further ethicscommittee approved research (Please tick appropriate box). Yes No I understand that I am free to withdraw from the study at any time and this decision will not otherwise affect my child s treatment at the Hospital. NAME OF CHILD: (Please print) NAME OF PARENT OR GUARDIAN: (Please print) SIGNATURE OF PARENT OR GUARDIAN: Date: NAME OF WITNESS*: (Please print) SIGNATURE OF WITNESS*: Date: NAME OF INTERPRETER: (Please print) SIGNATURE OF INTERPRETER: Date: *Use when participant has had this consent form read to them (i.e., illiterate, legally blind, translated into foreign language).
183 ADOLESCENT INFORMATION FORM (Age yrs) Corner Hawkesbury Road and Hainsworth Street Locked Bag 4001 Westmead NSW 2145 Sydney Australia DX 8213 Parramatta Tel Fax ABN The peripheral neuropathy in children with mitochondrial disease study Investigators: Dr Manoj Menezes, Paediatric Neurologist, The Institute for Neuroscience and Muscle Research Tel: (02) , Prof. John Christodoulou, Director, Western Sydney Genetics Program Tel: (02) Why am I here? This is a research study. Only people who choose to take part are included in research studies. You are being asked to take part in this study because you have a mitochondrial disease. Please take time to read this form and understand what the study involves. Talk to your family about it and be sure to ask questions about anything you don t understand. We expect that 30 children will take part in this study. Why are they doing this study? This study is being done to find out if the nerves that carry messages to muscles, and help one feel, are involved in mitochondrial disease. This may help us with reaching a diagnosis in other children with mitochondrial disease and start therapy earlier for children whose nerves are affected. What will I have to do if I take part? There will be two parts to your visit that will together take around 45 minutes to complete. The doctor will test your strength, walking ability and sensation. You will then have a nerve conduction test, where the small electrode dots are placed on the skin of an arm or a leg and a small electrical impulse is given by a probe over the skin. The doctor may also occasionally need to test a muscle by gently inserting a small needle (like an acupuncture needle) into the muscle. Some children feel these tests are uncomfortable, and we can give you some nitrous oxide or laughing gas through a mask to help with the uncomfortable sensation, if this is what you would like us to do. You will need to have fasted, i.e. have had nothing to eat and drink, for 2 hours before having the nitrous oxide. There are no long term side effects from having the nerve conduction test or nitrous oxide. This test will only be performed once and no more involvement will be needed. Will the study help me? We cannot promise you that being in this research study will help you. The nerve conduction test will help identify if there is any nerve involvement as a part of your mitochondrial disease and this information will be sent to your doctor. Information from this study may help other people with similar health issues now or in the future. Do I have to take part in the research? No you don t. You don t have to be in this study if you don t want to. Please discuss your decision with your parents and doctor. No one will be angry if you decide to stop being in the study. You will still be treated for your mitochondrial disease. Do my parents or guardians know about this? This study was explained to your parents/guardian and they said that you could be in it. You can talk this over with them.
184 What will happen to the information collected? The information you tell us and collected from the nerve conduction test will only be used by the investigators to help us reach a diagnosis in other children with mitochondrial disease and start therapy for children whose nerves are affected. No-one else will be allowed to use this information. The information could be used with information from other young people in reports or papers about the research. You will not be able to be identified in these reports or papers. The information you tell us will be stored securely in locked cupboards, or in password-protected computer files, at The Children s Hospital at Westmead. It will be stored for 15 years and then destroyed. This sheet is for you to keep. For questions about the study please call Dr Manoj Menezes at This project has been approved by The Sydney Children s Hospital Network Human Research Ethics Committee. If you have any concerns about the conduct of this study, please do not hesitate to contact the Secretary of the Ethics Committee ( ) and quote the approval code 12/SCHN/195.
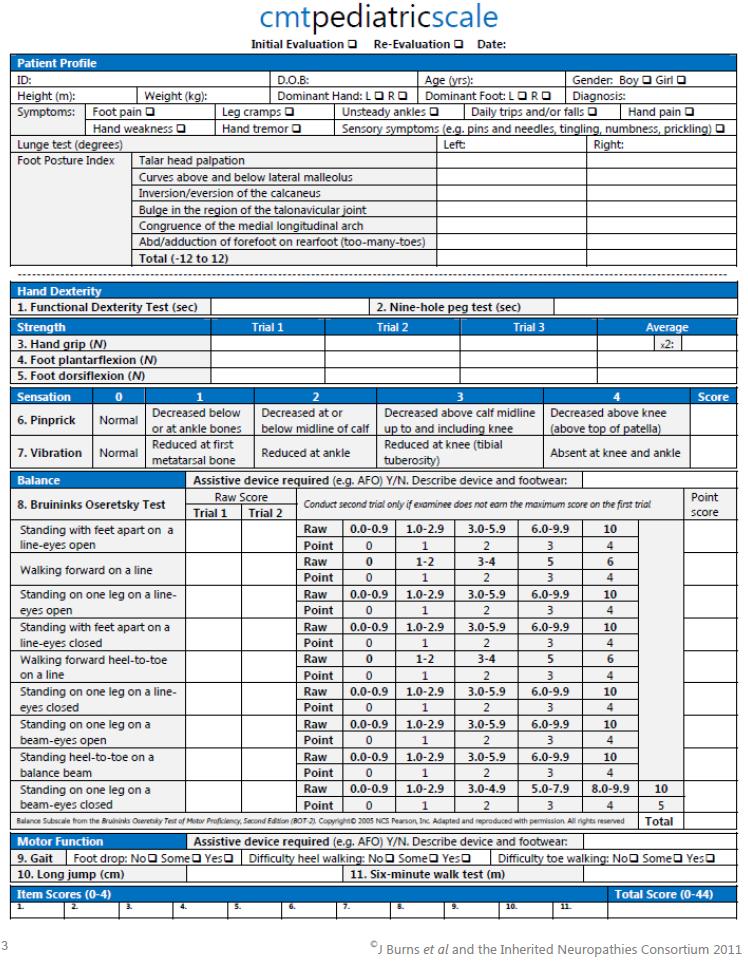
185
A CASE OF GIANT AXONAL NEUROPATHY HEMANANTH T SECOND YEAR POST GRADUATE IN PAEDIATRICS INSTITUTE OF SOCIAL PAEDIATRICS GOVERNMENT STANLEY HOSPITAL
 A CASE OF GIANT AXONAL NEUROPATHY HEMANANTH T SECOND YEAR POST GRADUATE IN PAEDIATRICS INSTITUTE OF SOCIAL PAEDIATRICS GOVERNMENT STANLEY HOSPITAL CASE HISTORY Nine year old male child Second born Born
A CASE OF GIANT AXONAL NEUROPATHY HEMANANTH T SECOND YEAR POST GRADUATE IN PAEDIATRICS INSTITUTE OF SOCIAL PAEDIATRICS GOVERNMENT STANLEY HOSPITAL CASE HISTORY Nine year old male child Second born Born
REQUISITION FORM NOTE: ALL FORMS MUST BE FILLED OUT COMPLETELY FOR SAMPLE TO BE PROCESSED. Last First Last First
 #: DEPARTMENT OF NEUROLOGY COLUMBIA COLLEGE OF PHYSICIANS & SURGEONS Room 4-420 630 West 168th Street, New York, NY 10032 Telephone #: 212-305-3947 Fax#: 212-305-3986 REQUISITION FORM NOTE: ALL FORMS MUST
#: DEPARTMENT OF NEUROLOGY COLUMBIA COLLEGE OF PHYSICIANS & SURGEONS Room 4-420 630 West 168th Street, New York, NY 10032 Telephone #: 212-305-3947 Fax#: 212-305-3986 REQUISITION FORM NOTE: ALL FORMS MUST
Nutritional Interventions in Primary Mitochondrial Disorders
 Nutritional Interventions in Primary Mitochondrial Disorders Carolyn J Ellaway MBBS PhD FRACP CGHGSA Genetic Metabolic Disorders Service Sydney Children s Hospital Network Disciplines of Child and Adolescent
Nutritional Interventions in Primary Mitochondrial Disorders Carolyn J Ellaway MBBS PhD FRACP CGHGSA Genetic Metabolic Disorders Service Sydney Children s Hospital Network Disciplines of Child and Adolescent
Presentation and investigation of mitochondrial disease in children
 Presentation and investigation of mitochondrial disease in children Andrew Morris Willink Unit, Manchester Mitochondrial function Carbohydrate Fat Respiratory chain Energy Mitochondria are the product
