S6K1 mediates oncogenic glycolysis in Pten deficient leukemia
|
|
|
- Nigel Osborn Mitchell
- 5 years ago
- Views:
Transcription
1
2 S6K1 mediates oncogenic glycolysis in Pten deficient leukemia A dissertation submitted to the Graduate School of the University of Cincinnati in partial fulfillment of the requirements for the degree of Doctor of Philosophy (Ph.D.) in the Department of Cancer and Cell Biology of the College of Medicine by Preeti Tandon M.S. Bowling Green State University, 2004
3 ABSTRACT Hyperactive Akt signaling triggers glycolysis and apoptosis resistance in human cancer. Because sustained glycolysis is required for Akt dependent apoptosis resistance, we investigated the downstream signaling components that mediate Akt dependent increases in glycolysis in cells deficient for Pten, a negative regulator of the PI3K/Akt pathway. Genetic inactivation of the ribosomal protein S6 Kinase 1 (S6K1) in Pten-deficient cells prevented glycolysis, triggered Bax translocation and committed cells to apoptosis. Pharmacological S6K1 inhibition using a small molecule kinase inhibitor recapitulated the effects of genetically inactivating S6K1. Inactivation of S6K1 was associated with decreased expression of the pro-glycolytic HIF1α transcription factor. Restoring HIF1α expression was sufficient to restore both glycolysis and cell survival in S6K1-deficient cells. Conversely, inhibiting HIF1α expression in Pten deficient cells resulted in decreased glycolysis and cell survival, mimicking the loss of S6K1. In vivo, S6K1 deficiency delayed the development of lethal disease in a Pten deficient mouse model of leukemia. Thus, together the data suggest that S6K1 is a useful target for counteracting the metabolic program that supports apoptosis resistance in Pten-deficient cancers. iii
4 iv
5 ACKNOWLEDGEMENTS I would first and foremost like to thank Dr David Plas for being my mentor. Your guidance and advice have been instrumental in my progress and development as a scientist. You have been a great teacher and I have learnt a lot from you over the years. Thank you for everything- all the impromptu quizzes during lab meetings, making sure that we always did the best experiment and for encouraging us to never say no to an opportunity to present. You have been an excellent role model and the best advisor that anyone could ever ask for. I would like to thank members of my thesis committee Dr. George Thomas, Dr. James Mulloy, Dr. Angela Drew and Dr. Maria Czyzyk Krzeska for their support, encouragement and critical review of this research. I would also like to thank all of the past and present members of the Plas lab for making it an enjoyable experience. Thank you all for your technical assistance, valuable scientific discussions and most importantly your friendship and support. I especially want to thank Shikha for being a great friend and a wonderful co-worker. A special thanks to my parents for their unwavering support and love. Thank you for inspiring me to be the best at whatever I do. Thanks mom for the unlimited supply of scrumptious food that you sent my way that allowed me to utilize those kitchen hours in the lab to do more experiments. Thank you papa for never letting me give up. Thanks to my parents-in-law for their support and well wishes. I would also like to thank my sister, brother-in-law and their beautiful kids- Saahil and Saaz. Their smiling faces made me forget the disappointment of failed experiments. v
6 Lastly but most importantly, I would like to thank my husband, Ritesh, for his unconditional love and support. Thank you for believing in me when I found it difficult to believe in myself. I couldn t have done this without your constant encouragement, patience, love and strength. vi
7 Table of Contents List of Figures.9 Chapter I: Introduction 11 References 41 Figures..53 Chapter II: Requirement for ribosomal protein S6 Kinase 1 to mediate 57 glycolysis and apoptosis resistance induced by Pten deficiency Abstract.58 Introduction...59 Results Discussion..68 Materials and Methods..70 References..75 Figures 78 Chapter III: Analysis of an S6K1 inhibitor for counteracting glycolysis and 89 survival in Pten deficient cells Abstract 90 Introduction..91 Results..93 Discussion.95 Materials and Methods.96 References.98
8 Figures 99 Chapter IV: Conclusions and Discussion 102 References..117 Figures 120 8
9 List of Figures Chapter I Figure 1 53 Figure 2 54 Figure 3.55 Figure 4.56 Chapter II Figure 1.78 Figure 2.79 Figure 3.80 Figure Figure Supp. Figure 1 83 Supp. Figure 2 84 Supp. Figure 3 85 Supp. Figure 4 86 Supp. Figure Supp. Figure 6 88 Chapter III Figure 1 99 Figure Figure
10 Chapter IV Figure Figure Figure Figure Figure Figure Figure
11 CHAPTER I Introduction 11
12 Cancer Cell Metabolism: The Warburg Effect Dysregulated cellular metabolism is a distinguishing feature of transformed cells. More than 80 years ago, Otto Warburg showed that tumor cells metabolize glucose to lactate at a much higher rate than normal cells despite the presence of adequate oxygen- a phenomenon now known as the Warburg Effect or aerobic glycolysis (1). This effect has since been observed in different tumor types and is considered an essential feature of cancer cells. Although the Warburg effect is a hallmark of cancer, its regulatory mechanism remains obscure. Identifying factors that regulate cancer cell metabolism will enhance our understanding of cancer development and progression and provide novel approaches for cancer therapy. A key question in cancer biology is why cancer cells preferentially activate glycolysis, which yields only 2 ATP per molecule of glucose, instead of glucose oxidation, which yields up to 36 ATP. Warburg reasoned that defects in mitochondrial respiration cause increased aerobic glycolysis in cancer cells (2). The discovery of oncogenic mutations in oxidative phosphorylation (OXPHOS) genes, such as, fumarate hydratase (FH), succinate dehydrogenase (SDH) and isocitrate dehydrogenase (IDH1 and IDH2) validated Warburg s hypothesis (3-5). However, these mutations are rare and only occur in a small subset of cancers (6). The majority of tumors retain the ability to consume oxygen at rates comparable to normal cells, suggesting other mechanisms underlying the metabolic reprogramming seen in tumor cells (7). An alternative explanation is that glycolysis confers proliferative advantages upon cancer cells. In addition to requiring ATP, cancer cells also require building blocks such as nucleotides, lipids, proteins and fatty acids to sustain uncontrolled cell proliferation. By preventing glucose oxidation via OXPHOS for maximal ATP production, cancer cells can divert glucose carbons 12
13 into macromolecular precursors such as acetyl co-a for fatty acid synthesis, ribose for nucleotide synthesis and 3-phosphoglycerate (3-PG) for amino acid synthesis (8-10). These changes in metabolic destinations of glucose can be regulated by altered expression of different isoforms of the glycolytic enzyme, pyruvate kinase (PK). Pyruvate kinase catalyzes the penultimate step in glycolysis, the conversion of phosphoenolpyruvate (PEP) to pyruvate. Cancer cells preferentially express pyruvate kinase M2 (PKM2) in place of PKM1, which is expressed in non-proliferating adult tissues (11). The PKM2 isoform is less active in converting PEP to pyruvate and generating ATP than PKM1 (11). PKM2 expression triggers the accumulation of the upstream metabolite PEP, which can now be incorporated into biosynthetic processes to support cell proliferation. Furthermore, PEP can donate phosphate via a phosphotransferase reaction to the metabolic enzyme phosphoglycerate mutase1 (PGAM1), producing phospho- PGAM1 and pyruvate (12). Phosphorylation of PGAM1 increases the mutase function of the enzyme, triggering a positive feedback loop whereby PEP increases the activity of upstream glycolytic pathway enzymes. Thus, pyruvate production by this alternative pathway may provide an additional mechanism to promote the redistribution of metabolites upstream of PGAM1 into biosynthetic pathways. Another survival advantage of switching from oxidative to aerobic metabolism is decreased production of reactive oxygen species (ROS). Mitochondrial respiration is the major source of ROS production in cells. Excessive ROS production can be harmful to cells as it can irreversibly damage proteins, nucleic acids and lipids triggering senescence and/or cell death (13, 14). Cancer cells in which pyruvate is forced to enter the mitochondria to undergo oxidative phosphorylation, as a result of lactate dehydrogenase A (LDH-A) inhibition, die from oxidative stress and are partially rescued by antioxidant N-acetylcysteine (15). These studies emphasize 13
14 that a critical aspect of actively suppressing mitochondrial respiration is evasion of ROS induced apoptosis. The powerful growth advantage provided by increased biosynthesis and decreased apoptosis renders cancer cells addicted to aerobic glycolysis. This increased dependency of cancer cells on glycolysis provides a biochemical basis to preferentially kill malignant cells by inhibiting glycolysis. Glucose limitation to tumors might be envisioned as an aspect of antiangiogenic therapy, but the more common approach to limiting glycolysis in tumors has been through the administration of direct inhibitors of glycolysis such as 2-deoxyglucose (2DG) and 3-bromopyruvate. These compounds have been shown to induce cytotoxicity and oxidative stress in cancer cells [Reviewed in (16)]. However, toxicity and inefficacy in clinical trials has limited the direct application of these inhibitors as potential chemotherapeutics. Thus, better therapeutic targeted approaches are needed to circumvent this limitation. Induction of glycolytic metabolism in cancer cells is often driven by genetic changes, which could provide more specific and effective targets for therapy. Mutations in oncogenes or tumor suppressor genes trigger activation of signaling pathways that reprogram metabolism to confer survival and proliferative advantage on cells. Thus, developing strategies to target these signaling pathways may prove efficacious in suppressing the metabolism that they activate. The best described signal transduction pathway that induces glycolytic metabolism is the phosphatidylinositol-3 kinase (PI3K) /Akt pathway. 14
15 PI3K-Akt Pathway PI3K-Akt pathway is one of the most important pathways in cancer metabolism, survival and growth. The different components of the pathway are described below (Figure 1). PI3K: The PI3K family of lipid kinases phosphorylates the 3 OH group of phosphotidylinositols (17). PI3Ks can be divided into three classes according to substrate specificity and lipid products (18). Class 1A PI3K is the most well studied class and is frequently deregulated in cancer. Class IA PI3Ks are heterodimers consisting of a p110 catalytic subunit and a p85 regulatory subunit. There are 3 p110 catalytic isoforms- p110α, p110β and p110δ and 3 isoforms of p85- p85α, p85β and p55γ [Reviewed in (18)]. Class 1A PI3Ks are activated downstream of receptor tyrosine kinases (RTKs). In response to growth factor stimulation and subsequent receptor activation, PI3K is recruited to the membrane via direct interaction of its regulatory subunit with the tyrosine phosphorylated receptor or via adaptor proteins associated with the receptors. This interaction relieves the inhibitory effect of p85 on p110. Once activated, p110 catalyzes the production of 3,4,5 phosphatidylinositol trisphosphate (PIP3) (Figure 1), which provides docking sites for proteins that contain pleckstrin homology (PH) domains. Akt and phosphoinositde dependent kinase 1 (PDK1) are prototypic PH-domain regulated kinases that respond to PIP3. AKT: Akt belongs to the AGC family of serine/threonine kinases (19). There are 3 isoforms of Akt- Akt1, Akt2 and Akt3 [Reviewed in (18)]. Analysis in gene-targeted animals indicates functional redundancy among the three isoforms in normal physiology (20, 21). Mice deficient in single Akt isoforms display a range of phenotypes: small body size and increased thymocyte cell death (Akt1 -/- ); impaired glucose homeostasis (Akt2 -/- ); reduced size and development in the central nervous system (Akt3 -/- ). Akt activation is initiated following translocation to the 15
16 membrane mediated by docking of its PH domain with the membrane located PIP3 (Figure 1). Once recruited to the membrane, Akt is phosphorylated at T308 and S473 by PDK1 and mammalian target of rapamycin complex 2 (mtorc2), respectively (22-24) (Figure 1). Phosphorylation at both these residues is required for full Akt activation. Akt activation triggers phosphorylation of a large repertoire of proteins leading to regulation of a wide range of cellular processes involved in survival, protein synthesis, metabolism and growth. An evolutionarily conserved substrate of Akt is the tuberous sclerosis complex 2 (TSC2) protein. TSC2 is a GTPase activating protein (GAP) for the small G protein Ras homolog enriched in brain (Rheb) (25) (Figure 1). Akt phosphorylation of TSC2 inhibits its GAP activity, increases Rheb GTP loading and stimulates mtorc1 activation (26). Akt can also regulate mtorc1 activation via transcriptional regulation of tuberous sclerosis complex 1 (TSC1), an obligate partner of TSC2. FOXOs induce the transcription of TSC1, which suppresses mtorc1 activation. Akt phosphorylation and inactivation of FOXOs is proposed to reduce the production and activity of TSC1/TSC2, resulting in increased mtorc1 signaling (27). Overall, activation of Akt promotes mtorc1 signaling. mtor: mtor integrates signals from many diverse inputs such as growth factors and nutrients, and in response, phosphorylates multiple effectors that regulate processes important for growth such as protein translation, autophagy and cell proliferation. The mtor kinase functions within two distinct multiprotein complexes- mtorc1 and mtorc2, which are distinguished by their partner proteins, downstream targets and differing sensitivities to rapamycin (28). Rapamycin is a bacterial macrolide that functions as an allosteric inhibitor of mtor. Within the cell, rapamycin binds to its receptor FKBP12 (FK506-binding protein of 12 kda) to form a complex that interacts with the FRB (FKBP12 rapamycin binding) domain of mtor and inhibits mtor 16
17 functions (29). Acute rapamycin treatment inhibits signaling by mtorc1 but not mtorc2. However, prolonged exposure to rapamycin can inhibit mtorc2 activity by disrupting the assembly of new mtorc2 complexes in some cell types (30). mtorc1 functions downstream of Akt and consists of the catalytic subunit mtor, regulatory associated protein of mtor (raptor), PRAS40, mammalian lethal with Sec 13 protein 8 (mlst8 or GβL) and DEP- domaincontaining mtor- interacting protein (DEPTOR). mtorc2 function is placed upstream of Akt and is comprised of mtor, rapapmycin insensitive companion of mtor (Rictor), mammalian stress activated protein kinase interacting protein (msin1), protein observed with Rictor-1 (Protor1), mlst8 and Deptor [Reviewed in (31, 32)]. The precise function of most of the mtor interacting proteins remains incompletely understood. The two best characterized substrates downstream of mtorc1 are the eukaryotic translation initiation 4E-binding protein 1 (4EBP1) and the ribosomal protein S6 kinase 1 (S6K1) (33) (Figure 1). These proteins function in parallel to modulate protein translation. 4EBP1: 4EBP1 functions to inhibit protein translation. Unphosphorylated 4EBP-1 inhibits mrna translation by binding and inactivating eukaryotic initiation factor 4E (eif4e). Phosphorylation of 4EBP-1 by mtorc1 reduces its affinity for eif4e and the two proteins dissociate. eif4e is now able to associate with components of eif4f to initiate translation (34). More recent studies demonstrated a regulatory role of 4EBP in cell proliferation. In these studies, pharmacologic or genetic inactivation of the mtorc1 pathway decreased cell proliferation, an effect that was rescued by co-depletion of 4EBPs (4EBP1-3). Furthermore, ablation of 4EBPs in mouse embryonic fibroblasts (MEFs) increased cell proliferation without affecting cell size or survival. This study provided the first evidence that 4EBPs mediate mtorc1 effects on cell proliferation but not cell growth or cell survival (35). 17
18 S6K1: S6K1 was initially identified as the kinase responsible for phosphorylation of the 40S ribosomal subunit S6 isolated from mitogen stimulated Swiss mouse 3T3 cells (36). S6K1 has two isoforms- p70s6k1 and p85s6k1 that arise from the same mrna transcript by alternative translational start sites (37, 38). p70s6k1 isoform is predominantly cytoplasmic, while p85s6k1 localizes in the nucleus due to the presence of a 23 amino acid sequence at the amino terminus that contains the nuclear localization signal (39). In addition to these isoforms, overexpression of the splicing factor SF2/ASF in mammalian cells promotes the expression of an alternatively spliced isoform of S6K1- S6K1 isoform-2 or p31s6k1. The mrna sequence of p31s6k1 is identical to that encoding p70s6k1 and p85s6k1 up to exon 6 but encodes a protein with a different C-terminus. NIH 3T3 cells overexpressing SF2/ASF or S6K1 isoform 2 undergo transformation and production of S6K1 isoform-2 is necessary for SF2/ASF mediated transformation (40). The mechanisms underlying the transforming ability of S6K1 isoform-2 remain unclear but seem to be independent of its kinase activity as the kinase domain is severely truncated in this isoform. More recently, genetic disruption of S6K1 in mice led to the identification of S6K1 homolog- S6K2 (41). S6K1 and S6K2 are encoded by different genes and share greater than 80% homology at the amino acid level. Similar to S6K1, S6K2 utilizes alternative translational start sites to produce 2 isoforms- p56s6k2 and p54s6k2 (42). S6K1 is activated by phosphorylation at multiple serine and threonine residues in various domains of the kinase in response to a variety of mitogenic stimuli. S6K1 consists of five domains- N- terminal domain, kinase catalytic domain, linker domain, autoinhibitory domain and the C-terminal domain (Figure 2). S6K1 activation is initiated by phosphorylation of a set of four proline directed serine/threonine (S/T-P) sites in the autoinhibitory domain, namely, S411, S418, T421 and S424 (43) (Figure 2). These sites are critical for S6K1 activation as mutating these 18
19 sites to alanine decreases S6K1 activity by greater than 5-fold whereas replacing the same residues with acidic amino acids increases the basal kinase activity (44). All these sites show a basal level of phosphorylation in quiescent cells, which is elevated upon mitogenic stimulus. Mulitple proline directed kinases including Cdk1, Erk1/2, Jnk1/2 have been proposed to phosphorylate these sites (45). Phosphorylation at S/T-P sites allows a second set of sites to be phosphorylated in response to mitogens. These sites include T389 and S404 in the linker region and T229 in the activation loop (46) (Figure 2). All three sites are rapamycin sensitive, with highest sensitivity displayed by T389 followed by S404 and then T229. Similar to S/T-P sites, T389 and T229 are critical regulators of S6K1 activation, as substituting these residues with alanine completely abrogates kinase activity. In contrast, S404 seems to play a less important role in activating S6K1 as replacing S404 with neutral or acidic amino acids has little effect on S6K1 activity. In addition to the above residues, mitogen stimulation induces phosphorylation of S371 in the linker domain of S6K1 (Figure 2). S371 phosphorylation contributes to S6K1 activation as well as regulates T389 phosphorylation in cells stimulated with serum or insulin (47). Phosphorylation at S371 and T389 is mediated by mtorc1 whereas T229 phosphorylation is regulated by PDK1 (48, 49). In summary, S6K1 activation is a multistep process that begins with phosphorylation of the S/T-P sites in the autoinhibitory domain. This phosphorylation induces conformational change that results in phosphorylation of T389 and S371 by mtor. This is followed by phosphorylation of T229 in the activation loop by the constitutively active PDK1 resulting in full activation of S6K1. Once activated, S6K1 regulates a diverse array of cellular processes 19
20 including growth, protein translation, survival and glucose homeostasis by directly phosphorylating numerous substrates. Growth: Genetic studies in Drosophila and mice provided the evidence for the involvement of S6K1 in the regulation of cell growth. Drosophila expresses only one S6K gene, deletion of which results in the death of most flies at the larval stage or during early pupation. A few S6K -/- flies that are able to survive are much smaller than their wild type counterparts (50). This reduction in body size is due to a reduction in cell size and not cell number, indicating that ds6k functions to regulate cell growth and not cell proliferation (50). In contrast to flies, mammals possess two genes encoding homologous S6Ks- S6K1 and S6K2. Disruption of S6K1 in mice also produces a small size phenotype which is attributable to a defect in cell size and not cell proliferation. Despite reduced body weight, S6 phosphorylation is unperturbed in S6K1-/- mice due to compensatory upregulation of S6K2 expression (41). Consistent with this, S6 phosphorylation is significantly reduced in S6K1 -/- /S6K2 -/- mice suggesting that S6K2 is the primary S6 kinase (51). Thus, S6K2 can carry out S6 phosphorylation in the absence of S6K1 but fails to regulate cell growth, suggesting non overlapping functions of S6K1 and S6K2. S6K1 mediates its effects on cell growth by activating protein translation. Protein translation: Initial studies implicated S6K1 mediated S6 phosphorylation in translational control of 5 -TOP mrnas (52). These mrnas contain an oligopyrimidine tract at their 5 terminus and encode for ribosomal proteins and elongation factors. However, subsequent studies showed that 5 -TOP mrnas are subject to normal translational control in S6K1 -/- /S6K2 -/- MEFs and embryonic stem cells (51). Furthermore, mutating S6K1 phosphorylation sites in S6 to alanine did not alter the translational activation of 5 -TOP mrnas, suggesting that S6K1 phosphorylation of S6 is dispensable for efficient translation of 5 -TOP mrnas (53). 20
21 More recently, several S6K1 targets have been identified that regulate protein translation. S6K1 regulates translation initiation by phosphorylating eif4b at Ser422 (54) (Figure 3). eif4b stimulates eif4a helicase activity to unwind inhibitory secondary structure in the 5 untranslated region of eukaryotic mrnas (55). This allows the 40S ribosomal subunit to bind to single stranded mrna and initiate translation. Substituting Ser422 with alanine diminishes eif4b activity suggesting that eif4b phosphorylation is important for its function (54). S6K1 also contributes to translation initiation by phosphorylating PDCD4, an inhibitor of eif4a. S6K1 mediated phosphorylation of PDCD4 triggers its degradation by the ubiquitin ligase, βtrcp (56). Thus, S6K1 greatly enhances eif4a activity in cells by activating its positive regulator (eif4b) and inhibiting its negative regulator (PDCD4) (Figure 3). S6K1 has also been shown to regulate splicing dependent translation via its association with SKAR (Figure 3). SKAR is a S6K1 specific interacting protein involved in mrna processing. SKAR gets deposited at the exon junction complex during splicing and recruits S6K1 to cap-binding complex 80 (CBP80)- bound mrna ribonulceoprotein (mrnp) on newly synthesized mrnas. Once recruited, S6K1 phosphorylates its substrates and drives pioneer round of translation of newly spliced mrnas (57). Interestingly, SKAR has previously been described as a S6K1 substrate, however, phosphorylation of SKAR has not been associated with its designated function (58). In addition to initiation, S6K1 exerts its control over translation elongation by phosphorylating eef2k. eef2k phosphorylates and inhibits eef2 function. S6K1 phosphorylation of eef2k inhibits its activity leading to eef2 dephosphorylation and subsequent activation (59) (Figure 3). Thus, by phosphorylating several substrates, S6K1 regulates multiple steps of protein synthesis. Deregulation at any of these steps can lead to uncontrolled growth and cancer development. 21
22 Survival: S6K1 and Akt share the same consensus phosphorylation motif- RXRXXS/T suggesting that Akt substrates could be phosphorylated by S6K1 as well. One such example is the pro-apoptotic protein Bad. Akt phosphorylates the BH3 protein Bad on Ser-136 (60, 61). Bad phosphorylation on Ser-136 creates a binding site for proteins (62), which sequester Bad in the cytoplasm and prevent Bad from triggering death through interactions with the prosurvival Bcl-2 family proteins. S6K1 induces phosphorylation at Bad Ser136 in response to IGF- 1. IGF-1 induced Bad phosphorylation is abolished in S6K1 -/- embryonic stem cells. Interestingly, rapamycin can inhibit IGF-1-mediated cell survival but not IL-3-dependent cell survival, suggesting that the pro-survival effects of S6K1 may vary depending on cell type and/ or growth factor receptor (63). Glucose homeostasis: Mice deficient in S6K1 exhibit impaired glucose homeostasis due to insufficient insulin secretion in response to glucose load. This defect is associated with reduced pancreatic β cell size which results in decreased circulating levels of insulin. Despite the fact that S6K1 -/- mice are glucose intolerant and display hypoinsulinemia, they maintain normal fasting glucose levels suggesting hypersensitivity to insulin. This increased insulin sensitivity is due to the loss of a negative feedback loop from S6K1 to insulin receptor substrate-1 (IRS-1). S6K1 signals not only downstream of mtor but also upstream as a negative regulator. S6K1 inhibits insulin signaling by phosphorylating IRS-1 at Ser307 and Ser636/Ser639. This S6K1 mediated attenuation of insulin signaling might have evolved to prevent insulin resistance under conditions of nutrient satiation. Consistent with this model, S6K1 -/- mice maintained on a high fat diet (HFD), conditions that promote insulin resistance, remain insulin sensitive. Wild type mice when fed HFD accumulate fat and show increased S6K1 activity. In contrast, S6K1 -/- mice are resistant to diet induced obesity due to increased lipolysis and β-oxidation (64). These studies 22
