Nature Biotechnology: doi: /nbt.1904
|
|
|
- Brittany Hart
- 5 years ago
- Views:
Transcription
1 Supplementary Information Comparison between assembly-based SV calls and array CGH results Genome-wide array assessment of copy number changes, such as array comparative genomic hybridization (acgh), is widely accepted by the scientific community for copy number variations (CNV) detection as CNVs, in principle, result from structural variation events. Array CGH typically has a resolution at the 10kbp scale without exact breakpoints defined, which does not overlap well with the length spectrum coverage and breakpoint features of our assembly-based method. Nevertheless, it is still interesting to see the comparison between the two technologies. We performed acgh between any two of the three genomes (the YH genome, the anonymous reference genome, and a Promega female sample ( to sort out any putative aberrant copy number changes that are specific to YH genome. In total, 144 CNVs, including 42 multi-probe and 102 single-probe signals were called. Using a reciprocal overlap threshold of 50%, we found 20 (47.6%) multi-probe CNVs and 11 (10.7%) single-probe CNVs were called in acgh had SVs from our assemblybased approaches (Supplementary Dataset). Of note, 19 (61%) out of all 31 overlapping CNVs actually had multiple SV events within their genomic ranges. For example, a copy number loss on chromosome X called by acgh actually involved 60 different deletions and insertion events called from the assembly (Figure S8). This indicates that acgh would not only have ambiguous breakpoints but also aggregates multiple signals into a misleading average result, losing the internal details. Comparison between SV call sets with other studies The HuRef assembly 6 resolved by conventional Sanger sequencing had a better contiguity than both assemblies in our study. Therefore, it would be interesting to see in which SV category we still have a margin for improvement. A comparison between call sets (Table S4a) showed that our assembly-based SVs have a larger reciprocal overlap with HuRef SV calls than those of any other methods, which indicates that our methods have better power to discover SVs. The overlap rate in small indels is much higher than that of large SVs, which could be because: 1) large SVs are more
2 individual-specific than small indels as they confer higher deleterious impact with stronger negative selection; and/or 2) current assembly contiguity needs to be improved to include large SVs within a contig. We also compared our call sets with Pang et al. 7 s call sets from a range of technologies (Table S4b). In the Pang et al. call sets, those called from SR mapping had the highest overlap with our call sets, rather than those called from PEM or arrays. Considering the fact that indels called by the SR method are smaller than those from other methods, this again suggests that large indels are less likely to overlap between individuals than small ones.
3 Supplementary figures Figure S1. A gapped alignment plot to indicate a deletion between NCBI36 Chromosome 1, segment from coordination 246,118,102 to 246,124,262, and YH whole genome de novo assembly scaffold6804, breakpoint at 8,953. Scaffold Chormosome 1

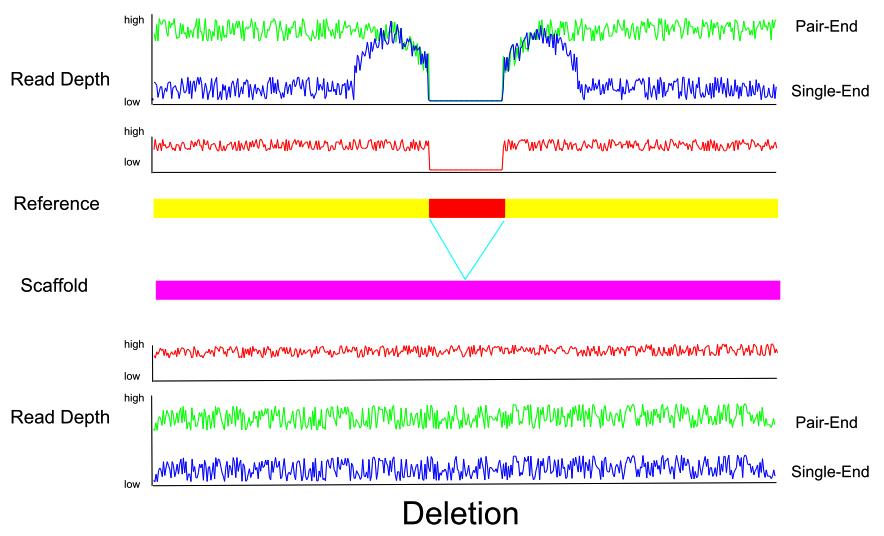
4 Figure S2. Illustration of how read pair and read depth changes on sites of insertion or deletion in reference and assembly. Well-assembled sequence should always achieve a good pair-wise alignment result and read depth since a PE read would be aligned as two single-end reads around indels in a reference and the read depth of deleted regions in the reference should be very low. a. b.
5
 at chromosome 10 with repetitive sequences (gray block) and several other events, including insertions")
6 Figure S3. A case of complex structural variation in YH genome. The figure illustrate a ~22kb inversion (pink line, as a cross between assembly and reference) at chromosome 10 with repetitive sequences (gray block) and several other events, including insertions ranging from 1bp to ~18kbp (green line and block), and deletions (violet line and block) among a hyper-mutation region (with over 10 insertion and deletion events spacing less than 200bp). Read depth (uppermost line chart) and PE reads alignment (medium curve chart) show the difficulty for this SV event to be detected by previous approaches including RD, PEM and SR.

7 Figure S4. The SV distribution of the whole genome and regions with significantly different numbers of SVs between YH and NA18507 genome. Histograms show the number of SVs in a 1-Mb bin on chromosomes. Regions with significant difference between two genomes are marked as purple (YH higher than NA18507) and green (NA18507 higher than YH) on the right of chromosomes.
8 Figure S5. Stacked histogram showing the portion of SVs of different length ranges overlap with unique and repetitive annotated regions in NCBI human reference genome build 36.
9 Figure S6. Venn graph showing the amount of affected gene features among those genes overlapping with SVs. CDS (Green); 3-UTR (Red); 5-UTR (Blue); Intron (Yellow). Numbers indicated are the numbers of genes with one or several gene features affected in YH genome; followed by that of the NA18507 genome. 890:;71&/4!"#$% &"#$% 23/023/ 3&022 20! 20! 330!5 &/0!1 /0/ 507 4/3304!!& & &/5 6/042 '() *+,-.+
10 Figure S7. The frequency of structural variations (x-axis) detected in coding sequences showed a negative correlation with their length (y-axis). Mean length (bp) Frequency
11 Figure S8. Comparison between array CGH signals and assembly-based SV calls showed that acgh signals are averaged from multiple smaller-scaled SV events. Insertion Deletion 91.45Mb 11438bp 38282bp 92.18Mb
12 Supplementary tables Table S1. Primers, sequences of randomly selected structural variations and Sanger capillary sequencing results for PCR validation. Table S1_PCR validation.xls Table S2. (a) Summary of Fosmid sequences validation results. (b) Details including chromosome and coordination of Fosmid sequences validation results. Table S2_Fosmid validation.xls Table S3. Structural variations predicted on the YH and NA18507 genome were, respectively, compared to sets of variants discovered by alternative approaches. Before the slash (/) are the numbers of overlapping variants of NA18507 genome, after are the numbers of overlapping variants of YH genome. Hyphen (-) means not applicable. The criteria FxOy extends x bp as flanking sequence at both sides of the breakpoints of identified variants for comparison, and require the length of the intersection between the validated and the identified variants to overlap by at least y bp of the length of the union of the intervals. DIP 1, small indels found as gaps in the paired-end alignment between the Fosmid end sequences and the reference; ESP 2, large structural variants that were found by analyzing discordant Fosmid clone-end alignment; Three separate sets of structural variants (maximum parsimony structural variation (MPSV) weighted, MPSV unweighted and probabilistic) predicted by Variation Hunter 3 ; MoDIL 4, the set of variants predicted by MoDIL utility. BreakDancer 5, a merged set with variants predicted by BreakDancerMini and BreakDancerMax; The dbsnp version 130 (v130) set refers to homozygous indels that are 30 bp or shorter in dbsnp version 130. The BreakSeq-YRI set refers to predicted variants in NA18507 by BreakSeq and a breakpoints library.
