HORMONAL REGULATION OF HES-1 IN BREAST CANCER CELLS
|
|
|
- Ellen Hoover
- 5 years ago
- Views:
Transcription

1 From the Department of Biosciences and Nutrition Karolinska Institutet, Stockholm, Sweden HORMONAL REGULATION OF HES-1 IN BREAST CANCER CELLS Patrick Müller Stockholm 2008
2 All previously published papers were reproduced with permission from the publisher. Published by Karolinska Institutet. Printed by Larserics Digital Print AB Patrick Müller, 2008 ISBN
3 VERITATE, SCIENTIA, LABORE
4
5 ABSTRACT Hairy and Enhancer of Split homolog 1 (HES-1) is a transcriptional repressor belonging to the basic helix-loop-helix family of proteins, and has been shown to have a pivotal role as a regulator of neurogenesis, myogenesis and hematopoiesis. HES-1 has several well conserved domains, from Drosophila to mammals, and is expressed in a variety of tissues, in both embryos and adults. The most described function of HES-1 is as the primary effector protein in Notch signaling, as an inhibitor of differentiation, where expression of HES-1 maintains precursor cells undifferentiated and in a proliferative state. In breast cancer cells, exogenous expression of HES-1 abolishes the estrogen-induced proliferation, whereas endogenous HES-1 expression is repressed in response to estrogen. These background data suggest that breast cancer cells need to repress HES-1 expression in order to proliferate in response to estrogen. The general aim of this thesis was to determine the role of HES-1 in estrogen- and retinoic acid-regulated proliferation of human breast cancer cells. In study I, we show that HES-1 is a mediator of retinoic acid-induced inhibition of proliferation in the human breast cancer cell line MCF-7. Increasing levels of retinoic acid caused increasing expression of HES-1, followed by decreasing proliferation. By stably expressing a dominant negative HES-1 in MCF-7 cells the endogenous activity of HES-1 was inhibited and the cells become unresponsive to retinoic acid. In study II, we show that exogenous expression of HES-1 causes cell cycle arrest in the G 1 phase of the cell cycle. We found the cell cycle factor E2F-1, critical for G 1 to S-phase progression, to be regulated by HES-1, via a cis-element in the 5 regulatory region of the E2F-1 gene. HES-1 was shown to mediate the negative regulation on E2F-1 expression induced by retinoic acid, as indicated by the lack of the negative effect when expressing the dominant negative HES-1. We also found that HES-1 antagonizes the effect of another mitogen, heregulin-β1. The heregulin-β1 induced E2F-1 expression and the subsequent proliferation were inhibited by overexpression of HES-1. In study III, we investigated the mechanism by which HES-1 is regulated in response to estrogen and retinoic acid. There is a retinoic acid response element 18 kb upstream of the HES-1 gene that was bound by retinoic acid receptor α and activated transcription in response to retinoic acid. Results from a whole-genome estrogen receptor α (ERα) binding site mapping experiment, show ERα binding 23 kb downstream of HES-1 gene. ERα binds and regulates transcription via a novel cis-element, containing one consensus estrogen response element and a half site. ChIP assays revealed that ERα, coactivators and corepressors were recruited to this cis-element in response to estrogen. Importantly, a decrease in acetylation of histone H3 and H4 and a decrease in recruitment of RNA POL II were observed, indicating a repression of HES-1 expression in response to estrogen. In study IV, we show that retinoic acid mediates the induction of HES-1 gene expression via the transcription factor SOX9. We found SOX9 bound to a cis-element 3.7 kb upstream of HES-1 gene in response to retinoic acid. This cis-element regulated transcription induced by retinoic acid. In conclusion, these results establish HES-1 as a mediator of the cell cycle arrest induced by retinoic acid, and suggest HES-1 to be an important regulator of proliferation of breast cancer cells.
6 LIST OF PUBLICATIONS I. Patrick Müller, Silke Kietz, Jan-Åke Gustafsson and Anders Ström. The antiestrogenic effect of all-trans-retinoic acid on the breast cancer cell line MCF-7 is dependent on HES-1 expression. J Biol Chem Aug 9;277(32): II. III. IV. Johan Hartman, Patrick Müller, James S Foster, Jay Wimalasena, Jan-Åke Gustafsson and Anders Ström. HES-1 inhibits 17β-estradiol and heregulin-β1- mediated upregulation of E2F-1. Oncogene Nov 18;23(54): Patrick Müller, Kenneth W Merrell, Caroline Rönnlund, Chin-Yo Lin, Jan- Åke Gustafsson and Anders Ström. Estrogen-dependent downregulation of HES-1 gene expression in breast cancer cells is mediated via a 3 distal element. Submitted manuscript Patrick Müller, Justin D. Crofts, Ben S. Newman, Laura C. Bridgewater, Chin-Yo Lin, Jan-Åke Gustafsson and Anders Ström. SOX9 mediates the retinoic acid-induced HES-1 gene expression in human breast cancer cells. Submitted manuscript
7 CONTENTS CANCER...1 Epidemiology...1 Etiology...2 Breast Cancer...3 ESTROGENS...4 Biosynthesis of Estrogen...4 Estrogen Function...6 A Flashback...6 Estrogen Receptor...6 ER Structure...7 ER Action...8 ER Coregulators...9 The Cell Cycle...10 Estrogen and the Cell Cycle...11 Anti-estrogens...11 RETINOIC ACID...13 Metabolism of RA...13 RA Function...15 RA Receptor...16 RAR Coregulators...17 RAR and Proliferation...18 Synthetic Retinoids...19 SOX SOX9 Action...21 SOX9 in Campomelic Dysplasia and Sex Reversal...23 SOX9 and the Cell Cycle...23 HES Transcriptional Repression by HES Regulation of HES Notch signaling...28 HES-1 Action...30 Aims of the Study...31 Results and Discussion...32 Paper I...32 Paper II...34 Paper III...35 Paper IV...37 Concluding Remarks...38 Acknowledgements...Fel! Bokmärket är inte definierat. References...41 Endnotes...51
8 LIST OF ABBREVIATIONS 9cRA 9-cis retinoic acid 13cRA 13-cis retinoic acid AF Activation function AMH Anti-müllerian hormone AP-1 Activator protein 1 atra All-trans retinoic acid bhlh Basic helix-loop-helix CBP Creb-binding protein CD Campomelic dysplasia CDK Cyclin-dependent kinase ChIP Chromatin immunoprecipitation CRABP Cellular retinoic acid-binding protein DBD DNA binding domain EGF Epidermal growth factor ER Estrogen receptor ERE Estrogen response element HAT Histone acetylase HDAC Histone deacetylase HERP Hairy and Enhancer of Split related protein HES Hairy and Enhancer of Split homolog HMG High mobility group LBD Ligand binding domain MAPK Mitogen-activated protein kinase MASH Mammalian achaete-scute homolog NcoR Nuclear receptor corepressor NICD Notch intracellular domain NPAT Nuclear protein, ataxia-telangiectasia locus NR Nuclear receptor P/CAF P300/CBP-associated factor prb Retinoblastoma protein RA Retinoic acid RAR Retinoic acid receptor RARE Retinoic acid response element RBP-jκ Recombination signal-binding protein 1 for jκ RIP140 Receptor-interacting protein 140 RXR Retinoid X receptor SF-1 Steroidogenic factor 1 SMRT Silencing mediator for retinoid and thyroid hormone receptors SRC Steroid receptor coactivator SRY Sex determining region Y STRA6 Stimulated by retinoic acid gene 6 homolog
9 CANCER Cancer is a diverse class of diseases that differ widely in their causes and biology. Principally, cancer is unrestricted growth of abnormal cells. The Latin word cancer or karkinos in Greek literally means crab, this applying to the swollen veins that surround cancerous tumors that resemble the legs of a crab sticking out from its shell. This observation was made by the ancient Greek physician Hippocrates, father of medicine ( B.C.). There are around 200 anatomically different cancers, but when cancers are subdivided according to the molecules underlying the disease, there are approximately different cancers. Since most cases of cancer occur in people with no family history of the disease, it is not considered an inherited disease. The risk for developing cancer, however, can be influenced by the inheritance of certain genetic changes. EPIDEMIOLOGY The World Health Organization estimates that cancer kills more than 7 million people every year. Although the incidence of cancer has been declining by approximately 2% worldwide each year since 1992, it is estimated that 10 million new cases of cancer occur annually (1). The cancer incidence is higher for all cancers combined in the western world (North America, Europe and Australia) than in Latin America and Asia. Earlier cigarette smoking patterns, differences in diet, lack of physical activity, prevalence of obesity but also increased diagnosis, are possible explanations for this. Breast and prostate cancer in women and men, respectively, are the most common causes of cancer mortality in the western world, whereas in China, cancer of the liver and stomach are the major forms (Table 1). In 2003, one in four deaths in the USA, 22.7% or persons, was due to cancer, making it the second leading cause of death (2). In 2006 in Sweden, cancers were reported, with were diagnosed for the first time persons died in cancer in 2005, and today, people living in Sweden have been diagnosed with cancer (3). Furthermore, there is an overall increase in the incidence of cancer of 1.7% in Swedish men and 1.1% increase in women, which is partly explained by an ageing population but also by the introduction of screening activities and improved diagnostic practices. Site Sweden USA China All sites All sites Breast Prostate Stomach Liver Table 1. Cancer death rates per population in 2002 (2). The death rates are ageadjusted to the World Health Organization world standard population. 1
10 ETIOLOGY There are many causes of cancer, and these can be divided into six classes (Table 2, a-f). Inherited mutated tumor suppressor genes, including p53, BRCA and prb increase the risk of developing cancer substantially (a). The implication of hormones as causative agents of cancer was established more than 75 years ago (b) (4). Excessive exposure to the sun increases the risk for skin cancers and radon induces lung and skin cancer (c) (5-7). Infections from certain viruses can also cause cancer (d) (8). For example, the DNA viruses, Hepatitis B and human papilloma can cause liver and cervical cancer, respectively, the human herpes virus-8 causes Kaposi s sarcoma, and the Epstein-Barr virus can cause various types of human cancer. Also, hepatitis C and human T lymphotropic virus type 1 are RNA viruses with the capability to cause liver cancer and adult T-cell leukemia, respectively (8). Finally, the ulcer-associated bacterium Helicobacter Pylori is linked to gastric cancer (9). HIV infection indirectly increases the risk for developing cancer, as HIV impairs the immune system and thus makes the infected individual susceptible to infection with oncogenic viruses. For example, co-infection of HIV-1 with human herpes virus-8 increases the incidence of Kaposi s sarcoma to times (e) (10). Certain subtypes of human papilloma virus are associated with cervical cancer, especially in individuals with an inadequate T-cell response, as in AIDS patients and renal transplants patients receiving immunosuppressive therapy (8). Despite the very strong association of lung cancer and tobacco smoking, shown as early as 1950s, 1.3 billion people worldwide are daily smokers, with an annual death rate of 1.2 million (11). Also, asbestos is well established as a carcinogen causing lung cancer (f) (12). Cause (a) Heredity (b) Hormonal imbalance (c) Ionizing radiation (d) Infectious disease (e) Dysfunctional immune system (f) Chemical carcinogens Examples Mutated BRCA, prb, p53 Hyperestrogenic status UV light, radon gas Oncogenic DNA and RNA viruses, bacteria HIV, human papilloma virus Tobacco smoking, asbestos fibers Table 2. The causes of cancer can be divided into six classes. Differences in the incidence of cancer in different countries can often be explained by particular dietary habits. For instance, the rates of mortality from gastric cancer are higher in Japan compared to many western countries whereas the mortality rate from breast and pancreas cancer is only a fraction in Japan versus many western countries. Furthermore, Japanese immigrants in the USA, experience lower rates of mortality from stomach cancer than the Japanese people, however, they experience an increase in the rates of mortality from cancers in breast, prostate, colon, pancreas and ovary (13). This change in risk of developing cancer when moving from one location to another suggests that certain cancers are not determined primarily by heredity, but may involve cultural, behavioral or environmental factors. 2
11 BREAST CANCER Breast cancer is the most common malignancy in women with 1 million new cases per year worldwide (14). It is more common in Western countries than in Asia and Latin America. Possible explanations include differences in diet, body size, physical activity and reproductive patterns. Breast cancer is also the most common cancer among women in Sweden 2006, with an incidence of 29.4% of all cancers, an annual increase of 1.7% the last 2 decades (Table 3) (3). A comparison of table 1 with 3A reveals that approximately 13% of breast cancers resulted in mortality. However, the death rates from breast cancer has fallen in western countries; in the USA the death rate decreased between 1990 and 1996 with an average of 1.8% per year, with a pronounced decrease among white young women (15). The overall decline in the European union between 1988 and 1998 was 16.0% although Eastern Europe (former non-market economy European countries) shows more of an inconsistent pattern (16). Elevated hormone levels have been associated with an increased risk for breast cancer, including hormonal replacement therapy, alcohol consumption, post-menopausal obesity, early menarche and late menopause. However, several years after terminating hormonal replacement therapy, the risk continues to be elevated (17). Breast cancer history in the family is another established risk factor (14,18). Key genes involved in the familial forms of breast cancer include tumor suppressors p53, BRCA1 and BRCA2. Disruptive expression of BRCA1 and BRCA2 is estimated to account for approximately 5% of all breast cancer (19). Protective factors, causing decreased hormone levels, are prolonged lactation, physical exercise and young age at first pregnancy (20). The risk of developing breast cancer increases with age, where incidence rates double every 10 years until menopause. A B Site All sites All sites Breast New cases % of all cancers Total Table 3. Breast cancer incidence in Sweden per 100,000 (A), and in total (B) (3). The rates are adjusted to the population in January
12 ESTROGENS Estrogens belong to the family of hormones termed steroids. The major naturally occurring estrogens in humans are 17β-estradiol (E 2 ), and the two metabolites, estrone (E 1 ) and estriol (E 3 ). Estrogens are, like all steroids, molecules of hydrocarbon rings with a cholesterol backbone (Figure 1). Figure 1. Chemical structure of A) 17β-estradiol, B) estrone and C) estriol The primary sites of production are the ovaries, and in smaller amount the adrenal glands, adipose tissue, the brain and the testis (21). The ovaries in women with active menstrual cycles produce daily between 70 and 500 µg of 17β-estradiol (22). Estrogen is considered to be a female sex hormone, however, is also produced in males, where it has been found to be important for male reproduction due to its involvement in sperm maturation, with disrupted estrogen signaling in male mice resulting in infertility (23,24). In premenopausal women, estrogen synthesized in the ovaries is an endocrine factor, a circulating hormone, whereas estrogen synthesized at extragonadal sites, as in men and postmenopausal women acts locally as a paracrine or even as an intracrine factor (21,25). 17β-estradiol circulates in the blood bound to α-2-globulin, also known as sex hormone-binding globulin (SHBG), or to serum albumin. Estrogen-dependent breast carcinoma is self-supporting with estrogens, since it has been shown to have an intratumoral production of sex steroids (26). BIOSYNTHESIS OF ESTROGEN The production of estrogens and other steroids is through a multi-step enzyme-catalyzed process named steroidogenesis (Figure 2). Although the products of steroidogenesis are not only estrogens but also testosterone, cortisol, aldosterone and progesterone, the starting material is the same, the cholesterol molecule. Several oxidative enzymes located in both the mitochondria and endoplasmic reticulum are required for the biosynthesis of steroids. The rate-limiting step in this process is the transport of free cholesterol from the cytoplasm into the mitochondria. Cholesterol is then converted to pregnenolone by CYP11A1, an enzyme located in the inner membrane of the mitochondria. Pregnenolone is the immediate precursor for the synthesis of all steroid hormones. The last step in the synthesis of estrogens is catalyzed by the enzyme CYP19, also called aromatase, in which the androgen precursors, androstenedione or testosterone through aromatization of the A-ring, are converted to 17βestradiol or estrone. These two estrogens can then later be converted to estriol (27). Inside malignant breast tissues, the process of aromatization has been shown to be increased, leading to increased 17β-estradiol levels in the breast cancer cell and its environment (28). 4




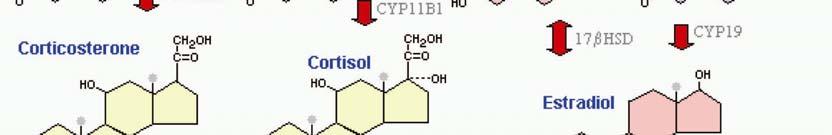
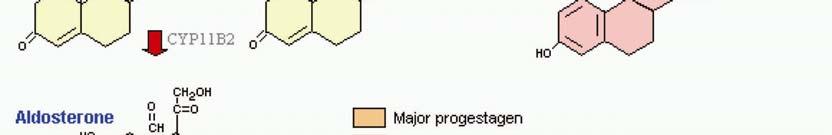

13 Figure 2. The biosynthesis of steroids starts with cholesterol and through several enzymecatalyzed steps results in progestagens, mineralcorticoids, glucocorticoids, androgens and estrogens. i 5
14 ESTROGEN FUNCTION Estrogens are vital molecules for the development of secondary female sex characteristics, reproductive functions, and the mammary gland. In both sexes, estrogens affect the brain, the liver, bone, cardiovascular system and several other organs (29). Cell proliferation and differentiation are pivotal mechanisms that are under the control of estrogen in both normal and malignant tissues, with estrogen implicated in certain pathological conditions, including breast and endometrial cancer. In fact, estrogen is now considered to be a classical etiological factor for both breast and endometrial cancer, with the US National Toxicology Program listing steroidal estrogens as carcinogens in 2002 (30). Estrogen is also used therapeutically not only as hormone replacement therapy and in contraception, but also to dampen menopausal symptoms, delay post-menopausal osteoporosis and other estrogen-deprived symptoms (31). A FLASHBACK The first link between ovarian hormonal production and breast cancer was made in 1896 by physician George Thomas Beatson. He made the revolutionary finding that all cancer tissue had vanished eight months after a bilateral ovariectomy on a premenopausal patient with a relapsed inoperable breast cancer (32). Then, William Stewart Halsted presented in 1907 that breast cancer disease is curable after removal through radical operation (33). Studies made in the 1920s showed that a chemical secreted from the corpus luteum, known as ovarian hormone or later estrone, was involved in tumor formation (34,35). In 1927, William S Murray showed that subcutaneous transplantation studies of ovarian tissue resulted in the development of spontaneous breast cancer (36). Lilienfeld and Johnson showed in 1955 that the greatest risk for developing female breast cancer is aging (37). Later, it was shown that longer exposure to estrogen was the increased risk (38). ESTROGEN RECEPTOR In 1958, Elwood Jensen found that estradiol was incorporated into a cytosolic component, then transported into the nucleus, where it exerted an effect. By using a radioactive marker, Jensen showed that only the estrogen-responsive tissues were able to concentrate injected estrogen from the blood, suggesting the existence of a binding component. This component was estrogen receptor (ER) (34,39). Further on, the cytosolic estrogen-binding component was isolated from rat uterus and subsequently characterized. By treating the estrogen receptor with proteolytic enzymes, binding of estrogen was abolished, indicating that estrogen receptor was a protein (40). By the end of 20 th century, it was well established that estrogen exerts its effects by binding to two types of related but distinct nuclear ligand-dependent transcription factors; estrogen receptor α (ERα) and estrogen receptor β (ERβ). Cloning of ERα was performed in 1985 from the breast cancer cell line MCF-7, and cloning of ERβ was succeeded ten years later first from rat prostate and later from human leukocytes (41-43). The two receptor types are expressed in a tissue- and cell specific manner and are encoded by different genes, the human ERα on chromosome 6 and the human ERβ on chromosome 14 (44,45) ( Figure 3). 6
 family: the N-terminal A/B domain with the")
 (D domain), the LBD with the hormone-dependent AF2 (E domain) and the C-terminal F domain (Figure 4) (46).")

