MD simulations suggest important surface differences between reconstituted and circulating spherical HDL
|
|
|
- Bruce Blake
- 5 years ago
- Views:
Transcription
1 MD simulations suggest important surface differences between reconstituted and circulating spherical HDL Jere P. Segrest, 1* Martin K. Jones, * and Andrea Catte * *Department of Medicine, Atherosclerosis Research Unit, and Center for Computational and Structural Dynamics, University of Alabama at Birmingham, Birmingham, AL 35294, USA 1 To whom correspondence should be sent: Jere P. Segrest, th Avenue South, Boshell Diabetes Building 630, Departments of Medicine and Biochemistry and Molecular Genetics, and Center for Computational and Structural Dynamics, Birmingham, AL 35294, Phone , Fax , segrest@uab.edu. Running title: MD simulations of spheroidal HDL Abbreviations: molecular dynamics (MD); all atom (AA); coarse grained (CG); molecular dynamics simulated annealing (MDSA); coarse grained molecular dynamics (CGMD); palmitoyloleoylphosphatidylcholine (POPC); cholesteryl ester (CE); unesterified cholesterol (UC); spheroidal HDL (shdl); discoidal HDL (dhdl); root mean-square deviation (RMSD) 1
2 Abstract Since spheroidal HDL particles (shdl) are highly dynamic, molecular dynamics (MD) simulations are useful for obtaining structural models. Here we use MD to simulate shdl with stoichiometries of reconstituted and circulating particles. The hydrophobic effect during simulations rapidly remodels discoidal HDL containing mixed lipids to shdl containing a CE/TG core. We compare the results of simulations of previously characterized reconstituted shdl particles containing two or three apoa-i created in the absence of PLTP with simulations of circulating human HDL containing two or three apoa-i without apoa-ii. We find that circulating shdl compared to reconstituted shdl with the same number of apoa-i per particle contain approximately equal volumes of core lipid but significantly less surface lipid monolayers. We conclude that in vitro reconstituted shdl particles contain kinetically trapped excess phospholipid and are less than ideal models for circulating shdl particles. In the circulation, phospholipid transfer via PLTP decreases the ratio of phospholipid to apolipoprotein for all shdl particles. Further, shdl containing two or three apoa-i adapt to changes in surface area by condensation of common conformational motifs. These results represent an important step toward resolving the complicated issue of the protein and lipid stoichiometry of circulating HDL. Supplementary key words: apoa-i, spheroidal HDL, Molecular Dynamics simulation, HDL subspecies 2
3 High density lipoproteins (HDL) are supramolecular assemblies of lipid and protein that represent a nanoscale soft form of condensed matter easily deformable by thermal fluctuations (1). Because the soft lipid environment in HDL modulates apoa-i conformation in dynamic ways, a detailed understanding of HDL structure-function requires new approaches. The common lipid-associating motif in apoa-i is the amphipathic α helix (2, 3). The first tangible experimental evidence for the conformation of apoa-i on the edge of discoidal HDL (dhdl) was determination of a 4 Å resolution solution phase x-ray structure for residues of lipid-free apoa-i reported by Borhani, et al in 1997 (4) that suggested an antiparallel double belt model. In the first experimental test of the belt model, Axelsen, et al (5) used polarized attenuated total internal reflection Fourier-transform infrared spectroscopy to conclude that the result unambiguously supported a belt model. Theoretical considerations of the geometric and physical chemical nature of the apoa-i double belt model resulted in the publication by our lab of an atomic resolution antiparallel double belt amphipathic helical model for dhdl (6) with a helix-helix registry termed LL5/5 oriented by salt bridges (6) surrounding the edge of a bilayer disc. Five laboratories subsequently have studied reconstituted dhdl using a variety of physical chemical methods and the results have been consistent with our double belt model (7-11). This model provided sound theoretical rationale for use of the lipid-free x-ray crystal structure as a valid model for molecular dynamics (MD) simulations of lipid-associated apoa-i. Comparison of the results of our MD simulations of the antiparallel full length apoa-i structure from an ensemble of sixteen MD simulations of 105 Å dhdl (12-15) with the high resolution C-truncated crystal structure of apoa-i by Mei and Atkinson (16) validates many of our key published MD findings (17). We previously published MD simulations of shdl using N-terminally truncated ( 43)apoA-I (18). We have also investigated the dynamics of the activation of LCAT by apoa-i using both all atom (AA) and coarse grained (CG) MD simulations of dhdl and shdl (12). More recently, we developed 3
4 methodology termed particle shrinkage that allows simulation of dhdl containing full length apoa-i (12-15). One of the major conundrums in performing MD simulations of shdl particles is deciding upon the surface lipid concentration. One can use experimentally determined stoichiometries for reconstituted shdl particles (19) or develop methods to calculate surface compositions of circulating shdl particles. In this paper we use both approaches to perform MD simulations on shdl particles containing two or three full length apoa-i by combining the particle shrinkage methodology with a technique we call surface loading (12). We show that circulating and reconstituted shdl particles with the same number of apoa-i appear to contain a constant molar ratio of core lipid to apoa-i, while circulating shdl with the same number of apoa-i have a lower ratio of surface lipid to apoa-i than comparable reconstituted particles. The likely explanation is that in vitro reconstituted particles, in the absence of PLTP, contain kinetically trapped phospholipid. Further, we find that shdl containing two or three apoa-i adapt to changes in surface area by condensation of common conformational motifs. METHODS Rationale for protein and lipid compositions of the simulated shdl particles Three basic categories of starting shdl models were simulated: i) Two de novo designed particles based loosely upon published protein and lipid compositional data from shdl particles reconstituted by sonication: S2-57 (2 apoa-i, 57 POPC, 16 CE and 6 UC) and S3-155 (3 apoa-i, 155 POPC, 58 CE, 45 UC and 14 TG) taken from a publication by Sparks, et al (19). ii) Two particles whose stoichiometries were taken directly from protein and lipid compositional data on shdl particles reconstituted in vitro using LCAT: S2-86 (2 apoa-i, 86 POPC, 12 CE, 6 UC and 10 TG, Silva, personal communication) and S3-94 (3 apoa-i, 94 POPC, 41 CE and 6 UC, Silva, et al (20)). iii) A series of particles whose stoichiometries were taken directly from compositional data of isolated native circulating shdl parti- 4
5 cles by Huang, et al (21): S2-18, S2-30a, S2-30b and S2-34 (2 apoa-i and progressively increasing POPC derived from the LpA-I particle, HDL[1]) and S3-41 (3 apoa-i, 41 POPC, 34 CE, 9 UC and 7 TG, derived from the LpA-I particle, HDL[4]). The initial stoichiometry of the S2-18 particle is derived from the compositional data of isolated native circulating shdl particles of Huang, et al (21) using data from their HDL[2]/HDL 3C. However, since HDL[1] is beyond the range of their density isolates, the stoichiometry of the S2-18 particle is incorrect; MD simulation produces a particle in which the core lipid is insufficiently covered by the surface protein and lipid. As discussed in the Results section, since the initial particle was obviously incorrect, we used a theoretical approach to modify the composition of the initial HDL[1] model until the final particle in the series, S2-34, fell precisely on the linear regression line shown in Fig. 5A. AA force field AA simulations were performed using NAMD (22) as described by Jones, et al (13). Each system was ionized and charge neutralized with NaCl to 0.15M with the Add Ions plug-in of VMD (23). The TIP3P water model was used (24). The CHARMM 22 (25, 26) and 27 (27, 28) force fields were used for protein and lipid molecules, respectively. AA MD simulations Our AA simulations began with a discoidal particle with a POPC:CE:UC:TG:apoA-I molar ratio of 160:0:24:0:2 that we described in Jones, et al (15). Through a series of centrally removed POPC, random deletion of UC, and random mutation of UC to CE, all followed by 5 ns of 310K and 1 atm equilibrations, we produced a spheroidal particle with a POPC:CE:UC:TG:apoA-I molar ratio of 57:16:6:0:2. We then equilibrated the 57:16:6:0:2 particle for an additional 5 ns of 310 K and 1 atm, and continued for another 5 ns at the same temperature and pressure after resolvation and reionization. These two particles, at 5 ns and 10 ns respectively, were each subjected four times to the MD simulated anneal- 5
6 ing (MDSA) 30 ns protocol, involving an initial temperature-jump to 500 K as described by Jones, et al (12), to produce an ensemble of eight particles. The first set of four particles was reported previously, but the second set is new to this paper. See flow diagram in Fig. S1A of the Supplemental Data. Spherical particles are named by the number of apoa-i and the number of POPC molecules they contain: e.g. S2-57 (also called 2-57) contains 2 apoa-i and 57 POPC; S3-155 (also called 3-155) contains 3 apoa-i and 155 POPC. They may also contain other lipids. CG force field All eight final structures of the AA 30 ns simulations described in the previous section were converted to CG using the MARTINI force field for lipid (29) and protein (30) molecules, respectively. Further details about CG parameters can also be found in our previous work (18). The protein secondary structure was assigned using as a reference the one generated by AA simulations (13). This approach was employed to give more flexibility to the apoa-i chains (12, 18) of the previously described CG simulations for particles of identical molar ratios. However, even after this change the majority of protein residues still had an α helical conformation (80%), and all the proline residues were in a β-turn conformation. CG MD simulations Creation of particles S2-57. After mapping the starting AA shdl particles with CG beads, all the particles were solvated with CG water and ionized with CG ions to get neutral systems with a physiological ionic strength of 0.15 M. These structures were subjected to 2000 steps of conjugate gradient energy minimization first without and then with CG water and ions. Then CGMD simulations of 19.6 µs were performed at 310 K and 1 atm. All CGMD simulations were performed using GROMACS (31). Nonbonded van der Waals and electrostatic interactions were truncated using a cutoff distance of 12 Å. Coordinate trajectories were updated every 10 ps of simulation. The pressure was held constant at 6
7 1 atm for all MD simulations using the Berendsen barostat (32). In all CGMD simulations the effective time, which is the time of simulation multiplied by four (29, 33), is used. Creation of particles S The starting structure for all six particles with a POPC:CE:UC:TG:apoA-I molar ratio of 155:58:45:14:3 was an AA disk with 566 POPC and both an apoa-i double belt and an apoa-i hairpin on the edge. After converting to CG, two similar separate reduction, modification, and CGMD sequences were performed, to explore conformational space, labeled as D 1 1 to S 1 1 and D 2 1 to S 2 1 in Fig. S1B. Then two different branching simulation protocols including 4 μs T-jumps where protein was heated to 380 K were performed to obtain the six particles S 1 2, S 1 3, S 1 4, S 2 3, S 2 5, and S 2 6. See flow diagram in Fig. S1B. From a topological standpoint, at least one hairpin is required in a disc trimer. There is crosslinking data by Silva, et al (34) suggesting the presence of both a double belt and a hairpin structure in the trimer. The only other possibility for the trimer is the presence of three hairpins in various head to tail and head to head orientations. We simulated such starting structures and the results were uninteresting. Some have suggested that the timer could be a triple belt. Besides possibly being too thick for a bilayer, such a trimer would be unable to make a disc larger than 120 Å; trimer discs are in the 150 Å range (14, 15, 35). Creation of particles S3-94, S3-41a, S3-41b, and S2-86. Similar steps were performed to create four particles with POPC:CE:UC:TG:apoA-I molar ratios of 94:41:6:0:3, 41:34:9:7:3, 41:36:9:8:3, and 86:12:6:10:2, respectively. All particles except S2-86 were subjected to an initial 2 μs simulation followed by a 4 μs T-jump with protein heated to 380 K. Then all four were simulated for 10 μs. See flow diagrams in Fig. S1A. Creation of particles S2-34, S2-30b, and S2-30a. The starting structure for all three particles was an AA shdl particle with a POPC:CE:UC:TG:apoA-I molar ratio of 57:16:6:0:2 from the set of S
8 To obtain S2-34, this structure was subjected to the mutation of 5 CE molecules to TG to reach a POPC:CE:UC:TG:apoA-I molar ratio of 57:11:6:5:2. The energy minimized structure of this latter model was converted to CG and subjected to the manual insertion of 1 CE and 5 TG molecules followed by the deletion of 23 POPC molecules to obtain a POPC:CE:UC:TG:apoA-I molar ratio of 34:12:6:10:2 and then solvated with CG polarizable water and ions and simulated for 10.8 μs (Fig. S1A). To obtain S2-30b, an AA shdl particle from the set of S2-57 was subjected to the deletion of 22 POPC and 1 CE molecule, and to the mutation of 5 POPC molecules to TG molecules, in order to reach a POPC:CE:UC:TG:apoA-I molar ratio of 30:15:6:5:2. Then, the energy minimized structure of the latter model was converted to CG and simulated for 22.4 μs. To obtain S2-30a, from the energy minimized structure of particle S2-30b, 3 TG molecules were deleted to reach a POPC:CE:UC:TG:apoA-I molar ratio of 30:15:6:2:2. Then the structure was solvated with polarizable water and ions and simulated for 25.6 μs. (Fig. S1A). Root mean-square deviation (RMSD) The root mean-square deviation (RMSD) of protein α carbon atoms was calculated for all 57:16:6:2 particles over the entire duration of the eight AA and CG simulations, then an average RMSD was calculated for each ensemble. Fraction of total α helicity The fraction of all protein residues that were α helical were measured every 20 ps over the course of all eight 30 ns AA trajectories by using VMD s protein secondary structure determination program, STRIDE (36). Then, an average was calculated for all the simulated AA particles. The fraction of the time that each protein residue was α helical (fractional α-helicity per residue) was calculated over the last 20% of each 30 ns trajectory and averaged over the eight simulated structures. Radial density distributions 8
9 The radial density distribution plots describe the number densities of the atoms and CG beads for each component of the system (37). Two different distributions were calculated with respect to the center of mass of the particle for both the AA and CG simulations. The first measures the mole fraction of each molecular component (what fraction of the atoms or CG beads found at that distance belong to each component) and the second measures the distribution of each molecular component (the fraction of the total atoms or CG beads of each component that are found at that distance). To calculate the mole fractions, six to eleven frames over the last 20% of the simulation were selected and each atom or CG bead of each molecular component was measured from the center of mass. To calculate the distributions, all frames over the last 20% of the simulation were selected and each atom or CG bead of each molecular component was measured from the center of mass. Average Cα coordinate structures Structures created by averaging the Cα coordinates over all final structures were calculated for the AA and CG ensembles by using the measure avpos command in VMD after aligning over the Cα atoms containing the less solvent-accessible salt bridges (residues ). RESULTS Simulations of reconstituted shdl particles containing two apoa-i Particle S2-57. The first particle containing two apoa-i, S2-57, that we simulated consisted of 2 apoa-i, 57 POPC, 16 CE and 6 UC. This stoichiometry (with the addition of 6 UC) was derived from work by Sparks, et al (19) in which shdl was produced by sonication. We monitored the structural and dynamic behavior of each model shdl particle using two different MD methodologies: i) MDSA allows the exploration of an increased conformational space via AA simulations and an initial temperature-jump; ii) CGMD allows the exploration of a longer time scale by using CG simulations to decrease computational costs. The equilibration of these systems was monitored by measuring the RMSD of each 9