13 Table S3_Computational validation.xls Table S4. Comparison between SVs detected in YH genome, Levy et al. 6 and Pang et al. 7 Table S4_Compare to Levy and Pang.xlsx Table S5. Classification of those strongly conserved (dn/ds 0.1) genes containing SVs. Table S5_gene function.xls Supplementary Dataset Supplementary_aCGH.txt
14 References 1. Bentley, D.R. et al. Accurate whole human genome sequencing using reversible terminator chemistry. Nature 456, 53-9 (2008). 2. Kidd, J.M. et al. Mapping and sequencing of structural variation from eight human genomes. Nature 453, (2008). 3. Hormozdiari, F., Alkan, C., Eichler, E.E. & Sahinalp, S.C. Combinatorial algorithms for structural variation detection in high-throughput sequenced genomes. Genome Res 19, (2009). 4. Lee, S., Hormozdiari, F., Alkan, C. & Brudno, M. MoDIL: detecting small indels from clone-end sequencing with mixtures of distributions. Nat Methods 6, (2009). 5. Chen, K. et al. BreakDancer: an algorithm for high-resolution mapping of genomic structural variation. Nat Methods 6, (2009). 6. Levy, S. et al. The diploid genome sequence of an individual human. PLoS Biol 5, e254 (2007). 7. Pang, A.W. et al. Towards a comprehensive structural variation map of an individual human genome. Genome Biol 11, R52 (2010).
Genomic structural variation
 Genomic structural variation Mario Cáceres The new genomic variation DNA sequence differs across individuals much more than researchers had suspected through structural changes A huge amount of structural
Genomic structural variation Mario Cáceres The new genomic variation DNA sequence differs across individuals much more than researchers had suspected through structural changes A huge amount of structural
Structural Variation and Medical Genomics
 Structural Variation and Medical Genomics Andrew King Department of Biomedical Informatics July 8, 2014 You already know about small scale genetic mutations Single nucleotide polymorphism (SNPs) Deletions,
Structural Variation and Medical Genomics Andrew King Department of Biomedical Informatics July 8, 2014 You already know about small scale genetic mutations Single nucleotide polymorphism (SNPs) Deletions,
Supplementary note: Comparison of deletion variants identified in this study and four earlier studies
 Supplementary note: Comparison of deletion variants identified in this study and four earlier studies Here we compare the results of this study to potentially overlapping results from four earlier studies
Supplementary note: Comparison of deletion variants identified in this study and four earlier studies Here we compare the results of this study to potentially overlapping results from four earlier studies
Dr Rick Tearle Senior Applications Specialist, EMEA Complete Genomics Complete Genomics, Inc.
 Dr Rick Tearle Senior Applications Specialist, EMEA Complete Genomics Topics Overview of Data Processing Pipeline Overview of Data Files 2 DNA Nano-Ball (DNB) Read Structure Genome : acgtacatgcattcacacatgcttagctatctctcgccag
Dr Rick Tearle Senior Applications Specialist, EMEA Complete Genomics Topics Overview of Data Processing Pipeline Overview of Data Files 2 DNA Nano-Ball (DNB) Read Structure Genome : acgtacatgcattcacacatgcttagctatctctcgccag
CNV detection. Introduction and detection in NGS data. G. Demidov 1,2. NGSchool2016. Centre for Genomic Regulation. CNV detection. G.
 Introduction and detection in NGS data 1,2 1 Genomic and Epigenomic Variation in Disease group, Centre for Genomic Regulation 2 Universitat Pompeu Fabra NGSchool2016 methods: methods Outline methods: methods
Introduction and detection in NGS data 1,2 1 Genomic and Epigenomic Variation in Disease group, Centre for Genomic Regulation 2 Universitat Pompeu Fabra NGSchool2016 methods: methods Outline methods: methods
CNV Detection and Interpretation in Genomic Data
 CNV Detection and Interpretation in Genomic Data Benjamin W. Darbro, M.D., Ph.D. Assistant Professor of Pediatrics Director of the Shivanand R. Patil Cytogenetics and Molecular Laboratory Overview What
CNV Detection and Interpretation in Genomic Data Benjamin W. Darbro, M.D., Ph.D. Assistant Professor of Pediatrics Director of the Shivanand R. Patil Cytogenetics and Molecular Laboratory Overview What
Interactive analysis and quality assessment of single-cell copy-number variations
 Interactive analysis and quality assessment of single-cell copy-number variations Tyler Garvin, Robert Aboukhalil, Jude Kendall, Timour Baslan, Gurinder S. Atwal, James Hicks, Michael Wigler, Michael C.
Interactive analysis and quality assessment of single-cell copy-number variations Tyler Garvin, Robert Aboukhalil, Jude Kendall, Timour Baslan, Gurinder S. Atwal, James Hicks, Michael Wigler, Michael C.
Supplementary Figure 1. Spitzoid Melanoma with PPFIBP1-MET fusion. (a) Histopathology (4x) shows a domed papule with melanocytes extending into the
 Histopathology (4x) shows a domed papule with melanocytes extending into the") Supplementary Figure 1. Spitzoid Melanoma with PPFIBP1-MET fusion. (a) Histopathology (4x) shows a domed papule with melanocytes extending into the deep dermis. (b) The melanocytes demonstrate abundant
Supplementary Figure 1. Spitzoid Melanoma with PPFIBP1-MET fusion. (a) Histopathology (4x) shows a domed papule with melanocytes extending into the deep dermis. (b) The melanocytes demonstrate abundant
Nature Genetics: doi: /ng Supplementary Figure 1. PCA for ancestry in SNV data.