15 Figure 3. Distribution of ERα and ERβ in humans. ii ER STRUCTURE ERs have evolutionarily conserved structurally and functionally distinct domains like other members of nuclear receptor (NR) family: the N-terminal A/B domain with the hormoneindependent activation function 1 (AF1) that weakly activates transcription but synergizes with activation function 2 (AF2), the DNA-binding domain (DBD) (C domain), the hinge region that connects the DBD with the ligand-binding domain (LBD) (D domain), the LBD with the hormone-dependent AF2 (E domain) and the C-terminal F domain (Figure 4) (46). ERα and ERβ are 97% homologous in the DBD and 59% homologous in the LBD. The high homology between ERα and ERβ in the DBD suggests that they share similar DNA target sites, and the high homology in the LBD that they share common ligands. However, there is a lot of data supporting that ERβ counteracts the transcriptional activity exerted by ERα (45,47,48). AF1 of ERβ has shown to be weaker than AF1 of ERα (49). The E/F region contains the dimerization domain and a binding surface for coregulators (50). ERα and ERβ bind 17β-estradiol with approximately the same affinity, whereas the two metabolites, estrone and estriol, bind the receptors with high affinity but are weaker agonists than 17β-estradiol on ER (51). Figure 4. Structure and homology between ERα and ERβ. The domains A-F is made up of activation function domains (AF1 and AF2), DNA-binding domain (DBD) and ligand-binding domain. iii 7
16 ER ACTION Upon binding of a ligand to the cytosolic estrogen receptor, a multiprotein inhibitory complex containing heat-shock proteins dissociates from the receptor following a conformational change of the receptor (52,53). The part of the receptor that is changed upon ligand-binding is the highly conserved α-helix 12. It has been shown that different ligands induce different positioning of α-helix 12 (54). For instance, binding of an agonist to the ligand-binding pocket induces a shift in the α-helix 12 position feasible for interaction and recruitment of coactivators. On the contrary, binding to an antagonist inhibits this position of α-helix 12 and results in a inactive receptor (54). The conformational change of the receptor promotes dimerization and the subsequent modulation of transcriptional regulation. There are at least four distinct modes of action for the ERs. The classical pathway is known as the direct pathway, in which ligand-bound ERs bind to an estrogen response element (ERE) (Figure 5A). ERs have been shown to be tethered to DNA-bound transcription factors, either through AF1 or AF2 (Figure 5B). There also exist rapid non-genomic actions of ERs, where a signal cascade is activated following the activation of second messengers (Figure 5C). In addition to ligand-dependent activation of ERs, ligand-independent activation has also been shown (Figure 5D). In this case, growth factors may induce phosphorylation of ERs, thereby activating them (55). The overall response to estrogen in all four pathways is a modulation of transcriptional regulation by the activated ERs (52,56). Figure 5. Four different pathways of ER action: the A) direct, B) tethered, C) non-genomic pathway and D) via growth factor signaling. ER, estrogen receptor; GF, growth factor; P, phosphate; SM, second messenger; TF, transcription factor. iv 8
17 ERE The canonical ERE is made up of a 5-base pair (bp) palindrome with a 3-bp spacer: GGTCAnnnTGACC, where n can be any nucleotide (57). ER can bind as a monomer to an ERE half-site (TGACC) in the presence of specificity protein 1 (SP1), as in the case of progesterone receptor promoter (58,59). The ERE is a cis-regulatory element, generally believed to be located within the first 1000 bp upstream of the estrogen-responsive gene. However, recent publications show that only a fraction (<5%) of in vivo binding of ERα occurs within the promoter-proximal regions (60,61). In fact, 40% of the EREs are located within introns, and 20% downstream of genes. Only 20% are located within 100,000 bp upstream of the gene (61). ER tethering to SP1 in response to estradiol, and the subsequent induction of gene expression, has been shown on retinoic acid receptor α (RARα), insulinlike growth factor binding protein-1 (IGFBP-1) and B-cell lymphoma protein-2 (BCL-2) (62-65). Estradiol-bound ER enhances activator protein 1 (AP-1) trans-activity through interaction with the p160 coactivator steroid receptor coactivator 1 (SRC-1) (66-68). The very rapid non-genomic effects observed are suggested to involve mitogen-activated protein kinase (MAPK) activation, interaction with phosphatidylinositol-3-oh kinase, activation of plasma membrane-associated ERs or ERs in the mitochondria (69-72). The expression of most ER target genes, described in the literature, are upregulated in response to estradiol. However, the majority of the genes are downregulated in response to estradiol, as found in gene expression analyses (60,73). ER COREGULATORS The regulation of transcription is an intricate process, involving recruitment of many proteins to the regulatory regions of the target gene. Transcriptional regulation by ER involves recruitment of coactivators, corepressors, histone acetyltransferases (HATs) and histone deacetyltransferases (HDACs) among others (74-77). Additionally, basal transcription factors have shown to interact with ER (78). A simplified definition of NR coregulators is that they interact directly with the NRs and modulate their transcription; coactivators enhance transcription whereas corepressors decrease transcription (79). The coactivators physically interact with ER through the LXXLL (where L is leucine and X is any amino acid) motif present in the coactivator protein (80). The coactivators P300/CBP-associated factor (P/CAF), SRC-1, SRC-2, SRC-3 and creb-binding protein (CBP/p300) have intrinsic HAT activity, whereas the corepressors recruit proteins with HDAC activity (50). More than 19 distinct coactivators have been shown to interact with ERα (74). At the estrogen-responsive genes, the ERE-bound ER interacts with coactivators from the p160 family of co-activators, SRC-1, SRC-2 and SRC-3, followed by enhanced transcription (80-82). CBP and the closely related p300 have been shown to be ubiquitous integrative components in all transcription complexes, and interact with ERα in a ligand-dependent fashion (75,79). The corepressors N- CoR and SMRT have been shown to interact with antagonist-bound ERα and with agonistbound ERβ (83,84). The coregulator receptor-interacting protein 140 (RIP140) interacts with agonist-bound but not antagonist-bound ERα in vitro and in vivo (85,86). However, RIP140 competes with the coactivators SRC-1 for binding to NRs with a decreased NR activity as a result, suggesting a repressive role for RIP140 in receptor activation (87). RIP140 has also been shown to recruit the corepressors C-terminus-binding protein (CtBP), HDAC1 and HDAC3 supporting the theory of RIP140 being a corepressor (88,89). 9


.")

18 THE CELL CYCLE The cell cycle is divided into four phases: G 1, S, G 2 and M phase, where one cycle in one cell leads to one daughter cell. In the G 1 phase, the cell prepares for the S-phase by inducing expression of enzymes and other proteins required for the DNA replication that takes place in the S-phase. Progression through the G 1 phase of the cell cycle is controlled by orderly activation of cyclin-dependent kinase (CDK) 4/6 and CDK2. Binding of cyclins to CDKs results in activation of CDKs, forming active kinase complexes, whereas the activity of CDK-cyclin complexes is inhibited by CDK inhibitors. The CDK inhibitor p16 INK4A inhibits binding of D-type cyclins to CDK4/6 and the subsequent activation (90,91) (Figure 6). CDK2 activation by cyclin E is inhibited by p21 CIP and p27 KIP1, whereas p21 CIP and p27 KIP1 promotes binding of cyclin D to CDK4/6 at low levels but inhibits the same binding at high levels (92). The G 1 /S transition promoted by CDK2-cyclin E is upheld by the CDK4/6-cyclin D complex through sequestering away p21 CIP and p27 KIP1 from CDK2-cyclin E complexes (93). E2F transcription factors are essential for activating genes required for entering the S phase, including cyclin A and cyclin E. The retinoblastoma protein (prb) inhibits this activity of E2F factors, either by sequestration or transcriptional repression (94). When prb becomes phosphorylated, by CDK-cyclin complexes, it releases E2F factors followed by G 1 /S transition. In response to DNA damage or cell stress, p16 INK4A is upregulated by tumor suppressor p53 and the result is cell cycle arrest or apoptosis. Figure 6. The progression through G 1 phase of the cell cycle includes activations of cyclindependent kinases (CDKs) in complexes with cyclins, such as cyclin D-CDK4, cyclin E- CDK2, and inhibition of p16 INK4A, p21 CIP and p27 KIP1 CDK inhibitors. Phosphorylation of the retinoblastoma protein (prb) by the active CDKs results in active E2F protein and the subsequent S-phase entry. v 10
19 ESTROGEN AND THE CELL CYCLE Estrogen is well established as a potent mitogen of human breast cancer cells, with progression through G 1 of the cell cycle accelerated in response to estrogen. One early protein to be activated by estrogen is the cell cycle regulator c-myc. c-myc, in turn, regulates the essential proteins for G 1 progression; cyclin D and CKD4 (95,96). The cell cycle regulators that are pivotal for S-phase entry, cyclin E and E2F proteins, are also regulated by c-myc (97-100). c- Myc represses the expression of the CDK inhibitors p21 CIP and p27 KIP1 (101,102). Treatment with estradiol of growth arrested MCF-7 breast cancer cells induces c-myc protein at 2h followed by cyclin D1 protein and CDK4 activity at 3-4h and CDK2 activity at 4h. After 6 h estradiol treatment, phosphorylated prb is present. The expression of CDK inhibitors p21 CIP and p27 KIP1 are altered at 8h and 12h, respectively (103,104). Ectopic expression of c-myc or cyclin D1 is sufficient for S-phase entry suggesting distinct pathways for c-myc and cyclin D1 in response to estradiol (105). Cyclin D1 is suggested as a target of estradiol in MCF-7 cells, in which mrna is upregulated between 1-3h and protein 3-6h in response to estradiol (103,104,106). Cyclin E can functionally replace cyclin D1 in cyclin D1-knockout mice, and rescue phenotypic signs of deficient cyclin D1 expression (107). The activity of AP-1 is associated with breast cancer cell proliferation (108). The expression of the AP-1 components, the proto-oncogenes c-jun and c-fos, has been shown to be elevated in response to estradiol, whereas the activity of AP-1 is enhanced without the expression of its components being affected (66,67,109). Adding the pure estrogen antagonist fulvestrant (ICI 182,780) to MCF-7 cells elicits growth arrest in a state characteristic of G 0, through decreasing cyclin D1-CDK4 and cyclin E-CDK2 activity, which is dependent on p21 expression (110). ANTI-ESTROGENS It took almost a century of research to develop a drug directed against an endocrine target, from the work done by Beatson in 1896, to the findings of ICI 46474, known as tamoxifen. However, tamoxifen has been shown to have an agonistic effect on endometrium and thus may increase the risk for endometrial cancer (30). After surgery, 5 years tamoxifen treatment with a systemic adjuvant therapy, is responsible for prolonging the survival and preventing the relapse of many women with ER positive breast tumors (111). Due to the estrogenic properties of tamoxifen in the endometrium, new molecules were developed in the 80s; aromatase inhibitors for postmenopausal women and luteinizing hormone-releasing hormone agonists for premenopausal women became available for the treatment of endometrial cancers and advanced breast cancer. The first generation of aromatase inhibitors were toxic, due lack of specificity, but today there are several potent and specific inhibitors. There are several anti-estrogens presently available with high efficacy named selective estrogen receptor modulators (SERMs) (Figure 7). These SERMs act as antagonists in the breast but as agonists in the bone and cardiovascular system without affecting the endometrial proliferation. For example, fulvestrant is a pure estrogen-antagonist that downregulates both estrogen receptors and progesterone receptors, is active in tamoxifenresistant breast cancer cell lines and shows efficacy in clinical studies of premenopausal women (112,113). Also, the third generation of aromatase inhibitors, such as anastrozole and letrozole, decreases the amount of estrogen to an undetectable level (114). There are less serious side effects associated with anastrozole and letrozole than tamoxifen, and these agents are now considered to be more effective in treating metastatic breast cancer in postmenopausal women than tamoxifen. Raloxifene is an anti-estrogen in breast and 11
20 endometrial tissue but shows estrogenic effects on bone and lipid metabolism and has the same efficacy as tamoxifen in reducing the risk of breast cancer (114). Figure 7. The structure of four synthetic anti-estrogens: A) fulvestrant (ICI 182,780), B) raloxifene, C) tamoxifen and D) anastrozole The use of tamoxifen has concluded that inhibition of estrogen signaling can be used as a means to prevent and cure breast cancer. Through screening analyses, a myriad of compounds has shown anti-estrogenic and anti-tumorigenic properties with various efficacy. Among these, a derivative of vitamin A, retinoic acid, has been promising in the prevention of breast cancer. 12
21 RETINOIC ACID Retinoic acid (RA) is a member of a family of both synthetic and naturally occurring molecules, the retinoids. Retinoids are non-steroid hormones, derivatives of vitamin A with intracrine activity. Humans are unable to produce vitamin A, and therefore require a daily intake of animal products, fruits and vegetables to fulfill the need. Yellow, red, orange and purple colored fruits and vegetables contain carotenoids that can be converted to vitamin A in humans (115,116). Vitamin A deficiency is a public health problem in more than half of all countries, and it is estimated that 250 million preschool children are vitamin A deficient (WHO 2008). Clinical vitamin A deficiency is characterized by decreased resistance to infections and xerophthalmia, a disease with several ocular features that leads to the worst case of blindness (117,118). On the other hand, an overexposure to vitamin A, hypervitaminosis A, is associated with embryonic malformations and osteoporosis (119,120). Through regulation of cell proliferation, differentiation and apoptosis, retinoids are pivotal in the development and homeostasis of all tissues within the body ( ). Naturally occurring retinoids are made up of four isoprenoid units, joined in a head to tail manner, making retinoids hydrophobic (124). Several naturally occurring retinoids have been shown to be active in the human body; all-trans retinoic acid (atra), and two stereoisomers, 9-cis retinoic acid (9cRA) and 13-cis retinoic acid (13cRA) (Figure 8). Figure 8. Chemical structure of three naturally occurring vitamin A-derivatives: A) all-trans retinoic acid, B) 13-cis retinoic acid, C) 9-cis retinoic acid METABOLISM OF RA Approximately half of the intake of vitamin A comes from animal products in the form of retinol and retinyl esters, the other half from plant carotenoids (125,126). After ingestion, hydrolysis of retinyl esters and metabolism of β-carotene are converted to all-trans retinol prior to uptake in the enterocytes in the intestine. Here it is protected from oxidation to RA by cellular retinol binding protein (CRBP-II). All-trans retinol is re-esterified for storage as retinyl esters mainly in the liver, but also in the lungs, kidney and bone marrow (126,127). When there is a demand for biologically active retinoids, retinol is released into the circulatory system bound to plasma retinol-binding protein (RBP), which is taken up by many tissues for conversion to the biologically active forms (Figure 9). 13
: The RA precursor retinol is formed from dietary intake of vitamin A, and transported into the cell where retinol is metabolized to RA through retinal.")