10 system as described in the Methods section. The average RMSD of the ensemble of simulated particles over the entire time of simulation reaches a plateau at about 16 ns and 19.6 µs for AA and CG simulations, respectively (see Fig. S2A-B), suggesting equilibration. Changes in total α helicity. The fraction of all protein residues that were α helical over the course of all the eight 30 ns trajectories of AA simulations are plotted in Fig. S2C. Percent α helicity plateaus at approximately 72%, comparable to the experimental value reported by Rye, et al (38) for reconstituted shdl particles in vitro (69%). Structural features of AA and CG ensembles. During our MD simulations, driven by the hydrophobic effect, the CE on the surface of the original discs form a separate phase or CE droplet, converting the discs to shdl of approximately 68 Å in unhydrated diameter. Fig. 1 shows various views of the eight AA and CG shdl ensembles in which apoa-i has been aligned in its central domain, residues ; this domain represents the location of the six less solvent-accessible salt bridges involved in registration of the LL5/5 antiparallel double belt conformation (6, 13-15, 18, 39, 40). Figs. 1A-B are cross-eyed stereo images of the aligned shdl particles produced by AA and CG, respectively. It is obvious that, although produced by two very different MD methods, both ensembles of particles have similar, though dynamic, protein conformations. Cross-sectional views of the two aligned shdl ensembles shown in Figs. 1C-D show clearly how the hydrophobic effect generated by MD simu- lation drives the CE (magenta) to the particle center of each type of ensemble, while the POPC (cyan) and UC (brown) are driven to form surface monolayers. The radial distributions of molecular components of the AA and CG ensembles of shdl particles averaged over the last 20% of their trajectories are plotted as mole fractions and individual distributions of each component in Figs. 1E-H. The most important observation from these plots is that both methods of MD simulation produce similar molecular distributions: Both ensembles contain only CE in 10
11 their centers (Figs. 1E-F); in both ensembles the POPC glycerol and the UC hydroxyl moieties have overlapping radial distributions (Figs. 1G-H), results that have been reported for membrane bilayers but never, to the best of our knowledge, for lipoprotein particles. In both ensembles protein and POPC headgroups have overlapping radial distribution and, acyl chains, as well as UC, have similar distribution curves. Particle S2-86. The second shdl containing two apoa-i simulated by CGMD, S2-86 (Figs. 2A- C), was composed of 2 apoa-i, 86 POPC, 12 CE, 6 UC and 10 TG. This stoichiometry was derived from work by Silva, et al (20) in which a reconstituted apoa-i:popc disc was incubated with human LCAT and LDL as a phospholipid donor resulting in a large spherical particle, that in turn was converted to a smaller sphere, S2-86, by incubating with CETP and Intralipid. Figs. 2A-B illustrate cross-eyed stereo images of the intact particle and the particle in cross-section, respectively. Fig. 2C represents the radial distributions of molecular components of the S2-86 particle averaged over the last 20% of its trajectory plotted as mole fractions; the radial distributions of protein and the different lipid moieties are similar to those seen for the S2-57 particles in Figs. 1E-F, with one important exception. The core lipid, TG, present in this particle, occupies the very center of the particle, displacing CE. Central TG can also be seen in the cross-sectional view (Fig. 2B). Simulations of reconstituted shdl particles containing three apoa-i Particle S The first particle containing three apoa-i simulated by CGMD, S3-155, was composed of 3 apoa-i, 155 POPC, 58 CE, 45 UC and 14 TG. This stoichiometry represents a modification of a particle described by Sparks, et al (19) in which shdl was produced by sonication by, among other changes, the addition of UC and TG to the particle. Fig. 3A shows a schematic diagram of the starting structure, a large discoidal POPC particle whose edge was lined by three apoa-i arranged as a double belt dimer adjacent to a hairpin monomer. Fig. 3B shows a cross-eyed stereo image of the dis- 11
12 coidal particle after an initial removal of 60 central POPC followed by 5 ns MD simulation at 310 K. In this view the POPC surface monolayer has been rendered transparent to allow visualization of the entire apoa-i double belt-hairpin. An ensemble of six particles, approximately 80.9 Å in unhydrated diameter, was produced by a network of T-jumps (see the flow diagram, Fig. S1B). Fig. 3C is a cross-eyed stereo image of one representative example of the ensemble of S3-155 particles in which the POPC surface monolayer has been rendered transparent to allow visualization of the CE/TG core. Fig. 3D is a crosseyed stereo image of a cross-section of an aligned ensemble of the six particles the double belt region of apoa-i has been aligned in its central domain, residues ; this domain represents the location of the six less solvent-accessible salt bridges involved in registration of the LL5/5 antiparallel double belt conformation (6, 13-15, 18, 39, 40). This image highlights the distribution of surface and core lipids. The radial distribution of molecular components of the S3-155 particle were averaged over the last 20% of their trajectories and plotted as mole fractions or individual distributions of each component in Figs. 3E-F. The radial distributions of protein and lipid are similar to that seen in the previous simulations; again note the central displacement of CE by TG (Figs. 3D-E). Particle S3-94. The second shdl containing three apoa-i simulated by CGMD, S3-94 (Fig. 4A- C), was composed of 3 apoa-i, 94 POPC, 41 CE, 6 UC and 0 TG. This stoichiometry is derived from work by Huang, et al (21) in which a reconstituted apoa-i:popc disc was incubated with human LCAT and LDL as a phospholipid donor resulting in a large spherical particle, S3-94. Figs. 4A-B illustrate cross-eyed stereo images of the intact particle and the particle in cross-section, respectively The particle contains no core TG. The radial distribution of molecular components of the S3-94 particle were averaged over the last 20% of their trajectories and plotted as mole fractions in Fig. 4C. Once again, the radial distributions of protein and lipid are similar to that seen in the previous simulations. Simulations of native circulating shdl particles containing two and three apoa-i 12
13 Best fit volumetric analyses of the stoichiometry of native shdl particles. As described in the preceding paper (Segrest, J. P., M. C. Cheung, and M. K. Jones, 2013), we developed a volumetric analysis procedure for selecting native shdl protein and lipid stoichiometries (Huang, et al (21)) for best fits to dimensions determined by NDGGE for HDL isolated by immunoaffinity chromatography (41) (see Table II in the preceding paper for an example of the application of this volumetric method to the well-characterized S2-86 and S3-94 particles (20)). Seven particles HDL[1-7] are described in Table III in the preceding paper, HDL[1], HDL[4] and HDL[7] being predominantly LpA-I (particles with apoa-i but without apoa-ii). MD simulations of circulating shdl particles. On the assumption that HDL[4] is the best characterized of the seven particles with a stoichiometry of 3 apoa-i: 41 PL: 34 CE: 9 UC: 7 TG (Table III in (Segrest, J. P., M. C. Cheung, and M. K. Jones, 2013)), we created a shdl particle with this composition (named S3-41), simulated it by CGMD for 10 µs and compared the resulting particle to S3-94; Fig. S2D is an RMSD plot showing that the simulation reaches equilibrium at 5 µs. The final S3-41 particle has an unhydrated diameter of 74 Å, significantly smaller than the unhydrated diameter, 80.9 Å, of the S3-94 particle. Figs. 4D-E illustrate cross-eyed stereo images of the intact particle and the particle in cross-section, respectively. Although S3-41 has less surface lipid (PL + UC) than S3-94 (compare Figs. 4D-E with Figs. 4A-B), importantly it has the same core volume (48,339 Å 3 vs. 47,946, respectively) resulting in a much thinner layer of surface lipid (compare Fig. 4E with Fig. 4B). The radial distribution of molecular components of the S3-41 particle were averaged over the last 20% of their trajectories and plotted as mole fractions in Fig. 4F; the radial distributions of protein and lipid are similar to that found in the other simulations. In spite of the small particle size, TG displaces CE from the core center. HDL[4] compositionally falls in the middle of the five particles that Huang, et al (21) isolated for their analyses. This fact and the apoa-i-alone nature of the particle make it the best characterized of the 13
14 seven circulating particles. In contradistinction, HDL[1], the smallest of the circulating apoa-i-alone particles with a diameter of 76.4 Å and a low concentration, falls outside the density range of the five particles isolated by Huang, et al (21) for their analyses. When subjected to best fit volumetric analysis, its inaccurately calculated stoichiometry (Table III in (Segrest, J. P., M. C. Cheung, and M. K. Jones, 2013)) places it well outside the composition and size of circulating shdl (see Fig. 5A). We modified the HDL[1] particle by creating two shdl particles with increased surface lipid and decreasing core lipid, named S2-30b (30 POPC: 15 CE: 6 UC: 5 TG) and S2-30a (30 POPC: 15 CE: 6 UC: 2 TG), respectively, simulated them for more than 20 µs by CGMD and analyzed the properties of each particle. We then created a third particle, S2-34 (34 POPC: 12 CE: 6 UC: 10 TG), in which the core composition (with a volume of 29,898 Å 3 ) was identical to S2-86, and simulated it for 10 µs by CGMD. The resulting particle, shown in Figs 2D-E, has an unhydrated diameter of 65.3 Å, smaller than the unhydrated diameter, 74.2 Å, of the S2-86 particle. The radial distribution of molecular components of the S2-34 particle were averaged over the last 20% of their trajectories and plotted as mole fractions of each component in Fig. 2F; the radial distributions of protein and lipid are similar to that found in the other simulations; e. g., even in this small particle, TG displaces CE from the core center. Comparison of general properties of reconstituted versus native shdl particles. Using the volumetric analysis, we calculated the thickness of the surface lipid monolayer for the reconstituted and native shdl particles. In Fig. 5A we have plotted monolayer thickness versus particle surface to volume ratio (S/V = 3/radius) for the four reconstituted shdl particles S2-57, S2-86, S3-155 and S3-94 and for the six circulating shdl-like particles subjected to CGMD simulations S2-18, S2-30b, S2-30a, S2-34, S3-41a and S3-41b. The solid line is the linear regression line for the six native shdl particles (see Fig. 6 in (Segrest, J. P., M. C. Cheung, and M. K. Jones, 2013)) whose equation is y = -0.01x + 14
15 We have plotted the PL thickness measured by CGMD simulations versus the total monolayer thickness measured by volumetric analysis in Fig. 5B and the percent coverage of HDL particle surface by protein versus total monolayer thickness measured by volumetric analysis in Fig. 5C. When the four reconstituted shdl particles, S2-57, S2-86, S3-155 and S3-94, are plotted in Figs. 5A-C, they fall sufficiently far away from the plots of the native particles to show that all four reconstituted particles have surface lipid thicknesses greater than the largest of the native shdl particles (large open circle marked MAX). For example, for S3-94 to be converted to S3-41a (HDL[4]) and S2-86 converted to S2-34 (HDL[1]), the core volumes would not change but the number of surface PL would decrease from 94 to 41 and 86 to 34, for S3 and S2 particles, respectively. This reduction in surface PL reduces the monolayer thicknesses from 14.5 Å to 11.0 Å for S3 and from 16.5 Å to 11.9 Å for S2 (Table I and Fig. 5A, thick and thin dotted arrows, respectively). The fact that the monolayer thicknesses of the shdl particles reconstituted by the LCAT reaction in the absence of PLTP, S2-86 and S3-94, are identical despite the difference in particle diameters (80 versus 93 Å), suggests disequilibrium between the two particles. In the insert to Fig. 5B., the changes in PL monolayer thickness (PLm) of reconstituted particles, S3-94 and S2-86, and corresponding native particle, S3-41 and S2-34, respectively, are shown schematically. Our results suggest that shdl particles such as S2-86 and S3-94 reconstituted by LCAT and CETP are not optimal models for circulating shdl particles since they possess more surface lipids than do the circulating particles exposed to PLTP (and perhaps other proteins that remodel HDL). We suggest, instead, that shdl particles reconstituted in the absence of PLTP are metastable; i. e. the surface PL is kinetically trapped. In the absence of PLTP it is impossible for excess surface PL, due to an almost infinitesimally small critical micelle concentration, to be transferred between particles in any biologically relevant period of time. 15
16 At least three factors influence the PL monolayer thickness in HDL subspecies: i) Acyl chain tilt increases with decreasing radius and results in decreasing monolayer thickness. ii) The wedge effect of amphipathic helixes on monolayers induces increased monolayer curvature (42). iii) The wedge effect also induces monolayer thinning (43). The wedge effects likely predominate. The effect of the thinning of the polar lipid monolayer as native circulating HDL subspecies decrease in size has an uncertain effect on PL packing: As we demonstrated previously, LDL appears to have structural mechanisms built into apob that dampen changes in surface pressure between differentsized subspecies (43). Similarly, because of the tendency for different domains of apoa-i to pop off and on the HDL surface, packing density might increase, decrease or remain constant between HDL subspecies. Further, there are a number of reasons that it is difficult to accurately predict the packing (or surface area per PL) for the different HDL subspecies. First, as we have pointed out elsewhere (13), it is very difficult to measure surface area per PL molecule on MD simulated spheroidal structures such as HDL. Second, calculations of surface area per PL using the volumetric method of analysis of shdl particles is an even greater challenge predominantly because apoa-i in particular possesses regions with variable lipid affinity. Furthermore, UC has a condensing effect on the surface area of PL monolayers (44). A useful parameter for distinguishing between the structures of reconstituted and circulating shdl is quantification of the percent of the particle surface covered by protein. We have used an approximate method to calculate surface area coverage with protein with our volumetric calculations: in these calculations we assume that each PL has a surface area of 65 Å 2 that is condensed by 1% for each 2 mole % of UC and that the surface area per UC is constant at 27 Å 2 (45). This approximation allows an estimate of the percent surface area occupied by protein and these calculations have been plotted 16
