CARCINOGENESIS: THE MOLECULAR BASIS OF CANCER
|
|
|
- Corey Lewis
- 6 years ago
- Views:
Transcription
1 CARCINOGENESIS: THE MOLECULAR BASIS OF CANCER
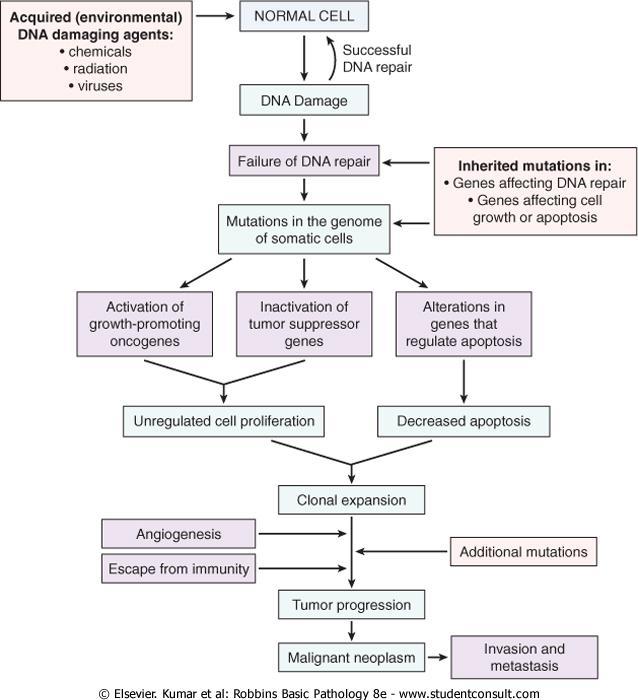
2 Nonlethal genetic damage lies at the heart of carcinogenesis. Mutation may be acquired by the action of environmental agents, such as chemicals, radiation, or viruses, or it may be inherited in the germ line.
3 Tumors are monoclonal
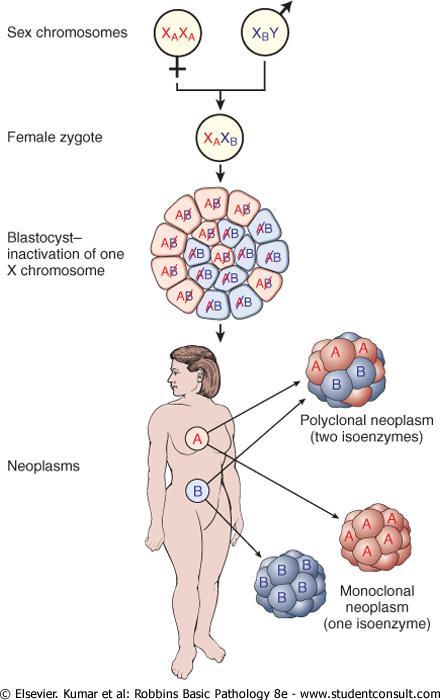
4
5 Four classes of normal regulatory genes are involved : 1-growth-promoting proto-oncogenes, 2-growth-inhibiting tumor suppressor genes, 3-genes that regulate apoptosis 4-genes involved in DNA
6
7 Mutant alleles of proto-oncogenes are called oncogenes. They are dominant because mutation of a single allele can lead to cellular transformation. Tumor suppressor genes are recessive.
8 Genes that regulate apoptosis may be dominant or recessive.
9 Carcinogenesis is a multistep process at both the phenotypic and the genetic levels resulting from the accumulation of multiple mutations.
10 Tumor progression
11 Tumor progression is associated with generation of subclones with different characteristics
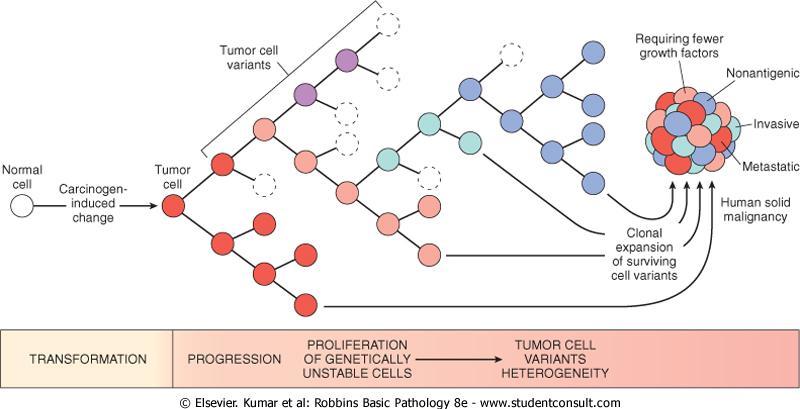
12
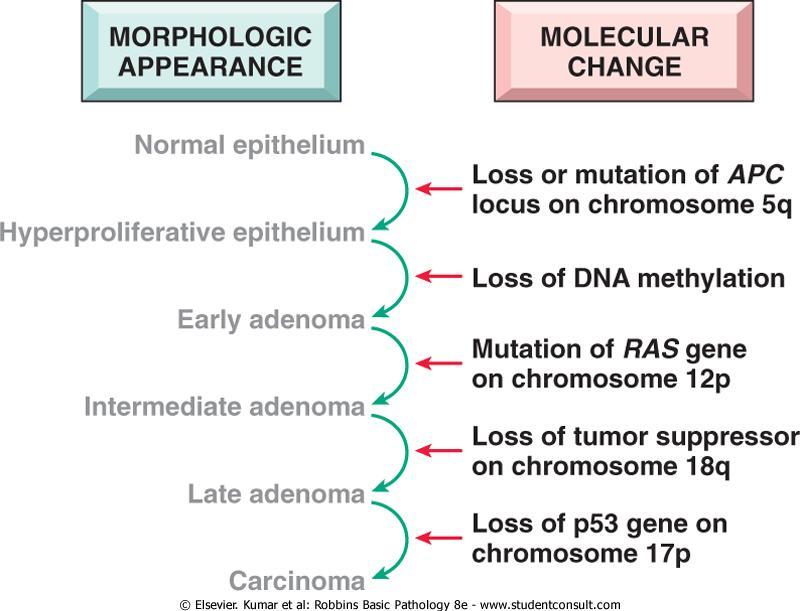
13
14 Features of malignent cells 1-Self-sufficiency in growth signals 2-Insensitivity to growth-inhibitory signals 3-Evasion of apoptosis 4-Limitless replicative potential (i.e., overcoming cellular senescence and avoiding mitotic catastrophe) 5-Development of sustained angiogenesis 6-Ability to invade and metastasize 7-Genomic instability resulting from defects in DNA repair
15 Self-Sufficiency in Growth Signals Genes that promote autonomous cell growth in cancer cells are called oncogenes. Oncogenes promote cell growth in the absence of normal growth-promoting signals. Their products, called oncoproteins Oncoproteins are devoid of important regulatory elements.
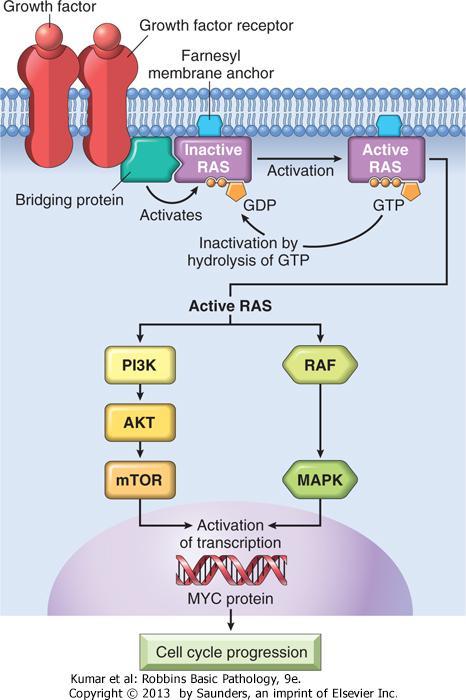
16 Growth Factors All normal cells require stimulation by growth factors to undergo proliferation. Types : 1- paracrine action. 2-autocrine action
17 Glioblastomas secrete PDGF and express its receptor. Many sarcomas make both TGF-α and its receptor. Fibroblast growth factors (e.g., hst-1 and FGF3) GIT and breast tumors. FGF-2 is expressed in human melanomas but not normal melanocytes.
18 HGF and its receptor c-met are both overexpressed in follicular carcinomas of the thyroid.
19 Growth Factor Receptors Mutant receptor proteins deliver continuous mitogenic signals to cells, even in the absence of the growth factor in the environment.
20 E.g overexpression involve EGF receptor family. ERBB1 the EGF receptor, is overexpressed in 80% of squamous cell carcinomas of the lung. In 50% or more of glioblastomas. In % of epithelial tumors of the head and neck.
21 HER2/NEU (ERBB2), is amplified in 25-30% of breast cancers and adenocarcinomas of the lung, ovary, and salivary glands. High level of HER2/NEU protein in breast cancer cells is a poor prognosis.
22 Treatment of breast cancer with anti- HER2/NEU antibody (Herciptin ) proved to be clinically effective.
23 Signal-Transducing Proteins Two important members in this category are 1-RAS gene 2-ABL gene
24 Approximately 30% of all human tumors contain mutated versions of the RAS gene.
25 RAS is a member of a family of small G proteins that bind guanosine nucleotides (guanosine triphosphate [GTP] and guanosine diphosphate [GDP]).
26 RAS proteins are inactive when bound to GDP. stimulation of cells by growth factors leads to exchange of GDP for GTP and subsequent activation of RAS.
27 Intrinsic guanosine triphosphatase (GTPase) activity hydrolyzes GTP to GDP, releasing a phosphate group and returning the protein to its quiescent inactive state.
28 GTPase-activating proteins (GAPs) prevent uncontrolled RAS activation by favoring hydrolysis of GTP to GDP.
29 The RAS gene is most commonly activated by point mutations. Point mutations can affect : 1-GTP-binding pocket 2-The enzymatic region essential for GTP hydrolysis.
30
31 mutations in RAS protein would be mimicked by mutations in the GAPs that fail to restrain normal RAS proteins. E.g mutation of neurofibromin 1, a GAP, is associated with familial neurofibromatosis type 1
32 The ABL proto-oncogene has tyrosine kinase activity. In chronic myeloid leukemia (CML) and acute lymphocytic leukemias.
33 ABL gene is translocated from its normal site on chromosome 9 to chromosome 22, where it fuses with part of the breakpoint cluster region (BCR) gene = Philadelphia (Ph) chromosome
34
35 The BCR-ABL hybrid protein has potent, unregulated tyrosine kinase activity, which activates several pathways, including the RAS-RAF cascade. Normal ABL protein localizes in the nucleus and promotes apoptosis of cells that suffer DNA damage.
36 The BCR-ABL gene cannot perform this function because it is retained in the cytoplasm as a result of abnormal tyrosine kinase activity.
37 A cell with BCR-ABL fusion gene is dysregulated in two ways: 1- inappropriate tyrosine kinase activity leads to growth autonomy. 2- impairment of apoptosis.
38 The patients with chronic myeloid leukemia respond to an inhibitor of the BCR-ABL fusion kinase called imatinib mesylate (Gleevec).
39 Nuclear Transcription Factors MYC, MYB, JUN, FOS, and REL oncogenes, function as transcription factors that regulate the expression of growth-promoting genes such as cyclins.
40 the MYC gene is involved most commonly in human tumors. The MYC proto-oncogene is expressed in virtually all cells the MYC protein is induced rapidly when quiescent cells receive a signal to divide.
41 In normal cells, MYC levels decline to near basal level when the cell cycle begins. MYC gene are associated with persistent expression or overexpression contributing to sustained proliferation.
