THE EFFECT OF STEARIC ACID ON BREAST CANCER DEVELOPMENT AND PROGRESSION LYNDA MICHELE EVANS
|
|
|
- Anna Miles
- 6 years ago
- Views:
Transcription
1 THE EFFECT OF STEARIC ACID ON BREAST CANCER DEVELOPMENT AND PROGRESSION by LYNDA MICHELE EVANS ROBERT W. HARDY, COMMITTEE CHAIR SUSAN L. BELLIS LISA M. SCHWIEBERT GENE P. SIEGAL DANNY R. WELCH ANNE WOODS A DISSERTATION Submitted to the graduate faculty of The University of Alabama at Birmingham, in partial fulfillment of the requirements for the degree of Doctor of Philosophy BIRMINGHAM, ALABAMA 2009
2 Copyright by Lynda Michele Evans 2009
3 THE EFFECT OF THE SATURATED FATTY ACID STEARATE ON BREAST CANCER DEVELOPMENT AND PROGRESSION LYNDA MICHELE EVANS CELLULAR AND MOLECULAR PHYSIOLOGY ABSTRACT Stearate is an 18-carbon saturated fatty acid that is found in many foods in the western diet including beef and chocolate. Cell culture studies indicate stearate may have various anti-cancer properties including inhibition of cancer cell proliferation and invasion, morphological changes, and induction of apoptosis. Animal studies have found that dietary stearate delays tumor development and decreases tumor incidence. To date, many of the mechanisms underlying these processes are unclear. In this study, evidence is presented showing stearate induces morphological changes in breast cancer cells. Inhibition of de novo diacylglycerol (DAG) generation and subsequent protein kinase C (PKC) activation inhibits stearate-induced cell rounding. Further examination of the individual PKC isozymes with pharmacologic inhibitors indicates that PKC may be directly involved in stearate-induced cell rounding. Similar results were obtained with caspase-3 activity assays where stearate treatment appears to induce apoptosis of breast cancer cells in a manner dependent on DAG and PKC. Stearate induces apoptosis in a time and dose dependent manner through a pathway mediated by both the extrinsic and intrinsic cascades. In vivo, dietary stearate decreases primary tumor size in athymic nude mice injected in the mammary fat pad with MDA-MB-435 breast cancer cells. Stearate also inhibits metastasis to the lungs through a mechanism independent of primary tumor size. Future studies are necessary to elucidate the mechanisms underlying the dietary stearate-induced decrease in primary tumor size and inhibition of metastasis. Taken iii
4 together, these results indicate stearate may be a potential preventative and/or adjuvant therapy for those at high risk for developing breast cancer. iv
5 DEDICATION For my parents, Julie Ann and Keith Dumire Evans it is through your love, compassion, understanding and guidance that I have succeeded. Thank you for never allowing me to quit especially during the times when the bridge seemed to be burning on both ends. Also, thank you to my grandmother, Ruth Ann Dumire Evans, for teaching me the importance of perseverance and faith. The road has been long, but with your love, it has not been lonely. v
6 ACKNOWLEDGEMENTS Without the help and support of many people, this dissertation would not have been possible. Thank you to my mentor, Dr. Robert Hardy. It is through your guidance, patience, and wisdom that I have completed this marathon. Thank you to Dr. Gene Siegal for guidance, advice and help designing and completing multiple experiments. Thank you to my committee members - Dr. Susan Bellis, Dr. Lisa Schwiebert, Dr. Gene Siegal, Dr. Danny Welch, and Dr. Anne Woods for their continuous support and guidance in the development of this project. Thank you to Dr. Arig Ibrahim Hashim, Eric Toline, Ashley Bonner, Patty Lott, and Dehzi Annie Wang for their assistance with the animal studies and immunohistochemistry. Thank to Dr. Stephanie Cowey for completion of the initial cell rounding studies (ECM studies and dose curve), invasion and migration assays, and the tail vein study. Thank you to Yekaterina Khotskaya, Grace Onorato, Catherine Paige, Kara Conlon, Theresa Henson, Erin McCoy, Kristin Hennessy, Kedar Vaidya, Suzanne McAlear, Lakisha Moore, Angelina Orozco, Amy Turk, Ritu Saxena, Edlira Bashari and Andrew Smith for their friendship, guidance and support throughout graduate school. vi
7 TABLE OF CONTENTS Page ABSTRACT... iii DEDICATION...v ACKNOWLEDGEMENTS... vi LIST OF TABLES...x LIST OF FIGURES... xi LIST OF ABBREVIATIONS... xiv CHAPTER 1 INTRODUCTION...1 Development and Progression of Breast Cancer...1 From Normal Breast to Invasive Breast Cancer The Pathology...2 Types of Invasive Breast Cancer...4 From Normal Breast to Invasive Breast Cancer The Molecular and Cellular Biology...4 From Invasive Breast Cancer to Distant Metastasis...7 Apoptosis as a Therapeutic Target...13 Defining Apoptosis...13 Caspases...14 The Extrinsic Cascade...14 The Intrinsic Cascade...20 The Extrinsic Cascade Activating the Intrinsic Cascade...22 Summary...22 Fatty Acids...22 Nomenclature...23 Metabolism...23 Dietary Fat as a Cancer Risk Factor...25 Meta-analyses of Dietary Fat Research...26 Large Multi-center Cohort Studies...26 Accuracy of Dietary Studies...31 Dietary Biomarkers of Fatty Acid Intake...32 Summary of Epidemiological Studies...32 Dietary Fat Studies in Rodent Mammals...33 Fatty Acids and Cancer Cells In Vitro...39 Conclusions Drawn from the Fatty Acid Studies...51 vii
8 Hypothesis, Objectives, and Significance THE EFFECTS OF STEARATE ON CELL MORPHOLOGY: THE INDUCTION OF ELL ROUNDING THROUGH A PROTEIN KINASE C DEPENDENT MECHANISM STEARATE PREFERENTIALLY INDUCES APOPTOSIS IN HUMAN BREAST CANCER CELLS...75 Abstract...76 Introduction...77 Materials and Methods...78 Materials...78 Cell Culture...79 Stearic Acid Preparation...80 Scratch Assay for Cell Migration...80 Cell Invasion Assay...81 Cytotoxicity Assay...81 Immunoblots...82 Caspase-3 Activity Assay...82 Statistical Analysis...83 Results...83 Stearate Inhibits Breast Cancer Cell Invasion and Migration...83 Stearate Preferentially Induces Apoptosis of Human Breast Cancer Cells...83 Stearate Induces Apoptosis through the de novo Synthesis of DAG and the Activation of PKC...85 Discussion...86 Acknowledgements STEARATE INDUCES APOPTOSIS THROUGH A MECHANISM DEPENDENT ON THE INTRINSIC AND EXTRINSIC CASCADES STEARATE REDUCES HUMAN BREAST CANCER METASTASIS BURDEN IN ATHYMIC NUDE MICE Abstract Introduction Materials and Methods Animals and Diets Cancer Cells Experimental Design Mammary Fat Pad Injections Tumor Measurement Tumor Excision viii
9 Necropsy Cell Treatment with Stearate Western Blotting Caspase-3 Activity Statistics Results Diets, Food Intake, and Weight Gain Mammary Tumor Analysis and Metastatic Burden of Low Fat, Safflower, Stearate (A) and Stearate (B) Diets Mammary Tumor Analysis and Metastatic Burden of Stearate (B) Subgroups Stearate Induces Apoptosis of MDA-MB-435 Cells In Vitro Discussion STEARATE REDUCES PRIMARY TUMOR BURDEN IN AN ORTHOTOPIC MODEL OF BREAST CANCER DISCUSSION, SUMMARY, AND CONCLUSIONS Cell Culture Studies Dietary Animal Studies The USDA Food Pyramid Dietary Fat Recommendations of the American Dietetic Association (ADA) and the Dieticians of Cancer (DC) Modulating the Human Diet to Increase Stearate Intake Summary Conclusions LIST OF REFERENCES APPENDIX A SUPPLEMENTAL DATA B The Origin of the MDA-MB-435 Cancer Cells C IACUC APPROVAL FORMS ix
10 LIST OF TABLES Table 1-1 Classification of Common Fatty Acids Composition of the diets x
11 LIST OF FIGURES Figure 1-1 Progression of Breast Cancer: From Normal Breast to Metastatic Disease Summary of TNF -TNFR1 Signaling Summary of Fas-FasL Signaling Summary of TRAIL-Induced Apoptosis Summary of Activation of the Intrinsic Cascade Fatty Acid Nomenclature Metabolism of Fatty Acids Omega-6 and Omega-3 Fatty Acid Metabolism A Current View of Dietary Fatty Acids and the Effect of Individual Fatty Acids on Breast Cancer Progression Stearate Induces Rounding of Human Breast Cancer Cells but Not Non-Cancerous Breast cells Independent of the Extracellular Matrix Inhibition of Mitochondrial -oxidation and de novo DAG Synthesis Partially Reverses Stearate-Induced Cell Rounding Inhibition of Protein Kinase C Partially Reverses Stearate Induced Cell Rounding Stearate Induces Phosphorylation of Multiple Novel and Classical PKC Isozymes Inhibition of PKC but not PKC Partially Reverses Stearate Induced Cell Rounding Stearate Decreases the Metabolic Activity of the Hs578t Breast Cancer Cells Stearate Inhibited Cell Migration and Invasion Stearate Decreased the Viability of the Human Breast Cancer Cells Stearate Induces Cleavage and Activation of Caspase-3 in the Hs578t xi
12 Cells Inhibition of Acyl-CoA Synthetase Inhibited Stearate-Induced caspase-3 Activity at 12 Hours Inhibition of PKC Inhibited Stearate-Induced Caspase-3 Activity at 12 Hours Stearate Induces Cleavage of Caspase-8 and Caspase Inhibition of Caspase-8 and Caspase-9 Reverses Stearate-Induced Caspase-3 Activity Stearate Causes a Decrease in Total Bid Experimental Timetable Food Intake and Weight Gain Tumor Weights and Volumes for the Low fat, Safflower, and Stearate (A) Groups Analysis of Lung Metastases from the Low fat, Safflower and Stearate (A) Groups Tumor Analysis of Animals on the Stearate (B) Diet Stearate Induced Apoptosis of MDA-MB-435 Breast Cancer Cells In Vitro Schematic of Mammary Fat Pad Injection Animals on the Stearate Diet consume Significantly More Food but there Is No Difference in Total Weight Gain Distribution of Plasma Fatty Acids for Animals on the Low Fat, Safflower, and Stearate Diets Tumors from Animals Fed the Stearate Diet are Smaller than Tumors from the Low Fat and Safflower Oil Diets Immunohistochemical Investigation of Tumors from the Three Diets Theoretical Diagram of Stearate-Induced Apoptosis in Human Breast Cancer Cells xii
13 A-1 Inhibition of NF B Reverses Stearate-Induced De-adhesion and Stearate- Induced Caspase-3 Activity A-2 Stearate Induces Cell Rounding in a Dose Dependent Manner A-3 Stearate Induces Translocation of PKC to the Plasma Membrane A-4 Stearate Does Not Prevent Tumors from Growing in the Lungs Following Tail Vein Injection xiii
14 LIST OF ABREVIATIONS ACC ACS ADH AIF ALA ALB acetyl CoA carboxylase acyl CoA Synthetase atypical ductule hyperplasia apoptosis inducing factor atypical lobule type A atypical lobule type B Apaf-1 apoptosis protease activating factor 1 CARD caspase recruitment domain CPT1 carnitine palmitoyal transferase 1 CRD DAG DCIS DcR DD DED DISC DH DHA DMBA DR EGF EGFR cysteine rich domain diacylglycerol ductal carcinoma in situ decoy receptor death domain death effector domain death inducing signaling complex ductal hyperplasia docosahexaenoic acid 7,12-dimethylbenz(a)anthracene death receptor epidermal growth factor epidermal growth factor receptor xiv
15 EndoG EPA ER EPIC FADD FAF-BSA FAS FasL FLIP IAP IBC IKK LCIS LDH MOMP NF B NMU NDGA OPG PARP PI3K PIDD PKC endonuclease G eicosapentaenoic acid estrogen receptor European Prospective Investigation into Nutrition and Cancer Fas associated death domain fatty acid free bovine serum albumin fatty acid synthase Fas ligand FLICE-like inhibitory protein inhibitor of apoptosis invasive breast cancer I B Kinase lobule carcinoma in situ lactate dehydrogenase mitochondrial outer membrane permeability nuclear factor B N-methyl-N-nitrosourea nordihydroguaiaretic acid osteoprotegrin poly-adp-ribosomal polymerase phosphatidyl inositol-3 kinase the p53 inducible protein with a DD protein kinase C xv
16 PR RAIDD RIP SODD tbid TNF TNFR TRADD progesterone receptor RIP-associated ICH-1/CD-3 homologous protein with a death domain receptor interacting kinase silencer of death domain truncated Bid tumor necrosis factor tumor necrosis factor receptor TNF -associated death domain protein TRAF2 TNF receptor associated factor 2 TRAIL TRAILR UL WHEL WHI WINS TNF -related apoptosis-inducing ligand TNF -related apoptosis-inducing ligand receptor unfolded lobules Women s Healthy Eating and Living Women s Health Initiative Women s Intervention Nutrition Study xvi
17 1 CHAPTER 1 INTRODUCTION In 2008 there were an estimated 182,640 new cases of breast cancer diagnosed in females in the United States and an estimated 40,480 deaths from breast cancer. While patients diagnosed at earlier stages show a positive prognosis and survival rate, those diagnosed after metastasis have an estimated 27% 5 year survival rate. (1). Clearly new therapies are needed to treat distant metastases. The purpose of this dissertation is to investigate the role of the saturated fatty acid stearic acid (stearate) as a potential preventative and/or therapeutic for breast cancer patients. Development and Progression of Breast Cancer Cancer is generally acknowledged to be a genetic disease centered on inappropriate and uncontrolled cell division. However, this definition does not fully define cancer. Many benign tumors form in the human body that are not and do not become cancerous. In order for a non-hematologic tumor to be considered malignant, it generally must express uncontrolled cell growth, degrade and invade through the basement membrane, and, in many cases, have the potential to grow at a distant organ. Cancer is now thought to be a developmental disease where cells lose their organ specificity and the necessity to die should they move away from that tissue (2). The development and progression of breast cancer results from distinct pathological changes in the breast tissue governed by molecular and cellular changes. A brief review of the
18 2 development of invasive breast cancer and progression to metastatic disease is found below. From Normal Breast to Invasive Breast Cancer - The Pathology In 1975, Wellings, Jensen, and Marcum published a study in which they examined and characterized the pathologic features of the breast thought to be precancerous legions that gave rise to invasive breast cancer. The female human breast is composed of a series of ducts that extend out from the nipple. A large duct becomes branched into an extralobular terminal duct (ETD) that leads into an intralobular terminal duct (ITD). The ITD is further branched into ductules. Together, the ITD and ductules comprise a lobule. Terminal duct lobular units (TDLU), the hypothesized region that the majority of breast cancer arise from, are composed of the lobule and ETD. (3). The ductules are composed of two cell layers an inner epithelial layer that is responsible for the production of milk proteins and an outer myoepithelial layer that is involved in milk ejaculation. (4,5). According to the Wellings-Jensen model of breast cancer development, the majority of breast cancers will arise in the ducts or lobules of patients. They observed two types of atypical lobules they hypothesized give rise to breast cancer designated as type A (ALA) and type B (ALB). ALA were more common, with many observed in the breast. They consisted of ductules that were fewer in number but larger than normal forms and had hyperplastic epithelial layers. ALB, on the other hand consisted of enlarged lobules with large, poorly defined ductules. It was hypothesized that ALA leads to ductal carcinoma in situ (DCIS) whereas ALB cause lobular carcinoma in situ (LCIS).
19 3 (3). Today, ALA are known as unfolded lobules (UL) and are distinct from normal lobules in that they are histologically larger than normal lobules because of abnormal proliferation of epithelial cells lining the ductules. (6) This is also generally known as ductal hyperplasia (DH) (7). The ULs give rise to atypical ductal hyperplasia (ADH) which can progress to DCIS. (6). It is estimated approximately 75% of invasive breast cancers arise from this route (8). Whereas ALA and ADH arise within the ductal network, ALB leads to atypical lobule hyperplasia which is characterized by abnormal epithelial cells filling the ducts and ductules. If the areas are largely distended by uniform but not hyperproliferative epithelial cells, this is known as a LCIS. LCIS can give rise to either invasive lobular carcinoma or invasive ductal carcinoma (6). Not every region of abnormal proliferation in the breast will give rise to invasive cancer. However, it has been found that women with DH, ADH and DCIS in their breast have a progressively increasing risk of developing invasive breast cancer, 2-fold, 5-fold and 10-fold, respectively. Furthermore, it is more likely that an invasive breast cancer (IBC) will be located adjacent to an area of ADH or DCIS. Genetic studies have shown that IBC has a tendency to develop loss of heterozygosity (LOH) at specific genomic alleles. The same regions of LOH are found in 50 and 80% of the ADH and DCIS, respectively, of a patient with breast cancer. (7). Interestingly, some TDLUs share the same LOH as an adjacent IBC, whereas TDLUs further out do not. This indicates that some of the mutations leading to the development of IBC may occur in normal looking epithelia. (9). Taken together these results show that breast cancer is a progressive disease that requires the accumulation of multiple pathological and molecular abnormalities before it
20 4 is characterized as IBC. Theoretically, the changes in the genome will mirror changes in the pathology of the breast. Types of Invasive Breast Cancer Invasive breast cancer is a very heterogeneous disease, with 5 common subtypes defended by gene array expression patterns luminal A, luminal B, Her2+, basal-like (triple negative), and unclassified/normal breast like. The luminal classes have gene expression profiles similar to the epithelial cells lining the lumens, where the luminal A are estrogen receptor (ER)+ and/or progesterone receptor (PR)+ and Her2- and the luminal B are ER+ and/or PR+ and Her2+. The basal-like breast cancers, also known as triple negative breast cancer, have gene profiles similar to the basal epithelial cells and are ER-/PR-/Her2- and cytokeratin 5/6+. The Her2+ cells lack ER and PR expression but are Her2/ERBB2+ and the unclassified are negative for all markers. (10, 11). Generally speaking, the luminal A have the best survival rates, followed by normal breast-like, and luminal B. Her2 + and the basal-like have the worst prognosis (12). From Normal Breast to Invasive Breast Cancer The Molecular and Cellular Biology As mentioned above, breast cancer is a very heterogeneous disease and can arise from a number of different pathways of genomic mutations. But, generally speaking, several different classes of genes control the transition from normal to malignant breast. Interestingly, the majority of mutations appear to occur in the DCIS and IBC stages, not the normal or ADH breast tissue. Allelic imbalance for ADH, DCIS, and IBC were <5%,
21 5 20% and 25%, respectively. These results suggest that cells genomes become instable during DCIS. (13). Among the first mutations associated with progression from DH to ADH include up-regulation of Her2 and increases susceptibility to p53 mutations (7). Her2 is a member of the EGFR family of tyrosine kinases that can stimulate proliferation in normal cells. p53 is a tumor suppressor known as the guardian of the genome because it is activated upon DNA damage to the cell. When p53 functions properly, it will arrest the cell cycle and either allow for repair of the damaged DNA or induce apoptosis (7). As ADH becomes DCIS, more mutations become apparent, especially those involved in cell cycle regulation. Two proteins, cyclin D and cyclin E are associated with controlling the G1/S checkpoint (7). Cyclins are a family of proteins that bind and activate cyclin dependent kinases (CDKs). Active CDKs play a role in promoting the cell cycle phases. If there is DNA damage, a family of proteins known as cyclin dependent kinase inhibitors (CKI) bind to the CDKs and induce cell cycle arrest. To overcome inhibition by CKI, abnormal cells generally either overexpress cyclins (72% of DCIS overexpress cyclin D1) or they inactivate the CKIs. There is also an increase in p53 mutations, which can cause a decrease in thrombospondin, thereby promoting angiogenesis, and an increase in MDR-1, which promotes drug resistance. (7). Consistent with the increase in cyclins, DCIS have a greater proliferative rate than ADH. Interestingly, there is also an increase in the apoptotic rate. This increase in apoptotic rate also increases as the DCIS become higher grade (14). This is generally thought to be attributed to a decrease in the anti-apoptotic protein Bcl-2 - it is expressed in 96% of cells in the normal epithelium, 69% in DCIS, and 45% in IBC. Although it
22 6 seems counter-intuitive, p53 promotes cell apoptosis partially through the downregulation of Bcl-2. Therefore, in DCIS, where the rate of p53 mutations resulting in increased proliferation and/or decreased apoptosis is greater, there is a decrease in Bcl-2 expression (7). Oncogenes are generally defined as genes that increase cellular proliferation and decrease differentiation. These genes are activated through a number of different mechanisms including gene mutation, amplification, or rearrangement. (7). Generally speaking, two oncogenes have been shown to play a significant role in the development of IBC from DCIS. Ras is a widely studied oncogene that is known to control cell proliferation, transformation, and migration, among other functions. Ras is widely expressed in the human breast although activity levels are generally low in normal breast tissue, DH, AHD and DCIS. However, IBCs have high levels of ras activity. When there are mutations in ras, this tends to make the IBCs more aggressive. The overexpression and mutations associated with ras occur late in tumor progression, suggesting that it is not involved in initiation of the cancer cells, but rather in progression. c-myc has also been shown to be upregulated in 4-6% of DCIS but 16% of IBC and is weakly associated with lymph node metastasis. Once again, it is thought c-myc is involved in breast cancer progression not initiation. (7). Although the genes and proteins listed above are nowhere near the total number known to play a role in the development of invasive breast cancer, they offer a glimpse into understanding this complex process.
23 7 From Invasive Breast Cancer to Distant Metastases To date, there are generally 6 recognized steps of the hematogenous metastatic cascade (as determined by transport through the vasculature). These steps include invasion from the primary tumor, intravasation into the blood vessels, survival in the blood stream, extravasation out of the vessel, colonization of the secondary site, and proliferation at the secondary site (15, 16). For the purposes of this dissertation, the term metastasis is defined by the criteria and explanation put forth by Welch (17). More specifically, a metastasis is a growth of cells outside of the vasculature at a distant and/or discontinuous site from the primary tumor. (17). As tumors are generally recognized to be a heterogeneous growth of cancer cells, it is worth noting that not all cells within a tumor are metastatic. With that being said, before a cell metastasizes it must acquire several different features to promote and facilitate its survival and growth at a secondary site. (18). Although most models support metastatic capability as being a late stage acquisition, recent arguments have been made stating that tumor cells acquire the mutations necessary to metastasize early in tumorigenesis. Along with this thought, it was suggested that the genes necessary for metastasis to occur must have a beneficial role in primary tumor development. (19). This argument, if accurate, questions several previously established ideas of metastatic development. First, it would argue that some tumors are highly aggressive from the start and have an increased propensity to metastasize, second that metastasis-specific genes are a fallacy, and finally that small tumors can shed cells that will become metastases. (19).
24 8 In general, these hypotheses do not support the majority of the literature. In the clinics, the size of the tumor correlates to the metastatic potential of the tumor, including breast tumors (20-22). Furthermore, to date, over 20 genes have been identified that inhibit the metastatic cascade without affecting the primary tumor growth (22, 23). Finally, the idea that a metastatic phenotype exists solely in the cancer cell fails to acknowledge the role of a tumor s microenvironment in progression and metastasis (22). Although the extracellular matrix (ECM) and tumor microenvironment play a role in cancer cell division, they also play a large role in the first step of the metastatic cascade invasion from the primary tumor. Invasion from the primary tumor: the extracellular matrix and breast cancer cells. As mentioned previously, abnormal growth is not the only criteria necessary to be considered a malignant cell. In solid tumors, the cancer cells must invade through the basement membrane. The ECM and stromal cells surrounding the tumor cell do not display characteristics of normal ECM and stromal cells (2, 22). In a normal, developing embryo, a crosstalk exists between the ECM and epithelial cells to ensure tissues and organs develop appropriately. As an organism grows, the ECM under the epithelial cells will grow thin, exerting forces on the epithelial cells (2). These forces induce physical stresses that sensitize the cells to growth factors thereby promoting cell division. As cell division occurs, a new basement membrane is laid down under the proliferating cells and the organ develops normally with appropriate layers of epithelial cells and appropriate thickness of the ECM. In tumor formation,
25 9 however, the cells grow without the exertion of external forces. As a result, the epithelia become crowded and new ECM is not deposited. (2). During tumorigenesis, the ECM, in addition to the premalignant cells, is affected (2). Due to the abnormal axis mentioned above, the ECM disruption causes gaps and regions that are thicker than others in the basement membrane. This, in turn, affects cell to cell adhesions in addition to cell to connective tissue interactions. As these connections are lost, the cells are at a greater risk of becoming anchorage independent due a decrease in adhesion to the ECM and loss of distortions of the cytoskeleton that induce regulated proliferation. (2). In this manner, the ECM can promote a malignant phenotype. In order to invade through the basement membrane, cancer cells must break down the ECM and invade through the disrupted tissue (22). To break down ECM, it is generally accepted that the cancer cells increase their production of matrix metalloproteases (24). However, stromal cells also express and secrete proteases and some stromal cell-only proteases serve as prognostic factors in cancer progression (22). Intravasation. Once a metastatic cell invades the basement membrane, its next role is to intravasate into a blood vessel. This is generally accepted to be the rate limiting step in metastasis. (22). Using a metastatic and non-metastatic line derived from a rat mammary adenocarcinoma, Wyckoff et al. demonstrated that metastatic cells are more likely to move towards blood vessels in vivo and that non-metastatic cells were less likely to survive in the blood stream (25). However, to date the exact mechanisms underlying intravasation are poorly understood. It has been hypothesized that the process may be
26 10 mediated by endothelial cell adhesion molecules that can interact with the tumor cells (26). There is also evidence that process may be mediated or aided by macrophages (27). Survival in the blood stream. Once the breast cancer cells get into the blood stream, it is estimated that less than 0.01% of them will survive to form distant metastases. This low survival rate is generally attributed to cell death due to shear forces of the blood stream or detection by the immune system. (28). In order to survive, it is hypothesized that the tumor cells complex with platelets and leukocytes. The role of the leukocytes is not well understood, but it is thought the platelets shield the tumor cells from immune cells. Consistent with this hypothesis, decreasing platelet number decreases the number of metastases that form in vivo. Furthermore, the anti-coagulant heparin also decreases the number of metastases. (29). Adhesion of the platelets to the cancer cells may occur by several mechanisms. First, some cancer cells express high levels of a platelet aggregating factor known as Aggrus (28). Second, the tumor cells have sialylated fucosylated mucins, known to bind to the P-selectins expressed on the platelets. When P-selectin is inhibited, tumor cell survival in the circulation decreases. (29). Extravasation. The mechanism by which tumor cells extravasate from the blood stream is very similar to that used by leukocytes. More specifically, the tumor cell weakly adheres to the endothelium and through these weak interactions, rolls along the endothelium until it makes stronger cell contacts (30). Once those contacts are made, the tumor cell migrates through the vasculature and then adheres to the subendothelium and induces remodeling of the stromal cells to repair the surrounding vessel. Finally, the tumor cell
27 11 colonizes the secondary site (30). Some of the tumor cells undergo non-specific weak adhesion to endothelial cells following arrest in capillaries whereas other tumor cells begin to proliferate before they exit the vasculature. However, there is some evidence that tumor cells can selectively attach to the vasculature of specific organs through sitespecific receptors. This survives as one mechanism by which tumor cells can home to an organ. (15). Growth at the secondary site and organotropism. Once the tumor cells exit the blood vessel, they must arrest and grow at the secondary site if they are to successfully form a metastasis. The process is very inefficient. The majority of the cells that survive in the blood stream will not form metastases. (15). This is thought to be due to the inability of the cancer cell to survive at the secondary organ. In 1889, Stephen Paget hypothesized that growth of metastases is dependent on compatibility of a tumor cell (the seed) with the secondary organ (the soil). Since that time, numerous studies have been performed to support this hypothesis and it is now well accepted. (31). Before a tumor cell arrives at a potential secondary metastatic site, there is some evidence that the tumor cell has prepared the site for metastasis. Termed the premetastatic niche, the mechanisms underlying this phenomenon are still poorly understood. It has been shown that cancer cells secrete factors that stimulate the fibroblasts at a secondary site to secrete fibronectin to which haematopoietic bone marrow progenitor cells (HPCs) bind. (22, 32, 33). These cells are thought to prepare the so-called soil for the tumor cells. This includes recruitment of VEGFR-1+ HPCs that
28 12 aid in angiogenesis of the new tumor and cancer cells. Inhibition of the VEGFR-1+ HPCs prevents formation of the niche and inhibits metastasis. (32-34). As has been previously alluded to, cancer metastasis is not a random process. The majority of breast cancer cases will metastasize to the bone, liver, or lungs. The specificity, or so-called organotropism, of the breast cancer cells to metastasize to distinct organs is thought to be due to differences in terms of expression of integrins and other receptors. This hypothesis that individual cancer cells can home to a particular organ also implies that the individual metastatic cancer cells with the properties described above are heterogeneous in other means. (16). Consistent with this thought, breast cancer cells have been isolated in vivo that home to either the bone or to the brain. Depending on their secondary site preference, these tumor cells have differences in expression of many metabolic enzymes. (35). Overall, this suggests that many layers of heterogeneity exist within any given tumor metastatic cells vs. non-metastatic cells, cells that metastasize to one organ vs. cells that metastasize to another. Apoptosis as a Therapeutic Target Apoptosis is a form of programmed cell death that does not elicit an immune response in patients. Also known as cell suicide, many current chemotherapeutics target the apoptotic cascade to induce cancer cell death (36). A compound, such as TRAIL (see below), that induces apoptosis of cancer cells but not normal cells would be optimal for cancer treatment.
29 13 Defining Apoptosis Attempting to define the cell death pathways is very difficult given that the pathways overlap and many of the morphological and biochemical changes observed in a dying cell can also occur in a living, healthy cell that does not die. To date, the term apoptosis is used to define certain morphological changes associated with a specific type of cell death. These changes include cell rounding, retraction of pseudopods, nuclear fractionation, and in late stages, membrane blebbing. Apoptosis is generally associated with cell death due to caspase activation. However, inhibition of the caspases does not necessarily prevent cell death. Instead the cells resort to another form of cell death resembling either necrosis or a mix between necrosis and apoptosis. (37). For simplicity, this review will discuss apoptosis as it relates to caspase activation. Caspases Cysteinyl-aspartic-acid-proteases, known for short as caspases, are a family of cysteine proteases that cleave the C-terminal peptide bond of aspartic acid residues. (38). To date, 14 caspases have been discovered seven of which are involved in the apoptotic cascade (the remaining seven are involved in inflammation). Caspases are translated as proenzymes that are activated by proteolytic cleavage induced by either cell signaling or other caspases. (36). The apoptotic caspases are divided into two classes initiator and executioner caspases. The initiator caspases have a long pro-domain that contains either a death effecter domain (DED) as is the case for caspase-8 (also known as FLICE) and caspase- 10, or they contain a caspase recruitment domain (CARD) as is seen in caspase-2 and
30 14 caspase-9. (36, 39). These protein domains induce homophilic interactions between the pro-caspases and their adaptor proteins leading to the autolytic cleavage and activation of the initiator caspases. (38). Once the initiator caspases are active, they sequester, cleave and activate the executioner caspases caspase-3, caspase-6, caspase-7. Interestingly, the executioner caspases lack a known DED or CARD sequence. (36). Many of the socalled death receptors and adaptor molecules also contain another domain known as the death domain (DD). It works in a manner similar to the DED and CARD domains the DD between two proteins bind each other (40). To date, there are four known apoptotic mechanisms - the extrinsic pathway, the intrinsic pathway, the extrinsic pathway activating the intrinsic pathway, and the endoplasmic reticulum pathway. Whereas much is known about the first three, the endoplasmic reticulum pathway is not well understood and will not be covered extensively in this portion of the introduction. Brief reviews of the extrinsic and intrinsic pathways are found below. The Extrinsic Cascade The extrinsic cascade is controlled by members of the tumor necrosis factor (TNF) and tumor necrosis factor receptor (TNFR) superfamilies. The main ligand/receptor combinations involved in apoptotic cell death are TNF and TNFR1, Fas (CD95) and Fas ligand (FasL), and TRAIL and death receptor (DR) 4 and DR5. TNF can also activate TNFR-2, but this receptor is mainly expressed in the immune system and will not be discussed in detail here. (39). The TNF ligands are a family of mostly type II membrane proteins (although FasL and TNF exist in a soluble form as well as a
31 15 membrane bound form). The TNFR superfamily are type I membrane proteins characterized by a conserved cysteine in the extracellular ligand binding domain in addition to several cysteine rich domains (CRD). Upon activation of the death receptors by ligand binding, receptors interact between the CRDs to form trimeric or multimeric complexes linked by disulfide bonds (40,41). Upon receptor activation, various adaptor molecules bind to allow activation of the initiator caspases, caspase-8 or caspase-10. TNF -TNFR1. Unlike the FasL and TRAIL pathways that will be discussed below, the TNF -TNFR1 axis generally results in a pro-survival signal (40). Although possible, it is rare that this complex induces death. When TNFR1 is in its ligand-free, inactive form, a silencer of death domain (SODD) prevents the receptor from recruiting its adaptor molecules. Upon activation of the receptor by ligand binding, the SODD is released, exposing TNFR1 s DD (Figure 1-2). TNF -associated death domain protein (TRADD) binds to the TNFR1 DD and recruits two proteins. (39). The first protein is known as receptor interacting kinase, or RIP. RIP binds to TRADD through a DD on the C terminal of TRADD. Another protein, TNF receptor associated factor 2 (TRAF2), binds to the N-terminal DD of TRADD (40). The TNFR1-TRADD-TRAF2 complex can recruit I B Kinase (IKK) and induce activation of the classical nuclear factor B (NF B) cascade, resulting in a strong prosurvival signal through the transcription of many anti-apoptotic molecules. Taken together, this complex is generally referred to as Complex I and is a membrane bound complex. If the NF B cascade is inhibited, a different function of the TNF -TNFR1 axis is induced. Upon the inability to activate NF B, an adaptor protein known as Fas
32 16 associated death domain (FADD) will be recruited TRADD and RIP1 that have dissociated from TNFR1. This cytosolic complex, also known as Complex II, can recruit caspase-8 and induce apoptosis. Interestingly, if NF B is active, Complex II also consists of the caspase-8 inhibitor FLIP (FLICE-like inhibitory proteins). Cellular FLIP proteins have DED domains that bind FADD to prevent the binding of caspase-8. When NF B is not active, caspase-8 can cleave executioner caspases such as caspase-3 and induce cell death. (39, 42). In 1997, another adaptor molecule associated with apoptosis was discovered. Named RAIDD (RIP-associated ICH-1/CD-3 homologous protein with a death domain), this protein was thought to complex with RIP1 directly and subsequently TRADD and TNFR1 to induce cell death. (43). It was suggested this complex can then recruit and activate caspase-2 which will induce apoptosis (44). It appears, however, that RAIDD does not play a large role in TNFR1-mediated apoptosis as mice without caspase-2 or RIP1 do not show a defect in TNF induced-cell death (45). RIP1 and RAIDD can also induce apoptosis by forming a complex known as the PIDDosome. Upon genetoxic stress, the p53 inducible protein with a DD (PIDD) causes the adaptor molecule RAIDD to interact with it through a DD. RAIDD then recruits caspase-2 via its CARD domain. Caspase-2 is then presumably activated to induce apoptosis. Interestingly, the PIDDosome appears to induce apoptosis independent of caspase-3. (46). To date, the mechanism underlying the PIDDosome s ability to induce caspase-2 activity is unknown (47).
33 17 FasL-Fas. The FasL-Fas death receptor mediated pathway was the first to be discovered and described. Upon binding of the ligand to the receptor, Fas undergoes a trimerization and internalization through an endosomal pathway. This recruits the adaptor molecule FADD (Fas associated protein with a death domain) which interacts with the receptor through a DD domain (Figure 1-3) (39, 40). FADD also contains a DED domain that recruits pro-caspase-8. This complex of FasL-Fas-FADD-caspase-8 is known as the death inducing signaling complex (DISC). Two molecules of caspase-8 are recruited to the complex and their proximity allows for the auto-cleavage and activation of caspase-8. In the so-called type I cells, caspase-8 cleaves and activates caspase-3 which in turn executes apoptosis (39). Interestingly, another initiator caspase known as caspase-10 can also be recruited to the DISC. Using an entirely in vitro assay, cleaved, active caspase-10 can cleave and activate the executioner caspases, whereas the executioner caspases cannot activate caspase-10, suggesting it is an initiator caspase. Furthermore, structurally caspase-10 is very similar to caspase-8. (44). In vivo, however, caspase-10 cannot replace the action of caspase-8 in inducing apoptosis (39). Although both caspase-8 and caspase-10 are ubiquitously expressed, animals with a knockout of caspase-8 do not survive past embryonic day 12.5 (48). The role of caspase-10 at the DISC is currently unknown. (39). Regulation of the FasL-Fas mediated apoptotic pathway occurs in several different ways. First, FLIP can be associated with the DISC to inhibit caspase-8 activation as in TNF -TNFR1 signaling. Furthermore, FLIP association can induce activation of NF B and result in a pro-survival signal. (39).
