Basal Ganglia Involvement in Mitochondrial Acetoacetyl-CoA Thiolase deficiency (T2).
|
|
|
- Eric Jordan
- 6 years ago
- Views:
Transcription
1 Basal Ganglia Involvement in Mitochondrial Acetoacetyl-CoA Thiolase deficiency (T2). Stéphanie Paquay Robert Debré Hospital Reference Center For Metabolic Diseases Paris, France
2 Mitochondrial Acetoacetyl-CoA Thiolase Deficiency (MAT or T2) Key role of MAT in both isoleucine and ketone bodies metabolism (from Buhas et al 2013)
3 Mitochondrial Acetoacetyl-CoA Thiolase Deficiency (T2) Acute ketoacidotic episodes: ph < 7,1, bicarbonate < 7 mmol/l, variable glycemia massive ketone bodies production Specific metabolites in urine organic acid analysis: 2MAA, 2M3HB, TG Blood acylcarnitine analysis : C5:1, C5-OH Enzyme assay Molecular diagnosis : ACAT 1 gene
4 Objectives Extrapyramidal signs observed without prior ketoacidotic events within two T2-deficient patients diagnosed through presymptomatic screening in already affected families. Review of a French cohort Relationship between neurological involvement and T2 deficiency?
5 Methods 26 patients Enzymatic assay in Lyon ( ) Collected data: Age at diagnosis and at end of follow-up Parental consanguinity Psychomotor development, clinical and radiological features During acute episode : supportive care, ph, bicarbonate, anionic gap, glycemia, NH3, lactate, ketonemia/ketonuria Urine organic acids, blood acylcarnitines profile Enzymatic assay Molecular analysis 2 groups based on normal / abnormal neurological examination
6 Results N e u r o l o g i c a l involvement 6/26 Absence of metabolic crisis 2/26 Ketoacidotic crisis 4/26 Neurological signs prior to the first decompensation : 2/4 Secondary neurological involvement 2/4
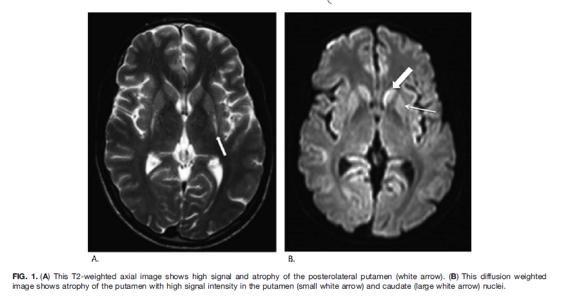
7 Clinical and radiological data for the 6/26 patients Presymptomatic with neurological diagnosis : abnormalities familial history Diagn Age End of Followup Consang Metabolic crisis # 1 5 m 8 y - - # 2 1 m 1 y + - # 3 2 m 3 y + + # 4 12 m 7 y - + Psychomotor Dvpt/Neurol. Examination Before acute E Psychomotor delay Axial hypotonia Psychomotor delay Axial hypotonia Dyskinesia # 5 24 m 13 y - + Normal After acute E Unchanged Unchanged Hypotonia Dyskinesia Dystonia # 6 11 m 19 y - + Normal Normal Evolution Motor delay Ataxia Dystonia Normal until 5 m. Hypotonia Dystonia Dystonia Improvement of dyskinesia Speech delay Dyspraxia Neurological sequelae 15 y Choreoathetosis Dystonia Brain MRI 6 y : T2/Flair hyperintensities in putamen 6 m : T2/Flair hyperintensities in putamen 11 m: T2/Flair hyperintensities in putamen 13 m : normal 28 m : T2 increased signal in putamen/ dentate nuclei 15 y : T2 high signal in pallidi and internal capsules

8 Results: 2 Patients with Neurological Signs Without Prior Ketoacidotic Events Patient #1: 8 years old Motor delay, frequent falls, ataxia, coordination disorders, Dystonic postures in upper limbs Other investigations unremarkable

9 Results: 2 Patients with Neurological Signs Without Prior Ketoacidotic Events Patient #2: 1-year old At the age of 5 months : axial hypotonia, dystonia, insufficient weight gain Other investigations unremarkable 9
10 Clinical and radiological data for the 6/26 patients with neurological abnormalities Neurological symptoms before the first crisis

11 Results: 2 Patients with Neurological Signs Before the First Ketoacidotic Episode Patient #3: 3 years old Psychomotor delay and axial hypotonia noted in the first months of life At the age of 5 months: bronchiolitis and first ketoacidotic event: ph 7,2, bicarbonate 9 mmol/l, glycemia 0,66 g/l. Rapid and favorable evolution with glucose infusion. Dystonic postures in upper right arm with closed fists. Other investigations unremarkable 11
12 Clinical and radiological data for the 6/26 patients with neurological abnormalities Neurological symptoms before the first crisis

13 Clinical and radiological data for the 6/26 patients with neurological abnormalities Neurological symptoms after the first crisis
14 Discussion T2 deficiency : Normal development or neurological sequelae after acute episode Our cohort : neurological findings and extrapyramidal signs in the absence of ketoacidotic events. What is known in the litterature?
15 Discussion 15
16 Physiopathological mechanisms : hypothesis? T2 : role in isoleucine metabolism >< SCOT! «Organic aciduria» 1. Toxicity of specific metabolites : role of 2-MAA et 2-MHB? Inhibition of energy metabolism by 2-methylacetoacetate and 2- methyl-3 hydroxybutyrate in cerebral cortex of developing rats». Rosa RB et al. J Inherit Metab 2005 Evidence that 2-methylacetoacetate induces oxidative stress in rat brain». Leipnitz G et al. Metab Brain Dis Cerebral vulnerability to oxidative stress Pathogenesis of CNS involvement in disorders of amino and organic acid metabolism. Kolker S et al, J Inherit Metab Dis
17 Discussion T2 deficiency : «An organic aciduria» Role of protein intake? T2 deficiency! accumulation of toxic isoleucine-derived acyl-coa esters in the brain mitochondria The protein intake could be responsible for an insidious cerebral toxicity Protein restriction prevents from neurological impairment? Usual practice «normal» protein intake + carnitine How much? An low-protein diet versus a «normal» protein intake? For how long? Vulnerability window during the 1 rst years? 17
18 Discussion Follow up : which monitoring tools? Correlation exists between the protein intake and body fluids excretion of T2 metabolites but intramitochondrial toxicity cannot be predicted based on the quantification of urine metabolites "Mild" genotype = detectable residual enzyme activity = "Mild" excretion but Our cohort : absence of genotype-phenotype correlation : great phenotypical variability within the same family 18
19 Conclusion 5/26 (19%) : neurological impairment and extrapyramidal signs without a clear relationship with a metabolic crisis T2 deficiency = in clinical practice not only as a ketolysis defect but also as an organic aciduria giving rise to a progressive and chronic cerebral intoxication Consider protein intake restriction : at least a «normal» protein diet + carnitine 19
20 Conclusion Neurological involvement in T2 deficiency as well as impact of protein intake on neurological impairment remain to be studied. Multicentric follow-up of newborns diagnosed through neonatal sreening could be useful to confirm the causality of this association Urine organic acid analysis could be performed in case of unexplained extrapyramidal signs since neonatal screening doesn t exclude the diagnosis 20
21 Acknowledgments M. Schiff Ch. Vianay-Saban P. de Lonlay D. Dobbelaere A. Fouilhoux N. Guffon F. Labarthe K. Mention G. Touati V. Valayannopoulos
Newborn Screen & Development Facts about the genetic diseases new since March 2006 (Excluding Cystic Fibrosis)
") Newborn Screen & Development Facts about the genetic diseases new since March 2006 (Excluding Cystic Fibrosis) 1) Argininosuccinic acidemia (ASA) a) Incidence: ~1 in 70,000 b) Deficiency in an enzyme of
Newborn Screen & Development Facts about the genetic diseases new since March 2006 (Excluding Cystic Fibrosis) 1) Argininosuccinic acidemia (ASA) a) Incidence: ~1 in 70,000 b) Deficiency in an enzyme of