42 MYC promotes cyclin-dependent kinases (CDKs) whose products drive cells into the cell cycle. Genes repressed by MYC repress the CDK inhibitors (CDKIs).
43 MYC promotes tumorigenesis by increasing expression of genes that promote progression through the cell cycle and repressing genes that slow or prevent progression through the cell cycle.
44 MYC gene overexpression occurs from (8;14) translocation in Burkitt lymphoma. MYC is also amplified in breast, colon, lung, and many other cancers. N-MYC and L-MYC genes are amplified in neuroblastomas and small-cell cancers of lung.
45 Cyclins and Cyclin-Dependent Kinases (CDKs) Progression of cells through the various phases of the cell cycle is controlled by CDKs. CDKs are activated by binding to cyclins.
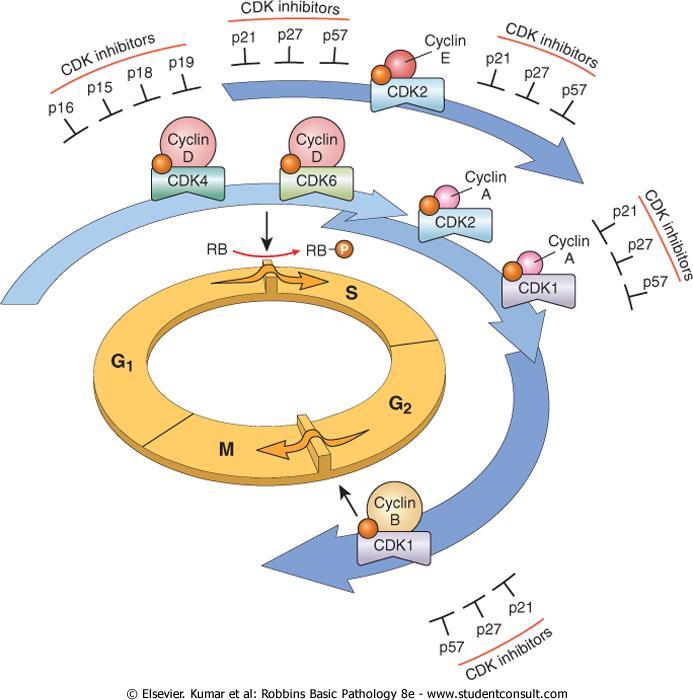
46
47 The CDK-cyclin complexes phosphorylate crucial target proteins that drive the cell through the cell cycle. On completion of this task, cyclin levels decline rapidly.
48 More than 15 cyclins have been identified; cyclins D, E, A, and B appear sequentially during the cell cycle and bind to one or more CDK.
49 The cyclin D genes are overexpressed in many cancers, including those affecting the breast, esophagus, liver, and a subset of lymphomas.
50 Amplification of the CDK4 gene occurs in melanomas, sarcomas, and glioblastomas.
51 CDK inhibitors (CDKIs) silence the CDKs and exert negative control over the cell cycle. One family of CDKIs, composed of three proteins : 1- p21 [CDKN1A], 2-p27 [CDKN1B], 3-p57 [CDKN1C], inhibits the CDKs broadly
52 The other family of CDKIs has selective effects on cyclin D/CDK4 and cyclin D/CDK6. The four members of this family : 1-p15 [CDKN2B] 2-p16 [CDKN2A] 3-p18 [CDKN2C] 4-p19 [CDKN2D] are sometimes called INK4 (A-D) proteins.
53 Germ-line mutations of p16(cdkn2a) are associated with: 25% of melanoma. 75% of pancreatic carcinomas 40% to 70% of glioblastomas 50% of esophageal cancers 20% of non-small-cell lung carcinomas, soft tissue sarcomas, and bladder cancers.
54 Insensitivity to Growth-Inhibitory Signals Retinoblastoma (RB) gene the first and prototypic cancer suppressor gene to be discovered. Retinoblastoma is an uncommon childhood tumor. Approximately 60% of retinoblastomas are sporadic, and 40% are familial.
55 To account for the sporadic and familial occurrence of an identical tumor, Knudson, in 1974, proposed his now famous two-hit hypothesis.
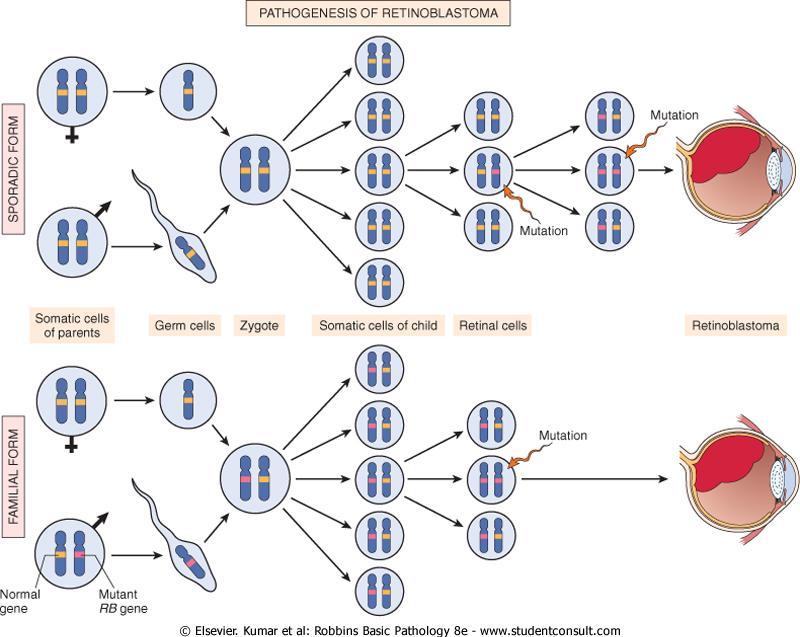
56
57 In familial cases, children inherit one defective copy of the RB gene in the germ line, the other copy is normal. Retinoblastoma develops when the normal RB gene is lost in retinoblasts as a result of somatic mutation.
58 In sporadic cases, both normal RB alleles are lost by somatic mutation in one of the retinoblasts. A retinal cell that has lost both of the normal copies of the RB gene becomes cancerous
59 RB gene mutation was discovered initially in retinoblastomas. Homozygous loss of this gene is a fairly common event in several tumors including : breast cancer. small-cell cancer of the lung. bladder cancer.
60 Patients with familial retinoblastoma also are at greatly increased risk of developing osteosarcomas and some soft tissue sarcomas.
61 RB Gene and Cell Cycle It exists in an active hypophosphorylated and an inactive hyperphosphorylated state. The importance of RB lies in its enforcement of G 1, or the gap between mitosis (M) and DNA replication (S).
62 Two gaps are incorporated into the cell cycle: 1-Gap 1 (G 1 ) between mitosis (M) and DNA replication (S) 2-Gap 2 (G 2 ) between DNA replication (S) and mitosis (M)
63 The transition from G 1 to S is believed to be an extremely important checkpoint in the cell cycle clock. Once cells cross the G 1 checkpoint they can pause the cell cycle for a time but they are obligated to complete mitosis.
64 In G 1 cells can exit the cell cycle : 1- temporarily, called quiescence 2- permanently, called senescence.
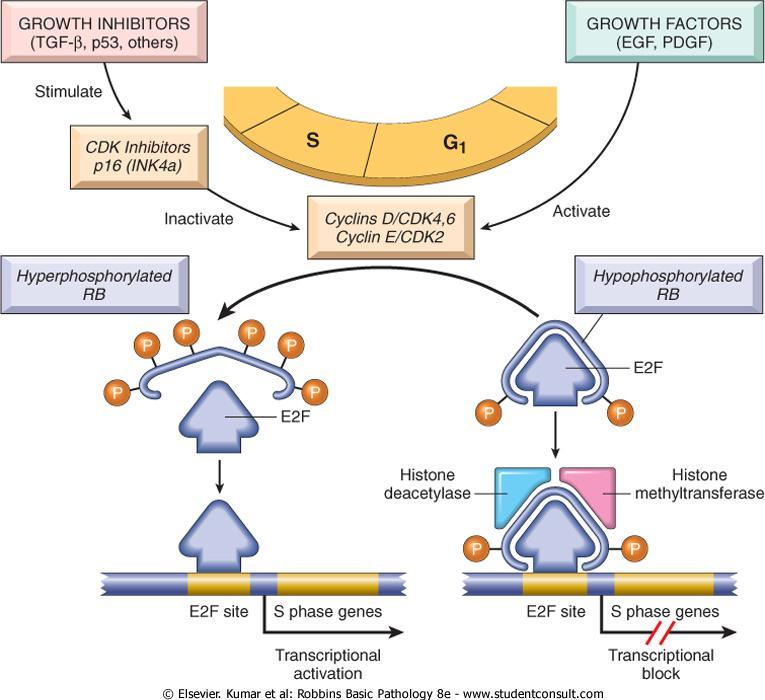
65
66 The initiation of DNA replication requires the activity of cyclin E/CDK2 complexes. The expression of cyclin E is dependent on the E2F family of transcription factors.
67 Early in G 1, RB is in its hypophosphorylated active form, and it binds to and inhibits the E2F family of transcription factors, preventing transcription of cyclin E.
68 Hypophosphorylated RB blocks E2F-mediated transcription in at least two ways : 1- it sequesters E2F preventing it from interacting with other transcriptional activators. 2- RB recruits chromatin remodeling proteins, such as histone deacetylases and histone methyltransferases, which bind to the promoters of E2F-responsive genes such as cyclin E. These enzymes modify chromatin at the promoters to make DNA insensitive to transcription factors.
69 Expression of cyclin E then stimulates DNA replication and progression through the cell cycle.
70 During the ensuing M phase the phosphate groups are removed from RB by cellular phosphatases, regenerating the hypophosphorylated (active ) form of RB.
71 Mutations in other genes that control RB phosphorylation can mimic the effect of RB loss such genes are mutated in many cancers that seem to have normal RB genes.
72 E.g mutational activation of CDK4 or overexpression of cyclin D would favor cell proliferation by facilitating RB phosphorylation and inactivation.
73 Cyclin D is overexpressed in many tumors because of gene amplification or translocation. Mutational inactivation of CDKIs also would drive the cell cycle by unregulated activation of cyclins and CDKs.
74 p53 Gene: Guardian of the Genome P53 induces neoplastic transformation by three interlocking mechanisms: 1-activation of temporary cell cycle arrest (termed quiescence). 2-induction of permanent cell cycle arrest (termed senescence). 3-triggering of programmed cell death (termed apoptosis).
75 p53 can be viewed as a central monitor of stress directing the stressed cells toward an appropriate response.
76 A variety of stresses can trigger the p53 response pathways including: 1-Anoxia. 2-Inappropriate oncogene expression (e.g MYC or RAS). 3-DNA damage.
77 In nonstressed normal cells p53 has a short halflife (20 minutes). P53 release from MDM2-p53 complex increases its half-life and becomes activated as a transcription factor.
78 Genes whose transcription is triggered by p53 are grouped into two broad categories: 1-those that cause cell cycle arrest 2-those that cause apoptosis.
79 If DNA damage can be repaired during cell cycle arrest the cell reverts to a normal state. If the repair fails p53 induces apoptosis or senescence.
80 The key initiators of the DNA-damage pathway are two related protein kinases: 1-ataxia-telangiectasia mutated (ATM). 2-ataxia-telangiectasia mutated related (ATR).
81 Once triggered both ATM and ATR phosphorylate a variety of targets including p53 and DNA repair proteins. Phosphorylation of these two targets leads to a pause in the cell cycle and stimulation of DNA repair pathways respectively.
82 p53-mediated cell cycle arrest occurs by p53- dependent transcription of the CDKI (p21). The p16 gene inhibits cyclin-cdk complexes and prevents phosphorylation of RB essential for cells to enter G 1 phase.