34 18 The second mechanism by which this pathway can be regulated is through the expression of a FasL decoy receptor (DcR). DcR3 has been shown to bind FasL with the same affinity as Fas, thereby acting as a competitive inhibitor (49). Because DcR3 lacks the transmembrane domain found in Fas, it is secreted from the cell. Interestingly, it has been proposed that overexpression of DcR3 in human cancers could lead to apoptotic resistance (50). One study examined the level of serum DcR3 receptors in normal and cancer patients and found the levels of DcR3 were significantly higher in the cancer patients. Although receptor expression strongly correlated with cancers of the gastrointestinal tract, one breast cancer patient out of 5 had elevated DcR3 levels. The authors stated 20% of breast cancer patients have increased DcR3 levels, however, analysis of more patient samples is necessary to draw a definitive conclusion. (51). TRAIL-DR4/DR5. TNF and FasL both have the ability to kill cancer cells. However, the use of these signaling pathways as a cancer therapeutic is currently not possible. Treatment of patients with TNF results in severe systemic toxicity, similar to sepsis, thought to be due to the strong pro-inflammatory effect of the ligand. Treatment of mice with a Fas-activating antibody resulted in lethal liver damage due to the induction of apoptosis in hepatocytes. (52,49). In the attempts to find a ligand less toxic than TNF and FasL, a third ligand with sequence homology to the other two was cloned and identified. Known as TNF -related apoptosis-inducing ligand (TRAIL), this ligand and its downstream apoptotic pathway shows promise as a potential chemotherapeutic as it induces apoptosis of tumor cells but not non-cancer cells. (52).
35 19 To date, five receptors have been identified that bind TRAIL. Death receptor 4 (DR4/ TRAIL-R1) and death receptor 5 (DR5/ TRAILR-2) induce apoptosis upon binding TRAIL (Figure 1-4). On the other hand, the remaining three receptors, decoy receptor 1 (DcR1/ TRAIL-R3), decoy receptor 2 (DcR2/TRAIL-R4), and osteoprotegrin (OPG) do not induce apoptosis upon ligand binding. DcR1 and DcR2 lack the cytoplasmic death domain found on DR4 and DR5. Conversely, while OPG can bind TRAIL, it has a very low affinity for the ligand in physiological settings. (39). When TRAIL binds its receptors, it does so as a homotrimer. The binding of TRAIL to its receptor induces a trimerization of the receptor. This activates the receptor, inducing the recruitment of FADD, as in Fas signaling. Once FADD binds to the DR4 or DR5, it can recruit pro-caspase-8, forming the DISC. Cleavage of caspase-8 and subsequent activation of caspase-3 will lead to cell death. Also, as in the apoptotic signaling activated by TNF and FasL, FLIP can be recruited to the complex and inhibit caspase-8 activation. (39, 52). When TRAIL was first discovered and shown to have a low toxicity for normal tissues but a high toxicity for cancerous tissues, it was thought that the normal cells must have high levels of decoy receptors whereas the cancer cells do not. This was found to be false, and, in fact, decoy receptor expression does not correlate to TRAIL sensitivity. (39). Currently, two antibodies designed to activate DR4 and DR5 are in clinical trials. Mapatumumab is a human monoclonal antibody currently in clinical trials that activates DR4. In phase-i clinical trials, the antibody was very well tolerated. Although no objective responses were noted for the anti-tumor activity of the patients treated, in one study of 49 patients, two of the patients who had progressive solid malignancy at the
36 20 start of the treatment experienced 9 months of stable disease whereas in another study of 41 patients, 12 experienced stable disease lasting for 1.9 to 29.4 months. (53). A recent phase 2 trial consisting of 29 patients with non-small cell lung cancer found the patients tolerated the antibody well and 29% had stable disease (54). Lexatumumab is another human monoclonal antibody targeted to DR5 that is currently in clinical development. As with mapatumumab, lexatumumab is generally well tolerated by patients. Of the 37 patients that received the treatment, twelve developed stable disease, 3 of whom had metastatic sarcoma. (55). The Intrinsic Cascade The intrinsic apoptotic cascade is activated by cellular stress such as UV radiation, DNA damaged, -radiation, viral factors, and chemotherapeutic agents (Figure 1-5; 56). Rather than being activated by a death receptor, as is the case with the extrinsic cascade, the intrinsic cascade is activated by mitochondrial outer membrane permeabilization (MOMP). MOMP is controlled by a family of proteins known as Bcl-2 proteins. To date, 20 members have been discovered and are divided into three classes based on their function and the number of homology domains they contain. The antiapoptotic proteins include Bcl-2, Bcl-Xl, Bcl-w, A1, and Mcl-1. These proteins promote cell survival by inhibiting a class of pro-apoptotic proteins known as the BH123 family. The BH123 group consists of the proteins Bax, Bak and Bok and they are responsible for permeabilizing the outer mitochondrial membrane. The so-called BH-3 only proapoptotic proteins include Bid, Bad, and Bim among others. The BH-3 only proteins trigger apoptosis by two mechanisms the first is to induce oligomerization of the
37 21 BH123 proteins and the second is to competitively bind the proteins, subsequently inducing release the BH123 proteins. (39). Once the BH123 proteins, specifically Bax and Bak, induce MOMP, water rushes into the mitochondria, disrupting the ion gradients and, consequently, mitochondrial membrane potential (39). Once this occurs, the mitochondrial outer membrane will rupture and release mitochondrial proteins into the cytosol. One of these proteins, cytochrome C, will bind to Apoptosis Protease Activating Factor-1 (Apaf-1). (56). When this binding occurs, an oligermization domain is uncovered. Oligermization of multiple Apaf-1/cytochrome C complexes forms a high molecular weight protein complex known as the apoptosome. The apoptosome, in turn, recruits pro-caspase-9 via a CARD domain. This induces cleavage and activation of the initiator caspase-9 which can cleave and activate executioners such as caspase-3 to induce cell death. (38). In addition to cytochrome C, several other mitochondrial proteins that are released into the cytoplasm are required to fully activate apoptosis. One of these proteins, SMAC/DIABLO, binds to and inhibits the Inhibitor of Apoptosis Proteins (IAPs). (56). Under normal circumstances, IAPs bind to cleaved caspases and prevent cleavage of cellular targets. When SMAC/DIABLO is released, it competitively binds the IAPs so that the active caspases can be released. (39). Other proteins, such as Apoptosis Inducing Factor (AIF) and Endonuclease G (EndoG) are also released into the cytoplasm. Interestingly, these proteins can directly induce chromatin condensation and proteolytic cleavage of the DNA. (38). These two proteins also may play a role in caspaseindependent cell death (57).
38 22 The Extrinsic Cascade Activating the Intrinsic Cascade In type I cells, activation of caspase-8 is sufficient to induce cell death. However, in the so-called type II cells, a positive feedback loop between the extrinsic and intrinsic cascades is required to induce cell death. The BH-3 protein Bid is a target of caspase-8. Upon cleavage, the truncated Bid (tbid) translocates to the mitochondria where it aids in MOMP. (48). It is thought the mitochondrial receptor for tbid is a phospholipid known as cardiolipin (56). Interestingly, cardiolipin sequesters cytochrome C to the inner mitochondrial space. The mechanism by which tbid can bind to cardiolipin is not known, but is thought to be due, in part, to constant remodeling of the mitochondrial membranes. (58). Summary of Apoptosis Apoptosis is a form of cell death controlled by a family of proteases known as caspases. The caspases can be activated by several mechanisms an extrinsic cascade that is dependent on death receptors and ligands, an intrinsic cascade involving the mitochondria, and a combination of the two cascades. Apoptosis is a promising cancer target as cells that undergo apoptosis do not illicit an immune response. Ideally, a potential chemotherapeutic would activate apoptosis specifically in cancer cells and not effect the surrounding normal cells. Fatty Acids Dietary fat has been associated with numerous diseases and conditions including cancer, diabetes, atherosclerosis, high cholesterol, and coronary heart disease (59-62).
39 23 However, the in vitro and in vivo studies suggest these pathologies are dependent on not only the amount of fat, but also the concentrations of the individual fatty acids in the diet. This section talks about how fatty acids are named and briefly discusses the metabolism of classes of fatty acids. Nomenclature Long chain fatty acids are carboxylic acids composed of a carboxyl head group followed by a long hydrocarbon chain (Figure 1-6A; 63). Saturated fatty acids such as palmitate (C16:0) and stearate (C18:0) have no double bonds along their hydrocarbon chains whereas unsaturated fatty acids have at least one double bond (Figure 1-6B). Unsaturated fatty acids are further characterized by the number and location of the double bonds. Unsaturated fatty acids can either be monounsaturated meaning they have one double bond or polyunsaturated meaning there are multiple double bonds. The final carbon on the hydrocarbon chain is known as the omega carbon. The number of carbons between the omega carbon and the first double bond determines the type of the unsaturated fatty acid. For example, the monounsaturated fatty acid oleate (C18:1) has its first double bond 9 carbons from the omega carbon and therefore is an omega-9 fatty acid. Linoleate (C18:2) is a polyunsaturated fatty acid whose first double bond is 6 carbons from the omega carbon and is therefore an omega-6 fatty acid. (63). Metabolism In biological systems, fatty acids serve several major roles they are a major component of the phospholipids and glycolipids, they are precursors to eicosanoids
40 24 (hormone-like lipid molecules), they can be esterified to form diacylglycerol, a 2 nd messenger signaling molecule, or triacylglycerols, energy stores used in times of famine and physiological stress, and they can affect protein function by covalently binding to the amino acid chain (acylation) (64). Fatty acids are either made endogenously or consumed in food. Humans lack the ability to make two fatty acids essential to normal physiological functions linoleate and linolenate (C18:3) and therefore must be consumed through the diet. Synthesis of fatty acids occurs when carbohydrate levels are high and fatty acid levels are low. The process is controlled by two enzymes acetyl CoA carboxylase (ACC) and fatty acid synthase (FAS). ACC converts acetyl CoA to malonyl CoA, one of the basic building blocks used by FAS to make fatty acids. (65). Approximately 80% of the fatty acids produced by FAS are palmitate, whereas stearate and myristate comprise 10% each (66). On the other hand, degradation of fatty acids occurs in the mitochondria and is known as mitochondrial -oxidation. -oxidation takes fatty acids and breaks them down into acetyl-coas making it essentially the reverse of fatty acid synthesis. The carnitine palmistry transferase 1 (CPT1) is mitochondrial outer membrane protein that, along with CPT2 on the inner mitochondrial membrane, transports fatty acids into the mitochondrial lumen. (67). CPT1 is inhibited by malonyl CoA whereas ACC is inhibited by insulin ensuring that the synthesis and degradation processes do not occur at the same time. (64). Figure 1-7 is a schematic of the fatty acid metabolism of a saturated fatty acid such as stearate. When stearate enters the cell (or is synthesized) it encounters an enzyme known as acyl-coa synthetase (ACS) (68). ACS converts hydrophobic fatty acids into
41 25 their hydrophilic Co-A derivatives. These hydrophilic molecules are the basic building blocks of phospholipids, phosphatidylinositides, acylglycerols, and sphingomyelins. This occurs through the formation of precursors such as lysophatidate (lysophatidic acid) and phosphatidate (phosphatidic acid). Several of these pathways will be investigated in chapters 2 and 3. These include de novo diacylglycerol synthesis (from phosphatidate), -oxidation, and de novo ceramide synthesis (from sphingosine). The omega-3 and omega-6 fatty acids follow a metabolism similar to palmitate s. However, they can also be used to synthesize eicosanoids, as depicted in Figure 1-8 (69). These molecules are discussed in greater detail later in the introduction and in chapter 5. Dietary Fat as a Cancer Risk Factor The role of dietary fat and breast cancer has remained a controversial area of research for over 65 years. Although animal and in vitro studies show a clear affect of fatty acids on breast cancer development and progression, human studies remain indecisive (59). In 1976, Carroll showed the average fat intake of a country was correlated to its mortality rate for breast cancer. Furthermore, he showed a positive correlation between breast cancer and fat intake derived from animal sources but no correlation for fat derived from vegetable sources. (70). Although the study did not control for cultural variables such as exercise, life span, or sun exposure, these results overall suggest the possibility that not only the amount of fat in the diet, but also the source of the fat may affect breast cancer development. Although saturated fatty acids are generally associated with an increased breast cancer risk, recent research suggests that individual saturated fatty acids are metabolized
42 26 differently in vivo. As a result, evidence is mounting that certain saturated fatty acids such as stearate and butyrate (C4:0) may have anti-cancer properties (71-77). Based on known epidemiological, in vivo, and in vitro data, I hypothesize that it is not the amount of fat in the diet, but rather the individual fatty acids that promote or inhibit breast cancer formation and progression. The purpose of the following review is to explain what is known about various classes of fatty acids (i.e. saturated vs. unsaturated) and the individual fatty acids in terms of breast cancer risk and development in humans, animals and cell culture with a focus on long chain fatty acids, especially stearate. Meta-analyses of Dietary Fat Research. Four meta-analyses have been performed looking at the effects of total fat, saturated fat, monounsaturated fat and polyunsaturated fat on breast risk. Of these analyses, two found no effect of dietary fat on breast cancer development - a metaanalysis of 21 cohort and case-control studies and a meta-analysis of 8 cohort studies (78, 79). A meta-analysis of 12 case-controlled studies found a positive association of total fat, saturated fat, and monounsaturated fat and breast cancer risk in post-menopausal women (80). A more recent analysis of 14 cohort and 31 case-controlled studies found similar results, with a positive association seen with total fat and saturated fat intake and breast cancer risk (81). Large Multi-center Cohort Studies. Several large scale studies have been arranged in recent years to investigate the role of dietary fat on breast cancer risk. Unlike the case-controlled studies that often
43 27 centered around one region, these studies were either national or continental studies. Such a large cohort of people may be more representative of society as a whole than the smaller scale studies. These include the Women s Health Initiative (WHI) in the United States and the European Prospective Investigation into Nutrition and Cancer (EPIC) in Europe that measured the risk of breast cancer following a reduction in dietary fat. Two other studies, the Women s Intervention Nutrition Study (WINS) and Women s Healthy Eating and Living (WHEL) in the United States, measured the effect of dietary fat on relapse and survival in patients previously diagnosed with breast cancer. The results of these studies that relate to breast cancer and fat intake are explained below. WHI. The Women s Health Initiative Dietary Modification Trial was the first large scale randomized trial to test the effects of a low fat diet on breast cancer risk. 48,835 postmenopausal women were enrolled at 30 sites around the United States between 1993 and Of those enrolled, 19,451 women were randomized into a dietary intervention group whereas 29,294 were in the comparison group. The women in the dietary intervention group were counseled to lower their fat consumption by 50% - from 40% of total energy intake to approximately 20% of total energy intake (82). Women were then followed for 8.1 years and the incidences of various cancers and heart diseases were recorded (83). The results for the WHI study were highly anticipated and to the surprise of the research community, no effect was seen in the risk of invasive breast cancer between the dietary intervention and comparison groups (82-84). There was a 9% decrease in breast cancer incidence in the intervention group, but the results were not significantly different from the control group (83). Perhaps even more surprising, the
44 28 women who were in the highest quartile of basal fat intake saw a decrease risk in invasive breast cancer (83). Interestingly, an unexpected, significant decrease in ovarian cancer was seen in the cohort of women on the lower fat diet (85). Since the release of the results and the media frenzy that followed declaring nutrition was not related to breast cancer, several issues have been raised about the study (86). The study design called for a 20% difference in fat intake between the intervention and control groups. In reality, the study saw a 10.7% difference after year one, 9.8% difference after year three and an 8.7% difference after year five. It is thought this inability to reach the targeted fat reduction may account for the unexpected results. Furthermore, the enrollment period needed to meet the participant goal of the study took longer than anticipated. As a result, the average follow-up period was 8.1 years as compared to the original goal of 9 years. Once again, this unanticipated decrease is thought to account for the lack of an effect of the low fat diet on breast cancer risk (82). EPIC. The EPIC study began in 1993 with data being collected for 23 centers in 10 countries around Europe. The study consisted of 521, 468 participants, 366,521 of whom were women. During the duration of the study, several papers were published examining the role of various nutrients on breast cancer development. Overall, of 319,826 women analyzed, 7119 developed breast cancer. Of those who developed breast cancer, no association was seen between cancer risk and total fat intake although a weak positive association was observed with saturated fat intake but not monounsaturated or polyunsaturated fatty acids. (87).
45 29 Consistent with no effect of polyunsaturated fatty acids, one study examined the fish consumption of 310,671 women, 4776 of whom developed invasive breast cancer. No association was found between total fish, lean fish, or fatty fish and breast cancer development. (88). Interestingly, analysis of individual centers had different effects in terms of individual fatty acids and breast cancer risk. This is despite there being no difference between centers in the Sieri study that analyzed fat intake in the entire study (87). Data from the Cambridge center found no association between saturated fat and breast cancer risk although women who consumed approximately 35 grams of fat a day had twice the risk of developing breast cancer than those who consumed 10 grams or less (89). On the other hand, data collected from 15,351 German women at the Potsdam location suggested the 137 cases of invasive breast cancer that developed were positively associated with total fat intake. Additionally, the breast cancer cases were positively correlated with saturated fat, monounsaturated fat, and polyunsaturated fat (both omega-3 and omega-6). It is worth noting, however, that dietary assessments were only performed at the beginning of the study. Therefore, any changes in diet that occurred between the beginning of the study and the time of diagnosis are not accounted for (90). WINS. Unlike the WHI and EPIC studies, the WINS study was designed to determine the effect of a low fat diet on breast cancer reoccurrence in patients with early staged, surgically removed breast tumors. (91) women aged 48 to 79 were recruited at 39 sites around the United States to participate. Of those enrolled, 975 were assigned to the dietary intervention group that aimed to reduce dietary fat to 15% of the diet where as 1462 were assigned to the control group and were not instructed to change their diet (92).
46 30 After one year, those patients on the low fat, intervention diet consumed significantly less total fat, and less saturated, monounsaturated, and polyunsaturated fat than their control counterparts. Furthermore, the patients in the intervention group also had fewer relapses than the control group. When the data was further stratified to look at the effect of a low fat diet on breast cancer hormone status, the effect of the low fat diet was greater in the estrogen negative subjects than the estrogen positive ones. (92). This study was among one of the first large scale studies to show that modifications in dietary fat could affect survival in patients already diagnosed with breast cancer. WHEL. The Women s Healthy Eating and Living Randomized Trial was designed in a manner similar to the WINS study. Participants were women aged 18 to 70 who had previously had a surgically removed primary breast tumor. They were enrolled at 7 sites around the United States between 1995 and The 3088 participants were divided into a dietary intervention group (n=1537) or a control group (n=1551). Those in the dietary intervention group were advised to take a diet high in fruits and vegetables and low in fat the fat intake goal was 15-20% of total caloric intake. Those in the control group were advised to follow the 5-a day plan. Those in the dietary intervention group had a significant lower fat intake throughout the experiment than those in the control group. Interestingly, no difference in breast cancer relapse or survival was observed between the two groups. (93). Although the study did not produce the expected results or mirror the WINS study, a sub-group of patients showed a different disease outcome. Hot flashes are often associated with breast cancer treatment and generally serve as a positive outcome. This
47 31 is thought to be due to a decrease in bio-available estrogens women were stratified according to hot flash status and further divided into either the control group or the dietary intervention group. No difference was observed in relapse time between the hot flash positive patients in the control or intervention groups. Conversely, women who had not had hot flashes and were assigned to the intervention group experienced fewer relapses compared to the women in the control group. This significant decrease is thought to be due to a dietary-induced decrease in bio-available estrogens. It is worth noting, however, that these results may be an artifact as those women in the intervention group with no hot flashes were more likely to have received anti-estrogens or had their ovaries removed compared to those women with no hot flashes in the control group. (94). These results point to the complexity of understanding the role of dietary fat, especially when other factors, such as hormonal status, are taken into account. Accuracy of Dietary Studies The variability observed in cohort and case controlled studies has often been attributed to methodological issues. Study design issues can include inaccurate dietary recall (breast cancer patients often report higher fat intake throughout life than controls), lack of control for other health factors such as alcohol intake, body size, and menopausal status. Additionally, many have questioned the accuracy and validity of food frequency questionnaires used to assess dietary habits. Such questionnaires often ask people to recall dietary patterns for short periods of time in their life and may not accurately represents one s lifetime dietary habits. Some questionnaires ask participants to assess dietary habits many years prior (i.e. middle aged women recalling adolescent dietary
48 32 habits) and such reports may be largely inaccurate. Finally, questions have been raised in cohort studies about the duration follow-up period. It is possible that participants are not assessed long enough to see a true effect between dietary fat and cancer. (59). Dietary Biomarkers of Fatty Acid Intake Due to the issues associated with dietary recall studies, many scientists are using biomarkers of fatty acid intake to draw conclusion concerning fat intake and breast cancer risk. These so-called biomarkers include the fatty acid composition of triglycerides, phospholipids and cholesterol esters of adipose tissue, plasma, and erythrocytes. A recent meta-analysis was published of 13 studies that examined the breast cancer risk in comparison to fatty acid content of either the plasma or adipose tissue. In this meta-analysis, it was shown that total fat correlated positively with breast cancer risk as did the levels of palmitate. On the other hand, stearate was negatively correlated with breast cancer risk (95). Similar results were obtained from a casecontrolled study measuring erythrocyte fatty acids relative to breast cancer risk based out of Shanghai, China. Palmitate concentration correlated to breast cancer risk whereas no effect was observed with stearate (96). Summary of Epidemiological Studies The role of dietary fat in breast cancer remains controversial and unclear. While some studies show a clear positive association, many others show no effect. This difference is often attributed to the error associated with current dietary recall studies. Studies measuring the fatty acid content of erythrocyte membranes and adipose tissue,
49 33 which are thought to be at least partially representative of diet, show that each fatty acid has a differential effect on breast cancer risk. For example, although total saturated fatty acids and palmitate have been associated with an increased risk, stearate has been found to either have no effect or to decrease the risk of breast cancer. Dietary Fat Studies in Rodent Models Despite the lack of a consistent effect of a high fat diet on breast cancer risk in epidemiological studies, rodent models have suggested that the amount, type and duration of fatty acid exposure plays a role in the development of breast cancer. These studies have shown the effect of fatty acids on spontaneous tumor development, carcinogen induced carcinoma, and models of metastasis. Primary Tumor Studies. In 1942, Albert Tannenbaum published a study in which DBA mice fed a high fat diet derived from hydrogenated cottonseed oil developed more spontaneous tumors than their control counterparts. When the diets were initiated earlier, the effect was greater, indicating that the duration of fatty acid exposure could affect tumorigenesis. (97). Unfortunately, the individual concentrations of fatty acids in the diets used by Tannenbaum were not reported. Nevertheless, this was the first study that suggested that not only the amount of fat in the diet but also the time of exposure could affect breast cancer risk. Since the initial studies, over 100 dietary fat studies have been performed in rodent models. Many of these have shown that the amount of fat in the diet correlates positively to breast cancer development. More specifically, fats derived from vegetable
50 34 oils have been shown to increase cancer in spontaneous models, chemically induced models and metastasis models. Similar results were observed with natural saturated fatty acid sources such as beef tallow and lard. A threshold effect has been proposed in which increasing the concentration of fatty acid past a certain percentage does not subsequently increase tumor yield. More studies are necessary to determine if this is accurate. (98). Along with the amount of fat in the diet, the type of fat has also been implicated in the development of breast cancer. A meta-analysis of the effect of specific fatty acids on breast cancer concluded that omega-6 polyunsaturated fatty acids greatly enhance tumor growth, saturated fatty acids weakly enhance tumor growth, omega-3 fatty acids prevent tumor growth, and monounsaturated fatty acids have no effect. (59). One of the major issues with dietary studies is that it is difficult to determine if the effect seen is due to an increase in one fatty acid or macronutrient or a decrease in another. To overcome this issue, Tinsley et al. analyzed the effect of 20 different diets composed of various concentrations of natural fats and oils to determine the effect of fat on spontaneous mammary tumor development in CH3 mice. Using linear regression, the role of individual fatty acids on breast cancer development was determined. Linoleate (C18:2) was the most significantly associated with increase in tumor incidence and palmitate (C16:0) was also associated with increased incidence. Laurate (C12:0), myristate (C14:0), oleate (C18:1) and linolenate (C18:3) had little effect on tumor development. Finally, stearate (C18:0) was substantially associated with a decrease in tumor incidence. (75). In a spontaneous model of mammary tumors in SHN mice, linoleate was shown to have no effect above control on tumor incidence. However, linolenate was shown to
51 35 decrease tumor incidence. (99). These results suggest the positive association seen with linoleate may be animal model dependent. Interestingly, despite evidence that saturated fats may induce tumor growth, a diet enriched with stearate has been shown to delay tumor development in A/St mice (71). When cleared mammary fat pads were injected with a precancerous cell line, animals on a 10% corn oil diet developed more tumors at a faster rate than animals fed a 10% hydrogenated cottonseed oil diet. The authors attributed this effect to the higher concentration of linoleate in corn oil than cottonseed oil (59.9% compared to undetectable levels). (100, 101). However, it is worth noting that the cottonseed oil diet was composed of 69.9% stearate whereas the corn oil diet is only 1.4% stearate (101). Therefore, it is possible that the decrease tumor incidence and rate in the cottonseed group is due to an increase in stearate not a decrease in linoleate. One of the common ways to study breast cancer development in rats is to treat them with a chemical carcinogen either 7,12-dimethylbenz(a)anthracene (DMBA) or N-methyl-N-nitrosourea (NMU) and wait for palpable mammary tumors to form. The tumors that form are either malignant or benign and the development of tumors appears to mirror human disease. (102). Using these models, many studies have been performed to determine the effect of dietary fat on carcinogen induced breast cancer development. The amount of fat the animals are exposed to also dictates the number of carcinogen-induced tumors. When animals were fed either a 5% fat diet or a 20% fat diet derived from corn oil ad libitum and injected with NMU, more animals developed tumors on the 20% fat diet. When diets were restricted to kilocalories/day, there was no difference in tumor incidence between the diets. This suggests that not only is the
52 36 amount of food in the diet a factor in determining tumor incidence, but also the amount of energy available to the animals. (103). As with the spontaneous models and human studies, the type of fat appears to affect the development of carcinogen-induced tumors. Animals fed a largely polyunsaturated diet (18.6% total fat 59% linoleate, 27% oleate) before and after DMBA administration had more tumors compared to animals fed a high saturated fat diet (18.6% total fat 37% oleate, 28% palmitate, 19% stearate). Interestingly, if animals were placed on the polyunsaturated diet before DMBA administration and switched to the high saturate diet after administration, they developed fewer tumors than those animals placed on the polyunsaturated diet throughout. Furthermore, when animals were fed a saturated diet before and a polyunsaturated diet after DMBA administration, then they developed more tumors than the animals fed the high saturated diet throughout. These results indicate that polyunsaturated fats affect the development of mammary tumors only when they are fed post-carcinogen injection, suggesting that fatty acids affect the development of breast cancer during the promotion phase rather than during the initiation phase. (104). In addition to the type of fat, the individual fatty acids also appear to play a role in carcinogenic tumor development. When animals were fed a 23% fat diet derived from corn oil, safflower oil, olive oil or coconut oil and injected with NMU, the latter two developed fewer tumors than the first two. The predominate fatty acid in safflower oil and corn oil is linoleate. Conversely, the predominant fatty acid in olive oil is oleate and in coconut oil is laurate. These results suggest that linoleate somehow promotes tumor formation. (105).
53 37 Instead of developing a high fat diet to test stearate s effects on carcinogenesis, Habib et al. injected stearate into NMU treated animals. Those injected with stearate had a decreased number of tumors, decreased latency period until first tumor development, and decreased tumor size. (106). These studies indicate that in addition to dietary modification, it may be possible to alter fatty acid concentrations in patient through injections of individual fatty acids. Metastasis Studies. In addition to affecting the development and growth of primary mammary tumors, fatty acids also have an effect on metastases. The best studied are the omega-3 and omega-6 fatty acids. To my knowledge, no metastasis studies, beyond those presented in this dissertation, have been performed analyzing the effect of saturated fatty acids breast cancer metastasis. Linoleate has been shown to enhance breast cancer metastasis in several xenograph mouse models. This effect appears to be dose dependent, as BALB/cAnN animals injected with the mouse mammary cancer line 4526 and fed increasing amounts of linoleate developed more metastatic lung nodules than their low fat counterparts. Furthermore, as the concentration of linoleate increased in the diet, it also increased in the primary tumors (107). Linoleate also enhances the metastasis of the MDA-MB-231 and MDA-MB-435 human breast cancer cells from the mammary fat pad to the lungs of athymic nude mice (see appendix B for a discussion of the origins of this cell line). The reasons behind this increase are not fully understood, although linoleate increases the invasion and type IV collagenase activity of these breast cancer cells in vitro. (108). As collagen IV is a major component of the basal lamina of the basement membrane, the
54 38 ability of the breast cancer cells to adhere could play a role in survival and invasion. Additionally, treatment of the animals with the cyclooxygenase inhibitor suppresses linoleate induced metastasis, suggesting that eicosanoids derived from linoleate may account for the increase in metastasis, perhaps through an increase in angiogenesis. (109). In support of this data, animals fed a diet of 20% safflower oil had a significant increase in blood vessel areas in tumors compared to animals fed a 20% flaxseed diet, which is rich in linolenate (110). In contrast to linoleate, the omega-3 fatty acids linolenate, eicosapentaenoic acid (EPA) and docosahexaenoic acid (DHA) have been shown to decrease the size of tumors that form from mammary fat pad injections. Animals were maintained on an 8% linoleate diet following injection of MDA-MB-435 cells into the mammary fat pad. Seven days before excision, the animals either remained on the linoleate diet or were placed on a 2,4, or 8% EPA or DHA diet. The EPA and DHA diets successfully decreased the number and volume of macroscopic lesions. Interestingly, when diets were changed after primary tumor removal, the EPA and DHA diets still decreased the number of micrometastases in the lung. (111). Feeding animals a diet composed of flaxseed oil resulted in decreased primary tumors in the mammary fat pads of animals injected with MDA-MB-435 cells and indicated a trend for fewer metastases in the lungs. Furthermore, the decrease in primary tumor size appears to be due to a decrease in epidermal growth factor receptor (EGFR) and proliferation (as determined by Ki67 staining). (112). The exact reason for the decrease in metastases is unknown, however there is some evidence that omega-3 fatty acids may inhibit angiogenesis (113).
55 39 Summary of Animal Studies. Animal studies have shown it is not only the amount of fat present in the diet, but also the individual fatty acids that affect breast cancer risk. Overall, the omega-6 fatty acid linoleate has been implicated in worsening the development and progression of breast cancer. On the other hand, omega-3 fatty acids such as EPA, DHA, and linolenate, have been shown to decrease tumor growth an inhibit metastases. Saturated fatty acids are generally thought to promote breast cancer although some evidence exists that stearate may inhibit tumor development and progression. Fatty Acids and Cancer Cells In Vitro Rather than focus on classes of fatty acids as was done for the human and animal studies, the following in vitro studies are divided up based on individual fatty acid. The major fatty acids found in the human plasma, and those with extensive breast cancer research are profiled. These include palmitate, stearate, oleate, linoleate, arachidonate, linolenate, EPA and DHA. See Table 1-1 for a summary of the characteristics of each fatty acid. In a healthy adult, non-esterified fatty acid concentration in the plasma range between 273 M to 763 M following a 12 hour fast. Palmitate and oleate are the most abundant in the plasma, followed by linoleate and stearate. Linolenate concentrations are below 5 M and arachidonate below 10 M, regardless if the patients have fasted. (114). Palmitate. Palmitate is the most abundant saturated fatty acid in the human body. It is also the most abundant saturated fatty acid in the diet. Palmitate can either be supplied exogenously or endogenously through activation of the fatty acid synthase (FAS).
56 40 When applied exogenously, treatment of human Hs578t breast cancer cells with 150 M palmitate conjugated to fatty acid free-bsa (FAF-BSA) for 6 hours inhibited epidermal growth factor (EGF)-induced proliferation (76). Furthermore, treatment of MDA-MB-231 breast cancer cells with palmitate completely inhibits cell growth (115). Treatment of the MDA-MB-231 cells with bromo-palmitate did not effect cell growth indicating that palmitate must be metabolized to inhibit growth (73). Approximately 6 hours post-treatment, palmitate caused a decrease in mitochondrial membrane potential and an increase in cytochrome C present in the cytosol suggesting the induction of the intrinsic apoptotic cascade. A concurrent increase in caspase-3 activity was observed approximately 8 hours post-treatment indicating palmitate was activating apoptosis. Although palmitate increased the concentration of the proapoptotic lipid ceramide, inhibition of ceramide synthesis with Fumonisin B1 did not reverse caspase-3 activity. Palmitate also caused a decrease in the levels of and an increase in the turnover of cardiolipin, the phospholipid that sequesters cytochrome C in the mitochondria. Palmitate-induced caspase-3 activity was reversed by inhibiting the formation of palmitoyl-coa with Triacsin C (this will also inhibit DAG formation please see Chapter 3) and by increasing mitochondrial -oxidation. This reversal is thought to be due to an increase in cardiolipin levels in palmitate treated cells. (73). In 1994, an overexpressed, poor prognostic breast cancer marker known as oncogenic antigen-519 was isolated, sequenced and identified as fatty acid synthase (FAS). For reasons that are not completely understood, it is thought that cancer cells require de novo synthesis of fatty acids. (66). Many studies have been performed showing inhibition of FAS leads to death of cancer cells (116). Recently, it was shown
57 41 that inhibition of acetyl-coa carboxylase and inhibition of FAS with sirna induced apoptosis of human breast cancer cells. When the cells lacking FAS or ACC were treated with palmitate, the decrease in cell viability was reversed, once again stressing the importance of fatty acids, and in this instance, palmitate, on breast cancer survival. (117). Stearate. Next to palmitate, stearate is the second most abundant saturated fatty acid in humans. However, to date, little research has been done investigating the effect of this fatty acid on breast cancer development and progression. Stearate has been shown to completely inhibit EGF-induced Hs578t breast cancer cell proliferation (76). Furthermore, stearate is known to induce translocation of Annexin II to detergent resistant membrane in the Hs578t cells. This is hypothesized to affect cellular signaling through modifications of lipid rafts and protein acylation. The effect this translocation has on the phenotype of the breast cancer cells is currently not known. (118). As with palmitate, stearate has been shown to induce apoptosis of the MDA-MB-231 breast cancer cells. However, the authors did not investigate the mechanism behind this apoptotic cascade so it is unknown if stearate affects cardiolipin synthesis as palmitate does (73). Finally, stearate demonstrated a preferential inhibition of the growth of DMBA-induced mammary cancer cells compared to non-cancer cells in vitro. (119). Oleate. In animal studies, oleate either causes slight increase in tumor formation or has no effect. However, epidemiological data from people on the so called high-olive oil Mediterranean diet, indicates they experience a decrease in breast cancer rates (120).
58 42 Because oleate is found in high concentrations in olive oil, it is hypothesized to be the major constituent associated with the results in the epidemiological studies. The effect of oleate on breast cancer cells in vitro, however, tells a story that is more closely related to the animal studies. In 1990 Rose and Connolly found they could promote the growth of the MDA- MB-231 breast cancer cells with very low concentrations of oleate (885 nm), however higher concentrations inhibited growth (8.85 M) (115). Furthermore, 34 M oleate was shown to have no effect on the MDA-MB-231 cell growth after 6 days of treatment. When cells were treated with doxorubicin, an oxidizing agent, and DHA, cell viability was greatly decreased, below that of doxorubicin alone. However, when cells were treated with oleate instead of DHA, the viability was of the cancer cells were equivalent to doxorubicin, indicating oleate does not appear to have chemotherapeutic properties. (121). Extended survival studies have shown that 24 hour treatment of serum starved MDA-MB-231 cells with 400 M oleate allowed the cells to survive for at least 10 days. The untreated cells began to die within 24 hours and were completely dead by day M oleate decreased the caspase-3 activity induced by serum starving. Additionally, the oleate was found to increase triacyglycerol levels in the MDA-MB-435 cells. When a panel of breast cell lines was tested, oleate promoted survival of the T-47D and MDA- MB-468 breast cancer cells in addition to the MDA-MB-231. Interestingly no effect was observed in the MCF-7 breast cancer cells or the MCF-10A non-cancerous breast epithelial cells. (122).
