Principles of phylogenetic analysis
|
|
|
- Lydia Walton
- 5 years ago
- Views:
Transcription
1 Principles of phylogenetic analysis Arne Holst-Jensen, NVI, Norway. Fusarium course, Ås, Norway, June 22 nd 2008
2 Distance based methods Compare C OTUs and characters X A + D = Pairwise: A and B; X characters 2X Simple approach, join most similar Cluster phylogeny! Evolutionary clock? Substitution rate More sophisticated, e.g. Neighbor-joining Build phylogeny on D min for total tree Starting point star-tree X 2X AB B Arne Holst-Jensen, NVI, Norway. Fusarium course, Ås, Norway, June 22 nd 2008
3 Parsimony analysis Construct tree with fewest changes A C A C = 1 change C A C = 2 changes (parallel) Find the shortest way through data! Gap handling, recoding, stepmatrixes Simple = presence / absence Recoding Arne Holst-Jensen, NVI, Norway. Fusarium course, Ås, Norway, June 22 nd 2008
4 Maximum likelihood analysis Describe model of evolution Substitution rates, base frequencies Create tree, map characters to tree Probability of tree (P t ) = sum of probabilities of characters across tree Determine probabilities of trees Compare probabilities ΔP t12 = P t1 P t2 significant? Given the evolutionary model Arne Holst-Jensen, NVI, Norway. Fusarium course, Ås, Norway, June 22 nd 2008
5 ML vs. Bayesian likelihood ML searches for best tree given the evolutionary model and observed data Kishino-Hasegawa test compares the probabilities of trees Bayesian analysis, MCMC simulation Create trees based on evolutionary model Prior probability Determine likelihood of data given model Optimal hypothesis = posterior probability max = Prior probability of tree x likelihood of data Determined for internal branches in treesample Arne Holst-Jensen, NVI, Norway. Fusarium course, Ås, Norway, June 22 nd 2008
6 Nice to know stuff Long branch attraction ti Support assessment Decay/Bremer support (parsimony) Consensus, Bootstrap / jackknife Confidence intervals Rooting of tree Outgroup to polarise, define ancestral states Midpoint Unrooted Arne Holst-Jensen, NVI, Norway. Fusarium course, Ås, Norway, June 22 nd 2008
7 Whole genome based phylogeny from a Fusarium point of view R K A H J
8
9 Fungal genomes Some are listed more than once! Less than 50 complete fungal genome sequences An overview of Genome Databases uk/nar/databases/c/ 32 publicly available fungal genome databases ( )
10 Fusarium genomes Three Fusarium genomes sequenced F. graminearum(2003) The second plant pathogenic fungus publicly available Size: ~40 MB Chromosomes: 4 Genes: F. verticillioides Size 41.8 MB Chromosomes: 12 Genes: F. oxysporum Size: 61.4 MB Chromosomes:? Genes:
 Expressed sequence tag library F.")
11 Fusarium genomes Two Fusarium genomes nominated candidates at F. proliferatum F. solani (Nectria haematococca) Expressed sequence tag library F. sporotrichioides tihi id 7517 ESTs
12 Whole genome based phylogeny y Most (all?) use protein sequences Too much information in DNA sequences Effectively impossible to establish phylogeny Strong selection criteria for proteins included in studies Must be represented in all isolates studied Many genes are not annotated BLASTP to find homologous sequences Excluding gene families with >1 representative
13 Whole genome based phylogeny y Best approach for reconstructing ti genome phylogenies? D Supertree methods vs. Concatenated methods
14 Supertree methods Supertrees are phylogenies assembled from smaller phylogenies that share some but not necessarily all taxa in common Supertrees can make novel statements about relationships of taxa that do not co-occur on any single input tree while still retaining hierarchical information from the input trees.
15 Supertree methods Conventional studies source data: measurable attribute of an organism basic unit: character can be viewed as a putative statement of relationship Supertrees source data: phylogenies hl basic unit: membership criterion / statement of relationship (branching topology) at best, can be viewed as a proxy for a shared derived character
16 Supertree p construction E F GH J KL Direct consensus-like techniques A B C K L C DE H I K AB C D E F GH I J K L optimization coding technique h i criterion it i Indirect
17 Supertree methods Direct Strict consensus supertrees MinCutSupertree (and variants) Semi-strict supertrees Indirect Most matrix representation (MR) supertrees Parsimony (MRP and variants) Compatibility (MRC) Minimum flip supertrees (MRF) Average consensus (MRD) Gene tree parsimony
18 Concatenated methods Constructs t multiple l concatenated t protein sequence alignments Maximum likelihood analysis on the concatenated protein sequence alignments from multiple protein families
19 Concatenated methods Multiple sequence alignments Each in principle coding for topology Concatenated sequence alignment Corresponding to one very long protein Phylogenetic analysis of concatenated sequence alignment
20 Whole genome based phylogeny y An example: A dataset of genes from 42 fungal genomes F. graminearum and F. verticillioides included
21 A fungal phylogeny based on 42 complete genomes.. Supertree method ClustalW on the 5316 protein families Manual adjustments of alignments not possible Only l used conserved alignments blocks Average length of alignment 697 sites reduced 214 sites Permutation tail probability test Better than random (P>0.001) 511 alignments failed 4805 alignments used in phylogenetic analysis
22 A fungal phylogeny based on 42 complete genomes.. Supertree method MultiPhyl protein substitution models Reconstruct t t ML phylogeny for each gene family 100 bootstrap replicates on all 4805 alignments! Results summarised: 70% majority-rule rule concensus These results used as input in supertree analysis Supertree analysis using Matrix representation with parsimony (MRP)
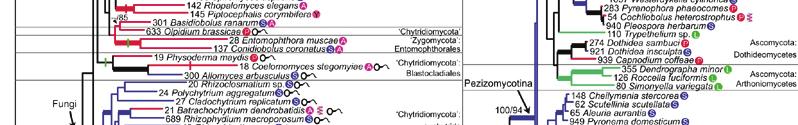
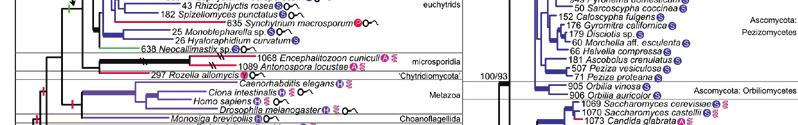
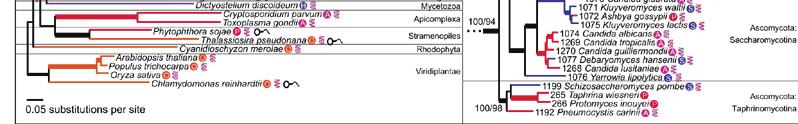
23 MSSA supertree derived from 4,805 fungal gene families. Bootstrap scores for all nodes are displayed. Rhizopus oryzae has been selected as an outgroup. The Basidiomycota and Ascomycota phyla form distinct clades. Subphyla and class clades are highlighted.
24 A fungal phylogeny based on 42 complete genomes.. Concatenated t method All proteins compared in FASTP to find orthologs Form multi-gene clusters of orthologs Only clusters with exactly one member per species 227 protein families Filtered out genes with no syntenic evidence 153 gene families used for further studies Individual gene families aligned, manually adjusted and concatenated together amino acid alignment! ML phylogeny
25 Maximum likelihood phylogeny reconstructed using a concatenated alignment of 153 universally distributed fungal genes. The concatenated alignment contains 42 taxa and exactly 38,000 amino acid positions.