 Supplementary Figure 1 PCA for ancestry in SNV data. (a) EIGENSTRAT principal-component analysis (PCA) of SNV genotype data on all samples. (b) PCA of only proband SNV genotype data. (c) PCA of SNV genotype
Supplementary Figure 1 PCA for ancestry in SNV data. (a) EIGENSTRAT principal-component analysis (PCA) of SNV genotype data on all samples. (b) PCA of only proband SNV genotype data. (c) PCA of SNV genotype
Figure S2. Distribution of acgh probes on all ten chromosomes of the RIL M0022
 96 APPENDIX B. Supporting Information for chapter 4 "changes in genome content generated via segregation of non-allelic homologs" Figure S1. Potential de novo CNV probes and sizes of apparently de novo
96 APPENDIX B. Supporting Information for chapter 4 "changes in genome content generated via segregation of non-allelic homologs" Figure S1. Potential de novo CNV probes and sizes of apparently de novo
Global variation in copy number in the human genome
 Global variation in copy number in the human genome Redon et. al. Nature 444:444-454 (2006) 12.03.2007 Tarmo Puurand Study 270 individuals (HapMap collection) Affymetrix 500K Whole Genome TilePath (WGTP)
Global variation in copy number in the human genome Redon et. al. Nature 444:444-454 (2006) 12.03.2007 Tarmo Puurand Study 270 individuals (HapMap collection) Affymetrix 500K Whole Genome TilePath (WGTP)
Victor Guryev. European Research Institute for the Biology of Ageing
 Victor Guryev European Research Institute for the Biology of Ageing September 29, 2014 Genomic resequencing in Medical diagnostics course Erasmus MC, Rotterdam /a /g Low coverage whole genome and deep
Victor Guryev European Research Institute for the Biology of Ageing September 29, 2014 Genomic resequencing in Medical diagnostics course Erasmus MC, Rotterdam /a /g Low coverage whole genome and deep
SUPPLEMENTARY INFORMATION
 Supplementary methods Genome Sequencing Paired-end and singleton 36 nucleotide-long reads were generated using Illumina GA and GAII instruments using standard protocols 1. Sequences were retained if of
Supplementary methods Genome Sequencing Paired-end and singleton 36 nucleotide-long reads were generated using Illumina GA and GAII instruments using standard protocols 1. Sequences were retained if of
DNA-seq Bioinformatics Analysis: Copy Number Variation
 DNA-seq Bioinformatics Analysis: Copy Number Variation Elodie Girard elodie.girard@curie.fr U900 institut Curie, INSERM, Mines ParisTech, PSL Research University Paris, France NGS Applications 5C HiC DNA-seq
DNA-seq Bioinformatics Analysis: Copy Number Variation Elodie Girard elodie.girard@curie.fr U900 institut Curie, INSERM, Mines ParisTech, PSL Research University Paris, France NGS Applications 5C HiC DNA-seq
Cytogenetics 101: Clinical Research and Molecular Genetic Technologies
 Cytogenetics 101: Clinical Research and Molecular Genetic Technologies Topics for Today s Presentation 1 Classical vs Molecular Cytogenetics 2 What acgh? 3 What is FISH? 4 What is NGS? 5 How can these
Cytogenetics 101: Clinical Research and Molecular Genetic Technologies Topics for Today s Presentation 1 Classical vs Molecular Cytogenetics 2 What acgh? 3 What is FISH? 4 What is NGS? 5 How can these
Applications of Chromosomal Microarray Analysis (CMA) in pre- and postnatal Diagnostic: advantages, limitations and concerns
 in pre- and postnatal Diagnostic: advantages, limitations and concerns") Applications of Chromosomal Microarray Analysis (CMA) in pre- and postnatal Diagnostic: advantages, limitations and concerns جواد کریمزاد حق PhD of Medical Genetics آزمايشگاه پاتوبيولوژي و ژنتيك پارسه
Applications of Chromosomal Microarray Analysis (CMA) in pre- and postnatal Diagnostic: advantages, limitations and concerns جواد کریمزاد حق PhD of Medical Genetics آزمايشگاه پاتوبيولوژي و ژنتيك پارسه
Agilent s Copy Number Variation (CNV) Portfolio
 Portfolio") Technical Overview Agilent s Copy Number Variation (CNV) Portfolio Abstract Copy Number Variation (CNV) is now recognized as a prevalent form of structural variation in the genome contributing to human
Technical Overview Agilent s Copy Number Variation (CNV) Portfolio Abstract Copy Number Variation (CNV) is now recognized as a prevalent form of structural variation in the genome contributing to human
Illuminating the genetics of complex human diseases
 Illuminating the genetics of complex human diseases Michael Schatz Sept 27, 2012 Beyond the Genome @mike_schatz / #BTG2012 Outline 1. De novo mutations in human diseases 1. Autism Spectrum Disorder 2.
Illuminating the genetics of complex human diseases Michael Schatz Sept 27, 2012 Beyond the Genome @mike_schatz / #BTG2012 Outline 1. De novo mutations in human diseases 1. Autism Spectrum Disorder 2.
Abstract. Optimization strategy of Copy Number Variant calling using Multiplicom solutions APPLICATION NOTE. Introduction
 Optimization strategy of Copy Number Variant calling using Multiplicom solutions Michael Vyverman, PhD; Laura Standaert, PhD and Wouter Bossuyt, PhD Abstract Copy number variations (CNVs) represent a significant
Optimization strategy of Copy Number Variant calling using Multiplicom solutions Michael Vyverman, PhD; Laura Standaert, PhD and Wouter Bossuyt, PhD Abstract Copy number variations (CNVs) represent a significant
Mutation Detection and CNV Analysis for Illumina Sequencing data from HaloPlex Target Enrichment Panels using NextGENe Software for Clinical Research
 Mutation Detection and CNV Analysis for Illumina Sequencing data from HaloPlex Target Enrichment Panels using NextGENe Software for Clinical Research Application Note Authors John McGuigan, Megan Manion,
Mutation Detection and CNV Analysis for Illumina Sequencing data from HaloPlex Target Enrichment Panels using NextGENe Software for Clinical Research Application Note Authors John McGuigan, Megan Manion,
Whole Genome and Transcriptome Analysis of Anaplastic Meningioma. Patrick Tarpey Cancer Genome Project Wellcome Trust Sanger Institute
 Whole Genome and Transcriptome Analysis of Anaplastic Meningioma Patrick Tarpey Cancer Genome Project Wellcome Trust Sanger Institute Outline Anaplastic meningioma compared to other cancers Whole genomes
Whole Genome and Transcriptome Analysis of Anaplastic Meningioma Patrick Tarpey Cancer Genome Project Wellcome Trust Sanger Institute Outline Anaplastic meningioma compared to other cancers Whole genomes
Global assessment of genomic variation in cattle by genome resequencing and high-throughput genotyping
 RESEARCH ARTICLE Open Access Global assessment of genomic variation in cattle by genome resequencing and high-throughput genotyping Bujie Zhan 1, João Fadista 1, Bo Thomsen 1, Jakob Hedegaard 1,2, Frank
RESEARCH ARTICLE Open Access Global assessment of genomic variation in cattle by genome resequencing and high-throughput genotyping Bujie Zhan 1, João Fadista 1, Bo Thomsen 1, Jakob Hedegaard 1,2, Frank
Below, we included the point-to-point response to the comments of both reviewers.
 To the Editor and Reviewers: We would like to thank the editor and reviewers for careful reading, and constructive suggestions for our manuscript. According to comments from both reviewers, we have comprehensively
To the Editor and Reviewers: We would like to thank the editor and reviewers for careful reading, and constructive suggestions for our manuscript. According to comments from both reviewers, we have comprehensively
Generating Spontaneous Copy Number Variants (CNVs) Jennifer Freeman Assistant Professor of Toxicology School of Health Sciences Purdue University
 Jennifer Freeman Assistant Professor of Toxicology School of Health Sciences Purdue University") Role of Chemical lexposure in Generating Spontaneous Copy Number Variants (CNVs) Jennifer Freeman Assistant Professor of Toxicology School of Health Sciences Purdue University CNV Discovery Reference Genetic
Role of Chemical lexposure in Generating Spontaneous Copy Number Variants (CNVs) Jennifer Freeman Assistant Professor of Toxicology School of Health Sciences Purdue University CNV Discovery Reference Genetic
CHROMOSOMAL MICROARRAY (CGH+SNP)
") Chromosome imbalances are a significant cause of developmental delay, mental retardation, autism spectrum disorders, dysmorphic features and/or birth defects. The imbalance of genetic material may be due
Chromosome imbalances are a significant cause of developmental delay, mental retardation, autism spectrum disorders, dysmorphic features and/or birth defects. The imbalance of genetic material may be due
Supplementary Figure 1. Estimation of tumour content
 Supplementary Figure 1. Estimation of tumour content a, Approach used to estimate the tumour content in S13T1/T2, S6T1/T2, S3T1/T2 and S12T1/T2. Tissue and tumour areas were evaluated by two independent
Supplementary Figure 1. Estimation of tumour content a, Approach used to estimate the tumour content in S13T1/T2, S6T1/T2, S3T1/T2 and S12T1/T2. Tissue and tumour areas were evaluated by two independent
SVIM: Structural variant identification with long reads DAVID HELLER MAX PLANCK INSTITUTE FOR MOLECULAR GENETICS, BERLIN JUNE 2O18, SMRT LEIDEN
 SVIM: Structural variant identification with long reads DAVID HELLER MAX PLANCK INSTITUTE FOR MOLECULAR GENETICS, BERLIN JUNE 2O18, SMRT LEIDEN Structural variation (SV) Variants larger than 50bps Affect
SVIM: Structural variant identification with long reads DAVID HELLER MAX PLANCK INSTITUTE FOR MOLECULAR GENETICS, BERLIN JUNE 2O18, SMRT LEIDEN Structural variation (SV) Variants larger than 50bps Affect
PSSV User Manual (V2.1)
") PSSV User Manual (V2.1) 1. Introduction A novel pattern-based probabilistic approach, PSSV, is developed to identify somatic structural variations from WGS data. Specifically, discordant and concordant
PSSV User Manual (V2.1) 1. Introduction A novel pattern-based probabilistic approach, PSSV, is developed to identify somatic structural variations from WGS data. Specifically, discordant and concordant
Using the Bravo Liquid-Handling System for Next Generation Sequencing Sample Prep
 Using the Bravo Liquid-Handling System for Next Generation Sequencing Sample Prep Tom Walsh, PhD Division of Medical Genetics University of Washington Next generation sequencing Sanger sequencing gold
Using the Bravo Liquid-Handling System for Next Generation Sequencing Sample Prep Tom Walsh, PhD Division of Medical Genetics University of Washington Next generation sequencing Sanger sequencing gold
Shape-based retrieval of CNV regions in read coverage data. Sangkyun Hong and Jeehee Yoon*
 254 Int. J. Data Mining and Bioinformatics, Vol. 9, No. 3, 2014 Shape-based retrieval of CNV regions in read coverage data Sangkyun Hong and Jeehee Yoon* Department of Computer Engineering, Hallym University
254 Int. J. Data Mining and Bioinformatics, Vol. 9, No. 3, 2014 Shape-based retrieval of CNV regions in read coverage data Sangkyun Hong and Jeehee Yoon* Department of Computer Engineering, Hallym University
Variant Classification. Author: Mike Thiesen, Golden Helix, Inc.