59 43 While 400 M was sufficient to induce survival of several breast cancer lines, Hardy et al. demonstrated 250 M and 500 M increased proliferation of the MDA-MB- 231, ZR-75-1, and MCF-7 breast cancer cells but not the T-47D. 100 M oleate was also able to reverse palmitate induced apoptosis in the MDA-MB-231 and T-47D cells in a dose dependent manner. The ability of oleate to prevent palmitate induced apoptosis and promote survival appears to be due to the activation of phosphatidylinositol-3-kinase (PI3K). (73). The reasons for the different responses between the cell lines in terms of survival studies and the proliferation study are unknown but may be due to heterogeneity of cell culture lines between laboratories. As mentioned previously fatty acid synthase is crucial for breast cancer cell survival and inhibition of the FAS complex results in cell death. One of the common ways to inhibit FAS is to use the pharmacologic inhibitor cerulenin. Cerulenin decreased the viability of the SK-Br3 cells, however, when breast cancer cells were treated with oleate in addition to cerulenin, a decrease in cell toxicity was observed (123). Furthermore, treatment of SK-Br3 breast cancer cells that overexpress FAS with oleate results in reduced FAS activity and expression levels. Once again, these results suggest oleate promotes cell survival. (123). If oleate is associated with an increase in breast cancer cell survival, why do people on the Mediterranean diet have a decreased risk for breast cancer? In addition to being rich in oleate, olive oil is also enriched with phenols that have been shown to have anti-cancer properties and therefore, the effects of olive oil may not be due to oleate (124). This points out one of the primary problems with dietary studies using oils they
60 44 contain many compounds besides fatty acids that may be carcinogenic or anticarcinogenic. Linoleate. Many in vivo studies have been performed with corn oil and safflower oil, two oils enriched in linoleate that show a tumor promoting effect. Consistent with the animal data, in vitro experiments indicate linoleate can stimulate proliferation of breast cancer cells. Rose and Connolly demonstrated 17.8 nm to 2.6 M induced growth of the MDA- MB-231 cells in a dose-dependent manner. The MCF-7 cells also responded to linoleate, however, the effect was maximal at 1.78 M. (125). When the effect of the linoleateinduced proliferation was analyzed over a course of 6 days, 2.6 M had the maximum effect. Treatment with the cyclooxygenase and 5-lipooxygenase inhibitor indomethacin reversed the pro-proliferation effect of linoleate. Treatment with the cyclooxygenase inhibitor piroxicam had no effect on linoleate-induced proliferation whereas the 5- lipooxygeanse inhibitors esculetin and nordihydroguaiaretic acid (NDGA) reversed the effect similar to indomethacin. These results suggest that the leukotrienes produced by 5- lipooxygenase and not the prostoglandins produced by cyclooxygenase are important in linoleate-induced proliferation and survival. (115). Consistent with these findings, 1.8 M 17.8 M linoleate was found to induce proliferation of the MCF-7 cells and activate phospholipase C and protein kinase C. However, there was no effect on prostaglandin E2 (PGE2) secretion indicating that cyclooxygenase activity was not involved. (126). Analysis of T47D cells treated with nm linoleate showed an increase in cells in the S-phase. Furthermore, microarray data indicated linoleate could modulate
61 45 expression of various proteins involved in cell cycle regulation and cell growth. For example, linoleate induced expression of the estrogen receptor which is known to be a growth stimulant when activated. On the other hand, linoleate decreased androgen receptor expression which is known to antagonize the effects of the estrogen receptor. (127). The effects of linoleate are not just specific to breast cancer cells. Epithelial cells isolated from normal breast tissue showed maximal growth stimulation when cultured with insulin, EGF and linoleate. This growth could be inhibited with indomethacin and the inhibition could be reversed with the addition of PGE2. Interestingly, epithelial cells isolated from fibroadenomas were unresponsive to linoleate. The normal epithelial cells showed an increase of linoleate being shuttled into the arachidonate pathway to increase prostaglandin expression compared to the fibroadenoma epithelial cells. (128). Unlike Rose s work with the breast cancer cell lines, these results indicate prostaglandins may play a role in at least some of the effects seen with linoleate. Linoleate does not only affect proliferation, it can also stimulate breast cancer cell invasion. Treatment of the MDA-MB-435 breast cancer cells with 891 nm and 1.78 M linoleate resulted in an increase in cell invasion through Matrigel compared to untreated cells. This effect was inhibited by indomethacin. (129). Type IV collagenase activity was induced by 2.6 M linoleate which could also be inhibited with indomethacin (130). These results indicate that either prostaglandins or leukotrienes may be involved in linoleate-induced invasion. Interestingly, the concentrations used to investigate the effects of linoleate on cancer cells are well below the physiological levels of non-esterified linoleate in the
62 46 plasma. Plasma concentrations range between 37 M and 117 M after a 12 hour fast (114). More studies are needed to determine the effects of linoleate on breast cancer using higher concentrations. Arachidonate. When linoleate enters into a cell, it is converted to arachidonate through a three step process. Arachidonate is then converted to eicosanoids, including the 2-series prostanoids, prostaglandins and thromboxanes, lipoxins, and the 4-series leukotrienes. As mentioned in the linoleate section, inhibition of 5-lipooxygenase results in an inhibition of leukotriene formation whereas inhibition of cyclooxygenase causes a decrease in prostaglandins and thromoxanes. (69). Since linoleate can be converted to arachidonate, much of the work studying arachidonate does so by studying its metabolism. However, this section, as with previous fatty acids discussed, will focus on the effects of arachidonate applied exogenously to breast cancer cells. One of the most striking features of treatment of the MDA-MB-435 cells with arachidonate is an increase in cell adhesion to type IV collagen. A large amount of work has been done investigating this adhesion phenomenon. Arachidonate (30 M) induced activation of the MAP kinase p38. Activation of the enzyme was confirmed by phosphorylation of its downstream target MAPKAPK2. The activation of p38 was dependent on the dose of arachidonate and inhibition of p38 reversed arachidonateinduced cell adhesion. (131). Arachidonate mediated cell adhesion was also dependent on the activation of PKC. Following the activation, PKC was cleaved by calpain. When this cleavage was inhibited, the cells did not adhere to the collagen IV. Interestingly, inhibition of p38 did
63 47 not prevent PKC phosphorylation and vice versa. However, inhibition of p38 did partially prevent PKC cleavage by calpain. (132). When arachidonate enters into the cell, it is converted to 15(S)-hydroxyeicosatetraenoic (15(S)-HETE) by 15- lipooxygenase. When 15(S)-HETE was added directly to the MDA-MB-435 cancer cells, it stimulated breast cancer cell adhesion in a dose dependent manner. Furthermore, treatment with 15(S)-HETE stimulated phosphorylation of p38, indicating it is the major eicosanoid responsible for arachidonate mediated adhesion. (133). Adhesion and migration are largely responsible for rearrangement of focal adhesion, induced by the activation of focal adhesion kinase (FAK). Treatment with arachidonate for 48 hours significantly increased migration of the MDA-MB-231 cell and 15 M arachidonate for 10 minutes significantly activated FAK in the MDA-MB-231 and MCF-7 cells. Additionally, treatment at the same concentration for 20 minutes activated FAK in the ZR-75 cells. No effect was observed in the MCF-10A breast epithelial cells. The activation of FAK could be prevented by pretreatment with NDGA indicating a lipooxygenase product was responsible for FAK activation. In addition to activating FAK, arachidonate also activated src in the MDA-MB-231, MCF-7 and ZR-75 cells. Finally, inhibition of the lipooxygenases or src prevented the increase in migration due to arachidonate. (134). In addition to affecting adhesion and migration, arachidonate has also been implicated in affecting cell proliferation. Treatment of T-47D breast cancer cells with 8.2 M and 16.4 M arachidonate stimulated growth and induced expression of cyclin D1 which has previously been associated with an increase in cell proliferation (135). On the other hand treatment of MDA-MB-231 cells with 1 M to 50 M arachidonate decreased
64 48 cell viability in a dose dependent manner. In the MDA-MB-231 cells, this decrease was associated with the activation of caspase-9 and caspase-3 indicating the decrease in viability is due to activation of the intrinsic apoptotic cascade. (136). It is worth noting however, that non-esterified fatty acid levels of arachidonate range from 3 M to 7 M after a 12 hour fast meaning the majority of concentrations used in the second study are supraphysiological (114). Linolenate. The effects of the essential -3 fatty acid alpha-linolenate on breast cancer cells have not been well studied and characterized with in vitro models. Relatively few studies treat with linolenate the majority of studies looking at the omega-3 fatty acids use DHA or EPA and these studies are discussed in the next section. Overall, however, linolenate appears to specifically inhibit breast cancer cell growth. Treatment of the MCF-7 cells with 6-30 M linolenate shows a dose dependent decrease in cell proliferation. Treatment of the MCF-10A with 6-24 M linolenate has no effect, although a decrease in viability is observed at 30 M. The effect was observed whether linolenate was supplied as a free fatty acid bound to albumin, or as an enriched phospholipid lysosome. (137). Linolenate also decreased the proliferation of the MDA- MB-231 breast cancer cells in a dose dependent manner using 25 M to 100 M concentrations (138). Physiological concentrations of non-esterified linolenate range between 1 M to 3 M after a 12 hour fast (114). DHA and EPA. DHA and EPA are generally referred to as the omega-3 fish oils as they are found in high concentration in oily fish such as salmon. When linolenate enters into a
65 49 cell, it is converted to EPA through the same pathway that converts linoleate to arachidonate. EPA can also be converted to DHA and vice versa. EPA is the precursor to the 3-series prostanoids, prostaglandins and thromboxanes, lipoxins, and the 5-series leukotrienes. Interestingly, these eicosanoids generally have the opposite effect as those derived from arachidonate, meaning they are generally anti-inflammatory and anticarcinogenic. (69). DHA and EPA have been shown to inhibit the growth of several different breast cancer cell lines at different concentrations. 100 M EPA and DHA have been shown to decrease cell growth after 5 days treatment. In addition, there was a dose dependent inhibition of colony growth. (139). In another study, DHA was found to inhibit MCF-7 growth in a dose dependent manner above 40 M. A time dependent increase in apoptotic was observed with 80 M treatment for 24, 48, and 72 hours. Consistent with this finding, the Bax/Bcl-2 ratio increased 48 to 72 hours after treatment indicating a possible mechanism for the induction of apoptosis. (140). DHA (25 M) also induced apoptosis in MDA-MB-231 cells through a mechanism dependent on ceramide formation (141). Multiple other mechanisms have been implicated to explain the apoptotic effect of DHA. Treatment of MDA-MB-231 cells with 200 M DHA caused an increase in the PPAR expression and a subsequent decrease in NF B activity. No effect was observed with EPA. (142). Furthermore, 30 M DHA has been shown to activate PPAR and induce caspase-3 activity in MCF-7 cells. Once again, no effect was seen with EPA, indicating it does not play a role in activating PPAR. (143). In addition to affecting proliferation and survival, DHA has been implicated in inhibiting MDA-MB-231 cell invasion (138). DHA has also been shown to sensitize
66 50 MCF-7 and MDA-MB-231 cells when used with the chemotherapeutic agent doxorubicin (121,144). To date, the mechanisms underlying these effects are largely unknown. In one study, the non-esterified concentrations of DHA and EPA were 0.55% and 0.03% of the total non-esterified fatty acids in the plasma, respectively (145). Assuming a plasma concentration between 273 M and 763 M, the concentration range for DHA would be 1.5 M to 4.2 M and EPA would be 0.08 M and 0.23 M (114). Therefore, the concentrations used in these in vitro studies are supraphysiological Summary of In Vitro Data. In some ways, the in vitro data can be as difficult to interpret as the animal and epidemiological studies. Different results are observed in the same cell lines, concentrations are inconsistent, and in many cases, not physiological. The majority of the studies focus on one fatty acid, so any effects that are the result of multiple fatty acids are unknown. Yet, despite these inconsistencies, many of the in vitro studies show results similar to the animal studies. When breast cancer cells were treated with exogenous palmitate, the cells underwent apoptosis (73). However, if FAS expression was decreased by sirna and the cells were treated with palmitate, it increased survival (117). Therefore, depending on FAS expression, palmitate can either induce or inhibit cytotoxicity. Stearate, on the other hand, shows consistent anti-cancer properties, including the inhibition of proliferation and induction of apoptosis. The omega-6 fatty acids linoleate and arachidonate appear to promote carcinogenesis by promoting both growth and invasion of cancer cells. Finally, the omega-3 fatty acids such as linolenate, DHA and EPA inhibit carcinogenesis by decreasing cell viability, proliferation, invasion, and increasing chemosensitivity.
67 51 Conclusions Drawn from the Fatty Acid Studies The Dietary Guidelines for Americans recommends the majority of dietary fats be composed of omega-3 and omega-6 fatty acids with limited intake of saturated fatty acids (146). However, as demonstrated here, not all unsaturated fatty acids prevent breast cancer and not all saturated fatty acids promote breast cancer. As a whole, the omega-6 fatty acids appear to promote carcinogenesis whereas the omega-3 fatty acids are noncarcinogenic. As for the saturated fatty acids, the role in breast cancer development is dependent on the individual fatty acids being studied. Figure 1-9 depicts the current fatty acid recommendations and the effects of individual fatty acids on breast cancer progression. Hypothesis, Objectives, and Significance To date, multiple epidemiological, animal and in vitro studies have been performed investigating the role of fatty acids on breast cancer development and progression. Despite 67 years of research, the exact role of fatty acids in breast cancer progression is unclear. Dietary studies suggest it is not only the amount and source of the fatty acid but also the concentration of individual fatty acids that affect breast cancer growth. Human studies are riddled with measurement errors and, as of yet, no studies have been performed by enriching the diet with one fatty acid and determining the effect on breast cancer risk. Furthermore, many of the animal studies were performed with diets consisting of various concentrations of oils, a feat that cannot be changed because animals require essential fatty acids to avoid illness. The oils, however, are composed of more than just fatty acids, and contain compounds which may confound test results.
68 52 Although cell studies have been performed using individual fatty acids, concentrations are not consistent and any synergistic or inhibitive effects of multiple fatty acids are rarely tested. Despite the difficulties in interpreting the data, using dietary means to improve breast cancer risk is a real and possible solution. Several fatty acids, such as linoleate and stearate have consistent results in at least animal and in vitro studies, suggesting the observed effect may have some merit. However, without modifying the fatty acid profiles of foods consumed on a daily basis, the exact roles of the individual fatty acids in breast cancer risk cannot be definitively determined. Based on the known epidemiological and experimental data, my general hypothesis is that increasing the amount of stearate in the diet will prevent breast cancer development and inhibit breast cancer progression. More specifically, I hypothesize stearate will decrease the viability of breast cancer cells in vitro and dietary stearate will inhibit metastasis in vivo. The goal of this study is to determine whether stearate may have potential anticancer activities in breast cancer development and metastasis. It has been estimated by Peto and Doll in 1981 and confirmed by Willet in 1993 that 50% of breast cancer deaths could be avoided through modification of the diet (147). Despite a large amount of epidemiological data that suggests decreasing the amount of fat in the diet does not significantly prevent cancer occurrence, high dietary fat is generally associated by the American public and researchers as increasing the risk of cancer. Based on in vivo and in vitro experiments performed, the amount and the type of fat present in the diet affect tumorigenesis. As fatty acids such as stearate, linolenate, EPA
69 53 and DHA appear to have cancer-inhibiting effects, a diet enriched in one or more of these fatty acids may have a beneficial effect in preventing breast cancer for those with a high risk and preventing progression to metastatic disease in patients already diagnosed with breast cancer. Although more safety and efficacy studies are needed to confirm the studies presented here, these results strongly implicate certain fatty acids as a potential adjuvant chemotherapeutic for breast cancer patients and as a chemopreventative for patients at high risk for developing breast cancer.

70 Figure 1-1: Progression of Breast Cancer: From Normal Breast to Metastatic Disease. A) A schematic of the Wellings-Jensen model of breast cancer progression. A normal duct experiences abnormal growth leading to hyperplasia. If the cells acquire mutations and begin to lose their polarity, they can progress to atypical ductal hyperplasia. If further mutations are acquired and the cells become malignant, but don t invade the basement membrane, ductal carcinoma in situ occurs. Once the cells invade the basement membrane, they are considered invasive breast cancer. B) For metastasis to occur, cancer cells invade the tissues surrounding the primary tumor and intravasate into a blood vessel. If the cells survive in the blood stream, usually with the help of platelets, they can then extravasate out of the vessel. Once the cells exit the blood vessel, they can colonize and grow at a secondary site. 54
71 55
72 Figure 1-2 Summary of TNF -TNFR1 Signaling. Under normal circumstances, upon activation, the TNFR1 activates a strong pro-survival through NF B. If the NF B activity is inhibited, then part of the signaling complex dissociates from the receptor and recruits and activates caspase-8 to induce apoptosis. If RAIDD is recruited to the receptor, apoptosis can be induced either through the release of RIP1 and RAIDD from the receptor and the subsequent recruitment of PIDD and caspase-2, or through the direct recruitment of capsase-2 to the receptor. 56
73 Figure 1-3: Summary of Fas-FasL Signaling. When FasL binds to its receptor Fas, this recruits FADD and caspase-8. Together, this is known as the DISC. Caspase-8 can then cleave and activate caspase-3 and cell death follows. However, if FLIP is recruited to the DISC to inhibit caspase-8 this can lead to the activation of NF B and a strong prosurvival signal. A decoy Fas receptor also exists. Known as DcR3, it can sequester FasL away from its receptor and therefore induce survival. 57
74 Figure 1-4: Summary of TRAIL-Induced Apoptosis. When TRAIL binds to DR4 or DR5, it recruits FADD and caspase-8. This induces activation of caspase-8 which then cleaves and activates caspase-3 to induce apoptosis. As with TNF and FasL, the complex can be inhibited by FLIP. Furthermore, if TRAIL binds to the decoy receptors DcR1 or DcR2, the apoptotic cascade is not activated and the cell survives. 58
75 Figure 1-5: Summary of Activation of the Intrinsic Cascade. Upon DNA damage, - radiation exposure or exposure to certain other radiations or factors, a form of mitochondrial dependent apoptosis is induced. This form of cell death is controlled by a family of proteins known as the Bcl-2 proteins. Under normal circumstances, the Bcl-2 anti-apoptotic proteins sequester the BH123 pro-apoptotic proteins. When the cascade is activated, the BH3 proteins sequester the anti-apoptotic proteins, thereby releasing the BH123 proteins. The BH123 proteins translocate to mitochondria where they induce permeablization of the outer mitochondrial membrane. This causes mitochondrial proteins to be released in the cytosol. One of these proteins, cytochrome C, binds to Apaf-1 and caspase-9 to form a complex known as the apoptosome. This induces activation of caspase-9 which then activates caspase-3 and cell death occurs. 59
76 Figure 1-6: Fatty Acid Nomenclature. A) Fatty acids are composed of a carboxyl head group followed by a hydrocarbon-like chain. The first carbon on the head group is known as the carbon, whereas the second is the carbon, etc. The final carbon is known as the carbon. Unsaturated fatty acids are classified based on the position of the first double bond from the carbon. B) Saturated fatty acids, such as stearate, have no double bonds. Monounsaturated fatty acids, such as oleate, have one double bond and polyunsaturated fatty acids like linoleate have more than one double bond. Oleate is an -9 because the double bond occurs 9 carbons from the omega carbon whereas linoleate is an

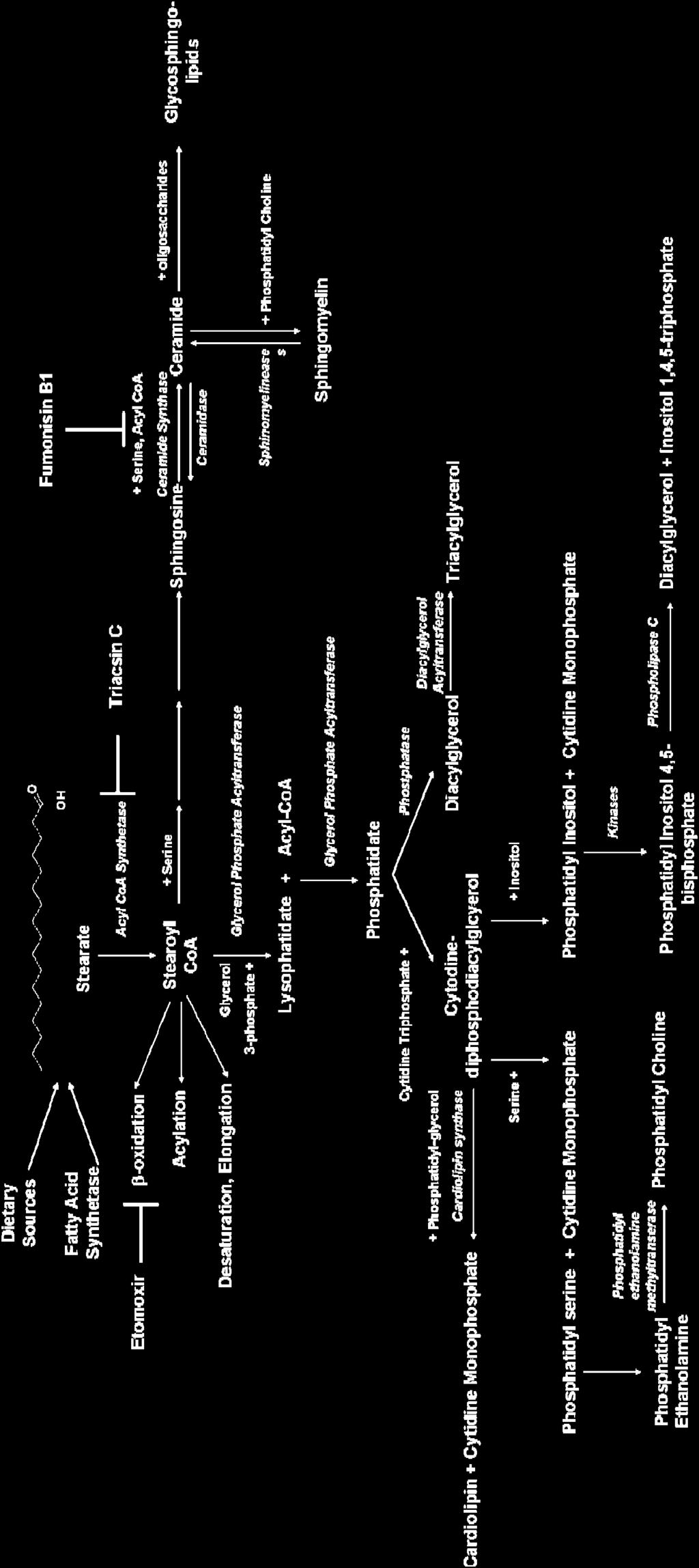
77 Figure 1-7: Metabolism of Stearate 61


78 Figure 1-8: Omega-6 and Omega-3 Fatty Acid Metabolism. Omega-3 and omega-6 fatty acids are converted into EPA and arachidonate, respectively. They are then converted to various eicosanoids, including prostaglandins, leukotrienes, and thromboxanes, among others. 62
79 63 Fatty Acid Common Name IUPAC Name Type Omega Class C16:0 Palmitate; hexadecanoic Saturated N/A Palmitic Acid acid C18:0 Stearate; Stearic Acid Octadecanoic acid Saturated N/A C18:1 Oleate; Oleic Acid C18:2 -Linoleic Acid; Linoleic Acid; Linoleate C18:3 -Linolenic Acid; Linolenic Acid; Linolenate C20:4 Arachidonic Acid; Arachidonate C20:5 Eicosapentaenoic acid; EPA C22:6 Docosahexaenoic acid DHA cis-9- octadecenoic acid cis, cis-9,12- octadecadienoic acid 9,12,15- octadecatrienoic acid 5,8,11,14- icosatetraenoic acid 5,8,11,14,17- icosapentaenoic acid 4,7,10,13,16,19- docosahexaenoic acid Monounsaturated Omega-9 Polyunsaturated Omega-6 Polyunsaturated Omega-3 Polyunsaturated Omega-6 Polyunsaturated Omega-3 Polyunsaturated Omega-6 Table 1-1: Classification of Long Chain Fatty Acids

80 Figure 1-9: A Current View of Dietary Fatty Acids and the Effect of Individual Fatty Acids on Breast Cancer Progression. 64
81 65 CHAPTER 2 THE EFFECTS OF STEARATE ON CELL MORPHOLOGY: THE INDUCTION OF CELL ROUNDING THROUGH A PROTEIN KINASE C DEPENDENT MECHANISM The extracellular matrix is important for several reasons. Once thought to be a static structure composed of large macromolecules such as collagens, laminins, and polysaccharides, the extracellular matrix is generally recognized now to play a large role in the development of normal and cancerous tissues (148). The ECM has been shown to affect cell to cell as well as cell to connective tissue interactions in addition to tissue and tumor morphology (2). Our laboratory previously demonstrated that stearate could induce morphological changes of human HT-1080 fibrosarcoma cells. This effect was greater when the cells were plated on laminin than other matrices such a collagen. (74). To determine if the breast cancer cells rounded preferentially on one of the matrices, cells were plated on collagen IV, osteopontin or laminin (albumin served as the negative control as it is a globular protein rather than a filamentous one). As shown in Figure 2-1, stearate induced rounding of the Hs578t and MDA-MB-435 breast cancer cells on all matrices tested. The rounding became statistically significant 12 hours after treatment with 50 M stearate conjugated to fatty acid free albumin (FAF-BSA). Interestingly, stearate did not induce rounding of the Hs578Bst myoepithelial cells, indicating the effects of stearate are specific to the cancer cells. Chapter 3 further confirms this specificity showing stearate does not induce cell death of the MCF-10A non-cancerous breast epithelial cells.
82 66 Stearate is known to enter into many metabolic pathways in the cells. Five major pathways include de novo diacylglycerol synthesis, protein acylation, ceramide synthesis, phospholipid formation, and -oxidation in the mitochondria. Molecular inhibitors were used to block selected pathways to determine whether one or more was responsible for stearate-induced changes in cell morphology. Fumonisin B1 was used to inhibit ceramide synthesis, Triacsin C was used to inhibit de novo diacylglycerol synthesis, and Etomoxir was used to inhibit -oxidation. As shown in Figure 2-2, Etomoxir and Triacsin C partially reversed the stearate-induced cell rounding in the Hs578t cells. Interestingly, when both inhibitors were tested simultaneously, no additive effect was observed (data not shown). These results suggest there may be some overlap between the metabolic pathways. Consistent with this concept, Triacsin C inhibits ACS the enzyme that converts fatty acids to their acyl-coa derivatives (68, 149). There is some evidence in the literature that it selectively inhibits the ACS isoforms that induce de novo synthesis of DAG (149). However, the Etomoxir target CPT1 binds acyl-coa proteins and translocates them into the mitochondria for -oxidation. Therefore, it is possible that DAG generation in addition to -oxidation is responsible for the induction of cell rounding. As DAG is a known activator of many PKC isozymes, rounding was quantitated in the presence of two PKC inhibitors. When cells were treated with GF X or Calphostin C prior to stearate treatment, the cell rounding was partially reversed suggesting the PKC isozymes are involved in the stearate-induced morphological changes (Figure 2-3). I hypothesize stearate induces de novo synthesis of DAG and therefore, stearate is most likely activating either a novel or classical PKC isozymes. To test this
83 67 hypothesis, Hs578t cells screened PKC isozyme expression. The cells have detectable levels of,,, and (data not shown). To determine which PKC isozymes were activated by stearate, cell lysates were immunoblotted for phosphorylated forms of the proteins. As shown in Figure 2-4, all detectable isoforms become phosphorylated by stearate. Pharmacologic inhibitors were used to attempt to discern the isozyme(s) responsible for cell rounding. GF X is not a potent inhibitor of the and isoforms, indicating their activation may not contribute significantly to cell rounding (150). To determine if the or isoforms were responsible for the morphological changes, cells were treated with either the PKC inhibitor 2,2,3,3,4,4 -Hexahydroxy-1,1 -biphenyl-6,6 -dimethanol dimethyl ether (HBDDE) or the myristilated PKC peptide inhibitor PKC V1-2 (151). As shown in Figure 2-5, HBDDE had no effect on stearate-induced cell rounding 24 hours after treatment. Interestingly, inhibition of PKC partially reversed cell rounding at 24 hours (Figure 2-5). These results indicate PKC may not be involved in cell rounding whereas PKC may be involved. Future studies inhibiting expression of these isozymes with sirna of a dominant negative construct are necessary to confirm these finding with the pharmacologic inhibitors. Cell rounding has been implicated in a number of different cellular functions such as apoptosis, anoikis, and proliferation (37, 152). To determine if stearate was affecting the health and viability of the cancer cells, metabolic activity was measured. As shown in Figure 2-6, treatment of the Hs578t cells with stearate for 12 hours significantly decreases the metabolic activity of the cells as determined by an MTT assay. Cells were tested at three different densities 5,000 cells/well (50-60% confluent), 10,000 cells/well
84 68 (90-100% confluent), and 50,000 cells/well (overgrown culture) and stearate decreased the metabolic activity at all three densities. However, it is worth noting that stearate was less affective at the higher densities. The reasons behind this decrease at higher densities are unknown, but may mimic the effects of stearate in a more physiological environment, such as in a tumor. If so, these results would argue that stearate would affect a malignant tumor, although with lower efficacy than would be observed on a cell culture plate.
85 Figure 2-1: Stearate Induces Rounding of Human Breast Cancer Cells but not Non- Cancerous Breast Cells Independent of the Extracellular Matrix. Cells were plated on tissue culture plastic that had previously been coated with either BSA, or 20 g/ml of murine osteopontin, human collagen IV or human laminin. Cells were allowed to adhere for 24 hours and then serum starved for 24 hours prior to counting the number of round and adherent cells. A round cell was defined as a cell with one or no processes. Cells were then treated with 50 M stearate or FAF-BSA. The number of adherent and round cells was counted prior to stearate treatment, and then 6, 12 and 24 hours post-stearate treatment. Stearate induced rounding of the Hs578t breast cancer cells (n=2; p<0.02 by ANOVA) and MDA-MB-435 breast cancer cells (n=3; p<0.02 by ANOVA) but not noncancerous Hs578Bst breast fibroblasts (n=2) on all matrices tested. 69
, the adherent and round cells were counted.")
86 Figure 2-2: Inhibition of Mitochondrial -oxidation and de novo DAG Synthesis Partially Reverses Stearate-Induced Cell Rounding. Hs578t cells were plated and serum starved for 24 hours. Prior to treatment with 50 M Fumonisin B1, 25 M Etomoxir, 5 M Triacsin C or vehicle (DMSO or H 2 O), the adherent and round cells were counted. One hour after inhibitor treatment, cells were treated with 50 M stearate or FAF-BSA. The number of round cells 12 hours after stearate treatment was determined. The graph represents the number of round cells induced by stearate (FAF-BSA background was subtracted out). As shown above, no effect was seen with Fumonisin B1 (n=2) indicating ceramide is not involved in stearate-induced cell rounding. Inhibition of -oxidation with Etomoxir partially reversed stearate-induced cell rounding (n=3; p<0.004 student s t-test). Inhibition of de novo diacylglycerol synthesis with Triacsin C also partially reversed stearate-induced cell rounding (n=4; p< Student s t-test). 70

87 Figure 2-3: Inhibition of Protein Kinase C Partially Reverses Stearate Induced Cell Rounding. Hs578t cells were plated and serum starved for 24 hours and the number of adherent and rounded cells were counted prior to treatment with 25 nm Calphostin C or 10 nm GF X. One hour later, cells were treated with FAF-BSA or 50 M stearate. Data shown are the percent of cells rounded by stearate 12 hours post-treatment. Inhibition of PKC with Calphostin C (n=2; P< student s t-test) or GF X (n=4; p< student s t-test) partially reversed stearate-induced cell rounding. 71

88 Figure 2-4: Stearate Induces Phosphorylation of Multiple Novel and Classical PKC Isozymes. Hs578t cells were serum starved for 24 hours prior to treatment with 50 M stearate for the times indicated. Cell lysates were collected and ppkc isozymes, -actin, or total PKC were detected by immunoblot. As shown above, stearate induces phosphorylation of PKC, PKC, and PKC 1 to 3 hours post-treatment. Stearate also induces phosphorylation of PKC 6 hours post-treatment. 72
 or PKC V1-2 (50 M).")
89 Figure 2-5: Inhibition of PKC but not PKC Partially Reverses Stearate Induced Cell Rounding. Cells were plated and serum starved for 24 hours and the adherent and rounded cells were counted prior to treatment with HBDDE (50 M) or PKC V1-2 (50 M). An hour later cells were treated with 50 M stearate or FAF-BSA. 24 hours later, the number of round and adherent cells was counted. The number of stearate-induced round cells is shown above. When PKC was inhibited by HBDDE, there was no effect on cell rounding (n=3). However, when PKC was inhibited with PKC V1-2, cell rounding was partially reversed (n=2; p< student s t-test). 73

90 Figure 2-6: Stearate Decreases the Metabolic Activity of the Hs578t Breast Cancer Cells. Cells were plated at the various densities and serum starved for 24 hours. The cells were then treated with FAF-BSA or 50 M stearate for 12 hours. After 12 hours, metabolic activity was estimated using the MTT assay. Stearate decreased the metabolic activity of the Hs578t breast cancer cells in cell density dependent manner. (n=4 in quadruplicate; *p<0.02 Student s t-test). 74
91 75 CHAPTER 3 STEARATE PREFERENTIALLY INDUCES APOPTOSIS IN HUMAN BREAST CANCER CELLS by LYNDA M. EVANS, STEPHANIE L. COWEY, GENE P. SIEGAL, ROBERT W. HARDY Accepted by Nutrition and Cancer Format adapted for dissertation
92 76 ABSTRACT Stearic acid (stearate) is an 18-carbon saturated fatty acid that has been shown to inhibit invasion and proliferation and induce apoptosis in various human cell types. The specificity of stearate-induced apoptosis for cancerous versus non-cancerous breast cells has not been examined and the mechanism underlying stearate-induced apoptosis is unknown. Morphological analysis, cell viability and caspase-3 activity assays demonstrated that stearate activated apoptosis preferentially in cancerous breast cells in a time and dose dependent manner. Inhibition of de novo diacylgycerol synthesis or protein kinase C (PKC) blocked stearate-induced caspase-3 activity indicating the involvement of a novel or classical PKC isozyme. To our knowledge, this is the first study showing that stearate induces apoptosis preferentially in breast cancer cells and implicates protein kinase C in the signaling cascade. These results raise the possibility of dietary stearate having a beneficial role in the prevention or treatment of breast cancer.