83 More than 70% of human cancers have a defect in this gene and the remaining malignant neoplasms have defects in genes up-stream or down-stream of p53.
84 Homozygous loss of the p53 gene is found in virtually every type of cancer including: 1-carcinomas of the lung. 2-carcinoma of colon. 3-carcinoma of breast.
85 Li-Fraumeni syndrome is due to inherited mutant p53. inheritance of one mutant allele predisposes individuals to develop malignant tumors because only one additional hit is needed to inactivate the second, normal allele.
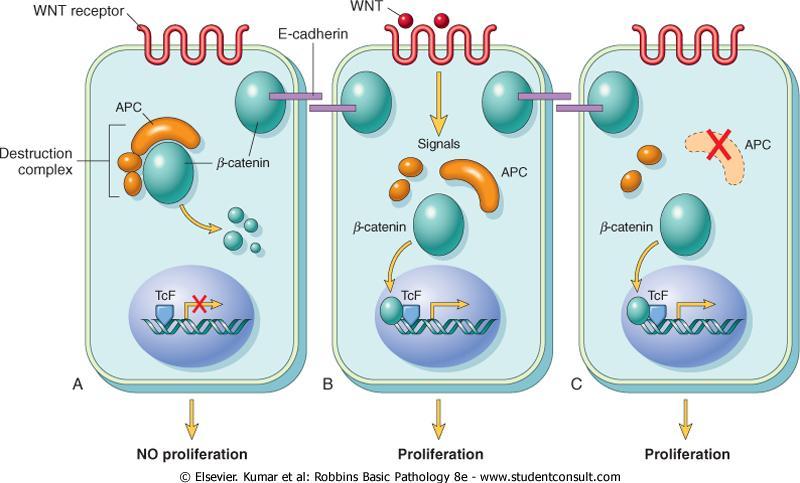
86 Patients with the Li-Fraumeni syndrome have a 25 X greater chance of developing a malignant tumor by age 50 compared with the general population. The most common types of tumors are: sarcomas, breast cancer, leukemia, brain tumors, and carcinomas of the adrenal cortex.
87 Transforming Growth Factor-β Pathway TGF-β is a potent inhibitor of proliferation in most normal epithelial, endothelial, and hematopoietic cells. It regulates cellular processes by binding to a complex composed of TGF-β receptors I and II.
88 Dimerization of the receptor upon ligand binding leads to a cascade of events that result in: 1-transcriptional activation of CDKIs. 2-suppression of growth-promoting genes such as MYC, CDK2, CDK4, and those encoding cyclins A and E.
89 Mutations affecting the type II receptor are seen in cancers of the colon, stomach, and endometrium. Mutational inactivation of SMAD4, 1 of the 10 proteins known to be involved in TGF-β signaling, is common in pancreatic cancers. In 100% of pancreatic cancers and 83% of colon cancers, at least one component of the TGF-β pathway is mutated.
90 Contact Inhibition NF2 and APC Cell-cell contacts in many tissues are mediated by transmembrane proteins called cadherins. E-cadherin mediates cell-cell contact in epithelial layers by mechanism not fully understood.
91 One mechanism that sustains contact inhibition is mediated by the tumor suppressor gene NF2.
92 NF2 product, neurofibromin-2, facilitates E-cadherin mediated contact inhibition. Homozygous loss of NF2 (neurofibromin-2) is associated with neurofibromatosis.
93 Adenomatous Polyposis Coli- β-catenin Pathway The APC gene and its cytoplasmic protein regulate the intracellular levels of β-catenin. β-catenin is a protein with many functions : 1- β-catenin binds to the cytoplasmic portion of E- cadherin, a cell surface protein that mediates intercellular interactions. 2- It can translocate to the nucleus and activate cell proliferation.
94 β-catenin is an important component of the socalled WNT signaling pathway that regulates cell proliferation.
95 WNT factor transmits signals that prevent the degradation of β-catenin. β-catenin acts as a transcriptional activator in conjunction with another molecule, called TcF.
96 In quiescent cells which are not exposed to WNT, cytoplasmic β-catenin is degraded by a destruction complex formed of products of APC gene and E-cadherin.
97
98 APC behaves as a typical tumor suppressor gene. Inheritance of mutated APC is associated with adenomatous polyposis coli. APC mutations are seen in 70-80% of sporadic colon cancers.
99 Colonic cancers that have normal APC genes show activating mutations of β-catenin that render them refractory to the degrading action of APC.
100 Evasion of Apoptosis There are 2 distinct programs that activate apoptosis: 1- the extrinsic pathway (death receptor CD95/Fas ). 2- the intrinsic pathway (DNA damage ).
101 Stimulation of either pathway results in activation of a normally inactive protease (caspase-8 or caspase-9) which initiates a proteolytic cascade involving "executioner" caspases that disassemble the cell in orderly fashion.
102 The cellular remains are then efficiently consumed by phagocytes without stimulating inflammation.
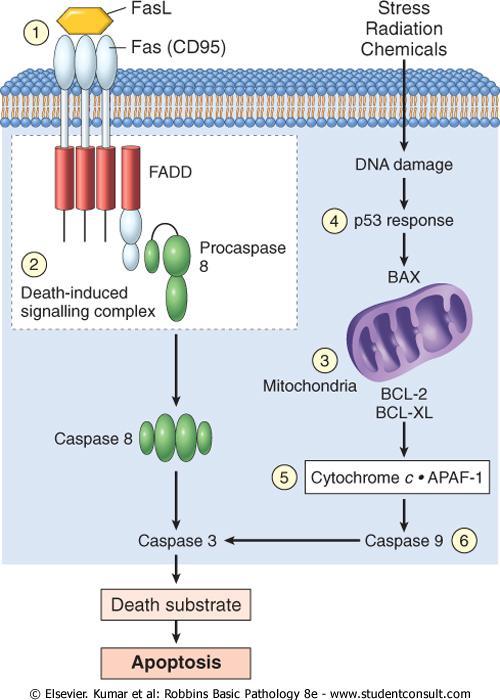
103
104 Extrinsic pathway of apoptosis TNF receptor (CD95,Fas)is bound to its ligand CD95L trimerization of the receptor and thus its cytoplasmic death domains α attract the intracellular adaptor protein FADD recruits procaspase 8 to form the deathinducing signaling complex.
105 Procaspase 8 is activated by cleavage into smaller subunits generating caspase 8. Caspase 8 then activates down-stream caspases such as caspase 3 (executioner caspase) that cleaves DNA and other substrates to cause cell death.
106 Intrinsic pathway of apoptosis It is triggered by a variety of stimuli including : 1- withdrawal of survival factors. 2- stress. 3- injury. Activation of this pathway leads to permeabilization of mitochondrial outer membrane, with resultant release of molecules such as cytochrome c, that initiate apoptosis.
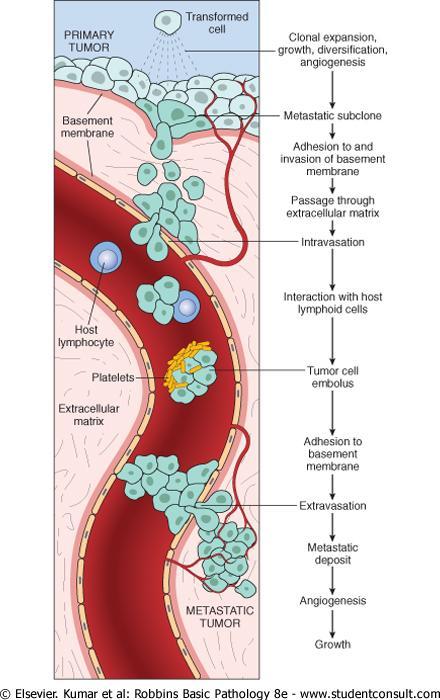
107 Cytochrome c leaks into the cytosol where it binds to APAF-1 activating caspase 9. caspase-9 can cleave and activate the executioner caspase.
108 The integrity of the mitochondrial outer membrane is regulated by pro-apoptotic and anti-apoptotic members of the BCL2 family of proteins. The pro-apoptotic proteins, BAX and BAK, are required for apoptosis and directly promote mitochondrial permeabilization.
109 Their action is inhibited by the anti-apoptotic members of this family exemplified by BCL2 and BCL-XL.
110 A third set of proteins (so-called BH3-only proteins) including BAD, BID, and PUMA, regulate the balance between the pro- and anti-apoptotic members of the apoptotic genes.
111 Malignent cells can escape apoptosis through different ways: 1-Reduced levels of CD95 may render the tumor cells less susceptible to apoptosis by Fas ligand (FasL). 2-Some tumors have high levels of FLIP, a protein that can bind death-inducing signaling complex and prevent activation of caspase 8.
112 3-Reduced release of cytochrome c from mitochondrion as a result of up-regulation of BCL2. 4- Reduced levels of pro-apoptotic BAX resulting from loss of p53. 5-Loss of APAF-1. 6-Up-regulation of inhibitors of apoptosis.
113 85% of B-cell lymphomas of the follicular type carry a characteristic t(14;18) translocation. BCL 2 (located at 18q21).
114 Ability to Invade and Metastasize The metastatic cascade can be subdivided into two phases: 1-invasion of ECM and vascular dissemination. 2-homing of tumor cells.
115 Invasion of Extracellular Matrix (ECM Human tissues are organized into a series of compartments separated from each other by two types of ECM: 1-basement membranes. 2-interstitial connective tissue.
116 ECM is composed of : 1-collagens. 2-glycoproteins. 3-proteoglycans.
117
118 Invasion of the ECM is an active process that requires four steps : 1-Detachment of tumor cells from each other. 2-Degradation of ECM. 3-Attachment to novel ECM components. 4-Migration of tumor cells.
119 E-cadherin function is lost in almost all epithelial cancers by : 1- mutational inactivation of E-cadherin genes. 2- by activation of β-catenin genes. 3-by inappropriate expression of the SNAIL and TWIST transcription factors which suppress E-cadherin expression.
120 Epithelial-to-mesenchymal transition (EMT) Loss of E-cadherin expression is the key event. EMT, and SNAIL and TWIST down-regulate E- cadherin expression. EMT has been documented mainly in breast cancers.
121 In EMT, carcinoma cells down-regulate certain epithelial markers (e.g., E-cadherin) and up-regulate certain mesenchymal markers (e.g., vimentin and smooth muscle actin).
122 The second step in invasion is local degradation of the basement membrane and interstitial connective tissue. Tumor cells may either secrete proteolytic enzymes themselves or induce stromal cells (e.g fibroblasts and inflammatory cells) to elaborate proteases.
123 Multiple different families of proteases are present : 1-matrix metalloproteinases (MMPs). 2-cathepsin D. 3-urokinase plasminogen activator.
124 MMPs also cause releasing ECM-sequestered growth factors. Cleavage products of collagen and proteoglycans also have chemotactic, angiogenic, and growth-promoting effects.
125 MMP-9 is a gelatinase that cleaves type IV collagen of the epithelial and vascular basement membrane and also stimulates release of VEGF from ECM.
126 Benign tumors of the breast, colon, and stomach show little type IV collagenase activity Malignant tumors overexpress this enzyme.
127 The levels of metalloproteinase inhibitors are reduced so that the balance is tilted greatly toward tissue degradation.
128 The third step in invasion involves changes in attachment of tumor cells to ECM proteins.
129 Normal epithelial cells have receptors, such as integrins, for basement membrane laminin and collagens that are polarized at their basal surface. Loss of adhesion in normal cells leads to induction of apoptosis.
130 Cleavage of the basement membrane proteins,collagen IV and laminin by MMP-2 or MMP-9 generates novel sites that bind to receptors on tumor cells and stimulate migration.