26 Phylogeny y High degree of concordance between supertree method and concatenated method Fusarium forms a monophyletic group with Trichoderma reesei as closest sister group The inference agreed with previous single gene The inference agreed with previous single gene phylogeny studies





27 Sordariomycetes Genome vs. multiple gene phylogeny y James et al., Nature 443: gene phylogeny of nearly 200 fungal species ()
28 Fungal phylogeny y High degree of overall congruence between the two phylogenetic methods a closer look at Sordariomycetes Supertree method 4805 protein families Bayesian analysis 6 genes
29 Phylogeny why, what & when? Arne Holst-Jensen, National Veterinary Institute, t Norway, arne.holst-jensen@vetinst.no
30 Phylogeny: y The evolutionary history and line of descent of a taxon Usually reconstructed based on available data (characters) Applicable also outside biology Arne Holst-Jensen, NVI, Norway. Fusarium course, Ås, Norway, June 22 nd 2008
31 Why? Evolutionary relationships Taxa Character evolution Systems biology Classification of taxa Identity verification Identify diagnostic features Prediction of features Arne Holst-Jensen, NVI, Norway. Fusarium course, Ås, Norway, June 22 nd 2008
32 What? Characters Phenotypic Genotypic Entities, often termed OTUs Operational taxonomic units But principle widely applicable Arne Holst-Jensen, NVI, Norway. Fusarium course, Ås, Norway, June 22 nd 2008
33 What, continued Character types: Two state: presence / absence Multistate, e.g.: DNA sequences (A, C, G, T, gaps) Very long, long, medium, short, very short Ordered, e.g.: Very long, long, medium, Unordered, e.g.: DNA sequences Polymorphic or missing characters Arne Holst-Jensen, NVI, Norway. Fusarium course, Ås, Norway, June 22 nd 2008
34 What, continued 2 Principles of phylogenetic analysis Distance based methods, e.g. N-J Minimise distance across global tree Parsimony P i based methods Minimise number of steps globally Maximum likelihood methods Probability of tree with evolutionary model Cluster analysis phylogenetic analysis! Arne Holst-Jensen, NVI, Norway. Fusarium course, Ås, Norway, June 22 nd 2008
35 When? Etblih Establish or test t evolutionary tree Appropriate data available Revise classification Develop diagnostics Predict features of OTU(s) Play with real-life computer game! Rationalise resource usage Arne Holst-Jensen, NVI, Norway. Fusarium course, Ås, Norway, June 22 nd 2008
36 Data retrieval Arne Holst-Jensen, NVI, Norway. Fusarium course, Ås, Norway, June 22 nd 2008
37 Arne Holst-Jensen, NVI, Norway. Fusarium course, Ås, Norway, June 22 nd


38 Arne Holst-Jensen, NVI, Norway. Fusarium course, Ås, Norway, June 22 nd 2008
39 Arne Holst-Jensen, NVI, Norway. Fusarium course, Ås, Norway, June 22 nd 2008



40 Arne Holst-Jensen, NVI, Norway. Fusarium course, Ås, Norway, June 22 nd 2008
41 Arne Holst-Jensen, NVI, Norway. Fusarium course, Ås, Norway, June 22 nd
42 Arne Holst-Jensen, NVI, Norway. Fusarium course, Ås, Norway, June 22 nd 2008
43 Arne Holst-Jensen, NVI, Norway. Fusarium course, Ås, Norway, June 22 nd 2008
44 Arne Holst-Jensen, NVI, Norway. Fusarium course, Ås, Norway, June 22 nd 2008
45 Arne Holst-Jensen, NVI, Norway. Fusarium course, Ås, Norway, June 22 nd 2008
46 Arne Holst-Jensen, NVI, Norway. Fusarium course, Ås, Norway, June 22 nd 2008
47 Arne Holst-Jensen, NVI, Norway. Fusarium course, Ås, Norway, June 22 nd 2008
48 Arne Holst-Jensen, NVI, Norway. Fusarium course, Ås, Norway, June 22 nd 2008 RK_EF1a_SRS.fas
49 Arne Holst-Jensen, NVI, Norway. Fusarium course, Ås, Norway, June 22 nd 2008
50 Arne Holst-Jensen, NVI, Norway. Fusarium course, Ås, Norway, June 22 nd 2008
51 Arne Holst-Jensen, NVI, Norway. Fusarium course, Ås, Norway, June 22 nd 2008
52 Arne Holst-Jensen, NVI, Norway. Fusarium course, Ås, Norway, June 22 nd 2008
53 Arne Holst-Jensen, NVI, Norway. Fusarium course, Ås, Norway, June 22 nd 2008
54 Arne Holst-Jensen, NVI, Norway. Fusarium course, Ås, Norway, June 22 nd 2008
55 Arne Holst-Jensen, NVI, Norway. Fusarium course, Ås, Norway, June 22 nd 2008
56 Arne Holst-Jensen, NVI, Norway. Fusarium course, Ås, Norway, June 22 nd 2008
57 Arne Holst-Jensen, NVI, Norway. Fusarium course, Ås, Norway, June 22 nd 2008
58 Arne Holst-Jensen, NVI, Norway. Fusarium course, Ås, Norway, June 22 nd 2008 New BlastN search
59 Arne Holst-Jensen, NVI, Norway. Fusarium course, Ås, Norway, June 22 nd 2008
60 Arne Holst-Jensen, NVI, Norway. Fusarium course, Ås, Norway, June 22 nd 2008
61 Arne Holst-Jensen, NVI, Norway. Fusarium course, Ås, Norway, June 22 nd 2008
Phylogenetic Methods
 Phylogenetic Methods Multiple Sequence lignment Pairwise distance matrix lustering algorithms: NJ, UPM - guide trees Phylogenetic trees Nucleotide vs. amino acid sequences for phylogenies ) Nucleotides:
Phylogenetic Methods Multiple Sequence lignment Pairwise distance matrix lustering algorithms: NJ, UPM - guide trees Phylogenetic trees Nucleotide vs. amino acid sequences for phylogenies ) Nucleotides:
Integrative Biology 200A PRINCIPLES OF PHYLOGENETICS Spring 2012
 Integrative Biology 200A PRINCIPLES OF PHYLOGENETICS Spring 2012 University of California, Berkeley Kipling Will- 1 March Data/Hypothesis Exploration and Support Measures I. Overview. -- Many would agree
Integrative Biology 200A PRINCIPLES OF PHYLOGENETICS Spring 2012 University of California, Berkeley Kipling Will- 1 March Data/Hypothesis Exploration and Support Measures I. Overview. -- Many would agree
The BLAST search on NCBI ( and GISAID
 Supplemental materials and methods The BLAST search on NCBI (http:// www.ncbi.nlm.nih.gov) and GISAID (http://www.platform.gisaid.org) showed that hemagglutinin (HA) gene of North American H5N1, H5N2 and
Supplemental materials and methods The BLAST search on NCBI (http:// www.ncbi.nlm.nih.gov) and GISAID (http://www.platform.gisaid.org) showed that hemagglutinin (HA) gene of North American H5N1, H5N2 and
Using Phylogenetic Structure to Assess the Evolutionary Ecology of Microbiota! TJS! iseem Call! April 2015!
 Using Phylogenetic Structure to Assess the Evolutionary Ecology of Microbiota! TJS! iseem Call! April 2015! How are Microbes Distributed In Nature?! A major question in microbial ecology! Used to assess
Using Phylogenetic Structure to Assess the Evolutionary Ecology of Microbiota! TJS! iseem Call! April 2015! How are Microbes Distributed In Nature?! A major question in microbial ecology! Used to assess
Estimating Phylogenies (Evolutionary Trees) I
 I") stimating Phylogenies (volutionary Trees) I iol4230 Tues, Feb 27, 2018 ill Pearson wrp@virginia.edu 4-2818 Pinn 6-057 Goals of today s lecture: Why estimate phylogenies? Origin of man (woman) Origin of
stimating Phylogenies (volutionary Trees) I iol4230 Tues, Feb 27, 2018 ill Pearson wrp@virginia.edu 4-2818 Pinn 6-057 Goals of today s lecture: Why estimate phylogenies? Origin of man (woman) Origin of
Name: Due on Wensday, December 7th Bioinformatics Take Home Exam #9 Pick one most correct answer, unless stated otherwise!