Presentation and investigation of mitochondrial disease in children
 Presentation and investigation of mitochondrial disease in children Andrew Morris Willink Unit, Manchester Mitochondrial function Carbohydrate Fat Respiratory chain Energy Mitochondria are the product
Presentation and investigation of mitochondrial disease in children Andrew Morris Willink Unit, Manchester Mitochondrial function Carbohydrate Fat Respiratory chain Energy Mitochondria are the product
Guideline for the diagnosis and management of isovaleryl-coa-dehydrogenase deficiency (isovaleric acidemia) - a systematic review -
 - a systematic review -") Guideline for the diagnosis and management of isovaleryl-coa-dehydrogenase deficiency (isovaleric acidemia) - a systematic review - Guideline development group International interdisciplinary guideline
Guideline for the diagnosis and management of isovaleryl-coa-dehydrogenase deficiency (isovaleric acidemia) - a systematic review - Guideline development group International interdisciplinary guideline
Organic acidaemias (OAs) & Urea cycle disorders (UCDs) PRESENTATION & MANAGEMENT
 & Urea cycle disorders (UCDs) PRESENTATION & MANAGEMENT") Great Ormond Street Hospital London 20/04/2018 Organic acidaemias (OAs) & Urea cycle disorders (UCDs) PRESENTATION & MANAGEMENT Spyros P. Batzios, MD, MSc, PhD OAs & UCDs How do they present? neonatal
Great Ormond Street Hospital London 20/04/2018 Organic acidaemias (OAs) & Urea cycle disorders (UCDs) PRESENTATION & MANAGEMENT Spyros P. Batzios, MD, MSc, PhD OAs & UCDs How do they present? neonatal
Fatty Acid Oxidation Disorders- an update. Fiona Carragher Biochemical Sciences, GSTS Pathology St Thomas Hospital, London
 Fatty Acid Oxidation Disorders- an update Fiona Carragher Biochemical Sciences, GSTS Pathology St Thomas Hospital, London An update. Overview of metabolism Clinical presentation and outcome Diagnostic
Fatty Acid Oxidation Disorders- an update Fiona Carragher Biochemical Sciences, GSTS Pathology St Thomas Hospital, London An update. Overview of metabolism Clinical presentation and outcome Diagnostic
ANATOMY OF A METABOLIC CRISIS: FAOD-style. Mark S. Korson, MD Tufts Medical Center Boston, MA
 ANATOMY OF A METABOLIC CRISIS: FAOD-style Mark S. Korson, MD Tufts Medical Center Boston, MA NORMAL PHYSIOLOGY Anabolic Eating well Calories eaten > body s needs BRAIN uses GLUCOSE MUSCLE uses GLUCOSE
ANATOMY OF A METABOLIC CRISIS: FAOD-style Mark S. Korson, MD Tufts Medical Center Boston, MA NORMAL PHYSIOLOGY Anabolic Eating well Calories eaten > body s needs BRAIN uses GLUCOSE MUSCLE uses GLUCOSE
Metabolic Changes in ASD. Norma J. Arciniegas, MD Simón E. Carlo, MD Instituto Filius
 Metabolic Changes in ASD Norma J. Arciniegas, MD Simón E. Carlo, MD Instituto Filius 12 patients 3 Autism: Ages 3/3/3.7 3 PDD: Ages 3/3/6 3 Asperger: Ages 6/7/15.1 3 Speech delay and Sensory Problems (SHL):
Metabolic Changes in ASD Norma J. Arciniegas, MD Simón E. Carlo, MD Instituto Filius 12 patients 3 Autism: Ages 3/3/3.7 3 PDD: Ages 3/3/6 3 Asperger: Ages 6/7/15.1 3 Speech delay and Sensory Problems (SHL):
The spectrum and outcome of the. neonates with inborn errors of. metabolism at a tertiary care hospital
 The spectrum and outcome of the neonates with inborn errors of metabolism at a tertiary care hospital Dr. Sevim Ünal Neonatology Division, Ankara Children s Hematology Oncology Research Hospital, Ankara,
The spectrum and outcome of the neonates with inborn errors of metabolism at a tertiary care hospital Dr. Sevim Ünal Neonatology Division, Ankara Children s Hematology Oncology Research Hospital, Ankara,
/04/ PEDIATRIC RESEARCH Vol. 56, No. 1, 2004 Copyright 2004 International Pediatric Research Foundation, Inc.
 0031-3998/04/5601-0060 PEDIATRIC RESEARCH Vol. 56, No. 1, 2004 Copyright 2004 International Pediatric Research Foundation, Inc. Printed in U.S.A. Mitochondrial Acetoacetyl-CoA Thiolase (T2) Deficiency:
0031-3998/04/5601-0060 PEDIATRIC RESEARCH Vol. 56, No. 1, 2004 Copyright 2004 International Pediatric Research Foundation, Inc. Printed in U.S.A. Mitochondrial Acetoacetyl-CoA Thiolase (T2) Deficiency:
Introduction to Organic Acidemias. Hilary Vernon, MD PhD Assistant Professor of Genetic Medicine Johns Hopkins University 7.25.
 Introduction to Organic Acidemias Hilary Vernon, MD PhD Assistant Professor of Genetic Medicine Johns Hopkins University 7.25.2014 A Brief Historical Overview Garrod, Archibald E. 1902. The Incidence of
Introduction to Organic Acidemias Hilary Vernon, MD PhD Assistant Professor of Genetic Medicine Johns Hopkins University 7.25.2014 A Brief Historical Overview Garrod, Archibald E. 1902. The Incidence of
Isovaleric Acidemia: Quick reference guide
 Isovaleric Acidemia: Quick reference guide Introduction Isovaleric acidemia (IVA) is an inborn error of the leucine pathway caused by defects of the isovaleryl-oadehydrogenase (IV). The clinical presentation
Isovaleric Acidemia: Quick reference guide Introduction Isovaleric acidemia (IVA) is an inborn error of the leucine pathway caused by defects of the isovaleryl-oadehydrogenase (IV). The clinical presentation
e-learning Fatty Acid Oxidation Defects Camilla Reed and Dr Simon Olpin Sheffield Children s Hospital
 e-learning Fatty Acid Oxidation Defects Camilla Reed and Dr Simon Olpin Sheffield Children s Hospital Fatty Acids Fatty acids are a major source of energy and body fat is an energy dense material. They
e-learning Fatty Acid Oxidation Defects Camilla Reed and Dr Simon Olpin Sheffield Children s Hospital Fatty Acids Fatty acids are a major source of energy and body fat is an energy dense material. They
Methylmalonic aciduria
 Methylmalonic aciduria Introductory information Written by: F. Hörster, S. Kölker & P. Burgard Reviewed & Revised for North America by: S. van Calcar Methylmalonic aciduria MMA 2 Methylmalonic aciduria
Methylmalonic aciduria Introductory information Written by: F. Hörster, S. Kölker & P. Burgard Reviewed & Revised for North America by: S. van Calcar Methylmalonic aciduria MMA 2 Methylmalonic aciduria
National Metabolic Biochemistry Network Guidelines for the investigation of hypoglycaemia in infants and children
 National Metabolic Biochemistry Network Guidelines for the investigation of hypoglycaemia in infants and children Aim To provide guidance on the biochemical investigation of hypoglycaemia in infants and
National Metabolic Biochemistry Network Guidelines for the investigation of hypoglycaemia in infants and children Aim To provide guidance on the biochemical investigation of hypoglycaemia in infants and
CLINICAL SIGNS SUGGESTIVE OF A NEUROMETABOLIC DISEASE. Bwee Tien Poll-The Amsterdam UMC The Netherlands
 CLINICAL SIGNS SUGGESTIVE OF A NEUROMETABOLIC DISEASE Bwee Tien Poll-The Amsterdam UMC The Netherlands FRAMEWORK OF PRINCIPALS 1. Problem-oriented clinical approach 2. Biomarkers in plasma, urine, CSF
CLINICAL SIGNS SUGGESTIVE OF A NEUROMETABOLIC DISEASE Bwee Tien Poll-The Amsterdam UMC The Netherlands FRAMEWORK OF PRINCIPALS 1. Problem-oriented clinical approach 2. Biomarkers in plasma, urine, CSF
THE ED APPROACH OF THE CHILD WITH SUSPECTED METABOLIC DISEASE
 THE ED APPROACH OF THE CHILD WITH SUSPECTED METABOLIC DISEASE Dr. Nadeem Qureshi M.D, FAAP, FCCM Associate Professor Pediatrics School of Medicine. St Louis University Attending Physician Pediatric Emergency
