An integrated map of genetic variation from 1092 human genomes
|
|
|
- Verity Warner
- 5 years ago
- Views:
Transcription
1 SUPPLEMENTAL METHODS AND MATERIALS Whole genome sequencing Alignment: Short insert paired-end reads were aligned to the GRCh37 reference human genome with 1000 genomes decoy contigs using BWA-mem(1). Somatic mutation calling: An integrated map of genetic variation from 1092 human genomes Substitution: Single base substitutions were called using CaVEMan (Cancer Variants through Expectation Maximisation) ( As described previously(2), the algorithm compares sequence data from each tumor sample to its own matched non-cancerous sample and calculates a mutation probability at each genomic locus. Copy number and cellularity information for CaVEMan were predicted with the Battenberg algorithm (2) using 1000 Genomes (3) loci within the NGS data. To improve specificity, a number of post-processing filters were applied as follows: 1. At least a third of the alleles containing the mutant must have base quality >= If mutant allele coverage >= 10X, there must be a mutant allele of at least base quality 20 in the middle 3rd of a read. If mutant allele coverage is < 10X, a mutant allele of at least base quality 20 in the first 2/3 of a read is acceptable. 3. The mutation position is marked by <3 reads in any sample in the unmatched normal panel. 4. The mutant allele proportion must be >5 times than that in the matched normal sample (or it is zero in the matched normal).
2 5. If the mean base quality is <20 then less than 96% of mutations carrying reads are in one direction. 6. Mutations within simple repeats, centromeric repeats, regions of excessive depth ( and low mapping quality were excluded. Additional unmatched normal filtering was performed using a set of unmatched normal samples. Mutations that were detected in >5% of the unmatched normal normal panel at >=5% mutant allele burden were excluded. Variant annotation was done in Ensembl v74 using VAGrENT(4). Small insertions and deletions: Small somatic insertions and deletions (indels) were identified using a modified version of Pindel ( To improve specificity, a number of post-processing filters were applied that required the following: 1) For regions with sequencing depth <200X, mutant variant must be present in at least 8% of total reads. 2) For regions with sequencing depth >=200X, mutant variant must be present in at least 4% of total reads. 3) The region with the variant should have <= 9 small (<4 nucleotides) repeats. 4) The variant is not seen in any reads in the matched normal sample or the unmatched normal panel. 5) The number of Pindel calls in the tumour sample is greater than 4 and either: a. The number of mutant reads mapped by BWA in the tumour sample is greater than 0 or
3 b. The number of mutant reads mapped by BWA in the tumour sample is equal to 0 but there are no repeats in the variant region and there are reads mapped by Pindel in the tumour sample on both the positive and negative strand. 6) Pindel SUM-MS score (sum of the mapping scores of the reads used as anchors) >=150 Additional unmatched normal filtering was performed using a set of unmatched normal samples (n=221). Mutations that were detected in >1% of the unmatched normal panel at >=1% mutant allele burden were excluded. Variant annotation was done in Ensembl v74 using VAGrENT(4). Structural rearrangements: Structural rearrangements were detected by an in house algorithm, BRASS (Breakpoints via assembly) [ which first groups discordant read pairs that span the same breakpoint and then using Velvet de novo assembler (6) performs local assembly within the vicinity to reconstruct and determine the exact position of the breakpoint to nucleotide precision. Copy number changes : Segmental copy number information was derived for each sample using the Battenberg algorithm as previously described(2). Briefly, the algorithm phases heterozygous SNPs with use of the 1000 genomes genotypes as a reference panel. The resulting haplotypes are corrected for occasional errors in phasing in regions with low linkage disequilibrium. After segmentation of the resulting b-allele frequency (BAF) values, t-tests are performed on the BAFs of each copy number segment to identify whether they correspond to the value resulting from a fully clonal copy number change. If not, the copy number segment is represented as a mixture of 2 different copy number states, with the fraction of cells bearing each copy number state estimated from the average BAF of the heterozygous SNPs
4 in that segment. The Battenberg algorithm could not be applied to chromosome X since BAFs are uninformative for male subjects. For this chromosome, logr values were segmented and segmented logr values were converted to copy number estimates as described previously(7). Without application of the Battenberg algorithm, the resolution of subclonal copy number states is not possible, so copy number segments are called as single integer values (corresponding to the copy number state of the dominant cancer clone) on chromosome X. Mutational signature analysis: Mutational signature analysis of the substitutions was performed using the R package DeconstructSigs(8). Small insertion/deletions were interrogated for the presence of either short tandem repeat or microhomology at the breakpoints as described previously(2). Complex indels were excluded from this analysis. Clonality analysis: For each mutation we calculated the cancer cell fraction as previously described(9), using the mutant allele burden, tumor purity and locus specific copy number in the tumor and matched normal. Subclones were identified by clustering the cancer cell fractions with Dirichlet Process-based clustering as described previously(10). Custom target sequencing Massively parallel sequencing of postcapture libraries was performed on an Illumina HiSeq2500 system as base pair (bp) paired-end reads for 74 primary tumor and 11 metastasis samples as well as for matching normal DNA. All samples were analysed using MSK-IMPACT, as previously described(11). This clinical sequencing platform is a hybridization capture-based next-
5 generation sequencing assay for targeted deep sequencing of all exons and selected introns of 341 or 410 oncogenes, TSGs, and members of pathways deemed actionable by targeted therapies (Supplemental Table 2). Sequence reads were aligned to the human reference genome GRCh37 using the Burrows-Wheeler Aligner software (BWA, v0.7.10)(1). The Genome Analysis Toolkit (GATK, v3.1.1) (12)was used for local realignment, duplicate removal and base quality recalibration. Somatic single nucleotide variants (SNVs) were identified using MuTect (v1.0) (13), and small insertions and deletions (indels) were detected using Strelka (v2.0.15) (14)and VarScan 2 (v2.3.7)(15). Sequence data were demultiplexed using BCL2FASTQ Conversion Software version (Illumina, San Diego, CA), aligned in paired-end mode to the hg19 b37 version of the human genome using BWA-MEM software (Burrows-Wheeler Aligner), and used MuTect and SomaticIndelDetector to identify point mutations/single nucleotide variants (SNVs) and small insertions/deletion (indels). In cases where variant calling was performed for a tumor without a normal match, variants with minor allele frequency >1% in the 1,000 genomes cohort were removed. SNVs and indels outside of the target regions, those with mutant allelic fraction (MAF) of <1% and/or those supported by 5 reads (16) were filtered out. We further excluded SNVs and indels for which the tumor MAF was <5 times that of the matched normal MAF, as well as SNVs and indels found at >5% global minor allele frequency of dbsnp. The mean coverage across tested samples was 371X. Probe sequences and concentrations were optimized to ensure maximally uniform and reproducible coverage across targets. As a result, more than 99% of exons were covered to greater than 20% of the median exon coverage for each sample (71X). To reduce false-positive findings, we included only variants with depth of unique sequencing coverage above 50X and
6 variant allele frequencies above 25%. Variants not annotated as common population polymorphisms (<1% population frequency in the 1000 genomes and NHLBI ESP cohorts but present at frequencies greater than 5% in our cohort were flagged as possible systematic assay artifacts. All putative mutations were reviewed manually using the Integrative Genomics Viewer (IGV)(17).
7 SUPPLEMENTAL FIGURES Supplemental Figure 1. Overview of the examined patient s sample cohort of chrcc. Pie charts depict the AJCC stages at the time of diagnosis for 38 metastatic chrcc (M-chRCC) patients and 41 non-metastatic ID-chRCC patients with no documented metastases at final data collection.
 and JHCHR7 (primary tumors).")

8 Supplemental Figure 2. Whole Genome Sequencing Analysis of Metastatic chrcc Cases in the exploratory cohort. Somatic alterations identified from WGS data for patients JHCHR5 (lung metastasis) and JHCHR7 (primary tumors). The uppermost track depicts inter- and intrachromosomal rearrangements as arcs whilst the second and third tracks show the genomic positions of the small insertion/deletions and substitutions, respectively. Inter-mutation distance for substitutions is plotted as a line in the third track. The bottom three tracks depict the total copy number (red, increase; blue, decrease;), logr and B allele frequency (BAF) in the tumor when compared to the normal. Rearrangements, indels and substitutions are coloured according to alteration type which is summarised in the bar plots in the side panel. See Supplemental method for details of the classification of insertion/deletions to micro homology or repeat-mediated type.