 Name: Due on Wensday, December 7th Bioinformatics Take Home Exam #9 Pick one most correct answer, unless stated otherwise! 1. What process brought 2 divergent chlorophylls into the ancestor of the cyanobacteria,
Name: Due on Wensday, December 7th Bioinformatics Take Home Exam #9 Pick one most correct answer, unless stated otherwise! 1. What process brought 2 divergent chlorophylls into the ancestor of the cyanobacteria,
CONSTRUCTION OF PHYLOGENETIC TREE USING NEIGHBOR JOINING ALGORITHMS TO IDENTIFY THE HOST AND THE SPREADING OF SARS EPIDEMIC
 CONSTRUCTION OF PHYLOGENETIC TREE USING NEIGHBOR JOINING ALGORITHMS TO IDENTIFY THE HOST AND THE SPREADING OF SARS EPIDEMIC 1 MOHAMMAD ISA IRAWAN, 2 SITI AMIROCH 1 Institut Teknologi Sepuluh Nopember (ITS)
CONSTRUCTION OF PHYLOGENETIC TREE USING NEIGHBOR JOINING ALGORITHMS TO IDENTIFY THE HOST AND THE SPREADING OF SARS EPIDEMIC 1 MOHAMMAD ISA IRAWAN, 2 SITI AMIROCH 1 Institut Teknologi Sepuluh Nopember (ITS)
Exploring HIV Evolution: An Opportunity for Research Sam Donovan and Anton E. Weisstein
 Microbes Count! 137 Video IV: Reading the Code of Life Human Immunodeficiency Virus (HIV), like other retroviruses, has a much higher mutation rate than is typically found in organisms that do not go through
Microbes Count! 137 Video IV: Reading the Code of Life Human Immunodeficiency Virus (HIV), like other retroviruses, has a much higher mutation rate than is typically found in organisms that do not go through
SUPPLEMENTAL INFORMATION
 SUPPLEMENTAL INFORMATION GO term analysis of differentially methylated SUMIs. GO term analysis of the 458 SUMIs with the largest differential methylation between human and chimp shows that they are more
SUPPLEMENTAL INFORMATION GO term analysis of differentially methylated SUMIs. GO term analysis of the 458 SUMIs with the largest differential methylation between human and chimp shows that they are more
Gene Ontology and Functional Enrichment. Genome 559: Introduction to Statistical and Computational Genomics Elhanan Borenstein
 Gene Ontology and Functional Enrichment Genome 559: Introduction to Statistical and Computational Genomics Elhanan Borenstein The parsimony principle: A quick review Find the tree that requires the fewest
Gene Ontology and Functional Enrichment Genome 559: Introduction to Statistical and Computational Genomics Elhanan Borenstein The parsimony principle: A quick review Find the tree that requires the fewest
To test the possible source of the HBV infection outside the study family, we searched the Genbank
 Supplementary Discussion The source of hepatitis B virus infection To test the possible source of the HBV infection outside the study family, we searched the Genbank and HBV Database (http://hbvdb.ibcp.fr),
Supplementary Discussion The source of hepatitis B virus infection To test the possible source of the HBV infection outside the study family, we searched the Genbank and HBV Database (http://hbvdb.ibcp.fr),
Going Nowhere Fast: Lentivirus genetic sequence evolution does not correlate with phenotypic evolution.
 Going Nowhere Fast: Lentivirus genetic sequence evolution does not correlate with phenotypic evolution. Brian T. Foley, PhD btf@lanl.gov HIV Genetic Sequences, Immunology, Drug Resistance and Vaccine Trials
Going Nowhere Fast: Lentivirus genetic sequence evolution does not correlate with phenotypic evolution. Brian T. Foley, PhD btf@lanl.gov HIV Genetic Sequences, Immunology, Drug Resistance and Vaccine Trials
Distinguishing epidemiological dependent from treatment (resistance) dependent HIV mutations: Problem Statement
 dependent HIV mutations: Problem Statement") Distinguishing epidemiological dependent from treatment (resistance) dependent HIV mutations: Problem Statement Leander Schietgat 1, Kristof Theys 2, Jan Ramon 1, Hendrik Blockeel 1, and Anne-Mieke Vandamme
Distinguishing epidemiological dependent from treatment (resistance) dependent HIV mutations: Problem Statement Leander Schietgat 1, Kristof Theys 2, Jan Ramon 1, Hendrik Blockeel 1, and Anne-Mieke Vandamme
(ii) The effective population size may be lower than expected due to variability between individuals in infectiousness.
 The effective population size may be lower than expected due to variability between individuals in infectiousness.") Supplementary methods Details of timepoints Caió sequences were derived from: HIV-2 gag (n = 86) 16 sequences from 1996, 10 from 2003, 45 from 2006, 13 from 2007 and two from 2008. HIV-2 env (n = 70) 21
Supplementary methods Details of timepoints Caió sequences were derived from: HIV-2 gag (n = 86) 16 sequences from 1996, 10 from 2003, 45 from 2006, 13 from 2007 and two from 2008. HIV-2 env (n = 70) 21
Global variation in copy number in the human genome
 Global variation in copy number in the human genome Redon et. al. Nature 444:444-454 (2006) 12.03.2007 Tarmo Puurand Study 270 individuals (HapMap collection) Affymetrix 500K Whole Genome TilePath (WGTP)
Global variation in copy number in the human genome Redon et. al. Nature 444:444-454 (2006) 12.03.2007 Tarmo Puurand Study 270 individuals (HapMap collection) Affymetrix 500K Whole Genome TilePath (WGTP)
Multiple sequence alignment
 Multiple sequence alignment Bas. Dutilh Systems Biology: Bioinformatic Data Analysis Utrecht University, February 18 th 2016 Protein alignments We have seen how to create a pairwise alignment of two sequences
Multiple sequence alignment Bas. Dutilh Systems Biology: Bioinformatic Data Analysis Utrecht University, February 18 th 2016 Protein alignments We have seen how to create a pairwise alignment of two sequences
Mapping the Antigenic and Genetic Evolution of Influenza Virus
 Mapping the Antigenic and Genetic Evolution of Influenza Virus Derek J. Smith, Alan S. Lapedes, Jan C. de Jong, Theo M. Bestebroer, Guus F. Rimmelzwaan, Albert D. M. E. Osterhaus, Ron A. M. Fouchier Science
Mapping the Antigenic and Genetic Evolution of Influenza Virus Derek J. Smith, Alan S. Lapedes, Jan C. de Jong, Theo M. Bestebroer, Guus F. Rimmelzwaan, Albert D. M. E. Osterhaus, Ron A. M. Fouchier Science
NJMerge: A generic technique for scaling phylogeny estimation methods and its application to species trees Supplementary Materials
 NJMerge: A generic technique for scaling phylogeny estimation methods and its application to species trees Supplementary Materials Erin K. Molloy 1[ 1 5553 3312] and Tandy Warnow 1[ 1 7717 3514] Department
NJMerge: A generic technique for scaling phylogeny estimation methods and its application to species trees Supplementary Materials Erin K. Molloy 1[ 1 5553 3312] and Tandy Warnow 1[ 1 7717 3514] Department
Evolutionary interactions between haemagglutinin and neuraminidase in avian influenza
 Ward et al. BMC Evolutionary Biology 2013, 13:222 RESEARCH ARTICLE Open Access Evolutionary interactions between haemagglutinin and neuraminidase in avian influenza Melissa J Ward 1*, Samantha J Lycett
Ward et al. BMC Evolutionary Biology 2013, 13:222 RESEARCH ARTICLE Open Access Evolutionary interactions between haemagglutinin and neuraminidase in avian influenza Melissa J Ward 1*, Samantha J Lycett
Rajesh Kannangai Phone: ; Fax: ; *Corresponding author
 Amino acid sequence divergence of Tat protein (exon1) of subtype B and C HIV-1 strains: Does it have implications for vaccine development? Abraham Joseph Kandathil 1, Rajesh Kannangai 1, *, Oriapadickal
Amino acid sequence divergence of Tat protein (exon1) of subtype B and C HIV-1 strains: Does it have implications for vaccine development? Abraham Joseph Kandathil 1, Rajesh Kannangai 1, *, Oriapadickal
BEAST Bayesian Evolutionary Analysis Sampling Trees
 BEAST Bayesian Evolutionary Analysis Sampling Trees Introduction Revealing the evolutionary dynamics of influenza This tutorial provides a step-by-step explanation on how to reconstruct the evolutionary
BEAST Bayesian Evolutionary Analysis Sampling Trees Introduction Revealing the evolutionary dynamics of influenza This tutorial provides a step-by-step explanation on how to reconstruct the evolutionary
Origins and evolutionary genomics of the novel avian-origin H7N9 influenza A virus in China: Early findings
 Origins and evolutionary genomics of the novel 2013 avian-origin H7N9 influenza A virus in : Early findings Jiankui He*, Luwen Ning, Yin Tong Department of Biology, South University of Science and Technology
Origins and evolutionary genomics of the novel 2013 avian-origin H7N9 influenza A virus in : Early findings Jiankui He*, Luwen Ning, Yin Tong Department of Biology, South University of Science and Technology