93 77 INTRODUCTION Stearic acid (stearate) is an 18-carbon saturated fatty acid found in relatively high concentrations in several foods in the western diet, including beef and chocolate. In vitro and in vivo studies suggest stearate may have unique properties, especially in terms of breast cancer development and neoplastic progression. For example, stearate has been shown to inhibit epidermal growth factor (EGF) receptor-mediated proliferation in Hs578t breast cancer cells (76), inhibit invasion of HT-1080 fibrosarcoma cells (74), and induce apoptosis of MDA-MB-231 breast cancer cells (73). In vivo, dietary stearate has been associated with a decrease in mammary tumor development and incidence in spontaneous carcinogenesis models (71, 75). A recent meta-analysis of 13 studies compared human erythrocyte membrane, serum and adipocyte fatty acid composition relative to breast cancer risk (95). The study found that stearate was associated with a decreased risk of breast cancer in post-menopausal women but showed no association with breast cancer risk in the remainder of cohort studies (95). A more recent casecontrol study from Shanghai, China, also reported that stearate had no association with breast cancer risk (96). These results indicate that dietary stearate either reduces or has no effect on breast cancer risk in humans. Taken together, in vitro, in vivo and epidemiological studies point to stearate as having potential for breast cancer prevention and treatment; however, little is known concerning the mechanism of stearate s action(s), including its induction of apoptosis. Apoptosis is a form of programmed cell death that is generally executed by a family of cysteine proteases known as caspases. Activation of the extrinsic cascade through the cleavage of caspase-8, or the intrinisic cascade through the cleavage of
94 78 caspase-9 leads to the cleavage and activation of executioner caspases such as caspase-3, resulting in cell death (Reviewed in 39). When stearate enters into a cell, it is converted to stearoyl-coenzyme A (CoA) by one of the acyl-coa synthetases (ACS). Based on the subcellular location of the synthetase, the stearoyl-coa can lead to the de novo generation of acyl-glycerols, phospholipids, ceramide, mitochondrial -oxidation, or protein acylation. In this manuscript, we investigated these pathways and found that de novo diacylglycerol (DAG) synthesis and protein kinase C (PKC) activation are necessary for stearate-induced caspase-3 activity. We also compared the ability of stearate to induce apoptosis in several breast cancer and non-cancer cell lines and found that stearate has a significant preferential effect on breast cancer cells. MATERIALS AND METHODS Materials Protease inhibitor cocktail, insulin, EGF, actin antibody, stearic acid, Etomoxir, Fumonisin B1, Triacsin C and diatomaceous earth were all obtained from Sigma-Aldrich (St. Louis, MO). GF X was obtained from Biomol (Plymouth Meeting, PA). Hs578t and Hs578Bst were obtained from the ATCC (Manassas, VA), MDA-MB-435 cells were a gift from Dr. Danny Welch (UAB, Birmingham, AL), MDA-MB-231 cells were a gift from Dr. Selvarangan Ponnazhagan (UAB, Birmingham AL), and MCF-10A cells were a gift from Dr. Andra Frost (UAB, Birmingham, AL). DMEM, fetal bovine serum (FBS), heat-inactivated FBS, DMEM:Ham F12, sodium pyruvate, glutamine, non-
95 79 essential amino acids, PBS, and penicillin/streptomycin were obtained from Cellgro (Mediatech, Inc. Herdon, VA). NEFA C was purchased from Wako Chemicals (Neuss, Germany). The BCA Protein Assay was obtained from Pierce Biotechnology (Rockford, IL). Cleaved caspase-3, cleaved PARP, and phospho-pkc (pan) ( II Ser 660) antibodies were obtained from Cell Signaling Technologies (Danvers, MA). Anti-mouse and antirabbit conjugated to horseradish peroxidase and ECL reagents were from Amersham Biosciences (Piscataway, NJ) and the caspase-3 activity assay was obtained from Invitrogen (Carlsbad, CA). Finally, the Matrigel coated Boyden chambers were from BD Biosciences (Franklin Lakes, NJ) and the lactate dehydrogenase assay was from Roche (Nutley, NJ). Cell Culture Human Hs578t breast cancer cells, MDA-MB-231 breast cancer cells, Hs578Bst myoepithelial breast cells, and MCF-10A epithelial cells were grown and maintained as recommended by the ATCC. MDA-MB-435 cells breast cancer cells were cultured in 5% FBS in DMEM:Ham F12 media supplemented with 2 mm glutamine, 1 mm sodium pyruvate, 0.2X non-essential amino acids, and 1% penicillin/streptomycin (5% CO 2 ). Hs578t cells were serum starved with DMEM without phenol red supplemented with 0.5% heat inactivated FBS, 1 mm sodium pyruvate, 1% penicillin/streptomycin. MDA- MB-231 breast cancer cells were serum starved with L-15 without phenol red supplemented with 0.5% heat inactivated FBS and 1% penicillin/streptomycin. MCF- 10A cells were serum starved with DMEM:F12 without phenol red supplemented with 0.5% heat inactivated FBS, 20 ng/ml EGF, 100ng/mL cholera toxin, 500ng/mL
96 80 hydrocortisone, 0.01mg/mL bovine insulin, and 1% penicillin/streptomycin. MDA-MB- 435 breast cancer cells were serum starved with DMEM:F12 without phenol red supplemented with 0.5% heat inactivated FBS, 1mM sodium pyruvate, 0.2% nonessential amino acids, and 1% penicillin/streptomycin. Stearic Acid Preparation Stearate was bound to fatty acid free-bsa (FAF-BSA) by the method of Spector and Hoak (153). Briefly, stearate was added to hexane, mixed with diatomaceous earth, and dried under a stream of nitrogen gas to form a powder. The powder was stirred with starvation media containing 1% FAF-BSA and then the diatomaceous earth was filtered out. The ph of the BSA-stearate mixture was adjusted to 7.4 with 1M HCl. The addition of the powder, filtration, and ph adjustment were repeated 3 more times. The BSA-fatty acid mixture was sterilized by filtration and the concentration of fatty acids was measured using the NEFA C enzymatic-colorimetric analysis. FAF-BSA was prepared the same way without the addition of stearate to the diatomaceous earth. Scratch Assay for Cell Migration Hs578t cells were plated on a 24-well plate in cell media containing 10% FBS. When the cells attained 90% confluence a 200 μl pipette tip was used to make one midhorizontal and one mid-vertical scratch across the cell layer in each well. The cells were gently washed 3 times with PBS and complete media was added with either FAF-BSA (control) or stearate (50 μm). The cells within a 1cm 2 area within the scratch region were
97 81 counted under a microscope in 4 fields of view before stearate treatment (time 0) and at 6, 12, and 24 hours after stearate treatment. Cell Invasion Assay Matrigel coated chambers were rehydrated using complete media for 2 hours. Hs578t cells were trypsinized from the plate, and diluted to 2.5 x 10 4 cells/ml in starvation media. Complete media was added to the lower well and 500 L of cells in starvation media were added to each invasion chamber. After two hours, 50 M stearate or FAF-BSA was added to the upper and lower chambers. After 24 hours of incubation, media was removed and cells were fixed with 3.7% paraformaldehyde, stained with 0.5% crystal violet, rinsed with distilled water and allowed to dry overnight. Five random areas on each insert were then counted at 100X magnification. Cytotoxicity Assays 40,000 cells/ well were plated on a 24 well plate. Wells were treated with stearate at the concentrations mentioned. 12 hours after stearate treatment, conditioned media was collect and cells and debris were removed by centrifugation. Lactate dehydrogenase (LDH) activity was measured in the conditioned media according to the manufacturer s instructions. Triton X was used as a positive control as it will lyse all cells in the well. Cells were treated with 2% Triton X for 10 minutes and conditioned media was collected as stated above. The values obtained with Triton X were taken as 100% cytoxicity and stearate s values were adjusted accordingly. Each experiment was performed in triplicate.
98 82 Immunoblots Hs578t cells were starved for 24 hours in starvation medium, and then treated with 50 M stearate for the times indicated. Cells were lysed using lysis buffer (25 mm HEPES; 150 mm NaCl; 1% NP-40; 10 mm MgCl 2 ; 1 mm EDTA; 2% Glycerol; 1 mm NaF; 1 mm Na 3 V0 4; 25 mm β-glycerophosphate; 1 mm tetrasodium pyrophosphate; 1:1000 PMSF [100 mm]; 1:1000 Protease Inhibitor Cocktail), equal amounts of protein were prepared by boiling the lysate with 6X sample buffer (300 mm Tris-HCl, ph 6.8; 12% SDS; 0.05% Bromophenol blue; 60% Glycerol; 12% -mercaptoethanol), and were separated using 10% SDS-PAGE gels. Proteins were then transferred to PVDF membrane and were probed with primary antibodies and incubated with secondary antibody conjugated to horseradish peroxidase. Caspase-3 Activity Assay Cells were plated to 60-70% confluence and were then serum starved for 24 hours. One hour before treatment with 50 M stearate or FAF-BSA, cells were treated with either 5 M Triacsin C, 25 M Etomoxir, 50 M Fumonisin B1, 10 nm GF X or DMSO (vehicle). After an additional 12 hours, cells were lysed and caspase- 3 activity was measured according to the manufacturer s protocol using the peptide Z- DEVD-AMC assay (Invitrogen). Cell lysates were transferred to a 96-well black plate and incubated with the peptide for 30 minutes. Fluorescence was measured using a fluorescent microplate reader.
99 83 Statistical Analysis Statistics were analyzed using either ANOVA (SigmaStat3.0) or student s t-test (Excel). Data presented are mean +/- standard error of the mean. RESULTS Stearate Inhibits Breast Cancer Cell Invasion and Migration Stearate has previously been demonstrated to markedly inhibit invasion of HT fibrosarcoma cells and induce morphological changes, such as retraction of cell processes and cell rounding in vitro, although the molecular mechanisms of these effects are currently not known (74). To determine whether or not stearate had a similar effect on breast cancer cells, we analyzed the effect of stearate on migration and invasion. Because albumin is the physiological carrier of free fatty acids in vivo, cells were treated either with FAF-BSA or 50 M stearate conjugated to BSA. Using an in vitro wound-healing assay, stearate significantly inhibited cell migration at 6, 12 and 24 hours compared to FAF-BSA (Figure 3-1A). To determine whether inhibition of cell motility by stearate would result in decreased cell invasion in vitro, we performed invasion assays using Matrigel coated chambers. We found that stearate completely blocked breast cancer cell invasion (Figure 3-1B). Stearate Preferentially Induces Apoptosis of Human Breast Cancer Cells Following treatment of the Hs578t breast cancer cells with stearate, we observed that stearate induced cell rounding 24 hours post-treatment. To determine if this effect
100 84 was specific to this cell line, we analyzed the MDA-MB-435 and MDA-MB-231 breast cancer cell lines 24 hours after stearate treatment and found that both cell lines rounded in the presence of stearate (Figure 3-2A and data not shown). When we analyzed the Hs578Bst myoepithelial breast cells and the MCF-10A breast epithelial cells, we found that even after 24 hours of stearate treatment, the morphology of the cells did not significantly change. These results indicated that the effects of stearate are preferential for the cancer versus the non-cancer cells. Because morphological changes such as cell rounding and cell shrinkage are hallmark characteristics of apoptosis, we next tested whether or not stearate was affecting the viability of the cancer cells. As stearate has been shown to induce death of the MDA- MB-231 breast cancer cells previously, they were used as a positive control (73). As shown in Figure 3-2B, stearate significantly decreased the viability of the three cancer cell lines tested. Twelve hours post-treatment, stearate decreased the viability of the Hs578t, MDA-MB-435, and MDA-MB-231 cells by 16.4%, 30.5%, and 21.5%, respectively. Stearate did not affect the viability of the MCF-10A cells, a finding that was consistent with the lack of stearate induced morphological changes in this cell line. To confirm the cells were undergoing cell death, cell media from the Hs578t cells was tested for lactate dehydrogenase activity (LDH). LDH is secreted into the media as cells die and activity was measured using colormetric assay. As shown in Figure 3-2C, stearate induced cytotoxicity in a dose dependent manner. Analysis of lysate from the Hs578t breast cancer cells confirmed that stearate is inducing caspase-dependent apoptosis as shown by the presence of cleaved caspase-3 (Figure 3-3A). Furthermore, the well-characterized caspase-3 target poly-adp-ribosomal
101 85 polymerase (PARP) is cleaved at the same time, indicating that caspase-3 is active (154). To confirm the immunoblot data, caspase-3 activity was measured using a fluorescent based assay. As shown in Figure 3-3B, stearate induced caspase-3 activity in a time dependent manner. Furthermore, we wanted to determine if stearate induced caspase-3 activity correlated with the cytotoxicity data. Cells were treated with varying concentrations of stearate for 12 hours and activity was increased in a dose dependent manner (Figure 3-3C). Stearate Induces Apoptosis in Human Breast Cancer Cells through the de novo Synthesis of DAG and the Activation of PKC To determine if stearate was being metabolized through a particular pathway to induce apoptosis, cells were treated with metabolic pathway inhibitors and stearate or FAF-BSA. When carnitine palmitoyltransferase-1 (CPT-1) was inhibited with Etomoxir, a partial reversal of caspase-3 activity was seen in the Hs578t cells, but not in the MDA- MB-435 cells (Figure 3-4). When cells were treated with Fumonisin B1 to inhibit ceramide synthesis, no effect was seen on either cell line. In contrast, when the ACS was inhibited with Triacsin C, stearate-induced caspase-3 activity was reversed completely in both cell lines. It is worth noting, however, that in the MDA-MB-435 cell lines, Triacsin C induced caspase-3 activity without stearate. The difference between the FAF-BSA/ Triacsin C group was not different from the stearate/ Triacsin C group. All other FAF- BSA and stearate combinations tested were significant. Finally, as DAG is a well-characterized activator of PKC, we tested whether or not stearate activated PKC and if PKC was involved in stearate-induced capase-3
102 86 activity. Cells were treated with 50 μm stearate and PKC activation was assessed using a pan-phosphorylation antibody. Stearate induced phosphorylation of PKC beginning at 1-3 hours after treatment (Figure 5A). Furthermore, when PKC was inhibited with the pan- PKC inhibitor GF X, stearate-induced caspase-3 activity was completely reversed indicating a direct role for PKC in stearate-induced apoptosis (Figure 3-5B). DISCUSSION Numerous studies have indicated that long chain saturated fatty acids including stearate and palmitate are capable of causing apoptosis in various cell types (73, ). However, these studies generally use elevated, pathophysiological concentrations of stearate. According to Staiger et al. stearate concentrations can reach ~192 M in fasting, healthy individuals. However, this includes total fatty acids, non-esterified and those found in phospholipids and acyglycerols. (157). When non-esterified plasma fatty acids where investigated in 8 men and 7 women, stearate concentrations ranged between 35 M at the start of the study to 66 M after 12 hours of fasting (114). Few studies have investigated concentrations of stearate less than 100 M. In one study, human granulosa cells were incubated for 3 days with 50 M stearate and there was no difference in cell viability between the stearate treated cells and controls while 100 M stearate significantly inhibited cell viability at 3 days (156). In another study, HT-29 human colon cancer cells were cultured with 30 M stearate for 15 days and cell growth did not differ from the other groups (79). Wicha et al. demonstrated a preferential ability of stearate to inhibit the growth of DMBA-induced rat mammary tumor cells compared to normal rat epithelial cells in vitro (119). It seems clear that stearate can induce apoptosis,
103 87 although this effect is cell type and concentration dependent. Our results concur with these general conclusions but are the first to directly show a preferential pro-apoptotic effect of stearate at physiological concentrations (50 M) for human breast cancer cell lines compared to non-cancerous breast cell lines. Interestingly, stearate completely inhibited invasion through Matrigel after 24 hours of treatment. Tryapn blue staining showed that approximately 50% of the cells are dead at 24 hours (unpublished data). This argues another potential mechanism underlying the inhibition of invasion. Little work has been done on the mechanism of stearate-induced apoptosis in breast cancer cells, although, some studies have been performed on palmitate-induced apoptosis. These studies using the MDA-MB-231 cell line concluded that fatty acid metabolism unrelated to -oxidation, ceramide synthesis, ROS production or changes in PI3-kinase activity affected palmitate-mediated apoptosis (73). Similar with these studies, we found inhibition of ceramide synthesis with Fumonisin B1 had no effect on stearateinduced caspase-3 activity in the Hs578t and MDA-MB-435 breast cancer cells. Stearate induced caspase-3 activity was partially blocked in the Hs578t cells by Etomoxir, an inhibitor of CPT-1, indicating a possible role of -oxidation in the induction of apoptosis. It was suggested that palmitate incorporation into diacylglycerol rather than triacylglycerol was related to fatty acid-induced apoptosis of the MDA-MB-231 breast cancer cells (73). In our studies, inhibition of DAG synthesis with Triacsin C completely reversed stearate-induced caspase 3 activity in both the Hs578t and MDA-MB-435 cells. Triacsin C has been shown previously to inhibit fatty acid-induced de novo synthesis of diacylglycerol (DAG), and stearate has been shown to induce DAG synthesis in cultured bovine aortic smooth muscle cells (165). Furthermore, Triacsin C has been reported
104 88 previously to inhibit de novo DAG synthesis without affecting phospholipid synthesis (166). Studies with rat homologues of ACS found that only certain isoforms were inhibited by Triacsin C. It has been hypothesized that there are different ACS isoforms located at distinct subcellular regions and that Triacsin C inhibits the ACS that feeds acyl-coas into the synthesis of DAG de novo. (Reviewed in 149). Because the PKC isozymes are well characterized downstream targets of DAG, we hypothesized that stearate was activating PKC through the generation of DAG to induce apoptosis. The PKC isozymes are a family of serine/threonine kinases that are subdivided into three classes. The classical isozymes (, I, II, ) are dependent on DAG and Ca 2+ for activation. The novel isozymes ( ) lack the Ca 2+ dependency and are dependent on DAG. Finally, the atypical isozymes ( and ) lack both the Ca 2+ and DAG requirements. The PKC isozymes as a class are already known to play a role in numerous cellular processes including apoptosis, proliferation, invasion and migration. In general, PKC and PKC have been portrayed as being anti-apoptotic whereas PKC is generally regarded as pro-apoptotic (Reviewed in 167). However, the exact role of PKC isoenzymes in apoptosis is known to depend on the specific isozymes present and the cell type being analyzed. For example PKC has been demonstrated to play an anti-apoptotic role in human colon cancer cells, being mediated by NF B and ciap-2 (168). We found that stearate activated PKC in the Hs578t cells and that inhibition of PKC with GF X was able to reverse caspase-3 activity. Identifying the specific PKC isoenzyme that mediates stearate-induced apoptosis in breast cancer cells is an ongoing area of research for our laboratory.
105 89 To date, very few studies have attempted to address the role of individual fatty acids in human breast cancer prevention studies. Saturates, as a whole class, are generally associated with an increase risk of breast cancer (80,81). However, using erythrocyte and adipose tissue compositions as a correlation to total fat intake, the individual saturates have varying effects on breast cancer. For example, palmitate positively correlates with breast cancer risk whereas stearate has a negative association (95). Although we found no such studies that found a positive correlation between stearate and breast cancer some studies found stearate to be neutral (no positive or negative effect). This may be related to the proportion of stearate in the diet compared to other dietary fatty acids. It is possible that by lowering the background of other dietary fatty acids stearate may become affective in vivo. Of course we cannot rule out other factors such as the environment and gene pools of the neutral studies. A number of human studies are necessary to determine the efficacy of stearate in breast cancer prevention and treatment. To date, no studies have been performed to determine if a diet rich in stearic acid could be an effective treatment. Along the same lines, minimal concentrations, optimal doses and mechanism of administration have not been monitored. Long chain saturated fatty acids have been linked to high cholesterol concentrations. Dietary stearate, unlike palmitate (C16:0), has not been shown to increase cholesterol and may even decrease low density lipoprotein cholesterol (LDLc) without affecting high density lipoprotein cholesterol (169). Several studies have claimed that a high stearic acid diet promotes coagulation (170). However, several more recent studies dispute this fact, finding stearate is not prothrombotic and may actually decrease thrombotic and athrogenic factors in vivo such as mean platelet volume and
106 90 plasma fatty acid concentrations (171,172). Interestingly, the opposite was observed with a diet high in palmitate (172). It is also worth noting a diet high in stearate does not appear to effect insulin sensitivity indicating that unlike other saturated fatty acids, it may not be involved in the development of type 2 diabetes (173). Thus, the growing body of literature indicating stearate may be a potential cancer preventative and therapeutic, suggest that clinical testing of stearate should be seriously considered. In summary, stearate inhibited breast cancer cell migration and invasion in vitro as well as preferentially induced apoptosis of human breast cancer cells. Inhibition of de novo DAG synthesis reversed stearate induced caspase-3 activation. Stearate activated PKC and inhibition of PKC also reversed caspase-3 activation. This is the first study we are aware of that has implicated PKC isozymes in stearate-induced apoptosis and supports in vivo studies indicating that stearate may inhibit breast cancer development. ACKNOWLEDGEMENTS The authors would like to thank A. Ibrahim-Hashim and E. Toline for help with the preparation of the manuscript. These studies were supported by the NIH/National Center for Complementary and Alternative Medicine (NCCAM), R21 AT and R21 AT and (RWH). Its contents are solely the responsibility of the authors and do not necessarily represents of the official views of the NCCAM, or the National Institutes of Health. This study was also made possible by a pre-doctoral fellowship from the National Cattlemen s Beef Association (LME/RWH).

107 Figure 3-1: Stearate Inhibited Cell Migration and Invasion. A) Hs578t cells were treated with FAF-BSA or 50 M stearate for 12 hours and then a scratch assay was performed. Representative images of the wound 24 hours after treatment with stearate or FAF-BSA are shown. Stearate significantly decreased the migratory ability of the cells 12 and 24 hours post treatment. (n=3; *p<0.001 by ANOVA). B) Hs578t cells were plated on Matrigel coated invasion chambers and treated with 50 M stearate. After 24 hours the number of cells that invaded through the Matrigel was counted. Stearate treatment abolished invasion of the Hs578t cells (n=3; *p< by ANOVA). Representative images of the crystal violet stained cells 24 hours post-treatment are shown. 91
 Stearate induced morphological changes of the breast cancer cells but not the noncancerous cells 24 hours post treatment.")
108 Figure 3-2: Stearate Decreased the Viability of the Human Breast Cancer Cells. A) Stearate induced morphological changes of the breast cancer cells but not the noncancerous cells 24 hours post treatment. B) Twelve hours post-treatment, stearate decreased the viability of the breast cancer cells but not the non-cancerous MCF-10A cells using the trypan blue exclusion assay (n=2 performed in triplicate; *p<0.005 by the Student s t-test). C) To confirm the decrease in cell viability, cytotoxicity induced by stearate 12 hours post-treatment was estimated by measuring lactate de-hydrogenase activity in the cell culture media. Stearate decreased the viability of the cancer cells in a dose dependent manner. (n=3 performed in triplicate; *p<0.04 compared to FAF-BSA, **p<0.02 by ANOVA) 92
109 Figure 3-3: Stearate Induces Cleavage and Activation of Caspase-3 in the Hs578t Cells. A) Beginning approximately 6 hours after stearate treatment, cleaved caspase-3 appeared and was present 24 hours post-treatment. Concurrent with cleaved caspase-3 was the presence of cleaved PARP, a well-characterized downstream target of caspase-3. The immunoblots are representative of 5 independent experiments. B) To confirm that stearate is activating caspase-3 in a time dependent manner, caspase-3 activity was measured 6, 12 and 24 hours after treatment. Stearate significantly increased caspase-3 activity approximately 12 hours post-treatment. (n=3-4; **p<0.03, *p<0.05 by ANOVA). C) Stearate induces caspase-3 activity in dose dependent manner (n=3-4; *p<0.04, **p<0.005 by ANOVA). 93
110 Figure 3-4: Inhibition of Acyl-CoA Synthetase Inhibited Stearate-Induced Caspase- 3 Activity at 12 hours. In the presence of the ACS inhibitor Triacsin C, stearate-induced caspase-3 activity was inhibited in the Hs578t and MDA-MB-435 cell lines (n=3; FAF- BSA vs. stearate for each inhibitor, *p<0.03 by the Student t-test). 94
111 Figure 3-5: Inhibition of PKC Inhibited Stearate-Induced Caspase-3 Activity at 12 Hours. A) Hs578t cells were treated with 50 M stearate for the times indicated, lysates were collected and immunoblotted with a pan phospho-pkc antibody. Stearate induced phosphorylation of PKC 1-3 hours post treatment (n=4). B) Hs578t cells were treated with the pan PKC inhibitor GF X for 1 hour prior to treatment with 50 M stearate. Inhibition of PKC blocked stearate-induced caspase-3 activity (n=5; * different compared to stearate treated controls p<0.02 but not different from non-stearate treated controls p>0.07 by the paired t-test). 95
112 96 CHAPTER 4 STEARATE INDUCES APOPTOSIS THROUGH A MECHANISM DEPENDENT ON THE INTRINSIC AND EXTRINSIC CASCADES Apoptosis is a form of programmed cell death that results in the activation of a family of cysteine proteases known as caspases. Overall, caspases can be divided into two categories initiators and executioners. Initiators, such as caspase-8 and caspase-9 are cleaved and activated due to cellular signaling and cleave executioner caspases such as caspase-3, caspase-6 and caspase-7. (39). Chapter 3 investigated the ability of stearate to induce apoptosis of human breast cancer cells but not non-cancerous breast cells. This chapter will investigate the molecular mechanisms upstream of stearateinduced caspase-3 activation. To determine if stearate was inducing apoptosis through the activation of intrinsic or extrinsic cascade, Hs578t breast cancer cells were treated with stearate for the times indicated, lysates were collected and probed by immunoblot for cleaved caspase-8 and cleaved caspase-9 (Figure 4-1). Stearate induced cleavage of both proteins indicating activation of both apoptotic signaling cascades. To determine if both cascades were necessary for caspase-3 activation, cells were treated with molecular inhibitors specific for caspase-8 or caspase-9, and one hour later, cells were treated with 50 M stearate. 12 hours after stearate treatment, cell lysates were collected and caspase-3 activity was measured using a fluorescent-based assay. As shown in Figure 4-2, inhibition of caspase- 8 with Z-IETD-FMK reversed stearate-induced caspase-3 activity. Additionally, inhibition of caspase-9 with Z-LEHD-FMK reversed stearate-induced caspase-3 activity.
113 97 These results indicate that both the intrinsic and extrinsic cascades are necessary for stearate-induced caspase-3 activity. Bid is a BH3 only, proapoptotic member of the BCL-2 protein family that is cleaved by caspase-8 upon activation of the extrinsic cascade. Truncated Bid then translocates to the mitochondria and activates the intrinsic cascade causing cleavage and activation of caspase-9. To determine if stearate could affect Bid levels, cell lysates were immunoblotted for total protein levels. As shown in Figure 4-3, stearate treatment caused a decrease in Bid levels after 12 hours. Although this is not a definitive experiment to show the involvement of Bid, the reduction in total Bid at 12 hours is suggestive of protein cleavage. Collectively, these studies suggest that stearate is inducing apoptosis through a mechanism dependent on both the extrinsic and intrinsic cascades. Total Bid protein levels appear to be affected by stearate and inhibition of caspase-8 and caspase-9 reverses stearate-induced caspase-3 activity. Stearate may be inducing apoptosis by activating the extrinsic cascade and then activating the intrinsic cascade. Future studies are necessary to identify the ligand responsible for activation of the extrinsic cascade and to confirm the involvement of Bid.

114 Figure 4-1: Stearate Induces Cleavage of Caspase-8 and Caspase-9. Hs578t cells were plated and serum starved for 24 hours prior to treatment with 50 M stearate. Cell lysates were collected and analyzed by immunoblot for the presence of cleaved caspase-8 and cleaved caspase-9. Stearate induced cleavage of both proteins. (n=3). 98
115 Figure 4-2: Inhibition of Caspase-8 and Caspase-9 Reverses Stearate-Induced Caspase-3 Activity. Cells were plated and serum starved for 24 hours prior to treatment with 20 M Z-IETD-FMK to inhibit caspase-8 or M Z-LEHD-FMK to inhibit caspase- 9. Caspase-8 inhibition reversed caspase-3 activity in stearate treated cells (n=4; p<0.04 Student s t-test). Caspase-9 inhibition also reversed stearate-induced caspase-3 activity (n=3; p<0.05 Student s t-test). 99
.")
116 Figure 4-3: Stearate Causes a Decrease in Total Bid. Cells were plated and serum starved prior to treatment with 50 M stearate. Total Bid protein levels were analyzed by western blot. Stearate induced a decrease in total Bid 12 hours post-treatment. (n=2). 100
117 101 CHAPTER 5 DIETARY STEARATE REDUCES HUMAN BREAST CANCER METASTASIS BURDEN IN ATHYMIC NUDE MICE by LYNDA M. EVANS, ERIC C. TOLINE, RENEÉ DESMOND, GENE P. SIEGAL, ARIG IBRAHIM HASHIM, ROBERT W. HARDY Clinical and Experimental Metastasis in press Format adapted for dissertation
118 102 ABSTRACT Stearate is an 18-carbon saturated fatty acid found in many foods in the western diet, including beef and chocolate. Stearate has been shown to have anti-cancer properties during early stages of neoplastic progression. However, previous studies have not investigated the effect of dietary stearate on breast cancer metastasis. In this study, we present evidence that exogenously supplied dietary stearate dramatically reduces the size of tumors that formed from injected human breast cancer cells within the mammary fat pads of athymic nude mice by approximately 50% and partially inhibits breast cancer cell metastasis burden in the lungs in this mouse model system. This metastatic inhibition appears to be independent of primary tumor size, as stearate fed animals that had primary tumors comparable in size to littermates fed either a safflower oil enriched diet or a low fat diet had reduced lung metastasis. Also stearate fed mice sub-groups had different primary tumor sizes but no difference in metastasis. This anti-metastasis effect may be due, at least in part, to the ability of stearate to induce apoptosis in these human breast cancer cells. Overall, this study suggests the possibility of dietary manipulation with selected long-chain saturated fatty acids such as stearate as a potential adjuvant therapeutic strategy for breast cancer patients wishing to maximize the suppression of metastatic disease.
119 103 INTRODUCTION The role that dietary fat plays in breast cancer development and progression has remained a controversial area for over 60 years. While epidemiological studies have produced conflicting results, in vivo studies consistently support a role for dietary fat in breast cancer, suggesting it is not only the amount of fat, but also the type, present in the diet that affects tumorigenesis, cancer growth and metastasis (Reviewed in (59, 98). To date, studies suggest the omega-6 unsaturated fatty acid linoleic acid (C18:2) promotes breast cancer growth and metastasis (108, 174, 175). In contrast, the omega-3 unsaturated fatty acids linolenic acid (C18:3), eicosapentaenoic acid (20:5; EPA) and docosahexaenoic acid (22:6; DHA) inhibit tumor development and metastasis (176, 177). The saturated fatty acid stearic acid (C18:0) also inhibits tumorigenesis although the effect on metastasis has not been tested. Stearic acid, or stearate, is an 18-carbon saturated fatty acid found in high concentrations in many foods in the western diet including beef, chocolate, and milk fats. Unlike other saturated fatty acids, such as palmitate (C16:0), stearate does not increase plasma low density lipoprotein cholesterol concentrations (169). Interestingly, stearate has been found to have anti-cancer properties both in vitro and in vivo, targeting proliferation, migration, and tumor invasion. In vitro, stearate has been shown to inhibit breast cancer cell proliferation, induce breast cancer cell apoptosis, and inhibit fibrosarcoma cell invasion (73, 74, 76). To date, the molecular mechanisms underlying these effects have yet to be determined. In vivo, dietary stearate has been shown to decrease tumor incidence and delay tumor development in spontaneous and chemically-induced mammary tumor carcinogenesis
120 104 models. Tinsley et al. fed CH3 mice diets composed of different concentrations of oils and, using linear regression, found that increased levels of dietary stearate correlated with decreased incidence of spontaneous mammary tumors (75). Similarly, when A/St mice were fed a 14% stearate/1% safflower oil diet, they developed fewer spontaneous tumors than the low fat diet, indicating that a diet composed of pure stearate, as opposed to an enriched oil, could effect tumorigenesis (71, 106). Habib et al. found that injecting stearate into rats treated with NMU decreased the average number of tumors/rat (106). So while stearate clearly effects the transformation and early neoplastic progression steps of cells, its role in an orthotopic cancer model of metastases using human cells derived from carcinomas has not previously been investigated. Metastasis is defined as tumor growth at a secondary site distant and discontinuous from the primary site (17). This growth is a multi-step process and for tumors which spread by lymphohematogenous routes such as breast cancer begin with growth and invasion of the extracellular matrix at the primary site, intravasion into the blood or lymphatic system, survival in this system during circulation, extravasion from the vessel, and colonization and growth at the secondary site. Inhibition of any of these steps results in an inhibition of secondary sites of tumor growth, i.e. metastatic foci. (Reviewed in 15). Based on known in vivo and in vitro data, we hypothesized that dietary stearate would inhibit breast cancer metastasis. Using the athymic nude mouse mammary fat pad injection model, we found that animals fed a high fat diet containing principally stearate demonstrate significantly decrease growth of primary tumors by approximately 50% and demonstrated a statistically significant decrease in macroscopic lung metastasis burden.
121 105 Further, we present evidence that this decrease in metastatic lung tumors is dependent on factors other than primary tumor size. Finally, we demonstrate that stearate in vitro induces cleavage of caspase-3 and PARP raising the possibility that stearate inhibits tumor growth and metastasis via apoptosis of the breast cancer cells. MATERIALS AND METHODS Animals and Diets 3-4 week old female athymic mice were purchased from Harlan Sprague Dawley, Inc. (Indianapolis, IN) and were maintained in microisolater cages in pathogen-free facilities. Animals were placed on one of three diets a low fat diet comparable to normal rodent chow, a 20% safflower oil diet, or a 17% stearic acid/3% safflower oil diet for 3 weeks prior to the injection of human breast cancer cells. The diets were prepared by Harlan-Teklad (Madison, WI) and dietary compositions are shown in Table 5-1. The animals were fed ad libitum and the amount of food consumed was recorded. Mice were anesthetized with 3% Isoflurane in 2.5% O 2 and weighed weekly. Cancer Cells MDA-MB-435 human breast cancer cells (obtained from Dr. Dan Welch; UAB) were grown and maintained in DMEM:F12 supplemented with 5% FBS, 2 mm glutamine, 1 mm sodium pyruvate, 0.2X non-essential amino acids and 1% penicillin/streptomycin (5% CO 2 ). Cells were grown to 80-90% confluence prior to preparation for injection. To detach cells from the plates, cells were washed with 1x PBS
122 106 and then treated with 3mM versene, ph 7.2. Cells were pelleted by centrifugation and resuspended in Hank s Buffered Saline Solution (HBSS). Cells were diluted to 1x10 7 cells/ml and were kept on ice until the time of injection to prevent clumping. Experimental Design The experimental timetable is shown in Figure 5-1. Briefly, animals were divided randomly into one of four groups a low fat diet group, a safflower oil diet group, and two stearate diet groups. All animals were placed on the diets three weeks prior to injection of cancer cells. The low fat and safflower oil diet exposed tumors were allowed to reach an approximate mean tumor diameter of 10mm 2-12mm 2 (~253.6mm mm 3 ) at which time the primary tumors were removed (~9 weeks post-injection). One stearate group followed the same pattern although the tumors were not as large at 9 weeks postinjection as the tumors from the two control groups. The second stearate group s tumors were allowed to grow to the size of the tumors in the safflower and low fat groups (10mm 2-12mm 2 ) or for a maximum of 13 weeks after injection. After removal of the primary tumors, the animals were allowed to develop metastases for four weeks, sacrificed and the lungs were collected. All in vivo procedures were approved by the Institutional Animal Care and Use Committee. Mammary Fat Pad Injections Animals were anesthetized with 3% Isoflurane in 2.5% O 2. A small incision was made between the right 2 nd and 3 rd mammary fat pads and 1x10 6 MDA-MB-435 cells suspended in HBSS were injected into the 2 nd mammary fat pad using a 27mm gauge
123 107 needle (final volume of 100 l). A single wound clip was used to close the incision and removed the following week. Tumor Measurement After the injection of the cells, mice were monitored weekly for the development of primary tumor masses. Once the tumors became visible (1-2 weeks post-injection), they were measured using a digital caliper. The tumor volume was estimated using the equation for a prolate ellipsoid where volume=π(4/3)(length/2)(width/2)((length + width)/4). Tumor Excision The animals were anesthetized with isoflurane. The skin overlying the mammary tumor area was cleaned with a betadine solution and an incision was made circumferentially around the tumor down to its base. The tumor was excised and weighed. The wound was closed using wound clips which were removed one week later. Necropsy Four weeks after the removal of the primary tumor, mice were anesthetized with a combination of ketamine and xylazine and then decapitated. A visual examination of the lymph nodes and liver found no metastasis. The lungs were dissected from the mice and stored in Bouin s solution prior to the counting of visible tumors on all surfaces of the lungs. The examiner was blinded to the identity of the samples prior to counting.