THE ED APPROACH OF THE CHILD WITH SUSPECTED METABOLIC DISEASE Dr. Nadeem Qureshi M.D, FAAP, FCCM Associate Professor Pediatrics School of Medicine. St Louis University Attending Physician Pediatric Emergency
Newborn Screening & Methods for Diagnosing Inborn Errors of Metabolism
 Newborn Screening & Methods for Diagnosing Inborn Errors of Metabolism Patricia Jones, PhD DABCC FACB UT Southwestern Medical Center Children s Medical Center Dallas, Texas Learning Objectives Justify
Newborn Screening & Methods for Diagnosing Inborn Errors of Metabolism Patricia Jones, PhD DABCC FACB UT Southwestern Medical Center Children s Medical Center Dallas, Texas Learning Objectives Justify
THIAMINE TRANSPORTER TYPE 2 DEFICIENCY
 THIAMINE TRANSPORTER TYPE 2 DEFICIENCY WHAT IS THE THIAMINE TRANSPORTER TYPE 2 DEFICIENCY (hthtr2)? The thiamine transporter type 2 deficiency (hthtr2) is a inborn error of thiamine metabolism caused by
THIAMINE TRANSPORTER TYPE 2 DEFICIENCY WHAT IS THE THIAMINE TRANSPORTER TYPE 2 DEFICIENCY (hthtr2)? The thiamine transporter type 2 deficiency (hthtr2) is a inborn error of thiamine metabolism caused by
Genomics & Modern Health Care Caring for the Special Children & Adults of Isolated Populations. Propionic Acidemia
 Genomics & Modern Health Care Caring for the Special Children & Adults of Isolated Populations Propionic Acidemia D. Holmes Morton MD Pediatrician, Clinic for Special Children Strasburg, Pennsylvania 17579
Genomics & Modern Health Care Caring for the Special Children & Adults of Isolated Populations Propionic Acidemia D. Holmes Morton MD Pediatrician, Clinic for Special Children Strasburg, Pennsylvania 17579
MR Imaging and Proton Spectroscopy in 3-Hydroxy-3-Methylglutaryl Coenzyme A Lyase Deficiency
 AJNR Am J Neuroradiol 19:78 82, February 1998 MR Imaging and Proton Spectroscopy in -Hydroxy--Methylglutaryl Coenzyme A Lyase Deficiency M. S. van der Knaap, H. D. Bakker, and J. Valk Summary: Three patients
AJNR Am J Neuroradiol 19:78 82, February 1998 MR Imaging and Proton Spectroscopy in -Hydroxy--Methylglutaryl Coenzyme A Lyase Deficiency M. S. van der Knaap, H. D. Bakker, and J. Valk Summary: Three patients
Clinical aspects of pterin disorders
 Clinical aspects of pterin disorders Thomas Opladen, MD University Children s Hospital Department of Inborn Errors of Metabolism Heidelberg Germany Introductory words Brain function depends on the capacity
Clinical aspects of pterin disorders Thomas Opladen, MD University Children s Hospital Department of Inborn Errors of Metabolism Heidelberg Germany Introductory words Brain function depends on the capacity
Cranial Ultrasonography in Maple Syrup Urine Disease
 Cranial Ultrasonography in Maple Syrup Urine Disease Giuseppe Fariello, Carlo Dionisi-Vici, Cinzia Orazi, Saverio Malena, Andrea Bartuli, Paolo Schingo, Enza Carnevale, Isora Saponara, and Gaetano Sabetta
Cranial Ultrasonography in Maple Syrup Urine Disease Giuseppe Fariello, Carlo Dionisi-Vici, Cinzia Orazi, Saverio Malena, Andrea Bartuli, Paolo Schingo, Enza Carnevale, Isora Saponara, and Gaetano Sabetta
Objectives By the end of lecture the student should:
 Objectives By the end of lecture the student should: Discuss β oxidation of fatty acids. Illustrate α oxidation of fatty acids. Understand ω oxidation of fatty acids. List sources and fates of active acetate.
Objectives By the end of lecture the student should: Discuss β oxidation of fatty acids. Illustrate α oxidation of fatty acids. Understand ω oxidation of fatty acids. List sources and fates of active acetate.
Urea Cycle Defects. Dr Mick Henderson. Biochemical Genetics Leeds Teaching Hospitals Trust. MetBioNet IEM Introductory Training
 Urea Cycle Defects Dr Mick Henderson Biochemical Genetics Leeds Teaching Hospitals Trust The Urea Cycle The urea cycle enables toxic ammonia molecules to be converted to the readily excreted and non toxic
Urea Cycle Defects Dr Mick Henderson Biochemical Genetics Leeds Teaching Hospitals Trust The Urea Cycle The urea cycle enables toxic ammonia molecules to be converted to the readily excreted and non toxic
For healthcare professionals Methylmalonic Acidurias
 www.e-imd.org For healthcare professionals Methylmalonic Acidurias Methylmalonic acidurias (MMAurias) comprise a group of inborn errors of metabolism characterized by an isolated accumulation of methylmalonic
www.e-imd.org For healthcare professionals Methylmalonic Acidurias Methylmalonic acidurias (MMAurias) comprise a group of inborn errors of metabolism characterized by an isolated accumulation of methylmalonic
SCAD and GA-II: Truths and Confusions
 SCAD and GA-II: Truths and Confusions Bill Rhead* Medical College of Wisconsin *MD, PhD GA-II Severe GA-II is as bad as: SCAD + MCAD + COMBINED! VLCAD + IVA + GA-I Severe GA-II is always fatal Mild
SCAD and GA-II: Truths and Confusions Bill Rhead* Medical College of Wisconsin *MD, PhD GA-II Severe GA-II is as bad as: SCAD + MCAD + COMBINED! VLCAD + IVA + GA-I Severe GA-II is always fatal Mild
o They are usually used in Forensic or Medico-legal practice, Commonly used are Blood Alcohol Concentration (BAC) and Expired Air
 and Expired Air") 1 ETHANOL: UNIVERSITY OF PNG SCHOOL OF MEDICINE AND HEALTH SCIENCES DIVISION OF BASIC MEDICAL SCIENCES DISCIPLINE OF BIOCHEMISTRY AND MOLECULAR BIOLOGY PBL SEMINAR OVERVIEW OF ALCOHOL (ETHANOL & METHANOL)
1 ETHANOL: UNIVERSITY OF PNG SCHOOL OF MEDICINE AND HEALTH SCIENCES DIVISION OF BASIC MEDICAL SCIENCES DISCIPLINE OF BIOCHEMISTRY AND MOLECULAR BIOLOGY PBL SEMINAR OVERVIEW OF ALCOHOL (ETHANOL & METHANOL)
Work-Up and Initial Management of Common Metabolic Emergencies in the Inpatient Setting
 Work-Up and Initial Management of Common Metabolic Emergencies in the Inpatient Setting Kristin Lindstrom, MD Division of Genetics and Metabolism Phoenix Children s Hospital AzAAP Pediatrics in the Red
Work-Up and Initial Management of Common Metabolic Emergencies in the Inpatient Setting Kristin Lindstrom, MD Division of Genetics and Metabolism Phoenix Children s Hospital AzAAP Pediatrics in the Red
Nutritional Interventions in Primary Mitochondrial Disorders
 Nutritional Interventions in Primary Mitochondrial Disorders Carolyn J Ellaway MBBS PhD FRACP CGHGSA Genetic Metabolic Disorders Service Sydney Children s Hospital Network Disciplines of Child and Adolescent
Nutritional Interventions in Primary Mitochondrial Disorders Carolyn J Ellaway MBBS PhD FRACP CGHGSA Genetic Metabolic Disorders Service Sydney Children s Hospital Network Disciplines of Child and Adolescent
Summary. Syndromic versus Etiologic. Definitions. Why does it matter? ASD=autism
 Summary It is becoming clear that multiple genes with complex interactions underlie autism spectrum (ASD). A small subset of people with ASD, however, actually suffer from rare single-gene Important to
Summary It is becoming clear that multiple genes with complex interactions underlie autism spectrum (ASD). A small subset of people with ASD, however, actually suffer from rare single-gene Important to
CHY2026: General Biochemistry. Lipid Metabolism