131 Locomotion is the final step of invasion. Migration is a complex, multistep process that involve the actin in the cytoskeleton. Such movement seems to be potentiated and directed by tumor cell-derived cytokines, such as autocrine motility factors.
132 Vascular Dissemination and Homing of Tumor Cells In the bloodstream, some tumor cells form emboli by aggregating and adhering to circulating leukocytes, particularly platelets.
133 Extravasation of free tumor cells or tumor emboli involves adhesion to the vascular endothelium.
134 The site of extravasation and the organ distribution of metastases generally can be predicted by the location of the primary tumor and its vascular or lymphatic drainage.
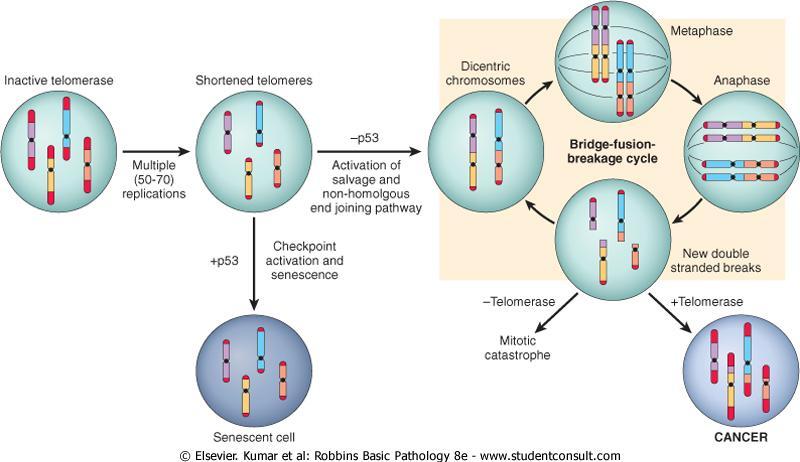
135 In many cases the natural pathways of drainage do not readily explain the distribution of metastases.
136 e.g.lung cancers tend to involve the adrenals with some regularity but almost never spread to skeletal muscle.
137 The mechanisms of site-specific homing involves : 1-the expression of adhesion molecules by tumor cells whose ligands are expressed preferentially on the endothelium of target organs.
138 2-chemokines and their receptors. chemokines participate in directed movement (chemotaxis) of tumor cells.
139 Human breast cancer cells express high levels of the chemokine receptors CXCR4 and CCR7. The ligands for these receptors (i.e chemokines CXCL12 and CCL21) are highly expressed only in those organs where breast cancer cells metastasize.
140 After extravasation, tumor cells are dependent on a receptive stroma for growth. The precise localization of metastases cannot be predicted with any form of cancer.
141 The prolonged survival of micrometastases without progression is well described in melanoma and in breast and prostate cancer.
142 Limitless Replicative Potential Most normal human cells have a capacity of 60 to 70 doublings. After this the cells lose the capacity to divide and enter senescence. This phenomenon is due to progressive shortening of telomeres at the ends of chromosomes.
143 Short telomeres are recognized by the DNA repair machinery leading to cell cycle arrest mediated by p53 and RB.
144 Cells in which the checkpoints are disabled by p53 or RB mutations the nonhomologous end-joining pathway is activated as a lasteffort to save the cell joining the shortened ends of two chromosomes.
145 This inappropriately activated repair system results in dicentric chromosomes that are pulled apart at anaphase resulting in new double-stranded DNA breaks.
146 The resulting genomic instability from the repeated bridge-fusion-breakage cycles eventually produces mitotic catastrophe characterized by massive cell death.
147
148 If during crisis a cell manages to reactivate telomerase, the bridge-fusion-breakage cycles cease and the cell is able to avoid death. During this period of genomic instability that precedes telomerase activation, numerous mutations could accumulate.
149 Passage through a period of genomic instability probably explains the complex karyotypes frequently seen in human carcinomas.
150 Telomerase, active in normal stem cells, is normally absent from, or at very low levels in most somatic cells. Telomere maintenance is seen in virtually all types of cancers. In 85-95% of cancers, this is due to upregulation of the enzyme telomerase.
151 Unregulated proliferation in incipient tumors leads to telomere shortening, followed by chromosomal instability and mutation accumulation. Reactivation of telomerase in these cells causes extension of telomeres and mutations become fixed contributing to tumor growth.
152 Grading of Cancer The grading of a cancer attempts to establish some estimate of its aggressiveness or level of malignancy. It is based on : 1-the cytologic differentiation of tumor cells. 2-the number of mitoses within the tumor. The cancer may be classified as grade I, II, III, or IV, in order of increasing anaplasia.
153 Criteria for the individual grades vary with each form of neoplasia. Difficulties in establishing clear-cut criteria have led in some instances to descriptive characterizations as : Well-differentiated Moderately-differentiated. Poorly-differentiated. Anaplastic tumors.
154 Staging of cancer Staging of cancers is based on : 1-the size of the primary lesion. 2-its extent of spread to regional lymph nodes. 3-the presence or absence of metastases.
155 This assessment is usually based on clinical and radiographic examination (CT scan & MRI) and in some cases surgical exploration.
156 Two methods of staging are currently in use: 1-the TNM system (T, primary tumor; N, regional lymph node involvement; M, metastases) 2-the AJC (American Joint Committee) system..
157 In the TNM system, T1, T2, T3, and T4 describe the increasing size of the primary lesion; N0, N1, N2, and N3 indicate progressively advancing node involvement; M0 and M1 reflect the absence or presence of distant metastases.
158 In the AJC method, the cancers are divided into stages 0 to IV, incorporating the size of primary lesions and the presence of nodal spread and of distant metastases. staging has proved to be of greater clinical value than grading.
159 Staging of breast carcinoma stage Tumor Lymph Nodes Metastasis Prognosis 0 DCIS or LCIS 0 M 0 92% I II III Invasive carcinoma 2 cm or less in diameter < or = 5cm > 5cm < or = 5 cm >5 cm Any size 0 M 0 87% < or or more Any number 10 or more M 0 M 0 Skin or chest wall involvement 5-yr survival 75% 46% IV Any size Any number M 1 13%
160 TNM Staging of Colon Cancers Tumor T0 none evident Tis = in situ (limited to mucosa) T1 = invasion of lamina propria or submucosa T2 = invasion of muscularis propria T3 = invasion through muscularis propria into subserosa or nonperitonealized perimuscular tissue T4 = invasion of other organs or structures Lymph Nodes (N) 0 = none evident 1 = 1 to 3 positive pericolic nodes 2 = 4 or more positive pericolic nodes 3 = any positive node along a named blood vessel
161 Distant Metastases (M) 0 = none evident 1 = any distant metastasis 5-Year Survival Rates T1 = 97% T2 = 90% T3 = 78% T4 = 63% Any T; N1; M0 = 66% Any T; N2; M0 = 37% Any T; N3; M0 = data not available Any M1 = 4%
162 Laboratory Diagnosis of Cancer 1-Morphologic Methods : (H&E stain) A-excision or biopsy. B-fine-needle aspiration. C-cytologic smears (Papanicolaou). D-Frozen sections.
163 Immunocytochemistry : Cytokeratin prostate-specific antigen (PSA) =prostate carcinoma. estrogen receptors =breast cancer.
164 Flow cytometry is used routinely in the classification of leukemias and lymphomas. Fluorescent antibodies against cell surface molecules and differentiation antigens are used to obtain the phenotype of malignant cells.
165 2-Tumor Markers : A-PSA Prostatic carcinoma can be suspected when elevated levels of PSA are found in the blood. PSA levels are often elevated in cancer. PSA levels also may be elevated in benign prostatic hyperplasia PSA test suffers from both low sensitivity and low specificity.
166 B-carcinoembryonic antigen (CEA). carcinomas of the colon, pancreas, stomach, and breast. C-α-fetoprotein. produced by : 1- hepatocellular carcinomas. 2-yolk sac remnants in the gonads. 3-teratocarcinomas. 4-embryonal cell carcinomas. 5-neural tube defect of the fetus. CEA and α-fetoprotein assays lack both specificity and sensitivity
167 3-Molecular Diagnosis
number Done by Corrected by Doctor Maha Shomaf
 number 21 Done by Ahmad Rawajbeh Corrected by Omar Sami Doctor Maha Shomaf Ability to Invade and Metastasize The metastatic cascade can be subdivided into two phases: 1-invasion of ECM and vascular dissemination:
number 21 Done by Ahmad Rawajbeh Corrected by Omar Sami Doctor Maha Shomaf Ability to Invade and Metastasize The metastatic cascade can be subdivided into two phases: 1-invasion of ECM and vascular dissemination:
Lecture 8 Neoplasia II. Dr. Nabila Hamdi MD, PhD
 Lecture 8 Neoplasia II Dr. Nabila Hamdi MD, PhD ILOs Understand the definition of neoplasia. List the classification of neoplasia. Describe the general characters of benign tumors. Understand the nomenclature
Lecture 8 Neoplasia II Dr. Nabila Hamdi MD, PhD ILOs Understand the definition of neoplasia. List the classification of neoplasia. Describe the general characters of benign tumors. Understand the nomenclature
number Done by Corrected by Doctor Maha Shomaf
 number 19 Done by Waseem Abo-Obeida Corrected by Abdullah Zreiqat Doctor Maha Shomaf Carcinogenesis: the molecular basis of cancer. Non-lethal genetic damage lies at the heart of carcinogenesis and leads
number 19 Done by Waseem Abo-Obeida Corrected by Abdullah Zreiqat Doctor Maha Shomaf Carcinogenesis: the molecular basis of cancer. Non-lethal genetic damage lies at the heart of carcinogenesis and leads
Emerging" hallmarks of cancer, a. Reprogramming of energy metabolism b. Evasion of the immune system, Enabling characteristics, a.
 HALLMARKS OF CANCER - Together dictate the malignant phenotype. 1. Self-sufficiency in growth signals 2. Insensitivity to growth inhibitory signals 3. Evasion of cell death 4. Limitless replicative potential
HALLMARKS OF CANCER - Together dictate the malignant phenotype. 1. Self-sufficiency in growth signals 2. Insensitivity to growth inhibitory signals 3. Evasion of cell death 4. Limitless replicative potential
Neoplasia 18 lecture 6. Dr Heyam Awad MD, FRCPath
 Neoplasia 18 lecture 6 Dr Heyam Awad MD, FRCPath ILOS 1. understand the role of TGF beta, contact inhibition and APC in tumorigenesis. 2. implement the above knowledge in understanding histopathology reports.
Neoplasia 18 lecture 6 Dr Heyam Awad MD, FRCPath ILOS 1. understand the role of TGF beta, contact inhibition and APC in tumorigenesis. 2. implement the above knowledge in understanding histopathology reports.
Molecular biology :- Cancer genetics lecture 11
 Molecular biology :- Cancer genetics lecture 11 -We have talked about 2 group of genes that is involved in cellular transformation : proto-oncogenes and tumour suppressor genes, and it isn t enough to
Molecular biology :- Cancer genetics lecture 11 -We have talked about 2 group of genes that is involved in cellular transformation : proto-oncogenes and tumour suppressor genes, and it isn t enough to
Cancer. The fundamental defect is. unregulated cell division. Properties of Cancerous Cells. Causes of Cancer. Altered growth and proliferation
 Cancer The fundamental defect is unregulated cell division. Properties of Cancerous Cells Altered growth and proliferation Loss of growth factor dependence Loss of contact inhibition Immortalization Alterated
Cancer The fundamental defect is unregulated cell division. Properties of Cancerous Cells Altered growth and proliferation Loss of growth factor dependence Loss of contact inhibition Immortalization Alterated
Disorders of Cell Growth & Neoplasia. Lecture 4 Molecular basis of cancer
 General Pathology VPM 152 Disorders of Cell Growth & Neoplasia Lecture 4 Molecular basis of cancer Enrique Aburto Apr 2010 Skin tumor in a 10-year-old Rottweiler. Considering the external appearance and
General Pathology VPM 152 Disorders of Cell Growth & Neoplasia Lecture 4 Molecular basis of cancer Enrique Aburto Apr 2010 Skin tumor in a 10-year-old Rottweiler. Considering the external appearance and
A class of genes that normally suppress cell proliferation. p53 and Rb..ect. suppressor gene products can release cells. hyperproliferation.