23 suggest that S6K1 may play a critical role in the development of diabetes and obesity and serves as a useful drug target in the treatment of these metabolic disorders. Overall, the Akt-mTORC1-S6K1 pathway activation plays a key role in promoting several processes that are important for cell growth and survival. Deregulation of any or several of these cellular processes due to aberrant pathway activation may contribute to the development or progression of cancer. The abnormal activation of PI3K/Akt/mTOR pathway is kept in check by the tumor suppressors - phosphatase and tensin homolog deleted on chromosome 10 (PTEN) and TSC proteins (Figure 1). As described earlier, the TSC proteins function downstream of Akt to inhibit mtorc1 activation. However, PTEN lies upstream of Akt and prevents aberrant Akt activation PTEN: PTEN negatively regulates the Akt pathway by opposing Class 1A PI3K activity. PTEN antagonizes PI3K activity through its intrinsic lipid phosphatase activity that reduces the cellular levels of PIP3 by converting PIP3 back to PIP2. PTEN is now known to be a phospholipid phosphatase but it was initially considered to be a protein phosphatase based on its sequence similarity with PTP family of enzymes. Studies demonstrated the ability of recombinant PTEN to dephosphorylate protein and peptide substrates phosphorylated on serine, threonine and tyrosine residues, establishing PTEN as a dual specificity protein phosphatase (65). However, unlike protein phosphatases, recombinant PTEN protein poorly dephosphorylated a number of artificial substrates and was found to be more active toward negatively charged multiply phosphorylated polymer of (Glu-Tyr) n. This observation suggested that PTEN prefers highly acidic substrates rather than Tyr or Ser/Thr phosphoproteins (65). In order to identify other physiologic substrates of PTEN, Maehama and Dixon transfected PTEN into 293 cells and analyzed changes in phospholipids (66). Overexpression of PTEN significantly reduced insulin induced PIP3 23
24 production without affecting PI 3-kinase activity. Furthermore, expression of a catalytically inactive mutant (C124S) of PTEN caused accumulation of PIP3 without insulin stimulation. In vitro, purified PTEN dephosphorylated PIP3 specifically at the D3 position of the inositol ring, indicating that PTEN is a phospholipid phosphatase (66) (Figure 1). The importance of the lipid phosphatase activity of PTEN came from studies examining the PTEN-G129E mutant (67). This mutation specifically abolishes the lipid phosphatase activity of PTEN while preserving its protein phosphatase activity (67). Expression of PTEN-G129E mutant in PTEN deficient prostate cancer cells failed to induce growth suppression mediated by wild type PTEN, indicating that lipid phosphatase and not protein phosphatase activity of PTEN is necessary for its tumor suppressive function (68). Functions of PTEN: Much knowledge about the functions of PTEN has come from mouse genetic studies. Homozygous deletion of PTEN results in early embryonic lethality between embryonic day E6.5 and E9.5, indicating that PTEN is essential during embryogenesis (69). Mice carrying heterozygous PTEN deletion are viable but show increased cancer incidence in multiple organs including the endometrium, thymus, liver, breast and gastrointestinal tract (69, 70). Additionally, adrenal tumors and hyperplasia of the prostate and lymph node have also been reported in a vast majority of heterozygous mice (70). Generation of conditional mouse models has provided further insight into the tissue specific functions of PTEN. Depending on the tissue type, PTEN inactivation can lead to fast, slow or no tumors. For example, prostate specific deletion of PTEN leads to development of prostate intraepithelial neoplasias (PINs) that progress to invasive and metastatic adenocarcinoma by 9 weeks of age (71). These tumors regress upon androgen ablation therapy but androgen independent tumors eventually arise, mimicking human disease progression. 24
25 Conversely, PTEN inactivation alone is insufficient to cause tumors in the central nervous system. PTEN deletion in adult mouse glial cells does not lead to glioma formation (72), however PTEN loss can cooperate with other genetic alterations to rapidly induce gliomas (73). This is consistent with the observation that PTEN mutations are rare in low grade tumors and are most commonly associated with glioblastoma mutliforme, the most malignant astrocytic tumor. PTEN control cell cycle progression: Cell culture studies demonstrated the function of PTEN in cell cycle control. Overexpression of wild type PTEN in PTEN deficient glioblastoma, breast and renal cancer cell lines resulted in accumulation of cells at G1 phase (68, 74, 75). This G1 arrest was accompanied by increased expression of cyclin-dependent kinase (CDK) inhibitor p27 Kip1 (76). p27 Kip1 induces G1 arrest by inhibiting CDK2/cyclin E activity which is required for entry into S phase. Consistent with this observation, p27 Kip1 expression and CDK2 activity were found to be downregulated in PTEN -/- ES cells and PTEN deficient cancer cells (76, 77). The inhibitory effect of PTEN on cell cycle progression could be effectively rescued by expression of constitutively active forms of PI3K or AKT, suggesting AKT signaling pathway as a key modulator of PTEN-sensitive cell proliferation (74, 78). However, PTEN overexpression failed to induce growth arrest in PTEN proficient glioblastoma cell lines such as LN18 and LN229 and retinoblastoma (Rb) deficient Saos-2 and C33A cells (79). Reconstitution of Rb in Saos-2 and C33A cells restored sensitivity to PTEN induced growth suppression (79). These studies suggest that the functional status of Rb and oncogenic signaling pathways other than AKT may influence the effects of PTEN on cell cycle progression. Contrary to the studies that established PTEN as a negative regulator of cell proliferation, recent data suggested that acute PTEN inactivation triggers p53 dependent cellular senescence. Combined inactivation of p53 and PTEN abrogated this senescence response and led to invasive 25
26 carcinoma of the prostate (80). Supporting this idea, concomitant mutations in both PTEN and p53 have been detected in several human tumors, indicating that combined loss of both tumor suppressors may be required to achieve maximal tumorigenic effect. PTEN control of apoptosis: In addition to regulating the cell cycle, PTEN controls various forms of programmed cell death (PCD). In mice, monoallelic inactivation of PTEN leads to lymph node hyperplasia due to defective apoptosis in B cells and macrophages (70). T lymphocytes isolated from these mice showed impaired activation induced cell death and Fas mediated apoptosis. PTEN deficient mouse embryonic fibroblasts exhibited a decreased sensitivity to a number of apoptotic stimuli and sensitivity to apoptosis was restored upon reconstitution of PTEN (81). In addition to apoptosis, PTEN overexpression can induce anoikis, a form of PCD that is initiated by disruption of cell-extracellular matrix interactions. This form of cell death has been observed in glioma and breast cancer cell lines over expressing PTEN (82, 83). PTEN control of genomic instability: Genome or chromosome instability is a hallmark of cancer. Nuclear PTEN has been shown to regulate genomic stability by physically associating with the centromeric protein CENP-C (84). Disruption of this association leads to centromere breakage and chromosomal translocations. Additionally, PTEN loss triggers Akt mediated cytoplasmic sequestration of CHK1 via phosphorylation and ubiquitination (85). DNA damage activates CHK1, which then leads to phosphorylation and inhibition of Cdc25a. As a result, cyclin/cdk complexes remain phosphorylated in an inactive state and cells undergo a transient arrest in G1, S or G2 phases. In PTEN-/- cells, nuclear exclusion of CHK1 prevents its checkpoint function and consequently promotes aneuploidy. 26
27 PTEN control of stem cell renewal: Pathways regulating stem cell renewal and maintenance are frequently altered in cancer. A recent study demonstrated that PTEN inactivation in hematopoietic cells led to depletion of hematopoietic stem cells (HSC) and enrichment of leukemia initiating cells resulting in transplantable leukemias in mice (86). PTEN loss triggered aberrant proliferation of HSCs leading to their exhaustion. In contrast, PTEN deletion in the nervous system increased the pool of self-renewing neural stem cells by promoting their deregulated proliferation. It is still unclear as to why proliferation driven by PTEN loss in some stem cell populations causes self-renewal while in others it causes exhaustion. The functions of PTEN have become more diverse since its discovery as a putative phosphatase. Many tumor suppressive effects of PTEN have been attributed to its ability to dephosphorylate PIP3 and thereby antagonize PI3K/Akt activation. However, a recent study indicated phosphatase independent functions of PTEN. In this study, nuclear PTEN increased the tumor suppressive activity of an E3 ubiquitin ligase complex involved in regulating cell cycle progression and senescence (87). This study has significantly contributed to our knowledge of PTEN function in the nucleus. However, future studies are needed to enhance our understanding of the roles of nuclear versus cytoplasmic PTEN, as well as, its phosphatase dependent versus independent functions and their relative contributions to tumor suppression. Post-translatoinal modifications of PTEN: Given the multitude of cellular processes regulated by PTEN, it is reasonable to assume that PTEN itself is tightly regulated. In addition to transcriptional, post-transcriptional and translational mechanisms, various post translational modifications regulate the stability, activity and subcellular localization of PTEN. These modifications include phosphorylation, acetylation and ubiquitination. 27
28 Phosphorylation: The C-terminal region of PTEN contains multiple serine and threonine residues that serve as putative phosphorylation sites for several kinases. Phosphorylation of these residues reduces the affinity of the catalytic and C2 domains for the membrane, thus, inhibiting PTEN activity. Phosphorylation also increases PTEN stability by preventing proteasomal degradation and proteolysis by caspases. In contrast, dephosphorylation of the C-terminal tail triggers rapid degradation of PTEN and increases its activity by inducing a conformational change that allows PTEN to interact with PIP3 at the membrane. Multiple kinases including casein kinase II (CK2), GSK3β, PICT-1 and ROCK have been implicated in phosphorylating the PTEN C-terminal tail (88). Acetylation: The histone acetyltransferase p300/cbp associated factor (PCAF) acetylates PTEN on lysines 125 and 128 in response to serum and growth factors (89). PCAF mediated acetylation of PTEN reduces its catalytic specificity towards PIP3 and triggers Akt activation. Thus, inhibiting acetylation may provide an effective means of restoring PTEN function in cells. Ubiquitination: PTEN ubiquitination is mediated by the E3 ubiquitin ligase Nedd4-1(90). Monoubiquitination at K289 promotes nuclear import of PTEN, whereas, polyubiquitination targets PTEN to the proteasome for degradation (91). The functional significance of ubiquitnation is further supported by the occurrence of K289E mutation in cowden disease patients. K289E mutant retains catalytic activity but fails to accumulate in nuclei of patient tissue due to an import defect. These studies suggest that nuclear compartmentalization of PTEN may comprise a key component of its tumor suppressive activity. 28
29 Post-translational regulation of PTEN may govern its diverse cellular and biological functions. Thus, enhancing or restoring PTEN function by targeting its regulatory machinery can be an effective cancer therapeutic approach. 29
30 Linking the PI3K/Akt pathway to human cancer Although the identification of Akt in the genome of an oncogenic virus clearly indicated a role for Akt in oncogenic transformation, the frequency and spectrum of cancers in which Akt is activated was better appreciated upon identification of mutations in upstream pathway components that regulate Akt. Akt activation frequently results from inactivation of PTEN. PTEN is the second most frequently mutated tumor suppressor gene, with the highest frequency of mutations found in endometrial, prostate, glioblastoma and skin cancers. In addition to somatic mutations, germline mutations in PTEN have been detected in multiple human autosomal dominant disorders that are characterized by the appearance of hyperplastic, disorganized and nonmalignant growths called hamartomas throughout the body. These syndromes include Cowden disease (CD), Bannayan-Zonana syndrome (BZS), Lhermitte-Duclos disease (LDD), Proteus syndrome (PS) and Proteus-like syndrome (PLS) (92). Besides mutations, PTEN expression is also reduced in response to micrornas (mirs), including mir-17, mir-19b, mir20a, mir-26a, mir-214, mir-221, and mir-222. These mirnas interfere with the production of PTEN, resulting in Akt activation. Frequent overexpression of the mir locus, which includes the PTEN-targeting mirs -17, -19b, and -20a, has been observed in B and T lymphomas, as well as carcinomas of the breast, lung, prostate and other tissues (93-95). As would be expected for loss of PTEN expression and increased Akt activation, overexpression of the mir locus or mir-214 is associated with cancer cell survival in the face of cytotoxic chemotherapeutics, such as hydroxyurea, gemcitabine, and cisplatin (96-98). Similar to overexpression of mirs to reduce PTEN levels, tumor cells can enhance mir targeting of PTEN by reducing the expression of mrna from the PTEN pseudogene PTENP1. PTENP1 mrna contains binding sites for the mirs that target 30
31 PTEN and can thus act as a sink for PTEN-targeting mirs. Reduced expression of PTENP1 correlates with increased Akt activation (99). Outside of PTEN, Akt signaling can be activated in cancer cells by mutations in the subunits of PI3K itself (100). Cancer-associated mutations have been detected in the gene encoding the p110α subunit of PI3K, known as PIK3CA (100). PIK3CA mutations are frequently located in two hotspots within or adjacent to the kinase domain. These mutations activate kinase activity by enhancing the affinity of PI3K for the plasma membrane and/or by impairing the negative regulatory function of the p85 subunit of PI3K (101). Mutations in the p85 regulatory subunits can also increase PIP3 levels and trigger Akt activation in cancer cells (102). This class of mutations has been detected most often in the p85α subunit, which is encoded by the PIK3R1 gene ( ). Rare mutations have been noted in similar locations of p85 homologues encoded by the PIK3R2, PIK3R4 and PIK3R5 genes (102). Altogether, mutations in PTEN, PIK3CA, and PIK3R1 have been shown to increase PIP3 and activate Akt (106). Although Akt is activated and probably contributes to the transformed phenotype in the majority of cells containing these mutations, a surprising finding is that Akt is required for cell survival in only a subset of cancer cell lines containing these mutations. Categorizing each cell line according to the level of Akt activation correlates with cellular addiction to Akt survival signals. In PTEN-deficient cells, Akt is highly phosphorylated and required for cell survival (107). In contrast to PTEN-deficient cells, a subset of PIK3CA-mutant cell lines have modest Akt phosphorylation, and shrna targeting Akt1 expression does not impair cell survival. Instead, SGK3, an Akt-related kinase that contains a PH domain, mediates survival in cells with low Akt activity (107). Other PIK3CA-mutant cell lines maintain high levels of Akt phosphorylation and require Akt for cell survival. Some breast cancers contain 31
32 both PTEN and PIK3CA mutations, reinforcing the idea that parallel signaling pathways can be induced by mutations in both genes (108). Oncogenic mutations in Akt itself have been observed in carcinomas of the breast, ovarian, and colorectal tissues (109). An E17K mutation in Akt1 that increases basal kinase activity by altering the conformation of the PH domain has been reported by multiple groups ( ). In addition, there is a report of an E17K mutation in Akt3 isolated from a melanoma tumor (112), and Akt2 amplification has been reported in ovarian and breast cancers (113). Mutations in Akt1 do not coincide with mutations in PTEN or PIK3CA, suggesting that these mutations may be functionally redundant (111). However, in vitro comparison of the transforming effects of overexpressing Akt1 E17K vs. PIK3CA in a breast epithelial cell line revealed differences in invasiveness (114). From this observation and the above-mentioned Aktindependent apoptosis resistance in PIK3CA-mutant cells, it appears likely that some PI3K prosurvival signals are transmitted by a pathway parallel to Akt, while Akt survival signaling is required in cells with PTEN inactivation or Akt1 mutants. More recently, human cancer genome database search led to the identification of six point mutaions in mtor- A8S (lung), M135T (malignant melanoma), M2011V (ovarian carcinoma), S2215Y (large intestine adenocarcinoma), P2476L (glioma) and R2505P (clear cell renal cell carcinoma) (115). Several of these mutations are located within or close to the kinase domain. Expression of S2215Y and R2505P mtor mutants in HEK293 cells conferred constitutive phosphorylation of mtorc1 substrates even under nutrient starvation conditions (115). Interstingly, no significant increase in Akt phosphorylation was detected with these mtor mutants, indicating that the mtor mutations may function to specifically activate mtorc1 and not mtorc2. 32
33 Mutations in components downstream of mtorc1 have been also been observed. Analysis of 372 primary breast tumors revealed amplification of S6K1 in 10% of tumors. S6K1 amplification co-existed with other gene amplifications; however 5 tumors displayed single gene amplifications for S6K1 (116). S6K1 has also been found to be mutated in colorectal cancer (104). The functional significance of these mutations, however, remains to be determined. 33
34 PI3K/Akt pathway activation induces the Warburg effect Activation of the Akt pathway induces aerobic glycolysis by affecting different regulatory steps in glucose metabolism. Beginning with glucose transport, Akt activation promotes Glut4 membrane translocation in muscle cells by phosphorylating and inhibiting the GTPase activating protein- AS160 (117); (118). AS160 is a GAP that promotes GTP hydrolysis by Rab family small G proteins, such as Rab8A. Rab8A in the GTP-bound form stimulates Glut4 vesicle translocation in muscle cells (118, 119). Similarly, in hematopoietic cells, constitutively active Akt is sufficient to promote growth factor-independent membrane translocation of Glut1 (120). In addition to enhancing glucose uptake, Akt also promotes association of hexokinase (HK) with the outer mitochondrial membrane, where HK may promote rapid glucose phosphorylation utilizing mitochondrial sources of ATP. Mitochondrial-bound HK is important for Akt mediated cell survival as pharmacologic inhibition of HK interaction with mitochondria induces apoptosis in constitutively active Akt expressing rat fibroblasts (121). The precise mechanism by which Akt promotes mitochondria-hk association remains unclear. Transcriptionally, Akt can regulate glycolytic gene expression by phosphorylating Foxo transcription factors (122). Akt phosphorylation of Foxo s triggers their ubiquitination and proteasomal degradation (123). Foxo1 has been shown to suppress glycolytic gene expression in hepatocytes (124, 125). Thus, Akt mediated inhibition of Foxo1 activates glycolytic gene expression. Downstream of Akt, mtorc1 signaling can drive increased glycolysis through activation of hypoxia inducible transcription factor 1α (HIF-1α) via mechanisms that remain poorly understood (Figure 1). HIF-1α expression is regulated by cellular oxygen levels (126). Under normoxic conditions, HIF-1α is continuously synthesized and rapidly degraded. HIF-1α 34
35 degradation is initiated by hydroxylation of two proline residues- Pro402 and Pro564 within the oxygen dependent degradation (ODD) domain of HIF-1α ( ). This hydroxylation process is mediated by three evolutionarily conserved prolyl hydroxylases PHD1, PHD2 and PHD3. Of these, PHD2 plays a predominant role in regulating cellular HIF-1α levels (131, 132). The Von Hippel-Lindau (VHL) tumor suppressor protein recognizes hydroxylated HIF-1α and targets it for polyubiquitination and degradation by the proteasome. In hypoxia, the activity of the PHD enzymes is inhibited and HIF-1α- pvhl interaction is prevented. As a result, HIF-1α ubiquitination and degradation is blocked and consequently the level of protein increases (133). Accumulated HIF-1α translocates to the nucleus, where it dimerizes with the constitutively expressed HIF-1β subunit to form the HIF1 complex (134). HIF1 binds to the hypoxia- response elements (HRE) within promoter regions of HIF1 target genes and activates transcription (135). Among the many genes induced by HIF1 are genes involved in glucose transport and glucose metabolism. HIF1 increases glucose uptake in cells by increasing the expression of glucose transporters- Glut1 and Glut3 (136, 137) (Figure 4). Once glucose is taken up by the cell, HIF1 promotes glucose entry into the glycolytic pathway by stimulating transcription of glycolytic genes. All 10 enzymes necessary for glycolysis are directly regulated by HIF1 such that the entire process is stimulated by HIF1 (138) (Figure 4). Under hypoxic conditions, HIF1 facilitates the conversion of pyruvate to lactate by transactivating LDHA enzyme (139) (Figure 4). The conversion of pyruvate to lactate provides the cell with a mechanism to regenerate cytosolic NAD +, required for further glycolysis. In addition to stimulating glycolysis, HIF1 actively suppresses mitochondrial respiration by directly upregulating the expression of pyruvate dehydrogenase kinase 1 (PDK1) (140, 141). PDK1 phosphorylates and inhibits pyruvate dehydrogenase (PDH), which catalyzes the conversion of pyruvate to acetyl-coa (142) (Figure 35
36 4). By inducing PDK1, HIF1 inhibits oxidative phosphorylation and reduces total cellular oxygen consumption. Overall, HIF1 activation promotes a metabolic switch from oxidative to glycolytic metabolism. Thus, HIF1 stabilization mediated by activation of Akt/mTOR pathway may contribute to altering metabolism and promoting the warburg effect in cancer cells. It is clear that Akt activation induces glycolysis through multiple mechanisms. Inhibiting Akt-activated glycolysis through interruptions in glucose availability or glycolytic function is sufficient to overcome the other pro-survival effects of Akt, suggesting that Akt dependent cell survival requires active glycolysis. (143). Thus, targeting the Akt pathway may provide an efficient means to alter tumor cell metabolism. 36
37 Targeting the Akt pathway The frequent deregulation of Akt signaling pathway in cancer has provided a strong rationale to target this pathway for cancer therapy. Emerging chemotherapeutic agents target Akt itself, its upstream activators and downstream effectors. Currently, approved chemotherapeutics within the pathway are rapamycin (sirolimus) and its derivatives everolimus and temsirolimus (144, 145). These compounds prevent mtorc1 from phosphorylating downstream substrates, particularly S6K1. Although beneficial for renal clear cell carcinoma, rapamycin and its derivatives have seen only limited application in cancer therapy, due to disappointingly low levels of cancer cell cytotoxicity and poor efficacy outside of renal cell carcinoma (146). The limited efficacy may be due to incomplete inhibition of mtorc1 (147, 148) or due to a compensatory induction of Akt in response to rapamycin, which has been observed in multiple cancers (149, 150). To more effectively inhibit mtorc1, new agents that interfere with the ATP-binding pocket of the mtor kinase subunit have been developed, termed active site TOR inhibitors, or astori (151). There are several advantages for this approach. First, interfering with mtor reduces the activity of all mtor-associated kinase complexes, including mtor complex 2 (mtorc2) (24). mtorc2 is required for Akt activation, and inactivation of mtorc2 suppresses Akt function. Thus, simultaneous inactivation of mtorc1 and mtorc2 with astori prevents the compensatory induction of Akt that plagues rapamycin and its derivatives in at least some cell types (30, 152). Also astori function as pan-inhibitors of mtorc substrate phosphorylation, rather than inhibiting a subset of targets as rapamycin does. An example of an mtor-specific inhibitor that inactivates both mtorc1 and mtorc2 is PP242. PP242 outperforms rapamycin in terms of potency in inducing apoptosis in transformed cells for 37
38 acute lymphoblastic leukemia and multiple myeloma ( ). Adding to the promise of astori, PP242 was shown to be simultaneously cytotoxic for transformed cells but permissive for the proliferation of non-transformed immune cells. Thus, PP242 may be more effective as a cytotoxic chemotherapeutic agent while avoiding immunosuppressive effects compared to rapamycin. Because the kinase domains of mtor and the p110 subunits of class I PI3Ks are similar, some inhibitors can target both enzymes at therapeutic concentrations. Heavily investigated dual PI3K/mTOR inhibitors are PI-103 and BEZ-235. A screen of multiple PI3K inhibitors for promoting cell cycle arrest in a panel of glioma cells showed that PI-103 was more effective than other inhibitors, despite similar efficacy in inhibiting Akt activation (156). Comparison with other PI3K inhibitors and rapamycin led to the conclusion that PI-103 s superior performance is linked to its dual ability to inhibit PI3K and mtor (156). PI-103 has not entered into clinical trial, but the PI-103 derivative GDC-0941 has been registered in a clinical trial at ClinicalTrials.gov (157). The BEZ-235 dual specific inhibitor is another well-studied agent that has recently entered clinical trial (158). Interestingly, a comparison of PI-103 (a dual specific inhibitor) and PP-242 (an mtor-selective astori) revealed a potential immune-suppressive action of the dual specific inhibitor, suggesting that mtor-selective compounds may be more attractive for chemotherapy (153). PI3K-selective inhibitors that suppress Akt-dependent survival without necessarily targeting mtor have also entered clinical trial, such as XL-187. Although some data favor the mtor-selective astori compounds, the relative merits of targeting PI3K alone, mtor alone, or PI3K/mTOR in combination will need to be thoroughly compared for effects on the phosphorylation of Akt substrates, glycolytic metabolism, and cytotoxic efficacy. 38