 CHY2026: General Biochemistry Lipid Metabolism Lipid Digestion Lipid Metabolism Fats (triglycerides) are high metabolic energy molecules Fats yield 9.3 kcal of energy (carbohydrates and proteins 4.1 kcal)
CHY2026: General Biochemistry Lipid Metabolism Lipid Digestion Lipid Metabolism Fats (triglycerides) are high metabolic energy molecules Fats yield 9.3 kcal of energy (carbohydrates and proteins 4.1 kcal)
Cardiac Disease in Organic Acidemias
 Cardiac Disease in Organic Acidemias Kathryn Chatfield, MD, PhD Assistant Professor of Pediatrics Division of Cardiology University of Colorado School of Medicine Children s Hospital Colorado Introduction
Cardiac Disease in Organic Acidemias Kathryn Chatfield, MD, PhD Assistant Professor of Pediatrics Division of Cardiology University of Colorado School of Medicine Children s Hospital Colorado Introduction
TEMPLE. Tools Enabling Metabolic Parents LEarning ADAPTED BY THE DIETITIANS GROUP. British Inherited Metabolic Diseases Group
 TEMPLE Tools Enabling Metabolic Parents LEarning ADAPTED BY THE DIETITIANS GROUP British Inherited Metabolic Diseases Group BASED ON THE ORIGINAL TEMPLE WRITTEN BY KOLKER AND BURGARD GA1 Information for
TEMPLE Tools Enabling Metabolic Parents LEarning ADAPTED BY THE DIETITIANS GROUP British Inherited Metabolic Diseases Group BASED ON THE ORIGINAL TEMPLE WRITTEN BY KOLKER AND BURGARD GA1 Information for
INBORN ERRORS OF METABOLISM (IEM) IAP UG Teaching slides
 IAP UG Teaching slides") INBORN ERRORS OF METABOLISM (IEM) 1 OBJECTIVES What are IEMs? Categories When to suspect? History and clinical pointers Metabolic presentation Differential diagnosis Emergency and long term management
INBORN ERRORS OF METABOLISM (IEM) 1 OBJECTIVES What are IEMs? Categories When to suspect? History and clinical pointers Metabolic presentation Differential diagnosis Emergency and long term management
ESPEN Congress Madrid 2018
 ESPEN Congress Madrid 2018 Inborn Errors Of Metabolism Urea Cycle Disorders Diagnosis And Care F. Feillet (FR) Urea cycle disorders, diagnosis and care F Feillet National reference centre for Inborn errors
ESPEN Congress Madrid 2018 Inborn Errors Of Metabolism Urea Cycle Disorders Diagnosis And Care F. Feillet (FR) Urea cycle disorders, diagnosis and care F Feillet National reference centre for Inborn errors
UK NATIONAL METABOLIC BIOCHEMISTRY NETWORK GUIDELINES FOR THE INVESTIGATION OF HYPERAMMONAEMIA
 UK NATIONAL METABOLIC BIOCHEMISTRY NETWORK GUIDELINES FOR THE INVESTIGATION OF HYPERAMMONAEMIA Hyperammonaemia results from defective catabolism of amino acids to urea. Recognition and treatment of hyperammonaemia,
UK NATIONAL METABOLIC BIOCHEMISTRY NETWORK GUIDELINES FOR THE INVESTIGATION OF HYPERAMMONAEMIA Hyperammonaemia results from defective catabolism of amino acids to urea. Recognition and treatment of hyperammonaemia,
Very-long-chain acyl-coa dehydrogenase deficiency
 Very-long-chain acyl-coa dehydrogenase deficiency Introductory information Written by: V. Prietsch & P. Burgard Reviewed & Revised for North America by: S. van Calcar Very-long-chain acyl-coa dehydrogenase
Very-long-chain acyl-coa dehydrogenase deficiency Introductory information Written by: V. Prietsch & P. Burgard Reviewed & Revised for North America by: S. van Calcar Very-long-chain acyl-coa dehydrogenase
Anaesthesia recommendations for patients suffering from. Glutaric acidemia type 1
 orphananesthesia Anaesthesia recommendations for patients suffering from Disease name: Glutaric acidemia type 1 Glutaric acidemia type 1 ICD 10: E72.3 - Disorders of lysine and hydroxylysine metabolism
orphananesthesia Anaesthesia recommendations for patients suffering from Disease name: Glutaric acidemia type 1 Glutaric acidemia type 1 ICD 10: E72.3 - Disorders of lysine and hydroxylysine metabolism
BASAL GANGLIA. Dr JAMILA EL MEDANY
 BASAL GANGLIA Dr JAMILA EL MEDANY OBJECTIVES At the end of the lecture, the student should be able to: Define basal ganglia and enumerate its components. Enumerate parts of Corpus Striatum and their important
BASAL GANGLIA Dr JAMILA EL MEDANY OBJECTIVES At the end of the lecture, the student should be able to: Define basal ganglia and enumerate its components. Enumerate parts of Corpus Striatum and their important
Part III => METABOLISM and ENERGY. 3.4 Lipid Catabolism 3.4a Fatty Acid Degradation 3.4b Ketone Bodies
 Part III => METABOLISM and ENERGY 3.4 Lipid Catabolism 3.4a Fatty Acid Degradation 3.4b Ketone Bodies Section 3.4a: Fatty Acid Degradation Synopsis 3.4a - Triglycerides (or fats) in the diet or adipose
Part III => METABOLISM and ENERGY 3.4 Lipid Catabolism 3.4a Fatty Acid Degradation 3.4b Ketone Bodies Section 3.4a: Fatty Acid Degradation Synopsis 3.4a - Triglycerides (or fats) in the diet or adipose
Fatty Acid Beta-Oxidation Disorders: A Brief Review
 Mini Review Received: July 12, 2015 Accepted: November 12, 2015 Published online: March 11, 2016 Fatty Acid Beta-Oxidation Disorders: A Brief Review Vijay A. Vishwanath Division of Pediatric Neurology,
Mini Review Received: July 12, 2015 Accepted: November 12, 2015 Published online: March 11, 2016 Fatty Acid Beta-Oxidation Disorders: A Brief Review Vijay A. Vishwanath Division of Pediatric Neurology,
Roles of Lipids. principal form of stored energy major constituents of cell membranes vitamins messengers intra and extracellular
 Roles of Lipids principal form of stored energy major constituents of cell membranes vitamins messengers intra and extracellular = Oxidation of fatty acids Central energy-yielding pathway in animals. O
Roles of Lipids principal form of stored energy major constituents of cell membranes vitamins messengers intra and extracellular = Oxidation of fatty acids Central energy-yielding pathway in animals. O
Analysis of Neurotransmitters. Simon Heales
 Analysis of Neurotransmitters Simon Heales HealeS@gosh.nhs.uk Tyrosine O 2 L-Dopa Dopamine HVA Tryptophan 5-HTP PLP Serotonin 5-HIAA Phenylalanine Tyrosine BH4 qbh2 BH2 CSF Sample Requirements Tube 1 Tube
Analysis of Neurotransmitters Simon Heales HealeS@gosh.nhs.uk Tyrosine O 2 L-Dopa Dopamine HVA Tryptophan 5-HTP PLP Serotonin 5-HIAA Phenylalanine Tyrosine BH4 qbh2 BH2 CSF Sample Requirements Tube 1 Tube
TITLE: NURSING GUIDELINES FOR THE MANAGEMENT OF CHILDREN WITH METHYLMALONIC ACIDURIA. Eilish O Connell, Clinical Education Facilitator, NCIMD
 NO. OF PAGES: Page 1 of 17 SUPERCEDES: N/A NURSING GUIDELINES FOR THE MANAGEMENT OF CHILDREN WITH METHYLMALONIC ACIDURIA NAME/ Eilish O Connell, Clinical Education Facilitator, NCIMD SIGNATURE: DATE: NAME/
NO. OF PAGES: Page 1 of 17 SUPERCEDES: N/A NURSING GUIDELINES FOR THE MANAGEMENT OF CHILDREN WITH METHYLMALONIC ACIDURIA NAME/ Eilish O Connell, Clinical Education Facilitator, NCIMD SIGNATURE: DATE: NAME/
Module : Clinical correlates of disorders of metabolism Block 3, Week 2
 Module : Clinical correlates of disorders of metabolism Block 3, Week 2 Department of Paediatrics and Child Health University of Pretoria Tutor : Prof DF Wittenberg : dwittenb@medic.up.ac.za Aim of this
Module : Clinical correlates of disorders of metabolism Block 3, Week 2 Department of Paediatrics and Child Health University of Pretoria Tutor : Prof DF Wittenberg : dwittenb@medic.up.ac.za Aim of this
Selective newborn screening of amino acid, fatty acid and organic acid disorders in the Kingdom of Bahrain.