 Variant Classification Author: Mike Thiesen, Golden Helix, Inc. Overview Sequencing pipelines are able to identify rare variants not found in catalogs such as dbsnp. As a result, variants in these datasets
Variant Classification Author: Mike Thiesen, Golden Helix, Inc. Overview Sequencing pipelines are able to identify rare variants not found in catalogs such as dbsnp. As a result, variants in these datasets
LTA Analysis of HapMap Genotype Data
 LTA Analysis of HapMap Genotype Data Introduction. This supplement to Global variation in copy number in the human genome, by Redon et al., describes the details of the LTA analysis used to screen HapMap
LTA Analysis of HapMap Genotype Data Introduction. This supplement to Global variation in copy number in the human genome, by Redon et al., describes the details of the LTA analysis used to screen HapMap
Ambient temperature regulated flowering time
 Ambient temperature regulated flowering time Applications of RNAseq RNA- seq course: The power of RNA-seq June 7 th, 2013; Richard Immink Overview Introduction: Biological research question/hypothesis
Ambient temperature regulated flowering time Applications of RNAseq RNA- seq course: The power of RNA-seq June 7 th, 2013; Richard Immink Overview Introduction: Biological research question/hypothesis
No mutations were identified.
 Hereditary High Cholesterol Test ORDERING PHYSICIAN PRIMARY CONTACT SPECIMEN Report date: Aug 1, 2017 Dr. Jenny Jones Sample Medical Group 123 Main St. Sample, CA Kelly Peters Sample Medical Group 123
Hereditary High Cholesterol Test ORDERING PHYSICIAN PRIMARY CONTACT SPECIMEN Report date: Aug 1, 2017 Dr. Jenny Jones Sample Medical Group 123 Main St. Sample, CA Kelly Peters Sample Medical Group 123
Supplementary Figure 1. Schematic diagram of o2n-seq. Double-stranded DNA was sheared, end-repaired, and underwent A-tailing by standard protocols.
 Supplementary Figure 1. Schematic diagram of o2n-seq. Double-stranded DNA was sheared, end-repaired, and underwent A-tailing by standard protocols. A-tailed DNA was ligated to T-tailed dutp adapters, circularized
Supplementary Figure 1. Schematic diagram of o2n-seq. Double-stranded DNA was sheared, end-repaired, and underwent A-tailing by standard protocols. A-tailed DNA was ligated to T-tailed dutp adapters, circularized
Supplementary Appendix
 Supplementary Appendix This appendix has been provided by the authors to give readers additional information about their work. Supplement to: Yatsenko AN, Georgiadis AP, Röpke A, et al. X-linked TEX11
Supplementary Appendix This appendix has been provided by the authors to give readers additional information about their work. Supplement to: Yatsenko AN, Georgiadis AP, Röpke A, et al. X-linked TEX11
Mosaic loss of chromosome Y in peripheral blood is associated with shorter survival and higher risk of cancer
 Supplementary Information Mosaic loss of chromosome Y in peripheral blood is associated with shorter survival and higher risk of cancer Lars A. Forsberg, Chiara Rasi, Niklas Malmqvist, Hanna Davies, Saichand
Supplementary Information Mosaic loss of chromosome Y in peripheral blood is associated with shorter survival and higher risk of cancer Lars A. Forsberg, Chiara Rasi, Niklas Malmqvist, Hanna Davies, Saichand
Vega: Variational Segmentation for Copy Number Detection
 Vega: Variational Segmentation for Copy Number Detection Sandro Morganella Luigi Cerulo Giuseppe Viglietto Michele Ceccarelli Contents 1 Overview 1 2 Installation 1 3 Vega.RData Description 2 4 Run Vega
Vega: Variational Segmentation for Copy Number Detection Sandro Morganella Luigi Cerulo Giuseppe Viglietto Michele Ceccarelli Contents 1 Overview 1 2 Installation 1 3 Vega.RData Description 2 4 Run Vega
BWA alignment to reference transcriptome and genome. Convert transcriptome mappings back to genome space
 Whole genome sequencing Whole exome sequencing BWA alignment to reference transcriptome and genome Convert transcriptome mappings back to genome space genomes Filter on MQ, distance, Cigar string Annotate
Whole genome sequencing Whole exome sequencing BWA alignment to reference transcriptome and genome Convert transcriptome mappings back to genome space genomes Filter on MQ, distance, Cigar string Annotate
Nature Genetics: doi: /ng Supplementary Figure 1
 Supplementary Figure 1 Multiple samples from five patients (P4, P8, P14, P15 and P17) with Barrett s esophagus and adjacent EAC show that the poor overlap is not a result of sampling bias. Bar graphs showing
Supplementary Figure 1 Multiple samples from five patients (P4, P8, P14, P15 and P17) with Barrett s esophagus and adjacent EAC show that the poor overlap is not a result of sampling bias. Bar graphs showing
Introduction. 8 These authors contributed equally to this work
 ARTICLE Complete Haplotype Sequence of the Human Immunoglobulin Heavy-Chain Variable, Diversity, and Joining Genes and Characterization of Allelic and Copy-Number Variation Corey T. Watson, 1,7 Karyn M.
ARTICLE Complete Haplotype Sequence of the Human Immunoglobulin Heavy-Chain Variable, Diversity, and Joining Genes and Characterization of Allelic and Copy-Number Variation Corey T. Watson, 1,7 Karyn M.