39 Targeting PI3K or mtor addresses the problem of Akt-dependent survival in tumor cells by inhibiting upstream and downstream elements of the Akt signaling pathway. In addition, inhibitors of Akt itself have been developed, falling into two classes: ATP-binding site kinase antagonists, and PIP3 mimetics that interfere with binding to the Akt PH domain. A number of kinase antagonists have been developed (reviewed in (159)), of which a promising example is GSK GSK has anti-tumor activity in xenograft models of breast, prostate, and ovarian cancers (160) and is cytotoxic for various leukemia cell lines (161). Initial clinical trials of this compound are underway. Targeting the Akt PH domain, compounds such as perifosine and phosphatidylinositol ether lipid analogues (PIAs) can prevent Akt phosphorylation/activation, and induce programmed cell death (162, 163). Although perifosine advanced to Phase 2 clinical trials, results failed to meet defined response targets for recurrent prostate or breast cancer (164, 165). A newer generation of PH domain antagonists have recently been shown to induce apoptosis, metabolic stress and autophagic cell death in PTEN-deficient glioblastoma (166). While targeting the Akt kinase domain may be the most specific way to reduce Aktinduced metabolism and survival in cancer cells, it is possible that this specificity will limit therapeutic efficacy. As discussed above, PI3K can activate SGK3 in a parallel pathway to mediate survival in cells with PI3K mutations. This would suggest that the PI3K, mtor, or PIP3 analogues may function better to suppress resistance to programmed cell death. Along these lines, the PIA compounds can activate the AMPK tumor suppressor pathway in parallel to suppressing Akt signaling (167). Important work remains to determine whether the benefits of targeting the pathway at multiple levels outweigh the risks of side effects associated with the inhibition of multiple signaling pathways. 39
40 Increased aerobic glycolysis is a hallmark of cancer, however, the mechanisms that contribute to this metabolic alteration remain largely unknown. Aberrant Akt activation occurs in a wide variety of human cancers. Constitutive Akt activity induces glycolysis in cells, rendering them addicted to glucose for maintenance of survival. Identifying regulators that coordinate Akt dependent increases in glycolysis may, therefore, provide novel therapeutic targets for anticancer treatment. In this thesis, we uncover the role of S6K1 in mediating glycolysis and survival induced by PTEN deficiency and define the mechanism of this regulation. 40
41 References 1. Warburg O, Wind F, Negelein E (1927) The metabolism of tumors in the body. J Gen Physiol 8: WARBURG O (1956) On the origin of cancer cells. Science 123: Baysal BE, Ferrell RE, Willett-Brozick JE et al (2000) Mutations in SDHD, a mitochondrial complex II gene, in hereditary paraganglioma. Science 287: Tomlinson IP, Alam NA, Rowan AJ et al (2002) Germline mutations in FH predispose to dominantly inherited uterine fibroids, skin leiomyomata and papillary renal cell cancer. Nat Genet 30: Zhao S, Lin Y, Xu W et al (2009) Glioma-derived mutations in IDH1 dominantly inhibit IDH1 catalytic activity and induce HIF-1alpha. Science 324: Frezza C, Gottlieb E (2009) Mitochondria in cancer: Not just innocent bystanders. Semin Cancer Biol 19: Weinhouse S (1976) The warburg hypothesis fifty years later. Z Krebsforsch Klin Onkol Cancer Res Clin Oncol 87: Vander Heiden MG, Cantley LC, Thompson CB (2009) Understanding the warburg effect: The metabolic requirements of cell proliferation. Science 324: Possemato R, Marks KM, Shaul YD et al (2011) Functional genomics reveal that the serine synthesis pathway is essential in breast cancer. Nature 476: Locasale JW, Grassian AR, Melman T et al (2011) Phosphoglycerate dehydrogenase diverts glycolytic flux and contributes to oncogenesis. Nat Genet 43: Christofk HR, Vander Heiden MG, Harris MH et al (2008) The M2 splice isoform of pyruvate kinase is important for cancer metabolism and tumour growth. Nature 452: Vander Heiden MG, Locasale JW, Swanson KD et al (2010) Evidence for an alternative glycolytic pathway in rapidly proliferating cells. Science 329: Johnson TM, Yu ZX, Ferrans VJ et al (1996) Reactive oxygen species are downstream mediators of p53-dependent apoptosis. Proc Natl Acad Sci U S A 93: Lu T, Finkel T (2008) Free radicals and senescence. Exp Cell Res 314: Le A, Cooper CR, Gouw AM et al (2010) Inhibition of lactate dehydrogenase A induces oxidative stress and inhibits tumor progression. Proc Natl Acad Sci U S A 107:
42 16. Pelicano H, Martin DS, Xu RH et al (2006) Glycolysis inhibition for anticancer treatment. Oncogene 25: Cantley LC (2002) The phosphoinositide 3-kinase pathway. Science 296: Liu P, Cheng H, Roberts TM et al (2009) Targeting the phosphoinositide 3-kinase pathway in cancer. Nat Rev Drug Discov 8: Hanks SK, Hunter T (1995) Protein kinases 6. the eukaryotic protein kinase superfamily: Kinase (catalytic) domain structure and classification. FASEB J 9: Peng J, Elias JE, Thoreen CC et al (2003) Evaluation of multidimensional chromatography coupled with tandem mass spectrometry (LC/LC-MS/MS) for large-scale protein analysis: The yeast proteome. J Proteome Res 2: Dummler B, Tschopp O, Hynx D et al (2006) Life with a single isoform of akt: Mice lacking Akt2 and Akt3 are viable but display impaired glucose homeostasis and growth deficiencies. Mol Cell Biol 26: Alessi DR, Deak M, Casamayor A et al (1997) 3-phosphoinositide-dependent protein kinase- 1 (PDK1): Structural and functional homology with the drosophila DSTPK61 kinase. Curr Biol 7: Alessi DR, James SR, Downes CP et al (1997) Characterization of a 3-phosphoinositidedependent protein kinase which phosphorylates and activates protein kinase balpha. Curr Biol 7: Sarbassov DD, Guertin DA, Ali SM et al (2005) Phosphorylation and regulation of Akt/PKB by the rictor-mtor complex. Science 307: Inoki K, Zhu T, Guan KL (2003) TSC2 mediates cellular energy response to control cell growth and survival. Cell 115: Inoki K, Li Y, Zhu T et al (2002) TSC2 is phosphorylated and inhibited by akt and suppresses mtor signalling. Nat Cell Biol 4: Khatri S, Yepiskoposyan H, Gallo CA et al (2010) FOXO3a regulates glycolysis via transcriptional control of tumor suppressor TSC1. J Biol Chem 285: Guertin DA, Sabatini DM (2007) Defining the role of mtor in cancer. Cancer Cell 12: Chen J, Zheng XF, Brown EJ et al (1995) Identification of an 11-kDa FKBP12-rapamycinbinding domain within the 289-kDa FKBP12-rapamycin-associated protein and characterization of a critical serine residue. Proc Natl Acad Sci U S A 92:
43 30. Sarbassov DD, Ali SM, Sengupta S et al (2006) Prolonged rapamycin treatment inhibits mtorc2 assembly and Akt/PKB. Mol Cell 22: Sabatini DM (2006) mtor and cancer: Insights into a complex relationship. Nat Rev Cancer 6: Wullschleger S, Loewith R, Hall MN (2006) TOR signaling in growth and metabolism. Cell 124: Hay N, Sonenberg N (2004) Upstream and downstream of mtor. Genes Dev 18: Ma XM, Blenis J (2009) Molecular mechanisms of mtor-mediated translational control. Nat Rev Mol Cell Biol 10: Dowling RJ, Topisirovic I, Alain T et al (2010) mtorc1-mediated cell proliferation, but not cell growth, controlled by the 4E-BPs. Science 328: Jeno P, Ballou LM, Novak-Hofer I et al (1988) Identification and characterization of a mitogen-activated S6 kinase. Proc Natl Acad Sci U S A 85: Reinhard C, Thomas G, Kozma SC (1992) A single gene encodes two isoforms of the p70 S6 kinase: Activation upon mitogenic stimulation. Proc Natl Acad Sci U S A 89: Grove JR, Banerjee P, Balasubramanyam A et al (1991) Cloning and expression of two human p70 S6 kinase polypeptides differing only at their amino termini. Mol Cell Biol 11: Reinhard C, Fernandez A, Lamb NJ et al (1994) Nuclear localization of p85s6k: Functional requirement for entry into S phase. EMBO J 13: Karni R, de Stanchina E, Lowe SW et al (2007) The gene encoding the splicing factor SF2/ASF is a proto-oncogene. Nat Struct Mol Biol 14: Shima H, Pende M, Chen Y et al (1998) Disruption of the p70(s6k)/p85(s6k) gene reveals a small mouse phenotype and a new functional S6 kinase. Embo J 17: Lee-Fruman KK, Kuo CJ, Lippincott J et al (1999) Characterization of S6K2, a novel kinase homologous to S6K1. Oncogene 18: Ferrari S, Bannwarth W, Morley SJ et al (1992) Activation of p70s6k is associated with phosphorylation of four clustered sites displaying Ser/Thr-pro motifs. Proc Natl Acad Sci U S A 89: Han JW, Pearson RB, Dennis PB et al (1995) Rapamycin, wortmannin, and the methylxanthine SQ20006 inactivate p70s6k by inducing dephosphorylation of the same subset of sites. J Biol Chem 270:
44 45. Mukhopadhyay NK, Price DJ, Kyriakis JM et al (1992) An array of insulin-activated, proline-directed serine/threonine protein kinases phosphorylate the p70 S6 kinase. J Biol Chem 267: Dennis PB, Pullen N, Kozma SC et al (1996) The principal rapamycin-sensitive p70(s6k) phosphorylation sites, T-229 and T-389, are differentially regulated by rapamycin-insensitive kinase kinases. Mol Cell Biol 16: Moser BA, Dennis PB, Pullen N et al (1997) Dual requirement for a newly identified phosphorylation site in p70s6k. Mol Cell Biol 17: Pullen N, Dennis PB, Andjelkovic M et al (1998) Phosphorylation and activation of p70s6k by PDK1. Science 279: Isotani S, Hara K, Tokunaga C et al (1999) Immunopurified mammalian target of rapamycin phosphorylates and activates p70 S6 kinase alpha in vitro. J Biol Chem 274: Montagne J, Stewart MJ, Stocker H et al (1999) Drosophila S6 kinase: A regulator of cell size. Science 285: Pende M, Um SH, Mieulet V et al (2004) S6K1(-/-)/S6K2(-/-) mice exhibit perinatal lethality and rapamycin-sensitive 5'-terminal oligopyrimidine mrna translation and reveal a mitogenactivated protein kinase-dependent S6 kinase pathway. Mol Cell Biol 24: Jefferies HB, Fumagalli S, Dennis PB et al (1997) Rapamycin suppresses 5'TOP mrna translation through inhibition of p70s6k. EMBO J 16: Ruvinsky I, Sharon N, Lerer T et al (2005) Ribosomal protein S6 phosphorylation is a determinant of cell size and glucose homeostasis. Genes Dev 19: Raught B, Peiretti F, Gingras AC et al (2004) Phosphorylation of eucaryotic translation initiation factor 4B Ser422 is modulated by S6 kinases. EMBO J 23: Rogers GW,Jr, Richter NJ, Lima WF et al (2001) Modulation of the helicase activity of eif4a by eif4b, eif4h, and eif4f. J Biol Chem 276: Dorrello NV, Peschiaroli A, Guardavaccaro D et al (2006) S6K1- and betatrcp-mediated degradation of PDCD4 promotes protein translation and cell growth. Science 314: Ma XM, Yoon SO, Richardson CJ et al (2008) SKAR links pre-mrna splicing to mtor/s6k1-mediated enhanced translation efficiency of spliced mrnas. Cell 133: Richardson CJ, Broenstrup M, Fingar DC et al (2004) SKAR is a specific target of S6 kinase 1 in cell growth control. Curr Biol 14:
45 59. Wang X, Li W, Williams M et al (2001) Regulation of elongation factor 2 kinase by p90(rsk1) and p70 S6 kinase. EMBO J 20: Datta SR, Dudek H, Tao X et al (1997) Akt phosphorylation of BAD couples survival signals to the cell- intrinsic death machinery. Cell 91: del Peso L, Gonzalez-Garcia M, Page C et al (1997) Interleukin-3-induced phosphorylation of BAD through the protein kinase akt. Science 278: Datta SR, Katsov A, Hu L et al (2000) proteins and survival kinases cooperate to inactivate BAD by BH3 domain phosphorylation. Mol Cell 6: Harada H, Andersen JS, Mann M et al (2001) p70s6 kinase signals cell survival as well as growth, inactivating the pro-apoptotic molecule BAD. Proc Natl Acad Sci U S A 98: Um SH, Frigerio F, Watanabe M et al (2004) Absence of S6K1 protects against age- and diet-induced obesity while enhancing insulin sensitivity. Nature 431: Myers MP, Stolarov JP, Eng C et al (1997) P-TEN, the tumor suppressor from human chromosome 10q23, is a dual-specificity phosphatase. Proc Natl Acad Sci U S A 94: Maehama T, Dixon JE (1998) The tumor suppressor, PTEN/MMAC1, dephosphorylates the lipid second messenger, phosphatidylinositol 3,4,5-trisphosphate. J Biol Chem 273: Myers MP, Pass I, Batty IH et al (1998) The lipid phosphatase activity of PTEN is critical for its tumor supressor function. Proc Natl Acad Sci U S A 95: Furnari FB, Huang HJ, Cavenee WK (1998) The phosphoinositol phosphatase activity of PTEN mediates a serum-sensitive G1 growth arrest in glioma cells. Cancer Res 58: Di Cristofano A, Pesce B, Cordon-Cardo C et al (1998) Pten is essential for embryonic development and tumour suppression. Nat Genet 19: Podsypanina K, Ellenson LH, Nemes A et al (1999) Mutation of Pten/Mmac1 in mice causes neoplasia in multiple organ systems. Proc Natl Acad Sci U S A 96: Wang S, Gao J, Lei Q et al (2003) Prostate-specific deletion of the murine pten tumor suppressor gene leads to metastatic prostate cancer. Cancer Cell 4: Backman SA, Stambolic V, Suzuki A et al (2001) Deletion of pten in mouse brain causes seizures, ataxia and defects in soma size resembling lhermitte-duclos disease. Nat Genet 29: Chow LM, Endersby R, Zhu X et al (2011) Cooperativity within and among pten, p53, and rb pathways induces high-grade astrocytoma in adult brain. Cancer Cell 19:
46 74. Ramaswamy S, Nakamura N, Vazquez F et al (1999) Regulation of G1 progression by the PTEN tumor suppressor protein is linked to inhibition of the phosphatidylinositol 3-kinase/Akt pathway. Proc Natl Acad Sci U S A 96: Weng LP, Smith WM, Dahia PL et al (1999) PTEN suppresses breast cancer cell growth by phosphatase activity-dependent G1 arrest followed by cell death. Cancer Res 59: Li DM, Sun H (1998) PTEN/MMAC1/TEP1 suppresses the tumorigenicity and induces G1 cell cycle arrest in human glioblastoma cells. Proc Natl Acad Sci U S A 95: Sun H, Lesche R, Li DM et al (1999) PTEN modulates cell cycle progression and cell survival by regulating phosphatidylinositol 3,4,5,-trisphosphate and Akt/protein kinase B signaling pathway. Proc Natl Acad Sci U S A 96: Li J, Simpson L, Takahashi M et al (1998) The PTEN/MMAC1 tumor suppressor induces cell death that is rescued by the AKT/protein kinase B oncogene. Cancer Res 58: Paramio JM, Navarro M, Segrelles C et al (1999) PTEN tumour suppressor is linked to the cell cycle control through the retinoblastoma protein. Oncogene 18: Chen Z, Trotman LC, Shaffer D et al (2005) Crucial role of p53-dependent cellular senescence in suppression of pten-deficient tumorigenesis. Nature 436: Di Cristofano A, Kotsi P, Peng YF et al (1999) Impaired fas response and autoimmunity in pten+/- mice. Science 285: Lu Y, Lin YZ, LaPushin R et al (1999) The PTEN/MMAC1/TEP tumor suppressor gene decreases cell growth and induces apoptosis and anoikis in breast cancer cells. Oncogene 18: Davies MA, Lu Y, Sano T et al (1998) Adenoviral transgene expression of MMAC/PTEN in human glioma cells inhibits akt activation and induces anoikis. Cancer Res 58: Shen WH, Balajee AS, Wang J et al (2007) Essential role for nuclear PTEN in maintaining chromosomal integrity. Cell 128: Puc J, Keniry M, Li HS et al (2005) Lack of PTEN sequesters CHK1 and initiates genetic instability. Cancer Cell 7: Yilmaz OH, Valdez R, Theisen BK et al (2006) Pten dependence distinguishes haematopoietic stem cells from leukaemia-initiating cells. Nature 441: Song MS, Carracedo A, Salmena L et al (2011) Nuclear PTEN regulates the APC-CDH1 tumor-suppressive complex in a phosphatase-independent manner. Cell 144: Tamguney T, Stokoe D (2007) New insights into PTEN. J Cell Sci 120:
47 89. Okumura K, Mendoza M, Bachoo RM et al (2006) PCAF modulates PTEN activity. J Biol Chem 281: Wang X, Trotman LC, Koppie T et al (2007) NEDD4-1 is a proto-oncogenic ubiquitin ligase for PTEN. Cell 128: Trotman LC, Wang X, Alimonti A et al (2007) Ubiquitination regulates PTEN nuclear import and tumor suppression. Cell 128: Eng C (2003) PTEN: One gene, many syndromes. Hum Mutat 22: Mavrakis KJ, Wolfe AL, Oricchio E et al (2010) Genome-wide RNA-mediated interference screen identifies mir-19 targets in notch-induced T-cell acute lymphoblastic leukaemia. Nat Cell Biol 12: He L, Thomson JM, Hemann MT et al (2005) A microrna polycistron as a potential human oncogene. Nature 435: Volinia S, Calin GA, Liu CG et al (2006) A microrna expression signature of human solid tumors defines cancer gene targets. Proc Natl Acad Sci U S A 103: Olive V, Bennett MJ, Walker JC et al (2009) mir-19 is a key oncogenic component of mir Genes Dev 23: Meng F, Henson R, Lang M et al (2006) Involvement of human micro-rna in growth and response to chemotherapy in human cholangiocarcinoma cell lines. Gastroenterology 130: Yang H, Kong W, He L et al (2008) MicroRNA expression profiling in human ovarian cancer: MiR-214 induces cell survival and cisplatin resistance by targeting PTEN. Cancer Res 68: Poliseno L, Salmena L, Zhang J et al (2010) A coding-independent function of gene and pseudogene mrnas regulates tumour biology. Nature 465: Samuels Y, Wang Z, Bardelli A et al (2004) High frequency of mutations of the PIK3CA gene in human cancers. Science 304: Zhao L, Vogt PK (2008) Helical domain and kinase domain mutations in p110alpha of phosphatidylinositol 3-kinase induce gain of function by different mechanisms. Proc Natl Acad Sci U S A 105: Jaiswal BS, Janakiraman V, Kljavin NM et al (2009) Somatic mutations in p85alpha promote tumorigenesis through class IA PI3K activation. Cancer Cell 16:
48 103. Parsons DW, Jones S, Zhang X et al (2008) An integrated genomic analysis of human glioblastoma multiforme. Science 321: Wood LD, Parsons DW, Jones S et al (2007) The genomic landscapes of human breast and colorectal cancers. Science 318: Cancer Genome Atlas Research Network (2008) Comprehensive genomic characterization defines human glioblastoma genes and core pathways. Nature 455: Ikenoue T, Kanai F, Hikiba Y et al (2005) Functional analysis of PIK3CA gene mutations in human colorectal cancer. Cancer Res 65: Vasudevan KM, Barbie DA, Davies MA et al (2009) AKT-independent signaling downstream of oncogenic PIK3CA mutations in human cancer. Cancer Cell 16: Stemke-Hale K, Gonzalez-Angulo AM, Lluch A et al (2008) An integrative genomic and proteomic analysis of PIK3CA, PTEN, and AKT mutations in breast cancer. Cancer Res 68: Carpten JD, Faber AL, Horn C et al (2007) A transforming mutation in the pleckstrin homology domain of AKT1 in cancer. Nature 448: Bleeker FE, Felicioni L, Buttitta F et al (2008) AKT1(E17K) in human solid tumours. Oncogene 27: Kim MS, Jeong EG, Yoo NJ et al (2008) Mutational analysis of oncogenic AKT E17K mutation in common solid cancers and acute leukaemias. Br J Cancer 98: Davies MA, Stemke-Hale K, Tellez C et al (2008) A novel AKT3 mutation in melanoma tumours and cell lines. Br J Cancer 99: Bellacosa A, de Feo D, Godwin AK et al (1995) Molecular alterations of the AKT2 oncogene in ovarian and breast carcinomas. Int J Cancer 64: Lauring J, Cosgrove DP, Fontana S et al (2010) Knock in of the AKT1 E17K mutation in human breast epithelial cells does not recapitulate oncogenic PIK3CA mutations. Oncogene 29: Sato T, Nakashima A, Guo L et al (2010) Single amino-acid changes that confer constitutive activation of mtor are discovered in human cancer. Oncogene 29: Barlund M, Monni O, Kononen J et al (2000) Multiple genes at 17q23 undergo amplification and overexpression in breast cancer. Cancer Res 60:
49 117. Kohn AD, Summers SA, Birnbaum MJ et al (1996) Expression of a constitutively active akt Ser/Thr kinase in 3T3-L1 adipocytes stimulates glucose uptake and glucose transporter 4 translocation. J Biol Chem 271: Sano H, Kane S, Sano E et al (2003) Insulin-stimulated phosphorylation of a rab GTPaseactivating protein regulates GLUT4 translocation. J Biol Chem 278: Ishikura S, Bilan PJ, Klip A (2007) Rabs 8A and 14 are targets of the insulin-regulated rab- GAP AS160 regulating GLUT4 traffic in muscle cells. Biochem Biophys Res Commun 353: Wieman HL, Wofford JA, Rathmell JC (2007) Cytokine stimulation promotes glucose uptake via phosphatidylinositol-3 kinase/akt regulation of Glut1 activity and trafficking. Mol Biol Cell 18: Majewski N, Nogueira V, Bhaskar P et al (2004) Hexokinase-mitochondria interaction mediated by akt is required to inhibit apoptosis in the presence or absence of bax and bak. Mol Cell 16: Brunet A, Bonni A, Zigmond MJ et al (1999) Akt promotes cell survival by phosphorylating and inhibiting a forkhead transcription factor. Cell 96: Plas DR, Thompson CB (2003) Akt activation promotes degradation of tuberin and FOXO3a via the proteasome. J Biol Chem 278: Gross DN, Wan M, Birnbaum MJ (2009) The role of FOXO in the regulation of metabolism. Curr Diab Rep 9: Zhang W, Patil S, Chauhan B et al (2006) FoxO1 regulates multiple metabolic pathways in the liver: Effects on gluconeogenic, glycolytic, and lipogenic gene expression. J Biol Chem 281: Semenza GL (2003) Targeting HIF-1 for cancer therapy. Nat Rev Cancer 3: Masson N, Willam C, Maxwell PH et al (2001) Independent function of two destruction domains in hypoxia-inducible factor-alpha chains activated by prolyl hydroxylation. EMBO J 20: Jaakkola P, Mole DR, Tian YM et al (2001) Targeting of HIF-alpha to the von hippellindau ubiquitylation complex by O2-regulated prolyl hydroxylation. Science 292: Ivan M, Kondo K, Yang H et al (2001) HIFalpha targeted for VHL-mediated destruction by proline hydroxylation: Implications for O2 sensing. Science 292: Yu F, White SB, Zhao Q et al (2001) HIF-1alpha binding to VHL is regulated by stimulussensitive proline hydroxylation. Proc Natl Acad Sci U S A 98:
50 131. Bruick RK, McKnight SL (2001) A conserved family of prolyl-4-hydroxylases that modify HIF. Science 294: Berra E, Benizri E, Ginouves A et al (2003) HIF prolyl-hydroxylase 2 is the key oxygen sensor setting low steady-state levels of HIF-1alpha in normoxia. EMBO J 22: Kallio PJ, Okamoto K, O'Brien S et al (1998) Signal transduction in hypoxic cells: Inducible nuclear translocation and recruitment of the CBP/p300 coactivator by the hypoxiainducible factor-1alpha. EMBO J 17: Wang GL, Jiang BH, Rue EA et al (1995) Hypoxia-inducible factor 1 is a basic-helix-loophelix-pas heterodimer regulated by cellular O2 tension. Proc Natl Acad Sci U S A 92: Wang GL, Semenza GL (1993) General involvement of hypoxia-inducible factor 1 in transcriptional response to hypoxia. Proc Natl Acad Sci U S A 90: Maxwell PH, Dachs GU, Gleadle JM et al (1997) Hypoxia-inducible factor-1 modulates gene expression in solid tumors and influences both angiogenesis and tumor growth. Proc Natl Acad Sci U S A 94: Chen C, Pore N, Behrooz A et al (2001) Regulation of glut1 mrna by hypoxia-inducible factor-1. interaction between H-ras and hypoxia. J Biol Chem 276: Iyer NV, Kotch LE, Agani F et al (1998) Cellular and developmental control of O2 homeostasis by hypoxia-inducible factor 1 alpha. Genes Dev 12: Firth JD, Ebert BL, Ratcliffe PJ (1995) Hypoxic regulation of lactate dehydrogenase A. interaction between hypoxia-inducible factor 1 and camp response elements. J Biol Chem 270: Kim JW, Tchernyshyov I, Semenza GL et al (2006) HIF-1-mediated expression of pyruvate dehydrogenase kinase: A metabolic switch required for cellular adaptation to hypoxia. Cell Metab 3: Papandreou I, Cairns RA, Fontana L et al (2006) HIF-1 mediates adaptation to hypoxia by actively downregulating mitochondrial oxygen consumption. Cell Metab 3: Patel MS, Korotchkina LG (2001) Regulation of mammalian pyruvate dehydrogenase complex by phosphorylation: Complexity of multiple phosphorylation sites and kinases. Exp Mol Med 33: Plas DR, Talapatra S, Edinger AL et al (2001) Akt and bcl-xl promote growth factorindependent survival through distinct effects on mitochondrial physiology. J Biol Chem 276:
51 144. Hudes G, Carducci M, Tomczak P et al (2007) Temsirolimus, interferon alfa, or both for advanced renal-cell carcinoma. New England Journal of Medicine 356: Motzer RJ, Escudier B, Oudard S et al (2008) Efficacy of everolimus in advanced renal cell carcinoma: A double-blind, randomised, placebo-controlled phase III trial. Lancet 372: Abraham RT, Gibbons JJ (2007) The mammalian target of rapamycin signaling pathway: Twists and turns in the road to cancer therapy. Clin Cancer Res 13: Thoreen CC, Kang SA, Chang JW et al (2009) An ATP-competitive mammalian target of rapamycin inhibitor reveals rapamycin-resistant functions of mtorc1. J Biol Chem 284: Choo AY, Yoon SO, Kim SG et al (2008) Rapamycin differentially inhibits S6Ks and 4E- BP1 to mediate cell-type-specific repression of mrna translation. Proc Natl Acad Sci U S A 105: Julien LA, Carriere A, Moreau J et al (2010) mtorc1-activated S6K1 phosphorylates rictor on threonine 1135 and regulates mtorc2 signaling. Mol Cell Biol 30: Dibble CC, Asara JM, Manning BD (2009) Characterization of rictor phosphorylation sites reveals direct regulation of mtor complex 2 by S6K1. Mol Cell Biol 29: Dowling RJ, Topisirovic I, Fonseca BD et al (2010) Dissecting the role of mtor: Lessons from mtor inhibitors. Biochim Biophys Acta 1804: O'Reilly KE, Rojo F, She QB et al (2006) mtor inhibition induces upstream receptor tyrosine kinase signaling and activates akt. Cancer Res 66: Janes MR, Limon JJ, So L et al (2010) Effective and selective targeting of leukemia cells using a TORC1/2 kinase inhibitor. Nat Med 16: Hsieh AC, Costa M, Zollo O et al (2010) Genetic dissection of the oncogenic mtor pathway reveals druggable addiction to translational control via 4EBP-eIF4E. Cancer Cell 17: Hoang B, Frost P, Shi Y et al (2010) Targeting TORC2 in multiple myeloma with a new mtor kinase inhibitor. Blood Fan QW, Knight ZA, Goldenberg DD et al (2006) A dual PI3 kinase/mtor inhibitor reveals emergent efficacy in glioma. Cancer Cell 9: Raynaud FI, Eccles SA, Patel S et al (2009) Biological properties of potent inhibitors of class I phosphatidylinositide 3-kinases: From PI-103 through PI-540, PI-620 to the oral agent GDC Mol Cancer Ther 8:
52 158. Serra V, Markman B, Scaltriti M et al (2008) NVP-BEZ235, a dual PI3K/mTOR inhibitor, prevents PI3K signaling and inhibits the growth of cancer cells with activating PI3K mutations. Cancer Res 68: Yap TA, Garrett MD, Walton MI et al (2008) Targeting the PI3K-AKT-mTOR pathway: Progress, pitfalls, and promises. Curr Opin Pharmacol 8: Rhodes N, Heerding DA, Duckett DR et al (2008) Characterization of an akt kinase inhibitor with potent pharmacodynamic and antitumor activity. Cancer Res 68: Levy DS, Kahana JA, Kumar R (2009) AKT inhibitor, GSK690693, induces growth inhibition and apoptosis in acute lymphoblastic leukemia cell lines. Blood 113: Castillo SS, Brognard J, Petukhov PA et al (2004) Preferential inhibition of akt and killing of akt-dependent cancer cells by rationally designed phosphatidylinositol ether lipid analogues. Cancer Res 64: Ruiter GA, Zerp SF, Bartelink H et al (2003) Anti-cancer alkyl-lysophospholipids inhibit the phosphatidylinositol 3-kinase-Akt/PKB survival pathway. Anticancer Drugs 14: Chee KG, Longmate J, Quinn DI et al (2007) The AKT inhibitor perifosine in biochemically recurrent prostate cancer: A phase II California/Pittsburgh cancer consortium trial. Clin Genitourin Cancer 5: Leighl NB, Dent S, Clemons M et al (2008) A phase 2 study of perifosine in advanced or metastatic breast cancer. Breast Cancer Res Treat 108: Miao B, Skidan I, Yang J et al (2010) Small molecule inhibition of phosphatidylinositol- 3,4,5-triphosphate (PIP3) binding to pleckstrin homology domains. Proc Natl Acad Sci U S A 107: Memmott RM, Gills JJ, Hollingshead M et al (2008) Phosphatidylinositol ether lipid analogues induce AMP-activated protein kinase-dependent death in LKB1-mutant non small cell lung cancer cells. Cancer Res 68:














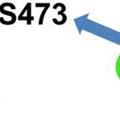


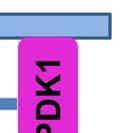



53 Figure 1 Figure 1. PI3K/Akt Pathway. Growth factor stimulation and subsequent receptor activation causes recruitment and activation of PI3K. PI3K converts PIP2 to PIP3. Akt and PDK1 bind PIP3 via their pleckstrin homology domains. Afterr translocation to the membrane, Akt is phosphorylated on T308 and S473 by PDK1 and mtorc2, respectively. Once fully activated, Akt phosphorylatess and suppresses TSC2 function leading to activation of mtorc1. mtorc1 mediatedd phosphorylation of S6K1 and 4EBP1 promotes translation and protein synthesis. Mechanisms by which mtorc1 activateshif1α remain elusive. 53





54 Figure 2 Figure 2. Schematic diagram of S6K1 structure depicting critical phosphorylation sites. NT, N-terminal domain; AI, Autoinhibitory domain; CT, C-terminal domain. 54











55 Figure 3 Figure 3. S6K1 regulation of protein translation. S6K1 phosphorylation of eif4b and PDCD4 increases helicase activity of eif4a resulting in increased translation initiation. S6K1 association with SKAR promotes its recruitment to exon junction complex on newly synthesized mrnas drivingg pioneer round of translation. S6K1 activates translation elongation by phosphorylating and inhibiting the activity of eef2k. 55





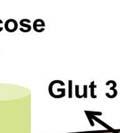


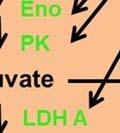



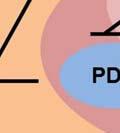
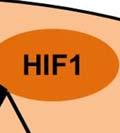
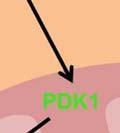



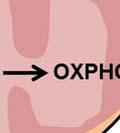

56 Figure 4 Figure 4. Regulation of glucose metabolism by HIF1.. HIF1 activates the transcription of genes encoding glucose transporters (GLUT ) 1 and 3, hexokinase (HK) I and II, glucosephosphate isomerase (GPI), phosphofructokinase (PFK), aldolase (ALD), triosephosphate isomerase (TPI), glyceraldehyde phosphate dehydrogenase (GAPDH), phosphoglyceratee kinase (PGK), enolase (Eno), pyruvate kinase (PK), lactate dehydrogenase A (LDH A) and pyruvate dehydrogenase kinase (PDK1). 56
57 CHAPTER II Requirement for ribosomal protein S6 Kinase 1 to mediate glycolysis and apoptosis resistance induced by Pten deficiency This work is published in The Proceedings of the National Academy of Sciences, 2011, 108,
58 Abstract Pten inactivation promotes cell survival in leukemia cells by activating glycolytic metabolism. We found that targeting S6K1 in Pten-deficient cells suppressed glycolysis and induced apoptosis. S6K1 knockdown decreased expression of HIF-1α, and HIF-1α was sufficient to restore glycolysis and survival of cells lacking S6K1. In the PTEN fl/fl Mx1-Cre + mouse model of leukemia, S6K1 deletion delayed the development of leukemia. Thus, S6K1 is a critical mediator of glycolytic metabolism, cell survival and leukemogenesis in Pten-deficient cells. 58
59 Introduction The Pten phosphatase is among the most commonly inactivated tumor suppressor proteins in human cancer. Spontaneous inactivating mutations in the Pten gene are found in cancers of the central nervous system, prostate, and endometrium at a frequency of 15-38% (1). In addition to mutations, Pten function can be reduced in cancer cells through epigenetic modifications, mirna regulation, subcellular translocation, and post-translational modification (2). Pten expression levels determine the tissue spectrum and aggressiveness of neoplastic tumors. In hematopoietic cells, heterozygous mice with one functional allele of Pten develop a lymphoproliferative autoimmune disease (3), while complete deletion in hematopoietic cells triggers aggressive lymphoid and myeloid leukemias (4, 5). Pten-deficiency contributes to the accumulation of tumor-initiating cells in cancers of hematopoietic, prostate, and brain tissues (4, 6, 7). Increased numbers of tumor-initiating cells indicate a need for targeted chemotherapeutic approaches to achieve long-term cancer remission in cancers associated with Pten inactivation. Loss of Pten triggers the accumulation of the lipid products of the class 1a phosphatidylinositol 3 kinases (PI3K) and activation of the Akt/PKB protein kinases. Among the three mammalian isoforms of the Akt kinases, Akt1 is required for oncogenesis in mice that are heterozygous for a null allele of Pten (8). Activation of Akt induces glycolytic metabolism and renders cells hypersensitive to interruptions in glycolysis, suggesting that Akt metabolic control can be targeted to induce apoptosis in cancer cells (9, 10). Rapamycin, an inhibitor of the mammalian target of rapamycin complex 1 (mtorc1), can prevent Akt-induced glycolysis (11). This indicates that substrates of mtorc1 are likely mediators for Akt-induced glycolysis, but the array of mtorc1 substrates that mediate glycolysis in Pten-deficient cells is not known. 59
60 The ribosomal protein S6 kinase 1 (S6K1) is an attractive target downstream of mtorc1 for activation of glycolysis in Pten-deficient cells. mtorc1 phosphorylation activates the protein kinase activity of S6K1, which in turn regulates protein translation by phosphorylating proteins that regulate translation initiation (12-14). S6K1 also functions in hormonal control of circulating glucose through effects in insulin-responsive tissues S6K1 -/- mice are glucose intolerant and exhibit increased blood glucose levels when fed a high fat diet (15). Because it can be inhibited using compounds selective for its ATP-binding pocket, S6K1 is a potential target for developing novel chemotherapeutics. We tested the potential for targeting S6K1 to reduce glycolytic metabolism and restore apoptosis in cellular and mouse models of Pten-deficient leukemogenesis. 60
61 Results S6K1 is required to maintain glycolysis and survival in Pten-deficient cells Pten inactivation induces Akt signaling, apoptosis resistance, and glycolytic metabolism in cancer cells. Loss of Pten is known to activate the protein kinase S6K1, but the role of S6K1 in regulating apoptosis resistance and glycolytic metabolism in carcinogenesis is not known. To determine the role of S6K1 in regulating apoptosis in Pten-deficient cells, we transduced IL-3- dependent hematopoietic progenitor FL5.12 cells with shrna expression vectors targeting Pten (shpten) and/or S6K1 (shs6k1) (Supplemental Figure 1A). S6K1 shrna and sirna reduced S6K1 expression without affecting the expression of the related kinase S6K2 (Supplemental Figure 1B & 1C). Pten-deficient cells but not controls have elevated Akt phosphorylation and resist the induction of apoptosis when cultured 48 hours in the absence of growth factor (Figure 1A, Supplemental Figure 1A). S6K1 knockdown abrogated cell survival in Pten-deficient cells, indicating a requirement for S6K1 to transmit pro-survival signals downstream of Akt (Figure 1A). S6K1 knockdown inhibited cell survival in PTEN-deficient cells to a similar degree as the mtorc1 inhibitor rapamycin, indicating that S6K1 is a major mediator of survival signals downstream of mtorc1 (Figure 1B). Next we determined the impact of S6K1 knockdown on metabolic parameters associated with Akt-dependent survival. S6K1 knockdown correlated with decreased mitochondrial membrane potential compared to cells with Pten knockdown alone, suggesting that glucosedependent cell survival may be compromised (Figure 1C) (10). We therefore measured the cellular glycolytic rate using a radiolabeled tracer assay that correlates tightly with the flux of glucose through the glycolytic pathway (16). In cells cultured in the absence of growth factor, 61
62 S6K1 knockdown reduced the rate of glycolysis in Pten-deficient cells (Figure 1D). Consistent with reduced glycolysis, Pten/S6K1-deficient cells produced less lactate as compared to Ptendeficient cells in the absence of growth factor (Supplemental Figure 2). Interestingly, S6K1 knockdown alone was sufficient to suppress glycolysis and accelerate cell death (Figures 1A, 1D). Metabolic decline preceded cell death because glycolysis and mitochondrial measurements were performed at three hours post-il-3 withdrawal, well before any decline in viability is evident (17). Recent work has shown that programmed cell death can proceed through distinct pathways, each with its own molecular mediators (18). To determine whether loss of S6K1 activated the apoptosis pathway for cell death, we measured the effects of S6K1 knockdown on subcellular localization of Bax and cytochrome c in Pten-deficient cells. In viable cells, Bax is maintained in a cytosolic location, while in apoptotic cells Bax is associated with the mitochondrial outer membrane (19). When apoptosis was induced by culturing cells in the absence of growth factor, Pten knockdown significantly reduced Bax translocation from the cytosol to mitochondria (Figure 2A lanes 7 and 8; Figure 2B). S6K1 knockdown restored Bax translocation in Pten-deficient cells, strongly indicating that S6K1 is required to prevent apoptosis induction in Pten-deficient cells. Bax translocation to the outer membrane is required for Mitochondrial Outer Membrane Permeability (MOMP) and the release of cytochrome c (20, 21). To determine if Bax translocation to mitochondria induced MOMP, we measured cytochrome c release to the cytosol in cells cultured in the absence of growth factor to induce cell death. S6K1 knockdown increased the fraction of cytochrome c in the cytosol in Pten- 62
63 deficient cells, demonstrating that S6K1 inactivation induces an apoptotic form of programmed cell death in Pten-deficient cells (Figure 2C). Decreased glycolysis in shpten/shs6k1 cells cultured in the absence of growth factor occurs as cells progress towards irreversible commitment to apoptosis. To rule out the possibility that S6K1 regulation of glycolysis is a consequence of cellular commitment to apoptosis, we measured glycolysis in knockdown cells overexpressing Bcl-xL. Bcl-xL expression prevented growth factor withdrawal-induced death (Supplemental Figure 3), permitting analysis of metabolic control in the absence of the effects of apoptosis commitment. In the absence of IL-3, S6K1 knockdown reduced glycolysis in Pten-deficient cells, demonstrating that S6K1 metabolic control is upstream of apoptosis commitment (Figure 2D). Together the data show that loss of S6K1 compromises the metabolic hallmarks of Akt-dependent survival, resulting in the induction of apoptosis. S6K1 induces glycolysis through HIF-1α Several reports have demonstrated increased expression of hypoxia inducible factor (HIF- 1α) in Pten-deficient cancer cell lines (22, 23). Because HIF-1α can regulate the transcription of glycolytic enzymes, we analyzed HIF-1α expression in Pten/S6K1-deficient cells. Consistent with previous reports, we observed a small increase in HIF-1α expression in shpten cells cultured under normoxic conditions, compared to vector control cells (Figure 3A). S6K1 knockdown in Pten-deficient cells suppressed HIF-1α expression under both normoxic and hypoxic (1% O 2 ) conditions, suggesting that limited HIF-1α expression could be responsible for decreased glycolysis. We confirmed this finding in Pten-deficient PC3 prostate cancer cells. S6K1 sirna 63
64 suppressed HIF-1α expression as compared to control sirna nucleofected cells (Figure 3B). We treated PC3 cells with dimethyloxallyl glycine (DMOG), a prolyl hydroxylase inhibitor that prevents the oxygen-dependent degradation of HIF-1α. Treatment with DMOG triggered rapid accumulation of HIF-1α in all cells, but the amount of HIF-1α stabilized by DMOG was significantly reduced in S6K1-knockdown PC3 cells. (Figure 3B). Reduced accumulation of HIF-1α in response to an inhibitor of oxygen-dependent degradation indicates that S6K1 regulates synthesis of HIF-1α. No change in HIF-1α mrna levels was observed upon S6K1 knockdown in PTEN-deficient cells (Supplemental Figure 4), consistent with a role for S6K1 in regulating HIF-1α production at the level of translation. Although there is strong evidence that increased HIF-1α correlates with Pten inactivation, it is not clear if HIF-1α is required for increased glycolysis and survival in Pten-deficient cells. To test if reduced HIF-1α abundance may account for decreased glycolysis in S6K1-knockdown cells, we compared the effects of HIF-1α and S6K1 knockdown in Pten-deficient cells. Depletion of HIF-1α reduced glycolysis to a level comparable to S6K1 knockdown (Figure 3C). Moreover, downregulation of HIF-1α and S6K1 together did not induce an additive decrease in glycolysis as compared to knockdown of either HIF-1α or S6K1 alone, suggesting that the two proteins function in the same pathway (Figure 3C). The phosphoglycerate kinase 1 (PGK-1) enzyme is a glycolytic enzyme whose expression is responsive to HIF-1α. Expression of PGK-1 in cells upon knockdown of S6K1 or HIF-1α was reduced to a similar extent, indicating that S6K1 regulates the expression of HIF-1α target genes in glycolysis (Figure 3D). Similar to glycolysis, knockdown of S6K1 or HIF-1α 64
65 triggered reduced apoptosis resistance, and there was little additive activity of combined S6K1/HIF-1α knockdown in a viability timecourse (Figure 3E). Thus, HIF-1α is necessary to promote glycolysis and apoptosis resistance in Pten-knockdown cells. Previous work demonstrated that limited glycolysis prevents survival of cells with inactivated Pten or constitutively active Akt (9, 10). To test whether decreased glycolysis contributes to limited survival in S6K1-knockdown cells, we restored glycolysis by expressing HIF-1α PP402,564AA, a mutant HIF-1α protein that lacks the proline hydroxylation sites in HIF-1α and thereby evades proteasomal degradation (24). Expression of the protein product was confirmed by intracellular flow cytometry (Supplemental Figure 5). We measured glycolysis in cells expressing the HIF-1α mutant and observed increased glycolysis relative to control cells, as expected (Figure 4A). Some variability was observed in the rate of glycolysis in cell lines expressing mutant HIF-1, suggesting additional layers of glycolysis regulation by a complex signaling network in response to Pten and/or S6K1 inactivation. Having increased glycolysis by expressing mutant HIF-1, experiments could now address whether elevated glycolysis correlated with viability in Pten/S6K1-knockdown cells. Survival in Pten/S6K1 double-knockdown cells expressing mutant HIF-1 was comparable to cells deficient in Pten alone (Figure 4B). This suggests that increased glycolysis contributes to the pro-survival effects of S6K1 in PTEN-deficient cells. Although expression of the HIF-1 mutant also increased viability in control cells, the level of cell survival mediated by HIF-1 itself did not approach that of Pten-knockdown cells. This suggests that increased glycolysis mediated by S6K1 and HIF-1 contributes to apoptosis resistance, but increased glycolysis 65
66 alone is not sufficient for cell survival. Pten-deficient cells also activate S6K1-independent prosurvival activities, such as Akt inactivation of FOXO transcription factors. S6K1 activation of glycolysis likely cooperates with parallel pro-survival mechanisms to mediate apoptosis resistance in Pten-deficient cells. S6K1-deficiency delays leukemogenesis induced by Pten deletion Decreased glycolytic metabolism and apoptosis resistance upon loss of S6K1 would be predicted to impair oncogenic transformation in Pten-deficient cells. We crossed Pten fl/fl mice expressing the Cre recombinase from the interferon-inducible Mx1-Cre transgene with S6K1 +/+ and S6K1 -/- mice. S6K1 -/- mice are viable but small in size, with no apparent alterations in hematopoiesis (25). Deletion of Pten in hematopoietic cells leads to myeloproliferative disease (MPD), acute lymphoblastic leukemia (ALL) and/or acute myeloid leukemia (AML) (4, 5). After injection with pipc to induce Pten deletion, mice were monitored for development of leukemia. Pten fl/fl Mx1-Cre + S6K1 +/+ mice developed a fatal combination of MPD and T-ALL with a mean survival of 35 days. Pten fl/fl Mx1-Cre + S6K1 -/- developed fatal disease with slower kinetics, increasing average lifespan to 46 days, an improvement of 32% (Figure 5A). The endpoints of the disease in Pten fl/fl S6K1 +/+ and Pten fl/fl S6K1 -/- mice did not differ significantly in the magnitude of splenomegaly or thymic enlargement (Supplemental Figure 6). We also found no significant difference in the frequency of cell subpopulations (CD4 +, CD8 +, TCRβ +, B220 +, Mac1 + and GR-1 + ) when assessed by flow cytometry (Supplemental Figure 6). To determine the role of S6K1 in regulating glycolysis in leukemogenesis, we analyzed the expression of PGK-1 in bone marrow progenitor cells that lack the expression of defined differentiation markers (Lineage-negative or Lin - cells). Lin - cells have been shown to contain leukemia-initiating cells 66
67 in Pten fl/fl mice (4). These cells isolated from Pten fl/fl S6K1 -/- mice showed greater than 90% reduction in PGK-1 expression as compared to Pten fl/fl S6K1 +/+ mice, suggesting that S6K1 loss may affect disease initiation and development by impairing glycolysis in this population (Figure 5B). Together the data reveal that S6K1 is required for oncogenic glycolytic metabolism and apoptosis resistance in PTEN-deficient neoplasia. 67
68 Discussion The results shown here identify S6K1 as a critical kinase that activates glycolysis to support cell survival and transformation in Pten-deficient cells by controlling the production of HIF-1α. Pten-deficient cells accumulate increased levels of HIF-1α, which requires mtorc1 signaling (22, 23). In response to increased mtorc1 signaling, HIF-1α translation is increased via mechanisms that include increased phosphorylation of the inhibitor of cap-dependent initiation 4EBP1 (26). Our studies reveal a similar role for S6K1 in regulating HIF-1α levels in Pten-deficient cells. Furthermore, we show that HIF-1α is required to induce glycolysis to sustain cell survival in Pten knockdown cells. To our knowledge, this is the first test of the requirement for HIF-1α to mediate increased glycolysis and apoptosis resistance in Pten-deficient cells. Importantly, HIF-1α reexpression was sufficient to restore glycolysis and apoptosis resistance in cells that lack S6K1, which strongly supports the regulation of glycolysis as a critical function of S6K1. S6K1-deficiency functioned similarly to rapamycin in PTEN-deficient leukemia, extending the lifespan of mice following deletion of Pten. One difference between targeting S6K1 and rapamycin treatment is that rapamycin suppressed leukemogenesis when coadministered with pipc in Pten fl/fl Mx1-Cre + mice, while the S6K1 -/- delayed disease development (4). It is possible that rapamycin suppressed leukemia because of an ability to suppress proliferation through the regulation of the 4EBP proteins (27). However, the antiproliferative effects of rapamycin-induced dephosphorylation of 4EBP-1 may be detrimental to cytotoxic chemotherapy, as some studies have shown that it is preferable to maintain cell cycle progression while treating with cytotoxic agents to maximize induction of apoptosis (28). 68
69 Several inhibitors are described that inactivate S6K1 downstream of Akt (29, 30). Whether direct inactivation of S6K1-induced glycolysis or combined inhibition of metabolism and cell cycle progression by targeting mtorc1 is preferable in cancer therapy will require direct comparison in future studies. 69
70 Materials and Methods Cell culture and viral transductions IL-3 dependent FL5.12 cells were cultured as described (31). PC3 cells were cultured in Dulbecco s Modification of Eagle s Medium (DMEM 1X) supplemented with 10% FBS (Hyclone), penicillin and streptomycin. Bcl-xL-expressing FL5.12 cells were previously described (10, 32). Constructs PCR reactions were used to prepare Pten shrna targeting sequences fused to the human U6 promoter; the resulting cassette was cloned into the pkd-gfp vector (33). Targeting sequences: shpten: GGGTGAATACAAAATACTCCGGTGTTTCGTCCTTTCCACAA; Scrambled: AAAAAAGGAGTATCTTGTACTCACCCTAACCTCGAGCTTA- AAAAAAGTCCTGCCTCGTAATAGCCGTACACCTCGAGCTGTACG- GCTATTACGAGGCAGGACGGTGTTTCGTCCTTTCCACAA. The S6K1 knockdown construct was from Open Biosystems, TRCN Bcl-xL and HIF-1α PP402,564AA constructs were expressed in the MIT retroviral vector. Retrovirus was prepared in 293 cells and lentiviral vectors were produced by the Viral Vector Core at the Translational Core Laboratories, Cincinnati Children s Hospital Research Foundation, in Cincinnati, Ohio Immunoblots Immunoblots were performed with the following antibodies: Akt ps473, S6K1 pt389, Akt, S6K1 and cytochrome c from Cell Signaling Technology; Pten, Actin, Bax, and FOXO3a from 70
71 Santa Cruz Biotechnology. HIF-1α blots were performed as described (34). Contrast, brightness, and levels adjustments were performed in Canvas and Photoshop software (ACDSee, Adobe). Average pixel density ± standard deviation for densitometry was determined using Quantity One software (BioRad). Flow cytometry For viability assays, FL5.12 cells were washed three times then plated in complete medium, lacking only IL-3, at a density of 2E5 cells/ml. Triplicate samples of each culture were resuspended in PBS containing 2µg/ml of propidium iodide and immediately analyzed on a flow cytometer. To assess mitochondrial membrane potential, cells were incubated for 30 minutes at 37 C with 20 nm tetramethylrhodamine ethyl ester (TMRE) and 5 µg/ml 4,6-diamidino-2- phenylindole, dihydrochloride (DAPI) with or without 5 µm chlorocarbonylcyanide phenylhydrazone (CCCP). TMRE fluorescence was measured using a FACSAria, with gating to remove DAPI + dead cells. The HIF-1α intracellular stain employed PE-conjugated anti-hif-1α or isotype control Ab (R&D Systems) after fixing in 4% paraformaldehyde and permeabilizing in 0.1% saponin. Nucleofection 7-10E6 FL5.12 cells were nucleofected with 10 ug sirna (Accell) using Nucleofector-II (Amaxa biosystems) and G-016 program. 24 hours post nucleofection, 1E6 cells were starved for IL-3 for 3 hours, following which glycolytic rates were measured. Cell viability and RNA 71
72 isolation experiments were also performed 24 hours post nucleofection. For PC3 cells, we used the same nucleofection kit with the program T-016. PC3 cells were nucleofected with 5-10 ug of S6K1 sirna in complete media. 24 hours post nucleofection, media was changed to 0.1% FBS containing DMEM. After hours, cells were treated with 5mM DMOG (Cayman Chemical Company) for 90 minutes, following which cells were harvested for protein. Quantitative Reverse Transcription- PCR RNA was isolated using Qiagen RNeasy mini kit coupled to DNase 1 digestion. 1 ug RNA was reverse transcribed using Taqman Reverse Transcription reagents (Applied Biosystems). Quantitative PCR was performed using TaqMan Gene Expression Master mix and HIF-1α,S6K1, PGK1 and S6K2 TaqMan probes (Applied Biosystems). Mitochondrial fractionation 1E7 cells were incubated on ice, resuspended in 250 µm Sucrose/20 mm MOPS containing protease and phosphatase inhibitors, sonicated, and cleared in a 500g spin. A 16,000g spin yielded a mitochondrial pellet and a cytosolic supernatant. Mitochondria were resuspended in Sucrose-MOPS buffer, and NP40 was added to both fractions to a final concentration of 1%. Glycolysis measurement Glycolysis was measured using 5-[ 3 H]-glucose in a protocol adapted from (35). 1E6 cells were cultured with 5 µci of 5-[ 3 H]-glucose and incubated for one hour at 37 C. 5-[ 3 H]-glucose is converted exclusively by glycolysis to 3 H-water during this incubation. After acid lysis, 3 H-water was separated from 5-[ 3 H]-glucose in a closed system consisting of an outer chamber filled with 72
73 1 ml of water and an inner chamber containing 3 H-water generated by glycolysis. After hours at room temperature, 3 H-water in the inner chamber equilibrated with the water in the outer chamber through evaporation and condensation. 3 H-water in the outer chamber was then measured by scintillation counting, and standardized to controls containing pure 3 H-water or pure 5-[ 3 H]-glucose. Data are expressed as nmol glucose converted/1 x10 6 cells/hour. Lactate production 4E6 cells were nucleofected with 5 μg sirna. 24 hours post nucleofection, cells were cultured in the absence of IL-3 for 7 hours and lactate secretion into media was measured using a Lactate Assay Kit (Biovision Inc), following the manufacturer s instructions. Mice Pten fl/fl mice on a mixed background (Jackson Laboratories) were crossed with Mx1-Cre and S6K1 -/- mice (gift from Sara Kozma/George Thomas laboratories). 6-8 week old mice were intraperitoneally injected twice with 12.5 µg of polyinosine-polycytidine (pipc) (Invivogen) per gram of body weight every other day. Mice were sacrificed upon observation of leukemiaassociated symptoms. Experiments were conducted in accordance with the animal care policies of the Institutional Animal Care and Use Committee of the University of Cincinnati. Isolation of Lin - bone marrow cells 6-8 week old mice were intraperitoneally injected with 12.5 μg of pipc per gram of mouse body weight for 2 consecutive days. Mice were sacrificed five days after the first pipc injection. BM was harvested from femurs and tibia and cells were pooled from 5 mice of each genotype. 73
74 Depletion of lineage-positive cells was performed using biotinylated antibodies specific for Ter119, CD3, GR1, Mac1 and B220 (BD Biosciences cat# ), and streptavidin-coated magnetic beads (Miltenyl Biotec), using an AutoMACS cell sorter. Statistical analysis Statistical calculations were t-tests performed using GraphPad Prism software. 74
75 Referencs 1. Forbes SA, et al. (2010) COSMIC (the Catalogue of Somatic Mutations in Cancer): a resource to investigate acquired mutations in human cancer. Nucleic Acids Res 38:D Keniry M & Parsons R (2008) The role of PTEN signaling perturbations in cancer and in targeted therapy. Oncogene 27: Di Cristofano A, et al. (1999) Impaired Fas response and autoimmunity in Pten+/- mice. Science 285: Yilmaz OH, et al. (2006) Pten dependence distinguishes haematopoietic stem cells from leukaemia-initiating cells. Nature 441: Guo W, et al. (2008) Multi-genetic events collaboratively contribute to Pten-null leukaemia stem-cell formation. Nature 453: Wang S, et al. (2006) Pten deletion leads to the expansion of a prostatic stem/progenitor cell subpopulation and tumor initiation. Proc. Natl. Acad. Sci. U. S. A. 103: Hambardzumyan D, et al. (2008) PI3K pathway regulates survival of cancer stem cells residing in the perivascular niche following radiation in medulloblastoma in vivo. Genes Dev. 22: Chen ML, et al. (2006) The deficiency of Akt1 is sufficient to suppress tumor development in Pten+/- mice. Genes Dev. 20: Gottlob K, et al. (2001) Inhibition of early apoptotic events by Akt/PKB is dependent on the first committed step of glycolysis and mitochondrial hexokinase. Genes Dev. 15: Plas DR, Talapatra S, Edinger AL, Rathmell JC, & Thompson CB (2001) Akt and Bcl-xL Promote Growth Factor-independent Survival through Distinct Effects on Mitochondrial Physiology. J. Biol. Chem. 276: Rathmell JC, Farkash EA, Gao W, & Thompson CB (2001) IL-7 enhances the survival and maintains the size of naive T cells. J. Immunol. 167: Dorrello NV, et al. (2006) S6K1- and betatrcp-mediated degradation of PDCD4 promotes protein translation and cell growth. Science 314: Holz MK, Ballif BA, Gygi SP, & Blenis J (2005) mtor and S6K1 mediate assembly of the translation preinitiation complex through dynamic protein interchange and ordered phosphorylation events. Cell 123: Raught B, et al. (2004) Phosphorylation of eucaryotic translation initiation factor 4B Ser422 is modulated by S6 kinases. EMBO J. 23:
76 15. Um SH, et al. (2004) Absence of S6K1 protects against age- and diet-induced obesity while enhancing insulin sensitivity. Nature 431: Sweet IR, Li G, Najafi H, Berner D, & Matschinsky FM (1996) Effect of a glucokinase inhibitor on energy production and insulin release in pancreatic islets. Am. J. Physiol. 271:E Vander Heiden MG, et al. (2001) Bcl-xL promotes the open configuration of the voltagedependent anion channel and metabolite passage through the outer mitochondrial membrane. J. Biol. Chem. 276: Degterev A & Yuan J (2008) Expansion and evolution of cell death programmes. Nat Rev Mol Cell Biol 9: Hsu YT, Wolter KG, & Youle RJ (1997) Cytosol-to-membrane redistribution of Bax and Bcl-X(L) during apoptosis. Proc. Natl. Acad. Sci. U. S. A. 94: Zong WX, Lindsten T, Ross AJ, MacGregor GR, & Thompson CB (2001) BH3-only proteins that bind pro-survival Bcl-2 family members fail to induce apoptosis in the absence of Bax and Bak. Genes Dev. 15: Cheng EH, et al. (2001) BCL-2, BCL-X(L) sequester BH3 domain-only molecules preventing BAX- and BAK-mediated mitochondrial apoptosis. Mol. Cell 8: Zhong H, et al. (2000) Modulation of hypoxia-inducible factor 1alpha expression by the epidermal growth factor/phosphatidylinositol 3-kinase/PTEN/AKT/FRAP pathway in human prostate cancer cells: implications for tumor angiogenesis and therapeutics. Cancer Res. 60: Zundel W, et al. (2000) Loss of PTEN facilitates HIF-1-mediated gene expression. Genes Dev. 14: Masson N, Willam C, Maxwell PH, Pugh CW, & Ratcliffe PJ (2001) Independent function of two destruction domains in hypoxia-inducible factor-alpha chains activated by prolyl hydroxylation. EMBO J. 20: Shima H, et al. (1998) Disruption of the p70(s6k)/p85(s6k) gene reveals a small mouse phenotype and a new functional S6 kinase. EMBO J. 17: Duvel K, et al. (2010) Activation of a Metabolic Gene Regulatory Network Downstream of mtor Complex 1. Mol. Cell 39: Dowling RJ, et al. (2010) mtorc1-mediated cell proliferation, but not cell growth, controlled by the 4E-BPs. Science 328:
77 28. Le DT, et al. (2004) Somatic inactivation of Nf1 in hematopoietic cells results in a progressive myeloproliferative disorder. Blood 103: Okuzumi T, et al. (2009) Inhibitor hijacking of Akt activation. Nat Chem Biol 5: Roca H, Varsos ZS, & Pienta KJ (2009) CCL2 is a negative regulator of AMP-activated protein kinase to sustain mtor complex-1 activation, survivin expression, and cell survival in human prostate cancer PC3 cells. Neoplasia 11: Plas DR & Thompson CB (2003) Akt Activation Promotes Degradation of Tuberin and FOXO3a via the Proteasome. J. Biol. Chem. 278: Vander Heiden MG, Chandel NS, Williamson EK, Schumacker PT, & Thompson CB (1997) Bcl-xL regulates the membrane potential and volume homeostasis of mitochondria. Cell 91: Fox CJ, et al. (2003) The serine/threonine kinase Pim-2 is a transcriptionally regulated apoptotic inhibitor. Genes Dev. 17: Schnell PO, et al. (2003) Regulation of tyrosine hydroxylase promoter activity by the von Hippel-Lindau tumor suppressor protein and hypoxia-inducible transcription factors. J. Neurochem. 85: Ashcroft SJ, Weerasinghe LC, Bassett JM, & Randle PJ (1972) The pentose cycle and insulin release in mouse pancreatic islets. Biochem. J. 126:












 were")






78 Figure 1 Figure 1. S6K1 is necessary for survival and glycolysis in Pten-deficient cells. A. S6K1 knockdown prevented cell survival of PTEN-knockdown cells. Standard deviation of triplicate viability measurements was <4% unless otherwise noted. B. Similarr to S6K1, 20 nm rapamycin reduced cell viability in shpten cells. C. Decreasedd mitochondrial membrane potential in Pten/S6K1 double-knockdown cells. Viable cells (DAPI-negative) were stained with the potentiometric dye tetramethylrh hodamine ethyl ester (TMRE) afterr culture in absence of IL-3. TMRE staining in Bcl-xL expr ressing cells is shown in both panels for reference. D. S6K1 knockdown reduces glycolysis in Pten-deficient cells. The rate of cellular glycolysiss was determined in a radiolabeled tracer assay after culture in the absencee of IL-3 for three hours. 78





")


")







, but")






79 Figure 2 Figure 2. S6K1-knockdown limits glycolysis to induce apoptosis. A. S6K1 knockdown restores Bax translocation in Pten-deficien nt cells upon IL-3 withdrawal. Cytosolic (C) and mitochondrial (M) fractions weree probed for Bax after culture ±IL-3 for 15 hours. Cytochrome c oxidase subunit IV (Cox IV) was used as a marker for the mitochondrial fraction. B. Bax abundance in cytosolic and mitochondrial fractions from A. was quantified as a ratio. Pten- of deficient cells maintained a relatively high cytosol:mitochondria ratio of Bax after withdrawal growth factor (IL-3), but S6K1 knockdown counteracted this effect. C. Cytochrome c is released in S6K1-deficient cells upon IL-3 withdrawal. Cytosolic and mitochondrial fractions were probed for cytochrome c after culture in the presence or absence of IL-3 for 18 hours. D. Glycolysis upon S6K1 knockdown is upstream of a cellular commitment to apoptosis. The rate of glycolysis was measured in cells expressing exogenous Bcl-xL to prevent the induction of apoptosiss after culture in the absence of growth factor. 79




. B.")


















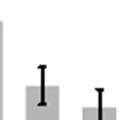
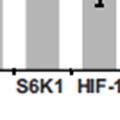

80 Figure 3 Figure 3. S6K1 regulates HIF-1α. (N). shpten/shs6k1 also exhibited reduced HIF-1α stabilization under hypoxic (1% O 2 ) conditions (H). B. S6K1 knockdown decreased HIF-1α expression in A. Endogenous HIF-1α is reduced in shpten/shs6k1 cells under normoxic conditions PC3 prostate cancer cells. DMOG treatment stabilized HIF-1αα expression but there was decreasedd HIF-1α accumulation in S6K1-depleted PC33 cells upon treatment with DMOG. C. shpten cells were nucleofected with control, sihif-1α or sis6k1. sihif-1α decreased glycolysis to a similar extent as observed with sis6k1. D. Comparable decreasee in mrna expression of the glycolytic enzyme PGK-1 upon knockdown of S6K1 or HIF-1. Data are the mean fold change in PGK-1 expression from three independent experiments ± standardd error. E. HIF-1α is required to sustain cell survival in shpten cells. Cells were cultured in the absence of growth factor to measure apoptosis resistance. 80


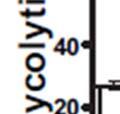










81 Figure 4 Figure 4: HIF-1α is sufficient to rescue glycolysis and survival in the absence of S6K1. A. Expression of HIF-1α PP402,564AA (+HIF-1α) increased glycolysis over control (dashed line) independent of S6K1. B. Increased viability in cells where glycolysis is supported by HIF-1α PP402,564AA independent of S6K1 expression. 81

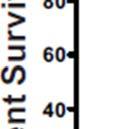
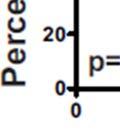





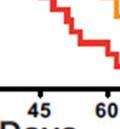



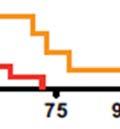




82 Figure 5 Figure 5: S6K1 loss delays Pten-deficient leukemia. A. Survival was compared in Pt S6K1 +/+ (n=24) and Pten fl/fl S6K1 -/- (n=14) mice after pipc injection. Mean survival for Pten fl/fl S6K1 +/+ mice was 35 days and 46 days for Pten fl/fl S6K11 -/- mice. p-value calculated by Log-rank test. B. PGK1 mrna expression was analyzed in pooled Lin - bone marrow cells from Pten fl/fl S6K1 +/+ mice (n=5) and Pten fl/fl S6K1 -/- mice (n=5) using qrt-pcr. Target gene expression in Pten fl/fl S6K1 +/+ BM cells was set to 1. ten fl/fl 82
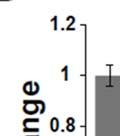


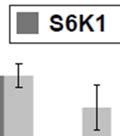
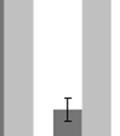

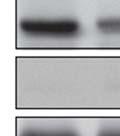


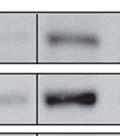





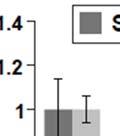

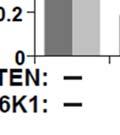


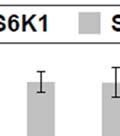
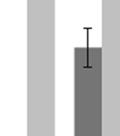
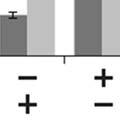
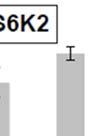

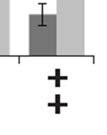
83 Supplemental Figure 1 Supplemental Figure 1. Pten and S6K1 knockdown. A. Cells were transduced with retroviral shrna constructs as indicated. Immunoblots of total protein lysates demonstrate effective knockdown of Pten and S6K1 protein levels, and reduced expression of phospho-s6k1. An additive increase in Akt phosphorylation was observedd in cells with combined inactivation of Pten and S6K1, as expected due to loss of the S6K1 negative feedback pathway (Radimerski et al. Genes & Development 2002). B. qrt-pcr analysis of S6K1 and S6K2 mrna in cells transduced with retroviral S6K1 shrna construct. C.. qrt-pcr analysis of S6K1 and S6K2 mrna in cells nucleofected with S6K1 sirna. 83








84 Supplemental Figure 2 Supplemental Figure 2. Decreased lactate production in S6K1 deficient cells. Lactate secretion into the media was measured in triplicate for each sample following IL-3 deprivation. Errorr bars indicate standard error. 84


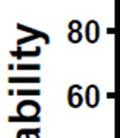
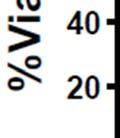

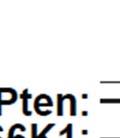

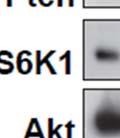











85 Supplemental Figure 3 Supplemental Figure 3. Bcl-xL blocked cell death in all cell lines at the 48 hour timepoint. A. Expression of Bcl-xL in FL5.12 cells expressing the indicated shrna constructs. B. Mean viability of triplicate samples is shown; the standard deviation was <2% for all samples. 85


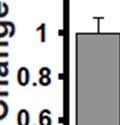



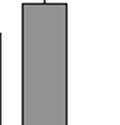

86 Supplemental Figure 4 Supplemental Figure 4. Representative qrt-pcr measurement of S6K1 and HIF-1α mrna after nucleofection with the indicated sirnas. Target gene expression in cells transfected with Non-Targeting sirna was set to 1. 86
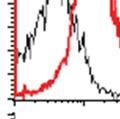
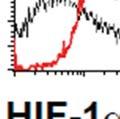




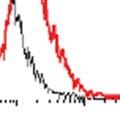
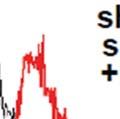
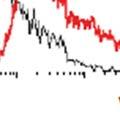
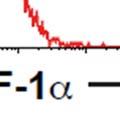

87 Supplemental Figure 5 Supplemental Figure 5. The indicated cell lines were transduced with control retroviral vector (Mock) or a vector driving the expression of HIF-1α PP402,564AA (HIF-1α). Expression of the HIF-1α protein was detected by intracellular flow cytometry using PE-conjugated anti-hif-1α (red) or an isotype control antibody (IgG ctrl. - black). 87