 Tumor Suppressor Genes A class of genes that normally suppress cell proliferation. p53 and Rb..ect Mutations that inactivate the tumor suppressor gene products can release cells from growth suppression
Tumor Suppressor Genes A class of genes that normally suppress cell proliferation. p53 and Rb..ect Mutations that inactivate the tumor suppressor gene products can release cells from growth suppression
Neoplasia 18 lecture 8. Dr Heyam Awad MD, FRCPath
 Neoplasia 18 lecture 8 Dr Heyam Awad MD, FRCPath ILOS 1. understand the angiogenic switch in tumors and factors that stimulate and inhibit angiogenesis. 2. list the steps important for tumor metastasis
Neoplasia 18 lecture 8 Dr Heyam Awad MD, FRCPath ILOS 1. understand the angiogenic switch in tumors and factors that stimulate and inhibit angiogenesis. 2. list the steps important for tumor metastasis
Cancer Genetics. What is Cancer? Cancer Classification. Medical Genetics. Uncontrolled growth of cells. Not all tumors are cancerous
 Session8 Medical Genetics Cancer Genetics J avad Jamshidi F a s a U n i v e r s i t y o f M e d i c a l S c i e n c e s, N o v e m b e r 2 0 1 7 What is Cancer? Uncontrolled growth of cells Not all tumors
Session8 Medical Genetics Cancer Genetics J avad Jamshidi F a s a U n i v e r s i t y o f M e d i c a l S c i e n c e s, N o v e m b e r 2 0 1 7 What is Cancer? Uncontrolled growth of cells Not all tumors
Cancer. The fundamental defect is. unregulated cell division. Properties of Cancerous Cells. Causes of Cancer. Altered growth and proliferation
 Cancer The fundamental defect is unregulated cell division. Properties of Cancerous Cells Altered growth and proliferation Loss of growth factor dependence Loss of contact inhibition Immortalization Alterated
Cancer The fundamental defect is unregulated cell division. Properties of Cancerous Cells Altered growth and proliferation Loss of growth factor dependence Loss of contact inhibition Immortalization Alterated
Karyotype analysis reveals transloction of chromosome 22 to 9 in CML chronic myelogenous leukemia has fusion protein Bcr-Abl
 Chapt. 18 Cancer Molecular Biology of Cancer Student Learning Outcomes: Describe cancer diseases in which cells no longer respond Describe how cancers come from genomic mutations (inherited or somatic)
Chapt. 18 Cancer Molecular Biology of Cancer Student Learning Outcomes: Describe cancer diseases in which cells no longer respond Describe how cancers come from genomic mutations (inherited or somatic)
1. The metastatic cascade. 3. Pathologic features of metastasis. 4. Therapeutic ramifications. Which malignant cells will metastasize?
 1. The metastatic cascade 3. Pathologic features of metastasis 4. Therapeutic ramifications Sir James Paget (1814-1899) British Surgeon/ Pathologist Paget s disease of Paget s disease of the nipple (intraductal
1. The metastatic cascade 3. Pathologic features of metastasis 4. Therapeutic ramifications Sir James Paget (1814-1899) British Surgeon/ Pathologist Paget s disease of Paget s disease of the nipple (intraductal
Multistep nature of cancer development. Cancer genes
 Multistep nature of cancer development Phenotypic progression loss of control over cell growth/death (neoplasm) invasiveness (carcinoma) distal spread (metastatic tumor) Genetic progression multiple genetic
Multistep nature of cancer development Phenotypic progression loss of control over cell growth/death (neoplasm) invasiveness (carcinoma) distal spread (metastatic tumor) Genetic progression multiple genetic
VIII Curso Internacional del PIRRECV. Some molecular mechanisms of cancer
 VIII Curso Internacional del PIRRECV Some molecular mechanisms of cancer Laboratorio de Comunicaciones Celulares, Centro FONDAP Estudios Moleculares de la Celula (CEMC), ICBM, Facultad de Medicina, Universidad
VIII Curso Internacional del PIRRECV Some molecular mechanisms of cancer Laboratorio de Comunicaciones Celulares, Centro FONDAP Estudios Moleculares de la Celula (CEMC), ICBM, Facultad de Medicina, Universidad
1.The metastatic cascade. 2.Pathologic features of metastasis. 3.Therapeutic ramifications
 Metastasis 1.The metastatic cascade 2.Pathologic features of metastasis 3.Therapeutic ramifications Sir James Paget (1814-1899) British Surgeon/ Pathologist Paget s disease of bone Paget s disease of the
Metastasis 1.The metastatic cascade 2.Pathologic features of metastasis 3.Therapeutic ramifications Sir James Paget (1814-1899) British Surgeon/ Pathologist Paget s disease of bone Paget s disease of the
Deregulation of signal transduction and cell cycle in Cancer
 Deregulation of signal transduction and cell cycle in Cancer Tuangporn Suthiphongchai, Ph.D. Department of Biochemistry Faculty of Science, Mahidol University Email: tuangporn.sut@mahidol.ac.th Room Pr324
Deregulation of signal transduction and cell cycle in Cancer Tuangporn Suthiphongchai, Ph.D. Department of Biochemistry Faculty of Science, Mahidol University Email: tuangporn.sut@mahidol.ac.th Room Pr324
Neoplasia 2018 lecture 4. Dr Heyam Awad MD, FRCPath
 Neoplasia 2018 lecture 4 Dr Heyam Awad MD, FRCPath ILOS To understand the concept of the hallmarks of cancer and that they are phenotypic changes needed in all cancer cells. To list the tumor enablers
Neoplasia 2018 lecture 4 Dr Heyam Awad MD, FRCPath ILOS To understand the concept of the hallmarks of cancer and that they are phenotypic changes needed in all cancer cells. To list the tumor enablers
Chapter 9, Part 1: Biology of Cancer and Tumor Spread
 PATHOPHYSIOLOGY Name Chapter 9, Part 1: Biology of Cancer and Tumor Spread I. Cancer Characteristics and Terminology Neoplasm new growth, involves the overgrowth of tissue to form a neoplastic mass (tumor).
PATHOPHYSIOLOGY Name Chapter 9, Part 1: Biology of Cancer and Tumor Spread I. Cancer Characteristics and Terminology Neoplasm new growth, involves the overgrowth of tissue to form a neoplastic mass (tumor).
Introduction. Cancer Biology. Tumor-suppressor genes. Proto-oncogenes. DNA stability genes. Mechanisms of carcinogenesis.
 Cancer Biology Chapter 18 Eric J. Hall., Amato Giaccia, Radiobiology for the Radiologist Introduction Tissue homeostasis depends on the regulated cell division and self-elimination (programmed cell death)
Cancer Biology Chapter 18 Eric J. Hall., Amato Giaccia, Radiobiology for the Radiologist Introduction Tissue homeostasis depends on the regulated cell division and self-elimination (programmed cell death)
General Pathology VPM 152. Disorders of Cell Growth & Neoplasia. Lecture 4 Molecular basis of cancer
 General Pathology VPM 152 Disorders of Cell Growth & Neoplasia Lecture 4 Molecular basis of cancer Enrique Aburto http://people.upei.ca/eaburto Winter 2015 Molecular Basis of Cancer Fundamental principles
General Pathology VPM 152 Disorders of Cell Growth & Neoplasia Lecture 4 Molecular basis of cancer Enrique Aburto http://people.upei.ca/eaburto Winter 2015 Molecular Basis of Cancer Fundamental principles
PATHOBIOLOGY OF NEOPLASIA
 PATHOBIOLOGY OF NEOPLASIA Department of Pathology Gadjah Mada University School of Medicine dr. Harijadi Blok Biomedis, 6 Maret 2009 [12] 3/17/2009 1 The pathobiology of neoplasia Normal cells Malignant
PATHOBIOLOGY OF NEOPLASIA Department of Pathology Gadjah Mada University School of Medicine dr. Harijadi Blok Biomedis, 6 Maret 2009 [12] 3/17/2009 1 The pathobiology of neoplasia Normal cells Malignant
Determination Differentiation. determinated precursor specialized cell
 Biology of Cancer -Developmental Biology: Determination and Differentiation -Cell Cycle Regulation -Tumor genes: Proto-Oncogenes, Tumor supressor genes -Tumor-Progression -Example for Tumor-Progression:
Biology of Cancer -Developmental Biology: Determination and Differentiation -Cell Cycle Regulation -Tumor genes: Proto-Oncogenes, Tumor supressor genes -Tumor-Progression -Example for Tumor-Progression:
oncogenes-and- tumour-suppressor-genes)
") Special topics in tumor biochemistry oncogenes-and- tumour-suppressor-genes) Speaker: Prof. Jiunn-Jye Chuu E-Mail: jjchuu@mail.stust.edu.tw Genetic Basis of Cancer Cancer-causing mutations Disease of aging
Special topics in tumor biochemistry oncogenes-and- tumour-suppressor-genes) Speaker: Prof. Jiunn-Jye Chuu E-Mail: jjchuu@mail.stust.edu.tw Genetic Basis of Cancer Cancer-causing mutations Disease of aging
Chapt 15: Molecular Genetics of Cell Cycle and Cancer
 Chapt 15: Molecular Genetics of Cell Cycle and Cancer Student Learning Outcomes: Describe the cell cycle: steps taken by a cell to duplicate itself = cell division; Interphase (G1, S and G2), Mitosis.
Chapt 15: Molecular Genetics of Cell Cycle and Cancer Student Learning Outcomes: Describe the cell cycle: steps taken by a cell to duplicate itself = cell division; Interphase (G1, S and G2), Mitosis.