Presentation and investigation of mitochondrial disease in children Andrew Morris Willink Unit, Manchester Mitochondrial function Carbohydrate Fat Respiratory chain Energy Mitochondria are the product
MITOCHONDRIAL DISEASE. Amel Karaa, MD Mitochondrial Disease Program Massachusetts General Hospital
 MITOCHONDRIAL DISEASE Amel Karaa, MD Mitochondrial Disease Program Massachusetts General Hospital Disclosures & Disclaimers United Mitochondrial Disease Foundation Research Grant North American Mitochondrial
MITOCHONDRIAL DISEASE Amel Karaa, MD Mitochondrial Disease Program Massachusetts General Hospital Disclosures & Disclaimers United Mitochondrial Disease Foundation Research Grant North American Mitochondrial
SELECTIVE VULNERABILITY (HYPOXIA AND HYPOGLYCEMIA)
") DEFICIENCY OF METABOLITE -HYPOXIA AND HYPOGLYCEMIA -HYPOVITAMINOSIS SELECTIVE VULNERABILITY (HYPOXIA AND HYPOGLYCEMIA) -SPECIFIC CELL TYPE NEURONS>OLIGODENDROCYTES>ASTROCYTES -SPECIFIC BRAIN REGION PYRAMIDAL
DEFICIENCY OF METABOLITE -HYPOXIA AND HYPOGLYCEMIA -HYPOVITAMINOSIS SELECTIVE VULNERABILITY (HYPOXIA AND HYPOGLYCEMIA) -SPECIFIC CELL TYPE NEURONS>OLIGODENDROCYTES>ASTROCYTES -SPECIFIC BRAIN REGION PYRAMIDAL
Guide to the use of nerve conduction studies (NCS) & electromyography (EMG) for non-neurologists
 & electromyography (EMG) for non-neurologists") Guide to the use of nerve conduction studies (NCS) & electromyography (EMG) for non-neurologists What is NCS/EMG? NCS examines the conduction properties of sensory and motor peripheral nerves. For both
Guide to the use of nerve conduction studies (NCS) & electromyography (EMG) for non-neurologists What is NCS/EMG? NCS examines the conduction properties of sensory and motor peripheral nerves. For both
Referring Physician Information Name: (Last, First, Middle):
:") Page 1 of 5 Patient Information Clinical Indication: Patient Name: (Last, First, Middle): DOB (M/D/Y): Sex: M F Guardian Name (for minor patients only): Address: City: State: ZIP: Phone: Ethnic Background
Page 1 of 5 Patient Information Clinical Indication: Patient Name: (Last, First, Middle): DOB (M/D/Y): Sex: M F Guardian Name (for minor patients only): Address: City: State: ZIP: Phone: Ethnic Background
An Introduction to mitochondrial disease.
 9 th September 2017 An Introduction to mitochondrial disease. Dr Andy Schaefer Consultant Neurologist and Clinical Lead NHS Highly Specialised Rare Mitochondrial Disease Service and Wellcome Trust Centre
9 th September 2017 An Introduction to mitochondrial disease. Dr Andy Schaefer Consultant Neurologist and Clinical Lead NHS Highly Specialised Rare Mitochondrial Disease Service and Wellcome Trust Centre
Table 1: Nerve Conduction Studies (summarised)
") Table 1: Nerve Conduction Studies (summarised) Sensory nerve conduction 1 week* 3 months Superficial radial sensory Normal, symmetric SNAP and CV No change Median to digit II Normal, symmetric SNAP and
Table 1: Nerve Conduction Studies (summarised) Sensory nerve conduction 1 week* 3 months Superficial radial sensory Normal, symmetric SNAP and CV No change Median to digit II Normal, symmetric SNAP and
Compound Action Potential, CAP
 Stimulus Strength UNIVERSITY OF JORDAN FACULTY OF MEDICINE DEPARTMENT OF PHYSIOLOGY & BIOCHEMISTRY INTRODUCTION TO NEUROPHYSIOLOGY Spring, 2013 Textbook of Medical Physiology by: Guyton & Hall, 12 th edition
Stimulus Strength UNIVERSITY OF JORDAN FACULTY OF MEDICINE DEPARTMENT OF PHYSIOLOGY & BIOCHEMISTRY INTRODUCTION TO NEUROPHYSIOLOGY Spring, 2013 Textbook of Medical Physiology by: Guyton & Hall, 12 th edition
Mitochondrial Diseases
 Mitochondrial Diseases Simon Heales SWIM Conference Taunton, November 29 th 2018 Respiratory Failure Cardiomyopathy Optic Atrophy / Retinitis Pigmentosa Seizures / Developmental delay Liver Failure Deafness
Mitochondrial Diseases Simon Heales SWIM Conference Taunton, November 29 th 2018 Respiratory Failure Cardiomyopathy Optic Atrophy / Retinitis Pigmentosa Seizures / Developmental delay Liver Failure Deafness
Motor and sensory nerve conduction studies
 3 rd Congress of the European Academy of Neurology Amsterdam, The Netherlands, June 24 27, 2017 Hands-on Course 2 Assessment of peripheral nerves function and structure in suspected peripheral neuropathies
3 rd Congress of the European Academy of Neurology Amsterdam, The Netherlands, June 24 27, 2017 Hands-on Course 2 Assessment of peripheral nerves function and structure in suspected peripheral neuropathies
MITOCHONDRIAL DISORDERS IN NEUROLOGY
 MITOCHONDRIAL DISORDERS IN NEUROLOGY Michio Hirano, MD Columbia University Medical Center New York, NY In the opening lecture of this course, Dr. Eric Schon will describe the molecular genetic and pathogenic
MITOCHONDRIAL DISORDERS IN NEUROLOGY Michio Hirano, MD Columbia University Medical Center New York, NY In the opening lecture of this course, Dr. Eric Schon will describe the molecular genetic and pathogenic
THIAMINE TRANSPORTER TYPE 2 DEFICIENCY
 THIAMINE TRANSPORTER TYPE 2 DEFICIENCY WHAT IS THE THIAMINE TRANSPORTER TYPE 2 DEFICIENCY (hthtr2)? The thiamine transporter type 2 deficiency (hthtr2) is a inborn error of thiamine metabolism caused by
THIAMINE TRANSPORTER TYPE 2 DEFICIENCY WHAT IS THE THIAMINE TRANSPORTER TYPE 2 DEFICIENCY (hthtr2)? The thiamine transporter type 2 deficiency (hthtr2) is a inborn error of thiamine metabolism caused by
A/Professor Arun Aggarwal Balmain Hospital
 A/Professor Arun Aggarwal Balmain Hospital Nerve Conduction Studies Test to evaluate the function of motor / sensory nerves Evaluate Paraesthesia (numbness, tingling, burning) Weakness of arms and legs
A/Professor Arun Aggarwal Balmain Hospital Nerve Conduction Studies Test to evaluate the function of motor / sensory nerves Evaluate Paraesthesia (numbness, tingling, burning) Weakness of arms and legs
Recent Advances in Neurology 2014: Neuromuscular Case Presentations
 Recent Advances in Neurology 2014: Neuromuscular Case Presentations Jeffrey W. Ralph, MD Associate Clinical Professor Patient #1: Young woman with severe polyneuropathy 25 year-old woman Normal motor and
Recent Advances in Neurology 2014: Neuromuscular Case Presentations Jeffrey W. Ralph, MD Associate Clinical Professor Patient #1: Young woman with severe polyneuropathy 25 year-old woman Normal motor and
A Practical Approach to Polyneuropathy SLOCUM DICKSON ANNUAL TEACHING DAY NOVEMBER 4, 2017
 A Practical Approach to Polyneuropathy SLOCUM DICKSON ANNUAL TEACHING DAY NOVEMBER 4, 2017 Disclosures Research support from Cytokinetics, Inc Catalyst, Inc Editorial fees from UptoDate. Objectives Describe
