FDA Advisory Committee Briefing Document. Medical Imaging Drugs Advisory Committee. Meeting to be held on February 14, 2013
|
|
|
- Cora Fitzgerald
- 5 years ago
- Views:
Transcription
1 FDA Advisory Committee Briefing Document Medical Imaging Drugs Advisory Committee Meeting to be held on February 14, 2013 New Drug Application Gadoterate meglumine Injection (Dotarem ), sponsored by Guerbet LLC This document was prepared on January 16, 2013 Table of Contents Page 1. Introduction 2 2. Draft Topics for Discussion 4 3. Regulatory Background 5 4. Pediatric Data Concerns Main Efficacy and Safety Data Summary Medical Officer s Draft Mid-Cycle Review slides Pharmacovigilance Review Review of Non-clinical Juvenile Animal Study Protocol Clinical Pharmacology Reviewer Notes Nonclinical Pharmacology/Toxicology Reviewer Notes 93 This document contains background information prepared by the Food and Drug Administration (FDA) for the panel members of the advisory committee. The FDA background package often contains assessments and/or conclusions and recommendations written by individual reviewers. Such conclusions and recommendations do not necessarily represent the final position of the individual reviewers, nor do they necessarily represent the final position of the Review Division or Office. Discussions by the committee as well as subsequent data submissions by the applicant may change all draft/preliminary review observations. We have brought this issue to this advisory committee in order to gain the committee s insights and opinions, and the background package may not include all issues relevant to the final regulatory recommendation and instead is intended to focus on issues identified by the Agency for discussion at the advisory committee. The FDA will not issue a final determination on the issues at hand until input from the advisory committee process has been considered and all reviews have been finalized. The final determination may be affected by issues not discussed at the advisory committee meeting.
2 1. Introduction The Medical Imaging Drugs Advisory Committee will meet to discuss the New Drug Application for gadoterate meglumine (Dotarem), a gadolinium-based contrast agent (GBCA) proposed for marketing in the United States with the following indication: for intravenous use with magnetic resonance imaging (MRI) in brain (intracranial), spine and associated tissues in adults and pediatric patients (from neonates to 17 years of age) to detect and visualize areas with disruption of the blood brain barrier (BBB) and/or abnormal vascularity. The gadoterate marketing application is brought for advisory committee discussion because of the proposed pediatric claim. If approved, gadoterate meglumine (referred to as gadoterate in this document) would be the ninth FDA-approved GBCA for magnetic resonance imaging (MRI) and the seventh agent approved specifically for imaging of the central nervous system (CNS) in adults; five GBCAs currently are approved for CNS imaging in certain pediatric patients. The proposed indication population for gadoterate importantly differs from all other GBCAs. The gadoterate population includes, pediatric patients (from neonates to 17 years of age). No GBCA is currently approved for use in pediatric patients less than two years of age for any indication. The NDA applicant s main phase 3 trial (050) achieved all primary endpoint objectives and the findings of the main supportive clinical study (051) are consistent with this demonstration of efficacy in adults. All data explorations of the main phase 3 study and the supportive study have, to date, consistently demonstrated success upon the primary endpoints. Safety findings within the adult population do not reveal any unique concerns. On the whole, the clinical data appear conducive to the ready construct of a risk-benefit perspective for use of gadoterate in adult patients. Forming a concise, over-all perspective on the pediatric experience appears more difficult, especially for patients less than two years of age. The pediatric clinical trial database supporting gadoterate use in pediatric patients is mainly derived from patients two year of age or greater (n = 130); only seven patients were less than two years of age. Three single site clinical trials of pediatric patients reported no safety concerns and improved anatomical visualization on contrasted MR images was reported by all the site investigators. The main phase 3 trial (050) included a subset of 38 pediatric patients (all greater than two years of age) who were not considered in the trial s primary endpoint analyses. No notable safety concerns were reported in this trial s pediatric subset population; improved anatomical visualization on contrasted MR ( paired ) images was demonstrated for the pediatric patients in a pattern similar to that observed in adults. Overall, the main limitations in the pediatric experience relate to an incomplete juvenile animal nonclinical study that FDA has requested, no pharmacokinetic data in pediatric patients and a very small clinical trial patient sample size within the subset of patients less than two years of age. Page 2 of 95
3 In addition to clinical trial data within the NDA, the applicant summarizes the post-marketing pharmacovigilance experience with gadoterate. Gadoterate has been marketed outside the United States for many years and the applicant estimates that more than 30 million people have been exposed to the drug. The applicant also estimates that approximately 52,000 pediatric patients aged less than two years have received gadoterate in the post-marketing experience outside the United States. The applicant s important pediatric use claim prompted the review division to assign a priority review timeline for this NDA. This review is ongoing and the committee needs to meet at a time point in advance of the completion due date for FDA inspectional findings as well as all primary review team documents. The FDA review team s preliminary observations are presented here as a summary of the strengths and weaknesses of the NDA data. The committee s perspective upon the summarized information is anticipated to importantly inform the FDA review team s on-going analysis and verification of the NDA data. Page 3 of 95
4 2. Draft Topics for Discussion The FDA review team requests the committee members to consider the data presented by the company in their briefing document, the strengths and limitations of these data as outlined in this summary document, the meeting day presentations and then comment upon the following: The sufficiency of the described data to support the market approval of gadoterate for the proposed CNS imaging in one or more of the following patient populations: o Adults o Pediatric patients aged two years o Pediatric patients of all ages (including patients less than two years of age) The types of data essential to assess the safety and efficacy of a GBCA when proposed for use in pediatric patients less than two years of age. In this document, we will generally refer to this population as infants and neonates. The important clinical considerations when MR imaging must be performed among infants and neonates, including the role for administration of a GBCA. The review team particularly desires to obtain comments from the radiologic experts on this committee regarding the practical considerations for MR imaging of infants and neonates such as individual and/or clinical site experience with imaging of these patients the risks and benefits. These considerations may prove useful in weighing the risks associated with a GBCA in infants and neonates due to renal immaturity that may cause delayed elimination of the GBCA as well as any potential developmental risks associated with the presence of the GBCA within the body at a time of critical organ development. The FDA team recognizes that many of the safety risks associated with MR imaging in neonates and infants may include procedural risks, such as the potential need for restraint, sedation and close monitoring to safely complete the imaging. Conceivably other important clinical matters may impact the risk-benefit consideration for MR imaging, in addition to the use of a GBCA among these patients. To support marketing of a GBCA for use among infants and neonates, FDA has, to date, largely emphasized the need for nonclinical data, PK information as well as pilot clinical safety and efficacy data. We encourage the advisors to consider other types of data that may be essential to support this use, such as the potential requirement for post-marketing registry studies that evaluate GBCA-exposed infants for a certain number of years in order to help detect developmental risks and other delayed reactions to the agent. Page 4 of 95
5 3. Regulatory Background GBCAs are MRI contrast agents used to improve the visualization of body structures or vasculature. To date, FDA has approved eight GBCAs (six for a CNS indication). The agents contain gadolinium, a paramagnetic metal which must remain chelated within the agent to avoid toxic effects from any unchelated, free gadolinium. FDA approved the first GBCA for use in MR imaging in The most recently approved GBCA, gadobutrol (Gadavist) was approved for CNS imaging in These agents are listed below with a column identifying some of the chemical structure features commonly used to categorize these drugs. Table 1. Currently Approved GBCAs and Gadoterate Trade Dose, adult Chemical Established name Indication Molar name mmol/kg structure Magnevist gadopentetate dimeglumine CNS, body Linear, ionic Prohance gadoteridol CNS Macrocyclic Omniscan gadodiamide CNS, body Linear, nonionic Optimark gadoversetamide CNS, liver Linear, nonionic Multihance gadobenate dimeglumine CNS, Renal and Aorto-Ileo- Femoral vessels Linear, ionic Eovist gadoxetate Liver Linear, ionic Ablavar gadofosveset Aorto-iliac vessels Linear, ionic Gadavist gadobutrol CNS Macrocyclic Dotarem Gadoterate meglumine CNS Macrocyclic a. Evolving Safety Concerns: When the first few GBCAs were approved, the agents were generally regarded as having very few clinical risks, particularly relative to the risks associated with the use of certain iodinated contrast agents in radiography. Clinical studies of all of these agents safety focused upon the detection of adverse reactions shortly following administration of the agents. It was only after the exposure of many millions of patients to GBCAs in the post-marketing experience, that a delayed risk for nephrogenic systemic fibrosis (NSF) was recognized. This experience underscores the potential for delayed reactions to GBCAs, an especially important consideration when a GBCA may be administered to a neonate/infant who may be at heightened risk for developmental problems or other toxicity related to the agent. Page 5 of 95
6 NSF, a scleroderma-like disease has been associated with the use of GBCAs predominantly among patients with severe renal insufficiency. NSF produces characteristic skin lesions and a fibrotic process within multiple body organs which may result in death. There is no generally accepted treatment or cure. Following FDA requests in 2007, manufacturers of marketed GBCAs revised their labels to include a boxed warning and other information intended to lessen the risk for NSF. The approved labeling text was applied to all the agents since FDA recognized some magnitude of risk for all the agents; the text explained the potential variation in the magnitude of the risk among the agents. Specifically, the text noted that "The extent of risk for NSF following exposure to any specific gadolinium-based contrast agent is unknown and may vary among the agents." Updated information, including data accumulated following the 2007 labeling alteration, was reviewed at an Advisory Committee in December, Based upon these data and the committee s advice, FDA has approved revisions of GBCA labels to distinguish two major subsets of GBCAs: a higher risk group that is contraindicated for use among the highest risk patient population (severe chronic renal failure or acute kidney injury) and a lower risk group that lacks this contraindication. This labeling change emphasizes some magnitude of NSF risk for all the GBCAs in the vulnerable population (especially patients with severe, chronic kidney disease or acute kidney injury, the highest risk population). Although the GBCA are viewed as a "class" based upon the same pharmacologic mechanism of action, the agents uniquely differ in multiple aspects (e.g., pharmacokinetics, pharmacodynamics, chemical structure, chelate-ion binding characteristics, etc). In this regard, FDA-approved labeling based upon a "GBCA class effect" does not mean that all GBCAs have identical risks and benefits nor does it mean that the magnitude of any individual risk (e.g., NSF) is the same for all members of the class. Instead, the NSF "class" risk indicates that the potential for the risk exists among all members of the drug class. Beyond the concept of a pharmacologic class, GBCAs have been categorized in various ways, e.g, by their molecular structure, thermodynamic stability, indication, etc. Categorizations are probably most often based upon the drugs' molecular structures and propensity for release of gadolinium. Based on molecular structure, three groups have been identified: linear, non-ionic; linear, ionic; and macrocyclic. Various in vivo and in vitro studies have strongly suggested that molecular structural features and gadolinium-binding properties help support the theoretical role of "transmetallation" in the pathogenesis of NSF. Transmetallation is a process that involves release of gadolinium following its dislocation from the chelation binding site and replacement by another metallic ion. Data appear to indicate that the "linear, non-ionic" agents, more so than the other molecular groups, tend to liberate gadolinium leading to free gadolinium in contact with body organs. The "linear ionic" agents are reported to have an intermediate propensity for liberation; the macrocyclic agents reportedly cage gadolinium and have the least propensity for transmetallation. The risk for NSF has been proposed to vary based upon the specific structural/transmetallation category (highest risk for linear, non-ionic and lowest risk for Page 6 of 95
7 macrocyclic). Gadoterate s chemical structure makes it a member of the class of macrocyclic agents and its physico-chemical stability measurements (as described in publications) suggest a relatively low propensity for the release of gadolinium. Most GBCAs are eliminated from the body via renal excretion. Similarly, most of an administered GBCA is eliminated from the body within 48 hours of administration of the agent to a subject with normal renal function. However, whether this elimination process completely removes the GBCA (or free gadolinium) from the body is unkown. In patients with renal insufficiency, a GBCA may readily remain within the body for a prolonged period of time, increasing the risk for NSF. Conceivably other patient-specific characteristics, such as concomitant medications, may impact the risk for NSF. These observations exemplify the many factors important to consider when estimating the risk for development of NSF. Additionally, data suggest that the dose of a GBCA (either as a single administration or the cumulative dose from repetitive administrations) represents an important consideration in the risk for NSF. Hence, the specific GBCA alone may not represent the single most important risk factor for NSF. Other considerations (dose, underlying renal function) appear to importantly impact the risk. In adults age does not appear to contribute to the risk of NSF. While relatively few cases of NSF have been reported in pediatric patients it is not known if the risk of NSF differs in pediatric patients compared to adults. To date, we have received no reports of NSF in infants; nevertheless, immature renal function in this subgroup (e.g. newborn, pre-term) raises concerns about increased risk for the condition. The FDA s approval of revised GBCA class labeling identified three agents (Omniscan, Optimark and Magnevist) as having higher risk for NSF, in comparison to other approved agents. This assessment was based upon post-marketing reports of NSF and considerations of in vitro dissociation properties as well as animal modeling results. Table 2 is excerpted from the December 2009 briefing materials to illustrate the number of NSF reports for the agents that were approved prior to Two agents (Ablavar, Eovist) were approved in 2008 and have not been cited in NSF reports. Table 2: Crude numbers of domestic reports of NSF for each GBCA* Product Year of FDA Approval Domestic NSF Reports in Domestic Single Agent NSF Reports in AERS** AERS Omniscan Magnevist Optimark Prohance *** Multihance *crude counts are generated from search terms and are not screened for duplicated submissions or submissions of unclear relevance. They do not take into account relative use of the products. **Adverse Event Reporting System (AERS) reports listing a single gadolinium agent. Reports with more than one gadolinium agent or unspecified gadolinium agents were excluded. ***although there are no domestic single-agent NSF reports for Prohance, AERS contains one foreign report of NSF in which a patient received only Prohance. Page 7 of 95
8 During the review of gadobutrol (Gadavist), FDA recognized two reports of NSF for the agent and noted, Considering the applicant s estimate of approximately five million gadobutrol patient exposures in the non-usa postmarketing experience with two single agent reports as well as the animal modeling data (see attachment) and in vitro dissociation data, the review team s preliminary findings suggest gadobutrol has an NSF risk similar to lower NSF risk agents (an excerpt from advisory committee briefing material). The current NDA applicant reports that, to date, no cases of NSF have unequivocally been attributed to gadoterate. This feature, in addition to the drug s putatively strong gadoliniumchelating strength, appears to place it also with an NSF risk similar to that of lower NSF risk agents. Some publications have suggested that the gadolinium-binding strength of gadoterate so far exceeds that of other tested agents, it may pose the least risk for free gadolinium release in the body. The NDA applicant has not provided source data from laboratory tests that evaluate the strength of gadolinium chelation by gadoterate in direct (same testing procedure) comparison to other agents; instead, published reports were provided in the NDA; hence, FDA cannot verify the accuracy of these published test results. b. FDA Advice Regarding GBCA Development for Neonates and Infants: On the next page, the nature of the clinical data supporting the pediatric use of GBCAs with CNS indications are shown in the table, based upon excerpts from approved labeling; also shown is the similar information for the gadoterate proposal. The first five agents shown in the table are the only drugs with approved pediatric claims and all these claims pertain to patients aged two years or more. The GBCA dose for all approved CNS pediatric indications is 0.1 mmol/kg (gadodiamide recommends a dose of 0.05 mmol/kg for renal imaging) and the proposed gadoterate dose is also 0.1 mmol/kg Page 8 of 95