Phylogenetic Tree Practical Problems
 Phylogenetic Tree Practical Problems Software Tools: MEGA A software package for constructing phylogenetic trees using neighbor-joining, UPGMA, and maximum parsimony. ClustalW A tool for constructing multiple
Phylogenetic Tree Practical Problems Software Tools: MEGA A software package for constructing phylogenetic trees using neighbor-joining, UPGMA, and maximum parsimony. ClustalW A tool for constructing multiple
Lecture 12. Immunology and disease: parasite antigenic diversity. and. Phylogenetic trees
 Lecture 12 Immunology and disease: parasite antigenic diversity and Phylogenetic trees Benefits of antigenic variation 2. Infect hosts with prior exposure Hosts often maintain memory against prior infections,
Lecture 12 Immunology and disease: parasite antigenic diversity and Phylogenetic trees Benefits of antigenic variation 2. Infect hosts with prior exposure Hosts often maintain memory against prior infections,
Structural Variation and Medical Genomics
 Structural Variation and Medical Genomics Andrew King Department of Biomedical Informatics July 8, 2014 You already know about small scale genetic mutations Single nucleotide polymorphism (SNPs) Deletions,
Structural Variation and Medical Genomics Andrew King Department of Biomedical Informatics July 8, 2014 You already know about small scale genetic mutations Single nucleotide polymorphism (SNPs) Deletions,
Maximum Likelihood ofevolutionary Trees is Hard p.1
 Maximum Likelihood of Evolutionary Trees is Hard Benny Chor School of Computer Science Tel-Aviv University Joint work with Tamir Tuller Maximum Likelihood ofevolutionary Trees is Hard p.1 Challenging Basic
Maximum Likelihood of Evolutionary Trees is Hard Benny Chor School of Computer Science Tel-Aviv University Joint work with Tamir Tuller Maximum Likelihood ofevolutionary Trees is Hard p.1 Challenging Basic
1 Supplementary Figures
 Supplementary Figures D. simulans (dsim) D. sechellia (dsec) D. melanogaster (dmel) S. cerevisiae (scer) S. paradoxus (spar) S. mikatae (smik) S. bayanus (sbay) S. castellii (scas) C. glabrata (cgla) K.
Supplementary Figures D. simulans (dsim) D. sechellia (dsec) D. melanogaster (dmel) S. cerevisiae (scer) S. paradoxus (spar) S. mikatae (smik) S. bayanus (sbay) S. castellii (scas) C. glabrata (cgla) K.
Adaptation vs Exaptation. Examples of Exaptation. Behavior of the Day! Historical Hypotheses
 Adaptation vs Exaptation 1. Definition 1: Adaptation = A trait, or integrated suite of traits, that increases the fitness (reproductive success) of its possessor. 2. However, traits can have current utility
Adaptation vs Exaptation 1. Definition 1: Adaptation = A trait, or integrated suite of traits, that increases the fitness (reproductive success) of its possessor. 2. However, traits can have current utility
Dr Rick Tearle Senior Applications Specialist, EMEA Complete Genomics Complete Genomics, Inc.
 Dr Rick Tearle Senior Applications Specialist, EMEA Complete Genomics Topics Overview of Data Processing Pipeline Overview of Data Files 2 DNA Nano-Ball (DNB) Read Structure Genome : acgtacatgcattcacacatgcttagctatctctcgccag
Dr Rick Tearle Senior Applications Specialist, EMEA Complete Genomics Topics Overview of Data Processing Pipeline Overview of Data Files 2 DNA Nano-Ball (DNB) Read Structure Genome : acgtacatgcattcacacatgcttagctatctctcgccag
Teaching Phylogeny and Direction of Viral Transmission using a Real HIV Criminal Case
 Tested Studies for Laboratory Teaching Proceedings of the Association for Biology Laboratory Education Volume 39, Article 24, 2018 Teaching Phylogeny and Direction of Viral Transmission using a Real HIV
Tested Studies for Laboratory Teaching Proceedings of the Association for Biology Laboratory Education Volume 39, Article 24, 2018 Teaching Phylogeny and Direction of Viral Transmission using a Real HIV
arxiv: v1 [cs.ce] 30 Dec 2014
![arxiv: v1 [cs.ce] 30 Dec 2014](/thumbs/88/115676320.jpg "arxiv: v1 [cs.ce] 30 Dec 2014") Fast and Scalable Inference of Multi-Sample Cancer Lineages Victoria Popic 1, Raheleh Salari 1, Iman Hajirasouliha 1, Dorna Kashef-Haghighi 1, Robert B West and Serafim Batzoglou 1* arxiv:141.874v1 [cs.ce]
Fast and Scalable Inference of Multi-Sample Cancer Lineages Victoria Popic 1, Raheleh Salari 1, Iman Hajirasouliha 1, Dorna Kashef-Haghighi 1, Robert B West and Serafim Batzoglou 1* arxiv:141.874v1 [cs.ce]
Network-assisted data analysis
 Network-assisted data analysis Bing Zhang Department of Biomedical Informatics Vanderbilt University bing.zhang@vanderbilt.edu Protein identification in shotgun proteomics Protein digestion LC-MS/MS Protein
Network-assisted data analysis Bing Zhang Department of Biomedical Informatics Vanderbilt University bing.zhang@vanderbilt.edu Protein identification in shotgun proteomics Protein digestion LC-MS/MS Protein
Annotation of Drosophila mojavensis fosmid 8 Priya Srikanth Bio 434W
 Annotation of Drosophila mojavensis fosmid 8 Priya Srikanth Bio 434W 5.1.2007 Overview High-quality finished sequence is much more useful for research once it is annotated. Annotation is a fundamental
Annotation of Drosophila mojavensis fosmid 8 Priya Srikanth Bio 434W 5.1.2007 Overview High-quality finished sequence is much more useful for research once it is annotated. Annotation is a fundamental
RNA Secondary Structures: A Case Study on Viruses Bioinformatics Senior Project John Acampado Under the guidance of Dr. Jason Wang
 RNA Secondary Structures: A Case Study on Viruses Bioinformatics Senior Project John Acampado Under the guidance of Dr. Jason Wang Table of Contents Overview RSpredict JAVA RSpredict WebServer RNAstructure
RNA Secondary Structures: A Case Study on Viruses Bioinformatics Senior Project John Acampado Under the guidance of Dr. Jason Wang Table of Contents Overview RSpredict JAVA RSpredict WebServer RNAstructure
aP. Code assigned: Short title: Remove (abolish) the species Narcissus symptomless virus in the genus Carlavirus, family Betaflexiviridae
 the species Narcissus symptomless virus in the genus Carlavirus, family Betaflexiviridae") This form should be used for all taxonomic proposals. Please complete all those modules that are applicable (and then delete the unwanted sections). For guidance, see the notes written in blue and the
This form should be used for all taxonomic proposals. Please complete all those modules that are applicable (and then delete the unwanted sections). For guidance, see the notes written in blue and the
README file for GRASTv1.0.pl
 README file for GRASTv.0.pl Genome Reduction Analysing Software Tool (GRAST). Produced by Christina Toft and Mario A. Fares Date 03/04/06 Reference and more information: Toft, C and Fares, MA (2006). GRAST:
README file for GRASTv.0.pl Genome Reduction Analysing Software Tool (GRAST). Produced by Christina Toft and Mario A. Fares Date 03/04/06 Reference and more information: Toft, C and Fares, MA (2006). GRAST:
Project PRACE 1IP, WP7.4
 Project PRACE 1IP, WP7.4 Plamenka Borovska, Veska Gancheva Computer Systems Department Technical University of Sofia The Team is consists of 5 members: 2 Professors; 1 Assist. Professor; 2 Researchers;
Project PRACE 1IP, WP7.4 Plamenka Borovska, Veska Gancheva Computer Systems Department Technical University of Sofia The Team is consists of 5 members: 2 Professors; 1 Assist. Professor; 2 Researchers;
OVERVIEW OF CURRENT IDENTIFICATION SYSTEMS AND DATABASES
 OVERVIEW OF CURRENT IDENTIFICATION SYSTEMS AND DATABASES EVERY STEP OF THE WAY 1 EVERY STEP OF THE WAY MICROBIAL IDENTIFICATION METHODS DNA RNA Genotypic Sequencing of ribosomal RNA regions of bacteria
OVERVIEW OF CURRENT IDENTIFICATION SYSTEMS AND DATABASES EVERY STEP OF THE WAY 1 EVERY STEP OF THE WAY MICROBIAL IDENTIFICATION METHODS DNA RNA Genotypic Sequencing of ribosomal RNA regions of bacteria
Evolution of influenza
 Evolution of influenza Today: 1. Global health impact of flu - why should we care? 2. - what are the components of the virus and how do they change? 3. Where does influenza come from? - are there animal
Evolution of influenza Today: 1. Global health impact of flu - why should we care? 2. - what are the components of the virus and how do they change? 3. Where does influenza come from? - are there animal
SUPPLEMENTARY INFORMATION
 Testing the accuracy of ancestral state reconstruction The accuracy of the ancestral state reconstruction with maximum likelihood methods can depend on the underlying model used in the reconstruction.