 Selective newborn screening of amino acid, fatty acid and organic acid disorders in the Kingdom of Bahrain. Jamal Golbahar PhD Associate Professor of Molecular Medicine, Department of Molecular Medicine,
Selective newborn screening of amino acid, fatty acid and organic acid disorders in the Kingdom of Bahrain. Jamal Golbahar PhD Associate Professor of Molecular Medicine, Department of Molecular Medicine,
Inborn Errors of Metabolism in the Emergency Department. Will Davies June 2014
 Inborn Errors of Metabolism in the Emergency Department Will Davies June 2014 Inborn Errors of Metabolism in the Emergency Department Overview Although individually rare, altogether they are 1:1000-2500
Inborn Errors of Metabolism in the Emergency Department Will Davies June 2014 Inborn Errors of Metabolism in the Emergency Department Overview Although individually rare, altogether they are 1:1000-2500
My Experiences and Understanding of VLCAD Deficiency and its Treatment Charles R. Roe, MD June 26, 2011 (Now retired)
") My Experiences and Understanding of VLCAD Deficiency and its Treatment Charles R. Roe, MD June 26, 2011 (Now retired) This disorder is characterized by the deficiency of the VLCAD enzyme that is required
My Experiences and Understanding of VLCAD Deficiency and its Treatment Charles R. Roe, MD June 26, 2011 (Now retired) This disorder is characterized by the deficiency of the VLCAD enzyme that is required
Manipulation of the Nutrient Sensors (AMPK/TOR) with Anaplerotic Diet Therapy (Triheptanoin) An Alternative to Diet Restriction
 with Anaplerotic Diet Therapy (Triheptanoin) An Alternative to Diet Restriction") Manipulation of the Nutrient Sensors (AMPK/TOR) with Anaplerotic Diet Therapy (Triheptanoin) An Alternative to Diet Restriction CharlesR.Roe,MD Institute of Metabolic Disease Baylor University Medical
Manipulation of the Nutrient Sensors (AMPK/TOR) with Anaplerotic Diet Therapy (Triheptanoin) An Alternative to Diet Restriction CharlesR.Roe,MD Institute of Metabolic Disease Baylor University Medical
For healthcare professionals Glutaric aciduria type I
 www.e-imd.org For healthcare professionals Glutaric aciduria type I Abstract Glutaric aciduria type I (synonym, glutaric acidemia type I) is a rare organic aciduria (estimated prevalence is 1 in 100-120,000
www.e-imd.org For healthcare professionals Glutaric aciduria type I Abstract Glutaric aciduria type I (synonym, glutaric acidemia type I) is a rare organic aciduria (estimated prevalence is 1 in 100-120,000
Inborn Errors of Metabolism. Metabolic Pathway. Digestion and Fasting. How is Expanded Newborn Screening Different? MS/MS. The body is a factory.
 Inborn Errors of Metabolism The body is a factory. Inborn errors of metabolism are rare genetic disorders in which the body cannot properly turn food into energy. The disorders are usually caused by defects
Inborn Errors of Metabolism The body is a factory. Inborn errors of metabolism are rare genetic disorders in which the body cannot properly turn food into energy. The disorders are usually caused by defects
Carnitine palmitoyl transferase 2 deficiency (CPT2) is a rare inherited disorder that occurs when
 is a rare inherited disorder that occurs when") CPT2 Deficiency Carnitine palmitoyl transferase 2 deficiency (CPT2) is a rare inherited disorder that occurs when the last step in the entry of fats into sac-like bodies called mitochondria is blocked.
CPT2 Deficiency Carnitine palmitoyl transferase 2 deficiency (CPT2) is a rare inherited disorder that occurs when the last step in the entry of fats into sac-like bodies called mitochondria is blocked.
THE GLUCOSE-FATTY ACID-KETONE BODY CYCLE Role of ketone bodies as respiratory substrates and metabolic signals
 Br. J. Anaesth. (1981), 53, 131 THE GLUCOSE-FATTY ACID-KETONE BODY CYCLE Role of ketone bodies as respiratory substrates and metabolic signals J. C. STANLEY In this paper, the glucose-fatty acid cycle
Br. J. Anaesth. (1981), 53, 131 THE GLUCOSE-FATTY ACID-KETONE BODY CYCLE Role of ketone bodies as respiratory substrates and metabolic signals J. C. STANLEY In this paper, the glucose-fatty acid cycle
A rough guide to Acylcarnitines
 A rough guide to Acylcarnitines Roy Talbot & Nigel Manning Roy.Talbot@sch.nhs.uk Dept. of Clinical Chemistry, Sheffield Children s Hospital Menu Acylcarnitines Basic Tandem MS theory SCADD MCADD LCHADD
A rough guide to Acylcarnitines Roy Talbot & Nigel Manning Roy.Talbot@sch.nhs.uk Dept. of Clinical Chemistry, Sheffield Children s Hospital Menu Acylcarnitines Basic Tandem MS theory SCADD MCADD LCHADD
Oxidation of Long Chain Fatty Acids
 Oxidation of Long Chain Fatty Acids Dr NC Bird Oxidation of long chain fatty acids is the primary source of energy supply in man and animals. Hibernating animals utilise fat stores to maintain body heat,
Oxidation of Long Chain Fatty Acids Dr NC Bird Oxidation of long chain fatty acids is the primary source of energy supply in man and animals. Hibernating animals utilise fat stores to maintain body heat,
New developments in Urea Cycle Disorders and its impact on patients
 New developments in Urea Cycle Disorders and its impact on patients Johannes Häberle University Children s Hospital Zurich, Division of Metabolism 16 June 2015 SFEIM 2015, Lille, France The urea cycle
New developments in Urea Cycle Disorders and its impact on patients Johannes Häberle University Children s Hospital Zurich, Division of Metabolism 16 June 2015 SFEIM 2015, Lille, France The urea cycle
Lecture 10 - Protein Turnover and Amino Acid Catabolism
 Lecture 10 - Protein Turnover and Amino Acid Catabolism Chem 454: Regulatory Mechanisms in Biochemistry University of Wisconsin-Eau Claire 1 Introduction 2 Proteins are degraded into amino acids. Protein
Lecture 10 - Protein Turnover and Amino Acid Catabolism Chem 454: Regulatory Mechanisms in Biochemistry University of Wisconsin-Eau Claire 1 Introduction 2 Proteins are degraded into amino acids. Protein
Senior Vice President, Medical Affairs and Health Outcomes, 150 South Saunders Road, Lake Forest, IL 60045, Horizon Pharma USA, Inc, USA
 Research Hyperammonemic crises in patients with urea cycle disorders on chronic nitrogen scavenger therapy with either sodium phenylbutyrate or glycerol phenylbutyrate Jeffrey D Kent 1, Robert J Holt,2
Research Hyperammonemic crises in patients with urea cycle disorders on chronic nitrogen scavenger therapy with either sodium phenylbutyrate or glycerol phenylbutyrate Jeffrey D Kent 1, Robert J Holt,2
The laboratory investigation of lactic acidaemia. J Bonham/T Laing
 The laboratory investigation of lactic acidaemia J Bonham/T Laing Reference range Typical ranges for blood lactate are: Newborn 0.3-2.2 mmol/l Nielsen J et al1 1994 1-12mo 0.9-1.8 mmol/l Bonnefont et al
The laboratory investigation of lactic acidaemia J Bonham/T Laing Reference range Typical ranges for blood lactate are: Newborn 0.3-2.2 mmol/l Nielsen J et al1 1994 1-12mo 0.9-1.8 mmol/l Bonnefont et al
Inborn Errors of Metabolism (IEM)
") Clinical Presentation Inborn Errors of Metabolism (IEM) Click on the following: - Clinical Pearl - link to movie clip - link to picture Investigations Blood Work Urine No Acidosis NH 4 + Metabolic Acidosis
Clinical Presentation Inborn Errors of Metabolism (IEM) Click on the following: - Clinical Pearl - link to movie clip - link to picture Investigations Blood Work Urine No Acidosis NH 4 + Metabolic Acidosis
A Turkish Patient With Succinyl-CoA:3-Oxoacid CoA Transferase Deficiency Mimicking Diabetic Ketoacidosis
 Original Article A Turkish Patient With Succinyl-CoA:3-Oxoacid CoA Transferase Deficiency Mimicking Diabetic Ketoacidosis Journal of Inborn Errors of Metabolism & Screening 2016, Volume 4: 1 5 ª The Author(s)
Original Article A Turkish Patient With Succinyl-CoA:3-Oxoacid CoA Transferase Deficiency Mimicking Diabetic Ketoacidosis Journal of Inborn Errors of Metabolism & Screening 2016, Volume 4: 1 5 ª The Author(s)
number Done by Corrected by Doctor Faisal Al-Khatib
 number 22 Done by Baraa Ayed Corrected by Yaseen Fatayer Doctor Faisal Al-Khatib 1 P a g e Today we are going to cover these concepts: Oxidation of odd number fatty acids Oxidation of very long fatty acids
number 22 Done by Baraa Ayed Corrected by Yaseen Fatayer Doctor Faisal Al-Khatib 1 P a g e Today we are going to cover these concepts: Oxidation of odd number fatty acids Oxidation of very long fatty acids
Most common is the congenital adrenogenital syndrome (AGS) or congenital adrenal hyperplasia (CAH) due to 21-hydroxylase deficiency.