9 Table 3. Approved GBCAs with Pediatric Labeling and Gadoterate Proposal Agent Pediatric Ages, Pediatric labeling Year approved Indication years Gadopentetate -Phase 3 trial included children & adults; showed dimeglumine improved lesion visualization in both populations (Magnevist) -No pediatric PK data but labeling notes, In a 1988 CNS/Head study with pediatric patients aged 2 months to < 2-2 years, the PK (body weight normalized clearance, neck/body etc) of gadopentetate were similar to adults. Gadoteridol (Prohance) 1992 Gadodiamide (Omniscan) 1993 CNS > 2 CNS/body 2 Gadobenate dimeglumine (Multihance) 2004 CNS 2 Gadobutrol (Gadavist) 2011 Gadoterate meglumine (Dotarem) CNS 2 CNS All -Pediatric safety database not detailed (combined with adult trial information) -Phase 3 trial in 103 children showed improved lesion visualization -No pediatric PK data -Pediatric safety database not detailed (combined with adult trial information) -Phase 3 trial in 97 children showed improved diagnostic information -No pediatric PK data -Pediatric safety database consisted of 97 children in phase 3 trial plus 144 children in published reports -Phase 3 trial in 92 children showed improved lesion visualization -PK data from 40 children showed elimination halflives similar to adults -Pediatric safety database consisted of 217 children in trials -No phase 3 trial data from children; efficacy inferred based on PK data -PK data from 130 children showed elimination half-lives similar to adults -Pediatric safety database consisted of 138 children in trials -Phase 3 trial included 37 patients aged 2 years or more; efficacy shown in the pediatric subset -No PK data -Pediatric safety database consists of 137 patients, including 7 < 2 years of age PK = pharmacokinetic Page 9 of 95
10 Since the recognition of the risk for GBCA-associated NSF, the FDA review team has expected at least three key sources of data to support GBCA use among neonates and infants: nonclinical information, and PK data as well as pilot clinical safety/efficacy data. When manufacturers have inquired about the data necessary to support use of a GBCA among neonates and infants, FDA has requested information from a juvenile animal nonclinical study (to help estimate toxicity risks), PK data (to help support the appropriateness of the proposed pediatric dosing as well as to provide labeling information related to the clearance of the drug as is essential to estimate the timing of any GBCA re-administration) in addition to pilot clinical safety and efficacy data. FDA has not requested clinical studies powered sufficiently for hypothesis testing of either safety or efficacy endpoints in pediatric patient populations. In considering the nature of data necessary to support pediatric labeling, FDA guidance has explicitly stated that the agency does not expect NDA applicants to, conduct separate safety and effectiveness studies in pediatric patients in every case. The Pediatric Research Equity Act (PREA) specifically notes that: If the course of the disease and the effects of the drug are sufficiently similar in adults and pediatric patients, the Secretary may conclude that pediatric effectiveness can be extrapolated from adequate and well-controlled studies in adults, usually supplemented with other information obtained in pediatric patients, such as pharmacokinetics studies. Action upon this ability to extrapolate information from adults is exemplified by FDA s review of the gadobutrol NDA, a situation where the applicant s phase 3 clinical trials did not include pediatric patients; however, the applicant provided PK data from 130 pediatric patients (aged two years or more). These PK data showed that, following administration of the drug to pediatric patients, the drug was distributed and eliminated in a manner similar to adults. Consequently, the FDA approved gadobutrol for use in both adults and pediatric patients aged two years or more. Importantly, the gadobutrol approval was accompanied with a post-marketing requirement to conduct a nonclinical juvenile animal study and a clinical study to be conducted among at least 40 pediatric subjects who were less than two years of age. Page 10 of 95
11 4. Pediatric Data Concerns In considering the pediatric information within the gadoterate NDA, the FDA review team highlights the following top-level pediatric data limitations: The application does not include data from the FDA-required juvenile animal nonclinical study that is intended to help assess the magnitude of risk for gadoterate toxicity, such as developmental problems and fibrosis-related toxicity. The applicant has initiated this juvenile animal study and FDA has been informed that the final study report will not be provided before the NDA review must be completed. The FDA recently completed a review of the non-clinical protocol (attached as an appendix to this document). The application lacks PK data from pediatric patients of any age. The supplied PK data were obtained in either healthy adult subjects or adult patients (including some patients with renal impairment). We encourage MIDAC members to consider the extent to which, if any, the PK data from adult patients with renal impairment can be extrapolated to neonates and infants. Marked limitations within the quality/thoroughness of the pediatric data. Of the 137 pediatric subjects in clinical trials, only 7 were aged less than two years. To exemplify an efficacy concern, the applicant supplies reports from three clinical trials that were conducted specifically in children; within each of the trials, the efficacy outcome was based solely upon a site investigator s impression of the imaging result (generally a comparison of a pre-contrast image to a post-contrast image), a design that appears highly conducive to bias. Regarding safety, less than half the pediatric subjects in clinical trials had laboratory evaluations (such as measures of renal function). Overall, the applicant s pediatric database consists of: 1) 137 pediatric patients enrolled in four clinical trials described in final trial reports. These trials consist of the Trial 050, the main phase 3 clinical trial as well as three single site, exploratory clinical trials conducted solely among pediatric patients (Trials 3-15, 3-16 and 3-29). The data are summarized in the Pediatric Clinical Trial Data section below. Only one trial (050) had any adverse reactions reported to gadoterate. 2) 2496 pediatric patients exposed to gadoterate in studies that the applicant calls postmarketing surveillance studies. This information is supplied in a very limited form in the NDA (for example, solely a published abstract for one study). The information is summarized in the Pediatric Post-marketing Surveillance Studies section below. Few or no adverse reactions to gadoterate were reported in this experience. 3) Within the spontaneous pharmacovigilance reporting experience, eight patients less than two years of age had adverse reactions; no fatalities were reported. The most frequently reported reporting terms were overdose (3 patients, including one described as serious ) followed by extravasation (2 patients). The only other serious event was a Page 11 of 95
12 report of a transient decrease in heart rate following gadoterate administration. 4) The sponsor also makes a claim that approximately 52,000 children less than two years of age were exposed to gadoterate worldwide between 2005 and The number is an extrapolation from one year medical services utilization data in France not directly linked to utilization of this specific drug in this age group and cannot be otherwise confirmed. a. Pediatric Clinical Trial Data Table 4. Summary of Pediatric Patients Exposed to Dotarem in Reported Clinical Trials Number of patients, aged: Trial 050 Trial 3-15 Trial 3-16 Trial 3-29 Total > 10 years through 10 years < 2 years Total Regarding efficacy, the three single site clinical trials all reported improved CNS visualization when gadoterate-contrasted images were compared to pre contrast MR images. However, these trials were all open-label and MR images were interpreted by a single reader at each clinical site a process highly conducive to bias. The subset of patients within the phase 3 trial 050 was reported to show improved image visualization in a pattern similar to that observed in adults (the primary endpoint population), as summarized later in this document. Regarding safety, within the full database of 137 pediatric patients in four clinical trials, seven patients were aged less than two years. Of the seven patients aged less than two years, only two patients underwent clinical monitoring that included blood testing. The nature of the evaluations in the four trials is summarized in the following table. No clinically significant laboratory (hematology, clinical chemistry) abnormalities were reported in any trials. Table 5. Number of Pediatric Patients Who Underwent Laboratory and other Clinical Evaluations Study Efficacy AE monitoring Laboratory tests PK PK = blood pharmacokinetic evaluation; AE = adverse event In the three single-site trials, no adverse reactions to gadoterate were reported. Within two of these clinical trials, no adverse events were reported for any patients (trials 3-15 and 3-29). In Trial 3-29, one adverse event (not attributed to gadoterate) was reported; a patient less than two years of age experienced transient vomiting after injection of oxybuterate (for sedation) and the gadoterate. Page 12 of 95
13 In the phase 3 trial (050), 8 of 38 (21%) exposed patients experienced at least one adverse event; all but one of these events was graded as mild or moderate in severity. One event was graded as serious but it was not attributed to gadoterate. Six patients had at least one adverse event attributed to gadoterate. Table 6. Summary of Treatment Emergent Adverse Events among 38 Pediatric Patients Exposed to Gadoterate in Trial 050 Event Number of Patients with the Event Headache 2 Dizziness 1 Nausea 1 Vomiting 1 Application site erythema 1 Asthenia 1 Blood detected in urine 1 Hypoxia 1 Pruritus 1 The one serious adverse event occurred in a 5 year old female who had severe hypoxia the day after receipt of gadoterate; the event was not considered to be related to gadoterate and the event resolved the same day. b. Pediatric Post-marketing Surveillance Studies Within the NDA, the information from the pediatric post-marketing studies is largely confined to summary reports published by medical journals. The applicant submits a final study report for only one of these studies (Mauer/2012). For all six of the studies, no patient-level data or other details are available to verify the study conduct or the accuracy of the comments made in the publications or the report. In general, the studies appeared to use a survey type design where patients were observed for adverse reactions shortly following gadoterate administration and subjective assessments were made of the MR images. Only the SECURE study information refers to extended follow-up to detect the occurrence of NSF. The SECURE study is ongoing; hence, a study report has not been submitted for review with this application. All of the studies were multicenter except for the study publication referred to as Emond/2011 (a single center experience) and most imaging was performed for CNS evaluations. Author/y ear Neiss/ 1991 Table 7. The Six post-marketing studies Conducted by Guerbet Study Information in NDA Number of patients/outcomes location France, Copy of the publication 4169 (including 305 children)/not Belgium, that appeared in a translated from French but the abstract is in Switzerland French journal English and reports that 0.8% of the patients had adverse reactions and none were life-threatening. The abstract concludes that, They [results] also bear out previous evidence on the outstanding Page 13 of 95
14 Briand/ 1992 Ishiguchi/ 2010 Emond/ 2011 Mauer/ 2012 SECURE study/ ongoing France Japan France Germany Multiple countries Copy of a abstract published by the European Society of Pediatric Radiology/1992 Copy of a published report that appeared in Drugs: 2010 Copy of a published report that appeared in Pediatr Radiol 2011 A study report that includes conclusions and summary tables. A copy of an abstract published in Pediatric Radiology 2012 safety profile of this MRI contrast agent, especially in children. 402 children (26 reported as 2 years of age or younger)/ No adverse reaction was reported in children of 15 or below The results confirm the advantages of injecting Gd-DOTA in children, as well as its excellent safety profile patients (including 41 children)/adverse reactions were reported in 32 patients (0.9%); most reactions were mild and no serious reactions were reported. 104 children/all children were < 2 years of age and were studies at a single clinical site; the publication notes, No immediate adverse event was reported. Also, Image quality was rated as excellent/good for Gd-DOTA-enhanced MRI in 102 (98%) children (including 672 children)/the report cites adverse reactions in 0.3% patients (predominantly nausea). No adverse events were observed in children below 2 years (n = 10). Also, Serious adverse events were observed in 0.01% of patients. The most common serious events were dyspnea, nausea and vomiting; anaphylactic shock was reported in one patient. Image quality was very good or good in 97.2% of all patients / As of November 16, 2011, the cutoff date for the interim safety analysis, this ongoing Post-Marketing Study (PMS) included data from 972 children (mean age 9.9 years; range 0 17 years; male 51.9%). Overall, No adverse events were observed. Page 14 of 95
15 a. Efficacy background: 5. Main Efficacy and Safety Data Summary The minimum regulatory expectation for a claim of GBCA CNS structural visualization efficacy relies upon an applicant demonstrating that the contrast agent improves visual anatomical delineation properties. This expectation is articulated within the 2004 FDA Guidance on Medical Imaging Agents which notes that, Ordinarily, the ability to locate and outline normal structures or distinguish between normal and abnormal anatomy can speak for itself with respect to the clinical value of the information and will not require additional information substantiating clinical usefulness. In general, new GBCA applicants have met the structural visualization efficacy expectation by demonstrating that the new GBCA increases the number of detected CNS lesions and/or improves the visualization of lesions, compared to non-contrasted images (some have also compared paired uncontrasted + contrasted to uncontrasted images). In the past, applicants have used a variety of relatively arbitrary scoring systems to assess a broad range of image features in which patients were enrolled into single arm studies and images interpreted by a central group of readers who were masked to clinical data. Guerbet designed the main phase 3 clinical trial (050) following this precedent. b. Clinical pharmacology: Four clinical studies examined various aspects of gadoterate pharmacokinetics. As summarized below, the data show that most gadoterate is eliminated via the kidneys following administration of the drug with a terminal half-life of approximately 1.6 hours in subjects with normal renal function. The elimination half-life is markedly prolonged among patients with severe renal impairment. Table 8. Gadoterate Pharmacokinetic Studies Study/Year Subjects Findings DGD-3-6/ 6 healthy adult males Gadoterate distributed in extracellular volume 1987 received a single IV dose Elimination half-life of 1.32 ± 0.2 hr of 0.1 mmol/kg Between 3 6 hrs post drug dosing 5/6 men had increased urine iron level and 1/6 had a low plasma iron level (both levels normalized by 48 hrs) Blood and urine levels of copper, zinc and manganese did not change Most gadoterate was eliminated in urine DGD-3-48/ Healthy adult males and females in two groups: After single 0.1 mmol/kg injection, 73 to 85% gadoterate recovered in urine over 48 hours Group A: 16 subjects received a single IV dose Single injection Tmax mean of 5 minutes and elimination mean half-life of 1.39 hr in women and 2.04 hr in men Page 15 of 95
16 DGD-3-28/ 1991 DGD / 2004 of 0.1 mmol/kg Group B: 16 subjects received an initial IV dose of 0.1 mmol/kg followed 20 minutes later by a second IV dose of 0.2 mmol/kg 4 healthy adult subjects and 8 adult patients with moderate or severe chronic renal failure (creatinine clearance between 30 and 60 ml/min (n = 4) and between 10 and 30 ml/min (n = 4) All received a single IV dose of 0.1 mmol/kg 40 adult patients received placebo and gadoterate ( triple dose, 0.1 mmol/kg followed by 0.2 mmol/kg) in cross-over design to assess EKG effects over time following the drug dose Urinary excretion may occur for several days at a very low level. After administration of a triple dose, gadoterate exposure is 3 times higher than after administration of a single 0.1 mmol/kg dose Triple dose Tmax mean of 5 minutes and elimination mean half-life of 1.69 hr in women and 1.87 hr in men Healthy volunteer mean elimination half-life of 1.62 hrs Patients with moderate renal failure had mean elimination half-life of 5.05 hrs Patients with severe renal failure had mean elimination half-life of hrs Gadoterate total mean clearance was 108 ml/min in healthy volunteers and was prolonged in patients with renal failure: total mean clearance of 40 ml/min in patients with moderate renal failure and 14 ml/min in patients with severe renal failure Areas under curves of plasma concentrations increased markedly in relation to the severity of renal failure. Gadoterate did not prolong QT or QTc by more than 5 ms compared to placebo c. Clinical efficacy data: Two clinical trials form the main efficacy database: Trial 050, a trial designed that received an FDA special protocol agreement designation, and Trial 051, a trial that was a reread of a previously conducted imaging trial. We refer to this trial as supportive of Trial 050. Trial 050 was designed to provide confirmatory evidence of gadoterate efficacy. This trial s major features are summarized below followed by a brief description of the supportive trial (051). Page 16 of 95