9
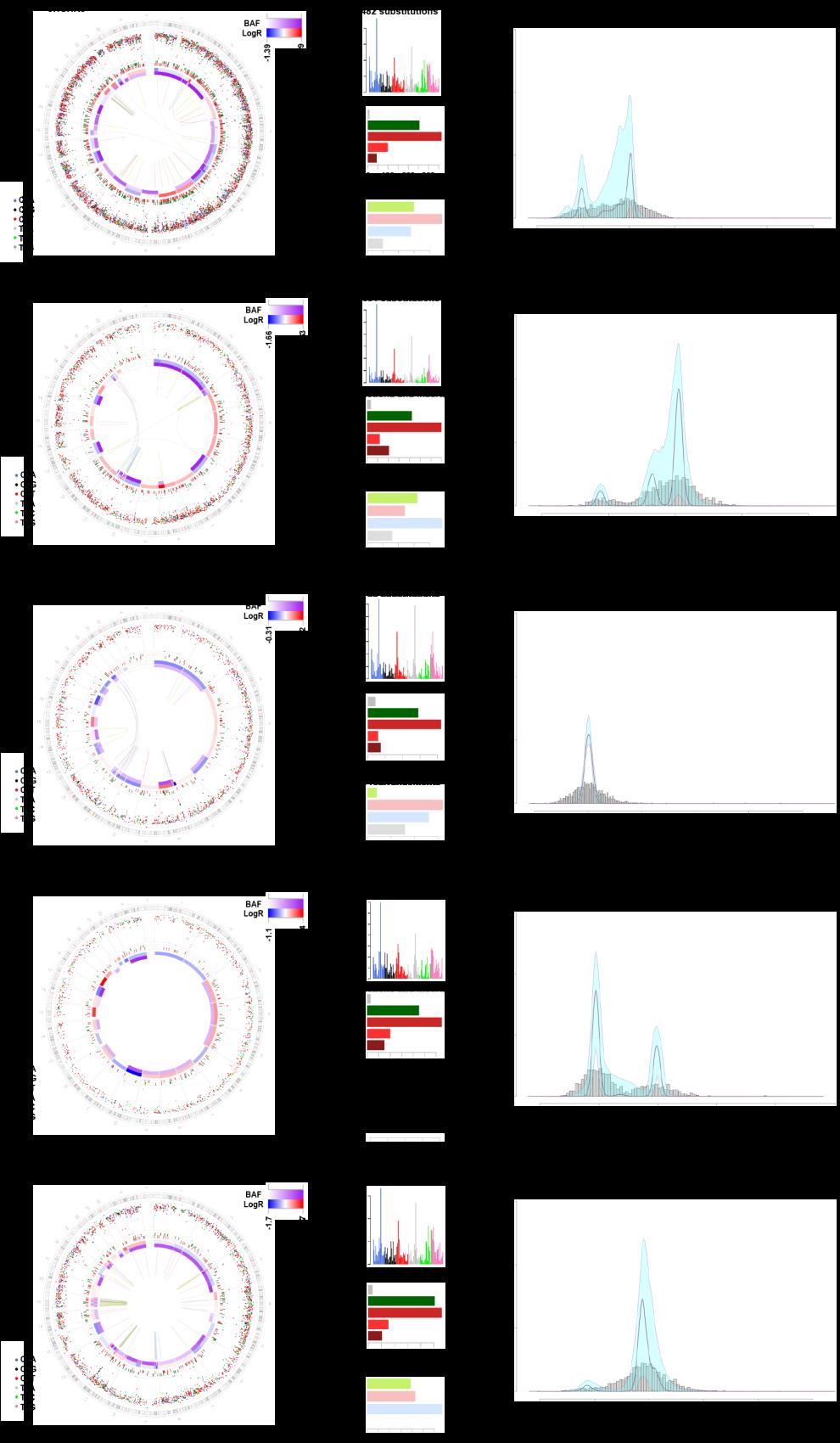
10 Supplemental Figure 3. Genomic Features revealed by WGS in the exploratory Cohort. Circos plots summarize the different types of alterations identified in the 5 patients that were whole-genome sequenced. Genomic rearrangements are plotted as arcs in the inner most track followed by the two tracks that show copy number changes as LogR and BAF. Genomic positions of the indels are shown in the fourth track. Inter-mutation distance for the substitutions is plotted in the outermost track. All mutations are coloured according to the alteration type summarized in the bar plots in the right panels. The second plot summarizes the results from the statistical analysis of the cancer cell fractions of the clonal and subclonal mutations by a Bayesian Dirichlet process-based clustering. Empiric histogram of substitutions is shown in grey together with the density from clustering in pale green and the fitted distribution in dark pink.


11 Supplemental Figure 4. Genomic alterations in the TCGA-KICH cohort by WES/WGS. Oncoprint of ploidy status and frequency of nonsynonymous mutations in 66 primary tumors. Asterisks denote samples excluded from the statistical analysis due to insufficient clinical data. The presence of sarcomatoid histology is denoted. Imbalanced chromosome duplication (ICD) status and loss of heterozygosity (LOH) at chromosome 17p were detected with FACETS. Mutation frequencies of individually specified genes are listed on the left. Mutation frequencies of individual genes in M- chrcc and ID-chRCC cases are shown as bar plots on the right. Gene mutation types are colorcoded. ICD status and LOH at chromosome 17p were detected with FACETS. Gene mutation types are color-coded.
12 Supplemental Figure 5. Patterns of 3 high-risk genomic features in 140 chrcc cases (33 M- chrcc, 41 ID-chRCC and 66 TCGA-KICH). PTEN mutations and ICD appear mutually exclusive (Fisher, P=0.0033).
13 SUPPLEMENTAL DATA FILES Supplemental Table 1. Clinical Data chrcc Supplemental Table 2. MSK-IMPACT List chrcc Supplemental Table 3. Mutations chrcc Supplemental Table 4. Primary Metastasis Pairs chrcc Supplemental Table 5. Influence of TP53, PTEN mutations and ICD on Survival in TCGA-KICH Supplemental Table 6. Influence of TP53, PTEN mutations, sarcomatoid status and ICD on survival in M-chRCC cohort Supplemental Table 7. TCGA KIRC-KICH Data
14 REFERENCES 1. Li H, and Durbin R. Fast and accurate long-read alignment with Burrows-Wheeler transform. Bioinformatics (Oxford, England). 2010;26(5): Nik-Zainal S, Van Loo P, Wedge DC, Alexandrov LB, Greenman CD, Lau KW, et al. The life history of 21 breast cancers. Cell. 2012;149(5): Abecasis GR, Auton A, Brooks LD, DePristo MA, Durbin RM, Handsaker RE, et al. An integrated map of genetic variation from 1,092 human genomes. Nature. 2012;491(7422): Menzies A, Teague JW, Butler AP, Davies H, Tarpey P, Nik-Zainal S, et al. VAGrENT: Variation Annotation Generator. Current protocols in bioinformatics. 2015;52: Raine KM, Hinton J, Butler AP, Teague JW, Davies H, Tarpey P, et al. cgppindel: Identifying Somatically Acquired Insertion and Deletion Events from Paired End Sequencing. Current protocols in bioinformatics. 2015;52: Zerbino DR, and Birney E. Velvet: algorithms for de novo short read assembly using de Bruijn graphs. Genome research. 2008;18(5): Van Loo P, Nordgard SH, Lingjaerde OC, Russnes HG, Rye IH, Sun W, et al. Allele-specific copy number analysis of tumors. Proceedings of the National Academy of Sciences of the United States of America. 2010;107(39): Rosenthal R, McGranahan N, Herrero J, Taylor BS, and Swanton C. DeconstructSigs: delineating mutational processes in single tumors distinguishes DNA repair deficiencies and patterns of carcinoma evolution. Genome biology. 2016;17: Bolli N, Avet-Loiseau H, Wedge DC, Van Loo P, Alexandrov LB, Martincorena I, et al. Heterogeneity of genomic evolution and mutational profiles in multiple myeloma. Nat Commun. 2014;5: Gundem G, Van Loo P, Kremeyer B, Alexandrov LB, Tubio JM, Papaemmanuil E, et al. The evolutionary history of lethal metastatic prostate cancer. Nature. 2015;520(7547): Cheng DT, Mitchell TN, Zehir A, Shah RH, Benayed R, Syed A, et al. Memorial Sloan Kettering-Integrated Mutation Profiling of Actionable Cancer Targets (MSK-IMPACT): A Hybridization Capture-Based Next-Generation Sequencing Clinical Assay for Solid Tumor Molecular Oncology. The Journal of molecular diagnostics : JMD. 2015;17(3): McKenna A, Hanna M, Banks E, Sivachenko A, Cibulskis K, Kernytsky A, et al. The Genome Analysis Toolkit: a MapReduce framework for analyzing next-generation DNA sequencing data. Genome research. 2010;20(9): Cibulskis K, Lawrence MS, Carter SL, Sivachenko A, Jaffe D, Sougnez C, et al. Sensitive detection of somatic point mutations in impure and heterogeneous cancer samples. Nature biotechnology. 2013;31(3): Saunders CT, Wong WS, Swamy S, Becq J, Murray LJ, and Cheetham RK. Strelka: accurate somatic small-variant calling from sequenced tumor-normal sample pairs. Bioinformatics (Oxford, England). 2012;28(14):
15 15. Koboldt DC, Zhang Q, Larson DE, Shen D, McLellan MD, Lin L, et al. VarScan 2: somatic mutation and copy number alteration discovery in cancer by exome sequencing. Genome research. 2012;22(3): De Mattos-Arruda L, Weigelt B, Cortes J, Won HH, Ng CK, Nuciforo P, et al. Capturing intra-tumor genetic heterogeneity by de novo mutation profiling of circulating cell-free tumor DNA: a proof-of-principle. Annals of oncology : official journal of the European Society for Medical Oncology. 2014;25(9): Robinson JT, Thorvaldsdottir H, Winckler W, Guttman M, Lander ES, Getz G, et al. Integrative genomics viewer. Nature biotechnology. 2011;29(1):24-6.