17 against total monolayer thickness (volumetric) in Fig 5C for both native (closed circles) and reconstituted (open circles). While HDL[4] and S2-86 each have the same unhydrated diameters, 74 and 74.2 Å, respectively, protein covers 86% of the surface of HDL[4] but only71% of the surface of S2-86. HDL[7] and S3-94 have unhydrated diameters of 98 and 81Å but very similar percent protein surface coverage, 75 and 69%, respectively. Comparison of reconstituted with circulating shdl particles with equal numbers of apoa-i clearly illustrates the structural difference between the two forms of HDL. Circulating HDL[4] and reconstituted S3-94 (18) both contain three apoa-i per particle but HDL[4] has greater protein surface coverage, 86% versus 69%. This difference reflects significant differences in condensation of apoa-i folding illustrated in Fig. 8. On the basis of two different assumptions from the ones we have used a fixed area of 55 Å 2 per PL and a surface area equivalent to a sphere with the hydrated, not the volumetric, HDL radii Huang, et al (21) also calculated the percent surface area occupied by protein for their isolated HDL subspecies equivalent to our HDL[2], HDL[3], HDL[5], HDL[6] and HDL[7]; their numbers have been plotted onto Fig. 5C (small open diamonds). In spite of significant differences in assumptions, both our calculations and those of Huang, et al (21) of the percent area occupied by protein suggests very protein-rich particle surfaces for native HDL subspecies. Our results suggest that shdl particles such as S2-86 and S3-94 reconstituted by LCAT and CETP are not optimal models for circulating shdl particles since they possess more surface lipids than do the circulating particles exposed to PLTP (and perhaps other proteins that remodel HDL). We suggest, instead, that shdl particles reconstituted in the absence of PLTP are metastable; i. e. the surface PL is kinetically trapped. In the absence of PLTP it is impossible for excess surface PL, due to an almost infinitesimally small critical micelle concentration, to be transferred between particles in any biologically relevant period of time. 17
18 Changes in protein conformation in reconstituted and circulating shdl particles containing two and three apoa-i Comparison of the average protein structures of the S2-57 and S3-155 shdl particles. The protein backbone in the final frames of each AA and CG ensemble of S2-57 were aligned between residues (green in Figs. 6D-E), the location of the less solvent-accessible salt bridges. The remarkable similarity between the protein structures in the AA and CG simulations is dramatically illustrated by the strong similarity between the two average structures created from the aligned ensembles (Figs. 6A-B). Overall, both average structures display clam-like conformations. In both average structures, the double belts hinge at approximately the same region, the sticky N-terminal domains (blue) self-associate (15) and the C-terminal helix 10 domains (red) are independent or promiscuous (15). The double belt in this average structure (red arrowhead) forms a clam-like structure similar to that created by S2-57 but, given its larger unhydrated diameter of 80.9 Å versus 68 Å, one that is more open. The surface above the open double belt is bridged by the hairpin monomer of apoa-i (white arrowhead). The overall coverage of the small and large shdl particles is similar. Each resembles a football helmet with a green face guard (aligned domain). A plot of the fraction helicity of each residue averaged over the last 20% of all eight AA simulations of S2-57 is shown in Fig. 7A. This plot shows two dramatic and symmetric decreases in helicity: hg1: residues (tandem repeat G3 and almost half of helix 1); hg2: residues (starts at the GGA sequence at the junction of helix 7 and 8 and ends at the end of helix 8). Validation of the hg1 domain, essentially a loop-helix-loop motif, is provided by the presence of a similar motif located at residues in the C-truncated apoa-i crystal structure (16). Interestingly, the dip in helicity at hg2 partially spares residues , an exception that includes the completely conserved residue pair, E191-18
19 Y192 (46) and the residue Y192 that Shao, et al (47) suggest acts as a preferred atherogenic oxidation target in apoa-i. From these results we derive the clam-fold model shown in Fig. 7B for the conformational adaptation of apoa-i from a largely planar double belt on the edge of dhdl particles to a clam-like structure that distributes itself around the surface of a shdl particle. During this conformational change, the double belt hinges the open circles in Fig. 7B at the domains noted in Fig. 7A; hg1, residues and hg2, residues These are approximately the same residues that create the more flattened hinges in S3-155 (open circles in Fig. 7C). Changes in protein conformation of shdl particles during transition between large and small particles. Fig. 8 compares cross-eyed stereo licorice representations of the protein conformations of the three simulated shdl particles containing two apoa-i (S2, left hand column) and the three containing three apoa-i (S3, right hand column). The three S2 particles, S2-86, S2-57 and S2-34, have an unhydrated diameter (d 0 ) of 74.2, 68.0 and 65.3 Å, respectively. The conformation of apoa-i in each particle represent variations on the clam-fold double belt conformation noted for S2-57 (Fig. 8, middle left panel). The largest, S2-86, represents an expanded version of the clam-fold structure in which the sticky N- terminal domains (blue) interact but in a more open fashion (Fig. 8, upper left panel). The smallest of the S2 particles, S2-34, forms a clam-fold structure condensed by twisting upon itself so that the sticky N-terminal pair (blue) end up to the opposite side of the particle from the helix 5 antiparallel pair (Fig. 8, lower left panel). The three S3 particles, S3-155, S3-94 and S3-41, have a d 0 of 93.1, 80.9 and 74.0 Å, respectively. Like the three S2 particles, the three S3 particles each represent variations on a common apoa-i conformational motif, the double belt-hairpin noted for the largest, S3-155 (Fig. 8, upper right panel). The middle S3 particle, S3-94, represents a condensed version of the double belt-hairpin (Fig. 8, middle right 19
20 panel) and the smallest, S3-41, represents a further condensation of the same basic double belt-hairpin structure (Fig. 8, lower right panel). With particle shrinkage, the double belt portion of the structures approaches the clam-fold conformation and, as the surface PL decreases, various portions of the double belts approach one another (red double arrow in S3-94 model). DISCUSSION Role of hydrophobic effect in MD simulations The concept of using a discoidal particle in which core lipids have been inserted by mutation into the disc bilayer is based upon the ability of MD simulations to simulate the hydrophobic effect. For CGMD, the hydrophobic effect is incorporated into the force fields for individual amino acid and lipid moieties. For example, a CG representation of water has a repulsive force toward hydrophobic moieties, such as CE. So as discs containing CE and TG are simulated by CGMD, these hydrophobic lipids are forced out of the polar monolayer by the force field and by phase separation, form a CE/TG core. Simulations by AAMD require no modified force field; rather the statistical thermodynamic process that leads to the hydrophobic effect comes directly into play. Water contact with CE/TG and other hydrophobic moieties (a low probability event by statistical thermodynamics) is minimized by phase separation of the CE/TG or for hydrophobic amino acids, by interaction with the acyl chains of surface PL resulting in water entering a higher probability state. Existing structural models of shdl Two basic structural topologies have been proposed for discoidal HDL: the picket fence and the double belt models. On the basis of numerous physical chemical studies, the double belt arrangement is now widely accepted as generally correct. Several variations on the double belt model have been proposed: these include the solar flare, the double superhelix, the belt buckle and the looped belt models. None of these models are directly relevant to the structure of shdl 20
21 The models for shdl can be divided into two basic types: particles containing dimeric apoa-i and those containing three or more apoa-i. The major issue so far for dimeric apoa-i has been whether the double belt model remains intact or separates into individual helical segments during conversion of discs to spheres; all experimental evidence so far suggests that the double belt remains intact. Two models have been proposed for shdl containing three or more apoa-i, the symmetric trefoil model and the asymmetric double belt-hairpin model. Simulations of shdl containing two apoa-i Reconstituted shdl particles. The striking similarity of the shdl structures for S2-57 a particle whose composition is derived from a sonication form of reconstitution from two dramatically different MD simulation methods, AA and CG, supports in a reasonably compelling way the conclusion that MD simulations provide robust virtual experimental imaging of the structure and dynamics of shdl. Both MD methods have strengths and weaknesses but not the same ones: i) The strength of AA is that it is all-atom; the principle weaknesses are a failure to robustly sample long simulation times or use of high temperature jumps to overcome that limitation. ii) The strength of CGMD is that it samples longer simulation times; the principle weakness is use of pseudoatoms to represent clusters of heavy atoms. Our results reported here support the concept first suggested computationally (18) and then ex- perimentally (20) that the intramolecular interactions between individual apoa-i chains do not change substantially from the LL5/5 double belt registry upon conversion from dhdl to shdl. Rather the double belt seems to adapt its conformation to associate with a curved spheroidal surface. For the double belt dimer, there are essentially no degrees of freedom in conformational space not explored using this technique. If the sticky N-termini remain in contact, there are only two possibilities: the double belt can either fold back upon itself to form the clam-fold structure or twist. Our MD simulations suggest that 21
22 both types of double belt conformational changes occur, with twisting following after the initial clamfold structure during particle shrinkage. Although there are conflicting reports as to how often dimeric apoa-i occurs in shdl (21, 48), a recent study by Gauthamadasa, et al (49) suggests that the dimer may be a significant oligomeric form of apoa-i in circulating HDL. Our volumetric analyses described in detail in the preceding paper (Segrest, J. P., M. C. Cheung, and M. K. Jones, 2013) support this possibility. The hinging of apoa-i double belts at or near hg1 (residues 33-52) and hg2 (residues ) have also been observed in dhdl particles of different sizes (14) and appears to be a general property of apoa-i double belts. Further, residues in the middle of hg2, partially helical in our AA shdl simulations, were also shown to be part of a loop-helix-loop motif in hg2 during MD simulations of dhdl as well (14). Circulating shdl particles. Our MD simulation studies of the S2-86 shdl particle, derived compositionally from published data on the composition of the S80 particle described by Huang, et al (21), along with our simulations of the native S2-34 and the S2-57 particle reconstituted by sonication, suggest that the clam-fold model (Fig. 7B) represents a general structural paradigm for double belt apoa-i on the surface of both S2 and S3 forms of shdl. Conversely, in the smallest of the S2 particles, S2-34 whose hydrated diameter is equal to that of the circulating HDL[1] particle the clam-fold structure has twisted upon itself, so that, unlike the intermediate-sized particle, S2-57, the N-N interactions have moved to the other side of the particle from the helix 5 antiparallel pairing. This twisting manner of folding or unfolding is virtually identical to the mechanism we proposed whereby dhdl adjusts to a varying PL content by incremental winding or unwinding of the twisted apoa-i double belt saddle shape on the discoidal particle (14). Simulations of shdl containing three apoa-i 22
23 The results of our MD simulations and analyses of shdl particles containing three apoa-i show a similar pattern of folding or unfolding of the apoa-i in the transitions from larger to smaller particles as the particles containing two apoa-i. Our simulated structures are quite similar to the structure proposed by Wu, et al (50) called the helical dimer-hairpin. In the absence of PLTP, however, the major caveat with any reconstitution method producing shdl particles is the issue of kinetically trapped excess surface lipid highlighted by our present paper; shdl particles, such as S3-94, containing three apoa-i per particle, reconstituted by LCAT alone, also possess more surface lipids than do the circulating particles exposed to PLTP because the surface PL of S3-94 is kinetically trapped. Our S3-41 model, on the other hand, more accurately reflects the lipid structure of the circulating apoa-i-alone shdl particle HDL[4] that is 85 Å in diameter (41). The major uncertainty with our studies of shdl containing three apoa-i is the starting model and the resulting apoa-i conformation on the shdl surface; a disc containing a double belt-hairpin perimeter subjected to our method that combines the particle shrinkage methodology (12-14) with surface loading (12) will likely always produce the same general type of surface conformation changes in apoa- I. For apoa-i trimers, not only can a starting discoidal model contain several different conformations, a double belt-hairpin or several combinations of three hairpins, but the method is incapable of producing a final trefoil structure as proposed by Davidson s lab (20, 21). One argument for the trefoil model has been that all three (or four) apoa-i must by necessity be in equilibrium. The trefoil model requires that the double belt separate into single strands and reassemble. If the trefoil model is correct, some additional metabolic process, such as the action of LCAT or the presence of apoa-ii, must be involved in overcoming the energy barrier required to split the double belt into single strands. TG favors the core center 23
24 Finally, our MD simulation studies yield one consistent finding regardless of the particle studied, TG displaces CE from shdl core centers. This may be due to the slight increase in polarity of CE versus TG; the low dielectric of the particle core would result in a slight increased attraction of CE for the charged particle surface compared to TG, resulting in an equilibrium that favors TG in the particle center. Another factor favoring TG over CE in the core center might be an increased flexibility of TG versus CE. Conclusions Our MD simulation models provide high resolution information about the probable structure and dynamics of circulating HDL, suggesting, in particular, that a large percent of the particle surface area, up to 85%, is covered by apolipoprotein. The high percent of surface area coverage by apolipoproteins can be tested by experimental methods, such as cryo-electron microscopy (51-53) and chemical crosslinking (21). NMR can be used to test the low percent surface area coverage by surface lipid predicted by our models. Measurement of the chemical shift of the choline N-methyl region of PL with high field 1 H NMR can distinguish between annular PL (within 7Å of the protein) and bulk PL (14, 54, 55). The recombinant shdl should have a greater percent of surface lipid in direct contact with the protein than circulating HDL particles (compare structures of recombinant shdl with circulating HDL in Figs. 2 and 4). These models also can serve as blueprints for comprehensively understanding the mechanisms involved in the biological functions of HDL, for example the role of apoa-i as a platform for attachment of the HDL proteome (56, 57), including apoa-ii. 24