 or congenital adrenal hyperplasia (CAH) due to 21-hydroxylase deficiency.") Newborn Screening Examination parameters: TSH-neonatal (hypothyreosis), 17-OH progesterone (AGS), galactose (galactosemia), galactose-uridyl transferase (galacto semia), biotinidase (biotinidase ), phenylalanine
Newborn Screening Examination parameters: TSH-neonatal (hypothyreosis), 17-OH progesterone (AGS), galactose (galactosemia), galactose-uridyl transferase (galacto semia), biotinidase (biotinidase ), phenylalanine
A Unusual Presentation of Propionic Acidemia with Thrombocytosis- A Case Report
 Article ID: WMC003015 ISSN 2046-1690 A Unusual Presentation of Propionic Acidemia with Thrombocytosis- A Case Report Corresponding Author: Dr. Rahul Sinha, Assistant Professor of Paediatrics, 167 Military
Article ID: WMC003015 ISSN 2046-1690 A Unusual Presentation of Propionic Acidemia with Thrombocytosis- A Case Report Corresponding Author: Dr. Rahul Sinha, Assistant Professor of Paediatrics, 167 Military
Tala Saleh. Razi Kittaneh ... Nayef Karadsheh
 Tala Saleh Razi Kittaneh... Nayef Karadsheh β-oxidation of Fatty Acids The oxidation of fatty acids occurs in 3 steps: Step 1: Activation of the Fatty acid FA + HS-CoA + ATP FA-CoA + AMP + PPi - The fatty
Tala Saleh Razi Kittaneh... Nayef Karadsheh β-oxidation of Fatty Acids The oxidation of fatty acids occurs in 3 steps: Step 1: Activation of the Fatty acid FA + HS-CoA + ATP FA-CoA + AMP + PPi - The fatty
Parkinson s Disease in the Elderly A Physicians perspective. Dr John Coyle
 Parkinson s Disease in the Elderly A Physicians perspective Dr John Coyle Overview Introduction Epidemiology and aetiology Pathogenesis Diagnosis and clinical features Treatment Psychological issues/ non
Parkinson s Disease in the Elderly A Physicians perspective Dr John Coyle Overview Introduction Epidemiology and aetiology Pathogenesis Diagnosis and clinical features Treatment Psychological issues/ non
LILY FOUNDATION. An Introduction to Ketogenic Diets. Susan Wood. Matthew s Friends. Registered Dietitian Adults & Paediatrics
 LILY FOUNDATION An Introduction to Ketogenic Diets Susan Wood Registered Dietitian Adults & Paediatrics Matthew s Friends What is a ketogenic diet? A MEDICAL treatment. Mimics fasting metabolism Alters
LILY FOUNDATION An Introduction to Ketogenic Diets Susan Wood Registered Dietitian Adults & Paediatrics Matthew s Friends What is a ketogenic diet? A MEDICAL treatment. Mimics fasting metabolism Alters
CSF Investigations in patients with seizures. Dr Simon Olpin Sheffield Children s Hospital
 CSF Investigations in patients with seizures Dr Simon Olpin Sheffield Children s Hospital Background Epileptic seizures common feature in many inherited metabolic disorders particularly those involving
CSF Investigations in patients with seizures Dr Simon Olpin Sheffield Children s Hospital Background Epileptic seizures common feature in many inherited metabolic disorders particularly those involving
Biochemistry and Management of Fatty Acid Oxidation Disorders: From Infancy to Adulthood
 Nutricia Metabolics Webinar Series Biochemistry and Management of Fatty Acid Oxidation Disorders: From Infancy to Adulthood April 20, 2016 Melanie Gillingham, PhD, RD Sandy van Calcar, PhD, RD Department
Nutricia Metabolics Webinar Series Biochemistry and Management of Fatty Acid Oxidation Disorders: From Infancy to Adulthood April 20, 2016 Melanie Gillingham, PhD, RD Sandy van Calcar, PhD, RD Department
Valproate overdose: what is the role of carnitine?
 Valproate overdose: what is the role of carnitine? Ruben Thanacoody National Poisons Information Service (Newcastle) Newcastle upon Tyne Hospitals NHS Trust Medical Toxicology Centre, Newcastle University
Valproate overdose: what is the role of carnitine? Ruben Thanacoody National Poisons Information Service (Newcastle) Newcastle upon Tyne Hospitals NHS Trust Medical Toxicology Centre, Newcastle University
Movement disorders in childhood: assessment and diagnosis. Lucinda Carr
 Movement disorders in childhood: assessment and diagnosis Lucinda Carr Movement disorders in childhood: Assessment Classification Causes Diagnosis Presentation of movement disorders in childhood: Concerns
Movement disorders in childhood: assessment and diagnosis Lucinda Carr Movement disorders in childhood: Assessment Classification Causes Diagnosis Presentation of movement disorders in childhood: Concerns
Package Insert. Elkar
 Package Insert Elkar Product Summary 1. Name of the medicinal product Elkar 500 mg tablets 2. Qualitative and quantitative composition Each tablet contains levocarnitine 500 mg. 3. Pharmaceutical form
Package Insert Elkar Product Summary 1. Name of the medicinal product Elkar 500 mg tablets 2. Qualitative and quantitative composition Each tablet contains levocarnitine 500 mg. 3. Pharmaceutical form
GENOTYPE-PHENOTYPE CORRELATIONS IN GALACTOSEMIA COMPLICATIONS COMPLICATIONS COMPLICATIONS LONG-TERM CHRONIC COMPLICATIONS WITH NO CLEAR CAUSE
 Galactosemia Deficiency: galactose-1-phosphate-uridyltransferase(galt) GENOTYPE-PHENOTYPE CORRELATIONS IN GALACTOSEMIA GALT D-galactose-1-phosphate UDPgalactose + + UDPglucose D-glucose-1-phosphate DIVISION
Galactosemia Deficiency: galactose-1-phosphate-uridyltransferase(galt) GENOTYPE-PHENOTYPE CORRELATIONS IN GALACTOSEMIA GALT D-galactose-1-phosphate UDPgalactose + + UDPglucose D-glucose-1-phosphate DIVISION
Metabolic Precautions & ER Recommendations
 Metabolic Precautions & ER Recommendations * To whom correspondence Sumit Parikh, should MD be addressed Center for Pediatric Neurology Cleveland Clinic Cleveland, OH UMDF 2010 The catabolic state Entering
Metabolic Precautions & ER Recommendations * To whom correspondence Sumit Parikh, should MD be addressed Center for Pediatric Neurology Cleveland Clinic Cleveland, OH UMDF 2010 The catabolic state Entering
MITO 101 Illness. Neurometabolic Clinic Children s Medical Center and UT Southwestern Medical Center Hospitals and Clinics
 1 MITO 101 Illness Juan M. Pascual, MD, PhD Division of Pediatric Neurology Departments of Neurology, Physiology and Pediatrics The University of Texas Southwestern Medical Center Neurometabolic Clinic
1 MITO 101 Illness Juan M. Pascual, MD, PhD Division of Pediatric Neurology Departments of Neurology, Physiology and Pediatrics The University of Texas Southwestern Medical Center Neurometabolic Clinic
Neurotransmitter Disorders.