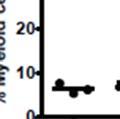



88 Supplemental Figure 6 Supplemental Figure 6. S6K1-deficiency does not impact leukemia phenotype. A. Size of thymus (A) and spleen (B) do not significantly differ byy S6K1 expression in leukemias induced by Pten-deletion (p= =0.30 and respectively, t-test). The frequency of CD4 single positive cells was not significantly altered in the thymus (C), nor was the frequency of GR1+/Mac1+ myeloid cells (D) in the spleen (p=0.32 and 0.53, respectively, t-test) ). 88
89 CHAPTER III Analysis of an S6K1 inhibitor for counteracting glycolysis and survival in Pten deficient cells 89
90 Abstract Constitutive Akt signaling promotes cancer cell survival through activation of glycolytic metabolism and repression of pro-apoptotic factors. We previously reported that S6K1 is required to sustain glycolysis and apoptosis resistance in Pten deficient cells, suggesting that S6K1 inhibitors may prove beneficial for treatment of Pten deficient cancers. We, therefore, investigated if pharmacological inhibition of S6K1 would recapitulate the phenotypic effects of S6K1 genetic depletion. We validated the inhibitor DG2 in Pten deficient hematopoietic cells. DG2 reduced glycolysis and induced apoptosis in Pten deficient cells, similar to S6K1 knockdown. These data indicate that S6K1 signaling can be efficiently targeted in Pten deficient cells using a small molecule kinase inhibitor. 90
91 Introduction Pten is one of the most frequently lost tumor suppressors in human cancers. Loss of Pten function triggers abnormal activation of Akt and its downstream effectors. Constitutive Akt activation induces glycolytic metabolism in both transformed and non-transformed cells (1, 2). Manipulating glycolytic metabolism by limiting the levels of glucose in the medium induces apoptosis in Akt activated cells, suggesting that Akt induced glycolysis is necessary for Akt mediated survival (1). This implies that regulators of Akt mediated glycolysis could prove useful targets in cancer therapy. Our previous studies showed that S6K1 is a critical regulator of Akt dependent glycolysis (3). Inactivating S6K1 impaired glycolysis in Pten knockdown IL-3 dependent hematopoietic progenitor FL5.12 cells. Decreased glycolysis induced cytochrome c release and commitment to apoptosis. S6K1 inactivation prevented expression of the pro-glycolytic transcription factor HIF- 1α leading to reduced glycolytic gene expression and diminished glycolysis. Restoring HIF-1α expression restored both glycolysis and apoptosis resistance in Pten/S6K1 deficient cells. Furthermore, S6K1 loss reduced the leukemic potential of Pten deficient cells, prolonging the lifespan of Pten fl/fl Mx1-Cre + mice (3). These data indicated that S6K1 may represent a useful therapeutic target for counteracting the metabolic program that supports tumorigenesis induced by Pten loss. Pharmacological inhibition of S6K1 can be achieved by small molecule compounds that are selective for its ATP binding pocket. Recently, Okuzumi et al developed and characterized a novel S6K1 inhibitor called DG2. DG2 inhibited S6K1 activity with an IC50 of 9.1nM. The inhibitor was found to be highly specific for S6K1 when tested against a panel of 220 highly 91
92 related kinases (4). In this study we investigated the effect of DG2 on the metabolic program that sustains survival in Pten deficient cells. Our results demonstrate that treatment with DG2 leads to inhibition of glycolysis and induction of apoptosis in Pten deficient cells. 92
93 Results DG2 suppresses S6K1 activity Aberrant S6K1 activation frequently results from Pten loss. We tested the effect of adding increasing concentrations of DG2 on the phosphorylation of the ribosomal protein S6, a well characterized S6K1 substrate, in Pten deficient hematopoietic cells. Phosphorylation at Ser 235/236 on S6 protein was inhibited by DG2 in a dose dependent manner such that it was significantly reduced by 0.25μM and strongly inhibited by 5μM inhibitor (Figure 1). Thus, DG2 treatment prevented S6K1 activity in Pten deficient FL5.12 cells. DG2 inhibits glycolysis in Pten deficient cells Pten loss activates glycolytic metabolism to promote survival in cancer cells. We previously identified S6K1 as a key pro-glycolytic effector downstream of Akt. We found that inactivating S6K1 reduced glycolysis and enhanced apoptosis in Pten depleted cells (3). To test if pharmacological inhibition of S6K1 would mimic the effects of S6K1 genetic inactivation, we assessed the effect of DG2 on glycolytic rates in Pten deficient cells (Figure 2). Similar to S6K1 knockdown, DG2 reduced the rate of glycolysis in Pten depleted FL5.12 cells. Moreover, treatment with DG2 did not further reduce glycolysis in S6K1 knockdown cells, suggesting that DG2 does not suppress glycolysis through off-target effects (Figure 2). DG2 induces apoptosis in Pten deficient cells Because S6K1 is required for Pten deficiency induced survival, we tested the effect of DG2 on cell viability (3). DG2 treatment abrogated cell survival in Pten knockdown cells and there was little additive decrease in apoptosis in S6K1 deficient cells treated with DG2 (Figure 93
94 3). Taken together, these results indicate that S6K1 inhibitor, DG2, induces apoptosis by suppressing glycolytic metabolism in cells deficient for Pten. 94
95 Discussion In these experiments we demonstrated that DG2 inhibition of S6K1 attenuated glycolytic metabolism in hematopoietic cells lacking Pten. Decreased glycolysis rendered Pten deficient cells sensitive to apoptosis induced by growth factor deprivation. Importantly, DG2 treatment recapitulated the biological effects of genetically inactivating S6K1 using RNAi. Furthermore, treating S6K1 knockdown cells with DG2 resulted in little additive decrease in glycolysis or apoptosis, confirming that DG2 exerts its effects by specifically inhibiting S6K1 signaling. Until now, researchers have employed rapamycin, an allosteric mtor inhibitor, to abrogate S6K1 activation. An important question is whether there would be a potential therapeutic benefit of targeting S6K1 over mtorc1, particularly in cancer. One potential advantage of specifically inhibiting S6K1 could be circumventing the immunosuppressive activity of rapamycin, which could be detrimental to its use as an anticancer agent. To the best of our knowledge, the immunophenotype of S6K1 knockout mouse has not been reported. Thus, it is unclear if DG2 will differ from rapamycin in this respect. Additionally, rapamycin has shown antitumor activity in cancers with elevated Akt signaling, especially in Pten deficient tumors (9, 10). However, the antitumor effects of rapamycin are mostly cytostatic rather than cytotoxic. Although, the molecular mechanisms that underlie the cytostatic effects of rapamycin remain elusive, it will be important to determine if specific inactivation of S6K1 signaling would elicit a cytotoxic response in these tumors. Future studies directly comparing the effects of rapamycin and DG2 will help us evaluate if specific inactivation of S6K1 would be more favorable than mtorc1 inhibition in cancer therapy. 95
96 Methods Cell lines and Constructs: IL-3 dependent FL5.12 cells were cultured as described (5). PCR reactions were used to prepare Pten shrna targeting sequences fused to the human U6 promoter; the resulting cassette was cloned into the pkd-gfp vector (6). Targeting sequences shpten:aaaaaaggagtatcttgtactcaccctaacctcgagcttagggtgaatacaa AATACTCCGGTGTTTCGTCCTTTCCACAA; Scrambled:AAAAAAGTCCTGCCTCGTAATAGCCGTACACCTCGAGCTGTACGGCTATT ACGAGGCAGGA S6K1 inhibitor: DG2 was a generous gift from Kevan M Shokat lab. DG2 was dissolved in DMSO and stored at C. Immunoblotting: Immunoblotting was performed as previously described (7). Immunoblots were performed with the following antibodies- S6K1, S6 ps235/236 from Cell Signaling Technology and Tubulin from Developmental Studies Hybridoma Bank. 96
97 Flow Cytometry: For viability assays, FL5.12 cells were washed three times, then plated in complete medium, lacking only IL-3, at a density of 2E5 cells per ml. Triplicate samples of each culture were resuspended in PBS containing 2 μg/ml propidium iodide and immediately analyzed on a flow cytometer. Nucleofection: Using Nucleofector II (Amaxa Biosystems) and the G-016 program, 7 10E6 FL5.12 cells were nucleofected with 10 μg sirna (Accell). At 24 h postnucleofection, 1E6 cells were starved for IL-3 for 3 h, following which glycolytic rates were measured. Cell viability experiments were performed 24 h post nucleofection. Glycolysis Measurement. Glycolysis was measured using 5-3 H-glucose in a protocol adapted from Ashcroft et al (8). 1E6 cells were cultured with 5 μci of 5-3 H-glucose and incubated for 1 h at 37 C. 5-3 H-glucose is converted exclusively by glycolysis to 3 H-water during this incubation. After acid lysis, 3 H-water was separated from 5-3 H-glucose in a closed system consisting of an outer chamber filled with 1 ml of water and an inner chamber containing 3 H-water generated by glycolysis. After h at room temperature, 3 H-water in the inner chamber equilibrated with the water in the outer chamber through evaporation and condensation. 3 H-water in the outer chamber was then measured by scintillation counting, and standardized to controls containing pure 3 H-water or pure 5-3 H-glucose. Data are expressed as nanomoles of glucose converted per cells per hour. 97
98 References 1. Plas DR, Talapatra S, Edinger AL et al (2001) Akt and bcl-xl promote growth factorindependent survival through distinct effects on mitochondrial physiology. J Biol Chem 276: Elstrom RL, Bauer DE, Buzzai M et al (2004) Akt stimulates aerobic glycolysis in cancer cells. Cancer Res 64: Tandon P, Gallo CA, Khatri S et al (2011) Requirement for ribosomal protein S6 kinase 1 to mediate glycolysis and apoptosis resistance induced by pten deficiency. Proc Natl Acad Sci U S A 108: Okuzumi T, Fiedler D, Zhang C et al (2009) Inhibitor hijacking of akt activation. Nat Chem Biol 5: Plas DR, Thompson CB (2003) Akt activation promotes degradation of tuberin and FOXO3a via the proteasome. J Biol Chem 278: Fox CJ, Hammerman PS, Cinalli RM et al (2003) The serine/threonine kinase pim-2 is a transcriptionally regulated apoptotic inhibitor. Genes Dev 17: Khatri S, Yepiskoposyan H, Gallo CA et al (2010) FOXO3a regulates glycolysis via transcriptional control of tumor suppressor TSC1. J Biol Chem 285: Ashcroft SJ, Weerasinghe LC, Bassett JM et al (1972) The pentose cycle and insulin release in mouse pancreatic islets. Biochem J 126: Podsypanina K, Lee RT, Politis C et al (2001) An inhibitor of mtor reduces neoplasia and normalizes p70/s6 kinase activity in pten+/- mice. Proc Natl Acad Sci U S A 98: Neshat MS, Mellinghoff IK, Tran C et al (2001) Enhanced sensitivity of PTEN-deficient tumors to inhibition of FRAP/mTOR. Proc Natl Acad Sci U S A 98:
99 Figure 1 DG2 (μm) ps6 Tubulin Figure 1. DG2 inhibits S6 phosphorylation. Cells were treated with the indicated concentrations of DG2 in the absence of IL-3 for 3 hours. 99
100 Figure 2 Glycolytic Rate NT DMSO 20uMDG2 sis6k1 Figure 2. DG2 suppresses glycolysis in Pten deficient cells. DG2 treatment inhibited glycolysis in Pten deficient cells to the same extent as S6K1 knockdown. The rate of cellular glycolysis was determined after the cells were cultured in the absence of IL-3 and presence of 20μM DG2 or DMSO as indicated. 100
101 Figure DMSO 20uMDG2 % Viability NT sis6k1 Figure 3. DG2 activates apoptosis in Pten deficient cells. Similar to S6K1 knockdown, 20 μm DG2 reduced cell viability in Pten knockdown cells. 101
102 CHAPTER IV Conclusions and Discussion 102
103 Pten loss is a frequent mechanism for constitutive Akt activation in human cancers. Akt activation promotes cell survival through coordinate inhibition of pro-apoptotic effectors and activation of glycolytic metabolism. An interruption in glycolytic metabolism renders Akt activated cells susceptible to apoptosis (1). This glucose dependent form of cell survival conferred by Akt may be therapeutically targeted to activate cell death programs in cancer cells. It is, therefore, important to understand the key mediators of Akt stimulated glycolysis. This study was aimed at elucidating the role of S6K1 in regulating glycolysis in Pten deficient cells. Herein, we used genetic (Chapter 2) and pharmacological (Chapter 3) approaches to demonstrate the novel function of S6K1 in modulating glycolytic metabolism and suppressing apoptosis resistance downstream of activated Akt. Our studies also defined the oncogenic role of S6K1 in a mouse model of Pten deficient leukemia. Together, the data indicate that S6K1 activation supports a metabolic program that is essential for Akt dependent survival and thus contribute to tumorigenesis in cells deficient for Pten. S6K1 mediates survival Several data demonstrate enhanced sensitivity of Pten deficient tumors to inhibition by rapamycin. Rapamycin treatment causes regression of uterine and adrenal medullary tumors that arise spontaneously in Pten +/- mice (2). Pten deficient prostate cancer xenografts are more sensitive to the pharmacological inhibition of mtor with the rapamycin analog, CCI-779, when compared with their wild type counterparts (3). Additionally, rapamycin suppresses acinar cell neoplasia induced by conditional inactivation of Pten and Apc tumor suppressors in the mouse salivary gland (4). Despite increased efficacy of rapamycin in Pten deficient tumors, little is 103
104 known about the molecular determinants of the drug response. We therefore sought to determine the function of S6K1 in Pten deficient cells. S6K1 loss suppressed apoptosis resistance, promoted Bax translocation and released cytochrome c in Pten deficient hematopoietic cells. The effect of disabling S6K1 was similar to that of rapamycin, indicating S6K1 as a major mediator of survival signals downstream of mtorc1. S6K1 selective inhibitor DG2 prevented survival of Pten deficient cells, providing pharmacological evidence of the requirement for S6K1 in apoptosis resistance. Akt activation supports cell survival by promoting glycolysis and maintaining a physiological mitochondrial membrane potential in the face of growth factor withdrawal. Pten loss appeared to mirror the phenotypic effects of constitutively active Akt expressing cells (1). Similar to activated Akt expressing cells, Pten deficient cells displayed increased mitochondrial membrane potential and high rates of glycolysis in the absence of growth factor. This enhanced rate of glycolysis was, however, suppressed in the absence of S6K1, establishing S6K1 as a proglycolytic effector downstream of activated Akt. Since Pten deficient cells rely on extrinsic sources of glucose for maintenance of elevated glycolysis and cell survival (Figure 1), an interruption of glucose metabolism following S6K1 loss rendered Pten deficient cells sensitive to apoptosis induced by cytokine withdrawal. The effect of S6K1 knockdown on glycolysis was recapitulated by DG2. Thus, S6K1 activation drives glycolytic metabolism to support survival in Pten knockdown cells upon growth factor withdrawal. Interestingly, S6K1 inactivation had little effect on glycolysis in cells cultured in the presence of cytokine (Figure 2), indicating engagement of an alternative pathway downstream of the IL-3 receptor, such as PIM family of kinases. Expression of the serine/threonine kinase Pim- 2 rapidly declines following IL-3 withdrawal, however, when constitutively expressed, Pim-2 104
105 promotes glycolysis and survival in IL-3 deprived cells independently of the Akt pathway. Thus, parallel pathways induced by IL-3 signal transduction may promote glycolytic metabolism in S6K1 depleted cells. However, S6K1 effects on glycolysis become apparent when these compensatory pathways are inhibited upon IL-3 withdrawal. Our studies reveal a role for S6K1 in suppressing apoptosis, however, the precise mechanisms by which S6K1 regulates anti-apoptotic effects remain elusive. S6K1 has been shown to repress apoptosis by phosphorylating the pro-apoptotic BH3 protein Bad at S136, thereby preventing Bad from binding and inactivating pro-survival Bcl-2 family proteins such as Bcl-xL (5). Interestingly, Akt can also phosphorylate Bad at S136, suggesting that the regulation of Bad phosphorylation can be controlled by either Akt or S6K1, depending on the availability of growth factors or nutrients, similar to what has been described for GSK-3 (6, 7). We tested Bad S136 phosphorylation in cells expressing wild type Bad or Bad S136A in shpten, and shpten/shs6k1 cells cultured in the absence of growth factor. S6K1 knockdown did not suppress S136 phosphorylation of Bad, potentially due to compensatory phosphorylation by Akt in the absence of PTEN (Figure 3). Because net Bad phosphorylation was unchanged upon S6K1 knockdown, Bad pro-apoptotic activity is unlikely to significantly contribute to the increased programmed cell death in cells deficient in S6K1. In addition to Bad, other pro-apoptotic Bcl-2 family proteins have also been implicated in suppressing Akt mediated cell survival. Shinjyo et al demonstrated that mtor mediated suppression of Bim expression is critical for survival of Ras transformed hematopoietic cells (8, 9). Inhibition of mtor signaling, through use of rapamycin, induced the expression of various Bim isoforms, which in turn associated with and inhibited the function of anti-apoptotic Bcl-2 proteins causing apoptosis in cells deprived of cytokine. Although mtorc1 substrates that 105
106 mediate this effect remain unclear, it will be important to investigate if Bim is subject to similar regulation by S6K1 in Pten depleted cells. Yet another mechanism by which constitutively active Akt bearing cells succumb to apoptosis involves the pro-apoptotic protein Puma. Zhao et al reported metabolic regulation of Puma in cells bearing activated Akt (9). Glucose deprivation resulted in upregulation of Puma and activation of apoptosis in Pten deficient human leukemic cells. Assuming that glycolysis inhibition by S6K1 loss mimics glucose deprivation, one may hypothesize activation of Puma in Pten/S6K1 deficient cells. This hypothesis, however, warrants further examination in future studies. Mechanism of S6K1 regulation of glycolysis In an effort to identify the mechanism of S6K1 regulation of glycolytic metabolism, we unraveled a novel link between S6K1 and the pro-glycolytic transcription factor HIF1α. mtorc1 signaling has previously been implicated in the activation of HIF1α in Pten deficient cancer cells (10, 11). We tested if mtorc1 effects on HIF1α were mediated by S6K1. In our studies, S6K1 deficiency resulted in a concomitant decline in HIF1α protein levels in Pten deficient hematopoietic and prostate cancer cells, indicating that activation of S6K1 is essential to stimulate increases in HIF1α expression. Rescue experiments with constitutively active HIF1α (HIF1α double proline mutant P402A/P564A which cannot be hydroxylated and hence degraded under normoxia) restored glycolysis and apoptosis resistance in Pten/S6K1 deficient cells, suggesting that S6K1 induces HIF1α to drive glycolysis and mediate survival in Pten deficient cells. Conversely, reducing HIF1α protein abundance prevented glycolytic gene expression, 106
107 suppressed glycolysis and activated apoptosis in Pten knockdown cells, similar to S6K1 deficiency. Thus, both S6K1 and HIF1α are necessary to drive the Warburg effect and hence apoptosis resistance in Pten deficient cells. Translational regulation of HIF1α by S6K1 Previous reports demonstrated both transcriptional and translational regulation of HIF1α by mtorc1 (12, 13). In our studies, the decline in HIF1α expression upon S6K1 loss appeared unrelated to gene transcription, as HIF1α mrna levels were unaltered upon S6K1 loss (Supplemental Figure 4, Chapter 2). Furthermore, inhibition of HIF1α degradation by prolyl hydroxylase inhibitor DMOG failed to rescue HIF1α expression in Pten/S6K1 knockdown cells, suggesting that S6K1 regulates HIF1α translation rather than HIF1α stability. In the future, HIF1α translational regulation by S6K1 will be tested more specifically using protein synthesis inhibitors and pulse chase labeling experiments. Parallel to our findings, Duvel et al recently reported regulation of HIF1α expression by 4EBP1 (12). These studies utilized reporter assays to demonstrate increased cap-dependent translation initiation of HIF1α in TSC2 -/- MEFS, an effect that was abrogated by overexpression of dominantly active 4EBP1. Although the authors did not observe any decrease in HIF1α translation initiation in the absence of S6K1, they did see a reduction in HIF1α protein levels in S6K1 knockdown cells. Since S6K1 control of translation extends beyond initiation, it is conceivable that S6K1 regulates translation elongation, possibly through eef2k phosphorylation, to alter cellular HIF1α protein levels (14). This possibility can be tested by investigating the phosphorylation status of eef2k in S6K1 deficient cells. If indeed eef2k 107
108 phosphorylation is decreased in the absence of S6K1, it will be important to determine if inhibiting eef2k expression rescues HIF1α protein levels in these cells (Figure 3, Chapter 1). Another possibility is that both S6K1 and 4EBP1 pathways contribute to HIF1α translation. Dissecting which pathway is critical in mediating mtorc1 dependent HIF1α translational control will require a side by side comparison of simultaneous inactivation of both pathways with inhibition of either pathway alone. These studies will reveal the relative contribution of each of these proteins towards translational regulation of HIF1α and provide rationale for targeting mtorc1 or S6K1 to reduce HIF1α abundance in cancer cells. Oncogenic functions of S6K1 The protumorigenic effects of S6K1 have been demonstrated by multiple groups in different types of cancers. Alliouachene et al showed that S6K1 is required for insulinoma formation induced by expression of constitutively active Akt1 in the mouse pancreas (15). More recently, Nardella et al reported that S6K1 genetic deletion impaired tumorigenesis driven by Pten heterozygous loss, with the most profound effects seen in adrenal gland tumors (16). Additionally, S6K1 has also been shown to be important for gliomagenesis, as S6K1 inhibition suppresses anchorage independent growth of human glioma cell lines (17). All the above findings suggest that S6K1 functions as an oncoprotein. Our study extends these previous findings by demonstrating the role of S6K1 in leukemogenesis driven by Pten loss. Pten deletion in the hematopoietic cells leads to myeloproliferative disease, acute myeloid leukemia (AML) and acute lymphoid leukemia (ALL) in mice (18). Our studies revealed that the genetic ablation of S6K1 delayed leukemia 108
109 progression and improved survival of Pten fl/fl Mx1-Cre + mice, without altering the disease phenotype. This is consistent with previous studies wherein rapamycin treatment prolonged the lifespan of Pten fl/fl Mx1-Cre + mice without affecting the type of leukemia that these mice developed (18). Even though S6K1 deletion did not affect the end points of the disease, it did reduce the expression of glycolytic gene PGK1 in a bone marrow progenitor cell population isolated from mice as early as five days after inducing Pten deletion. These data suggested that S6K1 ablation may prevent glycolysis and hence survival in this progenitor cell population shown to contain leukemia initiating cells, thereby delaying disease initiation and development. Consistent with this model, splenocytes isolated from Pten fl/fl Mx1-Cre + S6K1 -/- mice at this early time point formed fewer colonies in methylcellulose as compared to splenocytes derived from Pten fl/fl Mx1-Cre + S6K1 +/+ mice, indicating that S6K1 loss suppressed colony forming ability of Pten deficient cells (Figure 4). Altogether, our data suggest that S6K1 is necessary to mediate oncogenic metabolism and survival in Pten deficient neoplasia (Figure 5). Although S6K1 deficiency delayed leukemogenesis, it did not prevent the occurrence of leukemia in Pten deficient mice, suggesting that other pathways could compensate for the loss of S6K1. One possibility is that S6K1 deletion causes compensatory upregulation of S6K2. Elevated S6K2 mrna levels have been reported in multiple tissues of S6K1 deficient mice (19). Even though this possibility seems unlikely, as S6K1 inactivation in FL5.12 cells did not induce changes in S6K2 expression (Supplemental Figures 1B and IC, Chapter 2), it could provide a mechanistic explanation for why rapamycin treatment prevents leukemogenesis in Pten fl/fl Mx1- Cre + mice while S6K1 inactivation delays disease progression (18). Another possibility is the occurrence of oncogenic mutations that may induce parallel signaling pathways that can support glycolysis independent of S6K1, such as the PKC or Pim signaling pathways (20, 21). A third 109
110 possibility is that S6K1 loss delays the onset of secondary mutations. Hematopoietic specific deletion of Pten induces a tumor suppressor response characterized by increased expression of p53. Pten fl/fl Mx1-Cre + p53 +/- mice frequently acquire secondary mutations that result in loss of the wild type p53 allele thereby leading to accelerated leukemogenesis as compared to Pten fl/fl Mx1-Cre + p53 +/+ mice (22). Finally, S6K1 deletion may cause activation of alternative metabolic pathways that substitute for glycolysis to fuel survival. Recent studies have demonstrated that rapamycin resistance correlates with induction of carnitine palmitoyltransferase 1c (Cpt1c) which promotes fatty acid oxidation in breast cancer (23). Although Akt activated cells are highly addicted to glucose, pharmacological activation of fatty acid oxidation has been shown to permit glucose independent survival in these cells (24). In addition to fatty acid oxidation, glutaminolysis can confer survival and proliferative advantages to cells in the absence of glycolysis (25). Thus, S6K1 inhibition may activate metabolic pathways that promote glucose independent survival in leukemia. The best way to test this will be to perform in vivo metabolome analysis in S6K1/Pten deficient mice. S6K1- A therapeutic target in cancer The importance of S6K1 in mediating oncogenic glycolysis makes it a potential target for therapeutic intervention. Several ATP-competitive compounds have been developed that specifically block the activity of S6K1. However, the efficacy of these compounds for cancer chemotherapy remains unknown. The compound LYS6K2 blocks insulin induced phosphorylation of ribosomal protein S6 in primary rat hepatocytes at concentrations as low as 0.1μM (26). Even at higher concentrations, LYS6K2 did not prevent phosphorylation of other 110
111 kinases such as GSK3β and Erk1/2, indicating that it is highly specific for S6K1. Additionally, LYS6K2 treatment induced Akt phosphorylation, as expected, due to loss of the negative feedback loop. Similar to LYS6K2, PF compound has been reported to inhibit IGF induced S6 phosphorylation in HEK293 cells (27). In vitro specificity analysis revealed that PF did not inhibit the activity of closely related AGC family of kinases, including PDK1, Akt1, S6K2 and RSK1. However, PF treatment unexpectedly induced the phosphorylation of S6K1 at Thr389 in an mtorc1 dependent manner. This could result from enhanced Akt phosphorylation, which in turn would activate mtorc1 leading to increased S6K1 Thr389 phosphorylation. Surprisingly, PF did not promote Akt phosphorylation at concentrations that induced S6K1 Thr389 phosphorylation, suggesting Akt feedback independent mechanisms that mediate increases in S6K1 Thr389 phoshphorylation in the presence of PF Finally, the compound DG2 has been shown to block insulin induced S6 phosphorylation in L6 cells (28). DG2 exhibited minimal off-target effects when tested against a panel of 220 highly related kinases. Similar to PF , DG2 did not induce Akt phosphorylation in L6 cells. Although all these inhibitors prevent S6K1 activity, they exert differential effects on Akt activation. These conflicting data may be attributed to the different cell types used or to the specificity of the compound itself. So far, very little data exist regarding the effectiveness of these inhibitors in cell culture and/or animal based model systems. We tested the functional aspects of S6K1 signaling in response to DG2 (Chapter 3). Our studies provide evidence that DG2 reduces glycolysis and activates apoptosis in Pten deficient cells. These results are encouraging as they recapitulate the effects of genetically inactivating S6K1. However, the therapeutic efficacy of DG2 needs to be 111
112 tested in animal models of Pten deficient cancers. Future studies comparing different S6K1 inhibitors will reveal which compound is the most potent and safe in cancer treatment. Interrupting S6K1 mediated glycolysis to enhance cancer chemotherapy Pten loss is frequently associated with chemoresistance in multiple types of cancers. Pten inactivation promotes chemoresistance in prostate cancer cells via mechanisms that involve upregulation of Bcl-2 expression (29). In glioblastoma cells, Pten deficiency correlates with resistance to EGFR kinase inhibitor erlotinib, while Pten loss confers trastuzumab resistance in ErbB2 overexpressing breast cancers (30, 31). Since Pten is frequently inactivated in cancer and is an important determinant of the chemotherapeutic response, it is of paramount importance to develop novel strategies to sensitize Pten deficient cancer cells to chemotherapeutic agents. The ability of Pten deficient cells to promote survival depends on the maintenance of glucose metabolism. Our data demonstrate that preventing glycolytic metabolism by inactivating S6K1 renders Pten depleted cells sensitive to apoptosis. Thus, selective inhibition of glycolysis in Pten deficient cancer cells may provide a novel approach to overcome chemoresistance and potentiate the cytotoxicity of chemotherapeutic drugs. To test this hypothesis, we performed some preliminary experiments in Pten deficient human glioblastoma cell line-a172 and a Pten deficient human acute T-cell leukemia cell line- Jurkat. We chose these two tumor types as Pten loss has been shown to correlate with chemoresistance in both these types of cancer. Additionally, Pten null gamma secretase inhibitor (GSI) resistant T- cell leukemia cell lines exhibit increased glucose uptake and metabolism as 112
113 compared to Pten proficient GSI sensitive leukemic cell lines (32). Thus, inhibiting glycolysis may prove beneficial in resensitizing these leukemic cells to GSIs. Due to the limited availability of DG2, we chose rapamycin to inhibit S6K1 activation. Treatment with rapamycin attenuated glycolysis in both A172 and Jurkat cells, extending our previous findings of these effects in Pten deficient hematopoietic cells (Figure 6A and 6B). Future studies using chemotherapeutic drug arrays will assess if interruption of glycolytic metabolism will render these cancer cells susceptible to chemotherapy induced apoptosis. These data will provide new avenues for the treatment of cancer, particularly glioblastoma, where rapamycin by itself has proven inefficacious (33). Targeting mtorc1 versus S6K1 in cancer therapy Our studies demonstrate that both rapamycin, an allosteric mtor inhibitor, and DG2, an S6K1 selective inhibitor, suppress glycolysis to induce apoptosis in Pten deficient cells. These studies raise an interesting question about whether S6K1 will serve as a better therapeutic target than mtorc1, particularly in cancers where S6K1 is the major mediator of mtorc1 signal. There are potential disadvantages of using mtorc1 inhibitors in cancer chemotherapy that may be overcome by S6K1 inhibition. One limitation is the immunosuppressive activity of rapamycin and its analogs that may limit their therapeutic efficacy as anticancer agents. In addition to immunosuppression, several side effects such as hyperlipidemia, hypophosphatemia and hypercholesterolemia have been associated with mtorc1 inhibition (34). Targeting S6K1 specifically may prevent the deleterious side effects linked with mtorc1 inhibition. 113
114 Another potential drawback of rapamycin is feedback activation of Akt (35, 36). S6K1 activation, downstream of mtorc1, attenuates Akt signaling by phosphorylating and degrading IRS-1 (35). S6K1 inhibition by rapamycin prevents IRS-1 phosphorylation and subsequent degradation, leading to increased Akt activation and survival of tumor cells. Because S6K1 is the major player involved in this feedback mechanism, selectively inhibiting S6K1 is expected to have the same disadvantage as rapamycin. This assumption, however, may not be accurate given the recent findings of a novel mtorc1 substrate, the growth factor receptor-bound protein 10 (Grb10) that may also function to inhibit Akt activation (37, 38). These studies demonstrated that mtorc1 mediated phosphorylation and stabilization of Grb10 suppressed signaling by insulin and insulin like growth factor (IGF) while, rapamycin treatment triggered rapid degradation of Grb10 leading to increased Akt activation (37). The effects of rapamycin were recapitulated by Grb10 knockdown, suggesting that similar to S6K1, Grb10 participates in a negative feedback loop to suppress Akt activation. Although not tested, one may envisage that the magnitude of Akt signaling induced by rapamycin mediated inhibition of both Grb10 and S6K1 will be higher than S6K1 inactivation alone (Figure 7). Since the amplitude of Akt signaling correlates directly with survival, S6K1 inactivation alone may prove more beneficial than rapamycin treatment in anticancer therapy (1). Finally, the cytotoxic effects of conventional chemotherapeutic agents may be compromised in the presence of rapamycin. Rapamycin phosphorylation and inhibition of 4EBP1 prevents proliferation and induces cell cycle arrest (39). Since cytotoxic chemotherapy is often most effective against cells actively progressing through cell cycle, rapamycin mediated proliferation inhibition may prevent chemotherapy induced cell death. It remains to be seen if S6K1 inhibitors will enhance the therapeutic efficacy of current anticancer agents as compared to 114
115 mtorc1 inhibitors. Looking forward, there is great potential for chemotherapeutic exploitation of S6K1 inhibitors as single agents or in combination with cytotoxic chemotherapy. 115
116 Summary The data presented herein uncover a previously unknown mechanism by which Akt activation increases glycolytic metabolism. We demonstrate that S6K1 is a critical factor that promotes aerobic glycolysis in cells with oncogenic Akt activation. By inducing glycolysis, S6K1 supports apoptosis resistance in Pten deficient cells. We use loss of function approach to study the requirement for S6K1 to regulate glycolysis and survival. S6K1 inhibition using both genetic and pharmacological approaches suppresses glycolysis and activates apoptosis in cells knockdown for Pten. Furthermore, our studies delineate the mechanism of S6K1 modulation of glycolysis. S6K1 enhances glycolysis by inducing HIF1α expression. A decline in HIF1α abundance prevents glucose metabolism and survival downstream of S6K1, while restoration of HIF1α expression rescues glycolysis and apoptosis resistance in S6K1 depleted cells. These data provide the first evidence that HIF1α is necessary to drive the Warburg effect in Pten deficient cells. Additionally, these studies establish S6K1 as a novel HIF1α regulator. By genetic deletion of S6K1 in a Pten deficient mouse model of leukemia, we confirm that S6K1 is important for glucose metabolism and survival. Overall, we propose that selectively inhibiting S6K1 signaling may be an effective choice of therapy in cancers with activated Akt, particularly in cancers that harbor Pten mutations. 116
117 References 1. Plas DR, Talapatra S, Edinger AL et al (2001) Akt and bcl-xl promote growth factorindependent survival through distinct effects on mitochondrial physiology. J Biol Chem 276: Podsypanina K, Lee RT, Politis C et al (2001) An inhibitor of mtor reduces neoplasia and normalizes p70/s6 kinase activity in pten+/- mice. Proc Natl Acad Sci U S A 98: Neshat MS, Mellinghoff IK, Tran C et al (2001) Enhanced sensitivity of PTEN-deficient tumors to inhibition of FRAP/mTOR. Proc Natl Acad Sci U S A 98: Diegel CR, Cho KR, El-Naggar AK et al (2010) Mammalian target of rapamycin-dependent acinar cell neoplasia after inactivation of apc and pten in the mouse salivary gland: Implications for human acinic cell carcinoma. Cancer Res 70: Harada H, Andersen JS, Mann M et al (2001) p70s6 kinase signals cell survival as well as growth, inactivating the pro-apoptotic molecule BAD. Proc Natl Acad Sci U S A 98: Datta SR, Dudek H, Tao X et al (1997) Akt phosphorylation of BAD couples survival signals to the cell- intrinsic death machinery. Cell 91: Zhang HH, Lipovsky AI, Dibble CC et al (2006) S6K1 regulates GSK3 under conditions of mtor-dependent feedback inhibition of akt. Mol Cell 24: Shinjyo T, Kuribara R, Inukai T et al (2001) Downregulation of bim, a proapoptotic relative of bcl-2, is a pivotal step in cytokine-initiated survival signaling in murine hematopoietic progenitors. Mol Cell Biol 21: Zhao Y, Coloff JL, Ferguson EC et al (2008) Glucose metabolism attenuates p53 and pumadependent cell death upon growth factor deprivation. J Biol Chem 283: Hudson CC, Liu M, Chiang GG et al (2002) Regulation of hypoxia-inducible factor 1alpha expression and function by the mammalian target of rapamycin. Molecular and Cellular Biology 22: Zhong H, Chiles K, Feldser D et al (2000) Modulation of hypoxia-inducible factor 1alpha expression by the epidermal growth factor/phosphatidylinositol 3-kinase/PTEN/AKT/FRAP pathway in human prostate cancer cells: Implications for tumor angiogenesis and therapeutics. Cancer Res 60: Duvel K, Yecies JL, Menon S et al (2010) Activation of a metabolic gene regulatory network downstream of mtor complex 1. Mol Cell 39: Thomas GV, Tran C, Mellinghoff IK et al (2006) Hypoxia-inducible factor determines sensitivity to inhibitors of mtor in kidney cancer. Nat Med 12:
118 14. Wang X, Li W, Williams M et al (2001) Regulation of elongation factor 2 kinase by p90(rsk1) and p70 S6 kinase. EMBO J 20: Alliouachene S, Tuttle RL, Boumard S et al (2008) Constitutively active Akt1 expression in mouse pancreas requires S6 kinase 1 for insulinoma formation. J Clin Invest 118: Nardella C, Lunardi A, Fedele G et al (2011) Differential expression of S6K2 dictates tissuespecific requirement for S6K1 in mediating aberrant mtorc1 signaling and tumorigenesis. Cancer Res 71: Nakamura JL, Garcia E, Pieper RO (2008) S6K1 plays a key role in glial transformation. Cancer Res 68: Yilmaz OH, Valdez R, Theisen BK et al (2006) Pten dependence distinguishes haematopoietic stem cells from leukaemia-initiating cells. Nature 441: Shima H, Pende M, Chen Y et al (1998) Disruption of the p70(s6k)/p85(s6k) gene reveals a small mouse phenotype and a new functional S6 kinase. Embo J 17: Fox CJ, Hammerman PS, Cinalli RM et al (2003) The serine/threonine kinase pim-2 is a transcriptionally regulated apoptotic inhibitor. Genes Dev 17: Zhao Y, Altman BJ, Coloff JL et al (2007) Glycogen synthase kinase 3alpha and 3beta mediate a glucose-sensitive antiapoptotic signaling pathway to stabilize mcl-1. Mol Cell Biol 27: Lee JY, Nakada D, Yilmaz OH et al (2010) mtor activation induces tumor suppressors that inhibit leukemogenesis and deplete hematopoietic stem cells after pten deletion. Cell Stem Cell 7: Zaugg K, Yao Y, Reilly PT et al (2011) Carnitine palmitoyltransferase 1C promotes cell survival and tumor growth under conditions of metabolic stress. Genes Dev 25: Buzzai M, Bauer DE, Jones RG et al (2005) The glucose dependence of akt-transformed cells can be reversed by pharmacologic activation of fatty acid beta-oxidation. Oncogene 24: Choo AY, Kim SG, Vander Heiden MG et al (2010) Glucose addiction of TSC null cells is caused by failed mtorc1-dependent balancing of metabolic demand with supply. Mol Cell 38: Li S, Brown MS, Goldstein JL (2010) Bifurcation of insulin signaling pathway in rat liver: MTORC1 required for stimulation of lipogenesis, but not inhibition of gluconeogenesis. Proc Natl Acad Sci U S A 107:
119 27. Pearce LR, Alton GR, Richter DT et al (2010) Characterization of PF , a novel and highly specific inhibitor of p70 ribosomal S6 kinase (S6K1). Biochem J 431: Okuzumi T, Fiedler D, Zhang C et al (2009) Inhibitor hijacking of akt activation. Nat Chem Biol 5: Huang H, Cheville JC, Pan Y et al (2001) PTEN induces chemosensitivity in PTEN-mutated prostate cancer cells by suppression of bcl-2 expression. J Biol Chem 276: Wang MY, Lu KV, Zhu S et al (2006) Mammalian target of rapamycin inhibition promotes response to epidermal growth factor receptor kinase inhibitors in PTEN-deficient and PTENintact glioblastoma cells. Cancer Res 66: Nagata Y, Lan KH, Zhou X et al (2004) PTEN activation contributes to tumor inhibition by trastuzumab, and loss of PTEN predicts trastuzumab resistance in patients. Cancer Cell 6: Palomero T, Sulis ML, Cortina M et al (2007) Mutational loss of PTEN induces resistance to NOTCH1 inhibition in T-cell leukemia. Nat Med 13: Guo D, Prins RM, Dang J et al (2009) EGFR signaling through an akt-srebp-1-dependent, rapamycin-resistant pathway sensitizes glioblastomas to antilipogenic therapy. Sci Signal 2:ra Rodriguez-Pascual J, Cheng E, Maroto P et al (2010) Emergent toxicities associated with the use of mtor inhibitors in patients with advanced renal carcinoma. Anticancer Drugs 21: Um SH, Frigerio F, Watanabe M et al (2004) Absence of S6K1 protects against age- and diet-induced obesity while enhancing insulin sensitivity. Nature 431: Dibble CC, Asara JM, Manning BD (2009) Characterization of rictor phosphorylation sites reveals direct regulation of mtor complex 2 by S6K1. Mol Cell Biol 29: Yu Y, Yoon SO, Poulogiannis G et al (2011) Phosphoproteomic analysis identifies Grb10 as an mtorc1 substrate that negatively regulates insulin signaling. Science 332: Hsu PP, Kang SA, Rameseder J et al (2011) The mtor-regulated phosphoproteome reveals a mechanism of mtorc1-mediated inhibition of growth factor signaling. Science 332: Dowling RJ, Topisirovic I, Alain T et al (2010) mtorc1-mediated cell proliferation, but not cell growth, controlled by the 4E-BPs. Science 328:
120 Figure 1 % Viability at t=48 hrs mM Glucose 0.02mM Glucose Figure 1. Pten deficient cells require extracellular glucose to promote survival. Pten deficient cells were cultured in the presence of IL-3 and the indicated concentration of glucose for 48 hours and cell viability was measured. 120