25 Acknowledgements We would like to thank UAB Information Technology and Department of Mechanical Engineering for use of the commodity cluster Cheaha that they jointly maintain. This work was supported by National Institutes of Health grants HL34343 and HL
26 References 1. Zlotnick, A Viruses and the physics of soft condensed matter. Proceedings of the National Academy of Sciences of the United States of America 101: Segrest, J. P., R. L. Jackson, J. D. Morrisett, and A. M. Gotto, Jr A molecular theory of lipid-protein interactions in the plasma lipoproteins. FEBS Lett 38: Segrest, J. P., D. W. Garber, C. G. Brouillette, S. C. Harvey, and G. M. Anantharamaiah The amphipathic alpha helix: a multifunctional structural motif in plasma apolipoproteins. Adv Protein Chem 45: Borhani, D. W., D. P. Rogers, J. A. Engler, and C. G. Brouillette Crystal structure of truncated human apolipoprotein A-I suggests a lipid-bound conformation. Proc Natl Acad Sci U S A 94: Koppaka, V., L. Silvestro, J. A. Engler, C. G. Brouillette, and P. H. Axelsen The structure of human lipoprotein A-I. Evidence for the "belt" model. J Biol Chem 274: Segrest, J. P., M. K. Jones, A. E. Klon, C. J. Sheldahl, M. Hellinger, H. De Loof, and S. C. Harvey A detailed molecular belt model for apolipoprotein A-I in discoidal high density lipoprotein. J Biol Chem 274: Tricerri, M. A., A. K. Behling Agree, S. A. Sanchez, and A. Jonas Characterization of apolipoprotein A-I structure using a cysteine-specific fluorescence probe. Biochemistry 39: Davidson, W. S., and G. M. Hilliard The spatial organization of apolipoprotein A-I on the edge of discoidal high density lipoprotein particles: a mass specrometry study. J Biol Chem 278:
27 9. Martin, D. D., M. S. Budamagunta, R. O. Ryan, J. C. Voss, and M. N. Oda Apolipoprotein A-I assumes a "looped belt" conformation on reconstituted high density lipoprotein. Journal of Biological Chemistry 281: Bhat, S., M. G. Sorci-Thomas, R. Tuladhar, M. P. Samuel, and M. J. Thomas Conformational adaptation of apolipoprotein A-I to discretely sized phospholipid complexes. Biochemistry 46: Wu, Z., M. A. Wagner, L. Zheng, J. S. Parks, J. M. Shy, 3rd, J. D. Smith, V. Gogonea, and S. L. Hazen The refined structure of nascent HDL reveals a key functional domain for particle maturation and dysfunction. Nat Struct Mol Biol 14: Jones, M. K., A. Catte, L. Li, and J. P. Segrest Dynamics of activation of lecithin:cholesterol acyltransferase by apolipoprotein A-I. Biochemistry 48: Jones, M. K., A. Catte, J. C. Patterson, F. Gu, J. Chen, L. Li, and J. P. Segrest Thermal stability of apolipoprotein A-I in high-density lipoproteins by molecular dynamics. Biophys J 96: Gu, F., M. K. Jones, J. Chen, J. C. Patterson, A. Catte, W. G. Jerome, L. Li, and J. P. Segrest Structures of discoidal high density lipoproteins: a combined computational-experimental approach. J Biol Chem 285: Jones, M. K., F. Gu, A. Catte, L. Li, and J. P. Segrest "Sticky" and "promiscuous", the yin and yang of apolipoprotein A-I termini in discoidal high-density lipoproteins: a combined computational-experimental approach. Biochemistry 50: Mei, X., and D. Atkinson Crystal structure of C-terminal truncated apolipoprotein A-I reveals the assembly of high density lipoprotein (HDL) by dimerization. J Biol Chem 286:
28 17. Segrest, J. P., M. K. Jones, A. Catte, and S. P. Thirumuruganandham Validation of previous computer models and MD simulations of discoidal HDL by a recent crystal structure of apoa-i. J Lipid Res 53: Catte, A., J. C. Patterson, D. Bashtovyy, M. K. Jones, F. Gu, L. Li, A. Rampioni, D. Sengupta, T. Vuorela, P. Niemela, M. Karttunen, S. J. Marrink, I. Vattulainen, and J. P. Segrest Structure of spheroidal HDL particles revealed by combined atomistic and coarse-grained simulations. Biophys J 94: Sparks, D. L., S. Lund-Katz, and M. C. Phillips The charge and structural stability of apolipoprotein A-I in discoidal and spherical recombinant high density lipoprotein particles. J Biol Chem 267: Silva, R. A., R. Huang, J. Morris, J. Fang, E. O. Gracheva, G. Ren, A. Kontush, W. G. Jerome, K. A. Rye, and W. S. Davidson Structure of apolipoprotein A-I in spherical high density lipoproteins of different sizes. Proc Natl Acad Sci U S A 105: Huang, R., R. A. Silva, W. G. Jerome, A. Kontush, M. J. Chapman, L. K. Curtiss, T. J. Hodges, and W. S. Davidson Apolipoprotein A-I structural organization in high-density lipoproteins isolated from human plasma. Nat Struct Mol Biol 18: Kale, L., R. Skeel, M. Bhandarkar, R. Brunner, A. Gursoy, N. Krawetz, J. Phillips, A. Shinozaki, K. Varadarajan, and K. Schulten NAMD2: greater scalability for parallel molecular dynamics. J. Comp. Phys. 151: Humphrey, W., A. Dalke, and K. Schulten VMD: visual molecular dynamics. J Mol Graph 14: 33-38, Jorgensen, W. L., J. Chandrasekhar, and J. D. Madura Comparison of simple potential functions for simulating liquid water. J. Chem. Phys. 79:
29 25. Brooks, B. R., R. E. Bruccoleri, B. D. Olafson, D. J. States, S. Swaminathan, and M. Karplus CHARMM: a program for macromolecular energy, minimization, and dynamics calculations. Journal of computational chemistry 4: MacKerell, A. D., Jr., D. Bashford, M. Bellot, R. L. Dunbrack Jr., J. Evanseck, M. J. Field, S. Fischer, J. Gao, H. Guo, S. Ha, D. Joseph, L. Kuchnir, K. Kuczera, F. T. K. Lau, C. Mattos, S. Michnick, T. Ngo, D. T. Nguyen, B. Prodhom, W. E. Reiher III, B. Roux, M. Schlenkrich, J. Smith, R. Stote, J. Straub, M. Watanabe, J. Wiorkiewicz-Kuczera, D. Yin, and M. Karplus All-atom empirical potential for molecular modeling and dynamics studies of proteins. J. Phys. Chem. B. 102: Feller, S. E., and R. W. Pastor Length scales of lipid dynamics and molecular dynamics. In Pacific Symposium on Biocomputing. Pacific Symposium on Biocomputing Schlenkrich, M., J. Brickmann, A. MacKerell Jr, and M. Karplus An empirical potential energy function for phospholipids: criteria for parameter optimization and applications. In Biological Membranes: A Molecular Perspective from Computation and Experiment. KM Merz and B. Roux, editors. Birkhauser, Boston Marrink, S. J., H. J. Risselada, S. Yefimov, D. P. Tieleman, and A. H. de Vries The MARTINI force field: coarse grained model for biomolecular simulations. J Phys Chem B 111: Monticelli, L., S. K. Kandasamy, X. Periole, R. G. Larson, D. P. Tieleman, and S. J. Marrink The MARTINI coarse-grained force field: Extension to proteins. Journal of Chemical Theory and Computation 4:
30 31. Hess, B., C. Kutzner, D. van der Spoel, and E. Lindahl GROMACS 4: Algorithms for highly efficient, load-balanced, and scalable molecular simulation. Journal of Chemical Theory and Computation 4: Berendsen, H. J. C., J. P. M. Postma, W. F. van Gunsteren, A. DiNola, and J. R. Haak Molecular dynamics with coupling to an external bath. The Journal of Chemical Physics 81: Marrink, S. J., A. H. de Vries, and A. E. Mark Coarse grained model for semiquantitative lipid simulations. Journal of Physical Chemistry B 108: Silva, R. A., G. M. Hilliard, L. Li, J. P. Segrest, and W. S. Davidson A mass spectrometric determination of the conformation of dimeric apolipoprotein A-I in discoidal high density lipoproteins. Biochemistry 44: Li, L., J. Chen, V. K. Mishra, J. A. Kurtz, D. Cao, A. E. Klon, S. C. Harvey, G. M. Anantharamaiah, and J. P. Segrest Double belt structure of discoidal high density lipoproteins: molecular basis for size heterogeneity. J Mol Biol 343: Frishman, D., and P. Argos Knowledge-based protein secondary structure assignment. Proteins 23: Vuorela, T., A. Catte, P. S. Niemela, A. Hall, M. T. Hyvonen, S. J. Marrink, M. Karttunen, and I. Vattulainen Role of lipids in spheroidal high density lipoproteins. PLoS Comput Biol 6: e Rye, K. A., N. J. Hime, and P. J. Barter The influence of cholesteryl ester transfer protein on the composition, size, and structure of spherical, reconstituted high density lipoproteins. J Biol Chem 270:
31 39. Catte, A., J. C. Patterson, M. K. Jones, W. G. Jerome, D. Bashtovyy, Z. Su, F. Gu, J. Chen, M. P. Aliste, S. C. Harvey, L. Li, G. Weinstein, and J. P. Segrest Novel changes in discoidal high density lipoprotein morphology: a molecular dynamics study. Biophys J 90: Li, L., S. Li, M. K. Jones, and J. P. Segrest Rotational and hinge dynamics of discoidal high density lipoproteins probed by interchain disulfide bond formation. Biochim Biophys Acta 1821: Cheung, M. C., J. P. Segrest, J. J. Albers, J. T. Cone, C. G. Brouillette, B. H. Chung, M. Kashyap, M. A. Glasscock, and G. M. Anantharamaiah Characterization of high density lipoprotein subspecies: structural studies by single vertical spin ultracentrifugation and immunoaffinity chromatography. J Lipid Res 28: Campelo, F., H. T. McMahon, and M. M. Kozlov The hydrophobic insertion mechanism of membrane curvature generation by proteins. Biophys J 95: Hristova, K., W. C. Wimley, V. K. Mishra, G. M. Anantharamiah, J. P. Segrest, and S. H. White An amphipathic alpha-helix at a membrane interface: a structural study using a novel X-ray diffraction method. J Mol Biol 290: Rog, T., M. Pasenkiewicz-Gierula, I. Vattulainen, and M. Karttunen Ordering effects of cholesterol and its analogues. Biochimica Et Biophysica Acta-Biomembranes 1788: Rog, T., M. Pasenkiewicz-Gierula, I. Vattulainen, and M. Karttunen Ordering effects of cholesterol and its analogues. Biochim Biophys Acta 1788: Bashtovyy, D., M. K. Jones, G. M. Anantharamaiah, and J. P. Segrest Sequence conservation of apolipoprotein A-I affords novel insights into HDL structure-function. J Lipid Res 52:
32 47. Shao, B., M. N. Oda, C. Bergt, X. Fu, P. S. Green, N. Brot, J. F. Oram, and J. W. Heinecke Myeloperoxidase impairs ABCA1-dependent cholesterol efflux through methionine oxidation and site-specific tyrosine chlorination of apolipoprotein A-I. J Biol Chem 281: Colvin, P. L., E. Moriguchi, P. H. Barrett, J. S. Parks, and L. L. Rudel Small HDL particles containing two apoa-i molecules are precursors in vivo to medium and large HDL particles containing three and four apoa-i molecules in nonhuman primates. J Lipid Res 40: Gauthamadasa, K., C. Rosales, H. J. Pownall, S. Macha, W. G. Jerome, R. Huang, and R. A. G. D. Silva Speciated Human High-Density Lipoprotein Protein Proximity Profiles. Biochemistry 49: Wu, Z., V. Gogonea, X. Lee, R. P. May, V. Pipich, M. A. Wagner, A. Undurti, T. C. Tallant, C. Baleanu-Gogonea, F. Charlton, A. Ioffe, J. A. DiDonato, K. A. Rye, and S. L. Hazen The low resolution structure of ApoA1 in spherical high density lipoprotein revealed by small angle neutron scattering. J Biol Chem 286: Jones, M. K., L. Zhang, A. Catte, L. Li, M. N. Oda, G. Ren, and J. P. Segrest Assessment of the validity of the double superhelix model for reconstituted high density lipoproteins: a combined computational-experimental approach. J Biol Chem 285: Zhang, L., F. Yan, S. Zhang, D. Lei, M. A. Charles, G. Cavigiolio, M. Oda, R. M. Krauss, K. H. Weisgraber, K. A. Rye, H. J. Pownall, X. Qiu, and G. Ren Structural basis of transfer between lipoproteins by cholesteryl ester transfer protein. Nat Chem Biol 8: Zhang, L., H. Tong, M. Garewal, and G. Ren Optimized negative-staining electron microscopy for lipoprotein studies. Biochim Biophys Acta 1830:
33 54. Brouillette, C. G., J. L. Jones, T. C. Ng, H. Kercret, B. H. Chung, and J. P. Segrest Structural studies of apolipoprotein A-I/phosphatidylcholine recombinants by high-field proton NMR, nondenaturing gradient gel electrophoresis, and electron microscopy. Biochemistry 23: Brouillette, C. G., J. P. Segrest, T. C. Ng, and J. L. Jones Minimal size phosphatidylcholine vesicles: effects of radius of curvature on head group packing and conformation. Biochemistry 21: Shiflett, A. M., J. R. Bishop, A. Pahwa, and S. L. Hajduk Human high density lipoproteins are platforms for the assembly of multi-component innate immune complexes. J Biol Chem 280: Vaisar, T., S. Pennathur, P. S. Green, S. A. Gharib, A. N. Hoofnagle, M. C. Cheung, J. Byun, S. Vuletic, S. Kassim, P. Singh, H. Chea, R. H. Knopp, J. Brunzell, R. Geary, A. Chait, X. Q. Zhao, K. Elkon, S. Marcovina, P. Ridker, J. F. Oram, and J. W. Heinecke Shotgun proteomics implicates protease inhibition and complement activation in the antiinflammatory properties of HDL. J Clin Invest 117:
34 FIGURE LEGENDS Figure 1. Properties of aligned ensembles of S2-57 shdl particles formed after AA and CG simulations. The protein chains were aligned between residues , the domain containing the interhelical less solvent-accessible salt bridges. A-B. Cross-eyed stereo views from helix 5 side (green) of AA and CG ensembles, respectively. The protein is in licorice ribbon representation: prolines, yellow spacefilling; helix 5, green; helix 10, red; residues 1-43, blue; remainder of protein, silver; CE, magenta lines; POPC, cyan lines; UC, brown spacefilling; UC-OH, white spacefilling. C-D. Cross-eyed stereo views of cross-sections of aligned AA and CG ensembles, respectively. Graphic representation is the same as A- B except UC is in line format. E-F. Plots of radial mole fractions of components of MDSA and CGMD simulations, respectively, averaged over the last 20% of their trajectories. G-H. Plots of radial distributions of components of MDSA and CGMD, respectively, averaged over the last 20% of their trajectories. CE, magenta; UC, gold; POPC acyl chains, black; UC-OH, dashed gold; glycerol moiety of POPC, red; apoa-i, blue; POPC headgroups, cyan; solvent, dashed green. The black triangles on the x-axis represent the mean radius of gyration; the yellow triangles on the x-axis represent the radius calculated from molecular volumes of the individual components. Figure 2. Properties of S2-86 and S2-34 shdl particles formed after 10 µs CGMD simulations. A- B. Cross-eyed stereo images from helix 5 side of the S2-86 particle intact and in cross-section, respectively. The protein is in spacefilling mode with colors used in Fig. 1A; POPC, cyan CPK; CE, magenta CPK; UC, brown CPK; UC-OH, white CPK; TG, lime CPK. C. Plot of radial mole fractions of components of S2-86 particle, averaged over the last 20% of its trajectory. CE, magenta; UC, gold; POPC acyl chains, black; UC-OH, dashed gold; glycerol moiety of POPC, red; apoa-i, blue; POPC headgroups, cyan; solvent, dashed green. D-E. Cross-eyed stereo images from helix 5 side of S2-34 particle intact and in cross-section, respectively. The prolines are in spacefilling mode with colors used in Fig. 1A; POPC, cyan CPK; CE, magenta CPK; UC, brown CPK; UC-OH, white CPK; TG, lime CPK. F. Plot of 34
35 radial mole fractions of components of S2-34 particle, averaged over the last 20% of its trajectory. Color code as in C. Figure 3. Properties of S3-155 shdl particles created by CGMD simulations. A. Schematic of the starting double belt-hairpin disc. B. Cross-eyed stereo image of the discoidal particle after an initial removal of 60 central POPC followed by 5 ns MD simulation at 310 K. In this and C the POPC and UC surface monolayer are in cyan and brown, respectively, and are transparent to allow visualization of the entire apoa-i double belt-hairpin, the red arrowheads denote the helix 5 antiparallel pair, and the white arrowheads denote the helix 5 hairpin. C. Cross-eyed stereo image of a representative example of the ensemble of six S3-155 particles. CE, magenta spacefilling; UC, brown spacefilling; TG, lime spacefilling. D. Cross-eyed stereo image of a cross-section of the aligned ensemble of six particles the double belt region of apoa-i has been aligned in its central domain, residues POPC, cyan stick; UC, white stick; CE, magenta stick; TG, lime stick. E-F. Plots of radial mole fractions and radial distributions of components of the ensemble of CGMD simulations, respectively, averaged over the last 20% of their trajectories. TG, lime; CE, magenta; UC, gold; POPC acyl chains, black; UC-OH, dashed gold; glycerol moiety of POPC, red; apoa-i, blue; POPC headgroups, cyan; solvent, dashed green. Figure 4. Properties of S3-94 and S3-41 shdl particles formed after 10 µs CGMD simulations. A- B. Cross-eyed stereo images from helix 5 side of the S3-94 particle intact and in cross-section, respectively. The protein is in spacefilling mode with colors used in Fig. 1A; POPC, CE, UC, UC-OH, and TG are in CPK mode and in colors as in Fig. 2. C. Plot of radial mole fractions of components of S3-94 particle, averaged over the last 20% of its trajectory. CE, magenta; UC, gold; POPC acyl chains, black; UC- OH, dashed gold; glycerol moiety of POPC, red; apoa-i, blue; POPC headgroups, cyan; solvent, dashed green. D-E. Cross-eyed stereo images from helix 5 side of S3-41 particle intact and in cross-section, respectively. The protein is in spacefilling mode with colors used in Fig. 1A; POPC, CE, UC, UC-OH, 35