Whole Genome and Transcriptome Analysis of Anaplastic Meningioma. Patrick Tarpey Cancer Genome Project Wellcome Trust Sanger Institute
 Whole Genome and Transcriptome Analysis of Anaplastic Meningioma Patrick Tarpey Cancer Genome Project Wellcome Trust Sanger Institute Outline Anaplastic meningioma compared to other cancers Whole genomes
Whole Genome and Transcriptome Analysis of Anaplastic Meningioma Patrick Tarpey Cancer Genome Project Wellcome Trust Sanger Institute Outline Anaplastic meningioma compared to other cancers Whole genomes
Supplementary Figure 1. Estimation of tumour content
 Supplementary Figure 1. Estimation of tumour content a, Approach used to estimate the tumour content in S13T1/T2, S6T1/T2, S3T1/T2 and S12T1/T2. Tissue and tumour areas were evaluated by two independent
Supplementary Figure 1. Estimation of tumour content a, Approach used to estimate the tumour content in S13T1/T2, S6T1/T2, S3T1/T2 and S12T1/T2. Tissue and tumour areas were evaluated by two independent
Molecular Characterization of Tumors Using Next-Generation Sequencing
 Molecular Characterization of Tumors Using Next-Generation Sequencing Using BaseSpace to visualize molecular changes in cancer. Tumor-Normal Sequencing Data in BaseSpace To enable researchers new to next-generation
Molecular Characterization of Tumors Using Next-Generation Sequencing Using BaseSpace to visualize molecular changes in cancer. Tumor-Normal Sequencing Data in BaseSpace To enable researchers new to next-generation
Supplementary Materials for
 www.sciencetranslationalmedicine.org/cgi/content/full/7/283/283ra54/dc1 Supplementary Materials for Clonal status of actionable driver events and the timing of mutational processes in cancer evolution
www.sciencetranslationalmedicine.org/cgi/content/full/7/283/283ra54/dc1 Supplementary Materials for Clonal status of actionable driver events and the timing of mutational processes in cancer evolution
DNA-seq Bioinformatics Analysis: Copy Number Variation
 DNA-seq Bioinformatics Analysis: Copy Number Variation Elodie Girard elodie.girard@curie.fr U900 institut Curie, INSERM, Mines ParisTech, PSL Research University Paris, France NGS Applications 5C HiC DNA-seq
DNA-seq Bioinformatics Analysis: Copy Number Variation Elodie Girard elodie.girard@curie.fr U900 institut Curie, INSERM, Mines ParisTech, PSL Research University Paris, France NGS Applications 5C HiC DNA-seq
Dr Rick Tearle Senior Applications Specialist, EMEA Complete Genomics Complete Genomics, Inc.
 Dr Rick Tearle Senior Applications Specialist, EMEA Complete Genomics Topics Overview of Data Processing Pipeline Overview of Data Files 2 DNA Nano-Ball (DNB) Read Structure Genome : acgtacatgcattcacacatgcttagctatctctcgccag
Dr Rick Tearle Senior Applications Specialist, EMEA Complete Genomics Topics Overview of Data Processing Pipeline Overview of Data Files 2 DNA Nano-Ball (DNB) Read Structure Genome : acgtacatgcattcacacatgcttagctatctctcgccag
NGS in tissue and liquid biopsy
 NGS in tissue and liquid biopsy Ana Vivancos, PhD Referencias So, why NGS in the clinics? 2000 Sanger Sequencing (1977-) 2016 NGS (2006-) ABIPrism (Applied Biosystems) Up to 2304 per day (96 sequences
NGS in tissue and liquid biopsy Ana Vivancos, PhD Referencias So, why NGS in the clinics? 2000 Sanger Sequencing (1977-) 2016 NGS (2006-) ABIPrism (Applied Biosystems) Up to 2304 per day (96 sequences
Structural Variation and Medical Genomics
 Structural Variation and Medical Genomics Andrew King Department of Biomedical Informatics July 8, 2014 You already know about small scale genetic mutations Single nucleotide polymorphism (SNPs) Deletions,
Structural Variation and Medical Genomics Andrew King Department of Biomedical Informatics July 8, 2014 You already know about small scale genetic mutations Single nucleotide polymorphism (SNPs) Deletions,
Supplementary Methods
 Supplementary Methods Short Read Preprocessing Reads are preprocessed differently according to how they will be used: detection of the variant in the tumor, discovery of an artifact in the normal or for
Supplementary Methods Short Read Preprocessing Reads are preprocessed differently according to how they will be used: detection of the variant in the tumor, discovery of an artifact in the normal or for
Supplementary Tables. Supplementary Figures
 Supplementary Files for Zehir, Benayed et al. Mutational Landscape of Metastatic Cancer Revealed from Prospective Clinical Sequencing of 10,000 Patients Supplementary Tables Supplementary Table 1: Sample
Supplementary Files for Zehir, Benayed et al. Mutational Landscape of Metastatic Cancer Revealed from Prospective Clinical Sequencing of 10,000 Patients Supplementary Tables Supplementary Table 1: Sample
Supplementary Figure 1: Features of IGLL5 Mutations in CLL: a) Representative IGV screenshot of first
 Representative IGV screenshot of first") Supplementary Figure 1: Features of IGLL5 Mutations in CLL: a) Representative IGV screenshot of first intron IGLL5 mutation depicting biallelic mutations. Red arrows highlight the presence of out of phase
Supplementary Figure 1: Features of IGLL5 Mutations in CLL: a) Representative IGV screenshot of first intron IGLL5 mutation depicting biallelic mutations. Red arrows highlight the presence of out of phase
Multiple gene sequencing for risk assessment in patients with early-onset or familial breast cancer
 www.impactjournals.com/oncotarget/ Oncotarget, Supplementary Materials 2016 Multiple gene sequencing for risk assessment in patients with early-onset or familial breast cancer Supplementary Materials Supplementary
www.impactjournals.com/oncotarget/ Oncotarget, Supplementary Materials 2016 Multiple gene sequencing for risk assessment in patients with early-onset or familial breast cancer Supplementary Materials Supplementary
Advance Your Genomic Research Using Targeted Resequencing with SeqCap EZ Library
 Advance Your Genomic Research Using Targeted Resequencing with SeqCap EZ Library Marilou Wijdicks International Product Manager Research For Life Science Research Only. Not for Use in Diagnostic Procedures.
Advance Your Genomic Research Using Targeted Resequencing with SeqCap EZ Library Marilou Wijdicks International Product Manager Research For Life Science Research Only. Not for Use in Diagnostic Procedures.