22 Figure 9. Metabolism and action of retinoic acid (RA): The RA precursor retinol is formed from dietary intake of vitamin A, and transported into the cell where retinol is metabolized to RA through retinal. RA is then transported into the nucleus where it binds to RA receptors, resulting in transcriptional activation. In the absence of RA, RA receptors repress transcription. vi Retinol has no biological role itself, but instead serves as a precursor for the synthesis of the active retinoids. The naturally occurring active retinoids are either 11-cis-retinal, present in the retina of the eye, or the gene expression agents, the RAs (128). The retinol-rbp complex binds to the cell membrane-receptor STRA6, which mediates uptake of retinol into the cell (129). The conversion of retinol to retinaldehyde inside the cell is accomplished by retinol dehydrogenase (RODH) or alcohol dehydrogenase (ADH), and retinaldehyde to RA by retinaldehyde dehydrogenase (RALDH). The reactions are in the presence of cellular retinol binding proteins, but the molecular mechanisms are not fully elucidated (130). RALDH-2 is suggested to be an essential enzyme in the irreversible oxidation of retinalaldehyde to RA (131). Cytoplasmic RA is associated with cellular RA-binding protein (CRABP). To avoid any excess of local concentration of RA, an autoregulatory feedback is formed by the cytochrome P450 enzyme CYP26 (P450RAI) that metabolizes specifically atra, but not 9cRA or 13cRA (132). 14

23 RA FUNCTION Vision, reproduction, embryogenesis, bone remodeling, epithelial differentiation and the immune system are biological systems that are reliant on retinoids (Figure 10). There are indications that RA does not mediate every effect of vitamin A, as in the case of the visual mechanism of vitamin A. Due to its capacity to promote differentiation and regulate apoptosis, RA is a pivotal factor in the development of tissues and organs (133,134). RA has been shown to be essential in the development of CNS, reproduction, hematopoiesis and several other important biological processes, but is also implicated in pathological conditions: osteoporosis, rheumatoid arthritis, skin diseases and cancer (119, ). Figure 10. Vitamin A is important for several functions in humans. vii It is postulated that atra is the major biologically active RA. 13cRA is not as efficient in transactivation assays as atra, and many of the effects of 13cRA are thought to be mediated by atra after isomerization (142). Some questions have been raised regarding the physiological relevance of 9cRA, since the levels of 9cRA in mouse embryos and adult organs are very low and in some cases not detectable ( ). Also, in human liver, the level of atra is 25 times higher that that of 9cRA and 10 times higher than that of 13cRA. 9cRA is not detectable in mouse or rat liver whereas high levels of 13cRA are detected in mouse liver. The levels of atra in human liver are approximately three times higher than the levels in mouse or rat (143). 15
24 RA RECEPTOR In 1987, a nuclear receptor specific for RA was discovered (146,147). Shortly after this discovery of retinoic acid receptor α (RARα), RARβ and RARγ were identified (148,149). RA affects gene expression through two families of ligand-dependent nuclear receptors, RARs and RXRs, with 3 subtypes (α, β, γ) and multiple isoforms (Table 4) (150). As members of the steroid hormone receptor superfamily, RAR and RXR are structurally related, with an autonomous ligand-independent AF1 in the N-terminus that can synergize with AF2 at the C-terminus (151). The RARs are 97% homologous and RXRs are 60% homologous to each other in the DBD (152). The ligand-dependent AF2 is absolutely required for transcriptional activity. As for the ER, α-helix 12 is repositioned when bound to an agonist, causing dissociation of corepressors and association of coactivators (153). Gene Major isoforms Chromosomal location Ligand RARα α1, α2 17q21.1 atra, 9cRA RARβ β1, β2, β3, β4 3p24 atra, 9cRA RARγ γ1, γ2 12q13 atra, 9cRA RXRα α1, α2 9q34.3 9cRA RXRβ β1, β2 6q21.3 9cRA RXRγ γ1, γ2 1q22-q23 9cRA Table 4. RAR and RXR subtypes and isoforms, their chromosomal locations and known ligands. Despite its uncertain physiological relevance, 9cRA has been shown to bind with high affinity to RXRs and is up to 40-fold more potent than atra in transactivating RXRs (154). Also, 9cRa binds and activates both RARs and RXRs, whereas atra binds only RARs with high affinity. It is suggested that instead of 9cRA, unsaturated fatty acids like linoleic, linolenic and docosahexaenoic acids are the endogenous ligands for RXRs (145). 13cRA has not been shown to bind RARs in vitro studies (137). RAR and RXR can form a heterodimer and bind to DNA sequences termed RA response elements (RAREs), generally located within promoters of target genes (151). RAREs are composed of direct repeats (DR) of the consensus half-site sequence (A/G)G(G/T)TCA with 2-bp or 5-bp spacer, called DR2 and DR5, respectively (155). Each receptor monomer of the heterodimer binds to one of the two half-sites composing the RARE (156). In addition to the heterodimerization with RARs, the RXRs can homodimerize or heterodimerize with other nuclear receptors, such as the vitamin D and thyroid receptors (157). There are indications that there is a functional RA receptor redundancy, since knockout studies in mice show that disrupted expression of one receptor subtype causes limited abnormalities and survival of the mice, whereas in a double knockout of either two RAR subtypes or RARα and RXRα, the abnormalities are more severe and cause lethality (158). 16
25 RAR COREGULATORS RARs regulate transcription either in an activating or repressing fashion, either directly via an RARE or indirectly via modulation of transactivation of other transcription factors, as in the case with RA transrepression of AP-1 and NF-κB (159,160). The transcriptional activity of the RAR/RXR heterodimer is mediated through a large number of coregulatory proteins, including corepressors, coactivators and other cofactors. Upon recruitment to the RAR/RXR heterodimer, the coregulatory proteins affect transcription by directly or indirectly modifying the chromatin structure through histone acetyltransferases (HATs) or histone deacetylases (HDACs). Unliganded RAR/RXR heterodimers recruit the corepressors NCoR and SMRT, which in turn recruit histone deacetylases, with DNA of target gene less accessible to the basal transcription machinery as an outcome (161). Upon binding of an agonist, corepressors dissociates and there is a recruitment of coactivators followed by histone acetylases (162) (Figure 9). The RAR/RXR heterodimer has been shown to recruit several coactivators followed by transcriptional activation. Recruitment of the ATP-dependent chromatin remodeling complexes SWI/SNF by ligand-bound RAR/RXR is an important step in transcriptional activation of RA-regulated promoters (163). The ligand-dependent recruitment of the histone acetyltransferase PCAF is essential for RA-induced transcription, whereas binding of PCAF was abolished by binding of RAR antagonist (164). CBP and p300 are coactivators for RAR with intrinsic HAT activity (165,166). p300 interacts with the p160 member SRC-1 and is recruited to RAR in a ligand-dependent manner (165). All three LXXLL NR binding motifs of SCR-2 bind to RAR and are implicated in RAR-mediated transactivation (167). SRC-3 is recruited to RA-regulated promoters and interacts with ligand-bound RAR (168,169). In addition to these NR coactivators, the cellular RA-binding protein II (CRABP-II) enhances RAR transcriptional activity in a ligand-dependent manner. Upon binding of RA, CRABP-II undergoes nuclear localization and associates with RAR, and overexpression of CRABP-II in MCF-7 cells strongly enhances the sensitivity to RA-induced growth inhibition (158). Unliganded or antagonist-bound RAR interacts with the transcriptional repressors NCoR and SMRT, followed by a suppression of the basal transcriptional activity (170). NCoR and SMRT then recruit histone deacetylases of class I (171). RAR shows higher affinity to SMRT than to NCoR, whereas SMRT has weak affinity to RXR homodimer, although the affinity is greatly enhanced upon interaction with RAR (83,161). The N-terminus interacting domain 2 (ID2) but not ID1, defined for SMRT, interacts with RAR (172). RIP140 interacts with agonist-bound RAR and represses RA-induced transcription (173). Six out of the nine LXXLL RAR binding motifs in RIP140 interact with ligand-bound RAR (174). 17
26 RAR AND PROLIFERATION Proliferation of breast cancer cells requires signals from growth factors such as estrogens, heregulin, insulin-like growth factors (IGFs), epidermal growth factor (EGF) and transforming growth factor α (TGFα). RA is well established as an inhibitor of proliferation of estrogen-dependent breast cancer cells, mainly through preventing the G 1 - to S-phase cell cycle progression ( ). Sensitivity to RA is associated with expression of RARα (177). ERα positive cells express RARα, and thus are sensitive to RA, whereas the majority of the ERα negative cells cell lines do not express RARα, and thus are resistant to RA (178,179). It has been shown that induced RARα expression, either by ectopic expression of RARα or ERα, in ERα negative and RA resistant cells, restores sensitivity to RA (180). Possible mechanisms for regulation of cell proliferation by RA include regulation of AP-1 activity and cell cycle regulators. AP-1 Members of the jun family (c-jun, junb and jund) can homodimerize or heterodimerize with members of the fos family (c-fos, FosB, Fra1 and Fra2), resulting in a dimer designated AP- 1. Heterodimers are more stable and have a higher affinity to AP-1 response elements than homodimers (181). The activity of AP-1 is associated with breast cancer cell proliferation, with RA inhibiting AP-1 activity and AP-1-induced proliferation of breast cancer cells ( ). Although there is no clear picture of how RA inhibits AP-1, there are several possible explanations. RAR has been shown to interfere with the homo- and heterodimerization of AP-1 components (185). Expression of c-jun and c-fos is inhibited by RA and RAR (186,187). RA inhibits the JNK-induced but not the ERK-induced AP-1 activity (188). Competition for coactivators, such as SRC-1 and CBP, by RAR and AP-1 is another suggested mechanism (189,190). Activation of AP-1, followed by tethering of CBP and ERKs to an AP-1 responsive promoter, was inhibited by RA through alteration of AP-1 complex composition. Moreover, active AP-1 is composed of a jund and FosB heterodimer whereas treatment with RA results in an AP-1 composed of jund and fra-1 heterodimer, with an inactive AP-1 complex as a outcome (191). RA has been shown to inhibit the AP-1- mediated induction of the G 1 cell cycle regulators cyclin D1 and E2F gene expression (184). Cell Cycle Regulators The response to RA, in the context of cell cycle regulators causes a decrease in the expression of cyclin D1, D3, E, CDK2 and CDK4 and E2F1 (176, ). Phosphorylation of prb is abrogated resulting in inactive E2F1, and CDK2 and E2F1 are decreased in response to RA (176,192,193,196). The protein levels of CDK inhibitor p21 are increased, both total p21 and CDK2-bound but not CDK4-bound p21 (196). Both RA and tamoxifen inhibit cell cycle progression in G 1 phase in T47D breast cancer cells through decreased phosphorylation of prb. Estrogen treatment abolishes the inhibition of cell cycle progression by tamoxifen, but does not affect the inhibition induced by RA, showing different actions of tamoxifen and RA in inhibition of proliferation (193). The critical tumor suppressor gene p53 has been shown not to be implicated in the G 1 arrest in response to RA (197). 18
27 SYNTHETIC RETINOIDS The treatment of patients with advanced breast cancer using natural retinoids has been limited due to toxic side effects and the lack of clinical efficacy. In order to improve this, synthetic retinoids have been developed. A synthetic derivative of atra, 4-HPR, has been shown to be less toxic and more potent than atra. 4-HPR inhibits the proliferation of RAR-negative breast cancer cells, and it has been shown that 4-HPR mainly inhibits proliferation in an RAR-independent manner (198). Although 4-HPR has been shown to maintain prb in a hypophosphorylated state and to decrease the expression of cell cycle regulators, such as cyclin D1, CDK2 and CDK4, the main mechanism that 4-HPR inhibits proliferation of breast cancer cells is induction of apoptosis (199,200). In transactivation studies, 4-HPR is an activator of RARγ and RARβ but not RARα. It has also been shown that in transrepression studies, 4-HPR functions with RARα (201). The reduced toxicity shown by 4-HPR may be due to an accumulation of 4-HPR in the breast tissue (202). However, in patients with advanced breast cancer, 4-HPR shows limited therapeutic efficacy despite favorable preclinical data (203). 19
28 SOX9 The SRY-related high-mobility group (HMG) Box 9 (SOX9) belongs to the SOX family of proteins, which includes 30 vertebrate, 20 mouse and 20 human members, and are involved in a variety of developmental processes (204). The SOX proteins are defined by the presence of the HMG box amino acid sequence RPMNAFMVW (205). SOX9 is located at chromosome 17q24.3 and is composed of three exons encoding a 509-amino acid polypeptide. It has 70% amino acid homology to the sex-determining region Y (SRY) protein in the DBD, the HMG box. SOX9 activates transcription of its target genes by binding to specific DNA sites, generally located in the promoter of target genes leading to subsequent bending of DNA. DNA bending by SOX9 is proposed to be necessary for transcriptional activation, bringing proteins involved in SOX9 transcriptional activation into closer proximity (206,207). The binding of SOX9 to DNA is a critical event, since mutations in the HMG box that abolish DNA binding, inhibit SOX9 transcriptional activity (208). SOX9 binds the DNA sequence AACAAT, the consensus binding site for several HMG box containing proteins such as TCF-1, SRY, SOX2, SOX5, SOX6 and SOX17 ( ). The optimal SOX9 binding site in vitro is AGAACAATGG, where the 5 AG and 3 GG flanking residues enhance SOX9 binding but not SRY binding, suggesting a role for flanking residues in HMG box protein specificity (215). SOX9 has been shown to bind as a dimer in the promoter of genes involved in the development of cartilage ( ). Dimeric SOX9 binds to enhancers, cis-elements involved in transcriptional activation, in the promoter of the collagen gene COL11a2, at paired binding sites organized in the opposite direction to each other with a 3 or 4-bp spacer (217). The collagen gene COL9a1 has two interdependent enhancers in the promoter, and thus four SOX9 binding sites, and full activation of COL9a1 gene expression requires two interacting SOX9 dimers (219). In the promoter of genes involved in sex determination, such as anti-müllerian hormone (AMH) and steroidogenic factor 1 (SF-1), there is only one SOX9 binding site, thus suggesting that SOX9 binds as a monomer (216,218). 5 of the HMG box, lies a DNA-dependent SOX9 dimerization domain (218) (Figure 11). Downstream of the HMG box, there are two transcriptional activation domains, the PQA and PQS domains. The PQA domain is composed of proline, glutamine and alanine residues, whereas the PQS domain comprises mostly proline, glutamine and serine residues. The PQS domain is essential in transactivation of target genes, but for maximal activation the PQA domain is required as well (208,220). Figure 11. The known functional protein domains of SOX9 are the dimerization domain (DIMER), DNA-binding domain (HMG), and transactivation domains (PQA and PQS). 20
29 SOX9 protein sequence is highly conserved through vertebrate evolution, with the PQA domain being the only part of the protein in which a few variations in the amino acid composition between mammals and other vertebrates can be seen (207). The well conserved protein sequence of SOX9 between vertebrates suggests interactions with other cellular proteins, such as the interaction with SF-1 via the HMG box, or with the coactivator CBP/p300 via its carboxyl terminus activation domain of SOX9 (207,221,222). There are several putative phosphorylation sites within SOX9, and phosphorylation by protein kinase A (PKA) on serine 63 and 181 enhances the DNA binding and transcriptional activity of SOX9 (223). Phosphorylation induced by fibroblast growth factors (FGFs) enhances SOX9 expression through the MAPK pathway (224). Activation of the p38 MAPK pathway in human articular chondrocytes, increases SOX9 expression through stabilization of SOX9 mrna (225). SOX9 was identified in 1994, and through mutation analysis and DNA sequencing it was demonstrated that mutations in SOX9 can cause campomelic dysplasia (CD) and autosomal sex reversal (226). Mutations causing CD and sex reversal are missense mutations in the HMG box resulting in loss of DNA binding, or frameshift mutations in the PQA and PQS domains resulting in loss of transactivation (208). By the identification of a missense mutation outside the HMG box, in a non sex reversed patient with CD, that abolished dimerization, it was postulated that dimerization is required for chondrogenesis but not for sex determination (216). In addition to the well known role of SOX9 during chondrogenesis and testis formation, expression of SOX9 in the developing CNS, notochord, lungs, heart and urogenital system implicates other roles of SOX9 (209). SOX9 ACTION Other factors than SOX9 are most probably involved in SOX9 transactivation, since expression of SOX9 in the testis is not enough to induce expression of the collagen genes, the SOX9 target genes during chondrogenesis (209). Recently, it was shown that SOX9 activates transcription of COL2a1 by associating with Smad2/3 and the cofactors PGC-1α and CBP/p300, where the activity of p300 results in modified chromatin (222, ). SOX9 has also been shown to communicate with the general transcription machinery, through interactions with TRAP230, a component of the mediator complex (230). SOX9 and Sex Determination Maleness or femaleness in eutherian mammals is determined by the development of the testis and the ovaries, respectively. In 1959, it was shown that the presence of a Y chromosome results in male development, and that the Y chromosome encodes a male determining factor (231). The testis-determining gene SRY located on the Y-chromosome was identified in 1990 (232). The genes implicated in sex determination, other than SRY and SOX9, are far from being elucidated, although AMH, Wilms tumor gene 1 (WT1), SF-1, the zinc finger transcription factor GATA4, WNT4 and DAX1 genes are known to be involved. Male development is elicited by the transient expression of SRY in the bipotential gonads, with a strong upregulation of SOX9 expression as a result, followed by an import of cytoplasmic SOX9 into the nucleus (233) (Figure 12). The nuclear localization is suggested to be facilitated by the proteins importin β or by calmodulin ( ). 21

.")