Testing the accuracy of ancestral state reconstruction The accuracy of the ancestral state reconstruction with maximum likelihood methods can depend on the underlying model used in the reconstruction.
a-hV. Code assigned:
 This form should be used for all taxonomic proposals. Please complete all those modules that are applicable (and then delete the unwanted sections). For guidance, see the notes written in blue and the
This form should be used for all taxonomic proposals. Please complete all those modules that are applicable (and then delete the unwanted sections). For guidance, see the notes written in blue and the
Cahn - Ingold - Prelog system. Proteins: Evolution, and Analysis Lecture 7 9/15/2009. The Fischer Convention (1) G (2) (3)
 G (2) (3)") Chapter 4 (1) G Proteins: Evolution, and Analysis Lecture 7 9/15/2009 A V L I M P F W Chapter 4 (2) S (3) T N Q Y C K R H D E The Fischer Convention Absolute configuration about an asymmetric carbon related
Chapter 4 (1) G Proteins: Evolution, and Analysis Lecture 7 9/15/2009 A V L I M P F W Chapter 4 (2) S (3) T N Q Y C K R H D E The Fischer Convention Absolute configuration about an asymmetric carbon related
Research Strategy: 1. Background and Significance
 Research Strategy: 1. Background and Significance 1.1. Heterogeneity is a common feature of cancer. A better understanding of this heterogeneity may present therapeutic opportunities: Intratumor heterogeneity
Research Strategy: 1. Background and Significance 1.1. Heterogeneity is a common feature of cancer. A better understanding of this heterogeneity may present therapeutic opportunities: Intratumor heterogeneity
Department of Forest Ecosystems and Society, Oregon State University
 July 4, 2018 Prof. Christopher Still Department of Forest Ecosystems and Society, Oregon State University 321 Richardson Hall, Corvallis OR 97331-5752 Dear Prof. Christopher Still, We would like to thank
July 4, 2018 Prof. Christopher Still Department of Forest Ecosystems and Society, Oregon State University 321 Richardson Hall, Corvallis OR 97331-5752 Dear Prof. Christopher Still, We would like to thank
Classification Student Material
 Classification Student Material The Caminalcule creatures in the packet should be sorted on the basis of their similarity of their phenotypes (observable characteristics) as follows: 1. Start by identifying
Classification Student Material The Caminalcule creatures in the packet should be sorted on the basis of their similarity of their phenotypes (observable characteristics) as follows: 1. Start by identifying
A Universal Trend among Proteomes Indicates an Oily Last Common Ancestor. BI Journal Club Aleksander Sudakov
 A Universal Trend among Proteomes Indicates an Oily Last Common Ancestor BI Journal Club 11.03.13 Aleksander Sudakov Used literature Ranjan V. Mannige, Charles L. Brooks, and Eugene I. Shakhnovich. 2012.
A Universal Trend among Proteomes Indicates an Oily Last Common Ancestor BI Journal Club 11.03.13 Aleksander Sudakov Used literature Ranjan V. Mannige, Charles L. Brooks, and Eugene I. Shakhnovich. 2012.
Identification of mirnas in Eucalyptus globulus Plant by Computational Methods
 International Journal of Pharmaceutical Science Invention ISSN (Online): 2319 6718, ISSN (Print): 2319 670X Volume 2 Issue 5 May 2013 PP.70-74 Identification of mirnas in Eucalyptus globulus Plant by Computational
International Journal of Pharmaceutical Science Invention ISSN (Online): 2319 6718, ISSN (Print): 2319 670X Volume 2 Issue 5 May 2013 PP.70-74 Identification of mirnas in Eucalyptus globulus Plant by Computational
Host Dependent Evolutionary Patterns and the Origin of 2009 H1N1 Pandemic Influenza
 Host Dependent Evolutionary Patterns and the Origin of 2009 H1N1 Pandemic Influenza The origin of H1N1pdm constitutes an unresolved mystery, as its most recently observed ancestors were isolated in pigs
Host Dependent Evolutionary Patterns and the Origin of 2009 H1N1 Pandemic Influenza The origin of H1N1pdm constitutes an unresolved mystery, as its most recently observed ancestors were isolated in pigs
SMPD 287 Spring 2015 Bioinformatics in Medical Product Development. Final Examination
 Final Examination You have a choice between A, B, or C. Please email your solutions, as a pdf attachment, by May 13, 2015. In the subject of the email, please use the following format: firstname_lastname_x
Final Examination You have a choice between A, B, or C. Please email your solutions, as a pdf attachment, by May 13, 2015. In the subject of the email, please use the following format: firstname_lastname_x
Utilization of NCBI Pathogen Detection Tool in USDA FSIS
 Utilization of NCBI Pathogen Detection Tool in USDA FSIS Glenn Tillman, Ph.D. Branch Chief Microbiology: Characterization Branch FSIS Office of Public Health and Science Glenn.tillman@fsis.usda.gov 1 Background
Utilization of NCBI Pathogen Detection Tool in USDA FSIS Glenn Tillman, Ph.D. Branch Chief Microbiology: Characterization Branch FSIS Office of Public Health and Science Glenn.tillman@fsis.usda.gov 1 Background
Variant Classification. Author: Mike Thiesen, Golden Helix, Inc.
 Variant Classification Author: Mike Thiesen, Golden Helix, Inc. Overview Sequencing pipelines are able to identify rare variants not found in catalogs such as dbsnp. As a result, variants in these datasets
Variant Classification Author: Mike Thiesen, Golden Helix, Inc. Overview Sequencing pipelines are able to identify rare variants not found in catalogs such as dbsnp. As a result, variants in these datasets
YUMI YAMAGUCHI-KABATA AND TAKASHI GOJOBORI* Center for Information Biology, National Institute of Genetics, Mishima , Japan
 JOURNAL OF VIROLOGY, May 2000, p. 4335 4350 Vol. 74, No. 9 0022-538X/00/$04.00 0 Copyright 2000, American Society for Microbiology. All Rights Reserved. Reevaluation of Amino Acid Variability of the Human
JOURNAL OF VIROLOGY, May 2000, p. 4335 4350 Vol. 74, No. 9 0022-538X/00/$04.00 0 Copyright 2000, American Society for Microbiology. All Rights Reserved. Reevaluation of Amino Acid Variability of the Human
Reliable reconstruction of HIV-1 whole genome haplotypes reveals clonal interference and genetic hitchhiking among immune escape variants
 Pandit and de Boer Retrovirology 2014, 11:56 RESEARCH Open Access Reliable reconstruction of HIV-1 whole genome haplotypes reveals clonal interference and genetic hitchhiking among immune escape variants
Pandit and de Boer Retrovirology 2014, 11:56 RESEARCH Open Access Reliable reconstruction of HIV-1 whole genome haplotypes reveals clonal interference and genetic hitchhiking among immune escape variants
Study the Evolution of the Avian Influenza Virus
 Designing an Algorithm to Study the Evolution of the Avian Influenza Virus Arti Khana Mentor: Takis Benos Rachel Brower-Sinning Department of Computational Biology University of Pittsburgh Overview Introduction
Designing an Algorithm to Study the Evolution of the Avian Influenza Virus Arti Khana Mentor: Takis Benos Rachel Brower-Sinning Department of Computational Biology University of Pittsburgh Overview Introduction
Nature Methods: doi: /nmeth.3115
 Supplementary Figure 1 Analysis of DNA methylation in a cancer cohort based on Infinium 450K data. RnBeads was used to rediscover a clinically distinct subgroup of glioblastoma patients characterized by
Supplementary Figure 1 Analysis of DNA methylation in a cancer cohort based on Infinium 450K data. RnBeads was used to rediscover a clinically distinct subgroup of glioblastoma patients characterized by
Phylogenomics. Antonis Rokas Department of Biological Sciences Vanderbilt University.