17 Trial 050: Safety and efficacy evaluation of Dotarem in magnetic resonance imaging in patients with central nervous system (CNS) lesions (SENTIO Study) Trial 050 Design: The trial was to be a multicenter study of adults and pediatric patients (aged two year or more) who needed to undergo CNS MR imaging because of known or suspected lesions. The targeted enrollment was 396 subjects. The adult population was to form the main efficacy population adults were randomized (in a 2: 1 double-blind manner) to receive gadoterate or gadopentetate (Magnevist) following non-contrasted MR imaging. All pediatric patients were to receive gadoterate following an initial non-contrasted MR image, i.e., they were not randomized and their outcomes were not included in the primary endpoint analyses. The trial eligibility criteria excluded subjects with acute or chronic grade IV or V renal insufficiency, defined as an estimated glomerular filtration rate < 30 ml/min/1.73 m 2. Gadoterate and Magnevist were each to be administered as a single dose of 0.1 mmol/kg. The primary objective (endpoint) was to be determined solely within the population of adult patients who received gadoterate. Specifically, paired (non-contrast images plus contrasted images) were to be compared to the non-contrasted images in order to assess the extent to which the paired images improved CNS lesion visualization. All comparisons to Magnevist were secondary or exploratory endpoints with no hypothesis testing. Regarding the primary objective, three lesion visualization characteristics were each to be graded on a scale by three independent image readers (centralized readings), masked to clinical information. The lesion characteristics were: Border delineation Internal morphology visualization Degree of contrast enhancement The scale for grading each of these characteristics was a three point categorization: -0 unevaluable -1 seen imperfectly -2 seen completely Computations for the primary endpoints: Using the lesion scoring system, each patient was to have a patient score computed by adding up all lesion scores (up to a maximum of five lesions) within the subject for the modality (per subject paired sum versus per subject pre sum) then the within subject difference was to be calculated as the paired sum pre sum score. The lesions identified in the non-contrasted images were not matched to the lesions identified in the paired images. Examples are shown below: Examples of a patient score : Subject # Lesion # Score Pre Score Paired Subject s score Paired - Pre Page 17 of 95
18 = = = -1 The primary endpoint success hypothesis was that the sum of lesions in the paired MR images was at least on average 0.5 points higher than sum of lesion scores in pre MR images. The study was to be considered successful if two of the three independent readers simultaneously met the success hypothesis expectation for the paired images for all three components of the primary endpoint. These analyses were pre-specified to be confined to the full analysis set (FAS) population; this population was to consist of all adults who received gadoterate and who had valid assessments of the three primary endpoint lesion characteristics. Pre-specified rules described the handling of any missing data. As a secondary endpoint, gadoterate-contrasted images (alone) were to be compared to the noncontrasted images in analyses similar to those for the primary endpoint. Multiple other secondary endpoints were also to be analyzed (such as number of identified lesions, all comparisons to Magnevist outcomes and outcomes in the pediatric population). Safety evaluations were conducted at baseline and over a 48 hour period following GBCA administration, including clinical observations and clinical laboratory tests. Trial 050 Conduct: The study was conducted between 2010 and 2011 and included 52 clinical sites around the world. Major protocol violations occurred in approximately 7% of the adult population and mainly consisted of: -5 patients who did not receive the correct GBCA dose; -7 patients who did not receive any study drug dose (they withdrew participation) -4 patients who had missing images for the centralized (off site) image read -the remaining patients had insufficient safety evaluations and/or timing for signing of the study consent form. Page 18 of 95
19 Within the subset of 38 pediatric patients, seven had major protocol deviations, most related to the timing of safety evaluations (either too soon or too late) or failure to record the time of the evaluations. Overall, 416 patients were screened for the study and ultimately 402 patients formed the all included patient (AIP) population. The safety population consisted of subjects who received the study drug. The trial population distribution is shown below: Table 9. Trial 050 Population Distribution Population Adults Pediatrics Total Screened All Included (AIP) Safety Full Analysis (FAS)* 356 (Dot + Mag) 37 (all Dot) 393 *the FAS varied by each of the three independent readers (for adults, the FAS by reader was: n = 345, n = 347 and n = 354); Dot = gadoterate (Dotarem); Mag = gadopentetate (Magnevist); Randomization 2 Dot :1 Mag (adults) Baseline characteristics: The following tables summarize the baseline characteristics for adults and pediatric patients within the AIP groups. Table 10. Trial 050 Baseline Characteristics: Adult AIP Population Characteristic Dot Mag n = 245 n = 119 Age, median in years (range) 53 (19 85) 56 (19 94) Sex 54% female 55% female Caucasian 207 (85%) 95 (80%) History of renal disease or urinary disorder 13 (5%) 3 (3%) History of nervous system disorder 122 (50%) 49 (41%) Dot = gadoterate (Dotarem); Mag = gadopentetate (Magnevist) Trial 050 Efficacy Data: Table 11. Trial 050 Baseline Characteristics: Pediatric AIP Population Characteristic AIP, n = 38 Age, median in years (range) 8 (3 17) Sex 58% female Caucasian 26 (68%) History of renal disease or urinary disorder 0 History of nervous system disorder 19 (50%) Page 19 of 95
20 The primary and secondary imaging visualization objectives were achieved in both adults and pediatric patients, as summarized below. The gadoterate imaging outcomes were also similar to those for gadopentetate. As previously noted, the primary endpoints were to be analyzed within the adult FAS population that received gadoterate. Imputation of missing data was performed for some results within these analyses: 11 adults had imputed data for reader 1, 13 adults had imputed data for reader 2, and 22 adults had imputed data for reader 3. Endpoint Border delineation Internal morphology Contrast enhancement Table 12. Trial 050 Primary Endpoint Results: Patient Scores for Lesion Visualization, by Reader (mean, SD) Reader 1 Reader 2 Reader 3 Pre Paired Pre Paired Pre Paired n = 224 n = 230 n = 224 n = 230 n = 222 n = (1.23) (2.64) (1.43) (2.94) (1.29) (2.30) (1.05) (2.63) (1.24) (2.93) (1.13) (2.30) (0.20) (2.52) (0.15) (2.67) (0.13) (2.44) In Table 12, the paired imaging result is highlighted for ease of interpretation; all comparisons between the paired and the pre image results showed statistically significant differences for favoring the paired image results each reader for each endpoint (p < 0.001). The statistical tests (using regression models) were pre-specified in the study s statistical analytical plan. Improved image visualization for paired images was seen in exploratory analyses of subsets defined by gender, site, etc. An exploratory analytical examination of the distribution of visualization outcomes have also consistently shown improved visualization with gadoterate. The following table is excerpted from the FDA statistician s working notes. The table shows the distribution of patients by the lesions visualized by Reader 1 (a patient could have up to five lesions scored) using the FAS patient population. A similar distribution pattern was found for other readers and other coprimary endpoints. Highlighted is the distribution of patients with seen completely lesions, the highest visualization score. Table 13. FDA Exploratory Analysis of Trial 050 Primary Endpoint: Number of Patients by Five Largest Lesions Visualized by Reader 1 for Border Delineation Lesion score* First Second Third Fourth Fifth Total Number of Lesions Pre Pair Pre Pair Pre Pair Pre Pair Pre Pair Pre Pair Total Page 20 of 95
21 *0 = unevaluable; 1 = seen, but imperfectly, 2 = seen completely/perfectly The reported major lesion visualization outcomes (secondary endpoints) in the pediatric subset of patients also showed improved visualization for paired images as shown below. Table 14. Trial 050 Pediatric Efficacy Results: Patient Scores for Lesion Visualization, by Reader (mean, SD) Reader 1 Reader 2 Reader 3 Endpoint Pre n = 31 Paired n = 32 Pre n = 34 Paired n = 35 Pre n = 33 Paired n = 36 Border delineation 1.42 (1.09) 2.47 (1.52) 1.18 (1.03) 3.51 (2.50) 1.06 (0.66) 1.36 (1.10) Internal morphology 1.13 (0.88) 2.75 (1.50) 1.41 (0.78) 3.51 (2.48) 1.06 (0.56) 1.81 (1.09) Contrast enhancement (1.09) (2.03) (1.25) FDA s preliminary analyses also indicate improved visualization of the paired images compared to the pre-contrast images. The following table summarizes the pediatric imaging efficacy outcomes based upon a distribution of the number of patients with various scores for the first lesion listed within the dataset tabulations. Table 15. FDA Preliminary Analysis: Number of Pediatric Patients Categorized by First Lesion Visualization Scores* Border Delineation Patients with Lesion Reader 1 Reader 2 Reader 3 Score of: Pre Paired Pre Paired Pre Paired Total Patients Internal Morphology Patients with Lesion Reader 1 Reader 2 Reader 3 Score of: Pre Paired Pre Paired Pre Paired Total Patients Contrast Enhancement Patients with Lesion Reader 1 Reader 2 Reader 3 Score of: Pre Paired Pre Paired Pre Paired Total Patients Page 21 of 95
22 *Lesion scores: 0 = Unevaluable 1= Seen, but imperfectly, 2 = Seen completely/perfectly FDA obtained results generally similar to those in Table 15, when pediatric patients were analyzed by subsequent lesions (second, third, etc.) Trial 050 Safety: Regarding safety outcomes among adults, the occurrence of any adverse reactions to gadoterate was reported in 4% (9 of 240) patients. Any adverse reaction to gadopentetate was reported in 8% (9 of 117) patients. Reactions were graded as either mild or moderate in severity. One death was reported (unrelated to study drug). No serious reactions to the study drugs were reported. Pediatric safety was previously summarized in this document. Trial 051: A Supportive trial titled, Evaluation of MRI with Dotarem in the diagnosis or follow-up assessment of cerebral or spinal tumors Regulatory perspective: Trial 051 was originally conducted in ; a reread of images from this trial was completed in Trial 051 originally enrolled 151 adult patients who were undergoing surgical procedures to evaluate CNS lesions. The patients had baseline non-contrasted MR images followed by gadoterate-contrasted images. The original trial compared image outcomes to a histopathology truth standard of malignancy/benign tissue outcome. Images from the original trial were later reread in a manner similar to that used in Trial 050. The following questions illustrate FDA s perspective on Trial 051: Why does FDA regard Trial 051 as supportive and not a trial that provides independent confirmation of gadoterate efficacy? Trial 051 is a reread of a previously conducted trial that failed to achieve its primary endpoint; hence, imaging outcomes were known prior to development of the reread protocol. It is highly unlikely that FDA could recognize confirmatory, independent substantiation of imaging efficacy based upon a reread study of a trial that initially failed to achieve its primary endpoint. The original Trial 051 primary endpoint assessed the diagnostic accuracy of gadoterate in determining whether a lesion was malignant or benign, in comparison to non-contrasted images where all image outcomes were referenced to a tissue histopathology truth standard. How does Trial importantly differ from trial 050? While the reread design of Trial 051 largely paralleled that for Trail 050, the original design of Trial 051 necessitated that patients were highly likely to undergo surgery to obtain a histopathology truth standard; this was not a requirement in Trial 050. Unlike Trial 050, Trial 051 did not enroll any pediatric patients. Page 22 of 95
23 Does Trial 051 provide any useful efficacy information? FDA regards Trial 051 as providing useful supportive efficacy information since the reread protocol was designed in a manner to parallel the design of Trial 051 and the study results largely parallel those for Trial 050. Furthermore, the lesion visualization endpoints (secondary or exploratory outcomes in the original 051 protocol) were achieved in original Trial 051 analyses, an observation that enhances the interpretability of the reread findings. The Trial 051 primary endpoint results are shown in the table below (excerpted from the applicant s study report). All comparisons of paired to pre images were reported as statistically significant in favor of improved visualization for the paired image results (p < Table 16. Trial 051 Reread Primary Endpoint Results: Patient Scores for Lesion Visualization, by Reader (mean, SE) Endpoint Reader 1 Reader 2 Reader 3 Pre Paired Pre Paired Pre Paired Border delineation 0.93 (0.09) 1.88 (0.09) 1.41 (0.08) 2.13 (0.08) 0.32 (0.08) 1.37 (0.08) Internal morphology 1.09 (0.07) 1.32 (0.07) 1.33 (0.08) 2.34 (0.08) 0.66 (0.07) 1.39 (0.07) Contrast enhancement (0.07) 2.23 (0.07) (0.08) 2.32 (0.08) (0.09) 2.31 (0.09) The Overall Gadoterate Clinical Trial Safety Database Summary: Among the patients in the 49 clinical trials conducted by Guerbet, most (62%) were aged years, 32% were >65 years and 5% were less than 18 years of age. Overall, among the 2813 patients in clinical trials, 9.3% (263 out of 2813 patients) are reported to have experienced an adverse event and 3.9% were regarded as related (i.e., adverse reactions) to gadoterate. Of the 363 adverse events that occurred in 263 patients, most were of mild intensity (71%) and most (53%) were considered unrelated to gadoterate. Most of the adverse events either resolved spontaneously or with treatment (78.6%). Serious adverse events were reported in 23 patients but were not regarded by Guerbet as related to gadoterate. However, FDA notes that two patients experienced events that may meet serious criteria; the examination of these cases is on-going. One event concerns moderate hypersensitivity in a patient and another event concerns moderate renal failure in a patient. Eight patient deaths were reported (all reported as unrelated to gadoterate). The most common adverse reactions in the clinical trial database are shown below. Table 17. Adverse Reactions in 0.2% of the Gadoterate Clinical Trial Database Rate (%) Reaction n = 2813 Nausea 0.6 Headache 0.5 Page 23 of 95
24 Injection site pain 0.4 Injection site coldness 0.2 A burning sensation 0.2 The safety experience within the post-marketing surveillance studies as well as the routine pharmacovigilance experience shows an array of uncommonly reported adverse reactions, including serious hypersensitivity reactions to gadoterate. For example, one report describes a 65 year old man with known coronary artery disease who received gadoterate and immediately experienced anaphylactic shock that necessitated resuscitation. Overall, the nature of the events, as well as the reporting frequency, appears similar to that for other GBCAs. The following sections contain a draft of the lead medical officer s mid-cycle slides, the safety observations from FDA s Office of Surveillance and Epidemiology as well as notations from reviewers of the nonclinical data and the clinical pharmacology data. Medical Officer s Draft Mid-Cycle Review slides Are shown on the following pages. Page 24 of 95
25 1/23/2013 Clinical Midcycle Review NDA : Dotarem Injection (Gadoterate Meglumine) Barbara Stinson, PR Alex Gorovets, TL 1 NDA : Dotarem Injection Applicant: Guerbet LLC Submission Date: 2012 Sept 20 Review Designation: Priority PDUFA Date: 2013 Mar 20 2 Page 25 of 95 1
26 1/23/2013 Outline Proposed indication Gadolinium-based Contrast Agent (GBCA) approval precedents Dotarem background Dotarem clinical development Dotarem Phase 3 study summaries and Dotarem pediatric studies Safety summary 3 Proposed Indication Dotarem is a gadolinium-based contrast agent indicated for intravenous use with magnetic resonance imaging (MRI) in brain (intracranial), spine and associated tissues in adults and pediatric patients (from neonates to 17 years of age) to detect and visualize areas with disruption of the blood brain barrier (BBB) and/or abnormal vascularity. Intended population: Adults and pediatric patients from neonates to 17 years of age with known or suspected CNS disease 4 Page 26 of 95 2
27
28 1/23/2013 Visualization Precedent Ordinarily, the ability to locate and outline normal structures or distinguish between normal and abnormal anatomy can speak for itself with respect to the clinical value of the information and will not require additional information substantiating clinical usefulness. - FDA Guidance on Medical Imaging Agents, Primary Endpoint Precedent from Prior GBCA Approvals Added diagnostic information subjective assessment Increased numbers of lesions Improved visualization variety of relatively arbitrary scores 8 Page 28 of 95 4
29 1/23/2013 Outline Proposed indication Gadolinium-based Contrast Agent (GBCA) approval precedents Dotarem background Dotarem clinical development Dotarem Phase 3/peds study summaries Safety summary 9 Dotarem Marketed in 1989 in Europe Over 30 million exposed worldwide A macrocyclic GBCA Renal elimination, no metabolism 10 Page 29 of 95 5
30
31
32 1/23/2013 Outline Proposed indication Gadolinium-based Contrast Agent (GBCA) approval precedents Dotarem background Dotarem clinical development Dotarem Phase 3 study summaries and pediatric studies Safety summary 15 2 Phase 3 Trials - -Titles Phase 3 Study 050: Safety and efficacy evaluation of Dotarem in magnetic resonance imaging (MRI) in patients with central nervous system (CNS) lesions (SENTIO Study) Phase 3 Study 051: Evaluation of MRI with Dotarem in the diagnosis or follow up assessment of cerebral or spinal tumors. Rereading of MRI images 16 Page 32 of 95 8
33
34
35
36
37
38
39
40 1/23/2013 Dotarem Pediatric Studies 3 trials, N = 99, ages 1.2 mos-17 yrs, (mean ages 8 yrs, 10 yrs, 9 yrs) 7 subjects 0-23 months Efficacy for contribution of Dotarem and treatment modification, single investigator open label studies Study limitations: - No PK studies - Full safety evaluations for only 20 subjects, 2 under age 2 most were for AEs only - Dotarem administration method varied among all three studies and the 050 study -Efficacy bias 31 SAFETY SUMMARY 32 Page 40 of 95 16
41 1/23/2013 Sources of Safety Data 49 Phase 1-4 clinical trials: N = 2813; 2672 adults, 141* children,( 7 < 2 y.o.) Safety data pools: all indications, CNS indication, pivotal trials 6 Post marketing studies, 1 ongoing Global pharmacovigilance: to * 130 under age 17; 4 in other CNS trials 33 Dotarem Safety AEs, SAEs, Deaths, NSF 363 AEs in 263 (9.3%) subjects, 111 related (3.9%) 29 SAEs, 2 related, one moderate hypersensitivity and one renal failure in subject with low grade failure 8 deaths, none related (7 in the -051 study) No unconfounded cases of NSF 34 Page 41 of 95 17

42