 Neurotransmitter Disorders Simon.heales@gosh.nhs.uk Chemical Neurotransmission Neurotransmitters Substances that upon release from nerve terminals, act on receptor sites at postsynaptic membranes to produce
Neurotransmitter Disorders Simon.heales@gosh.nhs.uk Chemical Neurotransmission Neurotransmitters Substances that upon release from nerve terminals, act on receptor sites at postsynaptic membranes to produce
Providing the Right Fuels for FOD s. Elaina Jurecki, MS, RD Regional Metabolic Nutritionist Kaiser Permanente Medical Center Oakland, CA
 Providing the Right Fuels for FOD s Elaina Jurecki, MS, RD Regional Metabolic Nutritionist Kaiser Permanente Medical Center Oakland, CA Providing the Right Fuels for FOD s The Body s Use of Energy Fat
Providing the Right Fuels for FOD s Elaina Jurecki, MS, RD Regional Metabolic Nutritionist Kaiser Permanente Medical Center Oakland, CA Providing the Right Fuels for FOD s The Body s Use of Energy Fat
3 HYDROXY 3 METHYLGLUTARYL CoA (3 HMG CoA) LYASE DEFICIENCY RECOMMENDATIONS ON EMERGENCY MANAGEMENT OF METABOLIC DISEASES
 LYASE DEFICIENCY RECOMMENDATIONS ON EMERGENCY MANAGEMENT OF METABOLIC DISEASES") 3 HYDROXY 3 METHYLGLUTARYL CoA (3 HMG CoA) LYASE DEFICIENCY RECOMMENDATIONS ON EMERGENCY MANAGEMENT OF METABOLIC DISEASES Patient s name: Date of birth: Please read carefully. Meticulous and prompt treatment
3 HYDROXY 3 METHYLGLUTARYL CoA (3 HMG CoA) LYASE DEFICIENCY RECOMMENDATIONS ON EMERGENCY MANAGEMENT OF METABOLIC DISEASES Patient s name: Date of birth: Please read carefully. Meticulous and prompt treatment
UvA-DARE (Digital Academic Repository) Branched chain amino acids : facts and defects Loupatty, F.J. Link to publication
 Branched chain amino acids : facts and defects Loupatty, F.J. Link to publication") UvA-DARE (Digital Academic Repository) Branched chain amino acids : facts and defects Loupatty, F.J. Link to publication Citation for published version (APA): Loupatty, F. J. (2007). Branched chain amino
UvA-DARE (Digital Academic Repository) Branched chain amino acids : facts and defects Loupatty, F.J. Link to publication Citation for published version (APA): Loupatty, F. J. (2007). Branched chain amino
23.1 Lipid Metabolism in Animals. Chapter 23. Micelles Lipid Metabolism in. Animals. Overview of Digestion Lipid Metabolism in
 Denniston Topping Caret Copyright! The McGraw-Hill Companies, Inc. Permission required for reproduction or display. Chapter 23 Fatty Acid Metabolism Triglycerides (Tgl) are emulsified into fat droplets
Denniston Topping Caret Copyright! The McGraw-Hill Companies, Inc. Permission required for reproduction or display. Chapter 23 Fatty Acid Metabolism Triglycerides (Tgl) are emulsified into fat droplets
Strick Lecture 4 March 29, 2006 Page 1
 Strick Lecture 4 March 29, 2006 Page 1 Basal Ganglia OUTLINE- I. Structures included in the basal ganglia II. III. IV. Skeleton diagram of Basal Ganglia Loops with cortex Similarity with Cerebellar Loops
Strick Lecture 4 March 29, 2006 Page 1 Basal Ganglia OUTLINE- I. Structures included in the basal ganglia II. III. IV. Skeleton diagram of Basal Ganglia Loops with cortex Similarity with Cerebellar Loops
the fates of acetyl coa which produced by B oixidation :
 Ketone bodies the fates of acetyl coa which produced by B oixidation : 1) oxidized at the TCA cycle 2)synthesis of ketone bodies Ketone bodies : 1)acetoacetate 2) acetone 3) 3_hydroxybutyrate Naming acetonacetone:
Ketone bodies the fates of acetyl coa which produced by B oixidation : 1) oxidized at the TCA cycle 2)synthesis of ketone bodies Ketone bodies : 1)acetoacetate 2) acetone 3) 3_hydroxybutyrate Naming acetonacetone:
Hompes Method Lesson 29 Organic Acids Part One
 Hompes Method Lesson 29 Organic Acids Part One Health for the People Ltd not for reuse without expressed permission Organic Acids - Introduction The ultimate tool for laboratory evaluations in nutritional
Hompes Method Lesson 29 Organic Acids Part One Health for the People Ltd not for reuse without expressed permission Organic Acids - Introduction The ultimate tool for laboratory evaluations in nutritional
Contribution of Nutrients in Complex Inborn Errors of Metabolism: The Case of Methylmalonic Aciduria (MMA)
") Contribution of Nutrients in Complex Inborn Errors of Metabolism: The Case of Methylmalonic Aciduria (MMA) Charles P. Venditti, MD, PhD Head, Organic Acid Research Section No conflicts of interest to declare
Contribution of Nutrients in Complex Inborn Errors of Metabolism: The Case of Methylmalonic Aciduria (MMA) Charles P. Venditti, MD, PhD Head, Organic Acid Research Section No conflicts of interest to declare
680 La Revue de Santé de la Méditerranée orientale, Vol. 10, N o 4/5, 2004
 680 La Revue de Santé de la Méditerranée orientale, Vol. 10, N o 4/5, 2004 Case report Glutaric aciduria type 1 in a Kuwaiti infant H.A. Elsori, 1 K.K. Naguib 2 and M.S. Hammoud 3 Introduction Glutaryl-coenzyme
680 La Revue de Santé de la Méditerranée orientale, Vol. 10, N o 4/5, 2004 Case report Glutaric aciduria type 1 in a Kuwaiti infant H.A. Elsori, 1 K.K. Naguib 2 and M.S. Hammoud 3 Introduction Glutaryl-coenzyme
Acute Management of Sick Infants with Suspected Inborn Errors of Metabolism
 Indian J Pediatr (July 2011) 78(7):854 859 DOI 10.1007/s12098-011-0422-0 SYMPOSIUM ON PICU PROTOCOLS OF AIIMS Acute Management of Sick Infants with Suspected Inborn Errors of Metabolism Neerja Gupta Madhulika
Indian J Pediatr (July 2011) 78(7):854 859 DOI 10.1007/s12098-011-0422-0 SYMPOSIUM ON PICU PROTOCOLS OF AIIMS Acute Management of Sick Infants with Suspected Inborn Errors of Metabolism Neerja Gupta Madhulika
VL VA BASAL GANGLIA. FUNCTIONAl COMPONENTS. Function Component Deficits Start/initiation Basal Ganglia Spontan movements
 BASAL GANGLIA Chris Cohan, Ph.D. Dept. of Pathology/Anat Sci University at Buffalo I) Overview How do Basal Ganglia affect movement Basal ganglia enhance cortical motor activity and facilitate movement.