Challenges of CGH array testing in children with developmental delay. Dr Sally Davies 17 th September 2014
 Challenges of CGH array testing in children with developmental delay Dr Sally Davies 17 th September 2014 CGH array What is CGH array? Understanding the test Benefits Results to expect Consent issues Ethical
Challenges of CGH array testing in children with developmental delay Dr Sally Davies 17 th September 2014 CGH array What is CGH array? Understanding the test Benefits Results to expect Consent issues Ethical
PSSV User Manual (V1.0)
") PSSV User Manual (V1.0) 1. Introduction A novel pattern-based probabilistic approach, PSSV, is developed to identify somatic structural variations from WGS data. Specifically, discordant and concordant
PSSV User Manual (V1.0) 1. Introduction A novel pattern-based probabilistic approach, PSSV, is developed to identify somatic structural variations from WGS data. Specifically, discordant and concordant
Copy number variation detection and genotyping from exome sequence data
 Method Copy number variation detection and genotyping from exome sequence data Niklas Krumm, 1 Peter H. Sudmant, 1 Arthur Ko, 1 Brian J. O Roak, 1 Maika Malig, 1 Bradley P. Coe, 1 NHLBI Exome Sequencing
Method Copy number variation detection and genotyping from exome sequence data Niklas Krumm, 1 Peter H. Sudmant, 1 Arthur Ko, 1 Brian J. O Roak, 1 Maika Malig, 1 Bradley P. Coe, 1 NHLBI Exome Sequencing
Identification of regions with common copy-number variations using SNP array
 Identification of regions with common copy-number variations using SNP array Agus Salim Epidemiology and Public Health National University of Singapore Copy Number Variation (CNV) Copy number alteration
Identification of regions with common copy-number variations using SNP array Agus Salim Epidemiology and Public Health National University of Singapore Copy Number Variation (CNV) Copy number alteration
Introduction to genetic variation. He Zhang Bioinformatics Core Facility 6/22/2016
 Introduction to genetic variation He Zhang Bioinformatics Core Facility 6/22/2016 Outline Basic concepts of genetic variation Genetic variation in human populations Variation and genetic disorders Databases
Introduction to genetic variation He Zhang Bioinformatics Core Facility 6/22/2016 Outline Basic concepts of genetic variation Genetic variation in human populations Variation and genetic disorders Databases
p.r623c p.p976l p.d2847fs p.t2671 p.d2847fs p.r2922w p.r2370h p.c1201y p.a868v p.s952* RING_C BP PHD Cbp HAT_KAT11
 ARID2 p.r623c KMT2D p.v650fs p.p976l p.r2922w p.l1212r p.d1400h DNA binding RFX DNA binding Zinc finger KMT2C p.a51s p.d372v p.c1103* p.d2847fs p.t2671 p.d2847fs p.r4586h PHD/ RING DHHC/ PHD PHD FYR N
ARID2 p.r623c KMT2D p.v650fs p.p976l p.r2922w p.l1212r p.d1400h DNA binding RFX DNA binding Zinc finger KMT2C p.a51s p.d372v p.c1103* p.d2847fs p.t2671 p.d2847fs p.r4586h PHD/ RING DHHC/ PHD PHD FYR N
A Multi-Sample Based Method for Identifying Common CNVs in Normal Human Genomic Structure Using High- Resolution acgh Data
 A Multi-Sample Based Method for Identifying Common CNVs in Normal Human Genomic Structure Using High- Resolution acgh Data Chihyun Park 1, Jaegyoon Ahn 1, Youngmi Yoon 2, Sanghyun Park 1 * 1 Department
A Multi-Sample Based Method for Identifying Common CNVs in Normal Human Genomic Structure Using High- Resolution acgh Data Chihyun Park 1, Jaegyoon Ahn 1, Youngmi Yoon 2, Sanghyun Park 1 * 1 Department
cn.mops - Mixture of Poissons for CNV detection in NGS data Günter Klambauer Institute of Bioinformatics, Johannes Kepler University Linz
 Software Manual Institute of Bioinformatics, Johannes Kepler University Linz cn.mops - Mixture of Poissons for CNV detection in NGS data Günter Klambauer Institute of Bioinformatics, Johannes Kepler University
Software Manual Institute of Bioinformatics, Johannes Kepler University Linz cn.mops - Mixture of Poissons for CNV detection in NGS data Günter Klambauer Institute of Bioinformatics, Johannes Kepler University
Evaluation of MIA FORA NGS HLA test and software. Lisa Creary, PhD Department of Pathology Stanford Blood Center Research & Development Group
 Evaluation of MIA FORA NGS HLA test and software Lisa Creary, PhD Department of Pathology Stanford Blood Center Research & Development Group Disclosure Alpha and Beta Studies Sirona Genomics Reagents,
Evaluation of MIA FORA NGS HLA test and software Lisa Creary, PhD Department of Pathology Stanford Blood Center Research & Development Group Disclosure Alpha and Beta Studies Sirona Genomics Reagents,
2/10/2016. Evaluation of MIA FORA NGS HLA test and software. Disclosure. NGS-HLA typing requirements for the Stanford Blood Center
 Evaluation of MIA FORA NGS HLA test and software Lisa Creary, PhD Department of Pathology Stanford Blood Center Research & Development Group Disclosure Alpha and Beta Studies Sirona Genomics Reagents,
Evaluation of MIA FORA NGS HLA test and software Lisa Creary, PhD Department of Pathology Stanford Blood Center Research & Development Group Disclosure Alpha and Beta Studies Sirona Genomics Reagents,
Detection of aneuploidy in a single cell using the Ion ReproSeq PGS View Kit
 APPLICATION NOTE Ion PGM System Detection of aneuploidy in a single cell using the Ion ReproSeq PGS View Kit Key findings The Ion PGM System, in concert with the Ion ReproSeq PGS View Kit and Ion Reporter
APPLICATION NOTE Ion PGM System Detection of aneuploidy in a single cell using the Ion ReproSeq PGS View Kit Key findings The Ion PGM System, in concert with the Ion ReproSeq PGS View Kit and Ion Reporter
Lentiviral Delivery of Combinatorial mirna Expression Constructs Provides Efficient Target Gene Repression.
 Supplementary Figure 1 Lentiviral Delivery of Combinatorial mirna Expression Constructs Provides Efficient Target Gene Repression. a, Design for lentiviral combinatorial mirna expression and sensor constructs.
Supplementary Figure 1 Lentiviral Delivery of Combinatorial mirna Expression Constructs Provides Efficient Target Gene Repression. a, Design for lentiviral combinatorial mirna expression and sensor constructs.
Characterisation of structural variation in breast. cancer genomes using paired-end sequencing on. the Illumina Genome Analyser
 Characterisation of structural variation in breast cancer genomes using paired-end sequencing on the Illumina Genome Analyser Phil Stephens Cancer Genome Project Why is it important to study cancer? Why
Characterisation of structural variation in breast cancer genomes using paired-end sequencing on the Illumina Genome Analyser Phil Stephens Cancer Genome Project Why is it important to study cancer? Why
of TERT, MLL4, CCNE1, SENP5, and ROCK1 on tumor development were discussed.
 Supplementary Note The potential association and implications of HBV integration at known and putative cancer genes of TERT, MLL4, CCNE1, SENP5, and ROCK1 on tumor development were discussed. Human telomerase
Supplementary Note The potential association and implications of HBV integration at known and putative cancer genes of TERT, MLL4, CCNE1, SENP5, and ROCK1 on tumor development were discussed. Human telomerase
November 9, Johns Hopkins School of Medicine, Baltimore, MD,
 Fast detection of de-novo copy number variants from case-parent SNP arrays identifies a deletion on chromosome 7p14.1 associated with non-syndromic isolated cleft lip/palate Samuel G. Younkin 1, Robert
Fast detection of de-novo copy number variants from case-parent SNP arrays identifies a deletion on chromosome 7p14.1 associated with non-syndromic isolated cleft lip/palate Samuel G. Younkin 1, Robert
Table S1. Relative abundance of AGO1/4 proteins in different organs. Table S2. Summary of smrna datasets from various samples.