 Phylogenomics Antonis Rokas Department of Biological Sciences Vanderbilt University http://as.vanderbilt.edu/rokaslab High-Throughput DNA Sequencing Technologies 454 / Roche 450 bp 1.5 Gbp / day Illumina
Phylogenomics Antonis Rokas Department of Biological Sciences Vanderbilt University http://as.vanderbilt.edu/rokaslab High-Throughput DNA Sequencing Technologies 454 / Roche 450 bp 1.5 Gbp / day Illumina
I. Setup. - Note that: autohgpec_v1.0 can work on Windows, Ubuntu and Mac OS.
 autohgpec: Automated prediction of novel disease-gene and diseasedisease associations and evidence collection based on a random walk on heterogeneous network Duc-Hau Le 1,*, Trang T.H. Tran 1 1 School
autohgpec: Automated prediction of novel disease-gene and diseasedisease associations and evidence collection based on a random walk on heterogeneous network Duc-Hau Le 1,*, Trang T.H. Tran 1 1 School
Genomic structural variation
 Genomic structural variation Mario Cáceres The new genomic variation DNA sequence differs across individuals much more than researchers had suspected through structural changes A huge amount of structural
Genomic structural variation Mario Cáceres The new genomic variation DNA sequence differs across individuals much more than researchers had suspected through structural changes A huge amount of structural
White Paper Estimating Complex Phenotype Prevalence Using Predictive Models
 White Paper 23-12 Estimating Complex Phenotype Prevalence Using Predictive Models Authors: Nicholas A. Furlotte Aaron Kleinman Robin Smith David Hinds Created: September 25 th, 2015 September 25th, 2015
White Paper 23-12 Estimating Complex Phenotype Prevalence Using Predictive Models Authors: Nicholas A. Furlotte Aaron Kleinman Robin Smith David Hinds Created: September 25 th, 2015 September 25th, 2015
SiFit: inferring tumor trees from single-cell sequencing data under finite-sites models
 Zafar et al. Genome Biology (2017) 18:178 DOI 10.1186/s13059-017-1311-2 METHOD Open Access SiFit: inferring tumor trees from single-cell sequencing data under finite-sites models Hamim Zafar 1,2, Anthony
Zafar et al. Genome Biology (2017) 18:178 DOI 10.1186/s13059-017-1311-2 METHOD Open Access SiFit: inferring tumor trees from single-cell sequencing data under finite-sites models Hamim Zafar 1,2, Anthony
Principles and Practice of Phylogenetic Systematics. Biol Rich Strauss
 Principles and Practice of Phylogenetic Systematics Biol 6304-001 Rich Strauss 1 Phylogenetics Phylogenetics: the attempt to infer how organisms are related historically in terms of their evolutionary
Principles and Practice of Phylogenetic Systematics Biol 6304-001 Rich Strauss 1 Phylogenetics Phylogenetics: the attempt to infer how organisms are related historically in terms of their evolutionary
Benchmark datasets for phylogenomic pipeline validation
 Benchmark datasets for phylogenomic pipeline validation GenomeTrakr Meeting Sept. 2018 Ruth E. Timme, PhD Research Microbiologist GenomeTrakr data coordinator Validation for phylogenomics Phylogenomic
Benchmark datasets for phylogenomic pipeline validation GenomeTrakr Meeting Sept. 2018 Ruth E. Timme, PhD Research Microbiologist GenomeTrakr data coordinator Validation for phylogenomics Phylogenomic
Towards an open-source, unified platform for disease outbreak analysis using
 Towards an open-source, unified platform for disease outbreak analysis using XXIV Simposio Internacional De Estadística Bogotá 24-26th July 2014 Thibaut Jombart, Caitlin Collins, Anne Cori, Neil Ferguson
Towards an open-source, unified platform for disease outbreak analysis using XXIV Simposio Internacional De Estadística Bogotá 24-26th July 2014 Thibaut Jombart, Caitlin Collins, Anne Cori, Neil Ferguson
Intraseasonal Dynamics and Dominant Sequences in H3N2 Influenza
 Intraseasonal Dynamics and Dominant Sequences in H3N2 Influenza Nicole Creanza 1., Jason S. Schwarz 1. *, Joel E. Cohen 1,2 1 Laboratory of Populations, Rockefeller University, New York, New York, United
Intraseasonal Dynamics and Dominant Sequences in H3N2 Influenza Nicole Creanza 1., Jason S. Schwarz 1. *, Joel E. Cohen 1,2 1 Laboratory of Populations, Rockefeller University, New York, New York, United
Using Bayesian Networks to Analyze Expression Data. Xu Siwei, s Muhammad Ali Faisal, s Tejal Joshi, s
 Using Bayesian Networks to Analyze Expression Data Xu Siwei, s0789023 Muhammad Ali Faisal, s0677834 Tejal Joshi, s0677858 Outline Introduction Bayesian Networks Equivalence Classes Applying to Expression
Using Bayesian Networks to Analyze Expression Data Xu Siwei, s0789023 Muhammad Ali Faisal, s0677834 Tejal Joshi, s0677858 Outline Introduction Bayesian Networks Equivalence Classes Applying to Expression
It is well known that some pathogenic microbes undergo
 Colloquium Effects of passage history and sampling bias on phylogenetic reconstruction of human influenza A evolution Robin M. Bush, Catherine B. Smith, Nancy J. Cox, and Walter M. Fitch Department of
Colloquium Effects of passage history and sampling bias on phylogenetic reconstruction of human influenza A evolution Robin M. Bush, Catherine B. Smith, Nancy J. Cox, and Walter M. Fitch Department of
FINAL ANNOTATION REPORT: Drosophila virilis Fosmid 11 (48P14) Robert Carrasquillo Bio 4342
 Robert Carrasquillo Bio 4342") FINAL ANNOTATION REPORT: Drosophila virilis Fosmid 11 (48P14) Robert Carrasquillo Bio 4342 2006 TABLE OF CONTENTS I. Overview... 3 II. Genes... 4 III. Clustal Analysis... 15 IV. Repeat Analysis... 17 V.
FINAL ANNOTATION REPORT: Drosophila virilis Fosmid 11 (48P14) Robert Carrasquillo Bio 4342 2006 TABLE OF CONTENTS I. Overview... 3 II. Genes... 4 III. Clustal Analysis... 15 IV. Repeat Analysis... 17 V.
COMPARATIVE ANALYSIS OF BIOINFORMATICS TOOLS USED IN HIV-1 STUDIES
 The 10 th Conference for Informatics and Information Technology (CIIT 2013) COMPARATIVE ANALYSIS OF BIOINFORMATICS TOOLS USED IN HIV-1 STUDIES Daniel Kareski Navayo technologies Skopje, Macedonia Nevena
The 10 th Conference for Informatics and Information Technology (CIIT 2013) COMPARATIVE ANALYSIS OF BIOINFORMATICS TOOLS USED IN HIV-1 STUDIES Daniel Kareski Navayo technologies Skopje, Macedonia Nevena
Application of phylogeny reconstruction and character-evolution analysis to inferring patterns of directional microbial transmission
 Preventive Veterinary Medicine 61 (2003) 59 70 Application of phylogeny reconstruction and character-evolution analysis to inferring patterns of directional microbial transmission Tony L. Goldberg Department
Preventive Veterinary Medicine 61 (2003) 59 70 Application of phylogeny reconstruction and character-evolution analysis to inferring patterns of directional microbial transmission Tony L. Goldberg Department
Supplementary Online Content
 Supplementary Online Content Melo AS, Aguiar RS, Amorim MMR, et al. Congenital Zika virus infection: beyond neonatal microcephaly. JAMA Neurol. Published online October 3, 2016. doi:10.1001/jamaneurol.2016.3720.