Mutation Detection and CNV Analysis for Illumina Sequencing data from HaloPlex Target Enrichment Panels using NextGENe Software for Clinical Research
 Mutation Detection and CNV Analysis for Illumina Sequencing data from HaloPlex Target Enrichment Panels using NextGENe Software for Clinical Research Application Note Authors John McGuigan, Megan Manion,
Mutation Detection and CNV Analysis for Illumina Sequencing data from HaloPlex Target Enrichment Panels using NextGENe Software for Clinical Research Application Note Authors John McGuigan, Megan Manion,
Nature Biotechnology: doi: /nbt.1904
 Supplementary Information Comparison between assembly-based SV calls and array CGH results Genome-wide array assessment of copy number changes, such as array comparative genomic hybridization (acgh), is
Supplementary Information Comparison between assembly-based SV calls and array CGH results Genome-wide array assessment of copy number changes, such as array comparative genomic hybridization (acgh), is
Frequency(%) KRAS G12 KRAS G13 KRAS A146 KRAS Q61 KRAS K117N PIK3CA H1047 PIK3CA E545 PIK3CA E542K PIK3CA Q546. EGFR exon19 NFS-indel EGFR L858R
 KRAS G12 KRAS G13 KRAS A146 KRAS Q61 KRAS K117N PIK3CA H1047 PIK3CA E545 PIK3CA E542K PIK3CA Q546. EGFR exon19 NFS-indel EGFR L858R") Frequency(%) 1 a b ALK FS-indel ALK R1Q HRAS Q61R HRAS G13R IDH R17K IDH R14Q MET exon14 SS-indel KIT D8Y KIT L76P KIT exon11 NFS-indel SMAD4 R361 IDH1 R13 CTNNB1 S37 CTNNB1 S4 AKT1 E17K ERBB D769H ERBB
Frequency(%) 1 a b ALK FS-indel ALK R1Q HRAS Q61R HRAS G13R IDH R17K IDH R14Q MET exon14 SS-indel KIT D8Y KIT L76P KIT exon11 NFS-indel SMAD4 R361 IDH1 R13 CTNNB1 S37 CTNNB1 S4 AKT1 E17K ERBB D769H ERBB
AVENIO family of NGS oncology assays ctdna and Tumor Tissue Analysis Kits
 AVENIO family of NGS oncology assays ctdna and Tumor Tissue Analysis Kits Accelerating clinical research Next-generation sequencing (NGS) has the ability to interrogate many different genes and detect
AVENIO family of NGS oncology assays ctdna and Tumor Tissue Analysis Kits Accelerating clinical research Next-generation sequencing (NGS) has the ability to interrogate many different genes and detect
Supplementary Figure 1
 Supplementary Figure 1 Supplementary Fig. 1: Quality assessment of formalin-fixed paraffin-embedded (FFPE)-derived DNA and nuclei. (a) Multiplex PCR analysis of unrepaired and repaired bulk FFPE gdna from
Supplementary Figure 1 Supplementary Fig. 1: Quality assessment of formalin-fixed paraffin-embedded (FFPE)-derived DNA and nuclei. (a) Multiplex PCR analysis of unrepaired and repaired bulk FFPE gdna from
Genomic structural variation
 Genomic structural variation Mario Cáceres The new genomic variation DNA sequence differs across individuals much more than researchers had suspected through structural changes A huge amount of structural
Genomic structural variation Mario Cáceres The new genomic variation DNA sequence differs across individuals much more than researchers had suspected through structural changes A huge amount of structural
Statistical Analysis of Single Nucleotide Polymorphism Microarrays in Cancer Studies
 Statistical Analysis of Single Nucleotide Polymorphism Microarrays in Cancer Studies Stanford Biostatistics Workshop Pierre Neuvial with Henrik Bengtsson and Terry Speed Department of Statistics, UC Berkeley
Statistical Analysis of Single Nucleotide Polymorphism Microarrays in Cancer Studies Stanford Biostatistics Workshop Pierre Neuvial with Henrik Bengtsson and Terry Speed Department of Statistics, UC Berkeley
Genome. Institute. GenomeVIP: A Genomics Analysis Pipeline for Cloud Computing with Germline and Somatic Calling on Amazon s Cloud. R. Jay Mashl.
 GenomeVIP: the Genome Institute at Washington University A Genomics Analysis Pipeline for Cloud Computing with Germline and Somatic Calling on Amazon s Cloud R. Jay Mashl October 20, 2014 Turnkey Variant
GenomeVIP: the Genome Institute at Washington University A Genomics Analysis Pipeline for Cloud Computing with Germline and Somatic Calling on Amazon s Cloud R. Jay Mashl October 20, 2014 Turnkey Variant
Figure S4. 15 Mets Whole Exome. 5 Primary Tumors Cancer Panel and WES. Next Generation Sequencing
 Figure S4 Next Generation Sequencing 15 Mets Whole Exome 5 Primary Tumors Cancer Panel and WES Get coverage of all variant loci for all three Mets Variant Filtering Sequence Alignments Index and align
Figure S4 Next Generation Sequencing 15 Mets Whole Exome 5 Primary Tumors Cancer Panel and WES Get coverage of all variant loci for all three Mets Variant Filtering Sequence Alignments Index and align
The feasibility of circulating tumour DNA as an alternative to biopsy for mutational characterization in Stage III melanoma patients
 The feasibility of circulating tumour DNA as an alternative to biopsy for mutational characterization in Stage III melanoma patients ASSC Scientific Meeting 13 th October 2016 Prof Andrew Barbour UQ SOM
The feasibility of circulating tumour DNA as an alternative to biopsy for mutational characterization in Stage III melanoma patients ASSC Scientific Meeting 13 th October 2016 Prof Andrew Barbour UQ SOM
Myeloma Genetics what do we know and where are we going?
 in partnership with Myeloma Genetics what do we know and where are we going? Dr Brian Walker Thames Valley Cancer Network 14 th September 2015 Making the discoveries that defeat cancer Myeloma Genome:
in partnership with Myeloma Genetics what do we know and where are we going? Dr Brian Walker Thames Valley Cancer Network 14 th September 2015 Making the discoveries that defeat cancer Myeloma Genome:
Characterisation of structural variation in breast. cancer genomes using paired-end sequencing on. the Illumina Genome Analyser
 Characterisation of structural variation in breast cancer genomes using paired-end sequencing on the Illumina Genome Analyser Phil Stephens Cancer Genome Project Why is it important to study cancer? Why
Characterisation of structural variation in breast cancer genomes using paired-end sequencing on the Illumina Genome Analyser Phil Stephens Cancer Genome Project Why is it important to study cancer? Why
Golden Helix s End-to-End Solution for Clinical Labs
 Golden Helix s End-to-End Solution for Clinical Labs Steven Hystad - Field Application Scientist Nathan Fortier Senior Software Engineer 20 most promising Biotech Technology Providers Top 10 Analytics
Golden Helix s End-to-End Solution for Clinical Labs Steven Hystad - Field Application Scientist Nathan Fortier Senior Software Engineer 20 most promising Biotech Technology Providers Top 10 Analytics
BWA alignment to reference transcriptome and genome. Convert transcriptome mappings back to genome space
 Whole genome sequencing Whole exome sequencing BWA alignment to reference transcriptome and genome Convert transcriptome mappings back to genome space genomes Filter on MQ, distance, Cigar string Annotate
Whole genome sequencing Whole exome sequencing BWA alignment to reference transcriptome and genome Convert transcriptome mappings back to genome space genomes Filter on MQ, distance, Cigar string Annotate
Understanding DNA Copy Number Data
 Understanding DNA Copy Number Data Adam B. Olshen Department of Epidemiology and Biostatistics Helen Diller Family Comprehensive Cancer Center University of California, San Francisco http://cc.ucsf.edu/people/olshena_adam.php
Understanding DNA Copy Number Data Adam B. Olshen Department of Epidemiology and Biostatistics Helen Diller Family Comprehensive Cancer Center University of California, San Francisco http://cc.ucsf.edu/people/olshena_adam.php
Tumor mutational burden and its transition towards the clinic
 Tumor mutational burden and its transition towards the clinic G C C A T C A C Wolfram Jochum Institute of Pathology Kantonsspital St.Gallen CH-9007 St.Gallen wolfram.jochum@kssg.ch 30th European Congress
Tumor mutational burden and its transition towards the clinic G C C A T C A C Wolfram Jochum Institute of Pathology Kantonsspital St.Gallen CH-9007 St.Gallen wolfram.jochum@kssg.ch 30th European Congress
Analysis of Massively Parallel Sequencing Data Application of Illumina Sequencing to the Genetics of Human Cancers
 Analysis of Massively Parallel Sequencing Data Application of Illumina Sequencing to the Genetics of Human Cancers Gordon Blackshields Senior Bioinformatician Source BioScience 1 To Cancer Genetics Studies
Analysis of Massively Parallel Sequencing Data Application of Illumina Sequencing to the Genetics of Human Cancers Gordon Blackshields Senior Bioinformatician Source BioScience 1 To Cancer Genetics Studies
Mosaic loss of chromosome Y in peripheral blood is associated with shorter survival and higher risk of cancer
 Supplementary Information Mosaic loss of chromosome Y in peripheral blood is associated with shorter survival and higher risk of cancer Lars A. Forsberg, Chiara Rasi, Niklas Malmqvist, Hanna Davies, Saichand
Supplementary Information Mosaic loss of chromosome Y in peripheral blood is associated with shorter survival and higher risk of cancer Lars A. Forsberg, Chiara Rasi, Niklas Malmqvist, Hanna Davies, Saichand
AD (Leave blank) TITLE: Genomic Characterization of Brain Metastasis in Non-Small Cell Lung Cancer Patients
 TITLE: Genomic Characterization of Brain Metastasis in Non-Small Cell Lung Cancer Patients") AD (Leave blank) Award Number: W81XWH-12-1-0444 TITLE: Genomic Characterization of Brain Metastasis in Non-Small Cell Lung Cancer Patients PRINCIPAL INVESTIGATOR: Mark A. Watson, MD PhD CONTRACTING ORGANIZATION:
AD (Leave blank) Award Number: W81XWH-12-1-0444 TITLE: Genomic Characterization of Brain Metastasis in Non-Small Cell Lung Cancer Patients PRINCIPAL INVESTIGATOR: Mark A. Watson, MD PhD CONTRACTING ORGANIZATION:
SUPPLEMENTARY INFORMATION. Intron retention is a widespread mechanism of tumor suppressor inactivation.