A Practical Approach to Polyneuropathy SLOCUM DICKSON ANNUAL TEACHING DAY NOVEMBER 4, 2017 Disclosures Research support from Cytokinetics, Inc Catalyst, Inc Editorial fees from UptoDate. Objectives Describe
MYOCLONIC EPILEPSY WITH RAGGED RED FIBERS (MERRF) By- Promie Faruque
 By- Promie Faruque") MYOCLONIC EPILEPSY WITH RAGGED RED FIBERS (MERRF) By- Promie Faruque PHYSIOLOGY -MERRF is a rare panethnic mitochondrial disease which is caused by mutations in the mtdna -It mainly affects the muscle
MYOCLONIC EPILEPSY WITH RAGGED RED FIBERS (MERRF) By- Promie Faruque PHYSIOLOGY -MERRF is a rare panethnic mitochondrial disease which is caused by mutations in the mtdna -It mainly affects the muscle
The mitochondrion and its disorders
 100 PRACTICAL NEUROLOGY H O W T O U N D E R S T A N D I T The mitochondrion and its disorders Patrick F. Chinnery Department of Neurology, Regional Neurosciences Centre, Newcastle General Hospital and
100 PRACTICAL NEUROLOGY H O W T O U N D E R S T A N D I T The mitochondrion and its disorders Patrick F. Chinnery Department of Neurology, Regional Neurosciences Centre, Newcastle General Hospital and
Multifocal motor neuropathy: diagnostic criteria that predict the response to immunoglobulin treatment
 Multifocal motor neuropathy: diagnostic criteria that predict the response to immunoglobulin treatment 7 MMN RM Van den Berg-Vos, H Franssen, JHJ Wokke, HW Van Es, LH Van den Berg Annals of Neurology 2000;
Multifocal motor neuropathy: diagnostic criteria that predict the response to immunoglobulin treatment 7 MMN RM Van den Berg-Vos, H Franssen, JHJ Wokke, HW Van Es, LH Van den Berg Annals of Neurology 2000;
CLINICAL SIGNS SUGGESTIVE OF A NEUROMETABOLIC DISEASE. Bwee Tien Poll-The Amsterdam UMC The Netherlands
 CLINICAL SIGNS SUGGESTIVE OF A NEUROMETABOLIC DISEASE Bwee Tien Poll-The Amsterdam UMC The Netherlands FRAMEWORK OF PRINCIPALS 1. Problem-oriented clinical approach 2. Biomarkers in plasma, urine, CSF
CLINICAL SIGNS SUGGESTIVE OF A NEUROMETABOLIC DISEASE Bwee Tien Poll-The Amsterdam UMC The Netherlands FRAMEWORK OF PRINCIPALS 1. Problem-oriented clinical approach 2. Biomarkers in plasma, urine, CSF
CIDP + MMN - how to diagnose and treat. Dr Hadi Manji
 CIDP + MMN - how to diagnose and treat Dr Hadi Manji Outline Introduction CIDP Diagnosis Clinical features MRI Nerve conduction tests Lumbar puncture Nerve biopsy Treatment IV Ig Steroids Plasma Exchnage
CIDP + MMN - how to diagnose and treat Dr Hadi Manji Outline Introduction CIDP Diagnosis Clinical features MRI Nerve conduction tests Lumbar puncture Nerve biopsy Treatment IV Ig Steroids Plasma Exchnage
Charcot-Marie-Tooth Disease (CMT) Sometimes known as Hereditary Motor and Sensory neuropathy (HMSN) or Peroneal Muscular Atrophy (PMA)
 Sometimes known as Hereditary Motor and Sensory neuropathy (HMSN) or Peroneal Muscular Atrophy (PMA)") Version: 2 Published: January 2004 Updated: December 2009 Date of review: November 2011 Author: Dr.Hilton-Jones MD, FRCP, FRCPE Clinical Director, Oxford MDC Muscle and Nerve Centre, and revised by Dr.
Version: 2 Published: January 2004 Updated: December 2009 Date of review: November 2011 Author: Dr.Hilton-Jones MD, FRCP, FRCPE Clinical Director, Oxford MDC Muscle and Nerve Centre, and revised by Dr.
Diagnosis and Therapy in Neuromuscular Disorders: Diagnosis and New Treatments in Mitochondrial Diseases
 Diagnosis and Therapy in Neuromuscular Disorders: Diagnosis and New Treatments in Mitochondrial Diseases Shamima Rahman, Michael Hanna To cite this version: Shamima Rahman, Michael Hanna. Diagnosis and
Diagnosis and Therapy in Neuromuscular Disorders: Diagnosis and New Treatments in Mitochondrial Diseases Shamima Rahman, Michael Hanna To cite this version: Shamima Rahman, Michael Hanna. Diagnosis and
Making sense of Nerve conduction & EMG
 Making sense of Nerve conduction & EMG Drs R Arunachalam Consultant Clinical Neurophysiologist Wessex Neurological Centre Southampton University Hospital EMG/NCS EMG machine For the assessment of patients
Making sense of Nerve conduction & EMG Drs R Arunachalam Consultant Clinical Neurophysiologist Wessex Neurological Centre Southampton University Hospital EMG/NCS EMG machine For the assessment of patients
Genetics of Hereditary Spastic Paraplegia Dr. Arianna Tucci
 Genetics of Hereditary Spastic Paraplegia 1 Clinical Research Fellow Institute of Neurology University College London Hereditary spastic paraplegia: definition Clinical designation for neurologic syndromes
Genetics of Hereditary Spastic Paraplegia 1 Clinical Research Fellow Institute of Neurology University College London Hereditary spastic paraplegia: definition Clinical designation for neurologic syndromes
The Organism as a system
 The Organism as a system PATIENT 1: Seven-year old female with a history of normal development until age two. At this point she developed episodic vomiting, acidosis, epilepsy, general weakness, ataxia
The Organism as a system PATIENT 1: Seven-year old female with a history of normal development until age two. At this point she developed episodic vomiting, acidosis, epilepsy, general weakness, ataxia
A Hypothesis Driven Approach to the Neurological Exam
 A Hypothesis Driven Approach to the Neurological Exam Vanja Douglas, MD Assistant Clinical Professor UCSF Department of Neurology Disclosures None 1 Purpose of Neuro Exam Screen asymptomatic patients Screen
A Hypothesis Driven Approach to the Neurological Exam Vanja Douglas, MD Assistant Clinical Professor UCSF Department of Neurology Disclosures None 1 Purpose of Neuro Exam Screen asymptomatic patients Screen
High Yield Neurological Examination
 High Yield Neurological Examination Vanja Douglas, MD Sara & Evan Williams Foundation Endowed Neurohospitalist Chair Director, Neurohospitalist Division Associate Professor of Clinical Neurology UCSF Department
High Yield Neurological Examination Vanja Douglas, MD Sara & Evan Williams Foundation Endowed Neurohospitalist Chair Director, Neurohospitalist Division Associate Professor of Clinical Neurology UCSF Department
variant led to a premature stop codon p.k316* which resulted in nonsense-mediated mrna decay. Although the exact function of the C19L1 is still
 157 Neurological disorders primarily affect and impair the functioning of the brain and/or neurological system. Structural, electrical or metabolic abnormalities in the brain or neurological system can
157 Neurological disorders primarily affect and impair the functioning of the brain and/or neurological system. Structural, electrical or metabolic abnormalities in the brain or neurological system can
This is a repository copy of A novel mutation in the FGD4 gene causing Charcot-Marie-Tooth disease..