Supplementary Online Content Melo AS, Aguiar RS, Amorim MMR, et al. Congenital Zika virus infection: beyond neonatal microcephaly. JAMA Neurol. Published online October 3, 2016. doi:10.1001/jamaneurol.2016.3720.
PROTOCOL FOR INFLUENZA A VIRUS GLOBAL SWINE H1 CLADE CLASSIFICATION
 PROTOCOL FOR INFLUENZA A VIRUS GLOBAL SWINE H1 CLADE CLASSIFICATION January 23, 2017 1. Background Swine H1 viruses have diversified into three major genetic lineages over time. Recently, Anderson et al.
PROTOCOL FOR INFLUENZA A VIRUS GLOBAL SWINE H1 CLADE CLASSIFICATION January 23, 2017 1. Background Swine H1 viruses have diversified into three major genetic lineages over time. Recently, Anderson et al.
Dan Koller, Ph.D. Medical and Molecular Genetics
 Design of Genetic Studies Dan Koller, Ph.D. Research Assistant Professor Medical and Molecular Genetics Genetics and Medicine Over the past decade, advances from genetics have permeated medicine Identification
Design of Genetic Studies Dan Koller, Ph.D. Research Assistant Professor Medical and Molecular Genetics Genetics and Medicine Over the past decade, advances from genetics have permeated medicine Identification
Case-based reasoning using electronic health records efficiently identifies eligible patients for clinical trials
 Case-based reasoning using electronic health records efficiently identifies eligible patients for clinical trials Riccardo Miotto and Chunhua Weng Department of Biomedical Informatics Columbia University,
Case-based reasoning using electronic health records efficiently identifies eligible patients for clinical trials Riccardo Miotto and Chunhua Weng Department of Biomedical Informatics Columbia University,
Source and target enzyme signature in serine protease inhibitor active site sequences
 J Biosci., Vol 22, Number 5, December 1997, pp 555 565. Printed in India Source and target enzyme signature in serine protease inhibitor active site sequences BALAJI PRAKASH and Μ R Ν MURTHY* Molecular
J Biosci., Vol 22, Number 5, December 1997, pp 555 565. Printed in India Source and target enzyme signature in serine protease inhibitor active site sequences BALAJI PRAKASH and Μ R Ν MURTHY* Molecular
Review of The ancestral flower of angiosperms and its early diversification by H. Sauquet et al.
 Reviewers' comments: Reviewer #1 (Remarks to the Author): Review of The ancestral flower of angiosperms and its early diversification by H. Sauquet et al. This paper represents a significant landmark for
Reviewers' comments: Reviewer #1 (Remarks to the Author): Review of The ancestral flower of angiosperms and its early diversification by H. Sauquet et al. This paper represents a significant landmark for
Inter-country mixing in HIV transmission clusters: A pan-european phylodynamic study
 Inter-country mixing in HIV transmission clusters: A pan-european phylodynamic study Prabhav Kalaghatgi Max Planck Institute for Informatics March 20th 2013 HIV epidemic (2009) Prabhav Kalaghatgi 2/18
Inter-country mixing in HIV transmission clusters: A pan-european phylodynamic study Prabhav Kalaghatgi Max Planck Institute for Informatics March 20th 2013 HIV epidemic (2009) Prabhav Kalaghatgi 2/18
Gene Finding in Eukaryotes
 Gene Finding in Eukaryotes Jan-Jaap Wesselink jjwesselink@cnio.es Computational and Structural Biology Group, Centro Nacional de Investigaciones Oncológicas Madrid, April 2008 Jan-Jaap Wesselink jjwesselink@cnio.es
Gene Finding in Eukaryotes Jan-Jaap Wesselink jjwesselink@cnio.es Computational and Structural Biology Group, Centro Nacional de Investigaciones Oncológicas Madrid, April 2008 Jan-Jaap Wesselink jjwesselink@cnio.es
Mapping Evolutionary Pathways of HIV-1 Drug Resistance. Christopher Lee, UCLA Dept. of Chemistry & Biochemistry
 Mapping Evolutionary Pathways of HIV-1 Drug Resistance Christopher Lee, UCLA Dept. of Chemistry & Biochemistry Stalemate: We React to them, They React to Us E.g. a virus attacks us, so we develop a drug,
Mapping Evolutionary Pathways of HIV-1 Drug Resistance Christopher Lee, UCLA Dept. of Chemistry & Biochemistry Stalemate: We React to them, They React to Us E.g. a virus attacks us, so we develop a drug,
Understanding the Origins of a Pandemic Virus. Department of Biomedical Informatics, Columbia University College of Physicians and Surgeons,
 Understanding the Origins of a Pandemic Virus Carlos Xavier Hernández 1, Joseph Chan 1, Hossein Khiabanian 1, 2 1, 2*, Raul Rabadan 1 Center for Computational Biology and Bioinformatics, 2 Department of
Understanding the Origins of a Pandemic Virus Carlos Xavier Hernández 1, Joseph Chan 1, Hossein Khiabanian 1, 2 1, 2*, Raul Rabadan 1 Center for Computational Biology and Bioinformatics, 2 Department of
UvA-DARE (Digital Academic Repository)
") UvA-DARE (Digital Academic Repository) Superinfection with drug-resistant HIV is rare and does not contribute substantially to therapy failure in a large European cohort Bartha, I.; Assel, M.; Sloot, P.M.A.;
UvA-DARE (Digital Academic Repository) Superinfection with drug-resistant HIV is rare and does not contribute substantially to therapy failure in a large European cohort Bartha, I.; Assel, M.; Sloot, P.M.A.;
Table of content. -Supplementary methods. -Figure S1. -Figure S2. -Figure S3. -Table legend
 Table of content -Supplementary methods -Figure S1 -Figure S2 -Figure S3 -Table legend Supplementary methods Yeast two-hybrid bait basal transactivation test Because bait constructs sometimes self-transactivate
Table of content -Supplementary methods -Figure S1 -Figure S2 -Figure S3 -Table legend Supplementary methods Yeast two-hybrid bait basal transactivation test Because bait constructs sometimes self-transactivate
Journal: Nature Methods
 Journal: Nature Methods Article Title: Network-based stratification of tumor mutations Corresponding Author: Trey Ideker Supplementary Item Supplementary Figure 1 Supplementary Figure 2 Supplementary Figure
Journal: Nature Methods Article Title: Network-based stratification of tumor mutations Corresponding Author: Trey Ideker Supplementary Item Supplementary Figure 1 Supplementary Figure 2 Supplementary Figure
Drug Metabolism Disposition
 Drug Metabolism Disposition The CYP2C19 intron 2 branch point SNP is the ancestral polymorphism contributing to the poor metabolizer phenotype in livers with CYP2C19*35 and CYP2C19*2 alleles Amarjit S.
Drug Metabolism Disposition The CYP2C19 intron 2 branch point SNP is the ancestral polymorphism contributing to the poor metabolizer phenotype in livers with CYP2C19*35 and CYP2C19*2 alleles Amarjit S.
AutoOrthoGen. Multiple Genome Alignment and Comparison
 AutoOrthoGen Multiple Genome Alignment and Comparison Orion Buske, Yogesh Saletore, Kris Weber CSE 428 Spring 2009 Martin Tompa Computer Science and Engineering University of Washington, Seattle, WA Abstract
AutoOrthoGen Multiple Genome Alignment and Comparison Orion Buske, Yogesh Saletore, Kris Weber CSE 428 Spring 2009 Martin Tompa Computer Science and Engineering University of Washington, Seattle, WA Abstract
What can pathogen phylogenetics tell us about cross-species transmission?