43 1/23/2013 Other Safety Concerns No dose ranging studies; no PK studies in peds Special populations limited to 8 subjects with decreased renal clearance; elimination time with function (no dialysis but probably ok) ECG safety study-no safety signals Comprehensive monitoring lacking -Only 31% monitored with labs and vital signs -Inconsistent reporting of results -Reporting not always comparable (1980s & 1990s) Marked flow rate differences between studies (/min and /sec injections); 66.4% dosed at mmol/kg Subject compliance sometimes limited, e.g. for -051 study only 76% returned at 24 hours 37 Conclusions Two phase 3 studies achieved primary efficacy endpoints however the 051 study had a restricted enrollment and Dotarem was not contributory to therapeutic management for 2 of 3 readers Overall safety considerations similar to other GBCAs however safety assessment was comprehensive only for 31 % of subjects and only for the 050 trial Pediatric safety for <2 year group was insufficiently studied 38 Page 43 of 95 19
44 1/23/2013 Dotarem, Recommendations Approval for CNS Indication ages 2-17 and adults (-051 pivotal trial limited for efficacy and safety but acceptable as GBCA # 9) Do not recommend approval for <2 years based on insufficiency of safety and efficacy data in this age group 39 Page 44 of 95 20
45 Department of Health and Human Services Public Health Service Food and Drug Administration Center for Drug Evaluation and Research Office of Surveillance and Epidemiology Pharmacovigilance Review - Premarket Date: January 7, 2013 Reviewer(s): Michael E. Kieffer, Pharm.D., M.A., Division of Pharmacovigilance II Joseph M. Tonning, M.D., R.Ph., M.P.H., Division of Pharmacovigilance II Team Leader(s): Peter Diak, Pharm.D., M.P.H., Division of Pharmacovigilance II Allen Brinker, M.D., Division of Pharmacovigilance II Division Director(s): Min Chen, R.Ph., MS, Deputy Director, Division of Pharmacovigilance I Linda Scarazzini, M.D., R.Ph, Division of Pharmacovigilance I Product Name(s): Subject: Dotarem (gadoterate meglumine) All Adverse Events Application Type/Number: NDA Applicant/Sponsor: Guerbet OSE RCM #: Reference ID: Page 45 of 95
46 TABLE OF CONTENTS Executive Summary Introduction Background Regulatory History Proposed Product Labeling Methods and Materials FAERS Search Strategy Literature Search Results FAERS Reports Pediatric Cases Reported in FAERS (n=4) Pediatric Literature Review Adult Cases Reported in FAERS (n=47) Discussion/Conclusion Appendices Appendix A. Proposed Dotarem Labeling Regarding Pregnancy and Lactation Appendix B. Proposed Dotarem Label Warning and Precaution for Nephrogenic Systemic Fibrosis, Adverse Reactions and Postmarketing Sections Appendix C. FDA Adverse Event Reporting System (FAERS) Appendix D. AERS Case Numbers, AERS ISR Numbers and Manufacturer Control Numbers (n=51) Appendix F. Summary of Selected Cases (n=3) and Descriptive Review of All Cases Coded for Nephrogenic Systemic Fibrosis (n=10) Appendix G. Summary of Selected Cases (n=5) and Descriptive Listing of the Cases Grouped as Hypersensitivity Reactions (n=22) Appendix H. Case Summaries for Remaining Labeled Events Occurring in Adults (n=6) Appendix I. Case Summaries for Unexpected Adverse Events Per Dotarem s Proposed Labeling (N=9) Reference ID: Page 46 of 95
47 EXECUTIVE SUMMARY The Division of Medical Imaging Products (DMIP) requested that the Division of Pharmacovigilance II (DPV II) summarize postmarketing reports associated with the use of Dotarem found within the FDA Adverse Event Reporting System (FAERS) database and the medical literature in both adult and pediatric patients (0 to 17 years of age), including patients aged less than two years. If Dotarem receives approval, it will be the first GBCA indicated in the U.S. for patients aged less than two years. As of November 27, 2012, the FAERS database contained 51 cases associated with Dotarem (4 pediatric or 8% and 47 adult cases). There were no reports of pediatric deaths or NSF cases in the pediatric population. The pediatric cases consisted of one case each of: accidental overdose (without adverse event); premature birth (most likely not related to Dotarem use); heart rate decreased; and hypersensitivity reaction. Hypersensitivity reactions are described in the proposed Dotarem label s Warnings and Precautions section and bradycardia is listed in the Postmarketing Experience section. The majority of reported adverse events in the 47 adult cases are already included within the proposed Dotarem labeling to include: hypersensitivity reactions (n=22); NSF (n=10; all confounded); coma (n=1); convulsions (n=1); dizziness (n=1); fever and chills (n=1); and loss of consciousness (n=2). The remaining cases either reported related terms in the proposed labeling or were confounded by multiple drugs and/or underlying disease. These cases included: acute renal failure (n=3); thrombosis (n=2); acute cholestatic hepatitis with acute renal failure (n=1); agranulocytosis (n=1); intrathecal administration/medication error (n=1); and vagal reaction (n=1). A review of the literature did not elicit concerns of adverse events in children less than 2 years of age exposed to Dotarem. A PubMed search conducted on December 5, 2012, retrieved 13 articles with titles and abstracts related to the use of Dotarem in the pediatric population, eight of the articles addressing patients aged less than two years. The articles included approximately 1,203 pediatric patients administered Dotarem with about 177 of these patients aged less than two years. None of the articles mentioned specific adverse events in any of the pediatric patients studied. This review did not identify any new safety issues with Dotarem in either pediatric or adult populations. However, since the data provided from FAERS is limited with respect to quantity of reports, the appropriate final labeling for Dotarem should be made based on all available data to the Review Division. Reference ID: Page 47 of 95
48 1 INTRODUCTION 1.1 BACKGROUND Dotarem is an ionic macrocyclic gadolinium-based contrast agent (GBCA). The NDA was submitted on September 20, 2012, and the PDUFA action date is March 20, Dotarem s sponsor, Geurbet, is requesting United States marketing approval for the indication of intravenous use for magnetic resonance imaging (MRI) in brain (intracranial), spine and associated tissues in adults and pediatric patients (from neonates to 17 years of age) to detect and visualize areas with disruption of the blood brain barrier (BBB) and/or abnormal vascularity. No other GBCAs currently marketed in the US have been approved for use in children aged less than two years. A concern in this age group is that the reduced renal function of this patient population, compared to patients aged greater than 2 years, would lead to Dotarem retention and predispose them to the development of nephrogenic systemic fibrosis (NSF). Dotarem injection will contain mg/ml gadoterate meglumine (equivalent to 0.5 mmol/ml) when marketed, and will be available in vial and prefilled syringe REGULATORY HISTORY Dotarem is not currently approved for marketing in the United States. Thus, there is no requirement for the sponsor to submit postmarketing safety reports to the Agency. Dotarem s first global marketing approval was in France in Since then it has obtained marketing approval in many countries in Europe, Asia, Central and South America, and Africa. Using data collected from Europe, South Korea, Taiwan, Mexico, and Brazil and based on a 0.27% usage rate in France for children aged less than two years, the manufacturer estimates that 51,000 children (aged less than two years) have received Dotarem between 2005 and Based on the total liters of Dotarem sold and a formula based on normal doses and projected percentages of use for each indication, the sponsor estimates 30,532,599 people have received Dotarem since its first global marketing approval. 2 Dotarem has been investigated in 23 studies (n=1,329; United States proposed indication) involving magnetic resonance imaging (MRI) in patients with CNS lesions, suspected brain tumors or metastases, Alzheimer s disease, and for other neurological reasons. Three of these studies (DGD-3-15, DGD-3-16, and DGD-3-29) were specific for pediatric patients (n=141 children). Seven patients aged less than 2 years were included in these pediatric studies. Only one adverse effect was reported from these seven patients, which was vomiting determined by the study physician as unrelated to the Dotarem. Six postmarketing surveillance studies (n=234) were also conducted in children aged less than two and no related adverse effects were reported. The most common adverse reactions for all clinical trails involving Dotarem were nausea, headache, injection site pain, injection site coldness, and burning sensation. 1.3 PROPOSED PRODUCT LABELING 1 Proposed Dotarem Label - revision date currently not provided. 2 Integrated Safety Study, Dotarem, version 1, dated August 26, Reference ID: Page 48 of 95
49 The proposed labeling for Dotarem has the following information concerning the pediatric population: Adverse Reactions in Children (Section 6.1 of proposed label) During clinical trials, 141 children (7 aged < 24 months, 33 aged 2-5 years, 58 aged 6-11 years and 43 aged 12-17) received DOTAREM. Overall, 6 children (4.3%) reported at least one adverse reaction following DOTAREM administration. The most frequently reported adverse reaction was headache (1.5%). Most adverse events were mild in severity and transient in nature, and all patients recovered without treatment. Overall, in 241 children aged < 2 years old, from 6 post marketing studies (234 children) and 3 clinical trials (7 children), there were no adverse reactions reported. Adverse Reactions in Children Adverse events related to DOTAREM are uncommon in children. The expectedness of these events is similar to that of the events reported in adults. Pediatric Use (Section 8.4 of label) The safety and efficacy of DOTAREM at a single dose of 0.1 mmol/kg have been established in children from neonates to 17 years of age. No dosage adjustment is necessary in this population. Use of DOTAREM in children is supported by evidence from both adequate and well-controlled studies and post marketing studies. See Appendix A for proposed labeling regarding Dotarem use in pregnancy and during lactation. Proposed Boxed Warning WARNING: NEPHROGENIC SYSEMIC FIBROSIS (NSF) Gadolinium-based contrast agents (GBCAs) increase the risk for NSF among patients with impaired elimination of the drugs. Avoid use of GBCAs in these patients unless the diagnostic information is essential and not available with noncontrasted MRI or other modalities. NSF may result in fatal or debilitating fibrosis affecting the skin, muscle, and internal organs. The risk of NSF appears highest among patients with: - Chronic, severe kidney disease (GFR,30 ml/min/1.73m 2 ), or - Acute kidney injury. Screen patients for acute kidney injury and other conditions that may reduce renal function. For patients at risk for chronically reduced renal function (e.g. age > 60 year, hypertension, diabetes), estimate the glomerular filtration rate (GFR) through laboratory testing (5.1). For patients at risk for NSF, do not exceed the recommended DOTAREM dose and allow a sufficient period of time for elimination of the drug from the body prior to re-administration [see Warnings and Precautions (5.1)] Reference ID: Page 49 of 95
50
51 3 RESULTS 3.1 FAERS REPORTS The FAERS search retrieved 54 reports, which included 3 duplicate reports. Table 3 below summarizes the 51 cases included in this case series. Appendix D lists all the AERS case numbers, AERS ISR numbers and Manufacturer Control numbers for the 51 cases (three duplicates removed) in this case series. Table 3. Total number of FAERS reports (N=51; search from 1969 to November 27, 2012) All reports Serious Death Adults ( 17 years) Pediatrics (0-17 years) Age unknown (null values) Total Serious adverse drug experiences per regulatory definition (CFR ) include outcomes of death, lifethreatening, hospitalization (initial or prolonged), disability, congenital anomaly, and other serious important medical events. 3.2 PEDIATRIC CASES REPORTED IN FAERS (N-4) Table 4 summarizes the four FAERS cases from the Pediatric Case Series with Dotarem. Table 4. Descriptive characteristics of Pediatric Case Series from January 1, 1969 to November 27, 2012 (N=4) Age 0 1 month 1 1 month - <2 years years years years 0 Sex Male 2 Female 2 Country of reporter Foreign 4 France 3 Germany 1 Report type Expedited 4 Event date Dose (n=3) Dose 1 16mL(overdose) Dose 2 6 ml Dose 3 2 ml Reference ID: Page 51 of 95
52 Table 4. Descriptive characteristics of Pediatric Case Series from January 1, 1969 to November 27, 2012 (N=4) Indications (n=2) Cranial CT 1 MRI 1 Serious Hospitalized 4 Outcomes* MedDRA Preferred Terms (PTs)± Accidental overdose 1 Cough 1 Foetal growth restriction 1 Heart rate decreased 1 Hypersensitivity 1 Oropharyngeal discomfort 1 Small for dates baby 1 Premature baby 1 *Serious adverse drug experiences per regulatory definition (CFR ) include outcomes of death, life-threatening, hospitalization (initial or prolonged), disability, congenital anomaly, and other serious important medical events. ± Case may contain more than one PT No pediatric deaths were reported in this case series. No NSF cases were reported in pediatric patients within this case series. Below is a summary of the 4 pediatric cases: Case # (Germany). A 16 month-old male infant (weighing 10 kg) with no known allergies and no prior examinations with contrast media underwent a cranial computerized tomography study with Dotarem for first imaging and clarification of the symptom complex associated with neurofibromatosis type 1. Dotarem was administered manually and resulted in an accidental overdose of 16 milliliters (proposed label recommends 0.1 mmole/kg or 0.2 ml/kg of body weight). The infant was hydrated, his fluid intake and output monitored, and was admitted to the intensive care unit for monitoring purposes. Ultimately, no adverse events were reported as a result of the overdose. Case # (France). A female baby (weighing 1.27 kilograms) was delivered by cesarean section without neonatal defect, but prematurely at 31.5 weeks of gestation. The premature delivery was required due to the occurrence of preeclampsia in the mother resulting in fetal growth retardation. The mother received a magnetic resonance imaging study using Dotarem (dose not reported) as contrast at approximately two weeks into her pregnancy. The pregnancy was further complicated by the following factors: (1) the mother initially tried to abort the pregnancy with emergency contraceptive pills (Norlevo), (2) the mother had a history of hypertension treated with atenolol (continued during pregnancy with the dose doubled starting at the sixth month of pregnancy), and (3) the mother was diagnosed with multiple sclerosis treated with steroids during the fifth and six weeks of her pregnancy. The child was lost to follow-up and no further data is known. Case # (France). An 11 month-old male child received a magnetic resonance imaging (MRI) study with 2 mls of Dotarem as contrast. The patient received four patches of EMLA (lidocaine, prilocaine) to the elbow fold and hand for anesthesia prior to catheter insertion. During the MRI, the child s heart rate decreased from 110 beats per minute (bpm) to 65 bpm 40 Reference ID: Page 52 of 95
53 minutes later. He appeared pale and the MRI was stopped. Within a few minutes of stopping the MRI, his heart rate recovered without corrective treatment, and he recovered without sequelae. Case # (France). An eleven year old female, with a history of a previous well tolerated Omniscan administration, received 6 mls of Dotarem and experienced an allergic reaction with dry cough and pharyngeal discomfort. She was treated with an injection of polaramine (dexchlorpheniramine). She recovered without sequelae. PEDIATRIC CASE SUMMARY There is no evidence from these 4 cases that there are new pediatric safety concerns with Dotarem at this time, including any safety concerns in patients aged less than 2 years. 3.3 PEDIATRIC LITERATURE REVIEW PubMed was searched on December 5, 2012 for the MeSH term gadoterate. This term alone was searched to ensure as broad a search as possible, so that all articles coded with the gadoterate MeSH term were retrieved. This search retrieved 707 articles (all years and all languages). To comply with the request from DMIP to review literature in the <2 year-old age group, the titles and abstracts of these 707 articles were searched for the following text strings: pediatric, children, and infant. From this text string search, 13 relevant articles in English were retrieved (Appendix E). Of these 13 articles, only 8 mentioned gadoterate use in patients < 2 years of age. However, for completeness, 5 additional references 3 are included in Appendix E because these references mention gadoterate use in older pediatric patients > 2 years of age. Of these 13 references, all except the first reference (Emond and Brunelle) mention gadoterate in the context of its use as a diagnostic agent in a small number of pediatric patients. No specific adverse events are mentioned among these small patient groups. However, the first reference article in Appendix E (the Emond and Brunelle article) describes a larger post-marketing study of 104 neonates and infants who received gadoterate in a single pediatric hospital in France. This first reference is discussed separately below. Emond S, Brunelle F. Gd-DOTA administration at MRI in children younger than 18 months of age: immediate adverse reactions. Pediatr Radiol Nov;41(11): Epub 2011 Jul 24. (PMID ) This French post-marketing study was an observational, non-randomized, single-center, openlabel study. This study included 104 infants and neonates with an age range of 3 days to 18 months. The aim of the study was to gain further knowledge on the safety of gadoterate in children less than 18 months of age in routine clinical practice. Thus, adverse events were specifically being monitored. A standardized questionnaire was used to collect patient 3 In Appendix E: Secinaro et al (Reference 3); Merlini et al (Reference 5); Sebag et al (Reference 9); Hervé-Somma et al (Reference 12); and Romero et al (Reference 13) Reference ID: Page 53 of 95