 This is a repository copy of A novel mutation in the FGD4 gene causing Charcot-Marie-Tooth disease.. White Rose Research Online URL for this paper: http://eprints.whiterose.ac.uk/117077/ Version: Accepted
This is a repository copy of A novel mutation in the FGD4 gene causing Charcot-Marie-Tooth disease.. White Rose Research Online URL for this paper: http://eprints.whiterose.ac.uk/117077/ Version: Accepted
Distal chronic spinal muscular atrophy involving the hands
 Journal ofneurology, Neurosurgery, and Psychiatry, 1978, 41, 653-658 Distal chronic spinal muscular atrophy involving the hands D. J. O'SULLIVAN AND J. G. McLEOD From St Vincent's Hospital, and Department
Journal ofneurology, Neurosurgery, and Psychiatry, 1978, 41, 653-658 Distal chronic spinal muscular atrophy involving the hands D. J. O'SULLIVAN AND J. G. McLEOD From St Vincent's Hospital, and Department
Peripheral Neuropathies
 Peripheral Neuropathies ELBA Y. GERENA MALDONADO, MD ACTING ASSISTANT PROFESSOR UNIVERSITY OF WASHINGTON MEDICAL CENTER Objectives Definition Neurophysiology Evaluation of polyneuropathies Cases Summary
Peripheral Neuropathies ELBA Y. GERENA MALDONADO, MD ACTING ASSISTANT PROFESSOR UNIVERSITY OF WASHINGTON MEDICAL CENTER Objectives Definition Neurophysiology Evaluation of polyneuropathies Cases Summary
AII-type: Select the most appropriate answer
 AII-type: Select the most appropriate answer ( )1. Choose one best answer for the following pathologic pictures. A. choroid cyst B. choroid papilloma C. pontine glioma D. ependymoma E. metastatic tumor
AII-type: Select the most appropriate answer ( )1. Choose one best answer for the following pathologic pictures. A. choroid cyst B. choroid papilloma C. pontine glioma D. ependymoma E. metastatic tumor
Case Report Motor Neuron Syndrome as a New Phenotypic Manifestation of Mutation 9185T>C in Gene MTATP6
 Case Reports in Neurological Medicine, Article ID 701761, 4 pages http://dx.doi.org/10.1155/2014/701761 Case Report Motor Neuron Syndrome as a New Phenotypic Manifestation of Mutation 9185T>C in Gene MTATP6
Case Reports in Neurological Medicine, Article ID 701761, 4 pages http://dx.doi.org/10.1155/2014/701761 Case Report Motor Neuron Syndrome as a New Phenotypic Manifestation of Mutation 9185T>C in Gene MTATP6
Differential Diagnosis of Neuropathies and Compression. Dr Ashwin Pinto Consultant Neurologist Wessex Neurological Centre
 Differential Diagnosis of Neuropathies and Compression Dr Ashwin Pinto Consultant Neurologist Wessex Neurological Centre Outline of talk Mononeuropathies median and anterior interosseous nerve ulnar nerve
Differential Diagnosis of Neuropathies and Compression Dr Ashwin Pinto Consultant Neurologist Wessex Neurological Centre Outline of talk Mononeuropathies median and anterior interosseous nerve ulnar nerve
Biochemistry of cellular organelles
 Kontinkangas, L101A Biochemistry of cellular organelles Lectures: 1. Membrane channels; 2. Membrane transporters; 3. Soluble lipid/metabolite-transfer proteins; 4. Mitochondria as cellular organelles;
Kontinkangas, L101A Biochemistry of cellular organelles Lectures: 1. Membrane channels; 2. Membrane transporters; 3. Soluble lipid/metabolite-transfer proteins; 4. Mitochondria as cellular organelles;
Multifocal motor neuropathy: long-term clinical and electrophysiological assessment of intravenous immunoglobulin maintenance treatment
 Multifocal motor neuropathy: long-term clinical and electrophysiological assessment of intravenous immunoglobulin maintenance treatment 11 MMN RM Van den Berg-Vos, H Franssen, JHJ Wokke, LH Van den Berg
Multifocal motor neuropathy: long-term clinical and electrophysiological assessment of intravenous immunoglobulin maintenance treatment 11 MMN RM Van den Berg-Vos, H Franssen, JHJ Wokke, LH Van den Berg
Year 2004 Paper one: Questions supplied by Megan
 QUESTION 47 A 58yo man is noted to have a right foot drop three days following a right total hip replacement. On examination there is weakness of right ankle dorsiflexion and toe extension (grade 4/5).
QUESTION 47 A 58yo man is noted to have a right foot drop three days following a right total hip replacement. On examination there is weakness of right ankle dorsiflexion and toe extension (grade 4/5).