121 Figure 2 Figure 2. S6K1 knockdown does not suppress glycolysis in the presence of IL-3. Cells were cultured in the presence of IL-3. The rate of cellular glycolysis was determined using a radiolabeled tracer assay. Contributed by Shikha Khatri 121





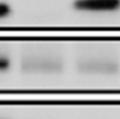
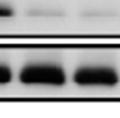



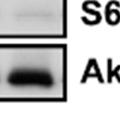
122 Figure 3 Figure 3. S6K1 knockdown does not alter Bad phosphorylation. Bad remains highly phosphorylated at S136 despite S6K1 knockdown. Indicated cell lines were transduced with empty vector, wildtype Bad or Bad S136A (Ala) expression constructs, then cultured in the absence of IL-3 for 2 hours. Contributed by Catherine A. Gallo 122








123 Figure 4 Figure 4. S6K1 losss inhibits colony formation. Pten + +/+ S6K1 +/+ Mx1-Cre +, Pten +/+ S6K1 -/- Mx1- Cre +, Pten fl/fl S6K1 +/+ + Mx1Cre + and Pten fl/fl S6K1 -/- Mx1Cre + were injected twice with pipc. Mice were sacrificed and their spleens were harvested onee day after the last injection. 100,000 splenocytes were plated in triplicate in methocult. Colony counts were determined after seven days. 123










124 Figure 5 Figure 5. Model for regulation of glycolysis and survival by S6K1. S6K1 promotes glucose metabolism and apoptosis resistance by inducing HIF1α expression. 124
125 Figure 6 A 150 Glycolytic Rate Vehicle Rapamycin B 250 Glycolytic Rate Vehicle Rapamycin Figure 6B. Contributed by John McGinty Figure 6. Rapamycin inhibits glycolysis in A) human glioblastoma A172 cells B) human T- ALL Jurkat cells. The rate of cellular glycolysis was determined after the cells were cultured in the presence of 20nM rapamycin or vehicle for 20hrs. 125