Basic tumor nomenclature
 Jonas Nilsson jonas.a.nilsson@surgery.gu.se Sahlgrenska Cancer Center Bilder gjorda av Per Holmfeldt och Jonas Nilsson Benign tumor Basic tumor nomenclature Malignant tumor = cancer Metastasis Carcinoma:
Jonas Nilsson jonas.a.nilsson@surgery.gu.se Sahlgrenska Cancer Center Bilder gjorda av Per Holmfeldt och Jonas Nilsson Benign tumor Basic tumor nomenclature Malignant tumor = cancer Metastasis Carcinoma:
The Hallmarks of Cancer
 The Hallmarks of Cancer Theresa L. Hodin, Ph.D. Clinical Research Services Theresa.Hodin@RoswellPark.org Hippocrates Cancer surgery, circa 1689 Cancer Surgery Today 1971: Nixon declares War on Cancer
The Hallmarks of Cancer Theresa L. Hodin, Ph.D. Clinical Research Services Theresa.Hodin@RoswellPark.org Hippocrates Cancer surgery, circa 1689 Cancer Surgery Today 1971: Nixon declares War on Cancer
1. Basic principles 2. 6 hallmark features 3. Abnormal cell proliferation: mechanisms 4. Carcinogens: examples. Major Principles:
 Carcinogenesis 1. Basic principles 2. 6 hallmark features 3. Abnormal cell proliferation: mechanisms 4. Carcinogens: examples Carcinogenesis Major Principles: 1. Nonlethal genetic damage is central to
Carcinogenesis 1. Basic principles 2. 6 hallmark features 3. Abnormal cell proliferation: mechanisms 4. Carcinogens: examples Carcinogenesis Major Principles: 1. Nonlethal genetic damage is central to
Cancer and Gene Alterations - 1
 Cancer and Gene Alterations - 1 Cancer and Gene Alteration As we know, cancer is a disease of unregulated cell growth. Although we looked at some of the features of cancer when we discussed mitosis checkpoints,
Cancer and Gene Alterations - 1 Cancer and Gene Alteration As we know, cancer is a disease of unregulated cell growth. Although we looked at some of the features of cancer when we discussed mitosis checkpoints,
Early Embryonic Development
 Early Embryonic Development Maternal effect gene products set the stage by controlling the expression of the first embryonic genes. 1. Transcription factors 2. Receptors 3. Regulatory proteins Maternal
Early Embryonic Development Maternal effect gene products set the stage by controlling the expression of the first embryonic genes. 1. Transcription factors 2. Receptors 3. Regulatory proteins Maternal
Oncogenes and Tumor Suppressors MCB 5068 November 12, 2013 Jason Weber
 Oncogenes and Tumor Suppressors MCB 5068 November 12, 2013 Jason Weber jweber@dom.wustl.edu Oncogenes & Cancer DNA Tumor Viruses Simian Virus 40 p300 prb p53 Large T Antigen Human Adenovirus p300 E1A
Oncogenes and Tumor Suppressors MCB 5068 November 12, 2013 Jason Weber jweber@dom.wustl.edu Oncogenes & Cancer DNA Tumor Viruses Simian Virus 40 p300 prb p53 Large T Antigen Human Adenovirus p300 E1A
Molecular Cell Biology. Prof. D. Karunagaran. Department of Biotechnology. Indian Institute of Technology Madras
 Molecular Cell Biology Prof. D. Karunagaran Department of Biotechnology Indian Institute of Technology Madras Module 9 Molecular Basis of Cancer, Oncogenes and Tumor Suppressor Genes Lecture 2 Genes Associated
Molecular Cell Biology Prof. D. Karunagaran Department of Biotechnology Indian Institute of Technology Madras Module 9 Molecular Basis of Cancer, Oncogenes and Tumor Suppressor Genes Lecture 2 Genes Associated
Test Bank for Robbins and Cotran Pathologic Basis of Disease 9th Edition by Kumar
 Link full download:https://getbooksolutions.com/download/test-bank-for-robbinsand-cotran-pathologic-basis-of-disease-9th-edition-by-kumar Test Bank for Robbins and Cotran Pathologic Basis of Disease 9th
Link full download:https://getbooksolutions.com/download/test-bank-for-robbinsand-cotran-pathologic-basis-of-disease-9th-edition-by-kumar Test Bank for Robbins and Cotran Pathologic Basis of Disease 9th
Disorders of Cell Growth & Neoplasia
 General Pathology VPM 152 Disorders of Cell Growth & Neoplasia Lecture 3 Rate of growth, local invasion, and metastasis. Molecular basis of cancer (normal cell-cycle and cellular proliferation). Enrique
General Pathology VPM 152 Disorders of Cell Growth & Neoplasia Lecture 3 Rate of growth, local invasion, and metastasis. Molecular basis of cancer (normal cell-cycle and cellular proliferation). Enrique
Chapter 12. Regulation of Cell Division. AP Biology
 Chapter 12. Regulation of Cell Division Coordination of cell division! Multicellular organism " need to coordinate across different parts of organism! timing of cell division! rates of cell division "
Chapter 12. Regulation of Cell Division Coordination of cell division! Multicellular organism " need to coordinate across different parts of organism! timing of cell division! rates of cell division "
p53 and Apoptosis: Master Guardian and Executioner Part 2
 p53 and Apoptosis: Master Guardian and Executioner Part 2 p14arf in human cells is a antagonist of Mdm2. The expression of ARF causes a rapid increase in p53 levels, so what would you suggest?.. The enemy
p53 and Apoptosis: Master Guardian and Executioner Part 2 p14arf in human cells is a antagonist of Mdm2. The expression of ARF causes a rapid increase in p53 levels, so what would you suggest?.. The enemy
Transformation of Normal HMECs (Human Mammary Epithelial Cells) into Metastatic Breast Cancer Cells: Introduction - The Broad Picture:
 into Metastatic Breast Cancer Cells: Introduction - The Broad Picture:") Transformation of Normal HMECs (Human Mammary Epithelial Cells) into Metastatic Breast Cancer Cells: Introduction - The Broad Picture: Spandana Baruah December, 2016 Cancer is defined as: «A disease caused
Transformation of Normal HMECs (Human Mammary Epithelial Cells) into Metastatic Breast Cancer Cells: Introduction - The Broad Picture: Spandana Baruah December, 2016 Cancer is defined as: «A disease caused
Regulation of Cell Division. AP Biology
 Regulation of Cell Division 2006-2007 Coordination of cell division A multicellular organism needs to coordinate cell division across different tissues & organs critical for normal growth, development
Regulation of Cell Division 2006-2007 Coordination of cell division A multicellular organism needs to coordinate cell division across different tissues & organs critical for normal growth, development
Neoplasia 2018 lecture 11. Dr H Awad FRCPath
 Neoplasia 2018 lecture 11 Dr H Awad FRCPath Clinical aspects of neoplasia Tumors affect patients by: 1. their location 2. hormonal secretions 3. paraneoplastic syndromes 4. cachexia Tumor location Even
Neoplasia 2018 lecture 11 Dr H Awad FRCPath Clinical aspects of neoplasia Tumors affect patients by: 1. their location 2. hormonal secretions 3. paraneoplastic syndromes 4. cachexia Tumor location Even
Prof. R. V. Skibbens
 Prof. R. V. Skibbens December 2, 2011 BIOS 10: BioScience in the 21 st Century Cell Cycle, Cell Division and Cancer (Part 2) Directionality The Cell Cycle clock goes in only one direction S-phase cells
Prof. R. V. Skibbens December 2, 2011 BIOS 10: BioScience in the 21 st Century Cell Cycle, Cell Division and Cancer (Part 2) Directionality The Cell Cycle clock goes in only one direction S-phase cells
Cancer genetics
 Cancer genetics General information about tumorogenesis. Cancer induced by viruses. The role of somatic mutations in cancer production. Oncogenes and Tumor Suppressor Genes (TSG). Hereditary cancer. 1
Cancer genetics General information about tumorogenesis. Cancer induced by viruses. The role of somatic mutations in cancer production. Oncogenes and Tumor Suppressor Genes (TSG). Hereditary cancer. 1
Apoptosis Oncogenes. Srbová Martina
 Apoptosis Oncogenes Srbová Martina Cell Cycle Control point Cyclin B Cdk1 Cyclin D Cdk4 Cdk6 Cyclin A Cdk2 Cyclin E Cdk2 Cyclin-dependent kinase (Cdk) have to bind a cyclin to become active Regulation
Apoptosis Oncogenes Srbová Martina Cell Cycle Control point Cyclin B Cdk1 Cyclin D Cdk4 Cdk6 Cyclin A Cdk2 Cyclin E Cdk2 Cyclin-dependent kinase (Cdk) have to bind a cyclin to become active Regulation
CELL BIOLOGY - CLUTCH CH CANCER.
 !! www.clutchprep.com CONCEPT: OVERVIEW OF CANCER Cancer is a disease which is primarily caused from misregulated cell division, which form There are two types of tumors - Benign tumors remain confined
!! www.clutchprep.com CONCEPT: OVERVIEW OF CANCER Cancer is a disease which is primarily caused from misregulated cell division, which form There are two types of tumors - Benign tumors remain confined
RAS Genes. The ras superfamily of genes encodes small GTP binding proteins that are responsible for the regulation of many cellular processes.
 ۱ RAS Genes The ras superfamily of genes encodes small GTP binding proteins that are responsible for the regulation of many cellular processes. Oncogenic ras genes in human cells include H ras, N ras,
۱ RAS Genes The ras superfamily of genes encodes small GTP binding proteins that are responsible for the regulation of many cellular processes. Oncogenic ras genes in human cells include H ras, N ras,
Genetics and Cancer Ch 20
 Genetics and Cancer Ch 20 Cancer is genetic Hereditary cancers Predisposition genes Ex. some forms of colon cancer Sporadic cancers ~90% of cancers Descendants of cancerous cells all cancerous (clonal)
Genetics and Cancer Ch 20 Cancer is genetic Hereditary cancers Predisposition genes Ex. some forms of colon cancer Sporadic cancers ~90% of cancers Descendants of cancerous cells all cancerous (clonal)
Regulation of Cell Division (Ch. 12)
") Regulation of Cell Division (Ch. 12) Coordination of cell division A multicellular organism needs to coordinate cell division across different tissues & organs critical for normal growth, development &
Regulation of Cell Division (Ch. 12) Coordination of cell division A multicellular organism needs to coordinate cell division across different tissues & organs critical for normal growth, development &
Cell cycle, signaling to cell cycle, and molecular basis of oncogenesis
 Cell cycle, signaling to cell cycle, and molecular basis of oncogenesis MUDr. Jiří Vachtenheim, CSc. CELL CYCLE - SUMMARY Basic terminology: Cyclins conserved proteins with homologous regions; their cellular
Cell cycle, signaling to cell cycle, and molecular basis of oncogenesis MUDr. Jiří Vachtenheim, CSc. CELL CYCLE - SUMMARY Basic terminology: Cyclins conserved proteins with homologous regions; their cellular
Carcinogenesis. Carcinogenesis. 1. Basic principles 2. 6 hallmark features 3. Abnormal cell proliferation: mechanisms 4. Carcinogens: examples
 Carcinogenesis 1. Basic principles 2. 6 hallmark features 3. Abnormal cell proliferation: mechanisms 4. Carcinogens: examples Major Principles (cont d) 4. Principle targets of genetic damage: 4 classes
Carcinogenesis 1. Basic principles 2. 6 hallmark features 3. Abnormal cell proliferation: mechanisms 4. Carcinogens: examples Major Principles (cont d) 4. Principle targets of genetic damage: 4 classes
Regulation of Cell Division
 Regulation of Cell Division Two HeLa cancer cells are just completing cytokinesis. Explain how the cell division of cancer cells like these is misregulated. Identify genetic and other changes that might
Regulation of Cell Division Two HeLa cancer cells are just completing cytokinesis. Explain how the cell division of cancer cells like these is misregulated. Identify genetic and other changes that might
CELL CYCLE MOLECULAR BASIS OF ONCOGENESIS
 CELL CYCLE MOLECULAR BASIS OF ONCOGENESIS Summary of the regulation of cyclin/cdk complexes during celll cycle Cell cycle phase Cyclin-cdk complex inhibitor activation Substrate(s) G1 Cyclin D/cdk 4,6
CELL CYCLE MOLECULAR BASIS OF ONCOGENESIS Summary of the regulation of cyclin/cdk complexes during celll cycle Cell cycle phase Cyclin-cdk complex inhibitor activation Substrate(s) G1 Cyclin D/cdk 4,6
Cancer. Throughout the life of an individual, but particularly during development, every cell constantly faces decisions.