The Internist s Approach to Neuropathy
 The Internist s Approach to Neuropathy VOLKAN GRANIT, MD, MSC ASSISTANT PROFESSOR OF NEUROLOGY NEUROMUSCU LAR DIVISION UNIVERSITY OF MIAMI, MILLER SCHOOL OF MEDICINE RELEVANT DECLARATIONS Financial disclosures:
The Internist s Approach to Neuropathy VOLKAN GRANIT, MD, MSC ASSISTANT PROFESSOR OF NEUROLOGY NEUROMUSCU LAR DIVISION UNIVERSITY OF MIAMI, MILLER SCHOOL OF MEDICINE RELEVANT DECLARATIONS Financial disclosures:
Auditory Neuropathy Spectrum Disorder. Yvonne S. Sininger PhD Professor of Head & Neck Surgery University of California Los Angeles
 Auditory Neuropathy Spectrum Disorder Yvonne S. Sininger PhD Professor of Head & Neck Surgery University of California Los Angeles 1 Financial Disclosure Information I have no relevant financial relationship
Auditory Neuropathy Spectrum Disorder Yvonne S. Sininger PhD Professor of Head & Neck Surgery University of California Los Angeles 1 Financial Disclosure Information I have no relevant financial relationship
Robert Barski. Biochemical Genetics St James s University Hospital, Leeds. MetBioNet IEM Introductory Training
 Robert Barski Biochemical Genetics St James s University Hospital, Leeds Lactate is produced as the fate of anaerobic metabolism of pyruvate. It is an important intermediary metabolite especially with
Robert Barski Biochemical Genetics St James s University Hospital, Leeds Lactate is produced as the fate of anaerobic metabolism of pyruvate. It is an important intermediary metabolite especially with
Peripheral neuropathies, neuromuscular junction disorders, & CNS myelin diseases
 Peripheral neuropathies, neuromuscular junction disorders, & CNS myelin diseases Peripheral neuropathies according to which part affected Axonal Demyelinating with axonal sparing Many times: mixed features
Peripheral neuropathies, neuromuscular junction disorders, & CNS myelin diseases Peripheral neuropathies according to which part affected Axonal Demyelinating with axonal sparing Many times: mixed features
Nerve Conduction Studies and EMG
 Nerve Conduction Studies and EMG Limitations of other methods of investigations of the neuromuscular system - Dr Rob Henderson, Neurologist Assessment of Weakness Thanks Peter Silburn PERIPHERAL NEUROPATHY
Nerve Conduction Studies and EMG Limitations of other methods of investigations of the neuromuscular system - Dr Rob Henderson, Neurologist Assessment of Weakness Thanks Peter Silburn PERIPHERAL NEUROPATHY
Treatment of multifocal motor neuropathy with interferon-β1a
 Treatment of multifocal motor neuropathy with interferon-β1a 12 MMN RM Van den Berg-Vos, LH Van den Berg, H Franssen, PA Van Doorn, ISJ Martina, JHJ Wokke Adapted from Neurology 2000; 54: 1518-1521. Chapter
Treatment of multifocal motor neuropathy with interferon-β1a 12 MMN RM Van den Berg-Vos, LH Van den Berg, H Franssen, PA Van Doorn, ISJ Martina, JHJ Wokke Adapted from Neurology 2000; 54: 1518-1521. Chapter
An Update on Auditory Neuropathy Spectrum Disorder in Children
 An Update on Auditory Neuropathy Spectrum Disorder in Children Gary Rance PhD The University of Melbourne Sound Foundations Through Early Amplification Meeting, Chicago, Dec 2013 Overview Auditory neuropathy
An Update on Auditory Neuropathy Spectrum Disorder in Children Gary Rance PhD The University of Melbourne Sound Foundations Through Early Amplification Meeting, Chicago, Dec 2013 Overview Auditory neuropathy
For convenience values outside the normal range are bolded. Normal values for the specified patient are stated below the tables.
 Case tudy 8 or convenience values outside the normal range are bolded. Normal values for the specified patient are stated below the tables. History: 60 year-ol man with a history of left hand weakness
Case tudy 8 or convenience values outside the normal range are bolded. Normal values for the specified patient are stated below the tables. History: 60 year-ol man with a history of left hand weakness
A family study of Charcot-Marie-Tooth disease
 Joturnal of Medical Genetics, 1982, 19, 88-93 A family study of Charcot-Marie-Tooth disease A P BROOKS* AND A E H EMERY From the University Department of Human Genetics, Western General Hospital, Edinburgh
Joturnal of Medical Genetics, 1982, 19, 88-93 A family study of Charcot-Marie-Tooth disease A P BROOKS* AND A E H EMERY From the University Department of Human Genetics, Western General Hospital, Edinburgh
Newborn Screen & Development Facts about the genetic diseases new since March 2006 (Excluding Cystic Fibrosis)
") Newborn Screen & Development Facts about the genetic diseases new since March 2006 (Excluding Cystic Fibrosis) 1) Argininosuccinic acidemia (ASA) a) Incidence: ~1 in 70,000 b) Deficiency in an enzyme of
Newborn Screen & Development Facts about the genetic diseases new since March 2006 (Excluding Cystic Fibrosis) 1) Argininosuccinic acidemia (ASA) a) Incidence: ~1 in 70,000 b) Deficiency in an enzyme of
Sensory Pathways & Somatic Nervous System. Chapter 15
 Sensory Pathways & Somatic Nervous System Chapter 15 How Does Brain Differentiate Sensations? Pain impulses make brain aware of injuries and infections. Impulses from eye, ear, nose and tongue make brain
Sensory Pathways & Somatic Nervous System Chapter 15 How Does Brain Differentiate Sensations? Pain impulses make brain aware of injuries and infections. Impulses from eye, ear, nose and tongue make brain
Exploding Genetic Knowledge in Developmental Disabilities. Disclosures. The Genetic Principle
 Exploding Genetic Knowledge in Developmental Disabilities How to acquire the data and how to make use of it Elliott H. Sherr MD PhD Professor of Neurology & Pediatrics UCSF Disclosures InVitae: clinical
Exploding Genetic Knowledge in Developmental Disabilities How to acquire the data and how to make use of it Elliott H. Sherr MD PhD Professor of Neurology & Pediatrics UCSF Disclosures InVitae: clinical
Childhood mitochondrial encephalomyopathies: clinical course, diagnosis, neuroimaging findings, mtdna mutations and outcome in six children
 Lu and Huang Italian Journal of Pediatrics 2013, 39:60 ITALIAN JOURNAL OF PEDIATRICS CASE REPORT Open Access Childhood mitochondrial encephalomyopathies: clinical course, diagnosis, neuroimaging findings,
Lu and Huang Italian Journal of Pediatrics 2013, 39:60 ITALIAN JOURNAL OF PEDIATRICS CASE REPORT Open Access Childhood mitochondrial encephalomyopathies: clinical course, diagnosis, neuroimaging findings,
Wrist, Elbow Hand. Surface Recording Technique, Study from Median Thenar (MT) Muscle
 Muscle") Surface ecording Technique, Study from Median Thenar (MT) Muscle Original Settings Sensitivity, duration of pulse, sweep speed, low-frequency filter, high- frequency filter, and the machine used were not
Surface ecording Technique, Study from Median Thenar (MT) Muscle Original Settings Sensitivity, duration of pulse, sweep speed, low-frequency filter, high- frequency filter, and the machine used were not
TOXIC AND NUTRITIONAL DISORDER MODULE
 TOXIC AND NUTRITIONAL DISORDER MODULE Objectives: For each of the following entities the student should be able to: 1. Describe the etiology/pathogenesis and/or pathophysiology, gross and microscopic morphology
TOXIC AND NUTRITIONAL DISORDER MODULE Objectives: For each of the following entities the student should be able to: 1. Describe the etiology/pathogenesis and/or pathophysiology, gross and microscopic morphology
419 Church Street East, Penrose, Auckland 1642 P O Box 12063, Penrose, Auckland
 CHARCOT-MARIE-TOOTH DISEASE What is Charcot-Marie-Tooth Disease? First described in 1886 by three physicians Jean-Marie Charcot, Pierre Marie, and Howard Henry Tooth, CharcotMarie-Tooth (CMT) is a group
CHARCOT-MARIE-TOOTH DISEASE What is Charcot-Marie-Tooth Disease? First described in 1886 by three physicians Jean-Marie Charcot, Pierre Marie, and Howard Henry Tooth, CharcotMarie-Tooth (CMT) is a group
Deformity and Pain in Foot of Neuromuscular Disease
 Deformity and Pain in Foot of Neuromuscular Disease CHA 의과학대학재활의학과김민영 (2012.11.10 제 4 회대한발의학회 ) Neuromuscular Diseases & Foot Deformity : almost Pain : infrequent Non-ambulatory, Ambulatory Neuromuscular
Deformity and Pain in Foot of Neuromuscular Disease CHA 의과학대학재활의학과김민영 (2012.11.10 제 4 회대한발의학회 ) Neuromuscular Diseases & Foot Deformity : almost Pain : infrequent Non-ambulatory, Ambulatory Neuromuscular
panel tests assessing multiple genes at the same time for the diagnosis of one or more related disorders
 NGS tests panel tests assessing multiple genes at the same time for the diagnosis of one or more related disorders UKGTN website lists 13 laboratories offering a total of 56 panel test UKGTN listed panel
NGS tests panel tests assessing multiple genes at the same time for the diagnosis of one or more related disorders UKGTN website lists 13 laboratories offering a total of 56 panel test UKGTN listed panel
Hereditary disorders of peroxisomal metabolism.