30 Figure 12. Model of the action by genes involved in sex determination. The presence of SRY suppresses female development and triggers male development, whereas the absence of SRY results in a suppression of male development and triggers female development. SRY binds to a SOX9 gonad-specific enhancer with SF-1 and cooperatively upregulates SOX9. After cessation of SRY expression, the levels of SOX9 are maintained through an auto-regulatory feedback (237). SOX9 expression is found exclusively within the Sertoli cells of the testis, both during development and in the adult testis (233). Certain genes required for the development of the ovaries, DAX1 and WNT4, are downregulated in response to SRY expression (238,239). DAX1 is in turn a negative regulator of SRY expression and has been shown to be induced by WNT4 (240). Gene duplication of WNT4 or DAX1 results in XY females, by antagonizing testis formation (240,241). SF-1 is crucial for the development of gonads, since mice with disrupted SF-1 expression lack gonads and heterozygous mutations in the SF-1 gene causes XY sex reversal in humans (242,243). SF-1 expression is dependent on LHX9 expression, indicating the importance of LHX9 during gonadogenesis, as shown by using LHX9 knock out mice (244). DAX1 negatively regulates SF-1, but SF-1 regulates DAX1, its own antagonist, in a positive manner (245). Mutations of WT-1 are associated with urogenital malformations (246). Secretion of the pivotal component of male sexual differentiation, AMH, by Sertoli cells, results in degeneration of the female Müllerian reproductive tract. AMH has been shown to be regulated by SOX9 and SF-1 and by GATA4 and WT-1 in synergy with SF-1 (221,247,248). It is postulated that SOX9 can substitute for SRY, shown by ectopical expression of SOX9 in XX gonads of mice resulting in testis formation and a male phenotype (249). Furthermore, duplication of SOX9 in an SRYnegative XX newborn infant resulted in female to male sex reversal, with abnormal male external genitalia (250). 22
31 SOX9 and Chondrogenesis The cartilage is a tissue that is resilient and pliant, and provides the template for the developing skeleton. The production and maintenance of the extracellular matrix of the cartilage, is made by specialized cells called chondrocytes (251,252). SOX9 plays a key role in the development of cartilage, the chondrogenesis, by regulating the expression of several extracellular matrix proteins such as the collagen genes type II COL2A1, type IX COL9A1, type XI COL11A2 ( ). In addition, one of the major structural components in cartilage matrix, aggrecan, a large chondroitin sulfate proteoglycan, has been shown to be regulated by SOX9 (256). Another key component of the cartilage extracellular matrix, the cartilage link protein (CRT-LP), is regulated by SOX9 via a cis-element (257). A transactivating complex including SOX9, SOX5 and SOX6 cooperatively induces COL2A1 expression, by binding to an enhancer containing HMG sites in the COL2A1 promoter (255). Using chromatin immunoprecipitation assay (ChIP), SOX9 was found in a complex with the coactivator p300 followed by an induction of COL2A1 expression (222). Additionally, SOX9 was found to interact with the transcription factor L-MAF in the induction of COL2A1 expression (258). Mutations of the components involved in chondrogenesis often result in skeletal abnormalities (226,259). SOX9 IN CAMPOMELIC DYSPLASIA AND SEX REVERSAL SOX9 haploinsufficiency results in a syndrome called campomelic dysplasia (CD) (226). The syndrome is rare, it is only seen in 1: births (260). In 75% of the CD cases, an XY male to female sex reversal is observed (261). CD is an autosomal dominant disorder, generally lethal in the first year of life. CD is characterized by bowing of shinbones (tibiae) and thighbones (femora), hypoplastic scapula, decreased number of ribs, degeneration of trachea resulting in respiratory distress and neonatal death (262). Chromosomal translocations, missense and frameshift mutations are common mutations of SOX9 gene seen in CD cases. SOX9 AND THE CELL CYCLE Expression of SOX9 in the M12 prostate cancer cells causes inhibition of cell proliferation, with cells arresting in G 0 /G 1 and G 1 phase. There was no measurable G2/M fraction and an increased apoptosis could be seen upon SOX9 expression (263). Overexpression of SOX9 in the rat chondrocytic cell line CFK2 resulted in accumulation of cells in the G 0 /G 1 phase, followed by an induction of cyclin- dependent kinase inhibitor p21 expression (264). SOX9 expression is also induced by RA receptor agonists, and mediates the RA-induced growth inhibition in the breast cancer cell line T47D (265). In SOX9 knock-in embryos, there was a strong reduction of cyclin D1 expression, hence proliferation of chondrocytes was decreased (266). 23
32 Other Roles of SOX9 SOX9 has been shown to induce neural crest differentiation in neural cells, as found by ectopical expression of SOX9 in chick embryos (267). A conditional knock-out of SOX9 in the mouse prostate, showed abnormal differentiation of the anterior prostate and a deficiency of ventral prostate development, implying a requirement of SOX9 in the development of the prostate (268). Not only the development, but also the maintenance of the normal prostate is regulated by SOX9 (269). During intestinal epithelial cell differentiation, SOX9 induces differentiation of Paneth cells, and when SOX9 was inactivated in the intestinal epithelial cells in mice, no Paneth cells were formed and a general hyperplasia and local crypt dysplasia in the intestine was observed (270,271). SOX9 has been shown to play a role during the development of the pancreas, by regulating the maintenance of the progenitor cell pool through stimulating their proliferation and survival. Inactivating SOX9 expression in the pancreas of mice strongly reduced the number of pancreatic cells positive for expression of the Notch effector protein, HES-1 (272). 24
33 HES-1 Hairy and Enhancer of Split homolog 1 (HES-1) belongs to the basic helix-loop-helix (bhlh) family of proteins. More than 240 proteins are members of this well conserved family, in organisms ranging from yeast to humans (273). bhlh proteins are shown to play essential roles in several developmental processes, such as neurogenesis, hematopoiesis and myogenesis ( ). This family of proteins can be divided into seven different classes, according to their binding specificities and biochemical properties (Table 5) (273). Class Characteristic Ex. I Transcriptional activators E proteins II Transcriptional activators MyoD III bhlh-lz proteins Myc IV bhlh-lz proteins Mad, Max V HLH without basic region Id VI Transcriptional repressors HES-1 VII bhlh-pas proteins AHR, Arnt Table 5. Classification of bhlh proteins The transcriptional regulators of bhlh proteins class I to IV have been shown to bind to DNA sequences called E-boxes, CANNTG (where N can be any of the four nucleotides) whereas class VI proteins, the transcriptional repressors, bind to N-boxes, CACNAG (273,277). The bhlh proteins bind to E- and N-boxes as homodimers or heterodimers. The binding to DNA is through direct contact between each basic domain of a dimer and a half site of the specific DNA sequences, where the basic domains are perpendicular to each other and interact with the major groove of DNA (278,279). The Class V proteins lack this basic region, which is necessary for DNA binding, and thus heterodimerization with class V proteins results in a complex unable to bind DNA, and these proteins act as dominant negative factors (280). The hydrophobic residues in the HLH domains promotes dimerization, and due to multiple interactions, the resulting dimer is stable, even in the absence of DNA (279). Class VI of the bhlh protein family, the transcriptional repressors, includes the Hairyrelated proteins. Besides the Drosophila proteins Hairy and the seven members of Enhancer of Split complex, HES and HERP are all Hairy-related proteins. The HES family consists of seven members, HES 1-7, which all act as transcriptional repressors, apart from HES-6 which is proposed to counteract HES-1 action (281). HES and Drosophila Hairy and Enhancer of Split genes share two common unique structural features; a proline residue in the basic domain and a four amino acid sequence, WRPW, in the C-terminus involved in transrepression of target genes ( ) (Figure 13). The proline residue has been shown to determine HES proteins binding preference to DNA for N-boxes, rather than E-boxes as for most other bhlh proteins (285). The proline residue in the DNA binding region is a hallmark for HES proteins and it is conserved from Drosophila Hairy and enhancer of split to human HES (283). In addition, Hairy-related proteins has a conserved domain called Orange domain, or helix 3 and 4 (H3/4), that is involved in repression (286). 25
34 Figure 13. HES functional protein domains, including bhlh, Orange and repression (r) domain. HES-1, the most studied HES member, is expressed in a wide variety of tissues of both embryos and adults (285). During mouse embryogenesis, HES-1 mrna is ubiquitously expressed: in the brain, liver, heart, pancreas and kidney with a high amount of mrna in the lung. Expression of HES-1 mrna is uniform at the embryonic days 10 to 18 (287). HES-1 is well conserved during the evolution, and exists in many species, such as zebra fish, African clawed frog and humans (Table 6A). Despite that all HES members have a bhlh region, an Orange domain and the tetrapeptide WRPW, the overall homology between HES-1 and other HES members is rather low (Table 6B). The HES-1 gene was identified in 1992, and is composed of 4 exons with the chromosomal location of 3q28, and the small HES-1 protein is made up of 280 amino acids in humans (285). HES-1 No. aa Homology % Homo sapiens Bos taurus Rattus norvegicus Mus musculus Xenopus laevis Danio rerio Gallus gallus HES No. aa Homology % Table 6. Homology of human HES-1 to other HES proteins (A), and to other species (B). TRANSCRIPTIONAL REPRESSION BY HES-1 HES-1 represses transcription in at least three different ways: DNA-dependent (active repression), DNA-independent (passive repression) or via the Orange domain. DNAdependent repression is through binding of the HES-1 homodimer to an N-box and the recruitment of the corepressor, the mammalian homolog to Groucho in Drosophila, the Transducin-like enhancer of split homolog (TLE). The corepressor TLE interacts with the tetrapeptide at the C-terminus of HES-1, the WRPW motif (288). The interaction of TLE with HES-1 induces phosphorylation of TLE by protein kinase CK2 followed by the recruitment of HDAC1 and the subsequent modulation of chromatin structure, which results in inhibition of transcription (289). The DNA-independent repression is through the formation of non-active heterodimers with bhlh transcriptional activators, such as E47, which prevents binding of these activators to the E-boxes (285). The Orange domain has been shown to be implicated in repression, via a not fully elucidated mechanism. It is proposed that the Orange domain either recruits a corepressor or exerts repression through stabilization of the DNA-dependent repression (286). 26
35 REGULATION OF HES-1 HES-1 has shown to be an immediate early gene, where serum treatment, without new protein synthesis, rapidly upregulates HES-1 expression (287). A rapid and transient induction of HES-1 mrna in PC12 pheochromocytoma cells is observed in response to nerve growth factor (NGF), basis fibroblast growth factor (bfgf) and EGF or in Rat-1 fibroblasts by EGF. After 5 hours of growth factor addition, HES-1 mrna levels are back to preinduced levels (290). There is no serum response element in HES-1 promoter region, but an AP-1 element, implying a possible HES-1 regulation by AP-1 (291). HES-1 was found in microarray analysis to be downregulated in response to estrogen (73). HES-1 has been shown to be regulated post-translationally, by NGF induced phosphorylation of PKC consensus sites in the DNA-binding domain of HES-1, which results in decreased HES-1 DNA binding (292). Negative feedback loop The regulation of both HES-1 mrna and protein is tightly controlled, with very short half lives of both mrna and protein, 24 and 22 min, respectively (293). HES-1 has shown to negatively regulate its own expression, via 3 N-boxes located within the 5 -untranslated region (5 -UTR) (291). These 3 N-boxes repress HES-1 expression synergistically, and not additively, where the number of N-boxes correlates well with fold repression, as shown by an in vitro transcriptional analysis (291). HES-1 oscillation HES-1 mrna oscillates with a periodicity of 2 hours in the vertebrate embryonic presomitic mesoderm (PSM) during somite segmentation (293,294). Expression of HES-1 appears as a wave front which sweeps across the PSM once during the formation of each somite (295). The segmentation clock, that drives the somite formation with the periodicity of 2 hours, is proposed to drive the periodic activation of Notch, a HES-1 regulator (295). Both HES-1 mrna and protein oscillate in a 2-hour cycle in a variety of cultured cells, including fibroblasts, neuroblastoma cells and teratocarcinoma cells after a single serum treatment (293). mrna oscillation precedes protein oscillation by 15 min, implying a time delay for protein degradation. After 6-12 hours the oscillation terminates, which corresponds to 3-6 cycles after a single serum treatment. The proposed mechanism for HES-1 oscillation is a negative feedback loop in combination with rapid degradation of HES-1 protein by the ubiquitin-proteasome pathway (Figure 14). The induced transcription of HES-1 and the subsequent translation are followed by HES-1 protein binding to its own promoter, which results in termination of transcription. HES-1 mrna and protein are rapidly degraded followed by a new cycle of transcription and translation (293). Overexpression of HES-1 yields no HES-1 mrna synthesis and treatment with a proteasome inhibitor shows a transient transcription of HES-1 mrna followed by a persistent suppression. Furthermore, overexpression of a dominant negative HES-1 or treatment with a inhibitor of translation results in high levels of HES-1 mrna, supporting the mechanism that HES-1 protein exerts negative feedback with oscillation of both mrna and protein as the result (293). 27
36 Figure 14. Model for HES-1 oscillation. HES-1 transcription (1) and translation (2) are induced by certain stimuli, followed by HES-1-mediated repression of transcription (3). HES- 1 protein is degraded (4) followed by a new round of transcription. viii NOTCH SIGNALING Notch signaling is involved in cell fate determination, where active signaling causes an inhibition of differentiation of precursor cells and maintaining them in an undifferentiated state (296). HES-1 is the primary effector protein of Notch pathway during several processes, such as the mammalian neural development and the T-cell maturation ( ). The Notch protein is a large membrane-spanning cell surface receptor for membrane-bound ligands, where Notch signaling is activated solely between cells that are in direct contact with each other (296,300). There are 4 mammalian Notch receptors, Notch 1-4, and 5 ligands, Delta 1, - 3, -4, Jagged 1, -2, and 6 known Notch target genes, HES-1, -5, -7 and HERP 1-3 (283). Notch signaling is activated upon ligand binding, by neighboring cells undergoing differentiation to Notch, expressed on neural precursor cells, where the proteolytic processed Notch intracellular domain (NICD) translocates to the nucleus and interacts with transcription factor RBP-Jκ (Figure 15) (299,301,302). The NICD interaction with RBP-Jκ converts RBP- Jκ from an transcriptional repressor to an transcriptional activator and induces the expression of Notch target genes, such as HES-1 (303). In neural development, HES-1 represses the transcription and activity of neuronal genes and in the differentiating cell, the neuronal genes promote neurogenesis by inducing neuronal specific genes (285, ). Expression of HES-1 in this cell is inhibited, by HDAC1-associated RBP-Jκ, and by the formation of an inactive heterodimer with HES-6 (281,303). 28
, where the intracellular part translocates to the nucleus and induces transcription of HES-1 in complex with RBP-Jκ (2).")