BASAL GANGLIA Chris Cohan, Ph.D. Dept. of Pathology/Anat Sci University at Buffalo I) Overview How do Basal Ganglia affect movement Basal ganglia enhance cortical motor activity and facilitate movement.
Further expansion of the neonatal screening panel in the Netherlands
 Further expansion of the neonatal screening panel in the Netherlands J.Gerard Loeber APHL-NBSGT, St.Louis (MO), USA 290216 Population Area Newborns 6.01 million 0.35:1 16.8 million 180,693 sq km 4.3:1
Further expansion of the neonatal screening panel in the Netherlands J.Gerard Loeber APHL-NBSGT, St.Louis (MO), USA 290216 Population Area Newborns 6.01 million 0.35:1 16.8 million 180,693 sq km 4.3:1
BIOTIN (BIOTINIDASE) DEFICIENCY Marc E. Tischler, PhD; University of Arizona
 DEFICIENCY Marc E. Tischler, PhD; University of Arizona") BIOTIN (BIOTINIDASE) DEFICIENCY Marc E. Tischler, PhD; University of Arizona BIOTIN (BIOTINIDASE) DEFICIENCY biotin in the body is recycled by its removal from carboxylase enzymes to which it is attached
BIOTIN (BIOTINIDASE) DEFICIENCY Marc E. Tischler, PhD; University of Arizona BIOTIN (BIOTINIDASE) DEFICIENCY biotin in the body is recycled by its removal from carboxylase enzymes to which it is attached
Post mortem investigation of Inherited Metabolic Disease - the last opportunity for a diagnosis -
 Post mortem investigation of Inherited Metabolic Disease - the last opportunity for a diagnosis - Dr Simon Olpin Lead Clinical Scientist in Inherited Metabolic Disease Sheffield Children s Hospital SIDS/SUDI
Post mortem investigation of Inherited Metabolic Disease - the last opportunity for a diagnosis - Dr Simon Olpin Lead Clinical Scientist in Inherited Metabolic Disease Sheffield Children s Hospital SIDS/SUDI
Companion to Biosynthesis of Ketones & Cholesterols, Regulation of Lipid Metabolism Lecture Notes
 Companion to Biosynthesis of Ketones & Cholesterols, Regulation of Lipid Metabolism Lecture Notes The major site of acetoacetate and 3-hydorxybutyrate production is in the liver. 3-hydorxybutyrate is the
Companion to Biosynthesis of Ketones & Cholesterols, Regulation of Lipid Metabolism Lecture Notes The major site of acetoacetate and 3-hydorxybutyrate production is in the liver. 3-hydorxybutyrate is the
Using the Organic Acids Test Part 3 Dr. Jeff Moss
 Using organic acids to resolve chief complaints and improve quality of life in chronically ill patients Part III Jeffrey Moss, DDS, CNS, DACBN jeffmoss@mossnutrition.com 413-530-08580858 (cell) 1 Summer
Using organic acids to resolve chief complaints and improve quality of life in chronically ill patients Part III Jeffrey Moss, DDS, CNS, DACBN jeffmoss@mossnutrition.com 413-530-08580858 (cell) 1 Summer
NEUROMETABOLIC DISORDER ARTICLE: CASE REPORT
 NEUROMETABOLIC DISORDER ARTICLE: CASE REPORT A Novel Mutation of Beta-ketothiolase Deficiency: The First Report from Iran and Review of Literature How to Cite This Article: Vakili R, Hashemian S. A Novel
NEUROMETABOLIC DISORDER ARTICLE: CASE REPORT A Novel Mutation of Beta-ketothiolase Deficiency: The First Report from Iran and Review of Literature How to Cite This Article: Vakili R, Hashemian S. A Novel
LIPID METABOLISM. Sri Widia A Jusman Department of Biochemistry & Molecular Biology FMUI
 LIPID METABOLISM Sri Widia A Jusman Department of Biochemistry & Molecular Biology FMUI Lipid metabolism is concerned mainly with fatty acids cholesterol Source of fatty acids from dietary fat de novo
LIPID METABOLISM Sri Widia A Jusman Department of Biochemistry & Molecular Biology FMUI Lipid metabolism is concerned mainly with fatty acids cholesterol Source of fatty acids from dietary fat de novo
SELECTIVE VULNERABILITY (HYPOXIA AND HYPOGLYCEMIA)
") DEFICIENCY OF METABOLITE -HYPOXIA AND HYPOGLYCEMIA -HYPOVITAMINOSIS SELECTIVE VULNERABILITY (HYPOXIA AND HYPOGLYCEMIA) -SPECIFIC CELL TYPE NEURONS>OLIGODENDROCYTES>ASTROCYTES -SPECIFIC BRAIN REGION PYRAMIDAL
DEFICIENCY OF METABOLITE -HYPOXIA AND HYPOGLYCEMIA -HYPOVITAMINOSIS SELECTIVE VULNERABILITY (HYPOXIA AND HYPOGLYCEMIA) -SPECIFIC CELL TYPE NEURONS>OLIGODENDROCYTES>ASTROCYTES -SPECIFIC BRAIN REGION PYRAMIDAL
ISOVALERIC ACIDAEMIA -ACUTE DECOMPENSATION (standard version)
") Contact Details Name: Hospital Telephone: This protocol has 5 pages ISOVALERIC ACIDAEMIA -ACUTE DECOMPENSATION (standard version) Please read carefully. Meticulous treatment is very important as there
Contact Details Name: Hospital Telephone: This protocol has 5 pages ISOVALERIC ACIDAEMIA -ACUTE DECOMPENSATION (standard version) Please read carefully. Meticulous treatment is very important as there
Peroxisomal Disorders
 Peroxisomal Disorders George Gray Birmingham Childrens Hospital Peroxisomal Disorders Peroxisomes are large single membrane bound organelles that are present in the cytoplasm of all cells. They are formed
Peroxisomal Disorders George Gray Birmingham Childrens Hospital Peroxisomal Disorders Peroxisomes are large single membrane bound organelles that are present in the cytoplasm of all cells. They are formed
BCH 4054 Spring 2001 Chapter 24 Lecture Notes
 BCH 4054 Spring 2001 Chapter 24 Lecture Notes 1 Chapter 24 Fatty Acid Catabolism 2 Fatty Acids as Energy Source Triglycerides yield 37 kj/g dry weight Protein 17 kj/g Glycogen 16 kj/g (even less wet weight)
BCH 4054 Spring 2001 Chapter 24 Lecture Notes 1 Chapter 24 Fatty Acid Catabolism 2 Fatty Acids as Energy Source Triglycerides yield 37 kj/g dry weight Protein 17 kj/g Glycogen 16 kj/g (even less wet weight)
Organic Acid Disorders
 Genetic Fact Sheets for Parents Organic Acid Disorders Screening, Technology, and Research in Genetics is a multi-state project to improve information about the financial, ethical, legal, and social issues
Genetic Fact Sheets for Parents Organic Acid Disorders Screening, Technology, and Research in Genetics is a multi-state project to improve information about the financial, ethical, legal, and social issues
PROTEIN METABOLISM: SPECIFIC WAYS OF AMINO ACIDS CATABOLISM AND SYNTHESIS
 PROTEIN METABOLISM: SPECIFIC WAYS OF AMINO ACIDS CATABOLISM AND SYNTHESIS SPECIFIC WAYS OF AMINO ACID CATABOLISM After removing of amino group the carbon skeletons of amino acids are transformed into metabolic
PROTEIN METABOLISM: SPECIFIC WAYS OF AMINO ACIDS CATABOLISM AND SYNTHESIS SPECIFIC WAYS OF AMINO ACID CATABOLISM After removing of amino group the carbon skeletons of amino acids are transformed into metabolic