124 108 Cell Treatment with Stearate Cells were grown to 60-70% confluence and serum starved for 24 hours prior to treatment with 50 M stearate conjugated to fatty acid free BSA (FAF-BSA) in a manner previously described (153). Fatty acid concentrations were measured using the NEFA-C kit from Wako Chemicals (Neuss, Germany). Western Blotting Following treatment, cells were washed with 1x PBS and lysed in lysis buffer composed of 25 mm HEPES; 150 mm NaCl; 1% NP-40; 10 mm MgCl 2 ; 1 mm EDTA; 2% Glycerol; 1 mm NaF; 1 mm Na 3 V0 4; 25 mm β-glycerophosphate; 1 mm tetrasodium pyrophosphate; 1:1000 PMSF (100 mm) and 1:1000 Protease Inhibitor Cocktail. Proteins were separated by SDS-PAGE and transferred to PVDF membranes. Standard western blotting protocols were followed to detect cleaved caspase-3 (Cell Signaling; Danvers, MA), cleaved PARP (Cell Signaling), or -actin (Sigma Aldrich; St. Louis, MO). Caspase-3 Activity Cells were grown to 60-70% confluence prior to serum starvation for 24 hours. After treatment with FAF-BSA or stearate for 12 hours, cells were washed with 1X PBS and caspase-3 activity was measured using the EnzCheck Capase-3 Activity Kit #1 (Invitrogen; Carlsbad, CA) according to the manufacturer s instructions.
125 109 Statistics All data represent means +/- the S.E.M. ANOVA calculations were carried out using the SigmaStat 3.1 software program and statistical significance was confirmed using Tukey tests. Tumor metastasis was examined by the Cochran-Armitage Test for trend. Differences in tumor weight were tested by the generalized linear models with Tukey adjustment for multiple comparisons. The analyses of subcutaneous tumor volume and body weight were performed using a repeated measures model with PROC MIXED (SAS Ver. 9.1). The effects of treatment group, time in days and the interaction of treatment group and time were evaluated by F tests. Curvature in the models was tested for by a quadratic term for time. The a priori planned comparisons of specific differences in predicted treatment means averaged over time and at the last timepoint were computed by t-statistics. For all analyses a two-sided p value of < 0.05 was deemed statistically significant. RESULTS Based on the known in vitro and in vivo data, we hypothesized that dietary stearate would inhibit breast cancer cell metastasis. To test this hypothesis, we injected MDA-MB-435 breast cancer cells into the mammary fat pad of athymic mice and monitored the animals for metastases to the lungs. Diets, Food Intake and Weight Gain With the help of Harlan Teklad, three experimental diets were developed - one low fat and two high fat. The low fat diet was 5% fat derived from corn oil and
126 110 comparable to normal rodent chow. The stearate diet was 17% pure stearic acid and 3% safflower oil to ensure the animals received adequate amounts of the essential fatty acids. Finally, a 20% safflower oil diet was used as a positive control, as linoleic acid, the predominant fatty acid in safflower oil, has been shown to promote metastasis in this model (108, 174, 175). Because the safflower oil and stearate diets were isocaloric, but not the low fat diet, we monitored food consumption and weight gain to ensure the animals did not have significant discrepancies in energy intake. As shown in Figure 5-2A, the low fat diet animals consumed the most kilocalories/day, followed by the stearate diet animals, and then the safflower oil diet ingesting mice. Despite differences in food intake, there was no overt difference in weight gain between the diets (Figure 5-2B). The slight decrease in weight at week 10 was mostly likely due to recovery from the tumor removal at week 9. The relatively large increase in mouse weight observed in week 1 is consistent with the normal nude mouse growth pattern (Harlan, data not shown). The drop off in mouse weight seen with all three diets from week is likely due to an overall decrease in health due to metastases however we cannot rule out other causes. To determine if there were differences in overall weight gain, the total weight gain at week 10, minus the weight of the tumor, was determined. No significant difference was observed between the three diets (Figure 5-2C). The stearate (B) group was nearly identical to the stearate (A) group and, therefore, was not shown.
127 111 Mammary Tumor Analysis and Metastatic Burden of Low fat, Safflower, Stearate (A), and Stearate (B) Diets Following injection of the MDA-MB-435 cancer cells, the length and width of the tumors was measured weekly and used to calculate the volume (Figure 5-3a). Between 1 and 3 weeks post-injection, no significant difference was seen in the mean tumor volumes of the four groups. Starting four weeks post-injection, the stearate groups began to become significantly smaller than the other two diets. This trend continued until the 9 th week post-injection. At that point, the tumors in the low fat, safflower and stearate (A) groups were excised. As described in the materials and methods and shown in Figure 5-1, the stearate (B) tumors were allowed to grow longer until the size reached that of the low fat and safflower tumors. Figure 5-3B depicts representative images of the tumors present in the three different diets. Primary tumor weights are shown in Figure 5-3C. The tumors from the low fat and safflower fed groups (black and grey solid bars) were significantly larger than the stearate (A) fed animals. The stearate (B) fed mice tumors were significantly different in weight than the safflower tumors, but not the stearate (A) or low fat fed mice tumors. As shown in Figure 5-4A, the stearate diet significantly decreased the number of lung metastases compared to the mice fed either the low fat or safflower diets. When the number of metastases was quantitated by diet, a larger number of stearate fed animals had lower lung tumor burdens as compared to the low fat and safflower diets, indicating stearate successfully inhibited metastases (Figure 5-4B). When the tumor volume to metastatic burden was calculated, no difference was observed indicating this effect may be independent of the primary tumor size (Figure 5-4C).
128 112 Mammary Tumor Analysis and Metastatic Burden of the Stearate (B) Subgroups When using the mammary fat pad model of metastasis, the size of the primary tumor appears to correlate to the number of metastases on the lung surface (178). Therefore, we hypothesized, if stearate decreases the size of the primary mammary fat pad tumor, any decrease in metastasis would be due to this decrease in primary tumor size rather than an effect on another part of the metastatic cascade. To test this hypothesis, a second group of athymic mice carrying human tumor loads was fed the stearate diet, but rather than removing the primary tumors at the time the tumors were removed from the low fat and safflower animals, the tumors were allowed to grow larger. One of the most striking results of this experiment was that stearate (B) animals displayed a much larger difference in the range of tumor volumes. Tumors in subgroups B-i and B-ii were removed as they reached approximately 10mm 2-12mm 2 in size (diagrammed in Figure 5-1). Since it appeared the tumors in the B-iii group reached a plateau 6 weeks post-injection, it was decided to remove the primary tumors 13 weeks post-injection. Tumor volumes for these groups are shown in Figure 5-5A. Interestingly, the tumor volumes were not statistically significant until 8 weeks post-injection. Week 11 post-injection was the only point in which all three groups were statistically significant. The tumors in B-i and B-ii appeared to continue growing, even in the presence of the stearate diet, whereas the B-iii reached a maximum volume at six weeks. Comparing the volume of the tumors at the times of removal for each sub group, B-iii tumors were significantly smaller than both B-i and B-ii tumors. No difference was observed between B-i and B-ii (Fig. 5B) and the tumor weight and volume of B-i and B-ii
129 113 groups were not different than tumors of the low fat and safflower groups. However, as with the tumor volume, the average weight of the B-iii tumors was significantly less than the other two groups. Finally, each stearate (B) subgroup was sacrificed four weeks after primary tumor removal (Figure 5-1) and the number of metastases was counted. There was no significant difference in the number of macroscopic metastases present between the subgroups; B-i, B-ii, and B-iii had 15 +/-7, 4 +/- 2, and 6 +/-3 average metastatic lesions per animal, respectively. The slightly larger average for the B-i group was most likely due to one mouse that had 52 macroscopic lung metastases. The number of metastatic tumors per animal was similar for the three subgroups irrespective of the fact that primary tumor weight was significantly lower in the B-iii subgroup compared to B-i and B-ii (Figure 5-5C). Thus, the stearate diet appears to decrease the number of metastases independent of primary tumor size. In addition, the primary tumor sizes for B-i and B-ii were similar to low fat and safflower groups but with significantly fewer metastases per animal, also indicating that stearate decreases metastasis independent of primary tumor size. Stearate Induces Apoptosis of MDA-MB-435 Breast Cancer Cells In Vitro As stearate has been shown to induce apoptosis of human breast cancer cells in vitro, we tested MDA-MB-435 cells for cleaved caspase-3 and cleaved PARP following treatment with stearate. As shown in Figure 5-6A, stearate induces cleavage of caspase-3 and its downstream target PARP. To determine what concentration of stearate induces caspase-3 activity, cells were treated with concentrations ranging from M.
130 114 Stearate begins to induce apoptosis of the MDA-MB-435 breast cancer cells at a concentration of approximately 25 M (Figure 5-6B). DISCUSSION Stearate has been found to inhibit invasion, inhibit proliferation and induce apoptosis of breast cancer and other cells, as well as induce cytotoxicity in a variety of normal and malignant cell lines (73, 74, 76, 179). This body of work indicates the possibility of stearate to functioning as an anti-metastatic agent in vivo. This is the first study to demonstrate that stearate does inhibit breast cancer metastasis in vivo and does so by approximately 50%. Interestingly stearate also inhibits the primary tumor size and weight by approximately 50% although stearate s inhibition of metastasis was independent of primary tumor size. While this does not necessarily rule out a relationship between the effects of stearate on the primary tumor and decreased metastasis it certainly increases the possibility that stearate affects another part of the metastatic cascade. In vitro, stearate induced apoptosis of the MDA-MB-435 breast cancer cells in a dose dependent manner (Figure 5-6). As 50 M can be considered a normal nonesterified stearate concentration in the plasma of humans, this suggests that physiological concentrations of stearate are sufficient to induce apoptosis of breast cancer cells. This may partially explain the decrease of tumor size and metastases seen in the stearate-fed mice compared to the low fat and high safflower fed mice. However, because the primary tumors have similar growth patterns up to 3 weeks post-injection, if stearate is affecting the viability of the cancer cells, it is most likely not acting through an initial selection of sub-populations of cells that are sensitive to the apoptotic effects of stearate.
131 115 The three week delay in dietary stearate decreasing primary tumor volumes may suggest changes in the tumor microenvironment such as an inhibition of angiogenesis. Compounds derived from arachidonic acid and those derived from EPA appear to have opposing roles in the cell. Studies have found that linoleic acid enhanced the development of tumors in animals that were injected in the mammary fat pad with MDA- MB-435 or MDA-MB-231 breast cancer cells. To date, the mechanism underlying this increase is unknown. However, it has been hypothesized that the increase in linoleic acid causes an increase in arachidonic acid, thereby increasing prostaglandin synthesis (Reviewed in 180, 181). Consistent with this hypothesis, treatment of animals on a high linoleic acid diet with indomethacin, a cyclooxygenase inhibitor, inhibited prostaglandin synthesis and linoleic acid induced metastasis (109). Omega-3 fatty acids such as EPA, and DHA have been shown to inhibit tumor angiogensis and breast cancer cell metastasis, perhaps due to a difference in lipid metabolism or an inhibition of arachidonic-derived eicosanoids (112, 113, 177, 182). To date, the role of saturated fatty acids on prostaglandin synthesis has not been investigated in breast cancer tumors. However, in vitro, 20 M stearate is sufficient to almost completely inhibit cyclooxgenase-1 activity and partially inhibit cyclooxygenase-2 activity (183). Furthermore, stearate has been shown to induce apoptosis of human endothelial cells, although the M concentrations tested are within the pathophysiological range (159, 184). These results indicate stearate may inhibit angiogenesis by inhibiting prostaglandin synthesis or affecting vessel viability in the mammary fat pad tumors, and, therefore, may cause a decrease in primary tumor size and inhibit metastasis.
132 116 Stearate could also be modulating the immune response of nude mice to the tumors, thereby accounting for the variation observed in the stearate (B) subgroups. These animals have been shown previously to display a large amount of heterogeneity concerning specific T-cell subpopulations (185). This suggests that although the mice are an inbred population, the immune response to the tumor may vary between mice. Interestingly, macrophages, cells known to be modulated by fatty acids, have been extensively studied for their cancer promoting or cancer inhibiting effects. Two lineages of macrophages have been uncovered that effect tumors in opposing manners. The M1 lineage is generally associated with tumor suppression and resistance whereas the M2 linage is associated with tumor promotion and angiogenesis (Reviewed in (186). Although much work has been done analyzing the effect of n-3 and n-6 fatty acids in regard to immune response, little is known concerning the saturated fatty acids. Stearate has been demonstrated previously to induce apoptosis of the murine macrophage cell line J774 (187). Dietary stearate has been associated with a decrease in natural killer T-cells in the spleen of mice, whereas palmitate, a 16-carbon saturated fatty acid has been associated with an increase in activity (188). NK cells are known to stimulate M2 macrophages through the release of certain cytokines (186). These results suggest that dietary stearate may inhibit the differentiation of monocytes into M2 macrophages, thereby inhibiting tumor progression. These data also suggest that the long chain saturated fatty acids have differing effects in vivo. Future studies are necessary to determine the cause of the decrease in tumor size. Interestingly, the animals in the stearate (B) group displayed a range in terms of tumor growth, but not macroscopic metastases. The tumors in the B-i and B-ii subgroups
133 117 reached a size comparable to the low fat and safflower groups although an additional 2-3 weeks of tumor growth were necessary for this to occur (9 weeks post-injection for the low fat and safflower animals versus 11 and 12 weeks post injection for B-i and B-ii, respectively). The tumors in the B-iii group reached a maximum volume approximately 6 weeks after cancer cell injection. The underlying cause of this plateau is unknown although stearate could be inhibiting proliferation or inducing apoptosis of the cancer cells. Stearate has been shown previously to inhibit epidermal growth factor dependent proliferation in the Hs578t human breast cancer cells and we present evidence here that the MDA-MB-435 breast cancer cells are sensitive to stearate induced apoptosis (76); Figure 5-6). Furthermore, as cancer cell masses are generally accepted as a heterogeneous population of neoplastic, there could be a selection occurring for stearateinsensitive cells. Further studies are necessary to test these hypotheses. Given the complexity of dietary studies, the effect seen with the high stearate diet may be due to a decrease in linoleate, the predominate fatty acid in safflower oil. Safflower oil is composed of 77% linoleic acid. The stearate diet was 3% safflower oil, meaning the diet was 2.3% linoleic acid as compared to the 15.5% in the safflower diet. Using the same mammary fat pad injection model with the MDA-MB-435 breast cancer cells, Rose et al. reported that significantly more macroscopic lung metastases in animals fed a diet composed of 12% linoleic acid than animals fed a diet with 2% linoleic acid, although no difference was observed in the growth rate of the primary tumor. (108, 175). These results would argue our effect may, at least in part, be due to a decrease in linoleic acid. However, corn oil is 56% linoleic acid, meaning that our low fat diet was composed of 2.8% linoleic acid. Given no significant difference was observed between
134 118 the low fat (2.8% linoleic acid) and safflower (15.5% linoleic acid) diets in terms of tumor volume or lung metastases, the effects seen with the stearate diet are most likely not due to a decrease in linoleic acid. Also, there was a significant reduction in primary tumor and metastasis seen with the stearate diet compared to the low fat diet that contained comparable amounts of linoleic acid. In summary, dietary stearate inhibited the growth of MDA-MB-435 human breast cancer cells in the mammary fat pad model system and partially reduced metastatic burden in the lung. The inhibition of metastasis was independent of the size of the primary tumor as animals that developed larger tumors also had an inhibition of metastasis. Physiological concentrations of stearate were also sufficient to induce apoptosis in vitro, providing a potential mechanism. There is also evidence in the literature to support other potential in vivo mechanisms, including inhibition of angiogenesis and modulation of the immune system, and these are on going areas of investigation in our laboratory. The degree of inhibition of metastasis by dietary stearate indicates that it may be a potential adjuvant therapeutic strategy for breast cancer patients to increase the suppression of metastatic disease.
135 Table 5-1: Composition of the diets. 119
136 Figure 5-1: Experimental Timetable. Nude mice were placed on either a low fat diet, a high linoleic diet (Safflower) or a high stearic acid diet (Stearate (A)) for 3 weeks prior to injection with cancer cells. Tumors were allowed to develop for 9 weeks and were removed. At 4 weeks post-tumor removal, animals were sacrificed and their lungs were analyzed for metastases. A fourth group of animals was placed on the high stearic acid diet (Stearate (B)) and were injected with cancer cells 3 weeks later. The tumors were allowed to grow to the size of the tumors in the low fat and safflower groups before removal. Due to variation in tumor growth within this stearate group, it was broken down into 3 smaller groups B-i, B-ii, and B-iii based on when the tumors were removed. After 4 weeks, animals were sacrificed and their lungs were analyzed for metastases. 120

137 Figure 5-2: Food Intake and Weight Gain. A) The average kcal consumed per animal per day was calculated. Of the three diets, the animals on the low fat diet ate the most, followed by the high stearate diet and then the safflower oil diet. Each group s intake was significantly different from the other two diets ( p value of stearate (A) vs. low fat <0.001; p value of low fat vs. safflower <0.001; *p value of stearate (A) vs. safflower <0.001 by ANOVA). B) The weight of the animals was measured once a week for the duration of the experiment. Although differences were observed at individual weeks in the experiment, overall no significant change was seen. (n=14-21 per diet per week; p value of stearate (A) vs. low fat <0.05; *p value of stearate vs. safflower <0.05 by ANOVA). The second stearate group was not different from the first and was therefore not represented on the graphs. C) The total weight gain of the animals at week 10 was determined. The weight does not include the weight of the tumors removed from the animals. No significant difference was observed between the three groups. 121
138 Figure 5-3: Tumor Weights and Volumes for the Low fat, Safflower, and Stearate (A) Groups. A) Mammary fat pad tumors of the animals were measured weekly after injection. Based on the estimated volumes, stearate began to decrease the average volume of the tumors approximately 4 weeks post-injection. The stearate (A) and stearate (B) primary tumors were not different. (n=20-21 animals per diet; p value of stearate (A) vs. low fat <0.009; p value of stearate (B) vs. low fat<0.021; *p value of stearate (A) vs. safflower <0.003; **p value of stearate (B) vs. safflower<0.015 by ANOVA). When the data were analyzed using a repeated measures model and the curvature of the data points were estimated, all diets were different at week 9 except low fat vs. safflower (p<0.05). B) Immediately prior to removal of the mammary fat pad tumors at week 9, photographs were taken of mice in the low fat, safflower, and stearate (A) groups. All three of these tumors were removed on the same day. Representative images are shown. Animals on the stearate (A) diet tended to have noticeably smaller 122
139 tumors compared to the other two diets. C) Following tumor removal, tumors were weighed and averages were calculated for each dietary group. Note: safflower oil, low fat and stearate A primary tumors were removed at 9 weeks post injection while the stearate B group tumors were removed and weighed at 11, 12 and 13 weeks post injection of cells. No difference was seen between the low fat and safflower oil treated animals, the low fat and stearate (B) animals, or the stearate (A) and stearate (B) animals. Differences were observed between all other diets (n=20-21; p value of low fat vs. stearate (A) <0.002; *p value of stearate (A) vs. safflower <0.001; **p value of stearate (B) vs. safflower <0.003 by ANOVA). 123
140 Figure 5-4: Analysis of Lung Metastases from the Low fat, Safflower and Stearate (A) Groups. A) The number of macroscopic lung metastases was counted following necropsy. Stearate significantly decreased the number of macroscopic lung metastases in both the stearate (A) and stearate (B) groups. (n=18-20 animals per diet; p value of stearate (A) vs. low fat <0.021; *p value of stearate (A) vs. safflower<0.024; p value of stearate (B) vs. low fat<0.022; **p value of stearate (B) vs. safflower<0.024 by Cochran- Armitage trend test). B) The animals on the low fat and safflower diets tended to have more metastatic tumors than those on the stearate diet. Interestingly, a large difference was not seen between the two groups of animals fed the stearate diet. C) When the tumor volume to metastases was calculated, no difference was observed. 124
141 Figure 5-5 Tumor Analysis of Animals on the Stearate (B) Diet. A) Tumor volumes were calculated weekly for each of the stearate (B) sub groups. Approximately week 8 post injection, 2c became statistically smaller than B-ii and around week 10 than 2b. At week 11 post injection, all groups were different. (n=7 animals per group; p value of B- i vs. B-iii <0.001; &p value of B-ii vs. B-iii <0.002; #p value of B-i vs. 2bii <0.02). B) After the tumors were removed, they were weighed. No difference was observed between B-i and B-ii. B-iii tumors were statistically smaller than both B-i and B-ii. C) There was no difference in the metastatic tumor number per animal between the three stearate (B) subgroups. 125
142 Figure 5-6 Stearate Induced Apoptosis of MDA-MB-435 Breast Cancer Cells In Vitro. A) Cells were treated with 50 M stearate for the times indicated and the presence of cleaved caspase-3 and cleaved PARP was determined by immunblot. As shown, stearate induces cleavage of the two proteins hours post-treatment indicating stearate is inducing apoptosis of the breast cancer cells (n=3). B) Cells were treated with M stearate for 12 hours and caspase-3 activity was measured using a fluorescence based assay. Stearate activated caspase-3 in a dose dependent manner (n=4; *p<0.031; **p<0.023; ***p<0.012 compared to 0 M Student t-test). 126
143 127 CHAPTER 6 DIETARY STEARATE REDUCES PRIMARY TUMOR BURDEN IN AN ORTHOTOPIC MODEL OF BREAST CANCER As shown in Chapter 5, dietary stearate reduces the size of the primary tumor that forms when MDA-MB-435 cells are injected into the mammary fat pad of athymic nude mice. To determine the cause of the stearate-induced decrease in primary tumor size, animals were placed on one of three diets a low fat diet, a high stearate diet or a high safflower oil diet (see Table 5-1 for diet compositions) and two weeks later injected with breast cancer cells. Tumors were allowed to grow for four weeks before they were removed. Animals were then sacrificed three weeks after tumor removal and soft organs and plasma were collected at the time of necropsy (see Figure 6-1 for experimental design). Initially, we wanted to investigate metastasis using this model, however we did not allow the primary tumors to grow large enough and, therefore, the lungs were completely devoid of tumors. After animals were placed on the diets, we monitored their daily food intake and weekly weight gain. As shown in Figure 6-2A, animals on the high stearate diet consumed more food than those on the control or safflower oil diets. However, as shown in Figure 6-2B, no difference was observed in weight gain. At the time of necropsy, plasma was collected and analyzed for fatty acid content. As shown in Figure 6-3, animals on the stearate diet had a higher percent distribution of stearic acid in their plasma than palmitic acid, where as the inverse was seen for animals on the low fat diet. Furthermore, animals
144 128 on the safflower oil diet had significantly higher distribution of linoleic acid in their plasma than animals on the other two diets. These results indicate the diets are successfully absorbed by the animals. After animals were injected with tumor cells, they were monitored for the development of primary tumors. Primary tumors were measured weekly post-injection. As shown in Figure 6-4A, animals on the stearate diet had significantly smaller tumors by week 4 post-injection than animals on the safflower and low fat diets. Furthermore, the stearate tumors weighed significantly less than tumors removed for animals on the other two diets (Figure 6-4B). To determine the cause of the smaller tumors from animals on the stearate diet, tumors were analyzed for proliferation, apoptosis, angiogenesis, and necropsy. In vitro, stearate is known to inhibit proliferation and induce apoptosis. Therefore, we hypothesized one of these mechanisms may be the reason behind the smaller tumors in the stearate group. First, tumor sections were stained for Ki67 to measure proliferation and the number of positive nuclei per 1000 cells was counted. As shown in Figure 6-5A, no difference was observed between the diets. Next, tumors sections were stained for cleaved caspase-3 and the number of positive cells in 1000 cells was counted to determine if stearate was preferentially inducing apoptosis of the tumor cells. As shown in Figure 6-5B, no difference was observed between the three diets. Because there was no difference in proliferation or apoptosis, we hypothesized that stearate may be decreasing angiogenesis. Sections were stained with CD-31 and the number of vessels in the three areas densest with vessels was counted for each tissue section. As shown in Figure 6-5C, stearate and safflower oil both decreased the number
145 129 of vessels in the tumor sections indicating they both inhibit angiogenesis. Because the safflower oil tumors are significantly larger than the stearate tumors, the decrease in vasculature is mostly likely not enough to explain the decrease tumors size. Finally, to determine if stearate was increasing tissue necrosis in the tumors, the amount of necrotic area per total tumor area in each tumor sections as determined on the H&E stained slides using Bioquant Software. As shown in Figure 6-5D, no difference was observed between the three diets although the p value between the control and safflower diets was 0.08 indicating safflower oil may slightly increase the viability of the tumor cells. Currently we are unsure why the stearate tumors are smaller. Because stearate is known to induce caspase-independent forms of cell death, tumor sections are being examined for other forms of cell death besides apoptosis (158).
146 Figure 6-1: Schematic of Mammary Fat Pad Injection 130
147 Figure 6-2: Animals on the Stearate Diet Consume Significantly More Food but There Is No Difference in Total Weight Gain. A) Animals on the stearate diet consumed significantly more food than animals fed the low fat or safflower oil diets. (*p<0.006 ANOVA). B) No difference was observed in weight gain between the three diets (n=20 animals/diet). 131
148 Figure 6-3: Distribution of Plasma Fatty Acids for Animals on the Low Fat, Safflower, and Stearate Diets. At the time of necropsy, plasma was collected and 3 animals/diet were analyzed for fatty acid distribution. Animals on the safflower oil and stearate diets should significantly more linoleate and stearate respectively than animals on the low fat diet (*p<0.03 ANOVA). 132
149 133 Figure 6-4: Tumors from Animals Fed the Stearate Diet are Smaller than Tumors from the Low Fat and Safflower Oil Diets. A) Tumors were measured beginning one week after the breast cancer cells were injected. By four weeks post-injection tumors on animals fed the stearate diet were significantly smaller than tumors on animals fed the low fat and safflower diets. (n=20 tumors/diet; *p value of stearate vs. control <0.05, p value of stearate vs. safflower <0.05 ANOVA). B) Four weeks after the injection of tumor cells, tumors were excised from the animals. At the time of tumor removal, tumors were weighed. Animals on the stearate diet has significantly smaller tumors than those on the low fat and safflower diets (n=20 tumors/diet; *p<0.05 ANOVA).
. B) Tumor sections were probed with an antibody to cleaved caspase-3 to determine the number of apoptotic cells.")
150 Figure 6-5: Immunohistochemical Investigation of Tumors from the Three Diets. A) Tumors sections were probed with an antibody to Ki67 and the number of positive nuclei was determined. No difference was observed between the three diets (n=6 sections/ diet). B) Tumor sections were probed with an antibody to cleaved caspase-3 to determine the number of apoptotic cells. No difference was observed between the three diets (n=6 sections/diet). C) Sections were probed with CD-31 and the number of vessels was counted. The safflower oil and stearate diets significantly decreased the number of tumor vessels. (n=9 sections/diet; *p<0.05). D) Bioquant was used to measure the % Necrotic Area/ Total Tumor Area on H&E stained sections. No difference was observed between the three diets although the p value for safflower vs. low was 0.08 indicating safflower oil may increase cell viability. (n=6 sections/diet). 134
151 135 CHAPTER 7 DISCUSSION, SUMMARY, AND CONCLUSIONS Despite six and a half decades of investigation, the effect of dietary fat on breast cancer development and progression remains unclear. In vitro and rodent studies indicate a clear role of fatty acids in breast cancer growth, invasion, migration and metastasis. Epidemiological studies, on the other hand, often produce conflicting, confusing results. This lack of consistency has caused some to conclude that breast cancer modulation with dietary fat may not be feasible. Based on the known epidemiological, animal, and cell culture studies done, however, I hypothesize that it is not the amount, source, or type of fatty acid present in the diet that affects breast cancer risk, but rather the amount of individual fatty acids. Furthermore, supporting the role for an effect of an individual fatty acid, exogenously supplied stearate appears to have various anti-cancer properties including the inhibition of proliferation, invasion and migration and induction of apoptosis in vitro and inhibition of metastasis in vivo. CELL CULTURE STUDIES Stearate induces apoptosis of the Hs578t and MDA-MB-435 breast cancer cells through a mechanism dependent on de novo DAG generation and PKC activation (Chapter 3). Stearate has been shown previously to induce apoptosis of the MDA-MB- 231 breast cancer cells, as does palmitate. Hardy et al., however, chose to focus on palmitate to elucidate the molecular mechanism and found that palmitate increased the
152 136 turnover of cardiolipin, the phospholipid that binds cyctochrome C to the inner mitochondrial membrane. (73). Although not investigated, it is plausible that stearate may also affect cardiolipin turnover. However, given that stearate and palmitate appear to be metabolized differently in vivo, stearate may be metabolized differentially in the breast cancer cells as well. Although this is the only paper to date that looks specifically at breast cancer cells, stearate s ability to induce apoptosis has been characterized in other cell lines. In nerve growth factor differentiated PC12 cells, stearate induced cleavage of caspase-8 and caspase-3 but not caspase-9, indicating stearate activated the extrinsic cascade. Further experimentation confirmed this finding and showed that stearate induced expression of both Fas and FasL (158). In the breast cancer cells, stearate activated the extrinsic and intrinsic cascade. Differential protein expression of Bid suggested that stearate was activating the extrinsic cascade and then the intrinsic cascade. Consistent with this finding, inhibition of either caspase-8 or caspase-9 reversed stearateinduced caspase-3 activity. Given that TNF generally elicits a pro-survival response and MDA-MB-435 and Hs578t cells are resistant to TRAIL-induced apoptosis up to 1 g/ml (MDA-MB-231 cells are sensitive to TRAIL and shown an effect at 50ng/mL), FasL-Fas is the most likely death receptor/ligand candidate induced by stearate (39, 189). The FasL promoter contains an NF B binding site suggesting that NF B activation is essential for transcription (190). Inhibition of NF B by peptide inhibitors reversed stearate-induced cell rounding after 12 hours of stearate treatment (Supplemental Figure A-1a). Furthermore, inhibition of I B Kinase (IKK), the enzyme responsible for the phosphorylation and degradation of I B so active NF B can translocate to the nucleus,
153 137 also reversed stearate induced cell rounding and stearate-induced caspase-3 activity (Supplemental Figures A-1b and A-1c). Although future studies are necessary to confirm the activation of FasL-Fas in stearate treated breast cancer cells, these results are very promising. As mentioned previously, treatment of cancer with a Fas-activating antibody is not possible because of extensive liver damage (52). Stearate, however, is not well incorporated into triacyglycerol or cholesterol esters in hepatoytes (191). This suggests that the toxicity associated with stearate activated Fas would be minimal in the liver. Furthermore, as stearate-induced apoptosis is specific to the cancer cells, this may serve as a potential way to selectively activate apoptotic signaling, whether through Fas or another death receptor. Through a mechanism that has not been investigated, protein kinase C appears to play a role in stearate-induced apoptosis. Inhibition of PKC with pharmacological inhibitors partially reversed stearate-induced caspase-3 activity. Because Triacsin C, a molecule known to prevent stearate induced de novo DAG synthesis also inhibited caspase-3 activity, stearate may be activating a classical or novel PKC that is dependent on DAG for proper activation. The PKC isozymes play a variety of different roles in apoptosis although, generally speaking, PKC is pro-apoptotic while PKC is antiapoptotic (192). Inhibition of stearate s metabolic activities with the pharmacologic inhibitors had identical results in terms of cell rounding and caspase-3 activation (Figure 2-2; Figure 3-4). Furthermore, cell rounding correlates to caspase-3 activity in terms of time, dose (Supplemental Figure A-2), and PKC inhibition with pan inhibitors. One of the criteria
154 138 for apoptosis put forth by the Nomenclature Committee on Cell Death 2009 is the rounding of cells (37). Therefore, cell rounding in the breast cancer cells is most likely due to the apoptosis. The ability of inhibition of PKC but not PKC to reverse cell rounding indicates PKC may be involved in stearate-induced apoptosis. PKC is generally characterized as an anti-apoptotic kinase as well as an oncogene. Over expression of PKC correlates with cellular transformation, colony growth in soft agar, and tumor growth in vivo. PKC has also been implicated in inhibiting Bax translocation to the mitochondria, thereby inhibiting MOMP. Expression of this isozyme also correlates with TRAIL resistance. (192).. There are very few studies suggesting PKC can aid in the induction of apoptosis. MCF-7 cells treated with Tamoxifen or Clomiphene show a strong activation of PKC as measured by translocation of the protein to the membrane. This translocation also correlates with the induction of apoptosis in the MCF-7 cell line. (193). Additionally, - radiation has been shown to induce translocation and activation of PKC and PKC to the particulate fraction (equivalent to the membrane fraction) of the cultured smooth muscle cell lysates. The -radiation also induced apoptosis of the smooth muscles and panpkc inhibitors and PKC V1-2 inhibited the induction of apoptosis. (194). Consistent with these findings, stearate also induces a translocation of PKC to the cell membrane and a subsequent activation (Supplemental Figure A-3; Figure 1-4). Taken together, these results indicate that PKC may play a pro-apoptotic role in stearateinduced apoptosis of human breast cancer cells. Because NF B is required for stearate-induced caspase-3 activity and cell rounding and PKC is necessary for cell rounding, there is a possibility the two proteins
155 139 may interact. In rabbit cardiomyocytes, activating of PKC increased NF B binding to DNA. PKC also increases the amount of p65 present in the nucleus, further confirming the isozyme may play a role in the activation of the classical NF B cascade. (195). PKC has been implicated in the activation of NF B-activating kinase (NAK). NAK phosphorylates IKK and induces activation of the kinase. The IKK complex can then phosphorylate I B, causing the activation of NF B. (196). Therefore, it is possible in stearate-treated cells that PKC and NF B are pro-apoptotic. Future studies are necessary to determine if this is accurate as it would define a novel role for PKC and NF B in cancer biology. IN VIVO DIETARY FAT STUDIES Nude mice fed a high stearate diet and injected with MDA-MB-435 cells into the mammary fat pad grow smaller primary tumors than their safflower and low fat diet counterparts. Stearate has been tested in spontaneous and carcinogenic models of mammary tumorigenesis, but this is the first example of dietary stearate affecting an orthotopic model. The spontaneous and carcinogen-induced models indicate stearate has a role in preventing cancer development. However, primary tumor reduction indicates stearate affects cancer cell viability in vivo. The mechanism by which this occurs is still under investigation, but it may involve a reduction in angiogenesis or a caspaseindependent cell death mechanism. In addition to inhibiting primary tumor growth, stearate also reduced the metastatic burden of the lungs. Allowing tumors to grow to the same size as the safflower and low fat diets did not effect tumor metastasis indicting the mechanism by which stearate is acting is independent of the primary tumor size. How is stearate
156 140 preventing metastasis? There are several potential mechanisms that could be occurring. Stearate could be activating a metastasis suppressor. Omega-6 and omega-3 fatty acids have been shown to differentially regulate expression of the nm-23 metastasis suppressor so it is plausible stearate could act via a similar mechanism (158). Stearate could also affect the invasion of cells from the primary tumor since stearate completely abolished breast cancer cell invasion after 24 hours (Figure 3-1). The role of fatty acids on breast cancer cell intravasation and extravasation has not been investigated, but it is possible stearate could inhibit one of these mechanisms. Furthermore, since there is a higher distribution of stearate in the plasma of the stearate diet fed animals, the cancer cells may not survive in the blood stream. Finally, stearate could prevent colonization or growth at the secondary site. The inhibition of colonization is most likely not the reason for the inhibition of metastasis. Tail vein injection of BALB/C mice with luciferase-tagged 4T1 (Luc-4T1) cells resulted in no difference in tumor number in the lungs between the low fat, safflower, and stearate diets (Supplemental Figure A-4a). Stearate appeared to induce death in the cell line 24 hours after treatment as shown by trypan blue exclusion (Figure A-4b). Future studies are needed to determine the pathway necessary for the decrease in tumor size and inhibition of metastasis. One of the most exciting aspects of the animal studies was the ability to enrich a diet with pure stearic acid without compromising the overall health of the animals. Diets were also composed of 3% safflower oil to ensure the animals received enough essential fatty acids. Such dietary modifications may be possible for sources of human food. This concept is discussed in detail below. Before discussing the enrichment of stearate, it is
157 141 reasonable to briefly review the food recommendations of the Food and Drug Administration (FDA), United States Department of Agriculture (USDA) and the America Dietetic Association (ADA). THE USDA FOOD PYRAMID In 1992, the USDA released a Food Guide Pyramid with the hopes of promoting overall American health and reducing the risk of chronic diseases associated with poor dietary habits. From its release, however, the pyramid has received great scrutiny. The base of the pyramid is 6 to 11 servings of complex carbohydrates. Fats, on the other hand, are to be eaten sparingly. This has lead to the misconception that carbohydrates are healthy while fats are not. Although the nutritionists who designed the pyramid were aware that not all fats promoted chronic diseases, such as increases in cholesterol and subsequent cardiovascular issues, they opted for a more simplistic view of food distribution. At the time, it was well understood that saturated fatty acids could increase cholesterol levels, and the thought was that by decreasing total fat, there would be a decrease in saturated fat intake. (158). Consistent with the notion that saturated fats are unhealthy, the FDA authorized a health claim in 1993 stating that dietary fat increases the risk for cancer (197). However, as has been explained throughout this dissertation, the exact role of dietary fat in cancer promotion is unclear. Certain fatty acids, such as the omega-3 fatty acids and stearate have anti-cancer properties. In recognizing that some omega-3 fatty acids can have positive health benefits, marketers of dietary supplements appealed the FDA ruling that dietary fat was not beneficial and asked to market supplements stating that omega-3 fatty acids may prevent coronary heart disease. In the
158 142 lawsuit Pearson v. Shalala, the marketers won with the support of a number of scientists who researched the subject. (198). A situation such as that listed above is useful in informing the public about the positive benefits of dietary fat. However, understanding the role of individual fatty acids in human health is a complicated one. For example, linoleate, which has been shown to promote breast cancer growth and metastasis lowers total and LDL cholesterol levels. So, from a heart disease prospective, linoleate is beneficial (158). As is discussed below, the lack of awareness concerning the negative health effects associated with linoleate could lead to a recommended concentration that is higher than necessary. Furthermore, the notion that saturated fats are unhealthy could result in a concentration of stearate that is below the effective concentration. DIETARY FAT RECOMMENDATIONS OF THE AMERICAN DIETETIC ASSOCIATION (ADA) AND THE DIETICIANS OF CANADA (DC) According to the ADA and DC, total fat intake should range between 20 and 35% of total energy intake. Within this recommendation, 3 to 10% of total fat should come from the omega-6 polyunsaturated fatty acids and 0.6 to 1.2% from omega-3 polyunsaturated fatty acids. Saturated fatty acids showed be consumed as little as possible and make up for no more than 10% of total fat, with recommendations as low as 3%. Monounsaturated fatty acids should be consumed to make up for the dietary fat percents not met by the other fatty acids. The report, however, acknowledges that stearate has no effect on cholesterol levels, although suggestions to increase stearate intake are not made. (199).