36 and TG are in CPK mode and in colors as in Fig. 2. F. Plot of radial mole fractions of components of S3-41 particle, averaged over the last 20% of its trajectory. Color code as in C. Figure 5. Linear regression plots of monolayer thickness measured by volumetric analysis versus surface to volume ratios for reconstituted shdl particles. A. Plot of monolayer thickness measured by volumetric analysis based upon molecular volumes versus surface to volume ratios assuming spherical shapes for: i) four reconstituted shdl particles, S2-57, S2-86, S3-155 and S3-94 ( ); ii) six CGMD simulated models of circulating shdl (gray circles); and iii) ( ) upper (d = Å) and lower (r = 13.5 Å) limits (labeled MAX and MIN, respectively) of monolayer thickness plotted along the linear regression line (solid diagonal) taken from Fig. 6 in the preceding paper (Segrest, J. P., M. C. Cheung, and M. K. Jones, 2013). The x and y intercepts of the linear regression line are indicated. The surface to volume ratio (S/V = (4πr 2 )/(4/3πr 3 ) = 3/radius) varies inversely with particle diameter and is a measure of the ratio of surface moieties (protein and polar lipid) to core lipid. The thick dotted arrow indicates the pathway of conversion of S3-94 to S3-41a. The thin dotted arrow indicates the pathway of conversion of S2-86 to S2-34. B. Plot of PL thickness measured by simulations versus total monolayer thickness measured by volumetric analysis for eight CGMD particles: S2-30b, S2-34, S2-57, S2-86, S3-41a, S3-41b, S3-94, and S The linear regression line y=0.8645x is shown. Insert: Crosssectional schematic diagram to scale showing changes of cores (black) and surface monolayers (gray) during conversion of S3-94 to S3-41 and S2-86 to S2-34 presumably driven by PLTP. C. Plot of percent coverage of HDL particle surface area versus total monolayer thickness measured by volumetric analysis for four reconstituted CGMD particles S2-57, S2-86, S3-94, and S3-155 (open circles) and seven native particles HDL[1]-HDL[7] (black circles). Two linear regression lines are shown for each group. 36
37 Measure of percent surface area occupied by protein for HDL[2], HDL[3], and HDL[5]-HDL[7] from Huang, et al (21) (small open diamonds). Figure 6. Cross-eyed stereo licorice images of the ensembles of the AA and CG simulations of S2-57 and the CG simulations of S A-B. Average structures of the AA and CG ensembles of S2-57. Aligned residues , green; residues 1-43, blue; helix 10, red; prolines, yellow spacefilling; remainder, silver. C. Average structure of the CG ensemble of six S3-155 simulations. Aligned residues , cyan; helix 5, green; residue 1-43, blue; helix 10, red; prolines, yellow spacefilling; remainder, silver. Red arrowhead, antiparallel helix 5 pair; white arrowhead, helix 5 hairpin. D-F. Aligned structures of the protein backbones of AA and CG ensembles for the S2-57 and the S3-155 ensemble, respectively. Aligned residues in the double belt, green; prolines, yellow spacefilling; remainder of the double belt and all of the hairpins, silver. Figure 7. Relationship of secondary and tertiary conformational features of the MD simulations. A. Fraction helicity for each residue averaged over the last 20% of the ensemble of eight AA S2-57 simulations. B. Schematic illustration of folding of discoidal double belt around hinges (open circles) to cover surface of shdl. The pairwise helix 5 domain is in green. C. Schematic illustration of folding of discoidal double belt and hairpin to cover surface of shdl. The pairwise helix 5 domain and the helix 5 hairpin are in green. The light blue arrow illustrates the dynamics of motion of the hairpin that swings in a right handed spiral from the helix 5 pair to the N-terminal domains with an increase in simulation time (see Fig. S3). Figure 8. Cross-eyed stereo licorice images of protein conformations of the S2 and S3 particles after CGMD simulations. The three S2 particles (left hand column) contain 2 apoa-i in a double belt and the three S3 particles (right hand column) contain 3 apoa-i in a double belt plus hair-pin. From top to bottom, the S2 and S3 particles show the effects of core and surface lipid reductions on protein confor- 37
38 mation. Color code: Residues 1-43, blue: helix 5, green; helix 10, red; prolines, yellow spacefilling. The unhydrated diameters calculated from volumes of lipid and protein components assuming a perfectly spherical shape is designated d 0. 38
39 Table I: Best fit volumetric analysis of simulated shdl particles COMPUTATIONAL MODELS Stoichiometry Diameter Monolayer thickness Object Origin Lipid (PC:CE:UC:TG) Apolipoprotein (A-I:A-II) NDGGE (Å) Calculated diameter (Å) Volumetric analysis MD simulation 2-57 Model 57:16:6:0 2: Model 155:58:45:14 3: S80 (2-86) Davidson 86:12:6:10 2:0 80 Å S93 (3-94) Silva 94:41:6:0 2:0 93 Å a HDL[1] 34:12:6:10 2: a b HDL[4] 41:34:9:7 3: ± a Modification of composition of HDL[1] to place it on the regression line in Fig. 5A. b Two S3-41 particles with differing lipid compositions were simulated. S3-41a is equivalent to HDL[4], S3-41b had a slightly greater lipid composition. 39



40 Fig. 1 A C B D 40
41 Fig. 1 E Mole fraction CE acyl water apoa-i PO4 UC glycerol Distance from center of mass (Å) F Mole fraction CE acyl UC apoa-i glycerol water PO4 Distance from center of mass (Å) UC-OH G UC-OH H Distribution Distribution Distance from center of mass (Å) Distance from center of mass (Å) 41


42 Fig. 2 A D B E 42
43 Fig. 2 C Mole fraction TG CE acyl apoa-i water F Mole fraction TG CE acyl apoa-i water UC glycerol PO4 UC glycerol PO4 Distance from center of mass (Å) Distance from center of mass (Å) 43





44 A B C Fig. 3 D 44
45 Fig. 3 E Mole fraction TG CE acyl apoa-i water F Distribution TG CE acyl UC glycerol UC-OH PO4 apoa-i UC glycerol PO4 Distance from center of mass (Å) Distance from center of mass (Å) 45




46 Fig. 4 A B D E 46
47 Fig. 4 C Mole fraction CE acyl apoa-i water Mole fraction F TG CE acyl apoa-i water UC glycerol PO4 UC glycerol PO4 Distance from center of mass (Å) Distance from center of mass (Å) 47
48 Fig. 5A MIN MAX 48
49 Total monolayer thickness (volumetric, Å) b 3-41a 2-30b y = x R² = PL m = 14.5 Å PLTP PL m = 11.0 Å S3-94 S3-41 PL m = 16.5 Å PLTP PL m = 11.9 Å S2-86 S2-34 Fig. 5B PL monolayer thickness (CGMD, Å)
50 Fig. 5C Total monolayer thickness (volumetric, Å) S3-155 S3-94 S2-86 S2-57 HDL[7] HDL[6] HDL[5] HDL[3] HDL[4] HDL[2] HDL[1] Percent coverage of HDL particle surface by protein 50





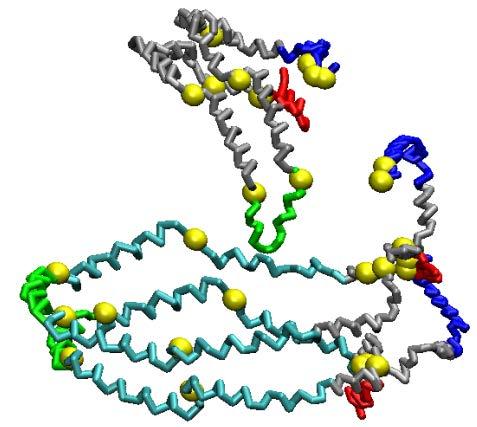

51 Fig. 6 A B D E C F 51




52 Fig. 7 A B C 52








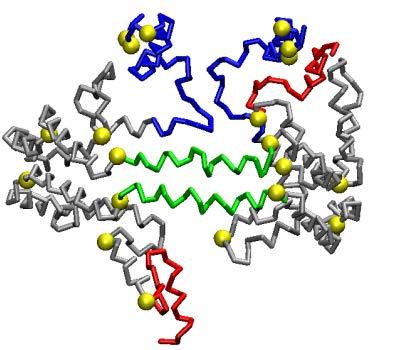
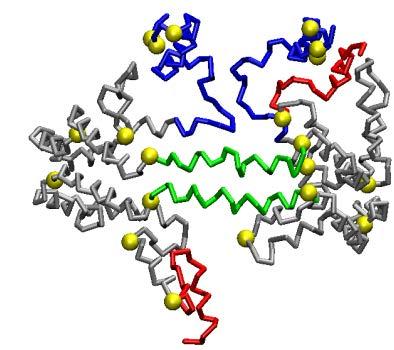
53 Fig. 8 S2-86 d 0 = 74.2 Å S3-155 d 0 = 93.1 Å S2-57 d 0 = 68.0 Å S3-94 d 0 = 80.9 Å S2-34 d 0 = 65.3 Å S3-41 d 0 = 74.0 Å 53
Novel Changes in Discoidal High Density Lipoprotein Morphology: A Molecular Dynamics Study
 Biophysical Journal Volume 90 June 2006 4345 4360 4345 Novel Changes in Discoidal High Density Lipoprotein Morphology: A Molecular Dynamics Study Andrea Catte,* James C. Patterson,* Martin K. Jones,* W.
Biophysical Journal Volume 90 June 2006 4345 4360 4345 Novel Changes in Discoidal High Density Lipoprotein Morphology: A Molecular Dynamics Study Andrea Catte,* James C. Patterson,* Martin K. Jones,* W.
Supplementary Materials for
 advances.sciencemag.org/cgi/content/full/4/3/eaaq0762/dc1 Supplementary Materials for Structures of monomeric and oligomeric forms of the Toxoplasma gondii perforin-like protein 1 Tao Ni, Sophie I. Williams,
advances.sciencemag.org/cgi/content/full/4/3/eaaq0762/dc1 Supplementary Materials for Structures of monomeric and oligomeric forms of the Toxoplasma gondii perforin-like protein 1 Tao Ni, Sophie I. Williams,
Transient β-hairpin Formation in α-synuclein Monomer Revealed by Coarse-grained Molecular Dynamics Simulation
 Transient β-hairpin Formation in α-synuclein Monomer Revealed by Coarse-grained Molecular Dynamics Simulation Hang Yu, 1, 2, a) Wei Han, 1, 3, b) Wen Ma, 1, 2 1, 2, 3, c) and Klaus Schulten 1) Beckman
Transient β-hairpin Formation in α-synuclein Monomer Revealed by Coarse-grained Molecular Dynamics Simulation Hang Yu, 1, 2, a) Wei Han, 1, 3, b) Wen Ma, 1, 2 1, 2, 3, c) and Klaus Schulten 1) Beckman
Molecular Belt Models for the Apolipoprotein A-I Paris and Milano Mutations
 Biophysical Journal Volume 79 September 2000 1679 1685 1679 Molecular Belt Models for the Apolipoprotein A-I Paris and Milano Mutations Anthony E. Klon,* Martin K. Jones, Jere P. Segrest,* and Stephen
Biophysical Journal Volume 79 September 2000 1679 1685 1679 Molecular Belt Models for the Apolipoprotein A-I Paris and Milano Mutations Anthony E. Klon,* Martin K. Jones, Jere P. Segrest,* and Stephen
Supplementary Figures
 Supplementary Figures Supplementary Figure 1. (a) Uncropped version of Fig. 2a. RM indicates that the translation was done in the absence of rough mcirosomes. (b) LepB construct containing the GGPG-L6RL6-
Supplementary Figures Supplementary Figure 1. (a) Uncropped version of Fig. 2a. RM indicates that the translation was done in the absence of rough mcirosomes. (b) LepB construct containing the GGPG-L6RL6-
SDS-Assisted Protein Transport Through Solid-State Nanopores
 Supplementary Information for: SDS-Assisted Protein Transport Through Solid-State Nanopores Laura Restrepo-Pérez 1, Shalini John 2, Aleksei Aksimentiev 2 *, Chirlmin Joo 1 *, Cees Dekker 1 * 1 Department
Supplementary Information for: SDS-Assisted Protein Transport Through Solid-State Nanopores Laura Restrepo-Pérez 1, Shalini John 2, Aleksei Aksimentiev 2 *, Chirlmin Joo 1 *, Cees Dekker 1 * 1 Department
Supplementary Information
 Coarse-Grained Molecular Dynamics Simulations of Photoswitchable Assembly and Disassembly Xiaoyan Zheng, Dong Wang,* Zhigang Shuai,* MOE Key Laboratory of Organic Optoelectronics and Molecular Engineering,
Coarse-Grained Molecular Dynamics Simulations of Photoswitchable Assembly and Disassembly Xiaoyan Zheng, Dong Wang,* Zhigang Shuai,* MOE Key Laboratory of Organic Optoelectronics and Molecular Engineering,
Double Belt Structure of Discoidal High Density Lipoproteins: Molecular Basis for Size Heterogeneity
 doi:10.1016/j.jmb.2004.09.017 J. Mol. Biol. (2004) 343, 1293 1311 Double Belt Structure of Discoidal High Density Lipoproteins: Molecular Basis for Size Heterogeneity Ling Li 1,2 *, Jianguo Chen 1,2, Vinod
doi:10.1016/j.jmb.2004.09.017 J. Mol. Biol. (2004) 343, 1293 1311 Double Belt Structure of Discoidal High Density Lipoproteins: Molecular Basis for Size Heterogeneity Ling Li 1,2 *, Jianguo Chen 1,2, Vinod
HDL surface lipids mediate CETP binding as revealed by electron microscopy and molecular dynamics simulation
 HDL surface lipids mediate CETP binding as revealed by electron microscopy and molecular dynamics simulation Meng Zhang 1, River Charles 1, Huimin Tong 1, Lei Zhang 1, Mili Patel 2, Francis Wang 1, Matthew
HDL surface lipids mediate CETP binding as revealed by electron microscopy and molecular dynamics simulation Meng Zhang 1, River Charles 1, Huimin Tong 1, Lei Zhang 1, Mili Patel 2, Francis Wang 1, Matthew
Interactions of Polyethylenimines with Zwitterionic and. Anionic Lipid Membranes
 Interactions of Polyethylenimines with Zwitterionic and Anionic Lipid Membranes Urszula Kwolek, Dorota Jamróz, Małgorzata Janiczek, Maria Nowakowska, Paweł Wydro, Mariusz Kepczynski Faculty of Chemistry,
Interactions of Polyethylenimines with Zwitterionic and Anionic Lipid Membranes Urszula Kwolek, Dorota Jamróz, Małgorzata Janiczek, Maria Nowakowska, Paweł Wydro, Mariusz Kepczynski Faculty of Chemistry,
Protein Structure and Function
 Protein Structure and Function Protein Structure Classification of Proteins Based on Components Simple proteins - Proteins containing only polypeptides Conjugated proteins - Proteins containing nonpolypeptide
Protein Structure and Function Protein Structure Classification of Proteins Based on Components Simple proteins - Proteins containing only polypeptides Conjugated proteins - Proteins containing nonpolypeptide
Lecture 15. Membrane Proteins I
 Lecture 15 Membrane Proteins I Introduction What are membrane proteins and where do they exist? Proteins consist of three main classes which are classified as globular, fibrous and membrane proteins. A
Lecture 15 Membrane Proteins I Introduction What are membrane proteins and where do they exist? Proteins consist of three main classes which are classified as globular, fibrous and membrane proteins. A
Biological Membranes. Lipid Membranes. Bilayer Permeability. Common Features of Biological Membranes. A highly selective permeability barrier
 Biological Membranes Structure Function Composition Physicochemical properties Self-assembly Molecular models Lipid Membranes Receptors, detecting the signals from outside: Light Odorant Taste Chemicals
Biological Membranes Structure Function Composition Physicochemical properties Self-assembly Molecular models Lipid Membranes Receptors, detecting the signals from outside: Light Odorant Taste Chemicals
review Sequence conservation of apolipoprotein A-I affords novel insights into HDL structure-function
 review Sequence conservation of apolipoprotein A-I affords novel insights into HDL structure-function Denys Bashtovyy, *, Martin K. Jones, *, G. M. Anantharamaiah, * and Jere P. Segrest 1, *, Department
review Sequence conservation of apolipoprotein A-I affords novel insights into HDL structure-function Denys Bashtovyy, *, Martin K. Jones, *, G. M. Anantharamaiah, * and Jere P. Segrest 1, *, Department
Supplementary Figure 1 (previous page). EM analysis of full-length GCGR. (a) Exemplary tilt pair images of the GCGR mab23 complex acquired for Random
. EM analysis of full-length GCGR. (a) Exemplary tilt pair images of the GCGR mab23 complex acquired for Random") S1 Supplementary Figure 1 (previous page). EM analysis of full-length GCGR. (a) Exemplary tilt pair images of the GCGR mab23 complex acquired for Random Conical Tilt (RCT) reconstruction (left: -50,right:
S1 Supplementary Figure 1 (previous page). EM analysis of full-length GCGR. (a) Exemplary tilt pair images of the GCGR mab23 complex acquired for Random Conical Tilt (RCT) reconstruction (left: -50,right:
Lipoproteins Metabolism
 Lipoproteins Metabolism LEARNING OBJECTIVES By the end of this Lecture, the student should be able to describe: What are Lipoproteins? Describe Lipoprotein Particles. Composition of Lipoproteins. The chemical
Lipoproteins Metabolism LEARNING OBJECTIVES By the end of this Lecture, the student should be able to describe: What are Lipoproteins? Describe Lipoprotein Particles. Composition of Lipoproteins. The chemical
Coarse grained simulations of Lipid Bilayer Membranes
 Coarse grained simulations of Lipid Bilayer Membranes P. B. Sunil Kumar Department of Physics IIT Madras, Chennai 600036 sunil@iitm.ac.in Atomistic MD: time scales ~ 10 ns length scales ~100 nm 2 To study
Coarse grained simulations of Lipid Bilayer Membranes P. B. Sunil Kumar Department of Physics IIT Madras, Chennai 600036 sunil@iitm.ac.in Atomistic MD: time scales ~ 10 ns length scales ~100 nm 2 To study
This exam consists of two parts. Part I is multiple choice. Each of these 25 questions is worth 2 points.