 SUPPLEMENTARY INFORMATION Intron retention is a widespread mechanism of tumor suppressor inactivation. Hyunchul Jung 1,2,3, Donghoon Lee 1,4, Jongkeun Lee 1,5, Donghyun Park 2,6, Yeon Jeong Kim 2,6, Woong-Yang
SUPPLEMENTARY INFORMATION Intron retention is a widespread mechanism of tumor suppressor inactivation. Hyunchul Jung 1,2,3, Donghoon Lee 1,4, Jongkeun Lee 1,5, Donghyun Park 2,6, Yeon Jeong Kim 2,6, Woong-Yang
MEDICAL GENOMICS LABORATORY. Next-Gen Sequencing and Deletion/Duplication Analysis of NF1 Only (NF1-NG)
") Next-Gen Sequencing and Deletion/Duplication Analysis of NF1 Only (NF1-NG) Ordering Information Acceptable specimen types: Fresh blood sample (3-6 ml EDTA; no time limitations associated with receipt)
Next-Gen Sequencing and Deletion/Duplication Analysis of NF1 Only (NF1-NG) Ordering Information Acceptable specimen types: Fresh blood sample (3-6 ml EDTA; no time limitations associated with receipt)
Identification of genomic alterations in cervical cancer biopsies by exome sequencing
 Chapter- 4 Identification of genomic alterations in cervical cancer biopsies by exome sequencing 105 4.1 INTRODUCTION Athough HPV has been identified as the prime etiological factor for cervical cancer,
Chapter- 4 Identification of genomic alterations in cervical cancer biopsies by exome sequencing 105 4.1 INTRODUCTION Athough HPV has been identified as the prime etiological factor for cervical cancer,
AVENIO ctdna Analysis Kits The complete NGS liquid biopsy solution EMPOWER YOUR LAB
 Analysis Kits The complete NGS liquid biopsy solution EMPOWER YOUR LAB Analysis Kits Next-generation performance in liquid biopsies 2 Accelerating clinical research From liquid biopsy to next-generation
Analysis Kits The complete NGS liquid biopsy solution EMPOWER YOUR LAB Analysis Kits Next-generation performance in liquid biopsies 2 Accelerating clinical research From liquid biopsy to next-generation
CNV Detection and Interpretation in Genomic Data
 CNV Detection and Interpretation in Genomic Data Benjamin W. Darbro, M.D., Ph.D. Assistant Professor of Pediatrics Director of the Shivanand R. Patil Cytogenetics and Molecular Laboratory Overview What
CNV Detection and Interpretation in Genomic Data Benjamin W. Darbro, M.D., Ph.D. Assistant Professor of Pediatrics Director of the Shivanand R. Patil Cytogenetics and Molecular Laboratory Overview What
Relationship between genomic features and distributions of RS1 and RS3 rearrangements in breast cancer genomes.
 Supplementary Figure 1 Relationship between genomic features and distributions of RS1 and RS3 rearrangements in breast cancer genomes. (a,b) Values of coefficients associated with genomic features, separately
Supplementary Figure 1 Relationship between genomic features and distributions of RS1 and RS3 rearrangements in breast cancer genomes. (a,b) Values of coefficients associated with genomic features, separately
Abstract. Optimization strategy of Copy Number Variant calling using Multiplicom solutions APPLICATION NOTE. Introduction
 Optimization strategy of Copy Number Variant calling using Multiplicom solutions Michael Vyverman, PhD; Laura Standaert, PhD and Wouter Bossuyt, PhD Abstract Copy number variations (CNVs) represent a significant
Optimization strategy of Copy Number Variant calling using Multiplicom solutions Michael Vyverman, PhD; Laura Standaert, PhD and Wouter Bossuyt, PhD Abstract Copy number variations (CNVs) represent a significant
LTA Analysis of HapMap Genotype Data
 LTA Analysis of HapMap Genotype Data Introduction. This supplement to Global variation in copy number in the human genome, by Redon et al., describes the details of the LTA analysis used to screen HapMap
LTA Analysis of HapMap Genotype Data Introduction. This supplement to Global variation in copy number in the human genome, by Redon et al., describes the details of the LTA analysis used to screen HapMap
Supplementary note: Comparison of deletion variants identified in this study and four earlier studies
 Supplementary note: Comparison of deletion variants identified in this study and four earlier studies Here we compare the results of this study to potentially overlapping results from four earlier studies
Supplementary note: Comparison of deletion variants identified in this study and four earlier studies Here we compare the results of this study to potentially overlapping results from four earlier studies
OncoPhase: Quantification of somatic mutation cellular prevalence using phase information
 OncoPhase: Quantification of somatic mutation cellular prevalence using phase information Donatien Chedom-Fotso 1, 2, 3, Ahmed Ashour Ahmed 1, 2, and Christopher Yau 3, 4 1 Ovarian Cancer Cell Laboratory,
OncoPhase: Quantification of somatic mutation cellular prevalence using phase information Donatien Chedom-Fotso 1, 2, 3, Ahmed Ashour Ahmed 1, 2, and Christopher Yau 3, 4 1 Ovarian Cancer Cell Laboratory,
COMPUTATIONAL OPTIMISATION OF TARGETED DNA SEQUENCING FOR CANCER DETECTION
 COMPUTATIONAL OPTIMISATION OF TARGETED DNA SEQUENCING FOR CANCER DETECTION Pierre Martinez, Nicholas McGranahan, Nicolai Juul Birkbak, Marco Gerlinger, Charles Swanton* SUPPLEMENTARY INFORMATION SUPPLEMENTARY
COMPUTATIONAL OPTIMISATION OF TARGETED DNA SEQUENCING FOR CANCER DETECTION Pierre Martinez, Nicholas McGranahan, Nicolai Juul Birkbak, Marco Gerlinger, Charles Swanton* SUPPLEMENTARY INFORMATION SUPPLEMENTARY
CRISPR/Cas9 Enrichment and Long-read WGS for Structural Variant Discovery
 CRISPR/Cas9 Enrichment and Long-read WGS for Structural Variant Discovery PacBio CoLab Session October 20, 2017 For Research Use Only. Not for use in diagnostics procedures. Copyright 2017 by Pacific Biosciences
CRISPR/Cas9 Enrichment and Long-read WGS for Structural Variant Discovery PacBio CoLab Session October 20, 2017 For Research Use Only. Not for use in diagnostics procedures. Copyright 2017 by Pacific Biosciences
p.r623c p.p976l p.d2847fs p.t2671 p.d2847fs p.r2922w p.r2370h p.c1201y p.a868v p.s952* RING_C BP PHD Cbp HAT_KAT11
 ARID2 p.r623c KMT2D p.v650fs p.p976l p.r2922w p.l1212r p.d1400h DNA binding RFX DNA binding Zinc finger KMT2C p.a51s p.d372v p.c1103* p.d2847fs p.t2671 p.d2847fs p.r4586h PHD/ RING DHHC/ PHD PHD FYR N
ARID2 p.r623c KMT2D p.v650fs p.p976l p.r2922w p.l1212r p.d1400h DNA binding RFX DNA binding Zinc finger KMT2C p.a51s p.d372v p.c1103* p.d2847fs p.t2671 p.d2847fs p.r4586h PHD/ RING DHHC/ PHD PHD FYR N
cn.mops - Mixture of Poissons for CNV detection in NGS data Günter Klambauer Institute of Bioinformatics, Johannes Kepler University Linz
 Software Manual Institute of Bioinformatics, Johannes Kepler University Linz cn.mops - Mixture of Poissons for CNV detection in NGS data Günter Klambauer Institute of Bioinformatics, Johannes Kepler University
Software Manual Institute of Bioinformatics, Johannes Kepler University Linz cn.mops - Mixture of Poissons for CNV detection in NGS data Günter Klambauer Institute of Bioinformatics, Johannes Kepler University
Research Strategy: 1. Background and Significance
 Research Strategy: 1. Background and Significance 1.1. Heterogeneity is a common feature of cancer. A better understanding of this heterogeneity may present therapeutic opportunities: Intratumor heterogeneity
Research Strategy: 1. Background and Significance 1.1. Heterogeneity is a common feature of cancer. A better understanding of this heterogeneity may present therapeutic opportunities: Intratumor heterogeneity
Nature Genetics: doi: /ng Supplementary Figure 1. Somatic coding mutations identified by WES/WGS for 83 ATL cases.