37 Figure 15. Notch signaling during neurogenesis. Binding of Notch to its ligand triggers cleavage of Notch (1), where the intracellular part translocates to the nucleus and induces transcription of HES-1 in complex with RBP-Jκ (2). HES-1 inhibits neural differentiation of a precursor cell (yellow cell) by inhibiting transcription and activity of neuronal activators (3). In the differentiating cell (green cell), the absence of Notch results in RBP-Jκ associates with corepressors and inhibits HES-1 expression (4). The absence of HES-1 results in expression of neuronal activators (5), such as HES-6, which maintain inactive HES-1 (6), Notch ligands maintain active Notch signaling in the neighboring cell (7). ix There are many indications that HES-1 can be regulated independently by Notch, as shown by Notch 1-, Notch 2- and RBP-Jκ- deficient mice, which have no alteration in HES-1 levels (307,308). At the neuroepithelial stage, during neurogenesis, HES-1 is expressed but not Notch, indicating Notch-independent regulation of HES-1 (309). Growth arrested human endothelial cells are associated with elevated levels of HES-1, induced by JNK signaling independently of Notch activation (310). In addition, despite the pivotal role of HES-1 during epidermal development, Notch signaling determines spinous cell fate in a HES-1 independent way (311). 29
38 HES-1 ACTION HES-1 is well established as a regulator of neurogenesis, myogenesis, eye morphogenesis, hematopoiesis and the development of several other tissues (285,298,312,313). Persistent expression of HES-1 inhibits mammalian neuronal development and retinal development (312,313). Overexpression of HES-1 downregulates endogenous CD4 expression, and CD4 promoter activity in CD4 + CD8 - T H cells via a N-box containing silencer, in the first intron of CD4 locus (314). CD4 expression is induced by c-myb, whereas interaction of HES-1 with c- Myb turns the complex into a repressor followed by a downregulation of CD4 expression (315). The expression of the glycogen-degrading enzyme Acid α-glucosidase (GAA) is regulated via a silencer within the first intron of the gene, by HES-1 in collaboration with the transcription factor Ying Yang 1 (YY1) in HepG2 cells (316). In human fibroblasts, on the contrary, this silencer functions as an enhancer when bound to HES-1 and YY1, giving HES- 1 a role as an transcriptional activator (317). The levels of cyclin-dependent kinase inhibitor p21 and transcription of the p21 promoter have been shown to be strongly repressed by ectopic expression of HES-1 (286). HES-1 is a negative regulator of MyoD and MASH1 transactivity in PC12 cells, by preventing the formation of MyoD/E47 and MASH1/E47 complexes, through sequestration of E47 (285). HES-1 has been suggested to be a potent regulator of proliferation, since overexpression of HES-1 inhibited cell cycle progression in several cell lines such as T47D, PC12, NIH 3T3, SHSY5Y, SHIN and SHEP1 (286,318,319). 30
39 AIMS OF THE STUDY General aim The aim of this thesis was to elucidate the role of HES-1 in estrogen- and retinoic acidregulated proliferation of human breast cancer cells. Specific aims Paper I: To determine whether HES-1 mediates the growth-inhibitory effect induced by retinoic acid in human breast cancer cells Paper II: To identify where in the cell cycle HES-1 exerts its effect, and what cell cycle factors HES-1 regulates Paper III: To identify cis-regulatory elements affecting HES-1 gene expression in response to estrogen or retinoic acid Paper IV: To investigate whether HES-1 is regulated by SOX9 in human breast cancer cells 31
40 RESULTS AND DISCUSSION PAPER I It is well established in human ERα positive breast cancer cell lines, such as MCF-7 and T47D that estrogen induces proliferation. Also, it has been shown that RA inhibits this estrogen-induced proliferation (176). In fact, breast cancer cells that respond to estrogen also respond to RA since breast cancer cells that express ERα express the functional receptor for RA, the RARα (177,178). The background knowledge used for these studies comes primarily from work undertaken previously in our laboratory, where it was shown that exogenous expression of HES-1 in estrogen-induced growth of T47D cells causes complete growth arrest (318). It was found that treatment of both T47D and MCF-7 cells with estrogen caused a strong downregulation of HES-1 protein, whereas in MDA-MB-231, an ERα negative cell line, HES-1 protein levels were unchanged in response to estrogen. Adding anti-estrogen to estrogen-treated T47D cells abolished the downregulation of HES-1 protein levels. These data suggested that ERα is involved in estrogen-mediated downregulation of the HES-1 protein. Furthermore, an inverse correlation between the HES-1 protein and the proliferation marker, PCNA protein, was found. The conclusion made was that HES-1 levels need to be downregulated for these cells to proliferate in response to estrogen, which gave HES-1 a role as a possible tumor suppressor in mammary epithelial cells (318). In a search for agents regulating HES-1, RA was found to induce HES-1 protein levels. Other laboratories have shown that exogenous expression of HES-1 in the rat pheochromocytoma cell line PC12, resulted in growth arrest, whereas in HeLa cells HES-1 overexpression induced growth (286,320). Moreover, in undifferentiated F9 embryonic carcinoma cells, RA downregulated HES-1, whereas in P19 embryonic carcinoma cells RA upregulated HES-1 (320,321). These conflicting results show that HES-1 regulation is cell-context dependent. In this study we wanted to investigate whether HES-1 is involved in RA induced growth inhibition, in the human breast cancer cell line MCF-7. We found that these cells proliferate in response to low levels of estrogen by approximately 50% increase of the cell number within 6 days. Theoretically, one would expect a more pronounced increase of the cell number since the calculated generation time of MCF-7 cells is estimated to be around 32 hours (American type culture collection). However, the observed lower increase in cell number in response to estrogen can possibly be explained by suboptimal culture conditions or degenerated cells. Adding RA to the estrogen-treated cells inhibited this estrogen-induced proliferation. 1 µm of RA was sufficient to completely inhibit the effect of estrogen. Analysis of the HES-1 expression under these conditions revealed a perfect inverse correlation between proliferation and HES-1 protein expression. Induction of proliferation, in response to estrogen, resulted in non-detectable HES-1 protein levels, whereas addition of increasing amounts of RA caused increasing inhibition of proliferation and thus increasing levels of HES-1 protein levels. To determine whether the correlation between HES-1 protein and the inhibition of proliferation were connected, we generated MCF-7 cells expressing a dominant negative HES-1. Dominant negative HES-1 was constructed with a missense mutation in the basic 32

.")
41 region and a deletion of C-terminus. The missense mutation resulted in a proline to alanine amino acid change and disrupted DNA binding, and deletion of C-terminus results in loss of corepressor recruitment. In co-immunoprecipitation assays, we show that ectopic expression of dominant negative HES-1 dimerizes with endogenous HES-1, in both transiently transfected COS-7 and the constructed MCF-7 stably expressing dominant negative HES-1 (MCF-7 dn-hes-1). The stronger protein band from endogenous wildtype HES-1 relative to exogenous dominant negative HES-1 is due to 2 epitopes for the HES-1 antibody, one at N- terminus and one at C-terminus, whereas dominant negative HES-1 has only 1 epitope. MCF-7 dn-hes-1 cell proliferation is enhanced compared to parental MCF-7 cells in response to estrogen, but importantly, they do not respond to RA, at any dose of RA added. HES-1 protein levels were higher in cells treated with the RARα specific agonist RO compared to the pan-agonist atra, implying regulation of HES-1 protein by RARα. Similar pattern of regulation was observed for HES-1 mrna as for HES-1 protein in response to estrogen and RA, although less pronounced. By treating MCF-7 cells for 7 hours with actinomycin D, an inhibitor of transcription, we found very low levels of HES-1 mrna compared to non-actinomycin D treatment, suggesting that regulation of HES-1 mrna in response to estrogen and RA is transcriptional and not on mrna stability. In conclusion, the correlation between inhibition of proliferation and HES-1 gene expression, and lack thereof while expressing dominant negative HES-1, indicates that HES-1 mediates the growthinhibitory effect induced by RA on MCF-7 cells (Figure 16). Figure 16. A model based on the conclusions from paper I. The estrogen-stimulated growth of MCF-7 cells is inhibited by RA. The inhibitory effect is mediated by HES-1. 33
42 PAPER II The purpose of this study was to determine whether HES-1 induces cell cycle arrest, and if so, what cell cycle factor HES-1 regulates. In the neuroblastoma cell lines PC12, SHSY5Y, SHEP1, and SHIN and in NIH 3T3 mouse fibroblasts, generating HES-1 overexpressing cell clones proved to be difficult since they fail to proliferate (286,319). By using a tetracyclineregulated HES-1 inducible system, this problem was overcome. Overexpression of HES-1 in PC12 and T47D cells resulted in cell cycle arrest in the G 1 phase, concomitant with decreased levels of the proliferation marker PCNA (286,318). By turning off HES-1 expression in the same cells proliferation was restored, showing that growth arrest was not due to apoptosis or nonspecific toxic effects. Although the cyclin-dependent kinase inhibitor p21 was shown to be downregulated in response to exogenous HES-1, this is unlikely to be the mechanism involved in cell cycle arrest, but probably in the inhibition of differentiation (286). In HeLa cells, overexpression of HES-1 resulted in transcriptional repression of cyclin dependent kinase inhibitor p27, resulting in enhanced proliferation (320). In this study, we used T47D breast cancer cells with tetracycline-regulated expression of HES-1 or the dominant version of HES-1 (dnhes-1). In the presence of tetracycline, exogenous HES-1 or dnhes-1 are not expressed but when tetracycline is withdrawn, they are expressed. These cells were synchronized in the G 1 phase of the cell cycle by using the pure anti-estrogen ICI 182,780. Firstly, we found that overexpression of HES-1 arrested T47D cells in the G 1 phase of the cell cycle, the phase where most of the cell cycle factors are regulated in response to a mitogen. In the absence of exogenous HES-1 expression, 15 hours estrogen treatment caused the majority of cells to exit G 1 and enter S-phase. But, expression of exogenous HES-1 caused the cells to be arrested in the G 1 phase. None of the D-cyclins or cyclin-dependent kinases were regulated by overexpression of HES-1 (data not shown) but the critical G 1 to S-phase progression factor E2F-1 was. When the E2F-1 promoter was activated, E2F-1 mrna and protein levels were enhanced in the absence of HES-1 expression compared to in the presence of exogenous HES-1 expression, in response to estrogen. Importantly, estrogen-induced activation of E2F-1 promoter, E2F-1 mrna and protein were downregulated in response to RA. Expression of dnhes-1 abolished this E2F-1 downregulation in response to RA, supporting the conclusion of paper I that HES-1 mediates the effect of RA. We found an N-box in the E2F-1 5 regulatory region, which was shown to be activated and bound by HES-1 in a reporter assay and an in vitro binding assay, respectively. Mutating this N-box abolished this activation and binding. Similarly to E2F-1, cyclin E and NPAT were regulated in the same way in response to RA and expression of exogenous HES-1 or dnhes- 1. This was expected, as cyclin E and NPAT are target genes for E2F-1 (322,323). Also, we showed proliferation of T47D cells in response to the mitogen heregulin-β1, which is a ligand for ErbB3 tyrosine kinase receptor. Similar to estrogen, heregulin-β1 induced E2F-1 mrna levels, which was antagonized by exogenous expression of HES-1. It is plausible that HES-1 inhibits the activity of all mitogens that affect E2F-1 expression. In conclusion, the results from paper II indicate that HES-1 inhibits estrogen- and heregulin-β1-induced proliferation of T47D breast cancer cells by the repression of E2F-1 gene expression (Figure 17). 34
43 Figure 17. Conclusions from paper II. The estrogen-stimulated cell cycle progression through induction of E2F-1 is inhibited by retinoic acid-mediated HES-1 gene expression. PAPER III The aim of study III, was to elucidate how HES-1 is regulated mechanistically in response to estrogen and RA. From the results in paper I, we hypothesized that HES-1 is regulated transcriptionally in response to estrogen and RA, There are no consensus estrogen response elements (ERE) or RA response elements (RARE) in HES-1 proximal promoter region. There are several AP-1 sites, and AP-1 has been shown to be regulated both by estrogen and RA (68,182). By using 1.5 Kb of HES-1 proximal promoter region in reporter assays, we neither found response to estrogen nor to RA (unpublished data). A cis-acting element regulating HES-1 in response to RA in P19 cells has been found, although this element was not active in breast cancer cells (321) (unpublished data). Recently, it was found that EREs can be located at much further distances than first believed. Less than 5% of the EREs were shown to be located within promoter-proximal regions, and 20% within 100 kb upstream and 20% downstream of the gene (60,61). We found a consensus RARE 18 kb upstream of the HES-1 gene by in silico analysis. This element was bound by RARα in vitro and also active in reporter assays in response to RA. Results from a whole-genome ERα binding site mapping experiment showed strong binding of ERα 23 kb downstream from HES-1 transcriptional start site (61). This putative ERE was also found in a genome-wide experiment of ERα binding sites performed by others (60). Interestingly, the ERE consists of one consensus ERα binding site and one half-site with 28 bp in between them, as shown in reporter assays. Deleting one or the other site abolished the responsiveness to estrogen in HEK 293 cells. We found an induction of reporter activity in response to estrogen in HEK 293 cells, in contrast to a repression of HES-1 gene expression in breast cancer cells, as shown in paper I and II. This may be due to lack of cofactors or other proteins in HEK 293 cells, or the presence of a repressor element in the deleted 20 kb between HES-1 gene and the putative ERE that was not included in the reporter construct. By using recombinant human ERα in vitro binding assays, we found binding to the response element, whereas no binding when either the half site or the consensus ERα binding site was deleted. 35

44 Through chromatin immunoprecipitation (ChIP) assays, we were able to monitor recruitment of ERα and cofactors to the ERE. We found an association of ERα with the ERE 23 kb downstream of HES-1 gene in response to 45 min and 2 hours estrogen treatment, but not at 12 and 24 hours. This does not correlate with the downregulation of HES-1 expression, since estrogen affects HES-1 expression both at 12 and 24 hours but not as early as 45 min or 2 hours. This lack of concordance between ChIP data and expression data has been noted for other genes by others as well. We observed an association of ERα with HES-1 5 promoterproximal region, suggesting physical interaction between the ERE and HES-1 promoter. Sequential ChIPs showed strong recruitment of NCoR and SMRT corepressors and RIP140, although RIP140 dislocates quickly, to the ERα-bound ERE in response to estrogen. Interestingly, SRC-1 and SRC-3 coactivators are also strongly recruited. Importantly, regardless of the recruitment of both corepressors and coactivators, the outcome was shown by decreased acetylation of histone H3 and H4 and decreased recruitment of RNA POL II to HES-1 promoter in response to estrogen. In conclusion, RA and estrogen regulates HES-1 via cis-elements located 18 kb upstream and 20 kb downstream of the HES-1 gene, respectively (Figure 18). Figure 18. A model based on the results from paper 3. Cis-regulatory elements upstream and downstream of HES-1 gene regulate HES-1 gene expression in response to retinoic acid and estrogen, respectively. 36
.")
45 PAPER IV In this study, we investigated whether SOX9 is a regulator of HES-1 gene expression. This was suggested recently, in mice with a pancreas-specific inactivation of SOX9, as it was found that the number of HES-1 expressing pancreatic cells was strongly decreased (272). Interestingly, SOX9 is a target for retinoids and it has been shown that SOX9 has a role in the retinoid-mediated growth inhibition of T47D breast cancer cells. By inactivating endogenous SOX9 through expression of a dominant negative SOX9, these cells became unresponsive to RA (265). In study IV, we found that exogenous expression of SOX9 in MCF-7 cells enhanced endogenous HES-1 mrna and protein levels. SOX9 and HES-1 mrna and protein levels were increased in response to RA, and exogenous expression of dominant negative SOX9 abolished most of this increase. Both SOX9 and HES-1 mrna levels were increased within 1 hour treatment with RA, but the increase over 6 hours was less for HES-1 mrna compared to SOX9 mrna. This was probably due to the negative feedback that HES-1 exerts. We showed that SOX9 binds in vitro to a cis-element 3.7 kb upstream of HES-1 gene. This putative HES-1 enhancer contains a paired SOX9 binding site and it was shown to bind dimeric SOX9, since dimeric SOX9 migrates slower than monomeric SOX9 in gel-shift assay. Importantly, SOX9 binds to this element in vivo in response to RA. The activity of the HES-1 enhancer was increased when cotransfection with SOX9 expression construct was performed in reporter assays, indicating SOX9 is indeed inducing HES-1 gene transcription via the HES-1 enhancer. In addition, the activity of the HES-1 enhancer was increased in response to RA, which could be abolished by the expression of dominant negative SOX9. In conclusion, this data indicates that RA induces HES-1 gene expression through SOX9 (Figure 19). Figure 19. Conclusion from paper IV. The enhanced HES-1 expression in response to retinoic acid is mediated through SOX9. 37
46 CONCLUDING REMARKS HES-1 is the main focus of this thesis, primarily the effect of RA and estrogen on HES-1. The study of HES-1 regulation was associated with several difficulties, due to the short half life of HES-1 and the negative feedback loop that causes the oscillation pattern. For example, using non-synchronized cells, if half of the cell pool has high HES-1 expression and other half has low, the results will be no change of HES-1 expression for the whole cell pool. Using synchronized cells, on the other hand, the status of HES-1 expression could be dependent on where in the oscillation the cells are. On the positive side, HES-1 protein is rather small, stable and potent. Although the major aims for this thesis were satisfactorily fulfilled, some issues still remain. Two questions are: firstly, does HES-1 mediate all growth-inhibitory effects induced by RA, and does HES-1 mediate other, maybe all, effects by RA? By comparing known effects of RA with known effects by HES-1 in the literature this could potentially be answered. However, available RA data are contradictory, and the same thing is true for the HES-1 data. For instance, the effects of RA on cell cycle regulators in MCF-7 cells seem to vary; one study shows an effect on cyclin D1 expression whereas another study shows no effect on cyclin D1 expression but instead on CDK2 activity (196,197). The different results probably depend on the fact that cultured cancer cells have an abnormal karyotype and are undergoing continuous mutations resulting in different results from study to study. Secondly, we have found two pathways for RA to regulate HES-1, one direct and one via SOX9. Are these two pathways working synchronously or differently, and is one pathway dominant or are they synergistic? As shown in paper IV, inactivating endogenous SOX9 by the expression of dominant negative SOX9 abolishes most of the response to RA, but not all. One way to address this question may be ChIP assays, by treating cells with RA and investigating whether recruitment of SOX9 and/or RA receptor is associated with recruitment of RNA POL II and whether SOX9 binding site and/or RARE is physically associated with HES-1 proximal promoter. Lastly, as shown in paper III, in response to estrogen there is recruitment of both corepressors and coactivators resulting in repression of HES-1 expression. But what happens if we also add RA? Expression analyses show that, in vitro, RA antagonizes estrogen in regulating HES-1. ChIP analysis for recruitment of cofactors to RARα and ERα in response to estrogen and RA would probably answer some of the questions. A plausible scenario following recruitment of coactivators to the RARE is a dismissal of ER corepressors, followed by recruitment of RNA POLII to HES-1 proximal promoter and induction of HES-1 expression, in response to RA in estrogen-treated cells. An overall summary of the results presented in this thesis is shown in figures
47 Figure 20. A summary model of estrogen treatment on breast cancer cells involving HES-1. Estrogen induces estrogen receptor α binding to a cis-regulatory element downstream of HES-1 gene followed by a repression of HES-1 expression. In response to estrogen, there is an induction of E2F-1 gene expression, promoting cell cycle progression as the result. 39



48 Figure 21. A summary model of retinoic acid-treatment on breast cancer cells involving HES-1. Retinoic acid induces HES-1 gene expression, either through retinoic acid receptor binding to a RARE and/or through SOX9 binding to a HES-1 enhancer. Induced HES-1 protein binds to an N-box in E2F-1 promoter and turns off E2F1 gene expression, resulting in cell cycle arrest. 40
INTERACTION DRUG BODY
 INTERACTION DRUG BODY What the drug does to the body What the body does to the drug Receptors - intracellular receptors - membrane receptors - Channel receptors - G protein-coupled receptors - Tyrosine-kinase
INTERACTION DRUG BODY What the drug does to the body What the body does to the drug Receptors - intracellular receptors - membrane receptors - Channel receptors - G protein-coupled receptors - Tyrosine-kinase
Biochemistry 673 Lecture 2 Jason Kahn, UMCP Introduction to steroid hormone receptor (nuclear receptor) signalling
 signalling") Biochemistry 673 Lecture 2 Jason Kahn, UMCP Introduction to steroid hormone receptor (nuclear receptor) signalling Resources: Latchman Lodish chapter 10, 20 Helmreich, chapter 11 http://www.nursa.org,
Biochemistry 673 Lecture 2 Jason Kahn, UMCP Introduction to steroid hormone receptor (nuclear receptor) signalling Resources: Latchman Lodish chapter 10, 20 Helmreich, chapter 11 http://www.nursa.org,
ANTIPROLIFERATIVE ACTION OF ESTROGEN RECEPTOR β AND HES-1 IN BREAST CANCER
 From the Department of Biosciences and Nutrition Karolinska Institutet, Stockholm, Sweden ANTIPROLIFERATIVE ACTION OF ESTROGEN RECEPTOR β AND HES-1 IN BREAST CANCER Johan Hartman Stockholm 2008 All previously
From the Department of Biosciences and Nutrition Karolinska Institutet, Stockholm, Sweden ANTIPROLIFERATIVE ACTION OF ESTROGEN RECEPTOR β AND HES-1 IN BREAST CANCER Johan Hartman Stockholm 2008 All previously
Design, Synthesis and Biological Screening of Focused Libraries of New Antiestrogens
 Design, Synthesis and Biological Screening of Focused Libraries of New Antiestrogens Presenter: Jelena Janjic, Dipl.Pharm. Graduate student Pharmaceutical Sciences Prof. Dr. Peter Wipf s group Dr. Billy
Design, Synthesis and Biological Screening of Focused Libraries of New Antiestrogens Presenter: Jelena Janjic, Dipl.Pharm. Graduate student Pharmaceutical Sciences Prof. Dr. Peter Wipf s group Dr. Billy
RAS Genes. The ras superfamily of genes encodes small GTP binding proteins that are responsible for the regulation of many cellular processes.