54 information. Variables recorded for each child included but were not limited to: demographics (age, sex, weight); risk factors for contrast agent reactions; volume of gadoterate administered; and overall tolerance to gadoterate. The volume of gadoterate injected per child ranged from 0.6 ml in a newborn (male, 3 days, 3 kg) to 4 ml in the heaviest/oldest child (female, 18 months, 20 kg), with a median of 2 ml. The children were observed in the hospital for at least 2 hours after gadoterate administration. No adverse events were reported among these children. The authors stated, We believe that our data are of significant clinical and research utility for centers assessing children with central nervous system disorders by MRI. Possible limitations of such studies are the small number of included patients and technical difficulties using MRI in children. Clinical trials in children are more challenging than those in adults. The authors further stated, Although difficult to conduct, more extensive clinical studies are warranted to assess long-term safety. This article mentioned two other references not retrieved by the PubMed search previously described. These 2 articles are summarized below. Briand Y, Neiss AC, Vitry A (1992) Efficacy and safety of the macrocyclic complex Gd-DOTA in children: results of a multicentre study. In: 29 th congress of the European Society of Pediatric Radiology, Budapest: R12. This study does not appear in PubMed and has not been published to our knowledge. However, the Emond article, when referring to this study, states Our study, conducted in France, included 402 patients (81% of the children were 15 years old or younger and 6.5% were 2 years old or younger). Our results confirmed the advantages of Gd-DOTA [gadoterate] injection in children as well as its favorable safety profile in terms of immediate adverse reactions. Herborn CU, Honold E, Wolf M, Kemper J, Kinner S, Adam G, Barkhausen J. Clinical safety and diagnostic value of the gadolinium chelate gadoterate meglumine (Gd-DOTA). Invest Radiol Jan;42(1): The purpose of this study was to assess the diagnostic value and safety of gadoterate. A total of 24,308 patients were intravenously injected with gadoterate for various diagnostic examinations. Demographic, clinical, imaging, and safety data were obtained from board certified radiologists in 61 radiologic institutions throughout Germany between January 2004 and October Patient data were collected through the use of standardized questionnaires completed by the radiologist. A detailed breakdown of patient age was not provided for the 24,308 patients studied. Regarding ages of the patients, the article has only this passing statement: Patients ranged in age from a few weeks to 103 years (mean, 51.8 years). However, the Emond article references this Herborn study and states that in the Herborn study 2.7% of the children were 18 years old or Reference ID: Page 54 of 95
55 younger and 0.008% were 2 years old or younger. Emond further states that in the Herborn study, Out of 24,000 patients, the overall incidence of reported adverse events was only 0.4%: one serious adverse event (anaphylactic shock) occurred in an adult patient and no adverse event in children younger than 2 years of age. The Herborn article confirms that adverse events were reported in 0.4% (n = 94) patients, but there is no breakdown by patient age. Herborn further states that of the 24,308 patients, 1 patient (0.004%) developed a serious adverse event considered to be life threatening. This event, anaphylactic shock, occurred in a 65 year-old man, confirming statements made by Emond when referring to the Herborn article that no adverse events occurred in pediatric patients. 3.4 ADULT CASES REPORTED IN FAERS (N=47) Table 5 summarizes the 47 cases reported in adults in the FAERS database. Table 5. Descriptive characteristics of Adult Case Series from January 1, 1969 to November 27, 2012 (47=number in case series) Age (n=42) (Years) Mean 52.6 Median 50 Range 18 to 78 Sex Male 24 Female 23 Country of reporter Foreign: 47 France 16 Germany 8 Chile 5 Belgium 4 Denmark 3 Italy 3 Great Britain 2 Japan 2 Argentina 1 Netherlands 1 Singapore 1 Spain 1 Report type Expedited 47 Event date Reference ID: Page 55 of 95
56 Table 5. Descriptive characteristics of Adult Case Series from January 1, 1969 to November 27, 2012 (47=number in case series) Unknown 8 Serious Outcomes*, ± Death 1 Life-threatening 10 Required Intervention 9 Hospitalized 47 Disability 8 Other serious 10 * Serious adverse drug experiences per regulatory definition (CFR ) include outcomes of death, life-threatening, hospitalization (initial or prolonged), disability, congenital anomaly, and other serious important medical events. ± There may be more than one outcome per case. Nephrogenic Systemic Fibrosis (n=10; Fatal [n=1], Non-fatal [n=9]) NSF is labeled within the proposed Dotarem label (Boxed Warnings). In nine of the ten cases, the patients have a history of chronic renal failure prior to the Dotarem exposure with one case not reporting a renal status (see Case # below). Determination of causality is confounded by the administration of at least one other concomitant gadolinium contrast agent in all ten cases. Three case summaries are included in Appendix F. Appendix F also includes an outline of all 10 ten cases of nephrogenic systemic fibrosis. Hypersensitivity Reactions (n=22) Hypersensitivity is labeled within the proposed Dotarem label (Warnings and Precautions section). Of the cases identified as describing hypersensitivity reaction within the adult case series, 14 patients were female and nine were male. The average age was 53.5 years (n=20), median age was 52 years, and the age range was 33 to 78 years. The outcome in all 22 cases was coded as serious. Four cases were coded with a preferred term (PT) referring to shock (anaphylactic shock-3; cardiogenic shock-1). Two cases were coded with the PT laryngeal edema, and one each was coded with the PT pharyngeal edema and angioedema. Six cases were coded with loss of consciousness (PT). Five representative cases are described in the Appendix G. Appendix G also includes an outline for all 22 cases grouped as hypersensitivity reactions. Appendix H contains the case summaries for the remaining labeled events (n=6) of coma (n=1); convulsions (n=1); dizziness (n=1); fever, chills (n=1); and loss of consciousness (n=2). Appendix I contains case summaries for the unexpected events in adults (n=9) occurring following Dotarem administration: acute renal failure (n=3); acute cholestatic hepatitis with acute renal failure (n=1); thrombosis (n=2); agranulocytosis (n=1); intrathecal administration (n=1); and vagal reaction (n=1). Reference ID: Page 56 of 95
57 SUMMARY OF ADULT CASE SERIES The FAERS search identified 47 adverse event reports (all coded as serious) associated with adult patients. The majority reported adverse events within the proposed Dotarem labeling (38/47, 81%) to include: hypersensitivity reactions (n=22); NSF (n=10; all confounded); coma (n=1); convulsions (n=1); dizziness (n=1); fever and chills (n=1); and loss of consciousness (n=2). Reported unlabeled adverse events (all coded as serious) include: acute renal failure (n=3; blood creatinine increase is labeled); thrombosis (n=2; superficial phlebitis is labeled); acute cholestatic hepatitis with acute renal failure (n=1); agranulocytosis (n=1); intrathecal administration/medication error (n=1); and vagal reaction (n=1; bradycardia labeled). These unlabeled and unexpected terms are not specifically listed in the proposed Dotarem labeling; however, many of these events will be described by related terms that will be contained in the proposed label. 4 DISCUSSION/CONCLUSION The FAERS case series contained 51 cases: four pediatric patients aged less than or equal to 17 years (three aged less than two years) and 47 adult patients. The pediatric literature search identified 13 articles that reported studying about 1,203 pediatric patients, of which around 177 were aged less than two years. Neither the FAERS nor the literature reviews identified any new safety signals with Dotarem in the pediatric population. There were no deaths or NSF cases in the pediatric case series. The most commonly reported adverse event in the adult case series were those related to hypersensitivity reactions (n=22). NSF was the next most frequently reported adverse event reported in adults (n=10). All cases of NSF associated with Dotarem use were confounded by coadministration of at least one other GBCA agent. Although there were nine cases that reported adverse events that are not listed in the proposed Dotarem labeling (unexpected), many of these events have related terms that will be contained in the proposed label (i.e., blood creatinine increase for acute renal failure n=3 (GBCAs are labeled for acute kidney injury [class labeling recommended by prior OSE review 4 ]), superficial phlebitis for thrombosis n=2, and bradycardia for vagal reaction n=1). The remaining three unexpected adverse reactions reported with Dotarem were single reports of a medication error due to intrathecal administration, hepatitis/renal failure, and agranulocytosis, The latter two cases were both confounded by the administration of multiple agents and/or underlying disease (i.e., HIV). This review did not identify any new safety issues with Dotarem in either pediatric or adult populations. However, since the data provided from FAERS is limited with respect to quantity of reports, the appropriate final labeling for Dotarem should be made based on all available data to the Review Division. 5 APPENDICES 4 Camilli S, Gelperin K, et al. Acute Renal Failure and Gadolonium-based Contrast Agents, dated January 11, Reference ID: Page 57 of 95
58 5.1 APPENDIX A. PROPOSED DOTAREM LABELING REGARDING PREGNANCY AND LACTATION Pregnancy (Section 8.1 of label) Pregnancy Category C There are no adequate and well-controlled studies conducted in pregnant women. DOTAREM Injection should be used during pregnancy only if the potential benefit justifies the potential risk to the fetus. DOTAREM Injection was not embryotoxic or teratogenic in rats and rabbits. A non-significant increase of incidence of incomplete or delayed ossification of some bones was observed in rats and rabbit fetuses born from female animals given daily dose levels starting from 4 mmol/kg/day in rats and 1 mmol/kg/day in rabbits from gestation day 6 to day 17 in rats or 19 in rabbits. These dose levels represented 6 and 3 times the human dose based on body surface area in rats and rabbits, respectively. Maternal toxicity was observed in rats at 10 mmol/kg/day (16 times the human dose based on body surface area) and in rabbits at 7 mmol/kg/day (23 times the human dose based on body surface area). Nursing Mothers (Section 8.3 of label) It is not known whether DOTAREM is excreted in human milk. Because many drugs are excreted in human milk, exercise caution when DOTAREM is administered to a nursing woman. NONCLINICAL DATA SHOW THAT DOTAREM IS EXCRETED INTO BREAST MILK IN VERY SMALL AMOUNTS (<0.1% OF THE DOSE INTRAVENOUSLY ADMINISTERED) AND THE ABSORPTION VIA THE GASTROINTESTINAL TRACT IS POOR. Reference ID: Page 58 of 95
59 5.2 APPENDIX B. PROPOSED DOTAREM LABEL WARNING AND PRECAUTION FOR NEPHROGENIC SYSTEMIC FIBROSIS, ADVERSE REACTIONS AND POSTMARKETING SECTIONS Nephrogenic Systemic Fibrosis Nephrogenic systemic fibrosis (NSF) has occurred with other GBCAs exhibiting a low stability in patients with severe kidney disease. Gadolinium-based contrast agents (GBCAs) increase the risk for nephrogenic systemic fibrosis (NSF) among patients with impaired elimination of the drugs. Avoid use of GBCAs among these patients unless the diagnostic information is essential and not available with non-contrast enhanced MRI or other modalities. The GBCA-associated NSF risk appears highest for patients with chronic, severe kidney disease (GFR < 30 ml/min/1.73m 2 ) as well as patients with acute kidney injury. The risk appears lower for patients with chronic, moderate kidney disease (GFR ml/min/1.73m2) and little, if any, for patients with chronic, mild kidney disease (GFR ml/min/1.73m 2 ). NSF may result in fatal or debilitating fibrosis affecting the skin, muscle and internal organs. Report any diagnosis of NSF following DOTAREM administration to Guerbet LLC ( ) or FDA (1-800-FDA-1088 or Screen patients for acute kidney injury and other conditions that may reduce renal function. Features of acute kidney injury consist of rapid (over hours to days) and usually reversible decrease in kidney function, commonly in the setting of surgery, severe infection, injury or drug-induced kidney toxicity. Serum creatinine levels and estimated GFR may not reliably assess renal function in the setting of acute kidney injury. For patients at risk for chronically reduced renal function (e.g., age > 60 years, diabetes mellitus or chronic hypertension), estimate the GFR through laboratory testing. Among the factors that may increase the risk for NSF are the use of low stability GBCAs, repeated or higher than recommended doses of a GBCA, and the degree of renal impairment at the time of exposure. Record the specific GBCA and the dose administered to a patient. For patients at highest risk for NSF, do not exceed the recommended DOTAREM dose and allow a sufficient period of time for elimination of the drug prior to re-administration. No unconfounded cases of NSF have been reported with DOTAREM. For patients receiving hemodialysis, physicians may consider the prompt initiation of hemodialysis following the administration of a GBCA in order to enhance the contrast agent s elimination. The usefulness of hemodialysis in the prevention of NSF is unknown [see Clinical Pharmacology (12) and Dosage and Administration (2)]. ADVERSE REACTIONS Clinical Studies Experience Because clinical trials are conducted under widely varying conditions, adverse reaction rates observed in the clinical trials of a drug cannot be directly compared to rates in the clinical trials of another drug and may not reflect the rates observed in clinical practice. The data described below reflect DOTAREM exposure in 2813 patients, representing 2672 adults and 141 children. Most patients received doses between 0.05mmol/kg and 0.3mmol/kg body weight. Overall, 54.6% of the patients were men. In clinical trials where ethnicity was recorded the distribution was 74% Caucasian, 12% Asian, 4% Black, and 10% others. The average age was 53 years (range from 0.1 to 97 years). Overall, 3.9% of patients reported at least one adverse reaction, primarily occurring immediately or several days following DOTAREM administration. In total, 149 adverse reactions were reported. Most adverse reactions were mild or moderate in severity and transient in nature. Table 2 lists adverse reactions that occurred in 0.2% patients who received DOTAREM. Table 2: Adverse Reactions ( 0.2%) Reaction Rate (%) n=2813 Nausea 0.6% Reference ID: Page 59 of 95
60 Headache 0.5% Injection Site Pain 0.4% Injection Site Coldness 0.2% Burning Sensation 0.2% Adverse reactions that occurred with a frequency <0.2% in patients who received DOTAREM include: feeling cold, rash, somnolence, fatigue, dizziness, vomiting, pruritus, paresthesia, dysgeusia, pain in extremity, anxiety, hypertension, palpitations, oropharyngeal discomfort, blood creatinine increased, blood lactate dehydrogenase increased, injection site inflammation, injection site extravasation, injection site pruritus, injection site warmth, and asthenia. Postmarketing Experience To date, it is estimated that more than 30 million doses of Dotarem have been administered worldwide. The following adverse reactions have been identified during post approval use of DOTAREM. Because these reactions are reported voluntarily from a population of uncertain size, it is not always possible to reliably estimate their frequency or establish a causal relationship to drug exposure. System Organ Class Cardiac Disorders Immune System Disorders Nervous System Disorders Musculoskeletal and Connective Tissue Disorders Gastrointestinal Disorders General Disorders and Administration Site Conditions Skin and Subcutaneous Tissue Disorders Vascular Disorders See Warnings and Precautions (5.1) Adverse Reaction bradycardia, tachycardia, arrhythmia hypersensitivity /anaphylactoid reactions including cardiac arrest, respiratory arrest, cyanosis pharyngeal edema, laryngospasm, bronchospasm, angioedema, conjunctivitis, ocular hyperemia, eyelid edema, lacrimation increased, hyperhidrosis, urticaria coma, convulsion, syncope, presyncope, parosmia, tremor muscle contracture, muscle weakness diarrhea, salivary hypersecretion malaise, fever nephrogenic systemic fibrosis (Most often in patients who also have received other GBCAs. No unconfounded cases of NSF have been reported with DOTAREM.) superficial phlebitis Reference ID: Page 60 of 95
61 5.3 APPENDIX C. FDA ADVERSE EVENT REPORTING SYSTEM (FAERS) The FDA Adverse Event Reporting System (FAERS) is a database that contains information on adverse event and medication error reports submitted to FDA. The database is designed to support the FDA s post-marketing safety surveillance program for drug and therapeutic biologic products. The informatic structure of the database adheres to the international safety reporting guidance issued by the International Conference on Harmonisation. Adverse events and medication errors are coded to terms in the Medical Dictionary for Regulatory Activities (MedDRA) terminology. The suspect products are coded to valid tradenames or active ingredients in the FAERS Product Dictionary (FPD). FDA implemented FAERS on September 10, 2012, and migrated all the data from the previous reporting system (AERS) to FAERS. Differences may exist when comparing case counts in AERS and FAERS. FDA validated and recoded product information as the AERS reports were migrated to FAERS. In addition, FDA implemented new search functionality based on the date FDA initially received the case to more accurately portray the follow up cases that have multiple receive dates. FAERS data have limitations. First, there is no certainty that the reported event was actually due to the product. FDA does not require that a causal relationship between a product and event be proven, and reports do not always contain enough detail to properly evaluate an event. Further, FDA does not receive reports for every adverse event or medication error that occurs with a product. Many factors can influence whether or not an event will be reported, such as the time a product has been marketed and publicity about an event. Therefore, FAERS data cannot be used to calculate the incidence of an adverse event or medication error in the U.S. population. Reference ID: Page 61 of 95
62 5.4 APPENDIX D. AERS CASE NUMBERS, AERS ISR NUMBERS AND MANUFACTURER CONTROL NUMBERS (N=51) Pediatric Case Series (N=4) AERS Case Numbers ISR Numbers Manufacturer Control Numbers DE-GUERBET FR-GUERBET FR-ASTRAZENECA-2010SE , FR-ASTRAZENECA-2010SE12076 Adult Case Series (N=47) AERS Case Numbers ISR Numbers Manufacturer Control Numbers FR-ABBOTT-11P DE-ROCHE , BE GB JP-GUERBET CH-GUERBET FR , FR-GUERBET , DE , DE-BAYER DE-GUERBET , , DK-BAYER GPV , , , , ,5298 FR-BAYER-FR , , , DE-SHR-DE , , CH-GUERBET CH-SHR-CH SG-PFIZER INC , CH DE-BAYER FR-GUERBET PHHY2011DE CH-GUERBET , GB JP-COVIDIEN/TYCO HEALTHCARE/MALLINCKRODT- T PHRM2004FR OSCN-NO-0912S BE-WATSON OMPQ-NO-1101S DK-BAYER FR FR-GUERBET FR , BE Reference ID: Page 62 of 95
63 AERS Case Numbers ISR Numbers Manufacturer Control Numbers DE-GUERBET FR FR IT-GUERBET FR-GUERBET ES-GUERBET , IT-GUERBET NL , FR-GUERBET AR IT-GUERBET FR BE FR Reference ID: Page 63 of 95
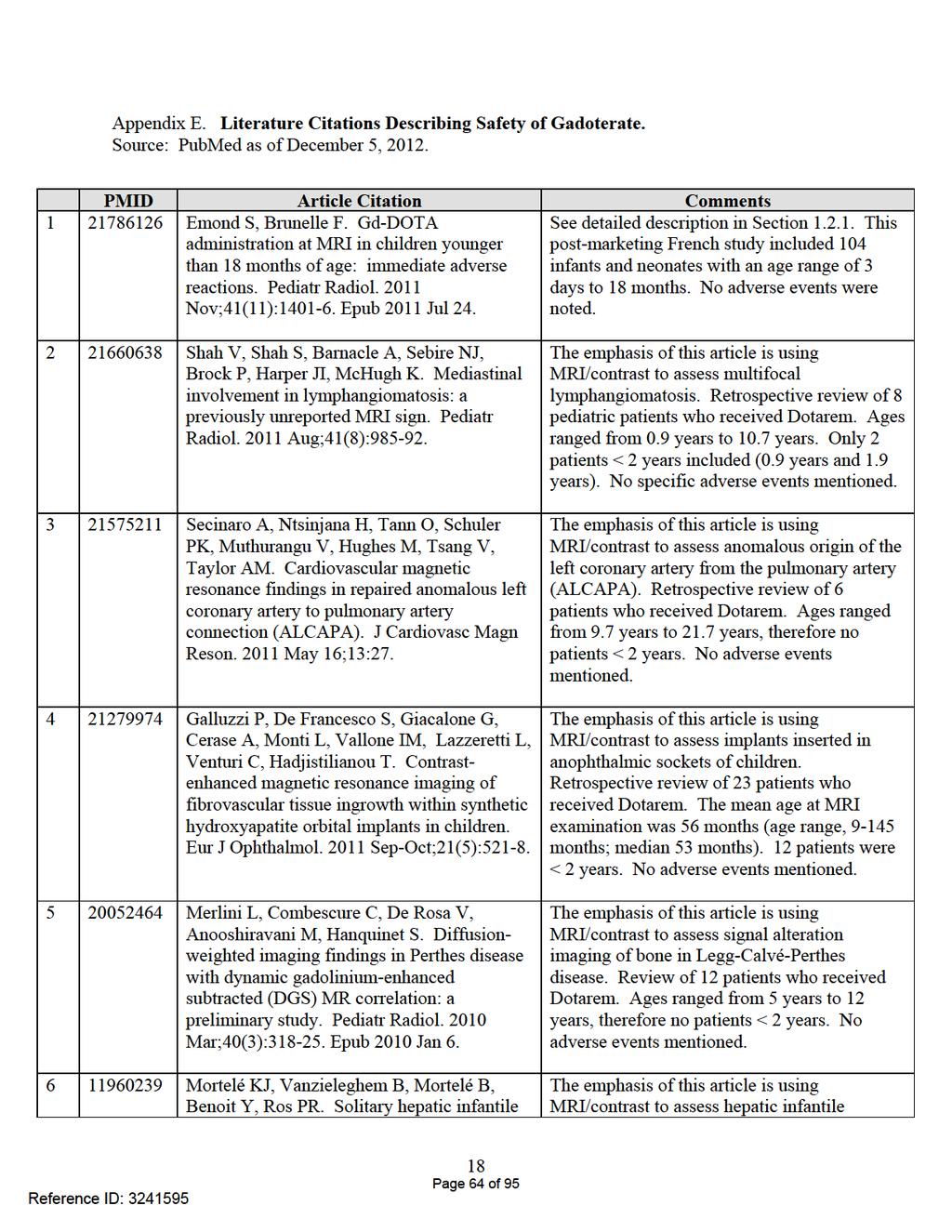
64
Gadolinium Based Contrast Agent Recommendations (Revised 06/07/2018)
") Gadolinium Based Contrast Agent Recommendations (Revised 06/07/2018) Introduction: Nephrogenic systemic fibrosis is the only known adverse health effect related to gadoliniumbased contrast agents (GBCAs).