 MBB 407/511 Molecular Biology and Biochemistry First Examination - October 1, 2002 Name Social Security Number This exam consists of two parts. Part I is multiple choice. Each of these 25 questions is
MBB 407/511 Molecular Biology and Biochemistry First Examination - October 1, 2002 Name Social Security Number This exam consists of two parts. Part I is multiple choice. Each of these 25 questions is
(B D) Three views of the final refined 2Fo-Fc electron density map of the Vpr (red)-ung2 (green) interacting region, contoured at 1.4σ.
 Three views of the final refined 2Fo-Fc electron density map of the Vpr (red)-ung2 (green) interacting region, contoured at 1.4σ.") Supplementary Figure 1 Overall structure of the DDB1 DCAF1 Vpr UNG2 complex. (A) The final refined 2Fo-Fc electron density map, contoured at 1.4σ of Vpr, illustrating well-defined side chains. (B D) Three
Supplementary Figure 1 Overall structure of the DDB1 DCAF1 Vpr UNG2 complex. (A) The final refined 2Fo-Fc electron density map, contoured at 1.4σ of Vpr, illustrating well-defined side chains. (B D) Three
Supplementary Materials. High affinity binding of phosphatidylinositol-4-phosphate. by Legionella pneumophila DrrA
 Supplementary Materials High affinity binding of phosphatidylinositol-4-phosphate by Legionella pneumophila DrrA Running title: Molecular basis of PtdIns(4)P-binding by DrrA Stefan Schoebel, Wulf Blankenfeldt,
Supplementary Materials High affinity binding of phosphatidylinositol-4-phosphate by Legionella pneumophila DrrA Running title: Molecular basis of PtdIns(4)P-binding by DrrA Stefan Schoebel, Wulf Blankenfeldt,
Lectures 11 12: Fibrous & Membrane Proteins. Lecturer: Prof. Brigita Urbanc
 Lectures 11 12: Fibrous & Membrane Proteins Lecturer: Prof. Brigita Urbanc (brigita@drexel.edu) 1 FIBROUS PROTEINS: function: structural microfilaments & microtubules fibrils, hair, silk reinforce membranes
Lectures 11 12: Fibrous & Membrane Proteins Lecturer: Prof. Brigita Urbanc (brigita@drexel.edu) 1 FIBROUS PROTEINS: function: structural microfilaments & microtubules fibrils, hair, silk reinforce membranes
Chapter 12: Membranes. Voet & Voet: Pages
 Chapter 12: Membranes Voet & Voet: Pages 390-415 Slide 1 Membranes Essential components of all living cells (define boundry of cells) exclude toxic ions and compounds; accumulation of nutrients energy
Chapter 12: Membranes Voet & Voet: Pages 390-415 Slide 1 Membranes Essential components of all living cells (define boundry of cells) exclude toxic ions and compounds; accumulation of nutrients energy
Protein structure. Dr. Mamoun Ahram Summer semester,
 Protein structure Dr. Mamoun Ahram Summer semester, 2017-2018 Overview of proteins Proteins have different structures and some have repeating inner structures, other do not. A protein may have gazillion
Protein structure Dr. Mamoun Ahram Summer semester, 2017-2018 Overview of proteins Proteins have different structures and some have repeating inner structures, other do not. A protein may have gazillion
High density lipoprotein metabolism
 High density lipoprotein metabolism Lipoprotein classes and atherosclerosis Chylomicrons, VLDL, and their catabolic remnants Pro-atherogenic LDL HDL Anti-atherogenic Plasma lipid transport Liver VLDL FC
High density lipoprotein metabolism Lipoprotein classes and atherosclerosis Chylomicrons, VLDL, and their catabolic remnants Pro-atherogenic LDL HDL Anti-atherogenic Plasma lipid transport Liver VLDL FC
Supplementary Figure 1 Preparation, crystallization and structure determination of EpEX. (a), Purified EpEX and EpEX analyzed on homogenous 12.
, Purified EpEX and EpEX analyzed on homogenous 12.") Supplementary Figure 1 Preparation, crystallization and structure determination of EpEX. (a), Purified EpEX and EpEX analyzed on homogenous 12.5 % SDS-PAGE gel under reducing and non-reducing conditions.
Supplementary Figure 1 Preparation, crystallization and structure determination of EpEX. (a), Purified EpEX and EpEX analyzed on homogenous 12.5 % SDS-PAGE gel under reducing and non-reducing conditions.
The role of Ca² + ions in the complex assembling of protein Z and Z-dependent protease inhibitor: A structure and dynamics investigation
 www.bioinformation.net Hypothesis Volume 8(9) The role of Ca² + ions in the complex assembling of protein Z and Z-dependent protease inhibitor: A structure and dynamics investigation Zahra Karimi 1 *,
www.bioinformation.net Hypothesis Volume 8(9) The role of Ca² + ions in the complex assembling of protein Z and Z-dependent protease inhibitor: A structure and dynamics investigation Zahra Karimi 1 *,
Supporting Information
 Electronic Supplementary Material (ESI) for Nanoscale. This journal is The Royal Society of Chemistry 018 Supporting Information Modulating Interactions between Ligand-Coated Nanoparticles and Phase-Separated
Electronic Supplementary Material (ESI) for Nanoscale. This journal is The Royal Society of Chemistry 018 Supporting Information Modulating Interactions between Ligand-Coated Nanoparticles and Phase-Separated
Apolipoprotein A-I structure in high-density lipoproteins
 Annals of Medicine. 2008; 40: 5 13 REVIEW ARTICLE Apolipoprotein A-I structure in high-density lipoproteins R. A. GANGANI D. SILVA, MATTHEW R. TUBB & W. SEAN DAVIDSON Department of Pathology and Laboratory
Annals of Medicine. 2008; 40: 5 13 REVIEW ARTICLE Apolipoprotein A-I structure in high-density lipoproteins R. A. GANGANI D. SILVA, MATTHEW R. TUBB & W. SEAN DAVIDSON Department of Pathology and Laboratory
We parameterized a coarse-grained fullerene consistent with the MARTINI coarse-grained force field
 Parameterization of the fullerene coarse-grained model We parameterized a coarse-grained fullerene consistent with the MARTINI coarse-grained force field for lipids 1 and proteins 2. In the MARTINI force
Parameterization of the fullerene coarse-grained model We parameterized a coarse-grained fullerene consistent with the MARTINI coarse-grained force field for lipids 1 and proteins 2. In the MARTINI force
Proteins and their structure
 Proteins and their structure Proteins are the most abundant biological macromolecules, occurring in all cells and all parts of cells. Proteins also occur in great variety; thousands of different kinds,
Proteins and their structure Proteins are the most abundant biological macromolecules, occurring in all cells and all parts of cells. Proteins also occur in great variety; thousands of different kinds,
Globular proteins Proteins globular fibrous
 Globular proteins Globular proteins Proteins are biochemical compounds consisting of one or more polypeptides typically folded into a globular or fibrous form in a biologically functional way. Globular
Globular proteins Globular proteins Proteins are biochemical compounds consisting of one or more polypeptides typically folded into a globular or fibrous form in a biologically functional way. Globular
Bioinformatics for molecular biology
 Bioinformatics for molecular biology Structural bioinformatics tools, predictors, and 3D modeling Structural Biology Review Dr Research Scientist Department of Microbiology, Oslo University Hospital -
Bioinformatics for molecular biology Structural bioinformatics tools, predictors, and 3D modeling Structural Biology Review Dr Research Scientist Department of Microbiology, Oslo University Hospital -
Interaction of Functionalized C 60 Nanoparticles with Lipid Membranes
 Interaction of Functionalized C 60 Nanoparticles with Lipid Membranes Kevin Gasperich Advisor: Dmitry Kopelevich ABSTRACT The recent rapid development of applications for fullerenes has led to an increase
Interaction of Functionalized C 60 Nanoparticles with Lipid Membranes Kevin Gasperich Advisor: Dmitry Kopelevich ABSTRACT The recent rapid development of applications for fullerenes has led to an increase
Nature Structural & Molecular Biology: doi: /nsmb Supplementary Figure 1
 Supplementary Figure 1 Design of isolated protein and RNC constructs, and homogeneity of purified RNCs. (a) Schematic depicting the design and nomenclature used for all the isolated proteins and RNCs used
Supplementary Figure 1 Design of isolated protein and RNC constructs, and homogeneity of purified RNCs. (a) Schematic depicting the design and nomenclature used for all the isolated proteins and RNCs used
Biology 5A Fall 2010 Macromolecules Chapter 5
 Learning Outcomes: Macromolecules List and describe the four major classes of molecules Describe the formation of a glycosidic linkage and distinguish between monosaccharides, disaccharides, and polysaccharides
Learning Outcomes: Macromolecules List and describe the four major classes of molecules Describe the formation of a glycosidic linkage and distinguish between monosaccharides, disaccharides, and polysaccharides
Levels of Protein Structure:
 Levels of Protein Structure: PRIMARY STRUCTURE (1 ) - Defined, non-random sequence of amino acids along the peptide backbone o Described in two ways: Amino acid composition Amino acid sequence M-L-D-G-C-G
Levels of Protein Structure: PRIMARY STRUCTURE (1 ) - Defined, non-random sequence of amino acids along the peptide backbone o Described in two ways: Amino acid composition Amino acid sequence M-L-D-G-C-G
Lecture Series 2 Macromolecules: Their Structure and Function
 Lecture Series 2 Macromolecules: Their Structure and Function Reading Assignments Read Chapter 4 (Protein structure & Function) Biological Substances found in Living Tissues The big four in terms of macromolecules
Lecture Series 2 Macromolecules: Their Structure and Function Reading Assignments Read Chapter 4 (Protein structure & Function) Biological Substances found in Living Tissues The big four in terms of macromolecules
H 2 O. Liquid, solid, and vapor coexist in the same environment
 Water H 2 O Liquid, solid, and vapor coexist in the same environment WATER MOLECULES FORM HYDROGEN BONDS Water is a fundamental requirement for life, so it is important to understand the structural and
Water H 2 O Liquid, solid, and vapor coexist in the same environment WATER MOLECULES FORM HYDROGEN BONDS Water is a fundamental requirement for life, so it is important to understand the structural and
Lipids and Membranes
 Lipids and Membranes Presented by Dr. Mohammad Saadeh The requirements for the Pharmaceutical Biochemistry I Philadelphia University Faculty of pharmacy Biological membranes are composed of lipid bilayers
Lipids and Membranes Presented by Dr. Mohammad Saadeh The requirements for the Pharmaceutical Biochemistry I Philadelphia University Faculty of pharmacy Biological membranes are composed of lipid bilayers
Molecular Graphics Perspective of Protein Structure and Function
 Molecular Graphics Perspective of Protein Structure and Function VMD Highlights > 20,000 registered Users Platforms: Unix (16 builds) Windows MacOS X Display of large biomolecules and simulation trajectories
Molecular Graphics Perspective of Protein Structure and Function VMD Highlights > 20,000 registered Users Platforms: Unix (16 builds) Windows MacOS X Display of large biomolecules and simulation trajectories
obtained for the simulations of the E2 conformation of SERCA in a pure POPC lipid bilayer (blue) and in a
 and in a") Supplementary Figure S1. Distribution of atoms along the bilayer normal. Normalized density profiles obtained for the simulations of the E2 conformation of SERCA in a pure POPC lipid bilayer (blue) and
Supplementary Figure S1. Distribution of atoms along the bilayer normal. Normalized density profiles obtained for the simulations of the E2 conformation of SERCA in a pure POPC lipid bilayer (blue) and
CHAPTER 4. Tryptophan fluorescence quenching by brominated lipids
 CHAPTER 4 Tryptophan fluorescence quenching by brominated lipids 102 4.1 INTRODUCTION The structure and dynamics of biological macromolecules have been widely studied with fluorescence quenching. The accessibility
CHAPTER 4 Tryptophan fluorescence quenching by brominated lipids 102 4.1 INTRODUCTION The structure and dynamics of biological macromolecules have been widely studied with fluorescence quenching. The accessibility
AFM In Liquid: A High Sensitivity Study On Biological Membranes
 University of Wollongong Research Online Faculty of Science - Papers (Archive) Faculty of Science, Medicine and Health 2006 AFM In Liquid: A High Sensitivity Study On Biological Membranes Michael J. Higgins
University of Wollongong Research Online Faculty of Science - Papers (Archive) Faculty of Science, Medicine and Health 2006 AFM In Liquid: A High Sensitivity Study On Biological Membranes Michael J. Higgins
Protein Secondary Structure
 Protein Secondary Structure Reading: Berg, Tymoczko & Stryer, 6th ed., Chapter 2, pp. 37-45 Problems in textbook: chapter 2, pp. 63-64, #1,5,9 Directory of Jmol structures of proteins: http://www.biochem.arizona.edu/classes/bioc462/462a/jmol/routines/routines.html
Protein Secondary Structure Reading: Berg, Tymoczko & Stryer, 6th ed., Chapter 2, pp. 37-45 Problems in textbook: chapter 2, pp. 63-64, #1,5,9 Directory of Jmol structures of proteins: http://www.biochem.arizona.edu/classes/bioc462/462a/jmol/routines/routines.html
Lecture Series 2 Macromolecules: Their Structure and Function
 Lecture Series 2 Macromolecules: Their Structure and Function Reading Assignments Read Chapter 4 (Protein structure & Function) Biological Substances found in Living Tissues The big four in terms of macromolecules
Lecture Series 2 Macromolecules: Their Structure and Function Reading Assignments Read Chapter 4 (Protein structure & Function) Biological Substances found in Living Tissues The big four in terms of macromolecules
CALCIUM CASEINATE. What Is Casein?