 ۱ RAS Genes The ras superfamily of genes encodes small GTP binding proteins that are responsible for the regulation of many cellular processes. Oncogenic ras genes in human cells include H ras, N ras,
۱ RAS Genes The ras superfamily of genes encodes small GTP binding proteins that are responsible for the regulation of many cellular processes. Oncogenic ras genes in human cells include H ras, N ras,
SAMPLE REPORT. Order Number: PATIENT. Age: 40 Sex: F MRN:
 Patient: Age: 40 Sex: F MRN: SAMPLE PATIENT Order Number: Completed: Received: Collected: SAMPLE REPORT Progesterone ng/ml 0.34 0.95 21.00 DHEA-S mcg/dl Testosterone ng/ml 48 35 0.10 0.54 0.80 430 Sex
Patient: Age: 40 Sex: F MRN: SAMPLE PATIENT Order Number: Completed: Received: Collected: SAMPLE REPORT Progesterone ng/ml 0.34 0.95 21.00 DHEA-S mcg/dl Testosterone ng/ml 48 35 0.10 0.54 0.80 430 Sex
Estrogen (ER) receptor biology: Implications for HRT? Dr. Christoph A. Meier Endocrine Unit University Hospital Geneva
 receptor biology: Implications for HRT? Dr. Christoph A. Meier Endocrine Unit University Hospital Geneva") Estrogen (ER) receptor biology: Implications for HRT? Dr. Christoph A. Meier Endocrine Unit University Hospital Geneva Estrogens prevent postmenopausal bone-loss HRT & osteoporosis bip & estrogen bip
Estrogen (ER) receptor biology: Implications for HRT? Dr. Christoph A. Meier Endocrine Unit University Hospital Geneva Estrogens prevent postmenopausal bone-loss HRT & osteoporosis bip & estrogen bip
Hormone. Free Androgen Index. 2-Hydroxyestrone. Reference Range. Hormone. Estrone Ratio. Free Androgen Index
 Hormonal Health PATIENT: Sample Report TEST REF: TST-12345 Hormonal Health 0.61 0.30-1.13 ng/ml DHEA-S 91 35-430 mcg/dl tient: SAMPLE TIENT e: x: N: Sex Binding Globulin 80 18-114 nmol/l Testosterone 0.34
Hormonal Health PATIENT: Sample Report TEST REF: TST-12345 Hormonal Health 0.61 0.30-1.13 ng/ml DHEA-S 91 35-430 mcg/dl tient: SAMPLE TIENT e: x: N: Sex Binding Globulin 80 18-114 nmol/l Testosterone 0.34
1st Year MB/BDS Plenary Lecture What is a Hormone?
 1st Year MB/BDS Plenary Lecture What is a Hormone? The term hormone (from the Greek for I arouse to activity or I excite) was first used by Starling in 1905. Hormones are just one type of first (or primary)
1st Year MB/BDS Plenary Lecture What is a Hormone? The term hormone (from the Greek for I arouse to activity or I excite) was first used by Starling in 1905. Hormones are just one type of first (or primary)
Chemical Classification of Hormones
 Steroid Hormones Chemical Classification of Hormones Hormones are chemical messengers that transport signals from one cell to another There are 4 major chemical classes of hormones steroid hormones - i.e.
Steroid Hormones Chemical Classification of Hormones Hormones are chemical messengers that transport signals from one cell to another There are 4 major chemical classes of hormones steroid hormones - i.e.
Cancer. The fundamental defect is. unregulated cell division. Properties of Cancerous Cells. Causes of Cancer. Altered growth and proliferation
 Cancer The fundamental defect is unregulated cell division. Properties of Cancerous Cells Altered growth and proliferation Loss of growth factor dependence Loss of contact inhibition Immortalization Alterated
Cancer The fundamental defect is unregulated cell division. Properties of Cancerous Cells Altered growth and proliferation Loss of growth factor dependence Loss of contact inhibition Immortalization Alterated
Exploring the Genome-Wide Impact of Estrogen Receptor Alpha and Estrogen Receptor Beta in Breast and Colon Cancer Cells
 From the Department of Biosciences and Nutrition Karolinska Institutet, Stockholm, Sweden Exploring the Genome-Wide Impact of Estrogen Receptor Alpha and Estrogen Receptor Beta in Breast and Colon Cancer
From the Department of Biosciences and Nutrition Karolinska Institutet, Stockholm, Sweden Exploring the Genome-Wide Impact of Estrogen Receptor Alpha and Estrogen Receptor Beta in Breast and Colon Cancer
Crosstalk between Adiponectin and IGF-IR in breast cancer. Prof. Young Jin Suh Department of Surgery The Catholic University of Korea
 Crosstalk between Adiponectin and IGF-IR in breast cancer Prof. Young Jin Suh Department of Surgery The Catholic University of Korea Obesity Chronic, multifactorial disorder Hypertrophy and hyperplasia
Crosstalk between Adiponectin and IGF-IR in breast cancer Prof. Young Jin Suh Department of Surgery The Catholic University of Korea Obesity Chronic, multifactorial disorder Hypertrophy and hyperplasia
Diabetes Mellitus and Breast Cancer
 Masur K, Thévenod F, Zänker KS (eds): Diabetes and Cancer. Epidemiological Evidence and Molecular Links. Front Diabetes. Basel, Karger, 2008, vol 19, pp 97 113 Diabetes Mellitus and Breast Cancer Ido Wolf
Masur K, Thévenod F, Zänker KS (eds): Diabetes and Cancer. Epidemiological Evidence and Molecular Links. Front Diabetes. Basel, Karger, 2008, vol 19, pp 97 113 Diabetes Mellitus and Breast Cancer Ido Wolf
Cancer. The fundamental defect is. unregulated cell division. Properties of Cancerous Cells. Causes of Cancer. Altered growth and proliferation
 Cancer The fundamental defect is unregulated cell division. Properties of Cancerous Cells Altered growth and proliferation Loss of growth factor dependence Loss of contact inhibition Immortalization Alterated
Cancer The fundamental defect is unregulated cell division. Properties of Cancerous Cells Altered growth and proliferation Loss of growth factor dependence Loss of contact inhibition Immortalization Alterated
Estrogens and progestogens
 Estrogens and progestogens Estradiol and Progesterone hormones produced by the gonads are necessary for: conception embryonic maturation development of primary and secondary sexual characteristics at puberty.
Estrogens and progestogens Estradiol and Progesterone hormones produced by the gonads are necessary for: conception embryonic maturation development of primary and secondary sexual characteristics at puberty.
Cancer and Gene Alterations - 1
 Cancer and Gene Alterations - 1 Cancer and Gene Alteration As we know, cancer is a disease of unregulated cell growth. Although we looked at some of the features of cancer when we discussed mitosis checkpoints,
Cancer and Gene Alterations - 1 Cancer and Gene Alteration As we know, cancer is a disease of unregulated cell growth. Although we looked at some of the features of cancer when we discussed mitosis checkpoints,
CANCER = Malignant Tumor = Malignant Neoplasm
 CANCER = Malignant Tumor = Malignant Neoplasm A tissue growth: Not necessary for body s development or repair Invading healthy tissues Spreading to other sites of the body (metastasizing) Lethal because
CANCER = Malignant Tumor = Malignant Neoplasm A tissue growth: Not necessary for body s development or repair Invading healthy tissues Spreading to other sites of the body (metastasizing) Lethal because
HORMONES (Biomedical Importance)
") hormones HORMONES (Biomedical Importance) Hormones are the chemical messengers of the body. They are defined as organic substances secreted into blood stream to control the metabolic and biological activities.
hormones HORMONES (Biomedical Importance) Hormones are the chemical messengers of the body. They are defined as organic substances secreted into blood stream to control the metabolic and biological activities.
BIOSYNTHESIS OF STEROID HORMONES
 BIOSYNTHESIS OF STEROID HORMONES Sri Widia A Jusman Department of Biochemistry & Molecular Biology FMUI sw/steroidrepro/inter/08 1 STEROID HORMONES Progestins (21 C) Glucocorticoids (21 C) Mineralocorticoids
BIOSYNTHESIS OF STEROID HORMONES Sri Widia A Jusman Department of Biochemistry & Molecular Biology FMUI sw/steroidrepro/inter/08 1 STEROID HORMONES Progestins (21 C) Glucocorticoids (21 C) Mineralocorticoids
Ch 11: Endocrine System
 Ch 11: Endocrine System SLOs Describe the chemical nature of hormones and define the terms proand prepro-hormone. Explain mechanism of action of steroid and thyroid hormones Create chart to distinguish
Ch 11: Endocrine System SLOs Describe the chemical nature of hormones and define the terms proand prepro-hormone. Explain mechanism of action of steroid and thyroid hormones Create chart to distinguish
mirna Dr. S Hosseini-Asl
 mirna Dr. S Hosseini-Asl 1 2 MicroRNAs (mirnas) are small noncoding RNAs which enhance the cleavage or translational repression of specific mrna with recognition site(s) in the 3 - untranslated region
mirna Dr. S Hosseini-Asl 1 2 MicroRNAs (mirnas) are small noncoding RNAs which enhance the cleavage or translational repression of specific mrna with recognition site(s) in the 3 - untranslated region
Multistep nature of cancer development. Cancer genes
 Multistep nature of cancer development Phenotypic progression loss of control over cell growth/death (neoplasm) invasiveness (carcinoma) distal spread (metastatic tumor) Genetic progression multiple genetic
Multistep nature of cancer development Phenotypic progression loss of control over cell growth/death (neoplasm) invasiveness (carcinoma) distal spread (metastatic tumor) Genetic progression multiple genetic
Nuclear Receptors. Estrogen and Thyroid Hormone Receptors and their Interaction. Estrogen Hormone Receptor.
 SZENT ISTVÁN UNIVERSITY FACULTY OF VETERINARY MEDICINE Nuclear Receptors Estrogen and Thyroid Hormone Receptors and their Interaction Lior Kerner Budapest, 2012 What are Nuclear Receptors? o Proteins located
SZENT ISTVÁN UNIVERSITY FACULTY OF VETERINARY MEDICINE Nuclear Receptors Estrogen and Thyroid Hormone Receptors and their Interaction Lior Kerner Budapest, 2012 What are Nuclear Receptors? o Proteins located
Vitamins. Nafith Abu Tarboush, DDS, MSc, PhD
 Vitamins Nafith Abu Tarboush, DDS, MSc, PhD natarboush@ju.edu.jo www.facebook.com/natarboush Vitamins Organic compounds required by an organism in tiny amounts as a vital nutrient Cannot be synthesized
Vitamins Nafith Abu Tarboush, DDS, MSc, PhD natarboush@ju.edu.jo www.facebook.com/natarboush Vitamins Organic compounds required by an organism in tiny amounts as a vital nutrient Cannot be synthesized
Animal and Veterinary Science Department University of Idaho. REGULATION OF REPRODUCTION AVS 222 (Instructor: Dr. Amin Ahmadzadeh) Chapter 5
 Chapter 5") Animal and Veterinary Science Department University of Idaho REGULATION OF REPRODUCTION AVS 222 (Instructor: Dr. Amin Ahmadzadeh) Chapter 5 I. DEFINITIONS A. Endocrine Gland B. Hormone Chemical messenger
Animal and Veterinary Science Department University of Idaho REGULATION OF REPRODUCTION AVS 222 (Instructor: Dr. Amin Ahmadzadeh) Chapter 5 I. DEFINITIONS A. Endocrine Gland B. Hormone Chemical messenger
Cancer genetics
 Cancer genetics General information about tumorogenesis. Cancer induced by viruses. The role of somatic mutations in cancer production. Oncogenes and Tumor Suppressor Genes (TSG). Hereditary cancer. 1
Cancer genetics General information about tumorogenesis. Cancer induced by viruses. The role of somatic mutations in cancer production. Oncogenes and Tumor Suppressor Genes (TSG). Hereditary cancer. 1
Cell cycle, signaling to cell cycle, and molecular basis of oncogenesis
 Cell cycle, signaling to cell cycle, and molecular basis of oncogenesis MUDr. Jiří Vachtenheim, CSc. CELL CYCLE - SUMMARY Basic terminology: Cyclins conserved proteins with homologous regions; their cellular
Cell cycle, signaling to cell cycle, and molecular basis of oncogenesis MUDr. Jiří Vachtenheim, CSc. CELL CYCLE - SUMMARY Basic terminology: Cyclins conserved proteins with homologous regions; their cellular
Identification of influential proteins in the classical retinoic acid signaling pathway
 Ghaffari and Petzold Theoretical Biology and Medical Modelling (2018) 15:16 https://doi.org/10.1186/s12976-018-0088-7 RESEARCH Open Access Identification of influential proteins in the classical retinoic
Ghaffari and Petzold Theoretical Biology and Medical Modelling (2018) 15:16 https://doi.org/10.1186/s12976-018-0088-7 RESEARCH Open Access Identification of influential proteins in the classical retinoic
WEIGHT GAIN DURING MENOPAUSE EMERGING RESEARCH
 MENOPAUSE WHEN DOES IT OCCUR? The cessation of the menstrual cycle for one year. WEIGHT GAIN DURING MENOPAUSE EMERGING RESEARCH Jan Schroeder, Ph.D. Chair of The Department of Kinesiology California State
MENOPAUSE WHEN DOES IT OCCUR? The cessation of the menstrual cycle for one year. WEIGHT GAIN DURING MENOPAUSE EMERGING RESEARCH Jan Schroeder, Ph.D. Chair of The Department of Kinesiology California State
Chapter 15: Signal transduction
 Chapter 15: Signal transduction Know the terminology: Enzyme-linked receptor, G-protein linked receptor, nuclear hormone receptor, G-protein, adaptor protein, scaffolding protein, SH2 domain, MAPK, Ras,
Chapter 15: Signal transduction Know the terminology: Enzyme-linked receptor, G-protein linked receptor, nuclear hormone receptor, G-protein, adaptor protein, scaffolding protein, SH2 domain, MAPK, Ras,
Principles of Genetics and Molecular Biology
 Cell signaling Dr. Diala Abu-Hassan, DDS, PhD School of Medicine Dr.abuhassand@gmail.com Principles of Genetics and Molecular Biology www.cs.montana.edu Modes of cell signaling Direct interaction of a
Cell signaling Dr. Diala Abu-Hassan, DDS, PhD School of Medicine Dr.abuhassand@gmail.com Principles of Genetics and Molecular Biology www.cs.montana.edu Modes of cell signaling Direct interaction of a
Computational Biology I LSM5191
 Computational Biology I LSM5191 Aylwin Ng, D.Phil Lecture 6 Notes: Control Systems in Gene Expression Pulling it all together: coordinated control of transcriptional regulatory molecules Simple Control:
Computational Biology I LSM5191 Aylwin Ng, D.Phil Lecture 6 Notes: Control Systems in Gene Expression Pulling it all together: coordinated control of transcriptional regulatory molecules Simple Control:
Synthesis of sex steroids
 Synthesis of sex steroids CH 3 NAD(P)H NAD(P)+ Dehidroepiandroszteron Androszténdion 17- -hidroxiszteroid dehidrogenáz CH 3 dehydroepiandrosterone Dehidroepiandroszteron (DHEA) 3SDH Koleszterin CH 3 aromatase
Synthesis of sex steroids CH 3 NAD(P)H NAD(P)+ Dehidroepiandroszteron Androszténdion 17- -hidroxiszteroid dehidrogenáz CH 3 dehydroepiandrosterone Dehidroepiandroszteron (DHEA) 3SDH Koleszterin CH 3 aromatase
Oncogenes and Tumor Suppressors MCB 5068 November 12, 2013 Jason Weber
 Oncogenes and Tumor Suppressors MCB 5068 November 12, 2013 Jason Weber jweber@dom.wustl.edu Oncogenes & Cancer DNA Tumor Viruses Simian Virus 40 p300 prb p53 Large T Antigen Human Adenovirus p300 E1A
Oncogenes and Tumor Suppressors MCB 5068 November 12, 2013 Jason Weber jweber@dom.wustl.edu Oncogenes & Cancer DNA Tumor Viruses Simian Virus 40 p300 prb p53 Large T Antigen Human Adenovirus p300 E1A
CELL CYCLE MOLECULAR BASIS OF ONCOGENESIS
 CELL CYCLE MOLECULAR BASIS OF ONCOGENESIS Summary of the regulation of cyclin/cdk complexes during celll cycle Cell cycle phase Cyclin-cdk complex inhibitor activation Substrate(s) G1 Cyclin D/cdk 4,6
CELL CYCLE MOLECULAR BASIS OF ONCOGENESIS Summary of the regulation of cyclin/cdk complexes during celll cycle Cell cycle phase Cyclin-cdk complex inhibitor activation Substrate(s) G1 Cyclin D/cdk 4,6
In The Name Of God. In The Name Of. EMRI Modeling Group
 In The Name Of God In The Name Of God EMRI Modeling Group Cells work together in functionally related groups called tissues Types of tissues: Epithelial lining and covering Connective support Muscle movement
In The Name Of God In The Name Of God EMRI Modeling Group Cells work together in functionally related groups called tissues Types of tissues: Epithelial lining and covering Connective support Muscle movement
Molecular biology :- Cancer genetics lecture 11
 Molecular biology :- Cancer genetics lecture 11 -We have talked about 2 group of genes that is involved in cellular transformation : proto-oncogenes and tumour suppressor genes, and it isn t enough to
Molecular biology :- Cancer genetics lecture 11 -We have talked about 2 group of genes that is involved in cellular transformation : proto-oncogenes and tumour suppressor genes, and it isn t enough to
Reproductive physiology
 Reproductive physiology Sex hormones: Androgens Estrogens Gestagens Learning objectives 86 (also 90) Sex Genetic sex Gonadal sex Phenotypic sex XY - XX chromosomes testes - ovaries external features Tha
Reproductive physiology Sex hormones: Androgens Estrogens Gestagens Learning objectives 86 (also 90) Sex Genetic sex Gonadal sex Phenotypic sex XY - XX chromosomes testes - ovaries external features Tha
number Done by Corrected by Doctor Maha Shomaf
 number 19 Done by Waseem Abo-Obeida Corrected by Abdullah Zreiqat Doctor Maha Shomaf Carcinogenesis: the molecular basis of cancer. Non-lethal genetic damage lies at the heart of carcinogenesis and leads
number 19 Done by Waseem Abo-Obeida Corrected by Abdullah Zreiqat Doctor Maha Shomaf Carcinogenesis: the molecular basis of cancer. Non-lethal genetic damage lies at the heart of carcinogenesis and leads
G-Protein Signaling. Introduction to intracellular signaling. Dr. SARRAY Sameh, Ph.D
 G-Protein Signaling Introduction to intracellular signaling Dr. SARRAY Sameh, Ph.D Cell signaling Cells communicate via extracellular signaling molecules (Hormones, growth factors and neurotransmitters
G-Protein Signaling Introduction to intracellular signaling Dr. SARRAY Sameh, Ph.D Cell signaling Cells communicate via extracellular signaling molecules (Hormones, growth factors and neurotransmitters
Chapter 3. Selective Estrogen Receptor Modulator and Antagonist Induced Transrepression of NF-kB Activity Mediated by Estrogen Receptor a.