 Supplementary files Table S1. Relative abundance of AGO1/4 proteins in different organs. Table S2. Summary of smrna datasets from various samples. Table S3. Specificity of AGO1- and AGO4-preferred 24-nt
Supplementary files Table S1. Relative abundance of AGO1/4 proteins in different organs. Table S2. Summary of smrna datasets from various samples. Table S3. Specificity of AGO1- and AGO4-preferred 24-nt
Supplementary Information. Supplementary Figures
 Supplementary Information Supplementary Figures.8 57 essential gene density 2 1.5 LTR insert frequency diversity DEL.5 DUP.5 INV.5 TRA 1 2 3 4 5 1 2 3 4 1 2 Supplementary Figure 1. Locations and minor
Supplementary Information Supplementary Figures.8 57 essential gene density 2 1.5 LTR insert frequency diversity DEL.5 DUP.5 INV.5 TRA 1 2 3 4 5 1 2 3 4 1 2 Supplementary Figure 1. Locations and minor
AVENIO family of NGS oncology assays ctdna and Tumor Tissue Analysis Kits
 AVENIO family of NGS oncology assays ctdna and Tumor Tissue Analysis Kits Accelerating clinical research Next-generation sequencing (NGS) has the ability to interrogate many different genes and detect
AVENIO family of NGS oncology assays ctdna and Tumor Tissue Analysis Kits Accelerating clinical research Next-generation sequencing (NGS) has the ability to interrogate many different genes and detect
Analysis with SureCall 2.1
 Analysis with SureCall 2.1 Danielle Fletcher Field Application Scientist July 2014 1 Stages of NGS Analysis Primary analysis, base calling Control Software FASTQ file reads + quality 2 Stages of NGS Analysis
Analysis with SureCall 2.1 Danielle Fletcher Field Application Scientist July 2014 1 Stages of NGS Analysis Primary analysis, base calling Control Software FASTQ file reads + quality 2 Stages of NGS Analysis
Multiplex target enrichment using DNA indexing for ultra-high throughput variant detection
 Multiplex target enrichment using DNA indexing for ultra-high throughput variant detection Dr Elaine Kenny Neuropsychiatric Genetics Research Group Institute of Molecular Medicine Trinity College Dublin
Multiplex target enrichment using DNA indexing for ultra-high throughput variant detection Dr Elaine Kenny Neuropsychiatric Genetics Research Group Institute of Molecular Medicine Trinity College Dublin
Nature Genetics: doi: /ng Supplementary Figure 1. Somatic coding mutations identified by WES/WGS for 83 ATL cases.
 Supplementary Figure 1 Somatic coding mutations identified by WES/WGS for 83 ATL cases. (a) The percentage of targeted bases covered by at least 2, 10, 20 and 30 sequencing reads (top) and average read
Supplementary Figure 1 Somatic coding mutations identified by WES/WGS for 83 ATL cases. (a) The percentage of targeted bases covered by at least 2, 10, 20 and 30 sequencing reads (top) and average read
Understanding DNA Copy Number Data
 Understanding DNA Copy Number Data Adam B. Olshen Department of Epidemiology and Biostatistics Helen Diller Family Comprehensive Cancer Center University of California, San Francisco http://cc.ucsf.edu/people/olshena_adam.php
Understanding DNA Copy Number Data Adam B. Olshen Department of Epidemiology and Biostatistics Helen Diller Family Comprehensive Cancer Center University of California, San Francisco http://cc.ucsf.edu/people/olshena_adam.php
Computational Identification and Prediction of Tissue-Specific Alternative Splicing in H. Sapiens. Eric Van Nostrand CS229 Final Project
 Computational Identification and Prediction of Tissue-Specific Alternative Splicing in H. Sapiens. Eric Van Nostrand CS229 Final Project Introduction RNA splicing is a critical step in eukaryotic gene
Computational Identification and Prediction of Tissue-Specific Alternative Splicing in H. Sapiens. Eric Van Nostrand CS229 Final Project Introduction RNA splicing is a critical step in eukaryotic gene
Hands-On Ten The BRCA1 Gene and Protein
 Hands-On Ten The BRCA1 Gene and Protein Objective: To review transcription, translation, reading frames, mutations, and reading files from GenBank, and to review some of the bioinformatics tools, such
Hands-On Ten The BRCA1 Gene and Protein Objective: To review transcription, translation, reading frames, mutations, and reading files from GenBank, and to review some of the bioinformatics tools, such
Multimarker Genetic Analysis Methods for High Throughput Array Data
 Multimarker Genetic Analysis Methods for High Throughput Array Data by Iuliana Ionita A dissertation submitted in partial fulfillment of the requirements for the degree of Doctor of Philosophy Department
Multimarker Genetic Analysis Methods for High Throughput Array Data by Iuliana Ionita A dissertation submitted in partial fulfillment of the requirements for the degree of Doctor of Philosophy Department
Nature Genetics: doi: /ng Supplementary Figure 1. Rates of different mutation types in CRC.
 Supplementary Figure 1 Rates of different mutation types in CRC. (a) Stratification by mutation type indicates that C>T mutations occur at a significantly greater rate than other types. (b) As for the
Supplementary Figure 1 Rates of different mutation types in CRC. (a) Stratification by mutation type indicates that C>T mutations occur at a significantly greater rate than other types. (b) As for the
MEDICAL GENOMICS LABORATORY. Next-Gen Sequencing and Deletion/Duplication Analysis of NF1 Only (NF1-NG)
") Next-Gen Sequencing and Deletion/Duplication Analysis of NF1 Only (NF1-NG) Ordering Information Acceptable specimen types: Fresh blood sample (3-6 ml EDTA; no time limitations associated with receipt)
Next-Gen Sequencing and Deletion/Duplication Analysis of NF1 Only (NF1-NG) Ordering Information Acceptable specimen types: Fresh blood sample (3-6 ml EDTA; no time limitations associated with receipt)
AVENIO ctdna Analysis Kits The complete NGS liquid biopsy solution EMPOWER YOUR LAB
 Analysis Kits The complete NGS liquid biopsy solution EMPOWER YOUR LAB Analysis Kits Next-generation performance in liquid biopsies 2 Accelerating clinical research From liquid biopsy to next-generation
Analysis Kits The complete NGS liquid biopsy solution EMPOWER YOUR LAB Analysis Kits Next-generation performance in liquid biopsies 2 Accelerating clinical research From liquid biopsy to next-generation
Multiple Copy Number Variations in a Patient with Developmental Delay ASCLS- March 31, 2016
 Multiple Copy Number Variations in a Patient with Developmental Delay ASCLS- March 31, 2016 Marwan Tayeh, PhD, FACMG Director, MMGL Molecular Genetics Assistant Professor of Pediatrics Department of Pediatrics
Multiple Copy Number Variations in a Patient with Developmental Delay ASCLS- March 31, 2016 Marwan Tayeh, PhD, FACMG Director, MMGL Molecular Genetics Assistant Professor of Pediatrics Department of Pediatrics
Introduction to LOH and Allele Specific Copy Number User Forum
 Introduction to LOH and Allele Specific Copy Number User Forum Jonathan Gerstenhaber Introduction to LOH and ASCN User Forum Contents 1. Loss of heterozygosity Analysis procedure Types of baselines 2.
Introduction to LOH and Allele Specific Copy Number User Forum Jonathan Gerstenhaber Introduction to LOH and ASCN User Forum Contents 1. Loss of heterozygosity Analysis procedure Types of baselines 2.