Gadolinium Based Contrast Agent Recommendations (Revised 06/07/2018) Introduction: Nephrogenic systemic fibrosis is the only known adverse health effect related to gadoliniumbased contrast agents (GBCAs).
Administration of Gadolinium Contrast in Adults Procedural Guideline
 Administration of Gadolinium Contrast in Adults Procedural Guideline This procedural guideline is designed to assist Radiologists by providing an analytical framework for the evaluation of gadolinium contrast
Administration of Gadolinium Contrast in Adults Procedural Guideline This procedural guideline is designed to assist Radiologists by providing an analytical framework for the evaluation of gadolinium contrast
Gadolinium-Based Contrast Media
 Gadolinium-Based Contrast Media Wm. Faulkner, B.S.,R.T.(R)(MR)CT, FSMRT, MRSO (MRSC ) Kristan Harrington, MBA, R.T.(R)(MR), MRSO (MRSC TM ) Gadolinium ( 64 Gd) 7 un-paired electrons chelate, any of a class
Gadolinium-Based Contrast Media Wm. Faulkner, B.S.,R.T.(R)(MR)CT, FSMRT, MRSO (MRSC ) Kristan Harrington, MBA, R.T.(R)(MR), MRSO (MRSC TM ) Gadolinium ( 64 Gd) 7 un-paired electrons chelate, any of a class
MR Contrast Agents. Why Use Contrast Agents in MRI? Why Use Contrast Agents in MRI? US Agents. Understanding and Embracing Change
 Why Use Contrast Agents in MRI? Improve disease detection and characterization Increase sensitivity to extent of disease MR Contrast Agents Understanding and Embracing Change Kristan Harrington, MBA, RT
Why Use Contrast Agents in MRI? Improve disease detection and characterization Increase sensitivity to extent of disease MR Contrast Agents Understanding and Embracing Change Kristan Harrington, MBA, RT
Please see Important Safety Information, including Boxed Warnings for Gadavist, Eovist and Magnevist on the following pages.
 Dear Valued Customer: As a valued partner we want to inform you that a technical issue occurred at our Berlin manufacturing center where final formulation and packaging of Bayer MRI (Magnetic Resonance
Dear Valued Customer: As a valued partner we want to inform you that a technical issue occurred at our Berlin manufacturing center where final formulation and packaging of Bayer MRI (Magnetic Resonance
Important Update - Availability of Bayer s MRI (Magnetic Resonance Imaging) Contrast agents
 Contrast agents") Important Update - Availability of Bayer s MRI (Magnetic Resonance Imaging) Contrast agents Certain batches of pharmaceutical products from our company s Supply Center in Berlin where final formulation
Important Update - Availability of Bayer s MRI (Magnetic Resonance Imaging) Contrast agents Certain batches of pharmaceutical products from our company s Supply Center in Berlin where final formulation
A Safety Update on the Gadolinium Chelates
 Control # 1029 A Safety Update on the Gadolinium Chelates Val M. Runge, MD Universitätsinstitut für Diagnostische, Interventionelle und Pädiatrische Radiologie University Hospital Bern Allergic Reactions
Control # 1029 A Safety Update on the Gadolinium Chelates Val M. Runge, MD Universitätsinstitut für Diagnostische, Interventionelle und Pädiatrische Radiologie University Hospital Bern Allergic Reactions
Seeing is Believing... Look Beneath the Surface
 References 1. Idee JM, Port C, Raynal I, et al. Clinical and geological consequences of transmetallation induced by contrast agents for magnetic resonance imaging: a review. Fundamental and Clinical Pharmacology.
References 1. Idee JM, Port C, Raynal I, et al. Clinical and geological consequences of transmetallation induced by contrast agents for magnetic resonance imaging: a review. Fundamental and Clinical Pharmacology.
INTRODUCTION CLINICAL EXPERIENCE CLINICAL EFFICACY PEDIATRIC EXPERIENCE DOSING SUMMARY
 1 2 DOTAREM Administered Doses 3 Molecular Structure of DOTAREM DOTAREM offers high thermodynamic and kinetic stability provided by its macrocyclic and ionic structure* 3 *The clinical significance of
1 2 DOTAREM Administered Doses 3 Molecular Structure of DOTAREM DOTAREM offers high thermodynamic and kinetic stability provided by its macrocyclic and ionic structure* 3 *The clinical significance of
PRODUCT MONOGRAPH DOTAREM. (gadoterate meglumine) Injection. For Intravenous Use
 Injection. For Intravenous Use") PRODUCT MONOGRAPH DOTAREM (gadoterate meglumine) Injection For Intravenous Use Gadolinium-Based Contrast Agent For Use with Magnetic Resonance Imaging (MRI) Guerbet LLC Date: August 26, 2013 120 W. 7 th
PRODUCT MONOGRAPH DOTAREM (gadoterate meglumine) Injection For Intravenous Use Gadolinium-Based Contrast Agent For Use with Magnetic Resonance Imaging (MRI) Guerbet LLC Date: August 26, 2013 120 W. 7 th
Please see Important Safety Information, including Boxed Warnings for Gadavist and Magnevist below.
 August 15 th, 2018 Dear Valued Customer: Thank you for your continued patience over the last several months as we have worked to resolve our contrast supply interruption. The purpose of this letter is
August 15 th, 2018 Dear Valued Customer: Thank you for your continued patience over the last several months as we have worked to resolve our contrast supply interruption. The purpose of this letter is
DIVISION OF MEDICAL IMAGING AND HEMATOLOGY PRODUCTS
 DIVISION OF MEDICAL IMAGING AND HEMATOLOGY PRODUCTS MEMORANDUM TO THE FILE i. NDA: 20-123, 22-066 Product: Omniscan ii. NDA: 19-596, 21-037 Product: Magnevist iii. NDA: 20-976, 20-937,20-975 Product: OptiMARK
DIVISION OF MEDICAL IMAGING AND HEMATOLOGY PRODUCTS MEMORANDUM TO THE FILE i. NDA: 20-123, 22-066 Product: Omniscan ii. NDA: 19-596, 21-037 Product: Magnevist iii. NDA: 20-976, 20-937,20-975 Product: OptiMARK
Gadolinium-Based Contrast Agents for Magnetic Resonance Imaging (marketed as Magnevist, MultiHance, Omniscan, OptiMARK, ProHance)
") Information for Healthcare Professionals Page 1 of 5 FDA ALERT [6/2006, updated 12/2006 and 5/23/2007: This updated Alert highlights FDA s request for addition of a boxed warning and new warnings about
Information for Healthcare Professionals Page 1 of 5 FDA ALERT [6/2006, updated 12/2006 and 5/23/2007: This updated Alert highlights FDA s request for addition of a boxed warning and new warnings about
University of Virginia Institutional Review Board - Health Sciences Research Guidelines for Researchers Using Gadolinium-Enhanced MRI in Research
 University of Virginia Institutional Review Board - Health Sciences Research Guidelines for Researchers Using Gadolinium-Enhanced MRI in Research Page 1 of 16 Table of Contents New Information:... 3 IRB-HSR
University of Virginia Institutional Review Board - Health Sciences Research Guidelines for Researchers Using Gadolinium-Enhanced MRI in Research Page 1 of 16 Table of Contents New Information:... 3 IRB-HSR
PRODUCT MONOGRAPH INCLUDING PATIENT MEDICATION INFORMATION DOTAREM. Gadoterate meglumine injection. (376.9 mg/ml, equivalent to 0.
 PRODUCT MONOGRAPH INCLUDING PATIENT MEDICATION INFORMATION DOTAREM Gadoterate meglumine injection (376.9 mg/ml, equivalent to 0.5 mmol/ml) For Intravenous Use Contrast Enhancement Agent for Magnetic Resonance
PRODUCT MONOGRAPH INCLUDING PATIENT MEDICATION INFORMATION DOTAREM Gadoterate meglumine injection (376.9 mg/ml, equivalent to 0.5 mmol/ml) For Intravenous Use Contrast Enhancement Agent for Magnetic Resonance
See 17 for PATIENT COUNSELING INFORMATION and Medication Guide. flow rate of approximately 2 ml/second for adults and 1-2 ml/second for pediatric
 HIGHLIGHTS OF PRESCRIBING INFORMATION These highlights do not include all the information needed to use DOTAREM safely and effectively. See full prescribing information for DOTAREM. DOTAREM (gadoterate
HIGHLIGHTS OF PRESCRIBING INFORMATION These highlights do not include all the information needed to use DOTAREM safely and effectively. See full prescribing information for DOTAREM. DOTAREM (gadoterate
DOTAREM (gadoterate meglumine) Injection for intravenous use Initial U.S. Approval: 2013
 Injection for intravenous use Initial U.S. Approval: 2013") HIGHLIGHTS OF PRESCRIBING INFORMATION These highlights do not include all the information needed to use DOTAREM safely and effectively. See full prescribing information for DOTAREM. DOTAREM (gadoterate
HIGHLIGHTS OF PRESCRIBING INFORMATION These highlights do not include all the information needed to use DOTAREM safely and effectively. See full prescribing information for DOTAREM. DOTAREM (gadoterate
Beyond NSF: Acute GBCA adverse reactions
 Source images Beyond NSF: Acute GBCA adverse reactions Martin R. Prince, MD, PhD, FACR Disclosures Patent Agreements: GE, Siemens, Philips, Hitachi, Toshiba, Bayer, Bracco, Mallinckrodt, Medrad, Nemoto,
Source images Beyond NSF: Acute GBCA adverse reactions Martin R. Prince, MD, PhD, FACR Disclosures Patent Agreements: GE, Siemens, Philips, Hitachi, Toshiba, Bayer, Bracco, Mallinckrodt, Medrad, Nemoto,
NSF Coming and Going
 NSF Coming and Going Martin R. Prince, MD, PhD Cornell and Columbia Universities Patent agreements: GE, Philips, Siemens, Hitachi, Medrad, Epix, Lantheus, Bayer, Bracco, Nemoto, Mallinckrodt and Topspins
NSF Coming and Going Martin R. Prince, MD, PhD Cornell and Columbia Universities Patent agreements: GE, Philips, Siemens, Hitachi, Medrad, Epix, Lantheus, Bayer, Bracco, Nemoto, Mallinckrodt and Topspins
Global reports and findings of NSF and its effects
 Safety Forum: MR Safety Update - SMRT 2008 Sunday May 8, 2:25 p.m. Global reports and findings of NSF and its effects Tim Leiner, MD PhD Assistant Professor of Radiology, Department of Radiology, Maastricht
Safety Forum: MR Safety Update - SMRT 2008 Sunday May 8, 2:25 p.m. Global reports and findings of NSF and its effects Tim Leiner, MD PhD Assistant Professor of Radiology, Department of Radiology, Maastricht
PATIENT CARE AND MRI SAFETY Module 7
 PATIENT CARE AND MRI SAFETY Module 7 1 Biological Considerations There are no reported adverse biological effects of extended exposure to MRI. However, several inconsequential and reversible effects of
PATIENT CARE AND MRI SAFETY Module 7 1 Biological Considerations There are no reported adverse biological effects of extended exposure to MRI. However, several inconsequential and reversible effects of
Information for Healthcare Professionals and other Stakeholders
 Information for Healthcare Professionals and other Stakeholders NEPHROGENIC SYSTEMIC FIBROSIS: AN UNCOMMON AND DEBILITATING DISEASE POSSIBLY ASSOCIATED WITH GADOLINIUM CHELATES Villepinte, February 2014
Information for Healthcare Professionals and other Stakeholders NEPHROGENIC SYSTEMIC FIBROSIS: AN UNCOMMON AND DEBILITATING DISEASE POSSIBLY ASSOCIATED WITH GADOLINIUM CHELATES Villepinte, February 2014
Gadolinium-Based MR Contrast Agents
 Gadolinium-Based MR Contrast Agents Wm. Faulkner, BS,RT(R)(MR)(CT), FSMRT, MRSO (MRSC ) Kristan Harrington, MBA, RT(R)(MR) ARRT, MRSO (MRSC ) Magnetic Resonance Imaging, Vol 3, pp27-35, 1985 0.35 T 35/1600
Gadolinium-Based MR Contrast Agents Wm. Faulkner, BS,RT(R)(MR)(CT), FSMRT, MRSO (MRSC ) Kristan Harrington, MBA, RT(R)(MR) ARRT, MRSO (MRSC ) Magnetic Resonance Imaging, Vol 3, pp27-35, 1985 0.35 T 35/1600
Reference ID:
 HIGHLIGHTS OF PRESCRIBING INFORMATION These highlights do not include all the information needed to use DOTAREM safely and effectively. See full prescribing information for DOTAREM. DOTAREM (gadoterate
HIGHLIGHTS OF PRESCRIBING INFORMATION These highlights do not include all the information needed to use DOTAREM safely and effectively. See full prescribing information for DOTAREM. DOTAREM (gadoterate
1 INDICATIONS AND USAGE 2 DOSAGE AND ADMINISTRATION. 1.1 Central Nervous System. 1.2 Extracranial/Extraspinal Tissues. 1.3 Body
 HIGHLIGHTS OF PRESCRIBING INFORMATION These highlights do not include all the information needed to use MAGNEVIST safely and effectively. See full prescribing information for MAGNEVIST. MAGNEVIST (gadopentetate
HIGHLIGHTS OF PRESCRIBING INFORMATION These highlights do not include all the information needed to use MAGNEVIST safely and effectively. See full prescribing information for MAGNEVIST. MAGNEVIST (gadopentetate
Annex III. Amendments to relevant sections of the product information
 Annex III Amendments to relevant sections of the product information Note: These amendments to the relevant sections of the product information are the outcome of the referral procedure. The product information
Annex III Amendments to relevant sections of the product information Note: These amendments to the relevant sections of the product information are the outcome of the referral procedure. The product information
Gadolinium-Based MR Contrast Agents
 Gadolinium-Based MR Contrast Agents Magnetic Resonance Imaging, Vol 3, pp27-35, 1985 Wm. Faulkner, BS,RT(R)(MR)(CT), FSMRT, MRSO (MRSC ) Kristan Harrington, MBA, RT(R)(MR) ARRT, MRSO (MRSC ) 0.35 T 0.35
Gadolinium-Based MR Contrast Agents Magnetic Resonance Imaging, Vol 3, pp27-35, 1985 Wm. Faulkner, BS,RT(R)(MR)(CT), FSMRT, MRSO (MRSC ) Kristan Harrington, MBA, RT(R)(MR) ARRT, MRSO (MRSC ) 0.35 T 0.35
Migraine: Developing Drugs for Acute Treatment Guidance for Industry
 Migraine: Developing Drugs for Acute Treatment Guidance for Industry U.S. Department of Health and Human Services Food and Drug Administration Center for Drug Evaluation and Research (CDER) February 2018
Migraine: Developing Drugs for Acute Treatment Guidance for Industry U.S. Department of Health and Human Services Food and Drug Administration Center for Drug Evaluation and Research (CDER) February 2018
flow rate of approximately 2 ml/second for adults and 1-2 ml/second for pediatric patients. The dose is delivered by manual or power injection.
 HIGHLIGHTS OF PRESCRIBING INFORMATION These highlights do not include all the information needed to use DOTAREM safely and effectively. See full prescribing information for DOTAREM. DOTAREM (gadoterate
HIGHLIGHTS OF PRESCRIBING INFORMATION These highlights do not include all the information needed to use DOTAREM safely and effectively. See full prescribing information for DOTAREM. DOTAREM (gadoterate
CONTRAINDICATIONS Clinically important hypersensitivity reactions to DOTAREM. (4)
") M120177AB HIGHLIGHTS OF PRESCRIBING INFORMATION These highlights do not include all the information needed to use DOTAREM safely and effectively. See full prescribing information for DOTAREM. DOTAREM (gadoterate
M120177AB HIGHLIGHTS OF PRESCRIBING INFORMATION These highlights do not include all the information needed to use DOTAREM safely and effectively. See full prescribing information for DOTAREM. DOTAREM (gadoterate
PRODUCT MONOGRAPH GADOVIST 1.0. gadobutrol injection 604 mg/ml (1.0 mmol/ml) For Intravenous Use
 For Intravenous Use") PRODUCT MONOGRAPH GADOVIST 1.0 gadobutrol injection 604 mg/ml (1.0 mmol/ml) For Intravenous Use Contrast Enhancement Agent for Magnetic Resonance Imaging (MRI) Bayer Inc. 77 Belfield Road Toronto, ON M9W
PRODUCT MONOGRAPH GADOVIST 1.0 gadobutrol injection 604 mg/ml (1.0 mmol/ml) For Intravenous Use Contrast Enhancement Agent for Magnetic Resonance Imaging (MRI) Bayer Inc. 77 Belfield Road Toronto, ON M9W
PRODUCT MONOGRAPH GADOVIST 1.0. gadobutrol injection 604 mg/ml (1.0 mmol/ml) For Intravenous Use
 For Intravenous Use") PRODUCT MONOGRAPH GADOVIST 1.0 gadobutrol injection 604 mg/ml (1.0 mmol/ml) For Intravenous Use Contrast Enhancement Agent for Magnetic Resonance Imaging (MRI) For Professional Use Only Bayer Inc. Date
PRODUCT MONOGRAPH GADOVIST 1.0 gadobutrol injection 604 mg/ml (1.0 mmol/ml) For Intravenous Use Contrast Enhancement Agent for Magnetic Resonance Imaging (MRI) For Professional Use Only Bayer Inc. Date
Clinical Study Synopsis
 Clinical Study Synopsis This Clinical Study Synopsis is provided for patients and healthcare professionals to increase the transparency of Bayer's clinical research. This document is not intended to replace
Clinical Study Synopsis This Clinical Study Synopsis is provided for patients and healthcare professionals to increase the transparency of Bayer's clinical research. This document is not intended to replace
Reference ID: MultiHance Injection, NDA , SAFETY LABELING CHANGES UNDER 505(o)(4) MultiHance PI to FDA_Revised Nov _CLEAN.