 Cancer Throughout the life of an individual, but particularly during development, every cell constantly faces decisions. Should it divide? Yes No--> Should it differentiate? Yes No-->Should it die? Yes-->Apoptosis
Cancer Throughout the life of an individual, but particularly during development, every cell constantly faces decisions. Should it divide? Yes No--> Should it differentiate? Yes No-->Should it die? Yes-->Apoptosis
CELL CYCLE REGULATION AND CANCER. Cellular Reproduction II
 CELL CYCLE REGULATION AND CANCER Cellular Reproduction II THE CELL CYCLE Interphase G1- gap phase 1- cell grows and develops S- DNA synthesis phase- cell replicates each chromosome G2- gap phase 2- cell
CELL CYCLE REGULATION AND CANCER Cellular Reproduction II THE CELL CYCLE Interphase G1- gap phase 1- cell grows and develops S- DNA synthesis phase- cell replicates each chromosome G2- gap phase 2- cell
Computational Systems Biology: Biology X
 Bud Mishra Room 1002, 715 Broadway, Courant Institute, NYU, New York, USA L#5:(October-18-2010) Cancer and Signals Outline 1 2 Outline 1 2 Cancer is a disease of malfunctioning cells. Cell Lineage: Adult
Bud Mishra Room 1002, 715 Broadway, Courant Institute, NYU, New York, USA L#5:(October-18-2010) Cancer and Signals Outline 1 2 Outline 1 2 Cancer is a disease of malfunctioning cells. Cell Lineage: Adult
Activation of cellular proto-oncogenes to oncogenes. How was active Ras identified?
 Dominant Acting Oncogenes Eugene E. Marcantonio, M.D. Ph.D. Oncogenes are altered forms of normal cellular genes called proto-oncogenes that are involved in pathways regulating cell growth, differentiation,
Dominant Acting Oncogenes Eugene E. Marcantonio, M.D. Ph.D. Oncogenes are altered forms of normal cellular genes called proto-oncogenes that are involved in pathways regulating cell growth, differentiation,
Dr Rodney Itaki Lecturer Anatomical Pathology Discipline. University of Papua New Guinea School of Medicine & Health Sciences Division of Pathology
 Neoplasia Dr Rodney Itaki Lecturer Anatomical Pathology Discipline University of Papua New Guinea School of Medicine & Health Sciences Division of Pathology General Considerations Overview: Neoplasia uncontrolled,
Neoplasia Dr Rodney Itaki Lecturer Anatomical Pathology Discipline University of Papua New Guinea School of Medicine & Health Sciences Division of Pathology General Considerations Overview: Neoplasia uncontrolled,
Cell Cycle and Cancer
 142 8. Cell Cycle and Cancer NOTES CELL CYCLE G 0 state o Resting cells may re-enter the cell cycle Nondividing cells (skeletal and cardiac muscle, neurons) o Have left the cell cycle and cannot undergo
142 8. Cell Cycle and Cancer NOTES CELL CYCLE G 0 state o Resting cells may re-enter the cell cycle Nondividing cells (skeletal and cardiac muscle, neurons) o Have left the cell cycle and cannot undergo
Test Bank for Robbins and Cotran Pathologic Basis of Disease 9th Edition by Kumar
 Link full download: http://testbankair.com/download/test-bank-for-robbins-cotran-pathologic-basis-of-disease-9th-edition-bykumar-abbas-and-aster Test Bank for Robbins and Cotran Pathologic Basis of Disease
Link full download: http://testbankair.com/download/test-bank-for-robbins-cotran-pathologic-basis-of-disease-9th-edition-bykumar-abbas-and-aster Test Bank for Robbins and Cotran Pathologic Basis of Disease
mirna Dr. S Hosseini-Asl
 mirna Dr. S Hosseini-Asl 1 2 MicroRNAs (mirnas) are small noncoding RNAs which enhance the cleavage or translational repression of specific mrna with recognition site(s) in the 3 - untranslated region
mirna Dr. S Hosseini-Asl 1 2 MicroRNAs (mirnas) are small noncoding RNAs which enhance the cleavage or translational repression of specific mrna with recognition site(s) in the 3 - untranslated region
CARCINOGENESIS THE MOLECULAR BASIS OF CANCER
 CARCINOGENESIS THE MOLECULAR BASIS OF CANCER We know that cancer is a genetic disease and that it is a multi-step process, therefore multiple genetic events (mutations) will occur in tumors. Given that
CARCINOGENESIS THE MOLECULAR BASIS OF CANCER We know that cancer is a genetic disease and that it is a multi-step process, therefore multiple genetic events (mutations) will occur in tumors. Given that
Lecture 1: Carcinogenesis
 Lecture 1: Carcinogenesis Anti-cancer (oncology agents): These are perhaps the most dangerous of drugs, other than the narcotic analgesics. This is due to their toxicities. Killing or inhibiting cancer
Lecture 1: Carcinogenesis Anti-cancer (oncology agents): These are perhaps the most dangerous of drugs, other than the narcotic analgesics. This is due to their toxicities. Killing or inhibiting cancer
Cell Cell Communication
 IBS 8102 Cell, Molecular, and Developmental Biology Cell Cell Communication January 29, 2008 Communicate What? Why do cells communicate? To govern or modify each other for the benefit of the organism differentiate
IBS 8102 Cell, Molecular, and Developmental Biology Cell Cell Communication January 29, 2008 Communicate What? Why do cells communicate? To govern or modify each other for the benefit of the organism differentiate
Prof. R. V. Skibbens. Cell Cycle, Cell Division and Cancer (Part 2)
") Prof. R. V. Skibbens November 22, 2010 BIOS 10: BioScience in the 21 st Century Cell Cycle, Cell Division and Cancer (Part 2) Directionality - clocks go in only one direction G1 doesn t have replication-inducing
Prof. R. V. Skibbens November 22, 2010 BIOS 10: BioScience in the 21 st Century Cell Cycle, Cell Division and Cancer (Part 2) Directionality - clocks go in only one direction G1 doesn t have replication-inducing
Oncogenes and Tumor. supressors
 Oncogenes and Tumor supressors From history to therapeutics Serge ROCHE Neoplastic transformation TUMOR SURESSOR ONCOGENE ONCOGENES History 1911 1960 1980 2001 Transforming retrovirus RSV v-src is an oncogene
Oncogenes and Tumor supressors From history to therapeutics Serge ROCHE Neoplastic transformation TUMOR SURESSOR ONCOGENE ONCOGENES History 1911 1960 1980 2001 Transforming retrovirus RSV v-src is an oncogene
Cancer and Oncogenes Bioscience in the 21 st Century. Linda Lowe-Krentz
 Cancer and Oncogenes Bioscience in the 21 st Century Linda Lowe-Krentz December 1, 2010 Just a Few Numbers Becoming Cancer Genetic Defects Drugs Our friends and family 25 More mutations as 20 you get older
Cancer and Oncogenes Bioscience in the 21 st Century Linda Lowe-Krentz December 1, 2010 Just a Few Numbers Becoming Cancer Genetic Defects Drugs Our friends and family 25 More mutations as 20 you get older
Cancer. Questions about cancer. What is cancer? What causes unregulated cell growth? What regulates cell growth? What causes DNA damage?
 Questions about cancer What is cancer? Cancer Gil McVean, Department of Statistics, Oxford What causes unregulated cell growth? What regulates cell growth? What causes DNA damage? What are the steps in
Questions about cancer What is cancer? Cancer Gil McVean, Department of Statistics, Oxford What causes unregulated cell growth? What regulates cell growth? What causes DNA damage? What are the steps in
BCHM3972 Human Molecular Cell Biology (Advanced) 2013 Course University of Sydney
 2013 Course University of Sydney") BCHM3972 Human Molecular Cell Biology (Advanced) 2013 Course University of Sydney Page 2: Immune Mechanisms & Molecular Biology of Host Defence (Prof Campbell) Page 45: Infection and Implications for Cell
BCHM3972 Human Molecular Cell Biology (Advanced) 2013 Course University of Sydney Page 2: Immune Mechanisms & Molecular Biology of Host Defence (Prof Campbell) Page 45: Infection and Implications for Cell
Oncogenes and tumour suppressor genes
 Cancer mutations disrupt cellular homeostasis Oncogenes and tumour suppressor genes Oncogenes: Gain of function mutations Proto-oncogene Tumour suppressor genes: loss of function mutations Normal cell
Cancer mutations disrupt cellular homeostasis Oncogenes and tumour suppressor genes Oncogenes: Gain of function mutations Proto-oncogene Tumour suppressor genes: loss of function mutations Normal cell
Tumor suppressor genes D R. S H O S S E I N I - A S L
 Tumor suppressor genes 1 D R. S H O S S E I N I - A S L What is a Tumor Suppressor Gene? 2 A tumor suppressor gene is a type of cancer gene that is created by loss-of function mutations. In contrast to
Tumor suppressor genes 1 D R. S H O S S E I N I - A S L What is a Tumor Suppressor Gene? 2 A tumor suppressor gene is a type of cancer gene that is created by loss-of function mutations. In contrast to
In vitro scratch assay: method for analysis of cell migration in vitro labeled fluorodeoxyglucose (FDG)
") In vitro scratch assay: method for analysis of cell migration in vitro labeled fluorodeoxyglucose (FDG) 1 Dr Saeb Aliwaini 13/11/2015 Migration in vivo Primary tumors are responsible for only about 10%
In vitro scratch assay: method for analysis of cell migration in vitro labeled fluorodeoxyglucose (FDG) 1 Dr Saeb Aliwaini 13/11/2015 Migration in vivo Primary tumors are responsible for only about 10%
Cell Cell Communication
 IBS 8102 Cell, Molecular, and Developmental Biology Cell Cell Communication January 29, 2008 Communicate What? Why do cells communicate? To govern or modify each other for the benefit of the organism differentiate
IBS 8102 Cell, Molecular, and Developmental Biology Cell Cell Communication January 29, 2008 Communicate What? Why do cells communicate? To govern or modify each other for the benefit of the organism differentiate
Development of Carcinoma Pathways
 The Construction of Genetic Pathway to Colorectal Cancer Moriah Wright, MD Clinical Fellow in Colorectal Surgery Creighton University School of Medicine Management of Colon and Diseases February 23, 2019
The Construction of Genetic Pathway to Colorectal Cancer Moriah Wright, MD Clinical Fellow in Colorectal Surgery Creighton University School of Medicine Management of Colon and Diseases February 23, 2019
Biochemistry of Cancer and Tumor Markers
 Biochemistry of Cancer and Tumor Markers The term cancer applies to a group of diseases in which cells grow abnormally and form a malignant tumor. It is a long term multistage genetic process. The first
Biochemistry of Cancer and Tumor Markers The term cancer applies to a group of diseases in which cells grow abnormally and form a malignant tumor. It is a long term multistage genetic process. The first
Aberrant cell Growth. Younas Masih New Life College of Nursing Karachi. 3/4/2016 Younas Masih ( NLCON)
") Aberrant cell Growth Younas Masih New Life College of Nursing Karachi 1 Objectives By the end of this session the learners will be able to, Define the characteristics of the normal cell Describe the characteristics
Aberrant cell Growth Younas Masih New Life College of Nursing Karachi 1 Objectives By the end of this session the learners will be able to, Define the characteristics of the normal cell Describe the characteristics
Division Ave. High School AP Biology
 Regulation of Cell Division 2008-2009 Coordination of cell division A multicellular organism needs to coordinate cell division across different tissues & organs u critical for normal growth, development
Regulation of Cell Division 2008-2009 Coordination of cell division A multicellular organism needs to coordinate cell division across different tissues & organs u critical for normal growth, development
III. Evasion of Cell Death - Accumulation of neoplastic cells may result from mutations in genes regulating apoptosis and of these candidates, is the
 III. Evasion of Cell Death - Accumulation of neoplastic cells may result from mutations in genes regulating apoptosis and of these candidates, is the role of BCL2 in protecting tumor cells from apoptosis.
III. Evasion of Cell Death - Accumulation of neoplastic cells may result from mutations in genes regulating apoptosis and of these candidates, is the role of BCL2 in protecting tumor cells from apoptosis.
Introduction to Cancer Biology
 Introduction to Cancer Biology Robin Hesketh Multiple choice questions (choose the one correct answer from the five choices) Which ONE of the following is a tumour suppressor? a. AKT b. APC c. BCL2 d.