MIR retrotransposon sequences provide insulators to the human genome
 Supplementary Information: MIR retrotransposon sequences provide insulators to the human genome Jianrong Wang, Cristina Vicente-García, Davide Seruggia, Eduardo Moltó, Ana Fernandez- Miñán, Ana Neto, Elbert
Supplementary Information: MIR retrotransposon sequences provide insulators to the human genome Jianrong Wang, Cristina Vicente-García, Davide Seruggia, Eduardo Moltó, Ana Fernandez- Miñán, Ana Neto, Elbert
Genome-Wide Analysis of Copy Number Variations in Normal Population Identified by SNP Arrays
 54 The Open Biology Journal, 2009, 2, 54-65 Open Access Genome-Wide Analysis of Copy Number Variations in Normal Population Identified by SNP Arrays Jian Wang 1,2, Tsz-Kwong Man 1,3,4, Kwong Kwok Wong
54 The Open Biology Journal, 2009, 2, 54-65 Open Access Genome-Wide Analysis of Copy Number Variations in Normal Population Identified by SNP Arrays Jian Wang 1,2, Tsz-Kwong Man 1,3,4, Kwong Kwok Wong
RASA: Robust Alternative Splicing Analysis for Human Transcriptome Arrays
 Supplementary Materials RASA: Robust Alternative Splicing Analysis for Human Transcriptome Arrays Junhee Seok 1*, Weihong Xu 2, Ronald W. Davis 2, Wenzhong Xiao 2,3* 1 School of Electrical Engineering,
Supplementary Materials RASA: Robust Alternative Splicing Analysis for Human Transcriptome Arrays Junhee Seok 1*, Weihong Xu 2, Ronald W. Davis 2, Wenzhong Xiao 2,3* 1 School of Electrical Engineering,
Comprehensive Chromosome Screening Is NextGen Likely to be the Final Best Platform and What are its Advantages and Quirks?
 Comprehensive Chromosome Screening Is NextGen Likely to be the Final Best Platform and What are its Advantages and Quirks? Embryo 1 Embryo 2 combine samples for a single sequencing chip Barcode 1 CTAAGGTAAC
Comprehensive Chromosome Screening Is NextGen Likely to be the Final Best Platform and What are its Advantages and Quirks? Embryo 1 Embryo 2 combine samples for a single sequencing chip Barcode 1 CTAAGGTAAC
Approach to Mental Retardation and Developmental Delay. SR Ghaffari MSc MD PhD
 Approach to Mental Retardation and Developmental Delay SR Ghaffari MSc MD PhD Introduction Objectives Definition of MR and DD Classification Epidemiology (prevalence, recurrence risk, ) Etiology Importance
Approach to Mental Retardation and Developmental Delay SR Ghaffari MSc MD PhD Introduction Objectives Definition of MR and DD Classification Epidemiology (prevalence, recurrence risk, ) Etiology Importance
Calling DNA variants SNVs, CNVs, and SVs. Steve Laurie Variant Effect Predictor Training Course Prague, 6 th November 2017
 1 Calling DNA variants SNVs, CNVs, and SVs Steve Laurie Variant Effect Predictor Training Course Prague, 6 th November 2017 Calling DNA variants SNVs, CNVs, SVs 2 1. What is a variant? 2. Paired End read
1 Calling DNA variants SNVs, CNVs, and SVs Steve Laurie Variant Effect Predictor Training Course Prague, 6 th November 2017 Calling DNA variants SNVs, CNVs, SVs 2 1. What is a variant? 2. Paired End read
SNP Array NOTE: THIS IS A SAMPLE REPORT AND MAY NOT REFLECT ACTUAL PATIENT DATA. FORMAT AND/OR CONTENT MAY BE UPDATED PERIODICALLY.
 SAMPLE REPORT SNP Array NOTE: THIS IS A SAMPLE REPORT AND MAY NOT REFLECT ACTUAL PATIENT DATA. FORMAT AND/OR CONTENT MAY BE UPDATED PERIODICALLY. RESULTS SNP Array Copy Number Variations Result: LOSS,
SAMPLE REPORT SNP Array NOTE: THIS IS A SAMPLE REPORT AND MAY NOT REFLECT ACTUAL PATIENT DATA. FORMAT AND/OR CONTENT MAY BE UPDATED PERIODICALLY. RESULTS SNP Array Copy Number Variations Result: LOSS,
PSE-HMM: genome-wide CNV detection from NGS data using an HMM with Position-Specific Emission probabilities
 Malekpour et al. BMC Bioinformatics (2017) 18:30 DOI 10.1186/s12859-016-1296-y METHODOLOGY ARTICLE PSE-HMM: genome-wide CNV detection from NGS data using an HMM with Position-Specific Emission probabilities
Malekpour et al. BMC Bioinformatics (2017) 18:30 DOI 10.1186/s12859-016-1296-y METHODOLOGY ARTICLE PSE-HMM: genome-wide CNV detection from NGS data using an HMM with Position-Specific Emission probabilities
SCALPEL MICRO-ASSEMBLY APPROACH TO DETECT INDELS WITHIN EXOME-CAPTURE DATA. Giuseppe Narzisi, PhD Schatz Lab
 SCALPEL MICRO-ASSEMBLY APPROACH TO DETECT INDELS WITHIN EXOME-CAPTURE DATA Giuseppe Narzisi, PhD Schatz Lab November 14, 2013 Micro-Assembly Approach to detect INDELs 2 Outline Scalpel micro-assembly pipeline
SCALPEL MICRO-ASSEMBLY APPROACH TO DETECT INDELS WITHIN EXOME-CAPTURE DATA Giuseppe Narzisi, PhD Schatz Lab November 14, 2013 Micro-Assembly Approach to detect INDELs 2 Outline Scalpel micro-assembly pipeline
Discovery of common Asian copy number variants using integrated high-resolution array CGH and massively parallel DNA sequencing
 Discovery of common Asian copy number variants using integrated high-resolution array CGH and massively parallel DNA sequencing Hansoo Park, Jong-Il Kim, Young Seok Ju, Omer Gokcumen, Ryan E. Mills, Sheehyun
Discovery of common Asian copy number variants using integrated high-resolution array CGH and massively parallel DNA sequencing Hansoo Park, Jong-Il Kim, Young Seok Ju, Omer Gokcumen, Ryan E. Mills, Sheehyun
Integrated detection and population-genetic analysis. of SNPs and copy number variation
 Integrated detection and population-genetic analysis of SNPs and copy number variation Steven A. McCarroll 1,2,*, Finny G. Kuruvilla 1,2,*, Joshua M. Korn 1,SimonCawley 3, James Nemesh 1, Alec Wysoker
Integrated detection and population-genetic analysis of SNPs and copy number variation Steven A. McCarroll 1,2,*, Finny G. Kuruvilla 1,2,*, Joshua M. Korn 1,SimonCawley 3, James Nemesh 1, Alec Wysoker
MSI positive MSI negative
 Pritchard et al. 2014 Supplementary Figure 1 MSI positive MSI negative Hypermutated Median: 673 Average: 659.2 Non-Hypermutated Median: 37.5 Average: 43.6 Supplementary Figure 1: Somatic Mutation Burden
Pritchard et al. 2014 Supplementary Figure 1 MSI positive MSI negative Hypermutated Median: 673 Average: 659.2 Non-Hypermutated Median: 37.5 Average: 43.6 Supplementary Figure 1: Somatic Mutation Burden
High-throughput transcriptome sequencing
 High-throughput transcriptome sequencing Erik Kristiansson (erik.kristiansson@zool.gu.se) Department of Zoology Department of Neuroscience and Physiology University of Gothenburg, Sweden Outline Genome
High-throughput transcriptome sequencing Erik Kristiansson (erik.kristiansson@zool.gu.se) Department of Zoology Department of Neuroscience and Physiology University of Gothenburg, Sweden Outline Genome
White Paper. Copy number variant detection. Sample to Insight. August 19, 2015
 White Paper Copy number variant detection August 19, 2015 Sample to Insight CLC bio, a QIAGEN Company Silkeborgvej 2 Prismet 8000 Aarhus C Denmark Telephone: +45 70 22 32 44 Fax: +45 86 20 12 22 www.clcbio.com
White Paper Copy number variant detection August 19, 2015 Sample to Insight CLC bio, a QIAGEN Company Silkeborgvej 2 Prismet 8000 Aarhus C Denmark Telephone: +45 70 22 32 44 Fax: +45 86 20 12 22 www.clcbio.com
SNP Array NOTE: THIS IS A SAMPLE REPORT AND MAY NOT REFLECT ACTUAL PATIENT DATA. FORMAT AND/OR CONTENT MAY BE UPDATED PERIODICALLY.