(4) MultiHance PI to FDA_Revised Nov _CLEAN.") MultiHance Injection, NDA 21-357, SAFETY LABELING CHANGES UNDER 505(o)(4) MultiHance PI to FDA_Revised Nov-18-2010_CLEAN.doc HIGHLIGHTS OF PRESCRIBING INFORMATION These highlights do not include all the
MultiHance Injection, NDA 21-357, SAFETY LABELING CHANGES UNDER 505(o)(4) MultiHance PI to FDA_Revised Nov-18-2010_CLEAN.doc HIGHLIGHTS OF PRESCRIBING INFORMATION These highlights do not include all the
DOTAREMÂ - gadoterate meglumineâ injectionâ Guerbet LLC
 DOTAREMÂ - gadoterate meglumineâ injectionâ Guerbet LLC ---------- HIGHLIGHTS OF PRESCRIBING INFORMATION These highlights do not include all the information needed to use DOTAREM safely and effectively.
DOTAREMÂ - gadoterate meglumineâ injectionâ Guerbet LLC ---------- HIGHLIGHTS OF PRESCRIBING INFORMATION These highlights do not include all the information needed to use DOTAREM safely and effectively.
Contrast-enhanced MRI: how do changing EU regulations impact daily practice?
 Safety considerations in contrast enhanced procedures Carlo Catalano Sapienza University of Rome Contrast-enhanced MRI: how do changing EU regulations impact daily practice? CE MRI: EU regulations - Initial
Safety considerations in contrast enhanced procedures Carlo Catalano Sapienza University of Rome Contrast-enhanced MRI: how do changing EU regulations impact daily practice? CE MRI: EU regulations - Initial
Radiology Reimbursement Information Setting: Hospital Outpatient
 Radiology Reimbursement Information Setting: Hospital Outpatient About DOTAREM: DOTAREM is a gadolinium-based contrast agent indicated for intravenous use with magnetic resonance imaging (MRI) in brain
Radiology Reimbursement Information Setting: Hospital Outpatient About DOTAREM: DOTAREM is a gadolinium-based contrast agent indicated for intravenous use with magnetic resonance imaging (MRI) in brain
Poster No.: B-229 Congress: ECR 2011 Scientific Paper. Authors: E. De Kerviler 1, S. Gaillard 2 ; 1 Paris/FR, 2 Villepinte/FR
 Pharmacovigilance based on spontaneous adverse event reporting of meglumine gadoterate (Gd-DOTA) after 15 million administrations and 20 years of clinical use Poster No.: B-229 Congress: ECR 2011 Type:
Pharmacovigilance based on spontaneous adverse event reporting of meglumine gadoterate (Gd-DOTA) after 15 million administrations and 20 years of clinical use Poster No.: B-229 Congress: ECR 2011 Type:
Contrast media Purpose of using contrast Contrast reaction Nephrotoxicity from contrast Nephrogenic systemic fibrosis When should contrast be used
 Contrast vs Non-Contrast When to Order Stephen McManus, M.D. Contrast media Purpose of using contrast Contrast reaction Nephrotoxicity from contrast Nephrogenic systemic fibrosis When should contrast be
Contrast vs Non-Contrast When to Order Stephen McManus, M.D. Contrast media Purpose of using contrast Contrast reaction Nephrotoxicity from contrast Nephrogenic systemic fibrosis When should contrast be
Report on the Investigation Results
 This English version is intended to be a reference material for the convenience of users. In the event of inconsistency between the Japanese original and this English translation, the former shall Report
This English version is intended to be a reference material for the convenience of users. In the event of inconsistency between the Japanese original and this English translation, the former shall Report
Annex II. Scientific conclusions
 Annex II Scientific conclusions 31 Scientific conclusions In accordance with Article 107k of Directive 2001/83/EC, the CHMP considered the PRAC recommendation adopted on 6 July 2017. Overall summary of
Annex II Scientific conclusions 31 Scientific conclusions In accordance with Article 107k of Directive 2001/83/EC, the CHMP considered the PRAC recommendation adopted on 6 July 2017. Overall summary of
PATIENT INFORMATION LEAFLET. DOTAREM mg/ml, solution for injection (Gadoteric acid)
") PATIENT INFORMATION LEAFLET DOTAREM 279.32 mg/ml, solution for injection (Gadoteric acid) Read all this leaflet carefully before you start using this medicine because it contains important information
PATIENT INFORMATION LEAFLET DOTAREM 279.32 mg/ml, solution for injection (Gadoteric acid) Read all this leaflet carefully before you start using this medicine because it contains important information
Protocol for iv. iodine and gadolinium contrast studies
 Protocol for iv. iodine and gadolinium contrast studies Royal College of Radiologists Standard The individual administering the contrast agent must ensure that the patient understands that it is to be
Protocol for iv. iodine and gadolinium contrast studies Royal College of Radiologists Standard The individual administering the contrast agent must ensure that the patient understands that it is to be
Guidance for Industry Migraine: Developing Drugs for Acute Treatment
 Guidance for Industry Migraine: Developing Drugs for Acute Treatment DRAFT GUIDANCE This guidance document is being distributed for comment purposes only. Comments and suggestions regarding this draft
Guidance for Industry Migraine: Developing Drugs for Acute Treatment DRAFT GUIDANCE This guidance document is being distributed for comment purposes only. Comments and suggestions regarding this draft
Information for Healthcare Professionals and other Stakeholders
 Information for Healthcare Professionals and other Stakeholders NEPHROGENIC SYSTEMIC FIBROSIS: AN UNCOMMON AND DEBILITATING DISEASE POSSIBLY ASSOCIATED WITH GADOLINIUM CHELATES Villepinte, April 2013 Any
Information for Healthcare Professionals and other Stakeholders NEPHROGENIC SYSTEMIC FIBROSIS: AN UNCOMMON AND DEBILITATING DISEASE POSSIBLY ASSOCIATED WITH GADOLINIUM CHELATES Villepinte, April 2013 Any
CONTRAINDICATIONS History of severe hypersensitivity reaction to EOVIST (4)
") HIGHLIGHTS F PRESCRIBING INFRMATIN These highlights do not include all the information needed to use EVIST safely and effectively. See full prescribing information for EVIST Injection. EVIST (gadoxetate
HIGHLIGHTS F PRESCRIBING INFRMATIN These highlights do not include all the information needed to use EVIST safely and effectively. See full prescribing information for EVIST Injection. EVIST (gadoxetate
FULL PRESCRIBING INFORMATION
 HIGHLIGHTS OF PRESCRIBING INFORMATION These highlights do not include all the information needed to use MAGNEVIST safely and effectively. See full prescribing information for MAGNEVIST. MAGNEVIST (gadopentetate
HIGHLIGHTS OF PRESCRIBING INFORMATION These highlights do not include all the information needed to use MAGNEVIST safely and effectively. See full prescribing information for MAGNEVIST. MAGNEVIST (gadopentetate
PRODUCT MONOGRAPH PRIMOVIST. gadoxetate disodium injection mg/ml (0.25 mmol/ml)
") PRODUCT MONOGRAPH PRIMOVIST gadoxetate disodium injection 181.43 mg/ml (0.25 mmol/ml) Intravenous contrast enhancement agent for magnetic resonance imaging (MRI) For Professional Use Only Distributed and
PRODUCT MONOGRAPH PRIMOVIST gadoxetate disodium injection 181.43 mg/ml (0.25 mmol/ml) Intravenous contrast enhancement agent for magnetic resonance imaging (MRI) For Professional Use Only Distributed and
Extracellular gadolinium-based contrast media: An overview
 European Journal of Radiology 66 (2008) 160 167 Extracellular gadolinium-based contrast media: An overview Marie-France Bellin, Aart J. Van Der Molen University Paris-Sud 11, Department of Radiology, University
European Journal of Radiology 66 (2008) 160 167 Extracellular gadolinium-based contrast media: An overview Marie-France Bellin, Aart J. Van Der Molen University Paris-Sud 11, Department of Radiology, University
Guidance for Industry
 Reprinted from FDA s website by Guidance for Industry E14 Clinical Evaluation of QT/QTc Interval Prolongation and Proarrhythmic Potential for Non-Antiarrhythmic Drugs Questions and Answers (R1) U.S. Department
Reprinted from FDA s website by Guidance for Industry E14 Clinical Evaluation of QT/QTc Interval Prolongation and Proarrhythmic Potential for Non-Antiarrhythmic Drugs Questions and Answers (R1) U.S. Department
E11(R1) Addendum to E11: Clinical Investigation of Medicinal Products in the Pediatric Population Step4
 Addendum to E11: Clinical Investigation of Medicinal Products in the Pediatric Population Step4") E11(R1) Addendum to E11: Clinical Investigation of Medicinal Products in the Step4 October 2017 International Council for Harmonisation of Technical Requirements for Pharmaceuticals for Human Use 1 Legal
E11(R1) Addendum to E11: Clinical Investigation of Medicinal Products in the Step4 October 2017 International Council for Harmonisation of Technical Requirements for Pharmaceuticals for Human Use 1 Legal
Amyotrophic Lateral Sclerosis: Developing Drugs for Treatment Guidance for Industry
 Amyotrophic Lateral Sclerosis: Developing Drugs for Treatment Guidance for Industry DRAFT GUIDANCE This guidance document is being distributed for comment purposes only. Comments and suggestions regarding
Amyotrophic Lateral Sclerosis: Developing Drugs for Treatment Guidance for Industry DRAFT GUIDANCE This guidance document is being distributed for comment purposes only. Comments and suggestions regarding
Appropriate Use of Recovery Groups in Nonclinical Toxicity Studies: Value in a Science-Driven Case-by-Case Approach
 Appropriate Use of Recovery Groups in Nonclinical Toxicity Studies: Value in a Science-Driven Case-by-Case Approach Veterinary Pathology 49(2) 357-361 ª The Author(s) 2012 Reprints and permission: sagepub.com/journalspermissions.nav
Appropriate Use of Recovery Groups in Nonclinical Toxicity Studies: Value in a Science-Driven Case-by-Case Approach Veterinary Pathology 49(2) 357-361 ª The Author(s) 2012 Reprints and permission: sagepub.com/journalspermissions.nav
Year in review. Vit Perlik Director of Regulatory Science and Clinical Development
 Year in review Vit Perlik Director of Regulatory Science and Clinical Development Content Year in review Covering September 2013 to September 2014 Where the regulation goes selection of events for illustration
Year in review Vit Perlik Director of Regulatory Science and Clinical Development Content Year in review Covering September 2013 to September 2014 Where the regulation goes selection of events for illustration
Policy #: 291 Latest Review Date: February 2013
 Effective for dates of service on or after April 1, 2013, refer to: https://www.bcbsal.org/providers/policies/carecore.cfm Name of Policy: Magnetic Resonance Angiography (MRA) of the Chest (excluding the
Effective for dates of service on or after April 1, 2013, refer to: https://www.bcbsal.org/providers/policies/carecore.cfm Name of Policy: Magnetic Resonance Angiography (MRA) of the Chest (excluding the
Gadolinium Deposition in the Brain: Summary of Known Science and Recommendations from the International Society for Magnetic Resonance in Medicine
 Gadolinium Deposition in the Brain: Summary of Known Science and Recommendations from the International Society for Magnetic Resonance in Medicine Vikas Gulani, MD, PhD 1-4*, Fernando Calamante, PhD 5,6,
Gadolinium Deposition in the Brain: Summary of Known Science and Recommendations from the International Society for Magnetic Resonance in Medicine Vikas Gulani, MD, PhD 1-4*, Fernando Calamante, PhD 5,6,
Safety Assessment in Clinical Trials and Beyond
 Safety Assessment in Clinical Trials and Beyond Yuliya Yasinskaya, MD Medical Team Leader Division of Anti-Infective Products Center for Drug Evaluation and Research FDA Clinical Investigator Training
Safety Assessment in Clinical Trials and Beyond Yuliya Yasinskaya, MD Medical Team Leader Division of Anti-Infective Products Center for Drug Evaluation and Research FDA Clinical Investigator Training
Instructions for the safe administration of gadolinium based contrast agents (GBCA). 1. PREADMINISTRATION PRECAUTIONS:
. 1. PREADMINISTRATION PRECAUTIONS:") POLICY NUMBER: RM 6-18 CATEGORY: Patient Care DATE: June 2016 NEXT REVIEW DATE: April 2017 SUBJECT: PURPOSE: POLICY: Intravenous Gadolinium Based Contrast Administration To prevent complications associated
POLICY NUMBER: RM 6-18 CATEGORY: Patient Care DATE: June 2016 NEXT REVIEW DATE: April 2017 SUBJECT: PURPOSE: POLICY: Intravenous Gadolinium Based Contrast Administration To prevent complications associated
Summary of product characteristics (SmPC)
") Summary of product characteristics (SmPC) What is it and what does it contain? An agency of the European Union Table of contents 1.What is the summary of product characteristics (SmPC)? 2.Where SmPC information
Summary of product characteristics (SmPC) What is it and what does it contain? An agency of the European Union Table of contents 1.What is the summary of product characteristics (SmPC)? 2.Where SmPC information
PRODUCT INFORMATION DOTAREM
 PRODUCT INFORMATION DOTAREM NAME OF THE MEDICINE DOTAREM (gadoteric acid) paramagnetic contrast medium for Magnetic Resonance Imaging (MRI). The structural formula of gadoteric acid is shown below: The
PRODUCT INFORMATION DOTAREM NAME OF THE MEDICINE DOTAREM (gadoteric acid) paramagnetic contrast medium for Magnetic Resonance Imaging (MRI). The structural formula of gadoteric acid is shown below: The
SUMMARY OF PRODUCT CHARACTERISTICS
 SUMMARY OF PRODUCT CHARACTERISTICS 1. NAME OF THE MEDICINAL PRODUCT DOTAREM 0.5 mmol/ml solution for injection in vials 2. QUALITATIVE AND QUANTITATIVE COMPOSITION 1 ml solution for injection contains
SUMMARY OF PRODUCT CHARACTERISTICS 1. NAME OF THE MEDICINAL PRODUCT DOTAREM 0.5 mmol/ml solution for injection in vials 2. QUALITATIVE AND QUANTITATIVE COMPOSITION 1 ml solution for injection contains
NDA NDA APPROVAL
 DEPARTMENT OF HEALTH AND HUMAN SERVICES Food and Drug Administration Silver Spring MD 20993 NDA 022200 NDA APPROVAL Amylin Pharmaceuticals, Inc. Orville Kolterman, M.D. Sr. Vice President, Research & Development
DEPARTMENT OF HEALTH AND HUMAN SERVICES Food and Drug Administration Silver Spring MD 20993 NDA 022200 NDA APPROVAL Amylin Pharmaceuticals, Inc. Orville Kolterman, M.D. Sr. Vice President, Research & Development
Incidence of Immediate Gadolinium Contrast Media Reactions
 Health Care Policy and Quality Original Research Prince et al. Immediate Gadolinium Contrast Media Reactions Health Care Policy and Quality Original Research Martin R. Prince 1 Honglei Zhang Zhitong Zou
Health Care Policy and Quality Original Research Prince et al. Immediate Gadolinium Contrast Media Reactions Health Care Policy and Quality Original Research Martin R. Prince 1 Honglei Zhang Zhitong Zou
File NDA SE5 046/047, NDA SE5 036/037, NDA SE5 020/021
 M E M O R A N D U M DEPARTMENT OF HEALTH AND HUMAN SERVICES PUBLIC HEALTH SERVICE FOOD AND DRUG ADMINISTRATION CENTER FOR DRUG EVALUATION AND RESEARCH DATE: 18 June 2007 FROM: TO: SUBJECT: Mitchell V.
M E M O R A N D U M DEPARTMENT OF HEALTH AND HUMAN SERVICES PUBLIC HEALTH SERVICE FOOD AND DRUG ADMINISTRATION CENTER FOR DRUG EVALUATION AND RESEARCH DATE: 18 June 2007 FROM: TO: SUBJECT: Mitchell V.