159 143 In recent years, numerous dietary studies and recommendations have shown that a high omega-6/omega-3 ratio promotes increases in viscosity of the blood, vasoconstriction of the blood vessels, and platelet aggregation. There is also a correlation between increased omega-6/omega-3 ratios and the risk of diabetes. (200). These negative health effects are often attributed to competition of linolenate and linoleate for the enzymes that lead to the formation of the eicosanoid precursors, EPA and arachidonate, respectively. As mentioned previously, many of the omega-3 derived eicosanoids are thought to have anti-inflammatory and anti-thrombotic effects whereas those derived from omega-6 fatty acids have pro-inflammatory, pro-thrombotic and carcinogenic effects (69). Furthermore, as the intake of omega-3 fatty acids increases, they have been shown to displace the omega-6 fatty acids in terms of not just eicosanoids, but also other lipid by-products such as phospholipids, and vice versa (201). The current ADA recommendations the lower limit of the omega6/omega3 ratio is 3/1 and the upper limit is 16/1. In 2000, the Nation Institute of Health hosted The Workshop on the Essentiality of and Recommended Dietary Intakes (RDIs) for Omega-6 and Omega-3 Fatty Acids. During this workshop, the attendees concluded humans should consume 2-3% energy from linolenate daily, 1% from linoleate and at least 0.3% DHA and EPA. (202). The recommended omega 6/omega 3 ratio based off these fat intakes ranges from 1.2/1 to 2.3/1. The ratio recommended by the workshop has been suggested by others to be the most beneficial in terms of human health (201). Based off of the known epidemiological and animal data about the risk of a high omega-6 fatty acid intake, it may be in the ADA s best interest to reduce the intake of omega-6 fatty acids. Also, recommending the decrease in saturated fatty acids may not be
160 144 the best way to promote for human health. While palmitate is associated with increased LDL cholesterol, the so-called bad cholesterol, chronic heart disease, and diabetes, these effects are not seen with stearate (169). Finally, based on the animal and in vitro studies, having monounsaturated fatty acids make up the difference in the diet could be detrimental to a person s health as they have been shown to promote tumor formation and breast cancer cell growth in experimental models ( , 128). The bottom line is the optimal concentrations of fat intake are not known. Not all unsaturated fatty acids are healthy and not all saturated fatty acids are unhealthy. More research is necessary to determine the optimal concentrations of individual fatty acids in the diet. MODULATING THE HUMAN DIET TO INCREASE STEARATE INTAKE Increasing the concentration of stearate in the diet could aid in several issues pertaining to breast cancer. First, an increase in stearate could help prevent breast cancer. The research presented in this dissertation strongly implicates stearate in preventing breast cancer development. Second, stearate could serve as an adjuvant therapy to treat breast cancer patients. Because other fatty acids, such as DHA, have been shown to increase the efficacy of chemotherapy, such studies are needed using a diet enriched in stearate. Thus it is important that combination studies be done with stearate and other dietary fatty acids to get optimum concentrations (i.e. reduce cancer promoting fats and increase stearate). To increase the concentration of stearate in the diet, one of several mechanisms could be employed. The high stearate diet discussed in chapters 5 and 6 is composed of a
161 145 very fine grain, high quality stearate as opposed to an oil enriched in stearate. By making stearate the predominant saturated fatty acid in the diet, many adverse health effects observed with other saturates may be avoided. Furthermore, the use of an enriched purestearate diet reduces the intake of other fatty acids that may inhibit the activity of stearate or induce carcinogenic processes. Such a form of stearate could be supplied as a supplement, such as in a vitamin, or as a powder to add to foods such as dietary fiber (i.e. Metamucil ). Another way to increase the amount of stearate in the diet is to increase the stearate present in food. Stearate is found in high concentrations in beef, milk fats, and chocolate. Studies have been performed enriching dairy cattle and sheep with stearate, either through duodenum infusion or dietary enrichment (203). Such studies indicate modulations of the lipid profiles of animals is possible (203, 204). Furthermore, research has indicated that stearate does not accumulate in the liver of diary cattle. Fatty liver in dairy cattle is a problem associated with decreased fertility of a lactating cow. (204, 205). Subsequently, the company MSC Specialty Nutrition has recently developed a product known as Energy Booster 100 that is enriched in stearate and is highly absorbable for the cattle. Such a supplement could be fed to the animals to increase the amount of stearate in their milk and muscle. SUMMARY Stearate induces apoptosis of human breast cancer cells but not non-cancerous breast cells through a mechanism dependent on de novo DAG synthesis, PKC activation, and NF B activity. Studies measuring cell rounding in response to stearate treatment that
162 146 correlate with caspase activity indicate that PKC may be involved in the induction of apoptosis. To induce apoptosis, stearate is activating the extrinsic cascade, possibly through the Fas-FasL pathway. The extrinsic cascade then appears to activate the intrinsic cascade through the cleavage of Bid. As diagram of the hypothetical stearateinduced apoptosis pathway is shown in Figure 7-1. Future studies are necessary to confirm the PKC isozyme(s) responsible for stearate induced apoptosis and to determine the pro-apoptotic role of NF B. In vivo, stearate decreases the size of mammary fat pad tumors formed by MDA- MB-435 breast cancer cells. The mechanism behind this decrease is not clear, but stearate does not appear to affect proliferation or apoptosis (Figure 6-5). Although a decrease in CD-31+ vasculature was observed in the stearate diet, the effect was also seen in the safflower diet indicating angiogenesis may not be the mechanism by which stearate is targeting. Stearate may be inducing a caspase-independent form of cell death. Future studies are necessary determine if another death cascade is activated in vivo. Stearate also decreased the number of macroscopic lesions in the lungs of mice injected in the mammary fat pad with the MDA-MB-435 breast cancer cells. The mechanism most likely does not involve an inhibition of colonization at the secondary site since there is no difference in tumor growth between the diets when Luc-4T1 cells are injected via tail vein. Future studies are necessary to determine the mechanism of inhibition of metastasis.
163 147 CONCLUSION The saturated fatty acid stearate appears to have numerous anti-cancer properties both in vitro and in vivo. Taken together with the epidemiological studies suggesting it may decrease breast cancer risk, stearate should be seriously considered for pre-clinical and possibly phase-i trials to attempt to prevent and treat breast cancer progression.
164 Figure 7-1: Theoretical Diagram of Stearate-Induced Apoptosis in Human Breast Cancer Cells. 148
165 149 LIST OF REFERENCES 1. Jemal, A., Siegel, R., Ward, E., Hao, Y., Xu, J., Murray, T., and Thun, M. J. Cancer statistics, CA Cancer J Clin, 58: 71-96, Ingber, D. E. Can cancer be reversed by engineering the tumor microenvironment? Semin Cancer Biol, 18: , Wellings, S. R., Jensen, H. M., and Marcum, R. G. An atlas of subgross pathology of the human breast with special reference to possible precancerous lesions. J Natl Cancer Inst, 55: , Arpino, G., Laucirica, R., and Elledge, R. M. Premalignant and in situ breast disease: biology and clinical implications. Ann Intern Med, 143: , Sims, A. H., Howell, A., Howell, S. J., and Clarke, R. B. Origins of breast cancer subtypes and therapeutic implications. Nat Clin Pract Oncol, 4: , Allred, D. C., Mohsin, S. K., and Fuqua, S. A. Histological and biological evolution of human premalignant breast disease. Endocr Relat Cancer, 8: 47-61, Singletary, S. E. A working model for the time sequence of genetic changes in breast tumorigenesis. J Am Coll Surg, 194: , Allred, D. C., Wu, Y., Mao, S., Nagtegaal, I. D., Lee, S., Perou, C. M., Mohsin, S. K., O'Connell, P., Tsimelzon, A., and Medina, D. Ductal carcinoma in situ and the emergence of diversity during breast cancer evolution. Clin Cancer Res, 14: , Deng, G., Lu, Y., Zlotnikov, G., Thor, A. D., and Smith, H. S. Loss of heterozygosity in normal tissue adjacent to breast carcinomas. Science, 274: , Sorlie, T., Tibshirani, R., Parker, J., Hastie, T., Marron, J. S., Nobel, A., Deng, S., Johnsen, H., Pesich, R., Geisler, S., Demeter, J., Perou, C. M., Lonning, P. E., Brown, P. O., Borresen-Dale, A. L., and Botstein, D. Repeated observation of breast tumor subtypes in independent gene expression data sets. Proc Natl Acad Sci U S A, 100: , 2003.
166 Carey, L. A., Perou, C. M., Livasy, C. A., Dressler, L. G., Cowan, D., Conway, K., Karaca, G., Troester, M. A., Tse, C. K., Edmiston, S., Deming, S. L., Geradts, J., Cheang, M. C., Nielsen, T. O., Moorman, P. G., Earp, H. S., and Millikan, R. C. Race, breast cancer subtypes, and survival in the Carolina Breast Cancer Study. Jama, 295: , Sorlie, T., Perou, C. M., Tibshirani, R., Aas, T., Geisler, S., Johnsen, H., Hastie, T., Eisen, M. B., van de Rijn, M., Jeffrey, S. S., Thorsen, T., Quist, H., Matese, J. C., Brown, P. O., Botstein, D., Eystein Lonning, P., and Borresen-Dale, A. L. Gene expression patterns of breast carcinomas distinguish tumor subclasses with clinical implications. Proc Natl Acad Sci U S A, 98: , Ellsworth, R. E., Ellsworth, D. L., Deyarmin, B., Hoffman, L. R., Love, B., Hooke, J. A., and Shriver, C. D. Timing of critical genetic changes in human breast disease. Ann Surg Oncol, 12: , Allred, D. C. and Mohsin, S. K. Biological features of premalignant disease in the human breast. J Mammary Gland Biol Neoplasia, 5: , Steeg, P. S. Tumor metastasis: mechanistic insights and clinical challenges. Nat Med, 12: , Lu, X. and Kang, Y. Organotropism of breast cancer metastasis. J Mammary Gland Biol Neoplasia, 12: , Welch, D. R. Do we need to redefine a cancer metastasis and staging definitions? Breast Dis, 26: 3-12, Eccles, S. A. and Welch, D. R. Metastasis: recent discoveries and novel treatment strategies. Lancet, 369: , Bernards, R. and Weinberg, R. A. A progression puzzle. Nature, 418: 823, Tubiana, M. and Koscielny, S. The rationale for early diagnosis of cancer--the example of breast cancer. Acta Oncol, 38: , Tresserra, F., Rodriguez, I., Garcia-Yuste, M., Grases, P. J., Ara, C., and Fabregas, R. Tumor size and lymph node status in multifocal breast cancer. Breast J, 13: 68-71, Duffy, M. J., McGowan, P. M., and Gallagher, W. M. Cancer invasion and metastasis: changing views. J Pathol, 214: , Horak, C. E., Lee, J. H., Marshall, J. C., Shreeve, S. M., and Steeg, P. S. The role of metastasis suppressor genes in metastatic dormancy. Apmis, 116: , 2008.
167 Alexandrova, A. Y. Evolution of cell interactions with extracellular matrix during carcinogenesis. Biochemistry (Mosc), 73: , Wyckoff, J. B., Jones, J. G., Condeelis, J. S., and Segall, J. E. A critical step in metastasis: in vivo analysis of intravasation at the primary tumor. Cancer Res, 60: , Dua, R. S., Gui, G. P., and Isacke, C. M. Endothelial adhesion molecules in breast cancer invasion into the vascular and lymphatic systems. Eur J Surg Oncol, 31: , Pollard, J. W. Macrophages define the invasive microenvironment in breast cancer. J Leukoc Biol, 84: , Tsuruo, T. and Fujita, N. Platelet aggregation in the formation of tumor metastasis. Proc Jpn Acad Ser B Phys Biol Sci, 84: , Varki, N. M. and Varki, A. Heparin inhibition of selectin-mediated interactions during the hematogenous phase of carcinoma metastasis: rationale for clinical studies in humans. Semin Thromb Hemost, 28: 53-66, Miles, F. L., Pruitt, F. L., van Golen, K. L., and Cooper, C. R. Stepping out of the flow: capillary extravasation in cancer metastasis. Clin Exp Metastasis, 25: , Fidler, I. J. and Poste, G. The "seed and soil" hypothesis revisited. Lancet Oncol, 9: 808, Kaplan, R. N., Rafii, S., and Lyden, D. Preparing the "soil": the premetastatic niche. Cancer Res, 66: , Psaila, B., Kaplan, R. N., Port, E. R., and Lyden, D. Priming the 'soil' for breast cancer metastasis: the pre-metastatic niche. Breast Dis, 26: 65-74, Kaplan, R. N., Riba, R. D., Zacharoulis, S., Bramley, A. H., Vincent, L., Costa, C., MacDonald, D. D., Jin, D. K., Shido, K., Kerns, S. A., Zhu, Z., Hicklin, D., Wu, Y., Port, J. L., Altorki, N., Port, E. R., Ruggero, D., Shmelkov, S. V., Jensen, K. K., Rafii, S., and Lyden, D. VEGFR1-positive haematopoietic bone marrow progenitors initiate the pre-metastatic niche. Nature, 438: , Chen, E. I., Hewel, J., Krueger, J. S., Tiraby, C., Weber, M. R., Kralli, A., Becker, K., Yates, J. R., 3rd, and Felding-Habermann, B. Adaptation of energy metabolism in breast cancer brain metastases. Cancer Res, 67: , 2007.
168 Vermeulen, K., Van Bockstaele, D. R., and Berneman, Z. N. Apoptosis: mechanisms and relevance in cancer. Ann Hematol, 84: , Kroemer, G., Galluzzi, L., Vandenabeele, P., Abrams, J., Alnemri, E. S., Baehrecke, E. H., Blagosklonny, M. V., El-Deiry, W. S., Golstein, P., Green, D. R., Hengartner, M., Knight, R. A., Kumar, S., Lipton, S. A., Malorni, W., Nunez, G., Peter, M. E., Tschopp, J., Yuan, J., Piacentini, M., Zhivotovsky, B., and Melino, G. Classification of cell death: recommendations of the Nomenclature Committee on Cell Death Cell Death Differ, 16: 3-11, Rupinder, S. K., Gurpreet, A. K., and Manjeet, S. Cell suicide and caspases. Vascul Pharmacol, 46: , Jin, Z. and El-Deiry, W. S. Overview of cell death signaling pathways. Cancer Biol Ther, 4: , Park, H. H., Lo, Y. C., Lin, S. C., Wang, L., Yang, J. K., and Wu, H. The death domain superfamily in intracellular signaling of apoptosis and inflammation. Annu Rev Immunol, 25: , Baker, S. J. and Reddy, E. P. Modulation of life and death by the TNF receptor superfamily. Oncogene, 17: , Micheau, O. and Tschopp, J. Induction of TNF receptor I-mediated apoptosis via two sequential signaling complexes. Cell, 114: , Duan, H. and Dixit, V. M. RAIDD is a new 'death' adaptor molecule. Nature, 385: 86-89, Cohen, G. M. Caspases: the executioners of apoptosis. Biochem J, 326 ( Pt 1): 1-16, Shearwin-Whyatt, L. M. and Kumar, S. Caspases in developmental cell death. IUBMB Life, 48: , Tinel, A. and Tschopp, J. The PIDDosome, a protein complex implicated in activation of caspase-2 in response to genotoxic stress. Science, 304: , Bao, Q. and Shi, Y. Apoptosome: a platform for the activation of initiator caspases. Cell Death Differ, 14: 56-65, Li, H. and Yuan, J. Deciphering the pathways of life and death. Curr Opin Cell Biol, 11: , 1999.
169 Ashkenazi, A. and Dixit, V. M. Apoptosis control by death and decoy receptors. Curr Opin Cell Biol, 11: , Bhardwaj, A. and Aggarwal, B. B. Receptor-mediated choreography of life and death. J Clin Immunol, 23: , Wu, Y., Han, B., Sheng, H., Lin, M., Moore, P. A., Zhang, J., and Wu, J. Clinical significance of detecting elevated serum DcR3/TR6/M68 in malignant tumor patients. Int J Cancer, 105: , Takeda, K., Stagg, J., Yagita, H., Okumura, K., and Smyth, M. J. Targeting deathinducing receptors in cancer therapy. Oncogene, 26: , Hotte, S. J., Hirte, H. W., Chen, E. X., Siu, L. L., Le, L. H., Corey, A., Iacobucci, A., MacLean, M., Lo, L., Fox, N. L., and Oza, A. M. A phase 1 study of mapatumumab (fully human monoclonal antibody to TRAIL-R1) in patients with advanced solid malignancies. Clin Cancer Res, 14: , Greco, F. A., Bonomi, P., Crawford, J., Kelly, K., Oh, Y., Halpern, W., Lo, L., Gallant, G., and Klein, J. Phase 2 study of mapatumumab, a fully human agonistic monoclonal antibody which targets and activates the TRAIL receptor-1, in patients with advanced non-small cell lung cancer. Lung Cancer, 61: 82-90, Plummer, R., Attard, G., Pacey, S., Li, L., Razak, A., Perrett, R., Barrett, M., Judson, I., Kaye, S., Fox, N. L., Halpern, W., Corey, A., Calvert, H., and de Bono, J. Phase 1 and pharmacokinetic study of lexatumumab in patients with advanced cancers. Clin Cancer Res, 13: , Khosravi-Far, R. and Esposti, M. D. Death receptor signals to mitochondria. Cancer Biol Ther, 3: , Kroemer, G. and Martin, S. J. Caspase-independent cell death. Nat Med, 11: , Esposti, M. D. Lipids, cardiolipin and apoptosis: a greasy licence to kill. Cell Death Differ, 9: , Lee, M. M. and Lin, S. S. Dietary fat and breast cancer. Annu Rev Nutr, 20: , Cerf, M. E. High fat diet modulation of glucose sensing in the beta-cell. Med Sci Monit, 13: RA12-17, Vidon, C., Boucher, P., Cachefo, A., Peroni, O., Diraison, F., and Beylot, M. Effects of isoenergetic high-carbohydrate compared with high-fat diets on human
170 154 cholesterol synthesis and expression of key regulatory genes of cholesterol metabolism. Am J Clin Nutr, 73: , Damjanovic, M. and Barton, M. Fat intake and cardiovascular response. Curr Hypertens Rep, 10: 25-31, Voet, D. a. V., J.G. Biochemistry, 3rd edition, p Hoboken, NJ: John Wiley & Sons, Inc., Berg, J. M., Tymoczko, J.L., Stryer, L. Biochemistry, 5th edition, p New York, NY: W. H. Freeman and Company, Voet, D. a. V., J.G. Biochemistry, 3rd edition, p Hoboken, NJ: John Wiley & Sons, Inc., Kuhajda, F. P., Jenner, K., Wood, F. D., Hennigar, R. A., Jacobs, L. B., Dick, J. D., and Pasternack, G. R. Fatty acid synthesis: a potential selective target for antineoplastic therapy. Proc Natl Acad Sci U S A, 91: , Voet, D. a. V., J.G. Biochemistry, 3rd edition, p Hoboken, NJ: John Wiley & Sons, Inc., Voet, D. a. V., J.G. Biochemistry, 3rd edition, p Hoboken, NJ: John Wiley & Sons, Inc., Larsson, S. C., Kumlin, M., Ingelman-Sundberg, M., and Wolk, A. Dietary longchain n-3 fatty acids for the prevention of cancer: a review of potential mechanisms. Am J Clin Nutr, 79: , Carroll, K. K. Experimental evidence of dietary factors and hormone-dependent cancers. Cancer Res, 35: , Bennett, A. S. Effect of dietary stearic acid on the genesis of spontaneous mammary adenocarcinomas in strain A/ST mice. Int J Cancer, 34: , Hardy, R., Wickramasinghe, N., Ke, S., and Wells, A. Fatty acids and breast cancer cell proliferation. Adv Exp Med Biol., 422: 57-69, Hardy, S., El-Assaad, W., Przybytkowski, E., Joly, E., Prentki, M., and Langelier, Y. Saturated fatty acid-induced apoptosis in MDA-MB-231 breast cancer cells. A role for cardiolipin. J Biol Chem, 278: , Singh, R. K., Hardy, R. W., Wang, M. H., Williford, J., Gladson, C. L., McDonald, J. M., and Siegal, G. P. Stearate inhibits human tumor cell invasion. Invasion Metastasis, 15: , 1995.
171 Tinsley, I. J., Schmitz, J. A., and Pierce, D. A. Influence of dietary fatty acids on the incidence of mammary tumors in the C3H mouse. Cancer Res, 41: , Wickramasinghe, N. S., Jo, H., McDonald, J. M., and Hardy, R. W. Stearate inhibition of breast cancer cell proliferation. A mechanism involving epidermal growth factor receptor and G-proteins. Am J Pathol, 148: , Williams, E. A., Coxhead, J. M., and Mathers, J. C. Anti-cancer effects of butyrate: use of micro-array technology to investigate mechanisms. Proc Nutr Soc, 62: , Byers, T. Nutritional risk factors for breast cancer. Cancer, 74: , Hunter, D. J., Spiegelman, D., Adami, H. O., Beeson, L., van den Brandt, P. A., Folsom, A. R., Fraser, G. E., Goldbohm, R. A., Graham, S., Howe, G. R., and et al. Cohort studies of fat intake and the risk of breast cancer--a pooled analysis. N Engl J Med, 334: , Howe, G. R., Hirohata, T., Hislop, T. G., Iscovich, J. M., Yuan, J. M., Katsouyanni, K., Lubin, F., Marubini, E., Modan, B., Rohan, T., and et al. Dietary factors and risk of breast cancer: combined analysis of 12 case-control studies. J Natl Cancer Inst, 82: , Boyd, N. F., Stone, J., Vogt, K. N., Connelly, B. S., Martin, L. J., and Minkin, S. Dietary fat and breast cancer risk revisited: a meta-analysis of the published literature. Br J Cancer, 89: , Prentice, R. L. and Anderson, G. L. The women's health initiative: lessons learned. Annu Rev Public Health, 29: , Prentice, R. L., Caan, B., Chlebowski, R. T., Patterson, R., Kuller, L. H., Ockene, J. K., Margolis, K. L., Limacher, M. C., Manson, J. E., Parker, L. M., Paskett, E., Phillips, L., Robbins, J., Rossouw, J. E., Sarto, G. E., Shikany, J. M., Stefanick, M. L., Thomson, C. A., Van Horn, L., Vitolins, M. Z., Wactawski-Wende, J., Wallace, R. B., Wassertheil-Smoller, S., Whitlock, E., Yano, K., Adams- Campbell, L., Anderson, G. L., Assaf, A. R., Beresford, S. A., Black, H. R., Brunner, R. L., Brzyski, R. G., Ford, L., Gass, M., Hays, J., Heber, D., Heiss, G., Hendrix, S. L., Hsia, J., Hubbell, F. A., Jackson, R. D., Johnson, K. C., Kotchen, J. M., LaCroix, A. Z., Lane, D. S., Langer, R. D., Lasser, N. L., and Henderson, M. M. Low-fat dietary pattern and risk of invasive breast cancer: the Women's Health Initiative Randomized Controlled Dietary Modification Trial. Jama, 295: , 2006.
172 Thacker, H. L. A low-fat dietary pattern intervention did not reduce incidence of breast cancer, colorectal cancer, or CVD in postmenopausal women. ACP J Club, 145: 6-7, Prentice, R. L., Thomson, C. A., Caan, B., Hubbell, F. A., Anderson, G. L., Beresford, S. A., Pettinger, M., Lane, D. S., Lessin, L., Yasmeen, S., Singh, B., Khandekar, J., Shikany, J. M., Satterfield, S., and Chlebowski, R. T. Low-fat dietary pattern and cancer incidence in the Women's Health Initiative Dietary Modification Randomized Controlled Trial. J Natl Cancer Inst, 99: , Yngve, A., Hambraeus, L., Lissner, L., Serra Majem, L., Vaz de Almeida, M. D., Berg, C., Hughes, R., Cannon, G., Thorsdottir, I., Kearney, J., Gustafsson, J. A., Rafter, J., Elmadfa, I., and Kennedy, N. The Women's Health Initiative. What is on trial: nutrition and chronic disease? Or misinterpreted science, media havoc and the sound of silence from peers? Public Health Nutr, 9: , Sieri, S., Krogh, V., Ferrari, P., Berrino, F., Pala, V., Thiebaut, A. C., Tjonneland, A., Olsen, A., Overvad, K., Jakobsen, M. U., Clavel-Chapelon, F., Chajes, V., Boutron-Ruault, M. C., Kaaks, R., Linseisen, J., Boeing, H., Nothlings, U., Trichopoulou, A., Naska, A., Lagiou, P., Panico, S., Palli, D., Vineis, P., Tumino, R., Lund, E., Kumle, M., Skeie, G., Gonzalez, C. A., Ardanaz, E., Amiano, P., Tormo, M. J., Martinez-Garcia, C., Quiros, J. R., Berglund, G., Gullberg, B., Hallmans, G., Lenner, P., Bueno-de-Mesquita, H. B., van Duijnhoven, F. J., Peeters, P. H., van Gils, C. H., Key, T. J., Crowe, F. L., Bingham, S., Khaw, K. T., Rinaldi, S., Slimani, N., Jenab, M., Norat, T., and Riboli, E. Dietary fat and breast cancer risk in the European Prospective Investigation into Cancer and Nutrition. Am J Clin Nutr, 88: , Engeset, D., Alsaker, E., Lund, E., Welch, A., Khaw, K. T., Clavel-Chapelon, F., Thiebaut, A., Chajes, V., Key, T. J., Allen, N. E., Amiano, P., Dorronsoro, M., Tjonneland, A., Stripp, C., Peeters, P. H., van Gils, C. H., Chirlaque, M. D., Nagel, G., Linseisen, J., Ocke, M. C., Bueno-de-Mesquita, H. B., Sacerdote, C., Tumino, R., Ardanaz, E., Sanchez, M. J., Panico, S., Palli, D., Trichopoulou, A., Kalapothaki, V., Benetou, V., Quiros, J. R., Agudo, A., Overvad, K., Bjerregaard, L., Wirfalt, E., Schulz, M., Boeing, H., Slimani, N., and Riboli, E. Fish consumption and breast cancer risk. The European Prospective Investigation into Cancer and Nutrition (EPIC). Int J Cancer, 119: , Gonzalez, C. A. The European Prospective Investigation into Cancer and Nutrition (EPIC). Public Health Nutr, 9: , Schulz, M., Hoffmann, K., Weikert, C., Nothlings, U., Schulze, M. B., and Boeing, H. Identification of a dietary pattern characterized by high-fat food choices associated with increased risk of breast cancer: the European Prospective
173 157 Investigation into Cancer and Nutrition (EPIC)-Potsdam Study. Br J Nutr, 100: , Copeland, T., Grosvenor, M., Mitchell, D. C., Smiciklas-Wright, H., Marsoobian, V., Blackburn, G., and Winters, B. Designing a quality assurance system for dietary data in a multicenter clinical trial: Women's Intervention Nutrition Study. J Am Diet Assoc, 100: , Chlebowski, R. T., Rose, D., Buzzard, I. M., Blackburn, G. L., Insull, W., Jr., Grosvenor, M., Elashoff, R., and Wynder, E. L. Adjuvant dietary fat intake reduction in postmenopausal breast cancer patient management. The Women's Intervention Nutrition Study (WINS). Breast Cancer Res Treat, 20: 73-84, Pierce, J. P., Natarajan, L., Caan, B. J., Parker, B. A., Greenberg, E. R., Flatt, S. W., Rock, C. L., Kealey, S., Al-Delaimy, W. K., Bardwell, W. A., Carlson, R. W., Emond, J. A., Faerber, S., Gold, E. B., Hajek, R. A., Hollenbach, K., Jones, L. A., Karanja, N., Madlensky, L., Marshall, J., Newman, V. A., Ritenbaugh, C., Thomson, C. A., Wasserman, L., and Stefanick, M. L. Influence of a diet very high in vegetables, fruit, and fiber and low in fat on prognosis following treatment for breast cancer: the Women's Healthy Eating and Living (WHEL) randomized trial. Jama, 298: , Gold, E. B., Pierce, J. P., Natarajan, L., Stefanick, M. L., Laughlin, G. A., Caan, B. J., Flatt, S. W., Emond, J. A., Saquib, N., Madlensky, L., Kealey, S., Wasserman, L., Thomson, C. A., Rock, C. L., Parker, B. A., Karanja, N., Jones, V., Hajek, R. A., Pu, M., and Mortimer, J. E. Dietary pattern influences breast cancer prognosis in women without hot flashes: the women's healthy eating and living trial. J Clin Oncol, 27: , Saadatian-Elahi, M., Norat, T., Goudable, J., and Riboli, E. Biomarkers of dietary fatty acid intake and the risk of breast cancer: a meta-analysis. Int J Cancer, 111: , Shannon, J., King, I. B., Moshofsky, R., Lampe, J. W., Gao, D. L., Ray, R. M., and Thomas, D. B. Erythrocyte fatty acids and breast cancer risk: a case-control study in Shanghai, China. Am J Clin Nutr, 85: , Tannenbaum, A. The genesis and growth of tumors III. effects of a high fat diet. Cancer Research, 2: , Welsch, C. W. Relationship between dietary fat and experimental mammary tumorigenesis: a review and critique. Cancer Res, 52: 2040s-2048s, Kamano, K., Okuyama, H., Konishi, R., and Nagasawa, H. Effects of a highlinoleate and a high-alpha-linolenate diet on spontaneous mammary tumourigenesis in mice. Anticancer Res, 9: , 1989.
174 Abraham, S., Faulkin, L. J., Hillyard, L. A., and Mitchell, D. J. Effect of dietary fat on tumorigenesis in the mouse mammary gland. J Natl Cancer Inst, 72: , Miyamoto-Tiaven, M. J., Hillyard, L. A., and Abraham, S. Influence of dietary fat on the growth of mammary ducts in BALB/c mice. J Natl Cancer Inst, 67: , Thompson, H. J. and Singh, M. Rat models of premalignant breast disease. J Mammary Gland Biol Neoplasia, 5: , Thompson, H. J., Meeker, L. D., Tagliaferro, A. R., and Roberts, J. S. Effect of energy intake on the promotion of mammary carcinogenesis by dietary fat. Nutr Cancer, 7: 37-41, Hopkins, G. J., West, C. E., and Hard, G. C. Effect of dietary fats on the incidence of 7,12-dimethylbenz(a)-anthracene-induced tumors in rats. Lipids, 11: , Cohen, L. A., Thompson, D. O., Maeura, Y., Choi, K., Blank, M. E., and Rose, D. P. Dietary fat and mammary cancer. I. Promoting effects of different dietary fats on N-nitrosomethylurea-induced rat mammary tumorigenesis. J Natl Cancer Inst, 77: 33-42, Habib, N. A., Wood, C. B., Apostolov, K., Barker, W., Hershman, M. J., Aslam, M., Heinemann, D., Fermor, B., Williamson, R. C., Jenkins, W. E., and et al. Stearic acid and carcinogenesis. Br J Cancer, 56: , Hubbard, N. E. and Erickson, K. L. Enhancement of metastasis from a transplantable mouse mammary tumor by dietary linoleic acid. Cancer Res, 47: , Rose, D. P., Connolly, J. M., and Liu, X. H. Effects of linoleic acid on the growth and metastasis of two human breast cancer cell lines in nude mice and the invasive capacity of these cell lines in vitro. Cancer Res, 54: , Connolly, J. M., Liu, X. H., and Rose, D. P. Dietary linoleic acid-stimulated human breast cancer cell growth and metastasis in nude mice and their suppression by indomethacin, a cyclooxygenase inhibitor. Nutr Cancer, 25: , Mukutmoni-Norris, M., Hubbard, N. E., and Erickson, K. L. Modulation of murine mammary tumor vasculature by dietary n-3 fatty acids in fish oil. Cancer Lett, 150: , 2000.
175 Rose, D. P., Connolly, J. M., and Coleman, M. Effect of omega-3 fatty acids on the progression of metastases after the surgical excision of human breast cancer cell solid tumors growing in nude mice. Clin Cancer Res, 2: , Chen, J., Stavro, P. M., and Thompson, L. U. Dietary flaxseed inhibits human breast cancer growth and metastasis and downregulates expression of insulin-like growth factor and epidermal growth factor receptor. Nutr Cancer, 43: , Hardman, W. E., Sun, L., Short, N., and Cameron, I. L. Dietary omega-3 fatty acids and ionizing irradiation on human breast cancer xenograft growth and angiogenesis. Cancer Cell Int, 5: 12, Mougios, V., Ring, S., Petridou, A., and Nikolaidis, M. G. Duration of coffeeand exercise-induced changes in the fatty acid profile of human serum. J Appl Physiol, 94: , Rose, D. P. and Connolly, J. M. Effects of fatty acids and inhibitors of eicosanoid synthesis on the growth of a human breast cancer cell line in culture. Cancer Res, 50: , Lupu, R. and Menendez, J. A. Targeting fatty acid synthase in breast and endometrial cancer: An alternative to selective estrogen receptor modulators? Endocrinology, 147: , Chajes, V., Cambot, M., Moreau, K., Lenoir, G. M., and Joulin, V. Acetyl-CoA carboxylase alpha is essential to breast cancer cell survival. Cancer Res, 66: , Zhao, H. and Hardy, R. W. Long-chain saturated fatty acids induce annexin II translocation to detergent-resistant membranes. Biochem J, 381: , Wicha, M. S., Liotta, L. A., and Kidwell, W. R. Effects of free fatty acids on the growth of normal and neoplastic rat mammary epithelial cells. Cancer Res, 39: , la Vecchia, C., Negri, E., Franceschi, S., Decarli, A., Giacosa, A., and Lipworth, L. Olive oil, other dietary fats, and the risk of breast cancer (Italy). Cancer Causes Control, 6: , Germain, E., Chajes, V., Cognault, S., Lhuillery, C., and Bougnoux, P. Enhancement of doxorubicin cytotoxicity by polyunsaturated fatty acids in the human breast tumor cell line MDA-MB-231: relationship to lipid peroxidation. Int J Cancer, 75: , 1998.