 CALCIUM CASEINATE A high quality milk protein that is Calcium Rich, manufactured from fresh pasteurized skimmed milk through precipitation of casein followed by neutralization and natural drying, which
CALCIUM CASEINATE A high quality milk protein that is Calcium Rich, manufactured from fresh pasteurized skimmed milk through precipitation of casein followed by neutralization and natural drying, which
Proteins. (b) Protein Structure and Conformational Change
 Protein Structure and Conformational Change") Proteins (b) Protein Structure and Conformational Change Protein Structure and Conformational Change Proteins contain the elements carbon (C), hydrogen (H), oxygen (O2) and nitrogen (N2) Some may also
Proteins (b) Protein Structure and Conformational Change Protein Structure and Conformational Change Proteins contain the elements carbon (C), hydrogen (H), oxygen (O2) and nitrogen (N2) Some may also
Chapter 3. Protein Structure and Function
 Chapter 3 Protein Structure and Function Broad functional classes So Proteins have structure and function... Fine! -Why do we care to know more???? Understanding functional architechture gives us POWER
Chapter 3 Protein Structure and Function Broad functional classes So Proteins have structure and function... Fine! -Why do we care to know more???? Understanding functional architechture gives us POWER
Reading for lecture 6
 Reading for lecture 6 1. Lipids and Lipid Bilayers 2. Membrane Proteins Voet and Voet, Chapter 11 Alberts et al Chapter 6 Jones, R.A.L, Soft Condensed Matter 195pp Oxford University Press, ISBN 0-19-850590-6
Reading for lecture 6 1. Lipids and Lipid Bilayers 2. Membrane Proteins Voet and Voet, Chapter 11 Alberts et al Chapter 6 Jones, R.A.L, Soft Condensed Matter 195pp Oxford University Press, ISBN 0-19-850590-6
Chapter VIII: Dr. Sameh Sarray Hlaoui
 Chapter VIII: Dr. Sameh Sarray Hlaoui Lipoproteins a Lipids are insoluble in plasma. In order to be transported they are combined with specific proteins to form lipoproteins: Clusters of proteins and lipids.
Chapter VIII: Dr. Sameh Sarray Hlaoui Lipoproteins a Lipids are insoluble in plasma. In order to be transported they are combined with specific proteins to form lipoproteins: Clusters of proteins and lipids.
Polarization and Circular Dichroism (Notes 17)
") Polarization and Circular Dichroism - 2014 (Notes 17) Since is vector, if fix molec. orient., E-field interact (absorb) with molecule differently when change E-orientation (polarization) Transitions can
Polarization and Circular Dichroism - 2014 (Notes 17) Since is vector, if fix molec. orient., E-field interact (absorb) with molecule differently when change E-orientation (polarization) Transitions can
SUPPLEMENTARY INFORMATION
 High-speed atomic force microscopy shows that annexin V stabilizes membranes on the second timescale Atsushi Miyagi, Chris Chipot, Martina Rangl & Simon Scheuring Supplementary Movies: Supplementary Movie
High-speed atomic force microscopy shows that annexin V stabilizes membranes on the second timescale Atsushi Miyagi, Chris Chipot, Martina Rangl & Simon Scheuring Supplementary Movies: Supplementary Movie
Chapter 1 Membrane Structure and Function
 Chapter 1 Membrane Structure and Function Architecture of Membranes Subcellular fractionation techniques can partially separate and purify several important biological membranes, including the plasma and
Chapter 1 Membrane Structure and Function Architecture of Membranes Subcellular fractionation techniques can partially separate and purify several important biological membranes, including the plasma and
Biological Molecules B Lipids, Proteins and Enzymes. Triglycerides. Glycerol
 Glycerol www.biologymicro.wordpress.com Biological Molecules B Lipids, Proteins and Enzymes Lipids - Lipids are fats/oils and are present in all cells- they have different properties for different functions
Glycerol www.biologymicro.wordpress.com Biological Molecules B Lipids, Proteins and Enzymes Lipids - Lipids are fats/oils and are present in all cells- they have different properties for different functions
Chapter 5 Structure and Function Of Large Biomolecules
 Formation of Macromolecules Monomers Polymers Macromolecules Smaller larger Chapter 5 Structure and Function Of Large Biomolecules monomer: single unit dimer: two monomers polymer: three or more monomers
Formation of Macromolecules Monomers Polymers Macromolecules Smaller larger Chapter 5 Structure and Function Of Large Biomolecules monomer: single unit dimer: two monomers polymer: three or more monomers
Proteins. Amino acids, structure and function. The Nobel Prize in Chemistry 2012 Robert J. Lefkowitz Brian K. Kobilka
 Proteins Amino acids, structure and function The Nobel Prize in Chemistry 2012 Robert J. Lefkowitz Brian K. Kobilka O O HO N N HN OH Ser65-Tyr66-Gly67 The Nobel prize in chemistry 2008 Osamu Shimomura,
Proteins Amino acids, structure and function The Nobel Prize in Chemistry 2012 Robert J. Lefkowitz Brian K. Kobilka O O HO N N HN OH Ser65-Tyr66-Gly67 The Nobel prize in chemistry 2008 Osamu Shimomura,
Lecture Series 2 Macromolecules: Their Structure and Function
 Lecture Series 2 Macromolecules: Their Structure and Function Reading Assignments Read Chapter 4 (Protein structure & Function) Biological Substances found in Living Tissues The big four in terms of macromolecules
Lecture Series 2 Macromolecules: Their Structure and Function Reading Assignments Read Chapter 4 (Protein structure & Function) Biological Substances found in Living Tissues The big four in terms of macromolecules
SUPPLEMENTARY INFORMATION. Computational Assay of H7N9 Influenza Neuraminidase Reveals R292K Mutation Reduces Drug Binding Affinity
 SUPPLEMENTARY INFORMATION Computational Assay of H7N9 Influenza Neuraminidase Reveals R292K Mutation Reduces Drug Binding Affinity Christopher Woods 1, Maturos Malaisree 1, Ben Long 2, Simon McIntosh-Smith
SUPPLEMENTARY INFORMATION Computational Assay of H7N9 Influenza Neuraminidase Reveals R292K Mutation Reduces Drug Binding Affinity Christopher Woods 1, Maturos Malaisree 1, Ben Long 2, Simon McIntosh-Smith
BIO 311C Spring Lecture 15 Friday 26 Feb. 1
 BIO 311C Spring 2010 Lecture 15 Friday 26 Feb. 1 Illustration of a Polypeptide amino acids peptide bonds Review Polypeptide (chain) See textbook, Fig 5.21, p. 82 for a more clear illustration Folding and
BIO 311C Spring 2010 Lecture 15 Friday 26 Feb. 1 Illustration of a Polypeptide amino acids peptide bonds Review Polypeptide (chain) See textbook, Fig 5.21, p. 82 for a more clear illustration Folding and
A. Lipids: Water-Insoluble Molecules
 Biological Substances found in Living Tissues Lecture Series 3 Macromolecules: Their Structure and Function A. Lipids: Water-Insoluble Lipids can form large biological molecules, but these aggregations
Biological Substances found in Living Tissues Lecture Series 3 Macromolecules: Their Structure and Function A. Lipids: Water-Insoluble Lipids can form large biological molecules, but these aggregations
G protein coupled receptor interactions with cholesterol deep in the membrane
 G protein coupled receptor interactions with cholesterol deep in the membrane Samuel Genheden a,b, Jonathan W. Essex a,c, & Anthony G. Lee c,d * a School of Chemistry, University of Southampton, Southampton,
G protein coupled receptor interactions with cholesterol deep in the membrane Samuel Genheden a,b, Jonathan W. Essex a,c, & Anthony G. Lee c,d * a School of Chemistry, University of Southampton, Southampton,
Supplementary Information A Hydrophobic Barrier Deep Within the Inner Pore of the TWIK-1 K2P Potassium Channel Aryal et al.
 Supplementary Information A Hydrophobic Barrier Deep Within the Inner Pore of the TWIK-1 K2P Potassium Channel Aryal et al. Supplementary Figure 1 TWIK-1 stability during MD simulations in a phospholipid
Supplementary Information A Hydrophobic Barrier Deep Within the Inner Pore of the TWIK-1 K2P Potassium Channel Aryal et al. Supplementary Figure 1 TWIK-1 stability during MD simulations in a phospholipid
Simulationen von Lipidmembranen
 Simulationen von Lipidmembranen Thomas Stockner Thomas.stockner@meduniwien.ac.at Summary Methods Force Field MD simulations Membrane simulations Application Oxidized lipids Anesthetics Molecular biology
Simulationen von Lipidmembranen Thomas Stockner Thomas.stockner@meduniwien.ac.at Summary Methods Force Field MD simulations Membrane simulations Application Oxidized lipids Anesthetics Molecular biology
Molecular Models of Nanodiscs
 This is an open access article published under an ACS AuthorChoice License, which permits copying and redistribution of the article or any adaptations for non-commercial purposes. pubs.acs.org/jctc Molecular
This is an open access article published under an ACS AuthorChoice License, which permits copying and redistribution of the article or any adaptations for non-commercial purposes. pubs.acs.org/jctc Molecular
Amino Acids and Proteins Hamad Ali Yaseen, PhD MLS Department, FAHS, HSC, KU Biochemistry 210 Chapter 22
 Amino Acids and Proteins Hamad Ali Yaseen, PhD MLS Department, FAHS, HSC, KU Hamad.ali@hsc.edu.kw Biochemistry 210 Chapter 22 Importance of Proteins Main catalysts in biochemistry: enzymes (involved in
Amino Acids and Proteins Hamad Ali Yaseen, PhD MLS Department, FAHS, HSC, KU Hamad.ali@hsc.edu.kw Biochemistry 210 Chapter 22 Importance of Proteins Main catalysts in biochemistry: enzymes (involved in
Sheet #5 Dr. Mamoun Ahram 8/7/2014
 P a g e 1 Protein Structure Quick revision - Levels of protein structure: primary, secondary, tertiary & quaternary. - Primary structure is the sequence of amino acids residues. It determines the other
P a g e 1 Protein Structure Quick revision - Levels of protein structure: primary, secondary, tertiary & quaternary. - Primary structure is the sequence of amino acids residues. It determines the other
Fluid Mozaic Model of Membranes
 Replacement for the 1935 Davson Danielli model Provided explanation for Gortner-Grendel lack of lipid and permitted the unit membrane model. Trans membrane protein by labelling Fry & Edidin showed that
Replacement for the 1935 Davson Danielli model Provided explanation for Gortner-Grendel lack of lipid and permitted the unit membrane model. Trans membrane protein by labelling Fry & Edidin showed that
0.5 nm nm acyl tail region (hydrophobic) 1.5 nm. Hydrophobic repulsion organizes amphiphilic molecules: These scales are 5 10xk B T:
 1.5 nm. Hydrophobic repulsion organizes amphiphilic molecules: These scales are 5 10xk B T:") Lecture 31: Biomembranes: The hydrophobic energy scale and membrane behavior 31.1 Reading for Lectures 30-32: PKT Chapter 11 (skip Ch. 10) Upshot of last lecture: Generic membrane lipid: Can be cylindrical
Lecture 31: Biomembranes: The hydrophobic energy scale and membrane behavior 31.1 Reading for Lectures 30-32: PKT Chapter 11 (skip Ch. 10) Upshot of last lecture: Generic membrane lipid: Can be cylindrical
Nafith Abu Tarboush DDS, MSc, PhD
 Nafith Abu Tarboush DDS, MSc, PhD natarboush@ju.edu.jo www.facebook.com/natarboush Protein conformation Many conformations are possible for proteins due to flexibility of amino acids linked by peptide
Nafith Abu Tarboush DDS, MSc, PhD natarboush@ju.edu.jo www.facebook.com/natarboush Protein conformation Many conformations are possible for proteins due to flexibility of amino acids linked by peptide
Quiz 8 Introduction to Polymers (Chemistry)
") 051117 Quiz 8 Introduction to Polymers (Chemistry) (Figures from Heimenz Colloid Sci.) 1) Surfactants are amphiphilic molecules (molecules having one end hydrophobic and the other hydrophilic) and are
051117 Quiz 8 Introduction to Polymers (Chemistry) (Figures from Heimenz Colloid Sci.) 1) Surfactants are amphiphilic molecules (molecules having one end hydrophobic and the other hydrophilic) and are
Nature Structural & Molecular Biology: doi: /nsmb Supplementary Figure 1
 Supplementary Figure 1 The UBL and RING1 interface remains associated in the complex structures of Parkin and pub. a) Asymmetric Unit of crystal structure of UBLR0RBR and pub complex showing UBL (green),
Supplementary Figure 1 The UBL and RING1 interface remains associated in the complex structures of Parkin and pub. a) Asymmetric Unit of crystal structure of UBLR0RBR and pub complex showing UBL (green),
theoretically molecular interactions to identify the major statistical factors contributing to the
 ANTONIJEVIC, TODOR, Ph.D. Theoretical Modeling of Physical Processes in Low Density Lipoprotein Nanostructures (2015) Directed by Dr. Joseph M. Starobin. 79 pp. The research work aims to analyze LDL structure
ANTONIJEVIC, TODOR, Ph.D. Theoretical Modeling of Physical Processes in Low Density Lipoprotein Nanostructures (2015) Directed by Dr. Joseph M. Starobin. 79 pp. The research work aims to analyze LDL structure
Interaction Between Amyloid-b (1 42) Peptide and Phospholipid Bilayers: A Molecular Dynamics Study
 Peptide and Phospholipid Bilayers: A Molecular Dynamics Study") Biophysical Journal Volume 96 February 2009 785 797 785 Interaction Between Amyloid-b (1 42) Peptide and Phospholipid Bilayers: A Molecular Dynamics Study Charles H. Davis and Max L. Berkowitz * Department