 The Boyd Orr Centre for Population and Ecosystem Health What can pathogen phylogenetics tell us about cross-species transmission? Roman Biek! Bovine TB workshop 3 Sep 2015 Talk outline Genetic tracking
The Boyd Orr Centre for Population and Ecosystem Health What can pathogen phylogenetics tell us about cross-species transmission? Roman Biek! Bovine TB workshop 3 Sep 2015 Talk outline Genetic tracking
Chapter 1. Introduction
 Chapter 1 Introduction 1.1 Motivation and Goals The increasing availability and decreasing cost of high-throughput (HT) technologies coupled with the availability of computational tools and data form a
Chapter 1 Introduction 1.1 Motivation and Goals The increasing availability and decreasing cost of high-throughput (HT) technologies coupled with the availability of computational tools and data form a
WGS Works! Shared Mission Different Roles APPLICATIONS SEQUENCING (WGS) Non-regulatory. Regulatory CDC. FDA and USDA. Peter Gerner-Smidt, MD ScD
 Non-regulatory. Regulatory CDC. FDA and USDA. Peter Gerner-Smidt, MD ScD") PUBLIC HEALTH FOOD SAFETY APPLICATIONS FOR WHOLE GENOME SEQUENCING (WGS) Peter Gerner-Smidt, MD ScD Chief, Enteric Diseases Laboratory Branch 4 th Asia-Pacific International Food Safety Conference, Penang,
PUBLIC HEALTH FOOD SAFETY APPLICATIONS FOR WHOLE GENOME SEQUENCING (WGS) Peter Gerner-Smidt, MD ScD Chief, Enteric Diseases Laboratory Branch 4 th Asia-Pacific International Food Safety Conference, Penang,
Molecular Epidemiology of HBV Genotypes Circulating In Acute Hepatitis B Patients In The Campania Region
 Molecular Epidemiology of HBV Genotypes Circulating In Acute Hepatitis B Patients In The Campania Region Caterina Sagnelli 1, Massimo Ciccozzi 2,3, Mariantonietta Pisaturo 4, Gianguglielmo Zehender 5,
Molecular Epidemiology of HBV Genotypes Circulating In Acute Hepatitis B Patients In The Campania Region Caterina Sagnelli 1, Massimo Ciccozzi 2,3, Mariantonietta Pisaturo 4, Gianguglielmo Zehender 5,
Among all organisms, humans are : Archaea... Bacteria... Eukaryotes... Viruses... Among eukaryotes, humans are : Protists... Plants... Animals...
 Among all organisms, Archaea..... Bacteria....... Eukaryotes... Viruses... Campbell & Reece, page 679 Among eukaryotes, Protists..... Plants........ Animals..... Fungi. Campbell & Reece, page 4 Among animals,
Among all organisms, Archaea..... Bacteria....... Eukaryotes... Viruses... Campbell & Reece, page 679 Among eukaryotes, Protists..... Plants........ Animals..... Fungi. Campbell & Reece, page 4 Among animals,
MutationTaster & RegulationSpotter
 MutationTaster & RegulationSpotter Pathogenicity Prediction of Sequence Variants: Past, Present and Future Dr. rer. nat. Jana Marie Schwarz Klinik für Pädiatrie m. S. Neurologie Exzellenzcluster NeuroCure
MutationTaster & RegulationSpotter Pathogenicity Prediction of Sequence Variants: Past, Present and Future Dr. rer. nat. Jana Marie Schwarz Klinik für Pädiatrie m. S. Neurologie Exzellenzcluster NeuroCure
Genetics and Genomics in Medicine Chapter 8 Questions
 Genetics and Genomics in Medicine Chapter 8 Questions Linkage Analysis Question Question 8.1 Affected members of the pedigree above have an autosomal dominant disorder, and cytogenetic analyses using conventional
Genetics and Genomics in Medicine Chapter 8 Questions Linkage Analysis Question Question 8.1 Affected members of the pedigree above have an autosomal dominant disorder, and cytogenetic analyses using conventional
Annotation of Chimp Chunk 2-10 Jerome M Molleston 5/4/2009
 Annotation of Chimp Chunk 2-10 Jerome M Molleston 5/4/2009 1 Abstract A stretch of chimpanzee DNA was annotated using tools including BLAST, BLAT, and Genscan. Analysis of Genscan predicted genes revealed
Annotation of Chimp Chunk 2-10 Jerome M Molleston 5/4/2009 1 Abstract A stretch of chimpanzee DNA was annotated using tools including BLAST, BLAT, and Genscan. Analysis of Genscan predicted genes revealed
A Network Partition Algorithm for Mining Gene Functional Modules of Colon Cancer from DNA Microarray Data
 Method A Network Partition Algorithm for Mining Gene Functional Modules of Colon Cancer from DNA Microarray Data Xiao-Gang Ruan, Jin-Lian Wang*, and Jian-Geng Li Institute of Artificial Intelligence and
Method A Network Partition Algorithm for Mining Gene Functional Modules of Colon Cancer from DNA Microarray Data Xiao-Gang Ruan, Jin-Lian Wang*, and Jian-Geng Li Institute of Artificial Intelligence and
Host-Specific Modulation of the Selective Constraints Driving Human Immunodeficiency Virus Type 1 env Gene Evolution
 JOURNAL OF VIROLOGY, May 1999, p. 3764 3777 Vol. 73, No. 5 0022-538X/99/$04.00 0 Copyright 1999, American Society for Microbiology. All Rights Reserved. Host-Specific Modulation of the Selective Constraints
JOURNAL OF VIROLOGY, May 1999, p. 3764 3777 Vol. 73, No. 5 0022-538X/99/$04.00 0 Copyright 1999, American Society for Microbiology. All Rights Reserved. Host-Specific Modulation of the Selective Constraints
aM (modules 1 and 10 are required)
") This form should be used for all taxonomic proposals. Please complete all those modules that are applicable (and then delete the unwanted sections). For guidance, see the notes written in blue and the
This form should be used for all taxonomic proposals. Please complete all those modules that are applicable (and then delete the unwanted sections). For guidance, see the notes written in blue and the
Section B. Comparative Genomics Analysis of Influenza H5N2 Viruses. Objective
 Section B. Comparative Genomics Analysis of Influenza H5N2 Viruses Objective Upon completion of this exercise, you will be able to use the Influenza Research Database (IRD; http://www.fludb.org/) to: Search
Section B. Comparative Genomics Analysis of Influenza H5N2 Viruses Objective Upon completion of this exercise, you will be able to use the Influenza Research Database (IRD; http://www.fludb.org/) to: Search
Large-scale identity-by-descent mapping discovers rare haplotypes of large effect. Suyash Shringarpure 23andMe, Inc. ASHG 2017
 Large-scale identity-by-descent mapping discovers rare haplotypes of large effect Suyash Shringarpure 23andMe, Inc. ASHG 2017 1 Why care about rare variants of large effect? Months from randomization 2
Large-scale identity-by-descent mapping discovers rare haplotypes of large effect Suyash Shringarpure 23andMe, Inc. ASHG 2017 1 Why care about rare variants of large effect? Months from randomization 2
SEQUENCE FEATURE VARIANT TYPES
 SEQUENCE FEATURE VARIANT TYPES DEFINITION OF SFVT: The Sequence Feature Variant Type (SFVT) component in IRD (http://www.fludb.org) is a relatively novel approach that delineates specific regions, called
SEQUENCE FEATURE VARIANT TYPES DEFINITION OF SFVT: The Sequence Feature Variant Type (SFVT) component in IRD (http://www.fludb.org) is a relatively novel approach that delineates specific regions, called
Learning Convolutional Neural Networks for Graphs
 GA-65449 Learning Convolutional Neural Networks for Graphs Mathias Niepert Mohamed Ahmed Konstantin Kutzkov NEC Laboratories Europe Representation Learning for Graphs Telecom Safety Transportation Industry
GA-65449 Learning Convolutional Neural Networks for Graphs Mathias Niepert Mohamed Ahmed Konstantin Kutzkov NEC Laboratories Europe Representation Learning for Graphs Telecom Safety Transportation Industry
Protein Reports CPTAC Common Data Analysis Pipeline (CDAP)
") Protein Reports CPTAC Common Data Analysis Pipeline (CDAP) v. 05/03/2016 Summary The purpose of this document is to describe the protein reports generated as part of the CPTAC Common Data Analysis Pipeline
Protein Reports CPTAC Common Data Analysis Pipeline (CDAP) v. 05/03/2016 Summary The purpose of this document is to describe the protein reports generated as part of the CPTAC Common Data Analysis Pipeline