176 Przybytkowski, E., Joly, E., Nolan, C. J., Hardy, S., Francoeur, A. M., Langelier, Y., and Prentki, M. Upregulation of cellular triacylglycerol - free fatty acid cycling by oleate is associated with long-term serum-free survival of human breast cancer cells. Biochem Cell Biol, 85: , Menendez, J. A., Mehmi, I., Atlas, E., Colomer, R., and Lupu, R. Novel signaling molecules implicated in tumor-associated fatty acid synthase-dependent breast cancer cell proliferation and survival: Role of exogenous dietary fatty acids, p53- p21waf1/cip1, ERK1/2 MAPK, p27kip1, BRCA1, and NF-kappaB. Int J Oncol, 24: , Waterman, E. and Lockwood, B. Active components and clinical applications of olive oil. Altern Med Rev, 12: , Rose, D. P. and Connolly, J. M. Stimulation of growth of human breast cancer cell lines in culture by linoleic acid. Biochem Biophys Res Commun, 164: , Park, Y., Allen, K. G., and Shultz, T. D. Modulation of MCF-7 breast cancer cell signal transduction by linoleic acid and conjugated linoleic acid in culture. Anticancer Res, 20: , Reyes, N., Reyes, I., Tiwari, R., and Geliebter, J. Effect of linoleic acid on proliferation and gene expression in the breast cancer cell line T47D. Cancer Lett, 209: 25-35, Balakrishnan, A., Cramer, S., Bandyopadhyay, G. K., Imagawa, W., Yang, J., Elias, J., Beattie, C. W., Das Gupta, T. K., and Nandi, S. Differential proliferative response to linoleate in cultures of epithelial cells from normal human breast and fibroadenomas. Cancer Res, 49: , Connolly, J. M. and Rose, D. P. Effects of fatty acids on invasion through reconstituted basement membrane ('Matrigel') by a human breast cancer cell line. Cancer Lett, 75: , Liu, X. H. and Rose, D. P. Stimulation of type IV collagenase expression by linoleic acid in a metastatic human breast cancer cell line. Cancer Lett, 76: 71-77, Paine, E., Palmantier, R., Akiyama, S. K., Olden, K., and Roberts, J. D. Arachidonic acid activates mitogen-activated protein (MAP) kinase-activated protein kinase 2 and mediates adhesion of a human breast carcinoma cell line to collagen type IV through a p38 MAP kinase-dependent pathway. J Biol Chem, 275: , 2000.
177 Kennett, S. B., Roberts, J. D., and Olden, K. Requirement of protein kinase C micro activation and calpain-mediated proteolysis for arachidonic acid-stimulated adhesion of MDA-MB-435 human mammary carcinoma cells to collagen type IV. J Biol Chem, 279: , Nony, P. A., Kennett, S. B., Glasgow, W. C., Olden, K., and Roberts, J. D. 15S- Lipoxygenase-2 mediates arachidonic acid-stimulated adhesion of human breast carcinoma cells through the activation of TAK1, MKK6, and p38 MAPK. J Biol Chem, 280: , Navarro-Tito, N., Robledo, T., and Salazar, E. P. Arachidonic acid promotes FAK activation and migration in MDA-MB-231 breast cancer cells. Exp Cell Res, 314: , Razanamahefa, L., Prouff, S., and Bardon, S. Stimulatory effect of arachidonic acid on T-47D human breast cancer cell growth is associated with enhancement of cyclin D1 mrna expression. Nutr Cancer, 38: , Bocca, C., Bozzo, F., Martinasso, G., Canuto, R. A., and Miglietta, A. Involvement of PPARalpha in the growth inhibitory effect of arachidonic acid on breast cancer cells. Br J Nutr, 100: , Grammatikos, S. I., Subbaiah, P. V., Victor, T. A., and Miller, W. M. n-3 and n-6 fatty acid processing and growth effects in neoplastic and non-cancerous human mammary epithelial cell lines. Br J Cancer, 70: , Horia, E. and Watkins, B. A. Comparison of stearidonic acid and alpha-linolenic acid on PGE2 production and COX-2 protein levels in MDA-MB-231 breast cancer cell cultures. J Nutr Biochem, 16: , Chamras, H., Ardashian, A., Heber, D., and Glaspy, J. A. Fatty acid modulation of MCF-7 human breast cancer cell proliferation, apoptosis and differentiation. J Nutr Biochem, 13: , Chiu, L. C., Wong, E. Y., and Ooi, V. E. Docosahexaenoic acid from a cultured microalga inhibits cell growth and induces apoptosis by upregulating Bax/Bcl-2 ratio in human breast carcinoma MCF-7 cells. Ann N Y Acad Sci, 1030: , Wu, M., Harvey, K. A., Ruzmetov, N., Welch, Z. R., Sech, L., Jackson, K., Stillwell, W., Zaloga, G. P., and Siddiqui, R. A. Omega-3 polyunsaturated fatty acids attenuate breast cancer growth through activation of a neutral sphingomyelinase-mediated pathway. Int J Cancer, 117: , 2005.
178 Horia, E. and Watkins, B. A. Complementary actions of docosahexaenoic acid and genistein on COX-2, PGE2 and invasiveness in MDA-MB-231 breast cancer cells. Carcinogenesis, 28: , Sun, H., Berquin, I. M., Owens, R. T., O'Flaherty, J. T., and Edwards, I. J. Peroxisome proliferator-activated receptor gamma-mediated up-regulation of syndecan-1 by n-3 fatty acids promotes apoptosis of human breast cancer cells. Cancer Res, 68: , Maheo, K., Vibet, S., Steghens, J. P., Dartigeas, C., Lehman, M., Bougnoux, P., and Gore, J. Differential sensitization of cancer cells to doxorubicin by DHA: a role for lipoperoxidation. Free Radic Biol Med, 39: , Conquer, J. A. and Holub, B. J. Effect of supplementation with different doses of DHA on the levels of circulating DHA as non-esterified fatty acid in subjects of Asian Indian background. J Lipid Res, 39: , Agriculture, U. S. D. o. H. a. H. S. a. U. S. D. o. Dietary Guidelines for Americans Vol. 2009, Willett, W. C. Diet, nutrition, and avoidable cancer. Environ Health Perspect, 103 Suppl 8: , Ghajar, C. M. and Bissell, M. J. Extracellular matrix control of mammary gland morphogenesis and tumorigenesis: insights from imaging. Histochem Cell Biol, 130: , Coleman, R. A., Lewin, T. M., Van Horn, C. G., and Gonzalez-Baro, M. R. Do long-chain acyl-coa synthetases regulate fatty acid entry into synthetic versus degradative pathways? J Nutr, 132: , Martiny-Baron, G., Kazanietz, M. G., Mischak, H., Blumberg, P. M., Kochs, G., Hug, H., Marme, D., and Schachtele, C. Selective inhibition of protein kinase C isozymes by the indolocarbazole Go J Biol Chem, 268: , Dhein, S., Weng, S., Grover, R., Tudyka, T., Gottwald, M., Schaefer, T., and Polontchouk, L. Protein kinase Calpha mediates the effect of antiarrhythmic peptide on gap junction conductance. Cell Commun Adhes, 8: , Murphy-Ullrich, J. E. The de-adhesive activity of matricellular proteins: is intermediate cell adhesion an adaptive state? J Clin Invest, 107: , Spector, A. A. and Hoak, J. C. An improved method for the addition of long-chain free fatty acid to protein solutions. Anal Biochem, 32: , 1969.
179 Rosen, A. and Casciola-Rosen, L. Macromolecular substrates for the ICE-like proteases during apoptosis. J Cell Biochem, 64: 50-54, Lu, Z. H., Mu, Y. M., Wang, B. A., Li, X. L., Lu, J. M., Li, J. Y., Pan, C. Y., Yanase, T., and Nawata, H. Saturated free fatty acids, palmitic acid and stearic acid, induce apoptosis by stimulation of ceramide generation in rat testicular Leydig cell. Biochem Biophys Res Commun, 303: , Mu, Y. M., Yanase, T., Nishi, Y., Tanaka, A., Saito, M., Jin, C. H., Mukasa, C., Okabe, T., Nomura, M., Goto, K., and Nawata, H. Saturated FFAs, palmitic acid and stearic acid, induce apoptosis in human granulosa cells. Endocrinology, 142: , Staiger, K., Staiger, H., Weigert, C., Haas, C., Haring, H. U., and Kellerer, M. Saturated, but not unsaturated, fatty acids induce apoptosis of human coronary artery endothelial cells via nuclear factor-kappab activation. Diabetes, 55: , Ulloth, J. E., Casiano, C. A., and De Leon, M. Palmitic and stearic fatty acids induce caspase-dependent and -independent cell death in nerve growth factor differentiated PC12 cells. J Neurochem, 84: , Artwohl, M., Roden, M., Waldhausl, W., Freudenthaler, A., and Baumgartner- Parzer, S. M. Free fatty acids trigger apoptosis and inhibit cell cycle progression in human vascular endothelial cells. Faseb J, 18: , Beeharry, N., Chambers, J. A., and Green, I. C. Fatty acid protection from palmitic acid-induced apoptosis is lost following PI3-kinase inhibition. Apoptosis, 9: , Paumen, M. B., Ishida, Y., Muramatsu, M., Yamamoto, M., and Honjo, T. Inhibition of carnitine palmitoyltransferase I augments sphingolipid synthesis and palmitate-induced apoptosis. J Biol Chem, 272: , Mishra, R. and Simonson, M. S. Saturated free fatty acids and apoptosis in microvascular mesangial cells: palmitate activates pro-apoptotic signaling involving caspase 9 and mitochondrial release of endonuclease G. Cardiovasc Diabetol, 4: 2, El-Assaad, W., Buteau, J., Peyot, M. L., Nolan, C., Roduit, R., Hardy, S., Joly, E., Dbaibo, G., Rosenberg, L., and Prentki, M. Saturated fatty acids synergize with elevated glucose to cause pancreatic beta-cell death. Endocrinology, 144: , Eitel, K., Staiger, H., Rieger, J., Mischak, H., Brandhorst, H., Brendel, M. D., Bretzel, R. G., Haring, H. U., and Kellerer, M. Protein kinase C delta activation
180 164 and translocation to the nucleus are required for fatty acid-induced apoptosis of insulin-secreting cells. Diabetes, 52: , Yu, H. Y., Inoguchi, T., Kakimoto, M., Nakashima, N., Imamura, M., Hashimoto, T., Umeda, F., and Nawata, H. Saturated non-esterified fatty acids stimulate de novo diacylglycerol synthesis and protein kinase c activity in cultured aortic smooth muscle cells. Diabetologia, 44: , Igal, R. A., Wang, P., and Coleman, R. A. Triacsin C blocks de novo synthesis of glycerolipids and cholesterol esters but not recycling of fatty acid into phospholipid: evidence for functionally separate pools of acyl-coa. Biochem J, 324 ( Pt 2): , Musashi, M., Ota, S., and Shiroshita, N. The role of protein kinase C isoforms in cell proliferation and apoptosis. Int J Hematol, 72: 12-19, Wang, Q., Wang, X., and Evers, B. M. Induction of ciap-2 in human colon cancer cells through PKC delta/nf-kappa B. J Biol Chem, 278: , Grundy, S. M. Influence of stearic acid on cholesterol metabolism relative to other long-chain fatty acids. Am J Clin Nutr, 60: 986S-990S, Mitropoulos, K. A., Miller, G. J., Martin, J. C., Reeves, B. E., and Cooper, J. Dietary fat induces changes in factor VII coagulant activity through effects on plasma free stearic acid concentration. Arterioscler Thromb, 14: , Tholstrup, T. Influence of stearic acid on hemostatic risk factors in humans. Lipids, 40: , Kelly, F. D., Sinclair, A. J., Mann, N. J., Turner, A. H., Abedin, L., and Li, D. A stearic acid-rich diet improves thrombogenic and atherogenic risk factor profiles in healthy males. Eur J Clin Nutr, 55: 88-96, Louheranta, A. M., Turpeinen, A. K., Schwab, U. S., Vidgren, H. M., Parviainen, M. T., and Uusitupa, M. I. A high-stearic acid diet does not impair glucose tolerance and insulin sensitivity in healthy women. Metabolism, 47: , Rose, D. P., Connolly, J. M., and Liu, X. H. Effects of linoleic acid and gammalinolenic acid on the growth and metastasis of a human breast cancer cell line in nude mice and on its growth and invasive capacity in vitro. Nutr Cancer, 24: 33-45, 1995.
181 Rose, D. P., Hatala, M. A., Connolly, J. M., and Rayburn, J. Effect of diets containing different levels of linoleic acid on human breast cancer growth and lung metastasis in nude mice. Cancer Res, 53: , Rose, D. P., Connolly, J. M., Rayburn, J., and Coleman, M. Influence of diets containing eicosapentaenoic or docosahexaenoic acid on growth and metastasis of breast cancer cells in nude mice. J Natl Cancer Inst, 87: , Rose, D. P. and Connolly, J. M. Effects of dietary omega-3 fatty acids on human breast cancer growth and metastases in nude mice. J Natl Cancer Inst, 85: , Price, J. E., Polyzos, A., Zhang, R. D., and Daniels, L. M. Tumorigenicity and metastasis of human breast carcinoma cell lines in nude mice. Cancer Res, 50: , Fermor, B. F., Masters, J. R., Wood, C. B., Miller, J., Apostolov, K., and Habib, N. A. Fatty acid composition of normal and malignant cells and cytotoxicity of stearic, oleic and sterculic acids in vitro. Eur J Cancer, 28A: , Rose, D. P. and Connolly, J. M. Regulation of tumor angiogenesis by dietary fatty acids and eicosanoids. Nutr Cancer, 37: , Harris, R. E., Robertson, F. M., Abou-Issa, H. M., Farrar, W. B., and Brueggemeier, R. Genetic induction and upregulation of cyclooxygenase (COX) and aromatase (CYP19): an extension of the dietary fat hypothesis of breast cancer. Med Hypotheses, 52: , Hardman, W. E. Dietary canola oil suppressed growth of implanted MDA-MB 231 human breast tumors in nude mice. Nutr Cancer, 57: , Fujimoto, Y., Yonemura, T., and Sakuma, S. Stearic acid potently modulates the activity of cyclooxygenase-1, but not cyclooxygenase-2, in the form of its CoA ester. Prostaglandins Leukot Essent Fatty Acids, 78: 81-84, Artwohl, M., Lindenmair, A., Sexl, V., Maier, C., Rainer, G., Freudenthaler, A., Huttary, N., Wolzt, M., Nowotny, P., Luger, A., and Baumgartner-Parzer, S. M. Different mechanisms of saturated versus polyunsaturated free fatty acid-induced apoptosis in human endothelial cells. J Lipid Res, MacDonald, H. R., Lees, R. K., Bron, C., Sordat, B., and Miescher, G. T cell antigen receptor expression in athymic (nu/nu) mice. Evidence for an oligoclonal beta chain repertoire. J Exp Med, 166: , Talmadge, J. E., Donkor, M., and Scholar, E. Inflammatory cell infiltration of tumors: Jekyll or Hyde. Cancer Metastasis Rev, 26: , 2007.
182 Lima, T. M., Kanunfre, C. C., Pompeia, C., Verlengia, R., and Curi, R. Ranking the toxicity of fatty acids on Jurkat and Raji cells by flow cytometric analysis. Toxicol In Vitro, 16: , Jeffery, N. M., Sanderson, P., Newsholme, E. A., and Calder, P. C. Effects of varying the type of saturated fatty acid in the rat diet upon serum lipid levels and spleen lymphocyte functions. Biochim Biophys Acta, 1345: , Brooks, A. D., Ramirez, T., Toh, U., Onksen, J., Elliott, P. J., Murphy, W. J., and Sayers, T. J. The proteasome inhibitor bortezomib (Velcade) sensitizes some human tumor cells to Apo2L/TRAIL-mediated apoptosis. Ann N Y Acad Sci, 1059: , Hsu, S. C., Gavrilin, M. A., Lee, H. H., Wu, C. C., Han, S. H., and Lai, M. Z. NFkappa B-dependent Fas ligand expression. Eur J Immunol, 29: , Kvilekval, K., Lin, J., Cheng, W., and Abumrad, N. Fatty acids as determinants of triglyceride and cholesteryl ester synthesis by isolated hepatocytes: kinetics as a function of various fatty acids. J Lipid Res, 35: , Yonekawa, H. and Akita, Y. Protein kinase Cepsilon: the mitochondria-mediated signaling pathway. Febs J, 275: , Lavie, Y., Zhang, Z. C., Cao, H. T., Han, T. Y., Jones, R. C., Liu, Y. Y., Jarman, M., Hardcastle, I. R., Giuliano, A. E., and Cabot, M. C. Tamoxifen induces selective membrane association of protein kinase C epsilon in MCF-7 human breast cancer cells. Int J Cancer, 77: , Claro, S., Kanashiro, C. A., Oshiro, M. E., Ferreira, A. T., and Khalil, R. A. alpha- and epsilon-protein kinase C activity during smooth muscle cell apoptosis in response to gamma-radiation. J Pharmacol Exp Ther, 322: , Li, R. C., Ping, P., Zhang, J., Wead, W. B., Cao, X., Gao, J., Zheng, Y., Huang, S., Han, J., and Bolli, R. PKCepsilon modulates NF-kappaB and AP-1 via mitogen-activated protein kinases in adult rabbit cardiomyocytes. Am J Physiol Heart Circ Physiol, 279: H , Tojima, Y., Fujimoto, A., Delhase, M., Chen, Y., Hatakeyama, S., Nakayama, K., Kaneko, Y., Nimura, Y., Motoyama, N., Ikeda, K., Karin, M., and Nakanishi, M. NAK is an IkappaB kinase-activating kinase. Nature, 404: , Nestle, M. Food Politics: How the Food Industry Influence Nutrition and Health, p Los Angeles: University of California Press, 2002.
183 Nestle, M. Food Politics: How the Food Industry Influence Nutrition and Health, p Los Angeles: University of California Press, Kris-Etherton, P. M., Innis, S., Ammerican Dietetic, A., and Dietitians of, C. Position of the American Dietetic Association and Dietitians of Canada: dietary fatty acids. J Am Diet Assoc, 107: , Simopoulos, A. P. Human requirement for N-3 polyunsaturated fatty acids. Poult Sci, 79: , Lands, W. E. Dietary fat and health: the evidence and the politics of prevention: careful use of dietary fats can improve life and prevent disease. Ann N Y Acad Sci, 1055: , Simopoulos, A. P., Leaf, A., and Salem, N., Jr. Workshop statement on the essentiality of and recommended dietary intakes for Omega-6 and Omega-3 fatty acids. Prostaglandins Leukot Essent Fatty Acids, 63: , Enjalbert, F., Nicot, M. C., Bayourthe, C., and Moncoulon, R. Effects of duodenal infusions of palmitic, stearic, or oleic acids on milk composition and physical properties of butter. J Dairy Sci, 83: , Sklan, D., Arieli, A., Chalupa, W., and Kronfeld, D. S. Digestion and absorption of lipids and bile acids in sheep fed stearic acid, oleic acid, or tristearin. J Dairy Sci, 68: , Rukkwamsuk, T., Geelen, M. J., Kruip, T. A., and Wensing, T. Interrelation of fatty acid composition in adipose tissue, serum, and liver of dairy cows during the development of fatty liver postpartum. J Dairy Sci, 83: 52-59, Ross, D. T., Scherf, U., Eisen, M. B., Perou, C. M., Rees, C., Spellman, P., Iyer, V., Jeffrey, S. S., Van de Rijn, M., Waltham, M., Pergamenschikov, A., Lee, J. C., Lashkari, D., Shalon, D., Myers, T. G., Weinstein, J. N., Botstein, D., and Brown, P. O. Systematic variation in gene expression patterns in human cancer cell lines. Nat Genet, 24: , Sellappan, S., Grijalva, R., Zhou, X., Yang, W., Eli, M. B., Mills, G. B., and Yu, D. Lineage infidelity of MDA-MB-435 cells: expression of melanocyte proteins in a breast cancer cell line. Cancer Res, 64: , Christgen, M. and Lehmann, U. MDA-MB-435: the questionable use of a melanoma cell line as a model for human breast cancer is ongoing. Cancer Biol Ther, 6: , Rae, J. M., Creighton, C. J., Meck, J. M., Haddad, B. R., and Johnson, M. D. MDA-MB-435 cells are derived from M14 melanoma cells--a loss for breast
184 168 cancer, but a boon for melanoma research. Breast Cancer Res Treat, 104: 13-19, You, H., Yu, W., Sanders, B. G., and Kline, K. RRR-alpha-tocopheryl succinate induces MDA-MB-435 and MCF-7 human breast cancer cells to undergo differentiation. Cell Growth Differ, 12: , 2001.
185 169 APPENDIX A SUPPLEMENTAL DATA Figure A-1: Inhibition of NF B Reverses Stearate-Induced De-adhesion and Stearate-Induced Caspase-3 Activity. A) Cells were plated and serum starved for 24 hours and the number of adherent and round cells was counted. Cells were then treated with 20 M control peptide or NF B inhibitor peptide. After 1 hour, cells were treated
186 with 50 M stearate of FAF-BSA for 12 hours. After 12 hours, the number of round and adherent cells was counted again. Data presented are the percent of stearate-induced round cells. The NF B inhibitor partially reversed the stearate-induced cell rounding. (n=1-2 counted in triplicate; *p<0.05 by Student s t-test). B) The experiment was performed as in part A, except cells were treated with 20 M wedelolactone. Inhibition of IKK partially reversed stearate-induced cell rounding. (n=4; p< by Student s t- test). C) Cells were plated and serum starved for 24 hours prior to treatment with 20 M wedelolactone for 1 hour. Cells were then treated with 50 M stearate for 12 hours and caspase-3 activity was measured using a fluorescent based assay. Inhibition of IKK partially reverses stearate-induced caspase-3 activity. (n=3; *p<0.03 by Student s t-test). 170

187 Figure A-2: Stearate Induces Cell Rounding in a Dose Dependent Manner. Cells were plated and serum starved for 24 hour and then the number of adherent and round cells was counted. Cells were then treated with various concentrations of stearate for 24 hours and the adherent and rounded cells were counted again. Stearate induced cell rounding in a dose dependent manner. (n=4 counted in triplicate; *p<0.03; **p<0.001 by ANOVA). 171

188 Figure A-3: Stearate Induces Translocation of PKC to the Plasma Membrane. Cells were serum starved for 24 hours prior to treatment with 50 M stearate for the times indicated. Cytosolic protein lysates and membrane protein lysates were collected and resolved by SDS-PAGE prior to immunoblotting for PKC. As show above, stearate induces translocation of PKC to the plasma membrane, thereby inducing the enzyme s activation. (n=1). 172
Introduction to pathology lecture 5/ Cell injury apoptosis. Dr H Awad 2017/18
 Introduction to pathology lecture 5/ Cell injury apoptosis Dr H Awad 2017/18 Apoptosis = programmed cell death = cell suicide= individual cell death Apoptosis cell death induced by a tightly regulated
Introduction to pathology lecture 5/ Cell injury apoptosis Dr H Awad 2017/18 Apoptosis = programmed cell death = cell suicide= individual cell death Apoptosis cell death induced by a tightly regulated
Molecular biology :- Cancer genetics lecture 11
 Molecular biology :- Cancer genetics lecture 11 -We have talked about 2 group of genes that is involved in cellular transformation : proto-oncogenes and tumour suppressor genes, and it isn t enough to
Molecular biology :- Cancer genetics lecture 11 -We have talked about 2 group of genes that is involved in cellular transformation : proto-oncogenes and tumour suppressor genes, and it isn t enough to
Apoptotic Pathways in Mammals Dr. Douglas R. Green
 Apoptotic Pathways in Mammals Douglas R. Green 1 Apoptosis A form of cell death that is defined morphologically, and features a number of biochemical events Programmed cell death Cell death that occurs
Apoptotic Pathways in Mammals Douglas R. Green 1 Apoptosis A form of cell death that is defined morphologically, and features a number of biochemical events Programmed cell death Cell death that occurs
Apoptosis Oncogenes. Srbová Martina
 Apoptosis Oncogenes Srbová Martina Cell Cycle Control point Cyclin B Cdk1 Cyclin D Cdk4 Cdk6 Cyclin A Cdk2 Cyclin E Cdk2 Cyclin-dependent kinase (Cdk) have to bind a cyclin to become active Regulation
Apoptosis Oncogenes Srbová Martina Cell Cycle Control point Cyclin B Cdk1 Cyclin D Cdk4 Cdk6 Cyclin A Cdk2 Cyclin E Cdk2 Cyclin-dependent kinase (Cdk) have to bind a cyclin to become active Regulation
Basement membrane in lobule.
 Bahram Memar, MD Basement membrane in lobule. Normal lobule-luteal phase Normal lobule-follicular phase Lactating breast Greater than 95% are adenocarcinomas in situ carcinomas and invasive carcinomas.
Bahram Memar, MD Basement membrane in lobule. Normal lobule-luteal phase Normal lobule-follicular phase Lactating breast Greater than 95% are adenocarcinomas in situ carcinomas and invasive carcinomas.
The death receptors: signaling and modulation
 The death receptors: signaling and modulation 1 1 The extrinsic cell death pathway 2 Nat Rev Drug Discov. 2008 Dec;7(12):1001-12. 2 Death receptors Belong to the tumor necrosis factor (TNF) receptor gene
The death receptors: signaling and modulation 1 1 The extrinsic cell death pathway 2 Nat Rev Drug Discov. 2008 Dec;7(12):1001-12. 2 Death receptors Belong to the tumor necrosis factor (TNF) receptor gene
Signaling Apoptosis. Scott André Oakes, M.D. Dept. of Pathology Univ. of Calif-San Francisco. Cyt c Release BAX/BAK. Apoptosome Formation
 Signaling Apoptosis Cyt c Release BAX/BAK Apoptosome Formation Caspase Activation Scott André Oakes, M.D. Dept. of Pathology Univ. of Calif-San Francisco Why do we need cell death? Sculpt Organs Control
Signaling Apoptosis Cyt c Release BAX/BAK Apoptosome Formation Caspase Activation Scott André Oakes, M.D. Dept. of Pathology Univ. of Calif-San Francisco Why do we need cell death? Sculpt Organs Control
p53 and Apoptosis: Master Guardian and Executioner Part 2
 p53 and Apoptosis: Master Guardian and Executioner Part 2 p14arf in human cells is a antagonist of Mdm2. The expression of ARF causes a rapid increase in p53 levels, so what would you suggest?.. The enemy
p53 and Apoptosis: Master Guardian and Executioner Part 2 p14arf in human cells is a antagonist of Mdm2. The expression of ARF causes a rapid increase in p53 levels, so what would you suggest?.. The enemy
Epigonal Conditioned Media from Bonnethead Shark, Sphyrna tiburo, Induces Apoptosis in a T-Cell Leukemia Cell Line, Jurkat E6-1
 Mar. Drugs 2013, 11, 3224-3257; doi:10.3390/md11093224 Article OPEN ACCESS marine drugs ISSN 1660-3397 www.mdpi.com/journal/marinedrugs Epigonal Conditioned Media from Bonnethead Shark, Sphyrna tiburo,
Mar. Drugs 2013, 11, 3224-3257; doi:10.3390/md11093224 Article OPEN ACCESS marine drugs ISSN 1660-3397 www.mdpi.com/journal/marinedrugs Epigonal Conditioned Media from Bonnethead Shark, Sphyrna tiburo,
Apoptosis Chapter 9. Neelu Yadav PhD
 Apoptosis Chapter 9 Neelu Yadav PhD Neelu.Yadav@Roswellpark.org 1 Apoptosis: Lecture outline Apoptosis a programmed cell death pathway in normal homeostasis Core Apoptosis cascade is conserved Compare
Apoptosis Chapter 9 Neelu Yadav PhD Neelu.Yadav@Roswellpark.org 1 Apoptosis: Lecture outline Apoptosis a programmed cell death pathway in normal homeostasis Core Apoptosis cascade is conserved Compare
#19 Apoptosis Chapter 9. Neelu Yadav PhD
 #19 Apoptosis Chapter 9 Neelu Yadav PhD Neelu.Yadav@Roswellpark.org Why cells decide to die? - Stress, harmful, not needed - Completed its life span Death stimulation or Stress Cell Survival Death Functions
#19 Apoptosis Chapter 9 Neelu Yadav PhD Neelu.Yadav@Roswellpark.org Why cells decide to die? - Stress, harmful, not needed - Completed its life span Death stimulation or Stress Cell Survival Death Functions
shehab Moh Tarek ... ManarHajeer
 3 shehab Moh Tarek... ManarHajeer In the previous lecture we discussed the accumulation of oxygen- derived free radicals as a mechanism of cell injury, we covered their production and their pathologic
3 shehab Moh Tarek... ManarHajeer In the previous lecture we discussed the accumulation of oxygen- derived free radicals as a mechanism of cell injury, we covered their production and their pathologic
1. The metastatic cascade. 3. Pathologic features of metastasis. 4. Therapeutic ramifications. Which malignant cells will metastasize?
 1. The metastatic cascade 3. Pathologic features of metastasis 4. Therapeutic ramifications Sir James Paget (1814-1899) British Surgeon/ Pathologist Paget s disease of Paget s disease of the nipple (intraductal
1. The metastatic cascade 3. Pathologic features of metastasis 4. Therapeutic ramifications Sir James Paget (1814-1899) British Surgeon/ Pathologist Paget s disease of Paget s disease of the nipple (intraductal
1.The metastatic cascade. 2.Pathologic features of metastasis. 3.Therapeutic ramifications
 Metastasis 1.The metastatic cascade 2.Pathologic features of metastasis 3.Therapeutic ramifications Sir James Paget (1814-1899) British Surgeon/ Pathologist Paget s disease of bone Paget s disease of the
Metastasis 1.The metastatic cascade 2.Pathologic features of metastasis 3.Therapeutic ramifications Sir James Paget (1814-1899) British Surgeon/ Pathologist Paget s disease of bone Paget s disease of the
Maram Abdaljaleel, MD Dermatopathologist and Neuropathologist University of Jordan, School of Medicine
 Maram Abdaljaleel, MD Dermatopathologist and Neuropathologist University of Jordan, School of Medicine The most common non-skin malignancy of women 2 nd most common cause of cancer deaths in women, following
Maram Abdaljaleel, MD Dermatopathologist and Neuropathologist University of Jordan, School of Medicine The most common non-skin malignancy of women 2 nd most common cause of cancer deaths in women, following
Cancer. The fundamental defect is. unregulated cell division. Properties of Cancerous Cells. Causes of Cancer. Altered growth and proliferation
 Cancer The fundamental defect is unregulated cell division. Properties of Cancerous Cells Altered growth and proliferation Loss of growth factor dependence Loss of contact inhibition Immortalization Alterated
Cancer The fundamental defect is unregulated cell division. Properties of Cancerous Cells Altered growth and proliferation Loss of growth factor dependence Loss of contact inhibition Immortalization Alterated
Disorders of Cell Growth & Neoplasia. Lecture 4 Molecular basis of cancer
 General Pathology VPM 152 Disorders of Cell Growth & Neoplasia Lecture 4 Molecular basis of cancer Enrique Aburto Apr 2010 Skin tumor in a 10-year-old Rottweiler. Considering the external appearance and
General Pathology VPM 152 Disorders of Cell Growth & Neoplasia Lecture 4 Molecular basis of cancer Enrique Aburto Apr 2010 Skin tumor in a 10-year-old Rottweiler. Considering the external appearance and
In vitro scratch assay: method for analysis of cell migration in vitro labeled fluorodeoxyglucose (FDG)
") In vitro scratch assay: method for analysis of cell migration in vitro labeled fluorodeoxyglucose (FDG) 1 Dr Saeb Aliwaini 13/11/2015 Migration in vivo Primary tumors are responsible for only about 10%
In vitro scratch assay: method for analysis of cell migration in vitro labeled fluorodeoxyglucose (FDG) 1 Dr Saeb Aliwaini 13/11/2015 Migration in vivo Primary tumors are responsible for only about 10%
Supplementary Figures
 Supplementary Figures Figure S1. Validation of kinase regulators of ONC201 sensitivity. Validation and screen results for changes in cell viability associated with the combination of ONC201 treatment (1
Supplementary Figures Figure S1. Validation of kinase regulators of ONC201 sensitivity. Validation and screen results for changes in cell viability associated with the combination of ONC201 treatment (1
Early cell death (FGF) B No RunX transcription factor produced Yes No differentiation
 B No RunX transcription factor produced Yes No differentiation") Solution Key - Practice Questions Question 1 a) A recent publication has shown that the fat stem cells (FSC) can act as bone stem cells to repair cavities in the skull, when transplanted into immuno-compromised
Solution Key - Practice Questions Question 1 a) A recent publication has shown that the fat stem cells (FSC) can act as bone stem cells to repair cavities in the skull, when transplanted into immuno-compromised
Cancer. Throughout the life of an individual, but particularly during development, every cell constantly faces decisions.
 Cancer Throughout the life of an individual, but particularly during development, every cell constantly faces decisions. Should it divide? Yes No--> Should it differentiate? Yes No-->Should it die? Yes-->Apoptosis
Cancer Throughout the life of an individual, but particularly during development, every cell constantly faces decisions. Should it divide? Yes No--> Should it differentiate? Yes No-->Should it die? Yes-->Apoptosis
Introduction: 年 Fas signal-mediated apoptosis. PI3K/Akt
 Fas-ligand (CD95-L; Fas-L) Fas (CD95) Fas (apoptosis) 年 了 不 度 Fas Fas-L 力 不 Fas/Fas-L T IL-10Fas/Fas-L 不 年 Fas signal-mediated apoptosis 度降 不 不 力 U-118, HeLa, A549, Huh-7 MCF-7, HepG2. PI3K/Akt FasPI3K/Akt
Fas-ligand (CD95-L; Fas-L) Fas (CD95) Fas (apoptosis) 年 了 不 度 Fas Fas-L 力 不 Fas/Fas-L T IL-10Fas/Fas-L 不 年 Fas signal-mediated apoptosis 度降 不 不 力 U-118, HeLa, A549, Huh-7 MCF-7, HepG2. PI3K/Akt FasPI3K/Akt
CLINICAL SIGNIFICANCE OF BENIGN EPITHELIAL CHANGES
 Papillomas. Papillomas are composed of multiple branching fibrovascular cores, each having a connective tissue axis lined by luminal and myoepithelial cells ( Fig. 23-11 ). Growth occurs within a dilated
Papillomas. Papillomas are composed of multiple branching fibrovascular cores, each having a connective tissue axis lined by luminal and myoepithelial cells ( Fig. 23-11 ). Growth occurs within a dilated
GMS 6644: Apoptosis. Introduction
 GMS 6644: Apoptosis Introduction (Feb. 15, 2006) Lei Xiao, Ph.D. Department of Anatomy & Cell Biology UF Shands Cancer Center ARB Rm R4-250, 846-1199, lxiao@ufl.edu Outline of the Lecture Different types
GMS 6644: Apoptosis Introduction (Feb. 15, 2006) Lei Xiao, Ph.D. Department of Anatomy & Cell Biology UF Shands Cancer Center ARB Rm R4-250, 846-1199, lxiao@ufl.edu Outline of the Lecture Different types
Transformation of Normal HMECs (Human Mammary Epithelial Cells) into Metastatic Breast Cancer Cells: Introduction - The Broad Picture:
 into Metastatic Breast Cancer Cells: Introduction - The Broad Picture:") Transformation of Normal HMECs (Human Mammary Epithelial Cells) into Metastatic Breast Cancer Cells: Introduction - The Broad Picture: Spandana Baruah December, 2016 Cancer is defined as: «A disease caused
Transformation of Normal HMECs (Human Mammary Epithelial Cells) into Metastatic Breast Cancer Cells: Introduction - The Broad Picture: Spandana Baruah December, 2016 Cancer is defined as: «A disease caused
609G: Concepts of Cancer Genetics and Treatments (3 credits)
") Master of Chemical and Life Sciences Program College of Computer, Mathematical, and Natural Sciences 609G: Concepts of Cancer Genetics and Treatments (3 credits) Text books: Principles of Cancer Genetics,
Master of Chemical and Life Sciences Program College of Computer, Mathematical, and Natural Sciences 609G: Concepts of Cancer Genetics and Treatments (3 credits) Text books: Principles of Cancer Genetics,
Deregulation of signal transduction and cell cycle in Cancer
 Deregulation of signal transduction and cell cycle in Cancer Tuangporn Suthiphongchai, Ph.D. Department of Biochemistry Faculty of Science, Mahidol University Email: tuangporn.sut@mahidol.ac.th Room Pr324
Deregulation of signal transduction and cell cycle in Cancer Tuangporn Suthiphongchai, Ph.D. Department of Biochemistry Faculty of Science, Mahidol University Email: tuangporn.sut@mahidol.ac.th Room Pr324
Mechanisms of Cell Death
 Mechanisms of Cell Death CELL DEATH AND FORMATION OF THE SEMICIRCULAR CANALS Carol M. Troy August 25, 2008 FROM: Fekete et al., Development 124: 2451 (1997) PHENOMENOLOGY OF CELL DEATH I. DEVELOPMENT A.