Inspections, Compliance, Enforcement, and Criminal Investigations
 Home Inspections, Compliance, Enforcement, and Criminal Investigations Enforcement Actions Warning Letters Inspections, Compliance, Enforcement, and Criminal Investigations Stanislaw R Burzynski, MD 12/3/13
Home Inspections, Compliance, Enforcement, and Criminal Investigations Enforcement Actions Warning Letters Inspections, Compliance, Enforcement, and Criminal Investigations Stanislaw R Burzynski, MD 12/3/13
Zinplava Swiss Risk Management Plan Summary V1.5. Swiss Summary of the Risk Management Plan (RMP) for. Zinplava. (Bezlotoxumab 1000mg)
 for. Zinplava. (Bezlotoxumab 1000mg)") Swiss Summary of the Risk Management Plan (RMP) for (Bezlotoxumab 1000mg) Concentrate for solution for infusion Version 1.5 (November 2016) The Risk Management Plan (RMP) is a comprehensive document submitted
Swiss Summary of the Risk Management Plan (RMP) for (Bezlotoxumab 1000mg) Concentrate for solution for infusion Version 1.5 (November 2016) The Risk Management Plan (RMP) is a comprehensive document submitted
SUMMARY OF PRODUCT CHARACTERISTICS
 SUMMARY OF PRODUCT CHARACTERISTICS 1. NAME OF THE MEDICINAL PRODUCT DOTAREM 0.5 mmol/ml (for multiple use) solution for injection in vials 2. QUALITATIVE AND QUANTITATIVE COMPOSITION 1 ml solution for
SUMMARY OF PRODUCT CHARACTERISTICS 1. NAME OF THE MEDICINAL PRODUCT DOTAREM 0.5 mmol/ml (for multiple use) solution for injection in vials 2. QUALITATIVE AND QUANTITATIVE COMPOSITION 1 ml solution for
Record of the Consultation on Pharmacogenomics/Biomarkers
 This English version of the record of the consultation has been published by PMDA. In the event of inconsistency between the Japanese original and this English translation, the former shall prevail. (Attachment
This English version of the record of the consultation has been published by PMDA. In the event of inconsistency between the Japanese original and this English translation, the former shall prevail. (Attachment
Clinical Endpoint Bioequivalence Study Review in ANDA Submissions. Ying Fan, Ph.D.
 Clinical Endpoint Bioequivalence Study Review in ANDA Submissions Ying Fan, Ph.D. 1 Disclaimer This presentation constitutes an informal communication that represents the best judgment of the speaker at
Clinical Endpoint Bioequivalence Study Review in ANDA Submissions Ying Fan, Ph.D. 1 Disclaimer This presentation constitutes an informal communication that represents the best judgment of the speaker at
IL PROBLEMA DEL GADOLINIO OGGI
 11-12 Maggio 2018 Rimini IL PROBLEMA DEL GADOLINIO OGGI Il punto di vista del Neuroradiologo pediatrico Dott.ssa Camilla Rossi Espagnet U.O. Neuroradiologia, Dipartimento Immagini, Ospedale Pediatrico
11-12 Maggio 2018 Rimini IL PROBLEMA DEL GADOLINIO OGGI Il punto di vista del Neuroradiologo pediatrico Dott.ssa Camilla Rossi Espagnet U.O. Neuroradiologia, Dipartimento Immagini, Ospedale Pediatrico
Clinical Study Synopsis
 Clinical Study Synopsis This Clinical Study Synopsis is provided for patients and healthcare professionals to increase the transparency of Bayer's clinical research. This document is not intended to replace
Clinical Study Synopsis This Clinical Study Synopsis is provided for patients and healthcare professionals to increase the transparency of Bayer's clinical research. This document is not intended to replace
The MOVE Trial Summary. Palovarotene FAQs. 1. What is the MOVE Trial?
 MOVE TRIAL FAQS Contents The MOVE Trial Summary... 1 1. What is the MOVE Trial?... 1 Palovarotene FAQs... 1 2. What is palovarotene?... 1 3. What is an Orphan designation?... 2 4. What is a Fast Track
MOVE TRIAL FAQS Contents The MOVE Trial Summary... 1 1. What is the MOVE Trial?... 1 Palovarotene FAQs... 1 2. What is palovarotene?... 1 3. What is an Orphan designation?... 2 4. What is a Fast Track
Sponsor/Company: sanofi-aventis Drug substance: Elitek/Fasturtec (rasburicase, SR29142)
") These results are supplied for informational purposes only. Prescribing decisions should be made based on the approved package insert in the country of prescription. Sponsor/Company: sanofi-aventis Drug
These results are supplied for informational purposes only. Prescribing decisions should be made based on the approved package insert in the country of prescription. Sponsor/Company: sanofi-aventis Drug
Bevacizumab for the treatment of recurrent advanced ovarian cancer
 Bevacizumab for the treatment of recurrent advanced ovarian cancer ERRATUM This report was commissioned by the NIHR HTA Programme as project number 11/40 Page 2 This document contains errata in respect
Bevacizumab for the treatment of recurrent advanced ovarian cancer ERRATUM This report was commissioned by the NIHR HTA Programme as project number 11/40 Page 2 This document contains errata in respect
Public Assessment Report for paediatric studies submitted in accordance with Article 45 and 46 of Regulation (EC) No1901/2006, as amended
 No1901/2006, as amended") Public Assessment Report for paediatric studies submitted in accordance with Article 45 and 46 of Regulation (EC) No1901/2006, as amended Gadopentetic acid (Gadopentate) Magnevist UK/W/049/pdWS/001 Rapporteur:
Public Assessment Report for paediatric studies submitted in accordance with Article 45 and 46 of Regulation (EC) No1901/2006, as amended Gadopentetic acid (Gadopentate) Magnevist UK/W/049/pdWS/001 Rapporteur:
Reference ID:
 HIGHLIGHTS OF PRESCRIBING INFORMATION These highlights do not include all the information needed to use TRIFERIC safely and effectively. See full prescribing information for TRIFERIC. TRIFERIC (ferric
HIGHLIGHTS OF PRESCRIBING INFORMATION These highlights do not include all the information needed to use TRIFERIC safely and effectively. See full prescribing information for TRIFERIC. TRIFERIC (ferric
Study Center(s): The study was conducted at 39 study sites in Japan.
: The study was conducted at 39 study sites in Japan.") SYNOPSIS Issue Date: 20 NOVEMBER 2012 Name of Sponsor/Company Janssen Pharmaceutical K. K. Name of Finished Product CONCERTA Name of Active Ingredient(s) Methylphenidate HCl Protocol No.: JNS001-JPN-A01
SYNOPSIS Issue Date: 20 NOVEMBER 2012 Name of Sponsor/Company Janssen Pharmaceutical K. K. Name of Finished Product CONCERTA Name of Active Ingredient(s) Methylphenidate HCl Protocol No.: JNS001-JPN-A01
Billing Guide. For Freestanding Imaging Centers Provided by Bayer HealthCare
 Billing Guide For Freestanding Imaging Centers Provided by Bayer HealthCare Introduction Bayer HealthCare Inc. offers this billing guide to freestanding imaging center customers as part of its comprehensive
Billing Guide For Freestanding Imaging Centers Provided by Bayer HealthCare Introduction Bayer HealthCare Inc. offers this billing guide to freestanding imaging center customers as part of its comprehensive
PFIZER INC. THERAPEUTIC AREA AND FDA APPROVED INDICATIONS: See USPI
 PFIZER INC. These results are supplied for informational purposes only. Prescribing decisions should be made based on the approved package insert. For publications based on this study, see associated bibliography.
PFIZER INC. These results are supplied for informational purposes only. Prescribing decisions should be made based on the approved package insert. For publications based on this study, see associated bibliography.
Sponsor Novartis. Generic Drug Name Vildagliptin/Metformin. Therapeutic Area of Trial Type 2 diabetes. Approved Indication Type 2 diabetes
 Clinical Trial Results Database Page 1 Sponsor Novartis Generic Drug Name Vildagliptin/Metformin Therapeutic Area of Trial Type 2 diabetes Approved Indication Type 2 diabetes Study Number CLMF237A2309
Clinical Trial Results Database Page 1 Sponsor Novartis Generic Drug Name Vildagliptin/Metformin Therapeutic Area of Trial Type 2 diabetes Approved Indication Type 2 diabetes Study Number CLMF237A2309
Clinical Study Synopsis
 Clinical Study Synopsis This Clinical Study Synopsis is provided for patients and healthcare professionals to increase the transparency of Bayer's clinical research. This document is not intended to replace
Clinical Study Synopsis This Clinical Study Synopsis is provided for patients and healthcare professionals to increase the transparency of Bayer's clinical research. This document is not intended to replace
The EU PIP - a step in Pediatric Drug Development. Thomas Severin Bonn,
 The EU PIP - a step in Pediatric Drug Development Thomas Severin Bonn, 13.01.2009 Agenda Implications for Industry Company Preparation Time of PIP Submission Content of the PIP The PIP Process and first
The EU PIP - a step in Pediatric Drug Development Thomas Severin Bonn, 13.01.2009 Agenda Implications for Industry Company Preparation Time of PIP Submission Content of the PIP The PIP Process and first
Benefits and Risks of Cancer Imaging
 Benefits and Risks of Cancer Imaging Jeffrey T. Yap, PhD http://catalyst.harvard.edu/ services/imagingconsulting.html Senior Diagnostic Physicist, Department of Imaging, DFCI Assistant Professor of Radiology,
Benefits and Risks of Cancer Imaging Jeffrey T. Yap, PhD http://catalyst.harvard.edu/ services/imagingconsulting.html Senior Diagnostic Physicist, Department of Imaging, DFCI Assistant Professor of Radiology,
Hepatobiliary Contrast Agents for Liver MRI
 Hepatobiliary Contrast Agents for Liver MRI Scott B. Reeder, MD, PhD International Society for Magnetic Resonance in Medicine Sociedad Mexicana de Radiologia e Imagen (SMRI) Mexico City June 4, 2014 Department
Hepatobiliary Contrast Agents for Liver MRI Scott B. Reeder, MD, PhD International Society for Magnetic Resonance in Medicine Sociedad Mexicana de Radiologia e Imagen (SMRI) Mexico City June 4, 2014 Department
Electrical impedance scanning of the breast is considered investigational and is not covered.
 ARBenefits Approval: 09/28/2011 Effective Date: 01/01/2012 Revision Date: Code(s): Medical Policy Title: Electrical Impedance Scanning of the Breast Document: ARB0127 Administered by: Public Statement:
ARBenefits Approval: 09/28/2011 Effective Date: 01/01/2012 Revision Date: Code(s): Medical Policy Title: Electrical Impedance Scanning of the Breast Document: ARB0127 Administered by: Public Statement:
General Considerations for Age-Appropriate Formulations: FDA Clinical Perspective
 General Considerations for Age-Appropriate Formulations: FDA Clinical Perspective Erica Radden, M.D. Medical Officer, Division of Pediatric and Maternal Health Office of New Drugs, FDA 1 Disclosure Statement
General Considerations for Age-Appropriate Formulations: FDA Clinical Perspective Erica Radden, M.D. Medical Officer, Division of Pediatric and Maternal Health Office of New Drugs, FDA 1 Disclosure Statement
Manual for Expedited Reporting of Adverse Events to DAIDS Version 2.0 January 2010
 Manual for Expedited Reporting of Adverse Events to DAIDS Version 2.0 January 2010 January 2010 Version 2.0 Table of Contents 1. INTRODUCTION...1 1.1 Scope... 1 1.2 Purpose... 1 1.3 Responsibilities...
Manual for Expedited Reporting of Adverse Events to DAIDS Version 2.0 January 2010 January 2010 Version 2.0 Table of Contents 1. INTRODUCTION...1 1.1 Scope... 1 1.2 Purpose... 1 1.3 Responsibilities...
Drug Regulatory Affairs. Lyxumia. Summary of the Risk Management Plan (RMP) for Lyxumia (lixisenatide)
 for Lyxumia (lixisenatide)") Drug Regulatory Affairs Lyxumia Summary of the Risk Management Plan (RMP) for Lyxumia (lixisenatide) Document version: 02 Document date: 08-Mar-2018 1 Summary of the risk management plan (RMP) for Lyxumia
Drug Regulatory Affairs Lyxumia Summary of the Risk Management Plan (RMP) for Lyxumia (lixisenatide) Document version: 02 Document date: 08-Mar-2018 1 Summary of the risk management plan (RMP) for Lyxumia
SUBJECT: ASSESSMENT OF LAB RESULTS PRIOR TO INJECTING GADOLINIUM-BASED CONTRAST MEDIA.
 ASSESSMENT OF PATIENTS: IMAGING SERVICES Policy #: 300.02 Effective Date: 6/29/09 Last Revision Date: 10/07 Page 1 of 5 SUBJECT: ASSESSMENT OF LAB RESULTS PRIOR TO INJECTING GADOLINIUM-BASED CONTRAST MEDIA
ASSESSMENT OF PATIENTS: IMAGING SERVICES Policy #: 300.02 Effective Date: 6/29/09 Last Revision Date: 10/07 Page 1 of 5 SUBJECT: ASSESSMENT OF LAB RESULTS PRIOR TO INJECTING GADOLINIUM-BASED CONTRAST MEDIA
PRAC recommendations on signals for update of the product information
 Thursday 22 January 2015 EMA/PRAC/62352/2015 Pharmacovigilance Risk Assessment Committee PRAC recommendations on signals for update of the product information Adopted at the 6-9 January 2015 PRAC 1. Atorvastatin,
Thursday 22 January 2015 EMA/PRAC/62352/2015 Pharmacovigilance Risk Assessment Committee PRAC recommendations on signals for update of the product information Adopted at the 6-9 January 2015 PRAC 1. Atorvastatin,
Study Number CAIN457C2302 (core study) and CAIN457C2302E1 (extension study)
 and CAIN457C2302E1 (extension study)") Clinical Trial Results Database Page 1 Sponsor Novartis Generic Drug Name Secukinumab Therapeutic Area of Trial Uveitis Approved Indication Investigational Study Number CC2302 (core study) and CC2302E1
Clinical Trial Results Database Page 1 Sponsor Novartis Generic Drug Name Secukinumab Therapeutic Area of Trial Uveitis Approved Indication Investigational Study Number CC2302 (core study) and CC2302E1
Review Report. Inavir Dry Powder Inhaler 20 mg
 Review Report June 27, 2016 Pharmaceuticals and Medical Devices Agency The following are the results of a review of the following pharmaceutical product submitted for marketing approval conducted by the
Review Report June 27, 2016 Pharmaceuticals and Medical Devices Agency The following are the results of a review of the following pharmaceutical product submitted for marketing approval conducted by the
Public Assessment Report for paediatric studies submitted in accordance with Article 45 of Regulation (EC) No1901/2006, as amended.
 No1901/2006, as amended.") Public Assessment Report for paediatric studies submitted in accordance with Article 45 of Regulation (EC) No1901/2006, as amended Omniscan UK/W/063/pdWS/001 Rapporteur UK Finalisation procedure (day 20)
Public Assessment Report for paediatric studies submitted in accordance with Article 45 of Regulation (EC) No1901/2006, as amended Omniscan UK/W/063/pdWS/001 Rapporteur UK Finalisation procedure (day 20)
The clinical trial information provided in this public disclosure synopsis is supplied for informational purposes only.
 The clinical trial information provided in this public disclosure synopsis is supplied for informational purposes only. Please note that the results reported in any single trial may not reflect the overall
The clinical trial information provided in this public disclosure synopsis is supplied for informational purposes only. Please note that the results reported in any single trial may not reflect the overall
Sponsor / Company: Sanofi Drug substance(s): SAR (iniparib)
: SAR (iniparib)") These results are supplied for informational purposes only. Prescribing decisions should be made based on the approved package insert in the country of prescription. Sponsor / Company: Sanofi Drug substance(s):
These results are supplied for informational purposes only. Prescribing decisions should be made based on the approved package insert in the country of prescription. Sponsor / Company: Sanofi Drug substance(s):
This document was created on 07 January 2005 and has been printed from
 1 Spontaneous Adverse Event Reporting Information Introduction This document provides information regarding spontaneous adverse event reports received concerning patients taking rosuvastatin, which relates
1 Spontaneous Adverse Event Reporting Information Introduction This document provides information regarding spontaneous adverse event reports received concerning patients taking rosuvastatin, which relates
Oncology Drug Development: A Regulatory Perspective Faculty Presenter
 2017 Society for Translational Oncology Fellows Forum Oncology Drug Development: A Regulatory Perspective Faculty Presenter Tatiana Prowell, MD, Breast Cancer Scientific Lead, FDA Office of Hematology
2017 Society for Translational Oncology Fellows Forum Oncology Drug Development: A Regulatory Perspective Faculty Presenter Tatiana Prowell, MD, Breast Cancer Scientific Lead, FDA Office of Hematology
Idelalisib Benefit assessment according to 35a Social Code Book V 1
 IQWiG Reports Commission No. A14-35 Idelalisib Benefit assessment according to 35a Social Code Book V 1 Extract 1 Translation of Assessment module I, Sections I 2.1 to I 2.9, and Assessment module II,
IQWiG Reports Commission No. A14-35 Idelalisib Benefit assessment according to 35a Social Code Book V 1 Extract 1 Translation of Assessment module I, Sections I 2.1 to I 2.9, and Assessment module II,
The clinical trial information provided in this public disclosure synopsis is supplied for informational purposes only.
 The clinical trial information provided in this public disclosure synopsis is supplied for informational purposes only. Please note that the results reported in any single trial may not reflect the overall
The clinical trial information provided in this public disclosure synopsis is supplied for informational purposes only. Please note that the results reported in any single trial may not reflect the overall
Considerations for Summary Review of Supplemental NDA/BLA Submissions in Oncology
 Considerations for Summary Review of Supplemental NDA/BLA Submissions in Oncology Considerations for Summary Review of Supplemental NDA/BLA Submissions in Oncology Rachel Sherman, MD Greenleaf Health LLC
Considerations for Summary Review of Supplemental NDA/BLA Submissions in Oncology Considerations for Summary Review of Supplemental NDA/BLA Submissions in Oncology Rachel Sherman, MD Greenleaf Health LLC