 SAMPLE REPORT SNP Array NOTE: THIS IS A SAMPLE REPORT AND MAY NOT REFLECT ACTUAL PATIENT DATA. FORMAT AND/OR CONTENT MAY BE UPDATED PERIODICALLY. RESULTS SNP Array Copy Number Variations Result: GAIN,
SAMPLE REPORT SNP Array NOTE: THIS IS A SAMPLE REPORT AND MAY NOT REFLECT ACTUAL PATIENT DATA. FORMAT AND/OR CONTENT MAY BE UPDATED PERIODICALLY. RESULTS SNP Array Copy Number Variations Result: GAIN,
Identification of genomic alterations in cervical cancer biopsies by exome sequencing
 Chapter- 4 Identification of genomic alterations in cervical cancer biopsies by exome sequencing 105 4.1 INTRODUCTION Athough HPV has been identified as the prime etiological factor for cervical cancer,
Chapter- 4 Identification of genomic alterations in cervical cancer biopsies by exome sequencing 105 4.1 INTRODUCTION Athough HPV has been identified as the prime etiological factor for cervical cancer,
Genome. Institute. GenomeVIP: A Genomics Analysis Pipeline for Cloud Computing with Germline and Somatic Calling on Amazon s Cloud. R. Jay Mashl.
 GenomeVIP: the Genome Institute at Washington University A Genomics Analysis Pipeline for Cloud Computing with Germline and Somatic Calling on Amazon s Cloud R. Jay Mashl October 20, 2014 Turnkey Variant
GenomeVIP: the Genome Institute at Washington University A Genomics Analysis Pipeline for Cloud Computing with Germline and Somatic Calling on Amazon s Cloud R. Jay Mashl October 20, 2014 Turnkey Variant
Supplementary Materials for
 www.sciencetranslationalmedicine.org/cgi/content/full/7/283/283ra54/dc1 Supplementary Materials for Clonal status of actionable driver events and the timing of mutational processes in cancer evolution
www.sciencetranslationalmedicine.org/cgi/content/full/7/283/283ra54/dc1 Supplementary Materials for Clonal status of actionable driver events and the timing of mutational processes in cancer evolution
Detection of copy number variations in PCR-enriched targeted sequencing data
 Detection of copy number variations in PCR-enriched targeted sequencing data German Demidov Parseq Lab, Saint-Petersburg University of Russian Academy of Sciences, current: Center for Genomic Regulation
Detection of copy number variations in PCR-enriched targeted sequencing data German Demidov Parseq Lab, Saint-Petersburg University of Russian Academy of Sciences, current: Center for Genomic Regulation
Implementation of the DDD/ClinGen OGT (CytoSure v3) Microarray
 Microarray") Implementation of the DDD/ClinGen OGT (CytoSure v3) Microarray OGT UGM Birmingham 08/09/2016 Dom McMullan Birmingham Women's NHS Trust WM chromosomal microarray (CMA) testing Population of ~6 million (10%)
Implementation of the DDD/ClinGen OGT (CytoSure v3) Microarray OGT UGM Birmingham 08/09/2016 Dom McMullan Birmingham Women's NHS Trust WM chromosomal microarray (CMA) testing Population of ~6 million (10%)
CITATION FILE CONTENT/FORMAT
 CITATION For any resultant publications using please cite: Matthew A. Field, Vicky Cho, T. Daniel Andrews, and Chris C. Goodnow (2015). "Reliably detecting clinically important variants requires both combined
CITATION For any resultant publications using please cite: Matthew A. Field, Vicky Cho, T. Daniel Andrews, and Chris C. Goodnow (2015). "Reliably detecting clinically important variants requires both combined
STATISTICAL METHODS FOR THE DETECTION AND ANALYSES OF STRUCTURAL VARIANTS IN THE HUMAN GENOME. Shu Mei, Teo
 Department of Medical Epidemiology and Biostatistics Karolinska Institutet, Stockholm, Sweden & Saw Swee Hock School of Public Health National University of Singapore, Singapore STATISTICAL METHODS FOR
Department of Medical Epidemiology and Biostatistics Karolinska Institutet, Stockholm, Sweden & Saw Swee Hock School of Public Health National University of Singapore, Singapore STATISTICAL METHODS FOR
Genome Structural Variation
 Genome Structural Variation Evan Eichler Howard Hughes Medical Institute University of Washington January 13 th, 2017, Genomics Workshop, Český Krumlov Genetic Variation Types Sequence Single base-pair
Genome Structural Variation Evan Eichler Howard Hughes Medical Institute University of Washington January 13 th, 2017, Genomics Workshop, Český Krumlov Genetic Variation Types Sequence Single base-pair
cn.mops - Mixture of Poissons for CNV detection in NGS data Günter Klambauer Institute of Bioinformatics, Johannes Kepler University Linz
 Software Manual Institute of Bioinformatics, Johannes Kepler University Linz cn.mops - Mixture of Poissons for CNV detection in NGS data Günter Klambauer Institute of Bioinformatics, Johannes Kepler University
Software Manual Institute of Bioinformatics, Johannes Kepler University Linz cn.mops - Mixture of Poissons for CNV detection in NGS data Günter Klambauer Institute of Bioinformatics, Johannes Kepler University
MPS for translocations
 MPS for translocations Filip Van Nieuwerburgh, Ph.D. Lab of Pharmaceutical Biotechnology NXTGNT massively parallel sequencing facility, Ghent University In collaboration with: Center for Medical Genetics,
MPS for translocations Filip Van Nieuwerburgh, Ph.D. Lab of Pharmaceutical Biotechnology NXTGNT massively parallel sequencing facility, Ghent University In collaboration with: Center for Medical Genetics,
Answers to Practice Items
 nswers to Practice Items Question 1 TEKS 6E In this sequence, two extra G bases appear in the middle of the sequence (after the fifth base of the original). This represents an insertion. In this sequence,
nswers to Practice Items Question 1 TEKS 6E In this sequence, two extra G bases appear in the middle of the sequence (after the fifth base of the original). This represents an insertion. In this sequence,
SUPPLEMENTARY INFORMATION
 doi:10.1038/nature10866 a b 1 2 3 4 5 6 7 Match No Match 1 2 3 4 5 6 7 Turcan et al. Supplementary Fig.1 Concepts mapping H3K27 targets in EF CBX8 targets in EF H3K27 targets in ES SUZ12 targets in ES
doi:10.1038/nature10866 a b 1 2 3 4 5 6 7 Match No Match 1 2 3 4 5 6 7 Turcan et al. Supplementary Fig.1 Concepts mapping H3K27 targets in EF CBX8 targets in EF H3K27 targets in ES SUZ12 targets in ES
Nature Genetics: doi: /ng Supplementary Figure 1. Details of sequencing analysis.
 Supplementary Figure 1 Details of sequencing analysis. (a) Flow chart showing which patients fall into each category and were used for analysis. (b) Graph showing the average and median coverage for all
Supplementary Figure 1 Details of sequencing analysis. (a) Flow chart showing which patients fall into each category and were used for analysis. (b) Graph showing the average and median coverage for all
Integrated detection and population-genetic analysis of SNPs and copy number variation
 8 Nature Publishing Group http://www.nature.com/naturegenetics Integrated detection and population-genetic analysis of SNPs and copy number variation Steven A McCarroll 4,, Finny G Kuruvilla 4,, Joshua
8 Nature Publishing Group http://www.nature.com/naturegenetics Integrated detection and population-genetic analysis of SNPs and copy number variation Steven A McCarroll 4,, Finny G Kuruvilla 4,, Joshua