 Hereditary disorders of peroxisomal metabolism http://www.cytochemistry.net/cell-biology/perox.jpg Peroxisomes single membrane organelles from less than 100 to more than 1000 per eukaryotic cell more than
Hereditary disorders of peroxisomal metabolism http://www.cytochemistry.net/cell-biology/perox.jpg Peroxisomes single membrane organelles from less than 100 to more than 1000 per eukaryotic cell more than
7 Medical Genetics. Hemoglobinopathies. Hemoglobinopathies. Protein and Gene Structure. and Biochemical Genetics
 SESSION 7 Medical Genetics Hemoglobinopathies and Biochemical Genetics J a v a d F a s a J a m s h i d i U n i v e r s i t y o f M e d i c a l S c i e n c e s, N o v e m b e r 2 0 1 7 Hemoglobinopathies
SESSION 7 Medical Genetics Hemoglobinopathies and Biochemical Genetics J a v a d F a s a J a m s h i d i U n i v e r s i t y o f M e d i c a l S c i e n c e s, N o v e m b e r 2 0 1 7 Hemoglobinopathies
Pediatric Aspects of EDX
 Pediatric Aspects of EDX Albert C. Clairmont, MD Associate Professor-Clinical The Ohio State University February 25, 2013 Objectives Overview of Pediatric Electrodiagnosis (EDX) Understand the different
Pediatric Aspects of EDX Albert C. Clairmont, MD Associate Professor-Clinical The Ohio State University February 25, 2013 Objectives Overview of Pediatric Electrodiagnosis (EDX) Understand the different
Electron transport chain chapter 6 (page 73) BCH 340 lecture 6
 BCH 340 lecture 6") Electron transport chain chapter 6 (page 73) BCH 340 lecture 6 The Metabolic Pathway of Cellular Respiration All of the reactions involved in cellular respiration can be grouped into three main stages
Electron transport chain chapter 6 (page 73) BCH 340 lecture 6 The Metabolic Pathway of Cellular Respiration All of the reactions involved in cellular respiration can be grouped into three main stages
MOTOR NEURONE DISEASE
 MOTOR NEURONE DISEASE Dr Arun Aggarwal Department of Rehabilitation Medicine, RPAH Department of Neurology, Concord Hospital. Motor Neurone Disease Umbrella term in UK and Australia (ALS in USA) Neurodegenerative
MOTOR NEURONE DISEASE Dr Arun Aggarwal Department of Rehabilitation Medicine, RPAH Department of Neurology, Concord Hospital. Motor Neurone Disease Umbrella term in UK and Australia (ALS in USA) Neurodegenerative
Fatty Acids Synthesis L3
 Fatty Acids Synthesis L3 The pathway for fatty acid synthesis occurs in the cytoplasm, whereas, oxidation occurs in the mitochondria. The other major difference is the use of nucleotide co-factors. Oxidation
Fatty Acids Synthesis L3 The pathway for fatty acid synthesis occurs in the cytoplasm, whereas, oxidation occurs in the mitochondria. The other major difference is the use of nucleotide co-factors. Oxidation
Metabolic Muscle Diseases
 Metabolic Muscle Diseases Dr Tim Hutchin, Birmingham Children s Hospital Jan 13, 2012 Metabolic myopathies are heterogeneous conditions with defects of muscle energy metabolism that result in predominantly
Metabolic Muscle Diseases Dr Tim Hutchin, Birmingham Children s Hospital Jan 13, 2012 Metabolic myopathies are heterogeneous conditions with defects of muscle energy metabolism that result in predominantly
Data Collection Worksheet
 Data Collection Worksheet To maximize consistency, the authors of the scale state that it is essential that clinicians adhere to the scale instructions. They also advise that the scales be administrated
Data Collection Worksheet To maximize consistency, the authors of the scale state that it is essential that clinicians adhere to the scale instructions. They also advise that the scales be administrated
Neurological Mitochondrial Cytopathies
 ORIGINAL ARTICLE M.M. Mehndiratta, P. Agarwal, M. Tatke,* M. Krishnamurthy Departments of Neurology and Pathology* G.B. Pant Hospital, New Delhi-110002, India. Summary The mitochondrial cytopathies are
ORIGINAL ARTICLE M.M. Mehndiratta, P. Agarwal, M. Tatke,* M. Krishnamurthy Departments of Neurology and Pathology* G.B. Pant Hospital, New Delhi-110002, India. Summary The mitochondrial cytopathies are
LOCALIZATION NEUROLOGY EPISODE VI HEARING LOSS AND GAIT ATAXIA
 LOCALIZATION NEUROLOGY EPISODE VI HEARING LOSS AND GAIT ATAXIA EPISODE VI HEARING LOSS APPROACH and DIAGNOSIS 2 Cochlea and Auditory nerve Pons (superior olive) lateral lemniscus Inferior colliculus Thalamus
LOCALIZATION NEUROLOGY EPISODE VI HEARING LOSS AND GAIT ATAXIA EPISODE VI HEARING LOSS APPROACH and DIAGNOSIS 2 Cochlea and Auditory nerve Pons (superior olive) lateral lemniscus Inferior colliculus Thalamus
Patterns of Single-Gene Inheritance Cont.
 Genetic Basis of Disease Patterns of Single-Gene Inheritance Cont. Traditional Mechanisms Chromosomal disorders Single-gene gene disorders Polygenic/multifactorial disorders Novel mechanisms Imprinting
Genetic Basis of Disease Patterns of Single-Gene Inheritance Cont. Traditional Mechanisms Chromosomal disorders Single-gene gene disorders Polygenic/multifactorial disorders Novel mechanisms Imprinting
10.1: Introduction. Cell types in neural tissue: Neurons Neuroglial cells (also known as neuroglia, glia, and glial cells) Dendrites.