 Supplementary Figure 1 Somatic coding mutations identified by WES/WGS for 83 ATL cases. (a) The percentage of targeted bases covered by at least 2, 10, 20 and 30 sequencing reads (top) and average read
Supplementary Figure 1 Somatic coding mutations identified by WES/WGS for 83 ATL cases. (a) The percentage of targeted bases covered by at least 2, 10, 20 and 30 sequencing reads (top) and average read
Performance Characteristics BRCA MASTR Plus Dx
 Performance Characteristics BRCA MASTR Plus Dx with drmid Dx for Illumina NGS systems Manufacturer Multiplicom N.V. Galileïlaan 18 2845 Niel Belgium Table of Contents 1. Workflow... 4 2. Performance Characteristics
Performance Characteristics BRCA MASTR Plus Dx with drmid Dx for Illumina NGS systems Manufacturer Multiplicom N.V. Galileïlaan 18 2845 Niel Belgium Table of Contents 1. Workflow... 4 2. Performance Characteristics
Using the Bravo Liquid-Handling System for Next Generation Sequencing Sample Prep
 Using the Bravo Liquid-Handling System for Next Generation Sequencing Sample Prep Tom Walsh, PhD Division of Medical Genetics University of Washington Next generation sequencing Sanger sequencing gold
Using the Bravo Liquid-Handling System for Next Generation Sequencing Sample Prep Tom Walsh, PhD Division of Medical Genetics University of Washington Next generation sequencing Sanger sequencing gold
Below, we included the point-to-point response to the comments of both reviewers.
 To the Editor and Reviewers: We would like to thank the editor and reviewers for careful reading, and constructive suggestions for our manuscript. According to comments from both reviewers, we have comprehensively
To the Editor and Reviewers: We would like to thank the editor and reviewers for careful reading, and constructive suggestions for our manuscript. According to comments from both reviewers, we have comprehensively
Breast and ovarian cancer in Serbia: the importance of mutation detection in hereditary predisposition genes using NGS
 Breast and ovarian cancer in Serbia: the importance of mutation detection in hereditary predisposition genes using NGS dr sc. Ana Krivokuća Laboratory for molecular genetics Institute for Oncology and
Breast and ovarian cancer in Serbia: the importance of mutation detection in hereditary predisposition genes using NGS dr sc. Ana Krivokuća Laboratory for molecular genetics Institute for Oncology and
Chromothripsis: A New Mechanism For Tumorigenesis? i Fellow s Conference Cheryl Carlson 6/10/2011
 Chromothripsis: A New Mechanism For Tumorigenesis? i Fellow s Conference Cheryl Carlson 6/10/2011 Massive Genomic Rearrangement Acquired in a Single Catastrophic Event during Cancer Development Cell 144,
Chromothripsis: A New Mechanism For Tumorigenesis? i Fellow s Conference Cheryl Carlson 6/10/2011 Massive Genomic Rearrangement Acquired in a Single Catastrophic Event during Cancer Development Cell 144,
Nature Genetics: doi: /ng Supplementary Figure 1. Mutational signatures in BCC compared to melanoma.
 Supplementary Figure 1 Mutational signatures in BCC compared to melanoma. (a) The effect of transcription-coupled repair as a function of gene expression in BCC. Tumor type specific gene expression levels
Supplementary Figure 1 Mutational signatures in BCC compared to melanoma. (a) The effect of transcription-coupled repair as a function of gene expression in BCC. Tumor type specific gene expression levels
Supplementary Figure 1. Schematic diagram of o2n-seq. Double-stranded DNA was sheared, end-repaired, and underwent A-tailing by standard protocols.
 Supplementary Figure 1. Schematic diagram of o2n-seq. Double-stranded DNA was sheared, end-repaired, and underwent A-tailing by standard protocols. A-tailed DNA was ligated to T-tailed dutp adapters, circularized
Supplementary Figure 1. Schematic diagram of o2n-seq. Double-stranded DNA was sheared, end-repaired, and underwent A-tailing by standard protocols. A-tailed DNA was ligated to T-tailed dutp adapters, circularized
PSSV User Manual (V2.1)
") PSSV User Manual (V2.1) 1. Introduction A novel pattern-based probabilistic approach, PSSV, is developed to identify somatic structural variations from WGS data. Specifically, discordant and concordant
PSSV User Manual (V2.1) 1. Introduction A novel pattern-based probabilistic approach, PSSV, is developed to identify somatic structural variations from WGS data. Specifically, discordant and concordant
DOES THE BRCAX GENE EXIST? FUTURE OUTLOOK
 CHAPTER 6 DOES THE BRCAX GENE EXIST? FUTURE OUTLOOK Genetic research aimed at the identification of new breast cancer susceptibility genes is at an interesting crossroad. On the one hand, the existence
CHAPTER 6 DOES THE BRCAX GENE EXIST? FUTURE OUTLOOK Genetic research aimed at the identification of new breast cancer susceptibility genes is at an interesting crossroad. On the one hand, the existence
PREPARED FOR: U.S. Army Medical Research and Materiel Command Fort Detrick, Maryland
 AD Award Number: W81XWH-10-1-0589 TITLE: Global Characterization of Protein Altering Mutations in Prostate Cancer PRINCIPAL INVESTIGATOR: Jay Shendure, M.D., Ph.D. CONTRACTING ORGANIZATION: University
AD Award Number: W81XWH-10-1-0589 TITLE: Global Characterization of Protein Altering Mutations in Prostate Cancer PRINCIPAL INVESTIGATOR: Jay Shendure, M.D., Ph.D. CONTRACTING ORGANIZATION: University
Global variation in copy number in the human genome
 Global variation in copy number in the human genome Redon et. al. Nature 444:444-454 (2006) 12.03.2007 Tarmo Puurand Study 270 individuals (HapMap collection) Affymetrix 500K Whole Genome TilePath (WGTP)
Global variation in copy number in the human genome Redon et. al. Nature 444:444-454 (2006) 12.03.2007 Tarmo Puurand Study 270 individuals (HapMap collection) Affymetrix 500K Whole Genome TilePath (WGTP)
A general framework for analyzing tumor subclonality using SNP array and DNA sequencing data
 Li and Li Genome Biology 2014, 15:473 METHOD A general framework for analyzing tumor subclonality using SNP array and DNA sequencing data Bo Li 1 and Jun Z Li 2 Open Access Abstract Intra-tumor heterogeneity
Li and Li Genome Biology 2014, 15:473 METHOD A general framework for analyzing tumor subclonality using SNP array and DNA sequencing data Bo Li 1 and Jun Z Li 2 Open Access Abstract Intra-tumor heterogeneity
ACE ImmunoID Biomarker Discovery Solutions ACE ImmunoID Platform for Tumor Immunogenomics
 ACE ImmunoID Biomarker Discovery Solutions ACE ImmunoID Platform for Tumor Immunogenomics Precision Genomics for Immuno-Oncology Personalis, Inc. ACE ImmunoID When one biomarker doesn t tell the whole
ACE ImmunoID Biomarker Discovery Solutions ACE ImmunoID Platform for Tumor Immunogenomics Precision Genomics for Immuno-Oncology Personalis, Inc. ACE ImmunoID When one biomarker doesn t tell the whole
Identifying driver mutations in sequenced cancer genomes: computational approaches to enable precision medicine
 Raphael et al. Genome Medicine 2014, 6:5 REVIEW Identifying driver mutations in sequenced cancer genomes: computational approaches to enable precision medicine Benjamin J Raphael 1,2*, Jason R Dobson 1,2,3,
Raphael et al. Genome Medicine 2014, 6:5 REVIEW Identifying driver mutations in sequenced cancer genomes: computational approaches to enable precision medicine Benjamin J Raphael 1,2*, Jason R Dobson 1,2,3,
High-Definition Reconstruction of Clonal Composition in Cancer
 Cell Reports Resource High-Definition Reconstruction of Clonal Composition in Cancer Andrej Fischer, 1, * Ignacio Vázquez-García, 1,2 Christopher J.R. Illingworth, 3 and Ville Mustonen 1, * 1 Wellcome
Cell Reports Resource High-Definition Reconstruction of Clonal Composition in Cancer Andrej Fischer, 1, * Ignacio Vázquez-García, 1,2 Christopher J.R. Illingworth, 3 and Ville Mustonen 1, * 1 Wellcome
The$mitochondrial$and$autosomal$mutation$landscapes$of$ prostate$cancer$ $supplementary$material$
 The$mitochondrial$and$autosomal$mutation$landscapes$of$ prostate$cancer$ $supplementary$material$ Johan Lindberg 1, Ian G. Mills 2-4, Daniel Klevebring 1, Wennuan Liu 5, Mårten Neiman 1, Jianfeng Xu 5,
The$mitochondrial$and$autosomal$mutation$landscapes$of$ prostate$cancer$ $supplementary$material$ Johan Lindberg 1, Ian G. Mills 2-4, Daniel Klevebring 1, Wennuan Liu 5, Mårten Neiman 1, Jianfeng Xu 5,
Supplementary Figure 1: Classification scheme for non-synonymous and nonsense germline MC1R variants. The common variants with previously established
 Supplementary Figure 1: Classification scheme for nonsynonymous and nonsense germline MC1R variants. The common variants with previously established classifications 1 3 are shown. The effect of novel missense
Supplementary Figure 1: Classification scheme for nonsynonymous and nonsense germline MC1R variants. The common variants with previously established classifications 1 3 are shown. The effect of novel missense
Analysis with SureCall 2.1
 Analysis with SureCall 2.1 Danielle Fletcher Field Application Scientist July 2014 1 Stages of NGS Analysis Primary analysis, base calling Control Software FASTQ file reads + quality 2 Stages of NGS Analysis