Introduction to Cancer Biology Robin Hesketh Multiple choice questions (choose the one correct answer from the five choices) Which ONE of the following is a tumour suppressor? a. AKT b. APC c. BCL2 d.
Cell cycle and Apoptosis. Chalermchai Mitrpant
 Cell cycle and Apoptosis 2556 Chalermchai Mitrpant Overview of the cell cycle Outline Regulatory mechanisms controlling cell cycle Progression of the cell cycle Checkpoint of the cell cycle Phases of the
Cell cycle and Apoptosis 2556 Chalermchai Mitrpant Overview of the cell cycle Outline Regulatory mechanisms controlling cell cycle Progression of the cell cycle Checkpoint of the cell cycle Phases of the
Biology is the only subject in which multiplication is the same thing as division
 The Cell Cycle Biology is the only subject in which multiplication is the same thing as division Why do cells divide? For reproduction asexual reproduction For growth one-celled organisms from fertilized
The Cell Cycle Biology is the only subject in which multiplication is the same thing as division Why do cells divide? For reproduction asexual reproduction For growth one-celled organisms from fertilized
TARGETED THERAPY FOR CHILDHOOD CANCERS
 TARGETED THERAPY FOR CHILDHOOD CANCERS AZIZA SHAD, MD AMEY DISTINGUISHED PROFESSOR OF PEDIATRIC HEMATOLOGY ONCOLOGY, BLOOD AND MARROW TRANSPLANTATION LOMBARDI CANCER CENTER GEORGETOWN UNIVERSITY HOSPITAL
TARGETED THERAPY FOR CHILDHOOD CANCERS AZIZA SHAD, MD AMEY DISTINGUISHED PROFESSOR OF PEDIATRIC HEMATOLOGY ONCOLOGY, BLOOD AND MARROW TRANSPLANTATION LOMBARDI CANCER CENTER GEORGETOWN UNIVERSITY HOSPITAL
Mohammed El-Khateeb. Tumor Genetics. MGL-12 May 13 th Chapter 22 slide 1 台大農藝系遺傳學
 Mohammed El-Khateeb Tumor Genetics MGL-12 May 13 th 2014 台大農藝系遺傳學 601 20000 Chapter 22 slide 1 Cancer Genetics Types of Genetic Alterations in Cancer Evidence that Mutations Cause Cancer Multistage Model
Mohammed El-Khateeb Tumor Genetics MGL-12 May 13 th 2014 台大農藝系遺傳學 601 20000 Chapter 22 slide 1 Cancer Genetics Types of Genetic Alterations in Cancer Evidence that Mutations Cause Cancer Multistage Model
An Overview of Molecular Cancer Pathogenesis, Prognosis, and Diagnosis
 1 An Overview of Molecular Cancer Pathogenesis, Prognosis, and Diagnosis John M. Cullen and Matthew Breen North Carolina State University, USA Fundamentals of cancer biology Cancer is a disease of the
1 An Overview of Molecular Cancer Pathogenesis, Prognosis, and Diagnosis John M. Cullen and Matthew Breen North Carolina State University, USA Fundamentals of cancer biology Cancer is a disease of the
Contents. Preface XV Acknowledgments XXI List of Abbreviations XXIII About the Companion Website XXIX
 Contents Preface XV Acknowledgments XXI List of Abbreviations XXIII About the Companion Website XXIX 1 General Aspects of Signal Transduction and Cancer Therapy 1 1.1 General Principles of Signal Transduction
Contents Preface XV Acknowledgments XXI List of Abbreviations XXIII About the Companion Website XXIX 1 General Aspects of Signal Transduction and Cancer Therapy 1 1.1 General Principles of Signal Transduction
Biochemistry of Carcinogenesis. Lecture # 35 Alexander N. Koval
 Biochemistry of Carcinogenesis Lecture # 35 Alexander N. Koval What is Cancer? The term "cancer" refers to a group of diseases in which cells grow and spread unrestrained throughout the body. It is difficult
Biochemistry of Carcinogenesis Lecture # 35 Alexander N. Koval What is Cancer? The term "cancer" refers to a group of diseases in which cells grow and spread unrestrained throughout the body. It is difficult
Principles of Genetics and Molecular Biology
 Cell signaling Dr. Diala Abu-Hassan, DDS, PhD School of Medicine Dr.abuhassand@gmail.com Principles of Genetics and Molecular Biology www.cs.montana.edu Modes of cell signaling Direct interaction of a
Cell signaling Dr. Diala Abu-Hassan, DDS, PhD School of Medicine Dr.abuhassand@gmail.com Principles of Genetics and Molecular Biology www.cs.montana.edu Modes of cell signaling Direct interaction of a
Cancer Biology Course. Invasion and Metastasis
 Cancer Biology Course Invasion and Metastasis 2016 Lu-Hai Wang NHRI Cancer metastasis Major problem: main reason for killing cancer patients, without it cancer can be cured or controlled. Challenging questions:
Cancer Biology Course Invasion and Metastasis 2016 Lu-Hai Wang NHRI Cancer metastasis Major problem: main reason for killing cancer patients, without it cancer can be cured or controlled. Challenging questions:
Cell Death and Cancer. SNC 2D Ms. Papaiconomou
 Cell Death and Cancer SNC 2D Ms. Papaiconomou How do cells die? Necrosis Death due to unexpected and accidental cell damage. This is an unregulated cell death. Causes: toxins, radiation, trauma, lack of
Cell Death and Cancer SNC 2D Ms. Papaiconomou How do cells die? Necrosis Death due to unexpected and accidental cell damage. This is an unregulated cell death. Causes: toxins, radiation, trauma, lack of
Follicular Lymphoma. ced3 APOPTOSIS. *In the nematode Caenorhabditis elegans 131 of the organism's 1031 cells die during development.
 Harvard-MIT Division of Health Sciences and Technology HST.176: Cellular and Molecular Immunology Course Director: Dr. Shiv Pillai Follicular Lymphoma 1. Characterized by t(14:18) translocation 2. Ig heavy
Harvard-MIT Division of Health Sciences and Technology HST.176: Cellular and Molecular Immunology Course Director: Dr. Shiv Pillai Follicular Lymphoma 1. Characterized by t(14:18) translocation 2. Ig heavy
Lecture 14 - The cell cycle and cell death
 02.17.10 Lecture 14 - The cell cycle and cell death The cell cycle: cells duplicate their contents and divide The cell cycle may be divided into 4 phases The cell cycle triggers essential processes (DNA
02.17.10 Lecture 14 - The cell cycle and cell death The cell cycle: cells duplicate their contents and divide The cell cycle may be divided into 4 phases The cell cycle triggers essential processes (DNA
Regulators of Cell Cycle Progression
 Regulators of Cell Cycle Progression Studies of Cdk s and cyclins in genetically modified mice reveal a high level of plasticity, allowing different cyclins and Cdk s to compensate for the loss of one
Regulators of Cell Cycle Progression Studies of Cdk s and cyclins in genetically modified mice reveal a high level of plasticity, allowing different cyclins and Cdk s to compensate for the loss of one
Introduction to pathology lecture 5/ Cell injury apoptosis. Dr H Awad 2017/18
 Introduction to pathology lecture 5/ Cell injury apoptosis Dr H Awad 2017/18 Apoptosis = programmed cell death = cell suicide= individual cell death Apoptosis cell death induced by a tightly regulated
Introduction to pathology lecture 5/ Cell injury apoptosis Dr H Awad 2017/18 Apoptosis = programmed cell death = cell suicide= individual cell death Apoptosis cell death induced by a tightly regulated
Src-INACTIVE / Src-INACTIVE
 Biology 169 -- Exam 1 February 2003 Answer each question, noting carefully the instructions for each. Repeat- Read the instructions for each question before answering!!! Be as specific as possible in each
Biology 169 -- Exam 1 February 2003 Answer each question, noting carefully the instructions for each. Repeat- Read the instructions for each question before answering!!! Be as specific as possible in each
Chapter 9. Cells Grow and Reproduce
 Chapter 9 Cells Grow and Reproduce DNA Replication DNA polymerase Addition of a nucleotide to the 3 end of a growing strand Use dntps as substrate Release of pyrophosphate Initiation of Replication Replication
Chapter 9 Cells Grow and Reproduce DNA Replication DNA polymerase Addition of a nucleotide to the 3 end of a growing strand Use dntps as substrate Release of pyrophosphate Initiation of Replication Replication
609G: Concepts of Cancer Genetics and Treatments (3 credits)
") Master of Chemical and Life Sciences Program College of Computer, Mathematical, and Natural Sciences 609G: Concepts of Cancer Genetics and Treatments (3 credits) Text books: Principles of Cancer Genetics,
Master of Chemical and Life Sciences Program College of Computer, Mathematical, and Natural Sciences 609G: Concepts of Cancer Genetics and Treatments (3 credits) Text books: Principles of Cancer Genetics,
Control of Cell Cycle. Unit 2 Part f III
 Control of Cell Cycle Unit 2 Part f III How often do cells divide and why? The timing and rate of cell division in different parts of the plant or animals are crucial to normal growth, development and
Control of Cell Cycle Unit 2 Part f III How often do cells divide and why? The timing and rate of cell division in different parts of the plant or animals are crucial to normal growth, development and
The mutations that drive cancer. Paul Edwards. Department of Pathology and Cancer Research UK Cambridge Institute, University of Cambridge
 The mutations that drive cancer Paul Edwards Department of Pathology and Cancer Research UK Cambridge Institute, University of Cambridge Previously on Cancer... hereditary predisposition Normal Cell Slightly
The mutations that drive cancer Paul Edwards Department of Pathology and Cancer Research UK Cambridge Institute, University of Cambridge Previously on Cancer... hereditary predisposition Normal Cell Slightly
Mitosis and the Cell Cycle
 Mitosis and the Cell Cycle Chapter 12 The Cell Cycle: Cell Growth & Cell Division Where it all began You started as a cell smaller than a period at the end of a sentence Getting from there to here Cell
Mitosis and the Cell Cycle Chapter 12 The Cell Cycle: Cell Growth & Cell Division Where it all began You started as a cell smaller than a period at the end of a sentence Getting from there to here Cell
NEOPLASIA. 3. Which of the following tumour is benign a. Chondrosarcoma b. Osteochondroma c. Chondroblastoma d. Ewing s tumour e.
 NEOPLASIA 1. malignant neoplasms a. are independent of hormonal influence b. are always composed of homogenous cell lines c. arise from differentiated cells by a process of anaplasia d. display abnormal
NEOPLASIA 1. malignant neoplasms a. are independent of hormonal influence b. are always composed of homogenous cell lines c. arise from differentiated cells by a process of anaplasia d. display abnormal
C) The graph should look exactly like the graph on the left (Mut1 cells + Mating Pheromone for 3 hours at 25 degrees). The cells arrest in G1.
 The graph should look exactly like the graph on the left (Mut1 cells + Mating Pheromone for 3 hours at 25 degrees). The cells arrest in G1.") 706-2000-Exam 4 Answer Key 1) The question asks you to explain peaks A and B in the top graph. The other two graphs were there to give you hints. The question did not ask for these other two graphs to
706-2000-Exam 4 Answer Key 1) The question asks you to explain peaks A and B in the top graph. The other two graphs were there to give you hints. The question did not ask for these other two graphs to
CYTOKINE RECEPTORS AND SIGNAL TRANSDUCTION
 CYTOKINE RECEPTORS AND SIGNAL TRANSDUCTION What is Cytokine? Secreted popypeptide (protein) involved in cell-to-cell signaling. Acts in paracrine or autocrine fashion through specific cellular receptors.
CYTOKINE RECEPTORS AND SIGNAL TRANSDUCTION What is Cytokine? Secreted popypeptide (protein) involved in cell-to-cell signaling. Acts in paracrine or autocrine fashion through specific cellular receptors.