 Dendrites.") 10.1: Introduction Copyright The McGraw-Hill Companies, Inc. Permission required for reproduction or display. Cell types in neural tissue: Neurons Neuroglial cells (also known as neuroglia, glia, and glial
10.1: Introduction Copyright The McGraw-Hill Companies, Inc. Permission required for reproduction or display. Cell types in neural tissue: Neurons Neuroglial cells (also known as neuroglia, glia, and glial
3) Approach to Ataxia - Dr. Zana
 Approach to Ataxia - Dr. Zana") 3) Approach to Ataxia - Dr. Zana Introduction Ataxia is derived from Greek word a -not, taxis -orderly, (not orderly/ not in order) Ataxia is the inability to make smooth, accurate and coordinated movements
3) Approach to Ataxia - Dr. Zana Introduction Ataxia is derived from Greek word a -not, taxis -orderly, (not orderly/ not in order) Ataxia is the inability to make smooth, accurate and coordinated movements
Exploring hereditary ataxia and spasticity in the era of whole exome sequencing
 Universiteit Antwerpen Faculteit Geneeskunde en Gezondheidswetenschappen Exploring hereditary ataxia and spasticity in the era of whole exome sequencing Proefschrift voorgelegd tot het behalen van de graad
Universiteit Antwerpen Faculteit Geneeskunde en Gezondheidswetenschappen Exploring hereditary ataxia and spasticity in the era of whole exome sequencing Proefschrift voorgelegd tot het behalen van de graad
Nerve Conduction Studies NCS
 Nerve Conduction Studies NCS Nerve conduction studies are an essential part of an EMG examination. The clinical usefulness of NCS in the diagnosis of diffuse and local neuropathies has been thoroughly
Nerve Conduction Studies NCS Nerve conduction studies are an essential part of an EMG examination. The clinical usefulness of NCS in the diagnosis of diffuse and local neuropathies has been thoroughly
In the past 13 years, a new chapter of human genetics, mitochondrial genetics, has opened
 ARTICLE The Other Human Genome Alan L. Shanske, MD; Sara Shanske, PhD; Salvatore DiMauro, MD In the past 13 years, a new chapter of human genetics, mitochondrial genetics, has opened up and is becoming
ARTICLE The Other Human Genome Alan L. Shanske, MD; Sara Shanske, PhD; Salvatore DiMauro, MD In the past 13 years, a new chapter of human genetics, mitochondrial genetics, has opened up and is becoming
Predominant Cerebellar Volume Loss as a Neuroradiologic Feature of Pediatric Respiratory Chain Defects
 AJNR Am J Neuroradiol 26:1675 1680, August 2005 Predominant Cerebellar Volume Loss as a Neuroradiologic Feature of Pediatric Respiratory Chain Defects Fernando Scaglia, Lee-Jun C. Wong, Georgirene D. Vladutiu,
AJNR Am J Neuroradiol 26:1675 1680, August 2005 Predominant Cerebellar Volume Loss as a Neuroradiologic Feature of Pediatric Respiratory Chain Defects Fernando Scaglia, Lee-Jun C. Wong, Georgirene D. Vladutiu,
Nerve Conduction Studies NCS
 Nerve Conduction Studies NCS Nerve conduction studies are an essential part of an EMG examination. The clinical usefulness of NCS in the diagnosis of diffuse and local neuropathies has been thoroughly
Nerve Conduction Studies NCS Nerve conduction studies are an essential part of an EMG examination. The clinical usefulness of NCS in the diagnosis of diffuse and local neuropathies has been thoroughly
AUTISM SCIENCE DIGEST: THE JOURNAL OF AUTISMONE ISSUE 01 APRIL 2011 REPRINTED WITH PERMISSION
 FRAN KENDALL, MD, has extensive experience in the diagnosis and management of children and adults with a wide array of inborn errors of metabolism, specifically mitochondrial and metabolic disorders. She
FRAN KENDALL, MD, has extensive experience in the diagnosis and management of children and adults with a wide array of inborn errors of metabolism, specifically mitochondrial and metabolic disorders. She
Peroxisomal Disorders
 Peroxisomal Disorders George Gray Birmingham Childrens Hospital Peroxisomal Disorders Peroxisomes are large single membrane bound organelles that are present in the cytoplasm of all cells. They are formed
Peroxisomal Disorders George Gray Birmingham Childrens Hospital Peroxisomal Disorders Peroxisomes are large single membrane bound organelles that are present in the cytoplasm of all cells. They are formed
Clinical Cell Biology Organelles in Health and Disease
 Department of Ophthalmology University of Kiel, University Medical Center Director: Prof. Dr. Johann Roider Clinical Cell Biology Organelles in Health and Disease Prof. Dr. Alexa Klettner Clinical cell
Department of Ophthalmology University of Kiel, University Medical Center Director: Prof. Dr. Johann Roider Clinical Cell Biology Organelles in Health and Disease Prof. Dr. Alexa Klettner Clinical cell
NEUROLOGY CLERKSHIP CORE CURRICULUM GUIDELINES
 NEUROLOGY CLERKSHIP CORE CURRICULUM GUIDELINES Endorsed by the following organizations - October 2000: American Academy of Neurology Association of University Professors of Neurology American Neurological
NEUROLOGY CLERKSHIP CORE CURRICULUM GUIDELINES Endorsed by the following organizations - October 2000: American Academy of Neurology Association of University Professors of Neurology American Neurological
Critical Illness Polyneuropathy CIP and Critical Illness Myopathy CIM. Andrzej Sladkowski
 Critical Illness Polyneuropathy CIP and Critical Illness Myopathy CIM Andrzej Sladkowski Potential causes of weakness in the ICU-1 Muscle disease Critical illness myopathy Inflammatory myopathy Hypokalemic
Critical Illness Polyneuropathy CIP and Critical Illness Myopathy CIM Andrzej Sladkowski Potential causes of weakness in the ICU-1 Muscle disease Critical illness myopathy Inflammatory myopathy Hypokalemic
Ahmed Abbas, Mark Cook, Liong Hiew Fu, Alistair Lewthwaite, Colin Shirley, Yusuf A. Rajabally
 2016, Elsevier. Licensed under the Creative Commons Attribution-NonCommercial-NoDerivatives 4.0 International http://creativecommons.org/licenses/by-nc-nd/4.0/ Accepted Manuscript A case of POEMS mimicking
2016, Elsevier. Licensed under the Creative Commons Attribution-NonCommercial-NoDerivatives 4.0 International http://creativecommons.org/licenses/by-nc-nd/4.0/ Accepted Manuscript A case of POEMS mimicking
Lactic Acidosis and Ascending Neuromuscular Syndrome. Mina Hosseinipour, M.D, MPH. University of North Carolina Project, Lilongwe Malawi
 Lactic Acidosis and Ascending Neuromuscular Syndrome Mina Hosseinipour, M.D, MPH. University of North Carolina Project, Lilongwe Malawi Introduction ART is rapidly scaling up in resource poor countries
Lactic Acidosis and Ascending Neuromuscular Syndrome Mina Hosseinipour, M.D, MPH. University of North Carolina Project, Lilongwe Malawi Introduction ART is rapidly scaling up in resource poor countries
Marah Bitar. Faisal Nimri ... Nafeth Abu Tarboosh
 8 Marah Bitar Faisal Nimri... Nafeth Abu Tarboosh Summary of the 8 steps of citric acid cycle Step 1. Acetyl CoA joins with a four-carbon molecule, oxaloacetate, releasing the CoA group and forming a six-carbon
8 Marah Bitar Faisal Nimri... Nafeth Abu Tarboosh Summary of the 8 steps of citric acid cycle Step 1. Acetyl CoA joins with a four-carbon molecule, oxaloacetate, releasing the CoA group and forming a six-carbon
Nerve. (2) Duration of the stimulus A certain period can give response. The Strength - Duration Curve
 Duration of the stimulus A certain period can give response. The Strength - Duration Curve") Nerve Neuron (nerve cell) is the structural unit of nervous system. Nerve is formed of large numbers of nerve fibers. Types of nerve fibers Myelinated nerve fibers Covered by myelin sheath interrupted
Nerve Neuron (nerve cell) is the structural unit of nervous system. Nerve is formed of large numbers of nerve fibers. Types of nerve fibers Myelinated nerve fibers Covered by myelin sheath interrupted
Neurophysiological Diagnosis of Birth Brachial Plexus Palsy. Dr Grace Ng Department of Paed PMH
 Neurophysiological Diagnosis of Birth Brachial Plexus Palsy Dr Grace Ng Department of Paed PMH Brachial Plexus Anatomy Brachial Plexus Cords Medial cord: motor and sensory conduction for median and ulnar
Neurophysiological Diagnosis of Birth Brachial Plexus Palsy Dr Grace Ng Department of Paed PMH Brachial Plexus Anatomy Brachial Plexus Cords Medial cord: motor and sensory conduction for median and ulnar
DSS-1. No financial disclosures
 DSS-1 No financial disclosures Clinical History 9 year old boy with past medical history significant for cerebral palsy, in-turning right foot, left clubfoot that was surgically corrected at 3 years of
DSS-1 No financial disclosures Clinical History 9 year old boy with past medical history significant for cerebral palsy, in-turning right foot, left clubfoot that was surgically corrected at 3 years of
Unit VIII Problem 5 Physiology: Cerebellum
 Unit VIII Problem 5 Physiology: Cerebellum - The word cerebellum means: the small brain. Note that the cerebellum is not completely separated into 2 hemispheres (they are not clearly demarcated) the vermis
Unit VIII Problem 5 Physiology: Cerebellum - The word cerebellum means: the small brain. Note that the cerebellum is not completely separated into 2 hemispheres (they are not clearly demarcated) the vermis
Fran D. Kendall, M.D.