Analysis with SureCall 2.1 Danielle Fletcher Field Application Scientist July 2014 1 Stages of NGS Analysis Primary analysis, base calling Control Software FASTQ file reads + quality 2 Stages of NGS Analysis
Cell-free tumor DNA for cancer monitoring
 Learning objectives Cell-free tumor DNA for cancer monitoring Christina Lockwood, PhD, DABCC, DABMGG Department of Laboratory Medicine 1. Define circulating, cell-free tumor DNA (ctdna) 2. Understand the
Learning objectives Cell-free tumor DNA for cancer monitoring Christina Lockwood, PhD, DABCC, DABMGG Department of Laboratory Medicine 1. Define circulating, cell-free tumor DNA (ctdna) 2. Understand the
Disclosure. Summary. Circulating DNA and NGS technology 3/27/2017. Disclosure of Relevant Financial Relationships. JS Reis-Filho, MD, PhD, FRCPath
 Circulating DNA and NGS technology JS Reis-Filho, MD, PhD, FRCPath Director of Experimental Pathology, Department of Pathology Affiliate Member, Human Oncology and Pathogenesis Program Disclosure of Relevant
Circulating DNA and NGS technology JS Reis-Filho, MD, PhD, FRCPath Director of Experimental Pathology, Department of Pathology Affiliate Member, Human Oncology and Pathogenesis Program Disclosure of Relevant
Single-strand DNA library preparation improves sequencing of formalin-fixed and paraffin-embedded (FFPE) cancer DNA
 cancer DNA") www.impactjournals.com/oncotarget/ Oncotarget, Supplementary Materials 2016 Single-strand DNA library preparation improves sequencing of formalin-fixed and paraffin-embedded (FFPE) DNA Supplementary Materials
www.impactjournals.com/oncotarget/ Oncotarget, Supplementary Materials 2016 Single-strand DNA library preparation improves sequencing of formalin-fixed and paraffin-embedded (FFPE) DNA Supplementary Materials
NGS ONCOPANELS: FDA S PERSPECTIVE
 NGS ONCOPANELS: FDA S PERSPECTIVE CBA Workshop: Biomarker and Application in Drug Development August 11, 2018 Rockville, MD You Li, Ph.D. Division of Molecular Genetics and Pathology Food and Drug Administration
NGS ONCOPANELS: FDA S PERSPECTIVE CBA Workshop: Biomarker and Application in Drug Development August 11, 2018 Rockville, MD You Li, Ph.D. Division of Molecular Genetics and Pathology Food and Drug Administration
underlying metastasis and recurrence in HNSCC, we analyzed two groups of patients. The
 Supplementary Figures Figure S1. Patient cohorts and study design. To define and interrogate the genetic alterations underlying metastasis and recurrence in HNSCC, we analyzed two groups of patients. The
Supplementary Figures Figure S1. Patient cohorts and study design. To define and interrogate the genetic alterations underlying metastasis and recurrence in HNSCC, we analyzed two groups of patients. The
SUPPLEMENTARY INFORMATION
 doi:10.1038/nature11396 A total of 2078 samples from a large sequencing project at decode were used in this study, 219 samples from 78 trios with two grandchildren who were not also members of other trios,
doi:10.1038/nature11396 A total of 2078 samples from a large sequencing project at decode were used in this study, 219 samples from 78 trios with two grandchildren who were not also members of other trios,
Nature Methods: doi: /nmeth.3115
 Supplementary Figure 1 Analysis of DNA methylation in a cancer cohort based on Infinium 450K data. RnBeads was used to rediscover a clinically distinct subgroup of glioblastoma patients characterized by
Supplementary Figure 1 Analysis of DNA methylation in a cancer cohort based on Infinium 450K data. RnBeads was used to rediscover a clinically distinct subgroup of glioblastoma patients characterized by
CNV detection. Introduction and detection in NGS data. G. Demidov 1,2. NGSchool2016. Centre for Genomic Regulation. CNV detection. G.
 Introduction and detection in NGS data 1,2 1 Genomic and Epigenomic Variation in Disease group, Centre for Genomic Regulation 2 Universitat Pompeu Fabra NGSchool2016 methods: methods Outline methods: methods
Introduction and detection in NGS data 1,2 1 Genomic and Epigenomic Variation in Disease group, Centre for Genomic Regulation 2 Universitat Pompeu Fabra NGSchool2016 methods: methods Outline methods: methods
Identifying Mutations Responsible for Rare Disorders Using New Technologies
 Identifying Mutations Responsible for Rare Disorders Using New Technologies Jacek Majewski, Department of Human Genetics, McGill University, Montreal, QC Canada Mendelian Diseases Clear mode of inheritance
Identifying Mutations Responsible for Rare Disorders Using New Technologies Jacek Majewski, Department of Human Genetics, McGill University, Montreal, QC Canada Mendelian Diseases Clear mode of inheritance
Cancer Genomics. Nic Waddell. Winter School in Mathematical and Computational Biology. July th
 Cancer Genomics Nic Waddell Winter School in Mathematical and Computational Biology 6th July 2015 Time Line of Key Events in Cancer Genomics Michael R. Stratton Science 2011;331:1553-1558 The Cancer Genome
Cancer Genomics Nic Waddell Winter School in Mathematical and Computational Biology 6th July 2015 Time Line of Key Events in Cancer Genomics Michael R. Stratton Science 2011;331:1553-1558 The Cancer Genome
Calling DNA variants SNVs, CNVs, and SVs. Steve Laurie Variant Effect Predictor Training Course Prague, 6 th November 2017
 1 Calling DNA variants SNVs, CNVs, and SVs Steve Laurie Variant Effect Predictor Training Course Prague, 6 th November 2017 Calling DNA variants SNVs, CNVs, SVs 2 1. What is a variant? 2. Paired End read
1 Calling DNA variants SNVs, CNVs, and SVs Steve Laurie Variant Effect Predictor Training Course Prague, 6 th November 2017 Calling DNA variants SNVs, CNVs, SVs 2 1. What is a variant? 2. Paired End read
De Novo Viral Quasispecies Assembly using Overlap Graphs
 De Novo Viral Quasispecies Assembly using Overlap Graphs Alexander Schönhuth joint with Jasmijn Baaijens, Amal Zine El Aabidine, Eric Rivals Milano 18th of November 2016 Viral Quasispecies Assembly: HaploClique
De Novo Viral Quasispecies Assembly using Overlap Graphs Alexander Schönhuth joint with Jasmijn Baaijens, Amal Zine El Aabidine, Eric Rivals Milano 18th of November 2016 Viral Quasispecies Assembly: HaploClique
NGS IN ONCOLOGY: FDA S PERSPECTIVE
 NGS IN ONCOLOGY: FDA S PERSPECTIVE ASQ Biomed/Biotech SIG Event April 26, 2018 Gaithersburg, MD You Li, Ph.D. Division of Molecular Genetics and Pathology Food and Drug Administration (FDA) Center for
NGS IN ONCOLOGY: FDA S PERSPECTIVE ASQ Biomed/Biotech SIG Event April 26, 2018 Gaithersburg, MD You Li, Ph.D. Division of Molecular Genetics and Pathology Food and Drug Administration (FDA) Center for
Introduction to LOH and Allele Specific Copy Number User Forum
 Introduction to LOH and Allele Specific Copy Number User Forum Jonathan Gerstenhaber Introduction to LOH and ASCN User Forum Contents 1. Loss of heterozygosity Analysis procedure Types of baselines 2.
Introduction to LOH and Allele Specific Copy Number User Forum Jonathan Gerstenhaber Introduction to LOH and ASCN User Forum Contents 1. Loss of heterozygosity Analysis procedure Types of baselines 2.
Nature Genetics: doi: /ng Supplementary Figure 1. SEER data for male and female cancer incidence from
 Supplementary Figure 1 SEER data for male and female cancer incidence from 1975 2013. (a,b) Incidence rates of oral cavity and pharynx cancer (a) and leukemia (b) are plotted, grouped by males (blue),
Supplementary Figure 1 SEER data for male and female cancer incidence from 1975 2013. (a,b) Incidence rates of oral cavity and pharynx cancer (a) and leukemia (b) are plotted, grouped by males (blue),
cn.mops - Mixture of Poissons for CNV detection in NGS data Günter Klambauer Institute of Bioinformatics, Johannes Kepler University Linz
 Software Manual Institute of Bioinformatics, Johannes Kepler University Linz cn.mops - Mixture of Poissons for CNV detection in NGS data Günter Klambauer Institute of Bioinformatics, Johannes Kepler University
Software Manual Institute of Bioinformatics, Johannes Kepler University Linz cn.mops - Mixture of Poissons for CNV detection in NGS data Günter Klambauer Institute of Bioinformatics, Johannes Kepler University
NGS in Cancer Pathology After the Microscope: From Nucleic Acid to Interpretation
 NGS in Cancer Pathology After the Microscope: From Nucleic Acid to Interpretation Michael R. Rossi, PhD, FACMG Assistant Professor Division of Cancer Biology, Department of Radiation Oncology Department
NGS in Cancer Pathology After the Microscope: From Nucleic Acid to Interpretation Michael R. Rossi, PhD, FACMG Assistant Professor Division of Cancer Biology, Department of Radiation Oncology Department
New Enhancements: GWAS Workflows with SVS
 New Enhancements: GWAS Workflows with SVS August 9 th, 2017 Gabe Rudy VP Product & Engineering 20 most promising Biotech Technology Providers Top 10 Analytics Solution Providers Hype Cycle for Life sciences
New Enhancements: GWAS Workflows with SVS August 9 th, 2017 Gabe Rudy VP Product & Engineering 20 most promising Biotech Technology Providers Top 10 Analytics Solution Providers Hype Cycle for Life sciences
Nature Genetics: doi: /ng Supplementary Figure 1. Rates of different mutation types in CRC.