Biophysical Journal Volume 96 February 2009 785 797 785 Interaction Between Amyloid-b (1 42) Peptide and Phospholipid Bilayers: A Molecular Dynamics Study Charles H. Davis and Max L. Berkowitz * Department
Supplementary Information: A Critical. Comparison of Biomembrane Force Fields: Structure and Dynamics of Model DMPC, POPC, and POPE Bilayers
 Supplementary Information: A Critical Comparison of Biomembrane Force Fields: Structure and Dynamics of Model DMPC, POPC, and POPE Bilayers Kristyna Pluhackova,, Sonja A. Kirsch, Jing Han, Liping Sun,
Supplementary Information: A Critical Comparison of Biomembrane Force Fields: Structure and Dynamics of Model DMPC, POPC, and POPE Bilayers Kristyna Pluhackova,, Sonja A. Kirsch, Jing Han, Liping Sun,
Lipids digestion and absorption, Biochemistry II
 Lipids digestion and absorption, blood plasma lipids, lipoproteins Biochemistry II Lecture 1 2008 (J.S.) Triacylglycerols (as well as free fatty acids and both free and esterified cholesterol) are very
Lipids digestion and absorption, blood plasma lipids, lipoproteins Biochemistry II Lecture 1 2008 (J.S.) Triacylglycerols (as well as free fatty acids and both free and esterified cholesterol) are very
Interactions between Bisphosphate. Geminis and Sodium Lauryl Ether
 Chapter 5 Interactions between Bisphosphate Geminis and Sodium Lauryl Ether Sulphate 110 5.1 Introduction The physiochemical and surface active properties of mixed surfactants are of more interest and
Chapter 5 Interactions between Bisphosphate Geminis and Sodium Lauryl Ether Sulphate 110 5.1 Introduction The physiochemical and surface active properties of mixed surfactants are of more interest and
Membranes & Membrane Proteins
 School on Biomolecular Simulations Membranes & Membrane Proteins Vani Vemparala The Institute of Mathematical Sciences Chennai November 13 2007 JNCASR, Bangalore Cellular Environment Plasma membrane extracellular
School on Biomolecular Simulations Membranes & Membrane Proteins Vani Vemparala The Institute of Mathematical Sciences Chennai November 13 2007 JNCASR, Bangalore Cellular Environment Plasma membrane extracellular
University of Groningen
 University of Groningen Role of Lipids in Spheroidal High Density Lipoproteins Vuorela, Timo; Catte, Andrea; Niemela, Perttu S.; Hall, Anette; Hyvonen, Marja T.; Marrink, Siewert; Karttunen, Mikko; Vattulainen,
University of Groningen Role of Lipids in Spheroidal High Density Lipoproteins Vuorela, Timo; Catte, Andrea; Niemela, Perttu S.; Hall, Anette; Hyvonen, Marja T.; Marrink, Siewert; Karttunen, Mikko; Vattulainen,
Molecular Dynamics Simulations of the Anchoring and Tilting of the Lung-Surfactant Peptide SP-B 1-25 in Palmitic Acid Monolayers
 Biophysical Journal Volume 89 December 2005 3807 3821 3807 Molecular Dynamics Simulations of the Anchoring and Tilting of the Lung-Surfactant Peptide SP-B 1-25 in Palmitic Acid Monolayers Hwankyu Lee,*
Biophysical Journal Volume 89 December 2005 3807 3821 3807 Molecular Dynamics Simulations of the Anchoring and Tilting of the Lung-Surfactant Peptide SP-B 1-25 in Palmitic Acid Monolayers Hwankyu Lee,*
Chapter 7: Membranes
 Chapter 7: Membranes Roles of Biological Membranes The Lipid Bilayer and the Fluid Mosaic Model Transport and Transfer Across Cell Membranes Specialized contacts (junctions) between cells What are the
Chapter 7: Membranes Roles of Biological Membranes The Lipid Bilayer and the Fluid Mosaic Model Transport and Transfer Across Cell Membranes Specialized contacts (junctions) between cells What are the
2. Which of the following amino acids is most likely to be found on the outer surface of a properly folded protein?
 Name: WHITE Student Number: Answer the following questions on the computer scoring sheet. 1 mark each 1. Which of the following amino acids would have the highest relative mobility R f in normal thin layer
Name: WHITE Student Number: Answer the following questions on the computer scoring sheet. 1 mark each 1. Which of the following amino acids would have the highest relative mobility R f in normal thin layer
review Structure of apolipoprotein B-100 in low density lipoproteins Jere P. Segrest, 1, *,, Martin K. Jones,*, Hans De Loof, and Nassrin Dashti, **
 review Structure of apolipoprotein B-100 in low density lipoproteins Jere P. Segrest, 1, *,, Martin K. Jones,*, Hans De Loof, and Nassrin Dashti, ** Departments of Medicine,* Biochemistry and Molecular
review Structure of apolipoprotein B-100 in low density lipoproteins Jere P. Segrest, 1, *,, Martin K. Jones,*, Hans De Loof, and Nassrin Dashti, ** Departments of Medicine,* Biochemistry and Molecular
Lane: 1. Spectra BR protein ladder 2. PFD 3. TERM 4. 3-way connector 5. 2-way connector
 kda 1 2 3 4 5 26 14 1 7 Lane: 1. Spectra BR protein ladder 2. PFD 3. TERM 4. 3-way connector 5. 2-way connector 5 4 35 25 15 1 Supplementary Figure 1. SDS-PAGE of acterially expressed and purified proteins.
kda 1 2 3 4 5 26 14 1 7 Lane: 1. Spectra BR protein ladder 2. PFD 3. TERM 4. 3-way connector 5. 2-way connector 5 4 35 25 15 1 Supplementary Figure 1. SDS-PAGE of acterially expressed and purified proteins.
Skeletal Muscle : Structure
 1 Skeletal Muscle : Structure Dr.Viral I. Champaneri, MD Assistant Professor Department of Physiology 2 Learning objectives 1. Gross anatomy of the skeletal muscle 2. Myofilaments & their molecular structure
1 Skeletal Muscle : Structure Dr.Viral I. Champaneri, MD Assistant Professor Department of Physiology 2 Learning objectives 1. Gross anatomy of the skeletal muscle 2. Myofilaments & their molecular structure
Supplementary Figure S1 Black silicon and dragonfly wing nanotopography.
 Supplementary Figure S1 Black silicon and dragonfly wing nanotopography. Representative low-magnification scanning electron micrographs of a) Black silicon (bsi) and b) Diplacodes bipunctata dragonfly
Supplementary Figure S1 Black silicon and dragonfly wing nanotopography. Representative low-magnification scanning electron micrographs of a) Black silicon (bsi) and b) Diplacodes bipunctata dragonfly
a) The statement is true for X = 400, but false for X = 300; b) The statement is true for X = 300, but false for X = 200;
 The statement is true for X = 400, but false for X = 300; b) The statement is true for X = 300, but false for X = 200;") 1. Consider the following statement. To produce one molecule of each possible kind of polypeptide chain, X amino acids in length, would require more atoms than exist in the universe. Given the size of
1. Consider the following statement. To produce one molecule of each possible kind of polypeptide chain, X amino acids in length, would require more atoms than exist in the universe. Given the size of
Apolipoprotein A-I: structure function relationships
 Apolipoprotein A-I: structure function relationships review Philippe G. Frank 1 and Yves L. Marcel 2 Lipoprotein & Atherosclerosis Group, University of Ottawa Heart Institute, 40 Ruskin Street, Ottawa,
Apolipoprotein A-I: structure function relationships review Philippe G. Frank 1 and Yves L. Marcel 2 Lipoprotein & Atherosclerosis Group, University of Ottawa Heart Institute, 40 Ruskin Street, Ottawa,
Chemical Surface Transformation 1
 Chemical Surface Transformation 1 Chemical reactions at Si H surfaces (inorganic and organic) can generate very thin films (sub nm thickness up to µm): inorganic layer formation by: thermal conversion:
Chemical Surface Transformation 1 Chemical reactions at Si H surfaces (inorganic and organic) can generate very thin films (sub nm thickness up to µm): inorganic layer formation by: thermal conversion:
Adaptable Lipid Matrix Promotes Protein Protein Association in Membranes
 Supporting information Adaptable Lipid Matrix Promotes Protein Protein Association in Membranes Andrey S. Kuznetsov, Anton A. Polyansky,, Markus Fleck, Pavel E. Volynsky, and Roman G. Efremov *,, M. M.
Supporting information Adaptable Lipid Matrix Promotes Protein Protein Association in Membranes Andrey S. Kuznetsov, Anton A. Polyansky,, Markus Fleck, Pavel E. Volynsky, and Roman G. Efremov *,, M. M.
MBB 694:407, 115:511. Please use BLOCK CAPITAL letters like this --- A, B, C, D, E. Not lowercase!
 MBB 694:407, 115:511 First Test Severinov/Deis Tue. Sep. 30, 2003 Name Index number (not SSN) Row Letter Seat Number This exam consists of two parts. Part I is multiple choice. Each of these 25 questions
MBB 694:407, 115:511 First Test Severinov/Deis Tue. Sep. 30, 2003 Name Index number (not SSN) Row Letter Seat Number This exam consists of two parts. Part I is multiple choice. Each of these 25 questions
Introduction to proteins and protein structure
 Introduction to proteins and protein structure The questions and answers below constitute an introduction to the fundamental principles of protein structure. They are all available at [link]. What are
Introduction to proteins and protein structure The questions and answers below constitute an introduction to the fundamental principles of protein structure. They are all available at [link]. What are
AP BIOLOGY: READING ASSIGNMENT FOR CHAPTER 5
 1) Complete the following table: Class Monomer Functions Carbohydrates 1. 3. Lipids 1. 3. Proteins 1. 3. 4. 5. 6. Nucleic Acids 1. 2) Circle the atoms of these two glucose molecules that will be removed
1) Complete the following table: Class Monomer Functions Carbohydrates 1. 3. Lipids 1. 3. Proteins 1. 3. 4. 5. 6. Nucleic Acids 1. 2) Circle the atoms of these two glucose molecules that will be removed
Q1: Circle the best correct answer: (15 marks)
") Q1: Circle the best correct answer: (15 marks) 1. Which one of the following incorrectly pairs an amino acid with a valid chemical characteristic a. Glycine, is chiral b. Tyrosine and tryptophan; at neutral
Q1: Circle the best correct answer: (15 marks) 1. Which one of the following incorrectly pairs an amino acid with a valid chemical characteristic a. Glycine, is chiral b. Tyrosine and tryptophan; at neutral
Supporting material. Membrane permeation induced by aggregates of human islet amyloid polypeptides
 Supporting material Membrane permeation induced by aggregates of human islet amyloid polypeptides Chetan Poojari Forschungszentrum Jülich GmbH, Institute of Complex Systems: Structural Biochemistry (ICS-6),
Supporting material Membrane permeation induced by aggregates of human islet amyloid polypeptides Chetan Poojari Forschungszentrum Jülich GmbH, Institute of Complex Systems: Structural Biochemistry (ICS-6),
Lecture 10 More about proteins
 Lecture 10 More about proteins Today we're going to extend our discussion of protein structure. This may seem far-removed from gene cloning, but it is the path to understanding the genes that we are cloning.
Lecture 10 More about proteins Today we're going to extend our discussion of protein structure. This may seem far-removed from gene cloning, but it is the path to understanding the genes that we are cloning.
BIOPHYSICS II. By Prof. Xiang Yang Liu Department of Physics,
 BIOPHYSICS II By Prof. Xiang Yang Liu Department of Physics, NUS 1 Hydrogen bond and the stability of macromolecular structure Membrane Model Amphiphilic molecule self-assembly at the surface and din the
BIOPHYSICS II By Prof. Xiang Yang Liu Department of Physics, NUS 1 Hydrogen bond and the stability of macromolecular structure Membrane Model Amphiphilic molecule self-assembly at the surface and din the
Lecture 4: 8/26. CHAPTER 4 Protein Three Dimensional Structure
 Lecture 4: 8/26 CHAPTER 4 Protein Three Dimensional Structure Summary of the Lecture 3 There are 20 amino acids and only the L isomer amino acid exist in proteins Each amino acid consists of a central
Lecture 4: 8/26 CHAPTER 4 Protein Three Dimensional Structure Summary of the Lecture 3 There are 20 amino acids and only the L isomer amino acid exist in proteins Each amino acid consists of a central
Supporting Information
 Composition and lipid spatial distribution of High Density Lipoprotein particles in subjects with Laxman Yetukuri, 1 Sanni Söderlund, 2 Artturi Koivuniemi, 3 Tuulikki Seppänen-Laakso, 1 Perttu S. Niemelä,
Composition and lipid spatial distribution of High Density Lipoprotein particles in subjects with Laxman Yetukuri, 1 Sanni Söderlund, 2 Artturi Koivuniemi, 3 Tuulikki Seppänen-Laakso, 1 Perttu S. Niemelä,
PROCEEDINGS OF THE YEREVAN STATE UNIVERSITY
 PROCEEDINGS OF THE YEREVAN STATE UNIVERSITY Physical and Mathematical Sciences 2018, 52(3), p. 217 221 P h y s i c s STUDY OF THE SWELLING OF THE PHOSPHOLIPID BILAYER, DEPENDING ON THE ANGLE BETWEEN THE
PROCEEDINGS OF THE YEREVAN STATE UNIVERSITY Physical and Mathematical Sciences 2018, 52(3), p. 217 221 P h y s i c s STUDY OF THE SWELLING OF THE PHOSPHOLIPID BILAYER, DEPENDING ON THE ANGLE BETWEEN THE
Nature Methods: doi: /nmeth Supplementary Figure 1. Salipro lipid particles.
 Supplementary Figure 1 Salipro lipid particles. (a) Gel filtration analysis of Saposin A after incubation with the indicated detergent solubilised lipid solutions. The generation of Saposin A-lipid complexes
Supplementary Figure 1 Salipro lipid particles. (a) Gel filtration analysis of Saposin A after incubation with the indicated detergent solubilised lipid solutions. The generation of Saposin A-lipid complexes