 Chapter 3 Selective Estrogen Receptor Modulator and Antagonist Induced Transrepression of NF-kB Activity Mediated by Estrogen Receptor a Submitted Chapter 3 Selective Estrogen Receptor Modulator and Antagonist
Chapter 3 Selective Estrogen Receptor Modulator and Antagonist Induced Transrepression of NF-kB Activity Mediated by Estrogen Receptor a Submitted Chapter 3 Selective Estrogen Receptor Modulator and Antagonist
Convergent and Divergent Mechanisms in Aging and Cancer
 Convergent and Divergent Mechanisms in Aging and Cancer Mariana S. De Lorenzo, PhD Department of Cell Biology & Molecular Medicine delorems@umdnj.edu LEARNING OBJECTIVES 1. To identify convergent and divergent
Convergent and Divergent Mechanisms in Aging and Cancer Mariana S. De Lorenzo, PhD Department of Cell Biology & Molecular Medicine delorems@umdnj.edu LEARNING OBJECTIVES 1. To identify convergent and divergent
Molecular Cell Biology - Problem Drill 19: Cell Signaling Pathways and Gene Expression
 Molecular Cell Biology - Problem Drill 19: Cell Signaling Pathways and Gene Expression Question No. 1 of 10 1. Which statement about cell signaling is correct? Question #1 (A) Cell signaling involves receiving
Molecular Cell Biology - Problem Drill 19: Cell Signaling Pathways and Gene Expression Question No. 1 of 10 1. Which statement about cell signaling is correct? Question #1 (A) Cell signaling involves receiving
Karyotype analysis reveals transloction of chromosome 22 to 9 in CML chronic myelogenous leukemia has fusion protein Bcr-Abl
 Chapt. 18 Cancer Molecular Biology of Cancer Student Learning Outcomes: Describe cancer diseases in which cells no longer respond Describe how cancers come from genomic mutations (inherited or somatic)
Chapt. 18 Cancer Molecular Biology of Cancer Student Learning Outcomes: Describe cancer diseases in which cells no longer respond Describe how cancers come from genomic mutations (inherited or somatic)
Genome of Hepatitis B Virus. VIRAL ONCOGENE Dr. Yahwardiah Siregar, PhD Dr. Sry Suryani Widjaja, Mkes Biochemistry Department
 Genome of Hepatitis B Virus VIRAL ONCOGENE Dr. Yahwardiah Siregar, PhD Dr. Sry Suryani Widjaja, Mkes Biochemistry Department Proto Oncogen and Oncogen Oncogen Proteins that possess the ability to cause
Genome of Hepatitis B Virus VIRAL ONCOGENE Dr. Yahwardiah Siregar, PhD Dr. Sry Suryani Widjaja, Mkes Biochemistry Department Proto Oncogen and Oncogen Oncogen Proteins that possess the ability to cause
Multimedia Appendix 6 Educational Materials Table of Contents. Intervention Educational Materials Audio Script (version 1)
") Multimedia Appendix 6 Educational Materials Table of Contents Intervention Educational Materials... 1 Audio Script (version 1)... 1 Text (version 1)... 5 Slides (version 1)... 17 Audio Script (version
Multimedia Appendix 6 Educational Materials Table of Contents Intervention Educational Materials... 1 Audio Script (version 1)... 1 Text (version 1)... 5 Slides (version 1)... 17 Audio Script (version
The Orphan Nuclear Receptors: Unrecognized Targets of Botanical Medicines?
 The Orphan Nuclear Receptors: Unrecognized Targets of Botanical Medicines? Kevin Spelman, PhD, MCPP These notes are designed as a primer for the lecture and do not necessarily represent lecture content.
The Orphan Nuclear Receptors: Unrecognized Targets of Botanical Medicines? Kevin Spelman, PhD, MCPP These notes are designed as a primer for the lecture and do not necessarily represent lecture content.
Signaling. Dr. Sujata Persad Katz Group Centre for Pharmacy & Health research
 Signaling Dr. Sujata Persad 3-020 Katz Group Centre for Pharmacy & Health research E-mail:sujata.persad@ualberta.ca 1 Growth Factor Receptors and Other Signaling Pathways What we will cover today: How
Signaling Dr. Sujata Persad 3-020 Katz Group Centre for Pharmacy & Health research E-mail:sujata.persad@ualberta.ca 1 Growth Factor Receptors and Other Signaling Pathways What we will cover today: How
REGULATORY MECHANISMS OF TRANSCRIPTION FACTOR FUNCTION
 Transcription Regulation And Gene Expression in Eukaryotes Cycle G2 (lecture 13709) RG Clerc 04.04.2012 REGULATORY MECHANISMS OF TRANSCRIPTION FACTOR FUNCTION Protein synthesized Protein phosphorylated
Transcription Regulation And Gene Expression in Eukaryotes Cycle G2 (lecture 13709) RG Clerc 04.04.2012 REGULATORY MECHANISMS OF TRANSCRIPTION FACTOR FUNCTION Protein synthesized Protein phosphorylated
VITAMINS-FAT SOLUBLE [LIPPINCOTT S ] Deeba S. Jairajpuri
![VITAMINS-FAT SOLUBLE [LIPPINCOTT S ] Deeba S. Jairajpuri](/thumbs/88/115610056.jpg "VITAMINS-FAT SOLUBLE [LIPPINCOTT S ] Deeba S. Jairajpuri") VITAMINS-FAT SOLUBLE [LIPPINCOTT S 381-394] Deeba S. Jairajpuri VITAMIN A othe term retinoids includes both natural and synthetic forms of vitamin A essential for vision, reproduction, growth and maintenance
VITAMINS-FAT SOLUBLE [LIPPINCOTT S 381-394] Deeba S. Jairajpuri VITAMIN A othe term retinoids includes both natural and synthetic forms of vitamin A essential for vision, reproduction, growth and maintenance
Genetics and Cancer Ch 20
 Genetics and Cancer Ch 20 Cancer is genetic Hereditary cancers Predisposition genes Ex. some forms of colon cancer Sporadic cancers ~90% of cancers Descendants of cancerous cells all cancerous (clonal)
Genetics and Cancer Ch 20 Cancer is genetic Hereditary cancers Predisposition genes Ex. some forms of colon cancer Sporadic cancers ~90% of cancers Descendants of cancerous cells all cancerous (clonal)
Receptors Functions and Signal Transduction- L4- L5
 Receptors Functions and Signal Transduction- L4- L5 Faisal I. Mohammed, MD, PhD University of Jordan 1 PKC Phosphorylates many substrates, can activate kinase pathway, gene regulation PLC- signaling pathway
Receptors Functions and Signal Transduction- L4- L5 Faisal I. Mohammed, MD, PhD University of Jordan 1 PKC Phosphorylates many substrates, can activate kinase pathway, gene regulation PLC- signaling pathway
Lecture #27 Lecturer A. N. Koval
 Lecture #27 Lecturer A. N. Koval Hormones Transduce Signals to Affect Homeostatic Mechanisms Koval A. (C), 2011 2 Lipophilic hormones Classifying hormones into hydrophilic and lipophilic molecules indicates
Lecture #27 Lecturer A. N. Koval Hormones Transduce Signals to Affect Homeostatic Mechanisms Koval A. (C), 2011 2 Lipophilic hormones Classifying hormones into hydrophilic and lipophilic molecules indicates
2015 AP Biology Unit #4 Quiz 1 Cell Communication, Cancer and The Cell Cycle Week of November
 Name: Class: Date: 2015 AP Biology Unit #4 Quiz 1 Cell Communication, Cancer and The Cell Cycle Week of 16-20 November Multiple Choice Identify the choice that best completes the statement or answers the
Name: Class: Date: 2015 AP Biology Unit #4 Quiz 1 Cell Communication, Cancer and The Cell Cycle Week of 16-20 November Multiple Choice Identify the choice that best completes the statement or answers the
The Endocrine System. I. Overview of the Endocrine System. II. Three Families of Hormones. III. Hormone Receptors. IV. Classes of Hormone Receptor
 The Endocrine System I. Overview of the Endocrine System A. Regulates long term metabolic processes B. Releases hormones from endocrine cells 1. Hormones are chemicals 2. Alter metabolism of cells 3. Release
The Endocrine System I. Overview of the Endocrine System A. Regulates long term metabolic processes B. Releases hormones from endocrine cells 1. Hormones are chemicals 2. Alter metabolism of cells 3. Release
By the name of Allah
 By the name of Allah Receptors function and signal transduction ( Hormones and receptors Types) We were talking about receptors of the neurotransmitters; we have 2 types of receptors: 1- Ionotropic receptors
By the name of Allah Receptors function and signal transduction ( Hormones and receptors Types) We were talking about receptors of the neurotransmitters; we have 2 types of receptors: 1- Ionotropic receptors
GENERAL CHARACTERISTICS OF THE ENDOCRINE SYSTEM FIGURE 17.1
 GENERAL CHARACTERISTICS OF THE ENDOCRINE SYSTEM FIGURE 17.1 1. The endocrine system consists of glands that secrete chemical signals, called hormones, into the blood. In addition, other organs and cells
GENERAL CHARACTERISTICS OF THE ENDOCRINE SYSTEM FIGURE 17.1 1. The endocrine system consists of glands that secrete chemical signals, called hormones, into the blood. In addition, other organs and cells
GENERAL SUMMARY Corpus luteum is a transient endocrine structure formed from the ruptured ovarian follicle. Its main function is to secrete P 4, a pro
 Corpus luteum is a transient endocrine structure formed from the ruptured ovarian follicle. Its main function is to secrete P 4, a pro-gestational hormone, essential for establishment and maintenance of
Corpus luteum is a transient endocrine structure formed from the ruptured ovarian follicle. Its main function is to secrete P 4, a pro-gestational hormone, essential for establishment and maintenance of
Cancer Biology How a cell responds to DNA Damage
 1 Cancer Biology How a cell responds to DNA Damage Jann Sarkaria Department of Oncology Mayo Clinic 2 EDUCATIONAL GOALS How proteins can transmit signals to each other. The definition of a tumor suppressor
1 Cancer Biology How a cell responds to DNA Damage Jann Sarkaria Department of Oncology Mayo Clinic 2 EDUCATIONAL GOALS How proteins can transmit signals to each other. The definition of a tumor suppressor
Cancer. October is National Breast Cancer Awareness Month
 Cancer October is National Breast Cancer Awareness Month Objectives 1: Gene regulation Explain how cells in all the different parts of your body develop such different characteristics and functions. Contrast
Cancer October is National Breast Cancer Awareness Month Objectives 1: Gene regulation Explain how cells in all the different parts of your body develop such different characteristics and functions. Contrast
Human Biochemistry. Hormones
 Human Biochemistry Hormones THE ENDOCRINE SYSTEM THE ENDOCRINE SYSTEM THE ENDOCRINE SYSTEM The ENDOCRINE SYSTEM = the organ system that regulates internal environment conditions by secreting hormones into
Human Biochemistry Hormones THE ENDOCRINE SYSTEM THE ENDOCRINE SYSTEM THE ENDOCRINE SYSTEM The ENDOCRINE SYSTEM = the organ system that regulates internal environment conditions by secreting hormones into
The Future of Cancer. Lawrence Tsui Global Risk Products Actuary Swiss Reinsurance Company Hong Kong. Session Number: WBR8
 Lawrence Tsui Global Risk Products Actuary Swiss Reinsurance Company Hong Kong Session Number: WBR8 Agenda Cancer the basics Cancer past and present Cancer the future CANCER THE BASICS Cancer the basics
Lawrence Tsui Global Risk Products Actuary Swiss Reinsurance Company Hong Kong Session Number: WBR8 Agenda Cancer the basics Cancer past and present Cancer the future CANCER THE BASICS Cancer the basics
One Day Hormone Check
 One Day Hormone Check DOB: Sex: F MRN: Order Number: Completed: Received: Collected: Salivary Hormone Results Estradiol pmol/l >3330.0 Testosterone pmol/l
One Day Hormone Check DOB: Sex: F MRN: Order Number: Completed: Received: Collected: Salivary Hormone Results Estradiol pmol/l >3330.0 Testosterone pmol/l
MECHANISM AND MODE OF HORMONE ACTION. Some definitions. Receptor: Properties of receptors. PRESENTED BY MBUNKUR GLORY NKOSI.
 MECHANISM AND MODE OF HORMONE ACTION. PRESENTED BY MBUNKUR GLORY NKOSI. OUTLINE. Introduction Some definitions Hormone secretion, transport, and clearance from the blood. Feedback control of hormone secretion.
MECHANISM AND MODE OF HORMONE ACTION. PRESENTED BY MBUNKUR GLORY NKOSI. OUTLINE. Introduction Some definitions Hormone secretion, transport, and clearance from the blood. Feedback control of hormone secretion.