 Supplementary Figure 1 Rates of different mutation types in CRC. (a) Stratification by mutation type indicates that C>T mutations occur at a significantly greater rate than other types. (b) As for the
Supplementary Figure 1 Rates of different mutation types in CRC. (a) Stratification by mutation type indicates that C>T mutations occur at a significantly greater rate than other types. (b) As for the
TITLE: - Whole Genome Sequencing of High-Risk Families to Identify New Mutational Mechanisms of Breast Cancer Predisposition
 AD Award Number: W81XWH-13-1-0336 TITLE: - Whole Genome Sequencing of High-Risk Families to Identify New Mutational Mechanisms of Breast Cancer Predisposition PRINCIPAL INVESTIGATOR: Mary-Claire King,
AD Award Number: W81XWH-13-1-0336 TITLE: - Whole Genome Sequencing of High-Risk Families to Identify New Mutational Mechanisms of Breast Cancer Predisposition PRINCIPAL INVESTIGATOR: Mary-Claire King,
Package ClusteredMutations
 Package ClusteredMutations April 29, 2016 Version 1.0.1 Date 2016-04-28 Title Location and Visualization of Clustered Somatic Mutations Depends seriation Description Identification and visualization of
Package ClusteredMutations April 29, 2016 Version 1.0.1 Date 2016-04-28 Title Location and Visualization of Clustered Somatic Mutations Depends seriation Description Identification and visualization of
VARIANT PRIORIZATION AND ANALYSIS INCORPORATING PROBLEMATIC REGIONS OF THE GENOME ANIL PATWARDHAN
 VARIANT PRIORIZATION AND ANALYSIS INCORPORATING PROBLEMATIC REGIONS OF THE GENOME ANIL PATWARDHAN Email: apatwardhan@personalis.com MICHAEL CLARK Email: michael.clark@personalis.com ALEX MORGAN Email:
VARIANT PRIORIZATION AND ANALYSIS INCORPORATING PROBLEMATIC REGIONS OF THE GENOME ANIL PATWARDHAN Email: apatwardhan@personalis.com MICHAEL CLARK Email: michael.clark@personalis.com ALEX MORGAN Email:
Evaluating somatic tumor mutation detection without matched normal samples
 Teer et al. Human Genomics (2017) 11:22 DOI 10.1186/s40246-017-0118-2 PRIMARY RESEARCH Open Access Evaluating somatic tumor mutation detection without matched normal samples Jamie K. Teer 1*, Yonghong
Teer et al. Human Genomics (2017) 11:22 DOI 10.1186/s40246-017-0118-2 PRIMARY RESEARCH Open Access Evaluating somatic tumor mutation detection without matched normal samples Jamie K. Teer 1*, Yonghong
MSI positive MSI negative
 Pritchard et al. 2014 Supplementary Figure 1 MSI positive MSI negative Hypermutated Median: 673 Average: 659.2 Non-Hypermutated Median: 37.5 Average: 43.6 Supplementary Figure 1: Somatic Mutation Burden
Pritchard et al. 2014 Supplementary Figure 1 MSI positive MSI negative Hypermutated Median: 673 Average: 659.2 Non-Hypermutated Median: 37.5 Average: 43.6 Supplementary Figure 1: Somatic Mutation Burden
No mutations were identified.
 Hereditary High Cholesterol Test ORDERING PHYSICIAN PRIMARY CONTACT SPECIMEN Report date: Aug 1, 2017 Dr. Jenny Jones Sample Medical Group 123 Main St. Sample, CA Kelly Peters Sample Medical Group 123
Hereditary High Cholesterol Test ORDERING PHYSICIAN PRIMARY CONTACT SPECIMEN Report date: Aug 1, 2017 Dr. Jenny Jones Sample Medical Group 123 Main St. Sample, CA Kelly Peters Sample Medical Group 123
Hands-On Ten The BRCA1 Gene and Protein
 Hands-On Ten The BRCA1 Gene and Protein Objective: To review transcription, translation, reading frames, mutations, and reading files from GenBank, and to review some of the bioinformatics tools, such
Hands-On Ten The BRCA1 Gene and Protein Objective: To review transcription, translation, reading frames, mutations, and reading files from GenBank, and to review some of the bioinformatics tools, such
AACR GENIE Data Guide
 AACR GENIE Data Guide About this Document Version of Data Data Access Terms of Access Introduction to AACR GENIE Human Subjects Protection and Privacy Summary of Data by Center Genomic Profiling at Each
AACR GENIE Data Guide About this Document Version of Data Data Access Terms of Access Introduction to AACR GENIE Human Subjects Protection and Privacy Summary of Data by Center Genomic Profiling at Each
Interactive analysis and quality assessment of single-cell copy-number variations
 Interactive analysis and quality assessment of single-cell copy-number variations Tyler Garvin, Robert Aboukhalil, Jude Kendall, Timour Baslan, Gurinder S. Atwal, James Hicks, Michael Wigler, Michael C.
Interactive analysis and quality assessment of single-cell copy-number variations Tyler Garvin, Robert Aboukhalil, Jude Kendall, Timour Baslan, Gurinder S. Atwal, James Hicks, Michael Wigler, Michael C.
A three-caller pipeline for variant analysis of cancer whole-exome sequencing data
 MOLECULAR MEDICINE REPORTS 15: 2489-2494, 2017 A three-caller pipeline for variant analysis of cancer whole-exome sequencing data ZE KUN LIU *, YU KUI SHANG *, ZHI NAN CHEN and HUIJIE BIAN Department of
MOLECULAR MEDICINE REPORTS 15: 2489-2494, 2017 A three-caller pipeline for variant analysis of cancer whole-exome sequencing data ZE KUN LIU *, YU KUI SHANG *, ZHI NAN CHEN and HUIJIE BIAN Department of
Supplemental Figure legends
 Supplemental Figure legends Supplemental Figure S1 Frequently mutated genes. Frequently mutated genes (mutated in at least four patients) with information about mutation frequency, RNA-expression and copy-number.
Supplemental Figure legends Supplemental Figure S1 Frequently mutated genes. Frequently mutated genes (mutated in at least four patients) with information about mutation frequency, RNA-expression and copy-number.
PSSV User Manual (V1.0)
") PSSV User Manual (V1.0) 1. Introduction A novel pattern-based probabilistic approach, PSSV, is developed to identify somatic structural variations from WGS data. Specifically, discordant and concordant
PSSV User Manual (V1.0) 1. Introduction A novel pattern-based probabilistic approach, PSSV, is developed to identify somatic structural variations from WGS data. Specifically, discordant and concordant
Next generation diagnostics Bringing high-throughput sequencing into clinical application
 Next generation diagnostics Bringing high-throughput sequencing into clinical application Leonardo A. Meza-Zepeda, PhD Translational Genomics Group Institute for Cancer Research Leonardo.Meza-Zepeda@rr-research.no
Next generation diagnostics Bringing high-throughput sequencing into clinical application Leonardo A. Meza-Zepeda, PhD Translational Genomics Group Institute for Cancer Research Leonardo.Meza-Zepeda@rr-research.no
CITATION FILE CONTENT/FORMAT
 CITATION For any resultant publications using please cite: Matthew A. Field, Vicky Cho, T. Daniel Andrews, and Chris C. Goodnow (2015). "Reliably detecting clinically important variants requires both combined
CITATION For any resultant publications using please cite: Matthew A. Field, Vicky Cho, T. Daniel Andrews, and Chris C. Goodnow (2015). "Reliably detecting clinically important variants requires both combined
Fluxion Biosciences and Swift Biosciences Somatic variant detection from liquid biopsy samples using targeted NGS
 APPLICATION NOTE Fluxion Biosciences and Swift Biosciences OVERVIEW This application note describes a robust method for detecting somatic mutations from liquid biopsy samples by combining circulating tumor
APPLICATION NOTE Fluxion Biosciences and Swift Biosciences OVERVIEW This application note describes a robust method for detecting somatic mutations from liquid biopsy samples by combining circulating tumor