Mechanisms of Cell Death CELL DEATH AND FORMATION OF THE SEMICIRCULAR CANALS Carol M. Troy August 25, 2008 FROM: Fekete et al., Development 124: 2451 (1997) PHENOMENOLOGY OF CELL DEATH I. DEVELOPMENT A.
Introduction. Cancer Biology. Tumor-suppressor genes. Proto-oncogenes. DNA stability genes. Mechanisms of carcinogenesis.
 Cancer Biology Chapter 18 Eric J. Hall., Amato Giaccia, Radiobiology for the Radiologist Introduction Tissue homeostasis depends on the regulated cell division and self-elimination (programmed cell death)
Cancer Biology Chapter 18 Eric J. Hall., Amato Giaccia, Radiobiology for the Radiologist Introduction Tissue homeostasis depends on the regulated cell division and self-elimination (programmed cell death)
Diseases of the breast (2 of 2) Breast cancer
 Breast cancer") Diseases of the breast (2 of 2) Breast cancer Epidemiology & etiology The most common type of cancer & the 2 nd most common cause of cancer death in women 1 of 8 women in USA Affects 7% of women Peak at
Diseases of the breast (2 of 2) Breast cancer Epidemiology & etiology The most common type of cancer & the 2 nd most common cause of cancer death in women 1 of 8 women in USA Affects 7% of women Peak at
Contents 1 The Windows of Susceptibility to Breast Cancer 2 The So Called Pre-Neoplastic Lesions and Carcinoma In Situ
 Contents 1 The Windows of Susceptibility to Breast Cancer... 1 1.1 Introduction... 1 1.2 Risk Factor and Etiological Agents... 2 1.3 The Concept of the Windows of Susceptibility to Carcinogenesis... 5
Contents 1 The Windows of Susceptibility to Breast Cancer... 1 1.1 Introduction... 1 1.2 Risk Factor and Etiological Agents... 2 1.3 The Concept of the Windows of Susceptibility to Carcinogenesis... 5
number Done by Corrected by Doctor Maha Shomaf
 number 19 Done by Waseem Abo-Obeida Corrected by Abdullah Zreiqat Doctor Maha Shomaf Carcinogenesis: the molecular basis of cancer. Non-lethal genetic damage lies at the heart of carcinogenesis and leads
number 19 Done by Waseem Abo-Obeida Corrected by Abdullah Zreiqat Doctor Maha Shomaf Carcinogenesis: the molecular basis of cancer. Non-lethal genetic damage lies at the heart of carcinogenesis and leads
BIOL 4374/BCHS 4313 Cell Biology Exam #1 February 13, 2001
 BIOL 4374/BCHS 4313 Cell Biology Exam #1 February 13, 2001 SS# Name This exam is worth a total of 100 points. The number of points each question is worth is shown in parentheses. Good luck! 1. (2) The
BIOL 4374/BCHS 4313 Cell Biology Exam #1 February 13, 2001 SS# Name This exam is worth a total of 100 points. The number of points each question is worth is shown in parentheses. Good luck! 1. (2) The
Problem Set 8 Key 1 of 8
 7.06 2003 Problem Set 8 Key 1 of 8 7.06 2003 Problem Set 8 Key 1. As a bright MD/PhD, you are interested in questions about the control of cell number in the body. Recently, you've seen three patients
7.06 2003 Problem Set 8 Key 1 of 8 7.06 2003 Problem Set 8 Key 1. As a bright MD/PhD, you are interested in questions about the control of cell number in the body. Recently, you've seen three patients
Breast pathology. 2nd Department of Pathology Semmelweis University
 Breast pathology 2nd Department of Pathology Semmelweis University Breast pathology - Summary - Benign lesions - Acute mastitis - Plasma cell mastitis / duct ectasia - Fat necrosis - Fibrocystic change/
Breast pathology 2nd Department of Pathology Semmelweis University Breast pathology - Summary - Benign lesions - Acute mastitis - Plasma cell mastitis / duct ectasia - Fat necrosis - Fibrocystic change/
Lecture 14 - The cell cycle and cell death
 02.17.10 Lecture 14 - The cell cycle and cell death The cell cycle: cells duplicate their contents and divide The cell cycle may be divided into 4 phases The cell cycle triggers essential processes (DNA
02.17.10 Lecture 14 - The cell cycle and cell death The cell cycle: cells duplicate their contents and divide The cell cycle may be divided into 4 phases The cell cycle triggers essential processes (DNA
Enzyme-coupled Receptors. Cell-surface receptors 1. Ion-channel-coupled receptors 2. G-protein-coupled receptors 3. Enzyme-coupled receptors
 Enzyme-coupled Receptors Cell-surface receptors 1. Ion-channel-coupled receptors 2. G-protein-coupled receptors 3. Enzyme-coupled receptors Cell-surface receptors allow a flow of ions across the plasma
Enzyme-coupled Receptors Cell-surface receptors 1. Ion-channel-coupled receptors 2. G-protein-coupled receptors 3. Enzyme-coupled receptors Cell-surface receptors allow a flow of ions across the plasma
#19 Apoptosis Chapter 9. Neelu Yadav PhD
 #19 Apoptosis Chapter 9 Neelu Yadav PhD Neelu.Yadav@Roswellpark.org Why cells decide to die? - Stress, harmful, not needed - Completed its life span Death stimulation or Stress Cell Survival Death Functions
#19 Apoptosis Chapter 9 Neelu Yadav PhD Neelu.Yadav@Roswellpark.org Why cells decide to die? - Stress, harmful, not needed - Completed its life span Death stimulation or Stress Cell Survival Death Functions
Cancer. The fundamental defect is. unregulated cell division. Properties of Cancerous Cells. Causes of Cancer. Altered growth and proliferation
 Cancer The fundamental defect is unregulated cell division. Properties of Cancerous Cells Altered growth and proliferation Loss of growth factor dependence Loss of contact inhibition Immortalization Alterated
Cancer The fundamental defect is unregulated cell division. Properties of Cancerous Cells Altered growth and proliferation Loss of growth factor dependence Loss of contact inhibition Immortalization Alterated
Test Bank for Robbins and Cotran Pathologic Basis of Disease 9th Edition by Kumar
 Link full download:https://getbooksolutions.com/download/test-bank-for-robbinsand-cotran-pathologic-basis-of-disease-9th-edition-by-kumar Test Bank for Robbins and Cotran Pathologic Basis of Disease 9th
Link full download:https://getbooksolutions.com/download/test-bank-for-robbinsand-cotran-pathologic-basis-of-disease-9th-edition-by-kumar Test Bank for Robbins and Cotran Pathologic Basis of Disease 9th
Biochemistry of Carcinogenesis. Lecture # 35 Alexander N. Koval
 Biochemistry of Carcinogenesis Lecture # 35 Alexander N. Koval What is Cancer? The term "cancer" refers to a group of diseases in which cells grow and spread unrestrained throughout the body. It is difficult
Biochemistry of Carcinogenesis Lecture # 35 Alexander N. Koval What is Cancer? The term "cancer" refers to a group of diseases in which cells grow and spread unrestrained throughout the body. It is difficult
T cell maturation. T-cell Maturation. What allows T cell maturation?
 T-cell Maturation What allows T cell maturation? Direct contact with thymic epithelial cells Influence of thymic hormones Growth factors (cytokines, CSF) T cell maturation T cell progenitor DN DP SP 2ry
T-cell Maturation What allows T cell maturation? Direct contact with thymic epithelial cells Influence of thymic hormones Growth factors (cytokines, CSF) T cell maturation T cell progenitor DN DP SP 2ry
The Hallmarks of Cancer
 The Hallmarks of Cancer Theresa L. Hodin, Ph.D. Clinical Research Services Theresa.Hodin@RoswellPark.org Hippocrates Cancer surgery, circa 1689 Cancer Surgery Today 1971: Nixon declares War on Cancer
The Hallmarks of Cancer Theresa L. Hodin, Ph.D. Clinical Research Services Theresa.Hodin@RoswellPark.org Hippocrates Cancer surgery, circa 1689 Cancer Surgery Today 1971: Nixon declares War on Cancer
Follicular Lymphoma. ced3 APOPTOSIS. *In the nematode Caenorhabditis elegans 131 of the organism's 1031 cells die during development.
 Harvard-MIT Division of Health Sciences and Technology HST.176: Cellular and Molecular Immunology Course Director: Dr. Shiv Pillai Follicular Lymphoma 1. Characterized by t(14:18) translocation 2. Ig heavy
Harvard-MIT Division of Health Sciences and Technology HST.176: Cellular and Molecular Immunology Course Director: Dr. Shiv Pillai Follicular Lymphoma 1. Characterized by t(14:18) translocation 2. Ig heavy
Cytokines, adhesion molecules and apoptosis markers. A comprehensive product line for human and veterinary ELISAs
 Cytokines, adhesion molecules and apoptosis markers A comprehensive product line for human and veterinary ELISAs IBL International s cytokine product line... is extremely comprehensive. The assays are
Cytokines, adhesion molecules and apoptosis markers A comprehensive product line for human and veterinary ELISAs IBL International s cytokine product line... is extremely comprehensive. The assays are
C-Phycocyanin (C-PC) is a n«sjfc&c- waefc-jduble phycobiliprotein. pigment isolated from Spirulina platensis. This water- soluble protein pigment is
 is a n«sjfc&c- waefc-jduble phycobiliprotein. pigment isolated from Spirulina platensis. This water- soluble protein pigment is") ' ^Summary C-Phycocyanin (C-PC) is a n«sjfc&c- waefc-jduble phycobiliprotein pigment isolated from Spirulina platensis. This water- soluble protein pigment is of greater importance because of its various
' ^Summary C-Phycocyanin (C-PC) is a n«sjfc&c- waefc-jduble phycobiliprotein pigment isolated from Spirulina platensis. This water- soluble protein pigment is of greater importance because of its various
SIBLINGs, cancer's multifunctional weapons
 SIBLINGs, cancer's multifunctional weapons 6/18/08 Akeila Bellahcène and Vincent Castronovo of the Metastasis Research laboratory of the University of Liège are among the first researchers to have discovered
SIBLINGs, cancer's multifunctional weapons 6/18/08 Akeila Bellahcène and Vincent Castronovo of the Metastasis Research laboratory of the University of Liège are among the first researchers to have discovered
Mousa. Israa Ayed. Abdullah AlZibdeh. 0 P a g e
 1 Mousa Israa Ayed Abdullah AlZibdeh 0 P a g e Breast pathology The basic histological units of the breast are called lobules, which are composed of glandular epithelial cells (luminal cells) resting on
1 Mousa Israa Ayed Abdullah AlZibdeh 0 P a g e Breast pathology The basic histological units of the breast are called lobules, which are composed of glandular epithelial cells (luminal cells) resting on
Disorders of Cell Growth & Neoplasia
 General Pathology VPM 152 Disorders of Cell Growth & Neoplasia Lecture 3 Rate of growth, local invasion, and metastasis. Molecular basis of cancer (normal cell-cycle and cellular proliferation). Enrique
General Pathology VPM 152 Disorders of Cell Growth & Neoplasia Lecture 3 Rate of growth, local invasion, and metastasis. Molecular basis of cancer (normal cell-cycle and cellular proliferation). Enrique
Neoplasia 18 lecture 8. Dr Heyam Awad MD, FRCPath
 Neoplasia 18 lecture 8 Dr Heyam Awad MD, FRCPath ILOS 1. understand the angiogenic switch in tumors and factors that stimulate and inhibit angiogenesis. 2. list the steps important for tumor metastasis
Neoplasia 18 lecture 8 Dr Heyam Awad MD, FRCPath ILOS 1. understand the angiogenic switch in tumors and factors that stimulate and inhibit angiogenesis. 2. list the steps important for tumor metastasis
Apoptotic cell signaling in cancer progression and therapyw
 Integrative Biology Dynamic Article Links Cite this: Integr. Biol., 2011, 3, 279 296 www.rsc.org/ibiology REVIEW ARTICLE Apoptotic cell signaling in cancer progression and therapyw Jessica Plati, a Octavian
Integrative Biology Dynamic Article Links Cite this: Integr. Biol., 2011, 3, 279 296 www.rsc.org/ibiology REVIEW ARTICLE Apoptotic cell signaling in cancer progression and therapyw Jessica Plati, a Octavian
34 Apoptosis Programmed cell death is vital to the health and development of multicellular organisms.
 Principles of Biology contents 34 Apoptosis Programmed cell death is vital to the health and development of multicellular organisms. Apoptosis is the reason we have separate fingers and toes. During embryonic
Principles of Biology contents 34 Apoptosis Programmed cell death is vital to the health and development of multicellular organisms. Apoptosis is the reason we have separate fingers and toes. During embryonic
CELL BIOLOGY - CLUTCH CH CANCER.
 !! www.clutchprep.com CONCEPT: OVERVIEW OF CANCER Cancer is a disease which is primarily caused from misregulated cell division, which form There are two types of tumors - Benign tumors remain confined
!! www.clutchprep.com CONCEPT: OVERVIEW OF CANCER Cancer is a disease which is primarily caused from misregulated cell division, which form There are two types of tumors - Benign tumors remain confined
Cancer as a Metabolic Disease
 Cancer as a Metabolic Disease On the Origin, Management and Prevention of Cancer Thomas N. Seyfried @WILEY "" Forword Preface xiii xv 1. Images of Cancer 1 How Cancer is Viewed 2 References 13 2. Confusion
Cancer as a Metabolic Disease On the Origin, Management and Prevention of Cancer Thomas N. Seyfried @WILEY "" Forword Preface xiii xv 1. Images of Cancer 1 How Cancer is Viewed 2 References 13 2. Confusion
Signaling Vascular Morphogenesis and Maintenance
 Signaling Vascular Morphogenesis and Maintenance Douglas Hanahan Science 277: 48-50, in Perspectives (1997) Blood vessels are constructed by two processes: vasculogenesis, whereby a primitive vascular
Signaling Vascular Morphogenesis and Maintenance Douglas Hanahan Science 277: 48-50, in Perspectives (1997) Blood vessels are constructed by two processes: vasculogenesis, whereby a primitive vascular
Virchow s Hypothesis lymphorecticular infiltration of cancer reflected the origin of cancer at sites of inflammation
 Virchow s Hypothesis 1863 lymphorecticular infiltration of cancer reflected the origin of cancer at sites of inflammation Barrett s esophagus/ Esophageal adenocarcinoma PSC / Cholangiocarcinoma Viral hepatitis
Virchow s Hypothesis 1863 lymphorecticular infiltration of cancer reflected the origin of cancer at sites of inflammation Barrett s esophagus/ Esophageal adenocarcinoma PSC / Cholangiocarcinoma Viral hepatitis
Crosstalk between Adiponectin and IGF-IR in breast cancer. Prof. Young Jin Suh Department of Surgery The Catholic University of Korea
 Crosstalk between Adiponectin and IGF-IR in breast cancer Prof. Young Jin Suh Department of Surgery The Catholic University of Korea Obesity Chronic, multifactorial disorder Hypertrophy and hyperplasia
Crosstalk between Adiponectin and IGF-IR in breast cancer Prof. Young Jin Suh Department of Surgery The Catholic University of Korea Obesity Chronic, multifactorial disorder Hypertrophy and hyperplasia
Breast Pathology. Breast Development
 Breast Pathology Lecturer: Hanina Hibshoosh, M.D. Reading: Kumar, Cotran, Robbins, Basic Pathology, 6th Edition, pages 623-635 Breast Development 5th week - thickening of the epidermis - milk line 5th
Breast Pathology Lecturer: Hanina Hibshoosh, M.D. Reading: Kumar, Cotran, Robbins, Basic Pathology, 6th Edition, pages 623-635 Breast Development 5th week - thickening of the epidermis - milk line 5th
Functional Limitations
 Regulation of the Cell Cycle Chapter 12 Pg. 228 245 Functional Limitations Various factors determine whether and when a cell divides. Two functional limitations for cell size limit growth or influence
Regulation of the Cell Cycle Chapter 12 Pg. 228 245 Functional Limitations Various factors determine whether and when a cell divides. Two functional limitations for cell size limit growth or influence
BREAST PATHOLOGY. Fibrocystic Changes
 BREAST PATHOLOGY Lesions of the breast are very common, and they present as palpable, sometimes painful, nodules or masses. Most of these lesions are benign. Breast cancer is the 2 nd most common cause
BREAST PATHOLOGY Lesions of the breast are very common, and they present as palpable, sometimes painful, nodules or masses. Most of these lesions are benign. Breast cancer is the 2 nd most common cause
Apoptosome dysfunction in human cancer
 Apoptosis 2004; 9: 691 704 C 2004 Kluwer Academic Publishers Apoptosome dysfunction in human cancer K. M. Hajra and J. R. Liu Department of Obstetrics and Gynecology, University of Michigan Medical School,
Apoptosis 2004; 9: 691 704 C 2004 Kluwer Academic Publishers Apoptosome dysfunction in human cancer K. M. Hajra and J. R. Liu Department of Obstetrics and Gynecology, University of Michigan Medical School,
PATHOBIOLOGY OF NEOPLASIA
 PATHOBIOLOGY OF NEOPLASIA Department of Pathology Gadjah Mada University School of Medicine dr. Harijadi Blok Biomedis, 6 Maret 2009 [12] 3/17/2009 1 The pathobiology of neoplasia Normal cells Malignant
PATHOBIOLOGY OF NEOPLASIA Department of Pathology Gadjah Mada University School of Medicine dr. Harijadi Blok Biomedis, 6 Maret 2009 [12] 3/17/2009 1 The pathobiology of neoplasia Normal cells Malignant
Major apoptotic mechanisms and genes involved in apoptosis
 Tumor Biol. (2016) 37:8471 8486 DOI 10.1007/s13277-016-5035-9 REVIEW Major apoptotic mechanisms and genes involved in apoptosis Yağmur Kiraz 1,2 & Aysun Adan 1 & Melis Kartal Yandim 2 & Yusuf Baran 1,2
Tumor Biol. (2016) 37:8471 8486 DOI 10.1007/s13277-016-5035-9 REVIEW Major apoptotic mechanisms and genes involved in apoptosis Yağmur Kiraz 1,2 & Aysun Adan 1 & Melis Kartal Yandim 2 & Yusuf Baran 1,2
Genetics and Cancer Ch 20
 Genetics and Cancer Ch 20 Cancer is genetic Hereditary cancers Predisposition genes Ex. some forms of colon cancer Sporadic cancers ~90% of cancers Descendants of cancerous cells all cancerous (clonal)
Genetics and Cancer Ch 20 Cancer is genetic Hereditary cancers Predisposition genes Ex. some forms of colon cancer Sporadic cancers ~90% of cancers Descendants of cancerous cells all cancerous (clonal)
Karyotype analysis reveals transloction of chromosome 22 to 9 in CML chronic myelogenous leukemia has fusion protein Bcr-Abl
 Chapt. 18 Cancer Molecular Biology of Cancer Student Learning Outcomes: Describe cancer diseases in which cells no longer respond Describe how cancers come from genomic mutations (inherited or somatic)
Chapt. 18 Cancer Molecular Biology of Cancer Student Learning Outcomes: Describe cancer diseases in which cells no longer respond Describe how cancers come from genomic mutations (inherited or somatic)
Src-INACTIVE / Src-INACTIVE
 Biology 169 -- Exam 1 February 2003 Answer each question, noting carefully the instructions for each. Repeat- Read the instructions for each question before answering!!! Be as specific as possible in each
Biology 169 -- Exam 1 February 2003 Answer each question, noting carefully the instructions for each. Repeat- Read the instructions for each question before answering!!! Be as specific as possible in each
Objectives. Abbas Chapter 11: Immunological Tolerance. Question 1. Question 2. Question 3. Definitions
 Objectives Abbas Chapter 11: Immunological Tolerance Christina Ciaccio, MD Children s Mercy Hospitals and Clinics February 1, 2010 To introduce the concept of immunologic tolerance To understand what factors
Objectives Abbas Chapter 11: Immunological Tolerance Christina Ciaccio, MD Children s Mercy Hospitals and Clinics February 1, 2010 To introduce the concept of immunologic tolerance To understand what factors
Test Bank for Robbins and Cotran Pathologic Basis of Disease 9th Edition by Kumar
 Link full download: http://testbankair.com/download/test-bank-for-robbins-cotran-pathologic-basis-of-disease-9th-edition-bykumar-abbas-and-aster Test Bank for Robbins and Cotran Pathologic Basis of Disease
Link full download: http://testbankair.com/download/test-bank-for-robbins-cotran-pathologic-basis-of-disease-9th-edition-bykumar-abbas-and-aster Test Bank for Robbins and Cotran Pathologic Basis of Disease
Part I Molecular Cell Biology
 1 Part I Molecular Cell Biology RNA Regulation: Advances in Molecular Biology and Medicine, First Edition. Edited by Robert A. Meyers. 2014 Wiley-VCH Verlag GmbH & Co. KGaA. Published 2014 by Wiley-VCH
1 Part I Molecular Cell Biology RNA Regulation: Advances in Molecular Biology and Medicine, First Edition. Edited by Robert A. Meyers. 2014 Wiley-VCH Verlag GmbH & Co. KGaA. Published 2014 by Wiley-VCH
PCB 3023 Exam 4 - Form A First and Last Name
 PCB 3023 Exam 4 - Form A First and Last Name Student ID # (U Number) A Before beginning this exam, please complete the following instructions: 1) Write your name and U number on the first page of this
PCB 3023 Exam 4 - Form A First and Last Name Student ID # (U Number) A Before beginning this exam, please complete the following instructions: 1) Write your name and U number on the first page of this
Genome of Hepatitis B Virus. VIRAL ONCOGENE Dr. Yahwardiah Siregar, PhD Dr. Sry Suryani Widjaja, Mkes Biochemistry Department
 Genome of Hepatitis B Virus VIRAL ONCOGENE Dr. Yahwardiah Siregar, PhD Dr. Sry Suryani Widjaja, Mkes Biochemistry Department Proto Oncogen and Oncogen Oncogen Proteins that possess the ability to cause
Genome of Hepatitis B Virus VIRAL ONCOGENE Dr. Yahwardiah Siregar, PhD Dr. Sry Suryani Widjaja, Mkes Biochemistry Department Proto Oncogen and Oncogen Oncogen Proteins that possess the ability to cause
VIII Curso Internacional del PIRRECV. Some molecular mechanisms of cancer
 VIII Curso Internacional del PIRRECV Some molecular mechanisms of cancer Laboratorio de Comunicaciones Celulares, Centro FONDAP Estudios Moleculares de la Celula (CEMC), ICBM, Facultad de Medicina, Universidad
VIII Curso Internacional del PIRRECV Some molecular mechanisms of cancer Laboratorio de Comunicaciones Celulares, Centro FONDAP Estudios Moleculares de la Celula (CEMC), ICBM, Facultad de Medicina, Universidad
mirna Dr. S Hosseini-Asl
 mirna Dr. S Hosseini-Asl 1 2 MicroRNAs (mirnas) are small noncoding RNAs which enhance the cleavage or translational repression of specific mrna with recognition site(s) in the 3 - untranslated region
mirna Dr. S Hosseini-Asl 1 2 MicroRNAs (mirnas) are small noncoding RNAs which enhance the cleavage or translational repression of specific mrna with recognition site(s) in the 3 - untranslated region
Early Embryonic Development
 Early Embryonic Development Maternal effect gene products set the stage by controlling the expression of the first embryonic genes. 1. Transcription factors 2. Receptors 3. Regulatory proteins Maternal
Early Embryonic Development Maternal effect gene products set the stage by controlling the expression of the first embryonic genes. 1. Transcription factors 2. Receptors 3. Regulatory proteins Maternal
UNIVERSITY OF MEDICINE AND PHARMACY CRAIOVA PhD SCHOOL. PhD THESIS
 UNIVERSITY OF MEDICINE AND PHARMACY CRAIOVA PhD SCHOOL PhD THESIS THE IMPORTANCE OF TUMOR ANGIOGENESIS IN CEREBRAL TUMOR DIAGNOSIS AND THERAPY ABSTRACT PhD COORDINATOR: Prof. univ. dr. DRICU Anica PhD
UNIVERSITY OF MEDICINE AND PHARMACY CRAIOVA PhD SCHOOL PhD THESIS THE IMPORTANCE OF TUMOR ANGIOGENESIS IN CEREBRAL TUMOR DIAGNOSIS AND THERAPY ABSTRACT PhD COORDINATOR: Prof. univ. dr. DRICU Anica PhD
C) The graph should look exactly like the graph on the left (Mut1 cells + Mating Pheromone for 3 hours at 25 degrees). The cells arrest in G1.
 The graph should look exactly like the graph on the left (Mut1 cells + Mating Pheromone for 3 hours at 25 degrees). The cells arrest in G1.") 706-2000-Exam 4 Answer Key 1) The question asks you to explain peaks A and B in the top graph. The other two graphs were there to give you hints. The question did not ask for these other two graphs to
706-2000-Exam 4 Answer Key 1) The question asks you to explain peaks A and B in the top graph. The other two graphs were there to give you hints. The question did not ask for these other two graphs to
MULTIPLE CHOICE. Choose the one alternative that best completes the statement or answers the question.
 Exam Name MULTIPLE CHOICE. Choose the one alternative that best completes the statement or answers the question. 1) All of the following are synthesized along various sites of the endoplasmic reticulum
Exam Name MULTIPLE CHOICE. Choose the one alternative that best completes the statement or answers the question. 1) All of the following are synthesized along various sites of the endoplasmic reticulum
Chapter 12. Regulation of Cell Division. AP Biology
 Chapter 12. Regulation of Cell Division Coordination of cell division! Multicellular organism " need to coordinate across different parts of organism! timing of cell division! rates of cell division "
Chapter 12. Regulation of Cell Division Coordination of cell division! Multicellular organism " need to coordinate across different parts of organism! timing of cell division! rates of cell division "
Overview of cell death signaling pathways
 Cancer Biology & Therapy ISSN: 1538-4047 (Print) 1555-8576 (Online) Journal homepage: http://www.tandfonline.com/loi/kcbt20 Overview of cell death signaling pathways Zhaoyu Jin & Wafik S. El-Deiry To cite
Cancer Biology & Therapy ISSN: 1538-4047 (Print) 1555-8576 (Online) Journal homepage: http://www.tandfonline.com/loi/kcbt20 Overview of cell death signaling pathways Zhaoyu Jin & Wafik S. El-Deiry To cite
Molecular Characterization of Breast Cancer: The Clinical Significance
 Molecular Characterization of : The Clinical Significance Shahla Masood, M.D. Professor and Chair Department of Pathology and Laboratory Medicine University of Florida College of Medicine-Jacksonville
Molecular Characterization of : The Clinical Significance Shahla Masood, M.D. Professor and Chair Department of Pathology and Laboratory Medicine University of Florida College of Medicine-Jacksonville
OVARY The surface of the ovary is covered with surface epithelium
 OVARY Cow The ovary, or female gonad, is: 1. an exocrine gland, producing oocytes 2. an endocrine gland, secreting hormones, i.e., estrogen and progesterone OVARY OVARY The surface of the ovary is covered
OVARY Cow The ovary, or female gonad, is: 1. an exocrine gland, producing oocytes 2. an endocrine gland, secreting hormones, i.e., estrogen and progesterone OVARY OVARY The surface of the ovary is covered
Prepared by Cyrus H. Nozad, MD, University of Tennessee and John Seyerle, MD, Ohio State University
 Allergy and Immunology Review Corner: Chapter 21 of Middleton s Allergy Principles and Practice, Seventh Edition, edited by N. Franklin Adkinson, et al. Chapter 21: Antigen-Presenting Dendritic Cells (Pages
Allergy and Immunology Review Corner: Chapter 21 of Middleton s Allergy Principles and Practice, Seventh Edition, edited by N. Franklin Adkinson, et al. Chapter 21: Antigen-Presenting Dendritic Cells (Pages
Chapt 15: Molecular Genetics of Cell Cycle and Cancer
 Chapt 15: Molecular Genetics of Cell Cycle and Cancer Student Learning Outcomes: Describe the cell cycle: steps taken by a cell to duplicate itself = cell division; Interphase (G1, S and G2), Mitosis.
Chapt 15: Molecular Genetics of Cell Cycle and Cancer Student Learning Outcomes: Describe the cell cycle: steps taken by a cell to duplicate itself = cell division; Interphase (G1, S and G2), Mitosis.
Programmed Cell Death (apoptosis)
") Programmed Cell Death (apoptosis) Stereotypic death process includes: membrane blebbing nuclear fragmentation chromatin condensation and DNA framentation loss of mitochondrial integrity and release of
Programmed Cell Death (apoptosis) Stereotypic death process includes: membrane blebbing nuclear fragmentation chromatin condensation and DNA framentation loss of mitochondrial integrity and release of
Cell cycle and apoptosis
 Cell cycle and apoptosis Cell cycle Definition Stages and steps Cell cycle Interphase (G1/G0, S, and G2) Mitosis (prophase, metaphase, anaphase, telophase, karyokinesis, cytokinesis) Control checkpoints
Cell cycle and apoptosis Cell cycle Definition Stages and steps Cell cycle Interphase (G1/G0, S, and G2) Mitosis (prophase, metaphase, anaphase, telophase, karyokinesis, cytokinesis) Control checkpoints
RAS Genes. The ras superfamily of genes encodes small GTP binding proteins that are responsible for the regulation of many cellular processes.
 ۱ RAS Genes The ras superfamily of genes encodes small GTP binding proteins that are responsible for the regulation of many cellular processes. Oncogenic ras genes in human cells include H ras, N ras,
۱ RAS Genes The ras superfamily of genes encodes small GTP binding proteins that are responsible for the regulation of many cellular processes. Oncogenic ras genes in human cells include H ras, N ras,
Getting TRAIL back on track for cancer therapy
 (2014) 21, 1350 1364 & 2014 Macmillan Publishers Limited All rights reserved 1350-9047/14 www.nature.com/cdd OPEN Review Getting TRAIL back on track for cancer therapy J Lemke 1,2, S von Karstedt 1, J
(2014) 21, 1350 1364 & 2014 Macmillan Publishers Limited All rights reserved 1350-9047/14 www.nature.com/cdd OPEN Review Getting TRAIL back on track for cancer therapy J Lemke 1,2, S von Karstedt 1, J
BY Mrs. K.SHAILAJA., M. PHARM., LECTURER DEPT OF PHARMACY PRACTICE, SRM COLLEGE OF PHARMACY
 BY Mrs. K.SHAILAJA., M. PHARM., LECTURER DEPT OF PHARMACY PRACTICE, SRM COLLEGE OF PHARMACY Cancer is a group of more than 100 different diseases that are characterized by uncontrolled cellular growth,
BY Mrs. K.SHAILAJA., M. PHARM., LECTURER DEPT OF PHARMACY PRACTICE, SRM COLLEGE OF PHARMACY Cancer is a group of more than 100 different diseases that are characterized by uncontrolled cellular growth,
Cell Quality Control. Peter Takizawa Department of Cell Biology
 Cell Quality Control Peter Takizawa Department of Cell Biology Cellular quality control reduces production of defective proteins. Cells have many quality control systems to ensure that cell does not build
Cell Quality Control Peter Takizawa Department of Cell Biology Cellular quality control reduces production of defective proteins. Cells have many quality control systems to ensure that cell does not build
Cancer Biology Course. Invasion and Metastasis
 Cancer Biology Course Invasion and Metastasis 2016 Lu-Hai Wang NHRI Cancer metastasis Major problem: main reason for killing cancer patients, without it cancer can be cured or controlled. Challenging questions:
Cancer Biology Course Invasion and Metastasis 2016 Lu-Hai Wang NHRI Cancer metastasis Major problem: main reason for killing cancer patients, without it cancer can be cured or controlled. Challenging questions:
Cancer Biology How a cell responds to DNA Damage
 1 Cancer Biology How a cell responds to DNA Damage Jann Sarkaria Department of Oncology Mayo Clinic 2 EDUCATIONAL GOALS How proteins can transmit signals to each other. The definition of a tumor suppressor
1 Cancer Biology How a cell responds to DNA Damage Jann Sarkaria Department of Oncology Mayo Clinic 2 EDUCATIONAL GOALS How proteins can transmit signals to each other. The definition of a tumor suppressor
LYMPHATIC DRAINAGE AXILLARY (MOSTLY) INTERNAL MAMMARY SUPRACLAVICULAR
 INTERNAL MAMMARY SUPRACLAVICULAR") BREAST LYMPHATIC DRAINAGE AXILLARY (MOSTLY) INTERNAL MAMMARY SUPRACLAVICULAR HISTOLOGY LOBE: (10 in whole breast) LOBULE: (many per lobe) ACINUS/I, aka ALVEOLUS/I: (many per lobule) DUCT(S): INTRA- or
BREAST LYMPHATIC DRAINAGE AXILLARY (MOSTLY) INTERNAL MAMMARY SUPRACLAVICULAR HISTOLOGY LOBE: (10 in whole breast) LOBULE: (many per lobe) ACINUS/I, aka ALVEOLUS/I: (many per lobule) DUCT(S): INTRA- or
Subject Index. Bcl-2, apoptosis regulation Bone marrow, polymorphonuclear neutrophil release 24, 26
 Subject Index A1, apoptosis regulation 217, 218 Adaptive immunity, polymorphonuclear neutrophil role 31 33 Angiogenesis cancer 178 endometrium remodeling 172 HIV Tat induction mechanism 176 inflammatory
Subject Index A1, apoptosis regulation 217, 218 Adaptive immunity, polymorphonuclear neutrophil role 31 33 Angiogenesis cancer 178 endometrium remodeling 172 HIV Tat induction mechanism 176 inflammatory
Mitosis and the Cell Cycle
 Mitosis and the Cell Cycle Chapter 12 The Cell Cycle: Cell Growth & Cell Division Where it all began You started as a cell smaller than a period at the end of a sentence Getting from there to here Cell
Mitosis and the Cell Cycle Chapter 12 The Cell Cycle: Cell Growth & Cell Division Where it all began You started as a cell smaller than a period at the end of a sentence Getting from there to here Cell
Cell Injury MECHANISMS OF CELL INJURY
 Cell Injury MECHANISMS OF CELL INJURY The cellular response to injurious stimuli depends on the following factors: Type of injury, Its duration, and Its severity. Thus, low doses of toxins or a brief duration
Cell Injury MECHANISMS OF CELL INJURY The cellular response to injurious stimuli depends on the following factors: Type of injury, Its duration, and Its severity. Thus, low doses of toxins or a brief duration
Multistep nature of cancer development. Cancer genes
 Multistep nature of cancer development Phenotypic progression loss of control over cell growth/death (neoplasm) invasiveness (carcinoma) distal spread (metastatic tumor) Genetic progression multiple genetic
Multistep nature of cancer development Phenotypic progression loss of control over cell growth/death (neoplasm) invasiveness (carcinoma) distal spread (metastatic tumor) Genetic progression multiple genetic
Principles of Genetics and Molecular Biology
 Cell signaling Dr. Diala Abu-Hassan, DDS, PhD School of Medicine Dr.abuhassand@gmail.com Principles of Genetics and Molecular Biology www.cs.montana.edu Modes of cell signaling Direct interaction of a
Cell signaling Dr. Diala Abu-Hassan, DDS, PhD School of Medicine Dr.abuhassand@gmail.com Principles of Genetics and Molecular Biology www.cs.montana.edu Modes of cell signaling Direct interaction of a
Cancer and Gene Alterations - 1
 Cancer and Gene Alterations - 1 Cancer and Gene Alteration As we know, cancer is a disease of unregulated cell growth. Although we looked at some of the features of cancer when we discussed mitosis checkpoints,
Cancer and Gene Alterations - 1 Cancer and Gene Alteration As we know, cancer is a disease of unregulated cell growth. Although we looked at some of the features of cancer when we discussed mitosis checkpoints,
Regulation of Cell Division. AP Biology
 Regulation of Cell Division 2006-2007 Coordination of cell division A multicellular organism needs to coordinate cell division across different tissues & organs critical for normal growth, development
Regulation of Cell Division 2006-2007 Coordination of cell division A multicellular organism needs to coordinate cell division across different tissues & organs critical for normal growth, development
Triple Negative Breast Cancer
 Triple Negative Breast Cancer Prof. Dr. Pornchai O-charoenrat Division of Head-Neck & Breast Surgery Department of Surgery Faculty of Medicine Siriraj Hospital Breast Cancer Classification Traditional
Triple Negative Breast Cancer Prof. Dr. Pornchai O-charoenrat Division of Head-Neck & Breast Surgery Department of Surgery Faculty of Medicine Siriraj Hospital Breast Cancer Classification Traditional