 Fran D. Kendall, M.D. VMP, LLC Biochemical Genetics Metabolic, Mitochondrial & Inherited Disorders To provide basic background information on Mitochondrial Disease, its clinical features, diagnosis, treatment,
Fran D. Kendall, M.D. VMP, LLC Biochemical Genetics Metabolic, Mitochondrial & Inherited Disorders To provide basic background information on Mitochondrial Disease, its clinical features, diagnosis, treatment,
Hereditary disorders of peroxisomal metabolism.
 Hereditary disorders of peroxisomal metabolism http://www.cytochemistry.net/cell-biology/perox.jpg Peroxisomes single membrane organelles from less than 100 to more than 1000 per eucaryotic cell more than
Hereditary disorders of peroxisomal metabolism http://www.cytochemistry.net/cell-biology/perox.jpg Peroxisomes single membrane organelles from less than 100 to more than 1000 per eucaryotic cell more than
Update on the Genetics of Ataxia. Vicki Wheelock MD UC Davis Department of Neurology GHPP Clinic
 Update on the Genetics of Ataxia Vicki Wheelock MD UC Davis Department of Neurology GHPP Clinic Outline Definitions Review of genetics Autosomal Dominant cerebellar ataxias Autosomal Recessive cerebellar
Update on the Genetics of Ataxia Vicki Wheelock MD UC Davis Department of Neurology GHPP Clinic Outline Definitions Review of genetics Autosomal Dominant cerebellar ataxias Autosomal Recessive cerebellar
Corporate Medical Policy
 Corporate Medical Policy Genetic Testing for the Diagnosis of Inherited Peripheral Neuropathy File Name: Origination: Last CAP Review: Next CAP Review: Last Review: genetic_testing_for_the_diagnosis_of_inherited_peripheral_neuropathy
Corporate Medical Policy Genetic Testing for the Diagnosis of Inherited Peripheral Neuropathy File Name: Origination: Last CAP Review: Next CAP Review: Last Review: genetic_testing_for_the_diagnosis_of_inherited_peripheral_neuropathy
Audit and Compliance Department 1
 Introduction to Intraoperative Neuromonitoring An intro to those squiggly lines Kunal Patel MS, CNIM None Disclosures Learning Objectives History of Intraoperative Monitoring What is Intraoperative Monitoring
Introduction to Intraoperative Neuromonitoring An intro to those squiggly lines Kunal Patel MS, CNIM None Disclosures Learning Objectives History of Intraoperative Monitoring What is Intraoperative Monitoring
diphenylhydantoin therapy1
 Journal of Neurology, Neurosurgery, and Psychiatry, 1975, 38, 1235-1239 Motor nerve conduction study in patients on diphenylhydantoin therapy1 S. CHOKROVERTY2 AND Z. A. SAYEED3 From the Neurology Service
Journal of Neurology, Neurosurgery, and Psychiatry, 1975, 38, 1235-1239 Motor nerve conduction study in patients on diphenylhydantoin therapy1 S. CHOKROVERTY2 AND Z. A. SAYEED3 From the Neurology Service
Introduction and aims of the study
 Introduction and aims of the study 1 Chapter 1 Motor neuron diseases include the most incapacitating and life-threatening illnesses but also rather benign disorders with only mild symptoms and slow progression.
Introduction and aims of the study 1 Chapter 1 Motor neuron diseases include the most incapacitating and life-threatening illnesses but also rather benign disorders with only mild symptoms and slow progression.
Related Policies None
 Medical policy MP 2.04.117 BCBSA Ref. Policy: 2.04.117 Last Review: 06/22/2017 Effective Date: 06/22/2017 Section: Medicine End Date: 06/26/2018 Related Policies None DISCLAIMER Our medical policies are
Medical policy MP 2.04.117 BCBSA Ref. Policy: 2.04.117 Last Review: 06/22/2017 Effective Date: 06/22/2017 Section: Medicine End Date: 06/26/2018 Related Policies None DISCLAIMER Our medical policies are
Electron Transport and oxidative phosphorylation (ATP Synthesis) Dr. Howaida Nounou Biochemistry department Sciences college
 Dr. Howaida Nounou Biochemistry department Sciences college") Electron Transport and oxidative phosphorylation (ATP Synthesis) Dr. Howaida Nounou Biochemistry department Sciences college The Metabolic Pathway of Cellular Respiration All of the reactions involved
Electron Transport and oxidative phosphorylation (ATP Synthesis) Dr. Howaida Nounou Biochemistry department Sciences college The Metabolic Pathway of Cellular Respiration All of the reactions involved
Median-ulnar nerve communications and carpal tunnel syndrome
 Journal of Neurology, Neurosurgery, and Psychiatry, 1977, 40, 982-986 Median-ulnar nerve communications and carpal tunnel syndrome LUDWIG GUTMANN From the Department of Neurology, West Virginia University,
Journal of Neurology, Neurosurgery, and Psychiatry, 1977, 40, 982-986 Median-ulnar nerve communications and carpal tunnel syndrome LUDWIG GUTMANN From the Department of Neurology, West Virginia University,
Clinical Summaries. CLN1 Disease, infantile onset and others
 Clinical Summaries CLN1 Disease, infantile onset and others The gene called CLN1 lies on chromosome 1. CLN1 disease is inherited as an autosomal recessive disorder, which means that both chromosomes carry
Clinical Summaries CLN1 Disease, infantile onset and others The gene called CLN1 lies on chromosome 1. CLN1 disease is inherited as an autosomal recessive disorder, which means that both chromosomes carry
Functions of Nervous System Neuron Structure
 Chapter 10 Nervous System I Divisions of the Nervous System Cell Types of Neural Tissue neurons neuroglial cells Central Nervous System brain spinal cord Peripheral Nervous System nerves cranial nerves
Chapter 10 Nervous System I Divisions of the Nervous System Cell Types of Neural Tissue neurons neuroglial cells Central Nervous System brain spinal cord Peripheral Nervous System nerves cranial nerves