126 Figure 7 Figure 7. Targetingg S6K1 versus targetingg mtorc1. Disrupting S6K1 signaling inhibits the negative feedback loop and activates Akt. mtorc1 inhibition inactivates both Grb10 and S6K1 resulting in greater Akt activation as compared to S6K1 inactivation alone. 126
A particular set of insults induces apoptosis (part 1), which, if inhibited, can switch to autophagy. At least in some cellular settings, autophagy se
, which, if inhibited, can switch to autophagy. At least in some cellular settings, autophagy se") A particular set of insults induces apoptosis (part 1), which, if inhibited, can switch to autophagy. At least in some cellular settings, autophagy serves as a defence mechanism that prevents or retards
A particular set of insults induces apoptosis (part 1), which, if inhibited, can switch to autophagy. At least in some cellular settings, autophagy serves as a defence mechanism that prevents or retards
Evidence for an Alternative Glycolytic Pathway in Rapidly Proliferating Cells. Matthew G. Vander Heiden, et al. Science 2010
 Evidence for an Alternative Glycolytic Pathway in Rapidly Proliferating Cells Matthew G. Vander Heiden, et al. Science 2010 Introduction The Warburg Effect Cancer cells metabolize glucose differently Primarily
Evidence for an Alternative Glycolytic Pathway in Rapidly Proliferating Cells Matthew G. Vander Heiden, et al. Science 2010 Introduction The Warburg Effect Cancer cells metabolize glucose differently Primarily
Enzyme-coupled Receptors. Cell-surface receptors 1. Ion-channel-coupled receptors 2. G-protein-coupled receptors 3. Enzyme-coupled receptors
 Enzyme-coupled Receptors Cell-surface receptors 1. Ion-channel-coupled receptors 2. G-protein-coupled receptors 3. Enzyme-coupled receptors Cell-surface receptors allow a flow of ions across the plasma
Enzyme-coupled Receptors Cell-surface receptors 1. Ion-channel-coupled receptors 2. G-protein-coupled receptors 3. Enzyme-coupled receptors Cell-surface receptors allow a flow of ions across the plasma
The PI3K/AKT axis. Dr. Lucio Crinò Medical Oncology Division Azienda Ospedaliera-Perugia. Introduction
 The PI3K/AKT axis Dr. Lucio Crinò Medical Oncology Division Azienda Ospedaliera-Perugia Introduction Phosphoinositide 3-kinase (PI3K) pathway are a family of lipid kinases discovered in 1980s. They have
The PI3K/AKT axis Dr. Lucio Crinò Medical Oncology Division Azienda Ospedaliera-Perugia Introduction Phosphoinositide 3-kinase (PI3K) pathway are a family of lipid kinases discovered in 1980s. They have
RAS Genes. The ras superfamily of genes encodes small GTP binding proteins that are responsible for the regulation of many cellular processes.
 ۱ RAS Genes The ras superfamily of genes encodes small GTP binding proteins that are responsible for the regulation of many cellular processes. Oncogenic ras genes in human cells include H ras, N ras,
۱ RAS Genes The ras superfamily of genes encodes small GTP binding proteins that are responsible for the regulation of many cellular processes. Oncogenic ras genes in human cells include H ras, N ras,
Post-translational modifications of proteins in gene regulation under hypoxic conditions
 203 Review Article Post-translational modifications of proteins in gene regulation under hypoxic conditions 1, 2) Olga S. Safronova 1) Department of Cellular Physiological Chemistry, Tokyo Medical and
203 Review Article Post-translational modifications of proteins in gene regulation under hypoxic conditions 1, 2) Olga S. Safronova 1) Department of Cellular Physiological Chemistry, Tokyo Medical and
KEY CONCEPT QUESTIONS IN SIGNAL TRANSDUCTION
 Signal Transduction - Part 2 Key Concepts - Receptor tyrosine kinases control cell metabolism and proliferation Growth factor signaling through Ras Mutated cell signaling genes in cancer cells are called
Signal Transduction - Part 2 Key Concepts - Receptor tyrosine kinases control cell metabolism and proliferation Growth factor signaling through Ras Mutated cell signaling genes in cancer cells are called
Phospho-AKT Sampler Kit
 Phospho-AKT Sampler Kit E 0 5 1 0 0 3 Kits Includes Cat. Quantity Application Reactivity Source Akt (Ab-473) Antibody E021054-1 50μg/50μl IHC, WB Human, Mouse, Rat Rabbit Akt (Phospho-Ser473) Antibody
Phospho-AKT Sampler Kit E 0 5 1 0 0 3 Kits Includes Cat. Quantity Application Reactivity Source Akt (Ab-473) Antibody E021054-1 50μg/50μl IHC, WB Human, Mouse, Rat Rabbit Akt (Phospho-Ser473) Antibody
Signal Transduction Cascades
 Signal Transduction Cascades Contents of this page: Kinases & phosphatases Protein Kinase A (camp-dependent protein kinase) G-protein signal cascade Structure of G-proteins Small GTP-binding proteins,
Signal Transduction Cascades Contents of this page: Kinases & phosphatases Protein Kinase A (camp-dependent protein kinase) G-protein signal cascade Structure of G-proteins Small GTP-binding proteins,
Signaling. Dr. Sujata Persad Katz Group Centre for Pharmacy & Health research
 Signaling Dr. Sujata Persad 3-020 Katz Group Centre for Pharmacy & Health research E-mail:sujata.persad@ualberta.ca 1 Growth Factor Receptors and Other Signaling Pathways What we will cover today: How
Signaling Dr. Sujata Persad 3-020 Katz Group Centre for Pharmacy & Health research E-mail:sujata.persad@ualberta.ca 1 Growth Factor Receptors and Other Signaling Pathways What we will cover today: How
mirna Dr. S Hosseini-Asl
 mirna Dr. S Hosseini-Asl 1 2 MicroRNAs (mirnas) are small noncoding RNAs which enhance the cleavage or translational repression of specific mrna with recognition site(s) in the 3 - untranslated region
mirna Dr. S Hosseini-Asl 1 2 MicroRNAs (mirnas) are small noncoding RNAs which enhance the cleavage or translational repression of specific mrna with recognition site(s) in the 3 - untranslated region
Cell cycle, signaling to cell cycle, and molecular basis of oncogenesis
 Cell cycle, signaling to cell cycle, and molecular basis of oncogenesis MUDr. Jiří Vachtenheim, CSc. CELL CYCLE - SUMMARY Basic terminology: Cyclins conserved proteins with homologous regions; their cellular
Cell cycle, signaling to cell cycle, and molecular basis of oncogenesis MUDr. Jiří Vachtenheim, CSc. CELL CYCLE - SUMMARY Basic terminology: Cyclins conserved proteins with homologous regions; their cellular
Growth and Differentiation Phosphorylation Sampler Kit
 Growth and Differentiation Phosphorylation Sampler Kit E 0 5 1 0 1 4 Kits Includes Cat. Quantity Application Reactivity Source Akt (Phospho-Ser473) E011054-1 50μg/50μl IHC, WB Human, Mouse, Rat Rabbit
Growth and Differentiation Phosphorylation Sampler Kit E 0 5 1 0 1 4 Kits Includes Cat. Quantity Application Reactivity Source Akt (Phospho-Ser473) E011054-1 50μg/50μl IHC, WB Human, Mouse, Rat Rabbit
Contents. Preface XV Acknowledgments XXI List of Abbreviations XXIII About the Companion Website XXIX
 Contents Preface XV Acknowledgments XXI List of Abbreviations XXIII About the Companion Website XXIX 1 General Aspects of Signal Transduction and Cancer Therapy 1 1.1 General Principles of Signal Transduction
Contents Preface XV Acknowledgments XXI List of Abbreviations XXIII About the Companion Website XXIX 1 General Aspects of Signal Transduction and Cancer Therapy 1 1.1 General Principles of Signal Transduction
609G: Concepts of Cancer Genetics and Treatments (3 credits)
") Master of Chemical and Life Sciences Program College of Computer, Mathematical, and Natural Sciences 609G: Concepts of Cancer Genetics and Treatments (3 credits) Text books: Principles of Cancer Genetics,
Master of Chemical and Life Sciences Program College of Computer, Mathematical, and Natural Sciences 609G: Concepts of Cancer Genetics and Treatments (3 credits) Text books: Principles of Cancer Genetics,
number Done by Corrected by Doctor Maha Shomaf
 number 19 Done by Waseem Abo-Obeida Corrected by Abdullah Zreiqat Doctor Maha Shomaf Carcinogenesis: the molecular basis of cancer. Non-lethal genetic damage lies at the heart of carcinogenesis and leads
number 19 Done by Waseem Abo-Obeida Corrected by Abdullah Zreiqat Doctor Maha Shomaf Carcinogenesis: the molecular basis of cancer. Non-lethal genetic damage lies at the heart of carcinogenesis and leads
G-Protein Signaling. Introduction to intracellular signaling. Dr. SARRAY Sameh, Ph.D
 G-Protein Signaling Introduction to intracellular signaling Dr. SARRAY Sameh, Ph.D Cell signaling Cells communicate via extracellular signaling molecules (Hormones, growth factors and neurotransmitters
G-Protein Signaling Introduction to intracellular signaling Dr. SARRAY Sameh, Ph.D Cell signaling Cells communicate via extracellular signaling molecules (Hormones, growth factors and neurotransmitters
Receptor mediated Signal Transduction
 Receptor mediated Signal Transduction G-protein-linked receptors adenylyl cyclase camp PKA Organization of receptor protein-tyrosine kinases From G.M. Cooper, The Cell. A molecular approach, 2004, third
Receptor mediated Signal Transduction G-protein-linked receptors adenylyl cyclase camp PKA Organization of receptor protein-tyrosine kinases From G.M. Cooper, The Cell. A molecular approach, 2004, third
Insulin Resistance. Biol 405 Molecular Medicine
 Insulin Resistance Biol 405 Molecular Medicine Insulin resistance: a subnormal biological response to insulin. Defects of either insulin secretion or insulin action can cause diabetes mellitus. Insulin-dependent
Insulin Resistance Biol 405 Molecular Medicine Insulin resistance: a subnormal biological response to insulin. Defects of either insulin secretion or insulin action can cause diabetes mellitus. Insulin-dependent
Cell Signaling part 2
 15 Cell Signaling part 2 Functions of Cell Surface Receptors Other cell surface receptors are directly linked to intracellular enzymes. The largest family of these is the receptor protein tyrosine kinases,
15 Cell Signaling part 2 Functions of Cell Surface Receptors Other cell surface receptors are directly linked to intracellular enzymes. The largest family of these is the receptor protein tyrosine kinases,
Glycolysis Part 2. BCH 340 lecture 4
 Glycolysis Part 2 BCH 340 lecture 4 Regulation of Glycolysis There are three steps in glycolysis that have enzymes which regulate the flux of glycolysis These enzymes catalyzes irreversible reactions of
Glycolysis Part 2 BCH 340 lecture 4 Regulation of Glycolysis There are three steps in glycolysis that have enzymes which regulate the flux of glycolysis These enzymes catalyzes irreversible reactions of
TIGAR's promiscuity Bolaños, Juan P.
 TIGAR's promiscuity Bolaños, Juan P. TIGAR [TP53 (tumour protein 53)-induced glycolysis and apoptosis regulator] is an important survival factor for cancer cells. The enzymatic activity supported by sequence
TIGAR's promiscuity Bolaños, Juan P. TIGAR [TP53 (tumour protein 53)-induced glycolysis and apoptosis regulator] is an important survival factor for cancer cells. The enzymatic activity supported by sequence
Introduction to Cancer Biology
 Introduction to Cancer Biology Robin Hesketh Multiple choice questions (choose the one correct answer from the five choices) Which ONE of the following is a tumour suppressor? a. AKT b. APC c. BCL2 d.
Introduction to Cancer Biology Robin Hesketh Multiple choice questions (choose the one correct answer from the five choices) Which ONE of the following is a tumour suppressor? a. AKT b. APC c. BCL2 d.
The functional investigation of the interaction between TATA-associated factor 3 (TAF3) and p53 protein
 and p53 protein") THESIS BOOK The functional investigation of the interaction between TATA-associated factor 3 (TAF3) and p53 protein Orsolya Buzás-Bereczki Supervisors: Dr. Éva Bálint Dr. Imre Miklós Boros University of
THESIS BOOK The functional investigation of the interaction between TATA-associated factor 3 (TAF3) and p53 protein Orsolya Buzás-Bereczki Supervisors: Dr. Éva Bálint Dr. Imre Miklós Boros University of
BIOL 4374/BCHS 4313 Cell Biology Exam #1 February 13, 2001
 BIOL 4374/BCHS 4313 Cell Biology Exam #1 February 13, 2001 SS# Name This exam is worth a total of 100 points. The number of points each question is worth is shown in parentheses. Good luck! 1. (2) The
BIOL 4374/BCHS 4313 Cell Biology Exam #1 February 13, 2001 SS# Name This exam is worth a total of 100 points. The number of points each question is worth is shown in parentheses. Good luck! 1. (2) The
Chapt 15: Molecular Genetics of Cell Cycle and Cancer
 Chapt 15: Molecular Genetics of Cell Cycle and Cancer Student Learning Outcomes: Describe the cell cycle: steps taken by a cell to duplicate itself = cell division; Interphase (G1, S and G2), Mitosis.
Chapt 15: Molecular Genetics of Cell Cycle and Cancer Student Learning Outcomes: Describe the cell cycle: steps taken by a cell to duplicate itself = cell division; Interphase (G1, S and G2), Mitosis.
AperTO - Archivio Istituzionale Open Access dell'università di Torino
 AperTO - Archivio Istituzionale Open Access dell'università di Torino From the nucleus to the mitochondria and backthe odyssey of a multitask STAT3 This is the author's manuscript Original Citation: From
AperTO - Archivio Istituzionale Open Access dell'università di Torino From the nucleus to the mitochondria and backthe odyssey of a multitask STAT3 This is the author's manuscript Original Citation: From
Transformation of Normal HMECs (Human Mammary Epithelial Cells) into Metastatic Breast Cancer Cells: Introduction - The Broad Picture:
 into Metastatic Breast Cancer Cells: Introduction - The Broad Picture:") Transformation of Normal HMECs (Human Mammary Epithelial Cells) into Metastatic Breast Cancer Cells: Introduction - The Broad Picture: Spandana Baruah December, 2016 Cancer is defined as: «A disease caused
Transformation of Normal HMECs (Human Mammary Epithelial Cells) into Metastatic Breast Cancer Cells: Introduction - The Broad Picture: Spandana Baruah December, 2016 Cancer is defined as: «A disease caused
Ch. 18 Regulation of Gene Expression
 Ch. 18 Regulation of Gene Expression 1 Human genome has around 23,688 genes (Scientific American 2/2006) Essential Questions: How is transcription regulated? How are genes expressed? 2 Bacteria regulate
Ch. 18 Regulation of Gene Expression 1 Human genome has around 23,688 genes (Scientific American 2/2006) Essential Questions: How is transcription regulated? How are genes expressed? 2 Bacteria regulate
What is the Warburg Effect
 What is the Warburg Effect Roles nutrients play in the biochemistry of a cell Thus, proliferating cells must acquire more nutrients, convert them into biosynthetic building blocks, and coordinate the reactions
What is the Warburg Effect Roles nutrients play in the biochemistry of a cell Thus, proliferating cells must acquire more nutrients, convert them into biosynthetic building blocks, and coordinate the reactions
Integration Of Metabolism
 Integration Of Metabolism Metabolism Consist of Highly Interconnected Pathways The basic strategy of catabolic metabolism is to form ATP, NADPH, and building blocks for biosyntheses. 1. ATP is the universal
Integration Of Metabolism Metabolism Consist of Highly Interconnected Pathways The basic strategy of catabolic metabolism is to form ATP, NADPH, and building blocks for biosyntheses. 1. ATP is the universal
Lecture 10. G1/S Regulation and Cell Cycle Checkpoints. G1/S regulation and growth control G2 repair checkpoint Spindle assembly or mitotic checkpoint
 Lecture 10 G1/S Regulation and Cell Cycle Checkpoints Outline: G1/S regulation and growth control G2 repair checkpoint Spindle assembly or mitotic checkpoint Paper: The roles of Fzy/Cdc20 and Fzr/Cdh1
Lecture 10 G1/S Regulation and Cell Cycle Checkpoints Outline: G1/S regulation and growth control G2 repair checkpoint Spindle assembly or mitotic checkpoint Paper: The roles of Fzy/Cdc20 and Fzr/Cdh1
BCHM3972 Human Molecular Cell Biology (Advanced) 2013 Course University of Sydney
 2013 Course University of Sydney") BCHM3972 Human Molecular Cell Biology (Advanced) 2013 Course University of Sydney Page 2: Immune Mechanisms & Molecular Biology of Host Defence (Prof Campbell) Page 45: Infection and Implications for Cell
BCHM3972 Human Molecular Cell Biology (Advanced) 2013 Course University of Sydney Page 2: Immune Mechanisms & Molecular Biology of Host Defence (Prof Campbell) Page 45: Infection and Implications for Cell
BIO360 Quiz #1. September 14, Name five of the six Hallmarks of Cancer (not emerging hallmarks or enabling characteristics ): (5 points)
: (5 points)") Name: BIO360 Quiz #1 September 14, 2012 1. Name five of the six Hallmarks of Cancer (not emerging hallmarks or enabling characteristics ): (5 points) 2. The controversial hypothesis that only a small subset
Name: BIO360 Quiz #1 September 14, 2012 1. Name five of the six Hallmarks of Cancer (not emerging hallmarks or enabling characteristics ): (5 points) 2. The controversial hypothesis that only a small subset
Regulation of Gene Expression in Eukaryotes
 Ch. 19 Regulation of Gene Expression in Eukaryotes BIOL 222 Differential Gene Expression in Eukaryotes Signal Cells in a multicellular eukaryotic organism genetically identical differential gene expression
Ch. 19 Regulation of Gene Expression in Eukaryotes BIOL 222 Differential Gene Expression in Eukaryotes Signal Cells in a multicellular eukaryotic organism genetically identical differential gene expression
Cell Quality Control. Peter Takizawa Department of Cell Biology
 Cell Quality Control Peter Takizawa Department of Cell Biology Cellular quality control reduces production of defective proteins. Cells have many quality control systems to ensure that cell does not build
Cell Quality Control Peter Takizawa Department of Cell Biology Cellular quality control reduces production of defective proteins. Cells have many quality control systems to ensure that cell does not build
p53 and Apoptosis: Master Guardian and Executioner Part 2
 p53 and Apoptosis: Master Guardian and Executioner Part 2 p14arf in human cells is a antagonist of Mdm2. The expression of ARF causes a rapid increase in p53 levels, so what would you suggest?.. The enemy
p53 and Apoptosis: Master Guardian and Executioner Part 2 p14arf in human cells is a antagonist of Mdm2. The expression of ARF causes a rapid increase in p53 levels, so what would you suggest?.. The enemy
Enzymes Part III: regulation II. Dr. Mamoun Ahram Summer, 2017
 Enzymes Part III: regulation II Dr. Mamoun Ahram Summer, 2017 Advantage This is a major mechanism for rapid and transient regulation of enzyme activity. A most common mechanism is enzyme phosphorylation
Enzymes Part III: regulation II Dr. Mamoun Ahram Summer, 2017 Advantage This is a major mechanism for rapid and transient regulation of enzyme activity. A most common mechanism is enzyme phosphorylation
BIO360 Fall 2013 Quiz 1
 BIO360 Fall 2013 Quiz 1 1. Examine the diagram below. There are two homologous copies of chromosome one and the allele of YFG carried on the light gray chromosome has undergone a loss-of-function mutation.
BIO360 Fall 2013 Quiz 1 1. Examine the diagram below. There are two homologous copies of chromosome one and the allele of YFG carried on the light gray chromosome has undergone a loss-of-function mutation.
Regulators of Cell Cycle Progression
 Regulators of Cell Cycle Progression Studies of Cdk s and cyclins in genetically modified mice reveal a high level of plasticity, allowing different cyclins and Cdk s to compensate for the loss of one
Regulators of Cell Cycle Progression Studies of Cdk s and cyclins in genetically modified mice reveal a high level of plasticity, allowing different cyclins and Cdk s to compensate for the loss of one
BCH Graduate Survey of Biochemistry
 BCH 5045 Graduate Survey of Biochemistry Instructor: Charles Guy Producer: Ron Thomas Director: Marsha Durosier Lecture 44 Slide sets available at: http://hort.ifas.ufl.edu/teach/guyweb/bch5045/index.html
BCH 5045 Graduate Survey of Biochemistry Instructor: Charles Guy Producer: Ron Thomas Director: Marsha Durosier Lecture 44 Slide sets available at: http://hort.ifas.ufl.edu/teach/guyweb/bch5045/index.html
Negative Regulation of c-myc Oncogenic Activity Through the Tumor Suppressor PP2A-B56α
 Negative Regulation of c-myc Oncogenic Activity Through the Tumor Suppressor PP2A-B56α Mahnaz Janghorban, PhD Dr. Rosalie Sears lab 2/8/2015 Zanjan University Content 1. Background (keywords: c-myc, PP2A,
Negative Regulation of c-myc Oncogenic Activity Through the Tumor Suppressor PP2A-B56α Mahnaz Janghorban, PhD Dr. Rosalie Sears lab 2/8/2015 Zanjan University Content 1. Background (keywords: c-myc, PP2A,
Introduction. Cancer Biology. Tumor-suppressor genes. Proto-oncogenes. DNA stability genes. Mechanisms of carcinogenesis.
 Cancer Biology Chapter 18 Eric J. Hall., Amato Giaccia, Radiobiology for the Radiologist Introduction Tissue homeostasis depends on the regulated cell division and self-elimination (programmed cell death)
Cancer Biology Chapter 18 Eric J. Hall., Amato Giaccia, Radiobiology for the Radiologist Introduction Tissue homeostasis depends on the regulated cell division and self-elimination (programmed cell death)
Molecular biology :- Cancer genetics lecture 11
 Molecular biology :- Cancer genetics lecture 11 -We have talked about 2 group of genes that is involved in cellular transformation : proto-oncogenes and tumour suppressor genes, and it isn t enough to
Molecular biology :- Cancer genetics lecture 11 -We have talked about 2 group of genes that is involved in cellular transformation : proto-oncogenes and tumour suppressor genes, and it isn t enough to
Biol403 MAP kinase signalling
 Biol403 MAP kinase signalling The mitogen activated protein kinase (MAPK) pathway is a signalling cascade activated by a diverse range of effectors. The cascade regulates many cellular activities including
Biol403 MAP kinase signalling The mitogen activated protein kinase (MAPK) pathway is a signalling cascade activated by a diverse range of effectors. The cascade regulates many cellular activities including
NFκB What is it and What s the deal with radicals?
 The Virtual Free Radical School NFκB What is it and What s the deal with radicals? Emily Ho, Ph.D Linus Pauling Institute Scientist Department of Nutrition and Food Management Oregon State University 117
The Virtual Free Radical School NFκB What is it and What s the deal with radicals? Emily Ho, Ph.D Linus Pauling Institute Scientist Department of Nutrition and Food Management Oregon State University 117
Under aerobic conditions, pyruvate enters the mitochondria where it is converted into acetyl CoA.
 Under aerobic conditions, pyruvate enters the mitochondria where it is converted into acetyl CoA. Acetyl CoA is the fuel for the citric acid cycle, which processes the two carbon acetyl unit to two molecules
Under aerobic conditions, pyruvate enters the mitochondria where it is converted into acetyl CoA. Acetyl CoA is the fuel for the citric acid cycle, which processes the two carbon acetyl unit to two molecules
Interrogating mtor Inhibition in Patients with HRPC
 Interrogating mtor Inhibition in Patients with HRPC Daniel George, John Madden, Andrew Armstrong, Mark Dewhirst, Nancy Major, and Phillip Febbo Divisions of Medical Oncology, Urology, Pathology, Radiology,
Interrogating mtor Inhibition in Patients with HRPC Daniel George, John Madden, Andrew Armstrong, Mark Dewhirst, Nancy Major, and Phillip Febbo Divisions of Medical Oncology, Urology, Pathology, Radiology,
Moh Tarek. Razi Kittaneh. Jaqen H ghar
 14 Moh Tarek Razi Kittaneh Jaqen H ghar Naif Karadsheh Gluconeogenesis is making glucose from non-carbohydrates precursors. Although Gluconeogenesis looks like Glycolysis in many steps, it is not the simple
14 Moh Tarek Razi Kittaneh Jaqen H ghar Naif Karadsheh Gluconeogenesis is making glucose from non-carbohydrates precursors. Although Gluconeogenesis looks like Glycolysis in many steps, it is not the simple
Dr. Mohnen s notes on GLUCONEOGENESIS
 Dr. Mohnen s notes on GLUCONEOGENESIS Note: Even though we did not get through all of these slides during lecture, I advise you to look them all through because they will be helpful to you as you learn
Dr. Mohnen s notes on GLUCONEOGENESIS Note: Even though we did not get through all of these slides during lecture, I advise you to look them all through because they will be helpful to you as you learn
Cell cycle and Apoptosis. Chalermchai Mitrpant
 Cell cycle and Apoptosis 2556 Chalermchai Mitrpant Overview of the cell cycle Outline Regulatory mechanisms controlling cell cycle Progression of the cell cycle Checkpoint of the cell cycle Phases of the
Cell cycle and Apoptosis 2556 Chalermchai Mitrpant Overview of the cell cycle Outline Regulatory mechanisms controlling cell cycle Progression of the cell cycle Checkpoint of the cell cycle Phases of the
Signal Transduction Pathways. Part 2
 Signal Transduction Pathways Part 2 GPCRs G-protein coupled receptors > 700 GPCRs in humans Mediate responses to senses taste, smell, sight ~ 1000 GPCRs mediate sense of smell in mouse Half of all known
Signal Transduction Pathways Part 2 GPCRs G-protein coupled receptors > 700 GPCRs in humans Mediate responses to senses taste, smell, sight ~ 1000 GPCRs mediate sense of smell in mouse Half of all known
PCB 3023 Exam 4 - Form A First and Last Name
 PCB 3023 Exam 4 - Form A First and Last Name Student ID # (U Number) A Before beginning this exam, please complete the following instructions: 1) Write your name and U number on the first page of this
PCB 3023 Exam 4 - Form A First and Last Name Student ID # (U Number) A Before beginning this exam, please complete the following instructions: 1) Write your name and U number on the first page of this
Signaling Through Immune System Receptors (Ch. 7)
") Signaling Through Immune System Receptors (Ch. 7) 1. General principles of signal transduction and propagation. 2. Antigen receptor signaling and lymphocyte activation. 3. Other receptors and signaling
Signaling Through Immune System Receptors (Ch. 7) 1. General principles of signal transduction and propagation. 2. Antigen receptor signaling and lymphocyte activation. 3. Other receptors and signaling
Review. Ageing 2: Cancer! Review: Mutations. Mutations 2/14/11. The Raw Material for Evolution. The Double Edged Sword
 Ageing 2: Cancer! Review: The force of natural selection declines with ageing due to increase in extrinsic mortality (= weakening of natural selection) and reduction in reproduction with age (selection
Ageing 2: Cancer! Review: The force of natural selection declines with ageing due to increase in extrinsic mortality (= weakening of natural selection) and reduction in reproduction with age (selection
Regulation of cell function by intracellular signaling
 Regulation of cell function by intracellular signaling Objectives: Regulation principle Allosteric and covalent mechanisms, Popular second messengers, Protein kinases, Kinase cascade and interaction. regulation
Regulation of cell function by intracellular signaling Objectives: Regulation principle Allosteric and covalent mechanisms, Popular second messengers, Protein kinases, Kinase cascade and interaction. regulation
TUMOR-SUPPRESSOR GENES. Molecular Oncology Michael Lea
 TUMOR-SUPPRESSOR GENES Molecular Oncology 2011 Michael Lea TUMOR-SUPPRESSOR GENES - Lecture Outline 1. Summary of tumor suppressor genes 2. P53 3. Rb 4. BRCA1 and 2 5. APC and DCC 6. PTEN and PPA2 7. LKB1
TUMOR-SUPPRESSOR GENES Molecular Oncology 2011 Michael Lea TUMOR-SUPPRESSOR GENES - Lecture Outline 1. Summary of tumor suppressor genes 2. P53 3. Rb 4. BRCA1 and 2 5. APC and DCC 6. PTEN and PPA2 7. LKB1
Propagation of the Signal
 OpenStax-CNX module: m44452 1 Propagation of the Signal OpenStax College This work is produced by OpenStax-CNX and licensed under the Creative Commons Attribution License 3.0 By the end of this section,
OpenStax-CNX module: m44452 1 Propagation of the Signal OpenStax College This work is produced by OpenStax-CNX and licensed under the Creative Commons Attribution License 3.0 By the end of this section,
5.0 HORMONAL CONTROL OF CARBOHYDRATE METABOLISM
 5.0 HORMONAL CONTROL OF CARBOHYDRATE METABOLISM Introduction: Variety of hormones and other molecules regulate the carbohydrates metabolism. Some of these have already been cited in previous sections.
5.0 HORMONAL CONTROL OF CARBOHYDRATE METABOLISM Introduction: Variety of hormones and other molecules regulate the carbohydrates metabolism. Some of these have already been cited in previous sections.
CHE 242 Exam 3 Practice Questions
 CHE 242 Exam 3 Practice Questions Glucose metabolism 1. Below is depicted glucose catabolism. Indicate on the pathways the following: A) which reaction(s) of glycolysis are irreversible B) where energy
CHE 242 Exam 3 Practice Questions Glucose metabolism 1. Below is depicted glucose catabolism. Indicate on the pathways the following: A) which reaction(s) of glycolysis are irreversible B) where energy
Regulation of PI3K effector signalling in cancer by the phosphoinositide phosphatases
 Review Article Regulation of PI3K effector signalling in cancer by the phosphoinositide phosphatases Samuel J. Rodgers, Daniel T. Ferguson, Christina A. Mitchell and Lisa M. Ooms Cancer Program, Monash
Review Article Regulation of PI3K effector signalling in cancer by the phosphoinositide phosphatases Samuel J. Rodgers, Daniel T. Ferguson, Christina A. Mitchell and Lisa M. Ooms Cancer Program, Monash
Glucose is the only source of energy in red blood cells. Under starvation conditions ketone bodies become a source of energy for the brain
 Glycolysis 4 / The Text :- Some Points About Glucose Glucose is very soluble source of quick and ready energy. It is a relatively stable and easily transported. In mammals, the brain uses only glucose
Glycolysis 4 / The Text :- Some Points About Glucose Glucose is very soluble source of quick and ready energy. It is a relatively stable and easily transported. In mammals, the brain uses only glucose
Metabolism. Metabolic pathways. BIO 5099: Molecular Biology for Computer Scientists (et al) Lecture 11: Metabolic Pathways
 Lecture 11: Metabolic Pathways") BIO 5099: Molecular Biology for Computer Scientists (et al) Lecture 11: Metabolic Pathways http://compbio.uchsc.edu/hunter/bio5099 Larry.Hunter@uchsc.edu Metabolism Metabolism is the chemical change of
BIO 5099: Molecular Biology for Computer Scientists (et al) Lecture 11: Metabolic Pathways http://compbio.uchsc.edu/hunter/bio5099 Larry.Hunter@uchsc.edu Metabolism Metabolism is the chemical change of
FACTORS AFFECTING SKELETAL MUSCLE PROTEIN SYNTHESIS IN THE HORSE
 University of Kentucky UKnowledge Theses and Dissertations--Animal and Food Sciences Animal and Food Sciences 2011 FACTORS AFFECTING SKELETAL MUSCLE PROTEIN SYNTHESIS IN THE HORSE Ashley Leigh Wagner University
University of Kentucky UKnowledge Theses and Dissertations--Animal and Food Sciences Animal and Food Sciences 2011 FACTORS AFFECTING SKELETAL MUSCLE PROTEIN SYNTHESIS IN THE HORSE Ashley Leigh Wagner University
Molecular Biology (BIOL 4320) Exam #2 May 3, 2004
 Exam #2 May 3, 2004") Molecular Biology (BIOL 4320) Exam #2 May 3, 2004 Name SS# This exam is worth a total of 100 points. The number of points each question is worth is shown in parentheses after the question number. Good
Molecular Biology (BIOL 4320) Exam #2 May 3, 2004 Name SS# This exam is worth a total of 100 points. The number of points each question is worth is shown in parentheses after the question number. Good
Multistep nature of cancer development. Cancer genes
 Multistep nature of cancer development Phenotypic progression loss of control over cell growth/death (neoplasm) invasiveness (carcinoma) distal spread (metastatic tumor) Genetic progression multiple genetic
Multistep nature of cancer development Phenotypic progression loss of control over cell growth/death (neoplasm) invasiveness (carcinoma) distal spread (metastatic tumor) Genetic progression multiple genetic
BIOL212 Biochemistry of Disease. Metabolic Disorders - Obesity
 BIOL212 Biochemistry of Disease Metabolic Disorders - Obesity Obesity Approx. 23% of adults are obese in the U.K. The number of obese children has tripled in 20 years. 10% of six year olds are obese, rising
BIOL212 Biochemistry of Disease Metabolic Disorders - Obesity Obesity Approx. 23% of adults are obese in the U.K. The number of obese children has tripled in 20 years. 10% of six year olds are obese, rising
Follicular Lymphoma. ced3 APOPTOSIS. *In the nematode Caenorhabditis elegans 131 of the organism's 1031 cells die during development.
 Harvard-MIT Division of Health Sciences and Technology HST.176: Cellular and Molecular Immunology Course Director: Dr. Shiv Pillai Follicular Lymphoma 1. Characterized by t(14:18) translocation 2. Ig heavy
Harvard-MIT Division of Health Sciences and Technology HST.176: Cellular and Molecular Immunology Course Director: Dr. Shiv Pillai Follicular Lymphoma 1. Characterized by t(14:18) translocation 2. Ig heavy
Problem Set 8 Key 1 of 8
 7.06 2003 Problem Set 8 Key 1 of 8 7.06 2003 Problem Set 8 Key 1. As a bright MD/PhD, you are interested in questions about the control of cell number in the body. Recently, you've seen three patients
7.06 2003 Problem Set 8 Key 1 of 8 7.06 2003 Problem Set 8 Key 1. As a bright MD/PhD, you are interested in questions about the control of cell number in the body. Recently, you've seen three patients
REGULATION OF ENZYME ACTIVITY. Medical Biochemistry, Lecture 25
 REGULATION OF ENZYME ACTIVITY Medical Biochemistry, Lecture 25 Lecture 25, Outline General properties of enzyme regulation Regulation of enzyme concentrations Allosteric enzymes and feedback inhibition
REGULATION OF ENZYME ACTIVITY Medical Biochemistry, Lecture 25 Lecture 25, Outline General properties of enzyme regulation Regulation of enzyme concentrations Allosteric enzymes and feedback inhibition
CHAPTER 16. Glycolysis
 CHAPTER 16 Glycolysis Net reaction of Glycolysis Converts: 1 Glucose Hexose stage 2 pyruvate - Two molecules of ATP are produced - Two molecules of NAD + are reduced to NADH Triose stage Glucose + 2 ADP
CHAPTER 16 Glycolysis Net reaction of Glycolysis Converts: 1 Glucose Hexose stage 2 pyruvate - Two molecules of ATP are produced - Two molecules of NAD + are reduced to NADH Triose stage Glucose + 2 ADP
TITLE: Overcoming Resistance to Inhibitors of the Akt Protein Kinase by Modulation of the Pim Kinase Pathway
 AWARD NUMBER: W81XWH-12-1-0560 TITLE: Overcoming Resistance to Inhibitors of the Akt Protein Kinase by Modulation of the Pim Kinase Pathway PRINCIPAL INVESTIGATOR: Andrew S. Kraft, MD CONTRACTING ORGANIZATION:
AWARD NUMBER: W81XWH-12-1-0560 TITLE: Overcoming Resistance to Inhibitors of the Akt Protein Kinase by Modulation of the Pim Kinase Pathway PRINCIPAL INVESTIGATOR: Andrew S. Kraft, MD CONTRACTING ORGANIZATION:
Tumor Metabolism. Hypoxia-inducible factor (HIF)
") Tumor Metabolism Kevin M. Brindle Department of Biochemistry, University of Cambridge Tennis Court Road, Cambridge CB2 1GA and Cancer Research UK Cambridge Research Institute, Li Ka Shing Centre, Robinson
Tumor Metabolism Kevin M. Brindle Department of Biochemistry, University of Cambridge Tennis Court Road, Cambridge CB2 1GA and Cancer Research UK Cambridge Research Institute, Li Ka Shing Centre, Robinson
Signal Transduction Pathway Smorgasbord
 Molecular Cell Biology Lecture. Oct 28, 2014 Signal Transduction Pathway Smorgasbord Ron Bose, MD PhD Biochemistry and Molecular Cell Biology Programs Washington University School of Medicine Outline 1.
Molecular Cell Biology Lecture. Oct 28, 2014 Signal Transduction Pathway Smorgasbord Ron Bose, MD PhD Biochemistry and Molecular Cell Biology Programs Washington University School of Medicine Outline 1.
Genome of Hepatitis B Virus. VIRAL ONCOGENE Dr. Yahwardiah Siregar, PhD Dr. Sry Suryani Widjaja, Mkes Biochemistry Department
 Genome of Hepatitis B Virus VIRAL ONCOGENE Dr. Yahwardiah Siregar, PhD Dr. Sry Suryani Widjaja, Mkes Biochemistry Department Proto Oncogen and Oncogen Oncogen Proteins that possess the ability to cause
Genome of Hepatitis B Virus VIRAL ONCOGENE Dr. Yahwardiah Siregar, PhD Dr. Sry Suryani Widjaja, Mkes Biochemistry Department Proto Oncogen and Oncogen Oncogen Proteins that possess the ability to cause
Signal Transduction: G-Protein Coupled Receptors
 Signal Transduction: G-Protein Coupled Receptors Federle, M. (2017). Lectures 4-5: Signal Transduction parts 1&2: nuclear receptors and GPCRs. Lecture presented at PHAR 423 Lecture in UIC College of Pharmacy,
Signal Transduction: G-Protein Coupled Receptors Federle, M. (2017). Lectures 4-5: Signal Transduction parts 1&2: nuclear receptors and GPCRs. Lecture presented at PHAR 423 Lecture in UIC College of Pharmacy,
CARBOHYDRATE METABOLISM 1
 CARBOHYDRATE METABOLISM 1 web 2017 József Mandl Strategy of metabolism 1 Strategy of metabolism to extract energy ( hydrogen ) from the environment to store the energy excess to store hydrogen CH 3 O 2
CARBOHYDRATE METABOLISM 1 web 2017 József Mandl Strategy of metabolism 1 Strategy of metabolism to extract energy ( hydrogen ) from the environment to store the energy excess to store hydrogen CH 3 O 2
Lecture 14 - The cell cycle and cell death
 02.17.10 Lecture 14 - The cell cycle and cell death The cell cycle: cells duplicate their contents and divide The cell cycle may be divided into 4 phases The cell cycle triggers essential processes (DNA
02.17.10 Lecture 14 - The cell cycle and cell death The cell cycle: cells duplicate their contents and divide The cell cycle may be divided into 4 phases The cell cycle triggers essential processes (DNA
Lecture: CHAPTER 13 Signal Transduction Pathways
 Lecture: 10 17 2016 CHAPTER 13 Signal Transduction Pathways Chapter 13 Outline Signal transduction cascades have many components in common: 1. Release of a primary message as a response to a physiological
Lecture: 10 17 2016 CHAPTER 13 Signal Transduction Pathways Chapter 13 Outline Signal transduction cascades have many components in common: 1. Release of a primary message as a response to a physiological
Cell Cycle. Trends in Cell Biology
 Cell Cycle Trends in Cell Biology Cell Cycle The orderly sequence of events by which a cell duplicates its contents and divides into two Daughter Cells Activities of a cell from one cell division to the
Cell Cycle Trends in Cell Biology Cell Cycle The orderly sequence of events by which a cell duplicates its contents and divides into two Daughter Cells Activities of a cell from one cell division to the
FIRST BIOCHEMISTRY EXAM Tuesday 25/10/ MCQs. Location : 102, 105, 106, 301, 302
 FIRST BIOCHEMISTRY EXAM Tuesday 25/10/2016 10-11 40 MCQs. Location : 102, 105, 106, 301, 302 The Behavior of Proteins: Enzymes, Mechanisms, and Control General theory of enzyme action, by Leonor Michaelis
FIRST BIOCHEMISTRY EXAM Tuesday 25/10/2016 10-11 40 MCQs. Location : 102, 105, 106, 301, 302 The Behavior of Proteins: Enzymes, Mechanisms, and Control General theory of enzyme action, by Leonor Michaelis
Oncogenes and Tumor Suppressors MCB 5068 November 12, 2013 Jason Weber
 Oncogenes and Tumor Suppressors MCB 5068 November 12, 2013 Jason Weber jweber@dom.wustl.edu Oncogenes & Cancer DNA Tumor Viruses Simian Virus 40 p300 prb p53 Large T Antigen Human Adenovirus p300 E1A
Oncogenes and Tumor Suppressors MCB 5068 November 12, 2013 Jason Weber jweber@dom.wustl.edu Oncogenes & Cancer DNA Tumor Viruses Simian Virus 40 p300 prb p53 Large T Antigen Human Adenovirus p300 E1A
Lecture #27 Lecturer A. N. Koval
 Lecture #27 Lecturer A. N. Koval Hormones Transduce Signals to Affect Homeostatic Mechanisms Koval A. (C), 2011 2 Lipophilic hormones Classifying hormones into hydrophilic and lipophilic molecules indicates
Lecture #27 Lecturer A. N. Koval Hormones Transduce Signals to Affect Homeostatic Mechanisms Koval A. (C), 2011 2 Lipophilic hormones Classifying hormones into hydrophilic and lipophilic molecules indicates
VIII Curso Internacional del PIRRECV. Some molecular mechanisms of cancer
 VIII Curso Internacional del PIRRECV Some molecular mechanisms of cancer Laboratorio de Comunicaciones Celulares, Centro FONDAP Estudios Moleculares de la Celula (CEMC), ICBM, Facultad de Medicina, Universidad
VIII Curso Internacional del PIRRECV Some molecular mechanisms of cancer Laboratorio de Comunicaciones Celulares, Centro FONDAP Estudios Moleculares de la Celula (CEMC), ICBM, Facultad de Medicina, Universidad
Lecture 15. Signal Transduction Pathways - Introduction
 Lecture 15 Signal Transduction Pathways - Introduction So far.. Regulation of mrna synthesis Regulation of rrna synthesis Regulation of trna & 5S rrna synthesis Regulation of gene expression by signals
Lecture 15 Signal Transduction Pathways - Introduction So far.. Regulation of mrna synthesis Regulation of rrna synthesis Regulation of trna & 5S rrna synthesis Regulation of gene expression by signals
Regulation of Citric Acid Cycle
 Paper : 04 Metabolism of carbohydrates Module : 30 Principal Investigator, Paper Coordinator and Content Writer Dr. Ramesh Kothari, Professor UGC-CAS Department of Biosciences Saurashtra University, Rajkot-5
Paper : 04 Metabolism of carbohydrates Module : 30 Principal Investigator, Paper Coordinator and Content Writer Dr. Ramesh Kothari, Professor UGC-CAS Department of Biosciences Saurashtra University, Rajkot-5
Final Review Sessions. 3/16 (FRI) 126 Wellman (4-6 6 pm) 3/19 (MON) 1309 Surge 3 (4-6 6 pm) Office Hours
 126 Wellman (4-6 6 pm) 3/19 (MON) 1309 Surge 3 (4-6 6 pm) Office Hours") Final Review Sessions 3/16 (FRI) 126 Wellman (4-6 6 pm) 3/19 (MON) 1309 Surge 3 (4-6 6 pm) Office ours 3/14 (WED) 9:30 11:30 am (Rebecca) 3/16 (FRI) 9-11 am (Abel) Final ESSENTIALS Posted Lecture 20 ormonal
Final Review Sessions 3/16 (FRI) 126 Wellman (4-6 6 pm) 3/19 (MON) 1309 Surge 3 (4-6 6 pm) Office ours 3/14 (WED) 9:30 11:30 am (Rebecca) 3/16 (FRI) 9-11 am (Abel) Final ESSENTIALS Posted Lecture 20 ormonal
1. Activated receptor tyrosine kinases (RTKs) phosphorylates themselves
 phosphorylates themselves") Enzyme-coupled receptors Transmembrane proteins Ligand-binding domain on the outer surface Cytoplasmic domain acts as an enzyme itself or forms a complex with enzyme 1. Activated receptor tyrosine kinases
Enzyme-coupled receptors Transmembrane proteins Ligand-binding domain on the outer surface Cytoplasmic domain acts as an enzyme itself or forms a complex with enzyme 1. Activated receptor tyrosine kinases
The elements of G protein-coupled receptor systems
 The elements of G protein-coupled receptor systems Prostaglandines Sphingosine 1-phosphate a receptor that contains 7 membrane-spanning domains a coupled trimeric G protein which functions as a switch
The elements of G protein-coupled receptor systems Prostaglandines Sphingosine 1-phosphate a receptor that contains 7 membrane-spanning domains a coupled trimeric G protein which functions as a switch
2013 W. H. Freeman and Company. 12 Signal Transduction
 2013 W. H. Freeman and Company 12 Signal Transduction CHAPTER 12 Signal Transduction Key topics: General features of signal transduction Structure and function of G protein coupled receptors Structure
2013 W. H. Freeman and Company 12 Signal Transduction CHAPTER 12 Signal Transduction Key topics: General features of signal transduction Structure and function of G protein coupled receptors Structure
What would you observe if you fused a G1 cell with a S cell? A. Mitotic and pulverized chromosomes. B. Mitotic and compact G1 chromosomes.
 What would you observe if you fused a G1 cell with a S cell? A. Mitotic and pulverized chromosomes. B. Mitotic and compact G1 chromosomes. C. Mostly non-compact G1 chromosomes. D. Compact G1 and G2 chromosomes.
What would you observe if you fused a G1 cell with a S cell? A. Mitotic and pulverized chromosomes. B. Mitotic and compact G1 chromosomes. C. Mostly non-compact G1 chromosomes. D. Compact G1 and G2 chromosomes.
Chapter 15: Signal transduction
 Chapter 15: Signal transduction Know the terminology: Enzyme-linked receptor, G-protein linked receptor, nuclear hormone receptor, G-protein, adaptor protein, scaffolding protein, SH2 domain, MAPK, Ras,
Chapter 15: Signal transduction Know the terminology: Enzyme-linked receptor, G-protein linked receptor, nuclear hormone receptor, G-protein, adaptor protein, scaffolding protein, SH2 domain, MAPK, Ras,
Part-4. Cell cycle regulatory protein 5 (Cdk5) A novel target of ERK in Carb induced cell death
 A novel target of ERK in Carb induced cell death") Part-4 Cell cycle regulatory protein 5 (Cdk5) A novel target of ERK in Carb induced cell death 95 1. Introduction The process of replicating DNA and dividing cells can be described as a series of coordinated
Part-4 Cell cycle regulatory protein 5 (Cdk5) A novel target of ERK in Carb induced cell death 95 1. Introduction The process of replicating DNA and dividing cells can be described as a series of coordinated
Chapter 10. Regulatory Strategy
 Chapter 10 Regulatory Strategy Regulation of enzymatic activity: 1. Allosteric Control. Allosteric proteins have a regulatory site(s) and multiple functional sites Activity of proteins is regulated by
Chapter 10 Regulatory Strategy Regulation of enzymatic activity: 1. Allosteric Control. Allosteric proteins have a regulatory site(s) and multiple functional sites Activity of proteins is regulated by
BY: RASAQ NURUDEEN OLAJIDE
 BY: RASAQ NURUDEEN OLAJIDE LECTURE CONTENT INTRODUCTION CITRIC ACID CYCLE (T.C.A) PRODUCTION OF ACETYL CoA REACTIONS OF THE CITIRC ACID CYCLE THE AMPHIBOLIC NATURE OF THE T.C.A CYCLE THE GLYOXYLATE CYCLE
BY: RASAQ NURUDEEN OLAJIDE LECTURE CONTENT INTRODUCTION CITRIC ACID CYCLE (T.C.A) PRODUCTION OF ACETYL CoA REACTIONS OF THE CITIRC ACID CYCLE THE AMPHIBOLIC NATURE OF THE T.C.A CYCLE THE GLYOXYLATE CYCLE
Cancer. The fundamental defect is. unregulated cell division. Properties of Cancerous Cells. Causes of Cancer. Altered growth and proliferation
 Cancer The fundamental defect is unregulated cell division. Properties of Cancerous Cells Altered growth and proliferation Loss of growth factor dependence Loss of contact inhibition Immortalization Alterated
Cancer The fundamental defect is unregulated cell division. Properties of Cancerous Cells Altered growth and proliferation Loss of growth factor dependence Loss of contact inhibition Immortalization Alterated
BIOLOGY 103 Spring 2001 MIDTERM LAB SECTION
 BIOLOGY 103 Spring 2001 MIDTERM NAME KEY LAB SECTION ID# (last four digits of SS#) STUDENT PLEASE READ. Do not put yourself at a disadvantage by revealing the content of this exam to your classmates. Your
BIOLOGY 103 Spring 2001 MIDTERM NAME KEY LAB SECTION ID# (last four digits of SS#) STUDENT PLEASE READ. Do not put yourself at a disadvantage by revealing the content of this exam to your classmates. Your
Ahmad Ulnar. Faisal Nimri ... Dr.Faisal
 24 Ahmad Ulnar Faisal Nimri... Dr.Faisal Fatty Acid Synthesis - Occurs mainly in the Liver (to store excess carbohydrates as triacylglycerols(fat)) and in lactating mammary glands (for the production of
24 Ahmad Ulnar Faisal Nimri... Dr.Faisal Fatty Acid Synthesis - Occurs mainly in the Liver (to store excess carbohydrates as triacylglycerols(fat)) and in lactating mammary glands (for the production of
Principles of Genetics and Molecular Biology
 Cell signaling Dr. Diala Abu-Hassan, DDS, PhD School of Medicine Dr.abuhassand@gmail.com Principles of Genetics and Molecular Biology www.cs.montana.edu Modes of cell signaling Direct interaction of a
Cell signaling Dr. Diala Abu-Hassan, DDS, PhD School of Medicine Dr.abuhassand@gmail.com Principles of Genetics and Molecular Biology www.cs.montana.edu Modes of cell signaling Direct interaction of a