Estrogen receptor co-regulators as prognostic and predictive markers of endocrine therapy in early breast cancer: the role of SMRT and p160 family
 Università degli Studi di Napoli Federico II Facoltà di Medicina e Chirurgia Dottorato in Morfologia Clinica e Patologica XXIII Ciclo 2007/2010 Coordinatore: Prof.ssa Stefania Montagnani Estrogen receptor
Università degli Studi di Napoli Federico II Facoltà di Medicina e Chirurgia Dottorato in Morfologia Clinica e Patologica XXIII Ciclo 2007/2010 Coordinatore: Prof.ssa Stefania Montagnani Estrogen receptor
Steroid Receptors in the Uterus and Ovary
 C H A P T E R 25 Steroid Receptors in the Uterus and Ovary April K. Binder, Wipawee Winuthayanon, Sylvia C. Hewitt Laboratory of Reproductive and Developmental Toxicology, National Institute of Environmental
C H A P T E R 25 Steroid Receptors in the Uterus and Ovary April K. Binder, Wipawee Winuthayanon, Sylvia C. Hewitt Laboratory of Reproductive and Developmental Toxicology, National Institute of Environmental
Mechanisms of Resistance to Endocrine Therapy in Breast Cancer: Focus on Signaling Pathways, mirnas and Genetically Based Resistance
 Int. J. Mol. Sci. 2013, 14, 108-145; doi:10.3390/ijms14010108 Review OPEN ACCESS International Journal of Molecular Sciences ISSN 1422-0067 www.mdpi.com/journal/ijms Mechanisms of Resistance to Endocrine
Int. J. Mol. Sci. 2013, 14, 108-145; doi:10.3390/ijms14010108 Review OPEN ACCESS International Journal of Molecular Sciences ISSN 1422-0067 www.mdpi.com/journal/ijms Mechanisms of Resistance to Endocrine
74. Hormone synthesis in the adrenal cortex. The glucocorticoids: biosynthesis, regulation, effects. Adrenal cortex is vital for life!
 74. Hormone synthesis in the adrenal cortex. The glucocorticoids: biosynthesis, regulation, effects. Adrenal cortex is vital for life! 5 g each Zona glomerulosa : Mineralocorticoids ALDOSTERON Zona fasciculata:
74. Hormone synthesis in the adrenal cortex. The glucocorticoids: biosynthesis, regulation, effects. Adrenal cortex is vital for life! 5 g each Zona glomerulosa : Mineralocorticoids ALDOSTERON Zona fasciculata:
BCHM3972 Human Molecular Cell Biology (Advanced) 2013 Course University of Sydney
 2013 Course University of Sydney") BCHM3972 Human Molecular Cell Biology (Advanced) 2013 Course University of Sydney Page 2: Immune Mechanisms & Molecular Biology of Host Defence (Prof Campbell) Page 45: Infection and Implications for Cell
BCHM3972 Human Molecular Cell Biology (Advanced) 2013 Course University of Sydney Page 2: Immune Mechanisms & Molecular Biology of Host Defence (Prof Campbell) Page 45: Infection and Implications for Cell
Hormone Balance - Female Report SAMPLE. result graph based on Luteal Phase. result graph based on Luteal Phase
 Patient Name: Patient DOB: Gender: Physician: Test Hormone Balance - Female Report SAMPLE Grote, Mary Jane Batch Number: B6437 2/16/1954 Accession Number: N52281 F Date Received: 2/3/2015 Any Lab Test
Patient Name: Patient DOB: Gender: Physician: Test Hormone Balance - Female Report SAMPLE Grote, Mary Jane Batch Number: B6437 2/16/1954 Accession Number: N52281 F Date Received: 2/3/2015 Any Lab Test
Regulation of P450. b. competitive-inhibitor but not substrate. c. non-competitive inhibition. 2. Covalent binding to heme or protein
 10. Inhibition of P450 Regulation of P450 a. competitive-2 chemicals metabolized by same isoform b. competitive-inhibitor but not substrate c. non-competitive inhibition 11. Induction of P450 1. Non-covalent
10. Inhibition of P450 Regulation of P450 a. competitive-2 chemicals metabolized by same isoform b. competitive-inhibitor but not substrate c. non-competitive inhibition 11. Induction of P450 1. Non-covalent
number Done by Corrected by Doctor مها شوماف
 number 15 Done by Ali Yaghi Corrected by Waseem Alhaj Doctor مها شوماف 1 P a g e Epidemiology Epidemiology is the study of the incidence of a disease. It can give us information about the possible causes
number 15 Done by Ali Yaghi Corrected by Waseem Alhaj Doctor مها شوماف 1 P a g e Epidemiology Epidemiology is the study of the incidence of a disease. It can give us information about the possible causes
Endocrine secretion cells secrete substances into the extracellular fluid
 Animal Hormones Concept 30.1 Hormones Are Chemical Messengers Endocrine secretion cells secrete substances into the extracellular fluid Exocrine secretion cells secrete substances into a duct or a body
Animal Hormones Concept 30.1 Hormones Are Chemical Messengers Endocrine secretion cells secrete substances into the extracellular fluid Exocrine secretion cells secrete substances into a duct or a body
Cell Signaling (part 1)
") 15 Cell Signaling (part 1) Introduction Bacteria and unicellular eukaryotes respond to environmental signals and to signaling molecules secreted by other cells for mating and other communication. In multicellular
15 Cell Signaling (part 1) Introduction Bacteria and unicellular eukaryotes respond to environmental signals and to signaling molecules secreted by other cells for mating and other communication. In multicellular
Biol403 MAP kinase signalling
 Biol403 MAP kinase signalling The mitogen activated protein kinase (MAPK) pathway is a signalling cascade activated by a diverse range of effectors. The cascade regulates many cellular activities including
Biol403 MAP kinase signalling The mitogen activated protein kinase (MAPK) pathway is a signalling cascade activated by a diverse range of effectors. The cascade regulates many cellular activities including
Reproductive Endocrinology
 Reproductive Endocrinology Reproductive Endocrinology Hypothalamic hormones Gonadotropin releasing hormone (GnRH) - stimulate release of FSH = follicle stimulating hormone LH = luteinizing hormone from
Reproductive Endocrinology Reproductive Endocrinology Hypothalamic hormones Gonadotropin releasing hormone (GnRH) - stimulate release of FSH = follicle stimulating hormone LH = luteinizing hormone from
AllinaHealthSystems 1
 Overview Biology and Introduction to the Genetics of Cancer Denise Jones, MS, CGC Certified Genetic Counselor Virginia Piper Cancer Service Line I. Our understanding of cancer the historical perspective
Overview Biology and Introduction to the Genetics of Cancer Denise Jones, MS, CGC Certified Genetic Counselor Virginia Piper Cancer Service Line I. Our understanding of cancer the historical perspective
Mechanisms of hormone drug resistance
 Mechanisms of hormone drug resistance Ljiljana Stamatović Institute for Oncology and Radiology of Serbia Tenth UMOS Conference, Belgrade, 16-17 th May 2015. Hormone receptor-positive breast cancer (HR+
Mechanisms of hormone drug resistance Ljiljana Stamatović Institute for Oncology and Radiology of Serbia Tenth UMOS Conference, Belgrade, 16-17 th May 2015. Hormone receptor-positive breast cancer (HR+
Eukaryotic transcription (III)
") Eukaryotic transcription (III) 1. Chromosome and chromatin structure Chromatin, chromatid, and chromosome chromatin Genomes exist as chromatins before or after cell division (interphase) but as chromatids
Eukaryotic transcription (III) 1. Chromosome and chromatin structure Chromatin, chromatid, and chromosome chromatin Genomes exist as chromatins before or after cell division (interphase) but as chromatids
Introduction. Cancer Biology. Tumor-suppressor genes. Proto-oncogenes. DNA stability genes. Mechanisms of carcinogenesis.
 Cancer Biology Chapter 18 Eric J. Hall., Amato Giaccia, Radiobiology for the Radiologist Introduction Tissue homeostasis depends on the regulated cell division and self-elimination (programmed cell death)
Cancer Biology Chapter 18 Eric J. Hall., Amato Giaccia, Radiobiology for the Radiologist Introduction Tissue homeostasis depends on the regulated cell division and self-elimination (programmed cell death)
Module J ENDOCRINE SYSTEM. Learning Outcome
 Module J ENDOCRINE SYSTEM Topic from HAPS Guidelines General functions of the endocrine system Chemical classification of hormones & mechanism of hormone actions at receptors. Control of hormone secretion
Module J ENDOCRINE SYSTEM Topic from HAPS Guidelines General functions of the endocrine system Chemical classification of hormones & mechanism of hormone actions at receptors. Control of hormone secretion
Cell Death and Cancer. SNC 2D Ms. Papaiconomou
 Cell Death and Cancer SNC 2D Ms. Papaiconomou How do cells die? Necrosis Death due to unexpected and accidental cell damage. This is an unregulated cell death. Causes: toxins, radiation, trauma, lack of
Cell Death and Cancer SNC 2D Ms. Papaiconomou How do cells die? Necrosis Death due to unexpected and accidental cell damage. This is an unregulated cell death. Causes: toxins, radiation, trauma, lack of
Natural Hormones Replacement An Evidence and Practice Based Approach
 Natural Hormones Replacement An Evidence and Practice Based Approach Andres Ruiz, PharmD, MSc, FACA President/Partner Stonegate Pharmacy PRESENTED BY THE AMERICAN COLLEGE OF APOTHECARIES 2830 SUMMER OAKS
Natural Hormones Replacement An Evidence and Practice Based Approach Andres Ruiz, PharmD, MSc, FACA President/Partner Stonegate Pharmacy PRESENTED BY THE AMERICAN COLLEGE OF APOTHECARIES 2830 SUMMER OAKS
Prof. R. V. Skibbens. Cell Cycle, Cell Division and Cancer (Part 2)
") Prof. R. V. Skibbens November 22, 2010 BIOS 10: BioScience in the 21 st Century Cell Cycle, Cell Division and Cancer (Part 2) Directionality - clocks go in only one direction G1 doesn t have replication-inducing
Prof. R. V. Skibbens November 22, 2010 BIOS 10: BioScience in the 21 st Century Cell Cycle, Cell Division and Cancer (Part 2) Directionality - clocks go in only one direction G1 doesn t have replication-inducing
Deregulation of signal transduction and cell cycle in Cancer
 Deregulation of signal transduction and cell cycle in Cancer Tuangporn Suthiphongchai, Ph.D. Department of Biochemistry Faculty of Science, Mahidol University Email: tuangporn.sut@mahidol.ac.th Room Pr324
Deregulation of signal transduction and cell cycle in Cancer Tuangporn Suthiphongchai, Ph.D. Department of Biochemistry Faculty of Science, Mahidol University Email: tuangporn.sut@mahidol.ac.th Room Pr324
Prof. R. V. Skibbens
 Prof. R. V. Skibbens December 2, 2011 BIOS 10: BioScience in the 21 st Century Cell Cycle, Cell Division and Cancer (Part 2) Directionality The Cell Cycle clock goes in only one direction S-phase cells
Prof. R. V. Skibbens December 2, 2011 BIOS 10: BioScience in the 21 st Century Cell Cycle, Cell Division and Cancer (Part 2) Directionality The Cell Cycle clock goes in only one direction S-phase cells
A Genetic Program for Embryonic Development
 Concept 18.4: A program of differential gene expression leads to the different cell types in a multicellular organism During embryonic development, a fertilized egg gives rise to many different cell types
Concept 18.4: A program of differential gene expression leads to the different cell types in a multicellular organism During embryonic development, a fertilized egg gives rise to many different cell types
Estrogens & Antiestrogens
 Estrogens & Antiestrogens Menstrual cycle... Changes and hormonal events Natural estrogens: Estadiol >> Estrone > Estriol Ineffective orally Synthesis: From cholesterol ; role of aromatase enzyme in converting
Estrogens & Antiestrogens Menstrual cycle... Changes and hormonal events Natural estrogens: Estadiol >> Estrone > Estriol Ineffective orally Synthesis: From cholesterol ; role of aromatase enzyme in converting
5. Summary of Data Reported and Evaluation
 168 IARC MONOGRAPHS VOLUME 91 5. Summary of Data Reported and Evaluation 5.1 Exposure data The first oral hormonal contraceptives that were found to inhibit both ovulation and implantation were developed
168 IARC MONOGRAPHS VOLUME 91 5. Summary of Data Reported and Evaluation 5.1 Exposure data The first oral hormonal contraceptives that were found to inhibit both ovulation and implantation were developed
7.012 Problem Set 6 Solutions
 Name Section 7.012 Problem Set 6 Solutions Question 1 The viral family Orthomyxoviridae contains the influenza A, B and C viruses. These viruses have a (-)ss RNA genome surrounded by a capsid composed
Name Section 7.012 Problem Set 6 Solutions Question 1 The viral family Orthomyxoviridae contains the influenza A, B and C viruses. These viruses have a (-)ss RNA genome surrounded by a capsid composed
Lecture 8 Neoplasia II. Dr. Nabila Hamdi MD, PhD
 Lecture 8 Neoplasia II Dr. Nabila Hamdi MD, PhD ILOs Understand the definition of neoplasia. List the classification of neoplasia. Describe the general characters of benign tumors. Understand the nomenclature
Lecture 8 Neoplasia II Dr. Nabila Hamdi MD, PhD ILOs Understand the definition of neoplasia. List the classification of neoplasia. Describe the general characters of benign tumors. Understand the nomenclature
Cellular Signaling Pathways. Signaling Overview
 Cellular Signaling Pathways Signaling Overview Signaling steps Synthesis and release of signaling molecules (ligands) by the signaling cell. Transport of the signal to the target cell Detection of the
Cellular Signaling Pathways Signaling Overview Signaling steps Synthesis and release of signaling molecules (ligands) by the signaling cell. Transport of the signal to the target cell Detection of the
Innovative Nuclear Receptor Modulation Medicine
 Innovative Nuclear Receptor Modulation Medicine Dale C Leitman, M.D., Ph.D. Isaac Cohen, O.M.D., Ph.D. October 2015 Corporate Mission Iaterion, Inc. is Developing Novel Pharmaceutical Nuclear Receptor
Innovative Nuclear Receptor Modulation Medicine Dale C Leitman, M.D., Ph.D. Isaac Cohen, O.M.D., Ph.D. October 2015 Corporate Mission Iaterion, Inc. is Developing Novel Pharmaceutical Nuclear Receptor
VIII Curso Internacional del PIRRECV. Some molecular mechanisms of cancer
 VIII Curso Internacional del PIRRECV Some molecular mechanisms of cancer Laboratorio de Comunicaciones Celulares, Centro FONDAP Estudios Moleculares de la Celula (CEMC), ICBM, Facultad de Medicina, Universidad
VIII Curso Internacional del PIRRECV Some molecular mechanisms of cancer Laboratorio de Comunicaciones Celulares, Centro FONDAP Estudios Moleculares de la Celula (CEMC), ICBM, Facultad de Medicina, Universidad
Part II The Cell Cell Division, Chapter 2 Outline of class notes
 Part II The Cell Cell Division, Chapter 2 Outline of class notes 1 Cellular Division Overview Types of Cell Division Chromosomal Number The Cell Cycle Mitoses Cancer Cells In Vitro Fertilization Infertility
Part II The Cell Cell Division, Chapter 2 Outline of class notes 1 Cellular Division Overview Types of Cell Division Chromosomal Number The Cell Cycle Mitoses Cancer Cells In Vitro Fertilization Infertility
General Biology 1004 Chapter 11 Lecture Handout, Summer 2005 Dr. Frisby
 Slide 1 CHAPTER 11 Gene Regulation PowerPoint Lecture Slides for Essential Biology, Second Edition & Essential Biology with Physiology Presentation prepared by Chris C. Romero Neil Campbell, Jane Reece,
Slide 1 CHAPTER 11 Gene Regulation PowerPoint Lecture Slides for Essential Biology, Second Edition & Essential Biology with Physiology Presentation prepared by Chris C. Romero Neil Campbell, Jane Reece,
Chapter 20. Cell - Cell Signaling: Hormones and Receptors. Three general types of extracellular signaling. endocrine signaling. paracrine signaling
 Chapter 20 Cell - Cell Signaling: Hormones and Receptors Three general types of extracellular signaling endocrine signaling paracrine signaling autocrine signaling Endocrine Signaling - signaling molecules
Chapter 20 Cell - Cell Signaling: Hormones and Receptors Three general types of extracellular signaling endocrine signaling paracrine signaling autocrine signaling Endocrine Signaling - signaling molecules
Cell Signaling part 2
 15 Cell Signaling part 2 Functions of Cell Surface Receptors Other cell surface receptors are directly linked to intracellular enzymes. The largest family of these is the receptor protein tyrosine kinases,
15 Cell Signaling part 2 Functions of Cell Surface Receptors Other cell surface receptors are directly linked to intracellular enzymes. The largest family of these is the receptor protein tyrosine kinases,
Polyomaviridae. Spring
 Polyomaviridae Spring 2002 331 Antibody Prevalence for BK & JC Viruses Spring 2002 332 Polyoma Viruses General characteristics Papovaviridae: PA - papilloma; PO - polyoma; VA - vacuolating agent a. 45nm
Polyomaviridae Spring 2002 331 Antibody Prevalence for BK & JC Viruses Spring 2002 332 Polyoma Viruses General characteristics Papovaviridae: PA - papilloma; PO - polyoma; VA - vacuolating agent a. 45nm